Appendix I
I.1 Chemical formulae nomenclature
IUPAC recommended, unless otherwise stated; other chemical and trivial names are also given.
I.1.1 Nomenclature for a homologous series of compounds (Greek number/word system) (see Table I.1)
Table I.1 Nomenclature.
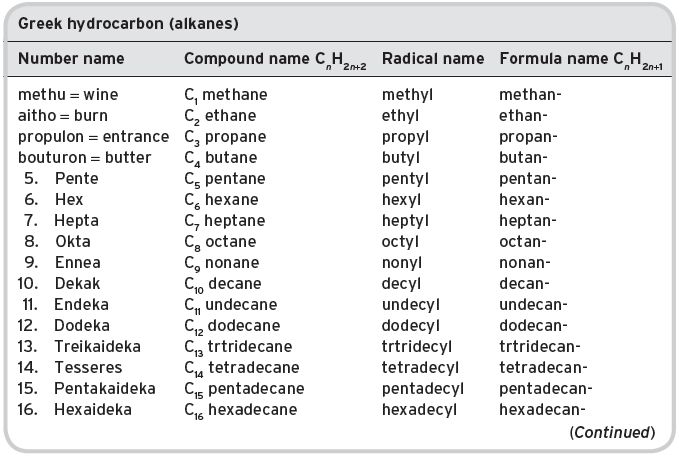

Unsaturated hydrocarbons (CnH2n+1) are more correctly named (methene), ethene, propene, etc.; their radicals, (methenyl), ethenyl, propenyl, etc. and their names in formulae, (methen-), ethen-, prop-1-en- (or −2-), etc.
I.1.2 System for substituent groups (derivatives) (see Table I.2)
Table I.2 Substituent groups.
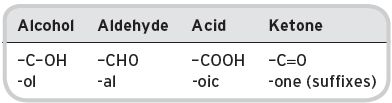
e.g. C4, butanol, butanal, butanoic, butanone. N.B. The same total carbon numbering, as in the initiating hydrocarbon, is retained in derivative alcohols, aldehydes, acids and ketones.
I.1.3 System for substituting in long-chain compounds
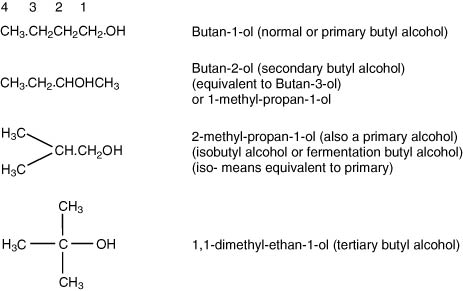
I.1.4 System for characterizing esters
A carboxylic acid (suffix, -oic) has its ending changed to -oate, with the name preceded by the name of the radical group of the esterifying alcohol, e.g.:
butanol (butyl alcohol) + butanoic acid = butyl butanoate (butanoic acid, butyl ester;
or butanoic ester of butanol)
Alternatively, esters can be named from the acylating group (i.e. from the acid), R.COO, by use of the suffix -oyl; thus caffeoyl quinic acid, or cinnamoyl tartaric acid (from the hydroxyl group).
I.1.5 System for characterizing unsaturated compounds
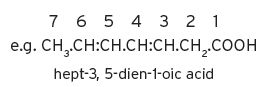
I.1.6 Systems for esters, thiols and thio-compounds
Esters: e.g. CH3OCH3 methoxy methane; then, diethoxy-, etc. (other names are dimethyl ether or oxide, or methyl oxymethane).
Thiols: e.g. CH3SH methan-thiol (other names are methyl thio-alcohol, methyl mercaptan, mercapto-methane).
Thio ether: e.g. CH3SCH3 dimethyl sulfane, dimethyl sulfide (other names are dimethyl thioether, thiobismethane).
I.1.7 Miscellaneous IUPAC recommendations
Stereochemistry (1) Z or E for cis/trans isomers. (2) Chiral orientation, use of R and S instead of D and L. (3) Optical activity, represented by (+) or (−) instead of d and el.
Linear hydrocarbon chains; numbering system, ω from end carbon atom.
I.1.8 Alternative chemical names (see Table I.3)
Table I.3 Alternative names.
| Chemical group | Example name | Other chemical name |
| Alcohols | Butan-1-ol | 1-Hydroxybutane |
| Aldehydes | Butan-1-al | Oxa-1-butane |
| Ketones (NB 1-one not possible) | Butan-2-one | Keto-2-butane (oxo-2-butane, methyl ethyl ketone) |
| Acids (carbon numbering system retained) | Decan-1-oic acid | 1-Decyclic acid |
| Acids | e.g. butan-1,4-dioic acid. Ethane dicarboxylic acid, dicarboxyethane. | |
| Alcohols | Methanol, carbinol, then methyl carbinol for ethanol, and so on. |
Carbon-numbering system not retained in alternative names, except in ring systems cyclohexane.
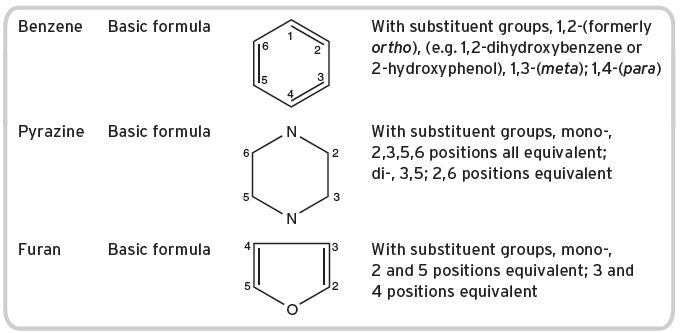
I.1.9 Numbering systems for ring compounds (see Table I.4)
Table I.4 Ring compounds.
I.1.10 Trivial and common names for derivative alkanes and other compounds
The names of organic volatile and other compounds in the tables in the text and Appendix II are given in both their IUPAC or chemical, and their traditional or trivial forms, as far as possible. Note, however:
(1) Saturated compounds (aldehydes, alcohols and acids). Common or trivial names accepted by IUPAC up to C4, e.g. butryric (butan-), C3 propionic (propan-), C2 acetic, acetaldehyde, (ethan-), C1 formic (methan-), but C5 valeric is pentan-, and so on up to C20. Isopentyl alcohol is acceptable for 3-methyl-butan-1-ol, but not isopentanol or isoamyl alcohol, etc. Oxo-, but not keto- is used for ketones. Radical names also used for all alcohols, e.g. propyl and hexyl alcohols.
(2) Unsaturated hydrocarbon radicals, e.g. C2 vinyl (acceptable for ethenyl), C3 allyl or acrylic (for prop-2-enyl).
(3) Trivial or traditional names recommended for complex hydroxy aliphatic acids, as in wine chemistry, such as tartaric, malic, citric, lactic, succinic and pyruvic acids.
(4) Use of the term to mean ‘acyl’ (–CO–) from the corresponding acid name (old nomenclature) is not generally recommended:
- Monoketones, e.g. acetyl methane, CH3COCH3, for propanone or dimethyl ketone; propionyl methane, for butan-2-one or methyl ethyl ketone.
- Diketones, e.g. diacetyl, CH3CO.CO.CH3, for butan-2,3-dione or biacetyl; acetylacetone, for pentan-2,3-dione or ethyl methyl ketone.
(5) Phenol is recommended for hydroxybenzene. For phenolic compounds, names such as catechin are generally retained. Cinnamic acid names are based upon propenoic acid derivatives.
(6) Terpenes, common or trivial names, such as limonene and α-terpineol, and furanones, such as furaneol (see information for particular cases but full chemical names generally recommended).
(7) Use of α, β, etc. for substituents in linear chains has been replaced by numbering system as in I.1.3 and I.1.5 above. The same applies to ring structures.
