Sulayman D. Dib-Hajj, Yang Yang, Joel A. Black, and Stephen G. Waxman
The voltage-gated sodium channel NaV1.7 is preferentially expressed in peripheral somatic and visceral sensory neurons, olfactory sensory neurons and sympathetic ganglion neurons. NaV1.7 accumulates at nerve fibre endings and amplifies small subthreshold depolarizations, poising it to act as a threshold channel that regulates excitability. Genetic and functional studies have added to the evidence that NaV1.7 is a major contributor to pain signalling in humans, and homology modelling based on crystal structures of ion channels suggests an atomic-level structural basis for the altered gating of mutant NaV1.7 that causes pain.
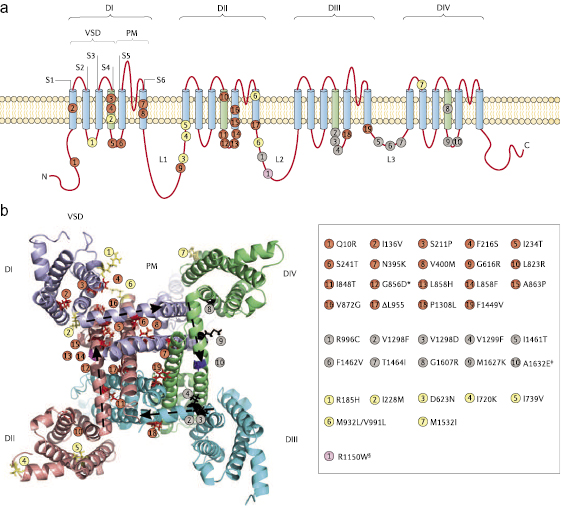
Voltage-gated sodium channels are essential for electrogenesis in excitable cells. Nine pore-forming α-subunits of such channels (referred to as channels hereinafter), NaV1.1–NaV1.9, have been identified in mammals.1 These isoforms share a common overall structural motif (figure 1). They are each composed of a long poly-peptide (1,700–2,000 amino acids) that folds into four homologous domains (DI–DIV) that are linked by three loops (L1–L3), with each domain having six transmembrane segments (S1–S6).2 The recent determination of the crystal structure of the homotetrameric bacterial sodium channel3 has provided insights into the atomic structure of mammalian sodium channels and the interactions between the voltage-sensing domain (VSD; encompassing S1–S4) and the pore module (PM; S5–S6) within each of the four homologous domains. Genetic, structural and functional studies have shown that NaV1.7 regulates sensory neuron excitability and is a major contributor to several sensory modalities, and have established the contribution of this sodium channel isoform to human pain disorders (figure 1).
The nine sodium channel isoforms display different kinetics and voltage-dependent properties.1 Their differential deployment in different types of neurons endows these cells with distinct firing properties. Sodium channels associate with multiple protein partners that regulate channel trafficking and gating,4–7 allowing sodium channel properties to be modulated in a cell-type-specific manner (for examples, see refs. 8–10), highlighting the need to study these channels within their native neuronal background whenever practicable. For example, the pathogenic G616R variant of NaV1.7 displays gating abnormalities within dorsal root ganglion (DRG) neurons that are not seen when these channels are expressed in HEK 293 cells.11 Methods are now available that allow the expression and functional profiling of sodium channels in peripheral neurons, which more closely mimic the in vivo environment of such channels.12
In humans, gain-of-function mutations in SCN9A, which encodes NaV1.7, lead to severe neuropathic pain, whereas loss-of-function mutations in this gene lead to an indifference to pain.13 Studies involving animal injury models and functional studies of neuronal excitability following expression of human mutant NaV1.7 have provided mechanistic insights into the role of this channel in the pathophysiology of pain. Additional studies have linked NAV1.7 to other sensory modalities, including olfaction,14,15 the afferent limb of the cough reflex16 and acid sensing.17
In this Review, we discuss functional and modeling studies of NaV1.7 that have yielded new insights into the structure–function relationship of gating mechanisms in this channel and its contribution to neuronal responses under normal and pathological conditions. We also explore strategies for targeting NaV1.7 in the treatment of pain and, finally, identify unanswered questions regarding the role of NaV1.7 in pain signaling.
Cellular and Subcellular Distribution
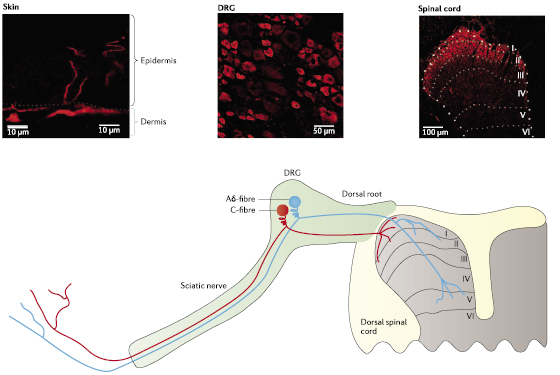
Three sodium channels—NaV1.7, NaV1.8 and NaV1.9—are preferentially expressed in peripheral neurons. NaV1.7 expression was first detected in somatosensory and sympathetic ganglion neurons,18 and has since been reported in myenteric neurons,19 olfactory sensory neurons (OSNs),14,15 visceral sensory neurons16,20 and smooth myocytes.21–23 NaV1.7 is expressed in both large and small diameter DRG neurons (figure 2), including functionally identified Aβ-fibres and C-fibres.24 NaV1.7 is also the predominant sodium channel isoform present in OSNs14,15 (box 1) and in nodose ganglion neurons.16 Measurable NaV1.7 levels have not been detected in the CNS18,25 (but see the discussion below on the purported role of NaV1.7 in epilepsy). NaV1.7 expression has also been detected within some non-excitable cells, including prostate and breast tumor cells,26,27 human erythroid progenitor cells28 and immune cells.29 NaV1.7 and NaV1.8 are both expressed at relatively high levels within functionally identified nociceptive neurons (nociceptors),24,30 in which their co-expression has important functional implications.10 Last, NaV1.7 is present peripherally within free nerve endings in the epidermis31 and centrally within superficial lamina of the dorsal horn in the spinal cord.32 The presence of NaV1.7 at nerve endings (figure 2) is consistent with its proposed role in amplifying weak stimuli.33
Figure 2 Pain signal transmission from peripheral terminals of DRG neurons that form synapses onto second-order neurons within the spinal cord. Dorsal root ganglion (DRG) neurons can be broadly classified into three types based on their soma size and the state of myelination of their axons: large diameter with heavily myelinated and rapidly conducting axons (Aβ-fibres; not shown here for simplicity); medium diameter with thinly myelinated and intermediate conducting axons (Aδ-fibres; cyan); and small diameter with unmyelinated and slowly conducting axons (C-fibres; red). Five voltage-gated sodium channels are reported to be expressed in DRG neurons,13 with NaV1.7 expressed in the majority of small unmyelinated neurons and in a notable population of medium and large diameter myelinated neurons (see middle panel; NaV1.7 expression is shown in red in this and other panels). Signals originating from the periphery are initiated by external stimuli (for example, thermal, mechanical or chemical stimuli) or injury- and inflammation-induced mediators (for example, cytokines or trophic factors), and are transduced by specific G protein-coupled receptors or acid- and ligand-gated ion channels at peripheral termini. Membrane depolarizations evoked by the graded receptor potential are integrated by voltage-gated sodium channels; when a threshold is reached, an action potential is initiated at these terminals and centrally propagated. NaV1.7 extends to the peripheral ends of these terminals (left panel) where it amplifies small depolarizing inputs. Although NaV1.7 is considered a peripheral sodium channel because it is expressed in peripheral neurons, it is present in central axonal projections of DRG neurons and their presynaptic terminals within the dorsal horn (right panel) where it may facilitate impulse invasion or evoked release of neurotransmitters that may include substance P, calcitonin-gene related peptide and glutamate.
Box 1 NaV1.7 contributes most of the sodium current in OSNs
NaV1.7 is the predominant sodium channel in olfactory sensory neurons (OSNs).14,15 Although an elaborate Ca2+-based and Cl–-based signaling amplification system in the OSN cilia can boost odorant receptor potential,133,134 the abundant expression of NaV1.7 in these cells14,15 and the ability of NaV1.7 to boost weak depolarizations, support a role for this sodium channel in the initiation of action potential firing along the peripheral olfactory neuraxis. Mouse and rat OSNs produce a tetrodotoxin (TTX)-sensitive current14,15,135 that is consistent with the predominant expression of NaV1.7 in these cells. Interestingly, the hyperpolarized activation and inactivation properties of this TTX-sensitive current are different from those recorded from NaV1.7 expressed in HEK 293 cells33,136 or dorsal root ganglion (DRG) neurons,11,35 and those in native rat DRG neurons.137–139 Ahn et al.14 reported identical sequences of the NaV1.7 cDNA in mouse OSN and DRG neuron samples. Together, these data suggest that post-translational modulation of NaV1.7 or interaction with OSN-specific channel partners may lead to altered gating properties of NaV1.7 in OSNs compared with DRG neurons.
Biophysical Properties
NaV1.7 produces a rapidly activating and inactivating, but slowly repriming (slow recovery from inactivation), current that is blocked by nanomolar concentrations of tetrodotoxin (TTX).34 The slow repriming nature of NaV1.7 makes it well-suited for low-frequency firing in C-fibres, but less well-suited to neurons that fire at a high frequency.33,35 Importantly, NaV1.7 is characterized by slow closed-state inactivation, allowing the channel to produce a substantial ramp current in response to small, slow depolarizations.33,35 The ability of NaV1.7 to boost subthreshold stimuli increases the probability of neurons reaching their threshold for firing action potentials. Thus, NaV1.7 is considered to be a threshold channel.36,37 NaV1.7 produces resurgent currents in DRG neurons,38,39 which are triggered by repolarization following a strong depolarization. Resurgent currents support burst firing in, for example, cerebellar Purkinje neurons.40,41 Production of a resurgent current by a given sodium channel isoform crucially depends on cell background; the same sodium channel that produces a robust resurgent current in one neuronal type may not generate such a current in a different neuronal type.40–42 Thus, it is not surprising that NaV1.7 produces a resurgent current only in a subset of DRG neurons.
