Design of Studies and Risk Management in Toxicologic Pathology
Addressing Risks in Product Discovery and Development
Ricardo Ochoa, Pre-Clinical Safety, Inc., Niantic, CT, United States
Abstract
This chapter discusses the issues of risk assessment, management, communication, and abatement that are important to the Toxicologic Pathologist to understand the significance of their contribution as it relates to the health of humans and the environment following exposure to products in development for use as pharmaceutical of chemical compounds. The division of the development stages is discussed along with the most important practices to make the contributions of the Toxicologic Pathologist useful to the client, ethically rigorous, and responsive to the needs of the public.
Keywords
Nomenclature; report production; risk; risk abatement; risk management; risk communication; adverse effects; reversibility; risk/benefit
Introduction
Risk is a generic term used to describe the probability, or chance, of a specific event occurring. For example, when gambling (e.g., games of “chance,” like blackjack), the risk applies to the possibility of winning offset by the potential for losing. Both the positive and negative outcomes depend to some extent on random chance, so both may be considered risks, though in many settings the term “risk” is applied to the negative consequence while the positive result is accorded an honorific like “benefit” or “opportunity.”
Toxicologic pathology is a key discipline in identifying, characterizing, and managing benefit versus risk (the “risk:benefit ratio”) when discovering and developing new products (e.g., chemicals, drugs, medical devices, modified cells and genes, and many other entities), and in particular the adverse (i.e., harmful) health outcomes that may afflict humans, animals, and their environments. An important additional consideration is to balance the health effects (beneficial and potentially hazardous) of the new product with the financial opportunities and risks faced by the firms developing and commercializing the new products.
Evaluating the health and financial risks involves a logical, measured approach to product development. The five principal aspects of the product development pathway are to identify possible negative effects (hazard identification), to determine their complete spectrum and the mechanism(s) and pathogeneses that lead to their initiation and progression (hazard characterization), to develop an interpretation concerning the likelihood that a risk will develop in a given setting (risk assessment), to devise means for reducing or ideally preventing negative outcomes from occurring (risk management), and to communicate with the consumer of the product what the risk is and how it compares to its benefits (risk communication). Risk of an adverse health effect can only be determined in the context of a potential exposure to a particular test article. The practice of toxicologic pathology is instrumental to successfully undertaking all of these tasks. For most industrial toxicological studies, hazard identification and dose–response assessment for xenobiotics (i.e., a foreign chemical substance) are the central work of the toxicologic pathologist, whereas exposure assessment and risk characterization also are important elements of environmental toxicologic pathology.
The toxicologic pathologist applies his expertise in many fashions during the product discovery and development process. Perhaps the most common roles are to determine morphologic (anatomic pathology) and biochemical (clinical pathology) alterations that are produced by exposure to a potential new product (termed a “test article”). Identification and characterization of such changes (an objective dataset) coupled with experience gained during the course of professional toxicologic pathology practice (a subjective, individual-specific or communal “database”), subsequently permits an interpretation to be made regarding whether or not an effect caused by a test article should be considered harmful (adverse) or incidental (nonadverse). As such, the toxicologic pathologist is in a unique position to help in the interpretation of data originating from nonclinical studies in many animal species that will have an often-decisive impact on the product development process with respect to management of health and financial risks.
This chapter concerns the nonclinical issues of health risks; the significance of toxicologic pathology findings as they impact the development process from the impact on human clinical risk; and provides strategies for management of these risks. The information in this chapter lies at the very core of professional toxicologic pathology practice.
Risk Assessment: The Process
Pathologists and toxicologists participate in evaluating risk on a daily basis. The pathologist, together with the input from colleagues in toxicology, initially identifies the potential adverse health effects of products in laboratory animals (of many species, but most typically rodents, rabbits, dogs, pigs, and nonhuman primates), defines the dose–response of the adverse effects in test animals, and then determines whether or not these effects are likely to express themselves in a given population (generally human patients with some disease). Estimation of risk is a multipronged analytical problem, involving a series of steps.
Hazard Identification
Hazard identification is a qualitative process utilized to discover potential adverse health effects (e.g., carcinogenicity, neurotoxicity, and developmental toxicity) for humans following exposure to a particular test article under a given set of conditions. The output of hazard identification is the detection of an abnormal finding, sometimes with a preliminary estimate of how the finding (or response) varies with the exposure level (or dose). Hazard statements are usually short (e.g., “Agent X is a known human carcinogen”). Adequate identification of a hazard incorporates numerous datasets, including structure–activity relationships; in vitro analyses; ADME (absorption, distribution, metabolism, and excretion) evaluations; genotoxicity; animal toxicity bioassays; and epidemiological (usually human or environmental) information. Part of the hazard identification is the detailed description of lesions that typify a reproducible treatment effect of the test article. Integration of these findings with knowledge about the pharmacology and clinical toxicology of the test article allows the pathologist to determine whether or not the treatment-associated effect is in fact a test article–related effect.
Hazard Characterization
Hazard characterization expands on the hazard identification step to more fully define “under what conditions the hazard is present.” Hazard characterization explores the complete constellation of changes that result from exposure to a test article. A common approach is to assess variations in structural changes (anatomic pathology data) and various biochemical and cellular alterations (clinical pathology data), and where possible to make mechanistic correlations between these types of findings under various exposure conditions: acute versus chronic, low-dose versus high-dose, different routes of exposure, different species, progression, regression or persistence of changes, etc. In modern practice, product registration packages for novel test articles emphasize inclusion of more extensive datasets than would be required for simple hazard identification.
Dose–Response Assessment
Dose–response assessment is a critical element of hazard characterization. Nonclinical toxicity studies are designed to evaluate the conditions under which exposure to the test article might induce an effect, and particularly an adverse effect (i.e., “toxicity”). It characterizes the relationship between variable levels of exposure to a test substance and the shifts in incidence and severity in any responses. Dose–response assessments incorporate both qualitative data (e.g., does a lesion exist, and if so what is its severity grade) and quantitative data (e.g., organ weights, cell numbers, and other end points). Conventional nonclinical studies in animals incorporate four dose groups (negative control, and low, medium, and high doses of the test article); in some cases, additional groups are included (e.g., additional doses of the test article, or a positive control cohort, or even a second negative control group). Dose–response relationships characterize development and progression of effects over a broad range of exposure periods, including acute, subchronic, and chronic effects. The intended goal of a dose–response assessment is to define a threshold of exposure above which the test article will cause adverse effects. Many calculated values have been defined to provide a numerical estimate of this threshold, including the “benchmark dose” (BMD, commonly used for chemicals) and the “no observed adverse effect level” (NOAEL, typically employed for drug candidates). Dose–responses may be linear or nonlinear.
Exposure Assessment
Exposure assessment is another aspect of hazard characterization, which is specific to agents that might be present in the environment. Such analyses entail the determination of the source, amount, and duration of potential exposure (usually to humans, but sometimes to wildlife and their habitat) to the agent of concern. In general, toxicologic pathologists do not obtain such data.
Risk Assessment: The Outcome
Risk assessment is the systematic integration of data regarding potential hazards and their relationship to exposure. This step is essential to predict the potential for adverse effects on human health. Risk assessments are based on results of many studies conducted in multiple species, in which each individual study examines the hazard posed by a specific exposure to a test article at a given level for a set period of time. Such analyses use the frequency and severity of effects seen in the exposed population under specific exposure conditions to estimate whether or not similar effects might be expected to develop in another species (usually humans) under a different set of exposure conditions. Such predictions culminate in calculations of a margin of safety (for chemicals) or therapeutic indices (for drugs, the difference between a minimally toxic dose and a lethal dose). Such assessments may be linked to a formal probability statement. For example, the lifetime exposure (cumulative dose) to a compound might result in a probability statement for developing cancer [Probability of cancer development is 10−6 (1 chance in 1,000,000)]. Alternatively, the assessment may rely on qualitative interpretation (a “weight of evidence” approach) of what is considered adverse, culminating in a simple statement of harm rather than a statistical calculation. In this case, not all observations are considered to have equal value (i.e., some outcomes are more undesirable than others), so the weight given to assess the risk depends on the potential extent of the negative health impact. For example, the outcome hepatocellular carcinoma is of greater concern than hepatic inflammation alone.
