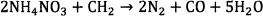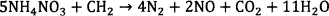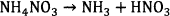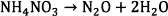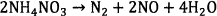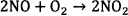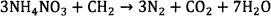
In recent years, the use of nitrogenous industrial explosives has come under intense scrutiny by environmental agencies due to emission of toxic fumes to the atmosphere during blasting of ammonium nitrate based explosives. Ammonium nitrate (AN) is a major ingredient in ammonium nitrate fuel oil (ANFO), and emulsion explosives, which are commonly applied in trench or surface mines blasting, and construction operations. It is well known that ammonium nitrate based explosives release toxic fumes (which contains NO and NO2 emissions) under certain blasting condition, and/or oxygen rich explosive formulations.
We investigated the effect of potassium on crystal structure, and thermal behaviour of ammonium nitrate. We observed multiphase structures in co-recrystallised ammonium nitrate containing low (2–4 mol %) and high (>25 mol %) concentrations of potassium in the crystallisation solution; mol % is based only on K+ and NH+4. Co-recrystallised salts were characterise using X-ray diffraction (XRD), Scanning electron microscopy – energy dispersive spectroscopy (SEM-EDS), Inductively coupled plasma – optical emission spectroscopy (ICP-OES), and Thermogravimetric analyser and differential scanning calorimeter (TGA/DSC).
In addition, we report concentration profiles of NO during thermal analyses of co-recrystallised potassium ammonium nitrates. The results obtained from thermal decomposition of the co-recrystallised potassium ammonium nitrate salts showed decrease in production of NO. and 3–5 mol % K was the optimal concentration when compared with pure AN.
Ammonium nitrate (AN) constitutes a major component in industrial explosives, used mainly in formulating ammonium nitrate and fuel oil (ANFO) explosives, as well as emulsion explosives. Detonation of commercial nitrogenous explosives results in rapid expanding hot gases, which under idea conditions produce water vapour (H2O), carbon dioxide (CO2) and nitrogen gas (N2)1–3. However, in the real world blasting of ammonium based explosives also generate toxic gases: mainly carbon monoxide (CO), and nitrogen oxides (NO, NO2, N2O5, etc.) 1–8 Detonation gases has the potential to either accumulate in muck pile or migrate out of the blasted area, so it is necessary to ventilate blast sites as soon as possible 7–8. Production of large volume of nitrogen oxides in blasting have adverse ecological effects: nitrogen oxides combine with water vapour to form nitric acid which raises the nitrate content in the soil and renders underground water acidic. CO danger lies with gas that remains in the soil after blast, which release to the environment during loading operation 7–8.
In recent years in Australia, the NOx emissions during blasting in mining operations have come under intense scrutiny of regulatory bodies and concerned citizens, as such emissions pose environmental and health risks; in particular, when orange clouds, characteristic of high levels of NO2, do not dissipate rapidly and drift into populated areas surrounding the mines. Thus, there is a great interest in industry for gaining improved understanding of the formation of NOx during blasting, and finding practical solutions to remedy the problem 8–14.
In general, ammonium nitrate based explosives which are formulated to form stoichiometry composition of AN and fuel oil (ANFO) or emulsion/water in gel explosive proceed according to the following overall reaction scheme 12.
(1)
Where CH2 represents as fuel oil or sometimes C can be used as in Reaction 215.
However, owing to factors like wicking of fuel oil of the explosive formulation among other factors, the explosive reaction may deviate from idea situations such that CO and NO may results. Attalla et al. (2008) listed Reactions 3 and 4 as possible mechanisms, which produce CO and NO gases 12.
Most researchers agreed that the initial step in decomposition of pure AN in the condensed phase involves an endothermic proton transfer from the ammonium ion (NH+4) to the nitrate ion (NO−3) to form nitric acid (HNO3) and ammonia (NH3), (Reaction 5) 16–21. Consensus also exists that the overall decomposition process of pure AN yields mostly H2O and N2O (Reaction 6)16–23.
Feick et al. (1954) confirmed that AN vapour is completely dissociated into Reaction 6 between 190 and 270 °C 24. It is also well appreciated that fast decomposition of ammonium nitrate between 200 to 230 °C results in release of NO gas (Reaction 7)25.
