Speciation and Genome Evolution
Jeffrey Feder, Scott P. Egan, and Patrik Nosil
OUTLINE
1. From beanbags to genomes
2. Geography and gene flow
3. Primary versus secondary geographic contact
4. Selection-recombination antagonism and genomic heterogeneity
5. Empirical data and patterns
6. Chromosomal rearrangements and speciation
7. Polyploidy and speciation
8. Sex chromosomes and speciation
9. Speciation and genomic architecture
Speciation involves the splitting of one group of interbreeding natural populations into two or more reproductively isolated groups. Therefore, to understand speciation one must understand how genetically based barriers to gene flow (i.e., reproductive isolation) evolve between populations. Progress has been made in discerning the importance of different factors, traits, and individual speciation genes in generating reproductive isolation. However, we are just beginning to understand how speciation genes are embedded and arrayed within the genome, and thus how genomes evolve collectively during population divergence. Although it is now clear that different regions of the genome often vary in their level of genetic divergence between populations, major questions remain about how genomic architecture facilitates or impedes speciation. This chapter reviews our current understanding of genome-wide patterns of genetic divergence during speciation. The focus is on the processes causing genomic divergence and on their consequences for speciation.
GLOSSARY
Chromosomal Inversion. A rearrangement of a chromosome in which a segment of the chromosome is reversed end to end in its orientation, causing the inverted region of the chromosome to have an inverse linear order of genes compared with the corresponding “collinear” arrangement.
Divergence Hitchhiking. A term used to describe the process by which physical linkage to a divergently selected gene(s) increases genomic divergence for adjacent regions along a chromosome.
Divergent Selection. Selection that acts in different directions between two populations, including the special case in which selection favors two extremes within a single population (i.e., disruptive selection).
Genome Hitchhiking. A term used to describe the process by which genetic divergence across the entire genome is facilitated, even for loci unlinked to those under selection, by a global reduction in average genome-wide gene flow caused by selection.
Genomic Island Of Divergence. A region of the genome, of any size, whose divergence exceeds neutral background expectations based on overall divergence across the genome.
Heterogeneous Genomic Divergence. A term used to describe the highly variable levels of genetic divergence between populations across different regions of the genome.
Isolation By Adaptation (IBA). A pattern of positive correlation between the degree of adaptive phenotypic divergence between populations and their level of molecular genetic differentiation, independent of geographic distance.
Linkage Disequilibrium. The nonrandom association (i.e., correlation) of alleles at two or more loci.
Recombination. In eukaryotes, a process by which a piece of DNA is broken and then joined to a different piece during gamete formation (i.e., meiosis) via chromosomal crossing-over, which leads to offspring that have different combinations of alleles from those of their parents.
Selection-Recombination Antagonism. A term coined by Joseph Felsenstein to describe how recombination breaks up associations between selected loci and loci that cause reproductive isolation, impeding genetic divergence across the genome, and constraining speciation with gene flow.
1. FROM BEANBAGS TO GENOMES
Speciation is a fundamental evolutionary process responsible for creating the great diversity of life on earth. Speciation occurs as one interbreeding population evolves into two or more reproductively isolated groups or taxa (see chapter VI.1 on the concepts of species and speciation). Defining speciation in this manner leads to a basic research question: How do genetically based barriers to gene flow that cause reproductive isolation evolve between populations? Identifying factors promoting population divergence and genetically characterizing traits that cause reproductive isolation are therefore central endeavors for students of speciation (see chapter VI.4 on different reproductive barriers and the role of natural selection in speciation). Another important question concerns how the genes that cause reproductive isolation are positioned relative to one another in the genome. Discerning the physical arrangement of such “speciation genes” is important, because when populations are not fully reproductively isolated and still interbreeding, this genomic architecture may facilitate or impede further divergence. Thus, the question of genome structure links the evolution of individual speciation genes to their collective consequences for speciation.
Our empirical and theoretical understanding of the genetics of speciation has been dominated by what Ernst Mayr described as “beanbag thinking”—a focus on identifying and characterizing individual genes that cause reproductive isolation. However, we are now capable of rapidly scanning large portions of the genome in both model and nonmodel organisms for genetic differentiation during speciation. This ability has enabled researchers to begin studying how speciation genes are embedded and arrayed within the genome and thus how genomes evolve collectively during population divergence. This shift in focus is due, in part, to the rapid technological advances in mass sequencing technologies (i.e., next-generation sequencing platforms) that allow the surveying of many more genomic regions at a fraction of the cost and time. Consequently, the field of evolutionary genomics is moving away from “beanbag” approaches and purely descriptive studies of individual genes and their individual effects toward a more predictive framework that tackles the causes and consequences of genome-wide patterns. In this chapter, we examine what is currently thought about how speciation genes are arrayed in the genome and how genome structure may influence speciation.
