Skeletal Muscle System
Brian R. Berridge1, Brad Bolon2 and Eugene Herman3, 1GlaxoSmithKline Research & Development, King of Prussia, PA, United States, 2GEMpath, Inc., Longmont, CO, United States, 3National Cancer Institute, Bethesda, MD, United States
Abstract
Skeletal muscle is a significant contributor to overall body mass and an important structural component of the mammalian body, primarily supporting conformation and locomotor activity. Like cardiac muscle, skeletal myocytes are energetically dynamic. Xenobiotic-induced injuries to the cellular components of the skeletal muscle system, though not as immediately life-threatening as injuries to the heart or other vital organs, can have profound effects on overall health and well-being. Accordingly an understanding of the components of the skeletal muscle system as a target of toxicity and their responses to injury is important. This chapter reviews the structure and function of skeletal muscle system components, contemporary methods for detecting and characterizing injuries, and current mechanistic understandings regarding how skeletal muscle injuries are initiated and sustained.
Keywords
Skeletal muscle; muscle; myotoxicity; myonecrosis; myopathy; regeneration; sarcolemma; statin; myodegeneration; neuromuscular; phospholipidosis
Skeletal muscle is a unique tissue with the role of support and movement of the body. The mass of the skeletal musculature is large, and constitutes 40%–50% of the total body weight. Recognition of gross pathologic alterations in muscular diseases is complicated by the normal variation in color and consistency observed among the muscles of various animal species and animals of differing ages. Detailed study of the skeletal musculature during routine necropsies generally is not performed. In fact, prosectors often overlook examination of the skeletal muscles in their haste to expose and observe the visceral organs.
Skeletal muscle fibers have unique structural features. Each fiber represents a syncytium formed during myogenesis by fusion of hundreds to thousands of individual myoblasts. The fibers are highly specialized, with approximately 80% of their cell volume occupied by contractile elements. Normal function of these fibers is dependent on normal nervous system function to initiate contraction, and further involves intricate control mechanisms to direct movement of calcium ions in and out of the sarcoplasmic reticulum (SR) during the contraction–relaxation cycle. Muscle fibers have high energy requirements, often needed on short notice; this metabolic feature may predispose these cells to injury from certain chemicals and drugs. Use of histochemical staining procedures allows recognition of several fiber types, each with unique metabolic and functional features that may render a specific population of fibers susceptible to certain insults. Critical nutrients needed to maintain the structural integrity of muscle fibers include adequate dietary intake of selenium, vitamin E, and protein. Thus the skeletal muscle fiber possesses a number of unique metabolic and structural features that will influence the pathological reactions observed in skeletal muscle diseases.
Structure and Function
Anatomy of Skeletal Muscle
Skeletal muscle arises from mesodermal somites in the embryo during the first trimester of gestation. The somites give rise to myotomes, sites where embryonic muscle cells or myoblasts aggregate, that roughly correspond to the segments of the vertebral column; each somite receives a spinal nerve. Skeletal muscles of the adult will often contain muscle fibers from several myotomes following migration and fusion of embryonic myoblasts, and thus will also receive nerve supply from several myotomes.
Myoblasts, the primitive mononuclear precursor cells of skeletal muscle fibers, elongate and fuse with each other to form myotubes. These cells rapidly evolve to form the early cytoplasmic components of mature muscle cells by production of thin (actin intertwined with troponin and tropomyosin) and thick (myosin) myofilaments and Z-band material that aggregate into sarcomeres; the sarcotubular membrane systems that propagate depolarizing ion gradients into the myofiber interior are also formed at this stage. Myotubes subsequently fuse with each other, and sarcomerogenesis continues as nuclei migrate to the subsarcolemmal positions. Finally, myofiber innervation occurs and individual fibers become organized into small groups for coordinated contractile function. Further growth of these fibers in width and length occurs during fetal life. Increased numbers of muscle fibers are produced by waves of growth in late gestation. This expansion is presumed to be the result of proliferation and activation of satellite cells to form new myofibers, which recapitulate the events of myoblast-associated muscle fiber development and maturation as just described.
Skeletal Muscle Fibers. Individual muscle fibers generally extend linearly from tendon to tendon in a muscle and do not branch or form syncytia. In cross-sections, skeletal myofibers have a polygonal or multifaceted shape in muscles of adults. Many factors, such as species, breed, age, sex, body weight, nutritional status, position and function of the muscle, and exercise, influence the diameter of muscle fibers. Measurements of fiber diameters in individual muscles will vary in size as a bell-shaped curve on a histogram. Differences in fiber size in various species are not directly related to body weight (typical fiber diameter: pig>human, cow, rabbit, dog, rat>mouse). Fiber size is greater in males than females, and tends to increase with age to maturity. Fiber size decreases with senescence.
The cellular features of skeletal muscle fibers are best appreciated in longitudinal sections. The fibers are bounded by the plasma membrane or sarcolemma, which is covered by an external lamina that is stained by the periodic acid-Schiff (PAS) reaction. Myofibers contain multiple thin, elongated nuclei, which are generally positioned immediately beneath the sarcolemma in a spiral pattern spaced 10–50 μm apart along the entire myofiber. At myotendinous junctions, however, muscle fibers have numerous centrally located nuclei. Nuclei of satellite cells are positioned between the sarcolemma and the external lamina.
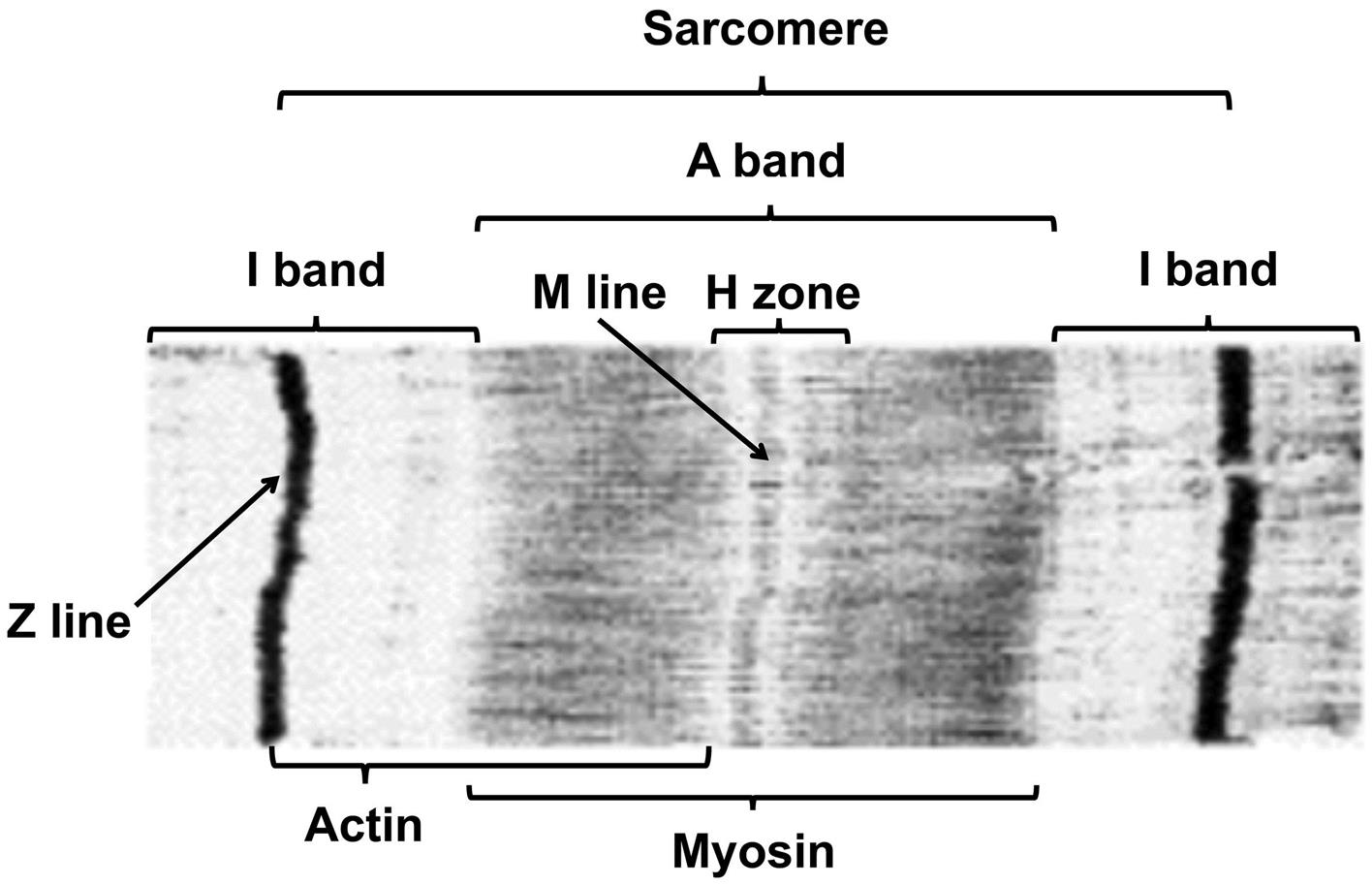
Skeletal muscle fibers contain hundreds of longitudinally aligned myofibrils composed of repeating sarcomeres. The characteristic transverse striations of skeletal muscle fibers results from parallel alignment of the bands in adjacent myofibrils (Figure 10.1). Bands are named according to their appearance in polarized light. The largest are A bands (anisotropic or birefringent, appear bright) and I bands (iostropic, appear dark). The I bands, composed of thin myofilaments (made of actin, troponin, and tropomyosin), are bisected by Z lines (disks, bands) that form the end of each sarcomere; the A bands, composed of thick myosin myofilaments, are bisected by the less-birefringent H bands. The banding pattern, named for the appearance in polarized light, is reversed when studied by light microscopy with phase contrast optics, light microscopy with conventional optics on sections stained with the usual cationic dyes, or transmission electron microscopy.
