Endocrine System
Matthew A. Wallig, University of Illinois at Urbana-Champaign, Urbana, IL, United States
Abstract
The endocrine glands are an important group of organs that are susceptible to chemical-induced changes that occur in short- and long-term preclinical toxicology studies. It is important to differentiate spontaneous and stress-related changes from compound-induced effects. There are multiple examples of drugs that are approved for human use that have been associated with degenerative or neoplastic changes in the endocrine glands of laboratory animals, but the weight of evidence regarding the pathogenesis and mode of action has demonstrated a lack of relevance for humans. The mode of action of chemicals on the toxicology of the endocrine glands can be direct or indirect. Therefore, studies on the modes of action require knowledge and experimental interrogation of physiological control and feedback mechanisms, hormone distribution and receptors on diverse target cells, the primary endocrine cells, and peripheral metabolism and excretion of hormones. In addition, there is a significant interspecies difference in the physiology and pathology of the endocrine glands, which makes it imperative to understand the mode of action of chemicals in order to compare preclinical toxicology findings in different species and predict human relevance. This chapter reviews the pathophysiology of the endocrine glands in the species typically used for preclinical toxicology studies and includes toxicologic mechanisms, classic examples of chemical-induced changes and their modes of action, and spontaneous diseases.
Keywords
Endocrine; adrenal cortex; zona glomerulosa; zona fasciculata; mineralocorticoid; glucocorticoid; aldosterone; cortisol; corticosterone; ACTH; hyperadrenocorticism; hypoadrenocorticism; hypercortisolism; adrenocortical hyperplasia; adrenal adenoma adrenal carcinoma; adrenal medulla; catecholamines; norepinephrine; epinephrine; pheochromocytoma; pituitary; adenohypophysis; pars distalis; pars intermedia; hypothalamic–pituitary axis; prolactin (PRL); gonadotropin (GSH) thyrotropin (thyroid-stimulating hormone; TSH); luteinizing hormone (LH); follicle-stimulating hormone (FSH); chromaffin; thyroid; colloid; thyroid-binding globulin (TBG); triiodothyronine (T3); thyroxine (T4); iodine; iodide; 5-deiodinase; goiter; goitrogenic; hypothyroidism; hyperthyroidism; pituitary–thyroid axis; C cell; calcium (Ca2+); parathyroid; chief cells; Vitamin D; parathyroid hormone (PTH); parathyroid hormone-related protein (PTHrP); humoral hypercalcemia of malignancy (HHM); hyperparathyroidism; hypoparathyroidism; oxyphil; phosphorus; pancreas; islet of Langerhans; α cell; β cell; diabetes mellitus (DM); Type a diabetes mellitus (T1DM); Type 2 diabetes mellitus (T2DM); insulin; insulitis; glucagon; atrophy; hypertrophy; hyperplasia; adenoma; carcinoma
Introduction
A review of the literature of chemically induced lesions of the endocrine organs indicates that the adrenal glands are the most commonly affected, followed in descending order by the thyroid, pancreas, pituitary, and parathyroid glands. In the adrenal glands, chemically induced lesions are found most frequently in the zona fasciculata (ZF), zona reticularis (ZR) and, to a lesser extent, in either the zona glomerulosa (ZG) or the medulla. In a survey of tumor types developing in carcinogenicity studies, conducted by the Pharmaceutical Manufacturers Association, endocrine tumors were observed frequently in rats. The thyroid gland was third in frequency (behind liver and mammary gland), followed by the pituitary gland (fourth), and adrenal gland (fifth).
In the following chapter, basic pharmacological and toxicological effects will be reviewed, with emphasis on the latter. Pharmacologic effects are defined as beneficial and desired drug-related changes with minimal side effects or morphological alterations (often reversible), whereas toxicologic effects are more severe adverse effects that often are irreversible.
Part 1: Adrenal Cortex
Structure and Function
Gross and Microscopic Anatomy
In mammals, the adrenal glands are flattened bilobed organs located in close proximity to the kidneys. They receive arterial blood from branches of the aorta or from the phrenic, renal, and lumbar arteries, resulting in a vascular plexus; perfusion occurs by separate sinusoids both to the capsule and to the entire gland, including cortex and medulla. Venous blood flow is derived from a sinusoidal network originating around the cells of the adrenal cortex with eventual flow into the medulla at its periphery. A venous tree is present within the medulla that ultimately flows into the adrenal vein by way of its larger branches.
Midsagittal sectioning of the adrenal gland reveals a clear separation between cortex and medulla. The cortex is firm and yellow and occupies approximately two-thirds of the entire cross-sectional diameter of the organ. In contrast, the medulla is soft, with prominent gray-tan coloration. The ratio of cortex:medulla is approximately 2:1 in healthy laboratory-reared animals.
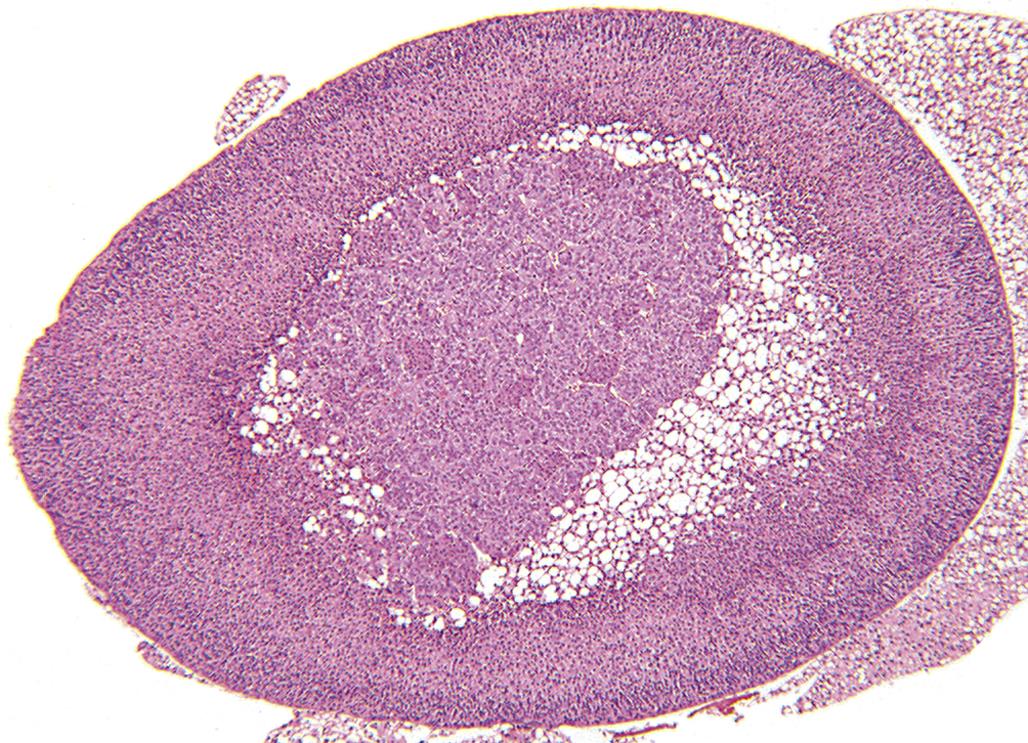
Defined regions or zones histologically characterize the cortex and include the zona glomerulosa (ZG, multiformis), zona fasciculata (ZF), and zona reticularis (ZR). Zones are not always clearly delineated, as illustrated in the normal rat adrenal cortex (Figure 20.1). The mineralocorticoid-producing ZG (15% of the cortex) contains cells aligned in a sigmoid pattern in relationship to the capsule. Cells of this zone secrete mineralocorticoids (e.g., aldosterone) essential for regulation of body potassium and sodium. The largest zone is the ZF (>70% of the cortex). Cells in this zone are arranged in long anastomosing cords or columns, separated by small capillaries/sinusoids. They are responsible for the secretion of glucocorticoid hormones (e.g., corticosterone in rat, mouse, and rabbit, or cortisol in dog, pig, monkey, and human), which promote the elevation of blood glucose in addition to many other effects. The innermost portion of the cortex is the ZR (15% of the cortex), which normally secretes minute quantities of adrenal sex hormones in some species.

There are structural and functional differences between species and sexes. Accessory cortical tissue is often seen in mice and cynomolgus monkeys, not to be mistaken for proliferative lesions. The marmoset has no obvious ZR, and in the dog the ZF and ZR are poorly demarcated. The mouse develops the x-zone, a prominent fourth cortical layer immediately adjacent to the medulla, which is especially prominent in females (Figure 20.2); this zone degenerates, leaving clumps of brown-yellow lipofuscin at the corticomedullary junction. Primates develop an area of cortical involution in late fetal life that regenerates after birth. The ZR is three times thicker in male Syrian hamsters than in females.
Ultrastructural Anatomy
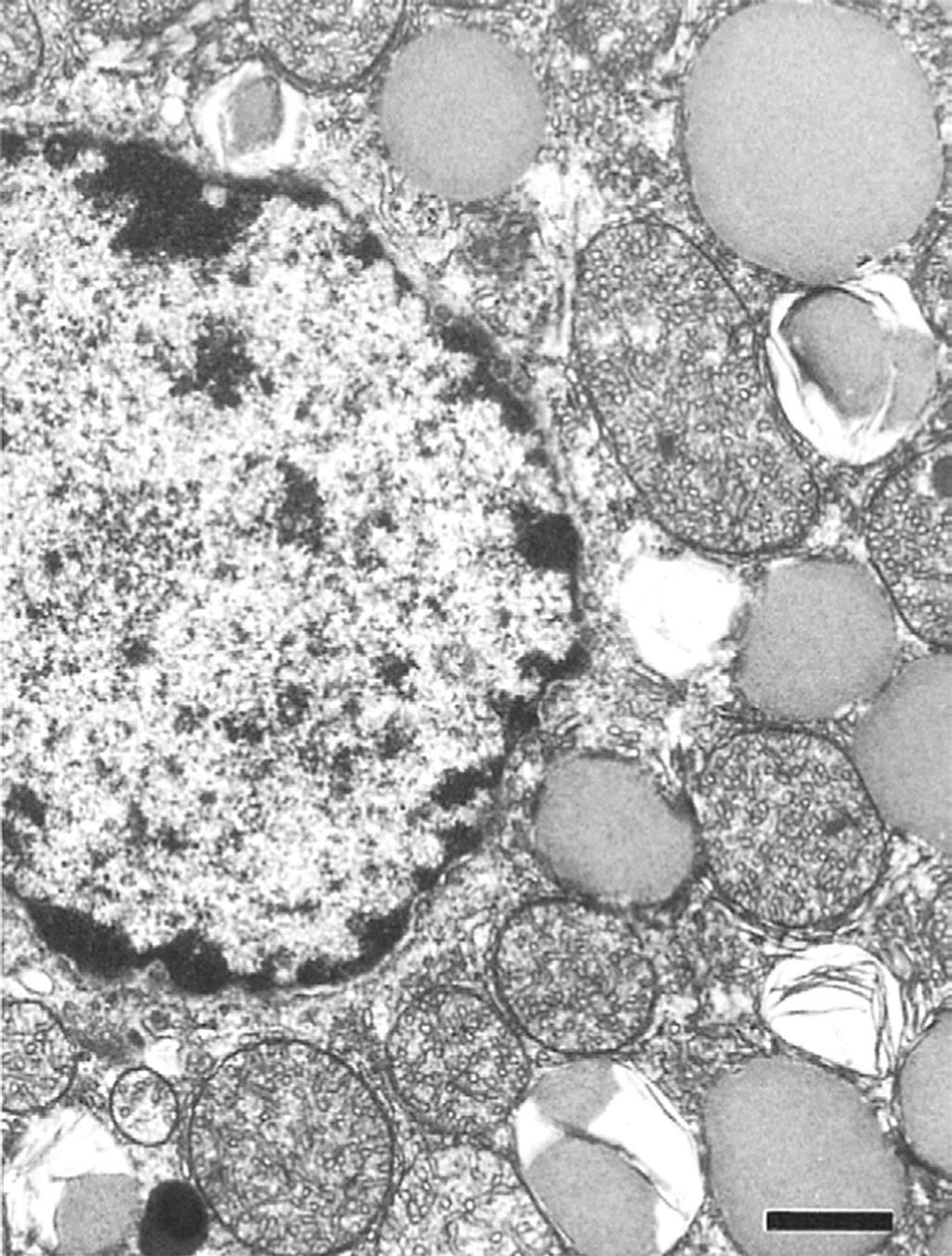
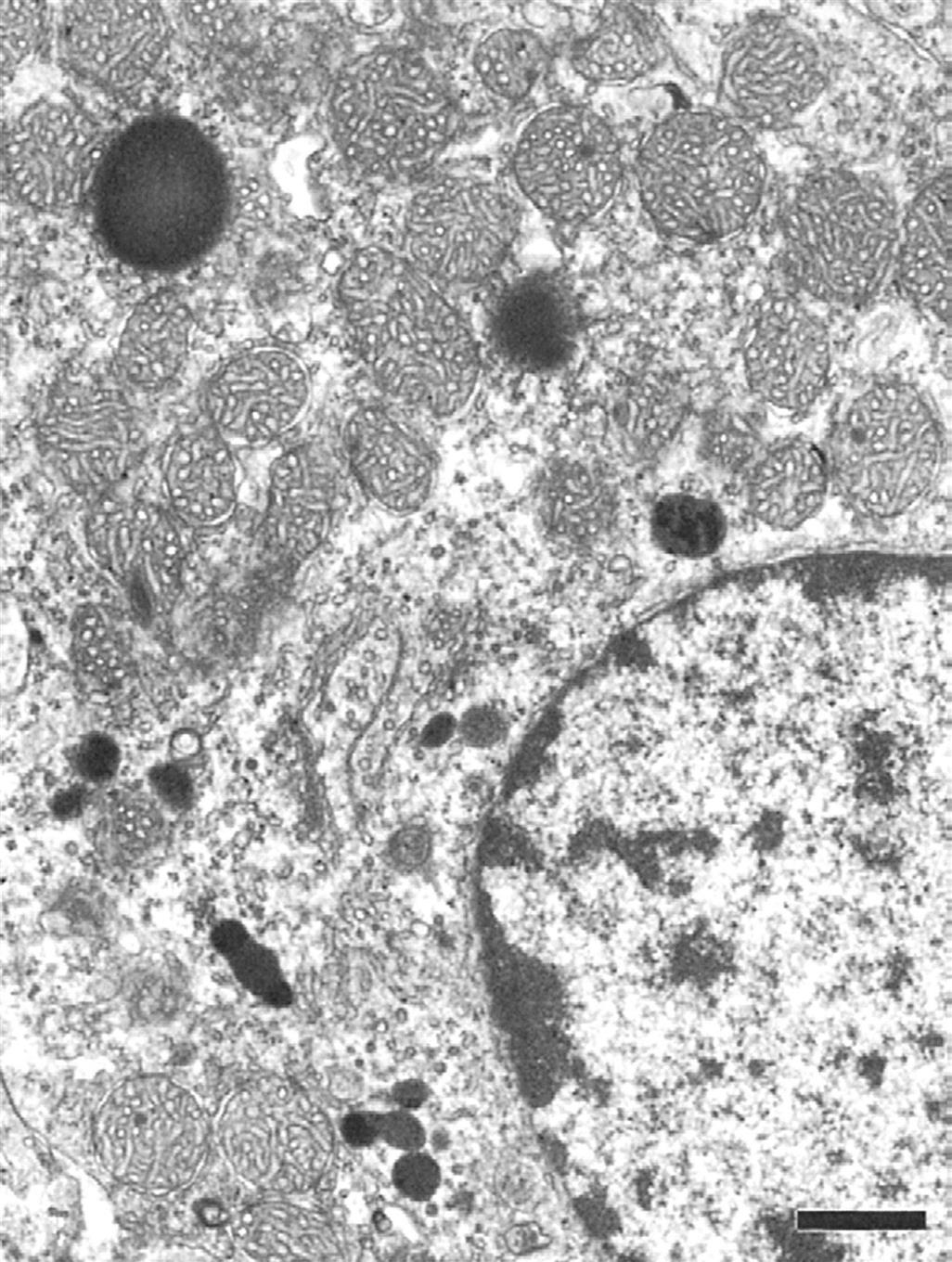
Adrenal cortical cells contain large cytoplasmic lipid droplets consisting of cholesterol and steroid precursors. The lipid droplets are in close proximity to the smooth endoplasmic reticulum and large mitochondria, which contain the specific hydroxylase and dehydrogenase enzyme systems required to synthesize the different steroid hormones. Unlike polypeptide hormone-secreting cells, there are no secretory granules in the cytoplasm because there is direct secretion without significant storage of preformed steroid hormones. The three cortical zones have unique ultrastructural characteristics in the rat adrenal cortex (Figures 20.3–20.5).

Physiological and Functional Considerations
All hormones produced by the adrenal cortex are steroids. Steroid-producing endocrine organs such as the adrenal cortex synthesize a major parent steroid compound with 1–4 additional carbon atoms added to the basic 17 carbon-containing steroid nucleus. Because steroid hormones are not stored in any significant amount, a continued rate of synthesis is required to maintain a normal secretory rate. This in turn requires continued stimulation of the adrenal cortex by pituitary derived adrenocorticotropic hormone (ACTH; see later) according to physiological requirements. In circulation, steroid hormones (e.g., cortisol or corticosterone) are bound to plasma proteins [e.g., transcortin (CBG, cortisol/corticosterone binding globulin), albumin]. Depending on the nature of the plasma proteins the binding affinity may be high or low, but nonetheless reversible, to allow the steroid to be in a free unbound state when interacting with target cells. Under normal conditions, 10% of the glucocorticoids are in a free unbound state and thus free to interact with target cells either to exert metabolic effects or to be transformed into an inactive metabolite. In conditions of elevated secretion of adrenal glucocorticoid, the free steroid fraction in the blood is increased and available to evoke a response in target cells and tissues.
Adrenal steroids are synthesized from cholesterol, which in turn is derived from acetate. A complex shuttling of steroid intermediates between mitochondria and endoplasmic reticulum characterizes specific synthetic processes. The specificity of mitochondrial hydroxylation reactions in terms of the target steroid and the substrate carbon that is hydroxylated is confined to a specific cytochrome P450 (CYP). The common biosynthetic pathway from cholesterol is the formation of pregnenolone, the basic precursor for the three major groups of adrenal steroids (Figure 20.6). Pregnenolone is formed after two hydroxylation reactions.
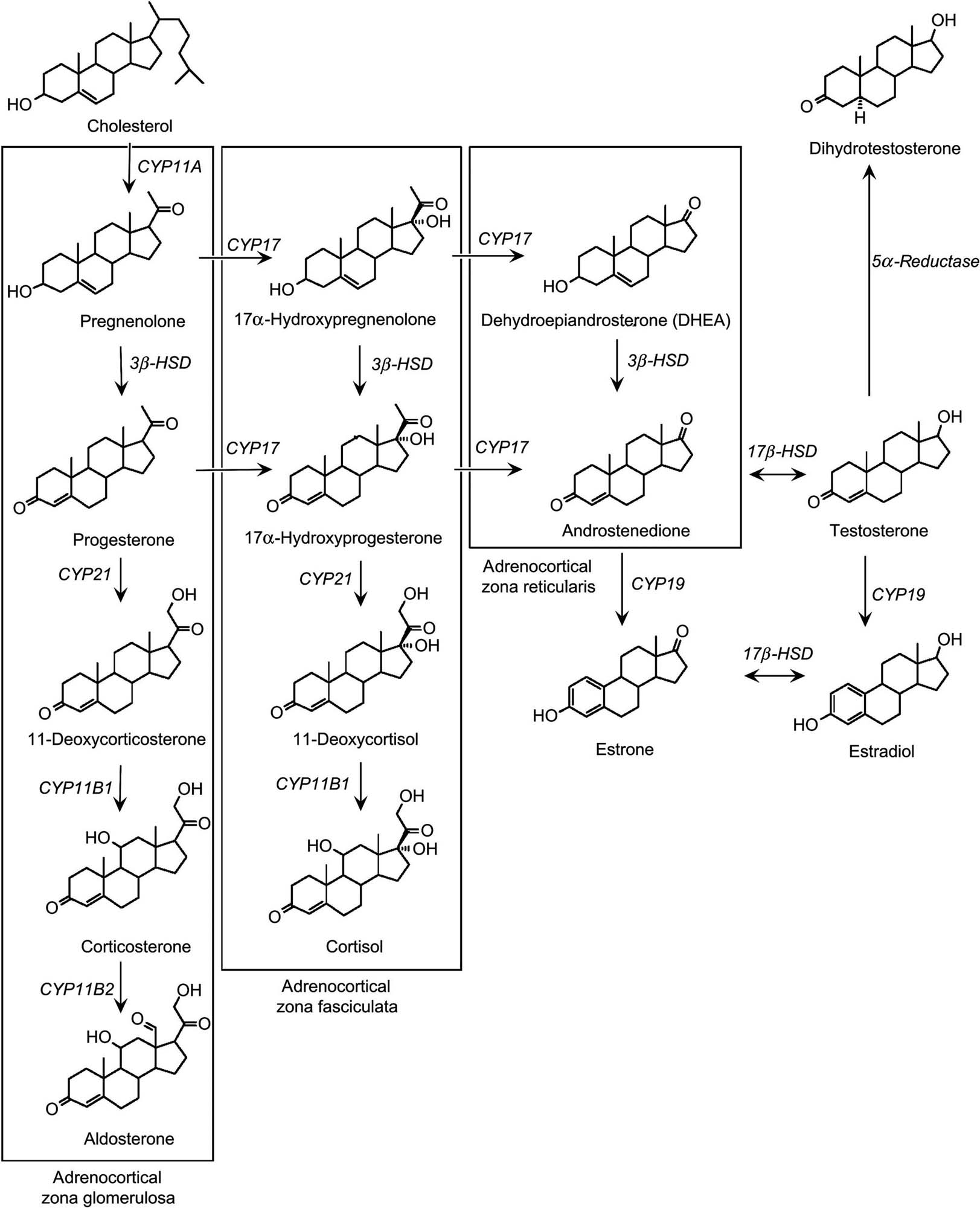
In the ZF, pregnenolone is first converted to progesterone by two microsomal enzymes, followed by hydroxylation reactions. The resulting steroid is cortisol, which is the major glucocorticoid in teleosts, hamsters, dogs, nonhuman primates, and humans. Corticosterone is the major glucocorticoid produced in amphibians, reptiles, birds, rats, mice, and rabbits. It is produced in a manner similar to the production of cortisol. However, rodents and the above mentioned species lack CYP17 needed to shunt progesterone into the cortisol pathway, and this is an important consideration for toxicology, as compounds that inhibit this enzyme may not be fully detected in rodent species. This may also account for species differences in adrenocortical toxicity between rodent and nonrodent species. Spironolactone and ketoconazole inhibit CYP17 as well as other enzymes in the steroidogenic pathway.
In the ZG, pregnenolone is converted to aldosterone by a series of enzymatic reactions similar to those in cortisol formation; however, the cells of this zone lack 17α-hydroxylase and thus, cannot produce cortisol. Therefore, the initial hydroxylation product is corticosterone. Some of the corticosterone is acted on by 18-hydroxylase to form 18-hydroxycorticosterone, which in turn interacts with 18-hydroxysteroid dehydrogenase to form aldosterone. Because 18-hydroxysteroid dehydrogenase is found only in the ZG, it is not surprising that only this zone has the capacity to produce aldosterone. In addition to the aforementioned steroid hormones, the adrenal cortex also produces small amounts of sex steroids, including progesterone, estrogens, and androgens. Thus, the adrenal cortex as a whole has all the necessary enzymes to synthesize the full range of steroids, differentially located across the various zones. Rodents do not produce adrenal estrogens and testosterone because they lack CYP17.
After their synthesis, secretion, and interaction with target cells, the adrenal steroid hormones are ultimately metabolized in peripheral tissues. Inactivation occurs in the liver by two main steps that include reduction or side chain removal and conjugation to glucuronic acid or sulfate. In the presence of liver disease, the turnover of steroid hormones, particularly cortisol, may be decreased and can result in abnormal adrenal function tests in patients or test animals without adrenal cortical lesions. Occasionally, peripheral tissues may activate steroid hormones (e.g., testosterone to dihydrotestosterone) or, as in the case of cortisol, convert the steroid to other less active forms of the hormone.
Mineralocorticoids (e.g., aldosterone) are the major steroids secreted from the ZG under the control of the renin–angiotensin II system. Mineralocorticoids have effects on ion transport by epithelial cells, particularly renal cells, resulting in the conservation of sodium (chloride and water) and loss of potassium. In the distal convoluted tubule of the mammalian nephron, a cation exchange exists that promotes the resorption of sodium from the glomerular filtrate and the secretion of potassium into the lumen.
Glucocorticoid hormones increase glucose production with a concomitant breakdown of proteins for purposes of gluconeogenesis. Glucocorticoids also suppress inflammation along with attenuation of fibroplasia and immunological responses. The suppression of the immunological responses is largely related to the stabilization of lysosomal membranes of phagocytic cells, inhibition of a number of lymphoid cell functions, and lysis of lymphocytes. The increase in blood glucose is an important physiological response in adverse situations, but the most important physiological effect of the glucocorticoids in stressful circumstances is to quench the inflammatory response to prevent it developing to the point where it overwhelms the animal.
The principal control for the production of steroids by the ZF and ZR is mediated by ACTH, a polypeptide hormone produced by corticotrophs in the pituitary adenohypophysis. ACTH release is largely controlled by the hypothalamus through the secretion of corticotropin-releasing hormone. An increase in ACTH production normally results in an increase in circulating levels of glucocorticoids, although it can cause weak stimulation of aldosterone secretion as well. Negative feedback control normally occurs when the elevated blood levels of cortisol act on the hypothalamus, anterior pituitary, or both to cause a suppression of ACTH secretion. This negative feedback mechanism can also be involved in adrenocortical toxicity in situations where a compound inhibits a critical enzyme in the glucocorticoid synthesis pathway, with loss of glucocorticoid production leading to less or no feedback inhibition of ACTH. The subsequent persistent overstimulation of the adrenal cortex by ACTH can produce marked adrenal hypertrophy.
In contrast to the normal negative feedback mechanism, abnormally high corticosteroid levels in the plasma, above physiological levels (due to exogenous administration of steroids or cortisol-producing adrenal lesions), will cause marked ACTH suppression. If the suppression is prolonged, secretory cells in the ZF and ZR will undergo atrophy, with a corresponding decrease in their future capability to synthesize and secrete corticosteroid hormones.
Evaluation of Toxicity
When a compound affects steroidogenesis, it is important to define the extent to which it impairs adrenal cortical functional reserve capacity. In some instances, clinical signs of hypoadrenocorticism may be observed in association with lower urinary and plasma corticosteroid levels. When such findings are not obvious, provocative testing for the evaluation of adrenal cortical functional reserves is essential to determine the extent of cortical damage. The most commonly used provocative test is the administration of ACTH to human patients or test animals (the dog is the ideal test animal for evaluation). ACTH has been shown to be a potent stimulus for the synthesis and secretion of naturally occurring glucocorticoids in noncompromised cells of the ZR and ZF.
In Vitro Assessment
In vitro studies are extremely useful in determining the specific cellular consequences of xenobiotic exposure on steroidogenesis. In many instances, the results of these in vitro assessments are helpful in correlating the development of adrenocortical degeneration to an inhibited pathway of steroidogenesis. By far, the best available in vitro model is the H295R cell line. This is a human adrenocortical carcinoma cell line that retains full adrenocortical steroidogenic enzyme capability and can secrete aldosterone, cortisol, and androgens, estrogens, and progestogens and their precursors in response to appropriate challenge. This system has been used to investigate the majority of chemicals reported to induce adrenocortical functional toxicity over the past decade, particularly to identify molecular sites of toxicity. It should be noted that rodent cell lines do exist but are deficient in at least CYP17, and the Y1 mouse cell line is only capable of producing progesterone because of deficiencies in numerous other steroidogenic enzymes. Rodent cell lines therefore have little use in establishing mechanisms of toxicity, at least in terms of human relevance.
Morphologic Evaluation
Following the death of a test animal, morphological evaluation commences with macroscopic observation of the adrenal glands to detect changes in size, color, and/or appearance (e.g., nodularity). Subsequent histological examination of adrenal tissue (including both cortex and medulla) on midsagittal sections stained with hematoxylin and eosin (H&E) is performed routinely. The most current terminology for describing various nonproliferative and proliferative lesions of the adrenal cortex, including representative images of common lesions, developed by the International Harmonization of Nomenclature for Diagnostic Criteria (INHAND) in Rats and Mice, will be available at the time of publication of this chapter on the Society of Toxicologic Pathology website (http://www.toxpath.org/inhand.asp).
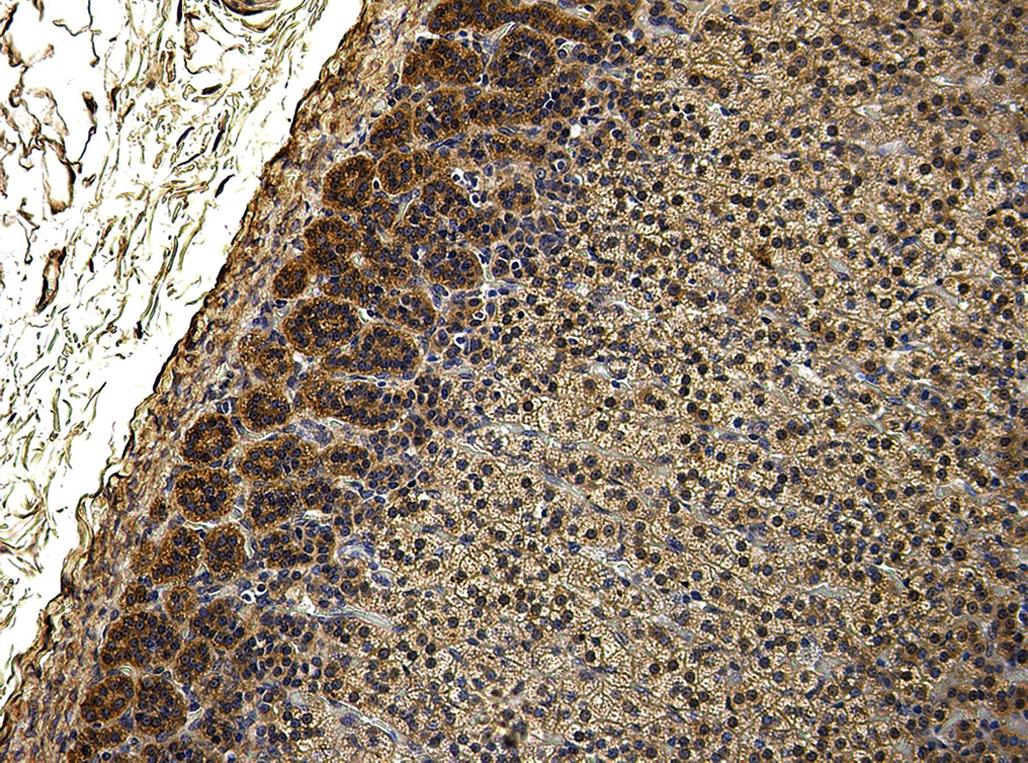
Microscopic study may be supplemented by the use of special stains and techniques to confirm or identify a particular pigment, cell type, or enzyme. Examples of useful stains are Congo red (amyloid), acid fast stains (ceroid pigment), Sudan black or Oil red O (lipid), Perl’s stain (iron), and immunohistochemistry for enzymes (e.g., aldosterone synthase in the ZG; Figure 20.7). Histomorphometric analysis can be used to assess subtle differences in cell size, width of the different cortical zones, and cortical:medullary ratio.
Use of Animals as Models
The adrenal cortex of animals is prone to develop proliferative and degenerative lesions, the etiology of which may be either spontaneous in nature or induced experimentally. Therefore, chemical testing using various domestic or laboratory animals is a valid means of assessing the toxic potential for humans exposed to various xenobiotic chemicals.
Species, Age, and Sex Variables
The correct choice of species of test animal is critical. There often is a variable species susceptibility to toxicity. This observation suggests that differences in metabolism play a role in the development of adrenal cortical toxicity and in the inhibition of steroidogenesis. As mentioned previously, the rodent is deficient in CYP17, and compounds inhibiting steroidogenesis and particularly cortisol production via this enzyme would not be adequately evaluated using the rodent only. However, dog and monkey could be used instead.
Likewise, the age of the test animal, to a lesser degree, may be a factor in the development of chemically induced adrenocortical lesions. For example, adrenal cortical necrosis was induced in rats at 50 days of age but not at 25 days by the administration of 7,12-dimethylbenz[a]anthracene.
Stress as a Variable in Animal Models
Although the rat, because of differences in steroidogenesis, is not the best model to fully evaluate adrenal toxicity in terms of human relevance, it often shows adrenal changes in toxicity studies. One of the most common findings in the rat is adrenocortical hypertrophy, which is indicative of ACTH stimulation (Figure 20.8). Adrenal hypertrophy may be a result of stress or indeed may result from functional impairment of the adrenal cortex and reduced capacity to secrete glucocorticoids. It is therefore important, in all cases of adrenocortical hypertrophy induced by excess ACTH stimulation, to establish the mechanism of ACTH elevation (i.e., stress versus adrenal toxicity/insufficiency). Atrophy of the thymus and other lymphoid tissues is a useful surrogate marker for adrenocortical competence, as this atrophy is induced by excess glucocorticoid secretion; similarly, the stress leukogram is useful evidence. Stress is a natural adaptive response designed to better equip the animal to survive, and it should result in both elevated ACTH and glucocorticoids. Elevation of the latter is diagnostic and proof of adrenocortical functional competence. Stress, however, should not produce irreversible histopathologic lesions in the adrenal cortex. By contrast, toxicity to the adrenal cortex may be obvious due to marked and often irreversible histopathological lesions (degeneration, necrosis, and fibrosis). However, in cases of pharmacotoxicological inhibition of steroidogenic enzymes there may be no histopathological lesions but grossly impaired glucocorticoid production, clearly a toxicological concern.
If findings are restricted to adrenocortical hypertrophy (i.e., no thymic atrophy, stress leukogram, or other supportive stress-related changes) then stress cannot be confirmed as the cause, and further work to test adrenocortical functionality should be undertaken.
Response to Injury
Disorders of Hyperfunction and Hypofunction
Chemicals can produce functional alterations of the adrenal cortex. Prolonged use of exogenous glucocorticoids can mimic a syndrome of excess adrenal cortical function. Abrupt cessation of steroid use may cause a patient to develop secondary adrenal cortical insufficiency because of the prolonged suppression of ACTH production and the trophic atrophy of cells in the adrenal cortex. More significant from a toxicologic point of view are the degenerative effects of chemicals on the adrenal cortex that result in primary adrenal cortical hypofunction. Disturbances in adrenal–cortical function have been best characterized in the dog. Because of this, and the fact that canine hyper- and hypoadrenocorticism are similar to the corresponding clinical syndromes in humans, the remaining discussion in this section will be principally confined to the dog.
Exogenous glucocorticoid hormone therapy (daily at high doses) often mimics naturally occurring cases of hypercortisolism (e.g., Cushing’s syndrome). Clinical observations include polyuria, polydipsia, an enlarged pendulous abdomen, muscular wasting, alopecia, thinning of the skin with cutaneous pigmentation and mineralization, and hepatomegaly. Osteoporosis is an important finding in human patients with cortisol excess but is not a common finding in the dog. Significant laboratory findings include an increase in alkaline phosphatase (steroid-induced isoenzyme), an eosinopenia with marked lymphopenia, and leukocytosis due to the increased formation of neutrophils. Dogs with hypercortisolism infrequently develop significant alterations in serum concentrations of sodium, potassium, or chloride, in contrast to those electrolyte imbalances seen in humans with Cushing’s syndrome. With the prolonged use of exogenous glucocorticoids, exogenous ACTH administration results in an inadequate release and blunted increase in blood levels of cortisol. In spontaneous cases of functional adrenal cortical hyperplasia that remains partially under the control of pituitary ACTH, a marked response to the exogenous ACTH challenge occurs, resulting in an exaggerated increase in blood cortisol levels.
