Conclusion
The first essential challenge that must be accomplished in B-cell development is the generation of B cells with a vast repertoire—many billions—of B-cell receptor specificities that are sufficient to ensure responses to virtually anything foreign that enters the body. The antibody diversity generated by gene rearrangements, junctional diversification, and different combinations of heavy and light chains (discussed in Chapter 6) is amplified by the fact that millions of new B cells are generated every day. B cells whose antibody specificities aren’t needed turn over, replaced by new B cells generated in the bone marrow by the processes of hematopoiesis and B-cell development.
Progression through the sequence of stages of hematopoiesis, commitment to the lymphoid lineage, and early B-cell development in the bone marrow resulting in the formation of immature B cells is driven by networks of transcription factors. Critically important is the key E2A → EBF1 → PAX5 transcription factor chain that constitutes a feed-forward regulatory network, with the end factor, PAX5, turning on the genes that determine the B lymphocyte phenotype that the cells retain until activated by antigen and other signals to differentiate into antibody-secreting plasma cells. The complex web of transcription factors modulates and is modulated by a variety of epigenetic changes that control the gene transcription and protein expression that characterize each stage.
The ordered and successful recombination of heavy and light-chain genes is programmed into and in some cases drives progression through the stages of B-cell development, with checkpoints along the way to ensure that the rearrangements are good ones that will eventually yield functional BCRs. So after the VH-DJ recombination, the µ heavy chain is tested for its ability to pair up and to associate with the surrogate light-chain polypeptide; if all goes well the resulting pre-BCR signals the pre-B cell to stop rearranging heavy-chain genes. The resulting heavy-chain allelic exclusion is important to ensuring that the B cell will not have a variety of mixed-chain BCRs that would have different specificities. Signals from the pre-BCR drive the pre-B cell to divide, generating a pool of cells expressing that good heavy chain, and then each of those cells will begin rearranging light-chain genes, starting (in the mouse) with κ before trying λ. All it takes is one productive rearrangement out of the four light-chain rearrangements possible, and the cell now expresses mIgM, marking it as an immature B cell. Expression of the receptor—without ligand binding—is sufficient to signal the cell to terminate light-chain rearrangements, ensuring that the cell expresses only one heavy chain and one light chain. This constitutes the second checkpoint in B-cell development.
Immature B cells now have to deal with the second challenge of B-cell development, making sure that they are not autoreactive. This is accomplished by the induction of apoptosis in response to strong signals coming from mIgM receptors that have been cross-linked by binding to multiple self antigens in their bone marrow surroundings. If they receive such a signal, before they die they are given the opportunity to rearrange any remaining light-chain genes (either on a different chromosome or using V and J segments flanking the rearranged and expressed light-chain gene). If their attempts at receptor editing fail to provide a light chain that does not form a self-reactive BCR, the cell undergoes apoptosis.
The surviving immature B cells now leave their bone marrow birthplace for the spleen, where they become T1 transitional cells and are tested for recognition of peripheral self antigens, which can also induce apoptosis. Passing that test, the cells become T2 B cells; if they then receive survival signals from BAFF they go on to traffic to the follicles of the spleen and lymph nodes where they constitute an army of mature B cells with diverse non-autoreactive BCR specificities, ready to respond to foreign invaders along with their helper T-cell partners. Other T2 B cells may be induced to become anergic by weakly stimulating self antigens such as soluble self proteins and be characterized as T3 cells.
While the follicular B cells (also called B-2 cells) described above are the major (or conventional) population of B cells in mice and probably humans as well, several other B-cell populations exist as variations on that theme. B-1 B cells and marginal zone (MZ) B cells seem to have been positively selected for low affinity recognition of some self antigens; they have non-random BCR specificities (in some cases using specific conserved V region genes) which seem to have evolved to recognize certain polysaccharide and lipid antigens that cross-react with microbial antigens. MZ B cells are derived from T2 B cells; instead of trafficking to lymphoid follicles they take up residence in the marginal zone of the spleen, where they are exposed to blood borne antigens entering through the marginal sinus. As they often do not require T-cell help, they can respond more quickly to antigen than can follicular B-2 cells.
B-1a (CD5+) B cells are the most unique population of B cells, as they are not derived from HSCs. Instead, most appear to have their origins in early embryonic progenitor cells that form before HSCs appear; the B-1a progenitors seed the pleural and peritoneal cavities where they self-renew throughout life. In addition to sharing specificities for polysaccharide and lipid antigens on self-components and bacteria, B-1a and MZ B cells both spontaneously produce natural antibodies that have some protective benefits. Thus, while B cells don’t have quite the functional diversity exhibited by T cells, there is now strong evidence for the existence of B-cell subsets with different developmental origins and functions.
REFERENCES
- Bankovich, A. J., et al. 2007. Structural insight into pre-B cell receptor function. Science 316:291.
- Bao, Y., and X. Cao. 2016. Epigenetic control of B cell development and B-cell–related immune disorders. Clinical Reviews in Allergy and Immunology 50:301.
- Cambier, J. C., S. B. Gauld, K. T. Merrell, and B. J. Vilen. 2007. B-cell anergy: from transgenic models to naturally occurring anergic B cells? Nature Reviews Immunology 7:633.