Application of histochemical stains (described later) to frozen sections of unfixed skeletal muscle will allow demonstration of various fiber-type populations that cannot be distinguished in paraffin-embedded sections stained with conventional histologic stains such as hematoxylin and eosin (H&E). The histochemical uniqueness of these fiber types correlates with differences in their physiologic features, such as contraction speed and fatigability; their biochemical and metabolic activities; their gross color; and their structure as revealed ultrastructurally (Table 10.1).
Table 10.1
Characteristics of Major Skeletal Muscle Fiber Types in Mammals
| Type I | Type IIA | Type IIB | |
| MORPHOLOGIC CHARACTERISTICS | |||
| Natural color | Dark | Dark | Pale |
| Glycogen content | Low | High | High |
| Myoglobin content | High | High | Low |
| Lipid globules | Numerous | Numerous | Few |
| Mitochondrial content | High | Intermediate | Low |
| PHYSIOLOGICAL FEATURES | |||
| Twitch speed | Slow | Fast | Fast |
| Fatigability | Resistant | Resistant | Susceptible |
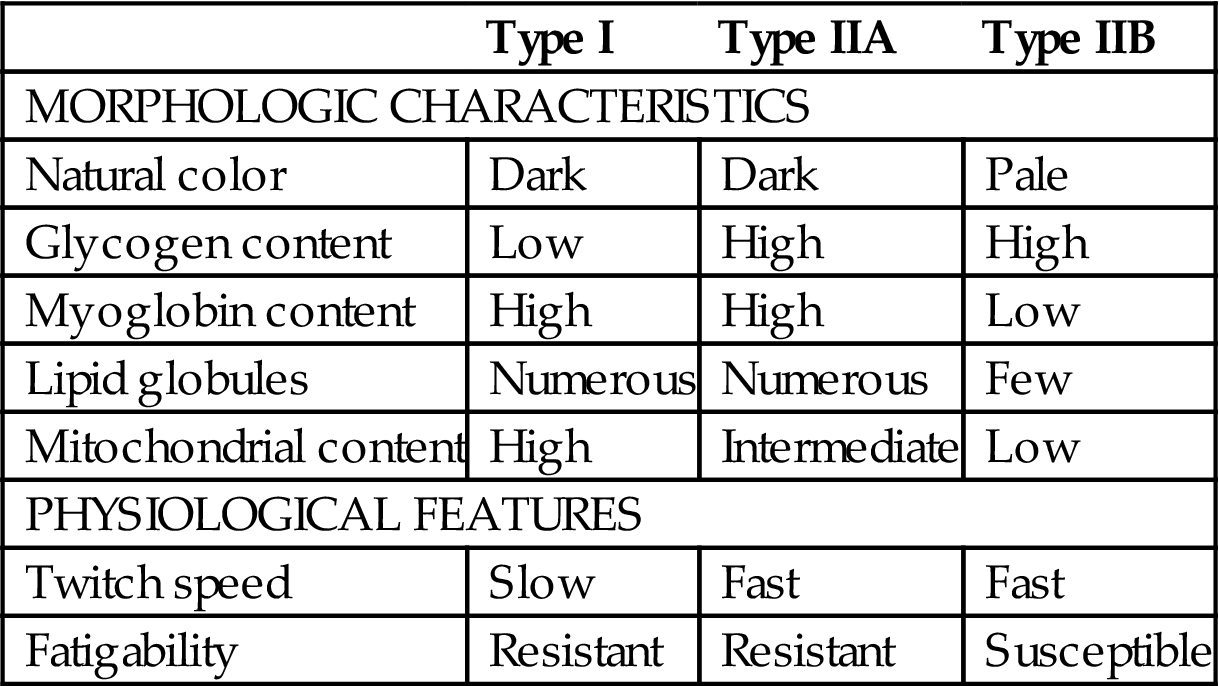
Table reproduced from Haschek, W.M., Rousseaux, C.G., Wallig, M.A. (Eds.) 2010. Fundamentals of Toxicologic Pathology, second ed. Academic Press, Table 12.9, p. 366 with permission.
Most skeletal muscles will have a mixture of all fiber types to produce the so-called “checkerboard” pattern that becomes evident with differential histochemical staining. Fibers innervated by the same nerve branches will have the same fiber type, although reinnervated fibers may show reversal of fiber types depending on which nerve endings reach the restored fiber. Some muscles will have a preponderance of one fiber type; e.g., the soleus is a “red” muscle high in type I (or “slow twitch”) fibers that are capable of sustained action or weight bearing, while the gastrocnemius is a “white” muscle high in type II (or “fast twitch”) fibers that are capable of sudden action and purposeful motion. The proportions of the various fiber types in a given muscle may vary with species, breed, age, and exercise, and also in certain muscular diseases.
Ultrastructurally the myofiber surface is covered by the sarcolemma, which in turn is encompassed by an external lamina. The elongated subsarcolemmal nuclei are surrounded by accumulations of mitochondria, lipid droplets, glycogen granules, elements of sarcoplasmic (endoplasmic) reticulum, and collections of Golgi apparatuses. The myofiber contains abundant contractile elements that are organized as many parallel myofibrils of 0.5–1.0 μm in diameter. Myofibrils are composed of repeating units, termed sarcomeres, of 2–3 μm in length (Figure 10.1). Sarcomeres end at dense Z lines that contain α-actinin, actin, and tropomyosin. Thin 6-nm-diameter myofilaments containing actin, troponin, and tropomyosin extend on both sides of the Z line to form the I bands. The middle half of the sarcomere contains thick (16 nm diameter) myofilaments that are composed of myosin and interdigitate with the adjacent thin filaments. The center of the sarcomere, with only thick filaments, is the H band and is bisected by the relatively dense M line, which represents interconnecting elements of the cytoskeleton. Fiber contraction results in shortening of sarcomeres due to sliding of bulbous heads of thick filaments along the length of thin filaments to produce narrowed I and H bands. Cross-sections of myofibrils show variable appearance depending on the location in the sarcomere, but the edges of the A band will have thick filaments surrounded by a hexagonal array of thin filaments. The sarcoplasm surrounding myofibrils contains elements of the transverse (T) tubular system and SR, mitochondria, lipid droplets, glycogen granules, and cytosol. The T tubules are invaginations of the sarcolemma that are often seen at the edge of the I band with two adjacent elements of SR to form a “triad.” The T-tubular system functions as a channel to allow rapid spread of an electrical impulse from the motor end plate (i.e., the junction between a nerve terminus and the surface of a myofiber) on the sarcolemma to the interior of the fiber to elicit release of calcium (Ca2+) stored in the SR. Increasing concentrations of cytoplasmic Ca2+ will bind to regulatory troponin proteins on the thin myofilament, thereby releasing binding sites on actin that will interact with myosin to initiate myofiber contraction.
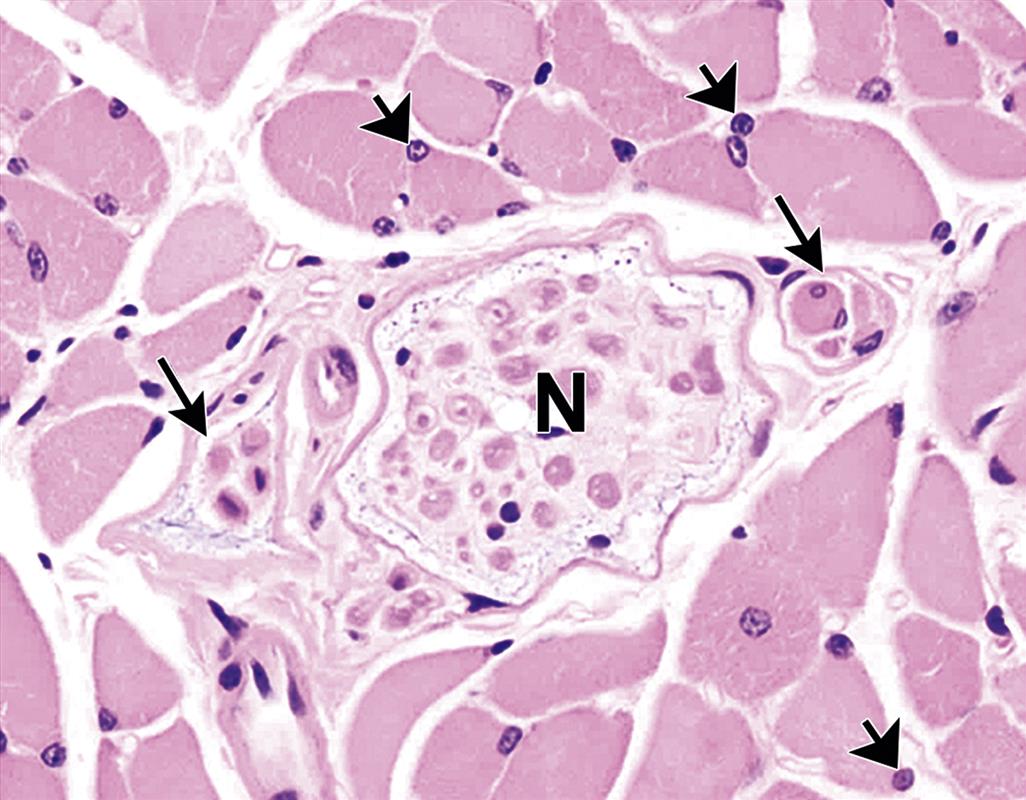
Satellite Cells. Satellite cells are thin cells with a nucleus and a scant amount of sarcoplasm that are interposed between the sarcolemma of myofibers and the external lamina (Figure 10.2). They are abundant in newborn animals; in muscle of mature animals, 3%–5% of nuclei in muscle fibers belong to satellite cells. These cells play an important role in normal development of myofibers and also in regeneration of damaged fibers during adult life by serving as stem cells that can be activated to undergo mitosis and subsequently differentiate to myoblasts, myotubes, and, eventually, mature myofibers.