I.1.11 General
Reformed systems for nomenclature of organic compounds were first considered and devised (e.g. the suffix -ol for alcohols) by an International Commission in Geneva in 1898, and taken up by the International Union of Pure and Applied Chemistry (IUPAC). Their recommendations are published from time to time (e.g. 1993). Register names (e.g. Chemical Abstracts and Macmillan Dictionary of Organic Compounds may have an inverted order for chemical names – substituent groups last).
I.2 Stereochemistry
Many pairs of chemical compounds, called isomers, each having the same molecular weight and linear representation of their constituent atoms/groups of atoms in their molecules, will show differences in actual molecular structure on account of different spatial distributions of attached component atoms. These compounds fall into one of two subdivisions, called (1) enantiomers (Gk. enantios – opposite) and (2) geometric isomers, in ways to be described. Some pairs of compounds may fall into both subdivisions. Both subdivisions may reflect colour/flavour and other physically measurable differences within the pairs. The second group may reflect some chemical reaction differences between the compounds, due to steric hindrance.
A further group of isomers, which have some important significance in wine chemistry, is known as tautomers (Gk. tautos or to auto – the same thing), and are characterized by a slight difference in linear representation of their molecules. Tautomerism refers to the ability of certain compounds to react in two different ways, dependent on their molecular structure, thus giving rise to two separate series of derivatives by which a compound can be identified.
I.2.1 Enantiomers
These compounds generally require the presence of at least one asymmetric carbon in the molecule. An asymmetric atom is an atom that is linked to four different monovalent atoms or groups. In this way, for a single asymmetric carbon atom, two spatially different forms of a given molecule are possible. These structural forms are not superimposable but are, in fact, mirror images. The two compounds will have identical chemical and physical properties, but they will differ markedly in their capacity to rotate the plane of polarized light, either to the left or to the right, to which they are exposed in suitable equipment. Numerous such pairs of compounds are familiar in wine chemistry, thus 2-hydroxypropanoic acid (lactic acid), CH3.C*HOH.COOH, with one asymmetric carbon atom (indicated by an asterisk) can exist in two different structures (a pair of enantiomers) and the molecule can be drawn as shown in I.I.
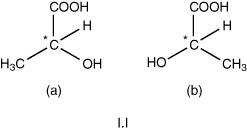
By an older convention, the structure (a) was accorded the designation D, now a small capital letter (D), and (b) L, now L. These designations refer solely to the arbitrarily stated orientation of the OH group, thus OH in (a) is shown on the right (Dextro-), and OH in (b) on the left (Laevo-). This type of designation is still used for the molecules of well known natural substances, for example, also, so-called ‘active’ amyl alcohol (2-methyl-butan-1-ol), CH3.CH2.C*H(CH3).CH2OH and especially for the aldo- and keto-hexoses, e.g. D-glucose, C6H12O6.
These long-chain sugar molecules will have several asymmetric carbon atoms (four in aldohexoses and three in ketohexoses) when looked at in their straight-chain form, and will have 16 and eight different spatial structures (eight and four pairs of enantiomers). In fact, the compounds are all known and have individual names. From these, dextro-glucose (based upon the configuration shown in Table 3.4) is of the most interest in wine chemistry. However, in its ring molecular structure, as glucose is believed to exist in aqueous solution, the molecule shows an additional asymmetric atom (No. 1, as shown also in Table 3.4), which gives the substance a characteristic optical rotation value. To the D- designation is added the symbol α for the main cyclic form, and β for the other. The molecular configuration represented by the D and L structures is again conventionally designated by the right-hand positioning of a particular hydroxyl group (i.e. situate at the No. 4 carbon atom in the chain) and similarly for α and β.
Optical activity
Optical activity is measured in 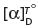 specific rotation units from experimental determinations, where D is the sodium line at 589 nm and temperature T°C; its value is calculated from α × 100/l × c, where l is the length in decimetres of liquid traversed, c is the number of grams substance in 100 ml solution, and α the angle of rotation observed, to the right or to the left. Its numerical value differs greatly for different compounds, dependent upon the groups attached (e.g. chromophores). It is not clearly related to the structural configurations described, of ‘handedness’. If the enantiomer rotates the plane of polarized light to the right, so-called dextrorotatory, it is designated as the + form (formerly lower case d); if to the left, laevorotatory (formerly lower case l). However, the molecular configuration (arbitrarily) is usually also quoted, as either D or L, in a straight-chain compound. In the simplest cases, D-structures will be associated with (+) optical dextro-rotation, and L- with (−). Whilst the main glucose is D-α, it is dextrorotatory, as is the D-β glucose but at a lower numerical value; fructose D-β is laevorotatory.
specific rotation units from experimental determinations, where D is the sodium line at 589 nm and temperature T°C; its value is calculated from α × 100/l × c, where l is the length in decimetres of liquid traversed, c is the number of grams substance in 100 ml solution, and α the angle of rotation observed, to the right or to the left. Its numerical value differs greatly for different compounds, dependent upon the groups attached (e.g. chromophores). It is not clearly related to the structural configurations described, of ‘handedness’. If the enantiomer rotates the plane of polarized light to the right, so-called dextrorotatory, it is designated as the + form (formerly lower case d); if to the left, laevorotatory (formerly lower case l). However, the molecular configuration (arbitrarily) is usually also quoted, as either D or L, in a straight-chain compound. In the simplest cases, D-structures will be associated with (+) optical dextro-rotation, and L- with (−). Whilst the main glucose is D-α, it is dextrorotatory, as is the D-β glucose but at a lower numerical value; fructose D-β is laevorotatory.
Chirality
This subject of enantiomerism is now more generally referred to as chirality (Gk. cheiros, meaning ‘hand’), a term first mentioned by Lord Kelvin in 1904. As is now increasingly recognized, the term has special significance to odour perception for volatile compounds. In non-volatile compounds, physical property differences can occur as a consequence of certain types of chiral difference of otherwise identical compounds and flavour/taste differences within pairs of enantiomers.
A detailed and clear description of chirality is given in the textbook, Organic Chemistry (Clayden et al., 2001). These authors start their chapter with the representation of the lactic acid molecule, drawn as either (a) or (b) shown in I.II.
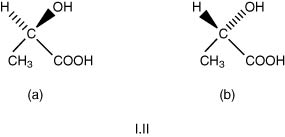
The plane of symmetry is conceived by drawing the carbon skeleton lying in the plane of the paper, with the other two groups H and OH either pointing back symbolized by  or pointing forward
or pointing forward 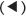 . Configuration (a) is called the R form (from R. rectus – right) and (b), the S form (S. sinister – left), which are the equivalents of the old D and L designations.
. Configuration (a) is called the R form (from R. rectus – right) and (b), the S form (S. sinister – left), which are the equivalents of the old D and L designations.
When there are two asymmetric carbon atoms (stereogenic or chiral centres), the structural definitions necessary become more complicated, thus for the most prevalent wine acid (2,3-dihydroxy-butan-dioic acid) tartaric acid, HOOC.C*HOH.C*HOH.COOH.
The following four structures, stereoisomers (22 = 4) are possible, and two so-called diastereoisomers (22–1 = 2) each with an expected pair of enantiomers, as follows: (H groups are omitted from the diagrams for convenience).
(1) With the OH group in the syn position (two enantiomers, one diastereo-isomer) (I.III)
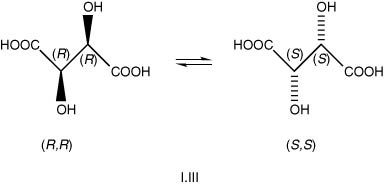
(2) With the OH groups in the anti-position (another diastereoisomer, no enantiomers) (I.IV).
