CHAPTER 12
How to design a study that everyone will believe: Random selection and allocation of patients to treatment conditions
Katie L. Tataris1, Mary Mercer2, and Prasanthi Govindarajan3
1 Section of Emergency Medicine, University of Chicago, Chicago, IL, USA
2 Department of Emergency Medicine, San Francisco General Hospital, San Francisco, CA, USA
3 Stanford University Medical Center, Stanford, CA, USA
“A clinical trial is a powerful tool for studying the efficacy of an intervention. While the concept of clinical trial dates back to 1926, only in the past few decades has this emerged as the preferred method of evaluating medical interventions” [1].
The issue of BMJ from which the quote is taken states the importance of a randomized control trial (RCT) and how observational findings should often be verified through a well-planned and executed RCT before practice change is implemented.
Why conduct a randomized control trial?
While other methods of study design exist, a well-designed, well-conducted RCT provides a better rationale for causality than observational designs. Observational studies are vulnerable to confounding, in which a false association could be shown when one fails to control for a variable related to the risk factor and outcome. For example, observational studies found evidence of lower rates of coronary heart disease (CHD) in postmenopausal women taking estrogen compared to women that not taking estrogen and advocated for use of hormone supplements in these women. However, follow up RCTs such as the Heart and Estrogen/progestin Replacement Study (HERS) and the Women’s Health Initiative study showed hormone replacement increased the overall risk of CHD events in postmenopausal women with established coronary artery disease, demonstrating the pitfalls of observational data [3]. Often in an observational study, factors that are known to confound a causal association are controlled for in the design or analytical phase. RCTs are superior in that they control for unmeasured confounders by balancing the known and unknown baseline variables between the groups. For example, when studying the association between community characteristics and survival in out-of-hospital cardiac arrest, there may be emergency medical services variables that could influence the outcome. In a RCT, these would be balanced between the intervention and control arm through the process of randomization, thereby eliminating their effect on the outcomes.
Planning a clinical trial
While several steps must occur before a Phase 3 trial is designed, this chapter describes a clinical trial that assesses the effectiveness of an intervention relevant to clinical practice in an acute care setting.
A well-planned and executed RCT can be carried out in the acute care environment. The key steps shown in Table 12.1 will help to design and execute the study, and in the writing up of the results according to the CONSORT guidelines. [4, 5]. CONSORT stands for Consolidated Standards of Reporting Trials and offers recommendations for reporting findings of RCTs.
Table 12.1 Phases of a clinical trial.
| Phase | Core elements |
| Planning | Protocol design Choosing interventions and controls Choosing randomization techniques Selecting outcomes Measuring baseline variables in the study population Defining inclusion (and exclusion) criteria Institutional Review Board (IRB) approval Acquiring funding |
| Preparatory | Establishing oversight infrastructure Specifying operations (manual of operations) Standardizing and testing study documents Training for participant investigators |
| Recruitment | Deploying inclusion and exclusion criteria Utilizing informed consent process (or exception from informed consent, where applicable) |
| Patient follow-up and trial termination | Interval-based follow-up Specific protocol for termination |
| Analysis | Statistical analysis Manuscript writing |
The planning phase
There are several steps for laying the groundwork for a successful trial. These include planning and protocol design, sample size calculation, recruitment of participating sites, implementation, and execution of the trial.
Choosing the intervention and controls
RCTs have an experimental arm and a control arm. Patients in the experimental arm are assigned to the intervention and controls are most commonly assigned to the non-intervention or “standard of care” arm. This arm may include a placebo or existing treatments and active therapies. Since patients are selected to receive the intervention, equivalent treatment, or placebo, it is important that the different treatments being studied show clinical equipoise. It is unethical if clinical patients are treated with agents that are considered clinically superior or inferior at the start of the trial.
Selecting outcomes
The impact of the intervention on outcome is of utmost importance to the research community. There should be a single primary outcome for each research question, as RCTs are usually powered to examine the primary outcome only. Any remaining outcome variables are classified as secondary outcomes. While secondary outcomes are important and informative (especially with regards to the design of a potential follow-up trial), conclusions pertaining to them are limited due to the nature of the study design. Also, outcomes can be simple or composite. A simple outcome include all-cause mortality while a composite outcome includes all cardiovascular deaths, non-fatal heart attacks and emergency surgery related to the intervention. Further, an outcome may be a direct measure of clinical status, such as mortality, or may be a measure of something else (i.e., a surrogate outcome) that implies clinical status, such as viral load in HIV patients or peak expiratory flow rate in asthmatics [8].