- Casola, S. 2007. Control of peripheral B-cell development. Current Opinions in Immunology 19:143.
- Dorshkind, K., and E. Montecino-Rodriguez. 2007. Fetal B-cell lymphopoiesis and the emergence of B-1-cell potential. Nature Reviews Immunology 7:213.
- Ghosn, E. E., and Y. Yang. 2015. Hematopoietic stem cell-independent B-1a lineage. Annals of the New York Academy of Sciences 1362:23.
- Goodnow, C. C. 1992. Transgenic mice and analysis of B-cell tolerance. Annual Review of Immunology 10:489.
- Hoek, K. L., et al. 2006. Transitional B cell fate is associated with developmental stage-specific regulation of diacylglycerol and calcium signaling upon B cell receptor engagement. Journal of Immunology 177:5405.
- Kee, B. L. 2009. E and ID proteins branch out. Nature Reviews Immunology 9:175.
- Koralov, S. B., et al. 2008. Dicer ablation affects antibody diversity and cell survival in the B lymphocyte lineage. Cell 132:860.
- Kurosaki, T., H. Shinohara, and Y. Baba. 2010. B cell signaling and fate decision. Annual Review of Immunology 28:21.
- Liao, D. 2009. Emerging roles of the EBF family of transcription factors in tumor suppression. Molecular Cancer Research 7:1893.
- Mackay, F., and P. Schneider. 2009. Cracking the BAFF code. Nature Reviews Immunology 9:491.
- Malin, S., et al. 2010. Role of STAT5 in controlling cell survival and immunoglobulin gene recombination during pro-B cell development. Nature Immunology 11:171.
- Melchers, F. 2015. Checkpoints that control B cell development. Journal of Clinical Investigation 125:2203.
- Monroe, J. G., and K. Dorshkind. 2007. Fate decisions regulating bone marrow and peripheral B lymphocyte development. Advances in Immunology 95:1.
- Nagasawa, T. 2006. Microenvironmental niches in the bone marrow required for B-cell development. Nature Reviews Immunology 6:107.
- Nemazee, D. 2006. Receptor editing in lymphocyte development and central tolerance. Nature Reviews Immunology 6:728.
- Nemazee, D. A., and K. Bürki. 1989. Clonal deletion of B lymphocytes in a transgenic mouse bearing anti-MHC class I antibody genes. Nature 337:562.
- Nutt, S. L., and B. L. Kee. 2007. The transcriptional regulation of B cell lineage commitment. Immunity 26:715.
- Pillai, S., and A. Cariappa. 2009. The follicular versus marginal zone B lymphocyte cell fate decision. Nature Reviews Immunology 9:767.
- Rothenberg, E. V. 2014. Transcriptional control of early T and B cell developmental choices. Annual Review of Immunology 32:283.
- Somasundaram, R., M. A. J. Prasad, J. Ungerback, and M. Sigvardsson. 2016. Transcription factor networks in B-cell differentiation link development to acute lymphoid leukemia. Blood 126:144.
- Srivastava, B., W. J. Quinn III, K. Hazard, J. Erikson, and D. Allman. 2005. Characterization of marginal zone B cell precursors. Journal of Experimental Medicine 202:1225.
- Tokoyoda, K., T. Egawa, T. Sugiyama, B. I. Choi, and T. Nagasawa. 2004. Cellular niches controlling B lymphocyte behavior within bone marrow during development. Immunity 20:707.
- Übelhart, R., M. Werner, and H. Jumaa. 2016. Assembly and function of the precursor B-cell receptor. Current Topics in Microbiology and Immunology 393:3.
- von Boehmer, H., and F. Melchers. 2010. Checkpoints in lymphocyte development and autoimmune disease. Nature Immunology 11:14.
- Whitlock, C. A., and O. Witte. 1982. Long-term culture of B lymphocytes and their precursors from murine bone marrow. Proceedings of the National Academy of Sciences USA 79:3608.
- Xiao, C., and K. Rajewsky. 2009. MicroRNA control in the immune system: basic principles. Cell 136:26.
- Yin, T., and L. Li. 2006. The stem cell niches in bone. Journal of Clinical Investigation 116:1195.
Useful Website
www.bio.davidson.edu/courses/immunology/Flash/Bcellmat.html An unusual animation of B-cell development.
STUDY QUESTIONS
- You wish to study the development of B-1 B cells in the absence of the other two major B-cell subsets. You have a recipient Rag1−/− mouse that you have already repopulated with T cells. What would you choose to be your source of B-1 progenitors and why? From which anatomical sites would you expect to harvest the B-1 B cells?
- Describe the phenotypic and functional differences between T1 and T2 immature B cells.
- Following expression of the pre-B-cell receptor on the progenitor B-cell surface, the B cell undergoes a few rounds of cell division. What purpose do these rounds of cell division serve in the development of the B-cell repertoire?
- Immature B cells bearing potentially autoimmune receptors can be managed in three ways to minimize the probability of disease. Describe these three strategies, noting whether they are shared by T-cell progenitors.
