ONE
VIVA PRIMORDIUM
IMAGINE A PLANET ABOUT THE size of our own, about as far from its star, rotating around its axis a bit faster, such that a day lasts about twenty hours. It is covered with a shallow ocean of salty water and has no continents to speak of—just some sporadic chains of basaltic black islands peeking up above the waterline. Its atmosphere does not have the same mix of gases as ours. It is a humid, toxic blanket of nitrogen, methane, and carbon dioxide.
There is no oxygen. There is no life.
Because this planet, our planet as it was 4 billion years ago, is a ruthlessly unforgiving place. Hot and volcanic. Electric. Tumultuous.
But that is about to change. Water is pooling next to warm thermal vents that litter one of the larger islands. Organic molecules cover all surfaces, having ridden in on the backs of meteorites and comets. Sitting on dry, volcanic rock, these molecules will remain just molecules, but when dissolved in pools of warm water, through cycles of wetting and drying at the pools’ edges, a special chemistry takes place.1 As the nucleic acids concentrate, they grow into polymers, the way salt crystals form when a seaside puddle evaporates. These are the world’s first RNA molecules, the predecessors to DNA. When the pond refills, the primitive genetic material becomes encapsulated by fatty acids to form microscopic soap bubbles—the first cell membranes.2
It doesn’t take long, a week perhaps, before the shallow ponds are covered with a yellow froth of trillions of tiny precursor cells filled with short strands of nucleic acids, which today we call genes.
Most of the protocells are recycled, but some survive and begin to evolve primitive metabolic pathways, until finally the RNA begins to copy itself. That point marks the origin of life. Now that life has formed—as fatty-acid soap bubbles filled with genetic material—they begin to compete for dominance. There simply aren’t enough resources to go around. May the best scum win.
Day in and day out, the microscopic, fragile life-forms begin to evolve into more advanced forms, spreading into rivers and lakes.
Along comes a new threat: a prolonged dry season. The level of the scum-covered lakes has dropped by a few feet during the dry season, but the lakes have always filled up again as the rains returned. But this year, thanks to unusually intense volcanic activity on the other side of the planet, the annual rains don’t fall as they usually do and the clouds pass on by. The lakes dry up completely.
What remains is a thick, yellow crust covering the lake beds. It is an ecosystem defined not by the annual waxing and waning of the waters but by a brutal struggle for survival. And more than that: it is a fight for the future—because the organisms that survive will be the progenitors of every living thing to come: archaea, bacteria, fungi, plants, and animals.
Within this dying mass of cells, each scrapping for and scraping by on the merest minimums of nutrients and moisture, each one doing whatever it can to answer the primal call to reproduce, there is a unique species. Let’s call it Magna superstes. That’s Latin for “great survivor.”
It does not look very different from the other organisms of the day, but M. superstes has a distinct advantage: it has evolved a genetic survival mechanism.
There will be far more complicated evolutionary steps in the eons to come, changes so extreme that entire branches of life will emerge. These changes—the products of mutations, insertions, gene rearrangements, and the horizontal transfer of genes from one species to another—will create organisms with bilateral symmetry, stereoscopic vision, and even consciousness.
By comparison, this early evolutionary step looks, at first, to be rather simple. It is a circuit. A gene circuit.
The circuit begins with gene A, a caretaker that stops cells from reproducing when times are tough. This is key, because on early planet Earth, most times are tough. The circuit also has a gene B, which encodes for a “silencing” protein. This silencing protein shuts gene A off when times are good, so the cell can make copies of itself when, and only when, it and its offspring will likely survive.
The genes themselves aren’t novel. All life in the lake has these two genes. But what makes M. superstes unique is that the gene B silencer has mutated to give it a second function: it helps repair DNA. When the cell’s DNA breaks, the silencing protein encoded by gene B moves from gene A to help with DNA repair, which turns on gene A. This temporarily stops all sex and reproduction until the DNA repair is complete.
This makes sense, because while DNA is broken, sex and reproduction are the last things an organism should be doing. In future multicellular organisms, for instance, cells that fail to pause while fixing a DNA break will almost certainly lose genetic material. This is because DNA is pulled apart prior to cell division from only one attachment site on the DNA, dragging the rest of the DNA with it. If DNA is broken, part of a chromosome will be lost or duplicated. The cells will likely die or multiply uncontrollably into a tumor.
With a new type of gene silencer that repairs DNA, too, M. superstes has an edge. It hunkers down when its DNA is damaged, then revives. It is superprimed for survival.