Also, it is well known that continuous oxidation of NO to NO2 occur in the presence of oxygen or ozone 26. Elsout et al. (1984) indicated that in the absence of ozone, 50 – 150 ppm of NO concentration would oxidise by oxygen as a consequence of Reaction 826:
Kinetic studies based on experimental data obtained after blasting of AN based explosives confirmed this reaction scheme for NO concentration up to 100 ppm 27. This results seem to confirmed that the orange cloud observed after blasting of ANFO and booster explosives is mainly due to oxidation of NO by O2 to NO2 11–12.27.
Some researchers have demonstrated production of nitrogen oxides and ammonia is due to early decomposition of AN in the explosive 28–29. Azarkovick et al. (1996) reported that fume production from ammonium nitrate based explosives can be minimized by adding substances that are capable of binding nitrogen oxides such as slaked lime (Ca(OH)2), chalk (CaCO3), and soda (Na2CO3)13. Sapko et al. (2002) demonstrated that urea, aluminium powder, and coal dust in ANFO blends were able to reduce nitrogen oxides production 11. Oxley et al. (2002) reported the effects of alkali and alkali earth salts on thermal decomposition of AN, and its decomposition gaseous products30. These authors ground different weight percentages of calcium, potassium, and sodium nitrate salts together with AN, and studied the thermal decomposition of the mixtures. Two observations of Oxley et al. (2002) are important. Firstly, when calcium, potassium, and sodium nitrates were added to AN, there was almost no change in the decomposition temperatures of the mixtures compared with the decomposition of pure AN. Secondly, a mixture containing 10 mol % potassium nitrate exhibited a lower N2O/N2 ratio compared with that of pure AN. The results indicated that the more stabilising the additives, the lower the N2O/N2 ratio.
In this research, attempts are made to synthesize ammonium nitrate salts doped with metal elements to scavenge excess oxygen which may be available for NOx formation during blasting or thermal decomposition. In particular, the use of potassium to scavenge excess oxygen that may be available for nitrogen oxides formation by forming metal oxides (e.g. K2O) or the metal ions may react directly with NOx to form metal salts (e.g. KNO3) during thermal decomposition of doped ammonium nitrate. We speculate that both of these phenomena would potentially decrease NOx emissions during thermal analyses of the solid solutions as it is reported that production of NOx during blasting of AN-based explosive is especially due to excess oxygen balance in the formulations 6–3.
Preparation of ammonium nitrate salts containing potassium was very important factor in formation of varies equilibrium phases of potassium ammonium nitrate salts. AN (crystalline 99.9 wt % pure (dry basis) was supplied by Dyno Nobel Asia Pacific Pty Ltd. Potassium nitrate (KN) salt was purchased from Ajax Chemicals (UNIVAR reagent 99.5 wt %). A total of 4.0 g of binary samples containing 1–75 mol % KN in balance of AN were ground together using Agate mortar and pestle in low relative humidity environment. In addition, similar weight of binary samples containing different compositions of pure AN and KN powders were dissolved in 1 mL of de-ionised water to form a supersaturated solution. Each supersaturated solution was placed in hot water bath at 80 °C while the solution was agitated every 15 minutes intermittently for complete dissolution. After heating the solutions for an hour at 80 °C, the solutions were quenched in ice-cold water bath at 1 °C to effect nucleation, growth, and precipitation of nitrate salts. Precipitated salts were filtered immediately. All precipitated samples were stored in a desiccator for at least 48 hours before characterisation.
Structural characterisation of pure AN, pure KN, ground AN-KN mixtures, and co-recrystallised AN-KN samples were analysed by Philips X’pert-MPD X-ray Diffraction system using Cu Kα1. XRD characterisation of samples was conducted at room temperature and atmospheric pressure conditions. Samples were scanned from 15 ° to 60 ° for a total duration of 15 minutes with a step size of 0.007 ° at 40 kV and 40 mA operating power. The divergence slit was fixed with divergence slit size of 0.25° and the specimen length was 10 mm. In addition, co-recrystallised samples were imaged using Philips XL30 SEM coupled with Oxford ISIS EDS for elemental analyses. Each sample was carbon coated using SPI Carbon Coating Unit prior to imaging. SEM images and EDS spectra of the nitrate salts were obtained by scanning samples at 15 kV. EDS characteristic energies of potassium are 3.314 and 3.590 keV for Kα1 and Kβ1 respectively. The concentrations of metal cations in the nitrate salts were quantitate using Varian 715-ES Inductively Coupled Plasma - Optical Emission Spectrometer (ICP-OES). Amounts of 250 mg of nitrate salts were dissolved in 500 mL of de-ionised water. Sample solutions were transferred via Varian SPS 3 sample preparation system for analyses in ICP-OES. High-Purity Standards ICP multi element standard solution containing eight metal cations in 2.0 % HNO3 was used for calibration.