Measuring baseline variables
Baseline variables are descriptive patient information and should be collected for patients in both arms of the study. Examples of baseline variables include age, gender, educational status, past medical conditions, and so on. Baseline variables that are known to influence outcome should definitely be recorded. For example, stroke severity is associated with functional status at the time of discharge. Therefore, the effect of an intervention on final outcome may be affected by stroke severity as measured by National Institute of Health Stroke Scale at the time of presentation. Therefore, having this as a baseline variable will help discover the true effect of the intervention on the outcome [9].
Participant selection
Patient selection is a requirement for meeting the study objectives and goals; it is accomplished objectively using the inclusion and exclusion criteria. Inclusion criteria are attributes that are present in a patient that qualify them to participate in a clinical trial. On the other hand, the presence of exclusion criteria disqualifies them from participating in a clinical trial. The advantages of having these criteria include reproducibility of patient enrollment at the different study sites, not subjecting patients at risk to the intervention and protecting the vulnerable population such as pregnant women, children, and so on [8].
Examples of inclusion criteria for a drug trial for Transient Ischemic Attack (TIA) would be a clinical diagnosis of TIA, objective criteria to stratify high risk TIA patients and minor stroke patients, and radiological confirmation of non-stroke causes. Exclusion criteria would include those with an allergy to the medication, those who may have risks such as bleeding or renal failure from the medication, those who cannot swallow pills, or those who cannot give informed consent.
While inclusion criteria help define the study population of interest, exclusions should be chosen sparingly in order to preserve the generalizability of a study’s findings. The most important reasons to exclude patients are if the participants would be harmed by the intervention, if there is certainty that there will be no benefit to this group, if age or other factors prevent them from being able to provide truly informed consent, or if the treatment regimen cannot be applied to this group as stated in the protocol.
Randomization and blinding
Once patients are screened and found to meet the inclusion criteria, they are randomized to the intervention or control group. Random allocation means that all participants have a defined probability of assignment to a particular intervention. Random allocation allows the baseline variables to be distributed equally across the groups and thus allows for balancing of groups. Random assignment of study participants is a critical part of the study design and must be performed methodically [8].
Randomization methods
- Simple randomization can be achieved by using the flip of a coin or a random number table or generator.
- This is the easiest method to randomize patients in an equal ratio into each intervention group.
- Advantage: easy to implement.
- Disadvantage: at any point in the randomization there could be an imbalance between the two groups.
- “Pseudorandomization” is a technique that assigns participants based on study entry on an even or odd day of the week, birth month, medical record number or other fixed patient/study characteristic. The problem with these methods is that the investigator knows in advance the group to which a particular patient will be assigned, which leads to selection bias and should be avoided.
-
Blocked randomization ensures an equal balance of patients in each arm of the study (stratifying by time).
- Balances patient characteristics over time.
- Divides potential patients into a number of groups or blocks; for example, for blocks of 4, 6, 8 there will be equal participation from each arm of the study (groups A and B) in each block.
- The order in which interventions are assigned in each block is randomized and the process is repeated for consecutive blocks until all participants are randomized.
- Advantages: The balance between the number of participants in each group is guaranteed during randomization, therefore increasing the power of the study. This is also useful with small sample sizes and if the trial is terminated before complete enrollment the groups will be balanced.
- Disadvantages: Assignments at the end of each block could be predicted and manipulated, but varying block size helps prevent this. Analysis of data may be more complicated, as many analytical methods assume simple randomization.
- Example: In a block size of four (Figure 12.1) there will be two control patients and two test patients. The order of the test and control patients will vary within the blocks but the number of patients is kept constant.
