Male Reproductive System
Dianne M. Creasy1 and Robert E. Chapin2, 1Dianne Creasy Consulting, Diss, Norfolk, United Kingdom, 2Retired, Pfizer Global R&D, Groton, CT, United States
Abstract
Much of the work of the pathologist in understanding male reproductive toxicity comes from trying to understand the initial site of toxicant action—that is, distinguishing the initial effects from the follow-on events. These changes can happen because of, or completely independent of, initial impacts on male reproductive hormones. Fortunately, the patterns of response and the holistic evaluation of a wide variety of different but complementary data allow for considerable confidence in assigning an initiating site of action. This chapter will review the anatomy and physiology as well as successful strategies for understanding toxicities in this complicated and sublime multiorgan system.
Keywords
Testis; epididymis; Sertoli cell; Leydig cell; germ cell; testosterone; luteinizing hormone; follicle stimulating hormone; efferent ducts; seminal vesicles; prostate; spermatogenesis; staging; mechanisms of toxicity; histology; toxicologic pathology; background pathology; organ weights; reproductive hormones; immaturity; rat; dog; monkey
Introduction
Recent concerns about falling sperm counts and the likely involvement of environmental endocrine disruptors in the declining reproductive health of human and wildlife populations has galvanized public awareness of the potential for environmental contaminants, occupational exposures, and therapeutic drugs to pose a significant hazard to male fertility. This whole field was underappreciated prior to 1977, when the salutary lesson of dibromochloropropane (DBCP)-induced testicular toxicity became public knowledge. The clear and profound effects produced by DBCP, and the fact that these effects had been noted in animals several years earlier and then overlooked, was a wake-up call about the potential vulnerability of the male reproductive system to environmental exposures.
From 1977 until the mid-1990s, the focus in human male reproductive health was primarily on occupational exposures to adult men and animal models of such exposures. Heavy metals such as cadmium, lead, and copper along with pesticides such as Kepone, ethylene dibromide, and carbaryl have all adversely impacted sperm measures (mostly count but also structure or motility) and/or fertility in men, mirroring their effects previously seen in animals. Solvents, such as the glycol ethers, were prominent tools in preclinical reproductive toxicology in the early 1980s, and two glycol ether congeners (2-ethoxyethanol and 2-methoxyethanol) were eventually found to affect sperm counts in occupationally exposed men, consistent with the effects noted earlier in animals.
Then, in the early 1990s, prenatal exposure to diethylstilbestrol was linked to malformations of the male reproductive tract in the offspring (its effects on females had been noted much earlier). It soon became clear that such effects could be produced by compounds which altered hormonal signaling in the fetus. Further work revealed that there were numerous ways to disrupt the endocrine system, from direct receptor interaction to reduced hormone synthesis. The pervasiveness of these endocrine disruptors has led to significant increases in regulatory oversight, and large new testing programs have been instituted by regulatory agencies. While the impacts of fetal and neonatal exposures will be touched on briefly, the bulk of this chapter will concentrate on the evaluation of tissue from adults after postpubertal exposures.
Male fertility can be impacted by disturbances at sites other than the testis, and it is necessary to evaluate the system as a whole and consider the functions of the individual components. In addition to the testis, this includes the excurrent ducts which transport, modify, and store sperm prior to ejaculation, the accessory sex glands, and the hypothalamic–pituitary–gonadal (HPG) axis, which provides overall regulation of the reproductive system.
Broadly speaking, reproductive embryology, anatomy, and physiology are similar between mammalian species, but there are detailed differences, some of which may have a significant impact on a species response to toxicants. Since the rat is the most common species studied in toxicologic pathology and for which most is known regarding reproductive physiology, it will be the primary species discussed in this chapter, but important species differences will be covered where relevant and when known.
Identification and evaluation of male reproductive toxicity requires integration of information from a variety of endpoints other than morphology. Male infertility can be mediated through alterations in sperm function that are not associated with any alterations in testicular or epididymal morphology, and so, an integrated assessment of as many endpoints as possible (organ weights, histomorphology, sperm parameters, hormonal measurements, and fertility endpoints) provides the most thorough and holistic evaluation. In most cases, multiple endpoints will be affected. The profile of changes identified often allows valuable insight into the possible site and mechanism of toxicity, and the likely relevance of the changes to man.
Morphologic evaluation of the testis is accepted to be the most sensitive method for identifying most male reproductive toxicants, but that sensitivity is directly proportional to the level of knowledge and understanding that the pathologist has of the spermatogenic process and spermatogenic organization within the seminiferous tubules. Subtle morphologic changes in germ-cell morphology or in germ-cell organization can result in dramatic effects on fertility, and frequently the only way a pathologist is able to recognize such subtle changes is by conducting a “stage-aware” evaluation. This means having a general knowledge of the shape and size of the different germ cell types as they progress through spermatogenesis, as well as which cell types occur in association with one another.
Light microscopic examination of the entire reproductive tract is the foundation of identifying male reproductive toxicity. Once identified, there is often a desire to provide a mechanistic explanation for the toxicity, but due to the complexity of cellular interactions within the testis and the interdependence of the various cell types, isolating and probing specific cell types outside of their normal in vivo environment is beset with problems. Even separating the individual cell types is technically challenging, and, certainly, once isolated and purified these cells are no longer the same as they were in situ. Such purified preparations are of limited usefulness in identifying the rich intercellular mechanisms of toxicity.
Structure, Function, and Cell Biology
Embryonic Development
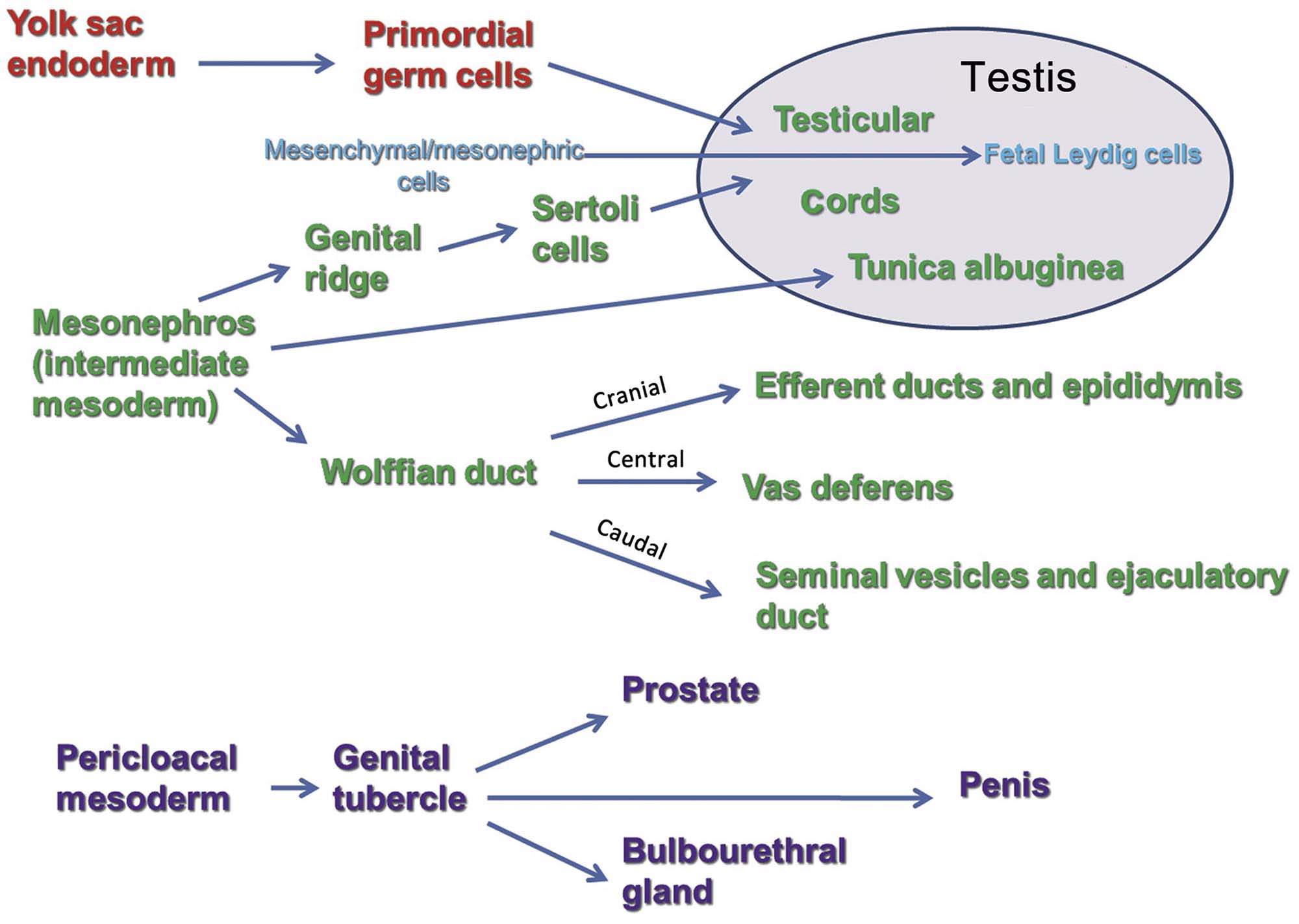
The primordial gonad develops as the genital ridge, which grows along the coelemic surface of the mesonephros. Primordial germ cells migrate from the yolk sac endoderm and combine with somatic (Sertoli) cells from the mesonephros to form the seminiferous (medullary) cords. Once these cords are formed, the fetal Leydig cells begin differentiating from mesenchymal cells, which are present in the interstitial tissue between the cords. This differentiation is thought to be under the control of the Sertoli cells (Figure 17.1).
Testosterone, secreted by fetal Leydig cells during the fetal and neonatal period is responsible for the masculinization of various processes and systems, including the central nervous system and sexual behavior. Around postnatal day 10, the fetal Leydig cells regress and are gradually replaced by a different generation of adult Leydig cells, which also differentiate from interstitial mesenchymal cells.
The excurrent duct system, comprising the rete testis, efferent ducts, epididymis, and vas deferens, develops from the Wolffian duct system. The seminal vesicles develop from lateral buds of this duct. However, the prostate and bulbourethral (Cowper’s) glands, along with the urethra and external genitalia, have a different embryologic origin, developing from the urogenital tubercle (Figure 17.1).
In mammalian species, the default phenotypic sex is the female; i.e., for a phenotypic male to develop, a whole series of events must be triggered and coordinated to form the male reproductive system and associated secondary sexual characteristics (Figure 17.2). The failure of certain genes or hormones to be expressed will normally result in a female phenotype. As the gonad emerges in the genital ridge in the embryo, it has the potential to become either a female or a male gonad. The critical event controlling maleness is the expression of the sex-determining gene (Sry), the expression of which is regulated by numerous proteins including Wt1, members of the insulin-receptor family, and a Gata4/Fog2 complex, amongst others. This in turn elicits a whole cascade of events to enable the fetal gonad to develop into a testis. In the fetal gonad, the development of the Sertoli cells plays a major role in how the reproductive system develops. The Sertoli cell synthesizes factors that drive the formation of an extensive vasculature and seminiferous cords. One of the most important of these is Müllerian-inhibiting substance which initiates the removal of the female structures (the Müllerian ducts) that would have formed the oviducts, uterus, and vagina. Sertoli cells also control the normal development of the testosterone-secreting Leydig cells, which then play an important role in the development of the epididymis, vas deferens, and seminal vesicles from the Wolffian ducts (Figure 17.2). Dihydrotestosterone, the testosterone metabolite produced locally from testosterone by the enzyme 5α-reductase, appears crucial for the development of the prostate and external genital structures from the urogenital sinus. The basic developmental processes are quite similar between species, but the timing of certain events show significant species differences. In rodents most of these events take place in late gestation (for the rat, gestation days 12–20), whereas in the human fetus most events occur in the first trimester of pregnancy. Some events show dramatic differences in timing—for example, descent of the testis into the scrotal sac occurs during gestation in the human and is a postnatal event in most other species. Table 17.1 illustrates the various time lines (where known) for some major developmental landmarks of the male reproductive tract in rats, dogs, monkeys, and humans.
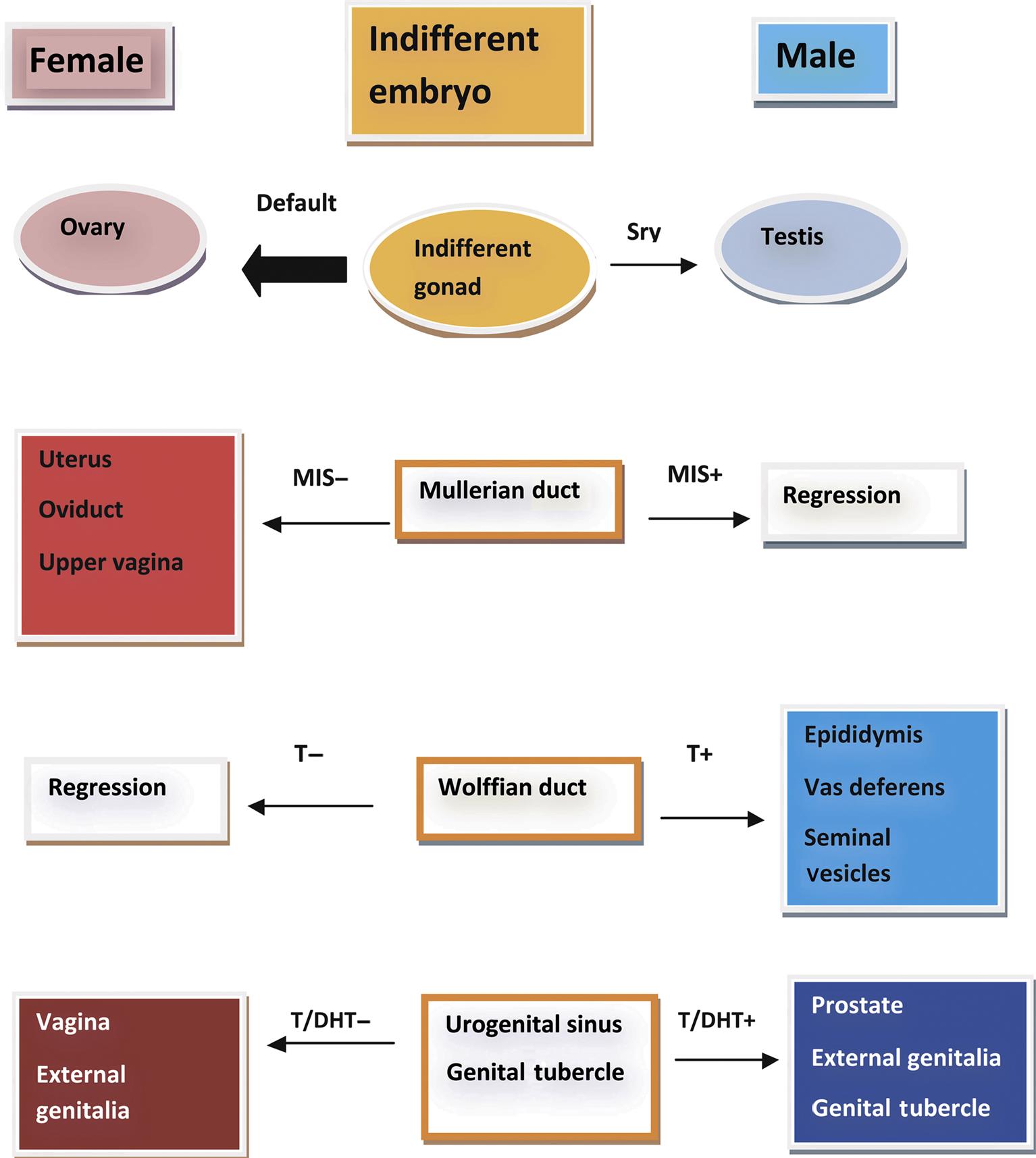
Table 17.1
Approximate Timelines and Hormonal Control for the Development of the Male Reproductive Organs in Rodents, Dogs, Primates, and Humans
| Rat | Dog | Monkey | Human | Event (hormone responsible) |
| GD 8–10 | GW 3 | GW 4–6 | GW 5–6 | Migration of germ cells, precursor Sertoli cells, and interstitial cells to indifferent gonad |
| GD 14 | GW 4 | GW 6 | GW 7–8 | Start of sex cord development, Sertoli cells identifiable |
| GD 15–20 | GW 5–6 | GW 7–9 | GW 8–9 | Fetal Leydig cell proliferation and androgen secretion begins, Wolffian duct development (T), Müllerian duct degeneration (MIS) |
| GD 19.5–PND 10 | N/A | GW 7–20 | GW 16–24 | Seminal vesicles develop (T) |
| GD 15.5–19.5 | GW 5–6 | GW 9–14 | GW 9 | External genitalia start to masculinize; continues through late gestation (DHT) |
| GD 15.5–PND 15 | GW 5–10 | Prostate develops from walls of urogenital sinus (DHT); most development occurs postnatally in rat | ||
| GD 19–PND 4 | GW 9–12 | GW 18–38 | Regression of fetal Leydig cells | |
| GD 21 | GW 9 | GW 22 | GW 40 | Birth |
| PND 13–15 | PNW 8 | PNM 32 | PNY 12 | Sertoli cells stop dividing |
| PND 22 | PND 3–42 | Birth and PNY 3 | GW 24–36 | Testis descent; in monkeys, testes descend at birth and again at ~3 years, returning to the inguinal canal in between (IL3) |
| PND 2–4 | PNM 4.5–5 | PNM 4–5 | PNY 1–10 | Spermatogonia begin to divide |
| PND 15 | PNM 5–6 | PNY 3–3.5 | PNY 11.8–12.2 | Tubular lumen develops |
| PND 15 | PNM 6–7 | PNY 3–3.5 | PNY 11.8–12.2 | Primary spermatocytes appear |
| PND 26 | PNM 6–8 | PNY 3.5–4 | PNY 11.8–14 | Spermatids appear |
| PND 42–45 | PNM 7–10 | PNY 3.5–5 | PNY 11.8–15.3 | Spermatozoa start to be shed |
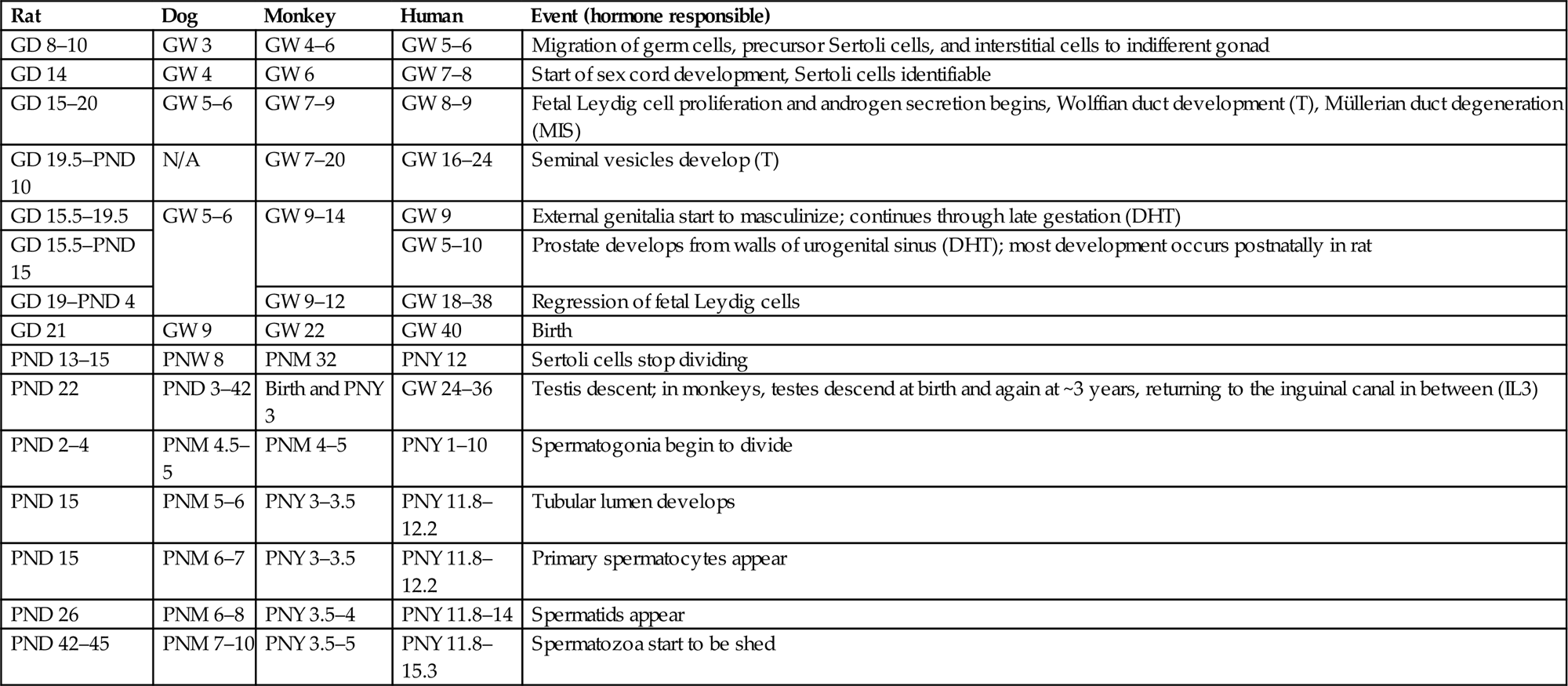
GD, gestation day; GW, gestation week; PND, postnatal day; PNW, postnatal week; PNM, postnatal month; PNY, postnatal year; T, testosterone; DHT, dihydrotestosterone; MIS, Müllerian duct inhibiting substance, IL3, insulin-like growth factor 3. Data have been assimilated from various sources.
Table reproduced from Handbook of Toxicologic Pathology (2013), third ed. (W. M. Haschek, C. G. Rousseaux, and M. A. Wallig, eds.), Academic Press, Table 59.1, p. 2498, with permission.
Postnatal Development of the Reproductive Tract
With the increased concern regarding juvenile toxicity, it is becoming a more common requirement for the pathologist to examine tissues from juvenile animals. The morphologic characteristics of the postnatal developing rat testis have been described by Picut et al. (2015) and a more thorough discussion of postnatal development is provided in Creasy and Chapin (2013). The rate of development and maturation of the reproductive tissues varies greatly between species. In rodents, the initiation of spermatogenesis begins immediately at birth and is slightly accelerated compared with the adult, so that sperm are starting to be released from the testis by 6–7 weeks of age in the rat and 4–5 weeks of age in the mouse. In contrast, larger animals such as the dog, monkey, and human go through a period of dormancy before spermatogenesis begins. The testes will not begin spermatogenesis prior to 5 months of age in the Beagle dog or 3 years of age in the cynomolgus monkey. Even though this is the age at which sperm starts to be produced, it should be appreciated that the efficiency of spermatogenesis and therefore the number of sperm and size of the reproductive organs will continue to increase over a longer period of time. In general, maturation follows a very consistent and predictable time line in rodents, with most animals reaching sexual maturity at the same age ±1 week, but there is significant individual variation in the rate of maturation in dogs and monkeys. In dogs, the age of sexual maturation can be anywhere between 7 and 12 months of age; in the cynomolgus monkey, it can be anywhere between 3.5 and 5.0 years of age. This has very important implications when attempting to evaluate the effects of test materials on sexual development and maturation in the small group sizes used for large animal studies. Table 17.1 provides an approximate timing for some major postnatal maturational changes in the testis of the rat, dog, monkey, and human.
Sertoli Cell Division and Maturation
In all species, Sertoli cells have a limited period in which they divide. In the rat, division starts at GD 12 and ceases (in vivo and in vitro) after approximately post natal day (PND) 13–15. This period of Sertoli cell division is critical, because the final sperm output is a function of the number of Sertoli cells in the testis. If division ceases too soon (due to juvenile or maternal hyperthyroidism, for example), it can result in smaller testes and reduced sperm output. Sertoli cell proliferation can also be reduced by too much estrogen, insufficient testosterone, and insufficient follicle-stimulating hormone (FSH). On the other hand, hypothyroidism delays the cessation of Sertoli divisions, which results in enlarged adult testis size and increased overall sperm output. Sertoli cell numbers are decreased in mice with inactivated androgen receptors, including the androgen receptor knockout and the testicular feminized mouse. Around this same time (PND 15) the Sertoli cell begins to produce seminiferous tubular fluid and androgen binding protein (ABP) and also undergoes significant maturational changes in its function and its regulation. FSH is the main regulatory hormone in the testis up until about PND 18, when its secretion peaks, after which time FSH levels decline and luteinizing hormone (LH) secretion increases with a commensurate increase in testosterone, androgen receptors, and ABP. By PND 25, testosterone is the major regulatory hormone in the testis.
In the rat, tight junctions between adjacent Sertoli cells, which will form the major part of the blood–tubule barrier, begin to develop 15–18 days after birth, which is coincident with the beginning of fluid secretion into the tubular lumen. Prior to this age, gap junctions join the Sertoli cells and substances permeate the tubular epithelium freely.
The dog and the cynomolgus monkey both have long postnatal quiescent periods (up to 4–5 months in the dog and 3–4 years of age in the monkey) with no activity in the seminiferous tubules other than the very slow final expansion of the stem cell spermatogonia and Sertoli populations.
Structure and Function of the Testis
Seminiferous Tubules
The testicular parenchyma is composed of convoluted seminiferous tubules separated by interstitial tissue and enclosed in a capsule. The seminiferous tubules are long, highly convoluted tubes that empty at both ends into the rete testis, which is a sac like reservoir. In the rat there are approximately 30 separate tubules that are arranged in a transverse circumferential organization. In the dog, monkey, and man the individual seminiferous tubules are grouped into lobules.
Leydig (Interstitial) Cells and Testicular Macrophages
Leydig cells comprise about 17% of the interstitial tissue in rats, in contrast to pigs, where Leydig cells make up around 50%. Mice have more and larger Leydig cells than rats, amounting to 35% of the volume of the interstitial tissue. Leydig cells are the major site for the synthesis of the predominant male steroid hormone, testosterone. Following secretion from the Leydig cell and entry into the peripheral circulation testosterone is metabolized further in the liver, androgen-dependent tissues (e.g., epididymis, seminal vesicles, and prostate) and a variety of peripheral tissues.
Testicular macrophages are a unique subset of the mononuclear phagocyte system that have close physical and functional interactions with the Leydig cells and participate in paracrine regulation and in the immuno-endocrinology of the testis.
Sertoli Cells
Structure
Sertoli cells provide the supporting framework in which the germ cells are embedded. The size and shape of the Sertoli cell are difficult to appreciate in conventional sections viewed by light microscopy. The germ cells are embedded into apical or lateral invaginations of the Sertoli cells (Figure 17.3). The Sertoli cell is connected to the germ cells by a variety of cell junctions, some of which are unique to this cell type. Adjacent Sertoli cells are joined at their basolateral aspect by specialized occluding junctions, which form the major component of the blood–testis barrier and help shield the nonself germ cells from the interstitial macrophages.