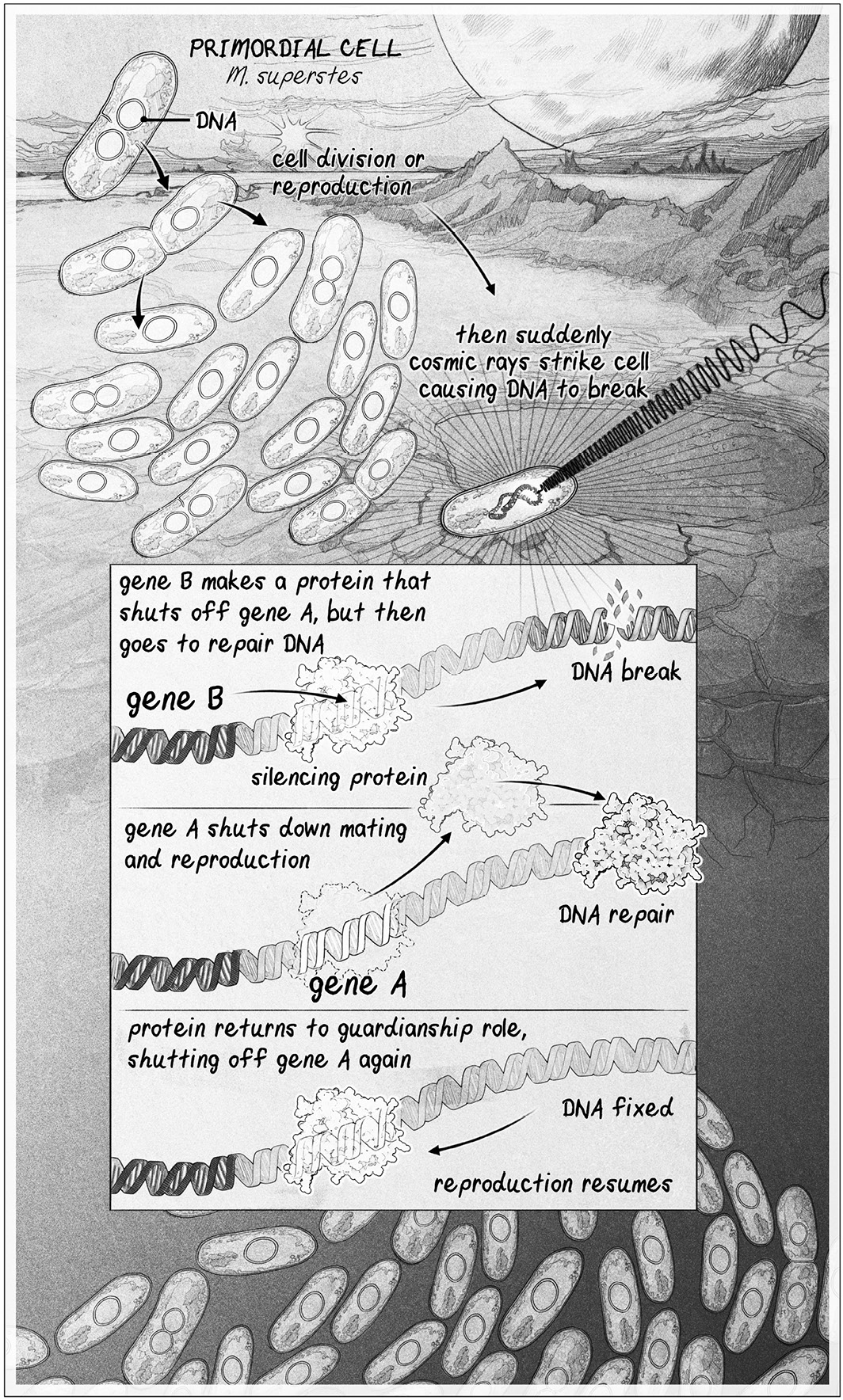
THE EVOLUTION OF AGING. A 4-billion-year-old gene circuit in the first life-forms would have turned off reproduction while DNA was being repaired, providing a survival advantage. Gene A turns off reproduction, and gene B makes a protein that turns off gene A when it is safe to reproduce. When DNA breaks, however, the protein made by gene B leaves to go repair DNA. As a result, gene A is turned on to halt reproduction until repair is complete. We have inherited an advanced version of this survival circuit.
And that’s good, because now comes yet another assault on life. Powerful cosmic rays from a distant solar eruption are bathing the Earth, shredding the DNA of all the microbes in the dying lakes. The vast majority of them carry on dividing as if nothing has happened, unaware that their genomes have been broken and that reproducing will kill them. Unequal amounts of DNA are shared between mother and daughter cells, causing both to malfunction. Ultimately, the endeavor is hopeless. The cells all die, and nothing is left.
Nothing, that is, but M. superstes. For as the rays wreak their havoc, M. superstes does something unusual: thanks to the movement of protein B away from gene A to help repair the DNA breaks, gene A switches on and the cells stop almost everything else they are doing, turning their limited energy toward fixing the DNA that has been broken. By virtue of its defiance of the ancient imperative to reproduce, M. superstes has survived.
When the latest dry period ends and the lakes refill, M. superstes wakes up. Now it can reproduce. Again and again it does so. Multiplying. Moving into new biomes. Evolving. Creating generations upon generations of new descendants.
They are our Adam and Eve.
Like Adam and Eve, we don’t know if M. superstes ever existed. But my research over the past twenty-five years suggests that every living thing we see around us today is a product of this great survivor, or at least a primitive organism very much like it. The fossil record in our genes goes a long way to proving that every living thing that shares this planet with us still carries this ancient genetic survival circuit, in more or less the same basic form. It is there in every plant. It is there in every fungus. It is there in every animal.
It is there in us.
I propose the reason this gene circuit is conserved is that it is a rather simple and elegant solution to the challenges of a sometimes brutish and sometimes bounteous world that better ensures the survival of the organisms that carry it. It is, in essence, a primordial survival kit that diverts energy to the area of greatest need, fixing what exists in times when the stresses of the world are conspiring to wreak havoc on the genome, while permitting reproduction only when more favorable times prevail.
And it is so simple and so robust that not only did it ensure life’s continued existence on the planet, it ensured that Earth’s chemical survival circuit was passed on from parent to offspring, mutating and steadily improving, helping life continue for billions of years, no matter what the cosmos brought, and in many cases allowing individuals’ lives to continue for far longer than they actually needed to.
The human body, though far from perfect and still evolving, carries an advanced version of the survival circuit that allows it to last for decades past the age of reproduction. While it is interesting to speculate why our long lifespans first evolved—the need for grandparents to educate the tribe is one appealing theory—given the chaos that exists at the molecular scale, it’s a wonder we survive thirty seconds, let alone make it to our reproductive years, let alone reach 80 more often than not.