Risk Assessment: The Elements
Product Discovery and Development Programs: General Features
The series of studies performed during product discovery and development programs are designed to optimize the quality of the products that enter the testing process and eventually become available for use. As the number of products that are explored for possible development increases, it becomes more critical to make decisions based on accurate data, and ideally to make “go—no go” decisions as early as possible during the development process. Obtaining accurate data is often not straightforward, especially in the pathology discipline where the data are based on the professional opinion of the pathologist making the interpretation of morphologic and clinical pathology findings.
Studies undertaken to support the eventual registration of new products [especially new chemical entities (NCEs) being developed as pharmaceuticals for intended human administration] collectively must provide information regarding many parameters. For NCEs, the most critical considerations addressed in nonclinical studies characterize the:
1. distribution of the target in various tissues;
2. distribution of the compound within various tissues (that contain or do not contain the target) in both animals and humans;
3. compound’s intrinsic toxicity for individual species (including any effects that occur in multiple test species); and
4. variations in toxicity based on changes in the:
Ultimately, the data are integrated to permit estimation of the initial dose (for NCEs) or margin of safety for humans, together with a statement of the risk–benefit ratio.
Product Discovery Studies
Discovery studies optimize the nature of the products being prepared for commercial use. Many institutions use toxicologic pathologists in discovery studies merely as special-purpose consultants to solely provide diagnoses for possible toxic effects. Inclusion of toxicologic pathologists as integral members of discovery teams (not as outside consultants) is vital to ensure the proper collection and processing of tissue and fluid samples (which is essential to making well-informed development decisions) as well as the identification and (as necessary) exclusion of potential lead candidates, and to document efficacy of the early candidates.
Studies that are carried out in animals during the discovery process are of two major categories: screening studies and exploratory toxicology studies. Due to low availability and high cost of novel test articles, these studies generally use rodents, unless they are inadequate for lack of therapeutic targets for the specific compound.
Screening studies are intended to select among several possible lead compounds to choose the ones with better likelihood of surviving development hurdles to become new products. Such studies often include demonstrating the efficacy of a test article in an animal model of a human condition; diseases in such animal models may be produced by surgical, infectious, or genetic modifications, or may arise spontaneously. The understanding of comparative pathophysiology, which is the core of the pathologist’s training, is instrumental in selecting the best animal model to use in the selection of candidates. The key result of screening studies is to show efficacy (i.e., that a test article can alleviate or even cure a disease). However, other important outcomes are to identify the optimal formulation and regimen (i.e., route of exposure) for delivering the compound, undesirable effects (toxicity) that can be eliminated, and biomarkers that can be used to follow the course of a disease and course of treatment.
The other main type of discovery study is the exploratory toxicology assay. These studies are intended to detect adverse effects of a test article, thereby allowing an analysis of its chances of successful development. These studies mimic, on a small scale, the safety assessment studies undertaken in development to satisfy regulatory requirements. While generally done in rodents, dogs or nonhuman primates may be utilized if prior in vitro or in vivo screens suggest that a species-specific response needs to be investigated.
Safety Assessment Studies
Regulatory animal studies most frequently used in pharmaceutical development may be divided into several categories (Table 7.1). Short-term tests explore such responses as acute toxicity (at high doses for NCEs) and mutagenic potential. A specialized short-term assay, the dose range-finding study, is undertaken in rodents (typically rats) and nonrodents (usually rabbits or dogs) as a means of establishing the spectrum of doses that will be employed in subsequent safety studies. Short-term studies may last for up to 4 weeks; dose range-finding studies generally are 2 weeks in length. Longer toxicity studies are conducted in rodents and nonrodents for weeks (“subchronic,” usually 13 weeks); months (“chronic,” typically 6 months in rodents, and 6–12 months in nonrodents); or life (“carcinogenicity bioassay” in rodents, 2 years). Special safety studies may be required for critical functions (e.g., developmental and reproductive toxicity, neurotoxicity). Most exploratory toxicology short-term studies are called “Dose-Range Finding Studies” and are undertaken outside of compliance with Good Laboratory Practices (GLP, a set of guidelines designed to ensure that study datasets used to make regulatory decisions are of highest quality). However, successful product approval generally requires that some short-term (2–4-week) studies and all longer-term studies used to assess safety must be conducted in compliance with GLP guidance.
Table 7.1
Accepted Duration of Animal Toxicology Studies to Support Human Clinical Trials of Various Durationsa
| Duration of clinical trial | Rodents | Nonrodents |
| Single dose | ||
| Unites States | 1–14 days | 1–14 days |
| European Community | 2 weeks | 2 weeks |
| Japan | 4 weeks | 2 weeks |
| Up to 2 weeks | 2 weeks (Japan: 4 weeks) | 2 weeks |
| Up to 1 month | 1 month | 1 month |
| Up to 3 months | 3 months | 3 months |
| Up to 6 months | 6 months | 6 months |
| >6 months | 6 months | Chronic (EC: 6 months; US and Japan: 6, 9, or 12 months) |
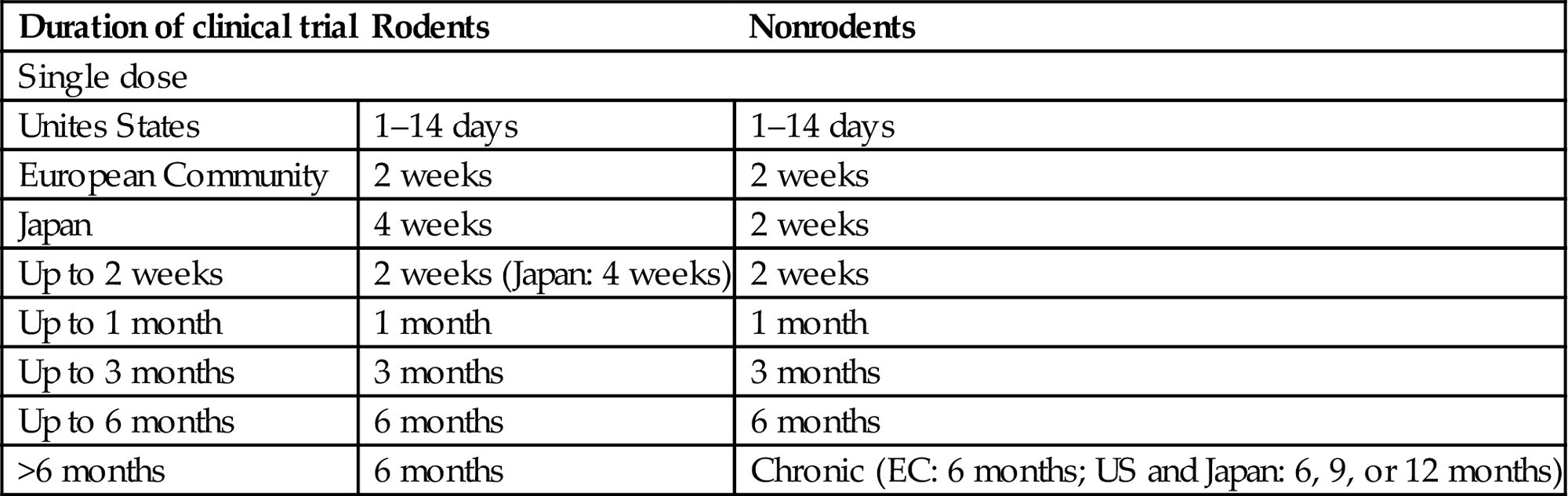
Abbreviations: EC, European Community; US, United States.
aGuidance reflects the consensus recognized by the ICH (International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use).