The designations R and S for multi-chiral centres in general are now determined by a somewhat complex set of rules to cover all cases, the so-called C-I-P rules of 1952, described in the book Organic Chemistry, but not necessary to examine here. However, the structures described above as (R,R) and (S,S), in Section (1), represent true enantiomers, that is, they are mirror images, but careful examination of the structures (R,S) and (S,R) in (2) will reveal that they are not true enantiomers, as they are superimposable. They have, in fact, the same molecular structure and are so-called achiral. The latter compound is known and is called meso-tartaric acid (Gk. mesos, middle). In addition, it has zero optical activity (internal compensation), compared with the RR compound, which has (+) and the SS (−). In practice, there will also be a so-called racemic mixture, which is an equi-mixture of the two optically active forms, resulting in zero optical activity. The meso compound has a lower melting point and a lower water solubility than either of the two enantiomers, which have equal numerical values of these physical properties. Pasteur, in his studies on vinification around 1848, was especially interested in the optical activity and other physical properties of tartaric acid. He also devised a method of separating the enantiomers in a racemic mixture. He made use of the selective destructive effect of certain yeasts and moulds on a d(+) ammonium tartrate over its l(−) enantiomer. He also found that a racemic mixture of sodium ammonium tartrate could be separated after recrystallization from aqueous solutions, since the crystals of the two enantiomers had a slightly different shape.
Recently it has been found that, though pairs of enantiomers in general have identical physical and chemical properties and behave identically, this is no longer true when they are placed in a chiral environment. This latter phenomenon, in fact, enables the separation of enantiomeric volatile compounds in GC equipment by use of a stationary phase, which is made chiral by bonding with enantiomerically pure compounds. Pasteur’s separations of tartaric acid salts were, probably, also chirally determined. More importantly, many pure compounds in an enantiomeric pair possess different odour/aroma characteristics in many instances, presumably related to the chirality of the olfactory organ receptors at the back of the nose. The effectiveness of compounds used as drugs in modern medicine also depends, in many instances, on their chiral structure. This discovery has given great impetus to developing methods of maximizing separation of enantiomers, and of so-called asymmetric synthesis of organic compounds. The slight differences of sweetness and acidity found within enantiomers of sugars and organic acids, perceived by the tongue, may well have a chiral basis.
Several volatile compounds in wine have been reported as having different odour characteristics in their R and S structures, though both enantiomers may not actually be present in particular wines. A compound with a single asymmetric carbon atom, such as oct-1-en-3-ol, CH3(CH2)4. *CHOH.CH:CH2, which is reportedly present in botrytized grapes, in its S form is said to have a ‘vegetable, mouldy’ flavour, whilst the R form has a ‘green mushroomy, meaty’ flavour, with a mushroom odour (quoted, Flament, 2001), though the differences in this instance may not be great. Chiral centres can be at a distance from each other in the molecule, but the lack of symmetry may be established. Limonene and carvone are clear examples, where (+) and (−) forms are reported to give compounds of markedly different odours and thresholds. Other examples will be found in the main text in Chapter 4, particularly amongst the flavanones and terpenes, though the presence of which enantiomer or both may not as yet have been clearly established. There does not appear to be much information in the literature on the different odours of the corresponding meso compounds.
Occurrence of enantiomers
In nature, biogenetic pathways appear to produce one form only; for example, through one important amino-acid precursor, alanine (and others) present in the laevorotatory, S form in plants, though R in bacterial cell walls. Malic acid (2-hydroxybutan-dioic acid), the ‘apple’ acid or ‘ordinary’ malic acid is laevo(−)rotatory. Tartaric acid, the ‘ordinary’ tartaric acid already found in grapes, is dextro(+)rotatory, and its formula in full, once d-tartaric acid, is now given as, {R,(R*,R*)}-2,3-dihydroxy-butandioic acid; though better, (2R,3R)(+)-2,3-with R* indicating the existence of the chiral centres as already described, or numerals before the R indicating position of the chiral centres.
Fermentation processes appear to produce the racemic form (as with the lactic acid bacteria to form dl-lactic acid). The optically active amyl alcohol (2-methyl-butan-1-ol) formed by the fermentation of sugars by Saccharomyces cerevisiae by conversion from 1-leucine is said to be laevorotatory, in contrast to the so-called fermentation amyl alcohol (3-methyl-butan-1-ol), which is inactive, non-chiral, deriving from isoleucine.
I.2.2 Geometrical (stereo-) isomers
So-called geometrical isomerism is found mainly in compounds with two carbon atoms linked by a double bond. When each of the two carbon atoms contain the same two but different groups, e.g. hydrogen/hydroxyl, they can be arranged and fixed in two separate ways, giving rise to so-called ‘cis’ and ‘trans’ forms, e.g. in general terms.
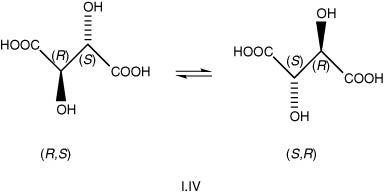
The terms cis and trans refer to the side contiguity of the atoms or groups, a and b, with cis meaning on the same side for each. According to the new IUPAC nomenclature, these are now called ‘Z’ (Ger. zusammen – together) and ‘E ‘(Ger. entgegen – opposite) respectively (I.V).

The compounds within a pair will have markedly different physical and chemical properties, and importantly, different flavour characteristics in many cases. Chemical reactivity differences can occur due to steric hindrance with some molecular structures. Examples have been described in the main text, such as for the unsaturated aldehydes/alcohols, hydroxycinnamic acids and 2-furanones; for example, the cis form of 2-nonenal has a known lower threshold value than the trans, and has a different odour, but both these features vary markedly with concentration. The ‘oak lactone’ has both geometric isomers but also optical activity variants in each, giving rise to four compounds with different flavour characteristics.
Biogenetic pathways usually produce only one form, e.g. linoleic and linolenic acids in vegetable oils are always in the cis form at each of their double bonds. Chemical transformations of these compounds, as in conjugation and hydrogenation, produce a quantity of trans forms. Unsaturated aliphatic aldehydes, like 2-nonenal and 4-heptenal, produced by the enzymatic oxidation of linoleic acid seem to be formed in both cis and trans forms. There seems, however, to be a tendency for similar groups to take up spatial positions, as far away as possible from each other, thus favouring the trans forms.
I.2.3 Tautomerism
Tautomerism has also been described as dynamic isomerism of two possible different molecular structures for the same molecule, though these will not be too dissimilar. Numerous examples are now known, falling into different chemical classes but the triad system, with three carbon atoms and in particular, the arrangement in the keto–enol system, C=C–OH, is of the greatest interest in wine chemistry. Compounds with this latter grouping and capacity for change, exhibit the properties of both ketones and hydroxyl compounds, thus for resorcinol (1,3-dihydroxybenzene, or m-hydroxy-phenol) we have the situation shown in I.VI.
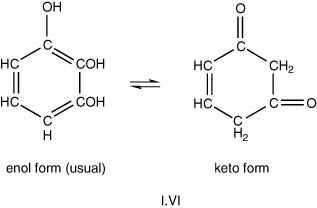
Similarly catechol (1,2-), hydroquinone (1,4-) and trihydroxybenzenes (1,3,5-, phloroglucinol) exhibit tautomerism, which is relevant to oxidative changes in wines.
A three-carbon system, as in –C=C–C, again with a mobile carbon atom, is again of interest to the wine industry, as in the following examples:
(1) Eugenol(2-methoxy-4(prop-2-enyl)phenol)↔iso-Eugenol[-(prop-1-enyl)].
(2) Linoleic and linolenic acids, to their conjugated forms.