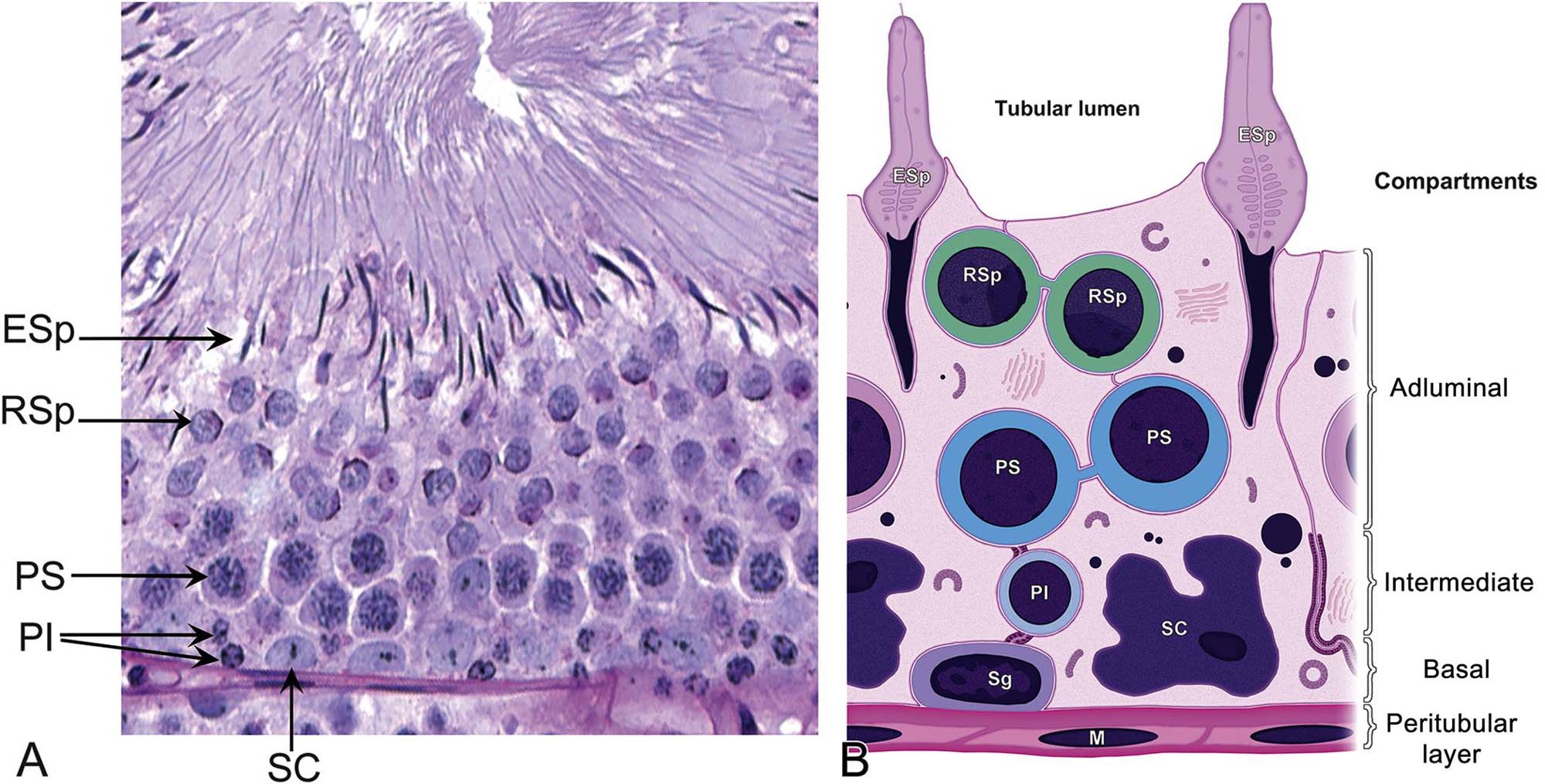
Normal Sertoli cell function is critical for the integrity of the seminiferous epithelium. Generally speaking, these functions include regulation of spermatogenesis, structural and metabolic support of the germ cells, sperm release, secretion of tubular fluid for sperm transport, and maintenance of a permeability barrier between interstitial and tubular compartments. Disturbance of any of these functions is likely to impair spermatogenesis and, in turn, sperm production.
Role in Structural Support and Germ Cell Translocation
The structural support and movement of germ cells is central to the process of spermatogenesis. As germ cells develop from spermatogonia to spermatids they are gradually moved up to the luminal surface, and then, as elongating spermatids, they are pulled back down towards the Sertoli cell nucleus before returning to the surface to be released. The cytoskeleton is thought to have a major role in this process along with the specialized junctions between the Sertoli and germ cells. Disruption of the cytoskeletal filaments and retraction of the lateral processes that surround germ cells are associated with sloughing of germ cells into the lumen.
Role in Metabolic Support of Germ Cells and Fluid Production
Germ cells in the adluminal compartment rely entirely on the Sertoli cell for their supply of nutrients and oxygen. Spermatocytes and spermatids are unable to utilize glucose and must be provided with lactate and/or pyruvate by the Sertoli cell as a substrate for energy production. Similarly, the metabolism of amino acids occurs within the Sertoli cell to produce α-keto acids that are utilized by the germ cells. In addition, the Sertoli cells secrete fluid which carries the newly released sperm to the epididymis for their acquisition of motility.
Role in Phagocytosis
Phagocytosis is another important function of the Sertoli cell. During spermatid development (spermiogenesis), the cytoplasmic volume of the spermatid is reduced by up to 70% and a large proportion of its organelles are discarded. This is accomplished by formation of the residual body, which contains redundant organelles, nucleic acids (various RNA species), and cytoplasm; it is shed from the spermatozoon at the time of spermiation, phagocytized by the Sertoli cell and merged with lysosomes while being moved down to the basal Sertoli cell cytoplasm, where digestion is completed. Apoptotic germ cells, which are a normal feature of spermatogenesis as well as a result of toxic injury, are also removed rapidly by phagocytosis.
Role in Blood–Testis Barrier Formation
Spermatogenesis occurs in a specialized and protected environment in the adluminal compartment of the seminiferous epithelium. The specialized conditions are maintained and protected by the presence of a blood–tubule barrier that shares some of the same exclusion properties as the blood–brain barrier. The main exclusion barrier is formed by the basolateral occluding cell junctions between adjacent Sertoli cells, situated at a level above the spermatogonia and below the spermatocytes (Figure 17.3). The interstitial compartment is exposed to all substances transported through the capillary endothelium, including toxicants. Although the peritubular cells may exclude some very large molecules, the basal compartment of the tubule, which contains the spermatogonia, is also readily accessible to virtually all bloodborne substances. In contrast, the adlumenal compartment, which contains all meiotic and postmeiotic cells, is only exposed to bloodborne substances that have been transported through the Sertoli cell. This might provide some protection against genetic damage or death of the gamete by toxicants.
Spermatogonia
Spermatogonia represent the stem-cell population of the germ cells. There are three classes of spermatogonia: stem cell spermatogonia, proliferative spermatogonia, and differentiating spermatogonia. Stem cell and proliferative spermatogonia are responsible for renewing their own cell number and producing a pool of spermatogonia that are committed to differentiation. Six mitotic divisions of these differentiating spermatogonia result in the main expansion of the spermatogonial population. As the cells undergo mitosis, cytokinesis is incomplete, leaving the descendent population of spermatocytes linked together in a syncytial arrangement. This is maintained throughout spermatogenesis and is believed to enable the synchronized development and differentiation of the individual populations of cells. It also underlies the generation of multinucleated giant cells after treatment with toxicants (below).
Spermatocytes
Preleptotene spermatocytes are formed from the final mitotic division of the differentiating spermatogonia. The cells then enter a 3-week-long meiotic prophase, passing through the leptotene, zygotene, pachytene and diplotene stages, and diakinesis, and the first meiotic division to produce secondary spermatocytes. The second meiotic division follows rapidly to produce the haploid spermatid.
Spermatids
The second meiotic division of the spermatocytes results in the formation of the haploid spermatid. The spermatid begins life as a conventional round cell, but the nucleus and cytoplasm undergo a number of extremely complex morphological modifications including the development of an acrosomal cap on the nucleus, profound shape changes, and condensation of the nucleus to form an elongated head, while the cytoplasm and mitochondria are totally rearranged to form a motile-tail section. The process of spermatid transformation is termed spermiogenesis.
In the final maturation phase of spermiogenesis, the cytoplasmic volume of the spermatid is greatly reduced by the condensation and expulsion of the spermatid cytoplasm including many of the redundant organelles and mRNA to form the residual body, which is shed to the Sertoli cell at the time of spermiation. One metabolic function retained by the spermatozoa is energy production by the sheath of mitochondria enveloping the tail to permit motility. Most of the other metabolic requirements of the mature spermatid within the testis are met by the Sertoli cells; outside the testis, the quiescent spermatozoon is sustained by the complex mixture of fluids secreted by the epididymis and accessory sex organs.
Spermatogenesis and the Spermatogenic Cycle
The process whereby primitive stem cell spermatogonia develop to form highly specialized spermatozoa is termed spermatogenesis. The process of spermatogenesis comprises a series of successive mitotic divisions by the spermatogonial population, meiosis by the spermatocytes, extensive cellular remodeling, and differentiation throughout the haploid–spermatid development (spermiogenesis) (Figure 17.4). Four generations of germ cells develop simultaneously within the seminiferous epithelium of the rat; their synchronous development gives rise to specific cellular associations that follow each other in a precisely defined sequence. One unit of repetition of the sequence of cellular associations is termed a cycle of the seminiferous epithelium (often abbreviated to spermatogenic cycle), whereas individual cell associations within this cycle are referred to as stages of the cycle (Figure 17.5A).
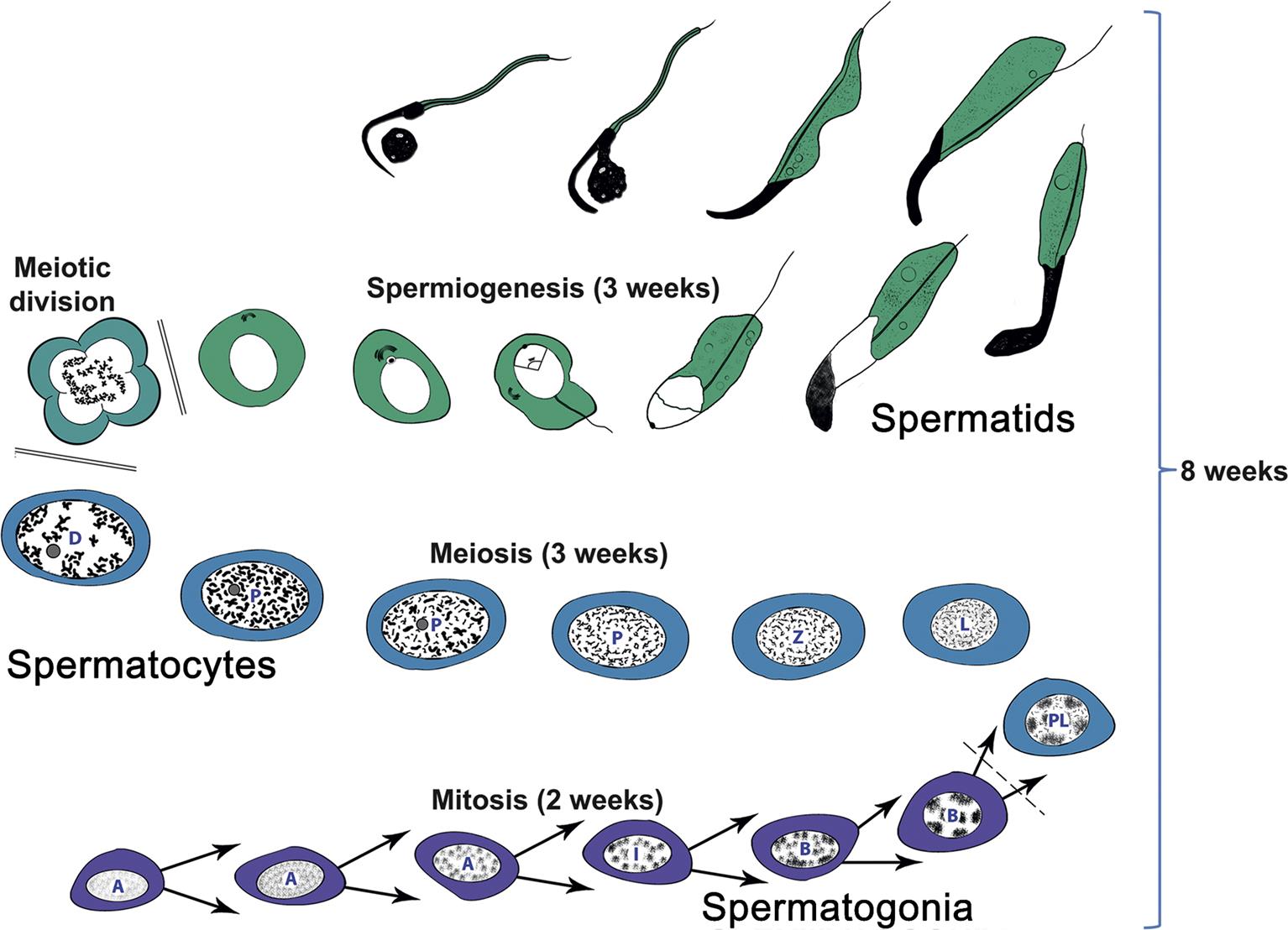

(B) Approximate staging of the spermatogenic cycle can be conducted in H&E stained sections by using the shape and position of the elongating spermatids in the epithelium rather than the detailed acrosome morphology of the round spermatids in a PAS section. In general, this is adequate for routine screening of testes for spermatogenic disturbances. Source: (A) Figure reproduced from Haschek and Rousseaux’s Handbook of Toxicologic Pathology (2013), third ed. (W.M. Haschek, C.G. Rousseaux and M.A. Wallig, eds.), Academic Press (Elsevier), Figure 59.9B, p. 2510 with permission. (B) Figure reproduced from Haschek and Rousseaux’s Handbook of Toxicologic Pathology (2013), third ed. (W.M. Haschek, C.G. Rousseaux and M.A. Wallig, eds.), Academic Press (Elsevier), Figure 59.9C, p. 2511 with permission.
Detailed classification of the rat spermatogenic cycle into stages is largely based on the developing morphology of the spermatid acrosome, which is visualized using periodic acid Schiff (PAS) stain. In the rat, 19 different steps of spermatid development, which are denoted by Arabic numerals (1–19) are used as a marker to identify 14 different cellular associations (or stages), referred to by roman numerals (I–XIV) (Figure 17.5A). (For more detailed guidance on staging, see Russell et al., 1990; Creasy, 1997; Creasy and Chapin, 2013) The number of stages into which the cycle is divided is arbitrary and depends on the classification criteria used by the originator of the particular scheme. The most commonly used schemes divide the mouse cycle into 12 stages, the dog cycle into 8 stages and the cynomolgus monkey cycle into 12 stages (Table 17.2). Maps similar to the one depicted in Figure 17.5A have now been devised for most of the common laboratory species. Although accurate staging of testes requires examination of the spermatid acrosome at high magnification in PAS-stained testes, an easier method of staging can be performed on H&E-stained sections of testes using the position and shape of the elongating spermatid as it moves from a luminal position to a basal position and back again (Figure 17.5B). This approximate staging of the cycle is generally adequate for most routine examinations and will allow the pathologist to readily distinguish between early, mid, and late stages of the spermatogenic cycle (Figure 17.6). Also see Creasy and Chapin (2013).
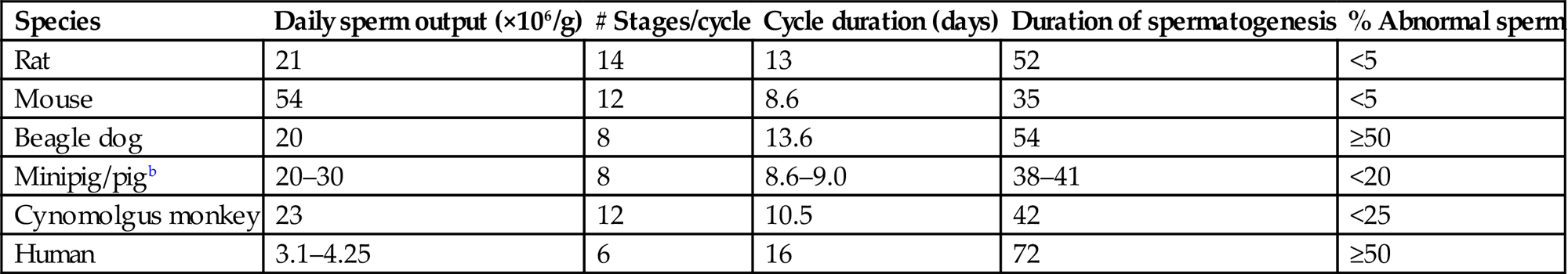
Table 17.2
Species Differences in the Spermatogenic Cycle and Sperm Outputa
| Species | Daily sperm output (×106/g) | # Stages/cycle | Cycle duration (days) | Duration of spermatogenesis | % Abnormal sperm |
| Rat | 21 | 14 | 13 | 52 | <5 |
| Mouse | 54 | 12 | 8.6 | 35 | <5 |
| Beagle dog | 20 | 8 | 13.6 | 54 | ≥50 |
| Minipig/pigb | 20–30 | 8 | 8.6–9.0 | 38–41 | <20 |
| Cynomolgus monkey | 23 | 12 | 10.5 | 42 | <25 |
| Human | 3.1–4.25 | 6 | 16 | 72 | ≥50 |
aValues are an approximation based on data derived from various sources.
bRelatively few data are available for the Göttingen minipig. The numbers provided are based on data published for the minipig, and domestic and wild boars.
Table reproduced from Handbook of Toxicologic Pathology (2013), third ed. (W. M. Haschek, C. G. Rousseaux, and M. A. Wallig, eds.), Academic Press, Table 59.2, p 2512, with permission.

Dynamics of the Spermatogenic Cycle
In all species, the entire process from stem cell spermatogonium to spermatozoal release spans approximately 4 cycles (depending when the start of spermatogenesis is defined). As each cycle is completed and the mature spermatozoa are released, another generation of spermatogonia divides and becomes committed to maturation (Figure 17.5A). The duration of the cycle and thus of spermatogenesis is fairly constant for a given species and strain of animal (Table 17.2). Once committed to undergoing spermatogenesis, it takes about 8 weeks in the rat (timing is species-specific) for spermatogonia to complete their development into spermatozoa that are ready to be released into the tubular lumen (Figure 17.5A and B). This process involves an individual spermatogonium passing through four spermatogenic cycles (each cycle lasting approximately 2 weeks) as it develops into a mature spermatid. The practical implications of this process are important when evaluating spermatogenesis. For example, take the development of one spermatogonium in a mid-stage (VII) tubule: on Day 1 it starts spermatogenesis as a type A spermatogonium. Two weeks later that segment of tubule will have passed through the other 14 stages of the spermatogenic cycle and will reenter stage VII, during which time the type A spermatogonium will have developed into a preleptotene spermatocyte. After another 2 weeks the tubule will reenter stage VII and the preleptotene spermatocyte will have developed into a mid-pachytene spermatocyte, and following another 2 weeks it will be a round spermatid. A further 2 weeks will see the cell become a fully mature elongated spermatid (step 19) that is about to be released into the lumen. This timing is important when evaluating cell degeneration and depletion in studies of different durations. Another important concept to bear in mind is the progressive expansion of each population of cells as it develops. Beginning with a single stem cell spermatogonium, by the time it has completed its multiple mitotic divisions, in theory it will produce 512 spermatocytes and, following another two meiotic divisions, there will be 4096 round spermatids for every one spermatogonium that enters the maturation sequence. Although there are regulatory processes that utilize apoptosis to reduce the population to an appropriate number that can be supported by each Sertoli cell, the important message is that there will be relatively few spermatogonia (they will be difficult to find), many more spermatocytes and a lot more spermatids, which will be about four times more numerous than spermatocytes. In practical terms, this means that it will be a lot easier to identify germ cell loss if it affects or has reached the spermatid population than if it is only affecting the spermatogonial or early spermatocyte population (see the “Germ Cell Depletion” section).
It is important to appreciate the dynamics of germ-cell development when evaluating toxicity because toxicity to a cell type at the beginning of a study will translate into depletion of a different cell type at the end of the study, and the cell type depleted will depend on the duration of dosing. Similarly, if certain cells have been affected by a drug during the dosing phase, the degree of recovery will depend on the duration of the recovery period and the cell type originally damaged by the drug. This progressive loss of more mature germ-cell types due to failed development of removed early germ cell types, explains why testes often appear to have more severe depletion of cells at the end of a 2- or 4-week recovery period than at the end of the dosing period (see “Recovery and Reversibility of Injury” section).
Structure and Function of the Rete Testis, Efferent Ducts, and Epididymis
When sperm are released into the lumen of the seminiferous tubule they are transported into a common collecting duct within the testis, termed the rete testis (Figure 17.7). Sperm then pass into a network of efferent ducts, which in the rat are located within the epididymal fat pad, and these in turn lead into a single highly coiled duct to form the epididymis. The epididymis is divided into four major parts: the initial segment, the caput (head), corpus (body), and cauda (tail) epididymis. The coiled epididymal duct then leads into a straight portion termed the ductus or vas deferens, which extends through to the urethra. Various glands empty their secretions into this duct.
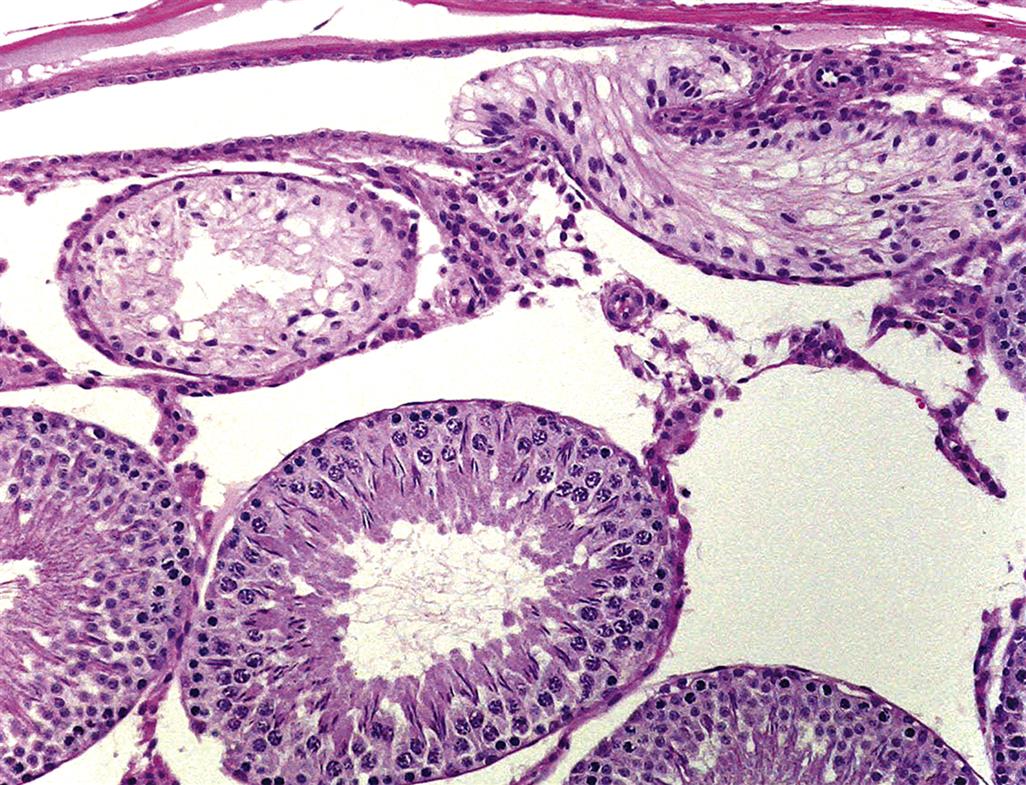
When sperm leave the testis, they are incapable of fertilization and are barely motile. They are transported out of the testis in seminiferous tubule fluid (STF) that is secreted by the Sertoli cell. Most of the fluid (up to 96%) and many of the proteins in the STF are reabsorbed by the rete testis and efferent ducts and the initial segment of the epididymis. During their passage through the epididymis the sperm are bathed in the secretions of the cells of the epididymal epithelium and thus, mature and acquire progressive forward motility and the ability to undergo capacitation and fertilize an oocyte. Maturation involves extensive remodeling of the sperm plasma membrane with the binding and incorporation of proteins synthesized by the epididymal epithelium. Once in the cauda epididymis, the sperm are stored for days (humans) or weeks (rodents) prior to being released by ejaculation through the vas deferens into the penile urethra.
Rete Testis and Efferent Ducts
The rete testis comprises a single or series of interconnected channels, lined by simple cuboidal or columnar epithelium, into which the seminiferous tubules open and which leads into the efferent ducts. In some species, such as the rat and mouse, the rete is situated in a subcapsular position at the cranial pole of the testis, but in other species, including the dog, rabbit, and monkey it forms a series of ducts running through the center of the testis.
The efferent ducts (ductuli efferentes) are a series of convoluted tubules that empty into the initial segment of the epididymis. In the case of the rodent, almost all of the long and tortuous efferent ducts (proximal and distal sections) are located within the epididymal fat pad, making it difficult to sample them, and so they are rarely examined. The final distal portion may occasionally be seen in standard sections of the rodent epididymis. In contrast, the efferent ducts of the dog are much shorter, being almost entirely located within the head of the epididymis and forming a large proportion of the epididymal head. This is also the case in man. In cynomolgus monkeys, the efferent ducts are enclosed within the same connective tissue capsule as the head of the epididymis but are located slightly proximal to the head and may or may not be sampled in a routine section of epididymis.
The main function of the efferent ducts is to transport sperm from the rete testis to the epididymis and to concentrate them by reabsorbing most of the fluid and the proteins from the accompanying STF. Over 90% of the fluid and protein is reabsorbed as the sperm suspension passes through the rat efferent ducts. Estrogen and endothelin 1 appear to be the major regulator for the fluid reabsorption. Active transport of water out of the lumen is accomplished in a similar manner to reabsorption of water from the renal nephron.
Epididymis
Superficially, the epididymis appears to be a histologically simple tissue comprising a convoluted, epithelial lined duct that carries sperm from the testis to the vas deferens. In reality it is surprisingly complex, both in the structural diversity of its epithelial cell types and in the number of critical functions it performs in the maturation and transport of sperm.
Functions of the epididymal epithelium include the synthesis and secretion of a wide range of proteins, and the transport of ions and small organic molecules across the epithelium to create a slightly basic luminal environment which is conducive to protein uptake into the sperm membranes. After early secretion into the lumen of the caput, the corpus is where the sperm incubate in the specialized luminal environment, followed by the selective absorption of luminal contents in the distal corpus and cauda. Thus, the epididymis provides the specialized environment required for the progressive maturation of sperm as they pass from the initial segment to the tail of the epididymis, during which time the sperm acquires the ability to fuse with, and fertilize, an oocyte. Newly motile sperm must then be maintained in a quiescent state by the caudal secretion of immobilin, which prevents sperm from swimming in the epididymis. This thick glycoprotein is stripped off the sperm by the process of ejaculation and the mixing with the proteases in the accessory organ secretions, resulting in ejaculated sperm which are motile.
Storage of the sperm prior to ejaculation is also an important function of the cauda epididymis. Depending on the species and the frequency of ejaculation, sperm reach the cauda 1–2 weeks after release from the seminiferous tubule. Sperm production is a continuous process; if the stored sperm are not removed by frequent ejaculation, they are voided in the urine.
Structure and Function of the Accessory Sex Organs
The accessory sex organs in rodents are the seminal vesicles, prostate, coagulating gland, bulbourethral gland, and preputial gland. They are located along the route of the urethra as it relays sperm from the vas deferens out through the penis. The glands secrete a variety of complex fluids that transport and sustain the sperm during their lengthy journey out of the male and through the female genital tract. Their structure is typical of active exocrine secretory glands, although the characteristics of the individual secretions are markedly different. Because the secretory activity of the accessory sex glands is extremely sensitive to androgen levels, weight change and altered cellular activity in the ventral prostate and seminal vesicles can be used as a good, and relatively rapid, indicator of altered circulating androgen levels. There are major species differences in the complement of accessory sex glands.
Prostate
The prostate is the only accessory sex gland that is present in the dog. In all species the glandular tissue forms multiple lobes around the urethra, but in the nonhuman primate the glandular tissue is only present on the posterior surface of the urethra and there are no anterior lobes. In the rodent, a discrete pair of ventral lobes and a smaller group of dorsal and lateral lobes are situated around the neck of the bladder. Also in rodents, a pair of anterior lobes, otherwise known as the coagulating glands, is situated along the inner curvature of the seminal vesicle. The prostatic fluid secretion constitutes 15%–30% of the ejaculate. It is a colorless fluid rich in proteolytic enzymes (e.g., acid phosphatase). The fluid also contains relatively high levels of zinc, inositol, transferrin, and citric acid.
Seminal Vesicles
The mucosa of the seminal vesicles has a honeycombed structure formed by complex folding. The seminal vesicle fluid is a viscous secretion constituting approximately 70% of the seminal fluid and about 50% of the ejaculate. The alkaline fluid is thought to neutralize the acid pH of the vagina; it contains citric acid as well as fructose and lactoferrin. Lactoferrin is one of the sperm-coating antigens and, as its name suggests, is also involved in iron binding and transport. In the rat, fluid from the coagulation gland, secreted immediately after ejaculation, mixes with vesicular fluid to form the copulatory plug within the vagina. This prevents loss of sperm and further copulation. The dog does not have seminal vesicles or coagulating glands. Primates have seminal vesicles but do not have coagulating glands.
Bulbourethral Glands
Bulbourethral glands (Cowper’s glands and Mery glands) are paired compound tubuloalveolar glands that secrete a mucoid material into the penile urethra during ejaculation. In rodents, it is secreted immediately before ejaculation of the sperm, to clear the urethra of urine and provide lubrication. It is not present in the dog.
Preputial Glands
Preputial glands are paired sebaceous glands located in the subcutaneous tissue of rodents near the tip of the penis in the mouse and along the ventral midline in the inguinal region of the rat. Ducts leading from the sebaceous acini are lined by squamous epithelium. The ductular secretion and the intracellular secretory granules are intensely eosinophilic, resembling keratin in appearance. Secretion from the preputial gland contains pheromones (aliphatic alcohols) and glucuronidase.