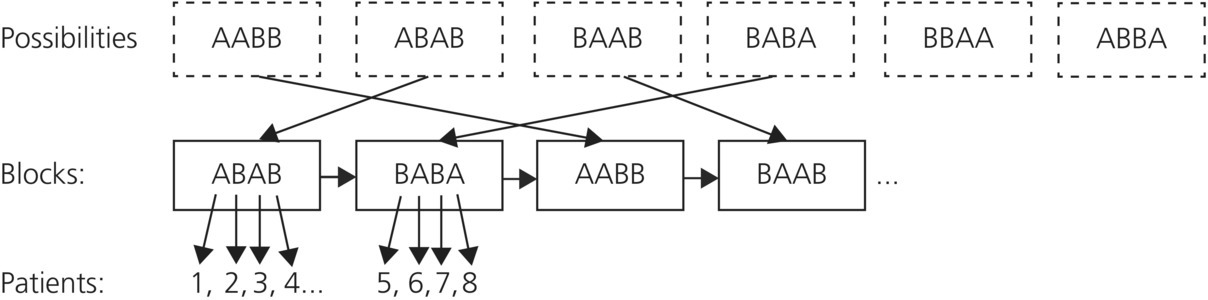
Figure 12.1 Blocked randomization example.
- Stratified randomization allows organization of participants by characteristic (age, sex) and then assignment of treatment randomly to each strata (stratifying by factors other than time).
- If a single factor is used, it is divided by two or more subgroups or strata (example: age 0–5 years, 6–10 years, 11–15 years). If several factors are used, that is, gender and race, a stratum is formed by selecting one subgroup (non-Hispanic whites and females) from each of them. The total stratum number is the product of the different subgroups (see table below for details).
- How does the randomization work? Measure characteristics or factors among participants, assign appropriate strata and randomize within strata.
- Within each strata, most clinical trials use blocked randomization to keep the group numbers balanced
- Advantage: The power of the study can be increased by using stratification in the analysis, as it reduces variability between group comparisons.
- Disadvantage: Increased stratification in small studies can limit data and so only important stratifying variables should be used.
- Example: An investigator is looking at the effect of household smoking on childhood asthma and wants to stratify on age, sex, and household smoke (Table 12.2).
- This design has 3 × 2 × 3 = 18 strata (Table 12.3).
- Participants between 0 and 5 years of age, male, and with no household smoke (stratum 1) would be assigned to groups A or B in the sequences ABBA, BABA, and so on.
- One variation on stratified randomization is Cluster Randomized Trials in which the group is randomized, not the individual. Examples would be hospitals, families, or towns. In some situations, the intervention can only be administered to one group for convenience or economic reasons, but are likely to randomize fewer groups and may have imbalance among group size.
Table 12.2 Factors in study of the effect of household smoking on childhood asthma.
| Age (years) | Sex | Household smoke |
| 0–5 | Male | None |
| 6–10 | Female | Occasional |
| 11–15 | Daily |
Table 12.3 Strata in study of the effect of household smoking on childhood asthma.
| Strata | Age (years) | Sex | Household smoke | Group assignment |
| 1 | 0–5 | M | None | ABBA, BABA, etc. |
| 2 | 0–5 | M | Occasional | |
| 3 | 0–5 | M | Daily | |
| 4 | 0–5 | F | None | |
| 5 | 0–5 | F | Occasional | |
| 6 | 0–5 | F | Daily | |
| 7 | 6–10 | M | None | |
| 8 | 6–10 | M | Occasional | |
| 9 | 6–10 | M | Daily | |
| 10 | 6–10 | F | None | |
| etc. |
Blinding is another important aspect of RCT. Concealing knowledge of the treatment assignments can help reduce bias. Physicians if not blinded to a weight loss drug intervention may consciously or unconsciously provide advice on lifestyle modification that is likely to influence the outcome. Similarly outcome assessors may show bias when abstracting outcomes from medical records if they are not blinded to the intervention.
The preparatory phase
This is the first step in the operational phase and involves creating a manual of operations, organizational oversight policies, reporting structure, data monitoring committee, and communications between sites [10].
Recruitment phase
Strategies for recruitment vary based on the clinical question and the setting, but the overall goal of this phase is to recruit the appropriate number of patients in a timely manner. Some methods of recruiting patients for a clinic-based trial for epilepsy include fliers and posters in doctor’s offices and support organizations for patients with seizure disorders. However, recruitment may not be possible for trials that include life threatening or time-sensitive conditions like seizures, acute intracranial hemorrhage, acute spinal cord injury, and so on. For these conditions, investigators inform the community about the ongoing trials through the process of community consultation and public disclosure, and patients who chose not to participate may opt out of these trials [11, 12].