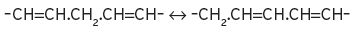
I.3 Chemistry of the oxidation of organic compounds
Oxygen (Gk. oxus – sharp) is an element in period 2, group 16 of the periodic table, with an atomic number of 8. Its atom has an electron distribution around the nucleus, described as 2,6,0, i.e. total of eight electrons, in the different energy shells, s, p and d. These quantum shells or orbitals are represented by the notation, 1s2, 2p2, 2d4. The electrons in the No. 2 levels have a total of six valency electrons, thus the oxygen atom normally only forms two paired bonds, :O:, i.e. a normal valency of two in compounds.
Molecular oxygen (O2, dioxygen) necessarily has shared orbitals from the two identical atoms but now with twelve electrons in the valence orbits. The molecular orbitals have a distribution designated as 1σ2 g, 2σu, 3σ2 g, 1π4 u, 2π2 g related to the corresponding atomic orbitals. The suffixes, g and u, refer to bonding capability, according to the orbital, σ or π. In the oxygen molecule, with the half-filled orbitals of 2πg, the outermost electrons have parallel spins in different orbitals. With filled orbitals, paired electrons have opposite spin. The numbers in superscript are again the number of electrons in each orbital. Molecular structure is a highly complex topic but the reader is referred to Inorganic Chemistry (Shriver et al., 1994) for detailed information.
The oxygen molecule is regarded as particularly interesting and different from others, in that the lowest energy configuration has two unpaired electrons in different orbitals, which makes it also diamagnetic (i.e. tending to move into magnetic fields). Further electrons present as in O2− and O2 − can be accommodated in the 2πg orbitals; the g orbitals are regarded as anti-bonding. These orbital configurations described do not, however, fully characterize energy levels. The oxygen molecule in the ground state is also characterized by the term O2 3∑, so-called triplet oxygen.
In this condition it has a radical character, enabling participation in chain reactions, as in some oxidative processes occurring in wines, which have been described in the text and are further discussed in the following sections. The oxygen molecule can also occur in two so-called singlet states. The first singlet state, O2 1∑ has paired electrons in the same two orbitals as in the ground state (but with higher energy); and another, represented as 1Δ, with electrons paired only in one orbital (with intermediate energy). The latter can survive long enough to participate in chemical reactions. It can be generated in solution by energy transfer from photo-excited molecules.
The other allotrope of oxygen is ozone (O3), which is a highly reactive substance in oxidation processes but of lesser interest in wine chemistry.
I.3.1 Auto- and enzymatic oxidation of lipids
The auto-oxidation of lipids, in particular the unsaturated acids, oleic, linoleic and linolenic and their glycerol esters, is a complex phenomenon but has been studied in detail by many investigators over recent decades. One proposed reaction mechanism, based upon a free radical chain process and applicable to commercial oils and fats, has been described by Belitz & Grosch (1987). It consists of three steps as follows:
(1) Initiation (by light/heat):
RH (unsaturated acid) → R• (fatty acid free radical)
(2) Propagation (i.e. a chain reaction):
R• + O2 (ground state) → ROO• (peroxide free radical)
ROO• + RHROOH (fatty acid peroxide) + R• (fatty acid free radical)
(3) Termination (in the absence of oxygen):
2ROO• → inactive dimer
2R• → inactive dimer
ROO• + R → inactive dimer
RH will be a typical alkyl compound, with a labile hydrogen atom, such as that of a methylene group (–CH2–), adjacent to two carbon atoms linked by a double bond. This is a three-carbon tautomeric system, one unit as in the oleic, two in the linoleic and three in the linolenic acid molecules. The mobility of the hydrogen atoms can be explained in terms of electron concentration.
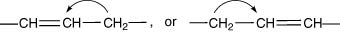
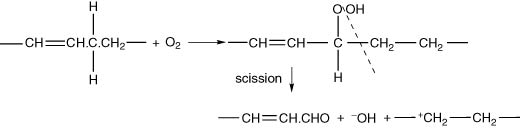
Apart from an inherent chain reaction, the other important feature of this process is the formation of relatively stable peroxides. However, they usually decompose with the formation of secondary reaction products, under the same influences of light/heat, metal catalysts and other initiating agents, as in Step 1 above. Several chemical groups of secondary products are possible, but of the greatest interest for wine are those arising from peroxide formation and chain scission, e.g. formation of unsaturated aldehydes (Fig. I.1).
The triad (C–C–C) in the initial radical form of RH will be subject to double-bond shifting in a dienoic or trienoic acid, so that we can find conjugation of the double bonds in the final reaction products. The usual Z (cis) double bond system affected by the methylene group is transformed into an E (trans) configuration. The other unaffected double bonds will, however, retain their original configuration (i.e. usually cis, in natural fatty acids).
Figure I.1 Oxygen attack at a methylene group adjacent to a double bond and subsequent reaction.
Retardation, though still in the presence of oxygen, can be effected by certain antioxidant compounds, particularly with edible fats containing oleic acid as the only fatty acid present; thus, for example, by the use of tocopherol, which has a phenolic hydrogen atom.
Peroxy free-radicals react preferentially, leading to a fat hydroperoxide, as is the case in a normal uninhibited oxidation reaction. However, a tocopherol free radical, which is produced simultaneously, has much less affinity to react with the oxygen molecules present, so the reaction chain is stopped. Some phenolic compounds found in grapes/wine, such as quercetin, are reported to be good antioxidants for oils and fats.
The decomposition reaction sequence described above was first used by Drawert (1974) to explain the formation of hexanal from linoleic acid, and of cis-hex-3-en-1-al and trans-hex-2-en-1-al from linolenic acid. The initial formation of the required hydroperoxide within plant tissues as in grapes was, however, considered to be oxygen activated by a lipoxygenase enzyme, and the scission by an ‘aldehydase’. Reduction to the corresponding saturated alcohols (taking place largely in the fermentation) to the unsaturated alcohols, was also described as being due to an ‘alcohol oxido-reductase’ (probably a dehydrogenase). However, with subsequent oxidation during maturation, there may be some reversion to the original aldehydes. The precise methylene group to be peroxidized will differ in different examples. Semmelroch & Grosch (1996) have provided similar schemes to show the production of 2-nonenal and 4-heptanal.
I.3.2 Oxidation–reduction of alkyl alcohols and aldehydes
Some explanatory mechanisms have been published for these reactions, which occur frequently in wines. Ethanol can be oxidized to ethanal by loss of hydrogen, and in reduction, vice versa, by gain,
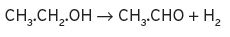
Typically, this reaction is said to take place in the presence of a dehydrogenase, together with a co-enzyme, nicotinamide adenine dinucleotide, NAD, thus
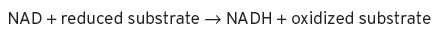
This co-enzyme is also used in the Krebs cycle for converting citric acid to α-keto-glutaric acid (2-oxobutandioic acid).
It is probable that these enzymes are no longer active after fermentation and clarifications. The proposed mechanism for the observed formation of ethanal from alcohol during maturation has been ascribed to the presence of phenolic substances and of ferric/cupric ions (Wildenradt & Singleton, 1974). In contrast, Ribéreau-Gayon et al. (2000) say that it is the procyanidins, polymerized during their oxidation that can then oxidize other components such as ethanol.
I.3.3 Oxidation of phenolic compounds
Many explanatory mechanisms have been published for the observed oxidation phenomena of phenolic compounds, which are particularly important in wines, as described in the main text of Chapter 3. It is generally considered that, of all the numerous compounds in wines, proteins, carbohydrates, etc., the phenolic compounds present, such as the tannins and anthocyanins contained in the grapes/must, are oxidized first, on exposure to oxygen, during vinification and subsequently. Although during the fermentation the fermenting yeasts will take up all the available oxygen, presumably the conditions in the fermenting must are non-oxidative. Oxygen-generated free radicals from other sources can be mopped up by phenolic substances, before undesirable chain reactions set in. Some of these substances, therefore, have well-publicized antioxidant roles, playing a part in different medical conditions. These mechanisms can be highly complex and indeed controversial in many cases.