But we do. Marvelously we do. Miraculously we do. For we are the progeny of a very long lineage of great survivors. Ergo, we are great survivors.
But there is a trade-off. For this circuit within us, the descendant of a series of mutations in our most distant ancestors, is also the reason we age.
And yes, that definite singular article is correct: it is the reason.
TO EVERYTHING THERE IS A REASON
If you are taken aback by the notion that there is a singular cause of aging, you are not alone. If you haven’t given any thought at all as to why we age, that’s perfectly normal, too. A lot of biologists haven’t given it much thought, either. Even gerontologists, doctors who specialize in aging, often don’t ask why we age—they simply seek to treat the consequences.
This isn’t a myopia specific to aging. As recently as the late 1960s, for example, the fight against cancer was a fight against its symptoms. There was no unified explanation for why cancer happens, so doctors removed tumors as best they could and spent a lot of time telling patients to get their affairs in order. Cancer was “just the way it goes,” because that’s what we say when we can’t explain something.
Then, in the 1970s, genes that cause cancer when mutated were discovered by the molecular biologists Peter Vogt and Peter Duesberg. These so-called oncogenes shifted the entire paradigm of cancer research. Pharmaceutical developers now had targets to go after: the tumor-inducing proteins encoded by genes, such as BRAF, HER2, and BCR-ABL. By inventing chemicals that specifically block the tumor-promoting proteins, we could finally begin to move away from using radiation and toxic chemotherapeutic agents to attack cancers at their genetic source, while leaving normal cells untouched. We certainly haven’t cured all types of cancer in the decades since then, but we no longer believe it’s impossible to do so.
Indeed, among an increasing number of cancer researchers, optimism abounds. And that hopefulness was at the heart of what was arguably the most memorable part of President Barack Obama’s final State of the Union address in 2016.
“For the loved ones we’ve all lost, for the family we can still save, let’s make America the country that cures cancer once and for all,” Obama said as he stood in the House of Representatives chamber and called for a “cancer moon shot.” When he placed then Vice President Joe Biden—whose son Beau had died of brain cancer a year earlier—in charge of the effort, even some of the Democrats’ staunch political enemies had trouble holding back the tears.
In the days and weeks that followed, many cancer experts noted that it would take far more than the year remaining to the Obama-Biden administration to end cancer. Very few of those experts, however, said it absolutely couldn’t be done. And that’s because, in the span of just a few decades, we had completely changed the way we think about cancer. We no longer submit ourselves to its inevitability as part of the human condition.
One of the most promising breakthroughs in the past decade has been immune checkpoint therapy, or simply “immunotherapy.” Immune T-cells continually patrol our body, looking for rogue cells to identify and kill before they can multiply into a tumor. If it weren’t for T-cells, we’d all develop cancer in our twenties. But rogue cancer cells evolve ways to fool cancer-detecting T-cells so they can go on happily multiplying. The latest and most effective immunotherapies bind to proteins on the cancer cells’ surface. It is the equivalent of taking the invisible cloak off cancer cells so T-cells can recognize and kill them. Although fewer than 10 percent of all cancer patients currently benefit from immunotherapy, that number should increase thanks to the hundreds of trials currently in progress.
We continue to rail against a disease we once accepted as fate, pouring billions of dollars into research each year, and the effort is paying off. Survival rates for once lethal cancers are increasing dramatically. Thanks to a combination of a BRAF inhibitor and immunotherapy, survival of melanoma brain metastases, one of the deadliest types of cancer, has increased by 91 percent since 2011. Between 1991 and 2016, overall deaths from cancer in the United States declined by 27 percent and continue to fall.3 That’s a victory measured in millions of lives.
Aging research today is at a similar stage as cancer research was in the 1960s. We have a robust understanding of what aging looks like and what it does to us and an emerging agreement about what causes it and what keeps it at bay. From the looks of it, aging is not going to be that hard to treat, far easier than curing cancer.
Up until the second half of the twentieth century, it was generally accepted that organisms grow old and die “for the good of the species”—an idea that dates back to Aristotle, if not further. This idea feels quite intuitive. It is the explanation proffered by most people at parties.4 But it is dead wrong. We do not die to make way for the next generation.
In the 1950s, the concept of “group selection” in evolution was going out of style, prompting three evolutionary biologists, J. B. S. Haldane, Peter B. Medawar, and George C. Williams, to propose some important ideas about why we age. When it comes to longevity, they agreed, individuals look out for themselves. Driven by their selfish genes, they press on and try to breed for as long and as fast as they can, so long as it doesn’t kill them. (In some cases, however, they press on too much, as my great-grandfather Miklós Vitéz, a Hungarian screenwriter, proved to his bride forty-five years his junior on their wedding night.)
If our genes don’t ever want to die, why don’t we live forever? The trio of biologists argued that we experience aging because the forces of natural selection required to build a robust body may be strong when we are 18 but decline rapidly once we hit 40 because by then we’ve likely replicated our selfish genes in sufficient measure to ensure their survival. Eventually, the forces of natural selection hit zero. The genes get to move on. We don’t.
Medawar, who had a penchant for verbiage, expounded on a nuanced theory called “antagonistic pleiotropy.” Put simply, it says genes that help us reproduce when we are young don’t just become less helpful as we age, they can actually come back to bite us when we are old.