Adapted from Ochoa, R., 2013. Pathology issues in the design of toxicology studies. In: Haschek, W.M., Rousseaux, C.G., Wallig, M.A., Bolon, B., Ochoa, R., Mahler, B.W. (Eds.), Haschek and Rousseaux's Handbook of Toxicologic Pathology, third ed. Academic Press (Elsevier), San Diego, CA, pp. 595–618 (Table 19.1).
With the exception of some biological products, safety assessment generally requires that product candidates be tested in both rodents and nonrodents. The rodents usually are outbred rats, where their genetic heterogeneity (mimicking that in human populations) and relatively larger size render them preferable to inbred mouse strains. The nonrodents typically are dogs (purpose-bred Beagles), nonhuman primates, rabbits, and, increasingly, minipigs. The choice of model depends on the nature of the test article. For example, nonhuman primates often are employed to evaluate biomolecules [fully human or humanized (i.e., chimeric test articles having a human-origin active domain attached to a backbone of animal derivation)] intended for administration to humans based on the phylogenetic proximity of simians to humans, while rodent studies are often not recommended for use with human biomolecules. The rationale is that conserved molecular properties among these primate groups will minimize rejection reactions and aberrant target interactions of heterologous proteins. However, claims that only primate studies will better predict human responses may be accepted by regulatory agencies only after presentation of relevant justification.
Many individual countries have their own regulatory guidelines for conducting safety studies, which will vary based on the type of product being developed. For example, the guidance provided by the U.S. Environmental Protection Agency (US EPA) for developing agricultural chemicals differs from that given by the U.S. Food and Drug Administration (US FDA) for developing pharmaceutical agents. Within the US FDA, guidance varies with the nature of the potential product (NCE vs medical device vs cell therapy, etc.). The trend toward product registration in multiple geographic regions has led to efforts toward standardizing regulatory requirements around the globe. Examples of such collaborative international rule-making include the ICH (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use) and the OECD (Organisation for Economic Co-operation and Development).
Wide consensus exists that nonclinical safety assessment studies support human clinical trials of equal length. Some exceptions exist to these rules, particularly when the risk–benefit analysis reveals that exposure to the potential harmful effects of a compound is less detrimental to the patient than the withdrawal of a drug providing obvious benefit. Such exceptions often are made for compounds being developed to treat life-threatening diseases for which there is no alternative treatment, such as terminal cancer or lethal genetic diseases. In contrast, decreased tolerance for risk is present when dealing with diseases considered “minor” or cosmetic, or for which there are safe alternatives (e.g., asthma, diabetes).
Experimental Design for Discovery and Safety Studies
Studies undertaken during product discovery and development generally should be designed in advance. Discovery studies may be planned informally, using an outline, while GLP-type safety studies are conducted according to signed protocols that detail all aspects of the experiment. Specific sections of standard study protocols describe the test article and control materials, the animal demographics, methods, and how data quality will be assured. The toxicologic pathologist will be instrumental in ensuring the accuracy of tissue and fluid sampling as well as the analytical battery to be used (i.e., anatomic pathology and clinical pathology endpoints). In addition, the pathologist should review other portions of the study protocol (e.g., choices of species/strain, sex, and age) so that obvious errors are avoided. For example, acute studies in nonhuman primates may be designed in which the test subjects are immature (younger than 3.5 years), which may confound the assessment of toxic responses in the gonads.
Perhaps the key consideration for developing NCE products is defining the dose range that will be tested in the nonclinical setting. In general, doses for animal studies are chosen that are a multiple of the effective dose as predicted from in vitro and efficacy discovery studies, where doses were escalated until intolerance was observed. From this starting point, doses are decreased and given over several days (often varying between 5 and 14 days, depending on compound availability). The dose range for safety studies in animals typically will exceed the proposed human dose by a large amount (ideally with a high dose that is 20-fold or greater than the intended human dose).
In general, GLP-type safety studies have the following design features with respect to the toxicologic pathology examination. Typical pathology endpoints are organ weights; clinical chemistry and hematologic parameters (comparable to the test panels performed in the diagnostic pathology setting) in blood samples; and histopathologic changes in specimens from all major organs as well as demonstrably unique organ regions (e.g., in brain, specimens are taken specifically from multiple cerebral zones that serve distinct functions). Special pathology procedures (electron microscopy, urinalysis, blood smears, etc.) may be undertaken if necessary, either as part of the original study protocol (based on prior experience with the test article or a structurally related compound) or by post hoc protocol amendment (due to in-life clinical signs seen during the course of the study). Where possible, baseline data collection (prior to initial treatment) is obtained for noninvasive (clinical pathology values, total body weight) or minimally invasive (e.g., needle biopsies) parameters; thus, each animal also can serve as its own control as an additional means of evaluating biological responses. The full constellation of pathology endpoints generally is examined at the end of the treatment period (the “terminal necropsy”) and also after a treatment-free period (the “recovery necropsy”).
The number of animals per treatment group is impacted by several factors. The group sizes for scheduled necropsies depend on the timing with respect to the final treatment. For terminal necropsies, the numbers of animals for all groups (including the control cohort) usually are set at 5 males for rodents and 2 males for nonrodents for discovery studies, and 10 per sex for rodents and 4–5 per sex for nonrodents for regulatory studies. For recovery necropsies, these sizes are halved (i.e., five per sex for rodents, and two per sex for nonrodents). The group sizes in rodent studies typically are increased for longer studies so that early, unexpected (unscheduled) deaths do not deplete the animal numbers so much that statistical calculations are thwarted. For example; initial group sizes in 2-year carcinogenicity studies in rodents usually are set at 50–65 per sex per treatment dose for a final per group count of 25 at termination.
Toxicologic pathologists are essential members of the discovery and development program, particularly through their advocacy to avoid major experimental errors. For instance, pathologists can ensure that sampling practices will permit acquisition of both morphologic and clinical pathology parameters, and avoid interference from other sampling on these parameters. Failure to collect these data, including during discovery studies, in the belief that they are either irrelevant or too expensive to obtain can make interpretation of the results difficult or impossible. Pathologists also can argue against the common practice of returning animals from discovery studies to the colony for reuse, since residual changes from the prior experiment complicate the interpretation of changes observed in later studies.
Toxicologic Pathology Issues in Risk Assessment
Diagnoses
Diagnoses of structural lesions (for anatomic pathology) or cellular and chemical changes (for clinical pathology) are the interpretations of a pathologist. Such interpretations represent a combination of objective criteria (e.g., appearance of a lesion) and subjective opinion (based on the training and unique experiences of the individual). Toxicologic pathologists are divided into those who believe that the diagnoses for different variations of the condition should be consolidated (the “lumpers”) and those who advocate that every feature should be represented individually in the text description and data tables (the “splitters”). Generally, diagnoses should be the synthesis of different descriptive components placed into perspective. When the pathologist provides multiple diagnoses to a common pathological response, the diagnosis may be obscured and the data tables greatly expanded, making interpretation difficult. However, occasionally splitting the diagnosis will clarify the interpretation of a response, and in those situations splitting is appropriate. A good example of this dilemma is the use of multiple diagnoses for different components of chronic progressive nephropathy (CPN) in rats, which can encompass varying degrees of regenerative (basophilic) tubules, proteinaceous content in tubules, interstitial fibrosis, interstitial lymphocytic infiltration, and glomerular sclerosis, among others. The separation of these components into individual diagnoses may make the detection of another renal effect difficult by masking a potential compound-related effect on the behavior of a background lesion.