The patient follow-up and termination phase
Patient follow-up entails a standardized protocol for follow-up intervals based on the study design and outcome measures, as well as a protocol for terminating patient involvement in the study. Follow up may be conducted in person, by phone, or by correspondence depending on the time intervals needed [8, 13]. Some examples of follow-up include 90-day in-person follow-up of functional status in a study evaluating early management of systolic blood pressure in acute intracranial hemorrhage. In addition to baseline variables, investigators should always record the name, phone number, and e-mail addresses of participants in order to maintain contact through the follow-up phase.
The analysis phase
The final phase of a study includes the tasks of statistical analysis and manuscript preparation. One needs to be familiar with the intent to treat and per protocol types of analysis. Intention-to-treat analysis is performed based on the study arm to which the patient was assigned and the treatment they were intended to have, regardless of whether they actually received that treatment. In per protocol analysis, statistical comparison of outcomes is based on what treatment the patient actually received rather than what was intended by the initial random assignment [8, 10]. Take, for example, a hypothetical study looking at the effectiveness of apple sauce in acute myocardial infarction. In an intention-to-treat analysis, a patient who was initially assigned to the apple sauce arm would still be included in the apple sauce cohort for outcomes analysis, even if he did not receive it. In a per protocol analysis, the same patient would be included in the non-apple sauce cohort for analysis based on the actual treatment they received. Intent-to-treat analysis is considered superior as it maintains the integrity of randomization.
Ethical aspects of research
The first step in any research process is to obtain an approval from the institutional review board (Chapter 8). The goal of any ethical review is to respect study participants through autonomy, minimize risks, maximize benefit, and ensure a fair selection of study patients [14]. A critical component of ethical principles is obtaining informed consent in research. However, there are situations that one may encounter in the emergency setting where informed consent may not be practically possible. This includes disease processes such as status epilepticus, traumatic brain injury and out-of-hospital cardiac arrest. Under these circumstances, FDA recommendations allow for exception from informed consent [12].
References
- 1 Friedman, L.M., Furberg, C.D., and Demets, D.L. (1998) Fundamentals of Clinical Trials, 3rd edn. Springer, NY.
- 2 Smith, G.C. and Pell, J.P. (2003) Parachute use to prevent death and major trauma related to gravitational challenge: a systematic review of randomized control trials. BMJ, 327(7429):1459–1461.
- 3 Rossouw, J.E, Anderson, G.L., Prentice, R.L., et al. (2002) Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA, 288:321–333.
- 4 Hulley, S., Grady, D., Bush, T., et al. (1998) Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. JAMA, 280(7):605–613.
- 5 CONSORT (n.d.) The CONSORT flow diagram. http://www.consort-statement.org/consort-statement/flow-diagram0/ (last accessed (3 May 2015).
- 6 Thayne, R.C., Thomas, D.C., Neville, J.D., and Van Dellen, A. (2005) Use of an impedance threshold device improves short-term outcomes following out-of-hospital cardiac arrest. Resuscitation, 67(1):103–108.
- 7 Aufderheide, T.M., Nichol, G., Rea, T.D., et al. (2011) A trial of an impedance threshold device in out-of-hospital cardiac arrest. N Engl J Med, 365:798–806.
- 8 Hulley, S.B., Cummings, S.R., Browner, W.S., et al. (2013) Designing Clinical Research, 4th edn. Lippincott Williams & Wilkins, Philadelphia, PA.
- 9 Meinert, C.L. (1981) Organization of multicenter clinical trials. Control Clin Trials, 1(4):305–312.
- 10 Klimt, C.R. (1981) The conduct and principles of randomized clinical trials. Control Clin Trials, 1(4):283–293.
- 11 Prout, T.E. (1981) Patient recruitment techniques in clinical trials. Control Clin Trials, 1(4): 313–318.
- 12 FDA (Food and Drug Administration) (2010) Exception from Informed Consent for Studies Conducted in Emergency Settings: Regulatory Language and Excerpts from Preamble – Information Sheet. Guidance for Institutional Review Boards and Clinical Investigators. http://www.fda.gov/RegulatoryInformation/Guidances/ucm126482.htm (3 May 2015).
- 13 Klimt, C.R. (1981) Terminating a long-term clinical trial. Control Clin Trials, 1(4):319–325.
- 14 Prout, T.E. (1981) The ethics of informed consent. Control Clin Trials, 1(4):429–434.