Twenty years later, Thomas Kirkwood at Newcastle University framed the question of why we age in terms of an organism’s available resources. Known as the “Disposable Soma Hypothesis,” it is based on the fact that there are always limited resources available to species—energy, nutrients, water. They therefore evolve to a point that lies somewhere between two very different lifestyles: breed fast and die young, or breed slowly and maintain your soma, or body. Kirkwood reasoned that organisms can’t breed fast and maintain a robust, healthy body—there simply isn’t enough energy to do both. Stated another way, in the history of life, any line of creature with a mutation that caused it to live fast and attempt to die old soon ran out of resources and was thus deleted from the gene pool.
Kirkwood’s theory is best illustrated by fictitious but potentially real-life examples. Imagine you are a small rodent that is likely to be picked off by a bird of prey. Because of this, you’ll need to pass down your genetic material quickly, as did your parents and their parents before them. Gene combinations that would have provided a longer-lasting body were not enriched in your species because your ancestors likely didn’t escape predation for long (and you won’t, either).
Now consider instead that you are a bird of prey at the top of the food chain. Because of this, your genes—well, actually, your ancestors’ genes—benefited from building a robust, longer-lasting body that could breed for decades. But in return, they could afford to raise only a couple of fledglings a year.
Kirkwood’s hypothesis explains why a mouse lives 3 years while some birds can live to 100.5 It also quite elegantly explains why the American chameleon lizard, Anolis carolinensis, is evolving a longer lifespan as we speak, having found itself a few decades ago on remote Japanese islands without predators.6
These theories fit with observations and are generally accepted. Individuals don’t live forever because natural selection doesn’t select for immortality in a world where an existing body plan works perfectly well to pass along a body’s selfish genes. And because all species are resource limited, they have evolved to allocate the available energy either to reproduction or to longevity, but not to both. That was as true for M. superstes as it was and still is for all species that have ever lived on this planet.
All, that is, except one: Homo sapiens.
Having capitalized on its relatively large brain and a thriving civilization to overcome the unfortunate hand that evolution dealt it—weak limbs, sensitivity to cold, poor sense of smell, and eyes that see well only in daylight and in the visible spectrum—this highly unusual species continues to innovate. It has already provided itself with an abundance of food, nutrients, and water while reducing deaths from predation, exposure, infectious diseases, and warfare. These were all once limits to its evolving a longer lifespan. With them removed, a few million years of evolution might double its lifespan, bringing it closer to the lifespans of some other species at the top of their game. But it won’t have to wait that long, nowhere near that. Because this species is diligently working to invent medicines and technologies to give it the robustness of a much longer lived one, literally overcoming what evolution failed to provide.
CRISIS MODE
Wilbur and Orville Wright could never have built a flying machine without a knowledge of airflow and negative pressure and a wind tunnel. Nor could the United States have put men on the moon without an understanding of metallurgy, liquid combustion, computers, and some measure of confidence that the moon is not made of green cheese.7
In the same way, if we are to make real progress in the effort to alleviate the suffering that comes with aging, what is needed is a unified explanation for why we age, not just at the evolutionary level but at the fundamental level.
But explaining aging at a fundamental level is no easy task. It will have to satisfy all known laws of physics and all rules of chemistry and be consistent with centuries of biological observations. It will need to span the least understood world between the size of a molecule and the size of a grain of sand,8 and it should explain simultaneously the simplest and the most complex living machines that have ever existed.
It should, therefore, come as no surprise that there has never been a unified theory of aging, at least not one that has held up—though not for lack of trying.
One hypothesis, proposed independently by Peter Medawar and Leo Szilard, was that aging is caused by DNA damage and a resulting loss of genetic information. Unlike Medawar, who was always a biologist, who built a Nobel Prize–winning career in immunology, Szilard had come to study biology in a roundabout way. The Budapest-born polymath and inventor lived a nomadic life with no permanent job or address, preferring to spend his time staying with colleagues who satisfied his mental curiosities about the big questions facing humanity. Early in his career, he was a pioneering nuclear physicist and a founding collaborator on the Manhattan Project, which ushered in the age of atomic warfare. Horrified by the countless lives his work had helped end, he turned his tortured mind toward making life maximally long.9
The idea that mutation accumulation causes aging was embraced by scientists and the public alike in the 1950s and 1960s, at a time when the effects of radiation on human DNA were on a lot of people’s minds. But although we know with great certainty that radiation can cause all sorts of problems in our cells, it causes only a subset of the signs and symptoms we observe during aging,10 so it cannot serve as a universal theory.
In 1963, the British biologist Leslie Orgel threw his hat into the ring with his “Error Catastrophe Hypothesis,” which postulated that mistakes made during the DNA-copying process lead to mutations in genes, including those needed to make the protein machinery that copies DNA. The process increasingly disrupts those same processes, multiplying upon themselves until a person’s genome has been incorrectly copied into oblivion.11
Around the same time that Szilard was focusing on radiation, Denham Harman, a chemist at Shell Oil, was also thinking atomically, albeit in a different way. After taking time off to finish medical school at Stanford University, he came up with the “Free Radical Theory of Aging,” which blames aging on unpaired electrons that whiz around within cells, damaging DNA through oxidation, especially in mitochondria, because that is where most free radicals are generated.12 Harman spent the better part of his life testing the theory.
I had the pleasure of meeting the Harman family in 2013. His wife told me that Professor Harman had been taking high doses of alpha-lipoic acid for most of his life to quench free radicals. Considering that he worked tirelessly on his research well into his 90s, I suppose, at the very least, it didn’t hurt.