Toxicologic pathology evaluation is undertaken to discriminate genuine treatment effects from incidental findings (“background pathology” or “normal abnormalities”). Accordingly, certain findings in pathology samples are not diagnosed. Examples include minimal elevations in serum activities of hepatocellular leakage enzymes [e.g., alanine (ALT) and aspartate (AST) aminotransferases] in many animal species and small foci of extramedullary hematopoiesis (EMH) in the liver and spleen of rodents. Such changes fall within the expected range of interindividual variability [“within normal limits” (WNL)] and do not impact organ function. Thus, ignoring such minor findings is a means of avoiding the expansion of pathology data tables with incidental changes that do not indicate the presence of an adverse effect. However, if the degree of such changes is increased in a dose-related fashion (e.g., EMH fills the parenchyma or leads to bulging of the splenic contours), such findings should be accorded a formal diagnosis so that their potential biological importance may be assessed. Of course, variability among pathologists in whether or not these changes are recorded may impact the historical control data tables, which are occasionally used to interpret the incidence and severity of these types of changes if the findings in the concurrent control group are confusing, inconclusive, or skewed.
Validation of Diagnoses
Masked (Blinded) Evaluation of Slides
Information regarding the treatment group, clinical signs, necropsy findings, clinical pathology, and any other relevant information should be taken into consideration when making diagnoses. Therefore, the initial evaluation of the changes in toxicology studies should not use masking (coded or “blind” reading), as the ancillary information is critical to arrive at a specific diagnosis. The most important role of the pathologist follows the clinical model, where all the available information is used to achieve an accurate diagnosis. Performing a masked (blinded) assessment becomes useful when a subtle change is identified and one wants to decide (1) whether or not the treated animals have a higher incidence or severity of the change relative to the controls and (2) to establish a NOAEL (i.e., at what dose the adverse finding is not present) or NOEL (“no observed effect level,” the lowest dose at which no treatment-related change—adverse or not—is seen). In such situations, the findings seen during the unblinded initial evaluation are used to define the grading criteria that will be utilized during the subsequent blinded examination. Blinding the slide assessment for that organ allows the pathologist to remove the possible unintentional bias of knowing which animals were treated and which animals were controls. Of course, review of specific slides in special-purpose studies, such as efficacy or mechanistic endpoint, can and should be done blindly in order to eliminate observer bias. In regulatory toxicology studies, masking (blinding) is a second-tier mechanism to clarify subtle changes in the diagnosis.
Peer Review
Because of the subjective nature of the diagnostic interpretation, the toxicologic pathology community has developed the process of formal peer review of histology slides and study data. Pathology peer review verifies and improves the accuracy and quality of pathology diagnoses and interpretations. Peer review is conducted at the discretion of the sponsoring organization, and may be planned in advance (by inclusion in the original study protocol) or undertaken retrospectively (by amending the protocol). In general, pathology peer review is recommended when important risk assessment or business decisions are based on data from nonclinical studies.
The peer review pathologist reviews sufficient slides and pathology data to assist the study pathologist in refining pathology diagnoses and interpretations before study completion. Consultations with additional experts or a formal (documented) pathology working group may be used to resolve discrepancies. The importance of pathology peer review to assuring data quality has resulted in the establishment of recommended (best) practices, which provide standardization while retaining flexibility.
Quality Assurance
Delivering defective samples to the pathologist (e.g., hemolyzed blood samples; slides with artifacts and with incomplete tissues or tissues that are unfit for evaluation due to staining, cutting, or overall processing defects) increases the time for analysis and reporting while obscuring evaluation. The pathologist, in such situations, has the further role of quality controller for the necropsy team and processing laboratory. For example, slides with defects identified by the toxicologic pathologist should be recut or reprocessed before microscopic evaluation of the tissue, while hemolysis may require remedial training of the necropsy team prior to the next study.
Adverse Events
Risk assessment requires data from toxicology studies that identify the nature of any adverse events and the doses at which they occur. Defining adverse events is core to a toxicologic pathologist’s job, though decisions regarding whether or not an effect is adverse is far from clear-cut when real data are presented. Many morphological changes (lesions) are routinely recognized as adverse (e.g., cancer, blindness, reproductive failure). Unfortunately, most treatment-related morphological changes are not obviously adverse, but nonetheless need to be addressed with respect to why they are or are not considered adverse. For example, the presence of increased liver weights as an expression of enhanced metabolic activity in hepatocytes to detoxify a NCE (i.e., “hepatic enzyme induction”) is a common event in rodent toxicology studies but seldom is observed in nonrodents (or human patients). Regardless, most toxicologic pathologists do not consider this treatment-associated effect to be adverse, even though liver cytosolic aminotransferases (e.g., ALT, AST) and other biomarkers of hepatic injury may leak into the circulation. This change becomes adverse in rodents when it interferes with critical functions or leads over time to neoplasia. As this adaptive response progresses and hepatocytes become swollen diffusely, adversity is indicated by an increased probability of liver rupture during handling of the animal and eventually increased susceptibility to liver neoplasia, both benign and malignant. A related finding is thyroid neoplasia, which is a secondary response to enhanced hepatocyte metabolism of thyroid hormone (T4) that leads to increased secretion of thyroid stimulating hormone by the pituitary gland and ultimately follicular cell hyperplasia in the thyroid gland. So, when do such findings become “adverse”?
First, an event is considered to be adverse only when there is a negative health impact (harm) to the animal that suffers the event. The presence of a detectable biomarker that demonstrates that a response has occurred is not necessarily proof of adversity. Second, the event may be adverse in one species, but not in other species where the compound is used. An example of such a finding is α2μ-globulin nephropathy, in which exposure to certain agents causes accumulation of hyaline droplets in renal proximal convoluted tubules only of male rats leading to renal dysfunction and neoplasia. Therefore, the designation of adversity is limited to the specific conditions of a given study in which the adverse event is found, and is not relevant to other species, lengths of treatment, doses, etc. Third, it is unacceptable to report that a finding is not adverse in a test species based on the known lack of applicability to human beings (as is the case with enzyme-induced liver hypertrophy and α2μ-globulin nephropathy, which are limited to rodents and may be adverse in these animals). Finally, data regarding morphological changes should not be extrapolated based on an assumed (known) course of future lesion progression.
Reversibility (Recovery)
In general, a reversible lesion is considered to have a lower risk than a nonreversible one, which may also be progressive. In many cases, the pathologist can address whether or not the given change is generally accepted as reversible and can even suggest what amount of time would be required for its reversal. Recovery must be assessed in the context of the treatment and the incidences of incidental findings. For example, some treatments may modify the pattern of age-related background changes (e.g., CPN, thymic involution), but such effects may not be reversible as the animals continue to age throughout the study.
For certain changes, the potential for reversibility is not very well understood (e.g., some new compound-induced changes through novel mechanisms), so there is a need to document whether and when the change resolves. How to document the reversibility of findings in toxicology studies that have a limited preplanned time course becomes an issue. One common approach is to increase the number of animals in each treatment group [generally by 50% (five animals per sex in rodent studies and two animals per sex in nonrodent studies)], with the extra animals allocated to the recovery necropsy.
Determining the numbers of animals dedicated to assessment of reversibility is tricky. If the morphological change occurs in 100% of the animals in the high-dose group, a few additional animals will suffice. In contrast, if the incidence is low (e.g., 1 out of 10 animals shows the change) and there is no reliable biomarker of the compound-induced effect, reversibility cannot be determined using only a small number of additional animals as there may not be any evidence that the animals expressed the lesion at the end of dosing. Hence, one cannot say that the change “reversed,” as one cannot determine if it was there in the first place. The practice varies among institutions; some study directors explore reversibility only in the high-dose and control groups, while others use recovery animals in all dose groups to find out at what dose level the change becomes reversible. In general, the reversibility groups are kept without dosing for a specified time following compound exposure—usually the same duration as dosing for short-term (4-week) studies, and shorter than dosing (one-third to one-half) for longer studies.
Deciding on the appropriate length of time to conduct the recovery necropsy can impact the evaluation of reversibility. Even short-term studies might have changes that require an extended time for recovery; hence, a 2-week recovery period may be insufficient to determine whether or not reversibility occurs at all, let alone occurs completely. If there is a biomarker that can be used to predict the presence of a morphological alteration, the length of the recovery period may be adjusted during the course of the study, until the biomarker comes down to normal levels. If no biomarker exists, and if the time of recovery necropsy is too early, lack of reversibility (or at best incomplete reversibility) may be evident. Risk managers often interpret this scenario as lack of reversibility. General practice for assessing reversibility is to have the pathologist review only tissues identified as targets during tissue evaluation for the terminal necropsy (i.e., encompassing the treatment period, and ending in the scheduled terminal necropsy), as the main study has already identified that there are no test article–related effects in nontarget organs.