2. GEOGRAPHY AND GENE FLOW
The geographic context under which populations diverge is a critical consideration for the role that genome architecture may play in speciation. When populations are completely geographically isolated in allopatry, there is no migration of individuals between populations. Hence, genetic differences can readily accumulate anywhere in the genome between populations by natural selection, sexual selection, and genetic drift. These differences can cause reproductive isolation as an inadvertent by-product if the populations come into contact. In cases where speciation occurs completely in allopatry, genome structure may not be critical to speciation, as the position of genes in the genome may not greatly affect their overall potential to diverge between populations. Thus, given enough time, divergence across the entire genome is inevitable in allopatry. Consequently, although much has been learned about specific reproductive barriers and individual speciation genes from studying allopatric taxa, it is difficult to ascribe any special significance to a particular genetic change or genetic architecture in such systems. Indeed, allopatric Drosophila fruit fly taxa, which have been the focus of much study, typically differ by hundreds of genes contributing to sterility and inviability distributed throughout the genome.
In contrast, genomic architecture could be very important for taxa that overlap geographically and exchange migrants and genes during the speciation process, either throughout the process (i.e., sympatric or parapatric) or during secondary contact between formerly allopatric populations before speciation is complete. Here, populations do not evolve completely independently of one another. Factors causing populations to diverge must overcome gene exchange due to migration and hybridization (interbreeding) if speciation is to proceed or, in cases of secondary contact, to continue. Thus when populations overlap and gene flow occurs during the speciation process, aspects of genomic architecture such as the distribution of genes within and among chromosomes (physical linkage relationship), and recombination rates among genes, may be important for enhancing the efficacy of selection and genetic drift in differentiating populations (see chapter VI.6 for additional discussion of gene flow and hybridization).
3. PRIMARY VERSUS SECONDARY GEOGRAPHIC CONTACT
Another important consideration for understanding the role genomic architecture may play in speciation is the timing of geographic overlap and the timing of the evolution of reproductive isolation. “Speciation with gene flow” can include speciation events during primary geographic contact (i.e., sympatric or parapatric speciation), where reproductive isolation evolved initially in the face of gene flow, and speciation events with secondary geographic contact, where reproductive isolation evolved partially in allopatry but then continues in sympatry. The timing of onset of divergence can vary widely among different instances of speciation, and in most cases of incipient speciation with gene flow present, it is very difficult to determine whether gene flow was present throughout divergence or only recently. The important concept here is that regardless of the starting point, when populations along the speciation continuum come into geographic contact and experience gene flow, genomic architecture can play an important role, albeit one dependent on the timing of the onset of gene flow. In cases of primary contact, genomic architecture can be critical to the initial stages of divergence; in cases of secondary contact, genomic architecture can help maintain (partial) reproductive isolation that evolved initially in allopatry.
4. SELECTION-RECOMBINATION ANTAGONISM AND GENOMIC HETEROGENEITY
Classic theoretical work by Joseph Felsenstein regarding the roles that selection, gene flow, and recombination play in shaping patterns of genetic divergence during speciation was described in his paper “Skepticism towards Santa Rosalia, or Why Are There So Few Kinds of Animals?” which clarified the potential importance of genomic architecture in affecting speciation with gene flow. This insightful paper developed the term selection-recombination antagonism to describe the tug-of-war between divergent selection—which builds up associations between loci adapting populations to different habitats and loci causing assortative mating—and gene flow and recombination, which break these favorable combinations apart. Gene flow and recombination impede divergence because they cause individuals adapted to a particular habitat to not breed true with other individuals possessing the same suite of locally adapted genes, and thus to generate genetically mixed offspring that do not exclusively possess the locally adapted phenotypes of their parents. The implication of selection-recombination antagonism was therefore that if different genes affect assortative mating and local adaptation, little progress could be made toward speciation unless the genes were fortuitously very tightly linked on a chromosome, such that recombination between them was very limited.