Each co-recrystallised nitrate salt sample was put in a 100 μL platinum crucible and placed inside quartz tube for thermal analysis. The quartz reactor was connected with two mass flow meter/controller (Brooks 4800 series model and Bronkhorst EL-Flow select model) to monitor and control the flow rates of O2 and He gases during thermal decomposition nitrate salts. The background gas was either He (99.9995 %) or oxygen-rich mixture (21 vol % O2 and 79 vol % He) flowing at 100 mL/min. All the MCFs were turned on for 30 minutes to warm up and reach equilibrium temperature before flowing gases. In addition, all the MFCs were recalibrated with bubble-flow meter before taken any measurements. A typical sample size of 50 mg of each nitrate salt was decomposed in the quartz reactor by heating gradually at a rate of 10 °C/min from 25 to 400 °C.
Thermo Scientific Chemiluminescence NO-NO2-NOx analyser model 42i-HL was couples to the thermal analyses setup to identify and quantify trace amount of NO in the decomposition gases. In addition, evolved gases from thermal decomposition of each co-crystallised salt was injected and analysed in Shimadzu IRAffinity-1 Fourier Transform Infrared Spectrophotometer. SKC FlexFoil Plus 5 L sample bag with single polypropylene septum fitting were used to collect gas products for analyses in FT-IR. The FT-IR measurements were made in the wavelength range of 400–4000 cm−1, a resolution of 0.5 cm−1, and a number of 32 scans.
X-ray powder diffraction (XRD) pattern of recrystallised AN is shown in Figure 1. At 2θ of 28.98 °, the 100 % peak, which corresponds to (111) surface and d-spacing 3.085 Å, matched very well with crystalline AN phase IV as can be seen in Figure 1. It was also observed that all other peaks characteristic of AN phase IV also matched with those displayed by this sample. AN phase IV has orthorhombic structure that is stable between −18 and 55 °C when dry or between - 18 and 32 °C in the presence of moisture31–33. Addition of 1 mol % KNO3 to AN during co-recrystallisation shows no visible modification of the XRD pattern compared with pure AN phase IV except the 100 % peak corresponded to (020) at 2θ of 32.83 ° as shown in Figure 2. This amount of K in the sample is probably at the detection limit of the XRD analysis. However, it stands to reason to suggest that the crystal lattice of AN could accommodate 1 mol % content of K+ ions as the ionic radius of K+ (0.133 nm) is only slightly smaller than that NH+4 (0.143 nm). Hence, NH+4 could undergo substitution by minor amount of K+ in the crystal structure without much distortion. Usually small inclusions in pure phase cause peak broadening in XRD pattern of the main component due to stress in the crystal structure which is observed as a small shift in the d-spacing. This is because a number of parallel planes of small inclusions may prevent diffraction to occur at precise angles. However, there was no visible shift of the XRD peaks of this sample compared with pure AN phase IV. Further analysis of a co-recrystallised sample containing 1 mol % KNO3 and 99 mol % AN by EDS indicated the presence of K in the sample. In addition, ICP-OES measurements of this sample confirmed the presence of K. This led us to a conclusion that co-recrystallisation of 1 mol % KNO3 and 99 mol % AN probably resulted in a complete solid solution of potassium ammonium nitrate without much distortion of the AN phase IV crystal structure.
Figure 1. XRD pattern of recrystallised AN salt.

Figure 2. XRD pattern of recrystallised AN salts containing low concentrations of K.
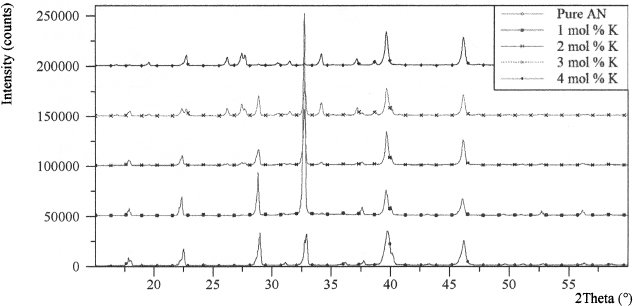
Figure 2 shows an XRD spectrum of a co-recrystallised sample containing 2 mol % K. The spectrum illustrates two crystalline phases of AN; that is, AN phase IV with a small trace of AN phase III. AN phase III has an orthorhombic structure which is stable between 32–84 °C but is less dense, by about 3.70 %, than AN phase IV 31–34. Co-recrystallisation of 2 mol % KNO3 and 98 mol % AN commences to change the crystal structure of AN from phase IV to III in order to accommodate the increased modal concentration of K+ ions. This is evident in the XRD peaks at 28 of 22.78 °, 27.46°, and 27.73 ° which correspond to (111), (210), and (021) planes of AN phase III.