Severity Grading
Toxicologic pathologists use severity grades to score changes observed. Such grades are semiquantitative in nature and generally follow a 4-point to 6-point scale, in which a term for lesion severity (ideally based on a set of specific descriptive criteria) is matched to a number (to permit statistical calculations). Possible ways to define such scales are:
• 4-point scale: WNLs (Within Normal Limits) (0), minimal (1), modest or moderate (2), and marked (3) changes;
• 5-point scale: WNLs (0), or minimal (1), mild (2), moderate (3), or marked or severe (4) changes; or
• 6-point scale: WNLs (0), or minimal (1), mild (2), moderate (3), marked (4), or severe (5) changes.
The only way that these diagnostic terms have any relevance is if the pathologist follows an a priori system that is preferably written and somewhat universally understandable before tissue evaluations start. The choice of scale (including the terms used for each grade) typically is decided by the pathologist, the 5-point scale is probably the one that is used most. For commonly seen lesions (especially if they occur reliably in a given animal model), pathologists are encouraged to either follow recognized scales gleaned from the literature or base their own scales on objectively defined criteria tempered by prior experience—and ideally publish them, or at least define the scale in the study report.
The severity grades should be independent of the study, and applicable to any lesions observed. For example, minimal severity may be defined as a change that is barely discernible from background morphology or that involves less than 10% of the organ surface and is not expected to affect the functionality of the organ. In contrast, a severe score may be defined as a visually apparent change that involves 75% or more of the organ surface and is expected to affect the functionality of the organ. The other severity grades are placed somewhere in between these two extremes, and should be divided into logical sections using recognizable structural attributes (the character of the finding, the number of affected cells, etc.).
As there is no universal system of severity scores to date, it is important to ask colleagues for the systems that they use when faced with devising a novel scoring scheme of your own. It is also essential to remember that these agreed-on severity grades constitute ranges of morphological features, which may include changes that are visibly distinguishable within the different grades, but that are not distinct enough to warrant a change in the severity grade. The adoption of severity grades based on the changes present in the individual study, by adopting the highest score for the most severe lesion observed in that study, leads to complications because the scale would be unique in different studies of the same compound, and therefore not universally understandable. The severity grade descriptors and the criteria used to define them should be part of an institutional standard operating procedure (SOP) or listed in the pathology report.
What Constitutes a Risk—Is the Risk Real?
When encountering an adverse effect, the development management team should make sure that the effect is “real” before making conclusions regarding safety and efficacy. Poorly designed studies can endanger the development plan and may lead to radical and perhaps rash decisions by the team. The following factors need to be considered when determining the implications of treatment-related effects.
Statistical Versus Biological Significance
Absolute reliance on statistical calculations does not address the biological relevance of the statistically significant effect recorded using multiple stars in pathology data tables. Given the large number of statistical analyses performed in nonclinical studies, and given the inherent physiological variation among members of the heterogeneous sample population, it is not unusual to see statistical significance in parameters that may have limited biological significance. These findings may be a statistical artifact (Type I error erroneous rejection of a “false positive” result). Statistical artifacts often are seen in clinical pathology and organ weight datasets.
Is the “No-Effect” Outcome Real?
The toxicologic pathologist needs to remember that the absence of evidence does not constitute evidence of absence. Failure to show a statistical difference for a biologically evident effect, referred to as a Type II error (erroneous acceptance of a “false negative” result), may occur if the power of the study (i.e., animal numbers in each treatment group) was inadequate. Therefore, the process of analyzing data from a study should first check that the computer-generated statistical probability for a given finding is consistent with its potential biological impact. This exercise requires an understanding of the design, execution, and objective results of the study, ultimately culminating in a part-objective/part-subjective segregation of the biologically important results from the spurious ones.
Inconsistent Nomenclature
Since pathology is an interpretive science, there is a high degree of variability in the application of diagnostic terms. Using incorrect nomenclature when describing treatment-related findings can either raise a false risk or ignore a true one. Misdiagnosis of findings can occur due to inexperience of the pathologist. Diagnostic drift (i.e., assigning two or more diagnoses or inconsistent severity to identical lesions during the course of a study) is an error sometimes made even by experienced pathologists. Pathologists should employ considerable care in avoiding these problems when evaluating nonclinical studies including masked review.
The discipline of toxicologic pathology provides two solutions to this problem. The first is the production of consensus terminology. The most consequential effort in this regard is the INHAND initiative (International Harmonization of Nomenclature and Diagnostic Criteria for Lesions), in which pathologists with organ-specific expertise from around the globe have defined descriptive terms and criteria for nonproliferative and proliferative lesions. The second solution is the introduction of pathology peer review as a routine component of the safety assessment process.
Splitting Diagnoses
The perils of dividing complex lesions (e.g., CPN or murine progressive cardiomyopathy) into separate diagnoses for each of the specific components (e.g., renal tubular degeneration, renal tubular regeneration, interstitial inflammation, interstitial fibrosis) have been mentioned earlier, but it warrants mentioning again. The practice may lead to identification of phantom issues that either will require correction during data interpretation or may obscure possible treatment-related effects characterized by specific induction of a related change (e.g., isolated interstitial inflammation or fibrosis in the absence of CPN).
Compound-Related Versus Compound Effects
Not all changes observed in toxicologic studies are a toxic reaction due to the compound. A compound can have an effect that is deleterious to the animal, but this effect is an expected exaggeration of the intended “on-target” biological effect (referred to as “exaggerated pharmacology”). In terms of toxic off-target responses, some effects are primary (caused by the compound directly), while others are secondary (caused by complications from a primary effect).
Exaggerated Pharmacology
Since toxicological studies are designed to expose individuals to doses of compounds at many multiples of intended therapeutic doses, the organism may show adverse effects due to excessive activation of the test article’s molecular target. Such predictable but heightened effects are not expected in the clinical setting, or at least not with the same severity. Of course, product developers are accustomed to extrapolating safety margins for anticipated physiological reactions, using data from nonclinical studies to estimate the potential response of tissues having a comparable molecular target following treatment of patients with the test article in the clinical setting. A possible problem occurs when such pharmacologic changes in humans may be new and/or unexpected and/or persistent. For example, it is very difficult to find a NOEL in glucocorticoids because these compounds impact endocrine and immune organ function at therapeutic doses. When given to excess (in terms of either dose or treatment length), these pharmacologic effects are adverse even though they are an expected consequence of glucocorticoid exposure.
Primary Toxicity
These effects can be ascribed to direct action of the test article. Off-target actions are effects on tissues or molecular pathways where the compound acts at an unintended site. The concept of an off-target effect implies that the test article interacts with a new target that is not the originally intended one, that the distribution of the original target was not well understood, or that the target has a different distribution in the toxicology test species. These off-target effects can be identified early during development by assessing expression of the pharmacologic target. Such expression studies (usually done for the functional protein) are essential components of selecting among possible lead candidates.
Secondary Effects
These outcomes are a very common reason for adverse compound effects, some of which can be devastating. These effects are indeed compound-related, but they are a downstream effect from an initial impact of the compound on systems that lead to a cascade of follow-on effects. It is not unusual that secondary effects will lead to major organ dysfunction and mortality, yet frequently they are confused with direct (primary) effects of the test article. Perhaps the most common secondary effect of compound exposure is “stress,” which is often definitely related to test article concentration and occurs in a dose-dependent fashion. Consequences of compound-related stress are deficient immunologic function (via the lympholytic action of glucocorticoids in the circulation and lymphoid organs), listlessness, and a general feeling of discomfort, which may depress the appetite, lower food consumption, and promote weight loss. These outcomes may be mediated by release of cytokines and chemokines. Once listlessness and low appetite manifest, the situation can intensify as indicated by increasing severity and incidence in the affected groups related to the release of endogenous glucocorticoids and other hormones (e.g., catecholamines), which in turn result in catecholamine-induced focal myocardial necrosis (termed “murine progressive cardiomyopathy”) or even sepsis and death.