The chemical reaction properties of the simple mono-, di-, and tri-hydroxybenzenes (phenols), which are the basic building blocks of the more complex polyphenolic compounds, mostly present in wines, need to be understood first of all.
(1) Oxidative. Dihydroxybenzenes, of which there are three: (a) hydroquinine (p-hydroxyphenol) is very easily oxidized to p-quinone (1,4-diketobenzene), but is of more interest as a photograph developer than in wine; (b) catechol (o-hydroxyphenol) of greater significance in wines, also oxidizes to a quinone (ortho-) (1,2-diketobenzene), easily reverting back, however, in acid solutions, but in the course of oxidation, hydrogen peroxide is produced, which is available for oxidation of other substances; and (c) resorcinol (m-hydroxyphenol) does not form a quinone on oxidation. There are three possible trihydroxybenzenes: (d) pyrogallol (pyrogallic acid, 2,3-dihydroxyphenol), an easily oxidizable substance, e.g. turning alkaline solutions brown, explaining its use in early oxygen gas analyses; (e) phloroglucinol (3,5-dihydroxyphenol), which is also a tautomeric substance, reacting also in a non-benzenoid tri-keto form; and (f) hydroxy-hydroquinone (2,4-dihydroxyphenol), of lesser interest. The oxidation characteristics of all these substances will be different when present in combined form.
(2) Colouring reactions. These phenols are generally colourless in aqueous solution, but catechol is noted for the formation of red compounds with alkaline ferric chloride solutions. Cyanidins and other polyphenolics use colouring reagents in their traditional analysis (see main text).
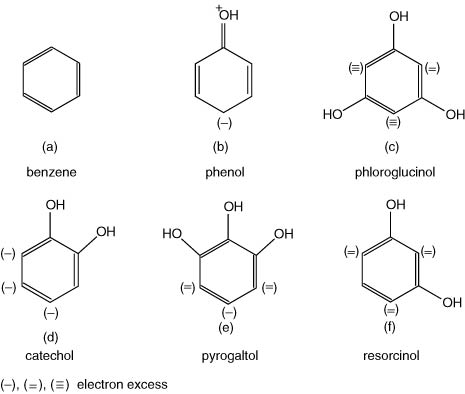
(3) Substitution reactions. The ease of substitution of hydrogen atoms in the benzene ring of phenolic compounds is commonly associated with the type of electron redistribution in the ring, as a result of the effect of the substituent hydroxyl groups. It is well known that, in a simple phenol (monohydroxybenzene), there is a so-called mesomeric effect (Gk. mesos, middle) in a carbon chain to give an electron excess at the o- (1,2) and p- (1,4) positions or nodes, together with a net positive charge at the oxygen atom of the hydroxyl group (Fig. I.2). Thus, substitution reactions are easier on phenol than benzene and in particular at the 2, 4 and 6 positions. This effect does not occur, however, at the meta-position (3). Ribéreau-Gayon et al. (2000) describe this effect, particularly with the trihydroxybenzene (phloroglucinol), with pyrogallol, and with the dihydroxybenzene (catechol), when the electron excess becomes additive, at the unsubstituted adjacent ortho-positions. In this way, electrophilic reagents (not necessarily compounds with a net positive charge) are facilitated in their substitutive attack at these nodes.
Figure I.2 Mesomeric effects in phenols.
In actual polyphenolic compounds with pyran rings, as in wines, there will be positions with mobile or labile hydrogen atoms in their molecules, susceptible to oxidative attack, in a similar fashion as lipids discussed in Section I.3.2. Use of these concepts can explain the results of oxidation of a flavan-3-ol or monomeric catechin in producing a wide range of intermediate compounds but ultimately additional polymeric substances. One such mechanism was well described in a Doctoral thesis of De Freitas (1995), quoted by Ribéreau-Gayon et al. (2000). These polymeric substances are increasingly brown with increasing size, and eventually they form precipitates.
Flavan-3-ol (I.VII) has a molecular formula, C15H14O6, with a molecular weight of 290.
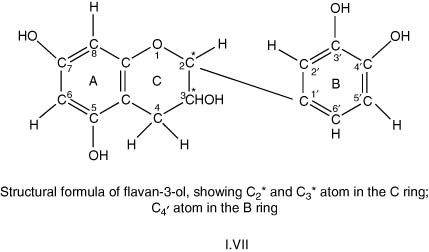
The proposed scheme includes the following sequences:
(1) Mobile hydrogen at C2 in the C-ring is removed by an O2 molecule to form first a flavan radical and a –OOH radical. The flavan radical then either reacts:
(a) with further oxygen to form a peroxy flavan radical which converts to a hydroperoxide in a chain reaction, which transforms into polymeric forms; or,
(b) with a peroxy radical, removes a hydrogen atom at C4 in the B benzene ring (catechol). This latter radical then converts into an o-quinone (keto-C4), and then again transforms into polymeric forms.
(2) Homolytic breakdown occurs at the hydroxyl group at C4 in the B ring producing flavan and hydrogen radicals. This flavan radical will give rise to a range of compounds, quinones and compounds with linked units, all transformable again into polymeric substances.
The mechanism of the final polymerizing stages is not described, though di-ortho-quinones seem to be important immediate precursors. There is some difference in speed of oxidative reaction, dependent upon the exact molecular and chiral structure of flavan-3-ol; thus (−)-epicatechin (R at both C2 and C3) is said to be more rapidly oxidizable than (+)-catechin (R at C2 and S at C3), possibly due to steric hindrance.
Oxidation of procyanidins
Red wine contains substantial quantities of so-called procyanidins that are already polymers of catechins (C) or epicatechins (eC) in several different structures (types, eight B dimers, C trimers and higher polymers), as first described in Chapter 3. The interflavan linkage is of two types, firstly a C4–C8 bond with the following combination of units as shown below.

Secondly, a C4–C6 bond, with procyanidins named from B5 to B8 and a similar sequence of units. The ease of oxidation of procyanidin dimers with a C4–C8 interflavan bond (types B1 to B4) is stated (Ribéreau-Gayon et al., 2000) to depend upon the upper structural unit, whilst those with a C4–C6 linkage (types B5 to B8) depend upon the lower unit. Catechin (+) in B3, B4 (upper unit) and in B5, B7 (lower unit) molecules make them more oxidizable than those containing (−)-epicatechin in B1, B2 and in B8, though the reason is not entirely clear. These procyanidins in acid solution are, however, also unstable even in the absence of oxygen to form further polymeric forms, usually higher; thus a B2 dimer will produce a C1 trimer. The B1 form is the most prevalent dimer in grapes. Acid hydrolysis can involve tautomeric enol-keto change and then the formation of a reactive cationic form. It is also stated that: (1) for the same basic unit (C or eC) oxidizability increases with the degree of polymerization; thus, C1 trimer > B2 > monomer, for eC-; (2) oxidizability increases with increased substitution in the B ring, thus gallocatechin > epicatechin. However, oxidizability is not defined, and some of the information appears to conflict with values of relative redox potential determined in model solutions for some of these substances, described in Section 3.8.3 (Chapter 3).
Even higher polymers are possible due to interaction of procyanidins with ethanal, itself formed by their presence during oxidation. Certain of the higher polymers may not have actual linkages, but are so-called ‘disorganized’ polymers. Anthocyanins can also form complexes with procyanidins or tannins. All these possible changes are especially related to changes in the basic taste characteristics of bitterness and astringency, particularly during maturation, as discussed in Chapter 3.
