A common admonition at rounds on the wards of teaching hospitals is “when you hear hoofbeats, think of horses,” to which the attending physician might add “and don’t limit your thoughts to zebras.” Zebras, in this setting, refer to rare diseases, while horses stand for common disorders. Our demonstration that gain-of-function changes in mutant NaV1.7 channels cause inherited erythromelalgia (Cummins, Dib-Hajj, and Waxman 2004; Dib-Hajj et al. 2005) and, following that, discovery that other gain-of-function mutations of NaV1.7 cause pain in paroxysmal extreme pain disorder (PEPD) (Fertleman et al. 2006), had shown that hyperactivity of NaV1.7 channels can produce disorders characterized by intense pain. But these were very rare diseases, zebras. Might NaV1.7 be a player in chronic pain within broader populations?
Peripheral neuropathy has increasingly been recognized as a source of pain in large numbers of patients. Diabetic neuropathy, for example, occurs in about one-half of people with diabetes and often causes pain that is not relieved by existing medications. Cancer chemotherapy can trigger the onset of painful neuropathy, and exquisite pain can occur in postherpetic neuralgia which follows shingles and with the neuropathies that occur as a complication of certain inflammatory disorders. Worldwide, millions of people suffer from pain that accompanies peripheral neuropathy.
Our peripheral nerves contain large-, medium-, and small-diameter axons, and neuropathy can affect any of these groups of nerve fibers. Peripheral axons carry information from our body surface and organs via nerves such as the sciatic nerve to the spinal cord, and they vary in size—axons that relay information about muscle tension, involved in deep tendon reflexes, are about 10 microns in diameter, while nerve fibers that convey information about pain are much smaller, usually with a diameter of less than one micron or 1/1,000th of a millimeter. Because of their diminutive size the small-diameter axons had been less well studied.
“Small fiber neuropathy” is a form of peripheral neuropathy that affects small-diameter, unmyelinated and thinly myelinated axons within peripheral nerve, including pain-signaling nerve fibers. We now know that the clinical picture of small fiber neuropathy is dominated by pain, often with a burning quality, which is usually first felt within the territories innervated by the longest nerves (Gorson and Ropper 1995; Hoeijmakers, Faber, et al. 2012; Holland et al. 1998; Lacomis 2002). Patients with small fiber neuropathy typically come to the doctor complaining of burning pain in the feet or the hands. This characteristic of peripheral neuropathy is widely taught to medical students as a “stocking-glove” pattern of clinical abnormality.
Although small fiber neuropathy is not rare—Faber, Merkies, and their colleagues estimated a prevalence of about 50 per 100,000, or 1 in 2,000 (Peters et al. 2013)—it was not widely recognized as a clinical entity until the 1990s. The reason was that, until then, it was difficult to diagnose. In its pure form small fiber neuropathy specifically affects small-diameter nerve fibers within peripheral nerves, sparing the large-diameter nerve fibers that support deep tendon reflexes and perception of vibration, which were assessed in the routine neurological examination. Because their large nerve fibers were not affected, patients with small fiber neuropathy presented to clinicians with symptoms such as pain but did not manifest objective signs such as loss of reflexes when tested with the neurologist’s hammer or impaired vibration sensibility when tested with a tuning fork. Since their neurological examinations were often normal, the complaints of patients with small fiber neuropathy—which occurred without physical signs of disease of the nervous system that can be seen by the physician—were, in the past, often dismissed as being of little consequence, or as having a psychological origin.
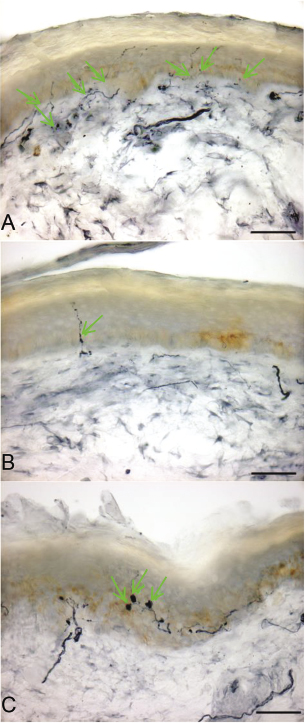
A crucial step forward in recognition of small fiber neuropathy as a disease came with the development of techniques for assessing the number, and health, of small nerve fibers. A breakthrough, led by clinical investigations at Johns Hopkins, came with recognition that the tips of the tiny nerve fibers, most distant from the DRG neurons that give rise to them, terminate within the skin where they can be assessed by microscopic examination of biopsies (Kennedy and Wendelschafer-Crabb 1999; McCarthy et al. 1995). A relatively noninvasive method for skin biopsy uses a small punch to remove a core of skin 3 mm across, about the size of a large pencil lead. Following removal, the skin sample is preserved, cut into sections, stained, and examined in the microscope, where the number of small nerve fibers can be counted and compared with normal, healthy controls (usually age-matched and gender-matched) so that loss of nerve fibers can be assessed. Injured nerve fibers can also be identified since they form abnormal bulb-like swellings (figure 9.1).
Figure 9.1 Small fiber neuropathy can be diagnosed using skin biopsy to demonstrate degeneration of the endings of nerve fibers within the skin. Panel A shows a skin biopsy from a normal individual in whom multiple nerve fibers within the skin (green arrows) can be seen. Panel B shows a skin biopsy from a patient with small fiber neuropathy, in whom there is a depletion of nerve fibers. Panel C shows swellings of nerve fibers (green arrows) in the skin, from another patient with small fiber neuropathy. These are predegenerative changes. Scale bars: 50 μm. From Hoeijmakers, Faber et al. (2012).