In hypoadrenocorticism, caused by natural disease or experimentally by the administration of xenobiotic chemicals, clinical signs often are not pathognomonic and nonspecific. However, there is generally an abnormal electrocardiogram with spiked T waves and flattening of the P wave. These electrocardiographic alterations appear to be due to the prominent increase in serum potassium in dogs with hypoadrenocorticism and subnormal secretion of aldosterone.
Plasma and urinary 17-hydroxycorticosteroids often are at low levels in the resting state. Stimulation tests, including the response to exogenous ACTH challenge, usually reveal a subnormal (“blunted”) increase in plasma cortisol due to the reduced number of cortical cells that can respond to the challenge.
Effects During Embryogenesis
It is well documented that synthetic and naturally occurring corticosteroids are potent teratogens in laboratory animals. The principal induced defect is cleft lip or palate; however, there is a paucity of information regarding the direct effect of chemicals on the development of the adrenal cortex. Adrenal aplasia has occurred in a subset of white Danish rabbits when thalidomide was given to their dams.
Morphologic Alterations
Macroscopic lesions of chemically affected adrenal glands are characterized by either enlargement or reduction in size that often is bilateral. Initially, cortical hypertrophy or swelling due to impaired steroidogenesis or hyperplasia due to long-term stimulation often is seen when the adrenal is increased in size. Similar gross findings may be the result of medullary hyperplasia or pheochromocytoma. In contrast, small adrenal glands often are indicative of degenerative changes, resulting in atrophy. Midsagittal longitudinal sections of the glands will reveal a disproportionately wider cortex relative to the medulla, or vice versa, resulting in an abnormal cortical:medullary ratio.
Nonneoplastic Lesions
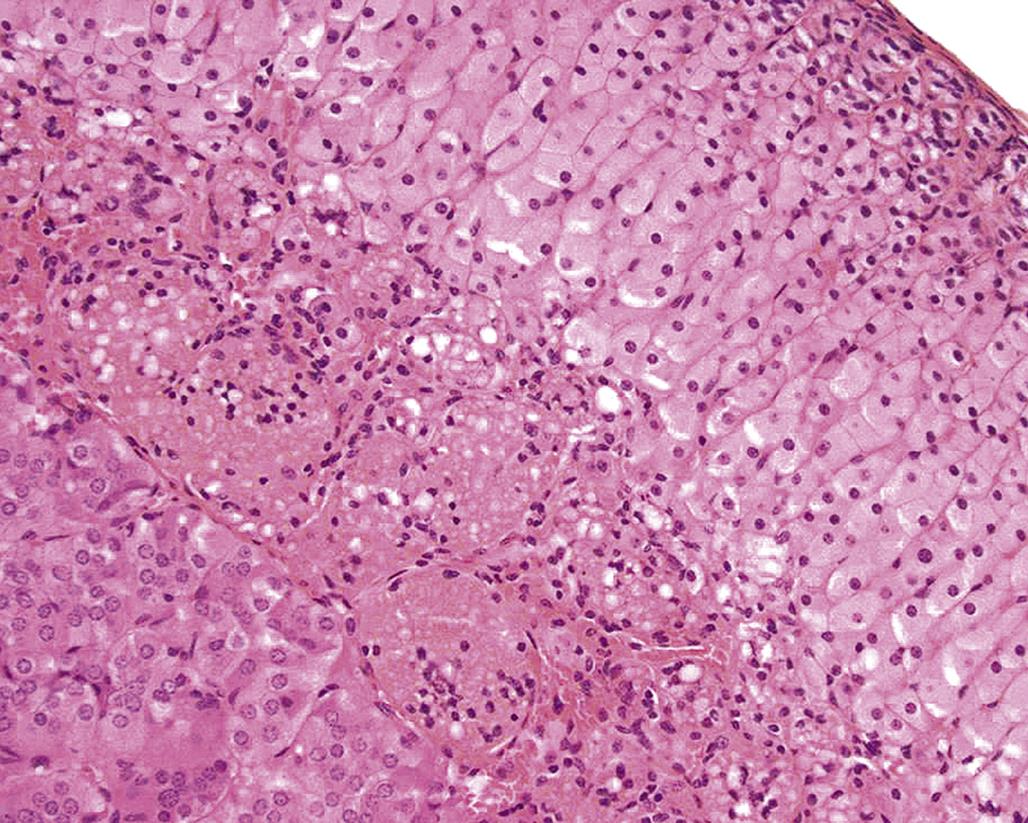
Histologically, nonneoplastic lesions of the adrenal cortex induced by chemical agents are characterized by changes ranging from acute progressive degenerative to reparative in nature. These may be exacerbations of normal spontaneous findings or directly compound-induced lesions. There is delayed involution of the x-zone in mice after treatment with luteinizing hormone (LH), acetonitrile and the antifungal, ketoconazole (see Figure 20.2). There can be a treatment-related increase in the pigment lipofuscin (formed from lipid products resulting from altered fat metabolism) in the ZR in rats, which increases normally with age, but also can be increased after dosing with estrogens, antithyroid agents, the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, and corticosteroids. It can also be increased after feeding diets high in saturated fats and with Vitamin E deficiency. This pigment can be demonstrated with fuschinophilic stains (Ziehl-Neelson and periodic acid-Schiff), which can help to differentiate it from hemosiderin (a brown blood-breakdown product), which can, in turn, be demonstrated using Perl’s stain (for iron) (Figure 20.9).
Early degenerative lesions characterized by enlarged-cortical cells filled with cytoplasmic vacuoles (often lipid) may result in a diffuse hypertrophy of the cortex. Lipid vacuolation is commonly seen in the ZF in normal unstressed animals, with numerous lipid droplets, ~0.5 μm diameter, containing esterified cholesterol. This increases with age and in response to reproductive and physiological changes (Figure 20.10).
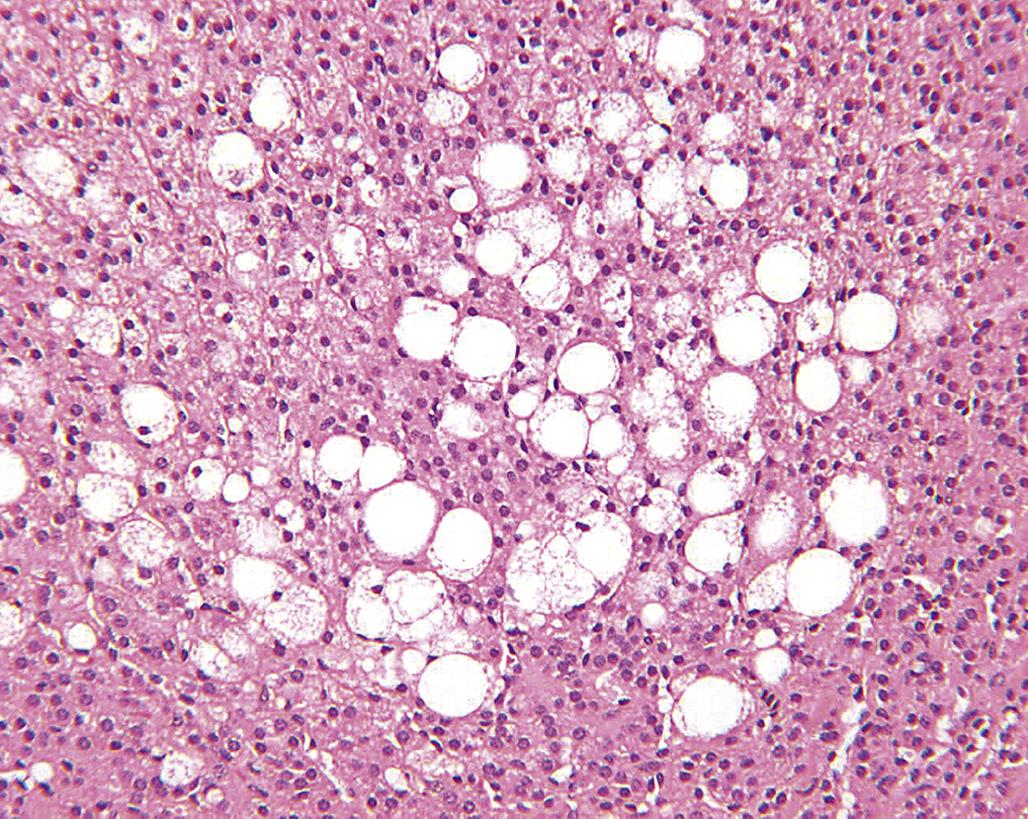
However, excessive coalescing lipid droplets have been observed in rats treated with the antibacterial compound, alpha-(1,4-dioxido-3-methylquinoxalin-2-yl)-N-methylnitrone (DMNM). This vacuolar type of degeneration is a reflection of impaired steroidogenesis, resulting in excess storage of unmetabolized steroid precursors. More destructive lesions may be observed in the form of hemorrhage and/or necrosis, often in association with an inflammatory response. At the same time, one area of the cortex (e.g., the ZG) may undergo hypertrophy while another area has degenerative lesions (e.g., vacuolar degeneration of the ZF). The ZG remains functional and there are no clinically significant signs of hypoadrenocorticism, and chronic regenerative changes may develop subsequently. Usually, the adrenal cortex will be shrunken or atrophic with fibrosis and areas of multinodular hyperplasia. In the case of chronic DMNM toxicity in rats, fibrosis and occasionally hemosiderin pigment may be found in the ZR. Cortical nodules of presumably functional remnants of the ZF and discernible areas of the ZG were found in rats that survived treatment for 90 days (Figure 20.11).
While many adrenocorticolytic agents affect the adrenal cortex initially at the ZR and inner ZF, some chemicals such as DMNM can cause a progressive degeneration of the adrenal cortex. Occasionally, the effect of a chemical is limited to a specific zone of the adrenal cortex and may be species specific. For example, the compound PD 132301-2, when administered to monkeys, induces a narrow band of degeneration or necrosis of cortical cells in the mid to outer ZF (Figure 20.12). In contrast, all three cortical zones are affected in the adrenal glands of dogs treated with this compound. Xenobiotic chemicals that cause degeneration of the adrenal cortex are summarized in Table 20.1, together with the predilection site of their effects and the most significant lesions. Ultrastructural alterations of adrenal–cortical cells associated with chemical injury are quite diverse in nature (Table 20.1). The ZR and ZF typically are most severely affected, although eventually the lesions involve the ZG. These alterations may be classified as follows: endothelial damage (e.g., acrylonitrile), mitochondrial damage (e.g., DMNM, o,p′-DDD, amphenone), endoplasmic reticulum changes (e.g., triparanol), lipid aggregation (e.g., aniline), lysosomal phospholipid aggregation (e.g., chlorophentermine), and possible secondary effects due to embolization by medullary cells (e.g., acrylonitrile).

Table 20.1
Examples of Chemically Induced Microscopic and Ultrastructural Changes of the Adrenal Cortex
| Compound | Initial predilection site | Histology | Ultrastructure |
| Nafenopin | Zona fasciculata | Hypertrophy | SERa and peroxisome proliferation |
| Acrylonitrile | Zona reticularis | Hemorrhage | Damage to vascular endothelium; embolization of medullary cells and cell fragments of capillaries |
| Aminoglutethimide | All zones; more marked in outer zona fasciculata | Vacuolar degeneration; increased lipid | Mitochondrial hypertrophy and cavitation |
| o,p′-DDD | Zona reticularis and fasiculata | Vacuolar degeneration; cytotoxic cellular atrophy | Mitochondrial vacuolization; SER dilation |
| α-(1,4-Dioxido-3-methylquinoxalin-2-yl)-N-methylnitrone | Zona reticularis and fasiculata | Granular and vacuolar degeneration; cytotoxic cellular atrophy | Mitochondrial vacuolization; SER dilation |
| Triparanol | All zones; most marked in zona fasciculata | Increased eosinophilia and inclusions | Decreased lipid droplets; mitochondrial alterations; SER hypertrophy; lysosomal formation |
| Cysteamine (1-mercaptoethylamine) | Zona reticularis and fasiculata | Hemorrhage and necrosis | Retrograde emboli of medullary cells |
| Amphenone | Zona reticularis and fasiculata | Fatty degeneration | Mitochondrial alterations |
| 7,12-Dimethylbenzanthracene | Zona reticularis and fasiculata | Necrosis; hemorrhage; calcification | Mitochondrial alterations, including variation in size |
| Corticosteroids, (e.g., prednisolone, dexamethasone) | Zona reticularis and fasiculata | Atrophy | Increased lipid droplets surrounded by membranous “whorls”; increased myelin figures and lysosomes |
| Propylthiouracil | Zona reticularis | Ceroid degeneration | Lipid and mitochondrial degeneration |
| Carbon tetrachloride | Zona reticularis and fasiculata | Necrosis | Swelling of SER |
| Tamoxifen | Zona reticularis and fasiculata | Degeneration and necrosis; lipid droplets | Necrosis; lipid droplets in macrophages; few lysosomal inclusions |
| Spironolactone | Zona glomerulosa | Hypertrophy and inclusions | Lipid droplets surrounded by whorls of SER (“spironolactone bodies”); mitochondrial alterations |
| Hexadimethrine bromide (polybrene) | Zona glomerulosa | Necrosis and infarction | Protein-containing vacuoles and (polybrene) hyalin bodies; microthrombi |
| RO1-8307, a sulfated mucopolysaccharide | Zona glomerulosa | Condensation | |
| Captopril | Zona glomerulosa | Atrophy | Decreased mitochondria and SER |
| 1,1′-Thio-diethylidene-ferrocene (MDL 80, 478) | All zones | Granular and vacuolar degeneration; hyperplastic zona glomerulosa | Mitochondrial vacuolation; increased lipid droplets |
| Aniline | All zones | Hypertrophic cortical cells laden with lipid droplets | Increased lipid droplets; hypertrophic SER; mitochondrial degeneration |
| Chlorophentermine | All zones | Nothing remarkable | Increased lysosomal alterations in the form of lamellated cytoplasmic inclusions |
| Triaryl phosphate | All zones | Cytoplasmic lipid droplet | Increased number/size cytoplasmic droplets |
| PD 132301-2 | Zona fasciculata | Coarse vacuolation | SER aggregation; changes of autophagosomes |

aSmooth endoplasmic reticulum.
Table modified from Handbook of Toxicologic Pathology, second ed. W. M. Haschek, C. G. Rousseaux, and M. A. Wallig, eds. (2002) Academic Press, Vol. 2, Table II, pp. 693–694.
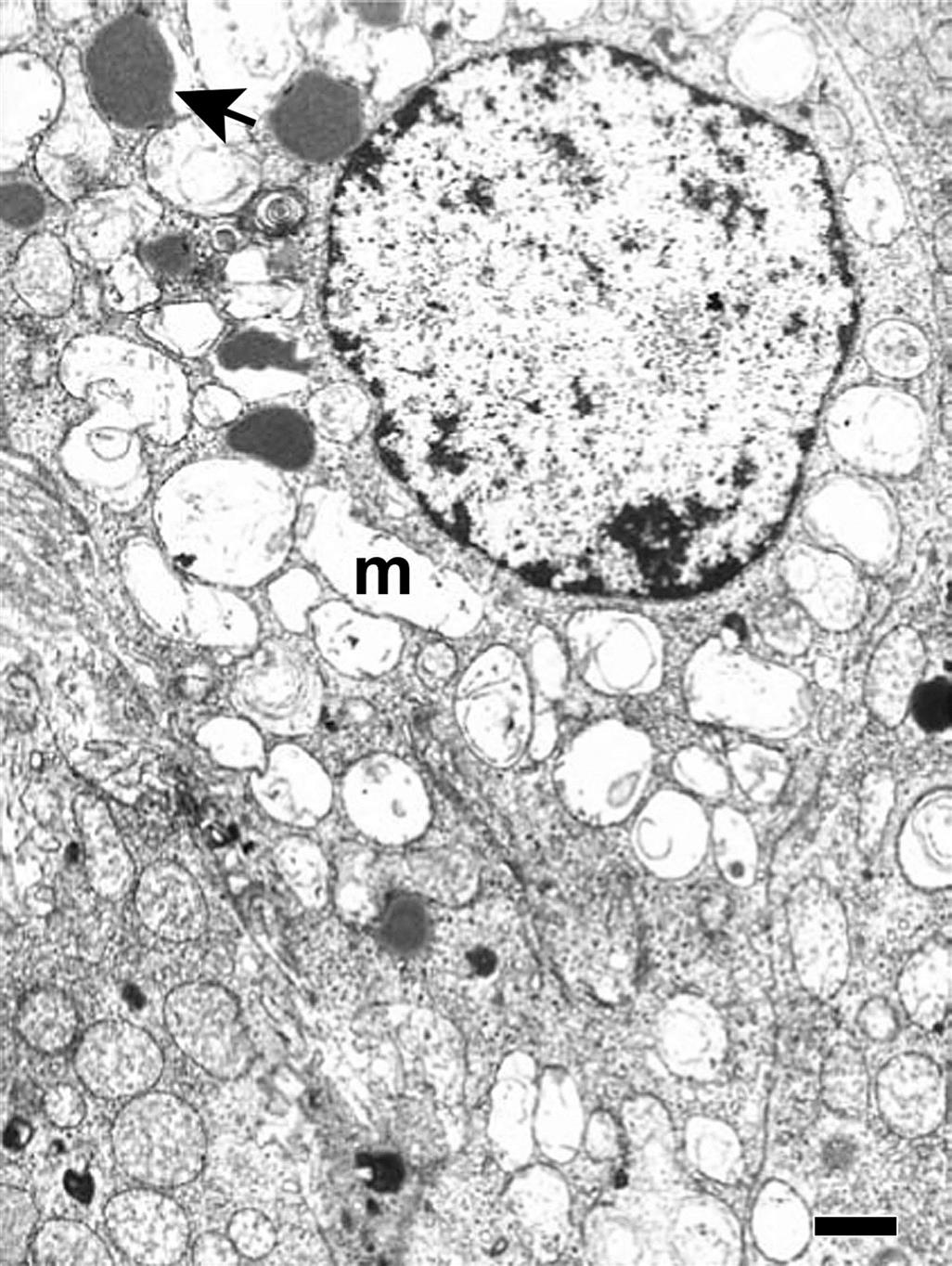
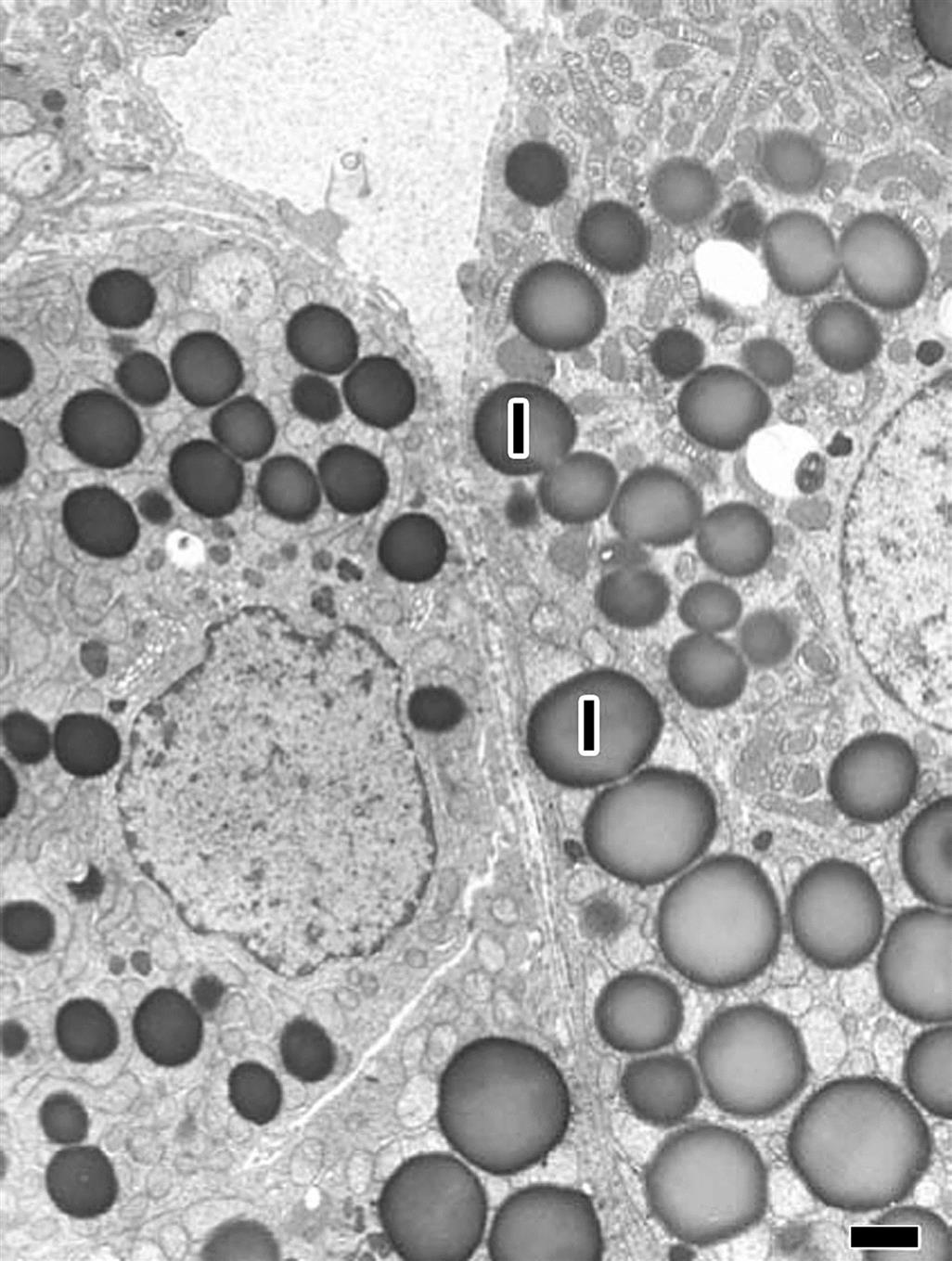
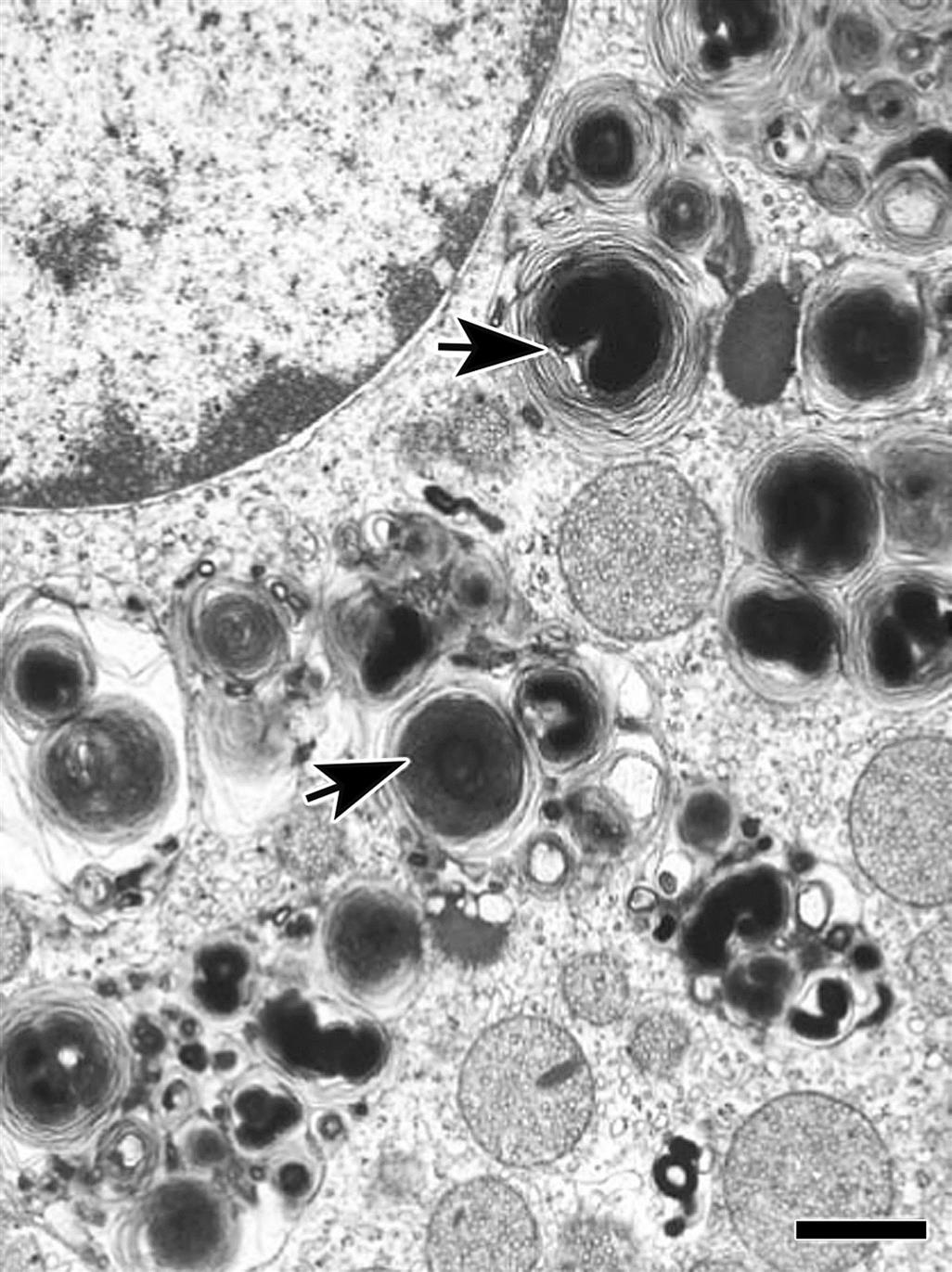
Mitochondrial damage with vacuolization (Figure 20.13) and accompanying changes in the endoplasmic reticulum and autophagocytic responses are among the most common ultrastructural changes observed following chemical injury in the adrenal cortex. Because mitochondria and smooth endoplasmic reticulum form an intimate network in cortical cells and important hydroxylases and dehydrogenase enzymes are found in these organelles, it is not surprising that many agents altering the ultrastructural morphology inhibit steroidogenesis. Similarly, increased lipid droplets (Figure 20.13) and lysosomal phospholipidosis (Figures 20.14 and 20.15) are compatible with altered steroid biosynthesis as a result of chemical inhibition of steroid precursors (e.g., cholesterol). The increased accumulation of lipid and severe mitochondrial vacuolization correspond to light microscopic findings of marked cytoplasmic vacuolar and granular degeneration. More severe ultrastructural injury may result with some parenchymal cells of the cortex having an electron-dense cytoplasm, chromatolysis, and disruption of the plasma membranes (necrosis). Frequently, macrophages containing cholesterol clefts, numerous lipid droplets, and membranous debris can be observed among the necrotic cells.
Proliferative Lesions
Spontaneous hyperplasia of the adrenal cortex is common in the rat, rabbit, golden hamster, mouse, dog, cat, horse, and baboon. Spontaneous proliferative lesions may be found in all zones of the adrenal cortex but are present most frequently in the ZF of adult rats. In these proliferative foci or larger capsule-distorting nodules, there is no change in architectural arrangement of cells and no obvious compression of adjacent cells; however, the cellular aggregates are cytologically different from normal cells surrounding them in terms of size and tinctorial properties. Large eosinophilic nodules in the rat often show vacuolated/cystic degeneration and hemorrhage which can replace many of the large eosinophilic adrenocortical cells (Figure 20.16). Cortical adenomas and, to a lesser extent, cortical carcinomas have been reported in moderately high incidence in certain strains of rat (e.g., Osborne Mendel, WAG/Rij, BUF, and BN/Bi strains). The incidence often increases markedly in rats over 18 months of age. A high incidence of cortical adenomas and fewer carcinomas have also been reported in the laboratory hamster (e.g., BIO 4.24 and BIO 45.5 strains). Subcapsular cortical cell proliferation and adenoma are common in laboratory mice; however, neoplasms arising deep within the adrenal cortex are rare but may be induced by gonadectomy. Naturally occurring adrenal cortical tumors are found infrequently in domestic animals, except in adult dogs and castrated male goats.
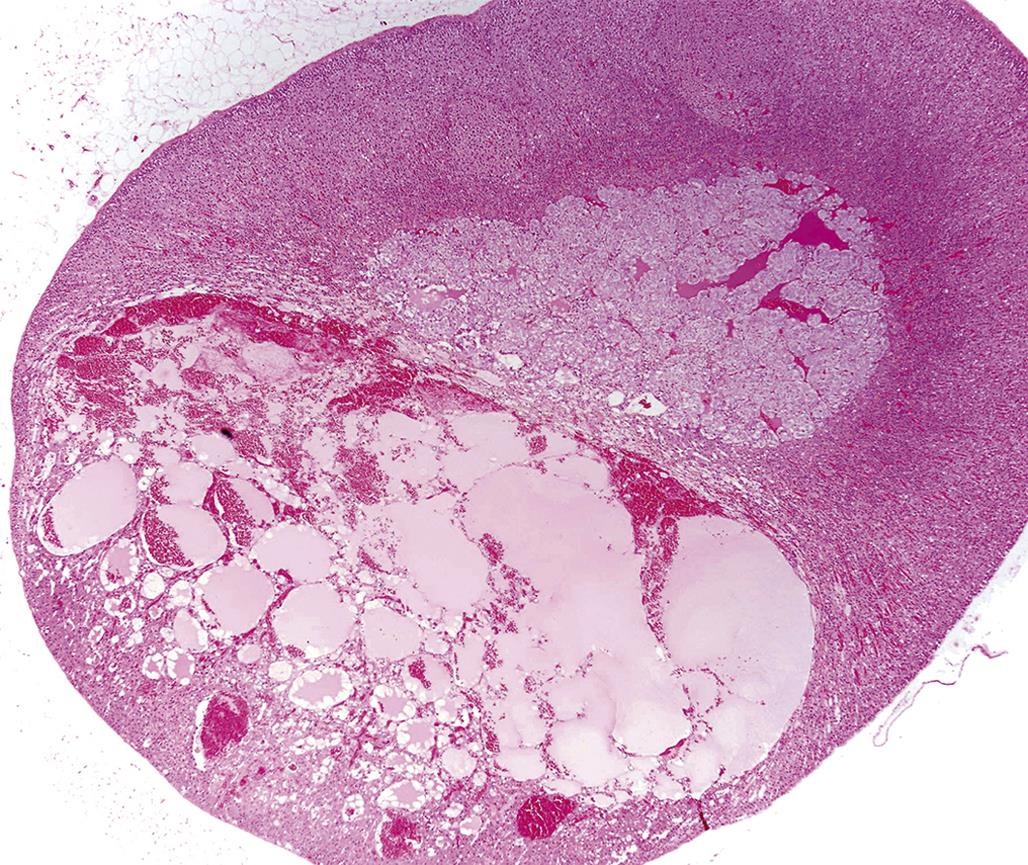
Less frequently reported are chemically induced proliferative lesions of the adrenal cortex. Unlike the diffuse hyperplasia and hypertrophy associated with the adrenal cortical response to excess ACTH stimulation, chemically induced hyperplasia usually is nodular in type and often multiple in distribution. Each focus or larger nodule is an oval-to-spherical lesion of variable size consisting of enlarged normal or vacuolated cells.
A summary of chemicals causing adrenocortical neoplasms is present in Table 20.2. Most of the reported tumors tend to be benign (adenoma), although an occasional one may be malignant (carcinoma). The ZR and ZF are more prone to develop tumors, whereas the ZG is spared unless invaded by an expanding tumor in the adjacent zones of the cortex. Tumorigenic agents of the adrenal cortex have a diverse chemical nature and use (Table 20.2). Several of the compounds have a steroid nucleus and are natural or synthetic estrogens or androgens.
Table 20.2
Examples of Compounds Producing Tumors of the Adrenal Cortexa
| Compound | Chemical properties | Type of lesion | Animal affected |
| Aflatoxin/stilbesterol | Fungal metabolite/steroid | Adenoma and hyperplasia | Rat |
| Cholesterol | Steroid | Microadenomas | Rabbit |
| Dibromochloropropane | Soil fumigant | Adenoma | Rat |
| 7,12-Dimethylbenzanthracene | Organic solvent | Adenoma; eosinophilic and basophilic foci | Rat |
| Estrone, estriol, diethylstilbesterol | Natural and synthetic estrogens | Adenocarcinoma | Rat |
| Formic acid 2-[4-(5-nitro-2-furyl)-2-thiazolyl]hydrazide | Antimicrobial | Adenoma | Hamster |
| Parathion | Insecticide | Adenoma; carcinoma | Rat |
| Tetrachlorovinphos | Insecticide | Adenoma | Rat |
| Testosterone | Hormone | Adenoma | Hamster |
| Urethane | Organic solvent; intermediate in organic synthesis | Adenoma | Rat |
| Linoleic acid | Unsaturated fatty acid | Carcinoma | Rat |
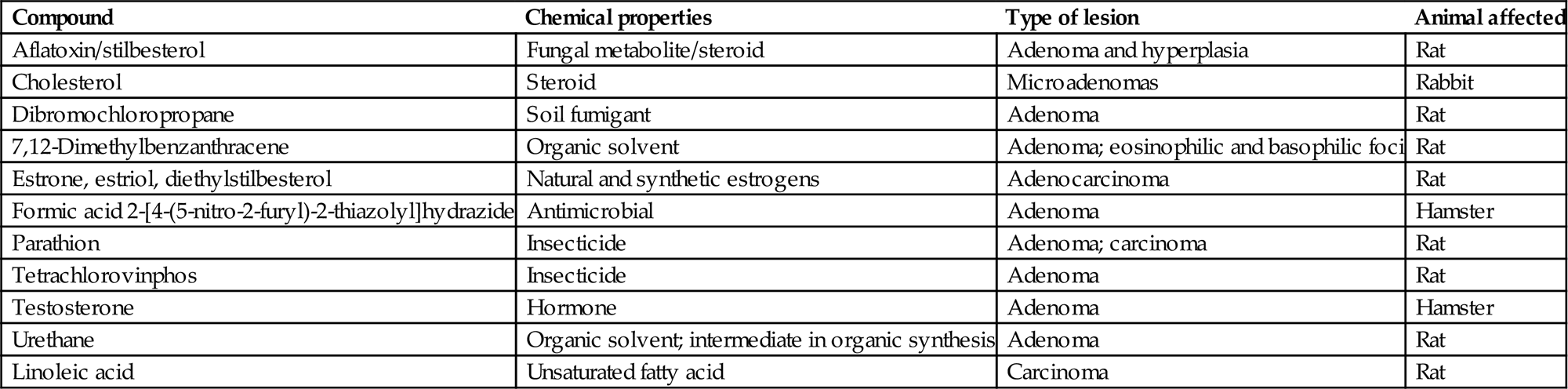
aPredilection sites are the zona reticularis and fasiculata.
Table modified from Handbook of Toxicologic Pathology, second ed. W. M. Haschek, C. G. Rousseaux, and M. A. Wallig, eds. (2002) Academic Press, Vol. 2, Table III, p. 697.