Motor End Plates. Motor end plates (i.e., neuromuscular junctions) represent a complex and intimate attachment site of the motor nerve fiber on the surface of the skeletal muscle fiber. The end of the nerve fiber is unmyelinated and branches into axon terminals that invaginate into a synaptic cleft, which is characterized by a thickened zone of subsarcolemmal sarcoplasm with numerous nuclei. The axon terminal has abundant synaptic vesicles containing the neurotransmitter acetylcholine (ACh). Release of ACh with binding to receptors on the postsynaptic sarcolemmal membrane results in increased sodium (Na+) and potassium (K+) conductance, depolarization of the sarcolemma, mobilization of intracellular Ca2+ stores, and myofiber contraction. Motor end plates generally are recognized in muscle tissue only by use of special techniques such as metallic impregnation, intravital dyes, histochemical procedures, or transmission electron microscopy.
Muscle Spindles. Muscle spindles are fusiform sensory receptors approximately 0.5–3.0 mm in length oriented longitudinally near the edges of muscle fasciculi. Their function is to detect changes in the length of muscle bellies; they regulate muscle contraction via the stretch reflex by activating motor neurons in the central nervous system to counter muscle stretch. The muscle spindle has a thick fibrous capsule and contains multiple small, variably sized, intrafusal muscle fibers, nerve fibers, specialized nerve endings, and blood vessels (Figure 10.2).
Connective Tissue. The interstitial connective tissue of skeletal muscle is subdivided into the epimysium (surrounds the entire muscle), perimysium (surrounds large angular fascicles divided into primary fascicles of 10–100 fibers), and endomysium (surrounds individual muscle fibers). The endomysium contains capillaries, nerve fibers, fibroblasts, and collagen fibrils. Larger amounts of collagen fibrils and larger blood vessels and nerves are in the perimysium.
Tendons are thick bands of dense fibrous connective tissue that connect muscles to bones. The collagen type I fibers in tendons, which are secreted by fibroblast-like tenocytes, are oriented parallel to their length and the lines of tension that are applied to the tendon during contraction of the muscle. Tendons not only serve in locomotion, as the anchor through which skeletal muscles fulfill their role, but also provide additional stability to nearby joints (as an adjunct to ligaments, which connect bone to bone). Basic functional and structural attributes of tendons as well as their responses to toxic insult are covered elsewhere in this volume (see Chapter 23: Bone and Joints).
Physiology and Function of Skeletal Muscle
The unique structural differentiation of skeletal muscle fibers is closely integrated with their highly developed, specialized contractile function for locomotion and maintenance of posture by conversion of chemical energy into mechanical energy. Further specialization in form and function is provided by the differentiation of myofibers into various fiber types, each of which is specifically suited for certain physiologic applications.
The functional unit of the neuromuscular system is the motor unit, consisting of (1) nerve cell bodies in the spinal cord ventral horns or brainstem, (2) axons of these neurons that course to the muscles and terminate as motor end plates, and (3) the group of histochemical type-specific skeletal muscle fibers that are innervated by the neuron. The number of muscle fibers supplied by a neuron of a motor unit varies widely depending on the degree of refined movement needed by the muscle. For example, 10 fibers are innervated by a single neuron in extrinsic eye muscles, which require exquisite control, while 2000 fibers are supplied per neuron in large limb muscles where actions are performed by large cooperating groups of fibers.
Contraction of muscle is the result of sarcomere shortening by interdigitation of thin and thick myofilaments. According to the sliding filament hypothesis, the force of contraction is generated by the movement of cross-bridges formed by heads that project from myosin molecules in thick myofilaments to actin molecules (the main portion of thin myofilaments), leading to migration of the myosin-based filaments along the actin-based filaments. The chemical energy for contraction is supplied by high-energy phosphate compounds (i.e., adenosine triphosphate (ATP) and creatine phosphate (CP)). In type I (slow twitch) fibers, these compounds are largely generated by mitochondrial oxidative phosphorylation via the electron transport system following the oxidation of fatty acids and glucose via the citric acid (or Krebs) cycle, while in type II (fast twitch) fibers they are produced mainly by sarcoplasmic anaerobic glycolysis and glycogenolysis. Thus the metabolic differences of the various fiber types are correlated with differences in their functional features, such as speed of contraction and resistance to fatigue.
Evaluation of Skeletal Muscle Toxicity
The techniques for assessing the toxic effects of xenobiotics on skeletal muscle vary from detection of biochemical constituents in the blood to sophisticated direct evaluation of isolated muscle tissue. Routine toxicity studies rely on assessment of muscle histopathology and analysis of serum creatine kinase (CK) activity.
Biochemical Evaluation. Certain enzymes present in high concentrations in the sarcoplasm can leak into the blood and serve as biomarkers of myotoxicity. The most sensitive enzyme is CK. This enzyme is released from muscle cells in a number of myopathies. Three isoenzymes (MM, MB, and BB) can be detected in blood; MM is the main isoenzyme found in skeletal muscle. A greater than 10-fold increase in CK concentration is usually indicative of muscle origin following myofiber injury. Smaller increases in CK occur at times during exercise or in response to localized damage resulting from intramuscular injections. CK levels usually are not elevated in those instances where the myopathy is due to denervation or myoneural junctional disorders. The serum values for other intramuscular constituents also may be increased following damage to skeletal muscle, including the enzymes alanine (ALT) and aspartate (AST) aminotransferase, lactic dehydrogenase (LDH), and aldolase as well as 3-methylhistidine (a component of actin in all types of muscle fibers, and of heavy and light myosin chains of the fast-twitch fibers). Hypo- or hyperkalemia can lead to episodes of flaccid muscle paralysis.
Other proteins may be indicative of skeletal muscle toxicity when found in circulation as well. The most abundant protein in skeletal muscle is myoglobin, a low molecular weight, oxygen-binding heme protein that constitutes up to 5%–10% of all the cytoplasmic proteins found in muscle cells. In blood, myoglobin is bound primarily to plasma globulins. If the plasma concentration exceeds the plasma-binding capacity (1.5 mg/dL in humans), myoglobin begins to appear in the urine. Myoglobin is rapidly released after muscle damage and thus can be a useful biomarker in the early phases of injury. Because myoglobin has a short half-life (2–3 hours) and undergoes rapid renal clearance, changes in serum concentrations usually occur over a shorter time course than do changes in serum levels of CK (i.e., myoglobin is elevated sooner and returns to normal at a more rapid rate). Dip stick urine tests are not specific as they cannot discern between myoglobin and hemoglobin. Troponins are important regulatory proteins present in both cardiac and skeletal muscles. Cardiac troponin isoforms (e.g., I, T) are widely used as biomarkers of cardiac muscle injury and can be measured in serum or plasma with a number of commercially available assays. Assays for skeletal muscle troponin isoforms are less specific and thus are not used as widely.
Electrophysiological Evaluation. Electromyography (EMG) involves placing two electrodes on the skin or inserting needle electrodes into the muscle to measure the summated electrical potentials of action potentials traveling along muscle fibers. Detection of muscle abnormalities depends on the analysis of spontaneous or evoked electrical potentials; properly calibrated, the readouts can distinguish muscle-centered from nerve-centered anomalies. A disadvantage of EMG is that recorded changes may be difficult to quantify. Electrophysiological analysis of isolated whole muscles or individual muscle cells may be used to evaluate the excitability of muscle cell membranes in response to chemical exposure in vitro.
Gross Examination. Complete examination of the skeletal muscles is rarely done at necropsy. The large mass of the skeletal musculature (40%–50% of body weight) precludes complete dissection of each individual muscle. In necropsies of animals without any historical evidence of clinical disease of the skeletal musculature, the prosector should at least inspect the muscles exposed during the necropsy procedure. In animals with clinical suspicion of muscle disease, a more detailed examination of the musculature should be carried out, with particularly careful inspection of those muscles presumed to be responsible for the clinical signs. Gross alterations in color, shape, size, and consistency should be sought by viewing the uncut surface and cut sections of the muscles. Prosectors should avoid over-interpretation of gross findings such as color and size of muscles, which may be a function of the normal variation associated with species, age, particular muscle, and state of nutrition seen among animals.
Following somatic death, skeletal muscle will generally remain in relaxation for 2–4 hours until muscle glycogen stores are metabolized and sufficient energy reserves are no longer present to maintain the relaxed state. Rigor mortis then ensues as stiffness of the muscles and immobility of the joints, and will persist for at least 24–48 hours. Subsequently, muscle rigor will dissipate as autolysis occurs. The onset of rigor mortis will be rapid if the muscle is kept at low pH and high temperature, while rigor will be delayed if abundant muscular glycogen stores at the time of death and will be weak if the animal was debilitated at the time of death.
Microscopic Evaluation. Histologic analysis of skeletal muscle sections can reveal changes associated with xenobiotics causing (1) myopathic or neurogenic alterations, (2) specific muscular structural alterations, or (3) specific muscular metabolic alterations. A number of pathologic muscular alterations can be detected in routine paraffin sections of muscle. Muscle fiber necrosis and regeneration are common features of myotoxicity. If necrosis is substantial and chronic, regeneration may fail, resulting in progressive myofiber loss and replacement by fat and fibrous tissue. Degeneration of muscle fibers without overt necrosis causes structural alteration of myofibrils and sarcoplasm. Pathologic changes may be restricted to one type of fiber in the muscle.
Sample collection must be done carefully to avoid hypercontraction artifacts that are inevitable in fresh muscle collected in biopsy samples and in samples taken from animals soon after death. To prevent contraction artifacts, isolated longitudinal strips of muscle are dissected and clamped or ligated to a strip of wood (e.g., tongue depressor) at isometric length (i.e., in a slightly stretched position) and then excised. Placing the clamped sample in physiologic saline solution for 20 minutes will further decrease problems with contraction artifacts.