Roles in Multiple Sensory Modalities
Pain
As stated above, NaV1.7 is expressed in both large and small diameter DRG neurons,13 including 85% of functionally identified nociceptors.24 These observations, together with its properties as a threshold channel, suggested that NaV1.7 contributes to pain signaling. The recent discovery of gain-of-function SCN9A mutations in human pain disorders solidified the status of NaV1.7 as having a central role in pain signaling,13 and its involvement in pathological pain signaling is discussed below.
Olfaction
The initial discovery that global knockout of Scn9a in mice is neonatally lethal, probably because the newborn mice do not feed,43 and the subsequent discovery that humans with homozygous SCN9A-null mutations are anosmic44,45 suggested that NaV1.7 is important in olfaction. Nassar et al.43 noted the absence of milk in the stomach of Scn9a–/– pups. As no hand-feeding was attempted to rescue these mice, the most parsimonious explanation for the observed neonatal lethality is anosmia, leading to a loss in the ability to suckle. In agreement with this observation, NaV1.7 is the predominant sodium channel isoform present in presynaptic OSNs in rodents.14,15 Knockout of Scn9a in OSNs in mice blocks odorant-induced synaptic transmission to mitral cells in the olfactory glomeruli and leads to weight loss in mice,15 providing compelling evidence that NaV1.7 has a central role in the sense of smell.
Cough Reflex
Two types of coughs involve the vagus nerve: aspiration-induced cough and irritating, itchy urge-to-cough. Aspiration-induced cough is mediated by the stimulation of touch-sensitive Aδ-fibres and occurs even in unconscious subjects, whereas irritating, itchy urge-to-cough is mediated by C-fibre stimulants, including acidic compounds, and occurs only in conscious animals.46 Nodose ganglion neurons produce both TTX-sensitive and TTX-resistant currents,47 but action potentials in the vagus nerves of rats or guinea pigs are completely blocked with 1 μM TTX,16,48 suggesting a crucial role for TTX-sensitive channels in the cough reflex. Recent data suggest that NaV1.7 produces almost all of the TTX-sensitive current in the majority of nodose ganglion neurons in guinea pigs, and adeno-associated virus (AAV)-mediated short hairpin RNA (shRNA) knockdown of NaV1.7 expression in these neurons markedly increases the rheobase and attenuates the firing of both Aδ-fibres and C-fibres.16 In agreement with this finding, selective knockdown of NaV1.7 expression in nodose ganglion neurons suppresses citric acid-induced coughing in guinea pigs, without having any effect on the rate of aspiration.16 Whether knocking down NaV1.7 expression has a similar effect on aspiration-induced cough remains untested.
Acid Sensing
Naked mole rats do not develop pain-related behaviors when they are exposed to acid or capsaicin, despite the presence of transient receptor potential vanilloid subfamily member 1 (TRPV1) channels in their nociceptors.49 This mystery has recently been resolved by the identification of a variant amino acid sequence in the outer vestibule of their NaV1.7 channels.17 In almost all mammalian orthologs of NaV1.7, the extracellular linker between S5 and S6 in DIV includes a KKV tripeptide sequence. This tripeptide sequence is replaced by EKE in the naked mole rat and by EKD in the microbat, which also lacks acid-induced pain-related behaviors. Interestingly, these two species live in large colonies in which high concentrations of CO2 can be generated. Such high levels of CO2 can cause tissue acidification and acid-induced pain in other animals. The EKE substitution in human NaV1.7 enhances acid-induced blockade of this channel, consistent with a failure to induce firing of action potentials in naked mole rat nociceptors.17 The corresponding tripeptide sequence in human NaV1.6, the other TTX-sensitive channel in adult nociceptors, is DKE, suggesting that it might be more sensitive to acidic conditions than NaV1.7.
Putative Role in Epilepsy
One study reported the presence of SCN9A variants in patients with seizures, including those with Dravet syndrome (Online Mendelian Inheritance in Man (OMIM) database #607208); these variants were present in the control population used in the study at >1% allele frequency.50 However, the function of NaV1.7 in CNS neurons and its role, if any, in the pathophysiology of seizures has not been established, although a knock-in mouse expressing one of these variants was reported to exhibit seizures. Importantly, neither patients with small-fibre neuropathy (SFN)39 carrying the same NaV1.7 variants reported by Singh et al.,50 nor patients with other gain-of-function SCN9A mutations associated with inherited erythromelalgia (IEM; also known as familial erythromelalgia and primary erythermalgia; OMIM #133020)13,51 have reported seizures. Thus, the contribution of SCN9A mutations, if any, to epilepsy remains incompletely understood.
Roles in Pain States
NaV1.7 in Acquired Pain Conditions
NaV1.7 has an important role in pain signaling.13,51 Axotomy of peripheral axons can produce a neuroma in which ectopic impulses arise, causing spontaneous pain.52 Application of TTX at concentrations that block only TTX-sensitive channels ameliorates neuropathic pain behavior in a rat axotomy model,53 suggesting that these channels contribute to spontaneous pain. Although the TTX-sensitive sodium channel NaV1.3 has been implicated in ectopic firing and spontaneous pain,13 NaV1.7 accumulates at nerve endings within neuromas together with activated mitogen-activated protein kinase 1 (MAPK1; also known as ERK2) and MAPK3 (also known as ERK1) in humans54 and in rats.31 MAPK1 and MAPK3 phosphorylate NaV1.7 at four sites within L1, producing a graded hyperpolarizing shift of channel activation. The extent of graded hyperpolarizing shift in NaV1.7 activation depends on the number of phosphorylated residues.55 Together with the finding that MAPK1 and MAPK3 exert a pro-excitatory effect on DRG neurons,55 these data suggest that NaV1.7 can contribute to injury-mediated DRG neuron excitability.
NaV1.7 expression levels and TTX-sensitive current density are increased in DRG neurons in response to inflammation.56 The increase in NaV1.7 expression levels is more robust than that of NaV1.3—the other TTX-sensitive channel that is upregulated under these conditions.56,57 NaV1.7 levels in DRG neurons are also increased in diabetic rats,58,59 a change that is predicted to contribute to hyperexcitability associated with pain. A direct contribution of NaV1.7 to pathological DRG neuronal hyperexcitability is further supported by knockdown and knockout studies in rodents. Knockdown of NaV1.7 attenuates complete Freund’s adjuvant-induced thermal hyperalgesia60 and diabetic pain.61 Conditional knockout of NaV1.7 expression in mouse DRG neurons, where NaV1.8 is expressed, abrogates inflammation-induced and burn injury-induced thermal hyperalgesia, but does not impair mechanical allodynia or hyperalgesia (neuropathic pain).43,62,63 However, a recent report62 provided evidence suggesting that knocking out NaV1.7 expression in both DRG and sympathetic neurons abrogated neuropathic pain (box 2).
Box 2 NaV1.7 in sympathetic neurons and pain signaling
The contribution of NaV1.7 to electrogenesis in sympathetic neurons and the contribution of these neurons to pain are not well understood. Although NaV1.7 is normally expressed in sympathetic neurons,18 individuals with NaV1.7-related complete insensitivity to pain (CIP) do not report sympathetic deficits,66 suggesting that the role of NaV1.7 in these neurons might be redundant. Gain-of-function mutant NaV1.7 in patients with severe pain can depolarize the resting membrane potential of dorsal root ganglion (DRG) neurons and sympathetic neurons. The resulting effect renders DRG neurons hyperexcitable and sympathetic neurons hypoexcitable,10 suggesting that severe pain may still occur even when sympathetic neuron excitability is reduced. However, studies in mice suggest that functional features of both sensory and sympathetic neurons, which are dependent on NaV1.7, contribute to the manifestation of pain symptoms.43,62 Minett et al.62 reported that knocking out Scn9a (the gene encoding NaV1.7) in DRG neurons alone does not cause a total loss of pain, whereas knocking out the expression of this channel in both sensory and sympathetic neurons recapitulates features of human CIP. Future studies are needed to investigate the role of NaV1.7 in sympathetic neurons and pain signaling.
NaV1.7 in Inherited Pain Disorders
The co-segregation of a familial mutation and disease symptoms in more than one generation provides a compelling case for a direct link between a target gene and a disease. Recently, mutations in SCN9A that alter the functional properties of NaV1.7 in a pro-excitatory manner have been shown to produce familial pain disorders that follow a Mendelian inheritance pattern (inherited sodium channelopathies). These findings provided a causative link in these pain disorders and confirmed that NaV1.7 has a central role in pain signaling in humans. Dominantly inherited gain-of-function missense mutations in SCN9A are found in individuals with IEM64 and paroxysmal extreme pain disorder (PEPD; previously known as familial rectal pain; OMIM #167400).65 By contrast, recessively inherited loss-of-function mutations in SCN9A are linked to complete insensitivity (indifference) to pain (CIP; OMIM #243000).66 Functional characterization of these gain-of-function mutations has elucidated the patho-physiological basis for DRG neuron excitability in these disorders, establishing a mechanistic link to pain.