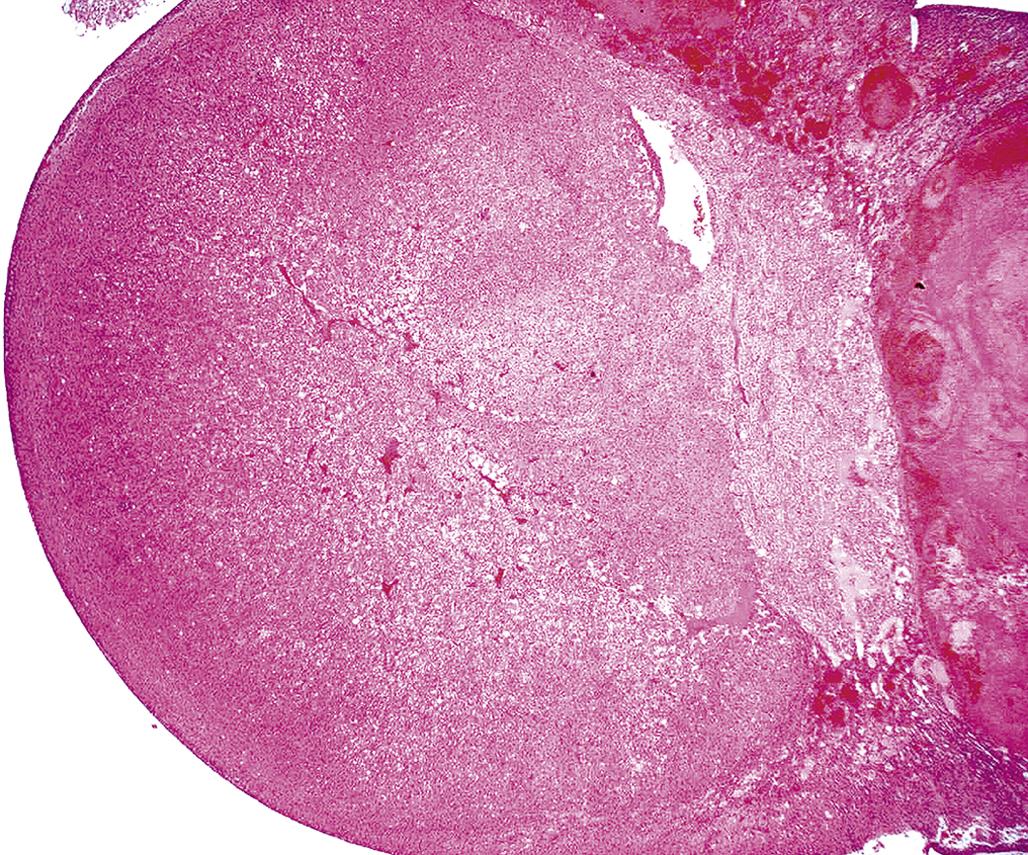
An adrenal cortical adenoma has characteristics similar to nodular hyperplasia, with the exception that adjacent parenchymal cells are compressed and often atrophic due to pressure. Cortical adenomas show loss of the cellular radial architecture but are well differentiated and circumscribed and may be partially encapsulated by a thin band of fibrous connective tissue (Figure 20.17).
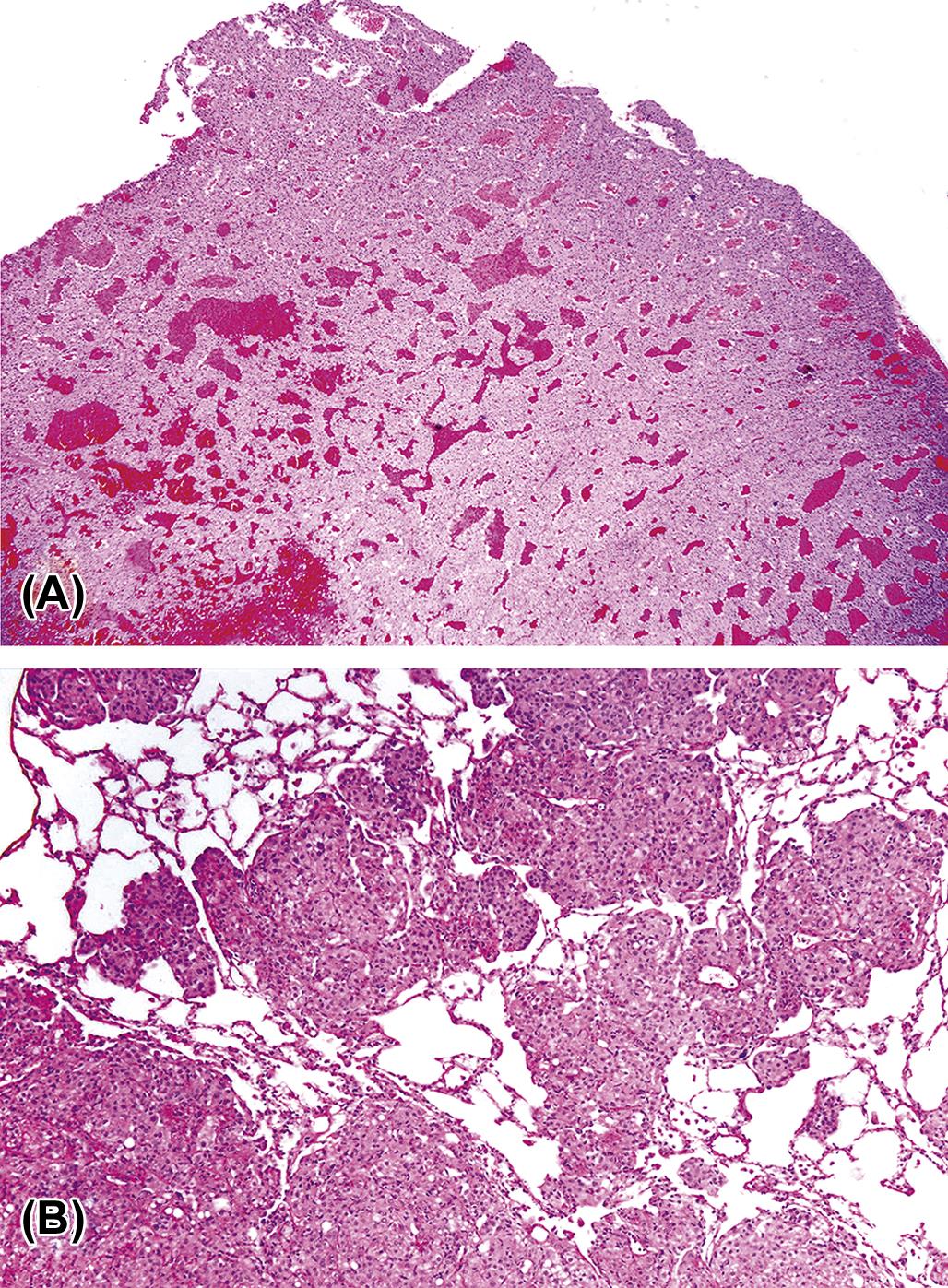
Adrenal cortical carcinomas are composed of large polyhedral or pleomorphic cells with an eosinophilic or vacuolated cytoplasm. Tumor cells have prominent nucleoli and variable numbers of mitotic figures, and form different histologic patterns, including sheets, lobules, and cords. The invasive nature of the malignant cells is also apparent by penetration through the capsule, obliteration of the normal architecture of the affected gland, and metastasis to distant sites. Blood-filled spaces and localized areas of necrosis are common in cortical carcinomas (Figure 20.18).
Mechanisms of Toxicity
As mentioned previously, the adrenal is the most common target organ for toxicity in the endocrine system. First, there is the dependence of the adrenal cortex on the trophic support of hormones from the pituitary and hypothalamus, and also hormones from other endocrine tissues such as adrenomedullary neuropeptides where, for example, adrenomedullin has a role in aldosterone and cortisol secretion. Toxicity in these other sites therefore could ultimately influence the adrenal cortex. Additionally, the adrenal cortex has both anatomic and molecular characteristics that convey vulnerability to toxic insult, and the following factors predispose the adrenal cortex to toxic insult in vivo.
1. Functional dependence on the hypothalamus and pituitary and peripheral hormone-carrier molecules (e.g., CBG)
The large number of potential toxicological targets such as enzymes, receptors, and biochemical functional mediators (e.g., adrenomedullin) of major concern is the sequentially dependent steroidogenic steps in cortisol/corticosterone or aldosterone production and secretion, which are at the end of the pathway and therefore have the highest probability of effect from upstream toxicity.
2. High vascularity and disproportionately large blood volume received per unit mass of adrenal tissue ensuring high exposures to toxicants.
3. The high content of unsaturated fatty acids in adrenocortical cell membranes that are susceptible to lipid peroxidation both directly and indirectly (see below).
4. Lipophilicity due to rich cholesterol and steroid content favoring deposition of lipophilic compounds.
5. The high content of CYP enzymes present in the adrenal cortex that can produce—(1) reactive metabolites of toxicants that then mediate toxicity and (2) hydroxylation reactions that may generate free radicals which then damage adrenocortical cells and membrane (as above).
Classes of chemicals known to be toxic for the adrenal cortex include short chain (three or four carbon) aliphatic compounds, lipidosis inducers, and amphiphilic compounds. It would also appear that hormones, especially exogenous steroids, have a direct effect on the adrenal cortex. The most potent aliphatic compounds are of three-carbon length with electronegative groups at both ends. These compounds frequently produce necrosis, particularly in the ZF and ZR. Examples include acrylonitrile, 3-aminopropionitrile, 3-bromopropionitrile, 1-butanethiol, and 1,4-butanedithiol. By comparison, lipidosis inducers can cause the accumulations (often coalescing) of neutral fats, which may be of sufficient quantity to cause a loss of organellar function and cellular destruction. The ZR and ZF appear to be the principal targets of xenobiotic chemicals. Examples of the compounds causing lipidosis include aminoglutethimide, amphenone, anilines, and imidazole antimycotic drugs. Biologically active cationic amphiphilic compounds tend to produce a generalized phospholipidosis that involves primarily the ZR and ZF. They cause microscopic and subcellular phospholipid-rich inclusions. These compounds affect the functional integrity of lysosomes, which appear ultrastructurally to be enlarged and filled with membranous lamellae or myelin figures. Examples of compounds known to induce these types of effect include chloroquine, triparanol, and chlorphentermine.
Another class of compounds that affects the adrenal cortex is certain hormones, particularly natural and synthetic steroids. Some of these steroid hormones (corticosteroids) may cause functional inactivity and morphological atrophy during prolonged exogenous use (Figure 20.19). Other steroid hormones (natural and synthetic estrogens and androgens) have been reported to cause proliferative lesions in the adrenal cortex of laboratory animals.

The final class of compounds represents a miscellaneous group of chemicals that affect hydroxylation and other functions of mitochondrial and microsomal fractions (smooth endoplasmic reticulum). Examples of these compounds include o,p′-DDD and -alpha-(l,4-dioxido-3-methylquinoxalin-2-yl)-N-methylnitrone (DMNM). Additional compounds in this category cause their effects by means of CYP metabolism and the activation of toxic metabolites. An example is the activation of carbon tetrachloride, resulting in lipid peroxidation and covalent binding to cellular macromolecules of the adrenal cortex. For another example, aminoglutethimide downregulates the ACTH receptor, and inhibits CYP11A1 (cholesterol side chain cleavage) and CYP11B1 (CYP11β/18), which is the terminal enzyme in cortisol synthesis.
Many of the chemicals that cause morphological changes in the adrenal glands can also affect adrenal cortical function (Table 20.3). Chemically induced changes in adrenal gland function result either from blockage of the effects of the adrenocorticoids at peripheral sites or from inhibition of synthesis and/or secretion (steroidogenesis). In the former case, many antisteroidal compounds (antagonists) act by competing with or binding to steroidal receptor sites, thereby either reducing the available receptor sites or altering their functional activity. Cortexolone (11α-deoxycortisol, an antiglucocorticoid) and spironolactone (an antimineralocorticoid) are two examples of peripherally acting hormone antagonists. Pharmacologically, many of these antagonists are beneficial for either diagnostic or therapeutic purposes.
Table 20.3
Examples of Pharmacological Inhibition of Adrenal Steroid Biosynthesis, Secretion, or Function
| Compound | Steroid or conversion site inhibited | Mechanism of action |
| Aminoglutethimide | Cholesterol to pregnenolone | Competitive inhibition of 20α-hydroxylase |
| o,p′-DDD | Cholesterol to pregnenolone; 11-deoxycortisol to cortisol | Partial 11β-hydroxylase inhibition |
| DMNM | Cholesterol to pregnenolone? | Unknown |
| Triparanol | Desmosterol (24-dehydrocholesterol) to cholesterol | Inhibited reduction of 24, 25 bond |
| Cyanoketone | δ5-3β-Ol steroids to δ4-3-oxo steroids | 3β-Hydroxysteroid dehydrogenase inhibition |
| Trilostane | Δ5-3β-Ol steroids to Δ4-3-oxo steroids | 3β-Hydroxysteroid dehydrogenase inhibition |
| Su-9055 | Cortisol; aldosterone | Inhibition of 17α-hydroxylase; interference of oxidation at C18 |
| Su-8000 | Cortisol; aldosterone | Inhibition of 17α-hydroxylase; interference of oxidation at C18 |
| Metapyrone | 11-Deoxycortisol to cortisol | Inhibition of 11β-hydroxylase; inhibition of other hydroxylation reactions depending on species |
| SKF 12185 | 11-Deoxycortisol to cortisol | Inhibition of 11β-hydroxylase |
| Carbon tetrachloride | Nonspecific inhibition | Inhibition of cytochrome P450 portion of microsomal enzymes 17α- and 21-hydroxylases |
| Cadmium | Nonspecific inhibition | Inhibition of NADPH-cytochrome P450 reductase portion of 21-hydroxylase; other microsomal as well as mitochondrial hydroxylases may also be affected |
| Amphenone | Nonspecific inhibition | Inhibition of 20α-, 11β-, 17α-, and 21-hydroxylases? |
| Cortexolone (11-deoxycortisol) | Competitive binding to glucocorticoid receptors | Diminished translocation of glucocorticoid—receptor complex to nucleus of target cell |
| ROl-8307/heparinoids | Aldosterone | Inhibition of 18-oxidation |
| Spironolactone | Aldosterone | Competitive inhibition of peripheral receptor sites, resulting in sodium diuresis; possible direct effects on synthesis and secretion |
| Captopril | Aldosterone; inactivation of renin–angiotensin system | Inhibition of angiotensin-converting enzyme |
| Triaryl phosphate | Cholesterol ester to cholesterol | Neutral cholesterol ester hydrolase inhibitor |
| PD 132301-2 | Esterification of cholesterol | Inhibition of acyl-CoA; cholesterol acyltransferase |
| 2,3,7,8-Tetrachlorodibenzo-p-dioxin | Cholesterol side chain cleavage | Cytochrome P450s |
Table modified from Handbook of Toxicologic Pathology, second ed. W. M. Haschek, C. G. Rousseaux, and M. A. Wallig, eds. (2002) Academic Press, Vol. 2, Table I, p. 688.
Most chemicals affecting adrenal function appear to do so by altering steroidogenesis. A study of the effects of these chemicals on the histology and ultrastructure of adrenal cortical cells often can give insight into possible selection sites of inhibition of steroidogenesis. For example, chemicals causing increased lipid droplets may be involved in inhibiting the utilization of steroid precursors, including the conversion of cholesterol to pregnenolone. Chemicals that affect the fine structure of mitochondria and smooth endoplasmic reticulum would be expected to impair the activity of 11α-hydroxylase and 17α- and 2l-hydroxylases, respectively. The previously mentioned examples of impaired steroidogenesis would result in lesions found primarily in the ZR and ZF. However, atrophy of the ZG may reflect specific inhibition of aldosterone synthesis or secretion, either directly (inhibition of 18Q1-hydroxylation) or indirectly (inactivation of the renin–angiotensin system II), by chemicals such as spironolactone and captopril, respectively. The inhibition of steroidogenesis in some situations is nonspecific, as many hydroxylation reactions are affected, such as with carbon tetrachloride and cadmium intoxications.
Part 2: Adrenal Medulla
Structure and Function
Anatomy
The medulla constitutes approximately 10% of the volume of the adrenal gland. In the normal rodent gland and in most other species the medulla is sharply demarcated from surrounding cortex. The bulk of the medulla is composed of chromaffin cells, which are sites of synthesis and storage of catecholamines. In the rat and mouse, norepinephrine and epinephrine are stored in separate cell types that can be distinguished ultrastructurally after fixation in glutaraldehyde and postfixation in osmium tetroxide. Norepinephrine-containing granules appear highly electron-dense, whereas epinephrine granules are less dense with finely granular matrices. In immature rat adrenals, granules of varying densities may be found in the same cell types. Human adrenal medullary cells contain both norepinephrine and epinephrine within a single cell.
In addition to chromaffin cells, the adrenal medulla contains variable numbers of ganglion cells. A third cell type has also been described, and has been designated the small granule containing cell or small intensely fluorescent cell. These cells appear morphologically intermediate between chromaffin cells and ganglion cells. They possibly may function as interneurons.
Adrenal medullary cells also contain serotonin and histamine, but it has not been determined if these products are synthesized in situ or taken up from the circulation. A number of neuropeptides, including encephalin, neurotensin, and neuropeptide Y, are also present in rat chromaffin cells. Another peptide, adrenomedullin, has been shown to modulate function of the adrenal cortex; this peptide has hypotensive actions and as such can influence the renin–angiotensin–aldosterone axis functionally. Pharmacotoxicological effects of a compound on the adrenal medulla can therefore have indirect effects on adrenocortical function.
Biochemistry and Physiology
The catecholamine biosynthetic pathways are in general well known. Tyrosine is acted on by tyrosine hydroxylase to produce L-DOPA (L-3,4-dihydroxyphenylalanine), which is converted to dopamine by aromatic L-amino acid (AA) (DOPA) decarboxylase. In turn, dopamine is acted on by dopamine β-hydroxylase to form norepinephrine, which is converted to epinephrine by phenylethanolamine-N-methyltransferase. Tyrosine hydroxylase is the rate-limiting step. The conversion of tyrosine into L-DOPA and dopamine occurs within the cytosol. Dopamine then enters the chromaffin granule, where it is converted to norepinephrine. Norepinephrine leaves the granule to be converted into epinephrine in the cytosol, and epinephrine then reenters the granule. Neuropeptides and chromogranin A proteins are synthesized in the rough endoplasmic reticulum and packaged into granules in the Golgi apparatus.
The role of innervation in regulating the functions of chromaffin cells has been studied extensively. During adult life, stresses such as insulin-induced hypoglycemia or reserpine-induced depletion of catecholamines produce a reflex increase in splanchnic nerve discharge, resulting in both catecholamine secretion and transsynaptic induction of catecholamine biosynthetic enzymes. These effects become apparent during the first week of life, following an increase in the number of nerve terminals in the adrenal medulla. Other environmental influences, including growth factors, extracellular matrix, and a variety of hormonal signals that generate cyclic AMP, may also regulate the function of chromaffin cells.
Physiological effects of adrenaline include an increase in systolic blood pressure and heart rate, diversion of blood to limb muscle beds/away from gut, a decrease in gut motility, bronchodilation, reduced mucus secretion, piloerection, and mydriasis. Noradrenaline only stimulates α and β1 receptors so does not cause bronchodilation, which is a β2 response, and although it also increases systolic (and diastolic) blood pressure leading to an increased mean arterial pressure, it antagonizes the effects of adrenaline on heart rate and decreases heart rate. Many of these physiological responses become pronounced in pathological conditions affecting the medulla, particularly pheochromocytoma, where excess catecholamine release typically results in hypertension and other distinctive symptoms.
Testing for Toxicity
Morphologic Evaluation
A variety of techniques may be used for the demonstration of catecholamines in tissue sections. The chromaffin reaction is the oxidation of catecholamines by potassium dichromate solution, and results in the formation of a brown-to-yellow pigment that may be seen both grossly and microscopically. The chromaffin reaction as performed traditionally possesses a very low-level of sensitivity, and should not be used for the routine demonstration of catecholamines. Similarly, both argentaffin and argyrophil reactions, which have been used extensively in the past for the demonstration of chromaffin cells, also possess low sensitivity and specificity. Fluorescence techniques using formaldehyde or glyoxylic acid can be used to demonstrate catecholamines at the cellular level. These aldehydes form highly fluorescent derivatives with catecholamines, which can be visualized by ultraviolet microscopy.
Immunohistochemistry provides an alternative approach for the localization of catecholamines in chromaffin cells and other cell types. Antibodies are now available that permit epinephrine- and norepinephrine-containing cells to be distinguished even in routinely fixed and embedded tissue samples. Tumor cells show positivity for general neuroendocrine markers, including Chromogranin A, synaptophysin, and CD56. Such antibodies are useful for differentiating pheochromocytoma from an adrenal cortical tumor. Catecholamine biosynthetic enzymes can also be demonstrated by immunohistochemical procedures (e.g., tyrosine hydroxylase) and can differentiate a paraganglioma from other neuroendocrine tumors.
Response to Injury
Characteristics of Proliferative Lesions
The adrenal medulla undergoes a series of proliferative changes ranging from diffuse hyperplasia to benign and malignant neoplasia. The latter neoplasms have the capacity to invade locally and to metastasize to distant sites. Diffuse hyperplasia, more common in the rat and strain-dependent, is characterized by symmetric expansion of the medulla with maintenance of the usual sharp demarcation of the cortex and the medulla. The medullary cell cords often are widened, but the ratio of norepinephrine to epinephrine cells is similar to that of normal glands.
Focal hyperplastic lesions are often juxtacortical but may occur within any area of the medulla. The small nodules of hyperplasia in general are not associated with compression of the adjacent medulla (Figure 20.20); however, the larger foci often are associated with medullary compression. Foci of adrenal medullary hyperplasia are typically composed of small cells with round to ovoid nuclei and scant cytoplasm.
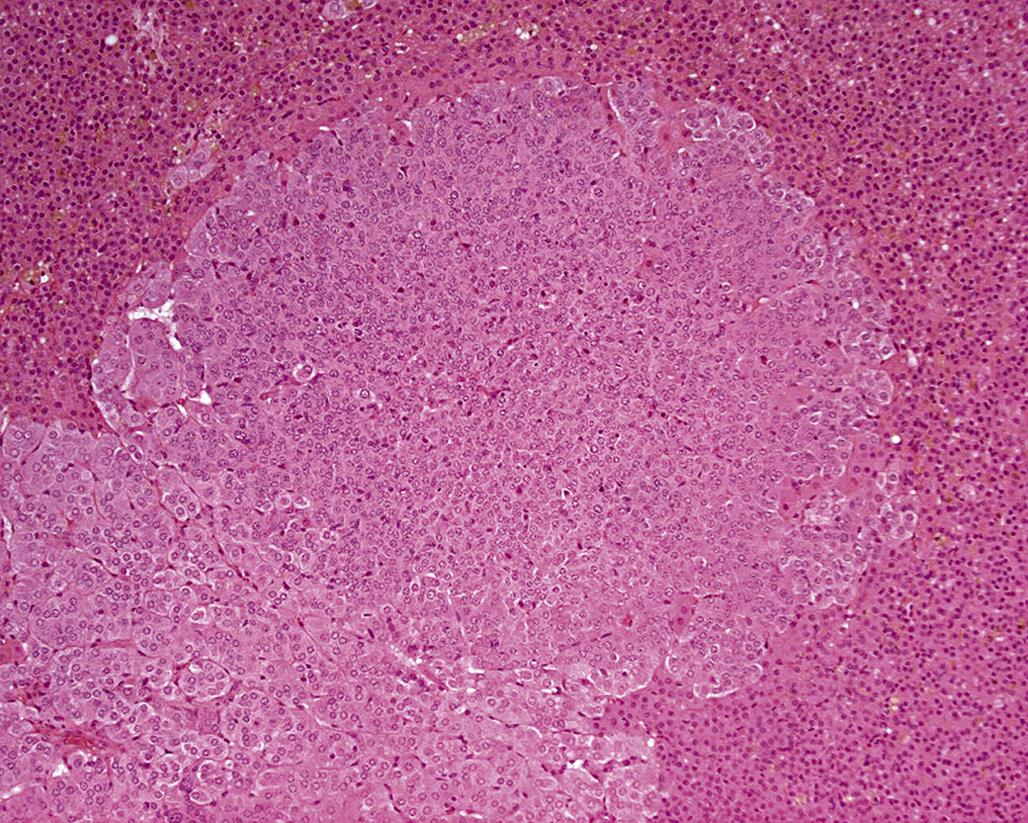
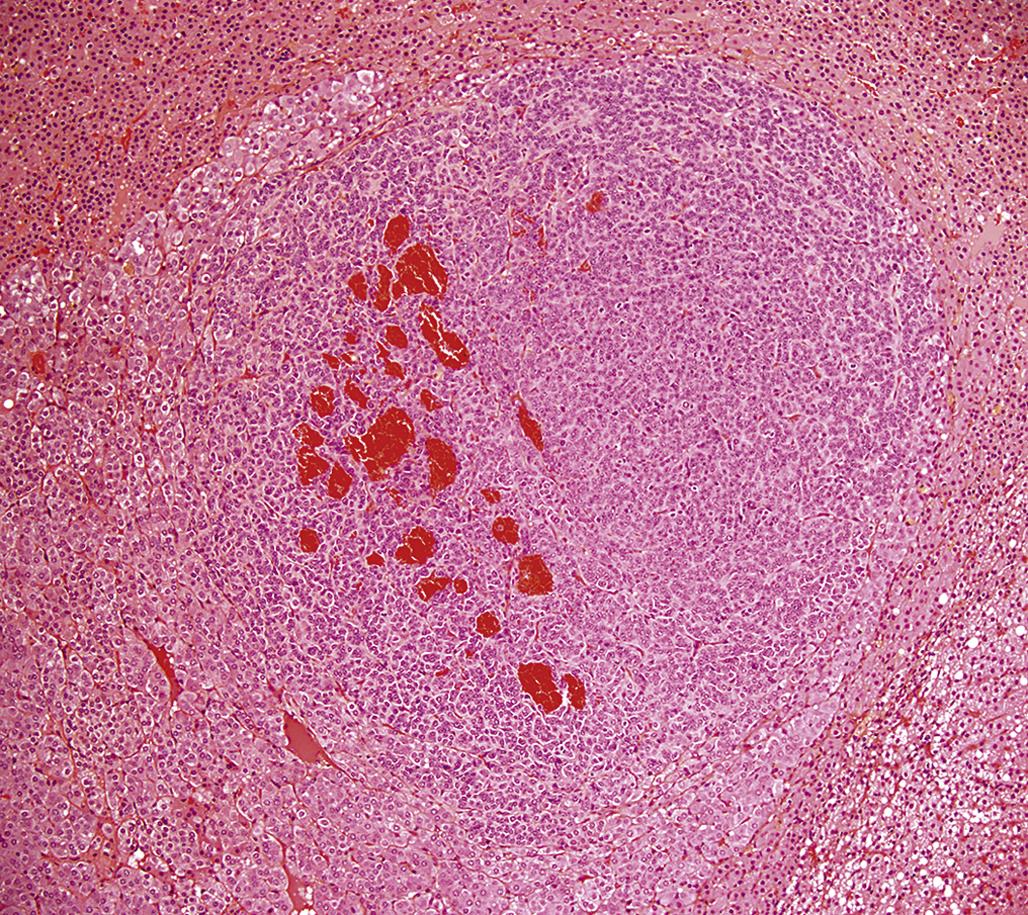
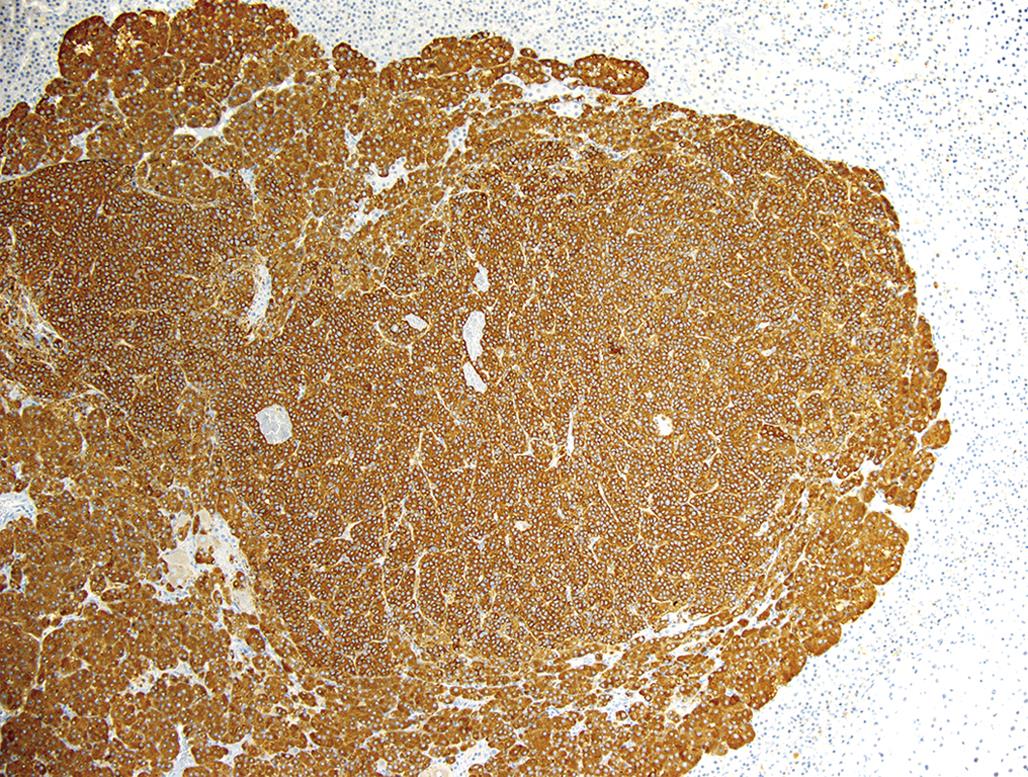
Larger adrenal medullary proliferative lesions are accepted generally as pheochromocytomas. These lesions may be composed of relatively small cells similar to those found in smaller proliferative foci. Alternatively, they may be composed of larger cells or a mixture of small and large cells (Figure 20.21). Even in the larger medullary lesions, the chromaffin reaction often is equivocal but catecholamines may be demonstrated both biochemically and histochemically, and the cells generally are synaptophysin positive (Figure 20.22). Invasion of the capsule of the adrenal with or without distant metastases occurs in malignant pheochromocytomas. Detailed criteria for evaluation of proliferative lesions of adrenal medulla have been developed by INHAND for rats and mice and will be available at the time of publication of this chapter on the Society of Toxicologic Pathology website (http://www.toxpath.org/inhand.asp).
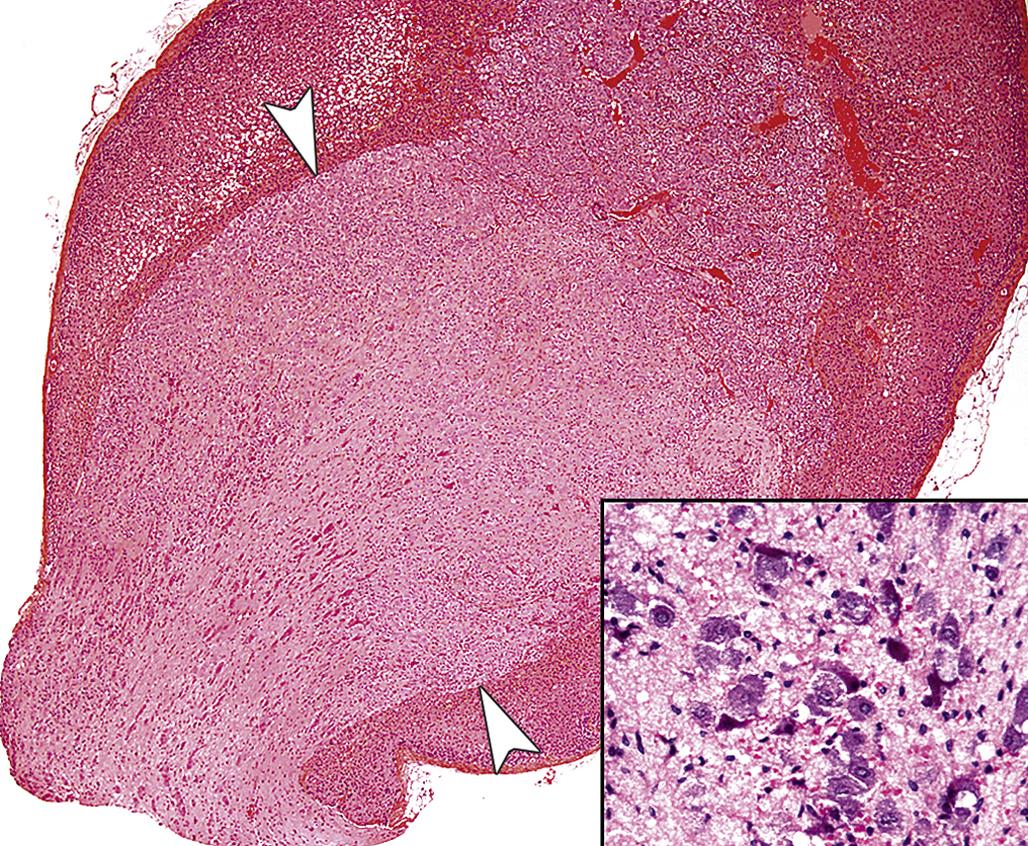
In contrast to the high frequency of pheochromocytomas, neuroblastomas and ganglioneuromas occur infrequently in the adrenal medulla in rodents and chemical exposure linked to the formation of these tumors has rarely if ever been reported. Neuroblastomas develop as a centrally located expansive mass that compresses the surrounding cortex and are composed of small cells with round to ovoid hyperchromatic nuclei and scanty cytoplasm. Cells comprising neuroblastomas resemble lymphocytes and tend to form pseudorosettes. Ganglioneuromas usually are small benign tumors arising in the medulla and compressing the surrounding cortex. They are composed of multipolar sympathetic ganglion cells and neurofibrils with a prominent fibrous connective tissue stroma (Figure 20.23).
Occurrence of Proliferative Lesions
Proliferative lesions occur with high frequency in many strains of laboratory rats. The incidence of these lesions varies with strain, age, sex, diet, exposure to drugs, and a variety of environmental agents. Studies from the National Toxicology Program (NTP) historical data base of 2-year-old F344 rats have reported that the incidence of pheochromocytomas was 17% and 3.5% for males and females, respectively. Malignant pheochromocytomas were detected in 1% of males and 0.5% of females. In addition to F344 rats, other strains with high incidences of pheochromocytoma include Wistar, New England Deaconess Hospital, Long–Evans, and Sprague-Dawley.
Pheochromocytomas are considerably less common in Osborne-Mendel, Charles River, Holtzman, and WAG/Rij rats. Most studies have revealed a higher incidence in males than in females. Crossbreeding of animals with high and low frequencies of adrenal medullary proliferative lesions results in first generation (F1) animals with an intermediate tumor frequency. Pheochromocytomas are less common in the mouse than in most strains of rats.
There is a striking relationship between age and the frequency, size, and bilateral occurrence of adrenal medullary nodules in the rat. In the Long–Evans strain, medullary nodules have been found in less than 1% of animals under 12 months of age. The frequency increases to almost 20% in 2-year-old animals and to 40% in animals between 2 and 3 years of age. The mean tumor size increases progressively with age, as does the frequency of bilateral and multicentric occurrence.
Mechanisms of Toxicity
Proliferative lesions of the medulla, particularly in the rat, have been reported to develop as a result of a variety of different mechanisms. Ionizing radiation has been linked in some studies to adrenal medullary tumors. This has been disputed by others, however.
Some data are available on the relationship of anterior pituitary hormones to the development of adrenal medullary lesions. For example, the long-term administration of growth hormone (GH) is associated with the development of pheochromocytomas as well as tumors in other sites. Some authors have suggested that prolactin (PRL)-secreting pituitary tumors, which occur commonly in many rat strains, may play a role in the development of proliferative medullary lesions. GH and PRL effects may be related because the two hormones are approximately 40% homologous; GH, when present in high concentrations in the circulation, can bind to PRL receptors. Estrogens and thyrotropin-releasing hormone (TRH) also would be expected to stimulate PRL release. Evidence for a role of pituitary hormones in the development of medullary lesions is provided by data suggesting that hypophysectomy eliminates the development of these lesions in a susceptible strain.
Both nicotine and reserpine have been implicated in the development of adrenal medullary proliferative lesions. Both agents may act by a shared mechanism, as nicotine directly stimulates nicotinic acetylcholine receptors, whereas reserpine causes a reflex increase in the activity of cholinergic nerve endings in the adrenal. A short dosing regimen of reserpine administration in vivo stimulates the proliferation of chromaffin cells in the adult rat, and the mechanism may involve a reflex increase in neurogenic stimulation via the splanchnic nerve. Treatment with antithyroid drugs such as propylthiouracil (PTU) also may affect chromaffin cells by a similar mechanism, as hypothyroid rats have increased sympathetic activity. An additional component of the action of reserpine occurs through the depletion of hypothalamic dopamine stores.
Several other drugs have been reported to increase the incidence of adrenal medullary proliferative lesions. These include zomepirac sodium (a nonsteroidal antiinflammatory drug), isoretinoin (a retinoid), and gemfibrozil (a hypolipidemic drug). However, the mechanisms responsible for the stimulation of adrenal medullary proliferation by these drugs are unknown.
Chlorinated compounds such as chlorinated paraffins, P-chloroaniline hydrochloride, 4-chloro-M-phenylenediamine, P-dichlorobenzene, hexachloroethane, 4,4′-methylenedianiline dihydrochloride, pentochlorophenol, and 1,1,2-trichloroethane comprise almost half the compounds on the NTP list for chemicals producing treatment-related pheochromocytoma.
