To be able to detect typical imaging findings in congenital metabolic diseases
To be able to detect typical imaging findings in acquired metabolic diseases
To understand the use of the different imaging modalities, mainly CT and MRI in metabolic diseases
Metabolic diseases lead to changes in the imaging studies that we need to be aware of to trigger further investigation as the exact differential diagnosis might be challenging
Toxic changes to the brain and spine are common and need to be identified in both an emergency and non-emergent situation
MRI is the imaging method of choice and functional imaging techniques like MR spectroscopy and perfusion methods may add important additional information
9.1 Most Common Congenital Metabolic Disorders Presenting or Persisting into Adulthood
9.1.1 X-Linked Adrenoleukodystrophy
This is a disorder of peroxisomal fatty acid beta oxidation leading to accumulation of long chain fatty acids, which can manifest itself in childhood (typically around 7 years of age) or in adult life. Adult onset cerebral ALD accounts for about 5% and usually presents with psychiatric features followed by dementia, ataxia, and seizures. As an X-linked disorder ALD is more frequent in males but around 15–20% of heterozygote females carriers become symptomatic.
The typical MRI appearance are T2 and FLAIR hyperintense white matter abnormalities staring in the parieto-occipital regions with early involvement of the splenium of the corpus callosum and corticospinal tracts. These signal abnormalities correspond to areas of demyelination. Active demyelination at the edge of the lesions is associated with contrast enhancement and restricted diffusion on DWI.
Adrenomyeloneuropathy (AMN) is a later onset adult phenotype presenting with slowly progressive spastic paraparesis, bladder and bowel dysfunction, sexual dysfunction, and peripheral neuropathy. Typical imaging appearances include increased T2/FLAIR signal in the posterior limbs of the internal capsules, brainstem and cerebellar white matter which may be followed by spinal cord atrophy [1].
9.1.2 Globoid Cell Leukodystrophy (Krabbe Disease)
Globoid cell leukodystrophy (GLD) has been linked to a mutation in the GALC gene on chromosome 14, leading to a deficiency of galactosylceramide β-galactosidase that causes accumulation of sphingolipids in the lysosomes.
Late adolescent and adult forms (10%) present with slowly progressive gait abnormalities or spastic paresis. Other features include cognitive decline, seizures, and cortical blindness.
MRI shows predominantly posterior T2/FLAIR hyperintense white matter changes with sparing of the U fibres and involvement of the splenium of the corpus callosum, extending along the corticospinal tracts into the posterior limbs of the internal capsules and the pyramidal tracts.
9.1.3 Metachromatic Leukodystrophy (MLD)
Several variants have been described, all of which have deficient activity of arylsulfatase A, which results in defective degradation of sulfatides in the lysosomes.
Approximately 20% of MLD present in adulthood often with psychiatric symptoms, followed by spastic paraparesis, cerebellar ataxia, and cognitive decline.
MRI demonstrates symmetrical areas of T2 prolongation in the periventricular white matter with sparing of the subcortical U fibres. Early involvement of the peritrigonal white matter, corpus callosum, and cerebellar white matter is common, showing typically a “tigroid” pattern of radiating stripes. As opposed to ALD, there is no contrast enhancement of the lesion edge.
9.1.4 Mitochondrial Encephalomyopathy with Lactic Acidosis and Stroke-like Episodes (MELAS)
Mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS) syndrome is a typical example of a mitochondrial disorder. About 40% of patients present in late childhood or early adulthood.
Clinical features are led by encephalomyopathy, lactic acidosis, and stroke-like episodes; other features include seizures, hearing loss, and neuropsychiatric dysfunction.
MRI demonstrates T2/FLAIR hyperintense areas involving cortical and deep grey matter, not confined to any of the major vascular territories. On diffusion-weighted imaging, ADC values can be elevated or decreased, depending on the acuity of symptoms.
9.1.5 Fabry Disease (Galactocerebrosidase Deficiency)
Fabry disease is a lysosomal storage disease related to a deficiency of a-galactosidase. It is an X-linked inherited disorder with varied clinical presentations and an estimated prevalence of one in 50,000 persons.
The deposition of globotriaosylceramide-3 in the endothelium and smooth muscles leads to involvement of multiple organ systems, including the blood vessels, heart, and kidneys.
Macro and microvascular complications are the leading CNS manifestations, with the first cerebrovascular event usually occurring around 40 years of age.
CT and MRI features are those of macrovascular disease (acute or chronic infarcts and parenchymal haemorrhage), and signs of microvascular disease (hyperintense foci in the periventricular and subcortical white matter and cerebral microhaemorrhages).
Characteristic MRI features of Fabry’s disease are T1 hyperintensity within the pulvinar nuclei of the thalamus (the “pulvinar sign”) and ectasia of the basilar artery on MRA, which has been found to be one of the best indicators of the presence of the disease in adults.