Morphologic analysis of skeletal muscle typically is undertaken using conventional tissue preparation techniques. For routine light microscopy, the clamped sample is fixed in neutral buffered 10% formalin, embedded in paraffin, sectioned at 4–5 μm, and stained with H&E. Other stains frequently used to evaluate skeletal muscle structures include Gomori or Masson’s trichrome (for connective tissue) and phosphotungstic acid hematoxylin (PTAH, to demonstrate myofibrils and cross-striations of muscle fibers). Application of lipid stains (e.g., oil red O) to frozen sections may be useful. Each muscle sample should have longitudinal and cross-sections prepared for microscopic study. Tissue samples to be used for high-resolution light microscopy or transmission electron microscopy are immersed in buffered 3% glutaraldehyde or Trump’s universal fixative, post-fixed in 1% osmium tetroxide, and then embedded in either methacrylate (“soft plastic”) or epoxy (“hard plastic”) resins.
Myofiber isotype characterization requires special histochemical techniques applied to fresh-frozen muscle sections. Key morphologic endpoints include myofiber dimensions, the distribution and quantity of mitochondria, lipid and glycogen content, myofibrillar enzymes, and the distribution of the fiber types. Muscle units differ both in size and in certain biochemical properties of their fibers. Two primary types of muscle fibers are characterized (Table 10.1): type I (slow twitch, fatigue resistant) and type II (fast twitch, fatigable); classically, the type II fibers have been further classified as types 2A (fast twitch oxidative) and 2B (fast twitch glycolytic), although modern fiber typing techniques have shown that this scheme is an over-simplification. Special histochemical stains used for categorizing fiber types include myosin ATPase activity at three different pH levels, to differentiate fast- and slow-contracting myofibers, and NADH-tetrazolium reductase (NADH-TR) activity and succinic dehydrogenase (SDH) activity, both of which are indicators of mitochondrial oxidative potential. Analysis of myosin heavy chain (myHC) composition also can be used to define fiber types, although some fibers are hybrids containing multiple myHC isoforms. Fiber-type differentiation may be useful in evaluating myotoxic effects since some agents injure only one fiber type.
Ultrastructural examination is useful for characterizing subcellular changes leading to myofiber degeneration or death. Vacuolation at the light microscopic level may represent accumulations of either neutral (nonpolar) lipids (i.e., accretion of potential energy substrates) or polar phospholipids (i.e., accrual of undigested membrane constituents), or even mitochondrial swelling and degeneration. Swollen muscle fibers with a “lacy” cytoplasm at the light microscopic level might have cytoplasmic glycogen accumulation. Pale staining or loss of discrete cross-striations at the light microscopic level may reflect myofibril lysis. Though some of these changes may be characterized at the light microscopic level using special stains (e.g., lipid, glycogen), they are all best examined at the ultrastructural level to ensure that the components that can be stained are not present as well (e.g., lipid accumulation with mitochondrial swelling).
Numerous artifacts may be observed in muscle sections prepared for either light or electron microscopic evaluation. These artifacts include hypercontracted fibers from improper collection and fixation procedures, mitochondrial vacuolation, distention of elements of SR and glycogen loss from faulty fixation, and occurrence of knife marks, chatter, and wrinkles from sectioning problems.
Response to Injury
Skeletal muscle will respond to insults with a limited number of morphological reactions. In fact diseases of differing etiologies may exhibit similar types of lesions. Thus it may not be possible to render a specific etiologic diagnosis for a given skeletal muscle lesion even following careful microscopic study from a given case.
Many injuries of skeletal muscle heal by regeneration rather than by fibrosis—which is a distinguishing feature of repair in skeletal versus cardiac muscle. This is especially true for the common monophasic or polyphasic polyfocal myopathies (see subsequent text) such as those associated with nutritional deficiencies, metabolic disorders, and myotoxicities. In these diseases, although extensive muscle fiber necrosis may occur, the scaffolding of external lamina that surrounds the degenerated muscle fiber and the innervation and blood supply to the damaged muscle are preserved, permitting myofiber regeneration within the external lamina (which is often virtually complete). Regeneration is further promoted in these conditions by the short-term nature of the insult responsible for the muscle injury. In contrast, prolonged insults such as denervation or genetic derangements often induce muscular diseases in which regeneration is limited. In severe myopathic diseases (i.e., those in which muscle tissue is directly damaged), extensive disruption of endomysial connective tissues and “tubes” of external laminae from damaged myofibers may occur from trauma, hemorrhage, or infection. In such cases, the outcome of healing will be limited regeneration accompanied by extensive fibrosis and scarring.
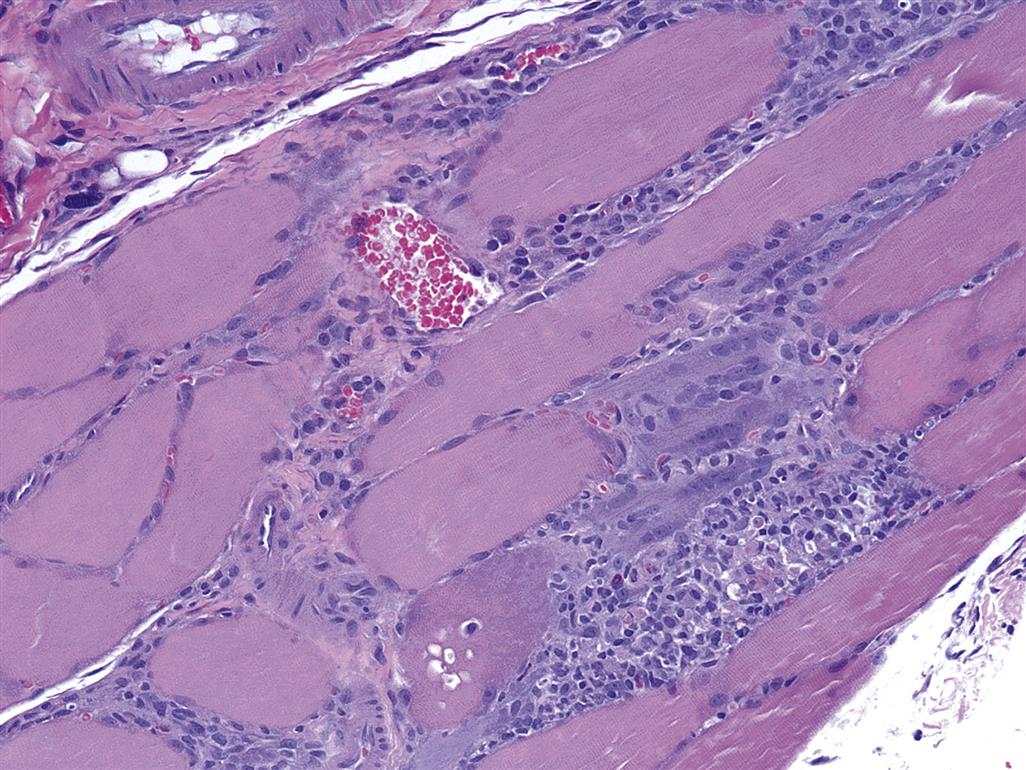
The cellular events of skeletal muscle regeneration are well characterized and center on the proliferation of mononucleated myogenic stem cells, termed myoblasts (Figure 10.3). Myoblasts arise from satellite cells, a population of resting and undifferentiated cells that persist in mature skeletal muscle and appear morphologically as very thin subsarcolemmal cells that lie between the sarcolemma and the external lamina of myofibers. For unknown reasons, satellite cells tend to be resistant to many insults that destroy mature myofibers. Following selective destruction of skeletal muscle fibers, the sarcoplasmic debris is removed rapidly by invasion of macrophages and phagocytic lysis (Figure 10.4). The persisting sarcolemmal “tubes” of external laminae rapidly become populated by elongated myoblasts with large vesicular nuclei and prominent basophilic sarcoplasm that reflects the numerous polyribosomes supporting intense synthesis of cellular proteins in these cells. Myoblasts fuse to form multinucleated cells termed sarcoblasts, which further elongate to form myotubes that rapidly bridge the gap of disrupted sarcoplasm in damaged myofibers. Myotubes have rows of centrally located nuclei and peripheral masses of forming contractile myofilaments that soon become oriented into sarcomeres and myofibrils with restoration of cross-striations in the immature myofibers (Figure 10.5). Subsequently the central nuclei will migrate to their normal subsarcolemmal location (as seen in mature fibers), and the regenerated muscle fibers may then be indistinguishable from adjacent fibers that have not suffered injury.


Pathologic Alterations in Skeletal Muscle
Degeneration and Necrosis. Unlike many tissues the difference between the reversible sublethal alterations of degeneration and the irreversible lethal changes of necrosis is difficult to detect by microscopic study. Skeletal muscle fibers are large multinucleated cells, and it is often not possible to view the entire length of the fiber in the plane of a tissue section to determine whether the sarcoplasmic damage involves the entire fiber or only a segment of the fiber. It seems likely that segmental degeneration occurs frequently, but necrosis of entire myofibers is uncommon. In any event the causes of both degeneration and necrosis are similar.
Specific morphologic types of skeletal muscle degeneration have been described. The most common type is so-called hyaline-type or waxy degeneration. Affected muscles may be detected grossly by diffuse pallor or scattered pale streaks (i.e., “white muscle disease;” Figure 10.6), especially if secondary calcification has occurred in damaged fibers. Microscopically, affected fibers appear swollen and hypereosinophilic with loss of cross-striations (Figures 10.5 and 10.7). The altered contractile material frequently becomes fragmented into large blocks or disks scattered along the “tube” of persisting external lamina of the degenerating muscle fiber. Within 24 hours the affected areas will be invaded by an occasional polymorphonuclear leukocyte and numerous macrophages. Macrophages are observed in the interstitium and also within injured muscle fibers. Ultrastructurally the affected myofibers show tangled masses of disrupted contractile material with damaged membranes of mitochondria, SR, and sarcolemma. The “tube” of external lamina persists to guide regenerative events and may be focally disrupted to allow entry of macrophages.