Environmental and dietary factors may be more important than genetic factors as determinants of the incidence of adrenal medullary proliferative disorders in rats. The incidence of adrenal medullary lesions can be reduced by lowering the carbohydrate content of the diet. Sugar alcohols, including mannitol, sorbitol, xylitol, and lacitol, have been reported to increase the incidence of all types of proliferative lesions of the adrenal medulla. In view of the fact that sugar alcohols are not mutagenic, it is likely that the increased incidence of adrenal medullary lesions represents a response to metabolic change.
Vitamin D is the most potent in vivo stimulus yet identified for chromaffin cell proliferation in the adrenal medulla. Vitamin D3 (5000, 10,000, or 20,000 IU/kg/day) increases proliferative indices (e.g., BrdU) substantially over the long term in the adrenal medullas of F344 rats. At higher doses, there are focal medullary proliferative lesions. The proliferative lesions are usually multicentric, bilateral, peripheral in location in the medulla and appear to represent a morphologic continuum rather than separate entities.
It has been suggested that at least three dietary factors may lead to hypercalcemia and increased incidence of adrenal medullary proliferative lesions. These are excessive intake of food associated with feeding ad libitum; excessive intake of Ca2+ and phosphorus, as commercial diets contain two to three times more Ca2+ and phosphorus than needed by young rats; and excessive intake of other food components (e.g., vitamin D and poorly absorbable carbohydrates), which predispose to increased Ca2+ absorption.
Part 3: Pituitary Gland
Structure and Function
Anatomy
The pituitary gland (hypophysis) may be divided into two major compartments: (1) the adenohypophysis (anterior lobe), composed of the pars distalis, pars tuberalis, and pars intermedia and (2) the neurohypophysis (posterior lobe), which includes the pars nervosa or infundibular process, infundibulum, infundibular stem, and tuber cinereum. In most animals and in human fetuses, the thin cellular zone between the adenohypophysis and neurohypophysis is referred to as the pars intermedia or intermediate lobe.
The pituitary lies within the sella turcica of the sphenoid bone. The gland receives its blood supply via the posterior and anterior hypophyseal arteries, which originate from the internal carotid arteries. Arteriolar branches penetrate the pituitary stalk, lose their muscular coat, and form a capillary plexus near the median eminence. These vessels drain into the hypophyseal portal veins, which supply the adenohypophysis. This hypothalamic–hypophyseal portal system transports hypothalamic-releasing and release-inhibiting hormones directly to the adenohypophysis for interactions with their specific target cells.
Functional Cytology
The neurohypophysis is joined to the hypothalamus via the infundibular stalk and is composed of densely packed bundles of nonmyelinated axons and capillaries that are supported by modified glial cells or pituicytes. The capillaries in the pars nervosa are termination sites for unmyelinated axons, which originate from the hypothalamic neurosecretory neurons. Axons that arise from neurons in the infundibular and tuberal regions (tuberohypophyseal tract) terminate predominantly in the median eminence. Axons arising from supraoptic and paraventricular nuclei (supraopticohypophyseal tract) terminate in the pars nervosa. Both oxytocin and vasopressin (antidiuretic hormone) are synthesized in supraoptic and paraventricular nuclei as large precursor molecules, which contain both active hormones and their associated neurophysins. As the biosynthetic precursor molecules travel along the axons in secretion granules from the neurosecretory neurons, the precursors are cleaved into the active hormones and their respective neurophysins. These secretory products can be detected immunocytochemically.
The adenohypophysis represents the largest portion of the pituitary gland. The cells within this lobe are responsible for the synthesis of at least six major hormones: growth hormone (GH), prolactin (PRL), ACTH, follicle-stimulating hormone (FSH), luteinizing hormone (LH), and thyroid-stimulating hormone (TSH) or thyrotropin. With H&E staining, cells of the anterior lobe have been divided into acidophils, basophils, and chromophobes, constituting approximately 40%, 10%, and 50% of the total cell population, respectively.
The GH-secreting acidophils (somatotrophs) constitute approximately 50% of the total cell population in the pars distalis. These cells are distributed throughout the adenohypophysis and are generally round to ovoid in shape. The GH-producing cells stain positively with orange C and eosin. Although most of the GH-synthesizing cells correspond to acidophils, some may be chromophobic, as they are in an actively synthesizing phase of the secretory cycle.
PRL-secreting acidophils (luteotrophs) compose 15%–25% of the total cell population. They tend to be concentrated in the dorsocephalic region of the adenohypophysis. Immunoperoxidase stains reveal that PRL cells have angular cell processes that may be closely applied to the cytoplasm of gonadotrophs. PRL cells, stain positively with erythrosine and carmoisine. However, the sparsely granulated cells with smaller granules often fail to stain with these dyes and appear chromophobic. Pregnancy and lactation are associated with hyperplasia of PRL-secreting cells. These so called “pregnancy cells” are chromophobic by routine staining techniques.
ACTH-producing cells (corticotrophs) are round to ovoid, chromophobic or lightly basophilic (in humans), and stain weakly positive with periodic acid-Schiff (PAS) stain. These cells are widely distributed within the pars distalis and form the bulk of the pars intermedia. Crooke’s hyaline change refers to the intracellular accumulation of an eosinophilic homogeneous material within the cytoplasm of these cells. Immunohistochemical staining reveals that this material represents a keratin-like moiety, which accumulates in conditions associated with glucocorticoid hormone excess.
Thyrotropin-producing (TSH) basophils or thyrotrophs account for about 5% of the cells of the adenohypophysis. These cells tend to occur in small clusters within the pars distalis and have a stellate to polygonal shape. They may be basophilic or chromophobic and are also PAS-positive. “Thyroidectomy cells” are enlarged and vacuolated TSH cells. Large PAS-positive lipofuscin granules often are found within these cells. Conditions, which lead to hypofunction of the thyroid, such as due thyroidectomy, administration of goitrogenic drugs or chemicals that inhibit the biosynthesis of triiodothyronine (T3) and thyroxine (T4) or exposure chemicals that increase the hepatic degradation of thyroid hormones, result in the development of thyroidectomy cells in the pituitary gland. Hypertrophy and hyperplasia of TSH cells under these conditions are due to a lack of negative feedback by T3 and, to a lesser extent, T4 on the hypothalamus and thyrotrophs.
Gonadotropin-producing basophils or gonadotrophs are relatively large round to oval cells, which account for approximately 10% of the cells in the pars distalis. These cells are responsible for the production of FSH and LH. They tend to be concentrated in the dorsocephalic region of the pars distalis. Immunoperoxidase stains have revealed that some cells contain both FSH and LH, whereas others contain only LH or FSH.
Gonadotroph cells undergo a series of changes following castration, resulting in the formation of “gonadectomy cells.” As a result of the lack of negative feedback by gonadal steroids, gonadotrophs are actively stimulated to synthesize and secrete FSH and LH. Gonadotrophs undergo hypertrophy, and the cytoplasm becomes vacuolated due to distention of endoplasmic reticulum with finely granular material in response to long-term stimulation by gonadotropin-releasing hormone (GnRH) from the hypothalamus. In some cells, a single large vacuole occupies most of the cytoplasm, producing cells with a “signet-ring” appearance.
In addition to specific hormone-secreting cells, a population of supporting cells is also present in the adenohypophysis. These cells have been referred to as stellate (follicular) cells and can be stained selectively with antibodies to S-100 protein. Stellate cells typically have elongate processes and prominent cytoplasmic filaments. These cells appear to provide a phagocytic or supportive function in addition to producing a colloid-like material.
The hypothalamus serves as the major regulator of the adenohypophysis. Each cell type within the adenohypophysis is under the control of a corresponding releasing hormone that is synthesized within nerve cell bodies of the hypothalamus. The releasing hormones are transported via axonal processes to the median eminence, where they are released into capillaries and are carried by the hypophyseal portal system to trophic hormone-producing cells in the adenohypophysis. Specific releasing factors have been identified for TSH, FSH, LH, ACTH, and GH. PRL secretion is stimulated by a number of factors, the most important of which appears to be TRH. TRH stimulates the release of PRL with many of the same dose–response characteristics as the stimulation of TSH release.
Multiple influences contribute to the control of adenohypophyseal hormone secretion. Dopamine serves as the major PRL inhibitory factor. This amine suppresses virtually all aspects of PRL secretion and also inhibits cell division and DNA synthesis of this cell type. Dopamine also suppresses ACTH production by corticotrophs in the pars intermedia of some species. A second hypothalamic release-inhibiting hormone is somatostatin (somatotropin release-inhibiting hormone, SRIH). This tetradecapeptide inhibits the secretion of both GH and TSH. In some situations, SRIH also inhibits the secretion of PRL and ACTH. The control of pituitary hormone secretion is also affected by negative feedback loops resulting from the interaction of end-organ hormones, adenohypophyseal hormones, and corresponding hypothalamic-releasing and release-inhibiting hormones.
Evaluation of Toxicity
Immunohistochemical evaluation is an important approach for the functional and morphological analysis of normal and abnormal pituitary glands. Hormones of the various types of adenohypophyseal cells in the rodent gland have crossreactions with antibodies to corresponding human pituitary hormones. However, higher levels of specificity may be achieved with the use of antibodies to the rat pituitary hormones. For most routine toxicological studies, formalin fixation provides adequate preservation of the pituitary hormones for immunohistochemical studies; however, some authors have recommended Bouin’s fixation for immunohistochemical staining of the pituitary because this fixative produces more precise localization of the hormones at the light microscopic level. In addition, in situ hybridization techniques have been employed for the identification of messenger RNAs encoding the hormonal peptides.
Response to Injury
Nonneoplastic Lesions
The term “cystoid degeneration” has been used to describe foci of cell loss in the adenohypophysis. Newer nomenclature, developed by the INHAND project (http://www.toxpath.org/inhand.asp), simply refer to these lesions as “cysts,” lined by cuboidal or pseudostratified columnar epithelium, or as “pseudocysts,” spaces not lined by epithelium. These cystic lesions often measure 50–150 mm in diameter. The edges of these foci are composed of cell types typical of the pars distalis. Occasional cell debris may be found within areas of cystoid degeneration, together with granular eosinophilic material. Occasionally the cysts represent remnants of Rathke’s pouch that normally regresses during embryogenesis. Cystic changes may also occur in hyperplastic foci and in neoplasms of the pituitary. The frequency of cystoid degeneration is increased by feeding diets containing diethylstilbestrol to female C3H HeN (MMTV+ mice).
Neoplastic Lesions
Craniopharyngioma
The craniopharyngioma is an uncommon, naturally occurring neoplasm in rodents. The tumor may originate from the craniopharyngeal duct epithelium of Rathke’s pouch and is composed of nests and cords of squamous cells with cysts lined by cuboidal or squamous cells that may be focally mineralized.
Pituicytoma
Primary neoplasms of the pars nervosa are extremely uncommon. The tumors that have been reported to occur at this site have been designated as pituicytomas. These tumors are composed of small, closely packed spindle cells arranged in cords and bundles. Most pituicytomas are quite small, but larger tumors may extend into the adenohypophysis and into the adjacent brain tissue. Metastases have not been reported.
Proliferative Lesions of the Adenohypophysis
The histopathological separation among nodular hyperplasia, adenoma, and carcinoma often is more difficult in endocrine glands, such as the pituitary, than in most organs of the body. However, criteria for their separation should be established and applied in a uniform manner. For trophic hormone-secreting cells of the adenohypophysis, there appears to be a continuous spectrum of proliferative lesions between diffuse or focal hyperplasia and adenomas. Prolonged stimulation of pituitary secretory cells predisposes to the subsequent development of a higher than expected incidence of tumors.
Focal (“nodular”) hyperplasia usually appears as multiple small areas that are well demarcated but not encapsulated. Cells making up an area of focal hyperplasia in the adenohypophysis closely resemble the cells of origin; however, the cytoplasmic area may be slightly enlarged and the nucleus more hyperchromatic than in normal cells.
Adenomas in rodents usually are solitary nodules that are larger than the multiple areas of focal hyperplasia, with diameters that cover at least half the width of the normal pituitary. They are sharply demarcated from the adjacent normal pituitary glandular parenchyma, and there is a thin, partial to complete, fibrous capsule. Adjacent parenchyma is compressed in at least one quadrant, depending on the size of the adenoma. Cells composing an adenoma may closely resemble the cells of origin morphologically and in their architectural pattern of arrangement; however, there are often histological differences such as multiple layers of cells lining follicles and vascular trabeculae or solid clusters of secretory cells subdivided into packets by a fine reticular stroma.
Carcinomas usually are larger than adenomas in the pituitary and generally result in a macroscopically detectable enlargement. Histopathological features suggestive of malignancy include invasion into adjacent structures (e.g., dura mater, sphenoid bone), formation of tumor cell thrombi within vessels, and, particularly, the establishment of metastases at distant sites. The growth of neoplastic cells subendothelially in highly vascular benign tumors should not be mistaken for vascular invasion. Malignant endocrine cells often are more pleomorphic (including oval or spindle shaped) than normal, but nuclear and cellular pleomorphism is not a consistent criterion to distinguish adenoma from carcinoma in rodents. Mitotic figures may be frequent in malignant cells; however, the degree of background stimulation of the endocrine gland must be taken into account.
Many neoplasms arising in the pituitary are functionally active, secrete an excessive amount of hormone either continuously or episodically and cause clinical syndromes of hormone excess. Quantitation of hormone levels in the serum of plasma in the basal or stimulated state and/or the measurement of the hormone or metabolites in the urine over a 24-hour period of excretion is essential to confirm that an endocrine tumor is functional and releasing hormone at an abnormally elevated rate. Morphologically, an endocrine tumor often can be interpreted as endocrinologically active if the rim of normal tissue around the tumor undergoes atrophy due to negative feedback inhibition by the elevated target hormone levels. The nonneoplastic secretory cells, especially their cytoplasm, become smaller than normal, and eventually the cells disappear. Functional pituitary neoplasms secreting an excess of a particular trophic hormone (e.g., ACTH or PRL) are associated with striking hypertrophy and hyperplasia of target cells in the adrenal cortex in chronic rodent studies.
Adenoma of Pars Intermedia
Adenomas of the pars intermedia are uncommon in the rat and mouse but common in the horse and dog. These lesions may vary considerably in size. Larger adenomas may compress the adenohypophysis and neurohypophysis, whereas smaller lesions may be of microscopic dimensions. These tumors are nonencapsulated but do compress adjacent normal tissue. The cells often have faintly basophilic cytoplasm with round to ovoid nuclei. Tumor cells in pigmented rats (e.g., Long–Evans) often contain melanin pigment similar to that found in the normal intermediate lobe.
Pituitary Carcinoma
Pituitary carcinomas are uncommon neoplasms compared with adenomas. They usually are endocrinologically inactive but can cause significant functional disturbances by destruction of the pars distalis and neurohypophyseal system, leading to adult-onset panhypopituitarism and diabetes insipidus. Carcinomas are large and invade extensively into the overlying brain, along the ventral aspect of the cranial cavity incorporating cranial nerves (II, III, IV), and locally into the adjacent sphenoid bone of the sella turcica, where they may cause osteolysis. Metastases occur infrequently to regional lymph nodes or to distant sites such as the spleen or liver. Carcinomas are highly cellular and often have large areas of hemorrhage and necrosis. Giant cells, nuclear pleomorphism, and mitotic figures are encountered more frequently than in adenomas.
Functional Characteristics of Pituitary Adenomas
The vast majority of pituitary adenomas in humans and rodents have been described as chromophobic in type; however, many of these tumors have been found to stain for PRL by immunohistochemistry. Of the PRL-producing tumors, most are sparsely granulated with low levels of PRL immunoreactivity by immunohistochemistry. Diffuse hyperplasia of the PRL cells has been noted adjacent to some adenomas.
The development of tumors and hyperplasia of PRL-secreting cells is accompanied by increasing serum levels of PRL. Occasionally, PRL cells within adenomas may be admixed with FSH/LH-, TSH-, or ACTH-positive cells. Infrequent adenomas composed of populations of ACTH or FSH/LH cells have also been reported. Pituitary nodules (focal hyperplasia and adenomas) composed primarily of gonadotrophs occur commonly in male rats of the Lobund-Wistar strain.
Functional (endocrinologically active) neoplasms arising in the pituitary gland of dogs most likely are derived from corticotroph (ACTH-secreting) cells in either the pars distalis or the pars intermedia. These neoplasms cause a clinical syndrome of cortisol excess (Cushing’s disease). The pituitary gland is consistently enlarged. Because the diaphragma sella is incomplete in the dog, the line of least resistance favors dorsal expansion of the gradually enlarging pituitary mass, with extension into the overlying brain leading to eventual compression or replacement of the hypothalamus and possible extension into the thalamus. Pituitary corticotroph adenomas are composed of well-differentiated, large or small, chromophobic cells supported by fine connective tissue septa. The cytoplasm of the neoplastic cells stains immunocytochemically for ACTH and melanocyte-stimulating hormone (MSH). Hormone-containing secretory granules can be demonstrated by electron microscopy. Bilateral enlargement of the adrenal glands occurs in dogs with functional corticotroph adenomas due to cortical hypertrophy/hyperplasia, primarily of the ZF and ZR.
Spontaneous Pituitary Adenomas
The high frequency of spontaneous pituitary adenomas in laboratory rats is a well-recognized phenomenon that must be considered in any long-term toxicological study. The incidence of these tumors is determined by many factors, including strain, age, sex, reproductive status, and diet. Studies from the NTP historical data base of 2-year-old F344 rats have shown that the incidence of pituitary adenomas was 21.7% and 44% for males and females, respectively. Corresponding figures for carcinomas arising in the adenohypophysis are 2.4% and 3.5% for males and females, respectively. Studies from 56 NTP rat studies completed between 1983 and 1987 revealed that the incidence of adenomas was 28.6% for males and 43.3% for females, while a diagnosis of pituitary hyperplasia was made in 10.4% of males and 10.3% of females. It should also be noted that numerous studies have called attention to the striking degree of strain variation in the incidence of pituitary tumors in rats, reported to range from 10% to more than 90%.
There is a striking relationship between age and development of pituitary adenomas. These lesions are uncommon in animals less than 1 year of age. When present in this age group, they are generally small and are present as single lesions. Pituitary adenomas are most common in rats at 2 years or more of age and tend to be larger and more frequently multicentric than in younger animals.
Rapid body growth rates and high levels of conversion of feed to body mass in early life or high protein intake in early adult life, predispose any strain of rat to the development of pituitary adenomas. In rats fed a low-protein diet (less than 12.7% crude protein), the overall tumor incidence, incidence of fatal tumors, number of multifocal tumors, and degree of cellular atypia within tumors are significantly lower than in rats fed a standard diet.
Mechanisms of Toxicity
Induction of Pituitary Tumors
Pituitary tumors can be induced readily by sustained uncompensated hormonal derangements leading to increased synthesis and secretion of pituitary hormones. In such a situation, the absence of feedback inhibition of the pituitary cell may lead to its unrestrained proliferation. This effect can be potentiated by the concurrent administration of ionizing radiation or chemical carcinogens.
Hyperplasia of the thyrotrophs occurs concurrently with cellular hypertrophy as a consequence of the absence of normal inhibitory feedback mechanisms. Foci of hyperplasia may progress to the formation of adenomas. The role of gonadectomy in pituitary tumor induction has been studied most intensively in mice. Pituitary tumors induced by gonadectomy in this species are markedly strain-dependent, and they may contain FSH, LH, or both.
The administration of estrogens is a reproducible method for inducing pituitary tumors in some experimental animals. The effect of exogenous estrogen on the rat pituitary includes stimulation of PRL secretion and the induction of PRL-secreting tumors. The administration of estrogens in susceptible strains results in increased serum PRL levels, increased numbers of PRL cells within the pituitary, and increased mitotic activity. There is considerable variation in the inductibility of pituitary tumors by estrogens in different rat strains. For example, F344 rats are more sensitive than Holtzman rats. The precise mechanisms of estrogen-induced PRL tumors are unknown.
The tumorigenic action of estrogen may not be due exclusively to its effect on the hypothalamus because estrogen can produce prolactinomas in pituitaries grafted beneath the renal capsule. Dopamine agonists, including lisuride and bromocriptine, antagonize the direct stimulatory effects of estrogens on the PRL cells. Other agents, including caffeine, have also been implicated in the development of pituitary adenomas. Additionally, the administration of N-methylnitrosourea is also associated with the development of pituitary adenomas in Wistar rats. The neuroleptic agent sulpiride has been reported to cause the release of PRL from the anterior pituitary in the rat and to stimulate DNA replication. The administration of clomiphene prevents the stimulation of DNA synthesis produced by sulpiride but does not affect PRL release from the gland. These findings have suggested that the intracellular PRL content of the anterior pituitary plays a role in the regulation of DNA synthesis through a mechanism mediated by estrogens.
Administration of salmon calcitonin for 1 year to Sprague-Dawley and Fisher 344 rats has been associated with an increased incidence of pituitary gland hyperplasia and adenomas. The pituitary tumors also produce the common α subunit of the glycoprotein hormones (LH, FSH, and TSH). This type of tumor has been shown to comprise a significant fraction of pituitary tumors in humans.
Studies have not determined whether the effects of calcitonin are direct or indirect. A striking feature of calcitonin-induced pituitary tumors and elevated serum α-subunit levels is the predilection for male compared with female rats. The basis for sex- and species-specific effects of calcitonin has not been determined. The relevance of the effects of calcitonin in the rat pituitary gland to human pathophysiology is uncertain.
Etiology and Pathogenesis of Spontaneous Pituitary Adenomas
Numerous hypotheses have been invoked to explain the high incidence of pituitary adenomas in certain inbred rat strains. Both hereditary factors and the levels of circulating sex steroids have been suggested as important etiological mechanisms. The hypothalamus has also been incriminated in the development of these tumors. Age-related hypothalamic changes may result in the diminished activity of dopamine, the major PRL inhibitory factor. In this regard, studies of aging male rats have demonstrated abnormal concentrations and metabolism of hypothalamic dopamine, norepinephrine, and serotonin and decreased hypothalamic PRL inhibitory activities. Supporting a role for dopamine in suppression of neoplastic development, dopamine has been found to suppress PRL secretion. The dopamine agonist, bromocriptine, has been reported to decrease serum PRL levels in female Long–Evans rats with spontaneously developing prolactinomas when compared to untreated age-matched controls, and the mean pituitary weight was reduced in rats treated for 44 days with bromocriptine compared to controls.
Part 4: Thyroid C Cells
Structure and Function
C cells, or parafollicular cells, of the thyroid gland, named after their major secretory product (calcitonin), are located within thyroid follicles between the basal aspects of the follicular cells and the basement membrane of the follicle. They also are present in parafollicular positions. In addition to calcitonin, C cells contain a variety of other peptides, including somatostatin and bombesin, and they are also positive for a wide variety of generic neuroendocrine markers, including chromogranin proteins and synaptophysin. In most mammalian species, C cells are concentrated within the central regions of the lobes, being most prominent at the levels of the parathyroid glands. By light microscopy C cells often have a clear or light appearance, but silver-positive cytoplasmic granules can be demonstrated by argyrophilic staining. At the ultrastructural level, the cells contain variable numbers of membrane-bound secretory granules in which calcitonin is stored prior to its release into the circulation. Although the secretory granules have considerable heterogeneity in terms of size and density, all of the granules contain immunoreactive calcitonin.
Calcitonin is a 32-AA peptide that is derived from a 141-AA precursor (procalcitonin). Ionized calcium (Ca2+) in plasma and extracellular fluids is the major physiologic stimulus for the secretion of calcitonin. The cell membrane Ca2+-sensing receptor, present in both parathyroid cells and C cells, contributes to the regulation of calcitonin secretion. Calcitonin interacts with specific receptors in target cells, principally in bone and kidney. Calcitonin inhibits osteoclast activity in bone, renal tubular resorption of Ca2+, and Ca2+ absorption by the intestines. Calcitonin secretion is increased in response to a high Ca2+ meal, often before a significant rise in plasma Ca2+ levels can be detected. An oral Ca2+ load stimulates the secretion of gastrin, cholecystokinin, and glucagon, and these hormones also serve as calcitonin secretagogues. Calcitonin also protects against Ca2+ loss from the skeleton during periods of Ca2+ mobilization, such as growth, pregnancy, and lactation.
Response to Injury
Proliferative Lesions of C Cells
Hyperplasia of C cells is not usually associated with any macroscopic abnormality in the thyroid gland. Two types of hyperplasia are evident microscopically: diffuse (Figure 20.24) and focal (nodular) (Figure 20.25). In diffuse hyperplasia, the numbers of C cells are increased throughout the thyroid lobes, to a point where they may be more numerous than follicular cells. Diffuse hyperplasia is typically associated with a physiological or pathologic response to chronic stimulation (e.g., persistent hypercalcemia) whereas nodular hyperplasia is generally considered to be a preneoplastic lesion. The relationship of C cells to follicular cells in cases of diffuse hyperplasia is similar to that noted in juvenile thyroid glands. Focal (nodular) hyperplasia of C cells often occurs concurrently with diffuse hyperplasia in the thyroid glands of rats. The follicles become progressively enlarged with increasing numbers of C cells. While normal and hyperplastic C cells are uniformly positive for calcitonin, relatively few also express somatostatin.
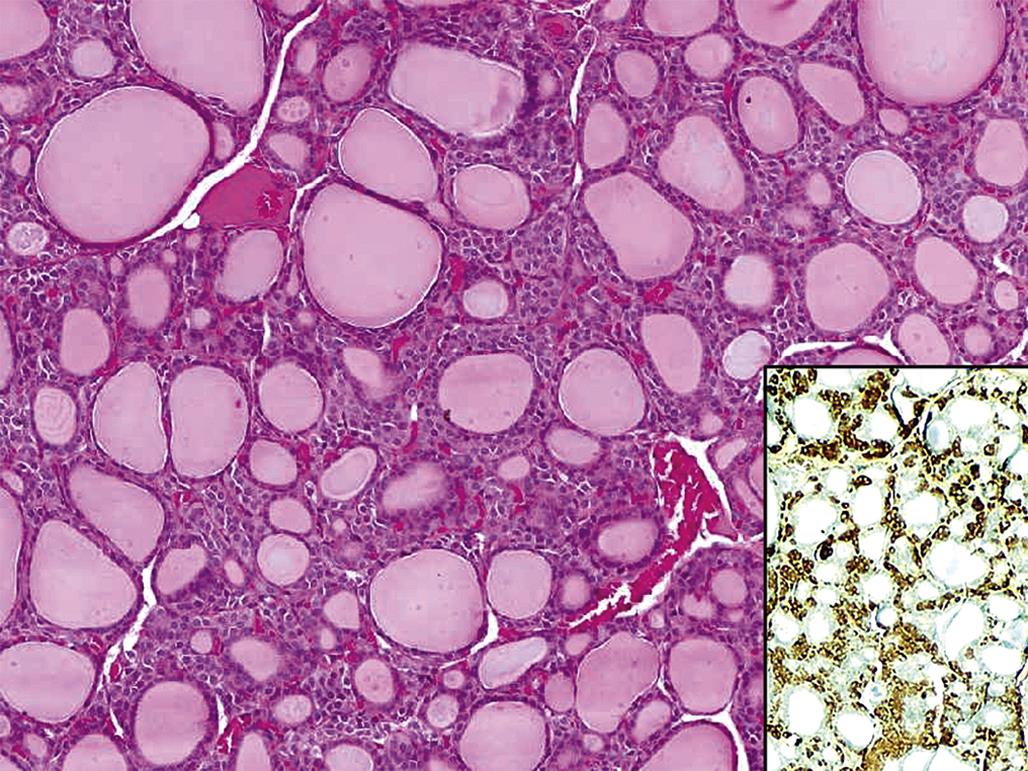
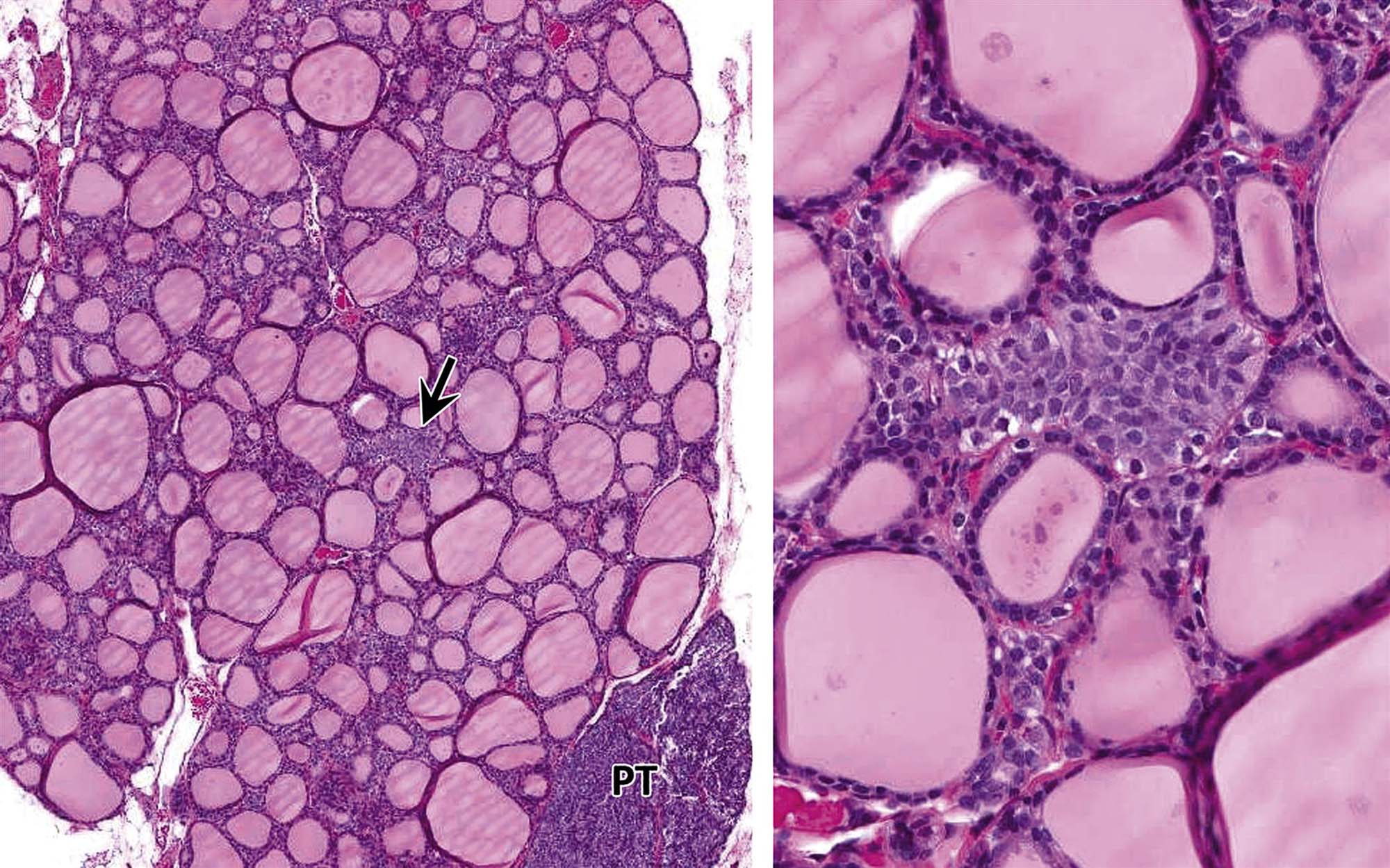
C cells are more numerous in rats (compared to mice) and increase in number with age. C cells in rats can be identified in routine H&E-stained tissue sections. Normal and diffuse hyperplasia of C cells in mice typically requires immunohistochemistry for calcitonin to identify the C cells.
At later stages of development, hyperplastic C-cell “follicles” often assume irregular, twisted, and elongate configurations. Occasional colloid-filled follicles are entrapped among the proliferating C cells. The histological distinction between focal hyperplasia and adenoma of C cells is often difficult to define. Typically, focal (nodular) hyperplasia is characterized by lesions measuring less than five average colloid-containing follicles without compression of the adjacent follicular cells; in addition, there is no compression of adjacent follicles. Cell boundaries are often indistinct.
C-cell adenomas are discrete expansive masses, or nodules of C cells larger than five average colloid-containing follicles (Figure 20.26). Adenomas are either well circumscribed, or partially encapsulated with varying degrees of compression of adjacent thyroid follicles. C cells have relatively abundant cytoplasm that is faintly eosinophilic with a round-to-ovoid nucleus with finely stippled chromatin. Adenomas may be subdivided into small cell groups by fine connective tissue septa and capillaries.
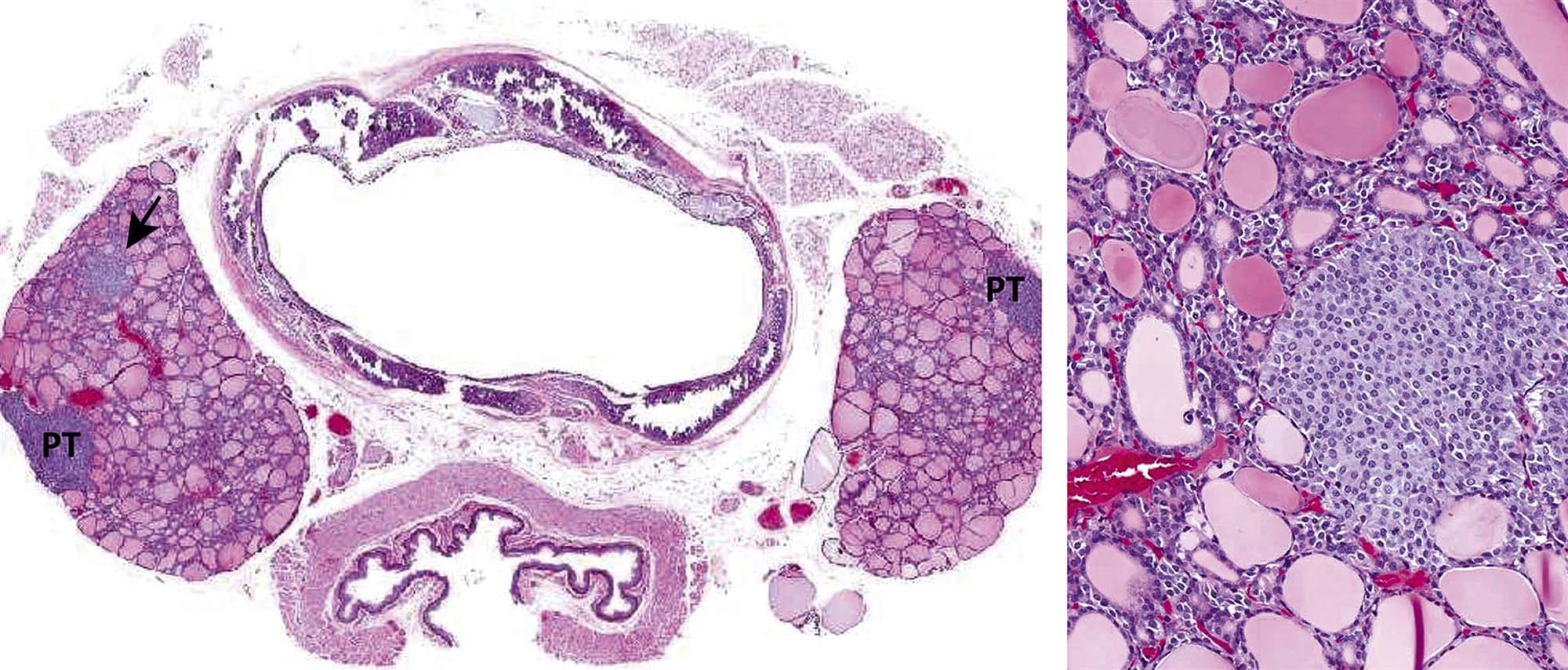
C-cell (medullary) carcinomas are characterized by infiltration of C cells into the adjacent thyroid stroma, extracapsular tissues, and lymphovascular channels (Figure 20.27). Areas of hemorrhage and necrosis may be particularly prominent in larger tumors. The component C cells are often more pleomorphic (cuboidal, oval, or spindle shaped) than those comprising focal hyperplasia or C-cell adenomas. Mitotic activity may be prominent in some cases, and foci of necrosis may be evident. Occasional tumors may contain large cells that may bear a striking resemblance to ganglion cells. Most C-cell carcinoma cells and some C-cell adenoma cells express somatostatin in addition to calcitonin. Amyloid deposits may be present between tumor cells, around blood vessels, and in the interstitium.