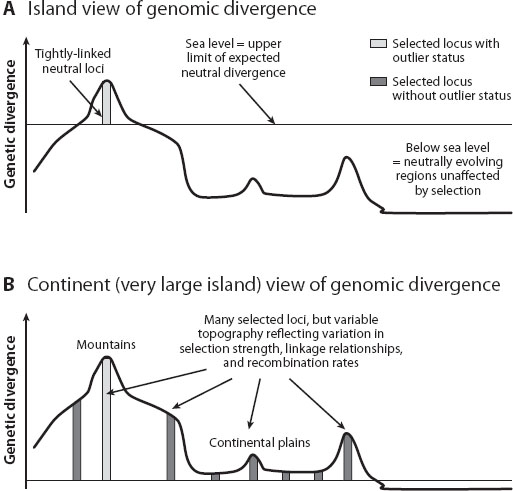
If the limitations imposed by the selection-recombination antagonism are pervasive in nature, then taxa speciating with gene flow should display a highly heterogeneous pattern of genomic divergence. A subset of the genome containing tightly linked genes under disruptive selection will display differentiation, while the homogenizing effects of gene flow and recombination should preclude divergence across the majority of the remaining genome (or in the case of secondary contact, eradicate existing levels of differentiation). Early empirical observations were consistent with this view of a highly heterogeneous genome (see further discussion) and led to the metaphor of genomic islands of speciation. A genomic island is any region of the genome that exhibits significantly greater differentiation between speciating taxa than predicted by neutral evolution, in which neutral patterns are based on overall divergence across the genome. The metaphor thus draws parallels between genetic differentiation observed along a chromosome, and the topography of oceanic islands (figure 1). In this case, isolated genomic islands contain putatively selected loci plus closely linked hitchhiking genes that rise above the statistical threshold predicted by neutrality, represented by sea level. In contrast, the ocean floor below sea level represents the majority of the neutrally evolving genome. Factors such as physical proximity between selected and other loci, rates of recombination, and strength of selection affect the height and the size of the genomic islands. Therefore, the metaphor of genomic islands offers a testable hypothesis for taxa speciating in the face of gene flow: if selection-recombination antagonism is common, the few genes under strong selection or those genes physically linked to loci experiencing strong divergent selection will exhibit strong differentiation and will tend to be clustered together in the genome, whereas gene flow will homogenize the remainder of the genome below sea level.
Figure 1. Illustration of the island (A) and continent (B) metaphors for genomic divergence. Note, these views are not mutually exclusive but represent parts of a continuum of genomic divergence among populations. (Figure from Michel et al. 2010 and reprinted with permission of the National Academy of Sciences of the United States of America.)
Despite the logic and heuristic appeal of the island metaphor, several aspects of Felsenstein’s model were not meant to be biologically realistic but rather were intended to highlight general points about the selection-recombination antagonism. For example, many organisms mate in preferred habitats rather than in a common mating pool, which can help alleviate the selection-recombination antagonism by reducing gene flow among populations with differing habitat preferences. The same is true when assortative mating is based on the phenotypic similarity of organisms rather than on their genotypes at assortative mating loci. Moreover, if selection is strong and migration is not random between populations, an association (i.e., linkage disequilibrium) is established between survival and habitat choice genes even in the absence of physical linkage. These considerations have led to the idea that multifarious selection, which is selection affecting multiple loci across the genome, could also kick-start speciation with gene flow. In this case, widespread selection could reduce gene flow sufficiently such that divergence could build up or be maintained across a larger extent of the genome.
5. EMPIRICAL DATA AND PATTERNS
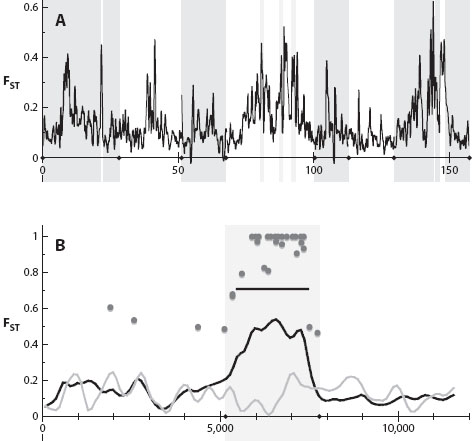
Researchers conduct “genome scans” to try to characterize patterns of genetic differentiation between speciating taxa and to identify genomic regions containing candidate speciation genes. For example, a genome scan might compare genome-wide patterns of genetic divergence among populations of an insect that inhabits different host plants (i.e., differing selective environments) and exhibits adaptive divergence and reproductive isolation associated with each environment. A genome scan study typically looks at genetic divergence across dozens to hundreds of thousands of molecular markers, such as amplified fragment length polymorphisms (AFLPs), microsatellites, single nucleotide polymorphisms (SNPs), or DNA sequences themselves, in a large number of individuals per population in each of two or more populations. These studies use the distribution of genetic divergence among markers to distinguish loci whose level of genetic divergence between populations exceeds neutral expectations. Such loci with unusually high levels of genetic divergence are deemed “outlier loci” and are interpreted as putatively being under divergent selection or closely linked to genes that are. The remainder of the genome (i.e., “nonoutlier loci”) is deemed to be neutrally evolving (at least statistically) and thus subject to homogenization among populations owing to gene flow. Genome scans thus have the potential to quantify the number of genomic regions under selection and their distribution across the genome (figure 2).