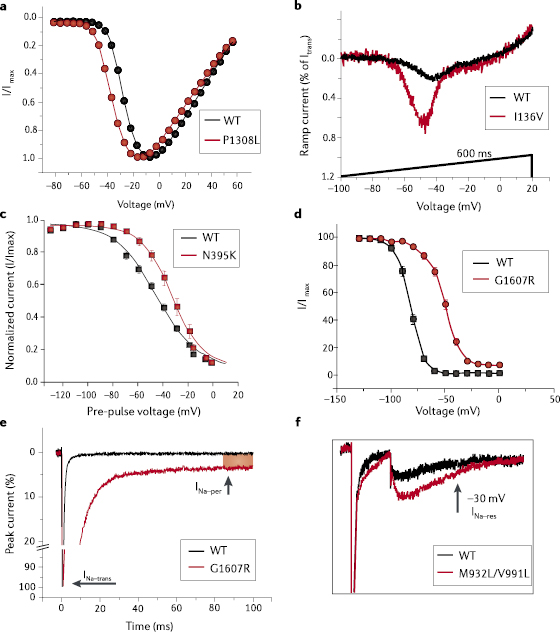
Pain in IEM is localized to the feet and hands, and symptoms of this condition usually appear in early childhood.13,51 Multiple families with IEM carry mutations in SCN9A that segregate with disease in affected individuals, providing strong genetic evidence for the pathogenicity of these mutations (figure 1). The familial IEM mutations in SCN9A that have been characterized to date all shift the voltage-dependence of NaV1.7 activation in a hyperpolarized direction (figure 3a), increase ramp current (figure 3b) and slow deactivation. IEM-linked SCN9A mutations can impair slow inactivation (figure 3c), thus enhancing DRG neuron hyperexcitability,67 whereas other IEM mutations enhance slow inactivation and therefore attenuate DRG neuron excitability.68
Another distinct set of mutations in SCN9A underlies PEPD, in which severe perirectal pain typically starts in infancy.65 The rectal pain is accompanied with skin flushing of the lower or upper body or face and can present in a harlequin pattern,69 which can alternate between the left and right sides of the body during different pain episodes.70 PEPD-linked SCN9A mutations produce different effects on NaV1.7 gating compared with IEM-associated mutations.13,65 PEPD-linked SCN9A mutations shift the voltage-dependence of steady-state fast inactivation toward a depolarizing direction (figure 3d) and, depending upon the specific mutation, make channel inactivation incomplete, which results in a persistent current (figure 3d,e). PEPD, but not IEM, mutant NaV1.7 manifests increased resurgent currents38 (figure 3f).
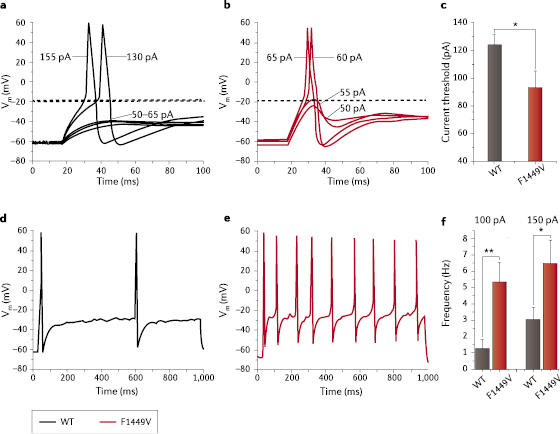
The IEM-linked SCN9A mutations studied to date13 lower the threshold for single action potentials (figure 4a–c) and increase the frequency of firing in DRG neurons (figure 4d–f), with many IEM-linked SCN9A mutations causing a depolarizing shift in resting potential.13 At the cellular level, PEPD mutant NaV1.7 lowers the threshold for single action potentials and increases the frequency of firing in DRG neurons, without altering the resting potential.71–73 Importantly, these functional profiles have been obtained by recordings from the somas of DRG neurons. It will be important, in the future, to assess the properties of these mutant channels and their effect on excitability near nerve terminals where NaV1.7 is thought to exert its influence as a threshold channel. A recent study from our group74 has begun to address this point, demonstrating a resting potential for DRG neurites close to −60 mV, a potential at which NaV1.7 channels are not strongly inactivated and are available for activation in fine diameter axons of DRG neurons. This study also demonstrated that action potential electrogenesis in DRG neurites in culture is driven by the sequential activation of TTX-sensitive and then TTX-resistant sodium currents.
De novo mutations in SCN9A in individuals with IEM, but without a family history of this disorder, produce similar functional changes in mutant NaV1.7 to those produced by familial mutations and render DRG neurons hyperexcitable, which is consistent with the pathogenicity of these mutant variants.75,76 However, the molecular genetic basis of delayed onset of pain in adult-onset IEM is not yet understood. As in IEM, de novo mutations in SCN9A in individuals with PEPD and no family history of this disorder have been identified; the effects of these de novo mutations on NaV1.7 gating is similar to those in familial PEPD, which is consistent with the pathogenicity of these mutations.70
The distinct and focal patterns of pain in IEM and PEPD are remarkable, considering that NaV1.7 is expressed in most DRG neurons (figure 2) and trigeminal neurons. An individual with a mixed phenotype that included symptoms of IEM and PEPD symptoms was found to carry the SCN9A mutation A1632E, which hyperpolarizes activation and depolarizes steady-state fast inactivation.71 Thus, NaV1.7-associated IEM and PEPD might be considered to be part of a clinical and physiological continuum that can produce IEM, PEPD and disorders that have characteristics of both of these conditions.
Recessively inherited SCN9A nonsense or splicing-defective mutations have been linked to NaV1.7-related CIP.66 Heterozygous parent carriers of these mutations are asymptomatic, indicating that the loss of one SCN9A allele does not lead to clinically manifested haploinsufficiency. Truncated NaV1.7 CIP fragments do not assemble into functional channels66,77 and do not act as dominant negative proteins,77 which reflects the normal pain experienced in the heterozygous carrier parents of patients with CIP. Although the first cases of NaV1.7-related CIP were from consanguineous families,66 later cases were identified in non-consanguineous marriages,44,45 indicating that there is a higher incidence of carriers of non-functional SCN9A alleles in the general population than was predicted from the initial reports. However, neither homozygous nonsense mutations nor compound heterozygous null mutations have been reported in healthy individuals. Patients with NaV1.7-related CIP do not experience any form of pain. Notably, they do not display motor, cognitive, sympathetic or gastrointestinal deficits, and have intact sensory modalities.66,77 An exception to this is that several patients have reported that they have an impaired sense of smell,44,45 although a recent study has described several members of a family with a non-sense SCN9A mutation, CIP and normal sense of smell.78
Although the expression of wild-type NaV1.7 at 50% of the normal protein level (that is, there is one functional allele) is sufficient for a normal pain phenotype (that is, there is no haploinsufficiency), the minimum level of functional NaV1.7 required to maintain the capacity to experience normal pain is not known. Interestingly, an individual with incomplete CIP (the patient retained some pain sensation) was found to carry compound heterozygous mutations in SCN9A, including a missense mutation (C1719R) affecting the S5–S6 extracurricular linker in DIV, and a one base-pair deletion in the 5′ splice donor site of exon 17 of SCN9A.79 Impaired splice donor sites, like most splice-site mutations, may cause exclusion of exon 17 and therefore lead to non-functional channels, which is consistent with the phenotype of impaired sensing. The reporting of some pain experience in this individual suggests that successful but inefficient exon 17 inclusion and production of functional NaV1.7 have occurred, but at levels that do not support full manifestation of pain.
Positive symptoms (pain) or negative symptoms (loss of pain sensing and anosmia) of patients with SCN9A-linked conditions can be explained by the effects of SCN9A gain-of-function and loss-of-function mutations, respectively, on nociceptors. The lack of an effect of SCN9A mutations on other sensory modalities is, however, not well understood. Although NaV1.7 is expressed in more than 50% of Aβ low-threshold mechanoreceptors,24 individuals with CIP have normal nerve conduction, tactile sense and vibration sense,66,77 suggesting that NaV1.7 function is redundant in these neurons. By contrast, normal proprioception in patients with CIP is consistent with the absence of NaV1.7 in muscle afferents.24 It is not fully understood why SCN9A gain-of-function mutations do not cause positive symptoms in carriers; for example, causing them to become ‘hyper-smellers’.
Functional Variants as Risk Factors
In agreement with the ‘common disease, common variant’ hypothesis,80 the R1150W variant of NaV1.7 has been associated with enhanced pain sensation.81,82 Estacion et al.81 demonstrated that the W1150 minor allele was present in 30% of people in an ethnically matched control population of Caucasian individuals of European descent. The W1150 variant of NaV1.7 induces hyperexcitability of DRG neurons, suggesting that carriers of this polymorphism might be predisposed to hyperalgesia. Indeed, a genome-wide association study found that the R1150W polymorphism is associated with an increased pain perception in patients with osteoarthritis, phantom limb pain or lumbar root pain, and that the effect is most strongly associated with C-fibre activation.82
About 30% of individuals with idiopathic SFN express functional mutant NaV1.7 channels arising from gain-of-function SCN9A missense variants,39 which may not be fully penetrant when found in families.83 People carrying these gain-of-function NaV1.7 variants are hypersensitive to pain, which reflects the expression of this channel in DRG neurons. These individuals also manifest profound autonomic dysfunction, which reflects the expression of NaV1.7 in sympathetic neurons.10,18 Gain-of-function attributes of NaV1.7 variants in SFN include depolarized fast inactivation (figure 3d) and/or slow inactivation (figure 3c), or an increase in the fraction of cells that produce resurgent currents (figure 3f). Surprisingly, however, individuals with SCN9A-null mutations do not manifest autonomic system deficits,66 suggesting that there is a redundant function for this channel in sympathetic neurons.
Does NaV1.7 Play a Role in the Dorsal Horn?
Based on studies in HEK 293 cells and DRG neuron somata, and on computer simulations, NaV1.7 is thought to act as a threshold channel that activates at relatively hyperpolarized potentials, thus amplifying small, slow depolarizations at potentials negative to an action potential threshold.36,84 This role, however, does not explain the total lack of pain sensation in patients with NaV1.7-related CIP even in response to the most intense stimulation, such as dental work or child-bearing labor. One possible theory is that NaV1.7 at central termini of primary afferents (figure 2) may play a part in synaptic transmission of pain signals.