Anthocyanins are also oxidizable in acid solutions, though malvidin 3-glucoside with two methoxy groups instead of hydroxyl, and only one hydroxyl group in the B-benzene ring, is stated more resistant to oxidation than cyanidin 3-glucoside (with two hydroxyl groups). The mesomeric effect will be lowered in the former compound. However, over time, all monomeric anthocyanins in matured wines disappear (Chapter 6).
Oxidation of non-flavanoid phenolic compounds
Caffeoyl-tartaric acid or caftaric acid [caffeic acid is 3-(3,4-dihydroxyphenyl)-prop-2-enoic acid or 3,4-dihydroxycinnamic acid], and p-coumaryl-tartaric acid or coutaric acid [coumaric acid is 3-(4-monohydroxyphenyl)-prop-2-enoic acid, or p-hydroxy-cinnamic acid] are both regarded as highly oxidizable, with the caffeic compound with two hydroxy groups, the most. Ester type linkage is through one of the enantio-hydroxy groups (said to be C2) of the tartaric acid (butan-2,3-diol-1,4-dioic acid). The mechanism of oxidation has been studied by Cheynier et al. (1989) quoted by Ribéreau-Gayon et al. (2000), also (1991) referenced by Jackson (2008) to experimental work in model solutions using polyphenol oxidase.
General
A review on phenolic compounds and polyphenol oxidase in relation to the browning of grapes and wines by Macheix et al. (1991) gives an excellent insight in the chemically complicated reactions involved. They conclude, after considering the available literature, that the following five points can be made:
(1) Coupled oxidation reactions occur, involving compounds not oxidized by polyphenol oxidase (grape enzyme triggering reactions which cause browning).
(2) Caftaric acid plays a special role, by starting coupled oxidation reactions.
(3) Glutathione limits browning by rapidly reacting with the caftaric acid quinone. The ratio between levels of caftaric acid and glutathione is thought to be one of the important parameters.
(4) Flavan-3-ols, in particular their monomers and dimers, are determining the extent of browning in wines.
(5) The mechanisms involved in oxidation are similar in enzymatically controlled and purely chemical reactions. Enzymatic catalysis occurs only during the first stage of oxidation, and reactions continue purely chemically, in particular the polymerization leading to brown end products. Enzymatic oxidation causes the rapid trigger to browning reactions.
The substrates most rapidly oxidized by polyphenol oxidase are (+)-catechin, (−)-epicatechin, caffeic acid, catechol and 4-methylcatechol. The caftaric acid quinone reacts most easily with glutathione to form 2,5-glutathionyl caftaric acid, which is not a substrate for polyphenol oxidase (see Macheix et al., 1991).
I.3.4 Oxidation–reduction (redox) potentials
Oxidation is always accompanied by reduction and vice versa, which can be expressed in the equation,
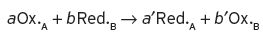
where Ox.A is the oxidized state of substance A, and Red.B is the reduced state of substance B, and so on; a, a′ are the number of unit molecules involved for substance A in the oxidized and reduced state respectively, and similarly for b and b′ for substance B. A so-called reaction quotient (Q) expresses the relationship between the molar concentrations [X] of these substances in their different states (as in Equation I.1).
(I.1) 
The Nernst equation expresses the relationship of this ratio Q with the electric potentials of the system, determinable from measurements in a suitable galvanic cell with appropriate electrodes (Equation I.2).
(I.2) 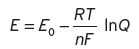
The reaction described above will proceed spontaneously if E > 0. E 0 is the so-called standard or normal potential, which is the value of E at equilibrium; as shown in Equation I.3, where K is the known equilibrium constant. R, T, nF, are described in Chapter 3. Whilst K in practice can range from 1034 to 10−34, the corresponding E 0 will only range +2 to −2.
(I.3) 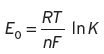
Redox potentials are conveniently thought of as half-reactions of so-called couples (Ox.A/Red.A), thus Ox.A + e− → Red.A, which is an expression in the so-called reduction mode, since a substance in the oxidized form has to gain one or more electrons in order to be oxidized. The standard reduction potential for this half reaction can be stated, say 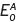 . Similarly, Ox.B + D → Red.B, with a standard potential of
. Similarly, Ox.B + D → Red.B, with a standard potential of 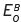 . For the moment, we can ignore the number of electrons which may be involved. By substracting, we have,
. For the moment, we can ignore the number of electrons which may be involved. By substracting, we have,
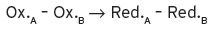
so the overall standard potential for the total reaction is given by, 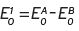 (Equation I.4).
(Equation I.4).
(I.4) 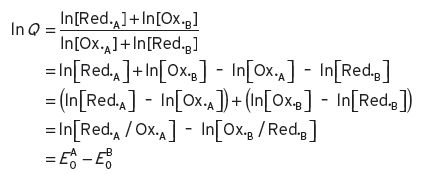
The potential of each couple is therefore given by Equation I.5.
(I.5) 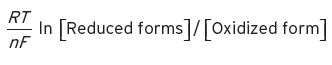
Numerical values for R, T, and F have been given in the main text: ln is the natural loge. This theoretical treatment of the subject follows that given by Shriver et al. (1994). Applying this treatment to actual oxidation reactions, such as the oxidation by dissolved oxygen of solutes such as Fe2+, we have two half-reactions,
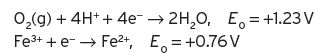
both in the reduction mode, with published standard potential values which, by difference, give the overall reaction as follows,
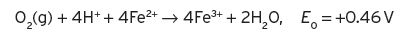
The oxidation of ferrous ions to ferric ions would be spontaneous, but the standard potential is not large and hence, in practice, the reaction would be slow. The E values at any given concentration of reactants can now be determined from the Nernst equation.
In the first half-reaction, above, Ribéreau-Gayon et al. (2000) state that
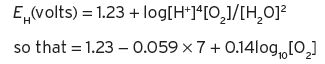
In distilled water, [O2] is a concentration in mole units (quantity, g/32). By multiplying through by 1000, E will be in millivolts and the oxygen concentration more conveniently in millimoles, for dissolved oxygen in water and wines. The figure 7 is the pH for distilled water only. The origin of the other two constants in the equation is not clear. The ratio used is oxidized/reduced with the positive sign instead of reduced/oxidized as in the original Nernst equation but the result is mathematically the same.
I.4 Estimation of partition coefficients of volatile compounds in air/water
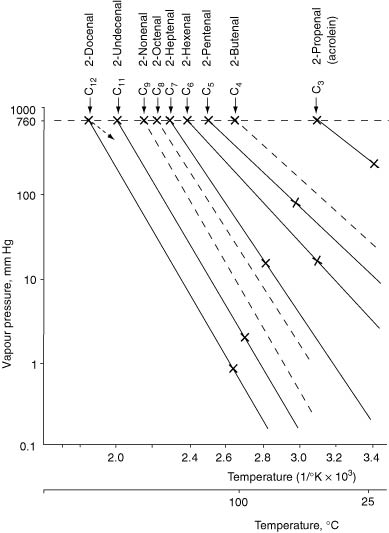
Estimates may be calculated, provided we first know the vapour pressure of the compound of interest at the required temperature. Vapour pressure data is widely available in tables. If necessary, available data can be graphically presented by plotting the logarithm of vapour pressure (P j s, in mmHg or kPa) against the reciprocal of the absolute temperature (1/T °K) over a range of values. A typical straight-line plot is shown in Figure I.3 for alkyl unsaturated aldehydes. Notably, the lines for each compound are not exactly parallel; equations are available to express relationships, such as log P = a.1/T + b, where a and b are constants. Vapour pressure at any other temperature may be found by interpolation or extrapolation from the plot. It will be noted also that the vapour pressures of the higher homologues at room temperature can be very low. Also needed are the vapour pressures of water over the same range of temperatures, but these are very accurately known.