Figure 2. Genome-wide differentiation (FST) between populations of the threespine stickleback (Gasterosteus aculeatus). Panel A shows the distribution of genetic differentiation across 8 of the 21 linkage groups for an overall comparison of oceanic and freshwater populations. Vertical shading indicates boundaries of linkage groups. Panel B highlights a close-up comparison on linkage group 21 of an overall comparison of oceanic and freshwater populations (in black) and among freshwater populations (in gray). The black bar represents bootstrap significant genetic differentiation of a genetic region; dots represent significant point estimates of differentiation from a G-test corrected for multiple testing. (Figures from Hohenlohe et al. 2010 and reprinted with permission of the Public Library of Science.)
Genome scan studies of populations diverging in the face of gene flow tend to find a few, often physically isolated, outlier loci and thus have generally been interpreted as supporting the island view of genomic divergence. For example, an AFLP-based genome scan performed by Scott Egan and colleagues in 2008 compared Neochlamisus leaf beetle populations undergoing speciation associated with adaptations to different host plants. In pairwise comparisons of populations on different host plants, roughly 5 percent of the AFLPs exhibited strong divergence, and these “outliers” appeared to be physically isolated based on linkage disequilibrium analysis. In contrast, evidence for more genomically widespread divergence, as expected for multifarious selection, is rarer. However, this may stem from limitations in relying on genome scans alone for detecting selection. Genome scans can predestine an island view, because only the most diverged regions will be identified as statistical outliers. Other loci affected by selection, but more weakly, will go unnoticed and be considered part of the mostly “undifferentiated” and neutral genome.
In contrast with genome scans of populations observed in the wild, manipulative experiments exposing individuals to different conditions or factors mirroring those experienced by populations in the wild might allow the detection of weak selection acting across numerous regions of the genome missed by traditional genome scans. For example, field experiments could be conducted in which experimental populations are created on novel and native (i.e., control) environments (e.g., herbivorous insects could be transplanted to their native host or to a host-plant species used by a close relative). Loci that showed consistent and statistically significant allele frequency changes across replicates in the novel environment, but not in the control, would likely be affected by selection in the novel environment, even if such frequency changes were slight. If properly replicated, experiments could thus detect weak selection and distinguish selection from genetic drift and experimental noise to a greater degree than observational genome scans alone.
In 2010, Andrew Michel and colleagues conducted an experimental test of the genomic islands hypothesis in apple- and hawthorn-infesting host-plant races of the fruit fly Rhagoletis pomonella, a model for speciation with gene flow. In the experiment, groups of individuals from the apple and hawthorn host races were raised under the overwintering conditions of their native host and the alternative host plant, which differ in their fruiting times and, thus, the prewinter period each host race experiences prior to undergoing a winter diapause. Contrary to expectations of the islands hypothesis, the researchers reported widespread genetic divergence and selection throughout the Rhagoletis genome, with the majority of loci (33 microsatellites and six allozymes) displaying within-generation responses to selection in the manipulative overwintering experiment. Additionally, the majority of loci showed associations with latitude, associations with an ecologically relevant trait (adult eclosion time), and significant host differences in nature despite levels of gene flow that are too high to allow divergence via genetic drift. These results were coupled with linkage disequilibrium (LD) analyses, which test for combinations of alleles appearing more often in a population than would be expected based on random combinations of alleles proportional to their frequency in the population. This LD analysis provided experimental evidence that divergence was driven by selection on numerous independent genomic regions and not on a few islands. Based on their findings, Michel and colleagues proposed that “continents” of multiple differentiated loci, rather than isolated islands of divergence, may characterize even the early stages of speciation (figure 1B).
The authors stress, however, that the “island” versus “continent” views of genomic divergence represent ends of a continuum, rather than mutually exclusive hypotheses. Importantly, standard outlier analyses in this same study system detected only two independent outlier gene regions. Thus, experimental data and biological information on gene flow in nature were critical for detecting weaker, yet widespread, divergence across the genome. Additional such studies combining genome scans and selection experiments are needed before general conclusions can be drawn about how selection affects genomic patterns of differentiation.