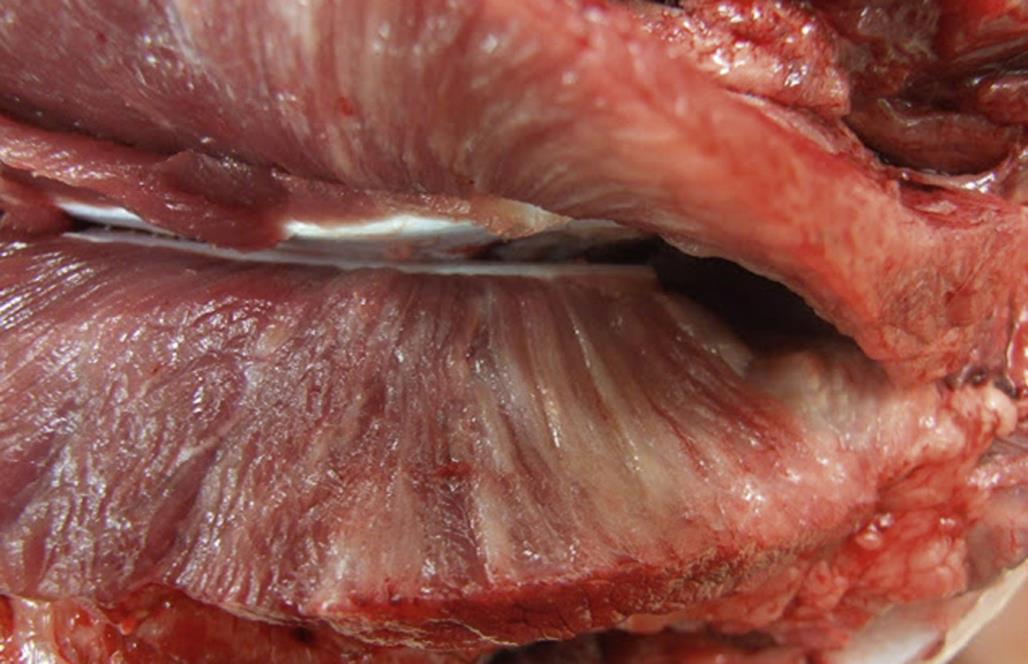

Another type of degeneration described in skeletal muscle is granular degeneration. The microscopic appearance differs from hyaline degeneration because the damaged sarcoplasm appears as small basophilic granules that fill the “tube” of external lamina and are identified as mineralized mitochondria by ultrastructural study. The causes of granular and hyaline degeneration are similar and include nutritional deficiencies (such as selenium/vitamin E deficiency), various myotoxic drugs and plants, and metabolic disorders such as azoturia and capture myopathy.
The spatial distribution and temporal pattern of degeneration and necrosis in skeletal muscle have been used to classify reactions as (1) monophasic monofocal, (2) monophasic polyfocal, (3) polyphasic monofocal, and (4) polyphasic polyfocal. Monophasic monofocal reactions result from an isolated, single mechanical injury such as external trauma or needle insertion. In monophasic polyfocal reactions (Figures 10.5 and 10.7) a single insult such as exposure to various myotoxic drugs or chemicals or various metabolic disorders may initiate widespread muscle lesions, but all the lesions are in the same phase of injury. Polyphasic monofocal reactions would be the result of repeated localized mechanical injury. Polyphasic multifocal reactions are frequent in muscular diseases of animals, and result from continued insults applied intermittently or continuously over a prolonged time (e.g., from nutritional deficiencies and from genetic disorders like muscular dystrophies); the lesions are widespread in the musculature, and various pathological reactions indicative of differently timed insults will occur concurrently, including necrosis (acute reaction), leukocyte invasion during resolution (often subacute response), and regeneration (subacute to chronic outcome). Xenobiotic-induced toxic myopathies tend to be monophasic polyfocal responses.
Other less common types of degeneration in skeletal muscle fibers include vacuolar or hydropic degeneration and fatty degeneration. Vacuolar or hydropic-type degeneration occurs with cortisol excess. The affected fibers have vacuolated, lace-like areas in the sarcoplasm. Fatty degeneration is uncommon. It is seen as a nonspecific response to injury, and results in abundant small spherical lipid vacuoles scattered between myofibrils. Similar appearing but less abundant lipid deposits are normally present in type I muscle fibers.
Table 10.2 summarizes the chemical and biological agents that have been associated with necrosis or degeneration of skeletal muscle in animals and humans. Many of these agents also produce myocardial injury.
Table 10.2
Toxic Chemical and Biological Agents That Cause Necrosis or Degeneration of Skeletal Muscle in Animals and Humans
| Causes | Skeletal muscle | Type I fiber selectivity | Species affected | |
| Necrosis | Degeneration | |||
| Ionophores—Monensin, lasalocid, narasin, A-204, maduramicin, salinomycin | + | + | + | Horse, cow, pig, sheep, dog, chicken, turkey, rat, mouse |
| Antivirals—Zidovudine (AZT) | − | + | ND | Rat, human |
| Quinolone antibacterials—Pefloxacin, levofloxacin | + | + | ND | Rat, human |
| Antimalarials—Emetine, chloroquine, quinine, plasmocid | + | + | + | Rat, human, rabbit, pig |
| Immunosuppressive and cytotoxic drugs—Vincristine, azathioprine, doxorubicin | + | + | ND | Rat, human |
| Corticosteroids—Cortisone, triamcinolone, fluorocortisone | + | + | − | Rabbit, human, dog |
| Antibiotics—Nitroxoline, thiabendazole | − | + | ND | Mouse, human |
| Local anesthetics—Bupivaccine, tetracaine, lignocaine | − | + | ND | Human, pig |
| Analgesics—Pentazocine, paracetamol, salicylates | + | + | ND | Human |
| Anesthetics—Halothane and others (via malignant hyperthermia) | + | + | + | Rat, human, pig |
| Agents producing hypokalemia—Carbonoxolone, licorice, corticosteroids, diuretics | − | + | ND | Human, rabbit |
| Hypocholesterolemic agents—Lovastatin, simvastatin, pravastatin, cerivastatin, rosuvastatin | + | + | − | Rat, human, rabbit |
| Hypolipidemic agents—Clofibrate | ± | ± | ± | Rat |
| Cationic amphiphilic drugs—Amiodarone, chlorphentermine, tamoxifen, chlorcyclizine | − | + | ND | Rat |
| Systemic toxic agents—Alcohol, oxygen, carbon monoxide, organophosphates | + | + | ND | Rat, human |
| FURTHER AGENTS ADMINISTERED SYSTEMICALLY | ||||
| p-Phenylenediamine, diphenylenediamine | + | + | ND | Rat |
| Iron dextran | + | + | ND | Pig |
| Brown FK | + | + | ND | Rat |
| Insulin | + | + | ND | Rabbit |
| Selenium | + | + | ND | Pig |
| ε-Aminocaproic acid | + | + | ND | Human |
| Iodide | + | + | ND | Rat |
| Iodpacetate, fluoroacetate | + | + | ND | Rat |
| Imidazole | + | + | ND | Rat |
| Dimethylsulfoxide | + | + | ND | Rat |
| Phencyclidine | + | + | ND | Rat, human |
| Cannabinoids | + | + | ND | Mouse |
| Colchicine | + | ND | − | Rat |
| Triethyltin sulfate, triethyltin bromide | − | + | ND | Rat |
| 2,4-Dichlorophenoxyacetate (2,4-D) | − | − | ND | Guinea pig |
| 6-Mercaptopurine | − | + | ND | Rat |
| Clenbuterol, salbutamol | + | + | ND | Rat, dog |
| Paraoxon, physostigmine, pyridostigmine, parathion | + | + | ND | Rat |
| 2,4-Dinitrophenol | − | + | ND | Rat |
| Acrylamide | − | + | ND | Rat |
| Rolziracetam | + | + | ND | Dog |
| Thyroxine | − | + | ND | Rabbit |
| Cassia occidentalis, C. obtusifolia | + | + | ND | Cow, horse, sheep, goat, rabbit |
| Karwinskia humboltiana | + | + | ND | Goat, sheep |
| Ageratina altissima (previously Eupatorium rugosum) | + | + | ND | Horse, cow |
| Gossypol | + | + | ND | Pig |
| Trigonella foenumgraecum | + | + | ND | Cow |
| Petiveria alliacea | + | + | ND | Cow |
| Lupine—Diaporthe toxica | + | + | ND | Sheep |
| Snake venom (sea snake, Australian mulga snake, tiger snake, prairie rattlesnake) | + | + | ND | Rat, mouse |
| Oriental hornet venom | − | + | ND | Human |
| Stonefish venom | − | + | ND | Mouse |
| Cicuta douglasii | + | + | ND | Sheep |
| Thermopsis montana | + | + | ND | Cow |
| Clostridium chauvoei, C. septicum, C. novyi, C. perfringens | + | + | ND | Cow, sheep, horse, pig |
| Uremic toxins | + | + | ND | Rat, human |

+, present; −, absent; ND, not determined.
Table modified from Haschek, W.M., Rousseaux, C.G., Wallig, M.A. (Eds.), 2013. Handbook of Toxicologic Pathology, third ed. Academic Press, Table 46.6, pp. 1649–1652 with permission.
Drug-Induced Neuromuscular Blockade. A number of drugs may interfere with neuromuscular transmission in human patients. Three clinical syndromes have been recognized: drug-induced myasthenic syndrome, which is uncommon; drug-induced aggravation or unmasking of existing myasthenia gravis; and postoperative respiratory depression from direct effect of the drug or from potentiation of muscle relaxants. Drugs implicated in these syndromes include aminoglycoside antibiotics (neomycin, kanamycin, streptomycin, gentamycin); polypeptide antibiotics (polymyxin B, colistins); other antibiotics (oxytetracycline, rolitetracycline, lincomycin, clindamycin); antirheumatic drugs (D-penicillamine, chloroquine); cardiovascular drugs (oxprenolol, practolol, quinidine, procainamide, propranolol); anticonvulsants (trimethadione, phenytoin); psychotropic drugs (lithium, chlorpromazine, promazine, phenelzine); anesthetics (diazepam, ketamine, propenidid, ether); and other drugs (busulfan, oral contraceptives, methoxyflurane, adrenocorticotropic hormone (ACTH), corticosteroids, thyroid hormones, anticholinesterases, oxytocin, aprotinin, procaine, lidocaine). These drug-induced clinical syndromes have not been associated with morphologic alterations in skeletal muscle. The mechanisms of these drug-induced neuromuscular blockades include (1) presynaptic local anesthetic-like action, (2) postsynaptic receptor blockade, (3) interference with ACh release, and (4) impairment of muscle membrane conductance.
