To familiarize radiologists with the revised classification of CNS tumors in terms of certain important mutation profiles including IDH (isocitrate dehydrogenase) mutation, 1p/19q-codeletion status, TP53 (tumor protein 53) mutation, BRAF mutation, and histone H3K27M-mutation and their influence on improving diagnostic accuracy, treatment strategies, and overall survival.
To provide an overview of the imaging phenotypes for the different glioma genotypes.
4.1 Introduction
The World Health Organization (WHO) Classification of Tumors of Central Nervous System (CNS) provided an update in 2016 nearly 10 years after the 2007 version to help more systematically categorize brain tumors. The revised system for the first time uniquely included molecular and genetic parameters of the individual tumor types, in addition to the histological features. Accordingly, each tumor is now identified by both its phenotype (based on histology), and genotype (based on its molecular and genetic parameters) [1].
4.2 Goals of the Revised Classification
The goals are multi-fold: (1) To resolve some of the confusion created by classifying brain tumors based only on histology, (2) Provide greater diagnostic accuracy, (3) Aid better treatment strategies, and (4) Allow for an improved assessment of the prognosis based on the specific tumor type.
4.3 Background
A classic example of uncertainty created in the past where tumors were classified based only on histology included the group “oligoastrocytoma.” These were tumors which exhibited features of both oligodendroglioma and astrocytoma on histology, and were therefore lumped together as oligoastrocytomas [2–4]. Accordingly, their management was not definitive which in turn influenced their prognosis. Categorizing this tumor group based on the underlying 1p/19q codeletion (genetic mutation) status allows them to be clearly distinguished almost always into either (1p/19q-codeleted) oligodendroglioma or (1p-19q-intact) astrocytoma [5–8]. Only a few tumors cannot be categorized into either group, and are known as oligoastrocytoma, NOS (not otherwise specified) [1, 9, 10]. This clear distinction allows more accurate diagnosis, which therefore influences more appropriate tumor specific treatment strategies, and a better predictor for the overall prognosis. Another perplexing prognostic feature was noted in terms of the overall survival of certain low grade gliomas, which surprisingly were noted to perform much worse than Grade III astrocytomas. This can now be explained based on their IDH-mutation status, with IDH-wildtype Grade I Gliomas performing much poorer than IDH-mutant Grade III astrocytomas [5, 6]. Thus, it is the mutation status which influences the overall prognosis much more than the histology.
Utilizing the background above, the discussion below will mention the salient features of how the Revised 2016 classification influences our understanding of brain tumors focusing on infiltrating gliomas in adults, gliomas in children, and the introduction of certain new tumor types. A description of all CNS tumor types included in the Revised 2016 Classification of CNS tumors is beyond the scope of this text.
4.4 Infiltrating Gliomas in Adults
Several mutations are associated with infiltrating gliomas in adults. Of these, some of the important ones include IDH mutation, 1p-19q codeletion, and TP53 mutation status [11].
The primary deterministic mutation includes the IDH-mutation status—presence suggests IDH-mutant, and absent an IDH-wildtype tumor [12].
4.5 IDH-Mutant gliomas
There are two types of IDH mutation, IDH1- and IDH2-mutated tumors. An overwhelming majority of such tumors are IDH1 mutated. Less than 3% of IDH-mutated tumors are exclusively IDH2-mutant tumors [13].
4.5.1 Clinical Relevance and Prognosis
IDH-mutant tumors are seen more commonly in the middle-aged population (30–60 years of age), than IDH-wildtype tumors which are more frequently seen in the older population (>60–65 years of age). The overall survival of IDH-mutant tumors is far better than IDH-wildtype tumors. In fact, as mentioned previously, low grade (grade I by histology) IDH-wildtype gliomas have an overall survival close to that of grade IV IDH-wildtype glioblastomas, but much worse than grade III IDH-mutant gliomas. It is the IDH-mutation status which is the driving force in terms of overall prognosis, much more than the histological grade. Furthermore, even among the Grade IV glioblastomas, it has been noted that IDH-mutant glioblastomas have an overall survival much better than IDH-wildtype glioblastomas. Supporting this is the fact that most IDH-mutant glioblastomas are the secondary type, while most IDH-wildtype glioblastomas are the de novo or primary type [14].