Divergence Hitchhiking and Physical Linkage
To fully understand patterns of genomic differentiation and their significance for speciation we must not only empirically characterize genome-wide patterns of genetic divergence but also understand the mechanism responsible for generating the patterns. One mechanism potentially linking evolutionary constraints imposed by the selection-recombination antagonism with the concept of speciation islands is called divergence hitchhiking. According to this mechanism, a gene under divergent selection creates a localized window of reduced gene flow around it in the genome, because migrants moving into the population will tend to have maladaptive gene combinations for specific loci under selection, but—importantly—other (e.g., neutral) genes will also be linked to the selected loci. If there is not enough time for these associations between selected and other loci to be broken down by recombination, genes of both types may be eliminated by natural selection. Thus, not only will effective gene flow be reduced for the selected locus but also for nearby sequences, facilitating the initiation and buildup of genomic islands, and potentially their subsequent growth, via aiding the establishment and divergence of new mutations between populations. In contrast, unlinked sequences that can freely recombine away from maladapted genes will more easily invade other populations and be subject to higher levels of gene flow. As a result, in the early stages of speciation with gene flow, only a few genomic islands containing a handful of genes under divergent selection may separate taxa. However, the reduction in effective gene flow around these loci may allow the buildup of larger regions of genetic differentiation in surrounding loci subject to divergent selection. Thus, the initial isolated islands may become progressively larger through time.
Genome Hitchhiking and Isolation by Adaptation (IBA)
Divergence hitchhiking has great intuitive appeal as a mechanism ameliorating the selection-recombination antagonism and explaining heterogeneous patterns of genetic divergence. However, the efficacy of divergence hitchhiking is tied to the size of the windows of reduced gene flow for genes surrounding those under divergent selection. In a 2010 theoretical study, Jeff Feder and Patrik Nosil found that under conditions of strong selection, low migration, and small population sizes, this window of reduced gene flow could be relatively pronounced. However, these conditions are rather restrictive, especially for earlier stages of speciation with gene flow, when migration rates are expected to be relatively high and population sizes not small. Thus, critical incipient stages of speciation may require strong multifarious selection acting on several different loci and traits to kick-start the process. Feder and Nosil did find, however, that once a few loci under strong selection become established, effective migration rates can be reduced sufficiently (on average) on a genomically global basis for newer, less well favored mutations to establish throughout the genome. This process of genome hitchhiking is therefore distinguished from divergence hitchhiking in that the former does not require tight physical linkage of new mutations with previously selected loci. Genome hitchhiking is consistent with the empirical results of Michel and colleagues in the Rhagoletis study discussed earlier and could play an important role in the evolutionary transition of partially reproductively isolated races to more fully differentiated species
Empirically, the reduction in effective gene flow due to genome hitchhiking is predicted to generate a positive association among population pairs between levels of adaptive divergence (a proxy for the strength of selection) and genetic differentiation. This pattern has been referred to as isolation by adaptation (IBA) and may occur even for neutral loci unlinked to genes under selection. Evidence for IBA has been found in nature. In a study by John Grahame and colleagues in 2006, intertidal snails adapted to different sympatric shore habitats exhibit partial reproductive isolation. The authors compared hundreds of putatively neutral loci. For a given geographic distance, they observed an increase in neutral genetic differentiation between habitats—relative to within-habitat comparisons—consistent with a general barrier to gene flow across the habitat transition. This study provided a direct example of IBA, since these two habitats are very close to each other, such that some gene flow between them occurs. In a 2009 review by Nosil and colleagues of 22 studies that could test for this pattern of IBA, 68 percent showed evidence for IBA of putatively neutral loci. This study suggests that the IBA may be common in nature, as it was found across many different types of organisms, including lizards, wolves, leaf beetles, stick insects, and flowering plants.
6. CHROMOSOMAL REARRANGEMENTS AND SPECIATION
As described earlier, the antagonism between selection and recombination can impede genomic divergence. It therefore follows that factors that reduce recombination, such as chromosome inversions, can facilitate genomic divergence. Consistent with this prediction, in the 2010 Rhagoletis study on apple and hawthorn host races conducted by Michel and colleagues, regions of the genome harboring chromosomal inversions had, on average, twice the genetic divergence of collinear regions. Similar findings have also been reported by groups working on Anopheles mosquitoes and Drosophila fruit flies. Inversions may therefore help create more elevated oceanic islands and broader, more mountainous continents of divergence between taxa. Similar arguments can be made for other features of the genome that result in reduced recombination, such as, for example, proximity of genes under selection to centromeres, as observed in the European rabbit.