Through the 1970s and 1980s, Harman and hundreds of other researchers tested whether antioxidants would extend the lifespan of animals. The results overall were disappointing. Although Harman had some success increasing the average lifespan of rodents, such as with the food additive butylated hydroxytoluene, none showed an increase in maximum lifespan. In other words, a cohort of study animals might live a few weeks longer, on average, but none of the animals was setting records for individual longevity. Science has since demonstrated that the positive health effects attainable from an antioxidant-rich diet are more likely caused by stimulating the body’s natural defenses against aging, including boosting the production of the body’s enzymes that eliminate free radicals, not as a result of the antioxidant activity itself.
If old habits die hard, the free-radical idea is heroin. The theory was overturned by scientists within the cloisters of my field more than a decade ago, yet it is still widely perpetuated by purveyors of pills and drinks, who fuel a $3 billion global industry.13 With all that advertising, it is not surprising that more than 60 percent of US consumers still look for foods and beverages that are good sources of antioxidants.14
Free radicals do cause mutations. Of course they do. You can find mutations in abundance, particularly in cells that are exposed to the outside world15 and in the mitochondrial genomes of old individuals. Mitochondrial decline is certainly a hallmark of aging and can lead to organ dysfunction. But mutations alone, especially mutations in the nuclear genome, conflict with an ever-increasing amount of evidence to the contrary.
Arlan Richardson and Holly Van Remmen spent about a decade at the University of Texas at San Antonio testing if increasing free-radical damage or mutations in mice led to aging; it didn’t.16 In my lab and others, it has proven surprisingly simple to restore the function of mitochondria in old mice, indicating that a large part of aging is not due to mutations in mitochondrial DNA, either, at least not until late in life.17
Although the discussion about the role of nuclear DNA mutations in aging continues, there is one fact that contradicts all these theories, one that is difficult to refute.
Ironically, it was Szilard, in 1960, who initiated the demise of his own theory by figuring out to how to clone a human cell.18 Cloning gives us the answer as to whether or not mutations cause aging. If old cells had indeed lost crucial genetic information and this was the cause of aging, we shouldn’t be able to clone new animals from older individuals. Clones would be born old.
It’s a misconception that cloned animals age prematurely. It has been widely perpetuated in the media and even the National Institutes of Health website says so.19 Yes, it’s true that Dolly, the first cloned sheep, created by Keith Campbell and Ian Wilmut at the Roslin Institute at the University of Edinburgh, lived only half a normal lifespan and died of a progressive lung disease. But extensive analysis of her remains showed no sign of premature aging.20 Meanwhile, the list of animal species that have been cloned and proven to live a normal, healthy lifespan now includes goats, sheep, mice, and cows.21
Because of the fact that nuclear transfer works in cloning, we can say with a high degree of confidence that aging isn’t caused by mutations in nuclear DNA. Sure, it’s possible that some cells in the body don’t mutate and those are the ones that end up making successful clones, but that seems highly unlikely. The simplest explanation is that old animals retain all the requisite genetic information to generate an entirely new, healthy animal and that mutations are not the primary cause of aging.22
It’s certainly no dishonor to those brilliant researchers that their theories haven’t withstood the test of time. That’s what happens to most science, and perhaps all of it eventually. In The Structure of Scientific Revolutions, Thomas Kuhn noted that scientific discovery is never complete; it goes through predictable stages of evolution. When a theory succeeds at explaining previously unexplainable observations about the world, it becomes a tool that scientists can use to discover even more.
Inevitably, however, new discoveries lead to new questions that are not entirely answerable by the theory, and those questions beget more questions. Soon the model enters crisis mode and begins to drift as scientists seek to adjust it, as little as possible, to account for that which it cannot explain.
Crisis mode is always a fascinating time in science but one that is not for the faint of heart, as doubts about the views of previous generations continue to grow against the old guard’s protestations. But the chaos is ultimately replaced by a paradigm shift, one in which a new consensus model emerges that can explain more than the previous model.
That’s what happened about a decade ago, as the ideas of leading scientists in the aging field began to coalesce around a new model—one that suggested that the reason so many brilliant people had struggled to identify a single cause of aging was that there wasn’t one.
In this more nuanced view, aging and the diseases that come with it are the result of multiple “hallmarks” of aging:
• Genomic instability caused by DNA damage
• Attrition of the protective chromosomal endcaps, the telomeres
• Alterations to the epigenome that controls which genes are turned on and off
• Loss of healthy protein maintenance, known as proteostasis
• Deregulated nutrient sensing caused by metabolic changes
• Mitochondrial dysfunction
• Accumulation of senescent zombielike cells that inflame healthy cells
• Exhaustion of stem cells
• Altered intercellular communication and the production of inflammatory molecules
Researchers began to cautiously agree: address these hallmarks, and you can slow down aging. Slow down aging, and you can forestall disease. Forestall disease, and you can push back death.
Take stem cells, which have the potential to develop into many other kinds of cells: if we can keep these undifferentiated cells from tiring out, they can continue to generate all the differentiated cells necessary to heal damaged tissues and battle all kinds of diseases.
Meanwhile, we’re improving the rates of acceptance of bone marrow transplants, which are the most common form of stem cell therapy, and using stem cells for the treatment of arthritic joints, type 1 diabetes, loss of vision, and neurodegenerative diseases such as Alzheimer’s and Parkinson’s. These stem cell–based interventions are adding years to people’s lives.
Or take senescent cells, which have reached the end of their ability to divide but refuse to die, continuing to spit out panic signals that inflame surrounding cells: if we can kill off senescent cells or keep them from accumulating in the first place, we can keep our tissues much healthier for longer.
The same can be said for combating telomere loss, the decline in proteostasis, and all of the other hallmarks. Each can be addressed one by one, a little at a time, in ways that can help us extend human healthspans.