Hormonal Regulation of Reproductive Function
Overall hormonal control of the testis is maintained by the hypothalamic–pituitary axis and is mediated by gonadotropin-releasing hormone (GnRH) secreted by the hypothalamus, which regulates secretion of the gonadotropins, LH and, FSH, by the pituitary (Figure 17.8). These major hormones are overlaid by paracrine, autocrine, and inacrine control mechanisms, which “fine tune” or modulate the endocrine effects.

Gonadotropin-Releasing Hormone
GnRH which is secreted in a pulsatile manner by the GnRH neurons located within the paraventricular nucleus of the hypothalamus, orchestrates the entire endocrine regulation of reproductive function by controlling gonadotropin (FSH and LH) secretion from the pituitary, which in turn regulates secretion of testosterone from the Leydig cells. Secretion of GnRH is regulated not only by negative feedback of the sex steroids but also by a myriad of other chemicals, many of which are involved in other pathways such as maintenance of energy status and regulation of the stress response. Recent research has provided strong evidence that the critical regulatory gatekeeper integrating these various inputs and controlling GnRH secretion involves the kisspeptin/GPR54 ligand–receptor complex, which is present in multiple hypothalamic nuclei adjacent to the GnRH secreting neurons. Kisspeptin/GPR54 also mediates the negative feedback of circulating sex steroids on the GnRH neurons and has recently been shown to be pivotal in the timing of pubertal onset in many species.
Follicle-Stimulating Hormone
FSH is produced and exported from the pituitary to act principally on the Sertoli cells. It is secreted in a pulsatile manner in response to GnRH, also referred to as luteinizing-hormone releasing hormone (LHRH), from the hypothalamus. Inhibin, secreted by the Sertoli cell, is believed to be involved in a feedback loop from the testis to the pituitary to inhibit FSH production. The action of FSH on immature and mature animals is profoundly different. FSH is often considered to be the hormone of puberty, as rising levels of FSH act as a trigger for testicular growth, junction formation between adjacent Sertoli cells and ABP secretion from the Sertoli cells, and generally initiates spermatogenesis and the expansion of the seminiferous tubules. Once this has occurred, the Sertoli cell switches its responsiveness from FSH to testosterone as many of the FSH-regulated functions in the immature animal are taken over by testosterone in the adult.
The primary impact and effects of FSH in the adult are poorly understood, although its importance seems to vary between species. Suppression of FSH in the adult rat has a negligible effect on spermatogenesis, whereas in nonhuman primates, it results in considerable suppression of spermatogenesis and sperm output.
Inhibin and Activin
Inhibin is a protein that is secreted by both testis and ovary and decreases FSH but not LH secretion by pituitary cells. In the male, inhibin is secreted by Sertoli cells and appears to have both endocrine (via its effect on the pituitary) and paracrine (other effects on cells within the testis) properties. Activins have a similar structure to inhibin but possess FSH-stimulating activity in pituitary cells. As well as regulation of FSH, dual opposing roles for activin and inhibin have been demonstrated for the regulation of steroidogenesis in the Leydig cell. The main source of circulating inhibin is the gonads, but activins are synthesized in a broad range of tissues. Although circulating activin has FSH-modulating abilities, the main role for activin is now considered to be as a paracrine rather than an endocrine regulator of reproductive function.
Luteinizing Hormone
LH, like FSH, is a glycoprotein hormone secreted in a pulsatile fashion by the pituitary under GnRH control. In rats it acts on Leydig cells and is the primary regulator of testosterone secretion, which is also secreted in a pulsatile manner. Circulating plasma testosterone (or its metabolites) completes the feedback loop via the kisspeptin/GPR54 complex, to modulate LH secretion.
Prolactin permits LH-stimulated testosterone secretion in the rat by increasing the number of LH receptors on the Leydig cell. The Leydig cells of other species do not appear to possess prolactin receptors, which has important implications with regard to species-specificity/sensitivity to chemically induced Leydig cell tumors.
Testosterone
The major androgenic steroid in males, testosterone, is synthesized primarily in the Leydig cells in rats and has both intratesticular effects (on spermatogenesis) and peripheral effects (on accessory sex organs as well as nonreproductive organs such as brain, muscle, bone, and skin). There is also significant testosterone metabolism in many peripheral tissues—for example, the aromatization of testosterone to estradiol which is an important reaction in nonrodent species.
Testosterone is not stored within the Leydig cell but is secreted into the interstitial fluid as it is synthesized. From here it is either (1) taken up by the Sertoli cells and bound to ABP, which is then secreted by the Sertoli cell and transported through the seminiferous epithelium into the STF and on into the epididymis; or (2) diffuses into the interstitial capillaries, where it binds quickly to steroid hormone binding globulin (SHBG) for transport through the body, where it has wide-ranging effects on all other tissues of the body. In the rat, ABP is synthesized by the Sertoli cell, whereas SHBG is synthesized in the liver. The main known effects of testosterone in supporting spermatogenesis are to stimulate STF production by the Sertoli cell, to regulate release of the mature spermatids from the Sertoli cell (spermiation) and to support the development of pachytene spermatocytes and later germ cell types through stage VII of the spermatogenic cycle.
The major stimulus for testosterone production comes from blood levels of LH from the pituitary. It is important to recognize that this connection varies widely across species: in dogs and primates, a pulse of LH is followed closely by a pulse of testosterone. In rats, this connection is much less predictable, and several LH pulses may elicit no rise in testosterone or there may be a significant delay before a testosterone pulse occurs.
Feedback inhibition of LH and hypothalamic GnRH is mediated through circulating levels of testosterone and its metabolites, dihydrotestosterone (DHT), and estradiol, but the relative importance of the various molecules is species-dependent. In the rat, testosterone is the main feedback molecule, whereas DHT is thought to have more importance in the mouse, and estrogen feedback is more important in the dog, monkey, and man.
Estrogen
Estrogen is an important metabolite of testosterone that plays a role in spermatogenesis and in fluid reabsorption in the efferent ducts. Aromatase, which irreversibly converts testosterone to estradiol, is expressed in Leydig cells, Sertoli cells, and germ cells, with the highest functional activity being present in spermatids. Estrogen receptors are also expressed in germ cells, Sertoli cells, and Leydig cells. Aromatase activity/estrogen has been shown to contribute to spermiogenesis and sperm motility. In addition, strogen is a major regulator of fluid reabsorption in the efferent ducts, with high concentrations of estrogen present in the epididymal fluid and high concentrations of estrogen receptor-α in the efferent ducts. Endothelin receptors are also present in high concentrations, and endothelin is also thought to be involved in the process of fluid resorption in humans. Reabsorption is achieved through an active sodium–chloride pump with aquaporins regulating membrane permeability.
Dihydrotestosterone
The major regulatory androgen in the epididymis (and in the accessory sex organs) is not testosterone, but the more potent 5-hydroxy metabolite DHT which is synthesized within the epididymal epithelium by 5α-reductase and binds to the androgen receptor with higher affinity than testosterone.
Androgen-Binding Protein
ABP is synthesized by the Sertoli cell in the testis, where ~80% is secreted into the luminal fluid and the other ~20% is secreted into the interstitial compartment and taken up into the systemic circulation. ABP binds testosterone and transports it in the STF to the epididymis, where it is taken up by a receptor-mediated process into the principal cells of the initial segment and caput epididymis. Once released from ABP, the testosterone is converted to DHT by the epithelial 5α-reductase. ABP is very similar in structure to SHBG, which is synthesized in the liver and binds testosterone in the peripheral blood. ABP has been identified in rat, dog, monkey, and human.
Paracrine Regulation of Testicular Function
As already discussed, testosterone is the major regulatory hormone within the testis, but its secretion is modified by a variety of substances produced by the Sertoli cells, the blood vessels, the peritubular cells, the interstitial macrophages, and the Leydig cells themselves. These paracrine and autocrine hormones provide a faster response time and allow more sensitive control of testosterone secretion as well as providing a cascade of cell-to-cell chemical communication. The list of these secreted “hormones” is growing continuously and rapidly and includes glycoprotein and steroid hormones, peptide growth factors, cytokines, proopiomelanocortin derivatives, and neuropeptides. The receptors for most of these molecules have been identified in vitro, although their physiological function and significance are unknown in most cases. The production of such a variety of pharmacologically potent molecules within the testis has obvious potential for being toxicologically important.
Regulation of Protein Secretion by the Sertoli Cell
The Sertoli cell secretes hundreds of different proteins and proteases, most of which have unknown functions. These include transport proteins, which bind metal ions, lipids, hormones, and vitamins; proteases and protease inhibitors; basement membrane glycoproteins; growth factors; and paracrine factors. Their production is stimulated by testosterone and the presence of germ cells in the adult and by FSH in the prepubertal animal. The Sertoli cell also secretes STF, which maintains patency of the tubules and transports sperm out of the testis to the epididymis. Secretion is also androgen regulated and is influenced markedly by the germ cell complement and if elongating spermatids are missing, STF production is inhibited and tubule diameter is decreased.
Regulation of Blood Flow and Interstitial Fluid
Interstitial fluid has a very specific composition, compared with plasma, and is responsible for providing the oxygen and nutrients required by the metabolically active Sertoli and germ cells, which are otherwise located in an avascular environment. Interstitial fluid is thought to be produced as a result of rhythmic alterations in arteriolar blood flow (vasomotion), which brings about movement of fluids from the blood through the unfenestrated capillaries into the interstitial space. Androgen receptors are present on the muscular wall of the small testicular arteries, and testosterone is able to regulate overall testicular blood flow as well as the volume of interstitial fluid and the degree of vasomotion.
Evaluation of Toxicity
The effects of toxicants on male reproductive function can occur through disturbances at one or more tissue sites, including hypothalamus, pituitary, testis, efferent ducts, epididymis, accessory sex organs, or penis. In most cases, effects at one site will cause knock-on (secondary) changes at other sites; therefore, in general, the more endpoints examined, the better the chance of detecting toxicity, the more information available for distinguishing the primary cause of toxicity versus the secondary consequences of toxicity, and the greater chance of detecting a single random change occurring in isolation with no biological consequence.
Physiologic Evaluation
Organ Weights
Although organ weights are a standard parameter measured in any regulatory toxicity study, they can provide unique and mechanistically important information for male reproductive tissues because the weight of the accessory sex organs is androgen-dependent (Table 17.3). It is particularly important that the seminal vesicles and prostate (including secretions) be sampled in all studies, since these provide a relatively sensitive indication of androgen status. More importantly, they smooth out, or integrate, the peaks and valleys of testosterone secretion, providing a much more reliable assessment of testosterone status than a single hormone assessment. The epididymis is also an androgen-dependent tissue, but nearly 50% of its weight reflects sperm content, which is a function of the efficiency of spermatogenesis. When measuring testis weight in rodents, it is important to use absolute rather than relative weight since the testis, like the brain, is conserved despite decreased body weight gain and even modest body weight loss (up to ~70% of normal body weight). Because of that fact, decreased body weight gains will generally lead to increase in relative testis weight (testis weight as a percentage of body weight). Decrease in absolute testis weight is generally due to decreased germ cell content and/or decreased fluid content. Increased absolute testis weight is generally due to increased STF content (Table 17.3).
Table 17.3
Rapid Reference Guide for Evaluation and Interpretation of Weight Changes in the Reproductive Tract
| Finding/observation | What to look for | Possible causes |
| Increased testis weight | Seminiferous tubular lumen dilatation | Increased seminiferous tubule fluid which may be due to obstruction of outflow, decreased emptying of tubules, decreased resorption of fluid by rete/epididymis, increased production by Sertoli cell |
| Increased interstitial fluid (interstitial edema) | Altered hemodynamics, injury to vascular endothelium, reduced lymphatic drainage | |
| Decreased testis weight | Germ cell depletion | Disruption of spermatogenesis through effects on germ cells, Sertoli cells, hormonal disturbance, or blood supply |
| Seminiferous tubule lumen contraction | Decreased production of seminiferous tubule fluid which may result from loss of elongating spermatids and/or decreased testosterone production | |
| Increased epididymal weight | Increased interstitial fluid | Altered hemodynamics, injury to vascular endothelium, reduced lymphatic drainage |
| Increased ductular fluid | Decreased resorption of fluid by rete, efferent ducts, and caput epithelium | |
| Sperm granulomas | Maybe spontaneous, but maybe induced by any agent causing inflammation or damage to the epididymal epithelial lining | |
| Decreased epididymal weight | Reduced sperm content and contraction of ductular lumen size | Disruption of spermatogenesis resulting in reduced sperm production or release from the testis |
| Decreased weight of seminal vesicles and/or prostate | Atrophic changes in the secretory epithelium and decreased secretory product | Reduced levels of circulating testosterone, inhibition of 5α-reductase, or disruption of androgen receptor binding |
| Increased weight of seminal vesicles and/or prostate | Increased secretory product | Androgenic activity of a treatment; hyperprolactinemia |
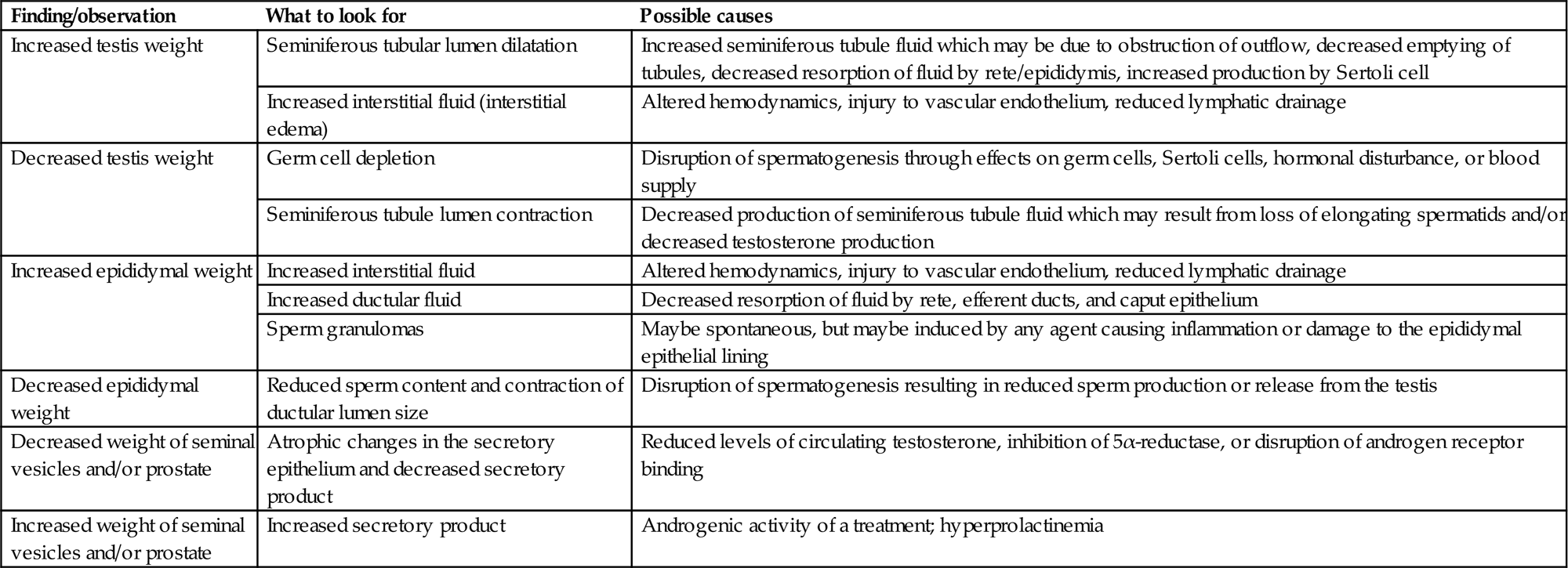
Table reproduced from Handbook of Toxicologic Pathology (2013), third ed. (W. M. Haschek, C. G. Rousseaux, and M. A. Wallig, eds.), Academic Press, Table 59.5, p. 2526, with permission.
Sperm Analysis and Spermatid Head Count
Sperm assessment is performed most often at the end of the study and after a mating trial which assesses the actual fertility of the male (Table 17.4). Measurement of sperm parameters (count, motility, morphology of epididymal sperm, and a count of testicular spermatid heads) can provide important information which can also add to mechanistic interpretations (Table 17.4). In many cases, the sperm measurements may serve to confirm and support the morphologic findings identified in the testis. Indeed, if spermatogenesis declines, all three sperm endpoints will decline. However, there are other situations, such as long-term, low-dose exposures, that result in spermatogenic disturbances that produce no visible lesion in the testis but where the sperm output is detectably compromised when measured as epididymal sperm count, and this will also impact fertility endpoints.
Table 17.4
Rapid Reference Guide for Evaluation and Interpretation of Sperm Parameters in the Reproductive Tract
| Finding/observation | What to look for | Possible causes |
| Decreased epididymal sperm count | Is there any evidence of testicular injury? Is there a similar decrease in testicular homogenization resistant spermatid (HRS) head count? Is there any decrease in accessory sex organ (ASO) weight? Are the other sperm parameters normal? | If there is no testicular injury and no decrease in HRS, the epididymis maybe the primary site of toxicity. If other sperm parameters are normal, decreased count maybe due to reduced (quicker) epididymal transit time. If ASO weight is decreased, low sperm count maybe due to low testosterone (T) |
| Increased epididymal sperm count | Increased (slower) epididymal transit time | |
| Decreased testicular spermatid head count (HRS) | Are there abnormalities in epididymal sperm parameters? Can you see degeneration/reduction in spermatogenesis or in the number of step 19 spermatids (stage VII/VIII tubules)? Is there any decrease in ASO weight? | Decreased numbers of HRS means reduced testicular spermatogenesis and should be accompanied by reduced epididymal sperm count. Any spermatogenic disturbance will usually result in decreased HRS. If ASO weight is decreased, low T could be the cause |
| Increased testicular spermatid head count (HRS) | Is there evidence of spermatid retention—i.e., presence of step 19 spermatids at the lumen of stage XI tubules or at the base of stage XII tubules? Is this correlated with a decrease in epididymal sperm count or changes in motility or morphology? | An increased count for HRS can only be caused by spermatid retention because spermatogenesis cannot increase its output of sperm. There may be a decrease in epididymal sperm count (but this may be less sensitive). Spermatid retention is usually associated with an increase in abnormal sperm and decreased motility |
| Decreased motility | Is there any evidence of testicular injury or changes in HRS? Is there any decrease in ASO weight? Are the other sperm parameters normal? | Decreased motility can be caused by testicular or epididymal toxicity. If it is associated with changes in other sperm parameters, it is likely of testicular origin. Frequently accompanies spermatid retention |
| Increased numbers of abnormal sperm | Is there any evidence of testicular injury or changes in HRS? Is there any decrease in ASO weight? Are the other sperm parameters normal? | An increase in the number of morphologically abnormal sperm is usually due to spermatogenic disturbance. It is a very sensitive indicator in the rat |

HRS, homogenization resistant spermatids; T, testosterone; ASO, accessory sex organs.
Table reproduced from Handbook of Toxicologic Pathology (2013), third ed. (W. M. Haschek, C. G. Rousseaux, and M. A. Wallig, eds.), Academic Press, Table 59.6, p. 2527, with permission.
Measuring the number, motility, and morphology of sperm in the epididymis or vas provides an integrated readout of the efficiency and overall health of testicular spermatogenesis as well as epididymal maturation and transport. It is unusual but not unheard of to see a selective effect on count, morphology, or motility in the absence of a change in at least one other measure. These sperm endpoints are all components of fertility, but none of them is sufficiently strongly correlated with, or drives, fertility that it may substitute for a functional assessment of how well the sperm work in a fertility test. Thus, while normal values in a treated group of animals offer some degree of assurance that an exposure did not produce severe damage to the male reproductive system, one should not be misled into assuming that fertility is normal in those animals.
Testicular spermatid head count (homogenization resistant spermatid head count) is an additional measurement that provides an indication of testicular sperm production. Contrast this with caudal epididymal sperm count which reflects both the production of sperm from the testis and storage of sperm by the epididymis. This is always a useful adjunct to the epididymal sperm data.
In rodents, sperm measurement is a terminal procedure conducted at necropsy, but since it is possible to collect ejaculates from most other species there is the potential for examining longitudinal samples, including predose, during dosing, and during recovery, thus providing regular monitoring of any significant effects on spermatogenesis and reversibility. For large animals (rabbits, dogs, and monkeys), sperm evaluation is also a useful hallmark of sexual maturity. A problem with sperm count is its degree of variability, resulting in large standard deviations and relatively wide ranges for normal background data. Variability exists not only between individual animals but within the same animal on different sampling occasions which is why multiple ejaculates are often collected per time point (either within 1 day or on adjacent days during a long study). Fortunately, both the number of morphologically abnormal sperm in rodents and their motility is fairly consistent from animal to animal, and therefore provides sensitive parameters for evaluation.
Sperm Count
Sperm count is generally taken from the cauda epididymis and since the function of the cauda is mostly to store sperm, this count reflects both sperm production by the testis and also the storage function of the epididymis. Reduced sperm count is generally due to reduced production but can also be caused by decreased transit time in the epididymis (i.e., the sperm move through more quickly, for example, with 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) exposure and some estrogenic compounds).
Sperm Motility
Sperm motility is a functional measurement of the sperm themselves. Sperm are sampled either from the cauda or the vas deferens and assessed for motility. In rats, motility is generally high and fairly consistent between animals: with control values in the 85%–96% range for rodents, dogs, and monkeys. Sperm motility can be affected by disturbances in testicular spermatogenesis or by effects on the epididymis.
Sperm Morphology
Sperm morphology, especially the head or nucleus, reflects the integrity and quality of the sperm. The number of abnormal sperm produced spontaneously, varies dramatically with species. In rodents (depending on the laboratory and strain), normal sperm values between 94% and 99.7% are common. In dogs, the percentage of normal sperm varies between 50% and 90%; in cynomolgus monkeys, the value is around 80%. For comparison, the number of normal sperm in normal fertile humans is generally around 50%. Increases in the number of abnormal sperm reflect disturbances in testicular spermatogenesis.
Testicular Spermatid Head Count
Testicular spermatid head count is a convenient way of quantifying spermatogenesis in the testis. It is much less labor-intensive than manual counting of testicular germ cells using a microscope. One testis is macerated sufficiently to destroy everything except the highly condensed, most mature heads of the elongating spermatid population (steps 16–19 in rats). This is a defined cell population that represents the final 6 days of spermatid development prior to release. It can therefore be used to provide a value for daily sperm production (when divided by ~6) or the number of spermatid heads per gram of tissue. Spermatid head count will decrease in response to anything that decreases spermatogenic efficiency (as long as the injury has had time to work through to the mature elongating spermatids). The count can also be increased if mature spermatids are not released, as in the case of low-testosterone induced spermatid retention. Integration of this measurement with that of epididymal sperm count provides powerful insight into the site of an effect and an integrated view of the toxicity of a treatment (Table 17.4).
Hormone Analysis
Measurement of hormones can be a powerful and informative tool if it is incorporated into a well-designed study that has adequate numbers of animals or samples to make the data statistically valid (reviewed by Chapin and Creasy, 2012). The most commonly measured hormones are testosterone, LH, FSH, and inhibin B. Additional hormones that can be useful in specific situations are estradiol and DHT, both metabolites of testosterone and important for specific functions of the reproductive tissues. In addition, prolactin which is secreted by the pituitary and has a regulatory role in the secretion of testosterone, may also provide useful information. Most of the reproductive hormones have a pulsatile secretory pattern, are affected by stress, and may also have circadian rhythms. Study design needs to take these factors into consideration if the results are to be trusted.
When to Measure Hormones
Hormone measurements can be useful to provide mechanistic support for the hypothesis that a reproductive toxicant is acting through a primary disturbance of hormonal regulation, and/or to identify potential biomarkers for that toxicity in man. Since relatively few toxicants act through a hormonally mediated mechanism, it is necessary to identify those that do before embarking on hormonal measurements. In theory, any disruption of spermatogenesis could result in a secondary response in hormone levels (e.g., an increase in testosterone or FSH levels to compensate for a decline in spermatogenesis); however, such hormonal responses are inconsistent in nature and magnitude, late in occurrence, and unlikely to provide a sensitive marker of testicular injury. Thus, it is essential for the pathologist to identify those cases of reproductive toxicity that are caused by a primary hormonal event which can be done using the morphologic profile (see “Patterns of Change Associated with Loss of Androgen Support” section).
One reason for embarking on hormone measurements is when Leydig cell tumors are encountered in a carcinogenicity study. In the rat, Leydig cell tumors are generally caused by any treatment that causes a prolonged increase in LH. This can occur through many different mechanisms, and there is a wide variety of drugs and chemicals that will trigger this response. Although such mechanisms have negligible risk for inducing Leydig cell tumors in man, their occurrence in rodents does reflect a hormonal disturbance which may have other toxicologic implications; therefore, it can be important to establish the cause of this disturbance.
Another, and arguably the most common, trigger for hormone measurements is when the major effects in a study are atrophy or weight loss in the accessory sex organs with or without altered spermatogenesis. If these changes are seen in the absence of significant body weight loss and moribundity, a mechanism involving decreased androgenic stimulation should be considered.
Fertility Assessment
A detailed review of fertility testing and evaluation of fertility endpoints is outside the scope of this chapter, but it is important to appreciate the basic concepts and the fact that fertility is an important endpoint for consideration by the toxicologic pathologist. Fertility is the final-integrated endpoint of the successful functioning of the male and female reproductive processes, and in that sense it sums all the related processes into a single integrated output.
Fertility can be impacted by morphological or biochemical injury in any of the reproductive tissues, by hormonal disturbances anywhere in the HPG axis, or by neurobehavioral alterations in sexual behavior (reduced libido means no or fewer litters). Due to the massive reserve capacity of spermatogenesis in rodents, fertility is relatively insensitive for identifying testicular toxicity, particularly when compared with morphology or sperm parameters. An often quoted observation is that rats can remain fertile (they can produce offspring) with less than 2% of their normal sperm numbers. Even though they may remain fertile, other fertility endpoints, such as total litter size, pup weight, or time to pregnancy, will show significant alterations. In comparison with rodents, humans have vastly less reserve to maintain fertility when reproductive function is compromised. This is due to the relative inefficiency of spermatogenesis (sperm output/gram of testis) in humans, which is significantly lower than most other species, and a high proportion of abnormal sperm (over 50%) produced as compared with less than 1% abnormal forms in rodents (Table 17.2). The fact that rodents have such high fecundity means that fertility (number of pregnant females per group) may remain high or unaffected in a group of 20 females, despite detectable histopathological changes or changes in sperm parameters. Aggregate data from many multigenerational studies have shown, however, a remarkably good correlation between overall fertility and sperm count and motility; even modest reductions in sperm count (e.g., 10%) in rodents will translate into reduced fertility in a population of rodents. Note that this may not be seen in a small group of 20 animals.
Morphologic Evaluation
Histopathology is often cited as being the most sensitive method for detecting disturbances in spermatogenesis, and indeed it is, if conducted on well-fixed tissue and by a pathologist with a clear understanding of spermatogenesis, the cell associations (stages) that make up the spermatogenic cycle and the dynamics of the overall process (Table 17.5). If these requirements are not met, important lesions will be missed, especially in short-term studies of 2–4 weeks. This can be critical for human safety, since in the case of pharmaceutical development, “first into man” clinical trials are often based on the results from such short-term studies.
Table 17.5
Rapid Reference Guide to Evaluation and Interpretation of Histopathologic Findings in the Reproductive Tract
| Finding/observation | What to look for | Possible causes |
| Testicular germ cell loss | Is a specific cell type(s) affected? Does the germ cell loss fit into a pattern of maturation depletion, or is it nonspecific? Is it focal or diffuse, is it partial or generalized? | The pattern of the germ cell loss will provide valuable clues as to the likely mechanism of injury, but this will also be very much influenced by the duration of the study (see main text for detail). The pathogenesis of germ cell loss is best investigated in a short time-course study |
| Loss of elongate and elongating spermatids | Degeneration of step 7 spermatids and pachytene spermatocytes in stage VII tubules | Disruption of testosterone secretion, which may be caused by direct effects on the Leydig cells or endocrine-mediated effects. Direct effects on elongating spermatids |
| Degeneration/apoptosis of germ cells | Is a specific cell type affected? Are the dying cells restricted to a specific tubular stage? Are the affected cells forming multinucleate aggregates? | The cause maybe direct toxicity to the affected germ cell, but may also be mediated through a stage-specific disturbance to the Sertoli cell. Apoptotic cells are rapidly removed. Multinucleate aggregates suggest a slow, nonspecific degenerative process |
| Germ cell exfoliation | Presence of exfoliated germ cells in the rete and epididymal lumens | Disruption of Sertoli/germ cell junctions leading to loss of adhesion. Disruption of Sertoli cell cytoskeletal fibers leading to sloughing of apical Sertoli cell cytoplasm and attached germ cells |
| Macro/microtubular vacuolation (in the absence of severe germ cell injury/loss) | Is this located in the basal Sertoli cell cytoplasm, or scattered as large vacuoles throughout tubule? Look for accompanying or additional focal germ cell loss (suggesting focal Sertoli cell damage) | Disturbance of Sertoli cell function leading to vacuolation of organelles or disturbance of fluid balance. NB: Do not confuse with osmotic induced fixation artifact |
| Necrosis and disorganization of tubular contents (including Sertoli cells) | Evidence of acute inflammatory infiltrate around affected tubules | Disturbance in hemodynamics or damage to the vascular endothelium leading to ischemic necrosis |
| Spermatid retention | Alteration in epididymal sperm parameters (morphology, motility and count) and possible increase in HRS. Are ASO weights reduced? | Reduced ASO weights → disturbance in testosterone secretion. No change in ASO weights implies disturbance in Sertoli cell function or in spermatid development |
| Dilated seminiferous tubule lumens | Blockage of efferent ducts or epididymal duct. Evidence of pressure-induced germ cell loss | Increased seminiferous tubule fluid due to obstruction of outflow, decreased emptying of tubules, decreased resorption of fluid by rete/epididymis, or increased production of fluid by Sertoli cell |
ASO, accessory sex organs; HRS, homegenization resistant spermatids.