Spontaneous C-Cell Lesions
C-cell tumors occur spontaneously in many rat strains. There is a striking correlation between the age of the animals and the extent of C-cell abnormalities. For example, approximately 80% of Long–Evans rats between 12 and 24 months of age have extensive diffuse and/or nodular C-cell hyperplasia, with C-cell adenomas or carcinomas occurring in 14%. By the age of 24–36 months, approximately 25% of the rats have evidence of C-cell adenomas or carcinomas. Rats with C-cell tumors also commonly have adrenal medullary hyperplasia, pheochromocytomas, and pituitary adenomas. In contrast to the high frequency of spontaneous C-cell adenomas and carcinomas in the rat, these tumors are rare in the mouse.
Mechanisms of Toxicity
Radiation administered to newborn rats is followed by a striking reduction in the numbers of C cells. The numbers of C cells increase after 1 year, but their distribution within the gland is often patchy, suggesting a clonal regrowth. Further studies have demonstrated that the incidence of C-cell tumors is increased after irradiation, but high levels of Ca2+ in drinking water do not appear to potentiate the carcinogenic effects of irradiation in C cells. However, it has been noted that a significant increase in C-cell tumor incidence has been associated with excess Vitamin D3 in the diet. These findings suggest that vitamin D or one of its metabolites may affect C-cell growth directly.
The administration of the glucagon-like peptide-1 (GLP-1) analogs, such as liraglutide, results in calcitonin release, upregulation of calcitonin gene expression, and C-cell proliferative lesions in rats and, to a lesser extent, in mice. In long-term rodent studies (104 weeks), C-cell tumors increased in a dose-dependent manner. In contrast to rodents, this effect does not appear to happen in humans and/or monkeys. Other pharmaceutical agents, such as the parathyroid hormone (PTH) analog teriparatide, have been associated with the development of C-cell tumors in rodents.
Part 5: Thyroid Follicular Cells
Structure and Function
Organogenesis
The thyroid gland originates as a thickened plate of epithelium in the floor of the pharynx. It is intimately related to the aortic sac in its development, and this association leads to the frequent occurrence of accessory thyroid parenchyma in the mediastinum. These nodules are usually 1–2 mm in size and may number from one to five. They are completely lacking in C cells, but their follicular structure and function are the same as that of the main thyroid lobes. This accessory thyroid tissue may undergo neoplastic transformation as, for example, in the adult dog.
Thyroid Histology
The thyroid gland is the largest of the organs that function exclusively as endocrine glands. The basic structure of the thyroid is unique for endocrine glands, consisting of follicles of varying size (20–250 μm) that contain colloid produced by follicular cells. Follicular cells are cuboidal to columnar, and their secretory polarity is directed toward the lumen of the follicles. An extensive network of inter- and intrafollicular capillaries provides the follicular cells with an abundant blood supply. Follicular cells have long profiles of rough endoplasmic reticulum and a large Golgi apparatus in their cytoplasm for the synthesis and packaging of substantial amounts of protein that are transported into the follicular lumen. The interface between the lumenal side of follicular cells and the colloid is modified by numerous microvillar projections.
Thyroid Hormone Synthesis
The biosynthesis of thyroid hormones is also unique among endocrine glands because the final assembly of the hormones occurs extracellularly within the follicular lumen. Essential raw materials, such as iodide, are trapped efficiently by follicular cells from plasma, transported rapidly against a concentration gradient to the lumen, and oxidized by a thyroid peroxidase in microvillar membranes and colloid to iodine (I2) (Figure 20.28). The assembly of thyroid hormones within the follicular lumen is made possible by a unique protein (thyroglobulin) synthesized by follicular cells.
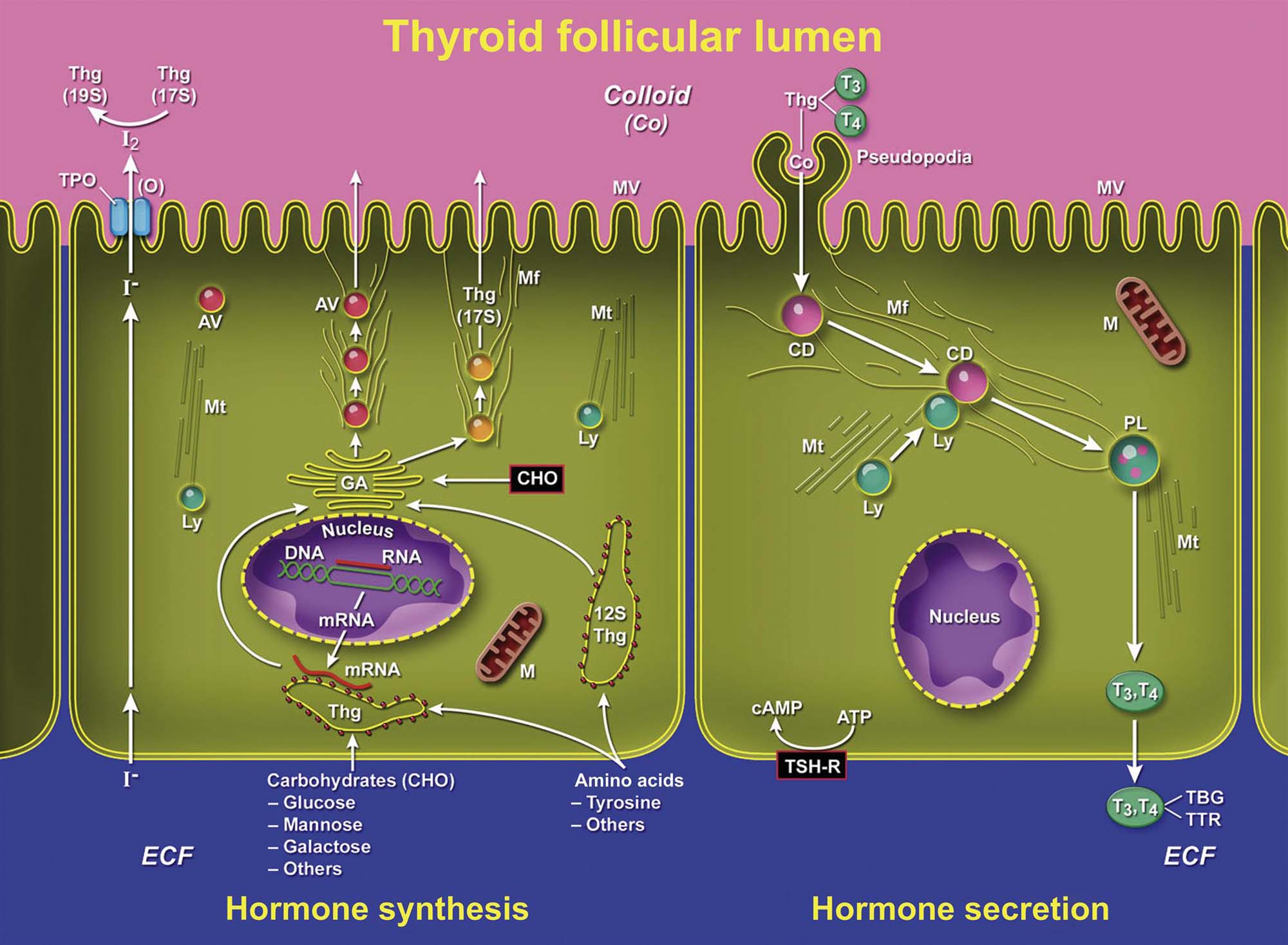
Thyroglobulin is a high molecular weight (600,000–750,000) glycoprotein synthesized in successive subunits on ribosomes of the endoplasmic reticulum in follicular cells. The constituent AA (tyrosine and others) and carbohydrates (i.e., mannose, fructose, and galactose) come from the circulation. Recently synthesized thyroglobulin (17S) leaving the Golgi apparatus is packaged into apical vesicles and extruded into the follicular lumen. The AA tyrosine, an essential component of thyroid hormones, is incorporated in the molecular structure of thyroglobulin. Iodine is bound to tyrosyl residues in thyroglobulin at the apical surface of follicular cells to form, successively, monoiodotyrosine (MIT) and diiodotyrosine (DIT) (Figure 20.29). The resulting MIT and DIT combine to form the two biologically active iodothyronines—T4 and T3—secreted by the thyroid gland.
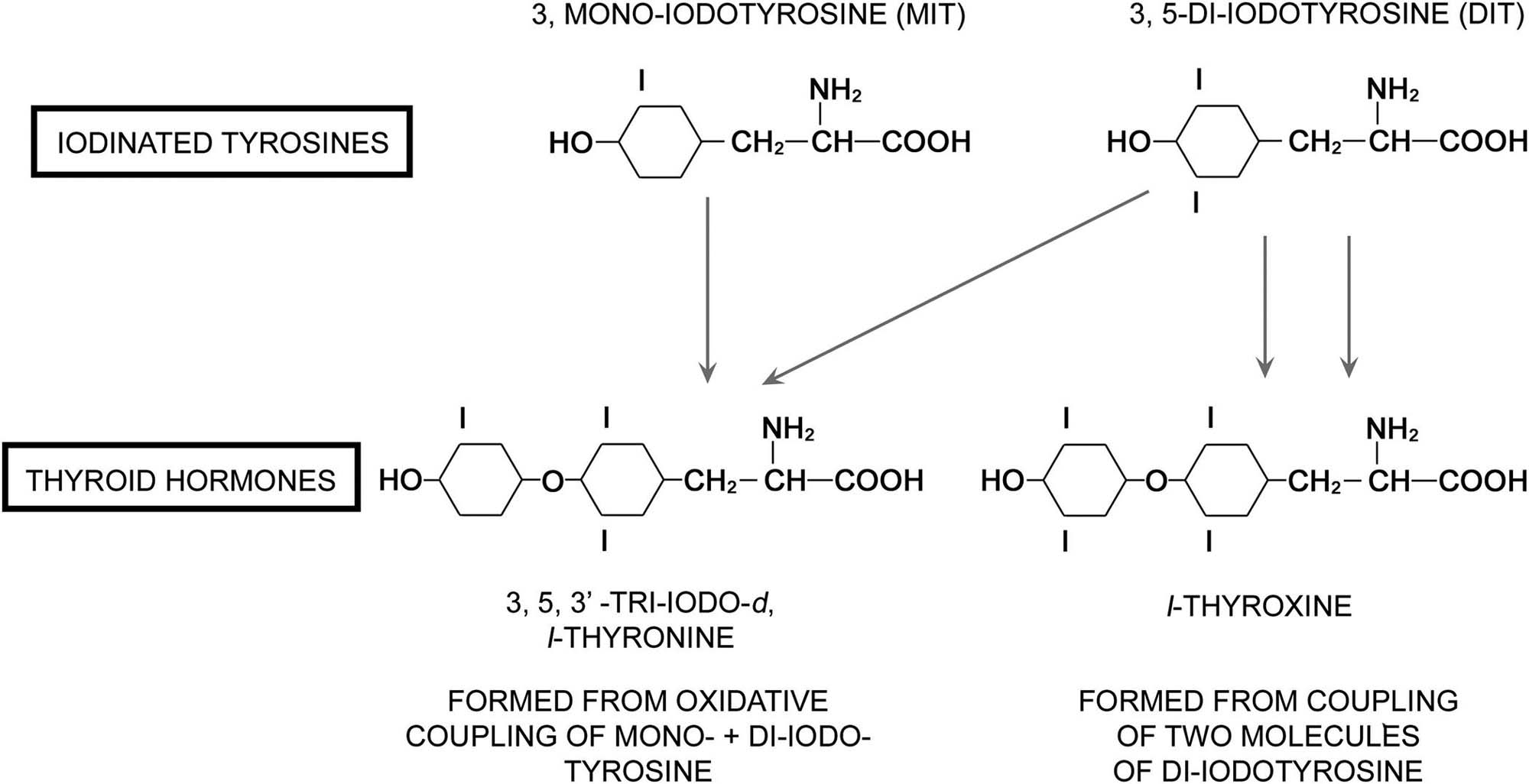
The mechanism of active transport of iodide is associated with a sodium iodide (Na+I−) symporter (NIS). The transport of iodide ion across the thyroid cell membrane is linked to the transport of Na+. The ion gradient generated by the Na+/K+-ATPase appears to be the driving force for the active cotransport of iodide. The transporter protein is present in the basolateral membrane of thyroid follicular cells (thyrocytes). Other tissues, such as the salivary gland, gastric mucosa, and lactating mammary gland, also have the capacity to actively transport iodide, albeit at a much lower level than the thyroid. NIS gene expression in the thyroid is upregulated by TSH. The functionally active iodine transport system has important toxicological implications. Radioiodine is used clinically to ablate residual tumor tissues as well as recurrent and metastatic thyroid cancer. Chemicals such as perchlorate and thiocyanate can selectively inhibit the NIS and active transport of iodide, thereby effectively blocking the ability of the gland to synthesize thyroid hormones.
The plasma T4 half-life in rats is considerably shorter (12–24 hours) than in man (5–9 days). In humans and monkeys, circulating T4 is bound primarily to T4-binding globulin (TBG), but this high-affinity binding protein is not present in rodents, birds, amphibians, or fish (Table 20.4).
Table 20.4
Thyroxine (T4) Binding to Serum Proteins in Selected Vertebrate Speciesa
| Species | T4-Binding globulin | Postalbumin | Albumin | Prealbumin (transthyretin) |
| Human | ++ | − | ++ | + |
| Monkey | ++ | − | ++ | + |
| Dog | + | − | ++ | − |
| Mouse | − | ++ | ++ | − |
| Rat | − | + | ++ | + |
| Chicken | − | − | ++ | − |

aDegree of T4 binding to serum proteins: + or ++. Absence of binding of T4 to serum protein: −.
Table modified from Döhler et al. (1979) The rat as model for the study of drug effects on thyroid function: consideration of methodological problems, Pharmacol. Ther. 5: 305–318.
The binding affinity of TBG for T4 is approximately 1000 times higher than for transthyretin (prealbumin). The percentage of unbound active T4 is lower in species with high levels of TBG than in animals in which T4 binding is limited to albumin, transthyretin, and postalbumin. Therefore, a rat without a functional thyroid requires about 10 times more T4 (20 μg/kg body weight) for full substitution than an adult human (2.2 μg/kg body weight). T3 is transported bound both to TBG and albumin in human, monkey, and dog but only to albumin in mouse, rat, and chicken (Table 20.5). In general, T3 is bound less avidly to transport proteins than T4, resulting in a faster turnover and shorter plasma half-life in most species. These differences in plasma half-life of thyroid hormones and binding to transport proteins between rats and humans may be one factor in the greater sensitivity of the rat thyroid to develop hyperplastic and/or neoplastic nodules in response to chronic TSH stimulation.
Table 20.5
Triiodothyronine (T3) Binding to Serum Proteins in Selected Vertebrate Speciesa
| Species | T3-Binding globulin | Postalbumin | Albumin | Prealbumin (transthyretin) |
| Human | + | − | + | − |
| Monkey | + | − | + | − |
| Dog | + | − | + | − |
| Mouse | − | + | + | − |
| Rat | − | − | + | − |
| Chicken | − | − | + | − |

aDegree of T3 binding to serum proteins: + or ++. Absence of binding of T3 to serum protein: −.
Table modified from Döhler et al. (1979) The rat as model for the study of drug effects on thyroid function: consideration of methodological problems, Pharmacol. Ther. 5: 305–318.
Thyroid Hormone Secretion
The elongation of microvilli and the formation of pseudopods on follicular cells initiate the secretion of thyroid hormones from stores within lumenal colloid. The pseudopods are increased by pituitary TSH, and they phagocytize a portion of adjacent colloid by endocytosis. Colloid droplets within follicular cells fuse with numerous lysosomal bodies that contain proteolytic enzymes and release T4 and T3, which diffuse out of the follicular cells and enter interfollicular capillaries. Iodinated tyrosines (MIT and DIT) released from the colloid droplets are deiodinated enzymatically, and the iodide generated under normal conditions either is recycled to the lumen to iodinate new tyrosyl residues in thyroglobulin or is released into the circulation.
Negative feedback control of thyroid hormone secretion is accomplished by the coordinated response of the adenohypophysis and certain hypothalamic nuclei to circulating and local tissue levels of T3. In the rat, 50% or more of the pituitary content of T3 is generated locally from circulating T4 by a 5′-deiodinase (type II). A decrease in thyroid hormone concentration in plasma is sensed by groups of neurosecretory neurons in the hypothalamus that synthesize and secrete TRH into the hypophyseal portal circulation. TRH binds to receptors on the plasma membrane of thyrotrophic basophils in the adenohypophysis and activates adenylyl cyclase. Intracellular accumulation of cyclic AMP in thyrotrophic basophils leads to release of TSH-containing secretory granules into pituitary capillaries.
TSH binds to the basilar aspect of the thyroid follicular cell, activates adenylyl cyclase, and increases the rate of synthesis and secretion of thyroid hormones. One of the initial responses by follicular cells to TSH is the formation of cytoplasmic pseudopodia (see above) resulting in the release of preformed hormone stored within the follicular lumen. If secretion of TSH is sustained (hours or days), thyroid follicular cells become more columnar and follicular lumens become smaller. Numerous PAS-positive colloid droplets are present in the lumenal aspect of the hypertrophied follicular cells.
Conversely, in response to an increase in circulating levels of thyroid hormones (T4 and T3), there is a corresponding decrease in circulating pituitary TSH. Thyroid follicles become enlarged and distended with colloid due to the decreased TSH-mediated endocytosis. Follicular cells lining the involuted follicles are low cuboidal and have few endocytic vacuoles at the interface with the colloid.
Degradation and Metabolism
Thyroid hormones are degraded primarily by conjugation in the liver. T4 is conjugated on the outer phenolic ring with glucuronic acid in a reaction catalyzed by T4 UDP glucuronosyltransferase (UGT), and conjugated T4 is excreted in the bile. T3 is conjugated, also on the outer ring, with sulfate in a reaction catalyzed by phenol sulfotransferase. Deamination and decarboxylation of the side chain at the other end of the thyroid hormone molecule and cleavage of the ether linkage are relatively minor pathways of degradation. A wide variety of drugs and chemicals can influence thyroid hormone metabolism by inducing one or more classes of hepatic microsomal enzymes that increase the degradation of thyroid hormones.
The stepwise monodeiodination of T4 in the liver, kidney, and elsewhere is also important in metabolism of thyroid hormones. The removal of an iodine molecule from the 5 position on the outer phenolic ring by 5′-deiodinase results in the formation of biologically active T3 (3,5,3′-triiodothyronine; Figure 20.30). However, if a molecule of iodine is removed from the 5 position of the inner phenolic ring of T4 by another enzyme, 5-deiodinase, reverse T3 (3,3′,5′-triiodothyronine) is produced, which is biologically inactive. Reverse T3 subsequently is degraded further by 5′-deiodinase and T3 by 5-deiodinase; both form DIT (T2). Chemicals such as FD&C Red No. 3 (erythrosine) and amiodarone disrupt thyroid hormone metabolism by inhibiting the 5′-deiodinase in liver (type I enzyme), thereby inhibiting the conversion of T4 to T3.
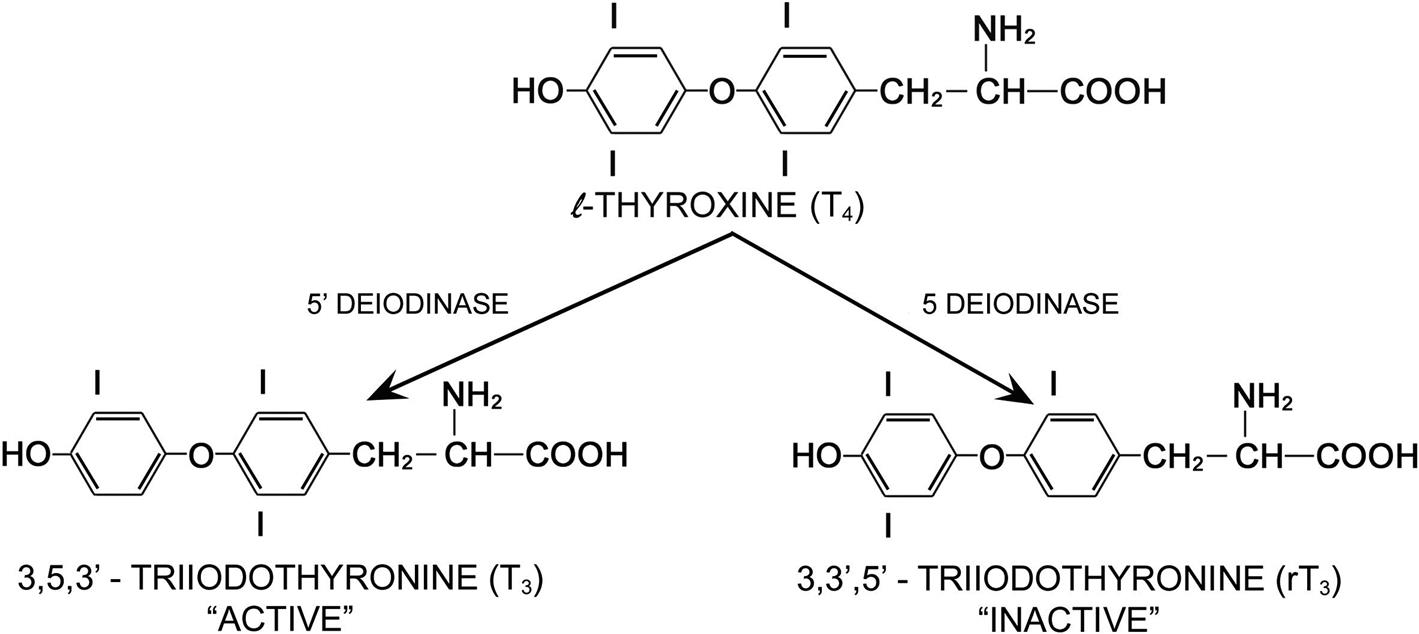
In dog plasma, T4 is bound to albumin and three globulin fractions, and T3 is bound to albumin and one globulin fraction. The overall binding affinity of the plasma proteins for T4 is lower in dogs than in humans. Most importantly, the affinity of the canine inter-α-globulin fraction for T4 is much less than that of the human TBG. As a result of this weaker binding, as well as an efficient enterohepatic excretory mechanism, the total T4 concentration is lower and the rate of thyroid hormone turnover is more rapid in dogs than in humans. Peak concentration usually occurs at about midday and the minimum at about midnight. Both the overall rates of turnover and the loss of hormone in the feces are much higher in dogs than in humans. Fecal wastage substantially reduces the efficiency of hormone utilization but also explains the remarkable tolerance of the dog for excess thyroid hormone.
Biological Action
T4 and T3, once released into the circulation, act on many different target cells in the body. The overall functions of T4 and T3 are similar, although much of the biological activity is the result of monodeiodination of T4 to T3 (3,5,3′-triiodothyronine) prior to interaction with target cells or by the target cells. Under certain conditions (protein starvation, neonatal animals, liver and kidney disease, and febrile illness) T4 is preferentially monodeiodinated to 3,3′,5′-triiodothyronine (reverse T3) (Figure 20.30). Because this form of T3 is biologically inactive, monodeiodination to reverse T3 provides a mechanism to attenuate the metabolic effects of thyroid hormones. T4 stimulates oxygen utilization and heat production by many different populations of body cells. It causes increased utilization of carbohydrates, increased protein catabolism, and greater oxidation of fats. The administration of T4 increases heart rate by a direct effect on heart muscle cells.
Normal function of the central nervous system (CNS) is dependent on T4. During periods of deficient T4 levels, the CNS fails to function normally and the animal is lethargic, dull, and mentally deficient. Myelin in fiber tracts is decreased, cortical neurons are smaller and fewer, and vascularity of the CNS is reduced. Neurons are permanently damaged by a deficiency of thyroid hormones in a young growing animal fed a severely deficient iodine diet. However, an excess of thyroid hormone production, as occurs in a hyperthyroid animal or human, stimulates CNS activity to the extent that the animal is nervous, irritable, and hyperactive.
The subcellular mechanism of action of thyroid hormones resembles that of steroid hormones because free hormone enters into target cells and binds to a cytosol-binding protein. Free T3 either binds to receptors on the inner mitochondrial membrane to activate mitochondrial energy metabolism, or binds to nuclear receptors and increases transcription of mRNA to facilitate new protein synthesis. The overall effects of thyroid hormones are to (1) increase the basal metabolic rate; (2) increase glycolysis, gluconeogenesis, and glucose absorption from the intestine; (3) stimulate new protein synthesis; (4) increase lipid metabolism and conversion of cholesterol into bile acids and other substances, activate lipoprotein lipase, and increase the sensitivity of adipose tissue to lipolysis by other hormones; (5) stimulate heart rate, cardiac output, and blood flow; and (6) increase neural transmission, cerebration, and neuronal development in young animals.
Evaluation of Toxicity
Thyroid Function Tests
Thyroid Hormones
The most sensitive and accurate method for measuring blood T4 and T3 levels is radioimmunoassay (RIA). The normal blood level of T4 in the dog ranges between 1.5 and 3.6 μg/dL (mean: 2.5 μg/dL) and T3 ranges between 48 and 154 ng/dL (mean: 95 ng/dL). In dogs with hypothyroidism, the T4 level is usually below 1.0 μg/dL and T3 is below 50 ng/dL. When the levels are border-line, clearer separation of animals with hypothyroidism from those that are euthyroid can be made by injecting exogenous TSH. In the euthyroid dog, the T4 level will at least double 8 hours after iv or im administration of TSH. In animals with hypothyroidism, the T4 and T3 levels do not change significantly after the injection of TSH. Serum T4 in the rat is higher than in the dog with a mean of approximately 4.5±0.2 μg/dL. Serum T3 in rats is more variable than circulating T4 levels with a range of 50–100 ng/dL, with T3 values tending to decrease with advancing age. The serum reverse T3 level in the normal rat usually ranges between 55 and 75 pg/mL but will increase dramatically and remain elevated following the administration of xenobiotics that inhibit the 5′-deiodinase. The range of values given for serum T4, T3, and reverse T3 should be considered only as a general guideline. A specific range of values for rats of different ages, strains, and sexes should be established in conjunction with the endocrine laboratory performing the assays on serum from a particular experiment. Whenever possible, samples at different intervals from an experiment should be frozen and all assays performed at a single run at the termination of the experiment to minimize interassay variations. Serum collection should be performed during the morning hours, if possible, and rats from control and experimental groups alternated during the collection interval.
Thyroid-Stimulating Hormone
In the evaluation of potential thyroid toxicity of various xenobiotics in rodents, an accurate quantitation of circulating levels of TSH is essential in order to determine whether proliferative lesions of follicular cells are mediated by a chronic hypersecretion of TSH. The immunoassay for TSH is highly species-specific, with considerable interanimal and interassay variation.
Xenobiotics that disrupt thyroid hormone synthesis, secretion, or peripheral metabolism often result in prompt increases in circulating TSH levels. It is important in the design of experiments to evaluate the effect of a xenobiotic on thyroid function to have several early collection intervals. TSH levels are higher in male than in female rats and castration decreases both the baseline serum TSH and the response to TRH injection. Commercial TSH assays are available to measure canine TSH and include immunoradiometric, luminescence, or enzyme-linked immunosorbent assay (ELISA) formats. The β subunit of canine TSH has an 89% homology with the human subunit, and circulating TSH levels in dogs are 10- to 20-fold lower than those in humans. The canine TSH assays have a low diagnostic sensitivity since some hypothyroid dogs do not have increased serum TSH concentrations.
Morphological and Morphometric Evaluation of Thyroid Follicles
Morphological and morphometric evaluations of the follicular cells are sensitive indicators of potential toxic effects of xenobiotics on the thyroid gland. Fixation of the thyroid lobes attached to the adjacent trachea in buffered formalin is the best procedure for routine studies. The combined weight of the right and left thyroid lobes can be expressed as a ratio to total body or brain weight. Changes in thyroid weight are a sensitive overall indicator of the effects of relatively mild goitrogenic chemicals on thyroid structure. Immunohistochemical procedures are particularly helpful with selected proliferative lesions to determine whether they are of follicular cell (thyroglobulin-positive) or C-cell (calcitonin-positive) origin.
Morphometric evaluation of thyroids is a valuable adjunct to histopathological evaluation and determination of thyroid weights to detect and quantitate subtle changes in follicular cells. This is particularly true for rats, where the histology of the normal thyroid (tall columnar follicular cells lining mainly small follicles containing small amounts of lightly eosinophilic colloid) reflects the high baseline TSH levels. Follicle diameter, colloid area, and follicular cell height can be measured from representative follicles in H&E-stained sections using image analysis software. Large, colloid-distended, inactive follicles at the periphery of the lobes are avoided.
Histological examination of a biopsy of the thyroid is a useful and reliable aid in the diagnosis of thyroid disease in larger animals when either the results of serum assays for T4 and T3 are equivocal or a nodule is palpated in the thyroid area.
Animals as Models
Much of the significant physiological and pathological data in the literature on thyroid function and structure has come from studies in animals. Although the basic hypothalamic–pituitary–thyroid axis functions in a similar manner in animals and humans, there are differences between species that are important when extrapolating animal data from toxicity and carcinogenicity studies for human risk assessment. Several of these differences have been summarized previously in this section on thyroid follicular cells.
Long-term perturbations of the pituitary–thyroid axis by various xenobiotics or physiological alterations (e.g., iodine deficiency, partial thyroidectomy) are more likely to predispose the laboratory rat to a higher incidence of proliferative lesions in the thyroid (e.g., hyperplasia and adenomas of follicular cells) than is seen in the human thyroid. This appears to be particularly true in the male rat, in which there is higher circulating TSH than in the female.
Response to Injury
Thyroid responses to injury histologically are primarily proliferative in nature, but nonproliferative lesions reflecting disorders of thyroid function also occur.
Species Differences in Response
Long-term perturbations of the pituitary–thyroid axis by xenobiotics or physiologic alterations (e.g., iodine deficiency, partial thyroidectomy, and natural goitrogens in food) are more likely to induce a higher incidence of proliferative lesions (e.g., hyperplasia and adenomas of follicular cells) in the rat, as a response to chronic TSH stimulation, than in the human. This is particularly true in the male rat, which has higher circulating levels of TSH than females. The greater sensitivity of the rodent thyroid to derangement by drugs, chemicals, and physiologic perturbations is also related to the shorter plasma half-life of T4 than in humans due to the considerable differences between these species in the transport proteins for thyroid hormones.
Histopathological Criteria of Proliferative Lesions
Uniform terminology and more specific criteria for the various proliferative lesions of thyroid follicles, including representative images of common lesions, developed by the INHAND in Rats and Mice project, will be available at the time of publication of this chapter on the Society of Toxicologic Pathology website (http://www.toxpath.org/inhand.asp). Diffuse thyroid enlargement with or without hyperplasia (i.e., “goiter”) will be discussed in a later section.
Follicular Cell Hyperplasia
Follicular “cystic” hyperplasia is characterized by single or multiple nodules in the thyroid formed by the coalescence of adjacent colloid-distended follicles. The follicular wall (or remnants) is lined by one or two layers of low-cuboidal epithelium with hyperchromatic nuclei that occasionally form papillary projections. Adjacent thyroid follicles are normal, with minimal evidence of compression and no encapsulation by fibrous connective tissue.
Focal (“nodular”) or multifocal areas of hyperplasia of thyroid follicular cells usually only enlarge the affected lobe(s) slightly. The areas of focal hyperplasia are not encapsulated and the adjacent thyroid parenchyma is not compressed. Hyperplastic nodules are composed of irregularly shaped, usually colloid-filled, follicles lined by more basophilic cuboidal follicular cells. The multiple nodules of follicular cell hyperplasia may coalesce to form macroscopically observable thyroid adenomas. Focal hyperplasia is different than the diffuse (TSH-mediated) type of follicular cell hyperplasia observed with iodine deficiency and with a wide variety of xenobiotic drugs and chemicals that disrupt thyroid hormone “economy” to result in increased secretion of TSH.
Follicular Cell Adenoma
An adenoma is a well-circumscribed, usually solitary, benign tumor formed by the autonomous proliferation of follicular cells within the thyroid lobe. Adjacent thyroid follicles are compressed to varying degrees, depending on the size of the adenoma. Adenomas usually do not have a complete fibrous capsule, but larger adenomas may be partially encapsulated by a thin layer of fibrous connective tissue. Neoplastic cells often are more hyperchromatic than the surrounding normal follicular cells and form variably sized colloid-containing follicles, line large cystic spaces, or solid sheets. Neoplastic cells may form papillary structures extending into larger cysts. Neoplastic cells vary from cuboidal to low columnar, and the nucleus:cytoplasm ratio often is high. There is no evidence of vascular invasion. Endocrinologically active thyroid adenomas result in the colloid involution of follicles in the rim of the surrounding thyroid due to the inhibition of TSH secretion by elevated blood T4 and T3 levels.
Follicular Cell Carcinoma
Thyroid carcinomas show evidence of intrathyroidal, capsular, or vascular invasion that often enlarges the affected thyroid lobe considerably (Figure 20.31). Malignant follicular cells form colloid-containing follicles of varying size, papillary projections extending into cystic spaces, or solid sheets of cells. The cells are pleomorphic, and nuclei are hyperchromatic with occasional mitotic figures. A desmoplastic reaction often accompanies the infiltration of neoplastic cells into the thyroid capsule. The perithyroidal connective tissue, as well as adjacent trachea and cervical muscles, may be invaded by larger carcinomas.
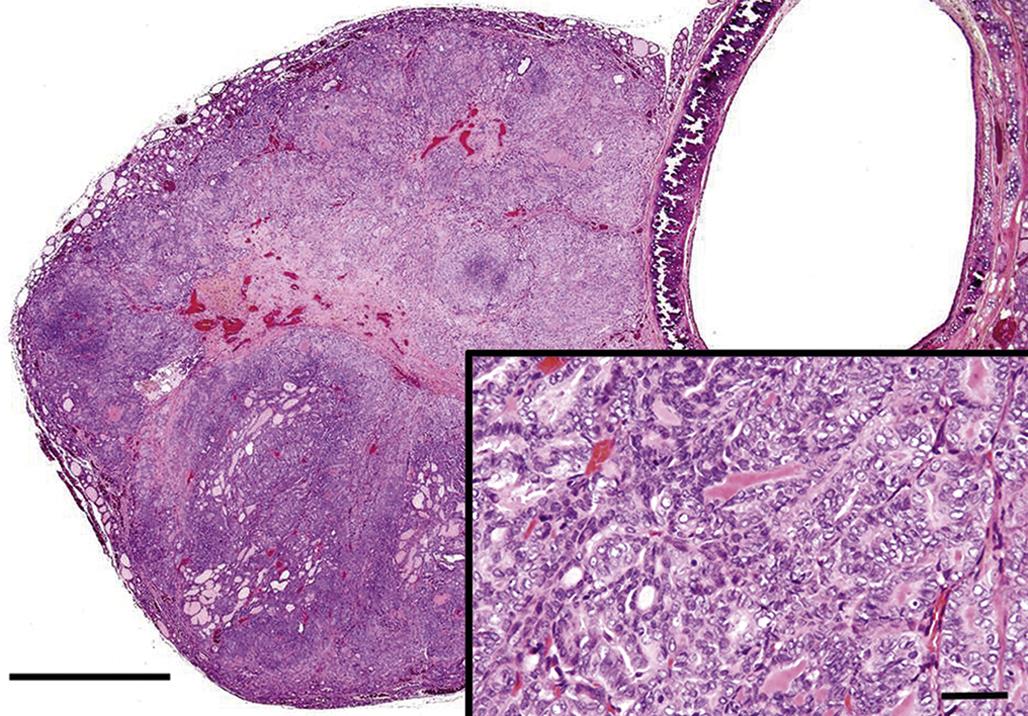
Disorders of Thyroid Function
Hypothyroidism
Hypothyroidism is a well-recognized clinical entity in dogs, and occurs in many purebred and mixed breed dogs. Subnormal function of the thyroid is recognized less frequently as a naturally occurring entity in other animal species. However, a large number of drugs, chemicals, dietary deficiencies, or excesses can disrupt one or more steps of thyroid hormone biosynthesis, secretion, metabolism, or degradation and result in subnormal blood levels of thyroid hormones. Hypothyroidism in dogs is often the result of primary diseases of the thyroid gland, especially idiopathic follicular atrophy (“follicular collapse”) and immune-mediated lymphocytic thyroiditis. Hypertrophy and hyperplasia of TSH-secreting basophils occur in the pars distalis of dogs with hypothyroidism.