Over the past quarter century, researchers have increasingly homed their efforts in on addressing each of these hallmarks. A broad consensus formed that this would be the best way to alleviate the pain and suffering of those who are aging.
There is little doubt that the list of hallmarks, though incomplete, comprises the beginnings of a rather strong tactical manual for living longer and healthier lives. Interventions aimed at slowing any one of these hallmarks may add a few years of wellness to our lives. If we can address all of them, the reward could be vastly increased average lifespans.
As for pushing way past the maximum limit? Addressing these hallmarks might not be enough.
But the science is moving fast, faster now than ever before, thanks to the accumulation of many centuries of knowledge, robots that analyze tens of thousands of potential drugs each day, sequencing machines that read millions of genes a day, and computing power that processes trillions of bytes of data at speeds that were unimaginable just a decade ago. Theories on aging, which were slowly chipped away for decades, are now more easily testable and refutable.
Although it is in its early days, a new shift in thinking is again under way. Once again we find ourselves in a period of chaos—still quite confident that the hallmarks are accurate indicators of aging and its myriad symptoms but unable to explain why the hallmarks occur in the first place.
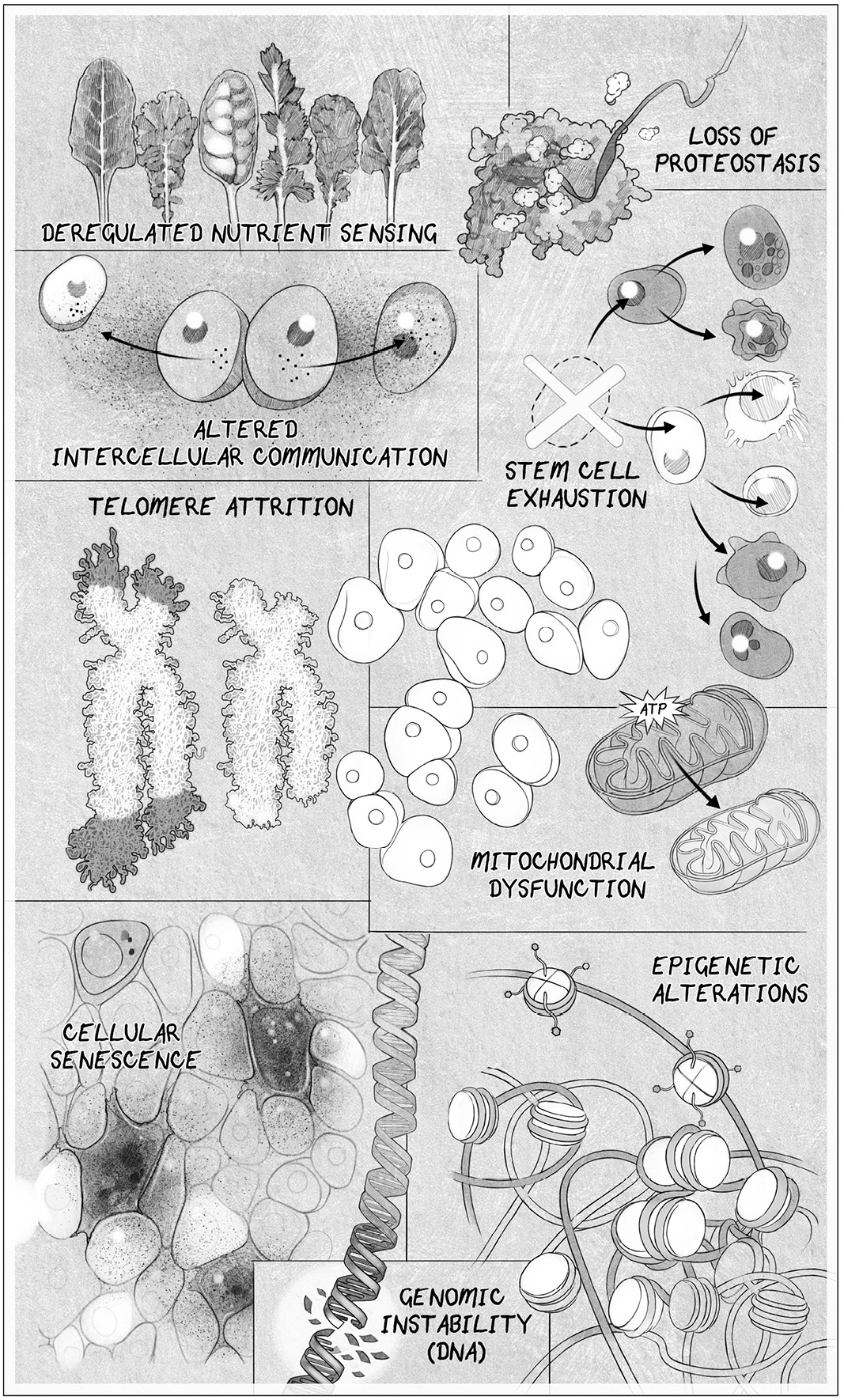
THE HALLMARKS OF AGING. Scientists have settled on eight or nine hallmarks of aging. Address one of these, and you can slow down aging. Address all of them, and you might not age.
It is time for an answer to this very old question.
Now, finding a universal explanation for anything—let alone something as complicated as aging—doesn’t happen overnight. Any theory that seeks to explain aging must not just stand up to scientific scrutiny but provide a rational explanation for every one of the pillars of aging. A universal hypothesis that seems to provide a reason for cellular senescence but not stem cell exhaustion would, for example, explain neither.
Yet I believe that such an answer exists—a cause of aging that exists upstream of all the hallmarks. Yes, a singular reason why we age.