The basic premise for a role of inversions in speciation is therefore that they reduce introgression across the regions of the genome they encompass by protecting favorable genotypic combinations from being broken up by recombination, by reducing crossing-over. Essentially, the favorable genes within the inversions are more favorable together in their natal habitat than they would be individually, and less favorable in the habitats of other populations than they would be alone. Hence, gene flow in the inversion is reduced. Moreover, in addition to preserving blocks of adaptively diverged genes, inversions also provide larger targets in the genome for divergence hitchhiking to work; by suppressing recombination among populations, inversions enlarge the area of the genome in which new favorable mutations could arise linked to already-diverged genes.
An example in which inversions were involved in the evolution of reproductive isolation associated with adaptation to different environments stems from field experiments with ecologically divergent ecotypes of Mimulus guttatus. In 2010, David Lowry and John Willis showed that traits involved in local adaptation and reproductive isolation between coastal and inland ecotypes are located within a chromosomal inversion. They then established Mimulus lab populations, where they incorporated this inverted gene region into the standard genetic background of the alternative plant ecotype, and used them in a field experiment to demonstrate that the inversion contributes to divergent adaptation and reproductive isolation. In a similar vein, Mohamed Noor and colleagues have shown in a series of studies that many of the genes contributing to the reproductive isolation of two Drosophila fruit fly sister species that overlap in the western United States reside in inversions.
In addition to direct experimental and gene mapping data, empirical patterns in nature also provide evidence that inversions can promote speciation. For example, Noor and colleagues reviewed the Drosophila speciation literature in 2001 and found that sympatric pairs of species contained more inversions than allopatric species. This pattern was interpreted to indicate that on secondary geographic contact of populations following a period of allopatry, inverted regions of the genome were more resistant to gene flow than collinear regions (i.e., regions with orthologous linear orientation) with free recombination, and thus the inversions remained differentiated while collinear regions exchanged alleles readily and homogenized.
We stress, however, that although inversions may facilitate divergence, they are not required for speciation. Indeed, theoretical work by Feder and Nosil in 2009 showed that there is no reason for the vast majority of loci contributing to reproductive isolation to reside in inversions; they should also be commonly found in collinear regions, particularly when selection is strong relative to migration. Genome scan studies seem to bear out this supposition, as outlier loci presumably associated with adaptive divergence are often widely distributed across the genome. A study by Strasburg and colleagues in 2009 underscores this point. The authors examined divergence between hybridizing sunflower species at 77 loci distributed across the genome and reported that divergence is not accentuated within inversions, except perhaps near chromosomal breakpoints, in contrast with the results from Rhagoletis, mosquitoes, and Drosophila. Moreover, the authors observed widespread adaptive divergence in collinear regions.
Nevertheless, recent theoretical advances have shown that the presence of inversions themselves can be an outcome of adaptive divergence and an indicator of speciation with gene flow. The recombination suppressing effects of inversions are often not strongly favored in allopatric populations, where directional selection can act unhindered by gene flow and recombination to generate locally adapted sets of genes. Moreover, most new inversions have detrimental consequences during meiosis. Thus, it may be rare for inversions to differentially fix between taxa in allopatry. Instead, it is gene flow between locally adapted populations that favors the recombination suppressing effects of inversions and their spread. This important point was made by Mark Kirkpatrick and Nick Barton in a 2006 paper showing that new inversions that originate in sympatric populations exchanging genes that fortuitously happen to trap a combination of genes all adapted to one habitat versus another can be selectively favored over collinear arrangements, and increase in frequency to differentiate populations. Jeff Feder and colleagues recently demonstrated how such adaptive spread of inversions in the face of gene flow is facilitated if the inversions arise in allopatry, such that they contain the perfect complement of locally adapted alleles, and then rise to high frequency when gene flow ensues on secondary contact. Such a “mixed mode” of geographic divergence, alternating between allopatry and sympatry, allows for the establishment of inversions under a wide range of conditions.