Table reproduced from Handbook of Toxicologic Pathology (2013), third ed. (W. M. Haschek, C. G. Rousseaux, and M. A. Wallig, eds.), Academic Press, Table 59.7, p. 2540, with permission.
Importance of Fixation
Testes fixed in conventional 10% neutral buffered formalin (NBF) causes severe shrinkage of the Sertoli and germ cells within the tubules. This seriously compromises the pathologist’s ability to detect subtle changes such as tubular vacuolation, shape changes in the head of the elongating spermatids, spermatid retention, and displacement of germ cells from their normal position within the seminiferous epithelium. Fixation of testes with Bouin’s or Modified Davidson’s fixative overcomes this problem and should be used for all studies if possible, or at least for studies of ≤13 weeks. The epididymis and accessory sex organs are best fixed in conventional NBF.
Testes need to be fixed whole to maintain the architectural integrity of the seminiferous tubules and prevent disruption of the delicate germ cell–Sertoli cell junctions. This includes large animal testes as well as rodents, and so penetration and protein crosslinking properties of the fixative are particularly important. Although formalin is a rapidly penetrating fixative, it is slow in its abilities to crosslink proteins, even when tissues are trimmed to an ideal thickness. When testes are fixed whole in formalin, it is likely that proteins are never fully crosslinked and are therefore susceptible to distortion during subsequent processing and paraffin embedding. Bouin’s and Modified Davidson’s both contain formalin, but they also contain acetic acid which causes swelling of cells, counteracting the shrinkage induced by formalin. Both Bouin’s and Modified Davidson’s provide excellent cellular and nuclear morphology of the germ and Sertoli cells; the main disadvantage of Bouin’s fixative is its tendency to cause excessive shrinkage of tubules and a graininess to the cytoplasm (in addition to the fact that the waste is explosive and requires special handling methods), whereas the main disadvantage of Modified Davidson’s is that it results in pale PAS staining of the acrosome. A recommended recipe for Modified Davidson’s is provided in Box 17.1.
Fixation of testes for immunohistochemistry (IHC) may require different fixatives and shorter fixation times, depending on the antigen being stained and antigen retrieval techniques employed. Trials are recommended to optimize for each antigen. Fixation of testes for electron microscopy and/or embedding in plastic (epoxy resin or glycol methacrylate resin) requires special fixation to provide optimal morphology. In the case of electron microscopy, perfusion fixation is essential if detailed ultrastructural evaluation is the goal. Successful perfusion of the testis can be challenging, and following specific published methodology relating to testicular perfusion is recommended. Perfusion fixation can also be used for glycol methacrylate embedding, although immersion fixation provides acceptable results. Surprisingly, NBF provides good results with this water-soluble embedding medium, and fixation is not improved when the fixative is perfused.
Importance of Sampling
Sampling of the testis should always incorporate part of the rete testis so that this can be evaluated for changes. In rodents, the testes can be sampled to provide a transverse or longitudinal section or one of each type can be taken from each testis. In large animals, the size of the testis may preclude obtaining an entire cross-section of testis unless using oversize slides. The section should at least contain a portion of the rete testis and also incorporate peripheral as well as the central regions of the tubule-containing lobules. Since the matrix of Sertoli cells and germ cells that constitutes the seminiferous epithelium is delicate and easily disrupted, it is important that the testes are handled gently at necropsy. Care should be taken to keep the capsule intact and not puncture or nick it when removing the epididymides. It is not necessary to puncture the capsule prior to immersion in fixative, even with nonrodents, since this often causes more damage than benefit.
Sampling of the epididymis should include all regions (initial segment, caput, corpus, cauda, and junction with the vas deferens). This is best achieved by taking a longitudinal section through the entire organ. Lesions are often region-specific in the epididymis and sperm density varies significantly depending on location, so consistent and complete sampling is important.
Sampling the accessory sex organs is relatively straightforward. In the case of rodents, it is important to ensure that the different lobes of the prostate are adequately sampled because they respond differentially to different endocrine-disrupting scenarios.
Stage-Aware Evaluation of Spermatogenesis
Whenever a pathologist examines a testis with the intent to identify changes in spermatogenesis, it needs to be done with an awareness of the stages of spermatogenesis. Failure to do this will result in important changes being missed. A stage-aware evaluation should not be considered a “special” evaluation; it should be the routine way that any toxicologic pathologist examines a testis. Ideally, a pathologist should have a thorough knowledge of spermatogenesis, as well as the cellular associations in each of the different stages of the spermatogenic cycle, in each of the species routinely evaluated, and a clear understanding of the dynamics of the process (reviewed by Creasy, 1997). In reality, most changes can be readily identified as long as the pathologist can recognize the different appearance and cell types within tubules that are at the beginning, in the middle, or at the end of the cycle (Figures 17.5B and 17.6). Having this level of knowledge allows detection of relatively subtle changes in any routine regulatory study using conventional H&E sections.
If there is a suspicion that a drug or chemical belongs to a class of known testicular toxicants, then a more detailed stage-aware evaluation maybe requested. In these situations it is recommended that the testes be stained with PAS-hematoxylin to stain the spermatid acrosome. This allows accurate placement of any tubule with respect to its precise stage within the spermatogenic cycle (Figure 17.5A). This is not the case with dog spermatogenesis, where the acrosome fails to stain with PAS.
The value of examining the testis in a stage-aware manner is that it allows recognition of when a cell that should be present is in fact missing, or, conversely, is present when it should not be. This can be surprisingly difficult to see unless the observer is familiar with the correct number of layers of cells and their general appearance in the different (early, mid, and late) phases of the cycle (Figures 17.9 and 17.10). Another advantage of examining the testis with a knowledge of stages is that it enables recognition of stage-specific cell degeneration (Figure 17.11B) or stage-specific Sertoli cell vacuolation. Stage-specificity of changes is only likely to be seen in short-term studies (≤4 weeks duration). Many testicular toxicants show stage-specific changes at the earliest time points, but as the affected tubule continues through consecutive stages of the spermatogenic cycle and the repeat dosing regimen continually damages more tubules in the sensitive stage, the appearance of stage-specificity is lost.
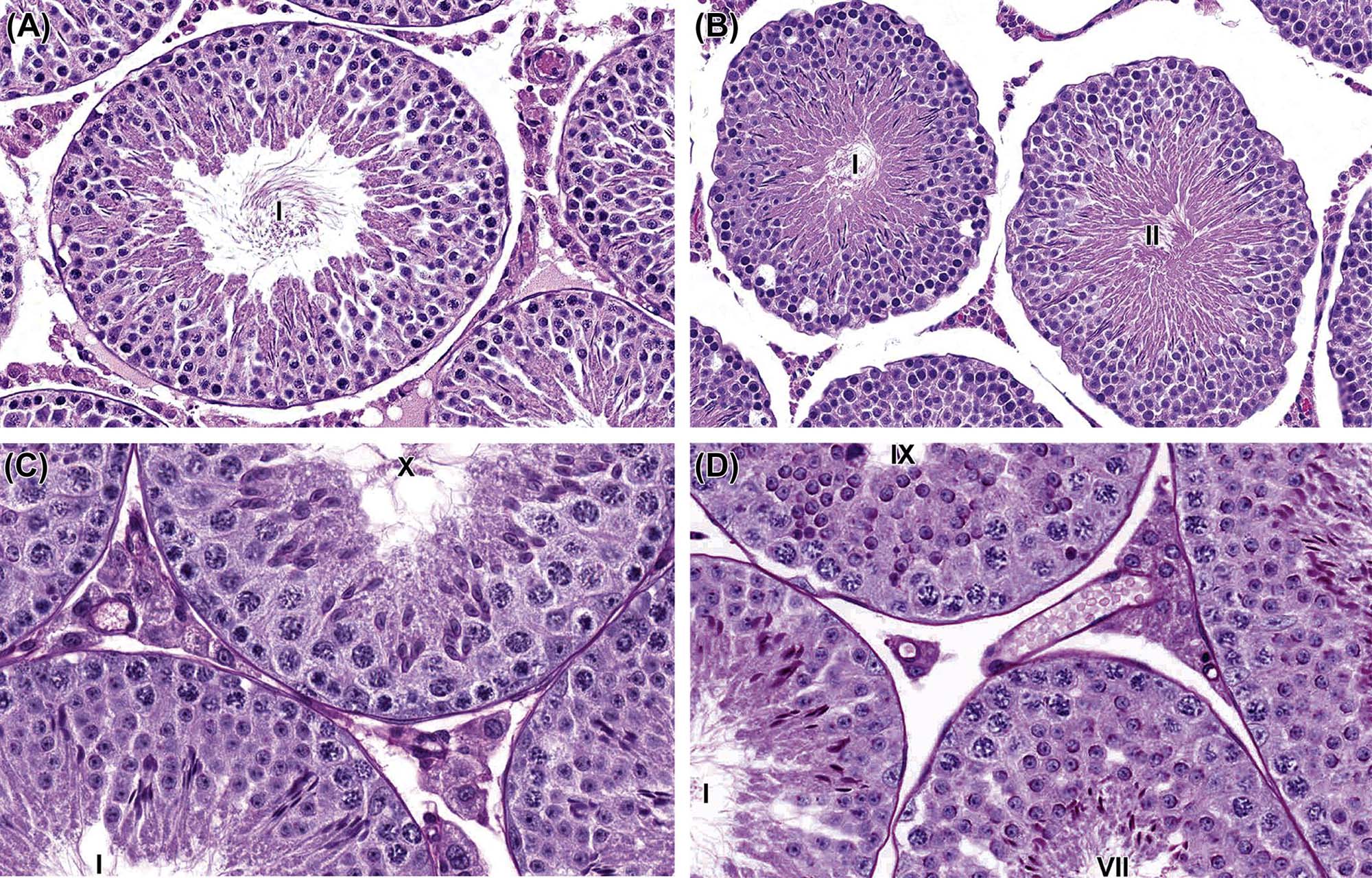
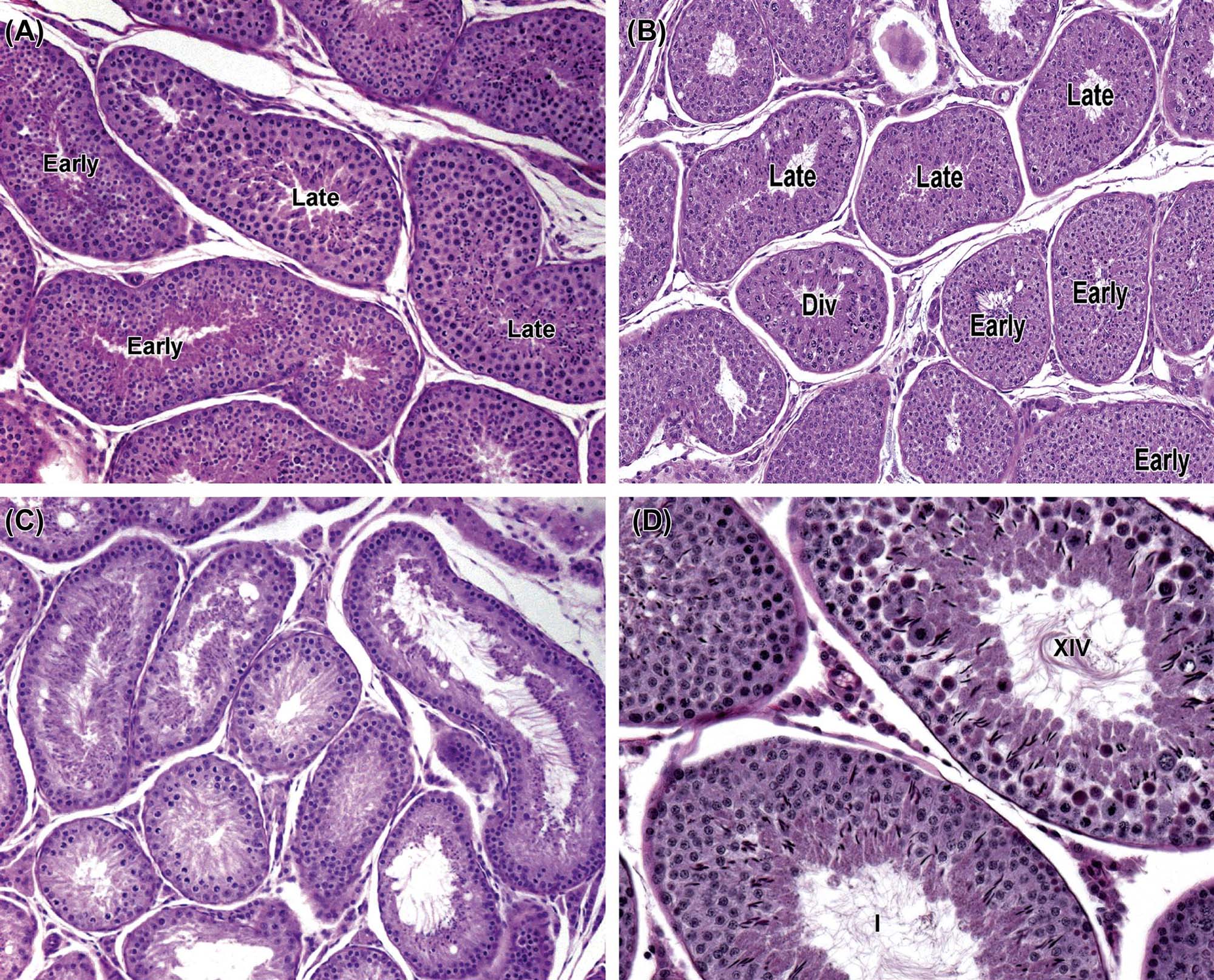

Using the same basic principles, a stage-aware evaluation can be conducted regardless of species, but the pathologist should be aware of the differences in cell associations between species. Staging maps are readily available for all the major species (see the “Further Reading” section) and should be referred to when examining testes. Using these maps, staging of tubules in the cynomolgus monkey is relatively straightforward as long as the monkey is mature. Staging spermatogenesis in the dog is much more difficult than in the rat or the monkey because the layering of the germ cells is less regular and the overall appearance of the seminiferous epithelium is less compact. Additional difficulties include the fact that the spermatid acrosome does not stain with PAS, the spatulate head of the elongating spermatid presents a variable profile in the same stage tubule depending on its plane of section, and mature spermatids maybe released from tubules (spermiation) in two consecutive stages (stage IV and V), resulting in significant numbers of “mid-stage” tubules that are missing their complement of elongating spermatids. Nevertheless, early mid- and late-stage tubules (Figure 17.10) can still be distinguished and can be examined to identify whether the main cell types are present in their expected proportions.
Nomenclature and Grading of Lesions
As with any organ or tissue, using consistent nomenclature and severity grading for findings is important. There is an added degree of complexity with respect to nomenclature and grading of changes in the testis because many of the early toxicant-induced disturbances in spermatogenesis are cell- and/or stage-specific and may only affect one cell type or only affect one or a few stages of the spermatogenic cycle.
Nomenclature
The recently published International Harmonization of Nomenclature and Diagnostic Criteria for Lesions in Rats and Mice nomenclature system for the male reproductive system in rodents (Creasy et al., 2012) recommends using cell- and stage-specific terminology when such changes are seen, and using generalized terminology (e.g., tubular degeneration/atrophy) for nonspecific changes (Figure 17.11). It is often important to capture the cell- and stage-specificity of a finding because it can provide important mechanistic information or important information for risk assessment. For example, a finding of “degenerating round spermatids and pachytene spermatocytes, stage VII/VIII” indicates that reduced testosterone is the probable cause of the degeneration, especially if the change was associated with reductions in accessory sex organ weight. Alternatively, if the change only involved elongating spermatids (e.g., “degeneration and depletion elongating spermatids”), this would signify that the toxicity is limited to the final stages of spermatid development, which would suggest it is likely to be a rapidly reversible change with only transient effects on fertility (although this would need to be tested). Cell- and stage-specific changes are most likely to occur in short-duration studies (≤4 weeks of dosing), and the longer the study duration, the more cell types and the more stages are likely to be affected. In these situations, nonspecific terminology (such as tubular degeneration/atrophy) is more appropriate.
Epididymal lesions are frequently also region-specific, and if this is the case the nomenclature should reflect the anatomic location because the information can have important mechanistic implications. Reductions in sperm content can even be region-specific depending whether the reduction in sperm output from the testis occurred within the past few days of dosing (low sperm in the caput, normal sperm in the cauda) or occurred over the previous 1–2 weeks of dosing (low sperm throughout the epididymis).
Severity Grading
Grading the severity of disturbances in testicular spermatogenesis can present a challenge. Severity grading for nonspecific testicular changes such as tubular degeneration/atrophy is generally not a problem and can be based on the proportion of affected tubules. However, severity grading for cell- and stage-specific changes needs to take into account the number of cells affected as well as the number of tubules affected. For example, spermatid retention only involves the retention of step 19 spermatids in stage IX–XI tubules, which constitute a very small proportion of the total tubular profiles but may involve a very high proportion of the step 19 spermatids. Should the change be considered minimal or severe? The decision has to be made on a case by case basis and explained in the text of the report if needed.
Special Techniques
Immunohistochemistry
The testis lends itself to IHC as well as any other tissue, although a word of caution is in order. We have the perception that the testis has its own isoforms of proteins; although the protein is present in the testis, it maybe as a splice variant which reduces or eliminates the ability of that protein to be recognized by the antibody which works perfectly well in other tissues. Investigators should be aware of the testis’ penchant for protein uniqueness, and guard their interpretations appropriately.
Staining for Proliferating and Apoptotic Cells
There are staining methods which will specifically highlight dividing cells (predominantly spermatogonia) and apoptotic cells. Proliferating cells can be stained using IHC methods. The most common methods include proliferating cell nuclear antigen (PCNA), Ki67 antigen, or administration of bromodeoxyuridine which will be taken up by the dividing cell and then visualized using subsequent immunohistochemical methods. PCNA is an antigen that is expressed for relatively long periods of time after cell division and is less specific than Ki67, which only stains recently divided cells. Both are technically easier than administering bromodeoxyuridine (BrDU). Since spermatogonia can be difficult to visualize and count, this relatively simple IHC method provides a good way of visualizing the spermatogonial population in different stages of the spermatogenic cycle and can be useful in confirming a suspected treatment-induced reduction or cessation in spermatogonial divisions.
Apoptotic cells can be stained using TUNEL (terminal deoxynucleotidyl transferase-mediated deoxyUTP-biotin nick end labeling) or Caspase 3 staining. TUNEL is a method which labels the ends of a DNA strand with a visualization tag. It was thought that this method labeled only that DNA cleaved by endonucleases between the histone solenoids. It is true that this method will visualize cells undergoing apoptosis in situ and will do it with significant sensitivity. However, it has also been shown to stain cells undergoing necrosis (the more disorderly cell death process), as well as cells undergoing DNA repair or damaged by mechanical or shearing forces. Thus, it might be appropriate to use TUNEL staining to identify DNA damage, but then to defer the decision on a specific mode of death to another technique, such as DNA electrophoresis, to look for the “ladder pattern” so characteristic of apoptosis. Methods are widely available to visualize cells stained with this method; the main message here is to remind us that TUNEL is not specific for apoptosis. If a conclusion about apoptosis is important, staining for Caspase 3 should be performed. Caspase 3 is a lysosomal enzyme involved in the apoptotic pathway and is more specific in the detection of apoptotic cells than TUNEL.
In Vitro Methods
Cell culture models are often used academically to investigate specific aspects of cell biology. Although there is increased effort to create engineered-tissue-culture models which would accommodate the long-term growth and maintenance of highly polarized epithelium characteristic of the seminiferous tubules (with the eventual production of mature sperm), no successful methods have been published outside of a few unreplicated academic reports. Thus, all in vitro models of testis or epididymis are short-term (a couple of days) or transformed cell lines which lack various metabolic capabilities. Cell lines can be a useful tool if they retain the features and pathways of interest to a project but this knowledge is required upfront.
Response to Injury
Organ Weight Changes
Table 17.3 provides guidance on the evaluation of organ weight changes. Growth and maintenance of the testis, like brain, is preserved despite body weight loss; thus, absolute rather than relative weight should be used for evaluation. As might be expected, a decrease in body weight gain will result in an increase in testis to body weight ratio when compared with controls. A decrease in absolute testis weight generally reflects a loss in germ cell number and STF, which will usually be reflected by a decrease in epididymal weight since sperm content is responsible for approximately 50% of its weight. An increase in the weights of testis or epididymis generally indicates increased fluid content. In the case of the testis, this is usually due to increased tubular fluid and is accompanied by obvious tubular dilation. It is most likely due to blockage of the efferent ducts, while in the epididymis, interstitial edema or possibly a sperm granuloma is the most likely cause.
In the rat, decreased weight of the seminal vesicles and ventral prostate is a relatively sensitive indicator of reduced androgen status, is more sensitive than morphological evaluation of atrophic changes and is less variable than measuring hormone levels. Indeed, these weights are the best integrative indicator of androgen status currently available. Most of the weight change is due to a reduction in the stored secretion, and therefore it is important that this is captured when the tissues are weighed—i.e., weigh the tissues with their stored secretions. The combined weight of the seminal vesicles, coagulating gland, and prostate can be used, or each organ can be weighed separately. In practice (and when allowed by the protocol), it is just as sensitive and much easier to lift the aggregate tissue mass by the bladder, cut the urethra immediately below the prostate, place the whole unit (bladder, prostate, and seminal vesicles) intact on the balance and then trim off the bladder. It is easy and fast and guarantees retention of the fluid.
In endocrine-disruptor screening studies, the ventral prostate is sometimes dissected and weighed separately since it is the most androgen-sensitive portion of the gland. Increased weight and secretory content of the accessory organs can occur if androgens are administered, or expulsion of secretion is inhibited, which has been reported with some alpha-adrenergic agents. Hyperprolactinemia has also been reported to cause enlargement of accessory sex organs in rodents.
Weight change of the accessory sex organs is not as sensitive a parameter in the dog and monkey because of the inter-animal variation, smaller group sizes, and the fact that comparatively little secretion is stored in these organs.
Morphologic Changes (Nonproliferative)
The most common appearance of the testis following dosing with any testicular toxicant is varying degrees of germ cell degeneration, depletion, and disorganization of the germ cell layers within the seminiferous epithelium (Table 17.5). This reflects the fact that the germ cells are exquisitely sensitive to any disturbance of their support, be it mechanical, biochemical, nutritive, or regulatory. So, whether a toxicant acts through the Sertoli cell to disrupt a biochemical pathway that regulates or supplies energy to the germ cells, or acts on the Leydig cells to decreases testosterone levels, or even on the vasculature to cause anoxia, it will be the germ cells that are most affected and show evidence of cell degeneration, death, and depletion. Although the Sertoli cells are very sensitive to disturbances due to their multiplicity of biochemical pathways, for the most part this is not reflected by morphological changes that are evident by light microscopy. It is not until they are severely damaged that morphological changes are recognizable. In fact, there are very few toxicants or injuries that actually kill Sertoli cells; one of the few examples is ischemia. This robustness maybe related to the fact that the Sertoli cell is incapable of division once it has passed its postnatal proliferative phase (day 15 rat, week 15 dog, and month 32 cynomolgus monkey), and because the Sertoli cell forms the blood–testis barrier that protects the seminiferous epithelium from the destructive/protective actions of the immune system. Once the Sertoli cell barrier is broken, the tubule is generally destroyed and replaced by granulation tissue and fibrosis. Apart from the few occasions where the Sertoli cell barrier is breached, inflammation is rarely seen as a response to injury.
Since all types of injury cause the same morphologic response, how can the pathologist gain insight into how and where the toxicant is acting? The answer is to look at the earliest morphological changes and concentrate on the pattern of toxicity (see “Morphological Patterns of Response to Different Types of Injury” section). Table 17.5 also provides a rapid reference guide on specific features to look for and how they can be interpreted. This may mean looking at the cell-specificity or stage-specificity of the early changes, as well as the character of the morphological changes (germ cell degeneration versus germ cell depletion versus tubular vacuolation or necrosis). It means taking into consideration the effects on the rest of the reproductive tissues (epididymis and accessory sex organs and sperm endpoints if available). This kind of detailed morphologic examination of early changes relies heavily on the pathologist’s knowledge of spermatogenesis and the ability to detect when a particular stage of the spermatogenic cycle is abnormal. This can be surprisingly subtle even when there are multiple cell types missing (Figures 17.9 and 17.10).
In reality, it is our evolving appreciation that almost all testicular toxicants probably produce disruption of spermatogenesis through a final common pathway due to effects on the Sertoli cell. This is because the Sertoli cell (also known as the nurse cell) provides almost all the needs of the germ cells and the regulation of spermatogenesis. Even testosterone, as a major regulator of spermatogenesis, is believed to have its main effects through interaction with androgen receptors on the Sertoli cell, which, in turn, controls functions such as the rate of spermatogonial differentiation, the processing of spermatocytes and round spermatids through androgen-dependent stages (stages VII and VIII), and the release of mature step 19 spermatids from the Sertoli cell cytoplasmic recesses (spermiation). Similarly, some spermatogonial toxicants act through disruption of the complex cascade of factors that involve the Sertoli cell stem cell factor (SCF)–c-kit system rather than a direct (cytotoxic) effect on the spermatogonia. Nevertheless, it is possible for the pathologist to distinguish between these different types of toxicity by characterizing the early pattern of changes, thereby providing important information that maybe useful for risk assessment, biomarker identification, or further mechanistic investigations. In addition, because of the central role the Sertoli cell plays in spermatogenesis, it is important to distinguish a toxicant that is producing its effects through disturbance of a biochemical pathway from one that is producing effects through overt structural damage of this critical cell.
Germ Cell Degeneration (Apoptosis)
Although the term “germ cell degeneration” suggests a potentially reversible effect on the germ cell, it is the recommended terminology that is applied to germ cells undergoing cell death (apoptosis). In the testis, apoptosis provides the important physiologic function of limiting the size of the germ cell population to numbers that can be supported adequately. The Sertoli cell regulates this function utilizing the FAS gene system. Germ cells express Fas, a transmembrane receptor protein that, when bound by Fas ligand (FasL) secreted by the Sertoli cell, transmits an apoptotic signal within the cell. All chemically induced germ cell death investigated so far also appears to occur through the process of apoptosis. This is true for both direct germ cell toxicity, as occurs with radiation, and indirect germ cell death resulting from Sertoli cell injury. It is even true for death that appears pyknotic (Figure 17.10D), such as the spermatocyte death induced by glycol ethers in rats. The precise mechanism triggering apoptosis appears to be different in each case. With Sertoli cell toxicants there is upregulation of Fas and FasL, but with germ cell toxicants there is only upregulation of Fas. Although DNA laddering electrophoresis might indicate that germ cell death occurs by apoptosis, for the bench pathologist looking at dying germ cells microscopically, the only dying cells that fit the classic morphologic appearance of apoptosis (DNA fragmentation and clumping at the periphery of the nucleus within an eosinophilic cytoplasm), are spermatogonia. These can often be seen undergoing normal attrition, particularly at the outside of stage XII tubules.