The XRD spectrum of recrystallised samples confirms that increasing K concentration to 3 mol % in the salt elevates the content of phase III in the multi-phase AN. Likewise, other researchers have demonstrated increased stability of AN doped with K as a consequence of the transformation of AN from crystalline phase IV to crystalline phase III32, 34–36.
When modal composition of K increases to 5 mol %, the XRD peak at 2θ of 27.48 °, and 27.79 ° matches well with the (102) and (210) surfaces that corresponds to 100 %, and 50.1 % peaks of K0.05(NH4)0.95NO3. K0.05(NH4)0.95NO3 displays orthorhombic structure similar to phase III of AN. Further increment in KNO3 concentration from 5 to 25 mol %, during preparation, reveals single phase Kx(NH4)1-xNO3 with no separate phase of potassium nitrate, according to the XRD results as presented in Figure 3 and Table I. From the overlaid spectra of Figure 3, the largest peak at 2θ of 34.59 ° corresponds to d-spacing of 2.59 Å in samples containing 10, 15, 20, and 25 mol % K. However, crystals obtained from the solutions of 30–75 mol % KNO3 in balance of AN reveal the presence of two or more potassium compounds in the XRD measurements as shown in Figure 4. This demonstrates that 30 mol % substitution of NH+4 by K+ exceeds the solid solubility limit of AN crystal structure, causing the precipitation of pure KNO3, when co-recrystallised with AN.
Figure 3. XRD patterns of co-recrystallised AN containing 5–25 mol % K.

Figure 4. XRD patterns of co-recrystallised AN containing 30–75 mol % K.
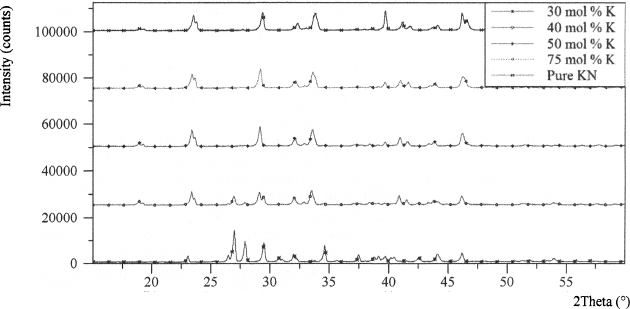
Table I. Summary of Powder XRD Results of Co-recrystallisation of KNO3 and AN
| Concentration of K (mol %) ** | Phase(s) Present in XRD | Comments |
| 0.00 | AN phase IV | Single phase |
| 1.00 | AN phase IV | Single phase; K detected by EDS and ICP-OES |
| 2.00 | AN phase III and IV | Multi-phase of AN |
| 3.00 | AN phase III and IV | Multi-phase of AN |
| 4.00 | AN phase III and IV | Multi-phase of AN |
| 5.00 | K0.05(NH4)0.95NO3 | Single phase |
| 10.00 | Kx (NH4)1-xNO3 | Single phase |
| 15.00 | Kx (NH4)1-xNO3 | Single phase |
| 20.00 | Kx (NH4)1-xNO3 | Single phase |
| 25.00 | Kx (NH4)1-xNO3 | Single phase |
| 30–75 | Kx (NH4)1-xNO3 and KNO3 | Multi-phase of AN and KNO3 |
Recrystallisation of pure AN from hot saturated solution by direct quenching in ice-cold water bath at 1 °C resulted in plate and needle-like shapes, consistently with the results of other researchers 37. SEM images could not be taken at magnification higher than 10,000 x. The samples seemed to evaporate at higher magnification even if the voltage was set as low as 5 kV. The SEM images of recrystallised pure AN displayed sheet-like surfaces with a few embedded needle-like crystals. However, the morphology of samples of AN containing K exhibited fused crystals with numerous cracks along the crystals boundaries, as illustrated in Figure 5. It appears that increasing concentration of K in solid solutions induces the formation of more distinct crystals with micro tree-like cracks in addition to crystal boundaries. This demonstrates the effect of rising stress on the crystal structure at increasing concentration of K, leading to separation of the material into two or more phases, once modal content of K exceeded 25 mol %.