Hypothyroidism may also develop secondary to longstanding pituitary or hypothalamic lesions that prevent the release of either TSH or TRH. The thyroid gland is moderately reduced in size and is composed of colloid-distended follicles lined by flattened follicular cells due to a lack of TSH-induced endocytosis of colloid and secretion of thyroid hormones. Somatostatin inhibits both GH and TSH secretion in the pars distalis of the pituitary gland. Long-acting somatostatin analogs, such as octreotide, have the potential to reduce TSH and serum thyroid hormone concentrations.
Many functional disturbances associated with hypothyroidism are due to a reduction in the basal metabolic rate. A gain in body weight without an associated change in appetite occurs frequently. The rate of lipid metabolism decreases in longstanding cases of hypothyroidism, and blood lipid levels often are elevated because of a decreased rate of intestinal cholesterol excretion and a decreased rate of hepatic conversion into bile acids and other compounds. The elevated blood cholesterol level predisposes to the development of atherosclerosis of small vessels (especially coronary arteries) and deposition of lipid in the liver. The cardiovascular system is further affected by the reduced level of thyroid hormones due to a slowing of the heart rate. The moderate slowing in the heart rate may be difficult to detect unless a normal resting heart rate has been measured previously.
The accumulation of excessive lipid in the liver often results in varying degrees of hepatomegaly with abdominal distension in animals with long-term hypothyroidism. Renal glomeruli may become plugged with lipid in hypothyroid animals with markedly elevated plasma cholesterol, resulting in progressive renal failure.
Abnormalities in reproduction are common when a breeding animal develops hypothyroidism. Lack of libido and reduction in sperm count may occur in males, whereas abnormal or absent estrus cycles with reduced conception rates often result in females. Obesity and changes in behavior resulting from hypothyroidism may have detrimental effects on reproduction.
Hyperthyroidism
Disturbances of growth in the thyroid gland, in which there is secretion of excess thyroid hormones, resulting in hyperthyroidism, are uncommon in animals except in adult to aged cats. Follicular cell adenomas, often developing in a thyroid with multinodular hyperplasia, are encountered more commonly than malignant thyroid tumors. A syndrome of hyperthyroidism has been recognized with increasing frequency since the early 1980s in aged cats associated with multinodular follicular cell hyperplasia, adenomas, or adenocarcinomas. Possible factors in the increasing incidence of functional thyroid lesions in cats include exposure to lawn herbicides or fertilizers, regular treatment with flea sprays or powders, and a canned food diet.
Neoplastic cells release both T4 and T3 at an uncontrolled rate, resulting in markedly elevated blood levels of both hormones. Follicles in the rim of the thyroid around a functional adenoma are enlarged markedly and distended by the accumulation of colloid. Follicular cells are low cuboidal and atrophic, with little endocytotic activity in response to the elevated levels of thyroid hormones.
The most common disturbance of function in hyperthyroidism is weight loss, despite a normal or increased appetite. Other clinical signs include polydipsia and polyuria, increased frequency of defecation, increased volume of stools, restlessness, and increased physical activity.
In contrast to cats, thyroid tumors in the dog only occasionally secrete sufficient thyroid hormone to produce clinical signs of hyperthyroidism. Dogs have a very efficient enterohepatic excretory mechanism for thyroid hormones that it is difficult to overload, either from endogenous production by a tumor or exogenous administration of hormone.
Therefore, the likelihood of developing clinical hyperthyroidism associated with thyroid neoplasms in animals depends on the capability of tumor cells to synthesize T4 and T3 (e.g., well-differentiated thyroid tumors that form follicles and produce colloid are more likely to synthesize thyroid hormones than poorly differentiated solid neoplasms), and the degree of elevation of circulating levels of T4 and T3, which depends on a balance between the rate of secretion of thyroid hormones by the tumor and the rate of degradation of thyroid hormones. Cats are very sensitive to phenol and phenol derivatives (such as T4). They have a poor ability to conjugate phenolic compounds with glucuronic acid and hence to excrete T4-glucuronide into the bile. The capacity to conjugate T3 to sulfate, an alternative pathway, is limited in all species and easily overloaded.
Diffuse Hyperplasia of Follicular Cells (“Goiter”)
Mechanisms of Goitrogenesis
Nonneoplastic and noninflammatory enlargement of the thyroid (“goiter”) develops in all laboratory animals, domestic mammals, birds, and other nonmammalian vertebrates. Certain forms of thyroid hyperplasia, especially nodular, may be difficult to differentiate from benign tumors (adenomas). The major pathogenic mechanisms responsible for the development of thyroid hyperplasia include iodine-deficient diets, goitrogenic compounds that interfere with thyroid hormone synthesis, dietary iodide excess, and genetic enzyme defects in the biosynthesis of thyroid hormones (Figure 20.32). All of these seemingly divergent factors result in inadequate T4 and T3 synthesis and decreased blood levels of thyroid hormones. The lowered blood levels are detected by the hypothalamus and pituitary to increase the secretion of TSH, which results in hypertrophy and hyperplasia of follicular cells in the thyroid gland.
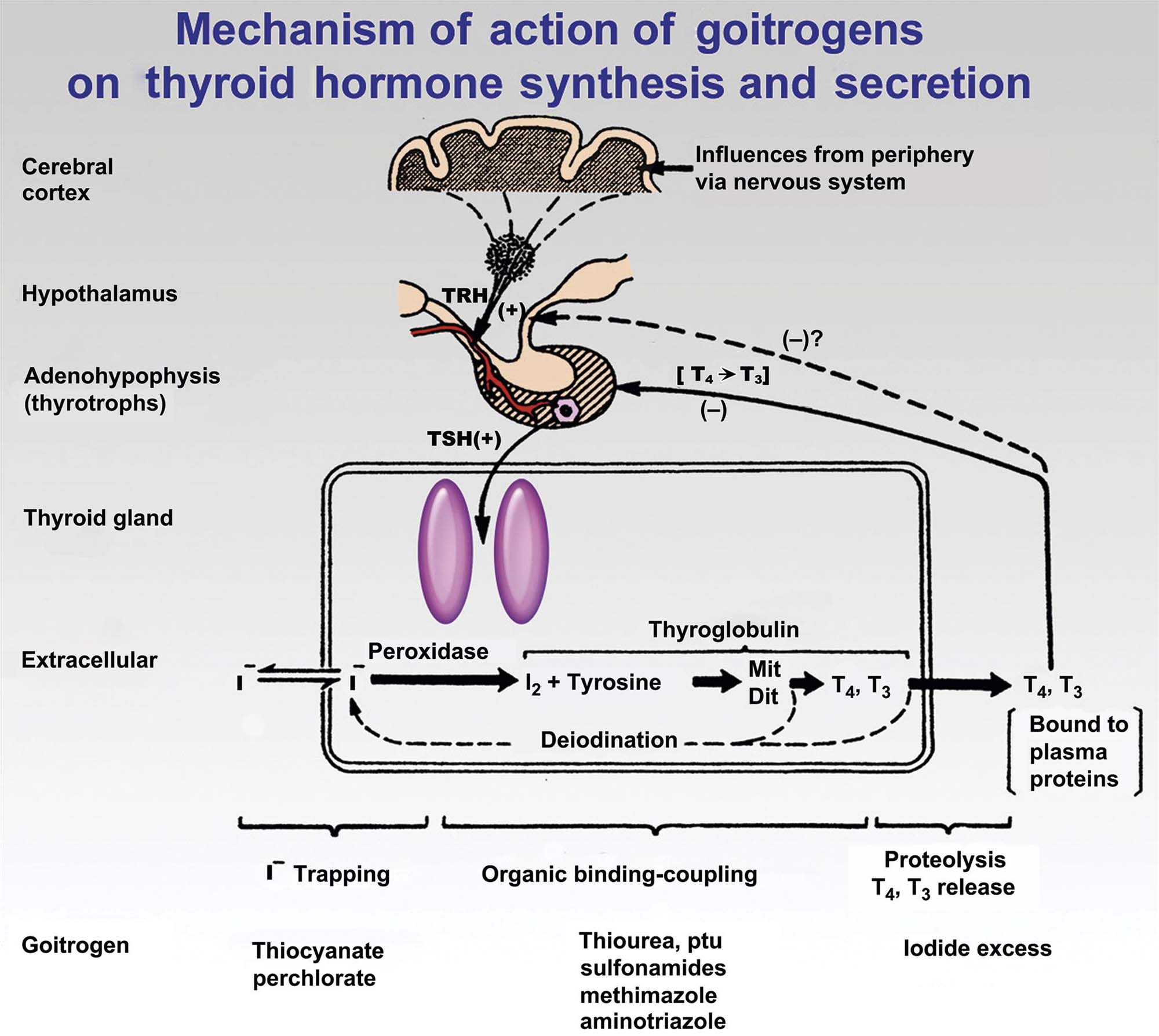
Diffuse Hyperplastic (Iodine-Deficient) Goiter
Dietary iodine deficiency leading to diffuse thyroid hyperplasia is still common in many areas throughout the world, although the widespread addition of iodized salt to animal and human diets has decreased deficiency substantially in recent decades. More often, marginal iodine-deficient diets containing certain goitrogenic compounds result in thyroid follicular cell hyperplasia and clinical evidence of goiter with hypothyroidism. These goitrogenic substances include thiouracil, sulfonamides, anions of the Hofmeister series, and a number of plants from the genus Brassica.
Both lateral lobes of the thyroid are enlarged uniformly in animals with diffuse hyperplastic goiter. The enlargements may be extensive and result in palpable swelling in the cranial cervical area. The affected lobes are firm and dark red because an extensive interfollicular capillary network develops under the influence of long-term TSH stimulation. The thyroid enlargements are the result of intense hypertrophy and hyperplasia of follicular cells, often with the formation of papillary projections into the lumens of follicles or multiple layers of cells lining follicles. Endocytosis of colloid usually proceeds at a rate greater than synthesis, resulting in the progressive depletion of colloid.
Under longstanding TSH stimulation, thyroid follicles become smaller than normal and there may be a partial collapse of follicles due to the lack of colloid. Hypertrophic-lining follicular cells are columnar with a deeply eosinophilic cytoplasm and small hyperchromatic nuclei that often are situated in the basilar part of the cell. The follicles are lined by either single or multiple layers of hyperplastic follicular cells that, in some follicles, form papillary projections into the lumen. Similar proliferative changes are present in ectopic thyroid parenchyma in the neck and mediastinum.
Colloid Goiter
Colloid goiter, or diffuse thyroid follicular dilatation, represents the involutional phase of diffuse hyperplastic goiter in young adults and reflects postproliferation atrophy of follicular cells with persistence of colloid already produced. The markedly hyperplastic follicular cells continue to produce colloid, but the endocytosis of colloid is decreased due to diminished pituitary TSH secretion in response to the return of blood T4 and T3 to the normal range. Colloid goiter may develop either after sufficient amounts of iodide have been added to the diet or after the requirements for T4 have diminished in an older animal. Both thyroid lobes are enlarged diffusely but are more translucent and lighter in color than with hyperplastic goiter. The differences in macroscopic appearances are the result of less vascularity in colloid goiter and the development of macrofollicles distended with colloid. Follicular cells lining the macrofollicles are flattened and atrophic, and the interface between the colloid and the lumenal surface of follicular cells is smooth.
Iodide-Excess Goiter
Although seemingly paradoxical, an excess of iodide in the diet also can result in thyroid hyperplasia in animals and humans. The thyroid glands of the young are exposed to higher blood iodide levels than those of the dam because the placenta concentrates iodide prior to birth and subsequently, after birth, the mammary gland also concentrates iodide. High blood iodide interferes with one or more steps of thyroxinogenesis, leading to lowered blood thyroid hormone levels and a compensatory increase in pituitary TSH secretion. Excess iodide appears primarily to block the release of T3 and T4 from thyroglobulin by interfering with the proteolysis of colloid by lysosomes, but it also interferes with the peroxidation of 2 I− to I2 and disrupts the conversion of MIT to DIT.
Nodular Goiter
Multinodular hyperplasia (“goiter”) in thyroid glands of old animals appears as white-to-tan nodules of varying size in one or both lobes. The affected lobes are moderately enlarged and irregular in contour. Nodular goiter in most animals (except cats) is endocrinologically inactive and encountered as an incidental lesion at necropsy.
Nodular goiter consists of multiple foci of hyperplastic follicular cells that are sharply demarcated from the adjacent thyroid parenchyma, but are not encapsulated, and there is minimal compression of adjacent parenchyma. Some hyperplastic cells form small follicles with little or no colloid. Other nodules are formed by larger irregularly shaped follicles lined by one or more layers of columnar cells that form papillary projections into the lumen. These changes appear to be the result of alternating periods of hyperplasia and colloid involution in the thyroid glands of old animals. The areas of nodular hyperplasia may be microscopic or grossly visible, causing modest enlargement of the thyroid.
Mechanisms of Toxicity
Direct Thyroid Effect
Inhibitors of Thyroid Hormone Synthesis
Blockage of Iodine Uptake
The initial step in the biosynthesis of thyroid hormones is the uptake of iodide from the circulation and subsequent transport against a gradient across follicular cells by the NIS into the lumen of the follicle. A number of anions act as competitive inhibitors of iodide transport in the thyroid, the most notable being perchlorate (ClO4−), thiocyanate (SCN−), and pertechnetate (Figure 20.32). Thiocyanate is a potent inhibitor of iodide transport and is a competitive substrate for thyroid peroxidase, but it does not appear to concentrate in the thyroid. Blockage of the iodide-trapping mechanism has a disruptive effect on the thyroid–pituitary axis similar to the effect of iodine deficiency. Blood levels of T4 and T3 decrease, resulting in a compensatory increase in the secretion of TSH by the pituitary gland. Hypertrophy and hyperplasia of follicular cells following sustained exposure result in an increased thyroid weight and the development of goiter.
Inhibition of Thyroid Peroxidase (Thyroperoxidase)
A wide variety of chemicals, drugs, and other xenobiotics affect the second step in thyroid hormone biosynthesis (Figure 20.32). Classes of chemicals that inhibit the organification of thyroglobulin include: (1) thionamides such as thiourea, thiouracil, PTU, methimazole, carbimazole, and goitrin; (2) aniline derivatives and related compounds such as sulfonamides, p-aminobenzoic acid, p-aminosalicylic acid, and amphenone; (3) substituted phenols such as resorcinol, phloroglucinol, and 2,4-dihydroxy-benzoic acid; and (4) miscellaneous inhibitors such as aminotriazole, tricyanoaminopropene, and antipyrine and its iodinated derivative, iodopyrine.
Many of these chemicals exert their action by inhibiting thyroid peroxidase, which results in a disruption both of the iodination of tyrosyl residues in thyroglobulin and of the coupling reaction of iodotyrosines (i.e., MIT and DIT) to form iodothyronines (T3 and T4). In rats, PTU has been shown to affect each step in thyroid hormone synthesis beyond iodide transport. The order of susceptibility to the inhibition by PTU is the coupling reaction (most susceptible), iodination of MIT to form DIT (moderately susceptible), and iodination of tyrosyl residues to form MIT (least susceptible). Thiourea differs from PTU and other thioamides because it does not inhibit guaiacol oxidation (the standard assay for peroxidase) and does not inactivate thyroid peroxidase in the absence of iodine. Its ability to inhibit organic iodinations is due primarily to the reversible reduction of active I2 to 2I−.
The goitrogenic effects of sulfonamides have been known since the reports of the action of sulfaguanidine on the rat thyroid in the early 1950s. Sulfamethoxazole and trimethroprim exert a potent goitrogenic effect in rats, resulting in marked decreases in circulating T3 and T4, a substantial compensatory increase in TSH, and increased thyroid weights due to follicular cell hyperplasia. The dog is also a sensitive species to the effects of sulfonamides, showing markedly decreased serum T4 and T3 levels, hyperplasia of thyrotrophic basophils, and increased thyroid weights. By comparison, the thyroids of monkeys and humans are resistant to the development of changes that sulfonamides produce in rodents (rats and mice) and dogs. Concentration of sulfonamide required to inhibit thyroid peroxidase in the monkey is about 500 times that required in the rat. Sensitive species (e.g., rat, mouse, and dog) are much more likely to develop follicular cell hyperplasia and thyroid tumors after long-term exposure to sulfonamides than are resistant species (e.g., nonhuman primate, human, guinea pig, and chicken).
Inhibitors of Thyroid Hormone Secretion
Blockage of Thyroid Hormone Release
Relatively few chemicals selectively inhibit the secretion of thyroid hormone from the thyroid gland (Figure 20.32). Excess iodine inhibits the secretion of thyroid hormone, and occasionally can cause goiter and hypothyroidism in animals and humans. Several mechanisms have been suggested for this effect of high iodide levels on thyroid hormone secretion, including a decrease in lysosomal protease activity, inhibition of colloid droplet formation, and inhibition of TSH-mediated increase in cAMP. Circulating levels of T4, T3, and rT3 are decreased by an iodide excess.
Lithium has also been reported to have a striking inhibitory effect on thyroid hormone release (Figure 20.32). The widespread use of lithium carbonate in the treatment of manic states occasionally results in the development of goiter with either euthyroidism or occasional hypothyroidism in human patients. Lithium inhibits colloid droplet formation stimulated by cAMP in vitro, and inhibits the release of thyroid hormones.
Xenobiotic- (or Metabolite-)Induced Thyroid Pigmentation or Colloid Alteration
The antibiotic minocycline produces a striking black discoloration of thyroid lobes in laboratory animals and humans with the formation of brown pigment granules within follicular cells. The pigment granules stain similarly to melanin and are best visualized on thyroid sections stained with the Fontana-Masson procedure. The pigment appears to be a metabolic derivative of minocycline, and administration of the antibiotic at a high dose to rats for extended periods may result in a disruption of thyroid function and development of thyroid enlargement (“goiter”).
Other xenobiotics [or metabolite(s)] selectively localize in the thyroid colloid of rodents, resulting in abnormal clumping and increased basophilia of the colloid. Brown to black pigment granules may be present in follicular cells, colloid, and macrophages in the interthyroidal tissues, resulting in a macroscopic darkening of both thyroid lobes. The altered colloid in the lumen of thyroid follicles appears to be less able to react with organic iodine in a stepwise manner or to be phagocytized by follicular cells and processed enzymatically to release active thyroid hormones. Serum T4 and T3 are decreased, serum TSH levels are increased by an expanded population of pituitary thyrotrophs, and thyroid follicular cells undergo hypertrophy and hyperplasia. The incidence of thyroid follicular cell tumors in 2-year carcinogenicity studies in rodents is increased significantly at the higher dose levels, usually with a greater effect in male than in female rats. Similar thyroid changes or functional alterations usually do not occur in dogs, monkeys, or humans.
Effect on Peripheral Metabolism of Thyroid Hormones
Inhibitors of 5′-Deiodinase
Fd&C Red No. 3 (Erythrosine)
FD&C Red No. 3 (erythrosine) is a red dye widely used as a color additive in foods, cosmetics, and pharmaceuticals. The Food and Drug Administration (FDA) peer review panel on Red No. 3 estimated the average quantity of the dye ingested in the United States to be 1.4 mg per person daily (0.023 mg/kg/day for a 60-kg person). FD&C Red No. is a tetraiodinated derivative of fluorescein, and iodine accounts for approximately 58% of the molecular weight of the dye.
Male Sprague-Dawley rats fed high 4% dietary concentrations of Red No. 3 beginning in utero and over their lifetime (30 months) developed a high incidence of thyroid follicular adenomas. Female rats fed similar amounts of the color additive did not have a significant increase in either benign or malignant thyroid tumors. Studies on the absorption, distribution, metabolism, and excretion of FD&C Red No. 3 confirmed earlier reports that the dye is absorbed poorly from the gastrointestinal tract and does not accumulate in the thyroid gland. Subsequent studies revealed that serum T4 concentrations in rats receiving 4% of the dye are increased from 1 through 6 months while serum T3 is reduced significantly. Circulating levels of reverse T3 increase approximately seven-fold after 1 month, and TSH levels are approximately three times baseline. Serum TSH levels in rats fed Red No. 3 and receiving daily T3 injections were decreased to undetectable levels. T4 metabolism is altered significantly in liver homogenates prepared from rats fed 4% Red No. 3 (Figure 20.33). Degradation of T4 is decreased accompanied by a drastic decrease in T3.
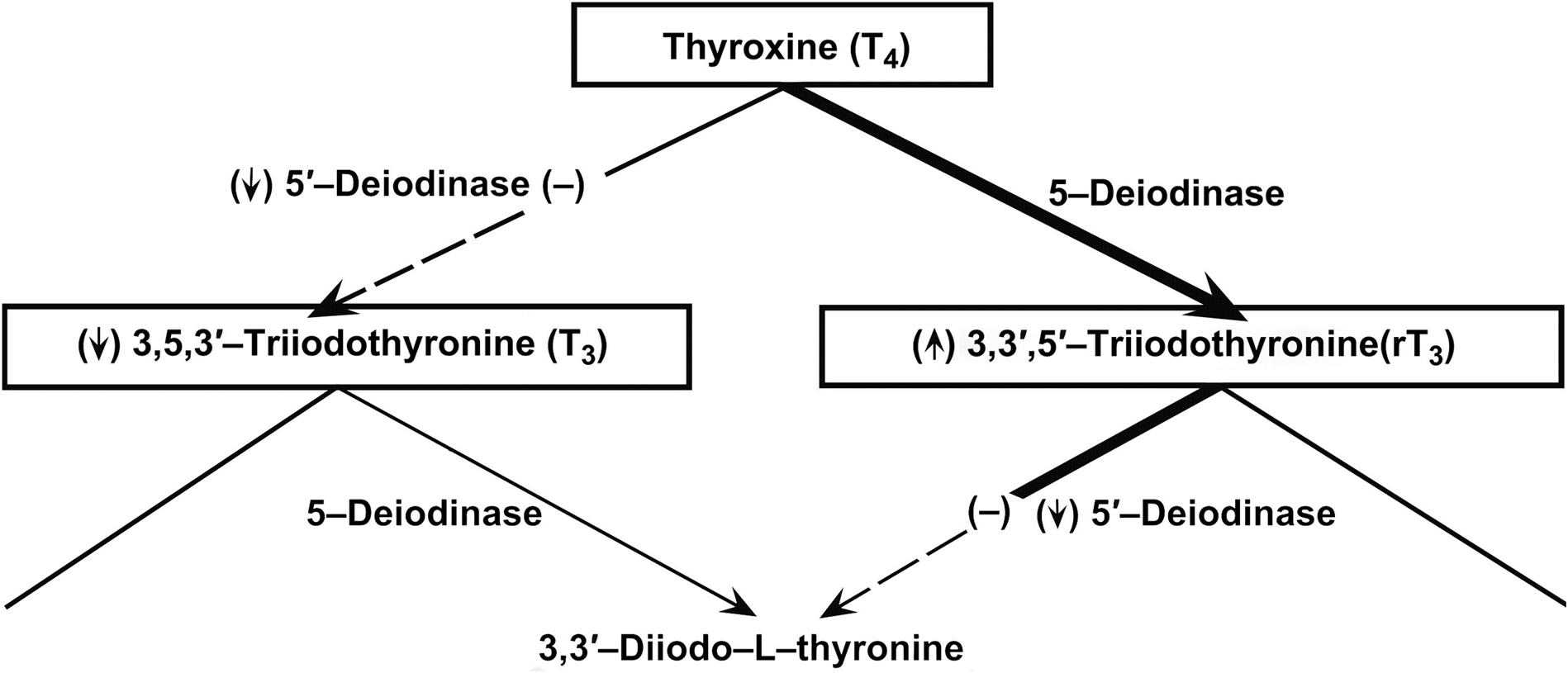
Putting these findings together, an inhibition of 5′-deiodinase in the liver and kidney by Red No. 3 would explain the lower circulating T3 levels. The monodeiodination of T4 by another enzyme (5-deiodinase) to reverse T3 and inhibition of the 5′-deiodinase [which is necessary to further degrade this inactive iodothyronine to 3,3′-diiodothyronine (T2)] explains the striking accumulation of serum reverse T3. The pituitary, sensing the lowered circulating levels of T3, compensates by increasing the secretion of TSH, which results in the morphological evidence of follicular cell stimulation.
Other compounds that are known to inhibit 5′-monodeiodinase in peripheral tissues and disrupt thyroid hormone economy by inhibiting the conversion of T4 to T3 include amiodarone and iopanoic acid. These xenobiotics (as well as FD&C Red No. 3) are iodinated compounds, but it is unlikely that their effects are due to the iodide component because even large doses of iodide (5 mg/day) do not affect T4 metabolism in rats.
Amiodarone
Amiodarone, an iodinated drug used to treat tachyarrhythmias, inhibits the in vivo 5′-monodeiodination of T4. It blocks the 5′-monodeiodinase (type I enzyme) in rat liver homogenates but not the 5′-deiodinase in the rat pituitary gland. Male and female Sprague-Dawley rats administered amiodarone in long-term carcinogenicity studies had an increased incidence of thyroid follicular cell hyperplasia and adenomas, whereas neither male nor female B6C3F1 mice in similar studies developed increased thyroid follicular cell hyperplasia or tumors. In shorter studies, male Sprague-Dawley rats receiving amiodarone had significant increases in serum TSH, T4, and T3, and significantly decreased serum rT3. These changes are similar to those following administration of Red No. 3. It is likely that amiodarone, like FD&C Red No. 3, produces thyroid follicular cell adenomas in rats through the TSH-mediated secondary mechanism.
Iopanoic Acid
Iopanoic acid is an iodinated radiographic contrast agent that inhibits both type I and type II 5′-monodeiodinase in rats. Iopanoic acid and FD&C Red No. 3 share the ability to inhibit the T4 to T3 conversion and increase serum TSH concentrations in rats. However, iopanoic acid inhibits both type I and type II 5′-monodeiodinase, whereas Red No. 3 inhibits only the type I enzyme in liver and kidney and not the type II enzyme in brain, pituitary, and brown adipose tissue. Iopanoic acid and FD&C Red No. 3 differ in their ability to affect human thyroid hormone economy. Iopanoic acid in doses used commonly in clinical radiographic studies results in increased serum concentrations of T4 and rT3, decreased serum T3 with a compensatory increase in basal serum TSH concentrations, and increased TSH response to TRH after 5–7 days of treatment.
Inducers of Hepatic Microsomal Enzymes
Hepatic microsomal enzymes play an important role in thyroid hormone economy because glucuronidation is the rate-limiting step in the biliary excretion of T4 and sulfation is the rate-limiting step in the excretion of T3. Long-term exposure of rats to a wide variety of different chemicals induces these enzyme pathways and results in chronic stimulation of the thyroid by disrupting the hypothalamic–pituitary–thyroid axis (Figure 20.34). The resulting chronic stimulation of the thyroid by increased circulating levels of TSH often results in a greater risk of developing tumors derived from follicular cells in 2-year or lifetime studies with these compounds in rats. Xenobiotics that induce liver microsomal enzymes and disrupt thyroid function in rats include CNS drugs [e.g., phenobarbital (PB), benzodiazepines], Ca2+ channel blockers (e.g., nicardipine, bepridil), steroids (spironolactone), retinoids, chlorinated hydrocarbons (e.g., chlordane, dichlorodiphenyltrichloroethane [DDT], 2,3,7,8-tetrachlorodibenzodioxin [TCDD]), and polyhalogenated biphenyls (e.g., polychlorinated biphenyls (PCB), polybrominated biphenyls [PBB]). Most of the hepatic microsomal enzyme inducers have no apparent intrinsic carcinogenic activity and produce little or no mutagenicity or DNA damage. Their promoting effect on thyroid tumors usually is greater in rats than in mice, with males developing a higher incidence of tumors more often than females. In certain strains of mice, these compounds alter liver cell turnover and promote the development of hepatic tumors.
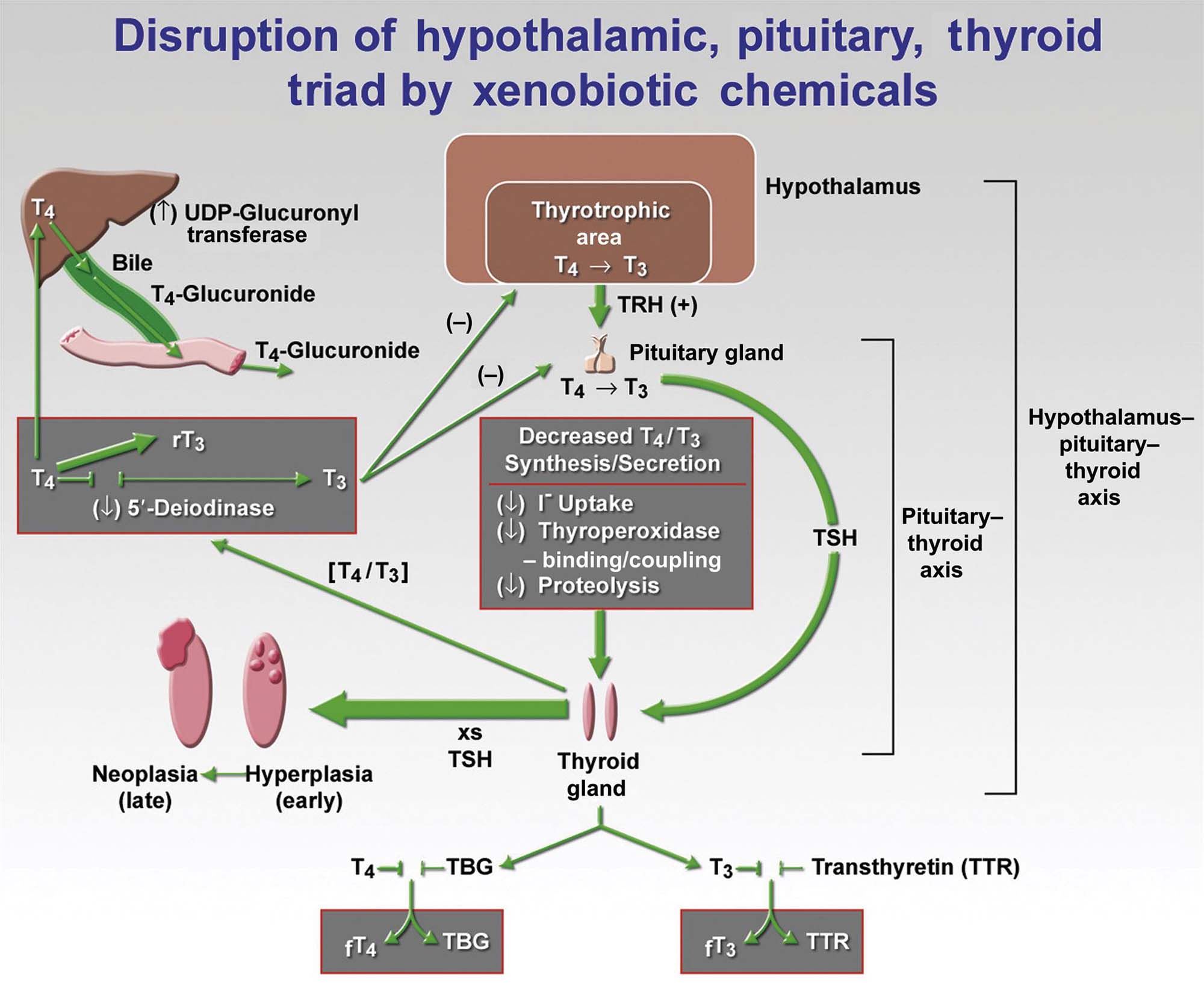
Many of the liver microsomal enzyme inducers in rats [such as PB, pregnenolone-16a-carbonitrile, 3-methylcholanthrene (3MC), and Arochlor 1254 (PCB)] that affect thyroid hormone economy typically induce the activity of T4-UGT, which is associated with increased excretion of T4-glucuronide and decreased serum T4 concentration. Activation of T3-UGT is more predictive of increased TSH in rats compared to T4-UGT.
Phenobarbital
PB has been studied extensively as the prototype for hepatic microsomal inducers that increase a similar spectrum of CYP isoenzymes. The activity of T4-UGT, the rate-limiting enzyme in T4 metabolism, also is increased by PB, with higher cumulative biliary excretion of T4. Most of the increase in biliary excretion is T4 glucuronide due to an increased metabolism of T4 in PB-treated rats. PB-treated rats develop a characteristic pattern of changes in circulating thyroid hormone levels. Plasma T3 and T4 are decreased markedly after 1 week and remain decreased for 4 weeks. By 8 weeks, T3 levels return to near normal due to compensation by the hypothalamic–pituitary–thyroid axis. Serum TSH values are elevated significantly throughout the first month but often decline somewhat after a new steady state is attained. Thyroid weights increase significantly. The prevailing hypothesis is that the promotion of thyroid tumors by PB in rats is not a direct effect of PB on the thyroid gland but an indirect effect mediated by TSH secretion from the pituitary secondary to the hepatic microsomal enzyme-induced increase of T4 excretion in the bile.
The activation of the thyroid gland during the treatment of rodents with substances that stimulate T4 catabolism is a well-known phenomenon. It occurs particularly with rodents because (1) UGT can be induced easily in rodent species, and (2) T4 metabolism takes place very rapidly in rats in the absence of TBG. Despite these findings in rodents, there is no convincing evidence that humans treated with drugs or exposed to chemicals that induce hepatic microsomal enzymes are at increased risk for the development of thyroid cancer. Epidemiologic studies of patients treated with therapeutic doses of PB have reported no increase in risk for the development of thyroid neoplasia. Likewise, there is no substantive evidence that humans treated with drugs or exposed to chemicals that induce hepatic microsomal enzymes are at increased risk for the development of liver cancer.
Polychlorinated Biphenyls
Another classic example of a class of chemical that induces hepatic microsomal enzymes and disrupts thyroid function is the PCB group. PCBs, commonly used industrial compounds, have caused widespread environmental contamination. The disease-producing capability of these compounds includes alterations in reproduction, growth, and development. PCBs cause a significant reduction in serum levels of thyroid hormones due to alterations in thyroid structure, in addition to the well-known induction of hepatic UGT and increased secretion of T4-glucuronide in the bile.
The most consistent thyroid lesions in follicular cells following chronic feeding of PCB to rats are the accumulation of numerous large colloid droplets and irregularly shaped lysosomal bodies in an expanded cytoplasm. Chronic PCB exposure results in a striking distension of many follicular cells with large lysosomal bodies and colloid droplets, blunt and abnormally branched microvilli, and mitochondrial vacuolation. The principal lesion that contributes to the altered thyroid function appears to be interference with the interaction between colloid droplets and lysosomal bodies, necessary for the enzymatic release of thyroid hormones.
Secondary Mechanisms of Thyroid Oncogenesis
Understanding the mechanism of action of xenobiotics on the thyroid gland provides a more rational basis to extrapolate findings from long-term rodent studies to safety assessment of a particular compound for humans. Many chemicals and drugs disrupt one or more steps in the synthesis, secretion, and peripheral metabolism of thyroid hormones, resulting in subnormal levels of T4 and T3, associated with a compensatory increased secretion of pituitary TSH. When tested in a highly sensitive species, such as rats, these compounds result early in follicular cell hypertrophy/hyperplasia and increased thyroid weight, and, in long-term studies, an increased incidence of thyroid tumors by a secondary (indirect) mechanism.
In the secondary mechanism of thyroid oncogenesis in rodents, the specific xenobiotic chemical or physiologic perturbation evokes another stimulus (e.g., chronic hypersecretion of TSH) that promotes follicular cell hypertrophy and then hyperplasia with development of proliferative lesions (initially focal hyperplasia, then adenomas, and less frequently carcinomas). Thresholds for a no effect on the thyroid gland can be established by determining the dose of xenobiotic that fails to elicit an elevation in the circulating level of TSH. Compounds acting by this indirect (secondary) mechanism usually have little or no evidence for mutagenicity or for producing DNA damage.