More drastic immunosuppression has pronounced effects on the ability of the organism to fight opportunistic bacteria or viruses, so that generalized infection (sepsis) and later death may follow. In these situations, it is imperative to determine whether or not the immunosuppressive effects are solely the result of stress or alternatively a component of immunotoxicity caused by the compound. Similarly, septicemic conditions can affect multiple organs such as liver, lungs, and kidneys, although these infected sites are not considered “target” organs of the test article.
Risk Perception and Acceptability
Perception of risk varies considerably among different populations and the conditions under which the perceived risk has occurred. Cultural norms in the society where the risk is assessed influence its perception, and the decisions that people take to decrease this risk. The accurate assessment of risk is often difficult because it requires the weighing of multiple factors that may combine in variable fashions to produce an adverse effect. Two of the most important characteristics in evaluating risk are assessment of its probability and of its severity. Ultimately, communities must decide what level of risk is acceptable. Negation of all risk is very difficult to achieve (if possible at all), and is associated with rapidly rising costs for relatively small gains.
Acceptability of the Risk
Perception of risk is a subjective, individual-specific process that is essential for survival. Scientists are often surprised by the reaction of various audiences to a potential risk. In some cases, findings that are thought to be of limited biologic relevance end up causing great concern from colleagues within the laboratory, regulators, or the general public. This diversity of perspective is better understood when we look at a number of factors that affect the ways in which different people under differing conditions perceive risk, which impacts whether or not they are willing or not to accept that risk. Some common attributes of the risk that influence the perception of its acceptability are outlined in the following sections.
Likelihood (Probability) of the Adverse Effect
The likelihood of a risk being realized as an adverse effect is one factor that affects how that risk is perceived. In many cases, risks that have a very small likelihood of affecting a person will be perceived to be less important than risks that are more likely to impact their health and life. However, some extremely unlikely effects may be deemed unacceptable due to the other factors discussed later. To further complicate the matter, some relatively large risks (e.g., dying of a smoking-related illness, or dying in a car accident) are perceived by many as being relatively acceptable to society as a whole since the decision to smoke or drive is an individual choice that incurs individual responsibility for the outcome.
The toxicologic pathologist is often ideally suited to help inform our communal understanding of the relative likelihood or severity of an adverse effect. In some instances, a specific morphologic diagnosis taken at face value may sound ominous, but when put in context with the relative severity of the effect, margin of safety, and other information, rational consideration permits the decision that the seemingly menacing risk is acceptable. An example of this scenario is use of Non-Steroidal Anti-inflammatory Drugs (NSAIDs). There has been mounting evidence that the use of these products for the long-term treatment of chronic pain may increase the probability of significant cardiovascular events. However, the risk is marginal and the prospects of living with crippling chronic pain make people disregard the small probability of increased cardiovascular events and continue taking the medication.
Permanent or Reversible Nature of the Adverse Effect
Different types of risk are usually perceived differently, and for obvious reasons (e.g., cancer vs transient nausea). Most people are more willing to accept the risk of effects that are relatively minor, and reversible or temporary (e.g., nausea). On the other hand, they are less likely to accept an increased risk of serious negative effects that are less reversible or lethal, and that a serious negative impact on the quality or length of life (e.g., cancer). This concern highlights the importance of understanding the reversibility of key test article–related toxicities.
Risk/Benefit
Knowledge of the severity of the adverse effect is required for accurate assessment and management of risk. There are multiple examples of acceptable risks in daily life (e.g., driving), and the development of new products is no exception. If the likelihood or impact of an adverse effect (risk) is of less concern than the test article’s ability to reduce or cure a disease (benefit), the existence of an adverse event may not prevent the approval and use of the treatment. The balance between risk and benefit will depend on the severity of any adverse event. For example, if the probability of a severe reaction is low (1/1000) relative to the devastation produced by a disease (say, 20%), the occurrence of the severe adverse reaction may be catastrophic to that one individual in a thousand. This possibility therefore creates ethical complications with respect to what risks should be taken by the many, and how to avoid the adverse event in the one.
Who Benefits From Acceptance of a Risk?
People who may benefit from accepting a risk tend to be more willing to tolerate that risk than people who do not benefit. This becomes particularly important when the people who face the risk (e.g., residents who live in an industrial area affected by factory emissions) are different from those who stand to reap the greatest financial benefit (i.e., the owners and operators of the factory). In this case, even very small risks may be completely unacceptable to nearby residents, due to the lack of perceived benefits. This concept highlights a key difference in the approach required for a toxicologic pathologist engaged in environmental risk assessment compared to a toxicologic pathologist involved in safety assessment for biomedical products (drugs, devices, cell therapies, etc.). In drug safety assessment, prescribers and patients typically are willing to accept certain risks given the benefit that the patients expect to receive by accepting the intentional exposure.
A fundamental challenge to risk communication is the fact that actual risks that bring harm are often completely different from the perceived risks about which the public is concerned. This dilemma has been highlighted by Covello and Sandman (2001) (see Further Reading), who define “Risk” as the sum of “Hazard” and “Outrage.” In their paradigm, “hazard” represents the data-driven assessment a scientist would use to identify and understand potential harm while “outrage” represents those factors, often emotional, that influence the public’s perception of risk. Positions held by the lay public, which trained scientists would view as irrational fears, may primarily be due to a lack of context for the general citizenry. For example, consider a molecule that, when inhaled in sufficient quantity, is fatal, and where accidental inhalation resulted in over 3000 deaths in 2007. Without appropriate context, the public might consider this molecule—common everyday water!—a highly toxic substance. Thus, to effectively communicate and manage risk, toxicologic pathologists need to understand that an individual’s perception of risk goes far beyond the data-driven, reasoned analysis that governs scientific dialogue. Understanding the emotional factors that influence how a risk is perceived will aid the toxicologic pathologist to more effectively communicate and manage risk.
Risk Communication
When communicating potential risk to different audiences, it is important the discourse be conducted as thoughtfully and rigorously as the experiments which provide the scientific basis for the risk assessment. The effectiveness and impact of a rigorous safety assessment program can be markedly diminished if the risks are not portrayed in a clear and concise manner. Communicating sensitive, technical information to a nonscientific audience can be one of the most difficult challenges that a scientist faces today. This is especially true for communicating with the general public given the public’s growing distrust of corporations, academic institutions, and government agencies charged with protecting the health and safety of our communities.
Risk communication has developed into a distinct discipline, so the toxicologic pathologist should have a basic understanding of certain key principles in this field. The following definition of “risk management” developed by the National Research Council (NRC) and US EPA provides useful insight into this field, which is relevant to individuals in both environmental risk assessment and safety assessment of novel biomedical products. According to these two groups, “risk communication is an interactive process of exchange of information and opinions among individuals, groups, and institutions.” The US EPA acknowledges specifically the key role of risk communication in the risk management process, stating that risk communication is “any purposeful exchange of information and interaction between interested parties regarding health, safety, or environmental risks.” In addition, the US EPA has defined “Seven Cardinal Rules of Risk Communication” outlining the critical steps to effective dialogue with the public. These points are:
1. accept and involve the public as a legitimate partner;
3. be honest, candid, and open;
4. coordinate and collaborate with credible sources;
5. speak clearly, concisely, with care and compassion;
6. plan carefully, and evaluate your efforts continually; and
While these steps are focused on information being delivered to the lay public, they are useful to consider when communicating risk to scientific audiences as well (including decision makers like managers and regulators).