Figure 5. SEM images of AN containing 10 mol % K (left), and 25 mol % K(right).

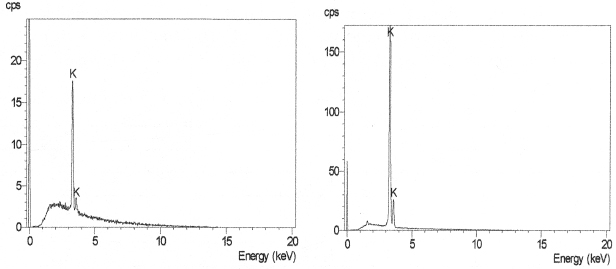
EDS analyses of single crystals of nitrate salts were not possible, but bulk qualitative EDS of recrystallised nitrate salts indicated the presence of potassium in all the potassium ammonium nitrate salts. Figures 6 illustrate the characteristic peaks Kα1 and Kβ1 of potassium, which appear at 3.314 and 3.590 keV respectively. As expected, the intensities of the characteristics potassium peaks of Kα1 and Kβ1 increase with increasing concentration of K in the co-recrystallised salts. Quantitative analysis of potassium concentrations in recrystallized salts were made using ICP-OES. ICP results indicated the presence of various K cations in all the recrystallized mixed nitrate salts. In addition, the data indicated that the ICP-OES concentration of K+ ions in the co-recrystallized nitrate salts were higher than estimated at high concentration (>10 mol % K). This is possibly due to leaching of NH4 ions from the aqueous solutions during preparation.
Figure 6. EDS spectra of K0.05(NH4)0.95NO3 (left), and K0.20(NH4)0.80NO3 (right).
Recrystallised nitrate salts prepared by freeze-drying were exposed to thermal treatment in a quartz reactor and NOx analyser was place downstream to identify and quantitate the decomposition gases. A typical in situ concentration profile of NO obtained during thermal analyses of pure ammonium nitrate in a gas mixture of 21 vol % O2 and 79 vol % He shown in Figure 7. Figure 7 also illustrates overlaid NO production curves from thermal decomposition of K0.05(NH4)0.95NO3, and K0.20(NH4)0.80NO3. The data shows that 2.19 mL of NO/g-salt was produced during thermal decomposition of pure AN. However, when pure AN was decomposed in the presence of oxygen-deficient helium atmosphere, 2.36 mL of NO /g-salt was produced. Thus there is not much difference in NO production during thermal decomposition of pure AN in both inert and oxygen-rich atmospheres. This is could be due to the fact that pure AN contains excess oxygen in its compounds, and the conversion of NO to NO2 is relatively slow at low temperatures in the absence of a catalyst. However, it was estimated that decomposition of K0.05(NH4)0.95NO3 in oxygen-rich gas mixture, and oxygen-deficient helium atmosphere produced 1.37 mL NO/g-salt and 2.04 mL NO/g-salt respectively.
Figure 7. A plot of concentration of NOx against real time during thermal decomposition of recrystallised pure AN, K0.5(NH4)0.95NO3, and K0.20(NH4)0.80NO3 in oxygen-rich atmosphere (21 vol % O2 and 79 vol % He).

Thermal decomposition of different co-recrystallised salts containing K is presented in Table II. It can be seen from Table II that NO production decreased from 2.19 mL/g-salt to 1.16 mL/g-salt for pure AN and continued up to AN containing 15 mol % K. The production of NO increased slightly after addition of 15 mol % K to AN. However, when the production of NO from each salt was compared with the total nitrogen content in the salt, the data seem to indicate that 3–5 mol % K is the optimum for destruction of NO during thermal decomposition as presented in Table II. Further investigation of the co-recrystallised salts was carried in DSC/TGA. We observed increased exothermicity of the decomposition reactions when the concentration of K in the co-crystallised salts was higher than (> 5 mol %). In addition to the co-recrystallised salts from solutions, several ground AN-KN binary compositions were thermally decomposed in the laboratory scale reactor. Comparison of NO productions from different ground salt mixtures and co-recrystallised salts are summarised in Table III. The results show that NO production is relatively low for co-recrystallised salts containing potassium when compared to ground mixtures containing the same mol % of K.