In humans with markedly altered changes in thyroid function and chronically elevated TSH levels, as in areas with a high incidence of endemic goiter due to iodine deficiency, there is little if any increase in the incidence of thyroid cancer. The relative resistance to the development of thyroid cancer in humans with elevated plasma TSH levels is in marked contrast to the response of the thyroid gland to chronic TSH stimulation in rats and mice.
Initiators of Thyroid Carcinogenesis
Specific chemicals and irradiation appear to have a direct effect on the thyroid gland, resulting in genetic damage that leads to cell transformation and tumor formation. Examples of thyroid initiators include 2-acetylaminofluorene, N-methyl-N-nitrosourea (MNU), N,N-bis(2-hydroxypropyl) nitrosamine (DHPN), MC, dichlorobenzidine, and polycyclic hydrocarbons. Chemicals in this group often increase the incidence of both benign and malignant thyroid tumors over controls. Iodine status may have a significant impact on development of thyroid tumors; for example, iodine deficiency is a strong promoter of MNU-initiated thyroid tumors in rats. A common component of permanent hair dye preparations, 2,4-diaminoanisole sulfate (2,4-DAAS), dramatically increases thyroid neoplasm incidence in both male and female F344 rats. Follicular cell carcinomas are the principal type of neoplasm induced by 2,4-DAAS in thyroids without a background of diffuse hyperplasia of follicular cells. The carcinomas (papillary, cystic, and solid) were invasive but did not metastasize. 2,4-DAAS has been found to be mutagenic but is not teratogenic. In the thyroid, it results in a dose-dependent accumulation of brown granules in follicular cells that are basic fuchsin positive; PAS, acid fast, and iron negative; and ultrastructurally different than lipofuscin. The thyroid pigment may represent a reaction product of 2,4-DAAS and iodine in the cytoplasm of follicular cells.
Part 6: Parathyroid Gland
Introduction
Parathyroid glands are present in all air-breathing vertebrates. Phylogenetically, parathyroids first appear in amphibians, coincident with the transition from an aquatic to a terrestrial life. It has been suggested that the appearance of parathyroid glands may have arisen from the need to protect against the development of hypocalcemia and the necessity to maintain skeletal integrity in terrestrial animals, which often are in a relatively low Ca2+-high phosphorus environment.
Ca+ plays a key role in many fundamental biological processes, including muscle contraction, blood coagulation, enzyme activity, neural excitability, hormone release, and membrane permeability, in addition to being an essential structural component of the skeleton. Therefore, the precise control of Ca2+ in extracellular fluids is vital to health. To maintain a constant concentration of Ca2+, despite variations in intake and excretion, endocrine control mechanisms have evolved that consist of the interaction of PTH, calcitonin, and the active form of Vitamin D (1,25-dihydroxyvitamin D or calcitriol).
Structure and Function
Embryology and Macroscopic Anatomy
Parathyroid glands in most animal species consist of two pairs of glands situated in the anterior cervical region. Rats are the exception because they have a single pair of parathyroid glands located close to the thyroid. Embryologically, parathyroids are of entodermal origin, derived from the third and fourth pharyngeal pouches in close association with the primordia of the thymus (Figure 20.35). The entodermal bud that forms the thyroid gland arises on the midline at the level of the first pharyngeal pouch. This gives rise to the thyroglossal duct that migrates caudally. In the dog and cat, both external and internal parathyroids are close to the thyroid gland. The external parathyroid (III) in the dog is 2–5 mm in length and is found in the loose connective tissue cranial and slightly lateral to the anterior pole of the thyroid. The internal parathyroid (IV) is smaller, flatter, and situated on the medial surface of the thyroid beneath the fibrous capsule.
Functional Cytology
Chief Cells
Parathyroids contain a single type of secretory cell concerned with the elaboration of a single hormone. Parathyroids are composed of chief cells in different stages of secretory activity or in transition to oxyphil cells in certain species (Figure 20.36). Chief cells interpreted to be in an inactive (resting or involuted) stage of their secretory cycle predominate in the parathyroid glands under normal conditions. Inactive chief cells are cuboidal and have uncomplicated interdigitations between contiguous cells. The relatively electron-transparent cytoplasm contains poorly developed organelles and infrequent secretory granules. The cytoplasm often has either numerous lipid bodies and lipofuscin granules or aggregations of glycogen particles. Chief cells in the active stage of the secretory cycle occur less frequently in the parathyroid glands of most species. The cytoplasm of active chief cells has an increased electron density due to the closer proximity of organelles and secretory granules, increased density of the cytoplasmic matrix, and loss of glycogen particles and lipid bodies.
Oxyphil Cells
The second cell type in the parathyroid glands of certain animal species and humans is the oxyphil cell (Figure 20.36). These cells are absent in parathyroids of the rat, chicken, and many species of lower animals. Oxyphil cells are observed either singly or in small groups interspersed between chief cells. They are larger than chief cells, and their abundant cytoplasmic area is filled with numerous large, often bizarre-shaped, mitochondria. Glycogen particles and free ribosomes are interspersed between the mitochondria. Granular endoplasmic reticulum, Golgi apparatuses, and secretory granules are poorly developed, suggesting that oxyphil cells do not have an active function in the biosynthesis of PTH. Associated with the marked increase in mitochondria, oxyphil cells have been shown histochemically to have higher oxidative and hydrolytic enzyme activity than chief cells. Cells are observed with cytoplasmic characteristics intermediate between those of chief and oxyphil cells suggesting that oxyphil cells are derived from chief cells as the result of aging or some other metabolic derangement.
Parathyroid Hormone
Biosynthesis and Chemistry
The biosynthesis and secretion of PTH is described in detail in Figure 20.37. The biologically active PTH secreted by chief cells is a straight chain polypeptide consisting of 84 AA residues with a molecular weight of 9500. Molecular fragments of PTH are formed in the peripheral circulation and in the liver. The immunoheterogeneity created by the multiple circulating fragments of PTH has caused significant problems in the development and application of highly specific RIA to diagnostic problems in human patients and animals.
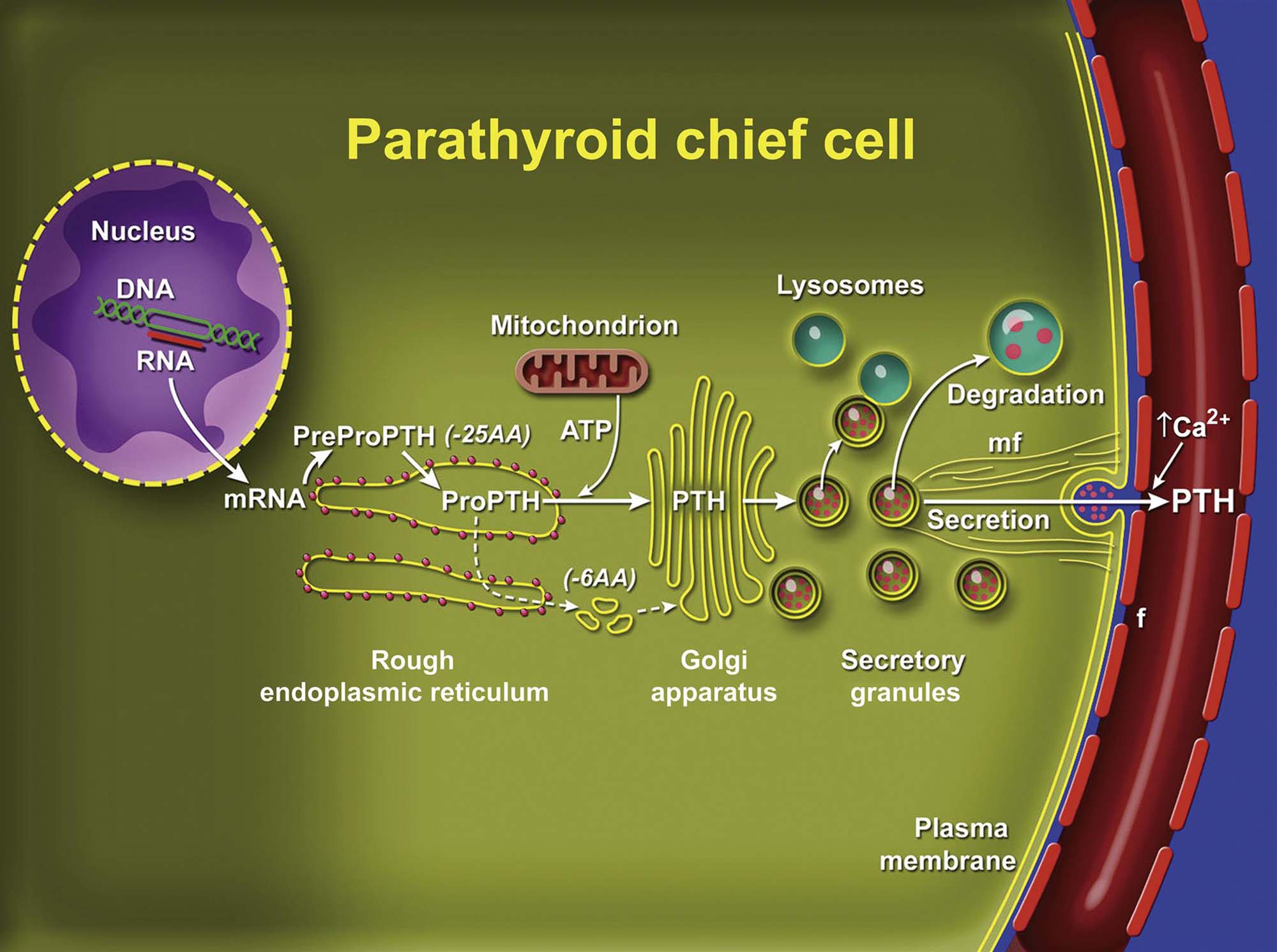
Storage and Secretion
Secretory (storage) granules have been demonstrated ultrastructurally within chief cells of the parathyroid glands. The paucity of secretory granules in certain species (e.g., rat) suggests that chief cells, in general, do not store a large concentration of preformed hormone. PTH in the cytoplasm and secretory granules can be stained by immunofluorescent or immunohistochemical techniques. Oxyphil cells do not stain for PTH.
Chief cells synthesize and secrete another major protein, chromogranin A (parathyroid secretory protein I). It is a 70-kDa protein composed of 430–448 AA that is costored and secreted with PTH. A similar molecule has been found in secretory granules of a wide variety of peptide hormone-secreting cells and in neurotransmitter secretory vesicles. Chromogranin A-derived peptides may act locally in an autocrine manner to inhibit the secretion of active hormone by endocrine cells, such as those of the parathyroid gland.
Chief cells in closely related species may vary considerably in the number of storage granules in their cytoplasm, for example, granules are infrequent in rats but numerous in mice. In response to the appropriate stimulus, secretory granules migrate peripherally in chief cells and their limiting membrane fuses with the plasma membrane of the cell. An internal cytoskeleton composed of microtubules and contractile microfilaments (mf) is important in the control of peripheral movement of secretory granules and liberation of secretory products. Colchicine, an inhibitor of intracellular transport and release of secretory products in a number of endocrine organs, also blocks the secretion of PTH from chief cells in rats.
Secretory cells in the parathyroid gland store small amounts of preformed hormone but are capable of responding to minor fluctuations in Ca2+ concentration rapidly by altering the rate of hormonal secretion and more slowly by altering the rate of hormonal synthesis. In contrast to most endocrine organs that are under complex controls involving both long and short feedback loops, parathyroids have a unique feedback controlled by the concentration of Ca2+ (and, to a lesser extent, magnesium) ion in the serum. If the blood Ca2+ is elevated by the intravenous infusion of Ca2+, there is a rapid and pronounced reduction in circulating levels of immunoreactive PTH (iPTH). Conversely, if the blood Ca2+ is lowered by ethylenediaminetetraacetic acid, there is a brisk and substantial increase in iPTH levels. The concentration of blood phosphorus has no direct regulatory influence on the synthesis and secretion of PTH; however, certain disease conditions with hyperphosphatemia are associated clinically with secondary hyperparathyroidism. An elevated blood phosphorus level may lead indirectly to parathyroid stimulation by virtue of its ability to lower blood Ca2+. Magnesium ion has an effect on parathyroid secretion rate similar to that of Ca2+, but its effect is not equipotent to that of Ca2+.
Recently synthesized and processed active PTH may be released directly in response to increased demand and bypass the storage pool of mature secretory granules in the cytoplasm of chief cells. Bypass secretion of PTH can be stimulated only by a low circulating concentration of Ca2+.
The major inhibitors of PTH synthesis and secretion are increased serum [Ca2+] and 1,25-dihydroxyvitamin D. Inhibition of PTH synthesis by 1,25-dihydroxyvitamin D completes an important endocrine feedback loop between the parathyroid chief cells and the renal epithelial cells because PTH stimulates renal production of 1,25-dihydroxyvitamin D.
Biological Actions
PTH is the principal hormone involved in the minute-to-minute fine regulation of blood Ca2+ in mammals. The action of PTH on bone is the mobilization of Ca2+ from skeletal reserves into extracellular fluids. The increase in blood Ca2+ results from an interaction of PTH with osteoblasts and osteoclasts in bone along with increased tubular reabsorption of Ca2+ in the kidney.
Bone
Osteoclasts are primarily responsible for the catabolic action of PTH on bone by increasing resorption. PTH stimulates increased numbers and activity of osteoclasts; however, osteoclasts do not have receptors for PTH. Receptors for PTH are present on osteoblasts. Isolated osteoclasts respond to PTH only with the concurrent presence of osteoblasts. PTH binds to the PTH receptor on osteoblasts and stimulates them to produce RANKL (receptor activator of NFκB ligand), which binds to its receptor, RANK, on osteoclast precursors and osteoclasts to increase the number and function of osteoclasts, respectively. PTH inhibits the production of osteoprotegerin (OPG) by osteoblasts. OPG is a secreted decoy receptor for RANKL, which blocks its action in bone. Therefore, the ratio of RANKL:OPG in the bone microenvironment regulates the level of osteoclastic bone resorption.
Kidney
PTH has a rapid and direct effect on renal tubular function, leading to decreased reabsorption of phosphorus and phosphaturia in the proximal tubule of the nephron. PTH binds to a receptor on the basolateral aspect of renal epithelial cells. The hormone stimulates adenylyl cyclase, increases intracellular cAMP, and inhibits phosphorus reabsorption across the brush border through the actions of protein kinases.
PTH also increases the reabsorption of Ca2+, which is of considerable importance in the maintenance of Ca2+ homeostasis. The effect of PTH on tubular reabsorption of Ca2+ appears to be due to a direct action on the distal convoluted tubule and is coupled to increases in intracellular cAMP. Transcellular transport of Ca2+ from the renal tubule lumen to the blood by the kidney involves the Ca2+-selective channels on the luminal (apical) side of the renal epithelial cells, intracellular calbindins to buffer Ca2+, and the Ca2+–Na+ exchanger (NCX) and Ca2+-ATPase (PMCA) on the basilar side of the epithelial cells. In addition to PTH, the cell membrane Ca2+ receptor, calcitriol, and calcitonin regulate renal Ca2+ reabsorption.
Subcellular Mechanism of Action
The receptor for the N-terminal portion of PTH, composed of 34 AA, is considered the most important region in terms of Ca2+ regulation. The receptor for N-terminal PTH is a seven transmembrane domain receptor that is expressed in renal epithelial cells, osteoblasts, and dermal fibroblasts and is also found on cells that are not associated with the actions of PTH. Binding of PTH to the receptor results in increased levels of cytoplasmic cAMP and Ca2+ by stimulation of the adenylyl cyclase and phosphatidyl inositol pathways.
Degradation
PTH is secreted continuously from chief cells primarily as the intact (1–84 AA sequence) molecule under normal conditions. In the liver (Küpffer cells) PTH is cleaved into the smaller (approximately one-third of the molecule) NH2-terminal (biologically active) portion and a larger (approximately two-thirds of the molecule) C-terminal (biologically inactive) portion. The kidney is also a major organ for the degradation and excretion of PTH. Biologically active PTH from the peritubular capillaries is degraded by specific proteases on the surface of renal tubular cells. In addition, both biologically active and inactive fragments are degraded intracellularly by lysosomal enzymes in renal tubular cells. C-terminal PTH has a longer circulating half-life compared to full-length and NH2-terminal PTH.
Evaluation of Toxicity
Morphologic Evaluation
The parathyroid gland is infrequently injured directly by xenobiotics. However, parathyroid function may be altered by a wide variety of chemicals that either elevate or lower the blood Ca2+ concentration. In response to hypocalcemia, chief cells undergo hypertrophy and eventually hyperplasia. On formalin- or Bouin’s-fixed sections, the expanded cytoplasmic area is lightly eosinophilic and vacuolated compared with chief cells in normal animals. Perivascular spaces are narrow in a hyperplastic parathyroid and there are few fat cells in the interstitium. In response to hypercalcemia, the cytoplasmic area of chief cells is decreased and more densely eosinophilic, often with a widening of intercellular and pericapillary spaces. If the hypercalcemia is prolonged, there is an overall reduction of glandular parenchyma with increased fibrous or adipose connective tissue in the interstitium. Subtle differences between treated and control groups can be best evaluated by the morphometric evaluation of parenchyma:interstitium and cytoplasmic:nuclear area of chief cells.
Ultrastructural evaluation of chief cells is a sensitive means of assessing morphologically whether a particular drug or chemical affects the parathyroid gland. Perfusion of the thyroid–parathyroid area with glutaraldehyde-based fixatives followed by postfixation with osmium tetroxide results in the best retention of structural detail. Morphometric studies at the ultrastructural level can be used to quantitate total cytoplasmic area and area occupied by a particular organelle (e.g., secretory granule).
In response to an acute lowering of blood Ca2+, a larger percentage of chief cells ultrastructurally will be in the active stage of synthesis and secretion than under steady-state conditions. This is indicated by a peripheral migration of secretory granules and alignment along the plasma membrane, aggregation of the endoplasmic reticulum into lamellar arrays, and enlargement of the Golgi apparatus associated with many small dense granules in the process of formation. Conversely, chief cells in response to hypercalcemia are predominantly in the inactive stage of the secretory cycle as evaluated by electron microscopy, with straight plasma membranes dispersed profiles of endoplasmic reticulum, small Golgi complexes with few granules, and often accumulations of either glycogen or lipid (depending on the species) in the cytoplasm.
Atrophic chief cells develop in response to sustained or more severe hypercalcemia. Their cytoplasm is more electron-dense and irregularly shrunken with widened intercellular spaces. Cytoplasmic organelles are poorly developed and may have early degenerative changes suggested by mitochondrial vacuolation with disruption of cristae, and distension of endoplasmic reticulum, with loss of ribosomes.
Assay of Circulating Parathyroid Hormone
The metabolism of PTH and the formation of multiple circulating forms of PTH have made the development of clinically useful immunoassays challenging. The principal circulating forms of PTH include intact PTH (1–84) and C-terminal peptides (e.g., PTH 35–84). Circulating N-terminal PTH is not present at biologically relevant concentrations, but administration of exogenous PTH (1–34) will nevertheless induce the typical actions of PTH on bone and kidney cells.
Intact serum PTH concentrations are best measured by two-site immunoradiometric assay or N-terminal RIA. Mouse- and rat-specific PTH assays are commercially available. Serum-intact PTH can be measured in dogs and cats with assays developed for human PTH due to the crossreactivity of the antisera used in the assays, but internal validation should be performed for preclinical toxicity studies.
Response of Parathyroid Chief Cells to Injury
Parathyroid Cysts
Small cysts are observed frequently as naturally occurring lesions within the parenchyma of the parathyroid or in the immediate vicinity of the glands in rats and dogs, and occasionally in other animal species. Parathyroid cysts usually are multiloculated, lined by a cuboidal to columnar (often partially ciliated) epithelium, and contain a densely eosinophilic proteinic material. Parathyroid (Kürsteiner’s) cysts develop from a persistence and dilatation of remnants of the duct that connects the parathyroid and thymic primordia during embryonic development. Similar cysts may be present in the anterior mediastinum when remnants of the embryonic duct are displaced with the caudal migration of the thymus. Parathyroid cysts are distinct from midline cysts derived from remnants of the thyroglossal duct. The latter are lined by multilayered thyroidogenic epithelium that often has colloid-containing follicles. They usually are located near the midline from the base of the tongue caudally into the mediastinum.
Proliferative Lesions of Parathyroid Chief Cells
Incidence
Proliferative lesions of the parathyroid gland include hyperplasia (diffuse and focal), adenomas, and carcinomas. Neoplasms of the parathyroid glands are uncommon in all species of laboratory and domestic animals, but occur at low incidence in rats, Syrian hamsters, dogs, and, rarely, mice. Parathyroid hyperplasia may be primary or secondary. Primary parathyroid hyperplasia or tumors may be functional (endocrinologically active) or non-functional. Primary hyperparathyroidism is the disorder that results from the excessive and autonomous secretion of PTH from adenomas or chief cell hyperplasia. Atrophy of the remaining normal parathyroid tissue is present with functional tumors and multinodular parathyroid hyperplasia.
Hyperplasia
Focal (“Nodular”) Hyperplasia
Chief cell hyperplasia may affect the parathyroid in a distinctly focal or multifocal distribution. In focal parathyroid hyperplasia there are single or multiple nodules in one or both glands where there is an increased number of closely packed chief cells often with an expanded cytoplasmic area (Figure 20.38). Focal areas of chief cell hyperplasia are poorly demarcated and not encapsulated from adjacent parenchyma. Chief cells within the nodules have a relatively uniform composition with a high cytoplasm:nucleus ratio and a slightly more hyperchromatic nucleus than adjacent normal chief cells. There may be slight compression of adjacent chief cells around larger focal areas of hyperplasia. Focal chief cell hyperplasia often is difficult to separate from chief cell adenoma using only morphological criteria. The presence of multiple nodules of varying sizes and uniform cellularity in one or both parathyroids with minimal compression and no encapsulation is more compatible with (multi)focal hyperplasia than chief cell adenoma.
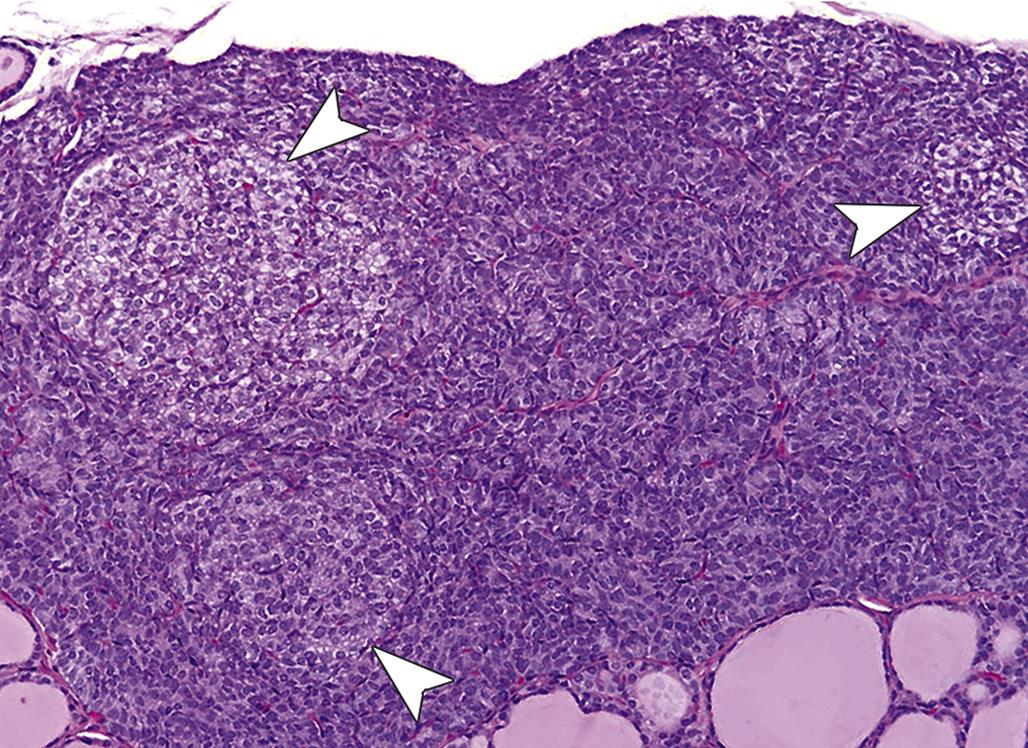
Diffuse Hyperplasia
Diffuse parathyroid hyperplasia, as seen with chronic renal failure and long-term dietary imbalances, results in a uniform enlargement of all parathyroid glands. In rats with chronic renal failure, diffusely hyperplastic parathyroid glands may be detected macroscopically as 1- to 2-mm pale nodules projecting from the surface of each thyroid lobe. The uniform enlargement of parathyroid glands is due to both hypertrophy and hyperplasia of chief cells. There is no peripheral rim of compressed atrophic parathyroid parenchyma as seen around a functional adenoma, but there is a uniform population of hyperplastic chief cells extending to the capsule of the gland (Figure 20.39). Chief cells are packed together closely, often with indistinct cell boundaries. The expanded cytoplasmic area of chronically stimulated chief cells is lightly eosinophilic, with occasional distinct vacuoles. A more prominent fibrovascular stroma in some diffusely hyperplastic parathyroids may result in a lobulated appearance. In other hyperplastic parathyroids, chief cells form distinct acinus-like structures in the gland.
Adenoma
Parathyroid adenomas in rats vary from microscopic in size to unilateral nodules several millimeters in diameter. Tumors of parathyroid chief cells do not appear to be sequelae of longstanding secondary hyperparathyroidism of either renal or nutritional origin. The unaffected parathyroid glands may be atrophic if the adenoma is functional, normal if the adenoma is nonfunctional, or enlarged if there is concomitant hyperplasia. In functional adenomas, the normal mechanism by which PTH secretion is controlled by the concentration of blood Ca2+ is lost and hormone secretion is excessive, despite an increased level of blood Ca2+.
Adenomas are solitary nodules that are sharply demarcated from adjacent parathyroid parenchyma (Figure 20.40). Because the adenoma compresses the rim of surrounding parathyroid to varying degrees, depending on its size, there may be a partial fibrous capsule, either from compression of existing stroma or from proliferation of fibrous connective tissue. Adenomas are usually nonfunctional in rats but may be functional in dogs, cats, and humans. Chief cells in nonfunctional adenomas are cuboidal or polyhedral and arranged in a diffuse sheet, lobules, or acini with or without lumens. Nuclei are round to oval and often vesicular, and there may be infrequent mitotic figures. Chief cells from functional adenomas often are closely packed into small groups by fine connective tissue septa and have lightly eosinophilic cytoplasm. There is a much lower density of cells in a functional parathyroid adenoma than in the adjacent rim with atrophic chief cells. Some parathyroid adenomas in the rat become cystic.
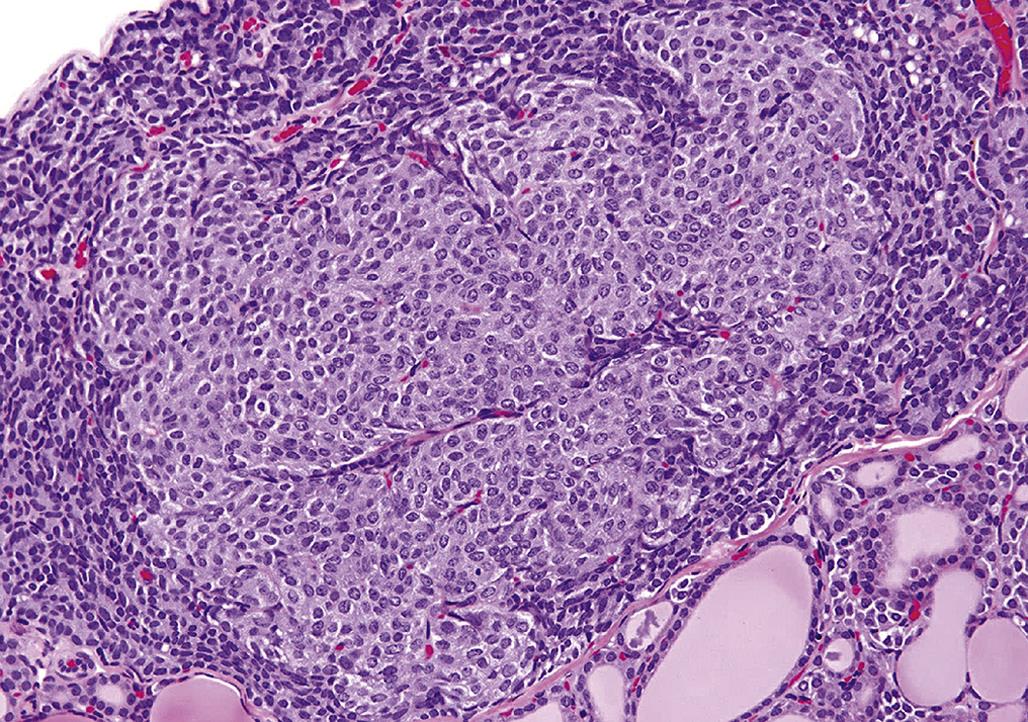
Carcinoma
Chief cell carcinomas are encountered rarely. Carcinomas result in a macroscopically detectable enlargement of one gland. Parathyroid carcinomas often are more fixed in position than chief cell adenomas due to invasion of adjacent thyroid or adjacent cervical skeletal muscle. Some of the enlargement may be due to central necrosis and hemorrhage in the carcinoma.
Malignant chief cells are arranged in solid sheets subdivided into lobules by a fine fibrous and richly vascular stroma, often palisading along blood sinusoids. Occasionally, they form acinar structures. There is usually complete incorporation of the affected gland and evidence of invasion through the parathyroid capsule. Evidence of vascular invasion and formation of tumor cell emboli are observed infrequently. Malignant chief cells may be more pleomorphic than those that constitute adenomas, but mitotic figures are infrequent. The cytoplasmic area stains lightly eosinophilic, and boundaries of adjacent chief cells are indistinct.
Multinucleated Syncytial Cells
Parathyroid glands of dogs, rats, and other animals occasionally develop a unique multinucleated syncytial giant cell. Syncytial cells are often more numerous near the periphery of the gland. The number of syncytial cells may account for up to one-half of the parenchyma of the gland. These cells appear to form by the fusion of the cytoplasmic area of adjacent chief cells. The mechanism by which syncytial cells form is uncertain, but it has been suggested that syncytial cells may be an artifact of immersion fixation with aldehydes due to membrane disintegration.
Mechanisms of Toxicity
Agents Influencing the Development of Proliferative Lesions
Age
There are relatively few chemicals or experimental manipulations reported in the literature that significantly increase the incidence of parathyroid tumors. Longstanding renal failure with intense diffuse hyperplasia does not appear to increase the development of chief cell tumors in animals. Parathyroid adenomas in F344 rats are an example of a neoplasm whose incidence increases dramatically when comparing 2-year studies to lifetime data, where the incidence of parathyroid adenomas increased in males from 0.1% at 2 years to 3.1% in lifetime studies. Corresponding data for female F344 rats was 0.1% at 2 years and 0.6% in lifetime studies.
Irradiation
Irradiation (131I) significantly increases the incidence of parathyroid adenomas in inbred Wistar albino rats of both sexes. However, tumor incidence can be modified by feeding diets with variable amounts of vitamin D, high dietary levels decrease the incidence whereas low levels increase it. Gonadectomy decreased the incidence of radiation-induced (131I) parathyroid adenomas in male rats but had little effect in females. X-irradiation of the thyroid–parathyroid region also increases the incidence of parathyroid adenomas.
Xenobiotics
Parathyroid adenomas have been encountered infrequently following the administration of a variety of chemicals in 2-year bioassay studies in F344 rats. In a study of the pesticide rotenone in F344 rats, there was an increased incidence of parathyroid adenomas in high-dose (75 ppm) males compared with low-dose (38 ppm) males, control males, or NTP historical controls. It is uncertain whether the increased incidence of this uncommon tumor was a direct effect of rotenone feeding or the increased survival time in high-dose males. Chief cell hyperplasia was not present in parathyroids that developed adenomas.
Modification of Parathyroid Function Associated with Metabolic Disorders
Renal Hyperparathyroidism
Secondary hyperparathyroidism as a complication of chronic renal failure is a metabolic state characterized by an excessive, but not autonomous, rate of PTH secretion. The secretion of hormone by the hyperplastic parathyroid gland usually remains responsive to fluctuations in blood Ca2+. The primary etiologic mechanism in this disorder is longstanding progressive renal disease. When the renal disease progresses to the point at which there is a significant reduction in the glomerular filtration rate, phosphorus is retained and progressive hyperphosphatemia develops (Figure 20.41). Hyperphosphatemia contributes to parathyroid stimulation by virtue of its ability to lower blood Ca2+ levels. Parathyroid stimulation associated with chronic renal disease also can be attributed to hypocalcemia. Impaired intestinal absorption of Ca2+ due to the acquired defect in Vitamin D metabolism associated with loss of tubular mass plays a role in the development of hypocalcemia. Thus, there is diminished production of 1,25-dihydroxyvitamin D3, in turn leading to diminished intestinal Ca2+ uptake. 1,25-Dihydroxyvitamin D3 also completes an important negative feedback loop with the parathyroid gland since it inhibits the synthesis and secretion of PTH. Decreased 1,25-dihydroxyvitamin D3 leads to release of chief cells from negative feedback and secondary hyperparathyroidism. All parathyroids are considerably enlarged as a result initially of hypertrophy of chief cells and subsequently due to hyperplasia as a compensatory mechanism.
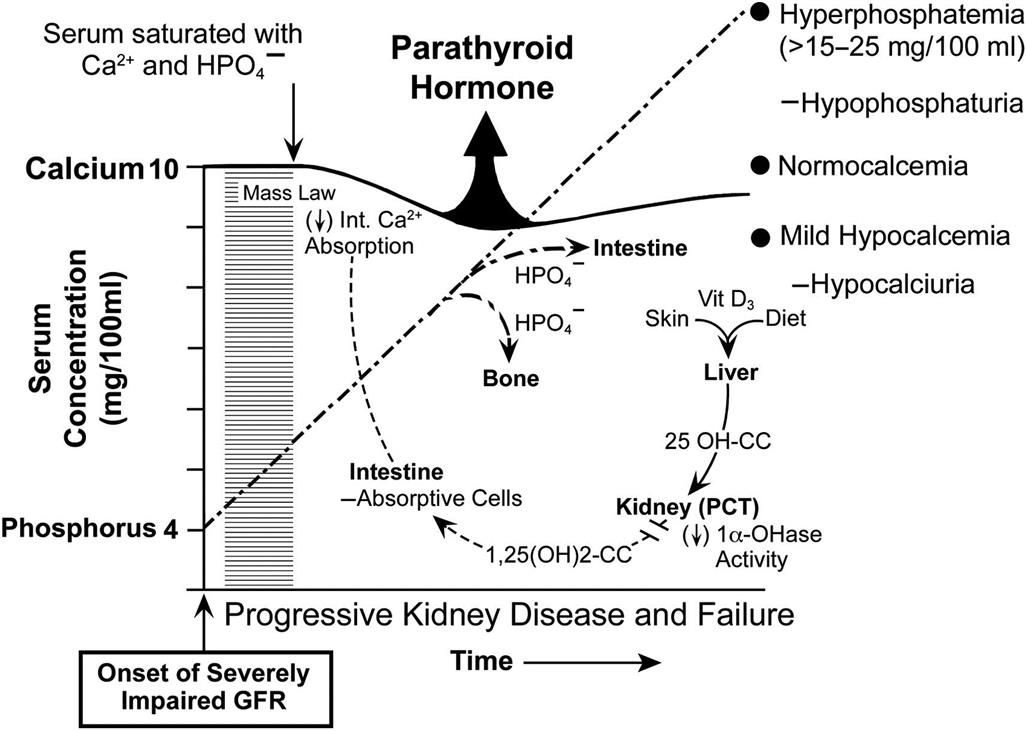
Nutritional Hyperparathyroidism
The increased secretion of PTH in nutritional hyperparathyroidism is a compensatory mechanism directed against a disturbance in mineral homeostasis induced by nutritional imbalances. The disease occurs in dogs, cats, monkeys, laboratory rodents, and others fed improper diets. Dietary mineral imbalances of etiologic importance in the pathogenesis are a low content of Ca2+, excessive phosphorus with normal or low Ca2+, and inadequate amounts of cholecalciferol (Vitamin D3) in New World nonhuman primates housed indoors without exposure to sunlight. The significant end result is hypocalcemia which results in parathyroid stimulation (Figure 20.42). A diet low in Ca2+ fails to supply the daily requirement, even though a greater proportion of ingested Ca2+ is absorbed, and hypocalcemia develops. Ingestion of excessive phosphorus results in increased intestinal absorption and elevation in blood phosphorus levels. Hyperphosphatemia does not stimulate the parathyroid gland directly but does so indirectly by virtue of its ability to lower blood Ca2+ levels and suppress the synthesis of 1,25-dihydroxycholecalciferol by the kidney. In response to nutritionally induced hypocalcemia, all parathyroid glands undergo cellular hypertrophy and hyperplasia.