In cases of testicular toxicity, one of the most obvious and common manifestations of degeneration is that of the “multinucleate giant cell” (see Figure 17.11A and B). These generally comprise round spermatids or occasionally pachytene spermatocytes that have fused together into one large cell. Recall the earlier notation that as spermatogonia proceed through their successive divisions, they remain connected to their siblings via cytoplasmic bridges (incomplete cytokinesis). These cytoplasmic bridges remain intact through the rest of spermatogenesis and are thought to allow for coordinated development between the progeny of a spermatogonial division. The multinucleate germ cells are believed to form when the cytoplasmic bridges between adjacent cells open and allow fusion of the cytoplasm. When round spermatids are involved, the nuclear acrosomic caps often appear to adhere to one another. Multinucleate giant cells appear to be a relatively slow form of germ cell death, since the nuclear and cytoplasmic characteristics often retain normal staining and morphology, are TUNEL negative and elicit very little response from the surrounding Sertoli cell. They can be seen as an occasional focal or diffuse background finding in rodents and dogs (Figure 17.11A), but when a dose-related increase is seen in number and incidence they are a good indication of toxicity. They can also be seen as a cell- and stage-specific change (Figure 17.11B).
Germ cells with eosinophilic, slightly shrunken cytoplasm and nuclei with condensed or pyknotic chromatin are a different manifestation of apoptosis, and probably represent a much more rapid type of cell death. Such cells are rapidly phagocytized by the surrounding Sertoli cell cytoplasm, and evidence of their demise is rapidly removed, generally within 24 hours. If this type of death occurs within a specific cell population (e.g., pachytene spermatocytes in stage I tubules), examination of the testis on the following day would show absence of stage II pachytene spermatocytes because the tubule will have progressed to the next stage and the pachytene spermatocytes will have been removed (Figure 17.9A and B).
Germ Cell Depletion
Once germ cells undergo apoptosis they are rapidly phagocytized by the Sertoli cell and all evidence of their previous existence disappears. If an entire generation (layer) of germ cells dies and the testis is examined soon afterwards, the resulting depletion can be difficult to detect (Figures 17.9 and 17.10). If the study is a repeat dose study and the same cell type is continually killed with each repeat dose, the depletion will probably appear relatively nonspecific by the end of the study (see the “Patterns of Change Associated with Germ Cell-Specific Toxicity” section). Death and depletion of a specific germ cell type suggests interference with a specific process, either within the affected germ cell or affecting a specific pathway in the Sertoli cell that is critical for the survival of that stage of germ cell development. More commonly, germ cell depletion occurs with a patchy, multifocal distribution and is accompanied by disorganization with some degeneration of the remaining germ cell layers. In this situation, the term “degeneration/atrophy” might be more appropriate (Figure 17.11). This pattern of change often suggests structural damage to the Sertoli cell, compromising its ability to support the survival of its entire cohort of cells.
Tubular Degeneration/Atrophy, Testis
Tubular degeneration/atrophy is a nonspecific term that can be used to describe any situation where there is a mixture of tubules containing degenerating germ cells, partial loss of germ cells, disorganization of germ cell layers, and tubules lined only by Sertoli cells. It is the most common pattern of change seen in repeat-dose studies with testicular toxicants (Figure 17.11A). If there is diffuse loss of all germ cells, leaving tubules lined only by Sertoli cells, the term “tubular atrophy” might be more appropriate (Figure 17.11C). When the change is diffuse, it is an end-stage lesion that provides no information about likely cause. Occasional atrophic or degenerate tubules (e.g., one to five tubular profiles per section of testis) is a relatively common background finding and represents a focal or segmental loss of germ cells from a part of one or a few seminiferous tubules. Tubules that are depleted of germ cells and lined only by Sertoli cells are often seen adjacent to the rete testis (Figure 17.7). In fact, these are sections through the tubulus rectus which is where the seminiferous tubule empties into the rete testis. These normal structures should not be mistaken for “atrophic tubules.”
Tubular Necrosis, Testis
Tubular necrosis differs from tubular degeneration/atrophy in the fact that it involves coagulative necrosis (rather than apoptosis) of the germ cells and generally involves disruption and necrosis of the Sertoli cells lining the tubule (Figure 17.12A). If there is significant disruption of the Sertoli cells, the blood–testis barrier will be breached, resulting in an inflammatory infiltrate around and invading the affected tubule. There are very few other cases where toxicant-induced testicular injury gives rise to an inflammatory response. Tubular necrosis is a relatively uncommon finding and most often occurs in response to ischemic injury. Necrosis of large areas, or of the entire testis (testicular necrosis), can be seen following torsion or infarction of the testis (Figure 17.12B).
Tubular Dilation, Testis
Enlarged testes containing seminiferous tubules with dilated lumens and thinned seminiferous epithelium (Figure 17.13A) can be seen as incidental lesions but can also be chemically induced (see with “Patterns of Change Associated Disturbance of Fluid Production, Resorption or Efferent Duct Obstruction” section). The finding is generally unilateral but can sometimes be bilateral. The change is generally due to efferent duct obstruction and, with time, will usually progress to total tubular atrophy, generally with dilated lumens (Figure 17.13B).
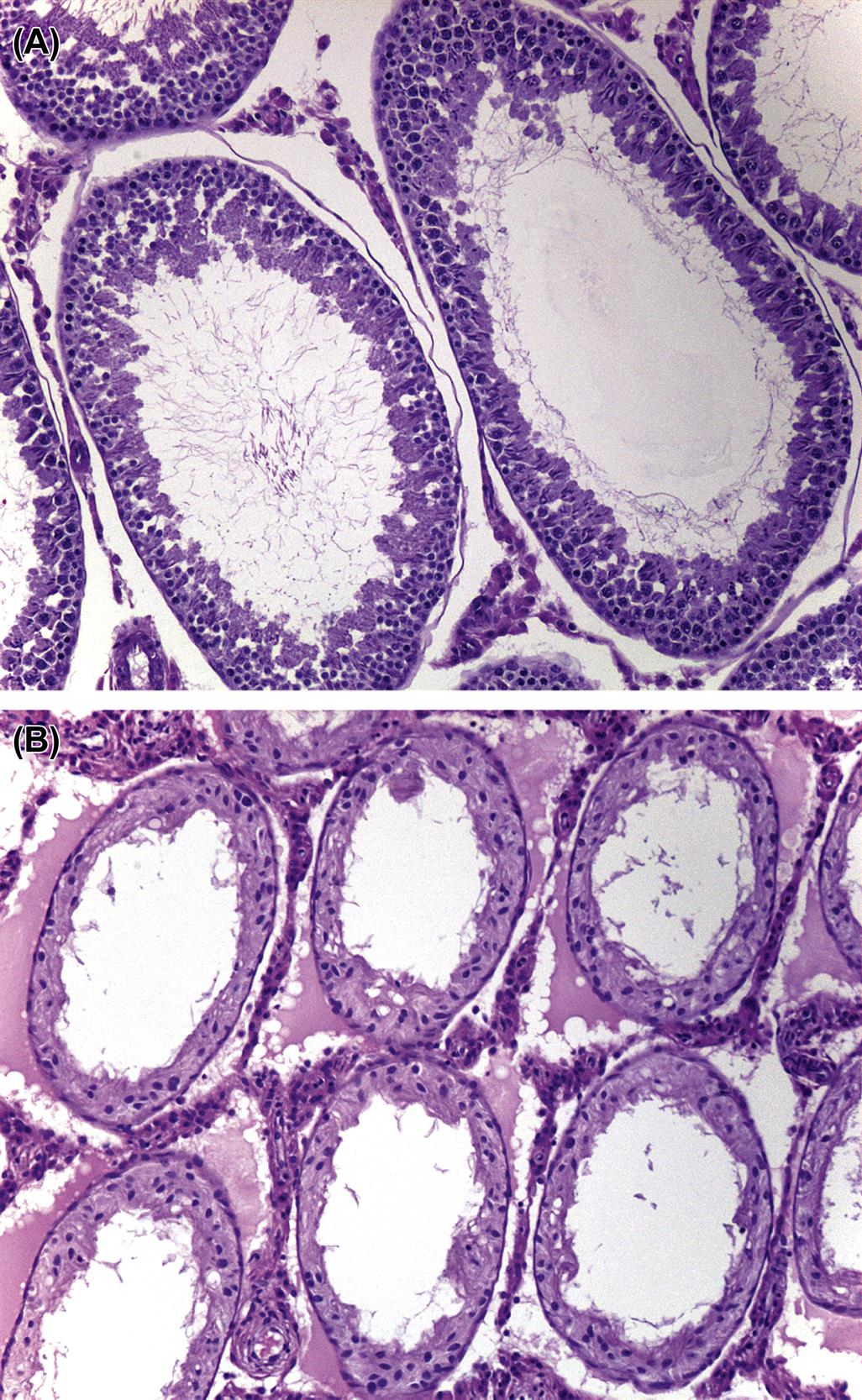
Tubular Vacuolation
Tubular vacuolation can represent a primary response of the Sertoli cell to toxicant-induced injury (Figure 17.14A and B) or it can be a secondary response due to loss of the space-occupying germ cell (Figure 17.14C). When it is a primary change, it can appear as microvesicular vacuolation in the basal Sertoli cell cytoplasm (Figure 17.14A) or as larger discrete vacuoles part way up the seminiferous epithelium (Figure 17.14B). The primary-response type of vacuole often displaces the germ cells lying adluminal to it.

For the larger primary-response vacuoles, it is usually difficult to identify whether they are intra- or intercellular. When examined, the basal vacuoles have been found to be intracellular and may represent expansion of cytoplasmic organelles (SER), phospholipididosis, or intracellular edema. These are generally an early toxicant-induced change associated with Sertoli cell disturbance and usually progress to tubular degeneration/atrophy with continued dosing. Occasional large vacuoles can be seen in normal testes, but they are generally few in number (Figure 17.14D).
Spermatid Retention
Step 19 mature spermatids should be released from the luminal Sertoli cell cytoplasm during Stage VIII of the spermatogenic cycle. If spermiation fails (which can occur due to disturbances in Sertoli cell processes and reduced testosterone levels or abnormal spermatid development), then step 19 spermatids fail to be released and are still seen at the tubular lumen in stage IX, X, and XI tubules (Figure 17.15A). By the time the tubule reaches stage XII, the spermatid heads have generally been pulled down into the basal Sertoli cell cytoplasm where they are phagocytized (Figure 17.15B). By Stage XIII, they have generally disappeared. Therefore, this subtle lesion will only be seen in a relatively small proportion of tubules within an affected testis. Although morphologically subtle, the finding is generally associated with significant changes in sperm parameters (particularly motility and morphology) and potential fertility. If there is significant spermatid retention, testicular spermatid head count should increase while epididymal sperm count decreases.

One aide memoire to help in identifying inhibited or delayed spermiation is that in a normal testis there is only a single generation of elongating or elongated spermatids in any tubule. A tubule that has two generations of elongating or elongated spermatids is an adversely affected tubule. A few tubules with mild spermatid retention may also be seen in normal testes.
Leydig Cell Atrophy, Testis
Leydig cells undergo atrophy in response to decreased LH stimulation or as a result of decreased testosterone synthesis (see the “Patterns of Change Associated with Loss of Androgen Support” section). Due to the variability in size of Leydig cells and the effects of fixation on their normal appearance, it can be quite difficult to evaluate size by qualitative examination unless the effect is marked. Image analysis combined with immunohistochemical markers can be used to quantify more subtle alterations in size/volume.
Cell Debris, Intraluminal, Epididymis
The presence of cells and cell debris admixed with sperm in the lumen of the epididymis generally reflects sloughing of germ cells from the testis (Figure 17.16). In the mature rat there are normally very few sloughed cells, making this a very sensitive indicator of spermatogenic disturbance in the testis. Sloughed cells are seen more frequently in the normal mouse and are quite common in the dog. They are also common in peripubertal animals of any species, which reflects the higher background level of germ cell degeneration and exfoliation during the first wave of spermatogenesis. In some cases the sloughed cells seen in peripubertal epididymal lumens also represent exfoliated epididymal cells. Thus, while most round cells in the epididymis of an animal treated with a testis-toxicant are exfoliated germ cells, be aware that in a prepubertal animal these round cells can also be of epididymal origin.
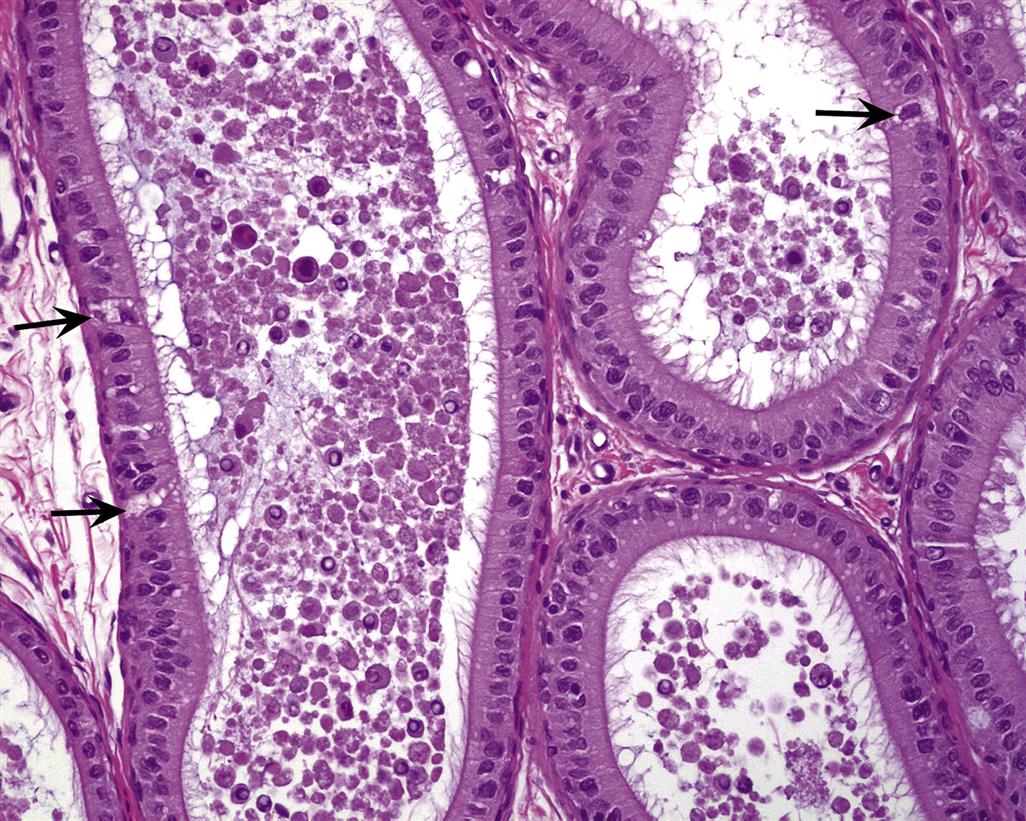
Sperm Granuloma, Epididymis
Sperm are antigenically foreign, due to the genetic exchange between chromosomes that occurred during meiosis. A granuloma is a foreign-body reaction whereby a tissue recruits many immune cells from the blood to surround and wall off foreign bodies with inflammatory cells and fibroblasts. The mechanism of formation of most epididymal granulomas is unknown. The most popular belief is that there is a rupture of the epididymal tubule, and eruption of the sperm from the break into the interstitium, followed by a responsive inflammation which forms a nodule encapsulating the sperm. This is supported by the fact that ligation of the vas deferens in rats invariably results in sperm granulomas developing at sites proximal to the ligation (and also occurs in a high proportion of men undergoing vasectomy). Moreover, the fact that sperm granulomas develop at the vas–cauda junction in rats that have had the sympathetic autonomic nervous supply chemically blocked using guanethidine has been explained on the basis of rupture due to the increasing pressure of sperm build-up in the cauda. Thus, there is some evidence for pressure-induced granuloma formation. However, this seems unlikely to be the only mechanism by which they form.
Consider this alternative explanation. The epididymis is filled with foreign bodies (sperm), which it must nurture and support and store … but they are foreign. To accommodate this, the epididymis must exist in a dynamic equilibrium between immune tolerance and activation. It would be possible, then, for a treatment to alter the internal biochemistry driving that balance. For example, a treatment that rendered the epididymis more tolerant would show no pathology, but an alteration that activated the epididymis would recruit neutrophils and macrophages from the circulation and would show interstitial inflammation. These recruited cells would eventually damage the interstitium and a tubule. The leakage of some sperm antigens would exacerbate the inflammatory stimulus and progress to ductular damage and granuloma formation. There are emerging data to support this “immune balance” theory, which is presented here primarily to broaden the thinking of those charged with investigating etiology. Although sperm granulomas can be quite large, in most cases there are sperm in the duct distal to the granuloma, which suggests continued flow of sperm down the epididymis. This implies that a granuloma does not necessarily obstruct the flow of sperm through the epididymal duct and therefore may not impact fertility. They can occur anywhere in the epididymis but are most frequently seen in the distal corpus or cauda.
Atrophy: Prostate or Seminal Vesicles
This change is characterized by decreased secretion and reduced size of the epithelial lining of the prostatic acini or seminal vesicular epithelium. It is pathognomonic of decreased androgen stimulation, which can be due to low testosterone, low conversion of testosterone to DHT, or interference with the androgen receptor in the affected tissue. As with Leydig cell atrophy, subtle changes can be difficult to recognize by qualitative morphologic evaluation, and organ weight is usually a much more sensitive endpoint. Atrophy of the accessory sex organs is a common age-related finding.
Morphologic Changes (Proliferative)
With the exception of Leydig cell hyperplasia and Leydig cell tumors, there are very few nonneoplastic or neoplastic proliferative lesions in the testis (For detailed review see Creasy et al., 2012). Leydig cell hyperplasia can occur as a focal or diffuse lesion. Focal Leydig cell hyperplasia and Leydig cell adenomas form a continuum of change, making it difficult to separate hyperplasia and adenoma (Figure 17.17A and B). In the absence of any significant morphological differences in cellular appearance, a size that is equal to or greater than the diameter of three seminiferous tubules is generally used as the main but arbitrary classification criterion in rats. Diffuse Leydig cell hyperplasia is generally a physiological response to hormone imbalance and may accompany severe atrophy of the seminiferous tubules. Diffuse hyperplasia (and tumors) can also be seen in response to estrogen administration in mice. One other relatively frequent proliferative lesion in the mouse is hyperplasia of the rete testis epithelium, which is a common age-related finding that can occasionally progress to adenoma. Although testicular tumors (Sertoli and Leydig cell tumors and, less commonly, seminomas) occur as spontaneous tumors in aging dogs, they are seen only occasionally in laboratory-maintained dogs, where the lifespan is normally restricted.

Reparative hyperplasia of the prostatic epithelium or, less commonly, the seminal vesicle epithelium often accompanies inflammatory lesions associated with urogenital infections in rodents, but tumors in the accessory sex organs are uncommon except in some transgenic mouse strains.
Proliferative Lesions of the Testis
Leydig Cell Hyperplasia and Tumors
Diffuse Leydig cell hyperplasia can develop in response to decreased spermatogenesis. The Leydig cells are generally increased in size as well as number, but it is important to distinguish real hyperplasia from an apparent increase in Leydig cell volume caused by shrinkage of the atrophic tubules. The spontaneous incidence of Leydig cell tumors in rodents is species- and strain-specific, but they are more common in the rat (up to 100% in aged F344 rats and generally <6% in Sprague Dawley and Wistar rats). In mice the incidence is generally lower (B6C3F1 <1%, CD1 <2%).
Prolonged disruption of the pituitary–gonadal hormone axis in rodents is very likely to result in Leydig cell tumors. In the rat, focal Leydig cell hyperplasia and Leydig cell tumors (Figure 17.17A and B) can be readily induced by a wide range of chemically diverse drugs and chemicals, including dopamine agonists, antiandrogens, LHRH analogs, peroxisome proliferators, and histamine receptor antagonists (Table 17.6). The proposed mechanism of action for these various classes of compounds is through interference with Leydig cell control mechanisms at a variety of points along the hypothalamic–pituitary–testicular axis. A major impetus for Leydig cell tumorigenesis in the rat is considered to be high circulating levels of LH. In human and mouse, persistent elevation in LH is not mitogenic; in the rat, they eventually cause Leydig cell mitosis and then hyperplasia, continuing to adenomas. Interestingly, a significant number of Leydig cell tumorigens have no effect on circulating levels of LH but do alter intratesticular testosterone (and other hormones), thereby affecting the paracrine feedback control of Leydig cell proliferation, presumably through local growth factors. Another interesting fact is that Leydig cell proliferation in the fetal testis occurs in the absence of circulating LH and is thought to be controlled by Sertoli cell factors.
Table 17.6
Chemicals Known to Produce Leydig Cell Hyperplasia and Tumors in Rats
| Chemical | Class/action | Putative mechanism of induction |
| Cimetidine | Histamine receptor antagonism | Androgen receptor antagonism, testosterone biosynthesis inhibition |
| Flutamide | Antiandrogen | Androgen receptor antagonism |
| Finasteride | 5α-Reductase inhibitor | Decreases androgen feedback |
| Buserelin/Leuprolide | GnRH agonists | Binds to Leydig cell LHRH receptors |
| Mesulergine | Dopamine agonists | Decreases prolactin which decreases Leydig cell LH receptor sensitivity |
| Isradipine | Calcium channel antagonist | Inhibition of testosterone biosynthesis |
| Gemfibrozil | PPAR-α/hypolipidemic | Decreases prolactin which decreases Leydig cell LH receptor sensitivity |
| Lansoprazole | Proton pump inhibitor | Inhibition of testosterone biosynthesis |
| Linuron | Phenylurea herbicide | Androgen receptor antagonism |
| Procymidone | Systemic plant fungicide | Androgen receptor antagonism |
Table reproduced from Handbook of Toxicologic Pathology (2013), third ed. (W. M. Haschek, C. G. Rousseaux, and M. A. Wallig, eds.), Academic Press, Table 59.8, p. 2561, with permission.
In contrast, the chemical induction of Leydig cell tumors in the mouse is less common and is generally associated with high circulating levels of estrogen or administrations of estrogenic compounds such as diethylstilbestrol. In general, the range of chemicals producing Leydig cell tumors in the rat is ineffective in mice. Furthermore, estrogen administration to the rat appears to be inhibitory to the development of spontaneous and chemically induced Leydig cell tumors, although in at least one case (ammonium perfluorooctonate) the major detectable hormonal change leading to Leydig cell tumors is an increase in plasma estradiol levels. Aromatase inhibitors such as formestane and letrozole reduce plasma estradiol levels by inhibiting the conversion of testosterone to estrogen. In the dog, but not rodents, this results in Leydig cell hypertrophy and hyperplasia. This is thought to be due to the differential sensitivity of the pituitary feedback mechanism to estrogens and androgens in the different species. The aromatization of testosterone plays a significant role in the control of gonadotropins in dogs, nonhuman primates, and man, whereas in rodents, testosterone and DHT are the main regulatory molecules.
The testicular tumor profile and the physiology of Leydig cell tumorigenesis in rodents and humans appear to be very different, and on this basis it has been argued that chemical induction of Leydig cell tumors in the rat is a species-specific effect with limited relevance for risk assessment to man (reviewed by Cook et al., 1999). Indeed, the antipsychotic dopamine agonists which produce Leydig cell tumors in rats by lowering prolactin levels, are taken by many for treatment of schizophrenia and there has been no observable increase in Leydig cell tumors, an otherwise extraordinarily rare tumor in humans. Similarly, the widely prescribed H2 histamine antagonist cimetidine which also produces Leydig cell tumors in rats through its antiandrogenic properties, also has shown no evidence of Leydig cell tumorigenesis in man. Nevertheless, claims of irrelevance need to be supported by careful investigations into the mechanism of hormonal disruption.
Proliferative Lesions of the Epididymis
Proliferative lesions of the epididymis are almost nonexistent. The epididymal epithelium does not respond with a true hyperplastic response. Cribriform change, where the epithelium folds in on itself and forms pseudoglandular structures, is sometimes considered a hyperplastic lesion, but because it is generally associated with collapse of the ductal lumen it is difficult to distinguish from epithelial folding. The only primary tumors that have been described in the epididymis are Leydig cell adenomas and histiocytic sarcomas; both occur in the mouse, but they are rare. Since Leydig cells are not normally found in the epididymis, the occurrence of Leydig cell adenomas is unexplained, but there are a number of reports describing these tumors in mice. The epididymis can also be a primary site for the development of histiocytic sarcoma in mice.
Proliferative Lesions of the Accessory Sex Organs
In contrast to man, where hyperplasia and neoplasia of the prostate are extremely common, spontaneous neoplasia of the prostate in other species is rare. Reactive (reparative) hyperplasia of the prostatic epithelium commonly accompanies inflammatory lesions resulting from urogenital infections. This is particularly common in mice and generally located in the dorsolateral lobes of the prostate. In more severe urogenital infections, the seminal vesicle epithelium may also respond. Hyperplasia of the accessory sex glands is not often seen as a response to inflammation in the dog or the monkey.
Focal hyperplasia in the absence of inflammation is most commonly seen in the ventral prostate of rats and mice. The diagnosis of hyperplasia in the rodent prostate is mainly based on the occurrence of multilayered normal epithelium in a few adjacent acini of otherwise normal glands without architectural disturbance. There appears to be a morphologic continuum between focal hyperplasia and benign adenoma of the prostate, and the distinction between the two is not always clear. Adenomas and adenocarcinomas of the ventral prostate and seminal vesicles are occasionally seen as a background finding in rodents, more commonly in some of the transgenic mouse strains. Dorsolateral prostatic adenomas appear to be rare as spontaneous lesions but have been chemically induced. Experimental models of prostate carcinogenesis have been developed in the rat using N-nitrosobis(2-hydroxypropyl)amine, N-methylnitrosourea, and 3,2′-dimethyl-4-aminobiphenyl. Invasive carcinomas of prostate and seminal vesicles can be induced when these carcinogens are coadministered with high doses of testosterone. Prostate carcinoma has also been induced by testosterone administration to a strain of rat (Noble rat) that has a genetic susceptibility to prostate cancer. Transgenic mouse models such as Transgenic Adenocarcinoma of Mouse Prostate have also been developed to study human prostate cancer.
Recovery and Reversibility of Injury
The reversibility of infertility is a very important consideration for risk assessment in male reproductive toxicology, and the potential for complete recovery is largely dependent on the site and the severity of the toxic insult. This puts additional importance on identifying the primary cell of toxicity and the pathogenesis of the lesion.
Following germ cell-specific damage or depletion (in the absence of significant Sertoli cell damage), the chances of regeneration of the entire germ cell population and recovery of functional spermatogenesis are good. Although many of the germ cell types are sensitive to physical and chemical disturbances, the renewing stem cell spermatogonial population is relatively resistant. This has been well illustrated using varying doses of radiation. The order of cell sensitivity to increasing doses of radiation shows that although the generations of differentiating spermatogonia are very sensitive to radiation, primitive stem cell spermatogonia require a much larger dose to cause death.
When choosing the duration of a recovery period, it is usually advisable to make it equal to the timing of the spermatogenic process for the test species (e.g., 8 weeks for the rat and dog, 5 weeks for the mouse, and 6 weeks for the cynomolgus monkey). However, if most of the spermatogonia have been affected it may be necessary to extend this to two or even three times the duration of spermatogenesis to allow sufficient time for the slowly dividing stem cell population to repeatedly cycle and replenish the differentiating spermatogonial population to allow recovery of the rest of spermatogenesis. When short-duration recovery periods (e.g., 2–4 weeks) are employed in studies where there has been germ cell-specific toxicity (particularly in studies of ≤28 days of dosing), it is quite common for the degree of germ cell depletion to appear more severe at the end of the recovery period than at the end of the dosing period. This can easily be misinterpreted as progression or irreversibility of the lesion, but this would be an incorrect conclusion. It is entirely predictable due to the progression of maturation depletion through the later (and more numerous) round and elongating spermatid population, which cannot be replenished until sufficient recovery time has elapsed to allow the spermatogonia and/or spermatocytes to replace them (see “Patterns of Change Associated with Germ Cell-Specific Toxicity” section). For example, if spermatogonia are the target cell in a 14-day study, the only cells that will be missing at the end of the dosing period will be spermatogonia and early spermatocytes. This will be detectable but will be a relatively subtle lesion (Figure 17.9C and D). If the animals complete a 4-week recovery period, that “hole” of missing cells will consist of absent spermatocytes and round spermatids as a result of “maturation depletion” caused by the loss of their precursor cells (Figure 17.10A–C). Although spermatogonia and early spermatocytes will probably have recovered, the overall severity of the germ cell depletion will appear greater and the weight and size of the testes will appear much less at the end of recovery than at the end of dosing. However, if the recovery period is extended to 8 weeks, full recovery will likely be witnessed, or, at least, significant evidence of recovery seen.