Table II. NO production during thermal decomposition of co-recrystallised potassium ammonium nitrate salts in oxygen-rich atmosphere (21 vol % O2 and 79 vol % He)
| Concentration of K in Sample (mol %) ** | mL of NO Production/g-salt | mg of NO / g of nitrogen in salt |
| 0 | 2.19 | 3.65 |
| 1 | 1.50 | 2.51 |
| 2 | 1.50 | 2.53 |
| 3 | 1.28 | 2.18 |
| 4 | 1.30 | 2.23 |
| 5 | 1.37 | 2.37 |
| 10 | 1.19 | 2.51 |
| 15 | 1.16 | 2.68 |
| 20 | 1.39 | 2.71 |
| 25 | 1.20 | 3.59 |
| 100 | -- | -- |
Table III. NO production during thermal decomposition of ground mixtures in oxygen-deficient atmosphere (99.9995 vol % He).
| Sample | mL of NO/g-salt |
| Pure AN | 2.36 |
| Co-recrystallised K0. 05(NH4)0.95NO3 | 2.04 |
| Ground mixture of 5 mol % KN in balance of AN | 2.29 |
| Ground mixture of 10 mol % KN in balance of AN | 2.17 |
| Ground mixture of 15 mol % KN in balance of AN | 2.14 |
Evolved gases obtained from thermal decomposition of recrystallised salts were analysed in FT-IR. GRAMS/ AI 8.0 Spectroscopy software with QASoft analyses utility was used for identification of species in the evolved gases. FT-IR spectra of gas products from K0.05(NH4)0.95NO3 is shown in Figure 8. NO, NO2, NH3, N2O, and H2O gases were identified as positive matches in the gas products. Also, trace amounts of CO2 were presence in the FT-IR spectra, a contribution from the background gas.
Figure 8. FT-IR spectra of gas products obtained from thermal decomposition of K0.5(NH4)0.95NO3 in oxygen-rich atmosphere (21 vol % O2 and 79 vol %He).

It was also observed that H2O spectra are sometimes inverted in the negative position. This happens when the moisture in the background gas is more than the moisture content in the gas products from thermal decomposition of the salts. In addition to the gas products from the decomposition experiments, substantial amount of solid residue were observed in the sample crucible and downstream of the heating zone of the quartz reactor. Metal ion concentrations in the solid residues were estimated using IC and ICP-OES. Data obtained from IC, ICP-OES, and DSC/TGA showed that KN was present in the solid residues after thermal decomposition of co-recrystallised salts. The reaction mechanism leading to the formation of the solid residue is not well understood at this point and further investigation is underway.
Attempts were made to prepare mixed ammonium nitrate salts for potential application in AN-based explosives to minimize the formation of NOx during decomposition or detonation. Co-recrystallised nitrate salts were prepared from pure ammonium nitrate, and potassium nitrate precursors using freeze-drying method. Powder XRD, SEM-EDX, and ICP-OES were used to characterise the co-recrystallised salts. Co-recrystallisation of 1 mol % KNO3 in balance of AN resulted in a single phase material, identified as AN phase IV. Increasing the concentration of KNO3 to between 2 and 4 mol % resulted in the appearance of an additional AN phase. Loadings of 5 to 25 mol % KNO3 formed solid solution of the form Kx(NH4)1-xNO3, which has orthorhombic structure similar to pure AN phase III. Attempts to increase KNO3 content in AN from 30 to 75 % led to the formation of two or more phases, solid solution of the form Kx(NH4)1-xNO3 plus pure KNO3.
Evolved gas analyses data obtained from thermal decomposition of AN containing potassium showed reduction in the production of NO when compared with pure AN. The data indicated that thermal decomposition of pure AN in oxygen-rich or oxygen-deficient atmospheres has the same effect on NO production. Also, production of NO from thermal decomposition of co-recrystallised salt containing 5 mol % K was lower than that obtained from ground mixture of the same K content. Thus AN containing K in its crystal structure resulted in lower production of NO compared with ground mixture of AN and KNO3 salts. In addition, there was up to 40 % reduction in NO production when K was added to AN crystal structure compared to pure AN; based on conversion of nitrogen content in the salt. Identification of gas products from thermal decomposition of mixed potassium ammonium nitrate salts indicated the presence of NO, NO2, N2O, O2, N2, NH3, and H2O. In addition, potassium nitrate residues were observed in the sample crucible and reactor after thermal decomposition of the co-recrystallised salts.