Challenges and Obstacles to Effective Risk Communication
To effectively communicate risk, it is important to be aware of various issues that complicate such communications. Overcoming these obstacles will help create a “common language” that can be understood by both the communicator and the intended audience(s).
Data Versus Information
Scientists are trained to deal effectively with data. Experiments are designed and conducted; the data are collected, organized, and analyzed; and the investigator reaches a conclusion about the meaning of the data. In other words, a “data” set is a collection of single observations that, when synthesized, provides “information.”
The public and other lay audiences are at a disadvantage compared to scientists when it comes to interpreting data. They often do not have the technical knowledge to synthesize data into information, let alone understand the meaning of a single data point. This lack of understanding can lead to misinterpretation of data, or to the inappropriate focusing of attention upon a single experimental result—both of which may lead to an incorrect interpretation regarding potential risk.
More data may be perceived by nonscientists to mean “more concern.” Therefore, the public should be provided with information in understandable language, and not just data. For a lay or public audience, it is important to avoid presenting excessive amounts of data when summarizing risks, particularly at the expense of providing integrated information. Effective communicators understand the needs of their audience, examine their data carefully, and present the data in a manner that provides information in language that is accessible to their audience. Accessibility of the language is key: information that is presented in unintelligible language does not impart knowledge, does not empower involvement in decision-making, and instead may be perceived to be a tactic designed to hide or conceal important risks from the audience. This dilemma can be a particular concern for toxicologic pathologists in that, for both scientific and lay audiences, the “language” of pathology (with its diverse array of diagnostic and descriptive terms that are assigned by subjective criteria) can often be confusing at best, and misleading at worst.
Public Expectations of Science
A scientist must communicate with many of the “lay” audiences, which may have unrealistic expectations regarding what science can provide and also preconceived notions regarding scientists in general. The media frequently report scientific failures and impending threats, such as the withdrawal of a marketed drug due to previously undiscovered side effects or the discovery of chemical contaminants in drinking water that may cause cancer.
At the same time, people seem to have serious doubts about the positive role of science in their lives. Many members of the lay public consider scientists to inaccessible (at best) and/or aloof. This impression is often a result of the inability, or unwillingness, on the part of scientists to learn and use clear, simple language to explain what they do, how they do it, and what they have found. It is not surprising that scientists have difficulty in meeting the public’s expectations of science: the public desires simple and absolute answers, while given complex interacting factors in the real-world science can generally only identify likely and unlikely outcomes but not what is guaranteed to happen. Thus, scientists resist distilling information down to simple “yes” or “no” answers, preferring instead to present probabilities and theories while recounting all the evidence in support and against them. This is an important disconnect in communication between technical experts and the public. The public understanding of the word “safe” as in categorizing the probability of a compound to produce adverse effects is simpler than the meaning of “safe” as understood by scientists. The public expectation is that a “safe” product has no deleterious effects, but the scientist knows that the word safe is just the beginning of a sentence that describes when, how, and in what circumstances the compound can be given to avoid adverse events.
Contradictory Expert Opinions
The scientific method is rooted in hypothesis testing, in healthy debate that validates interpretations of data through experimentation, and through reasoned argument about alternative explanations. Interpretations that withstand this rigorous scrutiny become established as the most plausible explanation, and become a platform for further expansion of knowledge. Unfortunately, the public rarely has an opportunity to see this constructive and (usually) collegial process, and do not comprehend how expert scientists can interpret the same objective dataset differently. This dynamic is particularly relevant to anatomic toxicologic pathologists in that histopathology is an interpretive science based on assigning subjective diagnostic terminology and thresholds; differences among pathology practitioners create confusion and, at times, distrust of the dataset and final interpretation. To forestall such objections, pathologists should seek to deliver peer-reviewed, consensus datasets rather than disparate subjective opinions on the effects of a test article.
Risk Management
Risk management encompasses the systematic scientific identification, evaluation, and prioritization of risks with respect to adverse health effects resulting from human or environmental exposure to hazardous agents or situations. The goal of risk management is the economical application of finite investigative and corrective resources to minimize, monitor, and control the probability and/or impact of the adverse events. The positioning of the toxicologic pathology findings is an important aspect of risk management.
Risk management practices differ depending on the context. In the case of pharmaceutical products, the risks are managed by making sure that the benefits outweigh the risks and that the individuals who are to be exposed to the NCEs understand the risks inherent to receiving the intended exposure (a process designated “informed consent”). A similar situation applies to the chemical industry, except that the potential risk is determined primarily by the likelihood that exposure will cause harm rather than the benefit offered by use of the product. The reason for the difference in regulatory approach to risk in these two industries is the management of exposure. Health products represent a risk that the individual takes knowingly and voluntarily, whereas environmental exposure to chemical hazards typically is involuntary and often unexpected.
The US FDA has defined risk management activities in the following progressive fashion:
• Risk Assessment: Estimation and evaluation of risk.
• Risk Confrontation: Determining an acceptable level of risk in a larger context.
• Risk Intervention: Risk control action.
• Risk Communication: Interactive process of exchanging information.
• Risk Management Evaluation: Measuring and ensuring effectiveness of risk management efforts.
Risk impact is evaluated as the probability (likelihood) of an event occurring, and the severity of that event if it occurs. An adverse event can be considered to have a high probability but low impact (when it will occur often but with little effect), or such events may have a low probability but high impact (when the effects of an event are high but the likelihood of it occurring is low). Differentiation between these two scenarios is essential in communicating risk, as they should be managed in different fashions.
Problems in Risk Management Caused by Toxicologic Pathology Data
Ineffective Reporting
Reports (both for the entire study and for its various component parts, including the pathology subreport) fail when they just enumerate findings based on the statistics and incidence tables without providing a cohesive interpretation for the reader. Frequently, the study report’s narrative may fail to address the relationship of different compound-related effects to each other and the potential mechanisms responsible for their evolution. Ineffectual reports may be worse than useless, because they not only fail to provide a usable interpretation but also may obscure findings of potential importance to the risk assessment and management activities.
Lack of Data Cohesiveness
It is essential to find the underlying causes and biological significance of the effects observed, not just provide a list of these effects. Failure to provide an expert evaluation of all relevant information to support interpretation of findings may result in inaccurate assessment of risks. The lack of cohesive interpretation regarding the biological relevance by a toxicologic pathologist may create difficulty for the development of the test article and cause confusion for scientists who are not trained in comparative pathology, whether these scientists are at drug and chemical companies, serving as managers, or in regulatory agencies. It is also important to expressly address the kinds of background lesions observed and their lack of relevance to the evaluation of the specific compound as a way to clear confusion from reviewers, who are often not versed in the normal laboratory animal findings.
Species Relationships: Is the Animal Finding Relevant to Humans?
Overprediction
Although animal studies are an important part of understanding the nature of toxic effects, their use to assess potential risk to humans has met with qualified success. In addition, assessment of human risk continues to be a challenge due to the increased complexity of the pharmaceutical and chemical compounds under development. The perils of extrapolation are not only based in the inadequacy of many animal models, which incompletely recapitulate human biological responses, but also in the need for accurate, enlightened, and unbiased evaluation of the data arising from animal models.
For example, it is clear that animal models overpredict carcinogenic risk to humans, particularly from exposure to nongenotoxic compounds (i.e., agents that cause parenchymal cells to proliferate). The ability to induce tumors is often based on mechanistic responses that are either not present in human subjects (e.g., α2μ-globulin nephropathy) or may occur at exposure levels, which exceed the human dose by such large multiples that it would be highly improbable that such compounds will be able to do harm to an individual. Compounds categorized as nongenotoxic carcinogens in rodents, which exhibit florid proliferative responses in liver, are considered to have a negligible risk of inducing neoplasia in humans, because human liver does not respond to exposure with excessive proliferation to the same extent as rodent liver. In contrast, both rodents and humans exhibit sensitivity for genotoxic carcinogens (i.e., agents that initiate mutations by damaging DNA).