Aging, quite simply, is a loss of information.
You might recognize that loss of information was a big part of the ideas that Szilard and Medawar independently espoused, but it was wrong because it focused on a loss of genetic information.
But there are two types of information in biology, and they are encoded entirely differently. The first type of information—the type my esteemed predecessors understood—is digital. Digital information, as you likely know, is based on a finite set of possible values—in this instance, not in base 2 or binary, coded as 0s and 1s, but the sort that is quaternary or base 4, coded as adenine, thymine, cytosine, and guanine, the nucleotides A, T, C, G of DNA.
Because DNA is digital, it is a reliable way to store and copy information. Indeed, it can be copied again and again with tremendous accuracy, no different in principle from digital information stored in computer memory or on a DVD.
DNA is also robust. When I first worked in a lab, I was shocked by how this “molecule of life” could survive for hours in boiling water and thrilled that it was recoverable from Neanderthal remains at least 40,000 years old.23 The advantages of digital storage explain why chains of nucleic acids have remained the go-to biological storage molecule for the past 4 billion years.
The other type of information in the body is analog.
We don’t hear as much about analog information in the body. That’s in part because it’s newer to science, and in part because it’s rarely described in terms of information, even though that’s how it was first described when geneticists noticed strange nongenetic effects in plants they were breeding.
Today, analog information is more commonly referred to as the epigenome, meaning traits that are heritable that aren’t transmitted by genetic means.
The term epigenetics was first coined in 1942 by Conrad H. Waddington, a British developmental biologist, while working at Cambridge University. In the past decade, the meaning of the word epigenetics has expanded into other areas of biology that have less to do with heredity—including embryonic development, gene switch networks, and chemical modifications of DNA-packaging proteins—much to the chagrin of orthodox geneticists in my department at Harvard Medical School.
In the same way that genetic information is stored as DNA, epigenetic information is stored in a structure called chromatin. DNA in the cell isn’t flailing around disorganized, it is wrapped around tiny balls of protein called histones. These beads on a string self-assemble to form loops, as when you tidy your garden hose on your driveway by looping it into a pile. If you were to play tug-of-war using both ends of a chromosome, you’d end up with a six foot-long string of DNA punctuated by thousands of histone proteins. If you could somehow plug one end of the DNA into a power socket and make the histones flash on and off, a few cells could do you for holiday lights.
In simple species, like ancient M. superstes and fungi today, epigenetic information storage and transfer is important for survival. For complex life, it is essential. By complex life, I mean anything made up of more than a couple of cells: slime molds, jellyfish, worms, fruit flies, and of course mammals like us. Epigenetic information is what orchestrates the assembly of a human newborn made up of 26 billion cells from a single fertilized egg and what allows the genetically identical cells in our bodies to assume thousands of different modalities.24
If the genome were a computer, the epigenome would be the software. It instructs the newly divided cells on what type of cells they should be and what they should remain, sometimes for decades, as in the case of individual brain neurons and certain immune cells.
That’s why a neuron doesn’t one day behave like a skin cell and a dividing kidney cell doesn’t give rise to two liver cells. Without epigenetic information, cells would quickly lose their identity and new cells would lose their identity, too. If they did, tissues and organs would eventually become less and less functional until they failed.
In the warm ponds of the primordial Earth, a digital chemical system was the best way to store long-term genetic data. But information storage was also needed to record and respond to environmental conditions, and this was best stored in analog format. Analog data are superior for this job because they can be changed back and forth with relative ease whenever the environment within or outside the cell demands it, and they can store an almost unlimited number of possible values, even in response to conditions that have never been encountered before.25
The unlimited number of possible values is why many audiophiles still prefer the rich sounds of analog storage systems. But even though analog devices have their advantages, they have a major disadvantage. In fact, it’s the reason we’ve moved from analog to digital. Unlike digital, analog information degrades over time—falling victim to the conspiring forces of magnetic fields, gravity, cosmic rays, and oxygen. Worse still, information is lost as it’s copied.
No one was more acutely disturbed by the problem of information loss than Claude Shannon, an electrical engineer from the Massachusetts Institute of Technology (MIT) in Boston. Having lived through World War II, Shannon knew firsthand how the introduction of “noise” into analog radio transmissions could cost lives. After the war, he wrote a short but profound scientific paper called “The Mathematical Theory of Communication” on how to preserve information, which many consider the foundation of Information Theory. If there is one paper that propelled us into the digital, wireless world in which we now live, that would be it.26
Shannon’s primary intention, of course, was to improve the robustness of electronic and radio communications between two points. His work may ultimately prove to be even more important than that, for what he discovered about preserving and restoring information, I believe, can be applied to aging.
Don’t be disheartened by my claim that we are the biological equivalent of an old DVD player. This is actually good news. If Szilard had turned out to be right about mutations causing aging, we would not be able to easily address it, because when information is lost without a backup, it is lost for good. Ask anyone who’s tried to play or restore content from a DVD that’s had an edge broken off: what is gone is gone.
But we can usually recover information from a scratched DVD. And if I am right, the same kind of process is what it will take to reverse aging.
As cloning beautifully proves, our cells retain their youthful digital information even when we are old. To become young again, we just need to find some polish to remove the scratches.
This, I believe, is possible.
A TIME TO EVERY PURPOSE
The Information Theory of Aging starts with the primordial survival circuit we inherited from our distant ancestors.
Over time, as you might expect, the circuit has evolved. Mammals, for instance, don’t have just a couple of genes that create a survival circuit, such as those that first appeared in M. superstes. Scientists have found more than two dozen of them within our genome. Most of my colleagues call these “longevity genes” because they have demonstrated the ability to extend both average and maximum lifespans in many organisms. But these genes don’t just make life longer, they make it healthier, which is why they can also be thought of as “vitality genes.”