The authors are grateful to Dr Jeff Gore of Dyno Nobel Asia Pacific Pty Ltd for his time and professional assistance. The first author would like to acknowledge the financial support from the University of Newcastle Postgraduate Scholarships (UNIPRS and UNRSC). We would like to thank Mr David Phelan and Ms Jennifer Zobec of Electron Microscope/X-Ray Unit, The University of Newcastle for their assistance with characterisation of the salts. The Australian Research Council and Dyno Nobel Asia Pacific Pty Ltd funded this study jointly.
* Corresponding author. Email: Bogdan.Dlugogorski@newcastle.edu.au
** mol % is based only on K+ and NH+4 and does not include the solution water
1C. E. Munroe. and C. Hall, A Primer on Explosives for Coal Miners, Bureau of Mines, US Department of The Interior, Bulletin 17 (Reprint of US Geological Survey Bulletin 423) (1911).
2S. Meyers, and E. S. Shanley, Industrial Explosives - A Brief History of Their Development and Use, Journal of Hazardous Materials, 23, 183–201 (1990).
3D. L. Lawrence, A Study of Post Blast Generation of Nitrogen Dioxide, Proceedings of the 22nd Annual Conference on Explosives and Blasting Technique, 1–12 (1995).
4A. G. Streng, Evaluation of Toxic After-Detonation Gases Formed by Industrial Explosives, Explosivstoffe, 19, 58–64 (1971).
5N. C. Karmakar, and S. P. Banerjee, A Review of Laboratory and Field Test Methods for Studying Fume Characteristic of Explosives, Journal of Mines, Metals and Fuels, Vol. 32, No. 1/2, 398–402 (1984).
6L. D. Santis, A Summary of Subsurface Carbon Monoxide Migration Incidents, Proceedings of the Twenty-Seventh Annual Conference on Explosives and Blasting Technique, 143–54 (2001).
7R. J. Mainiero, M. L Harris, and J. H. Rowland III, Dangers of Toxic Fumes from Blasting, NIOSH, U.S.A, (2007).
8Harris, M. L., Mainiero, R.J., Monitoring and removal of CO in blasting operations, Safety Science, 46, 1393–1405 (2008).
9J. H. Rowland III, R. J. Mainiero, and D. A. Hurd, Factors Affecting Fumes Production of an Emulsion and ANFO/Emulsion Blends, Proceedings of the 27th Annual Conference on Explosives and Blasting Technique, 133–44 (2001).
10J. H. Rowland III, and R. J. Mainiero, Factors Affecting ANFO Fumes Production, Proceedings of the 26th Annual Conference on Explosives and Blasting Technique, 163–74, (2000).
11M. J. Sapko, J. H. Rowland III, R. J. Mainiero, and I. A. Zlochower, Chemical and Physical Factors that Influence NOx Production During Blasting – Exploratory Study, Proceedings of the 28th Annual Conference on Explosives and Blasting Technique, 317–30 (2002).
12M. I. Attalla, S. J. Day, T. Lange, W. Lilley, and S. Morgan, NOx Emissions from Blasting Operations in Open-Cut Coal Mining, Atmospheric Environment, 42. 7874–83 (2008).
13A. E. Azarkovich, L. G. Boikhovitinov, and L. M. Pernik, Possibility of Minimizing Generation of Nitrogen Oxides In Blasting of Ammonium Nitrate Explosives, Int. J. of Rock Mechanics and Mining Sciences and Geomechanics, Vol. 33, 5, 222–25 (1995).
14C. R. Barnhart, Understanding the “Orange Smoke” Problem in Cast Blasting, Proceedings of the 29th Annual Conference on Explosives and Blasting Technique, 1–11 (2003).
15A. Miyake, H. Kobayashi, H. Echigoya, S. Kubota, Y. Wada, Y. Ogata, H. Arai, and T. Ogawa, Detonation Characteristics of Ammonium Nitrate and Activated Carbon Mixtures, Journal of Loss Prevention in the Process Industries, 20, 584–88 (2007).
16T. B. Brill, P. J. Brush, and D. G. Patil, Thermal Decomposition of Energetic Materials 58. Chemistry of AN and ADN near the Burning Surface Temperature, Combustion and Flame, 92, 178–86 (1993).