It is a known fact in glioma surgery that wider the resection, better is the overall survival. Knowing preoperatively that the tumor is an IDH-mutant type can influence the surgeon for a more complete surgical resection, including the FLAIR signal abnormality surrounding the enhancing mass [15]. The radiologist by suggesting the genetic profile preoperatively can accordingly influence the extent of surgical resection.
IDH-mutant tumors, seen more commonly in the middle-aged population (third to sixth decade of life), have a far better overall survival than IDH-wildtype tumors, which are seen more commonly in the older patients (>60–65 years of age).
4.5.2 Radiological Features
Both IDH1 and IDH2 mutations change the role of IDH in the citric acid cycle. This results in accumulation of 2-HG within tumor cells. 2-hydroxyglutarate (2-HG) can be detected on MR spectroscopy and is therefore considered to be the imaging hallmark of all IDH-mutant tumors [16]. However, reliable detection is challenging, and is possible only at some select centers with special MR spectroscopists on site [17–19].

A 69-year-old male with change in mental status. (a) Coronal T2WI demonstrates a heterogenous centrally necrotic mass in the left insular region extending to involve the frontal lobe. (b) Axial FLAIR image demonstrates FLAIR signal abnormality surrounding this lesion which shows indistinct margin with the adjacent brain. (c) Axial T1 post-contrast image demonstrates heterogenous but predominantly peripheral intense enhancement. (d) Corresponding axial DSC (dynamic susceptibility contrast) perfusion map demonstrates increased relative blood volume from the enhancing component of this lesion. Diagnosis: IDH-wildtype glioblastoma
4.6 1p/19q-Codeletion
IDH-mutant gliomas can subsequently be classified into those which are 1p/19q-codeleted tumors or 1p/19q-intact tumors. Of these, those gliomas which are 1p/19q-codeleted are the oligodendrogliomas, while those which are 1p/19q-intact are astrocytomas. Astrocytomas typically also show TP53 mutation, a mutation which is never seen in oligodendrogliomas, another distinguishing feature that separates these two entities. As mentioned previously, this 1p/19q-codeleted status and TP53 mutation help clearly separate the confusing oligoastrocytoma group into either oligodendroglioma or astrocytoma (which was not possible based on histological features alone), which helps better manage these patients.