Figure I.3 Logarithm of vapour pressure (Pj s, in mmHg or kPa) versus reciprocal of the absolute temperature (1/T°K) for some alkyl unsaturated aldehydes.
The so-called activity coefficient 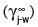 of the particular component at infinite dilution in water also needs to be known, but it can be estimated in either of two ways:
of the particular component at infinite dilution in water also needs to be known, but it can be estimated in either of two ways:
(1) Activity coefficient reflects the inherent characteristic of a compound not easily mixing with water and not forming a perfect solution, obeying Raoult’s Law. For this reason, the saturation water solubility (S t j,w) of the volatile compound (j) in water (w) expressed in molar units at a temperature of T°C, as a reciprocal value is equal to the dimensionless activity coefficient, i.e. 1/S = γ ∞. Data on the actual saturation solubilities of many volatile compounds in water is scarce, though generally available for room temperatures for well known compounds. The technique of obtaining reasonable accuracy in measurement at inevitably low solubilities needs care (Buttery, 1969).
(2) The Pierotti correlations (1959) showed that Equation I.6 applies to a wide range of compounds, where n 1 is the total number of carbon atoms in the molecule of compound. A, B and C are temperature dependent variables, values of which are available from Pierotti’s published tables.
(I.6) 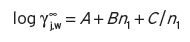
For normal alkanoate esters, A = −0.950, B = 0.64 and C = 0.260 at 20°C. However, account has also to be taken of secondary linkages between two radical groups, so that 1/n
1 is replaced by 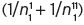 . For ethyl butanoate, with the molecular formula of CH3.CH2.CH2.CO.OC2H5, though n
1 is 4 + 2 = 6, n
1
1 is 4, but n
11
1 in fact is 3, to include also the carbon atom to the polar group, and is not 2. Once is known by either method, the value of K
j,a-w is determined from Equation I.7, where P
s
j is the vapour pressure of the pure substance at the same temperature. The value of the relative volatility (α∞) can be obtained from
. For ethyl butanoate, with the molecular formula of CH3.CH2.CH2.CO.OC2H5, though n
1 is 4 + 2 = 6, n
1
1 is 4, but n
11
1 in fact is 3, to include also the carbon atom to the polar group, and is not 2. Once is known by either method, the value of K
j,a-w is determined from Equation I.7, where P
s
j is the vapour pressure of the pure substance at the same temperature. The value of the relative volatility (α∞) can be obtained from 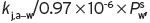 , where
, where 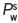 is the vapour pressure of pure water at the given temperature.
is the vapour pressure of pure water at the given temperature.
(I.7) 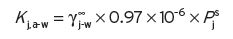
These calculations will now be illustrated for ethyl acetate (ethanoate), though there are some directly or indirectly determined values of K j,a-w for use. Estimated values may be usefully compared.
(1) The saturation solubility is reported by Lide (2001, loc. cit.) at 8.1% w/ws (8.1 parts per 100 parts solution) at 25°C, and the molecular weight is 88. The mole fraction solubility is therefore (8.1/88)/[(8.1/88) + (91.9/18)], i.e. 0.0176 so that 1/S = 56.8, say 57. The vapour pressure of ethyl acetate is 93 also at 25°C, so that K j,a-w at 25°C then calculates as 57 × 0.97 × 10−6 × 93 = 0.50 × 10−2.
(2) Pierotti correlations are only given for esters at a temperature of 20°C. For ethyl acetate, n 1 = 4; n 1 1 = 2 and n 11 1 = 3 (Equation I.8). Therefore, γj = 70, P = 70, and K = 70 × 70 × 0.97 × 10−6 = 0.47 × 10−2.
(I.8) 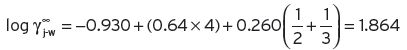
These figures may be compared with the data in Table I.5, for directly determined values at higher temperatures (Gretsch et al., 1991) and extrapolated at others from a straight line relationship of log K j = Constant/T. At 25°C, the values at × 102, 0.52 extrapolated from direct, and 0.50 estimated, from (1) above, at 20°C these values are 0.40 (direct) and 0.47 (from (2) above) respectively.
It is apparent that the solubility of ethyl acetate in water increases with a lowering of temperature, and decreases with increasing temperature. The expression T°K/log γ is approximately constant, indicating that the heat of solution of ethyl acetate in water is negative. The relative volatility value shows a slight decrease with increasing temperature. The value of K j,a-w, however, is markedly higher at higher temperatures, so that the higher the temperature of the wine being drunk, the greater will be the concentration of the ethyl acetate in the air head-space for a given concentration in the aqueous liquid phase (Table I.6). This may not be true for the other volatile components, which would have to be examined on an individual basis for their solubility–temperature characteristics. Currently, there are few direct determinations of K j for other alkanoates, except of the methyl alkanoates of limited wine interest, for comparison against estimates. These are presented in Table I.7.
Table I.5 Air–water partition coefficients for ethyl acetate (j) at different temperatures.
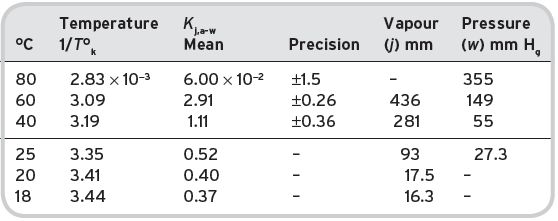
Table I.6 Effect of temperature on the physical properties of infinitely dilute solutions of ethyl acetate.
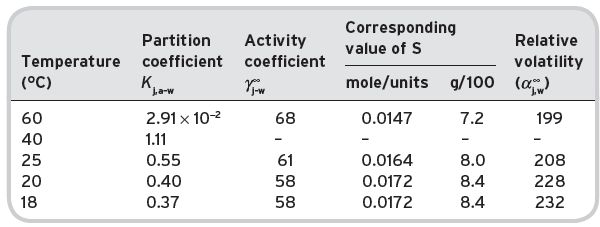
Other independent data, such as Chandrasekaran & King (1972) by indirect determination of K
j for ethyl acetate gives = 66 at 20°C and  but figures are based upon a higher vapour pressure figure of 74 mm.
but figures are based upon a higher vapour pressure figure of 74 mm.
The relevant physical properties for all other volatile compounds found in wines are presented in Tables II.1, II.2, II.3, II.4, II.5, II.6, II.7, II.8, II.9 and II.10, together with estimates where possible. Pierrotti correlations are available for alkyl ketones (n 1 1 and n 11 1 , applicable) and aldehydes (saturated and unsaturated), but not for branched chain alcohols (saturated only, but including secondary and tertiary variants).
I.5 Grape varieties and cultivars
To the non-geneticist and non-horticulturalist, the terms ‘variety’ and ‘cultivar’ may well cause some confusion, as they are frequently used interchangeably in certain books on wine.
Table I.7 Comparative data partition coefficients Kj,a-w for alkyl alkanoates determined by different methods.
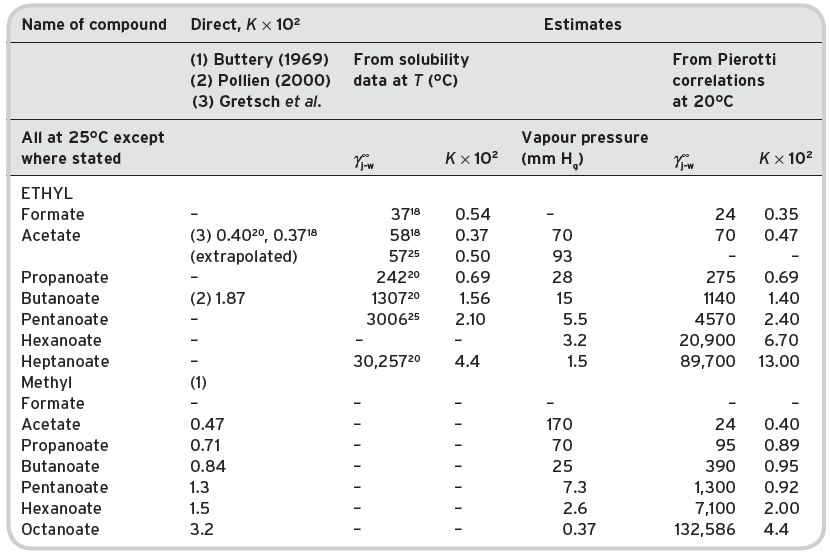
NB The data gives reasonable agreement between directly determined values for the methyl alkanoates, with a tendency to over-estimate for the higher homologues, especially the temperatures are slightly different (25°C v 20°C). There is little data for the ethyl alkanoates. The comparisons between estimates by the two methods are good for the lower homologues but not the higher homologues. There are practical difficulties in the determination of vapour pressure and solubilities for these compounds.