Underprediction
Some conditions are not easily addressed by animal models. For instance, the presence of hives and other cutaneous hypersensitivity reactions in humans are underpredicted by animal models. Similarly, developmental toxicology studies in rodents using thalidomide (marketed in Europe as an over-the-counter antinausea therapy for “morning sickness” of pregnancy) failed to predict the extreme sensitivity of the limb primordia in human embryos, leading to an epidemic of phocomelia (lack or underdevelopment of the proximal limbs). Instead, such species-specific differences in adverse reactions often are better addressed by specialized testing using human tissue samples or human volunteers. Notably, the regular battery of nonclinical toxicology studies utilized for development of compounds do not often alert the scientist to this and similar risks. The possibility that datasets derived from animal experimentation do not predict human biological responses emphasizes the critical importance of risk assessment and risk management.
Managing the Identified Risk—Remediation
Treating the Adverse Effect
If the effect is real and if there are health risks to the patient, understanding of the condition that is produced by the compound, regardless of how the product is to be used, is essential. One sometimes can avoid the initiation and progression of an adverse event by coadministration of the compound together with some treatment to mitigate or prevent the adverse effect. For example, coadministration of antiemetics with cancer treatments is used to remediate nausea and vomiting. Similarly, probiotics (i.e., living bacteria or yeast extracts) are commonly coadministered with oral antibiotics to prevent and treat the development of diarrhea that is associated with their use. Thorough understanding of the mechanisms present in the induction of the adverse event is necessary to formulate a treatment strategy for this kind of remediation.
Remediation of Biological Risk
Some severe adverse effects do not necessarily impede development and marketing of a product due to a perceived advantageous risk/benefit profile. The US FDA defines a “safe” product as “one with reasonable risks given the magnitude of the expected benefit and the available alternatives.” This definition means that there must be an acceptance of reasonable risks, based on the benefits that the patient and society will accrue from the use of the given substance; no pharmaceutical agent could be developed if one were to insist on avoiding all risks. The repurposing of thalidomide, a most feared teratogenic compound, for use in the treatment of erythema nodosum leprosum in nonpregnant human patients by careful monitoring and licensing of distribution, is a good example that a compound’s toxicity depends on the context in which exposure is to occur. Indeed, many compounds can be safely administered when the mechanisms of toxicity and the conditions of exposure are well understood.
Financial Risk and Toxicologic Pathology
The pharmaceutical, chemical, and food additive industries are businesses, and as such must produce a profit for survival. This financial motive must be integrated with the expectation that product development decisions must meet society’s need for efficacious and safe solutions regarding the health and other needs of its members. In order to attract operating capital, these industries need to convince their investors that the money they pay for their stock will produce more revenue than a similar amount of money invested in other opportunities. This reward is linked to success in developing new products, and in particular unique and/or better products that offer greater benefits or a better risk/benefit ratio to individuals and society as a whole. Discovery and development programs leading to approval of one new product may cost hundreds of millions or even billions of dollars, the cost of which must be recouped if more products are to be produced in the future. As the patents for old products expire, the reduced sales typically associated with competing products from other firms will impact the revenue that the companies can return to their investors. Although there is altruism in the search for new and better medications for patients, the bottom line is that companies engaged in product development cannot survive unless profits are made.
Risk of financial damage by errors of commission (i.e., taking an ineffective or mistaken action) often reflects the identification of false signals or the overconservative interpretation of the data. Errors of commission are not unusual, and typically are more prevalent in small companies and with test articles that are very early in development. Due to the fear of losing the compound later in development and interest in cutting financial losses early, compounds often are removed from the development pathway before enough data have been collected to rationally make a reasoned decision about its viability. For example, the history of the pharmaceutical industry is replete with compounds abandoned due to perceived risks, as well as with the ones that were successful because health risks were effectively managed. Many innovative “first-in-class” (FIC) compounds are at the verge of being dropped due to difficulties in the development program. Since the mechanism of activity is novel, FIC compounds generally face new problems that require considerable expenditure of funds and technical resources to be understood. Only by having a good grasp of these issues, gained in large part by toxicologic experimentation, can enough understanding be obtained to permit product development to proceed.
Risk of financial damage by errors of omission (i.e., not gathering certain data and/or taking timely action) corresponds to making the decision to avoid obtaining critical toxicity data in order to maintain the compound in the development portfolio. Obtaining relevant information for reasoned decision-making is necessary so that the compound may be placed in the appropriate relation to other competing development opportunities. However, gathering such data is risky; frequently, companies choose not to seek nonclinical data because it could be harmful to the compound’s success, or there is financial reward (i.e., bonus payments) in keeping several compounds in development at the same time. This approach consumes resources in the short term and, more critically, moves the critical decision regarding a compound’s fate to a later, and costlier, stage of development, thereby magnifying the cost of failure. Later in development, companies have a stronger commitment to work on elucidating and explaining away (or mitigating) adverse findings to ensure a path forward for late development compounds. This strategy reflects the prior financial cost, which cannot be lightly cast aside, but risk mitigation as an alternative to “killing” (ceasing development for) a compound is ethically and morally acceptable if sufficient efficacy and safety data are available to ensure that the molecule—a costly asset to the company—has a chance to continue in development and thereby benefit both society and the corporate coffers.
Societal costs occur when compounds helpful in solving health needs do not get developed, or when the public may be put at risk of adverse effects by exposure to potentially hazardous products. It is difficult to articulate and measure societal costs associated with halting development of a pharmaceutical candidate or chemical agent (e.g., herbicide, insecticide). Indeed, this cost is generally ignored.
The contribution of pharmaceuticals and chemicals to society is best understood by comparing the mortality rate of populations before and after the introduction of specific products. Obvious examples in the biomedical arena are the increase in longevity and the decrease in mortality due to cardiovascular disease and many infections that have been wrought since the introduction of compounds to treat hypercholesterolemia and routine childhood vaccination, respectively. Similarly, longevity and mortality have been positively impacted by the regular utilization of selective herbicides to increase crop production. It is clear from epidemiological studies that society at large has benefited substantially from the use of these products, certainly in comparison to the possible risks posed by unintended exposure of nontarget human and animal populations and their habitats. However, it is difficult to assess the societal cost of the lack of treatments for conditions for which there are no treatments or for which the treatments are inappropriate.
Summary
The critical role of the Toxicologic Pathologists in product development in the pharmaceutical and chemical industries is based on the training and experience of the pathologist to interpret morphologic and biochemical findings observed in animal studies that are preliminary to human or environmental exposure to the same compounds. Pathology is an interpretive science melding a practitioner’s objective observations with his subjective judgments, and as such does not have the rigor of the exact numerical determinations of other disciplines. This inherent flexibility in pathology interpretations creates an opportunity for errors and differences of opinion on the data resulting from these studies, and can produce damage to patients and product developers if the findings are not accurately evaluated. Understanding of the risks (health and financial) of the development process is of great benefit to the toxicologic pathologist and the compound development teams. The way that the pathologist perceives, interprets, and communicates the observed effect of the compound has a direct effect in regulatory acceptance or rejection of the compound for human experimentation or marketing authorization. This perception needs to be consistent with the highest standards of scientific discipline, and often is bolstered by seeking a consensus interpretation among multiple pathologists through rigorous peer review. Following the generally accepted nomenclature, application of the principles of scientific communication in presenting and interpreting data to the sponsors and regulators, and clear and comprehensive evaluation of the findings is the first step in the process. Participation in the early development programs to address efficacy in animal models, interaction with clinical pharmacologists to make sure that the nonclinical data are appropriately interpreted, addressing adverse events and developing hypothesis to be tested in elucidating the causes and mechanisms involved in the adverse event, and finally developing a strategy to manage and remediate the adverse events are important aspects of the practice of Toxicologic Pathology discussed in this chapter (Figure 7.1).

Acknowledgments
The author acknowledges the contributions of Drs. Colin Rouseaux, Stephen Durham, James Swenberg, and John Vahle for the materials from their chapters that are included in this chapter.