1p/19q-codeletion status in an IDH-mutant tumor is diagnostic of oligodendroglioma; 1p/19q-intact status with TP53 mutation is diagnostic of astrocytoma.
4.6.1 Clinical Relevance and Prognosis
It has been shown in two large randomized control trials that chemotherapeutic agents including procarbazine, lomustine, and vincristine (PCV) when added to radiation therapy significantly improve the overall survival in patients with 1p/19q-deleted tumors when compared with radiation therapy alone [22, 23]. This is now the standard of care for all 1p/19q-codeleted oligodendrogliomas.
4.6.2 Radiological Features

A 48-year-old man with seizures. (a) Axial T2WI demonstrates a heterogenous mass involving the right frontal lobe. (b) Corresponding axial T1 post-contrast image demonstrates heterogenous but minimal enhancement. (c) Axial CT scan from the same patient demonstrates multiple arcs of calcification within this mass. Diagnosis: Oligodendroglioma, IDH-mutant, 1p/19a codeleted tumor
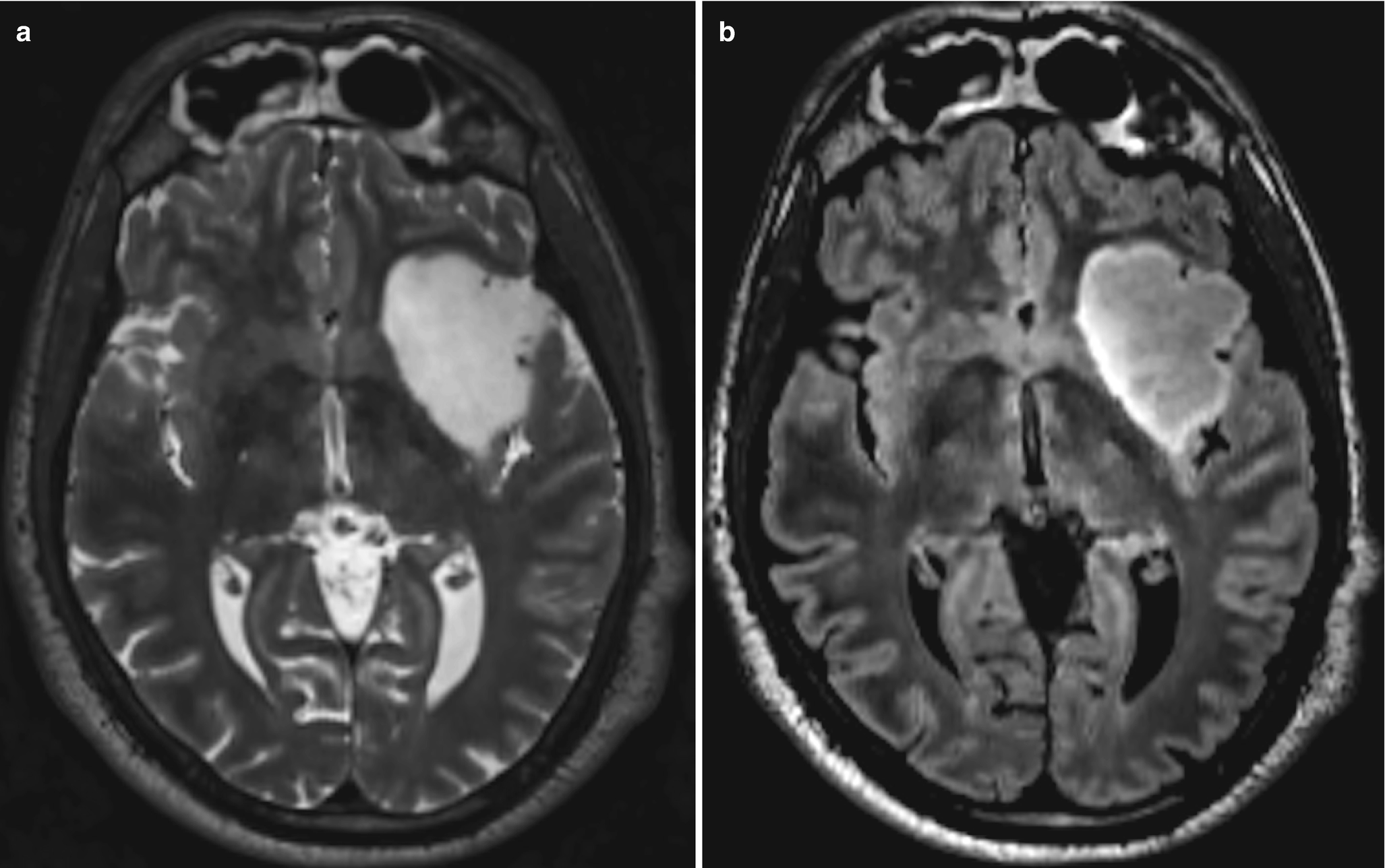
A 34-year-old man with headache. (a) Axial T2WI demonstrates a well-defined expansile mass involving the left insula, which appears predominantly bright in its signal intensity when compared to the gray matter. (b) Corresponding axial FLAIR image demonstrates the mass to be predominantly hypointense to the gray matter. Diagnosis: Diffuse astrocytoma, IDH-mutant, 1p/19q-noncodeleted (intact) tumor
4.7 Gliomatosis Cerebri
Gliomatosis cerebri as a specific tumor subtype was included in the 2007 version of the WHO Classification of CNS tumors. This term is deleted from the 2016 Update. A diffusely infiltrating non-enhancing tumor extending to involve three or more lobes is no longer to be considered as gliomatosis cerebri. It is recognized as a diffuse glioma type, with its subtype dependent on further genetic, molecular testing and histological evaluation [1].
Gliomatosis cerebri as a tumor type is no longer recognized.
4.7.1 Gliomas in Children
Gliomas in children have been known to behave differently than those seen in the adult population. This is related to the fact that mutations seen commonly in gliomas in adults including IDH mutation and 1p/19q-codeletion occur only uncommonly in children [27]. In fact, the two common mutation types commonly seen in children, include BRAF mutation and histone H3-K27 m-mutation.