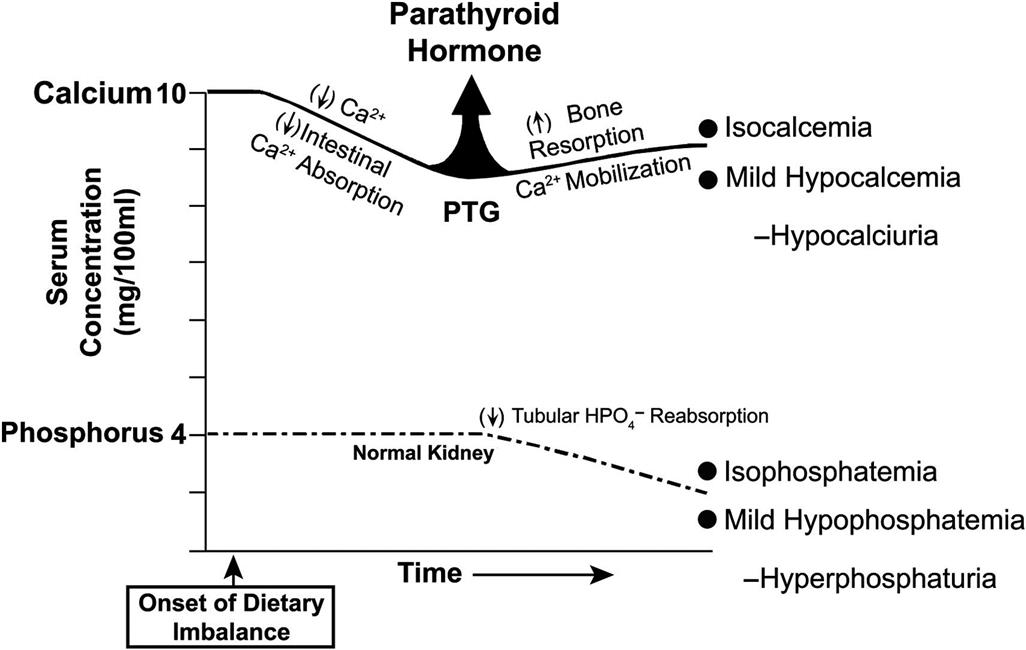
Hypoparathyroidism
Hypoparathyroidism is a metabolic disorder in which either subnormal amounts of PTH are secreted by pathologic parathyroids, or the hormone secreted is unable to interact normally with target cells. Hypoparathyroidism often is associated with diffuse lymphocytic parathyroiditis, resulting in the extensive degeneration of chief cells and replacement by fibrous connective tissue. In the early stages of lymphocytic parathyroiditis, there is infiltration of the gland with lymphocytes and plasma cells and nodular regenerative hyperplasia of the remaining chief cells. Lymphocytic parathyroiditis appears to develop by immune-mediated mechanisms. The functional disturbances of hypoparathyroidism primarily are the result of increased neuromuscular excitability and tetany. Bone resorption is decreased because of a lack of PTH, and blood Ca2+ levels diminish and serum phosphate levels increase progressively.
Primary Hyperparathyroidism
In primary hyperparathyroidism, PTH is produced in excess by a functional tumor in the gland. The normal control of PTH secretion by the concentration of blood Ca2+ is lost in primary hyperparathyroidism. Hormone secretion is autonomous, and the parathyroid produces excessive hormone despite the increased blood Ca2+. PTH acts initially on cells in the renal tubules to promote the excretion of phosphorus and the retention of Ca2+. A prolonged increased secretion of PTH results in accelerated bone resorption and increased renal production of 1,25-dihydroxyvitamin D3. The lesion in the parathyroid gland responsible for the excessive secretion of PTH usually is an adenoma composed of active chief cells.
Cancer-Associated Hypercalcemia
There are three mechanisms by which tumors can induce hypercalcemia: hematologic cancers present in the bone marrow induce hypercalcemia by causing the local destruction of bone; solid tumors can widely metastasize to bone and result in local bone loss associated with the tumor metastases; and humoral hypercalcemia of malignancy (HHM), in which solid tumors that have no or few metastases to bone induce their effects distant from the site of the tumor by the elaboration of one or more factors.
HHM is a syndrome associated with diverse malignant neoplasms in dogs, humans, and cats, and rarely in other animals, although it has been reported in rats and mice. Characteristic clinical findings with HHM include hypercalcemia, hypophosphatemia, hypercalciuria (often with decreased fractional Ca2+ excretion), increased fractional excretion of phosphorus, increased nephrogenous cAMP, and increased osteoclastic bone resorption. Hypercalcemia is induced by humoral effects on bone, kidneys, and possibly the intestine.
Malignant neoplasms that are commonly associated with HHM in animals include the adenocarcinoma derived from apocrine glands of the anal sac in dogs, some T-cell lymphomas of dogs, and miscellaneous carcinomas that induce HHM sporadically. Excessive secretion of biologically active PTH-related protein (PTHrP) by cancer cells plays a central role in the pathogenesis of hypercalcemia in most forms of HHM.
PTHrP is produced by a number of normal tissues in neonates and adults, where it functions primarily as a paracrine factor. For example, PTHrP expression by chondrocytes regulates the growth and differentiation of chondrocytes in the growth plate and, indirectly, the length of the long bones. In the fetus, PTHrP regulates Ca2+ balance and placental Ca2+ transport. Genetic disruption of PTHrP leads to death after birth.
In HHM, PTHrP binds to the common N-terminal PTH/PTHrP (PTH1) receptor in bone and kidney, where it stimulates adenylyl cyclase and increases intracellular Ca2+ in bone and kidney. Increased osteoclastic bone resorption with Ca2+ release is a consistent finding in HHM. The kidneys play a critical role in the pathogenesis of hypercalcemia, as renal Ca2+ reabsorption is stimulated by PTHrP. In some forms of HHM, there are increased serum 1,25-dihydroxyvitamin D3 levels, which may increase Ca2+ absorption from the intestine.
Part 7: Endocrine Pancreas
Introduction
The endocrine pancreas consists of a diffusely distributed system of endocrine cells organized mainly into “islets of Langerhans” (islets) but also occurring as small clusters of cells or individual cells in or adjacent to exocrine pancreatic ductules, exocrine acini, or within interstitial connective tissue. Islets are well defined and easily discerned. They range widely in size (100–200 μm in diameter). Islets are not randomly distributed throughout the pancreas but are concentrated in different lobes of the pancreas depending on the species. Each islet consists of multiple different endocrine cells that have separate functions, yet interact in a paracrine manner and by electrical gap-junction coupling. The most numerous endocrine cell type (70%–80%) is the β cell, which is responsible for secretion of insulin and regulation of glucose and fat metabolism. Electrically coupled β cells secrete insulin more effectively than single cells. Other cell types include the α cell (secretes glucagon), δ cell (secretes somatostatin), the PP or F cell (secretes pancreatic polypeptide-PP), and the ε cell (secretes ghrelin). Isolated or small clusters of endocrine cells in the pancreas are usually β cells.
Structure and Function
Functional Cytology
Immunohistochemistry and immunofluorescence are effective techniques to identify the endocrine cell types in the pancreas. In addition, ultrastructure can be used to identify cell types and evaluate cellular pathology. The secretory granules, including their dense cores and submembranous spaces, have distinct sizes and shapes, depending on the cell type and species. For example, in rats, insulin-containing secretory granules have variably sized round dense cores and wide submembranous spaces. In dogs and other species, however, some of the β-cell dense cores of secretory granules are crystalline and elongate.
Islet cells are organized so that β cells have homologous contacts with other β cells and heterologous contacts with α cells. In rats and mice, islet cells are organized in a specific manner. β Cells are the most numerous endocrine cell type and are located in the central region of the islet (Figure 20.43A). However, a small percentage of large islets may have β cells concentrated in the periphery as well. Smaller islets are composed entirely of β cells. The α cells are located in the periphery or mantle of the islet (Figure 20.43B). Small numbers of δ cells and PP cells are also scattered in the periphery of the islets. This pattern is different in humans and nonhuman primates.
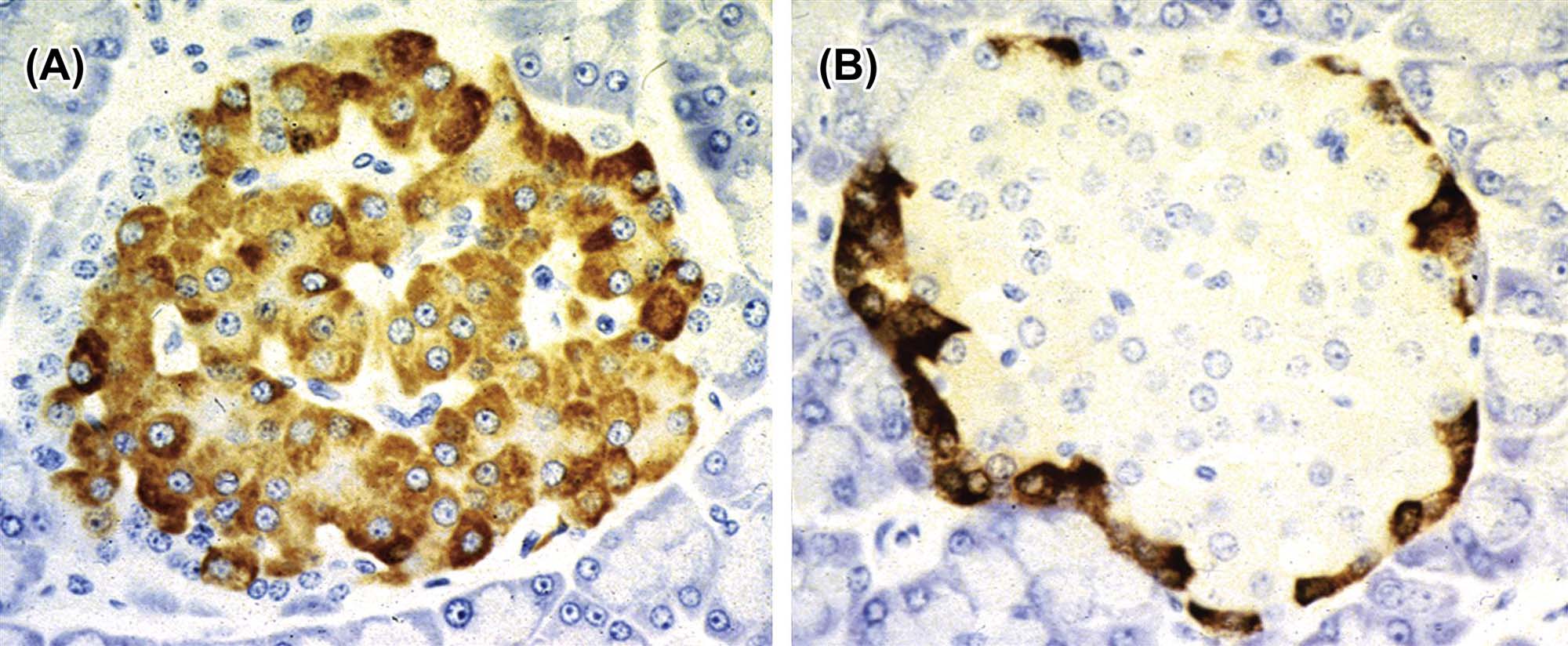
Physiology
A major role for the endocrine pancreas is to maintain a normal and stable concentration of blood glucose during different physiological states. Insulin decreases blood glucose by increasing the uptake of glucose by muscle, fat, and other cells and promoting the synthesis of glycogen. The liver and brain are not dependent on insulin for glucose uptake. Insulin secretion is regulated by blood glucose, GLP-1, gastric inhibitory peptide, GH, glucocorticoids, and the autonomic nervous system. In contrast, glucagon increases blood glucose by stimulating glycogenolysis in the liver and kidney. Glucagon secretion is stimulated by decreased blood glucose, catecholamines, insulin, and the sympathetic nervous system.
Evaluation of Toxicity
Morphologic Evaluation and Immunocytochemistry
Morphologic evaluation of the pancreatic islets can be assessed using standard sections stained with H&E. Individual cell types can be identified specifically using immunohistochemical stains for hormones, such as insulin, glucagon, somatostatin, and pancreatic polypeptide. Immunohistochemistry also can be used to identify inflammatory cell types or the deposition of collagen in regions of fibrosis. Quantitative pathology or stereology can be used to measure the effects of compounds on the pancreatic islets. However, correct nonbiased sampling is essential because there is normal variability in the size and distribution of islets in the different species.
Assay of Circulating Hormones or Chemicals
Measurement of circulating hormones is used to determine the function and clinical significance of islet cell degeneration, loss, hyperplasia, and neoplasia. Islet cell hyperplasia and neoplasia is usually associated with increased secretion of endocrine hormones. For example, functional β-cell or α-cell hyperplasia/tumors would be expected to be associated with hyperinsulinemia and hyperglucagonemia, respectively.
Glucose challenge or tolerance tests are used to measure the ability of the test subject to reduce serum glucose concentration effectively to an oral or intravenous glucose challenge. Usually, a 2-hour oral glucose tolerance test (with a baseline fasted sample) is adequate to diagnose a state of impaired glucose tolerance or diabetes mellitus (DM). The intravenous glucose challenge is useful for identification of prediabetic states.
Serum glycated hemoglobin (HbA1c) is measured to evaluate the average blood glucose concentration over a prolonged period of time. Effective therapeutics for diabetes are expected to reduce the HbA1c concentrations. Serum HbA1c concentrations can be used to predict the incidence and severity of cardiac, renal, and vascular complications associated with diabetes.
Animal Models
Most animal models that have been developed are associated with the study of DM (see below). The nonobese diabetic mouse is a strain that partially mimics the pathogenesis of Type I DM (T1DM) in humans with similar genetics and autoimmune inflammatory response (see below). Animal models for the study of Type 2 DM (T2DM) include the ob/ob mouse, db/db mouse, Zucker diabetic fatty rat, obese rhesus monkeys, sand rat, streptozotocin-induced adult diabetic animals, complete or partial pancreatectomy, insulin receptor knockout mouse, and mice with knockout of the insulin receptor in β cells.
Response to Injury
Functional Considerations
DM is the clinical condition where there is failure of control of blood glucose, with the development of hyperglycemia and hyperglucosuria; it is the consequence of islet dysfunction. T1DM is a chronic autoimmune disease that results in a primary lack of β cells and decreased insulin production and secretion. T1DM is usually seen in young animals or children and is due to a genetic lack or inflammatory destruction of β cells.
T2DM is the most common form of DM in humans. T2DM is a heterogeneous group of disorders and is associated with obesity, physical inactivity, and genetic predisposition. It is initially due to impaired insulin action from reduced sensitivity of insulin receptors to insulin. Insulin levels may be initially elevated but then are reduced over time when there is failure of the β cells. T2DM occurs in a wide variety of species; some strains of mice and rats are actually predisposed to developing T2DM with obesity. Obese dogs, however, are resistant to development of T2DM.
Islet Inflammation (Insulitis)
Inflammation is important in the pathogenesis of islet cell loss in both T1DM and T2DM. T1DM is due to autoimmune inflammation of the pancreatic islets. In the occult (early) phase there is a mixed population of leukocytes (lymphocytes and macrophages) in the islets that induce β-cell death. Microvascular leakage occurs in the early phases of the inflammatory response. In the overt phase, when DM is present, there are few β cells and little inflammation. Biomarkers of autoimmune insulitis are serum autoantibodies to a set of β-cell antigens (such as insulin, glutamic acid decarboxylase and protein tyrosine phosphatase-related molecules). In T2DM there are increased macrophages and lymphocytes in the islets associated with increased levels of cytokines (such as interleukin-1β) and chemokines. Fibrosis results from the chronic inflammation. Amyloidosis also may be seen in humans or cats. Hyperglycemia contributes to β-cell apoptosis possibly by induction of the Fas receptor by IL-1β (which is induced by high glucose concentrations). Fatty acids and oxidative stress are also involved in the pathogenesis of inflammation-induced damage to the pancreatic islets.
Islet Cell Amyloidosis
Some species are prone to developing amyloidosis of the islets, which can be confirmed using Congo red to stain the extracellular deposits. Amyloidosis can be exacerbated by inflammation and T2DM (Figure 20.44). The species affected include humans, cats, raccoons, Celebese apes, and the South American degu. The amyloid is formed from the peptide amylin (islet amyloid peptide), which has 37 AA and shares sequence homology with calcitonin gene-related peptide. Amylin is secreted along with insulin and is present in the submembranous clear zone region of the secretory granules. The species that are prone to islet amyloidosis have sequences of AA 20–29 of amylin that favor formation of a β-sheet configuration and amyloid formation in the interstitium of the islets. Overproduction of amylin, decreased catabolism, or aberrant enzymatic processing may also contribute to amyloid formation. Amyloidosis of the islet predisposes to the development of T2DM, but not all individuals with islet amyloidosis have DM.
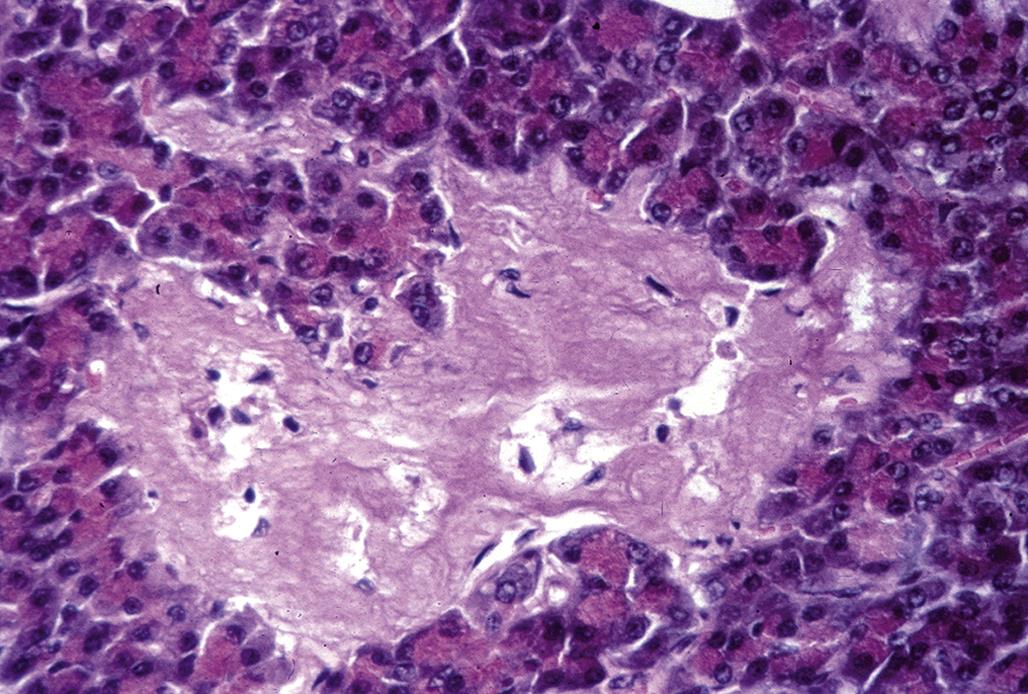
Islet Cell Degeneration and Loss
Degeneration of islet cells may occur with certain chemicals or drugs, islet inflammation, or β-cell dysfunction associated with chronic T2DM. Obesity and overeating are common causes of β-cell dysfunction, degeneration, and eventual loss due to apoptosis. In the early stages of obesity and metabolic syndrome there is peripheral insulin resistance with increased total insulin secretion, but the first phase of insulin secretion may be impaired. The pathophysiology of β-cell loss with T2DM is not well understood, but it is insidious. Potential causes of β-cell dysfunction include glucose toxicity, lipotoxicity, increased secretory demand due to insulin resistance, inflammation, and inflammatory cytokines (such as interleukin-1). By the time clinical signs are apparent, approximately 50% of the β-cell mass is gone. Islets in obese animals with T2DM have β-cell hypertrophy, vacuolation, cytoplasmic glycogen accumulation, apoptosis, islet congestion, mononuclear cell infiltration, and fibrosis with collagen deposition. Some islets may have irregular shapes with cellular hypertrophy and hyperplasia in addition to the degenerative, inflammatory and fibrotic changes. Eventually, enough β cells are lost so that the animals develop insulin-dependent DM.
Islet Cell Regeneration
Despite failure of β cells over time in conditions such as obesity and T2DM, islet β cells have a remarkable plasticity and capability to regenerate after injury. New β cells can develop from proliferation of differentiated β cells, neogenesis from uncommitted stem cells in the islets or pancreatic ducts, or by transdifferentiation from other islet cell types, such as α cells. β-Cell regeneration by the three potential mechanisms has been demonstrated in rodents and rodent models of β-cell injury, but the mechanisms and relevance are still controversial. In humans, α cells appear to be more capable of proliferation than β cells in response to disease or injury of the islets. Some of the regenerative α cells may be positive immunohistochemically for both insulin and glucagon. Proliferation of islet cells can be demonstrated using proliferation markers, such as Ki-67. It has been estimated that β cells survive for about 30 days in growing rats, after which they undergo apoptosis and are replaced by new cells. Proliferation may decrease with age.
Islet Cell Hyperplasia and Neoplasia
Islet cell hyperplasia and adenomas are common age-related lesions found in multiple species. Islet cell carcinomas occur less commonly. There is a continuum of islet cell hyperplasia with progression to adenoma and carcinoma. Proliferative lesions of the islets cells usually affect the β cells but can affect α cells or other cell types. Dietary restriction will reduce the background incidence of islet cell tumors in rats. Pancreatic islet cell hyperplasia is characterized by enlargement of multiple islets (up to 500 μm in diameter), but all islets are not typically involved (Figure 20.45). There is no compression of the adjacent pancreas, and there is lack of fibrous encapsulation. The islet cells usually appear normal but may be elongated. There is low mitotic activity. Adjacent islets may coalesce to form enlarged, irregularly shaped islets.
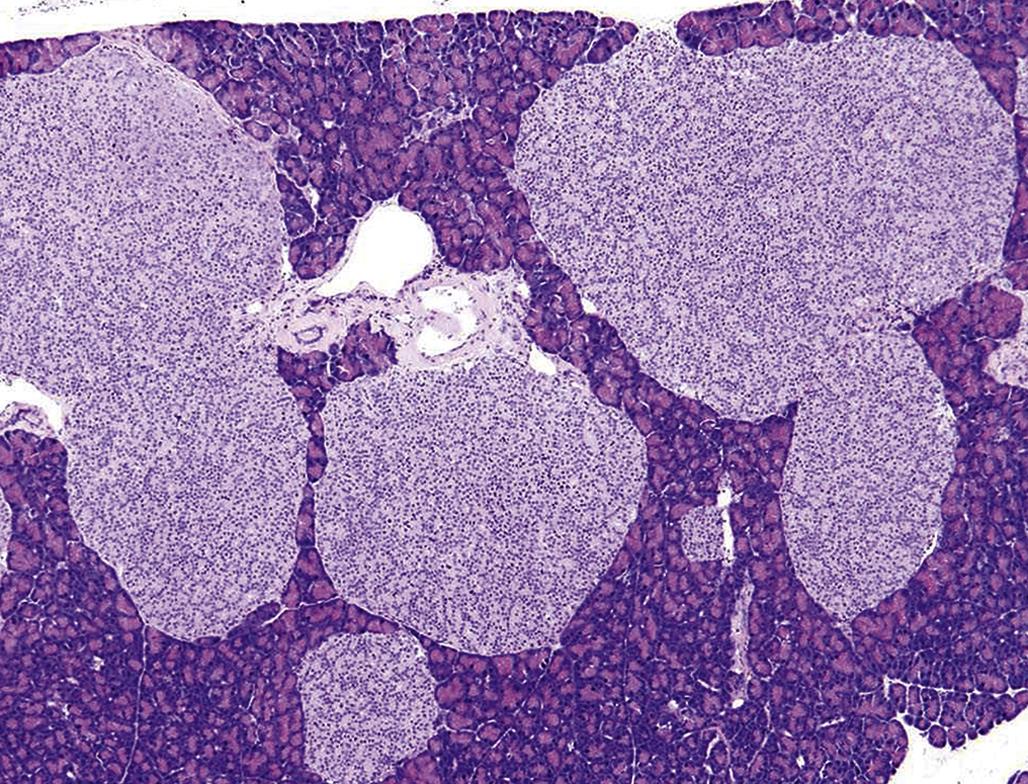
Adenomas are solitary and grow by expansion, inducing compression of adjacent pancreas; however, they are well differentiated (Figure 20.46). Adenomas may coalesce with adjacent hyperplastic islets. There may be a partial fibrous capsule. Islet cell carcinomas invade into the tumor capsule and adjacent pancreatic tissues (Figure 20.47). Distant metastases may be present in regional lymph nodes or the liver.
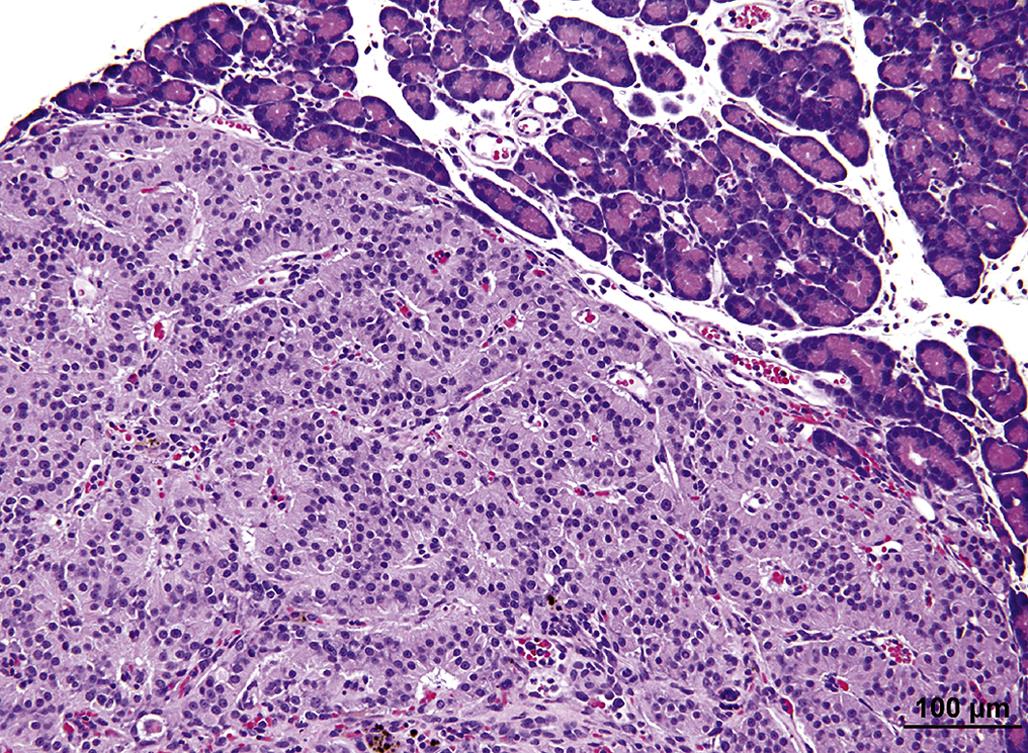
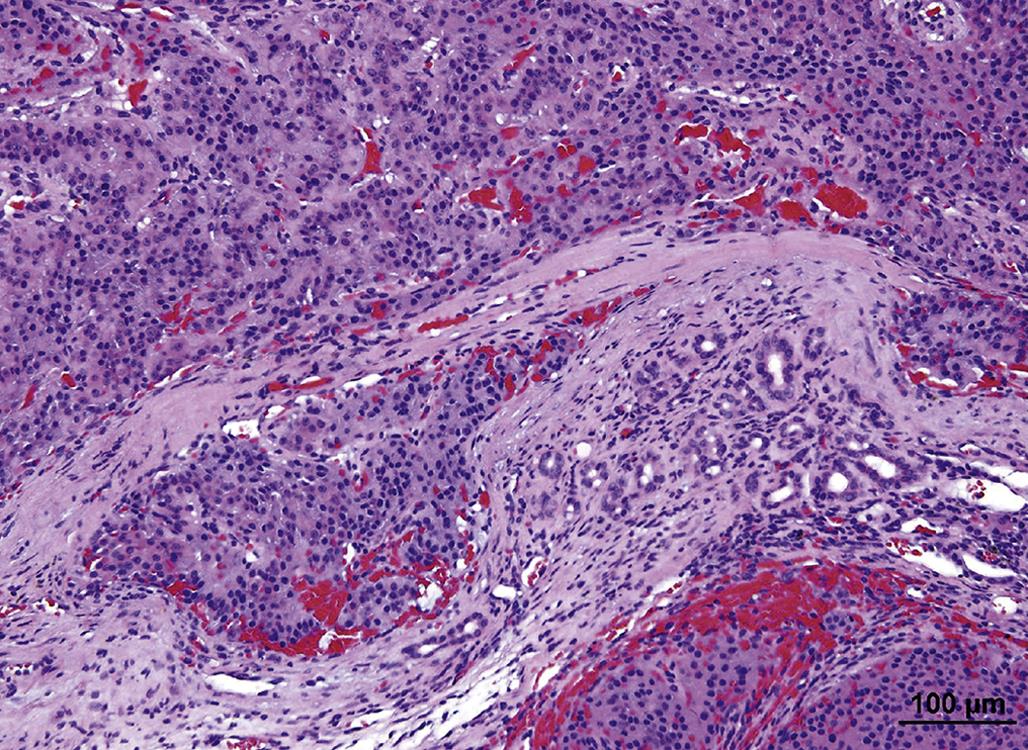
Mechanisms of Toxicity
Compounds that Cause Proliferation
Glucocorticoids increase blood glucose concentrations and promote insulin resistance. Prolonged administration of glucocorticoids to rats has the potential to increase the incidence of islet cell hyperplasia and neoplasia in 2-year studies. Male rats are particularly sensitive to glucocorticoid-induced islet cell tumors. This effect is not thought to have relevance to humans.
Glucagon receptor antagonists or antireceptor antibodies have the potential for treatment of T2DM due to their ability to decrease blood glucose and hepatic production of glucose. Animals with experimental DM and treated with functional or structural glucagon receptor antagonists develop α-cell hyperplasia with hyperglucagonemia. In addition, GLP-1 levels are increased, which have a beneficial effect in alleviating the DM. Plasma glucagon returns to normal quickly after cessation of antiglucagon receptor therapy even though regression of α-cell hyperplasia is slow.
Compounds that increase PRL secretion increase the incidence of pancreatic islet cell hyperplasia in rats and mice. PRL secretion is inhibited by dopamine. Therefore, dopamine antogonists or dopamine receptor antagonists will increase PRL. Sulpiride (dopamine receptor antagonist) has induced pancreatic islet cell tumors in rats. The combination of naproxen and metoclopramide (structurally related to sulpiride) has induced pancreatic islet carcinomas in male rats.
Nafarelin acetate (nasal solution) is a synthetic analog of GnRH, which increased pancreatic islet cell adenomas in male and female rats in 2-year dosage studies. Leuprolide acetate (depot dosage formulation) increased pancreatic islet cell adenomas in female rats in 2-year studies. The mechanism by which GnRH and its analogs induces islet cell tumors in rats is unknown. However, it has been reported that GnRH and its receptor are present in the rat pancreas and GnRH agonists increase insulin resistance.
Islet cell hyperplasia has been reported in transgenic mice that over express the EAT/mcl-1 gene that is related to Bcl-2. Bcl-2 and its analogs bind to the BAD and BAK proteins to inhibit apoptosis. The islet cell hyperplasia in EAT/mcl-1 mice is due to decreased apoptosis of β and α cells and resulted in a mild increase in serum insulin concentration. A few of the mice also developed islet cell adenomas.
Streptozotocin (Streptozocin) and Alloxan
Streptozotocin is a glucosamine-nitrosourea compound and alkylating agent that damages DNA and which is particularly toxic to islet β cells. Streptozotocin spontaneously degrades to carbonium ions ( ), which forms DNA adducts by alkylating purine and pyrimidine bases. It is approved by the FDA for use in patients with metastatic β-cell cancer. Streptozotocin is widely used to induce T1DM in animal models, such as rats and mice. A single dose of 50 mg/kg body weight in a rat will cause necrosis of β cells followed by β-cell loss and atrophy of the islets. Alloxan is a cyclic urea analog that is chemically similar to streptozotocin. It is selectively toxic to β cells and can induce T1DM. Alloxan is reduced to dialuric acid in the body and then autooxidized to H2O2 and hydroxyl free radicals, which can form hydroxyl adducts with DNA.
), which forms DNA adducts by alkylating purine and pyrimidine bases. It is approved by the FDA for use in patients with metastatic β-cell cancer. Streptozotocin is widely used to induce T1DM in animal models, such as rats and mice. A single dose of 50 mg/kg body weight in a rat will cause necrosis of β cells followed by β-cell loss and atrophy of the islets. Alloxan is a cyclic urea analog that is chemically similar to streptozotocin. It is selectively toxic to β cells and can induce T1DM. Alloxan is reduced to dialuric acid in the body and then autooxidized to H2O2 and hydroxyl free radicals, which can form hydroxyl adducts with DNA.
The DNA damage and strand breaks induced by streptozotocin or alloxan can be repaired by enzymes (such as poly[ADP]-ribose synthetase) that require nicotinamide adenine dinucleotide (NAD). Depletion of NAD also leads to β-cell damage and death. DNA damage eventually leads to development of β-cell neoplasms. Nicotinamide can be used to maintain NAD levels in β cells and reduce the toxicity of streptozotocin and alloxan.
Zinc
Zinc is an essential micronutrient and is associated with insulin production and metabolism and the normal function of β cells. Zinc is present in the secretory granules of β and α cells. Zinc deficiency predisposes to T1DM and T2DM, and zinc supplementation has a protective effect likely through antioxidant mechanisms and induction of metallothionein.
Summary
The endocrine glands are an important group of organs that are susceptible to chemical-induced changes that occur in short-term and long-term preclinical toxicology studies. It is important to differentiate spontaneous and stress-related changes from compound-induced effects. There are multiple examples of drugs that are approved for human use that have been associated with degenerative or neoplastic changes in the endocrine glands of laboratory animals, but the weight of evidence on the pathogenesis and mode of action has often demonstrated a lack of relevance for humans. Furthermore the mode of action of chemicals on the toxicology of the endocrine glands can be direct or indirect. Therefore, studies on the modes of action require knowledge and experimental interrogation of physiological control and feedback mechanisms, hormone distribution and receptors on diverse target cells, the primary endocrine cells, and peripheral metabolism and excretion of hormones. In addition, there are significant interspecies differences in the physiology and pathology of the endocrine glands, which makes it imperative to understand the mode of action of chemicals in order to compare preclinical toxicology findings in different species and predict human relevance. This chapter has reviewed the pathophysiology of the endocrine glands in the species typically used for preclinical toxicology studies, and includes toxicologic mechanisms, classic examples of chemical-induced changes and their modes of action, and spontaneous diseases.
Acknowledgments
The contributions of Drs. Thomas J. Rosol, Ronald A. DeLellis, Philip W. Harvey, and Catherine Sutcliffe for the original contribution in Handbook of Toxicologic Pathology, 3rd edition are gratefully acknowledged. Also acknowledged are Dr. Charles C. Capen (deceased), Professor Emeritus from The Ohio State University, for the original edition of this chapter in previous editions of Handbook of Toxicologic Pathology and Fundamentals of Toxicologic Pathology. In addition, the contributions of the Digital Imaging Unit and Dr. Ron A. Herbert of the National Toxicology Program Archives for some of the images are recognized.