Consistent with this hypothesis, Minett et al.62 showed that evoked release of substance P into the spinal cord in response to sciatic nerve stimulation, and synaptic potentiation of wide dynamic range neurons receiving input from primary afferents are attenuated in mice that had NaV1.7 knocked out in DRG neurons. NaV1.7 may have a role in facilitating the invasion of incoming action potentials from peripheral nociceptors into central pre-terminal exon branches or into terminals within the spinal cord. Alternatively, NaV1.7 may be involved within the terminals in the process of neurotransmitter release onto second-order dorsal horn neurons. Thus, we speculate that NaV1.7, deployed near presynaptic terminals in the dorsal horn,32 is important for release of neurotransmitters such as substance P. If this speculation is correct, then NaV1.7 inhibitors that act on both peripheral and central compartments might be needed for clinical efficacy.
Structural Features of NaV1.7
Our ability to understand the mechanistic bases of pathogenic SCN9A mutations and to develop rationally designed small-molecule inhibitors for the treatment of hyperexcitability disorders is limited by the lack of a high-resolution crystal structure of a eukaryotic sodium channel. High-resolution crystal structures of ion channels are necessary for a comprehensive understanding of the links between voltage-sensing and channel activation and inactivation, ion selectivity, and drug interactions. Our current understanding of these channel properties was derived from comparative sequence analysis, and from functional assays that measured ion conductance or fluorescence emission of tagged channels.2Atomic structural modeling following the determination of high-resolution crystal structures of potassium channels85–87 and, more recently, a bacterial sodium channel3 has advanced our understanding of the structure–function relationship of human SCN9A mutations, which is discussed below.
Lessons Learned from Potassium and Bacterial Sodium Channels
Crystallographic studies of potassium channels provided the first direct evidence for the structural basis for ion selectivity, pore gating and coupling of a voltage sensor to the pore components.85–87 These studies also yielded valuable insights into kinetics and sequence determinants of different gating mechanisms. Identification of the homotetramer bacterial voltage-gated sodium channel,88 with the monomer possessing the six transmembrane segment architecture of the homologous domains in the eukaryotic channels, facilitated the production of sufficient channel protein for crystallization and high-resolution structural studies. Intriguingly, bacterial voltage-gated sodium channels are most similar to DIII of human sodium channels.89 The first high-resolution crystal structure (resolved at 2.7 Å) of a pre-open conformation of the voltage-gated sodium channel from the bacterium Arcobacter butzleri (NaVAb)3 suggested that the S4 segments are in the activated position, but that the activation gate at the cytoplasmic end of the pore domain is closed. This study provided structural evidence for several of the gating steps of sodium channels and demonstrated a possible route for access of small hydrophobic pore-blocking molecules.
Because of the nature of eukaryotic sodium channels as four-domain polypeptides, which are linked by cytoplasmic loops with divergent lengths and sequences in the different members of the sodium channel family, there may be subtle yet important structural differences between these channels and the bacterial homotetrameric channels. Thus, caution is warranted in extrapolating from high-resolution crystal structures of a symmetrical homotetrameric bacterial sodium channel to eukaryotic single polypeptide multi-domain sodium channel isoforms. Moreover, individual channel mutations should optimally be assessed in their native isoform. For example, the S241L mutation within the DI S4–S5 linker of NaV1.7 produces a marked hyperpolarizing shift in its activation, steady-state fast and slow inactivation, compared with wild-type channels.90 By contrast, substitution of the corresponding residue in NaV1.4, S246L, hyperpolarizes steady-state fast and slow inactivation of the channel but, unlike S241L in NaV1.7, S246L had no effect on NaV1.4 activation,91 thus providing an example of an isoform-specific effect of conserved residues.
Atomic Structural Modeling of the Putative Activation Gate
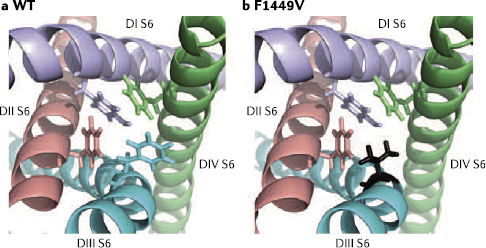
From a homology model of the NaV1.7 pore components, based on the crystal structure of the Streptomyces lividans potassium channel KcsA,85 we were able to identify a putative activation gate.92 This modeling approach identified an aromatic residue within the cytoplasm-proximal portion of each of the pore-lining S6 helices (DI Y405, DII F960, DIII F1449 and DIV F1752) that were predicted to form a hydrophobic ring at the cytoplasmic end of the pore that stabilizes the channel’s pre-open state. These aromatic residues in the four S6 helices form an energetically stable assembly due to extensive van der Waals bonds between their side chains, which is further strengthened by additional edge-face interaction with the adjacent aromatic residues.93 The hydrophobic ring is predicted to raise the energy barrier for the movement of S6, which is necessary to open the channel’s pore, thus stabilizing the closed or pre-open state of the channel. Although the activation gate at the narrow cytoplasmic vestibule of the channel in the NaVAb crystal structure consists of four methionine 221 residues (one from each monomer),3 modeling of NaV1.7 based on the NaVAb crystal structure recapitulates the activation gate that was previously identified based on the KcsA structure (figure 5).
Figure 5 A model of the putative activation gate of NaV1.7. The folded structure of the two S6 transmembrane segments presented here were based on the crystal structure of a bacterial sodium channel.3 The carboxy-terminal aromatic residue of each S6 is shown in stick representation for wild-type (WT; a) NaV1.7 and NaV1.7 with the F1449V mutation (b). The assembly of aromatic residues at the cytoplasmic C terminus of each of the S6 segments forms the putative activation gate of NaV1.7. The F1449V mutation in the homologous domain III (DIII) disrupts the hydrophobic ring and destabilizes the pre-open state of the channel.
Evidence for the formation of this hydrophobic block is provided by functional studies of the F1449V mutation in NaV1.7 that is found in patients with IEM. This mutation lowers the threshold for NaV1.7 activation.94 The F1449V substitution is predicted to destabilize interactions with the adjacent aromatic residues, thus reducing the energetic barrier for DIII S6 helix movement and facilitating bending motions associated with pore opening. The increased propensity of the DIII S6 helix to move would be expected to hasten channel activation. Support for this model of activation comes from studies of inwardly rectifying potassium (Kir) channels, which form a similar quadruple phenylalanine hydrophobic ring.95 Substitution of F168 in Kir6.2 (which is analogous to F1449 in NaV1.7) with smaller residues favors the channel’s open-state, whereas substitution of F168 with the aromatic amino acid tryptophan retains wild-type-like function.95
There are limitations on the use of the crystal structure of a homotetrameric voltage-gated ion channels (bacterial sodium channel and various potassium channels) for modeling the multi-domain mammalian NaV1.7, and our functional studies of the effect of F1449V on the activation gate provide an instructive example of such a limitation. Although the model suggested that phenylalanine or tyrosine residues at the carboxyl termini of the S6 segments in NaV1.7 stabilize the channel’s closed or pre-open state, functional analysis showed that these residues have different effects. Specifically, DII F960V and DIII F1449V markedly hyperpolarize channel activation, whereas DI Y405V and DIV F1752V do not alter channel activation.92 This may reflect the functional specialization of the four homologous, yet not identical, domains of eukaryotic sodium channels.
Dependence on Neuronal Background
Gating of wild-type or mutant sodium channels can be modulated in a cell-type-dependent manner, and this phenomenon can have important clinical implications. For example, resurgent sodium currents can be recorded from only a subset of small diameter DRG neurons transfected with NaV1.6 (ref. 42) or NaV1.7 (ref. 38), and NaV1.8 channels exhibit slow-inactivation properties that are differentially regulated in different subpopulations of (peptidergic and non-peptidergic) small diameter DRG neurons.9 For example, a single mutation in SCN9A, leading to L858H10 or I739V,96 renders DRG neurons hyperexcitable but superior cervical ganglion (SCG) neurons hypoexcitable. The latter phenomenon is related to the depolarization of the resting potential in both DRG and SCG neurons by the mutant NaV1.7, which leads to resting inactivation of all of the sodium channel isoforms in SCG neurons and hypoexcitability. The presence of NaV1.8, which is relatively resistant to inactivation by depolarization,97–99 in DRG neurons renders these neurons hyperexcitable in response to depolarization.10 These data demonstrate that sodium channel mutations can have a range of cell-background-dependent effects in different types of neurons.
Targeting NaV1.7 for Pain Treatment
The clear involvement of NaV1.7 in human pain, and the lack of serious cognitive, cardiac and adverse motor effects with a total loss of NaV1.7, as demonstrated in individuals with CIP, have fuelled intense efforts to develop NaV1.7-specific inhibitors or modulators for the treatment of pain. Despite these intensive efforts, progress has been slow.100 Nonetheless, the occasional reports of patients with IEM who respond to monotherapy using pan-sodium channel blockers,101,102 and the responsiveness of patients with PEPD to carbamazepine, suggest that small molecules may be developed to either inhibit or modulate NaV1.7 in a manner that can reduce excitability of DRG neurons and provide pain relief. Using the IEM NaV1.7 V400M carbamazepine-responsive mutation102 as a ‘seed’ for an atomic-level modeling and thermodynamic analysis, Yang et al.103 were able to predict carbamazepine-responsiveness of a second IEM mutation, NaV1.7 S241T, suggesting that, in the future, pharmacogenomic guided therapy may be possible. Alternative strategies may include the development of isoform-specific blockers or modulators of gating states of sodium channels that are differentially altered under pathological conditions; the development of compounds that weakly cross the blood–brain barrier to minimize CNS-related adverse effects; and gene therapy.