If the Sertoli cell is injured and the damage is mild, then full recovery may be possible. If Sertoli cells are destroyed or, as more often happens, permanently functionally compromised, regeneration is not possible because these cells are unable to replenish themselves in the adult. However, they are remarkably resistant to cell death and are often found as the only remaining cell type in the seminiferous epithelium when all germ cells have been lost. Toxicant-induced atrophy frequently manifests as an end-stage lesion where the only cells in the tubules are the Sertoli cells and the stem cell spermatogonia. For example, in the case of 2,5-hexanedione toxicity, where the Sertoli cell is believed to be the primary cell affected, the end-stage lesion comprises tubules that contain Sertoli cells and dividing spermatogonia, yet spermatogenesis never recovers. Investigations have suggested that this is due to the lack of a Sertoli cell-derived growth factor called SCF, which binds the c-kit receptor on spermatogonia. Reestablishment of SCF and reversibility of the lesion can be accomplished by administration of exogenous SCF or stimulation of endogenous SCF by treatment with the GnRH agonist, leuprolide.
Most effects of androgen deficiency, including spermatogenic disruption and atrophy of the epididymis and accessory sex organs, are also readily reversible with the reestablishment of normal hormone balance. An obvious exception to this is the Leydig cell tumor, which, once established, is androgen-independent and irreversible. Effects on the epididymis that result in granulomatous inflammation and sperm granulomas are generally progressive and irreversible. The presence of persistent inflammation in close proximity to viable sperm runs the additional risk of leukotriene-induced genotoxic damage to the sperm, as seen with methyl chloride.
Immaturity and Peripuberty as Confounding Factors for Identifying Toxicity
Regardless of mechanism, the most obvious feature of testicular toxicity is death and depletion of germ cells. If an animal is immature and at a stage where spermatogenesis has not started or has only barely started (5- to 6-month-old dogs or 3- to 4-year-old cynomolgus monkeys), then testicular toxicity, however severe, will go undetected. If spermatogenesis is progressing but the animal has not reached puberty or is on the cusp of puberty (7- to 8-month-old dogs and 3.5- to 4.5-year-old cynomolgus monkeys), then the testes will likely contain significant numbers of degenerating germ cells and have partially depleted seminiferous tubules which will be indistinguishable from a degenerative response to a testicular toxicant. This presents a real risk of producing a false positive result if a greater proportion of the peripubertal animals are in the high-dose group. Table 17.7 provides a summary of the approximate age that animals attain sexual maturity compared with the age of animal commonly used in routine regulatory studies.
Table 17.7
Age of Sexual Maturation Versus Typical Age of Animal Used in Toxicity Studies
| Species | Typical starting age for routine studies | Age of sexual maturation |
| Rat | Soon after weaning (6–7 weeks after acclimation) | 9–10 weeks |
| Mouse | Soon after weaning (6–7 weeks after acclimation) | 7–8 weeks |
| Dog | 4–6 months | 7–12 months |
| Minipig | 3–4 months | 3–4 months |
| Cynomolgus monkey | Young adults (often 2–3 years) | 4.5–5.5 years |
Table reproduced from Handbook of Toxicologic Pathology (2013), third ed. (W. M. Haschek, C. G. Rousseaux, and M. A. Wallig, eds.), Academic Press, Table 59.11, p. 2565, with permission.
Rodents
Rats are able to sire a litter from around 8 weeks of age, but the testes do not appear morphologically “normal” until around 10 weeks of age. Rats aged 8–9 weeks often have low numbers of elongating spermatids in the testes with occasional evidence of degenerating cells. In addition, the epididymis contains increased numbers of degenerating germ cells and has relatively few sperm in the cauda epididymis. These features can be mistaken as evidence of testicular toxicity and reduced spermatogenesis if, by chance, there is a higher incidence in the high-dose group. A similar situation exists in 5- to 6-week-old mice. To overcome this problem, rats should be at least 10 weeks of age by the end of the study and mice should be at least 8 weeks old.
Dogs
The use of immature and peripubertal dogs represents the most common cause of confusion to pathologists when evaluating testicular toxicity. Dogs vary in the age they reach sexual maturity; at 7 months of age one dog may have testes that appear fully mature with expanded epididymal ducts filled with sperm, while other dogs of the same age have testes with partially depleted seminiferous tubules and frequent degenerating germ cells, and epididymides that have contracted ducts containing large numbers of sloughed degenerate germ cells and no sperm (Figures 17.18 and 17.19). Spermatogonial proliferation and fluid secretion begins at around 5–6 months in the dog. This results in gradual lumen formation and progressive expansion of the tubules as secretion increases. In dogs of this age, it is common to see tubular (Sertoli cell) vacuoles accompanying the start of lumen formation. These are likely fluid-filled vacuoles related to the initiation of fluid production by the Sertoli cell and should not be considered abnormal (Figure 17.18B and C).

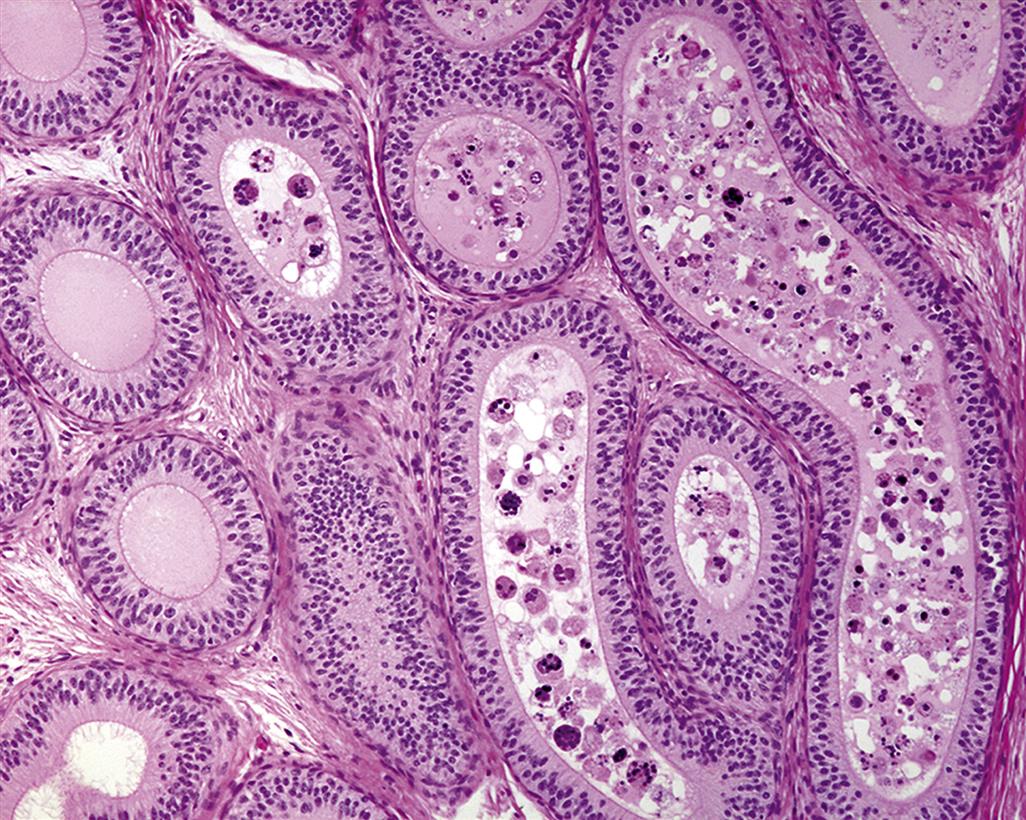
Most dogs have normal appearing testes with significant sperm content in the cauda epididymis by 10 months of age, but the efficiency of spermatogenesis and the size of the testes will continue to increase until the animals are 12–18 months of age. With this degree of variability, small group sizes, and the fact that maturing and peripubertal testes are indistinguishable from testes undergoing toxicant induced degeneration, there is a risk that immaturity will mask a toxicant induced lesion or that immaturity will be mistaken for testicular toxicity. At the very least, it will add an additional layer of complexity to the evaluation of a tissue that is already difficult to evaluate.
The variation in age of maturation is reflected by dramatic differences in testicular weight and testicular size from animals that are the same age and same body weight. Prostatic development cannot be used as an indicator of sexual maturity because this tissue often develops at a different rate from the testis, presumably due to differential development/expression of androgen receptors between the two organs. It should also be borne in mind that size of dog and age of sexual maturation vary with the source (supplier) of the dogs.
The best way to avoid confusion and avoid the possibility of missing an important toxicant induced lesion is to use dogs that are at least 10 months of age by study termination. If there is a suspicion that the test article may affect spermatogenesis, it would be prudent to ensure the animals are 12 months of age at termination to guarantee sexual maturity. The same rule should be applied to any juvenile study that is conducted in the dog. Dosing should be continued until the dogs are at least 10 months of age to avoid misinterpretation of changes in the developing testis.
Cynomolgus Monkeys
The majority of studies undertaken in cynomolgus monkeys utilize animals that are immature or are a mixture of immature, peripubertal, and mature. As with dogs, the animals mature over a relatively wide age range. There are numerous references regarding the age and body weight of male cynomolgus monkeys in relation to attainment of sexual maturity, and most put the minimum age at around 4.5–5 years and a body weight of approximately 5 kg. In studies where sexually mature monkeys are needed, minimal criteria should be based on age and body weight. Even then, additional measurements and endpoints, such as sperm evaluation, testicular volume, and testosterone measurements, are generally needed to gain a high degree of confidence that the animals are sexually mature. Due to the size, cost, and limited supply of mature cynomolgus monkeys, it is not practical to use them for routine regulatory toxicity studies. Accepting that, it should be borne in mind that studies conducted with immature or mixed maturity status animals will not be able to detect testicular toxicants.
Minipigs
Use of the Göttingen minipig as a preclinical species is becoming more common. Boars become sexually mature at 3–4 months of age with a body weight of 7–9 kg. A characteristic feature of pig testes is the large size and volume of Leydig cells in the interstitial compartment. This increases with age and can be quite variable in peripubertal animals (3–4 months of age) such that the more mature animals can appear to have Leydig cell “hyperplasia” when compared with the less mature animals. Similarly, the regularity and efficiency of spermatogenesis will continue increasing with age, and there maybe variability in pigs that have recently attained maturity.
Background Pathology as a Confounding Factor for Identifying Reproductive Toxicity
In addition to immaturity, background pathology can cause significant problems for detecting toxicity, particularly in the dog. This is rarely a problem when examining rodents, because they have relatively few background lesions and group sizes are large enough to be able to distinguish between test article-related changes and background changes.
Rodents
Spermatogenesis is very efficient and consistently normal between animals. Even though there may be an occasional animal that shows degenerative testicular changes, the incidence is low and the group sizes large enough that distinguishing them from test article-induced changes is generally not a problem. Unilateral or bilateral severe, diffuse tubular degeneration/atrophy is occasionally seen as a background finding in young adult rats and mice. When unilateral, the finding is likely associated with obstruction of the efferent ducts. The incidence is generally low, and should not amount to more than 1 or 2 rats in any one study of 40 males. If the number is significantly more than this, a relationship to the test article should be considered.
Dogs
Background pathology in dogs presents much more of a problem. Spermatogenesis in the Beagle dog is relatively inefficient, and it is very common to see tubules with partial depletion of one or more generations of germ cells (hypospermatogenesis) or focal areas of undeveloped tubules (hypoplasia). Almost all dogs will have scattered degenerating (multinucleate) germ cells and abnormal appearing, swollen spermatocytes (Figure 17.20). The incidence and severity of these lesions seem to vary with the cohort of dogs, suggesting a possible genetic component, but the high incidence of the findings combined with small group sizes make these changes problematic for distinguishing them from testicular toxicity (reviewed by Rehm, 2000).

The typical characteristics of segmental hypospermatogenesis in the dog are the absence of one or more generations of germ cells (e.g., pachytene spermatocytes and round spermatids) combined with the presence of earlier (spermatogonia) and later (elongating spermatids) germ cells in the tubule. Characteristically, there are very few actively degenerating germ cells associated with this lesion, and the pattern suggests failure of the spermatogonial division over two to three cycles in this part of the tubule. This is effectively “maturation depletion” as seen with spermatogonial toxicants, but tubules are affected in a segmental and patchy distribution rather than a diffuse manner. Another finding that seems largely unique to the dog is focal or segmental hypoplasia. The characteristics of this lesion are contracted tubules lined only by Sertoli cells, with the appearance of never having supported germ cells. This is different from atrophic tubules, which are usually lined by Sertoli cells with disorganized and vacuolated cytoplasm and evidence of residual germ cells or debris. Hypoplastic tubules generally form a wedge-shaped area which is often subcapsular. Segmental hypospermatogenesis and hypoplasia are specific diagnostic terms that have been defined and described in detail in the literature (see Suggested Reading). It is important to use the same terminology and diagnostic criteria for these lesions when seen in dog toxicity studies, so that the cited literature can be used to support any conclusion relating to their relationship to test article-administration. A final cautionary note is that hypospermatogenesis can also be caused or exacerbated by test article-exposure; thus, if the incidence and/or severity of this common background finding appears to be dose-related, a relationship to treatment should be considered.
The difference between immaturity, hypospermatogenesis, and tubular degeneration/atrophy is subtle, but it is extremely important for the interpretation of a study and for distinguishing test article-related testicular toxicity from either background degenerative changes or prepubertal degenerative changes. Table 17.8 lists the main features and a recommended approach for addressing this issue.
Table 17.8
Distinguishing Immaturity from Testicular Toxicity in Dogs and Primates
| Maturity status | Epididymal caudal duct expansion | Epididymal sperm content | Testis: presence of degenerate/multinucleate germ cells |
| Immature; normal | Contracted | None | None |
| Prepubertal; normal | Contracted | None | Few—frequent |
| Peripubertal; normal | Partial expansion | Few in corpus, none in cauda | Frequent |
| Mature; normal | Expanded | Plenty in distal corpus and cauda | Infrequent |
| Mature with testicular degeneration | Expanded | Some in distal corpus and cauda | Increased—frequent |
| Mature with hypospermatogenesis | Expanded | Usually normal but maybe low in severe hypospermatogenesis | Infrequent |

Table reproduced from Handbook of Toxicologic Pathology (2013), third ed. (W. M. Haschek, C. G. Rousseaux, and M. A. Wallig, eds.), Academic Press, Table 59.12, p. 2568, with permission.
Distinguishing testicular toxicity from background pathology and immaturity is difficult and ultimately comes down to a weight-of-evidence approach. This is where having multiple measures of the same thing can be most helpful (organ weights, sperm measures, and histology). Since hypospermatogenesis can be exacerbated by chemical exposure, an important part of that evidence is the background incidence and severity of hypospermatogenesis in the laboratory conducting the study. This calls for consistent grading and recording of the main background changes so that the historical control database is an accurate reflection of these changes.
Cynomolgus Monkeys
Relatively few studies employ mature cynomolgus monkeys, so there is comparatively little information on background lesions. Compared with the dog, there appear to be relatively few background degenerative changes and spermatogenesis appears more regular and efficient in this species. Focal tubular dilation with thinning of the seminiferous epithelium is quite commonly seen, especially in peripubertal animals. In addition, animals with focal areas of atrophic and vacuolated tubules are sometimes seen. A lesion that has become prevalent in the testes of cynomolgus monkeys in recent years is a congenital/developmental abnormality whereby varying proportions of the seminiferous tubules are replaced by dense collagen (Figure 17.21). This can be seen in immature and mature monkeys.
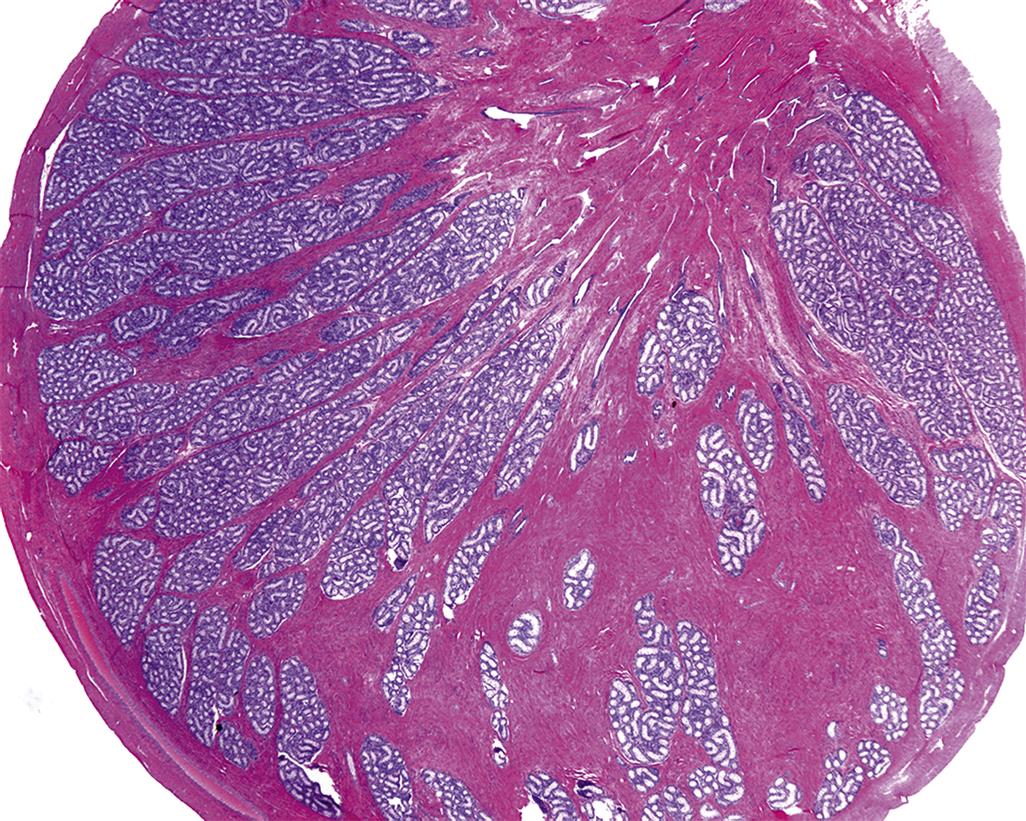
Minipigs
Tubular hypoplasia or tubular atrophy is a very common background change in adult minipig testes. It occurs in approximately 25% of untreated pigs. The affected tubules are generally contracted, devoid of germ cells, and lined only by Sertoli cells. In some cases they may contain a layer of spermatogonia and early spermatocytes, and in other cases there may be a patchy and partial depletion of germ cells. In most cases (approximately 60% of affected animals) the number of tubules affected is small (minimal severity) and the affected tubules are scattered throughout the testis, but in some cases a large proportion or the majority of tubules are affected. In these more severe cases, there is a reduction or absence of sperm in the epididymis. The change is generally bilateral and often associated with an apparent increase in the number of Leydig cells surrounding the affected tubules. If the incidence or severity appears to show a dose relationship it makes it very difficult to distinguish this common atrophic lesion from a test article-related effect. One criterion that maybe useful is whether the atrophic tubules are accompanied by a significant number of actively degenerating germ cells. Active germ cell degeneration is often a good indicator of ongoing toxicity and is less commonly associated with a longstanding background lesion. One final consideration when evaluating pig testes is the variable volume of the Leydig cell population. Since pigs are generally examined at an age where they have only just attained sexual maturity, the size of the Leydig cell compartment and the efficiency of spermatogenesis can vary significantly between different individuals. Differences should not necessarily be interpreted as test article-related findings.
Stress and Body Weight Loss as a Confounding Factor for Identifying Reproductive Toxicity
Reproductive success is tied to food availability, adequate metabolic reserves, and suitable environmental conditions. In general, activation of the hypothalamic–pituitary–adrenal axis by stress results in suppression of the HPG axis. Many of the neurotransmitters involved in energy homeostasis, such as leptin, ghrelin, cholecystokinin, and vasoactive intestinal peptide, as well as mediators of the stress response, such as corticotropin-releasing hormone, arginine-vasopressin, glucocorticoids, and β endorphins, are also major regulators of the GnRH neurons in the hypothalamus and will decrease reproductive capability in times of low food availability and/or stress (reviewed by Everds et al., 2013). In nonclinical studies, decreased food intake, decreased body weight, and nonspecific stress are common consequences of dosing animals near the maximum tolerated dose, and therefore effects on reproductive parameters are relatively common.
GnRH suppression will result in reduced testosterone secretion from the Leydig cells and reduced plasma testosterone levels, and this largely dictates the morphological changes seen in the reproductive tissues. In rodents, the most sensitive endpoint is a decrease in the weight of the accessory sex organs (prostate and seminal vesicles). Epididymal weight may also be decreased, but testis weight is generally unaffected except in mice. There are generally no detectable histopathological changes in any of the tissues. Exceptions include minimal tubular degeneration in the testes of mice and focal tubular degeneration in the testes of rats subjected to repeated immobilization in restraint tubes.
Dogs and monkeys are generally less susceptible to the effects of stress and body weight decrease, and few changes are seen. An exception to this is the effect of social stress on cynomolgus monkeys, particularly relating to hierarchical status. It is becoming common practice to house monkeys in social groupings during nonclinical safety assessment studies, but introduction of individuals to one another naturally leads to a ranking of dominant and subordinate individuals. High-ranking males will be those animals with the highest body weight, and they will also maintain the highest testosterone levels, while the low-weight monkeys will generally be the subordinate animals and will have low testosterone levels. Reductions in testicular size of up to 45% from baseline in subordinate males have been observed follow introduction to social groupings.
Mechanisms of Toxicity
Molecular and Biochemical
There have been numerous in-depth discussions of mechanisms of specific toxicants in recent years (see Boekelheide et al., 2005), so rather than duplicating these in detail a relatively high-level overview of some selected compounds will be provided, followed by a view of the emerging involvement of oxidative stress or damage in the male reproductive system. First, though, a frequently used mechanistic concept of “the target cell” will be considered.
Reporter Cell versus Target Cell
In the early 1980s, the term “target cell” was frequently used to indicate the cell type that showed the first visible changes during the development of a lesion in the testis (Table 17.9). This was done with the tacit understanding that there would be preceding biochemical changes which would be invisible to the pathologist, and these could be in any of the cells in the testis, not necessarily only in the first to be visibly affected. This subtlety is easily overlooked, and can lead to misguided investigational approaches and narrow interpretations regarding the nature of the toxicity. For example, a toxicant causing cell-specific death in the spermatocyte population may well be mediated through disturbances of cellular processes in the Sertoli cell that are critical to spermatocyte survival. Any mechanistic investigations into events leading up to cell death would benefit from looking not only at the spermatocytes but also at the cells required to keep the spermatocytes alive—the Sertoli cells.
Table 17.9
Site/Cell-Specific Changes Associated with Male Reproductive Toxicants
| Reporter/target cell | Toxicant | Effect |
| Leydig cell | Ethane dimethane sulfonate | Leydig cell necrosis with secondary germ cell death and depletion and atrophy of accessory sex organs |
| Lansoprazole (Prevacid) | Inhibition of testosterone synthesis and increased plasma clearance of testosterone resulting in Leydig cell hyperplasia and tumors in rats | |
| Sertoli cell | 2,5-Hexanedione, phthalate esters | Sertoli cell vacuolation with subsequent death or exfoliation of germ cells |
| Carbendazim, colchicine | Disruption of apical Sertoli cell cytoskeleton resulting in shedding of germ cells and Sertoli cell cytoplasm | |
| Spermatogonia | Busulfan, cyclophosphamide | Spermatogonial death with subsequent maturation depletion of later germ cells |
| Spermatocytes | Glycol ethers, nitroaromatics | Stage-specific death of pachytene spermatocytes, mediated through disturbances in Sertoli cell |
| Round spermatids | Ethyl methane sulfonate, methyl chloride | Damages the round spermatid and leads to depletion and abnormalities in the elongating spermatid head shape |
| Elongating spermatid | Boric acid, dibromoacetic acid | Retention and phagocytosis of step 19 spermatids, abnormalities in the released sperm |
| Testicular blood vessels | Cadmium chloride | Damages endothelium of testicular blood vessels causing ischemic necrosis of all testicular cell types |
| Epididymis | Phosphodiesterase 4 inhibitor, α-chlorohydrin | Inhibition of fluid reabsorption in efferent ducts resulting in sperm granulomas and secondary testicular atrophy |
| Methyl chloride | Epithelial degeneration resulting in sperm granulomas | |
| Epididymal sperm | Ornidazole, α-chlorohydrin | Inhibition of glycolysis resulting in loss of sperm motility |
| Vas deferens | Guanethidine | Inhibition of ejaculation due to adrenergic receptor blockade, resulting in rupture at vas epididymal junction and sperm granulomas |
| Prostate and seminal vesicles | Flutamide | Androgen receptor blockade resulting in secretory inhibition and atrophy |
| Finasteride | Inhibition of metabolism of testosterone to dihydrotestosterone resulting in secretory inhibition and atrophy | |
| α-Adrenergic receptor antagonists | Inhibit contraction/secretion of fluid from the seminal vesicles resulting in dilation |
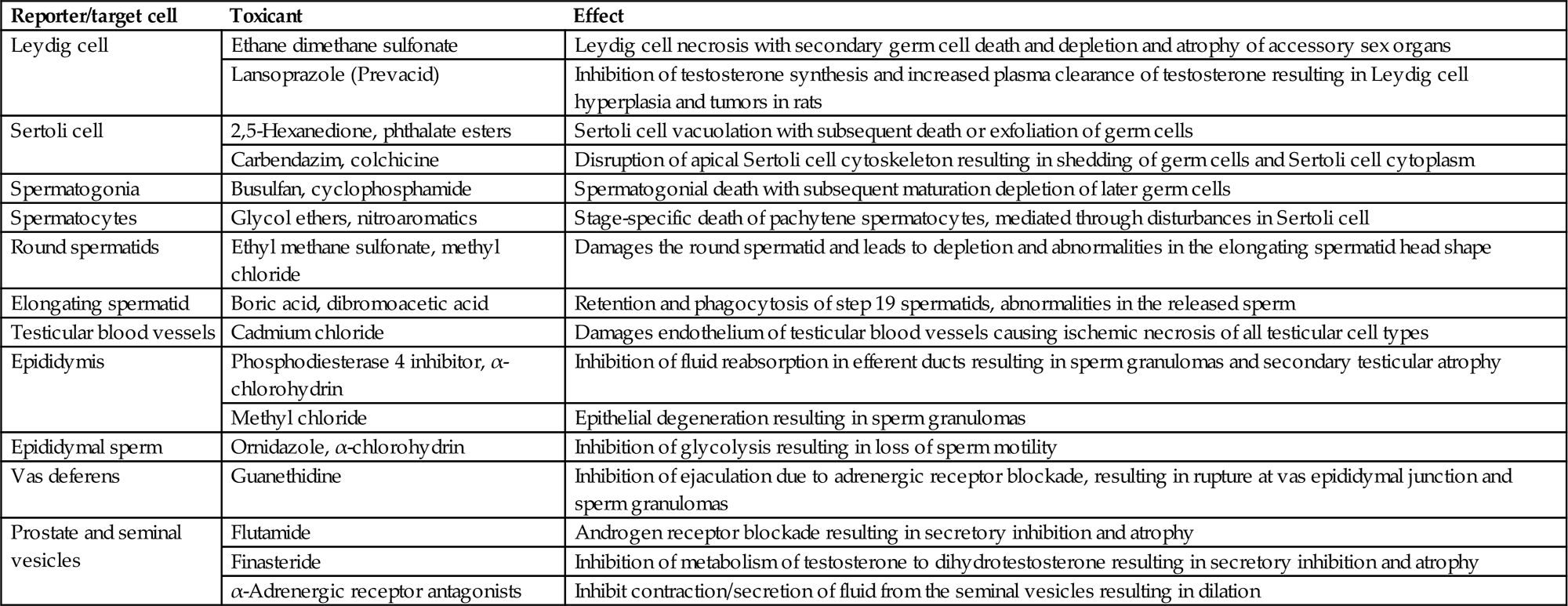
Table reproduced from Handbook of Toxicologic Pathology (2013), third ed. (W. M. Haschek, C. G. Rousseaux, and M. A. Wallig, eds.), Academic Press, Table 59.13, p. 2573, with permission.
Rather than “target cell,” we propose the use of the term “reporter cell.” This carries with it an implicit acknowledgment that the altered cell is merely reporting a lesion and does not imply that this cell is necessarily the main site of the biochemical events leading up to the lesion. Using “reporter cell” removes the potential confusion and assumptions associated with the term “target cell,” and additionally accommodates the complexity that is likely to be uncovered by toxicogenomics (see below).
In some cases the reporter cell and target cell are indeed the same cell, and in a very few cases this has been demonstrated. One such example is 2,5-hexanedione which was extensively investigated by Boekelheide et al., whose results implicate the Sertoli cell as the initial cell showing morphologic and biochemical evidence of injury, following exposure. In addition, the extensive sloughing of the adluminal third of the seminiferous epithelium caused by intratesticularly administered colchicine can plausibly be explained by the fact that colchicine targets microtubules, and the Sertoli cells are the site of microtubules in the epithelium. These two lesions are unlikely to be changed by the use of a “reporter cell” term. However, the involvement of the Sertoli cell in the germ cell death lesion seen after ethylene glycol monomethyl ether (EGME) exposure would not be so readily overlooked. We expect that the Sertoli cells will be found to play an etiologic role in other lesions of germ cells, and the reporter cell term should help remind us of the limits of our knowledge about how these lesions develop.