It is generally understood that the wine grape is the fruit of the vine of the Vitis genus and the vinifera species by the Linnean classification, with additional word ‘sativa’ to indicate a cultivated species, as distinct from the original wild state. The terms ‘variety’ and ‘cultivar’, or ‘c.v.’ are lower levels of botanical rank in determining characterizing differences between plants. To the wine chemist, characterization of a grape is most conveniently expressed by the name of the variety of the vine plant from which it was harvested. Plant variety is essentially determined by botanical features (taxonomy) and genetic make-up (ampelography, Gk. ampelos – vine), to which DNA profiling can now be included.
The domestic cultivation of vines has been practised now for many centuries, by initial selection from the wild, with differently appearing vines being given different varietal names, such as the familiar Chardonnay and Cabernet Sauvignon but many other locally given names. Pinot Noir is considered to be perhaps the variety to be first directly named, some 2000 years ago, and known to be planted in Burgundy in the fourth century AD (& Rand, 2001). Variety stability and difference has been largely sustained by the self-pollination characteristics of Vitis vinifera and by the modern use of clones, to be described. Mutations of genes, however, do occur over the centuries to give botanical sub-variants of the basic type; in fact, the variety Pinot Noir is particularly prone to this phenomenon.
To the horticulturalist in the vineyards, the term ‘cultivar’ gives an additional dimension of meaning over ‘variety’ to a particular vinestock or plant. A cultivar is variously defined as ‘plant material of particular genetic characteristics produced by horticultural techniques’. One such technique is the production of clones by vegetative reproduction, which is taking cuttings from the plant and rooting them, to produce a next generation of vinestock that will be genetically identical. Clones, for example, can be prepared from different plants of the Chardonnay variety that show slightly different agronomic differences (fruiting yield, etc.) more favourable than others. These clones will be described as still of the Chardonnay variety, but will be additionally named, thus the Mendoza Chardonnay clone (Clarke & Rand, 2001). The use of clones is the way in which replanting of vineyards is carried out, though only necessary after about thirty years of fruit bearing.
Another horticultural technique is that of crossing varieties by manual pollination methods, which was especially practised in Germany in the last century and earlier. Müller-Thurgau and Scheurebe (both intra-specific, Riesling × Silvaner) are described in the main text of Chapter 5 and are still referred to as varieties. Clarke & Rand (2001) are somewhat disparaging about the success in terms of quality of such crossing, compared with ‘natural’ varieties – though they are more enthusiastic about the South African Pinotage grapes (Pinot Noir × Cinsault). Some crossings have also been made to produce inter-specific hybrids, though of limited interest for wine grapes.
The production of disease resistant plants is also important in enology, by cloning techniques from favourable vinestock. Laboratory growing techniques of tissue culture can be used. The most effective horticultural technique to be used was that following the Phylloxera epidemic in the 1860s in France, when grafting of branches of existing disease-free vines in France on root-stock from America (albeit of a different Vitis species) was successful. Varietal differences were not substantially altered. Since Phylloxera is ubiquitous in most vine growing regions, most vines are still grafted on a so-called American rootstock or a hybrid, which confers resistance to Phylloxera attack of the entire vine.
Recent studies in DNA profiling of vines have revealed the true origins for some of the traditionally named varieties. There appears to have been post-crossings (presumably some by chance inter-pollination); for example, in Chardonnay, which is really a crossing of Pinot Noir with an old variety, ‘Gouais blanc’ (at some genetic distance away). An investigation of this history, together with that of other grape varieties, has been given by Bowers et al. (2000).
References
Belitz, H.-D. & Grosch, W. (1987) Food Chemistry. Chapter 3, pp. 128–200. Springer-Verlag, Berlin.
Bowers, J., Boursiquot, J.M., This, P., Chu, K., Johansson, H. & Meredith, C. (2000) Historical Genetics: The Parentage of Chardonnay, Gamay, and Other Wine Grapes of Northeastern France. Science, 285, 1562–1565.
Buttery, R.G., Guadagni, D.E. & Ling, L.C. (1969) Volatilities of aldehydes, ketones and esters in dilute solution. Journal of Agricultural and Food Chemistry, 17(2), 385–389.
Chandrasekaran, S.K. & King, C.J. (1972) Multicomponent diffusion and vapor-liquid equilibria of dilute organic components in aqueous sugar solutions. American Institute of Chemical Engineers Journal, 18, 513–520.
Clarke, O. & Rand, M. (2001) Grapes and Wines, Webster, London.
Clayden, D., Greeves, N., Warren, S. & Wothers, P. (2001) Organic Chemistry. Oxford University Press, Oxford.
Drawert, F. (1974) Wine making as a biotechnological sequence. In: Chemistry of Wine Making. Advances in Chemistry Series, 157, pp. 1–10. American Chemical Society, Washington.
Flament, I. (2001) Coffee Flavour Chemistry. John Wiley & Sons, Ltd, Chichester.
Gretsch, C., Grandjean, G., Maering, M., Liardon, K. & Westfall, S. (1995) Determination of the partition coefficients of coffee volatiles using static head-space. In: Proceedings of the 16th ASIC Colloquium (Kyoto), pp. 326–31, ASIC, Paris, France.
The International Union of Pure and Applied Chemistry (1993) A Guide to IUPAC Nomenclature of Organic Compounds, Recommendations 1993. R. Panico, W.H. Powell & J.C. Richter (Eds.), Blackwell Publishing Ltd., Oxford.
Jackson, R.S. (2008) Wine Science, Principles, Practice, Perception, 3nd edn. Academic Press, San Diego.
Lide, D. (ed.) (2001) Handbook of Chemistry and Physic, 81st edn., CRC Press, Boca Raton, FL.
Macheix, J.J., Sapis, J.C. & Fleuriet, A. (1991) Phenolic compounds and polyphenol-oxidase in relation to browning in grapes and wines. Critical Reviews in Food Science and Nutrition, 30, 441–486.
Pierotti, G.J., Deal, C.H. & Derr, E.L. (1959) Activity Coefficients and Molecular Structure. Industrial & Engineering Chemistry, 51, 95–102.
Ribéreau-Gayon, P., Glories, Y., Maujean, A. & Dubourdieu, D. (2000) Handbook of Enology, Vol I, Wine and Wine Making, John Wiley & Sons, Ltd, Chichester.
Ribéreau-Gayon, P., Glories, Y., Maujean, A. & Dubourdieu, D. (2000) Handbook of Enology, Vol II, The Chemistry of Wine, Stabilization and Treatments, John Wiley & Sons, Ltd, Chichester.
Semmelroch, P. & Grosch, W. (1996) Studies in character impact compounds in coffee beans. Journal of Agricultural and Food Chemistry, 44, 537–543.
Shriver, D.F., Atkins, P.W. & Langford, C.H. (1994) Inorganic Chemistry, 2 nd Edn. Oxford University Press, Oxford.