Small-Molecule Blockers
Several purportedly selective small-molecule inhibitors of NaV1.7 have been described and have shown efficacy in animal models of pain.104–107 These reports, however, lack documentation for selectivity against human sodium channel isoforms in a native neuronal environment. Reports detailing the efficacies of these compounds in animal models of pain should therefore be interpreted with caution, as these results could be due to inhibition of any of the neuronal sodium channel isoforms. A small-molecule blocker with robust selectivity for human NaV1.7 was recently developed.108 This orally bioavailable compound bound preferentially to the slow-inactivated state of the channel, and showed notable selectivity for NaV1.7 over other voltage-gated sodium channel isoforms (by 10-fold to 900-fold). The compound also showed 1,000-fold selectivity for NaV1.7 over potassium and calcium channels. These favorable properties suggest that this small-molecule blocker holds promise for future clinical studies.
State-Dependent Blockers
Local anesthetics, anticonvulsants and tricyclic compounds block sodium channels, mostly in a use-dependent fashion, and are among the first-line treatments that are currently available for neuropathic pain.109,110 However, these agents are not isoform-specific and only provide partial pain relief due in part to their limited therapeutic window that results from CNS-related adverse effects, such as dizziness or sedation.111,112 Despite these limitations, lidocaine derivatives and carbamazepine are effective in patients carrying certain SCN9A mutations that render NaV1.7 pharmacoresponsive,65,101,102 suggesting that personalized, genomically based therapeutics for pain is possible.
Patients with PEPD harbouring SCN9A mutations respond favorably to treatment with carbamazepine, which acts to counterbalance impaired fast inactivation of the mutant channel and hence reduces the persistent current caused by these mutations.65 Although most patients with NaV1.7-linked IEM do not respond to pharmacotherapy, a few have reported control of pain symptoms with lidocaine, mexiletine or carbamazepine. Successful lidocaine or mexiletine monotherapy was reported in a patient carrying the NaV1.7 V872G mutation, possibly resulting from enhanced lidocaine use-dependent block of the mutant channels.101 Three members of a family with IEM, carrying the mutation NaV1.7 V400M, reported control of their pain symptoms with carbamazepine.102 Preincubation of V400M channels with clinically relevant concentrations of carbamazepine induced a depolarizing shift in activation, which returned to wild-type voltages.102 This normalization of activation suggests that carbamazepine acts in an allosteric manner on the mutant NaV1.7 channel and induces a wild-type-like pre-open state.
Computer simulation studies67 and functional characterization of the NaV1.7 delL955 mutation68 suggest that enhancing the slow inactivation of NaV1.7 may allow an alternative approach to the treatment of pain. Lacosamide, a functionalized amino acid with sodium channel-blocking activity, showed beneficial effects in animal studies and clinical trials of epilepsy, in animal models of acute, inflammatory and neuropathic pain,113–116 and in initial clinical trials for diabetic neuropathic pain.117,118 Lacosamide’s blocking activity is unusual in that it involves selective enhancement of the slow inactivation of voltage-gated sodium channels, including NaV1.3, NaV1.7 and NaV1.8 (ref. 119). Interestingly, lacosamide induces substantially greater inhibition of NaV1.3, NaV1.7 and NaV1.8 when these channels are in an inactivated state.119 This feature of lacosamide might mean it would exhibit a better safety profile and greater tolerability than state-independent voltage-gated sodium channel blockers, as it might preferentially target injured depolarized neurons with hyperactive sodium channels.120 Although lacosamide has not been approved for the treatment of human neuropathic pain,121 targeting of the NaV1.7 slow-inactivated state might provide a viable drug-development option.
Natural Toxins
Natural peptide toxins might provide a source of isoform-specific inhibitors of sodium channels, because binding of these toxins to channels is regulated by multiple contact points, and minor sequence changes in the channel could have a profound effect on the affinity of the channel–toxin interaction. Venoms of a variety of snails are reservoirs of peptide toxins, and some of these have demonstrated sodium channel isoform selectivity.122–124 However, NaV1.7 is among the channels that are only weakly blocked by the conotoxins identified to date.122,123 By contrast, peptide toxins from tarantulas manifest preferential effect on NaV1.7. For example, ProTx-II is ~50-fold more selective for NaV1.7 than NaV1.5 (refs. 125,126). Huwentoxin-I and huwentoxin-IV are potent inhibitors of NaV1.7 and other neuronal TTX-sensitive channels, but are not effective against NaV1.4 (refs. 127,128). The exchange of two residues in the DII S3–S4 linker of NaV1.7 and NaV1.4 reverses the affinity of huwentoxins to these channels.128 Additionally, a charge-conserving substitution in KIIIA, a member of the μ-conotoxin subfamily, enhances the selectivity for NaV1.7 over NaV1.2 and NaV1.4 (ref. 129). It may therefore be possible to engineer peptide toxins with the desirable NaV1.7 isoform specificity.
Peptide toxins, however, have poor oral bioavailability and it is difficult to deliver them to nerve endings, implying that their use as therapeutic agents remains limited. However, modification of conotoxins by cyclization can enhance their stability in vivo without compromising their biological activity,130 and it may be possible to develop cyclized NaV1.7-specific peptide toxins when such molecules become available.
Gene Therapy
Advances in virus-mediated gene therapy have led to the initiation of Phase I trials for pain involving a herpes simplex virus (HSV) platform to transfer human preproencephalin (PENK) to DRG neurons.131 Local delivery of a gene product within the projection zone of an injured or diseased nerve associated with a focal pain syndrome (as in post-herpetic neuralgia or peripheral nerve injury) could be used to treat pain in a topologically defined manner, reducing systemic adverse effects. Animal studies have provided the proof-of-principle for this approach, showing that anti-NaV1.7 antisense constructs, delivered by a HSV virion, can attenuate pain behavior in mice following peripheral inflammation60 and in diabetic rats.61 We have recently succeeded in targeting another sodium channel, NaV1.3, in DRG neurons using RNA interference molecules (shRNA for gene knockdown) delivered using the non-virulent AAV platform,132 which is less immunogenic than other viral delivery platforms, suggesting that a similar strategy for targeting NaV1.7 using AAV-mediated delivery of shRNA may be successful.
Summary and Future Directions
NaV1.7 has proven to be a key player at the organismal level in human pain, at the cellular level as a major regulator of neuronal excitability and at the molecular level as a platform for discovering the contribution of specific residues to gating mechanisms. Studies of the rare monogenic disorders IEM, PEPD and CIP definitively show that NaV1.7 is critically important for human pain, and studies on SFN demonstrate a role for this channel in more common pain disorders. In addition to insights into the pathophysiology of pain gleaned from studying mutant NaV1.7 in its native neuron, modeling of mutant channels, based upon the crystal structures of the bacterial sodium channel and other ion channels, has led to identification of the putative activation gate of NaV1.7, and allows predictions of the dynamic interaction of the voltage-sensor and pore segments within the same domain and between different domains. Assessment of naturally occurring mutations in these studies could be especially informative, as they are already known to have large effects on gating properties of the channel. Finally, the relatively restricted expression pattern of NaV1.7, its central role in pain signaling in humans, and the minimal cognitive, cardiac, motor and sensory deficits in people totally lacking NaV1.7 have shown that this channel is a valid and indeed attractive target for drug development, and support the view that single target engagement for pain treatment might have therapeutic potential.
Nevertheless, despite progress in our understanding of NaV1.7 and its contribution to diverse sensory modalities, crucial questions remain unanswered. For example, why do patients with IEM or PEPD mutations manifest different pain topography despite the ubiquitous expression of NaV1.7 in sensory neurons? Why does skin flushing in some patients with PEPD alternate from side to side of the body? Why does the age-of-onset of IEM symptoms vary from infancy to adulthood? Why is there no evidence for compensatory changes that rescue nociception in CIP? In addition to missense or nonsense substitutions or loss-of-function mutations of splice sites in SCN9A, do synonymous or intronic insertions–deletions affect splicing efficiency or RNA stability and cause disease? What is the relative contribution of NaV1.7 to signal integration and transmission at peripheral and central termini of sensory and sympathetic neurons? Finally, why are the gating properties of the TTX-sensitive current in OSNs, which are mostly carried by NaV1.7, markedly different from those in HEK 293 cells and DRG neurons? These questions, and other related questions, will undoubtedly be answered in the near future.
Acknowledgments
The authors thank the members of their group for valuable discussions. Work in the authors’ laboratory is supported in part by grants from the Rehabilitation Research and Development Service and Medical Research Service, US Department of Veterans Affairs, and from the Erythromelalgia Association. The Center for Neuroscience and Regeneration Research is a collaboration between the Paralyzed Veterans of America and Yale University, Connecticut, USA.
About the Authors
Sulayman D. Dib-Hajj, Department of Neurology, and Center for Neuroscience and Regeneration Research, Yale University School of Medicine, New Haven, CT; Rehabilitation Research Center, Veterans Affairs Connecticut Healthcare System, West Haven, CT.
Yang Yang, Department of Neurology, and Center for Neuroscience and Regeneration Research, Yale University School of Medicine, New Haven, CT; Rehabilitation Research Center, Veterans Affairs Connecticut Healthcare System, West Haven, CT.
Joel A. Black, Department of Neurology, and Center for Neuroscience and Regeneration Research, Yale University School of Medicine, New Haven, CT; Rehabilitation Research Center, Veterans Affairs Connecticut Healthcare System, West Haven, CT.
Stephen G. Waxman, Department of Neurology, and Center for Neuroscience and Regeneration Research, Yale University School of Medicine, New Haven, CT; Rehabilitation Research Center, Veterans Affairs Connecticut Healthcare System, West Haven, CT.
Competing Interests Statement
The authors declare competing financial interests. See Web version for details.
Construction of a model of a folded protein based on the atom coordinates of a related member of the family whose high-resolution crystal structure is determined and additional constraints derived from studies of distant members of the superfamily.
inherited sodium channelopathies
Pathologies linked to mutations or functional variants in sodium channels that can be transmitted to progeny.
neuroma
A collection of demyelinated and dysmyelinated axon sprouts and connective tissue that result from abortive regeneration of transected axons.
neuropathic pain
Pain resulting from lesions or diseases of the somatosensory system.
nociceptors
Pain-sensing or damage-sensing neurons.
ramp current
Inward current due to transient channel activation in response to the small, slow depolarization of cell membranes.
repriming
Refolding of a channel after opening and inactivating to restore a closed, but available channel. The channel is refractory to additional stimulations during repriming.