Together, these genes form a surveillance network within our bodies, communicating with one another between cells and between organs by releasing proteins and chemicals into the bloodstream, monitoring and responding to what we eat, how much we exercise, and what time of day it is. They tell us to hunker down when the going gets tough, and they tell us to grow fast and reproduce fast when the going gets easier.
And now that we know these genes are there and what many of them do, scientific discovery has given us an opportunity to explore and exploit them; to imagine their potential; to push them to work for us in different ways. Using molecules both natural and novel, using technology both simple and complex, using wisdom both new and old, we can read them, turn them up and down, and even change them altogether.
The longevity genes I work on are called “sirtuins,” named after the yeast SIR2 gene, the first one to be discovered. There are seven sirtuins in mammals, SIRT1 to SIRT7, and they are made by almost every cell in the body. When I started my research, sirtuins were barely on the scientific radar. Now this family of genes is at the forefront of medical research and drug development.
Descended from gene B in M. superstes, sirtuins are enzymes that remove acetyl tags from histones and other proteins and, by doing so, change the packaging of the DNA, turning genes off and on when needed. These critical epigenetic regulators sit at the very top of cellular control systems, controlling our reproduction and our DNA repair. After a few billion years of advancement since the days of yeast, they have evolved to control our health, our fitness, and our very survival. They have also evolved to require a molecule called nicotinamide adenine dinucleotide, or NAD. As we will see later, the loss of NAD as we age, and the resulting decline in sirtuin activity, is thought to be a primary reason our bodies develop diseases when we are old but not when we are young.
Trading reproduction for repair, the sirtuins order our bodies to “buckle down” in times of stress and protect us against the major diseases of aging: diabetes and heart disease, Alzheimer’s disease and osteoporosis, even cancer. They mute the chronic, overactive inflammation that drives diseases such as atherosclerosis, metabolic disorders, ulcerative colitis, arthritis, and asthma. They prevent cell death and boost mitochondria, the power packs of the cell. They go to battle with muscle wasting, osteoporosis, and macular degeneration. In studies on mice, activating the sirtuins can improve DNA repair, boost memory, increase exercise endurance, and help the mice stay thin, regardless of what they eat. These are not wild guesses as to their power; scientists have established all of this in peer-reviewed studies published in journals such as Nature, Cell, and Science.
And in no small measure, because sirtuins do all of this based on a rather simple program—the wondrous gene B in the survival circuit—they’re turning out to be more amenable to manipulation than many other longevity genes. They are, it would appear, one of the first dominos in the magnificent Rube Goldberg machine of life, the key to understanding how our genetic material protects itself during times of adversity, allowing life to persist and thrive for billions of years.
Sirtuins aren’t the only longevity genes. Two other very well studied sets of genes perform similar roles, which also have been proven to be manipulable in ways that can offer longer and healthier lives.
One of these is called target of rapamycin, or TOR, a complex of proteins that regulates growth and metabolism. Like sirtuins, scientists have found TOR—called mTOR in mammals—in every organism in which they’ve looked for it. Like that of sirtuins, mTOR activity is exquisitely regulated by nutrients. And like the sirtuins, mTOR can signal cells in stress to hunker down and improve survival by boosting such activities as DNA repair, reducing inflammation caused by senescent cells, and, perhaps its most important function, digesting old proteins.27
When all is well and fine, TOR is a master driver of cell growth. It senses the amount of amino acids that is available and dictates how much protein is created in response. When it is inhibited, though, it forces cells to hunker down, dividing less and reusing old cellular components to maintain energy and extend survival—sort of like going to the junkyard to find parts with which to fix up an old car rather than buying a new one, a process called autophagy. When our ancestors were unsuccessful in bringing down a woolly mammoth and had to survive on meager rations of protein, it was the shutting down of mTOR that permitted them to survive.
The other pathway is a metabolic control enzyme known as AMPK, which evolved to respond to low energy levels. It has also been highly conserved among species and, as with sirtuins and TOR, we have learned a lot about how to control it.
These defense systems are all activated in response to biological stress. Clearly, some stresses are simply too great to overcome—step on a snail, and its days are over. Acute trauma and uncontrollable infections will kill an organism without aging that organism. Sometimes the stress inside a cell, such as a multitude of DNA breaks, is too much to handle. Even if the cell is able to repair the breaks in the short term without leaving mutations, there is information loss at the epigenetic level.
Here’s the important point: there are plenty of stressors that will activate longevity genes without damaging the cell, including certain types of exercise, intermittent fasting, low-protein diets, and exposure to hot and cold temperatures (I discuss this in chapter 4). That’s called hormesis.28 Hormesis is generally good for organisms, especially when it can be induced without causing any lasting damage. When hormesis happens, all is well. And, in fact, all is better than well, because the little bit of stress that occurs when the genes are activated prompts the rest of the system to hunker down, to conserve, to survive a little longer. That’s the start of longevity.
Complementing these approaches are hormesis-mimicking molecules. Drugs in development and at least two drugs on the market can turn on the body’s defenses without creating any damage. It’s like making a prank call to the Pentagon. The troops and the Army Corps of Engineers are sent out, but there’s no war. In this way, we can mimic the benefits of exercise and intermittent fasting with a single pill (I discuss this in chapter 5).
Our ability to control all of these genetic pathways will fundamentally transform medicine and the shape of our everyday lives. Indeed, it will change the way we define our species.
And yes, I realize how that sounds. So let me explain why.