- You suspect that a new transcription factor is expressed at the pre-pro-B-cell stage of development. How would you test your hypothesis? What is the status of heavy- and light-chain rearrangement at this stage of development and how would you test it?
- How would you determine whether a particular stage of B-cell development occurs in association with a stromal cell that expresses CXCL12?
- Describe the order in which B-cell receptor genes undergo rearrangement, indicating at what steps you might expect to see the B cell express one or both chains on the cell surface. In what sense(s) does this gene rearrangement process mimic the analogous progression in αβ T cells, and in what ways do the two processes differ?
- In addition to the experiment shown in Figure 9-9 and Table 9-4, Goodnow and colleagues set up another experimental group. In this case a transgene was created that allowed HEL to be expressed as a membrane-associated protein throughout the body, including in the bone marrow. These membrane HEL-transgenic mice were crossed to mice transgenic for the anti-HEL-specific antibody. What do you think happens to the B cells in the double-transgenic mice? Do you think B cells expressing an HEL-specific BCR would be found in the periphery? Do you think these mice would generate an anti-HEL antibody response after immunization with HEL?
ANALYZE THE DATA
- The two columns of data in the following figure are flow cytometric plots that describe the levels of antigens denoted on the x and y axes. Left: Antigens present on spleen (a) and bone marrow (b) from wild-type (genetically normal) animals. The plots represent all lymphocytes in the spleen (a) or B-cell progenitor and precursor cells in the bone marrow (b). Right: The same plots for animals in which the Dicer gene has been knocked out. As you will recall, Dicer is required for the maturation of microRNAs.

- For each pair of plots, describe the differences in the cell populations, indicating whether the differences reflect losses or gains in particular developing B-cell populations.
- At what point(s) in B-cell development do you think microRNAs are functioning?
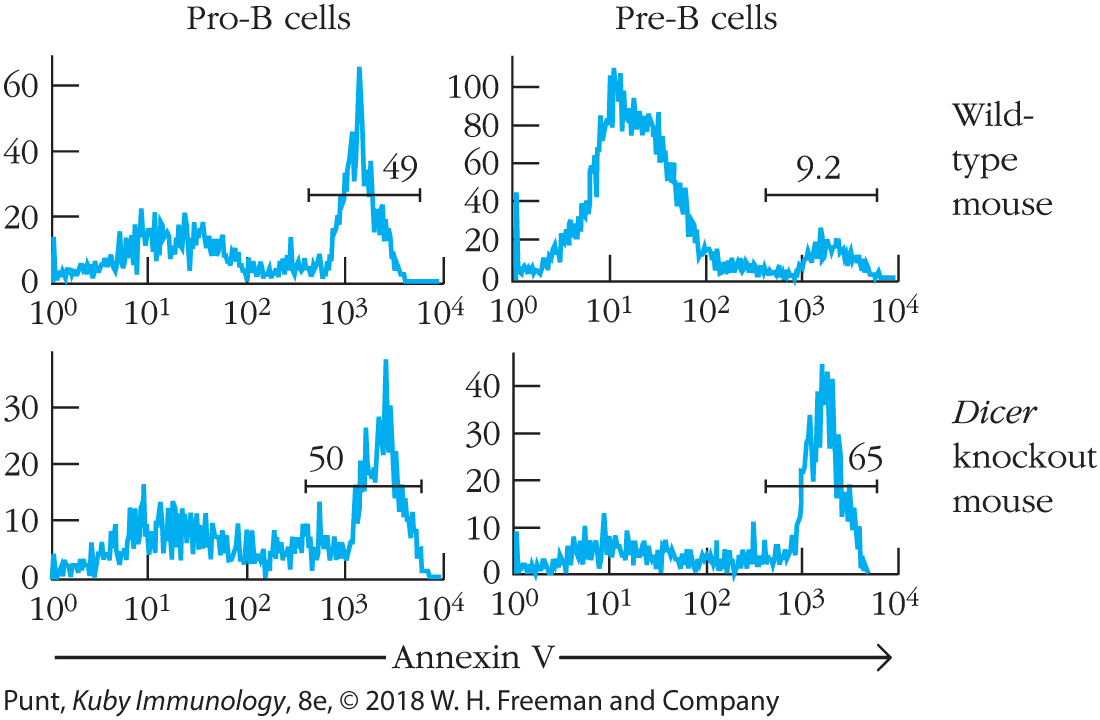
- The following figure is derived from the same article as the figure in the preceding question. In this case the data are expressed as histograms, in which the y axis represents the number of cells binding the molecule shown on the x axis, annexin A5. Annexin A5 binds to phosphatidylserine on the outer leaflet of cell membranes. Phosphatidylserine is found on the outer leaflet only in cells about to undergo apoptosis. The top two panels represent cells from a wild-type animal, and the bottom two panels represent cells from animals in which the Dicer gene has been knocked out.
- Does the presence of Dicer have an effect on the fraction of pro-B cells undergoing apoptosis? Explain your reasoning.
- Does the presence of Dicer have an effect on the fraction of pre-B cells undergoing apoptosis? Again explain your reasoning.
- Describe one function that you now think microRNAs fulfill in B-cell development.