9.1.6 Alexander Disease
Defects in the glial fibrillary acidic protein gene have been identified as the underlying cause of this disease, in which approximately 25% of patients present in adulthood, most commonly with bulbar dysfunction, pyramidal involvement, cerebellar ataxia, and sleep abnormalities.
The MRI findings of the infantile and juvenile form include extensive white matter change with frontal predominance, a periventricular rim with high signal on T1-weighted images and low signal on T2-weighted images, abnormalities of the basal ganglia and thalami, brainstem abnormalities and contrast enhancement.
In adult onset disease, MRI findings consist of atrophy and abnormal signal within the medulla and upper cord, which has been termed a “tadpole” appearance and may be accompanied by periventricular white matter change.
9.1.7 Primary Familial Brain Calcification
Primary familial brain calcification, idiopathic basal ganglia calcification, and bilateral strioapallidodentate calcinosis are terms used for what was formerly called Fahr disease. This is a group of genetic disorders with currently four known mutations that are inherited in an autosomal dominant pattern. Symptomatic patients present in the late 40s with a mostly Parkinsonian movement disorder, followed by cognitive decline, cerebellar features, and speech disorders. Symmetrical calcification involving the lentiform nuclei, caudate nuclei, thalami and dentate nuclei is mostly readily detected by CT. On MRI, the structures involved may show high signal on T1-weighted spin echo sequences and signal loss on susceptibility-weighted imaging.
9.2 Acquired Metabolic Disorders
These are mostly seen in young adults or adults and present with typical imaging findings.
9.2.1 Wernicke’s Encephalopathy
Wernicke’s encephalopathy (WE) is caused by a deficiency of vitamin B1 (thiamine), which may be due to alcohol abuse, malabsorption, poor nutrition, increased metabolism, or iatrogenic elimination (haemodialysis).
Thiamine depletion leads to failure of conversion of pyruvate to acetyl-CoA and α-ketoglutarate to succinate and the lack of Krebs cycle resulting in cerebral lactic acidosis with intra- and extracellular oedema, swelling of astrocytes, oligodendrocytes, myelin fibres, and neuronal dendrites.
The classic clinical triad of ocular dysfunctions (nystagmus, conjugate gaze palsy, ophthalmoplegia), ataxia, and confusion is observed only in 30% of cases. Treatment consists of thiamine infusion, which can prevent progression to Korsakoff’s dementia or death.
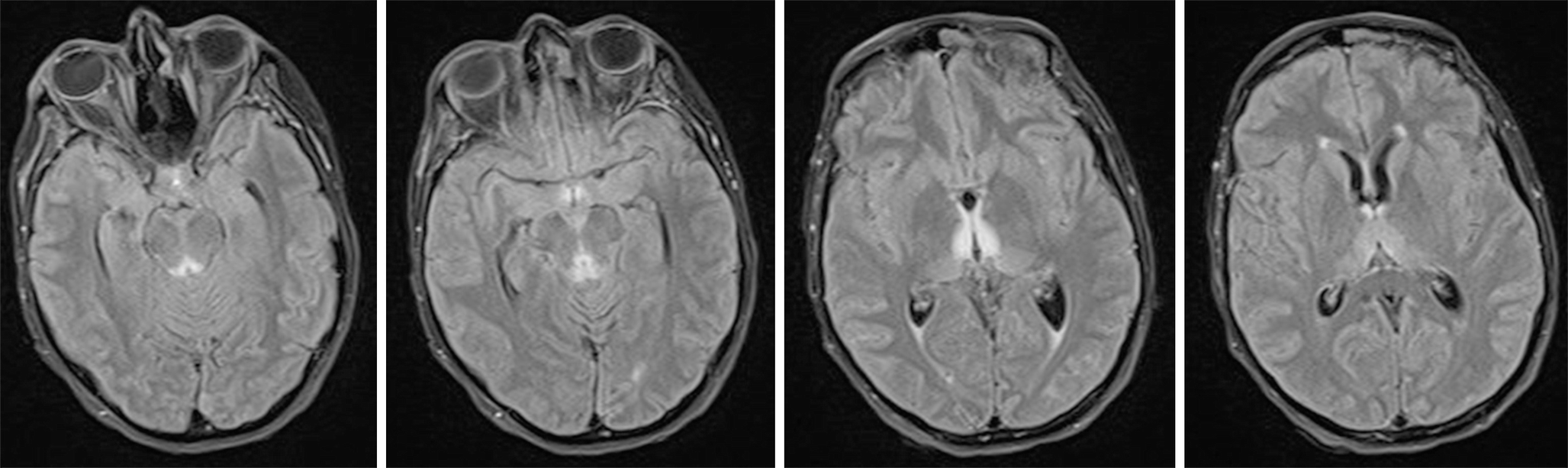
Typical FLAIR hyperintense tissue changes in the medial thalamus, the periaqueductal area, mammillary bodies, and tectal plate
Contrast enhancement occurs in 80% in the mammillary bodies (often prior to T2 hyperintensities) and in 50% in the peri-acqueductal regions.