7. POLYPLOIDY AND SPECIATION
Polyploidy describes the phenomenon in which there is a numerical increase in the whole set of chromosomes within an organism; polyploidy has been associated with speciation. Most species of multicellular eukaryotes are diploid, meaning that they have two sets of chromosomes—one set inherited from each parent—whereas polyploids have three or more complete sets of chromosomes. Polyploidy can occur via autopolyploidy, which describes the increase in ploidy within a species, or allopolyploidy, which describes the increase in ploidy resulting from hybridization between two distinct species. Polyploidy can arise via three pathways, regardless of whether it arises via hybridization or within a lineage: somatic doubling, meiotic nonreduction, and polyspermy. Somatic doubling occurs when irregularities arise during mitosis, and mitotic products fail to segregate to each pole, thus giving rise to a polyploid. If this doubling occurs in somatic tissue that gives rise to reproductive tissue or very early in development, a polyploid individual will result. Meiotic nonreduction occurs when a cell wall fails to form in late meiosis, generating diploid gametes, followed by fusion of two unreduced gametes fusing or one unreduced gamete and one normal gamete to form a triploid. This triploid in turn generates an unreduced triploid gamete, which fuses with a normal gamete, generating a polyploid. Polyspermy occurs when two sperm fertilize a single egg, yielding a triploid individual. Any of these mechanisms of polyploidy then relies on the event spawning a population of individuals that can reproduce.
Most eukaryotes have likely gone through a polyploidization event in their evolutionary history; thus polyploidy is a prominent feature in the evolution of eukaryotic life. However, the frequency of polyploidy seems to differ between animals and plants. While polyploidy has been clearly documented in animals, such as salmon and salamanders, it appears to be more common among plants, including ferns and flowering plants. In general, 47 to 70 percent of all plants are the descendants of polyploidy. More specifically, in 2009, Troy Wood and colleagues found that 31 percent of all speciation events in ferns and 15 percent of all speciation events in flowering plants were accompanied by an increase in ploidy. The important implication of polyploidy to speciation is that the polyploidization event itself can instantly generate some degree of reproductive isolation across the whole genome in sympatry, owing to meiotic difficulties experienced by the offspring of the diploid ancestor and the derived polyploid: their uneven number of sets of chromosomes causes varying degrees of sterility or inviability in the hybrids. The reduction in gene flow due to polyploidy is not always complete, and the tempo and magnitude of this reduction can vary among the different modes of polyploidy. However, if the level of gene flow between the parental and derived polyploidy is sufficiently reduced, divergent selection can then act to generate widespread genomic differentiation.
8. SEX CHROMOSOMES AND SPECIATION
Sex-determining chromosomes may also play an important role in the speciation process relative to the rest of the genome, termed autosomes (see chapter VI.8 for additional examples of the role sex chromosomes play in the speciation process). In a review of sex-linked speciation genes, Dorothy Pashley Prowell (1998) describes a common pattern in butterflies: a disproportionate association between traits that distinguish closely related butterfly species and the X chromosome. These traits also tend to be associated with prezygotic reproductive barriers, such as habitat preference. In a specific study using the fall army worm (Spodoptera frugiperda), Prowell found that of the nine traits that divided two ecologically divergent strains adapted to corn or grass, 33 percent were on the sex chromosomes compared with the 30 (or more) autosomal chromosomes. She posits that this association provides strong evidence that speciation genes are likely to be sex-linked in general across taxa. However, this prediction has yet to receive overwhelming support.
Additional data on the role of sex chromosomes in speciation comes from fish. Among some fish lineages, there has been an uncommon pattern of sex-chromosome turnover, such that many closely related species have different sex-chromosome systems. This finding has led to the idea that sex chromosomes may play an important role in the speciation process. Using threespine sticklebacks (Gasterosteus aculeatus) from Japan, Jun Kitano and colleagues (2009) provided strong evidence for the role of sex chromosomes in the speciation process. Here, the common sex chromosome in other populations has fused to a segment of another chromosome, forming a neo–sex chromosome. Using genetic mapping techniques, Kitano found that prezygotic (male courtship displays) and postzygotic (hybrid male sterility) traits map to the neo–sex chromosomes in this system, providing strong evidence for sex-linked genes in speciation. Again, the generality of these findings has yet to be determined.
9. SPECIATION AND GENOMIC ARCHITECTURE
We are just beginning to understand how speciation genes are embedded and arrayed within the genome, and thus how genomes evolve collectively during population divergence. Divergent selection can play multiple roles during the speciation process, such as affecting fitness-associated loci and loci physically linked to these loci, and facilitating genetic drift across the genome by reducing gene flow. Moreover, physical aspects of the genome may facilitate speciation through chromosomal rearrangements, genome duplications, and the evolution of sex chromosomes, often reducing effective recombination between populations. Thus, heterogeneity in genome divergence, polyploidy, chromosomal rearrangements, and the disproportionate role of sex-determining chromosomes in the evolution of reproductive isolation can all have important effects. Major questions therefore still remain about the generalities of genomic architecture during speciation and how this architecture either facilitates or impedes further divergence, especially for populations diverging with gene flow. Since speciation is usually not a discrete event (with the possible exception of polyploidy) but rather a continuous process, studying the genomic architecture along this continuum of divergence—using comparisons that vary in the degree to which speciation has progressed—will allow us to further address the role of the genome in speciation. In this regard, a major focus in the immediate future will be to determine how the selective effects of individual genes themselves, divergence hitchhiking, and genome hitchhiking contribute to speciation with gene flow.