Nitroaromatics
The testis is sensitive to multiple nitroaromatics, including nitrofurantoin, nitrofurazone, nitroimidazoles, di and trinitrotoluene, chlorodinitrobenzene, and 1,3-dinitrobenzene (DNB). Since the first description of its toxicity in 1981, DNB has been the subject of numerous in-depth investigations. Some of these studied key events in the development of this specific lesion. Others used DNB as a tool to explore the nature of response to toxic exposures and the testis’ recovery after varying degrees of insult. Others looked for differences between a single administration and multiple administrations, and added biochemical measures of effect, or evaluated fluid production or hormonal effects.
The first morphologically identifiable effects produced by DNB are ultrastructural vacuolation in the Sertoli cells and, in fixed tissue sections, the appearance of space between the Sertoli cells and germ cells, interpreted as retraction of Sertoli cells from the germ cells. Perhaps predictably, when mixed-cell cultures are made with Sertoli cells and germ cells and DNB is added to the cultures, this retraction translates to a release of the germ cells into the medium. This is dose-related and quantifiable. The Sertoli cells catabolize DNB in vitro, creating more polar metabolites through the reactive nitrosonitrobenzene, which produces a similar type of germ cell loss when applied to the cultures and appears more potent than the parent DNB. The hypothesis was put forward that the local production of this metabolite was responsible for the observed toxicity and is one of only three or four compounds whose testicular metabolism is thought to play a central role in the observed testicular toxicity. Collectively, these data above led to defining the Sertoli cell as the target of DNB’s toxicity, and its site of metabolism. Another important aspect of the toxicity caused by DNB is the role of the glutathione detoxification pathway in determining species sensitivity to DNB. The hamster is resistant to testicular toxicity when exposed to the same blood levels or cumulative exposure of DNB as is toxic to the rat. This is despite the fact that mitochondrial preparations of hamster testicular tubular mitochondria are more efficient in catabolizing DNB to its active metabolite. However, cellular levels of glutathione and ATP are depleted more rapidly in rat mitochondria than in hamster, resulting in oxidative stress within the Sertoli cell of the rat but not the hamster. This has been proposed as the probable cause of the differential species sensitivity.
In the case of DNB, toxicogenomics has led to several interesting insights, not least of which is the complexity of the response to a toxic insult. First, as expected, there are numerous significant gene expression changes that occur before any lesion is visible in the tissue. The earliest gene reductions (and by far the largest) were those enriched in pachytene spermatocytes, which were the site of the first visible change by light microscopy (spermatocyte apoptosis), even though the initial ultrastructural changes were seen in the Sertoli cell. Those authors focused on the cell adhesion genes and found that the genes coding for the cell-cell adhesion molecules (many of them Sertoli cell based) were all significantly increased at times when cells were beginning to be sloughed from the testis, and even trended insignificantly upwards at 4 hours after exposure, before there were any observable lesions. There were also changes in genes responding to oxidative stress and nitro metabolism, though these last were not explored. In this situation, we are left considering how to relate an upregulated Sertoli adhesion gene to the release and death and sloughing of germ cells. The most likely possibility is that this increase responds to an earlier effect on the function of those adhesions. If that is true, it demonstrates how genomics, as inclusive and insightful as it is, does not capture nongene-based functions of proteins. Adhesions malfunctioning because of an oxidative damage-induced change in intracellular signaling will not be revealed by an array analysis. This demonstrates the need for additional biochemical assays which evaluate the function of subcellular components. To obtain a full and balanced picture of the events in lesion development will require relating biochemical, genomic, and structural changes to each other over time.
Glycol Ethers
The glycol ethers were also used as investigative toxicants in the emerging field of male reproductive toxicology in the early 1980s. This included explorations of the structure–activity relationships, and many studies using the two most potent congeners (EGME and ethylene glycol monoethyl ether) as tool compounds to visualize various patterns of response. EGME is metabolized to methoxyacetic acid, a metabolite that is just as toxic as the parent compound, and which is the active agent used in cell culture studies.
Numerous pathogenesis studies identified the early and late pachytene spermatocytes (but not mid pachytene spermatocytes) and the dividing spermatocyte as the most sensitive germ cell for this family of toxicants, with cell death being stage-specific and occurring within 12 hours of a single dose. Numerous follow-on studies have used this compound as a tool, based on that observation and on the assumption of the spermatocyte being the specific target. Despite this collective tunnel vision, it is important to remember that several other studies have reported either small vacuoles at the ultrastructural level in Sertoli cells which resolved by 24 hours after a single administration, or early changes in Sertoli cell enzymes or receptors or kinases. The Leydig cells never appeared abnormal, although prostate weights were occasionally reduced in some studies.
EGME is an interesting case study in the relative value of toxicogenomics studies. Work in the 1980s and 1990s regarding various mechanisms had explored several separate avenues: (1) that methoxy acetic acid (MAA) could form an MAA–CoA metabolite which could disturb carbohydrate metabolism, (2) that calcium channel blockers and kinase inhibitors could prevent many of the testicular (and other) toxicities observed after EGME treatment, (3) that Sertoli cells were required for the full expression of the cell death program in the spermatocytes, and (4) that kinase activity plays a central role in the spermatocyte death. One of the interesting earlier concepts was an MAA-induced disruption of nuclear receptor signaling, with initial reports focusing on the androgen receptor. This helped to identify early responses in Sertoli cells. Subsequent work on this and another short-chain fatty acid (valproic acid) found early and specific dysregulation of multiple nuclear receptors through activating the mitogen-activated protein kinase (MAPK) and inhibiting histone deacetylase, although neither were sufficient to account for all the receptor-potentiating effects reported for MAA.
Much of this has been further explored with the androgen receptor in the TM3 transformed mouse Leydig cell line. Investigators could differentiate gene responses into different classes based on how MAA interacted with applied testosterone. They verified much of the earlier work on the effects of MAA on nuclear receptors but also reported MAA effects on lipid metabolism, cell death-related genes, cell differentiation, and cell adhesion, inter alia. The fact that such gene sets were upregulated in a transformed line of a cell type which shows no pathology or visible change after EGME exposure in vivo is perplexing. On the one hand, it is reassuring that genes presumably underlying (or at least consistent with) the in vivo lesion are altered in vitro. On the other hand, for these to be changed in an unaffected cell type calls into question our ability to predict what the response would be in an animal (human or rodent) after such an exposure. How would we be able to predict that spermatocytes would be the responding cell based on these data from a Leydig cell line? Although confusing, a possible way to view these data is that they may imply a future where pathway testing is done in any available cell type (or perhaps multiple cell types), and then a yet-to-be-discovered method is applied to identify which cell types will be the ones responding. It is encouraging to think that multiple cell types would give the same response, but daunting to consider how much more we have to learn in order to predict a real in vivo response. The other thing this calls into question is the current common practice of linking gene response data from affected cells to the lesions in those cells.
More recent toxicogenomics work with MAA has found significant response of oxidative stress responsive genes, which leads into our next proposal for considering mechanisms.
Oxidative Stress
Oxidative stress has been known as a mechanism of toxicity for decades, with initial work focusing on unstable epoxides formed by the metabolism of specific chemical structures. Its occurrence appears to be much more widespread than the initial focused work might imply.
All cells of obligate aerobes (this would include all major laboratory species as well as humans) use mitochondria to pass electrons from metabolic end-products (lactate and pyruvate, among others) down the electron transport chain and combine them with oxygen to form water. Not all electrons will be fully controlled, and if the estimated 2%–3% of these electrons are not fully reduced, their uncontrolled recombinations would produce reactive oxygen species (ROS). It has been estimated that, under normal metabolic conditions, the mitochondrion and membrane-bound NADPH oxidase are the two significant sources of oxidation damaging agents in the cell. A survey of the literature shows there is good evidence that oxidative stress is a regular feature of testicular toxicants or toxic exposures.
The following have been found to produce testis lesions and either upregulate oxidative stress responsive genes or have that lesion be ameliorated by antioxidant treatment: experimental diabetes, cadmium, cryptorchidism, sasanguasaponin, Di(2- ethylhexyl) phthalate (DEHP), chlorpyrifos, lead, TCDD, and EGME. Each of these reports has its shortcomings, but the strongest papers individually, and the whole of them collectively, lend support to the concept of oxidative damage as an early, even initiating, factor in testis damage. But the question must be asked: How might a general feature such as oxidative damage manifest as a lesion in one tissue and not others?
The mitochondria are the main source of ROS in the cells, and the mitochondria from different tissues express their own unique suite of proteins. Oxidative damage is more than lipid peroxidation and membrane damage. Oxidant molecules (superoxide and hydrogen peroxide much more than the hydroxyl radical) can impact signaling systems and either act as signaling molecules themselves or impact several critical intracellular control systems. Chief among these are MAPK, the Jun N-terminal kinases (JNKs), and members of the p38 family. In most other cell types, these proteins are key to cell survival or death, and help control cell migration and division … in short, they control many key cell outcomes. These are all found in the testis, and the excellent review by Wong and Cheng (2005) demonstrates how these help control not only cell life and death but also the junctions that attach the germ cells to the Sertoli cells as well as the junctions forming the blood–tubule barrier between adjacent Sertoli cells. Disrupted blood–testis barrier structure or function has been associated with severe testis lesions, and certainly sloughing of prematurely released germ cells is a key feature of many acute testicular toxicants. Given our evolving appreciation of the complexity of these junctions and their control by kinases and numerous intracellular activities, it appears very plausible that oxidation of, for example, a controlling cysteine residue on a MAPK or a JNK or p38 protein would then disassemble the junctions holding the germ cells to the Sertoli cells. Even if this was not followed by FasL synthesis and release, the loss of junctional attachment should result in the death of that cell, given the dependence of the germ cells on the Sertoli cell for nutrition and maintenance. Such release would then produce the familiar sight of an epididymal lumen with increased numbers of sloughed round germ cells in varying stages of death and degradation.
It is unlikely that all testicular toxicants would produce an increase in ROS, but the increase in oxygen consumption and mitochondrial activity which indicates compensated toxicity (the hormetic response) is seen for an astonishing number of toxic exposures and conditions. If this increased mitochondrial activity does lead to increased flow through the electron transport chain, and if some proportion of those are poorly controlled or produce ROS, it is easy to see how there would be oxidative damage and likely changes in intracellular signaling after exposures which do not produce epoxide intermediates. Moreover, the tissue-specific mitochondrial proteome provides a plausible mechanism to begin to explain why one tissue might respond to a toxicant and others would not. Altogether, the data appear more convincing that some degree of oxidative damage or stress is at least a component of, if not a cause of, many testis lesions.
Morphologic Patterns of Response to Different Types of Injury
It would be truly rewarding for the pathologist to be able to examine the reproductive tissues in a short-term or time-course study, identify the true target cell for toxicity, postulate a mechanism for injury based on the known pharmacology or biological activity of the test material, and utilize this in the overall risk assessment or mode of action (MOA) evaluation for exposure in man. In reality, complete understanding of any mechanism of testicular toxicity is rare and the information is difficult to generate (Tables 17.9–17.12). As discussed earlier, the complexity of cell–cell signaling in the testis is such that morphologic injury, even of a specific cell type within a specific stage of spermatogenesis, generally reflects a downstream effect of disturbed cell signaling in a different cell type. Thus, as suggested above, the use of the term “reporter cell” rather than “target cell” is probably a more valid concept.
Table 17.10
Patterns of Change Associated with Sertoli and Germ Cell Injury
| Injury | Pattern of change | ||
| Testis | Epididymis | Prostate/seminal vesicles | |
| Sertoli cell injury (early) | Foamy vacuolation in Basal Sertoli cell cytoplasm or solitary large discrete vacuoles. Focal drop out of germ cells. Spermatid retention. Disorganization of germ cell layers | Sloughed testicular Germ cells present in lumen (initially confined to caput, later in the cauda) | Normal |
| Sertoli cell injury (later/late) | Partial/patchy loss of germ cells. Progressive diffuse degeneration of germ cells (without cell-or stage-specificity). Sloughing of germ cells into lumen | Larger numbers of sloughed germ cells. Decreased sperm content | Normal |
| Germ cell injury (early) | Degeneration or depletion of a specific germ cell population(s) with remaining germ cell layers appearing normal (cell- and stage-specific) | Normal | Normal |
| Germ cell injury (later/late) | Progressive depletion of more mature germ cell layers (maturation depletion) | Decreased sperm content, but not until the maturation depletion has reached step 19 spermatids | Normal |
Table reproduced from Handbook of Toxicologic Pathology (2013), third ed. (W. M. Haschek, C. G. Rousseaux, and M. A. Wallig, eds.), Academic Press, Table 59.14, p. 2577, with permission.
Table 17.11
Patterns of Change Associated with Efferent Duct Obstruction and Anoxia/Ischemia
| Injury or lesion | Testis | Epididymis | Prostate/seminal vesicles |
| Efferent duct obstruction/disturbed fluid reabsorption | Mixture of tubular dilation and severe tubular atrophy. Frequently unilateral. Dilated testes are increased in weight. Rete testis maybe dilated. Atrophic tubules frequently have patent or slightly dilated lumens. Interstitial edema maybe present. Incidence of affected testes often sporadic with poor dose relationship | Sperm stasis and granulomatous inflammation in the efferent ducts. Epithelial apoptosis of the initial segment. Cribriform change in the caput epididymis. Epididymis associated with testis that has dilated tubules may or may not have sperm content. No sperm and generally no sloughed germ cells in epididymis of atrophic testis | Normal |
| Anoxia/ischemia (testis) | Initial anoxia causes increased apoptosis of spermatogonia and early spermatocytes. Early ischemia causes separation of germ cells (similar to autolysis). Later progresses to necrosis (coagulative) of germ cells and Sertoli cells. Generally, associated with an interstitial inflammatory response to necrotic tubules. Affected tubules often become mineralized and may proceed to fibrosis. May affect individual tubules or affect focal areas | Sloughed testicular germ cells and reduced sperm | Normal |

Table reproduced from Handbook of Toxicologic Pathology (2013), third ed. (W. M. Haschek, C. G. Rousseaux, and M. A. Wallig, eds.), Academic Press, Table 59.15, p. 2583, with permission.
Table 17.12
Patterns of Change Associated with Androgen Imbalance in Rodents
| Type of androgen imbalance | Pattern of change | ||
| Testis | Epididymis | Prostate/seminal vesicles | |
| Decreased testosterone (early/mild) | Degeneration of occasional pachytene spermatocytes and round spermatids in stage VII/VIII tubules. Spermatid retention. Leydig cell atrophy (if severe testosterone reduction) | Apoptosis of ductal epithelium (most prominent in the initial segment) | Decreased weight. May see increased epithelial apoptosis |
| Decreased testosterone (late/severe) | Progressive degeneration and depletion of elongating spermatids. Leydig cell tumors (rat) | Reduced sperm content, reduced weight, ductal atrophy | Reduced weight, reduced secretion, atrophy |
| Androgen agonist | Same effects as with decreased testosterone due to effects on hypothalamic pituitary feedback. Inverse dose relationship with most severe disruption of spermatogenesis at low doses and less severe effects at high dose | Reduced sperm content, reduced weight, ductal atrophy | Increased weight, increased secretion, enlargement |
| Androgen antagonist | Leydig cell hyperplasia (early), Leydig cell tumors (late) (rat) | Normal or slightly reduced weight | Reduced weight, reduced secretion, atrophy |
| Reduced DHT (5α-reductase inhibitor) | Normal (rat). Leydig cell hyperplasia and tumors (mouse) | Normal or slightly reduced weight | Reduced weight, reduced secretion, atrophy |
| Estrogen agonist | Progressive degeneration and depletion of elongating spermatids. Leydig cell hyperplasia and tumors (mouse) | Apoptosis of ductal epithelium (most prominent in the initial segment). Reduced sperm content, reduced weight, ductal atrophy | Reduced weight, reduced secretion, atrophy |
Table reproduced from Handbook of Toxicologic Pathology (2013), third ed. (W. M. Haschek, C. G. Rousseaux, and M. A. Wallig, eds.), Academic Press, Table 59.16, p. 2584, with permission.
Nevertheless, the terms “Sertoli cell toxicants” versus “germ cell toxicants” and “hormonally mediated toxicants” are commonly used to distinguish between different manifestations of toxicity within the testis (reviewed by Creasy, 2001). This classification is based largely on the reporter cell, as indicated above: the cell type that shows the earliest morphological changes, along with the characteristics of the lesion as it progresses with time (Table 17.9). Taking that one step further, toxicants can be classified into broad categories based on their overall pattern of morphologic change. This is useful since it allows the design of additional biochemical or molecular investigations, and because it provides useful information for risk assessment or MOA evaluation. There are a number of different patterns of change that the pathologist can recognize, but these depend on whether the lesion is very early in its development or has been developing for some time, or whether it has reached its end stage (see Tables 17.10–17.12 for a summary of the major features of the different patterns). If an end-stage or advanced stage of a lesion is present in the high-dose group of a study, it is often possible to see the early stages of lesion development in the lower-dose groups, because in many cases lower exposure levels take longer to begin exerting their effects—so in some cases the same information can be gained from a routine toxicity study without doing an actual time-course study. When considering a time-course study to identify early changes, the duration of dosing will depend on the toxicant and the dose level, and the speed of onset of the lesion. Changes can be seen within hours of a single dose for some toxicants but can take weeks to develop for others.
The following patterns of change are based largely on the responses seen in the rat which is an ideal species for detecting subtle changes. The same broad principles apply across species, but it is much more difficult to identify specific changes in the dog and monkey.
Patterns of Changes Associated with Sertoli Cell Injury
When the Sertoli cell is the primary site of morphological damage, the pattern of subsequent germ cell death and depletion is frequently multifocal and affects numerous or all different germ cell types when examined in a repeat-dose study (Table 17.10). However, if the earliest signs of the lesion are examined, it is common to see stage- and cell-specificity associated with the germ cell death. For example, DNB is a Sertoli cell toxicant that produces Sertoli cell vacuolation in stages VII and XI within 24 hours of dosing, which is followed by apoptosis of late pachytene spermatocytes in stages VI–XIII 24–48 hours after dosing. With repeat dosing and examination at a later time point, this stage- and cell-specificity will obviously expand through more cells and more stages.
Focal Germ Cell Loss
Each Sertoli cell supports a specific cohort of germ cells as they develop through spermatogenesis. If that Sertoli cell suffers significant injury, all of the germ cells it supports will likely die and disappear. In the earliest stages of lesion development, this leads to an appearance of focal loss or “dropout” of germ cells from that one Sertoli cell or a patchy partial loss of germ cells from multiple Sertoli cells in affected tubules (Figure 17.22). As the degree of damage progresses and more Sertoli cells become injured, a more generalized germ cell degeneration and depletion will develop, leading to a final end-stage lesion of severe tubular degeneration/atrophy (Figure 17.11A and C). Since almost all testicular toxicants produce the same end-stage lesion (severe tubular degeneration/atrophy), it is important to study the earlier changes to gain any insight into pathogenesis. In general, this pattern of change is associated with slow or incomplete recovery.
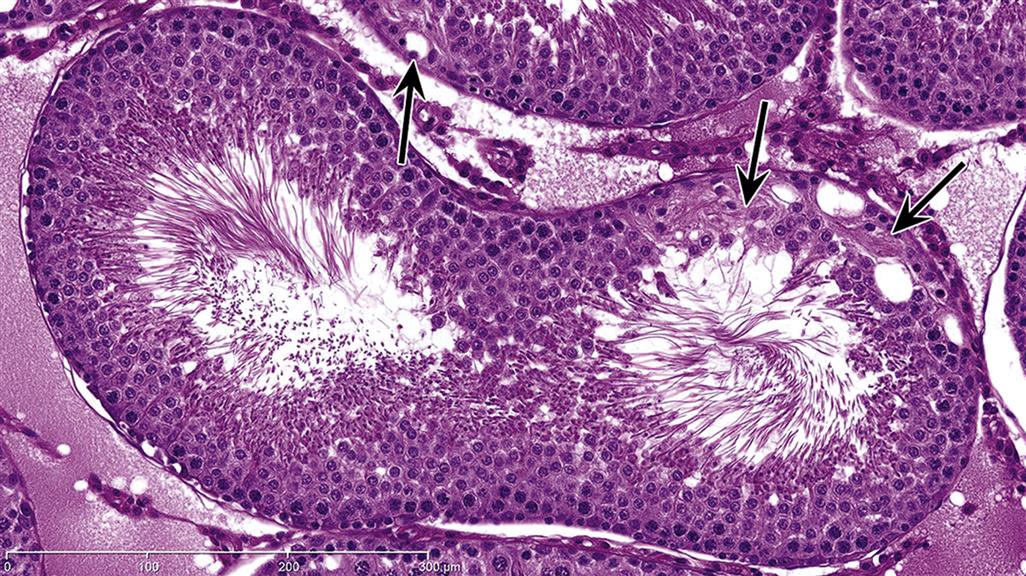
The other significant insight from such an early lesion is that not all Sertoli cells are equal. If they were a homogeneous population, they would all respond the same way. The fact that they do not must indicate considerable heterogeneity among the cells.
Vacuolation
Early evidence of Sertoli cell injury can also be reflected by macro- or microvesicular vacuolation of scattered Sertoli cells (Figure 17.14). Microvacuolation is generally present in the basal Sertoli cell cytoplasm and represents swelling and coalescence of membrane-bound organelles such as endoplasmic reticulum or vesicles. This may be accompanied by disorganization or displacement of the regular layering of germ cells. Although there maybe degeneration of germ cells, it is generally infrequent or develops subsequent to the vacuolation of the Sertoli cells. Macrovesicular vacuolation also occurs with some testicular toxicants, such as 2,5-hexanedione. This change is characterized by solitary or multiple large vacuoles at the base or part way up the tubular epithelium (Figure 17.14B). As an early change, this type of vacuolation is generally not associated with germ cell degeneration; however, if treatment continues, it inevitably leads to germ cell death and loss.
Germ Cell Sloughing and Shedding
When immature germ cells (spermatocytes and round spermatids) are admixed with sperm in the epididymis, they have been prematurely released from the seminiferous epithelium (Figure 17.16). In most cases the shed germ cells retain relatively normal nuclear and cytoplasmic characteristics, suggesting that they have lost contact with their Sertoli cell processes and been “passively” shed into the tubular lumen. Since the rat epididymis contains negligible round germ cells when there is normal spermatogenesis, this can be a very sensitive and very early indicator of spermatogenic disturbance and often is easier to see than the testicular change that is causing it. Care should be taken not to confuse apocrine cytoplasmic blebs, originating from the normal process of apocrine secretion by the efferent duct or epididymal epithelium, with sloughed testicular germ cells. Apocrine cytoplasmic blebs can be a prominent change, particularly in the dog epididymis.
The Sertoli cell supports and moves the germ cells up and down within the seminiferous epithelium, utilizing a well-developed cytoskeleton of microtubules and intermediate filaments. The germ cells share a variety of specialized junctions with the Sertoli cell, some of them unique to the testis, that hold the germ cells in place and embedded within the Sertoli cell cytoplasmic processes. If those junctions are broken or there is retraction of the cytoplasmic processes, the germ cells can be “cast loose” and shed into the lumen. This can be a very prominent change with some Sertoli cell toxicants, such as the phthalate esters, where retraction of the cytoplasmic processes that support and separate the germ cells has been shown to be an early change (Figure 17.23). Microtubule inhibitors, such as colchicine and carbendazim, cause shedding of the germ cells in sheets that are still attached to the apical cytoplasm of the Sertoli cell. This appears to be a wholesale sloughing of the apical third of the Sertoli cell and cytoplasm and germ cells, presumably due to disruption/dissolution of the cytoskeletal microtubules.
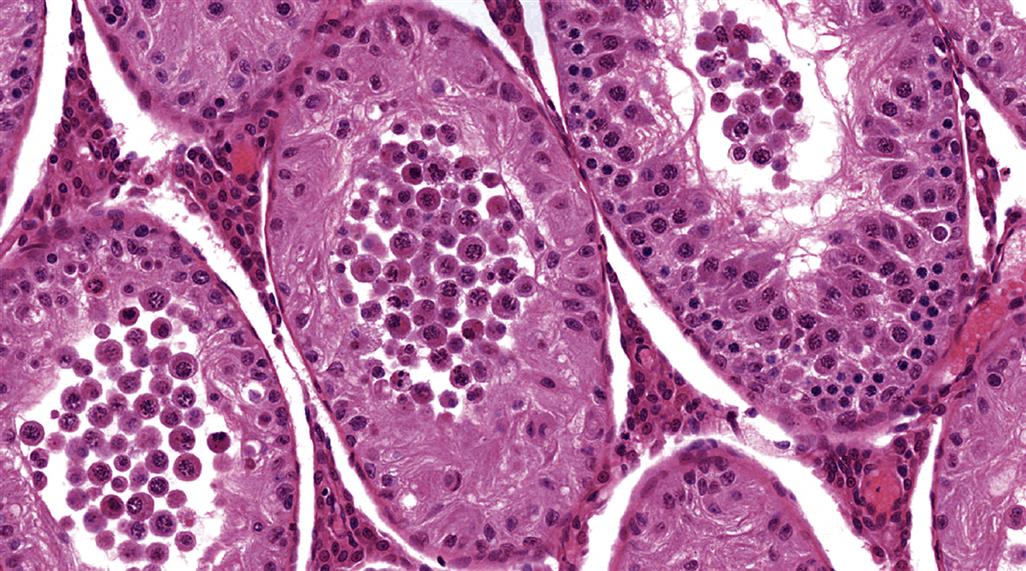
Tubular Contraction and Decreased Seminiferous Tubular Fluid
The Sertoli cells continuously secrete STF into the tubular lumen. This transports the sperm out of the tubule and into the rete and the efferent ducts, and is responsible for the patency of the tubular lumens. The size of the tubular lumen is directly related to the amount of fluid within the lumen, so if secretion is decreased the tubule will contract, and if secretion increases, or its exit from the testis is blocked, the tubule will dilate. Reduced secretion of STF has been demonstrated as an early event with a number of Sertoli cell toxicants, including the phthalate esters, 2,5-hexanedione, and indenopyridine exposure. Although qualitative microscopic evaluation is not very sensitive for detecting tubular contraction, particularly when there is differential shrinkage of tubules due to fixation artifact, the volume of STF can be measured. This is done by ligating the efferent ducts of one testis and comparing the weight difference between the ligated testis and the contralateral unligated testis after ≥15 hours. Decreased fluid content will also be reflected by a decrease in the weight of the testis; therefore, if there is a significant weight loss in the testis and no obvious germ cell loss, a disturbance in STF should be considered.
Patterns of Change Associated with Germ Cell-Specific Toxicity
When germ cells are the primary cell type damaged, the changes are generally cell- and stage-specific (Table 17.10). If the testes are examined early in lesion development, it is possible to identify apoptotic germ cells in the presence of normal-appearing Sertoli cells. More often, the testes are examined after repeat dosing, and since apoptotic cells are rapidly eliminated, the most characteristic feature of germ cell toxicity is a stage- and cell-specific depletion, which is commonly referred to as “maturation depletion.” The number of cell types depleted will depend on the cell type that has died as well as the duration of the dosing regimen. As an example, if a toxicant kills stage I pachytene spermatocytes, and this occurs on day 1 of dosing, by day 2 of the study most (but perhaps not all) of the necrotic cells will have been phagocytized and the tubule will have progressed to stage II, minus its pachytene spermatocyte population (Figure 17.9B). Moreover on day 2, the tubules that were in stage XIV on day 1 of dosing will now have entered stage I and their pachytene spermatocytes will become susceptible to toxicity and die (Figure 17.10D), and the process is repeated. If the testes are examined after 2 weeks of continuous dosing (one complete cycle of spermatogenesis), the death of the stage I pachytene spermatocytes 2 weeks previously will be reflected by the absence of step 1 round spermatids, as well as all of the developmental stages of pachytene spermatocytes in between (i.e., in stages II–XIV). The progress of maturation depletion over time is illustrated in Figure 17.24, and this pattern of germ cell depletion is the hallmark for recognizing germ cell toxicity.