Imaging abnormalities can regress with treatment, but the prognosis is usually poor once there is cortical involvement or T1 hyperintensity in the thalami and mammillary bodies indicating haemorrhagic change.
Chronic cases show atrophy of the mammillary bodies.
9.2.2 Subacute Combined Degeneration
This is a disorder of the spinal cord secondary to vitamin B12 deficiency, characterised by gliosis and spongiform degeneration of the posterior and lateral columns. Clinical presentations are in spastic paraparesis and spinal ataxia.
MRI demonstrates T2 hyperintensities in the posterior and lateral columns of the spinal cord, which can reverse after adequate B12 administration. Chronic non- or undertreated cases show spinal cord atrophy.
9.2.3 Osmotic Myelinolysis Syndrome
This entity includes central pontine myelinolysis (CPM) and extrapontine myelinolysis and occurs usually in patients with hyponatremia that has been corrected too quickly.
The traditionally proposed pathophysiological mechanism is a disruption of the blood–brain–barrier resulting in vasogenic oedema, fibre tract compression, and myelinolysis. Additional implicated mechanisms are cerebral dehydration, intramyelinic oedema, and oligodendrocyte degeneration. The most common damage is in the central pontine fibres (CPM). Extrapontine demyelination (in sites such as basal ganglia, thalami, lateral geniculate body, cerebellum and cerebral cortex) may occur together with CPM or in isolation (in approximately 10%).
Clinical symptoms of CPM include paralysis, dysphagia, dysarthria, and pseudobulbar palsy.
Typical MRI features of central pontine myelinolysis are T2/FLAIR hyperintensity in the central pons showing a symmetric “trident” or “bat-wing”-pattern, due to sparing of the peripheral fibres and the axons of the corticospinal tracts.
In the acute phase DWI shows restricted diffusion with a decreased ADC. This may occur within 24 h of symptom onset and precede the signal abnormalities seen on T2 or FLAIR images. The ADC values usually return to baseline within 3–4 weeks.
CPM can appear moderately hypointense in T1 and may infrequently show contrast enhancement. If the patient survives the acute phase, the pontine lesions can cavitate and appear markedly hypointense on T1 weighted images.
9.2.4 Hepatic Encephalopathy
The term hepatic encephalopathy includes a spectrum of neuropsychiatric abnormalities in patients with liver dysfunction. Most cases are associated with cirrhosis and portal hypertension or portal-systemic shunts, but the condition can also occur in acute liver failure.

Hepatic Encephalopathy—typical T1 hyperintense tissue changes in the basal ganglia based on manganese deposition
Magnetisation transfer imaging, fast FLAIR, and DWI sequences may demonstrate white matter abnormalities secondary to increased CNS ammonia concentration. These return to normal with restoration of liver function, and are thought to reflect mild diffuse interstitial brain oedema, which appears to play an essential role in the pathogenesis of hepatic encephalopathy.
In acute hepatic encephalopathy, bilateral symmetric T2-hyperintensities of the cortical grey matter are often associated with restricted diffusion. There may be additional involvement of the subcortical white matter, basal ganglia, thalami, and midbrain. These imaging abnormalities are thought to reflect cytotoxic oedema secondary to acute hyperammonemia.
9.2.5 Hypoglycemic Encephalopathy
An acute decrease in serum glucose levels can arise from an excess of exogenous or endogenous insulin and hypoglycaemia inducing drugs. This causes a decline of the cell membrane ATPase pump activity and release of excitatory neurotransmitters such as aspartate.
CT can demonstrate enhancing hypodensities in the basal ganglia, cerebral cortex, hippocampus, and substantia nigra.
MRI is more sensitive and shows T2 hyperintensities as well as restricted diffusion in the posterior limb of the internal capsules, hippocampi, the basal ganglia and cortical areas and splenium of the corpus callosum. These changes are likely to reflect cytotoxic oedema and extensive DWI changes in the basal ganglia and deep white matter are associated with poor clinical outcome.
9.2.6 Hyperglycaemic Encephalopathy
Hyperglycaemia occurs in uncontrolled diabetes mellitus and can lead to osmotic derangements the basal ganglia and subthalamic region.
Imaging findings are hyperdense changes on CT and T1 hyperintensities on MRI within the putamen and caudate nuclei, which may be uni- or bilateral. SPECT has been reported to show hypoperfusion in the basal ganglia. After correction of blood glucose, the imaging abnormalities usually regress [3].
Both congenital and acquired metabolic diseases present with typical imaging findings.
Even sometimes subtle, the course will enable to identify patterns that help in the differential process.
Only the combination between clinical information, laboratory findings, and imaging will allow to make a solid differential diagnosis.

Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.