Notes
1 Catterall WA, Goldin AL, Waxman SG. 2005. International Union of Pharmacology. XLVII. Nomenclature and structure–function relationships of voltage-gated sodium channels. Pharmacol Rev 57: 397–409. [A general review on the sodium channel subfamily of voltage-gated ion channels.]
2 Catterall WA. 2000. From ionic currents to molecular mechanisms: The structure and function of voltage-gated sodium channels. Neuron 26: 13–25.
3 Payandeh J, Scheuer T, Zheng N, Catterall WA. 2011. The crystal structure of a voltage-gated sodium channel. Nature 475: 353–358. [The first description of a high-resolution crystal structure of a homotetrameric bacterial voltage-gated sodium channel.]
4 Catterall WA. 2010. Signaling complexes of voltage-gated sodium and calcium channels. Neurosci Lett 486: 107–116.
5 Dib-Hajj SD, Waxman SG. 2010. Isoform-specific and pan-channel partners regulate trafficking and plasma membrane stability; and alter sodium channel gating properties. Neurosci Lett 486: 84–91.
6 Leterrier C, Brachet A, Fache MP, Dargent B. 2010. Voltage-gated sodium channel organization in neurons: Protein interactions and trafficking pathways. Neurosci Lett 486: 92–100.
7 Patino GA, Isom LL. 2010. Electrophysiology and beyond: Multiple roles of Na+ channel β subunits in development and disease. Neurosci Lett 486: 53–59.
8 Cummins TR, et al. 2001. NaV1.3 sodium channels: Rapid repriming and slow closed-state inactivation display quantitative differences after expression in a mammalian cell line and in spinal sensory neurons. J Neurosci 21: 5952–5961. [This study documents the effect of cell background on the biophysical properties of voltage-gated sodium channels and highlights the need to study these channels in their native cell types.]
9 Choi JS, Dib-Hajj SD, Waxman S. 2007. Differential slow inactivation and use-dependent inhibition of NaV1.8 channels contribute to distinct firing properties in IB4+ and IB4– DRG neurons. J Neurophysiol 97: 1258–1265.
10 Rush AM, et al. 2006. A single sodium channel mutation produces hyper- or hypoexcitability in different types of neurons. Proc Natl Acad Sci USA 103: 8245–8250. [This study demonstrates that the distinct cellular responses of DRG neurons to expression of mutant NaV1.7 channel depends on the presence or absence of another sodium channel, NaV1.8.]
11 Choi JS, et al. 2010. Alternative splicing may contribute to time-dependent manifestation of inherited erythromelalgia. Brain 133: 1823–1835.
12 Dib-Hajj SD, et al. 2009. Transfection of rat or mouse neurons by biolistics or electroporation. Nat Protoc 4: 1118–1126.
13 Dib-Hajj SD, Cummins TR, Black JA, Waxman SG. 2010. Sodium channels in normal and pathological pain. Annu Rev Neurosci 33: 325–347.
14 Ahn HS, et al. 2011. NaV1.7 is the predominant sodium channel in rodent olfactory sensory neurons. Mol Pain 7: 32.
15 Weiss J, et al. 2011. Loss-of-function mutations in sodium channel NaV1.7 cause anosmia. Nature 472: 186–190.
16 Muroi Y, et al. 2011. Selective silencing of NaV1.7 decreases excitability and conduction in vagal sensory neurons. J Physiol 589: 5663–5676.
17 Smith S, et al. 2011. The molecular basis of acid insensitivity in the African naked mole-rat. Science 334: 1557–1560.
18 Toledo-Aral JJ, et al. 1997. Identification of PN1, a predominant voltage-dependent sodium channel expressed principally in peripheral neurons. Proc Natl Acad Sci USA 94: 1527–1532. [The first study to report the major cellular distribution of NaV1.7.]
19 Sage D, et al. 2007. NaV1.7 and NaV1.3 are the only tetrodotoxin-sensitive sodium channels expressed by the adult guinea pig enteric nervous system. J Comp Neurol 504: 363–378.
20 Kwong K, et al. 2008. Voltage-gated sodium channels in nociceptive versus non-nociceptive nodose vagal sensory neurons innervating guinea pig lungs. J Physiol 586: 1321–1336.
21 Holm AN, et al. 2002. Sodium current in human jejunal circular smooth muscle cells. Gastroenterology 122: 178–187.
22 Jo T, et al. 2004. Voltage-gated sodium channel expressed in cultured human smooth muscle cells: Involvement of SCN9A.FEBS Lett 567: 339–343.
23 Saleh S, Yeung SY, Prestwich S, Pucovsky V, Greenwood IA. 2005. Electrophysiological and molecular identification of voltage-gated sodium channels in murine vascular myocytes. J Physiol 568: 155–169.
24 Djouhri L, et al. 2003. Sensory and electrophysiological properties of guinea-pig sensory neurones expressing NaV1.7 (PN1) Na+ channel α-subunit protein. J Physiol 546: 565–576. [This study demonstrates the presence of NaV1.7 in functionally identified nociceptors.]
25 Felts PA, Yokoyama S, Dib-Hajj S, Black JA, Waxman SG. 1997. Sodium channel α-subunit mRNAs I, II, III, NaG, Na6 and HNE (PN1) — different expression patterns in developing rat nervous system. Brain Res Mol Brain Res 45: 71–82.
26 Diss JK, et al. 2005. A potential novel marker for human prostate cancer: Voltage-gated sodium channel expression in vivo.Prostate Cancer Prostatic Dis 8: 266–273.
27 Fraser SP, et al. 2005. Voltage-gated sodium channel expression and potentiation of human breast cancer metastasis. Clin Cancer Res 11: 5381–5389.
28 Hoffman JF, Dodson A, Wickrema A, Dib-Hajj SD. 2004. Tetrodotoxin-sensitive Na+ channels and muscarinic and purinergic receptors identified in human erythroid progenitor cells and red blood cell ghosts. Proc Natl Acad Sci USA 101: 12370–12374.
29 Kis-Toth K, et al. 2011. Voltage-gated sodium channel NaV1.7 maintains the membrane potential and regulates the activation and chemokine-induced migration of a monocyte-derived dendritic cell subset. J Immunol 187: 1273–1280.
30 Djouhri L, et al. 2003. The TTX-resistant sodium channel NaV1.8 (SNS/PN3): Expression and correlation with membrane properties in rat nociceptive primary afferent neurons. J Physiol 550: 739–752.
31 Persson AK, Gasser A, Black JA, Waxman SG. 2011. NaV1.7 accumulates and co-localizes with phosphorylated ERK1/2 within transected axons in early experimental neuromas. Exp Neurol 230: 273–279.
32 Black JA, Frezel N, Dib-Hajj SD, Waxman SG. 2012. Expression of NaV1.7 in DRG neurons extends from peripheral terminals in the skin to central preterminal branches and terminals in the dorsal horn. Mol Pain 8: 82.
33 Cummins TR, Howe JR, Waxman SG. 1998. Slow closed-state inactivation: A novel mechanism underlying ramp currents in cells expressing the hNE/PN1 sodium channel. J Neurosci 18: 9607–9619. [This study shows that NaV1.7 can produce a robust ramp current, suggesting that NaV1.7 can amplify subthreshold depolarizations and act as a threshold channel.]
34 Klugbauer N, Lacinova L, Flockerzi V, Hofmann F. 1995. Structure and functional expression of a new member of the tetrodotoxin-sensitive voltage-activated sodium channel family from human neuroendocrine cells. EMBO J 14: 1084–1090. [The first report of the isolation and characterization of NaV1.7 as a TTX-sensitive sodium channel.]
35 Herzog RI, Cummins TR, Ghassemi F, Dib-Hajj SD, Waxman SG. 2003. Distinct repriming and closed-state inactivation kinetics of NaV1.6 and NaV1.7 sodium channels in mouse spinal sensory neurons. J Physiol 551: 741–750.
36 Rush AM, Cummins TR, Waxman SG. 2007. Multiple sodium channels and their roles in electrogenesis within dorsal root ganglion neurons. J Physiol 579: 1–14.
37 Dib-Hajj SD, Cummins TR, Black JA, Waxman SG. 2007. From genes to pain: NaV1.7 and human pain disorders. Trends Neurosci 30: 555–563.
38 Jarecki BW, Piekarz AD, Jackson JO, 2nd, Cummins TR. 2010. Human voltage-gated sodium channel mutations that cause inherited neuronal and muscle channelopathies increase resurgent sodium currents. J Clin Invest 120: 369–378.
39 Faber CG, et al. 2012. Gain of function NaV1.7 mutations in idiopathic small fiber neuropathy. Ann Neurol 71: 26–39. [This study was the first to show that patients with idiopathic SFN can harbour NaV1.7 variants; it also shows that these variants cause hyperexcitability of DRG neurons.]
40 Raman IM, Bean BP. 1997. Resurgent sodium current and action potential formation in dissociated cerebellar Purkinje neurons. J Neurosci 17: 4517–4526. [This study documents a state of open channel block, which permits the passing of a current upon hyperpolarization of the cell membrane to negative potentials immediately following a strong depolarizing pulse that fully activates and inactivates the channel.]
41 Raman IM, Sprunger LK, Meisler MH, Bean BP. 1997. Altered subthreshold sodium currents and disrupted firing patterns in Purkinje neurons of Scn8a mutant mice. Neuron 19: 881–891.
43 Nassar MA, et al. 2004. Nociceptor-specific gene deletion reveals a major role for NaV1.7 (PN1) in acute and inflammatory pain. Proc Natl Acad Sci USA 101: 12706–12711. [The first report showing that knockout of NaV1.7 in DRG neurons impairs acute and inflammatory pain.]