Drug-Induced Myotonic Syndrome. Myotonia is the failure of normal muscular relaxation following contraction. Myotonic syndrome occurs in human patients, rats, and goats administered 20,25-diazacholesterol and its analogs as hypocholesterolemic agents. Propranolol and suxamethonium may precipitate or exacerbate myotonia in human patients.
Drug-Induced Denervation Atrophy. Over 50 drugs used clinically in human patients have been reported to produce peripheral neuropathies. Damage to the nerve supply leads to myofiber atrophy of affected motor units and eventually progression to the distinctive morphologic alteration of “fiber group atrophy” (Figure 10.8). Neurogenic atrophy represents a loss of myofiber mass as an indirect response caused by biochemical or structural interruption that prevents nerve impulses from depolarizing the myofiber. Key morphologic attributes of neurogenic lesions include clusters of small angulated myofibers (i.e., foci of denervation atrophy) intermingled with large oval myofibers (i.e., those retaining an intact somatic nerve supply). Drugs implicated include antimicrobial agents (isoniazid, nitrofurantoin, sulfonamides, clioquinol, metronidazole, amphotericin B); antineoplastic agents (vincristine, procrabazine, nitrofurazone, cytosine arabinoside, podophyllir, chlorambucil); antirheumatic drugs (gold, colchicine, chloroquine, indomethacin, phenylbutazone); hypnotics and psychotropics (thalidomide, methoqualone, glutathimide); cardiovascular drugs (perhexiline, amiodarone, hydralazine, diisopyramide, clofibrate, digitalis); and other drugs (phenytoin, disulfiram, dapsone, ergotamine, methimazole, propylthiouracil, methylthiouracil).

Muscle Repair. Evidence of repair typically is a feature of myopathic lesions (i.e., those in which muscle tissue is directly injured), but usually is not present in neurogenic lesions (i.e., those in which muscle is affected secondarily as a consequence of primary damage at an extra-muscular site) (Figure 10.8). Successful restoration of muscle parenchyma leads to myofiber regeneration within intact external laminae (to restore necrotic muscle cells; Figures 10.4 and 10.7). More extensive myopathic lesions that result in both myofiber necrosis and interruption of external laminae may evoke inflammation (typically mononuclear cells), expansion of interstitial fibrous connective tissue, and sometimes mineralization of damaged myofibers. Over time the inflammatory reaction may recede, but fibrosis and mineralization represent permanent changes.
Mechanisms of Toxicity
Skeletal muscle accounts for a significant portion of total body weight. The normal structure and function of skeletal muscle can be altered by a variety of chemicals and drugs. Skeletal muscle susceptibility to potential toxicants appears in part to be related to high metabolic activity and the presence of multiple potential sites by which toxicants can interfere with crucial energy generating pathways. In addition, sensitivity to toxic agents may be enhanced because the high rate of blood flow increases exposure to many substances capable of binding to skeletal muscle. Skeletal muscle dysfunction or damage has been induced by both indirect and direct mechanisms.
Altered Neurogenic Function
Muscular atrophy can develop when interruptions occur in peripheral motor nerve function. Usually only some of the motor fibers supplied by the nerve undergo atrophy, while others remain relatively normal (Figure 10.8). The nerve damage may occur centrally in the neuron that innervates a motor unit, or peripherally in either the nerve fiber or at the motor end plate. For example, intramuscular injections to the hind limb may damage the sciatic nerve, leading to primary nerve fiber degeneration and secondary atrophy of muscles supplied by sciatic nerve branches. Botulinum toxin affects muscle function by blocking the release of ACh from presynaptic motor nerve terminals at the motor end plate (i.e., neuromuscular blockade). Magnesium also interferes with neuromuscular transmission by blocking the release of ACh. Muscle weakness also occurs after exposure to a variety of substances that are capable of causing or intensifying neuromuscular blockade. No significant morphologic changes appear during neuromuscular blockade, and the blockade is usually reversed when the causative substance is removed. However, severe prolonged neuromuscular blockade can induce neurogenic muscle atrophy. In some instances, xenobiotics have induced muscle necrosis by dramatically increasing motor nerve activity or by allowing significant amounts of ACh to accumulate at the myoneural junction. In rats injection of paraoxon causes a progressive myopathy, which ultimately leads to necrosis of muscle fibers; the myopathy is attributed to excessive concentrations of ACh at the neuromuscular junction. Acute rhabdomyolysis is a severe necrotizing myopathy associated with heroin addiction, amphetamine overdoses, and phencyclidine abuse. The muscle damage probably occurs as a result of extreme motor nerve excitation rather than direct myotoxicity.
Altered Immunologic Function
A xenobiotic may also induce myotoxicity indirectly by provoking immunologic reactions that lead to generalized muscle weakness. D-Penicillamine induces a variety of antibodies, especially an anti-acetylcholine receptor antibody. Experimentally, high doses of D-penicillamine given to guinea pigs over a prolonged period of time cause a mild level of neuromuscular blockade. The slow onset of muscular weakness and the identification of anti-acetylcholine receptor antibodies suggest that the pathogenesis is immune-mediated. Clinical use of D-penicillamine has been associated with adverse effects such as dermatomyositis (inflammation of the skin and muscles) and polymyositis. There is evidence to indicate that autoimmune reactions play a role in the etiology of these types of inflammatory myopathies. A form of myositis may also develop as part of a drug-induced, lupus-like reaction during treatment with procainamide. Rare adverse effects, such as myasthenia gravis and myositis, have been reported in patients given interferon-α. Evidence of dermatomyositis and polymyositis has been reported in some patients treated with statin drugs. These cases responded to steroid treatment, suggesting that statins either initiated immune-mediated skeletal muscle alterations or unmasked a latent subclinical autoimmune condition.
Direct Localized Injury
Some xenobiotics may exert localized toxicity when injected into or near the muscle. Focal damage (i.e., degeneration, necrosis, and hemorrhage) may occur following intramuscular injection of narcotic analgesics and oxytetracycline. Fibrosis of overlying connective tissue, fascia, and skin has been noted after frequent repeated intramuscular administration of agents such as pentazocine and meperidine. Experimentally, all local anesthetics have been found to induce injury to skeletal muscles with procaine causing the least and bupivacaine the most myotoxicity. Other xenobiotics can produce diffuse damage when given systemically. It is apparent that myopathic effects can occur when a substance directly alters critical structural or functional components of the muscle fiber.
Cell Membrane Alterations
The sarcolemma represents an important cellular component that is usually exposed to the highest concentration of a substance. Some xenobiotics may change the electrical properties of the membrane. Muscle weakness, which occurs when concentrations of potassium are reduced, is apparently the result of a decrease in membrane excitability. Severe hypokalemia may lead to muscle fiber necrosis. In contrast, increased membrane excitability is the likely mechanism by which a number of agents (lithium, cimetidine, salbutamol, danazol, and captopril) cause muscle cramping.
The use of lipid-lowering drugs (statins and fibrates) may elicit a myopathy in both clinical and experimental situations. The precise mechanism by which these types of agents induce muscle fiber necrosis has not been completely elucidated, but there is evidence that the function of certain sarcolemmal ion channels is affected. Ion channel alterations may occur because these drugs inhibit the biosynthesis of important cholesterol-related membrane components. In addition, the lipid-lowering drugs also have important intracellular effects, which likely contribute to the muscle toxicity.
The hypocholesterolemic agent 20,25-diazacholesterol (no longer in clinical use) interferes with the biosynthesis of cholesterol and, as a result, causes accumulation of the cholesterol precursor desmosterol in the serum, the sarcolemma, and the SR. The presence of excessive amounts of desmosterol in the sarcolemma leads to excessive chloride permeability and myotonia (i.e., inability to relax voluntary muscles). Myotonia also occurs following exposure to the herbicidal compound, 2,4-dichlorophenoxyacetic acid (2,4-D). This agent causes metabolic alterations by inhibiting glucose-6-phosphate dehydrogenase, an effect that leads to membrane and subcellular changes; alterations in ion transport are also provoked. Advanced lesions induced by this agent include vacuolization and muscle fiber necrosis.
Alcohol consumption has been associated with skeletal muscle myopathy. Many pathogenic mechanisms are thought to be responsible for this toxicity, one of which appears to involve alterations in membrane control of electrolyte homeostasis.
Monocarboxylic acids, capable of inhibiting chloride conductance, also produce myotonia. Monensin, a carboxylic acid ionophore that selectively interacts with the voltage-dependent sodium (Nav) channel, consistently causes both skeletal and cardiac muscle necrosis in a variety of animals. Evidence suggests that these effects are due to calcium overloading in myofibers.