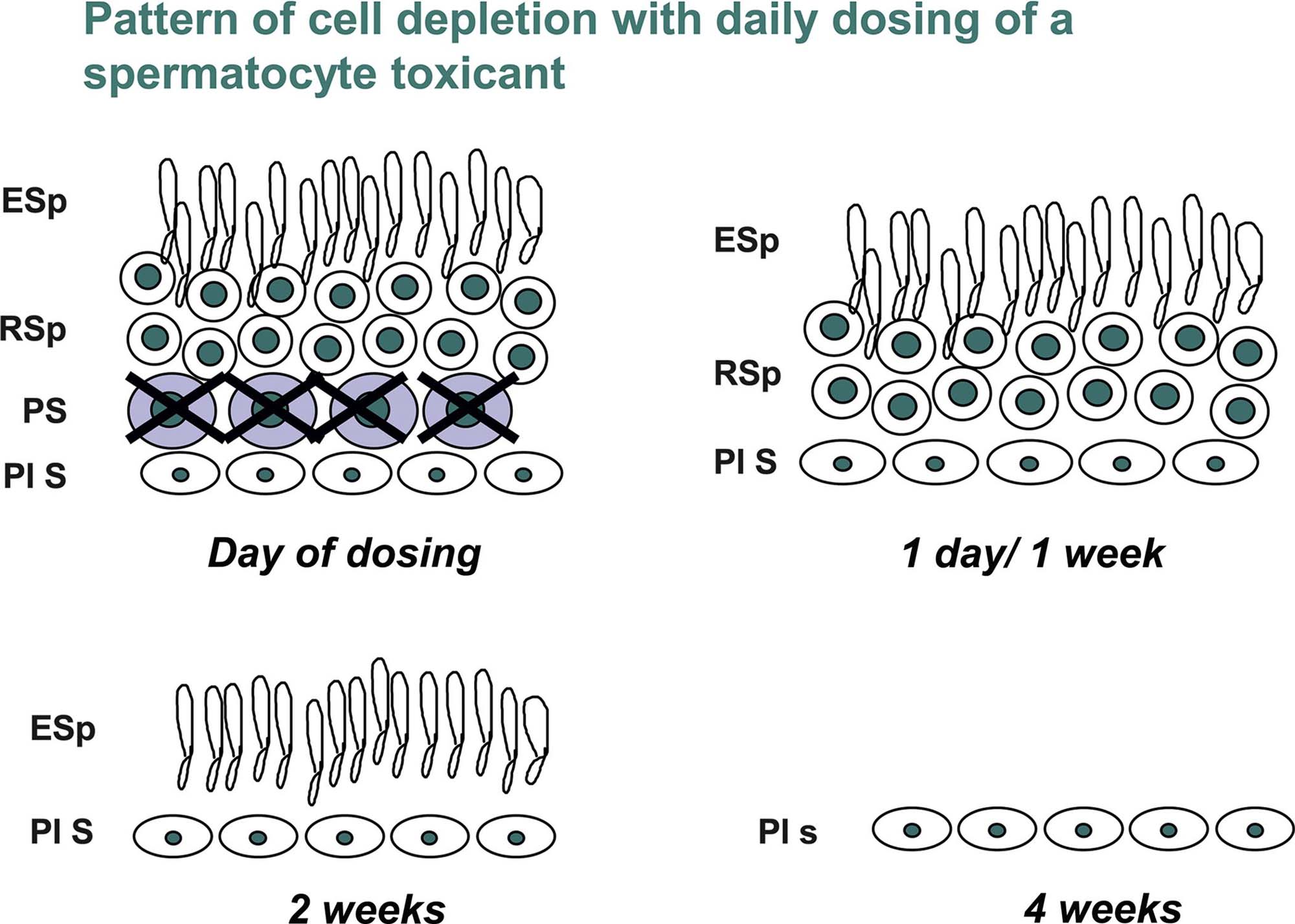
A feature of maturation depletion is that the effect becomes more obvious as the later developmental stages of germ cells (e.g., spermatids) are lost. This is because there are theoretically 4 spermatids for every primary spermatocyte, 2 spermatocytes for every B spermatogonium, and 32 B spermatogonia for every type A1 spermatagonium that enters spermatogenesis. The progressive expansion of the germ cell population means that a compound which targets stem cell spermatogonia will be very difficult to identify in conventional H&E sections during the first 2 weeks of dosing, because very few cells will be lost. By 4 weeks after the onset of dosing, depletion will have reached the much more numerous pachytene spermatocyte population and be more obvious, and by 6 weeks after the start of dosing it will be very obvious because a large proportion of the round spermatids will be missing, in addition to all the earlier cell types.
Another aspect of the dynamics of this progressive maturation depletion is the appearance of the testes at the end of dosing versus the end of recovery. At the end of dosing, maturation depletion may only have caused the loss of spermatogonia and a few stages of pachytene spermatocytes and be difficult to identify (Figure 17.10A and B), but following an additional 2- or 4-week recovery period the progressive depletion of the spermatid population will make the severity of germ cell loss appear much more severe (Figure 17.10C). In these situations, it is important to note which cells are missing at the end of dosing (spermatogonia and certain stages of spermatocytes) versus which cells are missing at the end of recovery (e.g., round and elongating spermatids) but with recovery of spermatogonia and early spermatocytes. Failure to do this will result in an erroneous conclusion that the damage is getting worse rather than recovering.
Degeneration and malformation of elongating spermatids is a form of germ cell-specific toxicity that is occasionally seen. In order to recognize this, the pathologist needs to have a good knowledge of the normal shape changes that the elongating spermatid head goes through as it moves through each of the stages of the cycle. A head malformation is most easily recognized in stages XI–XIII, where the head loses its long delicate curvature and becomes shortened, clubbed, and condensed (Figure 17.25). There also appears to be abnormal tail formation and retraction of the cytoplasmic coating around the developing flagellum of the tail. These changes are usually accompanied by the presence of cell debris in the head of the epididymis, which appears to consist of prematurely shed heads and tails of malformed elongating spermatids. It is possible that these deformed elongating spermatids are due to disturbances in the formation of the acrosome earlier in spermiogenesis. This has been described in the case of carbendazim.
Germ cell toxicity has been described for a number of toxicants; the best documented is probably EGME or its active metabolite MAA (see “Mechanisms of Toxicity of Glycol Ethers” section). At low doses, this chemical causes very specific death of stage I–V pachytene spermatocytes and stage XI–XIV pachytene and dividing spermatocytes (Figure 17.10D) within hours of dosing. Although there is no visible evidence of Sertoli cell injury, additional investigations indicate that the Sertoli cell is essential in mediating the germ cell toxicity, thus, supporting the concept of the reporter cell. As the dose level increases, pachytene spermatocytes in all stages become susceptible, and at even higher doses the toxicity expands into the spermatid population and the prepachytene spermatocyte population. This loss of cell- and stage-specificity as the dose level increases is a common phenomenon.
Patterns of Change Associated with Anoxia or Ischemia
In theory, the seminiferous epithelium should be very susceptible to reduced blood flow, because there are no capillaries within the epithelium and all oxygen and nutrients need to travel from the interstitial capillaries into the interstitial fluid and be transported through the Sertoli cells to reach the germ cells sequestered within the Sertoli cell processes (Table 17.11). An alternative view is that the tubules would likely be resistant to anoxia because they operate normally at such low oxygen levels (3–5 mm O2 intratubularly). In fact, in early experiments where the blood supply was ligated for varying lengths of time, total ligation for up to 60 minutes resulted in little or no damage but ligation for 90–105 minutes caused selective damage to the spermatogonia and preleptotene spermatocytes. The degree of damage observed depends on whether the testes are examined at the end of the ligation period or whether the blood flow is reinstated (reperfusion). If blood flow is cut off and then reestablished (as is the case for surgically treated testicular torsion in humans), there is often no obvious damage from the ischemic event arising from the torsion, but spermatogenesis generally is severely affected due to the subsequent apoptotic death of a large proportion of the germ cells. This delayed damage appears to be due to the generation of ROS by the leukocytes that infiltrate the ischemic tissue during the torsion phase.
The most sensitive cells to the injurious effects of reduced oxygen tension appear to be the spermatogonia and early spermatocytes, which undergo an increased rate of apoptosis. This is surprising because they are the closest cells to the oxygen-containing interstitial fluid and are outside the blood–testis barrier, whereas the pachytene spermatocytes and spermatids, which are metabolically active and inside the blood–testis barrier, which is further from the source of oxygen, appear less sensitive to reduced blood flow. Studies that examined the effects of graded reductions in testicular blood flow on spermatogenesis have demonstrated an increased level of apoptosis in the spermatogonia and early spermatocytes after 5 hours of blood flow reduced by 70% of its pretreatment level. At this reduced level of flow, the numbers of apoptotic germ cells were increased by approximately 1.8-fold of pretreatment values and kept increasing in direct correlation with a further decrease in blood flow up to 50% of pretreatment values. Further reductions in blood flow resulted in a more generalized disruption, necrosis, and exfoliation of the tubular epithelium rather than germ cell apoptosis (Figure 17.12A). Reduction in testicular blood flow was also accompanied by increased numbers of leukocytes in the blood vessels, with some migrating out into the interstitial space.
A particular pattern of ischemic necrosis is seen with reductions in blood flow produced by some kinds of toxicants. The change has been best described, following a subcutaneous injection of very large doses of human chorionic gonadotropin (hCG) to the rat. The lesion is characterized by a focal area of coagulative necrosis, associated with a leukocytic infiltrate and located specifically in the frontal lower pole of the testis. The mechanism of the hCG-induced necrosis appears to be due to stimulation of the Leydig cells by hCG to synthesize prostaglandins which cause prolonged (>12 hours) contraction and cessation of blood flow in the intratesticular arteries in the frontal lower pole of the testis, resulting in tubular necrosis in this part of the testis. Although the testicular arteries in the remainder of the testis initially showed the same contraction and reduced blood flow, they recovered much more rapidly and did not cause necrosis of the adjacent parenchyma.
A unique characteristic of blood flow in the testis is its pulsatile nature. This is called vasomotion, and, although its function is not entirely understood, it appears to be very important in the movement of oxygen and nutrients from the capillaries into the interstitial fluid. It has been shown to be reduced or ablated in a number of vascular-induced testicular lesions, including partial arterial obstruction (graded ischemia), hCG-induced testicular necrosis, 5HT-induced testicular vasoconstriction, and testicular torsion. It appears to be a critical component of blood flow when dealing with recovery of the seminiferous epithelium, in that recovery can be compromised if vasomotion is absent, even though blood flow may have returned to normal.
Patterns of Change Associated with Loss of Androgen Support
Recognition of testicular toxicity mediated by androgen withdrawal requires an integrated evaluation of the entire reproductive tract, because changes in the epididymis and accessory sex organs are generally more sensitive than the often subtle cell- and stage-specific changes in the testes (Table 17.12). In addition, there are a number of different mechanisms of reduced androgen status, and each will have a different pattern of effects.
Testicular Changes
Androgen status can be reduced by various pathways: (1) decreasing testosterone synthesis, (2) blocking the interaction of androgens with the androgen receptor (antiandrogens, and antagonists), or (3) inhibiting conversion of testosterone to the more potent androgen, DHT. In all cases there will be atrophic changes in the epididymis and accessory sex organs, but the nature of the changes seen in the testes depends on the nature of androgen disruption. Quantitatively normal spermatogenesis requires high levels of intratesticular testosterone, but in the rodent qualitatively normal spermatogenesis can continue despite moderate reductions in testosterone levels. In the dog, reduced testosterone appears to have a more dramatic effect on spermatogenesis.
The effects of testosterone withdrawal have been most fully described in the rat, following ethane dimethane sulfonate administration, which causes Leydig cell necrosis and reduces testosterone to castrate levels. The earliest changes are seen 4 days after destruction of the Leydig cells, and comprise a cell- and stage-specific apoptosis of occasional pachytene spermatocytes and round spermatids in stage VII and VIII tubules (Figure 17.26A). At later times, spermatocytes, and particularly elongating spermatids in later stages (IX–XIV), show degeneration (Figure 17.26B). This reflects the fact that these cells have passed through stages VII–VIII in the absence of testosterone, undergoing lethal damage in the process; however, degeneration is delayed to a later time when they undergo a critical event, which relies on their previous exposure to testosterone. The endstage lesion for severe testosterone insufficiency in the rat, which is seen after about 2 weeks of treatment, is a slight reduction in the numbers of mid and late pachytene spermatocytes and round spermatids, but an almost complete loss of elongating and maturation phase spermatids (Figure 17.26B). This is accompanied by a marked shrinkage of the tubule, which is partly due to a loss of germ cells but is also caused by a reduction in the secretion of androgen-dependent STF by the Sertoli cell. Testosterone withdrawal also leads to abnormal retention and phagocytosis of step 19 spermatids at stages IX–XII. Although spermatid retention appears to be a common finding with treatments that disturb testosterone levels, it is not unique to this class of compound.
The effects of low testosterone on the dog testis do not show the same cell- and stage-specificity as seen in the rat. With mild reductions in testosterone there appears to be an increased rate of germ cell degeneration and depletion, but with more marked testosterone reduction there is a total loss of spermatogenesis, leaving empty, shrunken seminiferous tubules.
Any morphologic changes in the Leydig cell will depend on the nature of the hormonal disturbance and the species. Increased LH stimulation of rodent Leydig cells will result in hypertrophy of the cell due to an increased volume of smooth endoplasmic reticulum and, if prolonged, may also lead to diffuse hyperplasia of the cell population. Conversely, reduced stimulation of the Leydig cell by LH, or inhibition of the steroidogenic enzymes, will lead to a reduction in the size of the cells and the volume of endoplasmic reticulum (Figure 17.26). In the dog, Leydig cell response is more variable, and Leydig cell vacuolation and hypertrophy is frequently seen accompanying decreased testosterone production.
Administration of exogenous androgens or androgen agonists has an interesting inverse dose–response relationship with regard to its effect on testicular spermatogenesis. Administration of testosterone or other androgen agonists will inhibit LH production from the pituitary, resulting in inhibition of Leydig cell steroidogenesis and decreased intratesticular testosterone levels. There is a marked differential in the concentrations of peripheral versus intratesticular testosterone levels, and, since spermatogenesis requires very high local testosterone concentrations to progress normally, spermatogenesis will decline in response to the locally reduced steroidogenesis. The higher the administered dose of testosterone, the more it is able to compensate for the decrease in testicular steroidogenesis, and the less effect it has on spermatogenesis. The net result is that the lower the dose of testosterone administered, the more severe the degenerative effect on spermatogenesis. The opposite is true for the effect on accessory sex organs, where the increased circulating levels of administered testosterone will be directly metabolized to DHT within the prostate and seminal vesicles and result in enlargement and increased weight of these tissues.
Androgen receptor antagonists (e.g., flutamide) generally have no detectable histopathologic effect on spermatogenesis, but they cause Leydig cell hyperplasia through their stimulatory effect on LH levels (blocked androgen receptor in the CNS leads to increased LH synthesis and release, which produces the Leydig cell hypertrophy). Conversely, 5α-reductase inhibitors (e.g., finasteride) have no detectable effect on spermatogenesis or on Leydig cells in the rat. However, both cause marked atrophy of the accessory sex organs and the epididymis, which are dependent on DHT for their activity and interaction of the androgen with the androgen receptor. In the mouse, finasteride does produce Leydig cell hyperplasia, which is thought to be due to the fact that DHT is more important than testosterone in feedback inhibition of gonadotropin secretion from the pituitary.
Epididymal and Accessory Sex Organ Changes
In general, the epididymis and especially the accessory sex organs are much more sensitive than the testis to reduced androgen status and rapidly respond with weight loss and epithelial apoptosis and atrophy (Figures 17.27–17.29). In the case of the rat ventral prostate, it loses over 90% of its weight within 4 weeks of androgen withdrawal, while the epididymis loses 80% of its weight in the same time period. The weight loss in the epididymis is due to epithelial apoptosis and atrophy (Figure 17.27), as well as reduced fluid and reduced numbers of sperm from the testis (approximately 50% of epididymal weight is made up of sperm and fluid). Following androgen withdrawal, the epididymal sperm lose their motility and fertilizing ability and die. The rat seminal vesicles are at least as responsive as the ventral prostate, and because of their much greater weight in controls have a larger dynamic range, as well as being easier to dissect if it is necessary to separate these organs from each other.


Withdrawal of androgen results in a wave of apoptosis that starts in the initial segment of the epididymis and moves down the rest of the epididymis with time (Figure 17.27). Many functions of the epididymis are androgen-dependent, including the energy-dependent transport of Na+ ions across the epididymal epithelium and the activity of the detoxification enzymes glutathione S transferase and gamma glutathione transferase. In the prostate and seminal vesicles, there is epithelial apoptosis and epithelial atrophy (thinning) and decreased secretory content which is roughly proportional to the degree of hormone loss.
Pattern of Change Associated with Disturbance of Fluid Production, Reabsorption or Efferent Duct Obstruction
Patency of the seminiferous tubule lumen and the movement of sperm out of the testis and into the efferent ducts rely on the secretion of STF into the tubular lumen by the Sertoli cell (Table 17.11). The production of STF is continuous, as is the reabsorption of most of the fluid in the efferent ducts. Significant amounts of fluid are produced by the testis in a day (up to 0.8 mL/testis/day in the rat, and approximately 40 mL/testis/day in the boar).
Decreased Fluid Production
Testosterone is a major regulator of STF secretion, but it has also been demonstrated that the presence of elongating spermatids also regulates secretion. Thus, if elongating spermatids are lost from the seminiferous tubules, STF secretion will decrease. This loss of secretory function will result in narrowing of the tubular lumen and general reduction in the diameter of the tubule (tubular atrophy) in addition to the underlying germ cell loss. Fluid content is a significant component of testis weight, and so decreased testis weight is likely due to reduced STF as well as decreased numbers of germ cells. If a decrease in absolute testis weight is seen in the absence of any significant germ cell loss, a decrease in STF should be considered as a possible cause.
Increased Fluid Production, or Decreased Tubular Emptying
An increase in absolute testis weight almost always reflects an increase in tubular fluid content and is generally accompanied by an increase in the diameter of the tubular lumen. Although increased fluid secretion by the Sertoli cell is a possible cause, there are no reports of this as a known chemically induced change. Another possibility is reduced emptying of the seminiferous tubules or relaxation of the encompassing peritubular cells. Endothelin antagonists cause increased testis weight and tubular dilation (Figure 17.30), and Endothelin-1 (ET1) has been suggested as a mediator of tubular contraction in the rat. Vasomotion as well as rhythmic contractions of the testicular capsule may also be involved in expulsion of fluid from the seminiferous tubules and interstitial lymph, and these could provide a potential target for disturbance of function. However, the most common known cause of increased fluid in the testis of rodents is obstruction of outflow in the efferent ducts. Although efferent duct obstruction and granulomas can be seen as a background incidental lesion in rats, they can also be caused by a chemically induced disturbance of fluid reabsorption. It is important to identify and elucidate the efferent duct as a target site because an effect on it often leads to severe testicular tubular atrophy, which may otherwise be considered a primary testicular lesion caused by the drug. If the efferent ducts become blocked or the secretion rate of STF exceeds the reabsorption rate, pressure build-up of fluid in the seminiferous tubules will cause severe damage to the testis. The sequence of changes that occurs has been demonstrated using a number of models; the simplest is efferent duct ligation.
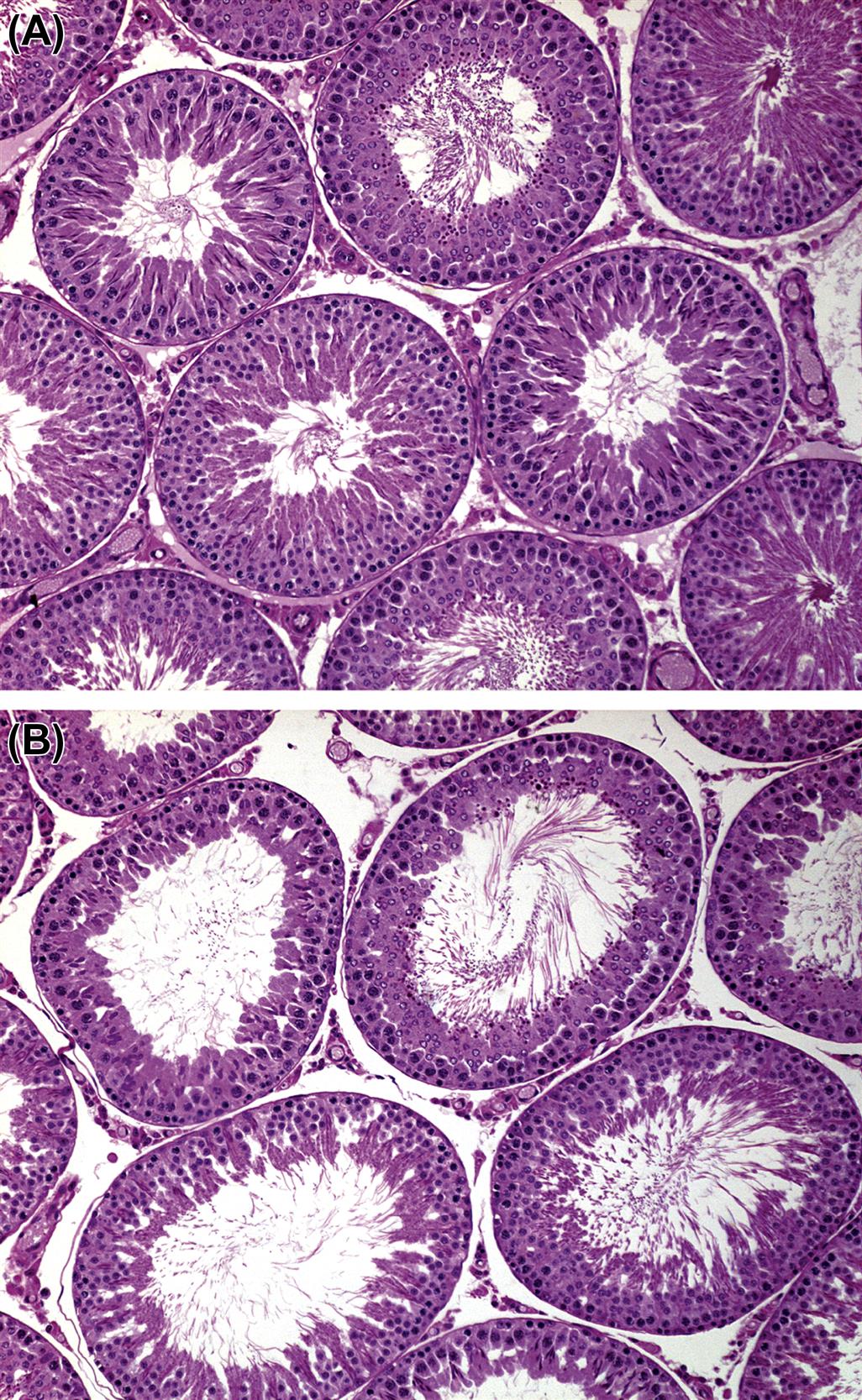
Efferent Duct Ligation
When the efferent ducts are ligated there is a rapid (within hours) build-up of fluid within the testis causing an increase in tubular luminal diameter, thinning of the tubular epithelium, and an increase in testicular size and weight (Figure 17.31A). This reaches a peak at 16–24 hours after the ligation and is then followed by a gradual shrinkage of testicular size and testis weight, due to the progressive degeneration and death of the germ cells from the tubules. By 28 days after ligation the testis has lost 50% of its original weight and the tubules are severely atrophic, having lost all of their organization and most of their germ cells. The end-stage appearance of the testis following this type of injury is tubules lined only by Sertoli cells and occasional spermatogonia, generally having a slightly dilated (rather than collapsed) lumen, possibly accompanied by a dilated rete testis and interstitial edema (Figure 17.31B). The severity of the dilation and atrophy can be modified by ligating only a proportion of the proximal ducts, thereby leaving the common duct and some of the proximal ducts patent.
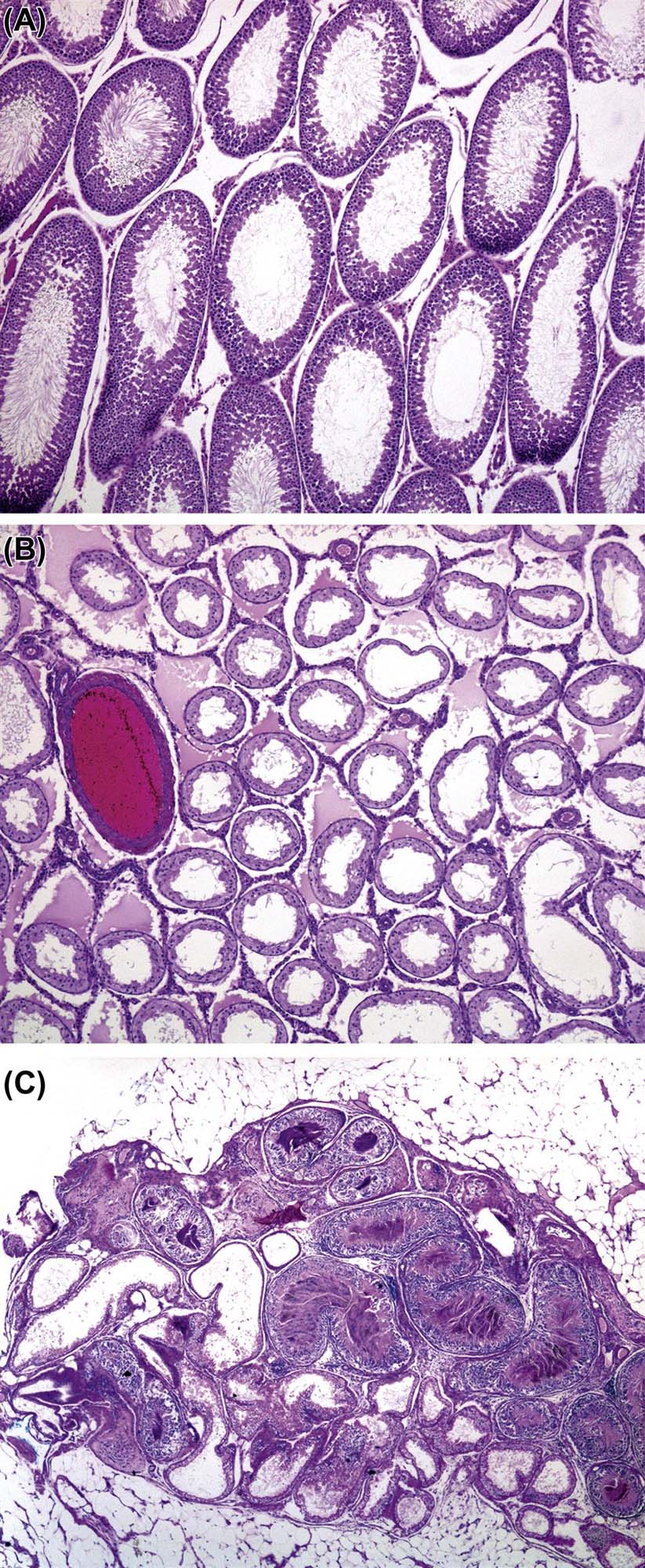
Chemically-Induced Efferent Duct Blockage
Efferent duct obstruction, with its secondary testicular effects, can also be induced by exposure to a variety of drugs and chemicals that disturb the process of fluid reabsorption, either by increasing or inhibiting it. If too much fluid is reabsorbed, the sperm become prematurely concentrated and sequestered within the thin tortuous loops of the efferent ducts. Due to the presence of leaky tight junctions between adjacent epithelial cells, the sequestered sperm will incite a granulomatous inflammatory response, which will permanently obstruct the flow of sperm and fluid through the ducts (Figure 17.31C). If there is inhibition of fluid reabsorption, the upstream ducts dilate with excess fluid and again appear to develop granulomatous inflammation and sperm stasis, possibly due to loss of junctional integrity, and cause occlusion of the ducts and backpressure of fluid to the testes. Inhibition of fluid reabsorption can occur through disturbance of the complex fluid/ion fluxes within the epithelium, or through disturbance of the vasculature that removes the fluid following reabsorption. In the case of a 5HT agonist, experimental evidence to support the latter explanation has been described.
It is important to recognize and distinguish tubular dilation and tubular degeneration/atrophy due to efferent duct blockage from other types of testicular toxicity, because the testicular damage is secondary to build-up of fluid pressure rather than a primary effect on spermatogenesis. Hallmarks of this type of toxicity include the phenomena that the lesions are frequently unilateral, some animals may have one dilated and one normal testis, others may have one atrophic and one normal testis and yet others may have one dilated and one atrophic testis. Another feature is that atrophic tubules often have a patent or slightly dilated lumen (rather than collapsed lumen) and the rete testis maybe dilated. Due to the fact that the testicular lesion (initially dilation, followed by atrophy) will only develop when there are enough efferent ducts obstructed to cause sufficient backpressure to the testis, the incidence maybe sporadic and not show a strong dose relationship (La et al., 2012). In addition, apoptosis of the epithelium of the initial segment and cribriform change in a specific segment of the caput epididymis are frequently seen when the efferent ducts are obstructed.