44 Goldberg Y, et al. 2007. Loss-of-function mutations in the NaV1.7 gene underlie congenital indifference to pain in multiple human populations. Clin Genet 71: 311–319.
45 Nilsen KB, et al. 2009. Two novel SCN9A mutations causing insensitivity to pain. Pain 143: 155–158.
46 Undem BJ, Carr MJ. 2010. Targeting primary afferent nerves for novel antitussive therapy. Chest 137: 177–184.
47 Schild JH, Kunze DL. 1997. Experimental and modeling study of Na+ current heterogeneity in rat nodose neurons and its impact on neuronal discharge. J Neurophysiol 78: 3198–3209.
48 Farrag KJ, Costa SK, Docherty RJ. 2002. Differential sensitivity to tetrodotoxin and lack of effect of prostaglandin E on the pharmacology and physiology of propagated action potentials. Br J Pharmacol 135: 1449–1456.
49 Park TJ, et al. 2008. Selective inflammatory pain insensitivity in the African naked mole-rat (Heterocephalus glaber). PLoS Biol 6: e13.
50 Singh NA, et al. 2009. A role of SCN9A in human epilepsies, as a cause of febrile seizures and as a potential modifier of Dravet syndrome. PLoS Genet 5: e1000649.
51 Drenth JP, Waxman SG. 2007. Mutations in sodium-channel gene SCN9A cause a spectrum of human genetic pain disorders. J Clin Invest 117: 3603–3609.
52 Devor M. 2006. Sodium channels and mechanisms of neuropathic pain. J Pain 7: S3–S12.
53 Lyu YS, Park SK, Chung K, Chung JM. 2000. Low dose of tetrodotoxin reduces neuropathic pain behaviors in an animal model. Brain Res 871: 98–103.
54 Black JA, Nikolajsen L, Kroner K, Jensen TS, Waxman SG. 2008. Multiple sodium channel isoforms and mitogen-activated protein kinases are present in painful human neuromas. Ann Neurol 64: 644–653. [This study demonstrates the presence of sodium channels NaV1.3, NaV1.7 and NaV1.8, and activated MAPK1, MAPK3 and MAPK12 within blind axon terminals of painful human neuromas.]
55 Stamboulian S, et al. 2010. ERK1/2 mitogen-activated protein kinase phosphorylates sodium channel NaV1.7 and alters its gating properties. J Neurosci 30: 1637–1647.
56 Black JA, Liu S, Tanaka M, Cummins TR, Waxman SG. 2004. Changes in the expression of tetrodotoxin-sensitive sodium channels within dorsal root ganglia neurons in inflammatory pain. Pain 108: 237–247.
57 Gould HJ, et al. 2004. Ibuprofen blocks changes in NaV1.7 and 1.8 sodium channels associated with complete Freund’s adjuvant-induced inflammation in rat. J Pain 5: 270–280.
58 Chattopadhyay M, Mata M, Fink DJ. 2008. Continuous δ-opioid receptor activation reduces neuronal voltage-gated sodium channel (NaV1.7) levels through activation of protein kinase C in painful diabetic neuropathy. J Neurosci 28: 6652–6658.
59 Chattopadhyay M, Mata M, Fink DJ. 2011. Vector-mediated release of GABA attenuates pain-related behaviors and reduces NaV1.7 in DRG neurons. Eur J Pain 15: 913–920.
60 Yeomans DC, et al. 2005. Decrease in inflammatory hyperalgesia by Herpes vector-mediated knockdown of NaV1.7 sodium channels in primary afferents. Hum Gene Ther 16: 271–277.
61 Chattopadhyay M, Zhou Z, Hao S, Mata M, Fink DJ. 2012. Reduction of voltage gated sodium channel protein in DRG by vector mediated miRNA reduces pain in rats with painful diabetic neuropathy. Mol Pain 8: 17.
62 Minett MS et al. 2012. Distinct NaV1.7-dependent pain sensations require different sets of sensory and sympathetic neurons. Nature Commun 3: 791. [This study suggests that knockout of NaV1.7 in neurons from DRG and sympathetic ganglia is needed to attenuate neuropathic pain.]
63 Shields SD, et al. 2012. Sodium channel NaV1.7 is essential for lowering heat pain threshold after burn injury. J Neurosci 32: 10819–10832.
64 Yang Y, et al. 2004. Mutations in SCN9A, encoding a sodium channel α subunit, in patients with primary erythermalgia. J Med Genet 41: 171–174. [This report identifies gain-of-function mutations in SCN9A in patients with IEM.]
65 Fertleman CR, et al. 2006. SCN9A mutations in paroxysmal extreme pain disorder: Allelic variants underlie distinct channel defects and phenotypes. Neuron 52: 767–774. [This study identifies and characterizes gain-of-function mutations in SCN9A in patients with PEPD.]
66 Cox JJ, et al. 2006. An SCN9A channelopathy causes congenital inability to experience pain. Nature 444: 894–898. [This study identifies and characterizes loss-of-function mutations in SCN9A that underlie CIP.]
67 Sheets PL, Jackson Ii JO, Waxman SG, Dib-Hajj S, Cummins TRA. 2007. NaV1.7 channel mutation associated with hereditary erythromelalgia contributes to neuronal hyperexcitability and displays reduced lidocaine sensitivity. J Physiol 581: 1019–1031.
68 Cheng X, et al. 2011. Deletion mutation of sodium channel NaV1.7 in inherited erythromelalgia: Enhanced slow inactivation modulates dorsal root ganglion neuron hyperexcitability. Brain 134: 1972–1986.
70 Choi JS, et al. 2011. Paroxysmal extreme pain disorder: A molecular lesion of peripheral neurons. Nat Rev Neurol 7: 51–55.
71 Estacion M, et al. 2008. NaV1.7 gain-of-function mutations as a continuum: A1632E displays physiological changes associated with erythromelalgia and paroxysmal extreme pain disorder mutations and produces symptoms of both disorders. J Neurosci 28: 11079–11088.
72 Dib-Hajj SD, et al. 2008. Paroxysmal extreme pain disorder M1627K mutation in human NaV1.7 renders DRG neurons hyperexcitable. Mol Pain 4: 37.
73 Cheng X, et al. 2010. Mutations at opposite ends of the DIII/S4–S5 linker of sodium channel NaV1.7 produce distinct pain disorders. Mol Pain 6: 24.
74 Vasylyev DV, Waxman SG. 2012. Membrane properties and electrogenesis in the distal axons of small dorsal root ganglion neurons in vitro.J Neurophysiol 108: 729–740.
75 Han C, et al. 2009. Early- and late-onset inherited erythromelalgia: Genotype–phenotype correlation. Brain 132: 1711–1722.
76 Harty TP, et al. 2006. NaV1.7 mutant A863P in erythromelalgia: Effects of altered activation and steady-state inactivation on excitability of nociceptive dorsal root ganglion neurons. J Neurosci 26: 12566–12575.
77 Ahmad S, et al. 2007. A stop codon mutation in SCN9A causes lack of pain sensation. Hum Mol Genet 16: 2114–2121.
78 Kurban M, Wajid M, Shimomura Y, Christiano AM. 2010. A nonsense mutation in the SCN9A gene in congenital insensitivity to pain. Dermatology 221: 179–183.
79 Staud R, et al. 2011. Two novel mutations of SCN9A (NaV1.7) are associated with partial congenital insensitivity to pain. Eur J Pain 15: 223–230.
80 Reich DE, Lander ES. 2001. On the allelic spectrum of human disease. Trends Genet 17: 502–510.
81 Estacion M, et al. 2009. A sodium channel gene SCN9A polymorphism that increases nociceptor excitability. Ann Neurol 66: 862–866. [This report identifies and characterizes a common variant of SCN9A that is associated with pain.]
82 Reimann F, et al. 2010. Pain perception is altered by a nucleotide polymorphism in SCN9A.Proc Natl Acad Sci USA 107: 5148–5153.
83 Estacion M, et al. 2011. Intra- and interfamily phenotypic diversity in pain syndromes associated with a gain-of-function variant of NaV1.7. Mol Pain 7: 92.
84 Choi JS, Waxman SG. 2011. Physiological interactions between NaV1.7 and NaV1.8 sodium channels: A computer simulation study. J Neurophysiol 106: 3173–3184.
85 Doyle DA, et al. 1998. The structure of the potassium channel: Molecular basis of K+ conduction and selectivity. Science 280: 69–77.
86 Jiang Y, et al. 2002. The open pore conformation of potassium channels. Nature 417: 523–526.
87 Long SB, Campbell EB, Mackinnon R. 2005. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science 309: 897–903.
88 Ren D, et al. 2001. A prokaryotic voltage-gated sodium channel. Science 294: 2372–2375.
89 Charalambous K, Wallace BA. 2011. NaChBac: The long lost sodium channel ancestor. Biochemistry 50: 6742–6752.
91 Tsujino A, et al. 2003. Myasthenic syndrome caused by mutation of the SCN4A sodium channel. Proc Natl Acad Sci USA 100: 7377–7382.
92 Lampert A, et al. 2008. A pore-blocking hydrophobic motif at the cytoplasmic aperture of the closed-state NaV1.7 channel is disrupted by the erythromelalgia-associated F1449V mutation. J Biol Chem 283: 24118–24127. [An atomic structural modelling of NaV1.7 based on the potassium channel KcsA crystal structure identifies a putative activation gate.]
93 Burley SK, Petsko GA. 1985. Aromatic–aromatic interaction: A mechanism of protein structure stabilization. Science 229: 23–28.
94 Dib-Hajj SD, et al. 2005. Gain-of-function mutation in NaV1.7 in familial erythromelalgia induces bursting of sensory neurons. Brain 128: 1847–1854. [The first demonstration that a gain-of-function familial mutation in SCN9A renders DRG neurons hyperexcitable, thus providing the pathophysiological basis for pain in these patients.]