4.8 BRAF Mutation
These tumors are usually well-circumscribed and carry an excellent prognosis.
4.9 Radiological Features
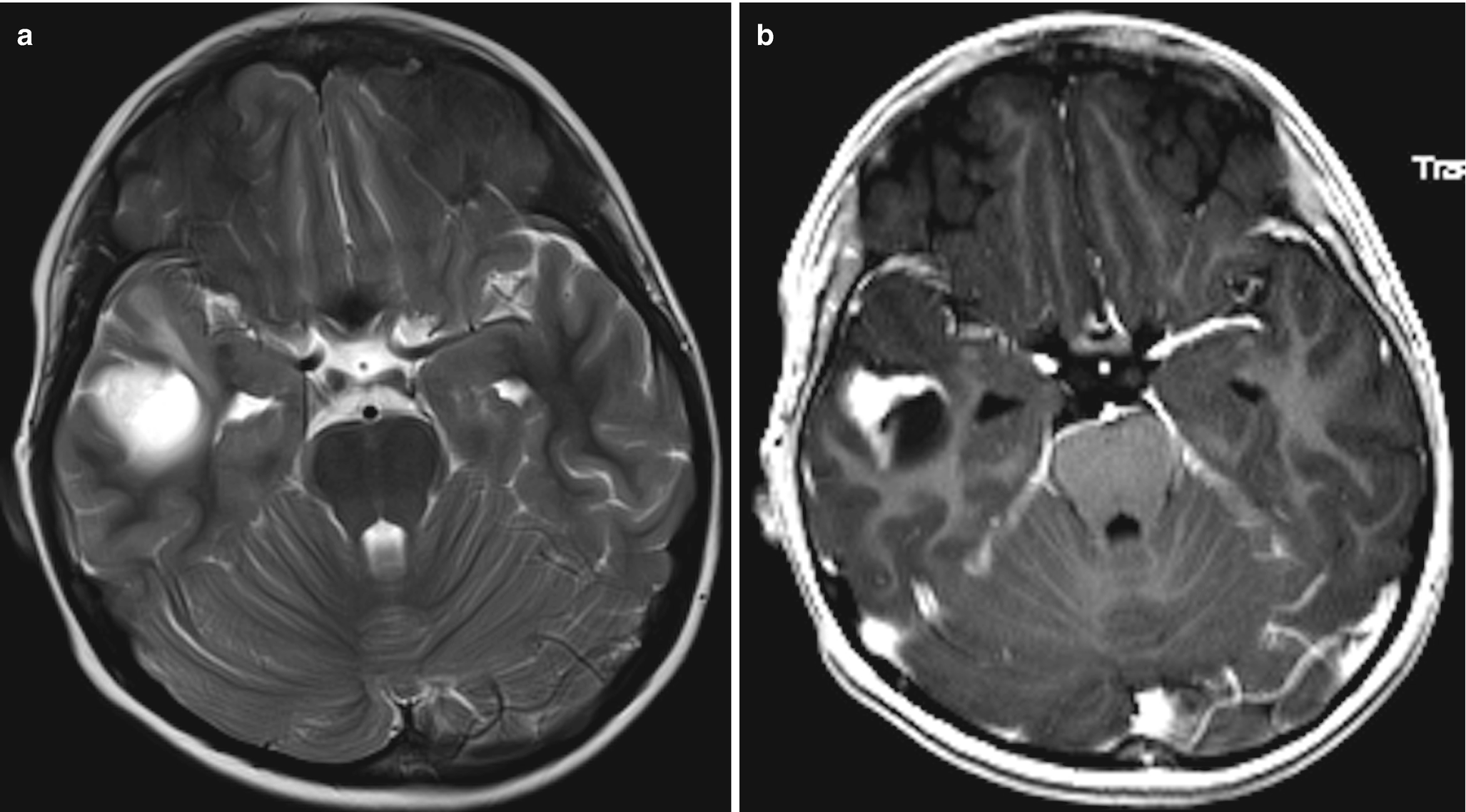
A 18-year-old boy with seizures. (a) Axial T2WI demonstrates a well-defined cystic appearing lesion in the right temporal lobe. (b) Corresponding axial T1 post-contrast image demonstrates a mural enhancing nodule along the lateral aspect of this lesion. Diagnosis: Pleomorphic xanthoastrocytoma, BRAF-mutant tumor
BRAF mutation is one of the most common mutations seen in the pediatric population, and includes tumor types such as pilocytic astrocytoma, pilomyxoid astrocytoma, and ganglioglioma.