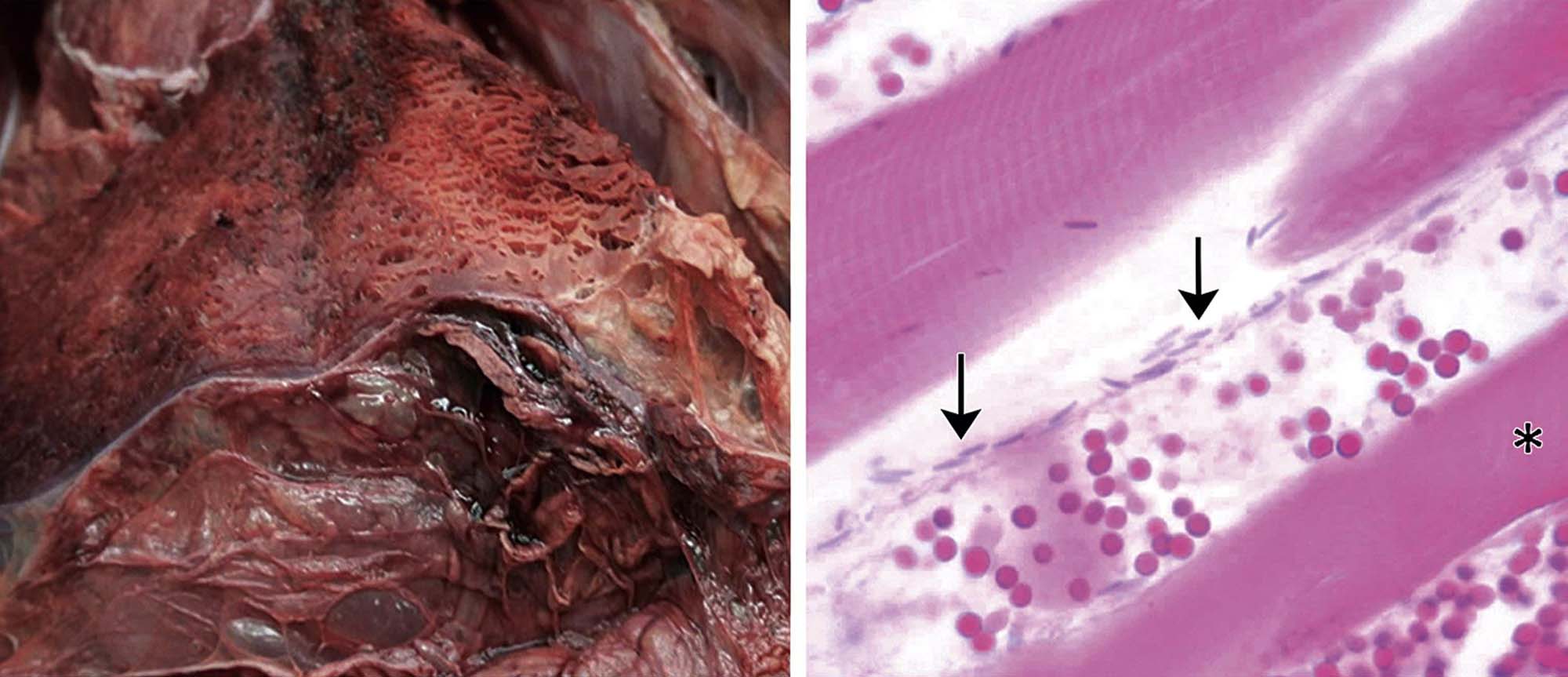
Injection of Type A Clostridium perfringens toxin (CPA) induces a unique myopathy. This toxin initially causes alterations in the sarcolemma. Subsequently, changes occur in the mitochondria and SR, and ultimately lesions are found in certain contractile components (I and Z bands, A-band filaments). These changes arise through the phospholipase activity of CPA, which breaks down cellular membranes. Clostridium chauvoei toxin A (CctA) is a potent cytotoxin that is a key hemolytic and necrotizing component responsible for inducing “blackleg,” an infectious hemorrhagic necrosis affecting the hind quarters of cattle (Figure 10.9) and other ruminants.
Sarcoplasmic Reticulum Alterations
Intraperitoneal injection of doxorubicin damages the diaphragm, an effect which is characterized by dilatation of the SR (appears as cytoplasmic vacuolization), myofibrillar degeneration, interstitial edema, and alteration of mitochondrial membranes. Exposure to doxorubicin has been associated with clinical and experimental evidence of respiratory muscle weakness. Vacuolar myopathic alterations have also been observed in rabbits treated chronically with doxorubicin. The vacuolization and other cellular changes noted in skeletal muscle are similar to those that are well known to be induced, either clinically or experimentally, in cardiac myocytes by anthracycline-type agents such as doxorubicin.
Microtubular Alterations
There are similarities in the overt myotoxic effects of colchicine and vincristine. Both of these agents interfere with the function of microtubules by interacting with tubulin and inhibiting microtubular polymerization. The most common adverse effects reported in patients after treatment with colchicine are generalized myopathy and painful neuromyopathy. Vincristine may initiate proximal muscle weakness and myalgias (i.e., muscle pain) that include denervation and varying degrees of muscle fiber necrosis. Both agents cause comparable vacuolar myopathy, which includes the aggregation of lysosomes and the appearance of autophagic vacuoles. These chemicals cause alterations in one or more other organ systems—an effect that may influence the pathogenesis of the myopathy. It is possible that the tubulin inhibitors share a mechanism with the cationic amphiphilic inducers of phospholipidosis (described below) by inhibiting cellular autophagy and inducing cellular toxicity.
Myofilament Alterations
Myofilament degeneration in certain striated muscles occurs following treatment with plasmocid. In the rat diaphragm, plasmocid initially causes selective loss of actin filaments together with muscle cell Z and I bands. These effects precede the onset of localized myofiber necrosis. It has been suggested that skeletal muscles that are in continual use, such as the masseter, external ocular muscles, and the diaphragm, are most likely to be injured by plasmocid. In cardiac muscle, plasmocid induces mitochondrial dysfunction prior to myocyte necrosis.
Emetine, a constituent of ipecac, produces a pattern of myotoxicity that is similar to the muscle fiber breakdown seen in certain human myopathies. In humans, high doses of emetine cause profound, predominately proximal, myopathy, and cardiomyopathy. In humans and animals, emetine has been found to induce atrophy in both types of striated muscle fibers. Emetine affects intermediate filaments, resulting initially in disruption of the Z discs and subsequently the breakdown of myofilaments in conjunction with accumulation of myofibrillar protein. Multifocal loss of muscle cell cross-striations has been reported to occur in animals treated with emetine. In some areas, loss of oxidative enzyme activity was associated with muscle necrosis. Emetine inhibits protein synthesis, but it is not clear what role this action plays in the myotoxicity. Clinically, some improvement in the myopathy has been reported when use of emetine is terminated.
Lysosomal Alterations
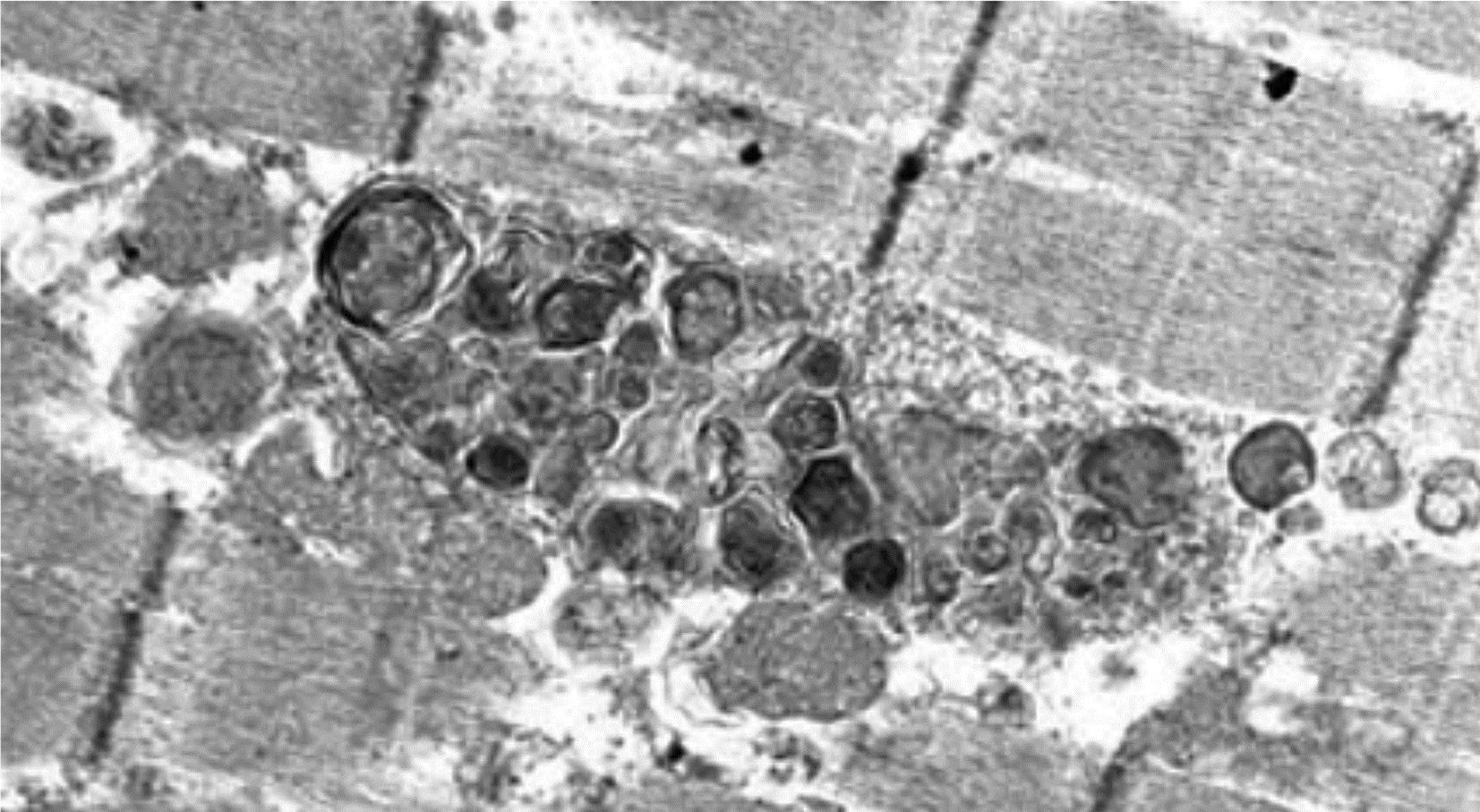
Cationic amphiphilic drugs (CADs) are those whose structure includes both a hydrophilic domain and also a hydrophobic domain that includes a primary or substituted positively charged amine group. These features can facilitate interactions with anionic phospholipids that are constituents of the membranes that surround the sarcolemma and organelles. Drug binding to these phospholipids leads to their intracellular accumulation, which can occur in a variety of cell types and organs including skeletal myofibers. Certain of these cationic amphiphilic agents with diverse pharmacologic actions cause a skeletal myopathy characterized ultrastructurally by lysosomal phospholipid accumulation in the form of large masses of concentric lamellar and curvilinear bodies (i.e., drug-induced phospholipidosis) (Figure 10.10). Type I (slow twitch) myofibers are more frequently affected. The antimalarial chloroquine is associated with this type of myopathy. Chloroquine-induced myopathy is mostly painless, is slowly progressive, and is more pronounced proximally and in the lower extremities. Cardiomyopathy can also occur. Examples of other amphiphilic agents associated with myopathy are amiodarone and perhexiline. The myopathy caused by these substances is part of a more general form of intracellular lipid accumulation, which, in addition to skeletal muscle, affects cells in other tissues such as the peripheral and central nervous systems. These agents are both water- and lipid-soluble, and therefore readily diffuse through the plasma membrane. After passing through the sarcolemma, they become adsorbed onto intracellular membranes and form stable drug–lipid complexes that progressively accumulate within the lysosomes of skeletal muscle cells and other tissues. Apparently, such drug–lipid complexes are resistant to degradation by lysosomal enzymes, which fosters the formation of the autophagic vacuoles that are filled with lamellar material.