17K. R. Brower, J. C. Oxley, and M. Tewari, Evidence for Homolytic Decomposition of Ammonium Nitrate at High Temperature. J. Phys. Chem., 93, 1029–33 (1989).
18J. C. Oxley, S. M. Kaushik, and S. N. Gilson, Thermal Decomposition of AN-based Composites, Thermochem. Acta, 153, 269–86 (1989).
19C. R. Richardson, and R. L. Hightower, Evaporation of Ammonium Nitrate Particles, Atmospheric Environment, Vol. 21, No. 4, 971–75 (1987).
20V. P. Sinditskii, V. Y. Egorshev, A. I. Levshenkov, and V. V. Serushkin, Ammonium nitrate: combustion mechanism and the role of additives, Propellants, Explosives, Pyrotechnics, 30 (4), 269–80 (2005).
21S. Vyazovkin, S. J. Clawson, C. A. Wight, Thermal Dissociation Kinetics of Solid and Liquid Ammonium Nitrate. Chem. Mater., 13, 960–66 (2005).
22H. L. Saunders, The Decomposition of Ammonium Nitrate by Heat, J. Chem. Soc., 121, 698–711 (1922).
23G. H. Madany, and G. Burnet, Inhibition of the Thermal Decomposition of Ammonium Nitrate, J. Agr. Food. Chem., Vol. 16, No. 1, 136–41 (1968).
24G. Feick, The Dissociation Pressure and Free Energy of Formation of Ammonium Nitrate, J. Am. Chem. Soc., 76, 5858–60 (1954).
25C. Oommen, and S. R. Jain, Ammonium nitrate: A Promising Rocket Propellant Oxidizer, Journal of Hazardous Materials, A67, 253–81 (1999).
26A. J. Elshout, and S. Beilke, Oxidation of Nitric Oxide to Nitrogen Dioxide in Flue Gas Plumes of Power Stations, Comm. Eur. Communities, (Rep.) Phys.-Chem, Behav. Atmos. Pollut., 9436, 535–43 (1984).
27R. J. Mainiero, J. H. Rowland III, M. L. Harris, and M. J. Sapko, Behaviour of Nitrogen Oxides in the Product Gases from Explosive Detonations, NIOSH, U.S.A, (2006)
28R. Gunawana, S. Freija, D. Zhanga, F. Beachb, and M. Littlefairb, A Mechanistic Study into the Reactions of Ammonium Nitrate with Pyrite, Chemical Engineering Science, 61, 5781–90 (2006).
29Y. Miron, T. C. Ruhe, and J. E. Hay, Reactivity of ANFO with Pyrite Containing Weathering Products-Evaluation of Additional Inhibitors, U.S. Bureau of Mines, Pittsburgh, 1–14 (1982).
30J. C. Oxley, and J. L. Smith, E. Rogers, and Y. Ming, Ammonium nitrate: Thermal Stability and Explosivity Modifiers, Thermochim. Acta, 384, 23–45 (2002).
31I. Dellien, A DSC Study of the Phase Transformations of Ammonium Nitrate, Thermochim. Acta, 55, 181–191 (1981):
32C. Oommen, and S. R. Jain, Phase Modification of Ammonium Nitrate by Potassium Salts, Journal of Thermal Analysis and Calorimetry, 55 (3), 903–18 (1999).
33H. Langfelderova, Study of the Influence of Experimental Conditions on the Course of the DSC Curve of Ammonium Nitrate (20–140 °C), Thermochim. Acta, 56, 385–89 (1982).
34M. J. Herrmann, and W. Engel, Phase Transitions and Lattice Dynamics of Ammonium Nitrate, Propellants, Explosives, Pyrotechnics, 22, 143–47 (1997).
35A. A. Vargeese, S. S. Joshi, and V. N. Krishnamurthy, Use of Potassium Ferrocyanide as Habit Modifier in the Size Reduction and Phase Modification of Ammonium Nitrate Crystals in Slurries, Journal of Hazardous Materials, 180, 583–89 (2010).
36H. B. Wu, and C. K. Chan, Effects of Potassium Nitrate on the Solid Phase Transitions of Ammonium Nitrate Particles, Atmospheric Environment, 42, 313–22 (2008).
37A. A. Vargeese, S. S. Joshi, and V. N. Krishnamurthy, Effect of Method of Crystallization on the IV—III and IV—II Polymorphic Transitions of Ammonium Nitrate, Journal of Hazardous Materials, 161, 373–79 (2009).