4.10 Histone H3-K27m-Mutant Tumors
These are diffuse midline gliomas (previously known as diffuse infiltrating pontine glioma) and carry an extremely poor prognosis. Location of this tumor type makes it difficult to biopsy these tumors or attempt a surgical resection [28, 29]. Radiation and chemotherapy are not particularly helpful.
4.10.1 Radiological Features
Brainstem (pons) is the most common location. Other common locations include thalami and spinal cord. As previously described this is a diffusely infiltrating lesion which results in secondary expansion of the structure involved. Only minimal enhancement is typically seen. Occasionally however, heterogenous enhancement and cyst(s) can be seen [30]. Leptomeningeal dissemination is seen in about one-third of all such patients.
H3K27M-mutant glioma now includes the previously known diffuse infiltrating pontine glioma, and carries a dismal prognosis.
4.11 Solitary Fibrous Tumor (SFT) and Hemangiopericytoma (HPC)
Both these tumors share the same genetic feature which includes genomic inversion at the 12q13 locus, fusing the NAB2 and STAT6 genes. Hence, these two previously distinct tumors are now combined as SFT/HPC tumor. Of these, the SFT/HPC Grade I is a slowly growing tumor with excellent prognosis, while SFT/HPC Grade II and III have a slightly poor prognosis, carry a high risk to recur following resection, and are associated with metastasis [27].
4.12 New Tumors and Patterns
4.12.1 Multinodular and Vacuolating Neuronal Tumor
This rare entity has received mention in the 2016 Revised CNS Tumor Classification. It is unclear if it is a distinct tumor or in the tumor-dysplasia category. It carries an excellent prognosis and is believed to be a “Leave-me-alone” lesion.
4.12.2 Radiological Features
It is known to occur anywhere in the brain, but commonly in the supratentorial compartment and especially in the frontal and temporal lobes. On morphological appearance, the lesion is seen as a cluster of FLAIR and T2 bright lesions typically in the subcortical white matter. Involvement of the overlying cortex and periventricular white matter has been reported. The lesion appears dark on T1WI and does not demonstrate contrast enhancement or diffusion restriction. No susceptibility is seen [31].
4.13 Diffuse Leptomeningeal Glioneuronal Tumor
This is a rare glioneuronal neoplasm mainly seen in children. It is largely localized to the leptomeningeal compartment. Oligodendroglioma like tumor cells are seen on histology.
4.13.1 Radiological Features
Diffuse, somewhat heterogenous leptomeningeal enhancement is noted. Frequently the basal cisterns are involved with associated extensive involvement of the subarachnoid space along the surface of the cord. Secondary hydrocephalus is commonly noted [32]. Parenchymal involvement can also be seen. When present, it is seen to involve the spinal cord and the brain stem.
4.14 Conclusion
The Revised 2016 classification of CNS tumors by including the genetic profile improves diagnostic accuracy of brain tumors. This allows neuro-oncologists and the surgeons to optimize treatment strategies targeted to the specific tumor type, thus allowing for a better prognosis, and improved overall survival. The neuroradiologist by identifying the imaging phenotype of the particular glioma genotype plays an important role in guiding the clinical team in their treatment planning.
IDH-mutated tumors are more solid in their imaging profile, and demonstrate less enhancement than IDH-wildtype counterparts.
1p/19q-codeleted tumors are more heterogenous in their imaging appearance, and exhibit calcification more frequently than their 1p/19q-intact (noncodeleted) counterparts.
BRAF-mutant tumors are seen more commonly in the pediatric population. These include pilocytic astroctyoma, pilomyxoid astrocytoma, and ganglioglioma in their molecular profile spectrum.
H3K27M-mutant tumors are also seen more commonly in the pediatric population, but are more diffuse and aggressive. These mutant tumors encompass the previously described diffuse infiltrating pontine glioma spectrum of tumors.

Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.