Altered Intracellular Calcium Concentration
A potentially serious drug-induced myopathic condition, characterized by muscular rigidity and myoglobinuria, hypermetabolism, and metabolic acidosis, occurs in genetically susceptible individuals undergoing routine surgery. Nonspecific observations obtained from muscle biopsy specimens include variability in fiber size, increased size of internal nuclei, “moth-eaten” fibers, and muscle cell necrosis. Malignant hyperthermia is commonly precipitated in these individuals by exposure to halothane and succinyl choline. Other halogenated anesthetic agents, nitrous oxide, muscle relaxants, and certain local anesthetic agents also may induce this reaction. A condition similar to the human syndrome has been described in certain breeds of pigs. Susceptible pigs are sensitive to stress and develop muscle rigidity, acidosis, and hyperkalemia. Death ensues due to heart failure if these pigs are subjected to stress. These sensitive pigs also react to halothane and depolarizing neuromuscular blocking agents. The malignant hyperthermia reaction appears to result from an excessive release of calcium by the ryanodine receptor (a SR calcium channel). Acute increases in this cation cause contracture of muscle fibers, decreased ATP levels, increased cellular metabolism and core body temperature, and secondary changes in a variety of cellular components.
Altered Protein Synthesis
Prolonged corticosteroid therapy is known to cause a skeletal muscle myopathy. Steroids that are fluorinated in the 9th position (triamcinolone, dexamethasone, and betamethasone) are most likely to be myotoxic, but continuous treatment with any of the corticosteroids will also lead to a myopathy. Clinically, steroids induce proximal weakness and atrophy of the lower and upper limbs. The clinical syndrome tends to be less severe than experimentally induced skeletal muscle alterations. According to some definitions, the steroids do not cause a true myopathy but only atrophy of type II fibers. Slow recovery of muscle function usually occurs when administration with the offending steroid is terminated. Corticosteroids bind to specific target cell receptors and eventually are transported into the nuclei, where they affect the transcription of certain genes. The mechanism of steroid-induced myopathy has been explored in a number of animal studies. These hormones are found to alter multiple myocyte functions, such as protein synthesis (decrease) and degradation (increase), carbohydrate metabolism, sarcolemmal excitability (reduce), and electrolyte homeostasis (yielding hypokalemia). However, the primary mechanism responsible for muscle fiber atrophy appears to be suppression of protein synthesis. This has been attributed to enhancement of the production of myostatin, a protein that inhibits muscle anabolism and promotes proteolysis. Activation of the ubiquitin–proteosome system may also play a role.
Altered Mitochondrial Function
Chronic treatment with nucleoside analog reverse transcriptase inhibitors (NRTIs) such as zidovudine (AZT) has been associated with skeletal and cardiac muscle pathologies. Other NRTI agents, such as zalcitabine (ddC), didanosine (ddI), and lamuvidine (3TC), cause neuropathy while stavudine (d4T) and fialurdine (FIAU) induce myopathy and/or neuropathy and lactic acidosis. Clinical signs of AZT myopathy include progressive proximal muscle weakness, myalgias, and fatigue. In some instances, it has been difficult to separate the effects of human immunodeficiency virus (HIV) disease on skeletal muscle from drug-induced myotoxicity. NRTIs exert clinical activity by competing with certain innate substrates of HIV reverse transcriptase. These agents lack specificity and also block the action of a key skeletal muscle mitochondrial DNA replication enzyme (mitochondrial DNA polymerase gamma). Characteristics of the AZT-induced myopathy include “ragged red” fibers (resulting from accumulation of abnormal mitochondria beneath the sarcolemma), multiple cytochrome c oxidase-negative fibers (indicating reduced mitochondrial capacity for ATP production), biochemical cytochrome oxidase defects, and structurally abnormal mitochondria (when viewed by electron microscopy). Animals given AZT and cells in culture exposed to NRTIs develop similar changes. AZT-induced myopathy is reversible following termination of drug treatment.
Mitochondrial dysfunction has also been implicated in the pathogenesis of statin myopathy. The hypocholesterolemic effect of these agents is related to inhibition of the key enzyme in cholesterol biosynthesis (3-hydroxy-methylglutaryl CoA (HMG-CoA) reductase), which converts HMG-CoA to mevalonate. Clinical expression of myotoxicity in human patients varies from asymptomatic elevations of serum CK activity (i.e., a key cytosolic enzyme found at high levels in skeletal myofibers) to myalgia, cramps, exercise intolerance, fatigability, and rarely muscle fiber necrosis as well as potentially fatal rhabdomyolysis.
Animal studies of the mechanism and lesions produced in skeletal muscle by statin treatment show selective damage to Type IIB fibers. Early ultrastructural alterations involve mitochondrial damage with subsarcolemmal accumulations of myelinoid whorls with fragments of mitochondria. Although multiple mechanisms of muscle fiber injury have been advanced, a central role of mitochondrial function impairment with resulting alterations in intrafiber calcium homeostasis is likely.
The histopathologic alterations produced in rats by treatment with statins showed scattered myofiber necrosis with or without infiltration of inflammatory cells and evidence of myofiber regeneration. Many muscles were affected, but some muscles including the soleus, tongue, masseter, diaphragm, and flexor carpi ulnaris were spared.
In addition to its role in cholesterol biosynthesis, mevalonate is a precursor for intermediates that are key substrates of coenzyme Q10 (ubiquinone), which plays a key role in the mitochondrial electron transport chain. Accordingly a proposed mechanism for statin myopathy has been reduction in coenzyme Q10 leading to impaired muscle energy metabolism. Clinical testing of that hypothesis has seen mixed results. Other proposals focused on mitochondrial energy metabolism include carnitine palmitoyltransferase II deficiency and carnitine abnormalities.
Skeletal muscle atrophy is a recognized consequence of advanced neoplasia (i.e., “cachexia of cancer”). This condition is attributed to induction of inflammatory cytokines. Evidence also suggests that the drugs used to treat cancer may be culprits. Clinically, patients may experience muscle pain and weakness, exercise intolerance, and persistent fatigue. Since several classes of cancer drugs, such as alkylating agents (cisplatin), antitumor antibiotics (e.g., anthracyclines), and antimetabolites (e.g., 5-fluorouracil, capecitabine, gemcitabine), act directly or indirectly but nonselectively to interfere with DNA or RNA replication or synthesis, mitochondria may be targets.
Altered Muscle Cell Differentiation
Information regarding the possible effect of exposure to xenobiotics during pregnancy on subsequent growth and development of skeletal muscle in the fetus is inconclusive. In tissue culture studies, some drugs seem to interfere with certain aspects of muscle cell differentiation. For example, prenatal exposure of rats to 6-mercaptopurine (a purine analog that disrupts nucleic acid synthesis in rapidly dividing cells) leads, after a latent period, to a continuous and progressive degeneration of muscle cells. These myotoxic effects are not seen when 6-mercaptopurine is administered during the postnatal period. Intrauterine exposure of pregnant rats to low doses of chloroacetonitrile causes fetal skeletal muscle tissue alterations in which the fibers were distorted, smaller in size, and widely separated by connective tissue. Skeletal muscle mitochondrial myopathy has been observed in fetal monkeys whose mothers were exposed to zidovudine (AZT) during pregnancy.
Summary
The skeletal muscle system is an important and potentially less appreciated target of xenobiotic-induced injury. However, its contribution to overall body mass and important roles in structural conformation and locomotion significantly impact general health and well-being. Though important elements of structure and physiology of skeletal and cardiac myocytes are shared, skeletal myocytes have an important regenerative capability that cardiomyocytes do not. Accordingly, mechanisms of xenobiotic-induced injury often are common between the two muscle types, but the consequences to the individual’s overall health typically are very different.
Contraction and relaxation of skeletal myocytes are initiated by changes in action potentials across cell membranes influenced by neuromuscular signals and facilitated by intracellular calcium gradients and mitochondrial energy production. All of these activities are important targets that may lead to skeletal muscle toxicity. Basic cellular maintenance functions like protein and nucleic acid synthesis as well as protein degradation and membrane production are also targets. The long-term consequences of xenobiotic-induced skeletal muscle injury are influenced by the severity and duration of injury as well as the impact on the regenerative capacity of the myocytes. Pathologists tasked with evaluating skeletal muscle for potential toxic effects will need to be able to differentiate one-time (i.e., monophasic) from intermittent or continuous (i.e., polyphasic) lesions, and to recognize the attributes of damaged tissue in the midst of repair.