FURTHER READING
Feder, J. L., S. P. Egan, and P. Nosil. 2012. The genomics of speciation-with-gene flow. Trends in Genetics 28: 342–350. Review of the emerging field of speciation genomics and description of a four-phase model of speciation defined by changes in the relative effectiveness of divergence and genome hitchhiking.
Feder, J. L., and P. Nosil. 2009. Chromosomal inversions and species differences: When are genes affecting adaptive divergence and reproductive isolation expected to reside within inversions? Evolution 63: 3061–3075. The results of this theoretical study show that genes contributing to reproductive isolation should be found not only in inverted regions of the genome but elsewhere as well. Inverted regions may retain suites of diverged genes for longer, but double recombination events and gene conversion will eventually break them down here, too.
Feder, J. L., and P. Nosil. 2010. The efficacy of divergence hitchhiking in generating genomic islands during ecological speciation. Evolution 64: 1729–1747. The results of this theoretical study show that divergence hitchhiking can generate large regions of neutral differentiation around loci under selection, but under limited conditions. However, they also demonstrate that when many loci are under selection, genome-wide divergence can occur.
Felsenstein, J. 1981. Skepticism towards Santa Rosalia, or why are there so few kinds of animals? Evolution 35: 124–138. Classic theoretical work regarding the roles that selection, gene flow, and recombination play in shaping paterns of genetic divergence during speciation.
Gavrilets, S. 2004. Fitness Landscapes and the Origin of Species. Princeton, NJ: Princeton University Press. Explores many of the theoretical issues about speciation discussed in this chapter.
Hohenlohe, P. A., S. Bassham, P. D. Etter, N. Stiffler, E. A. Johnson, and W. A. Cresko. 2010. Population genomics of parallel adaptation in threespine stickleback using sequenced RAD Tags. PLoS Genetics 6: e1000862. A high-density genome scan of genetic differentiation using >45,000 SNPs between ecologically divergent populations of the threespine stickleback. The results confirm genomic regions previously associated with speciation while also identifying additional genomic regions subject to divergent selection.
Kirkpatrick, M., and N. Barton. 2006. Chromosome inversions, local adaptation and speciation. Genetics 173: 419–434. Theoretical paper demonstrating that chromosomal inversions may rise to high frequency within populations owing to natural selection by protecting combinations of well-adapted alleles due to reduced recombination.
Lowry, D. B., and J. H. Willis. 2010. A widespread chromosomal inversion polymorphism contributes to a major life-history transition, local adaptation, and reproductive isolation. PLoS Biology 8: e1000500. One of the first experiments to show that chromosomal inversions play an important role in adaptation and speciation in nature, confirming theoretical predictions.
Michel, A. P., S. Sim, T.H.Q. Powell, M. S. Taylor, P. Nosil, and J. L. Feder. 2010. Widespread genomic divergence during speciation. Proceedings of the National Academy of Sciences USA 107: 9724–9729. An empirical counterexample to the island model of genomic divergence showing widespread divergence across all chromosomes among ecologically divergent insect host races.
Noor, M., K. L. Grams, L. A. Bertucci, and J. Reiland. 2001. Chromosomal inversions and the reproductive isolation of species. Proceedings of the National Academy of Sciences USA 98: 12084–12088. This paper was one of the first to argue for an important role of inversions in speciation. Evidence was presented for traits conferring reproductive isolation between two geographically overlapping species of Drosophila fruit flies mapping almost exclusively to two rearranged regions of the genome. A verbal argument was developed for how reduced recombination in inversions could help account for the observed pattern.
Nosil, P., D. J. Funk, and D. Ortiz-Barrientos. 2009. Divergent selection and heterogeneous genomic divergence. Molecular Ecology 18: 375–402. Review paper that explains the interplay of evolutionary forces generating reproductive isolation and heterogeneous patterns of genetic divergence across the genome and integrates them with the concept of ecological isolation by adaptation.
Rieseberg, L. H. 2001. Chromosomal rearrangements and speciation. Trends in Ecology & Evolution 16: 351–358. An argument is developed that rearrangements reduce gene flow more by suppressing recombination and extending the effects of linked isolation genes than by directly reducing fitness themselves.