95 Rojas A, Wu J, Wang R, Jiang C. 2007. Gating of the ATP-sensitive K+ channel by a pore-lining phenylalanine residue. Biochim Biophys Acta 1768: 39–51.
96 Han C, et al. 2012. Functional profiles of SCN9A variants in dorsal root ganglion neurons and superior cervical ganglion neurons correlate with autonomic symptoms in small fibre neuropathy. Brain 135: 2613–2628.
97 Akopian AN, Sivilotti L, Wood JN. 1996. A tetrodotoxin-resistant voltage-gated sodium channel expressed by sensory neurons. Nature 379: 257–262.
98 Akopian AN, et al. 1999. The tetrodotoxin-resistant sodium channel SNS has a specialized function in pain pathways. Nature Neurosci 2: 541–548. [Together with reference 97, these studies were the first to identify and characterize NaV1.8 from DRG neurons and demonstrates a role for this channel in pain.]
99 Sangameswaran L, et al. 1996. Structure and function of a novel voltage-gated, tetrodoxtoxin-resistant sodium channel specific to sensory neurons. J Biol Chem 271: 5953–5956.
100 England S, de Groot MJ. 2009. Subtype-selective targeting of voltage-gated sodium channels. Br J Pharmacol 158: 1413–1425.
101 Choi JS, et al. 2009. Mexiletine-responsive erythromelalgia due to a new NaV1.7 mutation showing use-dependent current fall-off. Exp Neurol 216: 383–389.
102 Fischer TZ, et al. 2009. A novel NaV1.7 mutation producing carbamazepine-responsive erythromelalgia. Ann Neurol 65: 733–741. [This study identifies the SCN9A mutation V400M in patients who responded to treatment with carbamazepine, and demonstrates that this mutation increases responsiveness to carbamazepine without altering the affinity of the channel to the drug.]
103 Yang Y, et al. 2012. Structural modelling and mutant cycle analysis predict pharmacoresponsiveness of a NaV1.7 mutant channel. Nature Commun 3: 1186. [Using V400M as a ‘seed’ SCN9A mutation, this atomic structural modelling and thermodynamic coupling analysis predicts and then confirms that a second SCN9A mutation, S241T, is responsive to carbamazepine.]
104 Williams BS, et al. 2007. Characterization of a new class of potent inhibitors of the voltage-gated sodium channel NaV1.7. Biochemistry 46: 14693–14703.
105 London C, et al. 2008. Imidazopyridines: A novel class of hNaV1.7 channel blockers. Bioorg Med Chem Lett 18: 1696–1701.
106 Bregman H, et al. 2011. Identification of a potent, state-dependent inhibitor of NaV1.7 with oral efficacy in the formalin model of persistent pain. J Med Chem 54: 4427–4445.
107 Chowdhury S, et al. 2011. Discovery of XEN907, a spirooxindole blocker of NaV1.7 for the treatment of pain. Bioorg Med Chem Lett 21: 3676–3681.
108 Chapman ML, et al. 2012. Characterization of a novel subtype-selective inhibitor of human NaV1.7 voltage-dependent sodium channels (PT 418). IASP 14th World Congress on Pain [online],http://www.abstracts2view.com/iasp/sessionindex.php.
109 Rice AS, Hill RG. 2006. New treatments for neuropathic pain. Annu Rev Med 57: 535–551.
110 Dworkin RH, et al. 2007. Pharmacologic management of neuropathic pain: Evidence-based recommendations. Pain 132: 237–251.
111 Sindrup SH, Jensen TS. 2007. Are sodium channel blockers useless in peripheral neuropathic pain? Pain 128: 6–7.
112 Gerner P, Strichartz GR. 2008. Sensory and motor complications of local anesthetics. Muscle Nerve 37: 421–425.
113 Beyreuther B, Callizot N, Stohr T. 2006. Antinociceptive efficacy of lacosamide in a rat model for painful diabetic neuropathy. Eur J Pharmacol 539: 64–70.
114 Beyreuther BK, et al. 2007. Antinociceptive efficacy of lacosamide in rat models for tumor- and chemotherapy-induced cancer pain. Eur J Pharmacol 565: 98–104.
115 Hao JX, Stohr T, Selve N, Wiesenfeld-Hallin Z, Xu XJ. 2006. Lacosamide, a new anti-epileptic, alleviates neuropathic pain-like behaviors in rat models of spinal cord or trigeminal nerve injury. Eur J Pharmacol 553: 135–140.
116 Stohr T, et al. 2007. Lacosamide, a novel anti-convulsant drug, shows efficacy with a wide safety margin in rodent models for epilepsy. Epilepsy Res 74: 147–154.
117 Doty P, Rudd GD, Stoehr T, Thomas D. 2007. Lacosamide. Neurotherapeutics 4: 145–148.
118 Rauck RL, Shaibani A, Biton V, Simpson J, Koch B. 2007. Lacosamide in painful diabetic peripheral neuropathy: A phase 2 double-blind placebo-controlled study. Clin J Pain 23: 150–158.
119 Sheets PL, Heers C, Stoehr T, Cummins TR. 2008. Differential block of sensory neuronal voltage-gated sodium channels by lacosamide [(2R)-2-(acetylamino)-N-benzyl-3-methoxypropanamide], lidocaine, and carbamazepine. J Pharmacol Exp Ther 326: 89–99.
120 Xu GY, Zhao ZQ. 2001. Change in excitability and phenotype of substance P and its receptor in cat Aβ sensory neurons following peripheral inflammation. Brain Res 923: 112–119.
121 Dworkin RH, et al. 2010. Recommendations for the pharmacological management of neuropathic pain: An overview and literature update. Mayo Clin Proc 85: S3–S14.
122 Wilson MJ, et al. 2011. μ-Conotoxins that differentially block sodium channels NaV1.1 through 1.8 identify those responsible for action potentials in sciatic nerve. Proc Natl Acad Sci USA 108: 10302–10307.
123 Lewis RJ, Dutertre S, Vetter I, Christie MJ. 2012. Conus venom peptide pharmacology. Pharmacol Rev 64: 259–298.
124 Dib-Hajj SD, et al. 2009. Voltage-gated sodium channels in pain states: Role in pathophysiology and targets for treatment. Brain Res Brain Res Rev 60: 65–83.
125 Middleton RE, et al. 2002. Two tarantula peptides inhibit activation of multiple sodium channels. Biochemistry 41: 14734–14747.
126 Smith JJ, Cummins TR, Alphy S, Blumenthal KM. 2007. Molecular interactions of the gating modifier toxin ProTx-II with NaV1.5: Implied existence of a novel toxin binding site coupled to activation. J Biol Chem 282: 12687–12697.
127 Peng K, Shu Q, Liu Z, Liang S. 2002. Function and solution structure of huwentoxin-IV, a potent neuronal tetrodotoxin (TTX)-sensitive sodium channel antagonist from Chinese bird spider Selenocosmia huwena.J Biol Chem 277: 47564–47571.
128 Xiao Y, et al. 2008. Tarantula huwentoxin-IV inhibits neuronal sodium channels by binding to receptor site 4 and trapping the domain II voltage sensor in the closed configuration. J Biol Chem 283: 27300–27313.
129 McArthur JR, et al. 2011. Interactions of key charged residues contributing to selective block of neuronal sodium channels by μ-conotoxin KIIIA. Mol Pharmacol 80: 573–584.
130 Clark RJ, Akcan M, Kaas Q, Daly NL, Craik DJ. 2012. Cyclization of conotoxins to improve their biopharmaceutical properties. Toxicon 59: 446–455.
131 Fink DJ, et al. 2012. Gene therapy for pain: Results of a phase I clinical trial. Ann Neurol 70: 207–212.
132 Samad OA, et al. 2012. Virus-mediated shRNA knockdown of NaV1.3 in rat dorsal root ganglion attenuates nerve injury-induced neuropathic pain. Mol Ther 21: doi:10.1038/mt.2012.169.
133 Firestein S. 2001. How the olfactory system makes sense of scents. Nature 413: 211–218.
134 Kaupp UB. 2010. Olfactory signalling in vertebrates and insects: Differences and commonalities. Nat Rev Neurosci 11: 188–200.
135 Rajendra S, Lynch JW, Barry PH. 1992. An analysis of Na+ currents in rat olfactory receptor neurons. Pflugers Arch 420: 342–346.
136 Cummins TR, Dib-Hajj SD, Waxman SG. 2004. Electrophysiological properties of mutant NaV1.7 sodium channels in a painful inherited neuropathy. J Neurosci 24: 8232–8236. [The first demonstration that mutations in SCN9A from patients with IEM manifest gain-of-function attributes.]
137 Blair NT, Bean BP. 2002. Roles of tetrodotoxin (TTX)-sensitive Na+ current, TTX-resistant Na+ current, and Ca2+ current in the action potentials of nociceptive sensory neurons. J Neurosci 22: 10277–10290.
138 Cummins TR, Waxman SG. 1997. Downregulation of tetrodotoxin-resistant sodium currents and upregulation of a rapidly repriming tetrodotoxin-sensitive sodium current in small spinal sensory neurons after nerve injury. J Neurosci 17: 3503–3514.
139 Elliott AA, Elliott JR. 1993. Characterization of TTX-sensitive and TTX-resistant sodium currents in small cells from adult rat dorsal root ganglia. J Physiol 463: 39–56.
140 Cheng X, Dib-Hajj SD, Tyrrell L, Waxman SG. 2008. Mutation I136V alters electrophysiological properties of the NaV1.7 channel in a family with onset of erythromelalgia in the second decade. Mol Pain 4: 1.
141 Han C, et al. 2012. NaV1.7-related small fiber neuropathy: Impaired slow-inactivation and DRG neuron hyperexcitability. Neurology 78: 1635–1643.