As another measure of the status of small nerve fibers, sensitivity to warmth, coolness, heat-induced pain, and cold-induced pain could be quantitatively assessed in human subjects using computerized techniques, in a test called quantitative sensory testing, or QST. The development of methods for assessment of small nerve fibers within skin biopsies, and QST, made it possible for small fiber neuropathy to be diagnosed on the basis of objective, quantifiable criteria (Bakkers et al. 2009; Herrmann et al. 1999; McArthur et al. 1998; Walk et al. 2007).
Our characterization of NaV1.7 mutations in patients with small fiber neuropathy began in mid-2010, when I started to work with Catharina Faber and Ingemar Merkies of Maastricht University. Working as a team, these peripheral neuropathy experts had assembled a cohort of patients with painful small fiber neuropathy. They now wanted to know whether there were disease-causing NaV1.7 mutations in these patients. Here we had an obvious scaffold for collaboration.
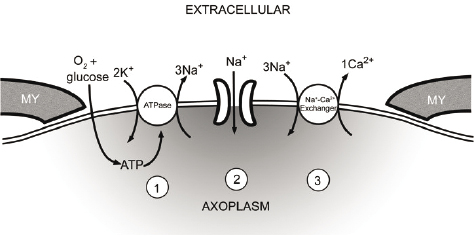
My interest in sodium channels in peripheral neuropathy was partly based on some clinical similarities—burning pain in the feet and hands—to inherited erythromelalgia. It was also partly propelled by work carried out nearly two decades previously in my laboratory by Peter Stys, then a research fellow and now a distinguished professor of neuroscience at the University of Calgary. In studies on small axons within the optic nerve that connects the eye and the brain in the rat, Stys had demonstrated that sodium channels play an important role in axonal injury. His work showed that in various forms of axonal injury, the channels provide a route for a small but persistent flow of sodium ions into the axons. This tiny but sustained sodium influx, in turn, triggers the activity of a molecule called the sodium-calcium exchanger (Stys, Waxman, and Ransom 1992). The sodium-calcium exchanger is an antiporter or see-saw molecule that normally carries a small amount of sodium from the outside of cells where its concentration is high, to the inside of cells where its concentration is normally low. As implied by its name, the sodium-calcium exchanger, in a swap for the influx of sodium, normally moves calcium ions out of cells, thereby maintaining the intracellular concentration of calcium at low levels where it cannot activate injurious enzymes. Stys’s experiments showed that, after various insults to axons in the optic nerve, a small but sustained influx of sodium ions increases the concentration of sodium within these fragile nerve fibers, forcing the sodium-calcium exchanger to work in a “reverse” mode where, instead of extruding calcium, it imports calcium into axons. The high levels of calcium then activate deleterious enzymes that lead to axonal demise (figure 9.2). Importantly, experiments we had done with Stys in the 1990s showed that blockade of sodium channels could protect axons so that they did not degenerate after various insults (Stys, Ransom, and Waxman 1992; Stys, Waxman, and Ransom 1992). These earlier studies established sodium channels as key players in axonal injury within the optic nerve, which is an extension of the brain. Now, twenty years later, it was logical to ask whether sodium channels might also play a role in axonal injury within peripheral nerves.
Figure 9.2 Diagram showing how, in injured axons (1) energy reserves (ATP) are depleted, leading to failure of the ATPase and collapse of ionic gradients; (2) Na+ ions enter through sodium channels; (3) the resultant increase in Na+ within the axon causes the Na+-Ca2+ exchanger to operate in reverse mode, carrying damaging quantities of Ca2+ into the axon. MY, myelin. Modified from Stys, Ransom, and Waxman (1992).
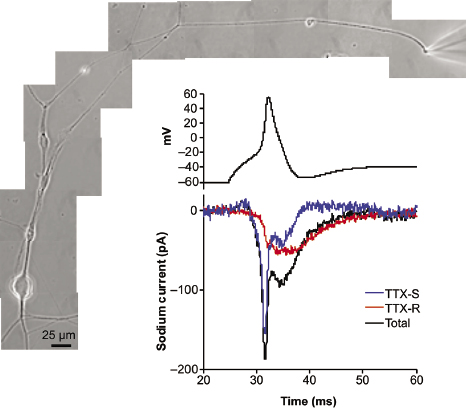
Joel Black, working in my lab, had shown that small nerve fibers in our peripheral nerves and their terminals in the skin contain NaV1.7 channels (figure 9.3A) (Black et al. 2012). I also knew, from the observations of Swedish pain researcher Anna-Karin Persson, who was working in my lab, that small nerve fibers in the skin contain the sodium-calcium exchanger, located in the same regions as NaV1.7 (figure 9.3B). And as shown in figure 9.4, Dymtro Vasylyev was able to use tiny microelectrodes in my laboratory to accomplish the challenging task of directly recording the electrical activity from small nerve fibers. These recordings showed us that NaV1.7 channels were not only present within these tiny axons, but were also inserted into the axon membrane in a normal manner so that they are functional (Vasylyev and Waxman 2012).
Figure 9.3 (A) A micrograph showing small-diameter nerve fibers within the skin, in which red staining indicates the presence of NaV1.7. Scale bar: 20 μm (Black et al. 2012). (B) A micrograph showing small nerve fibers in the skin, in which red staining indicates the presence of the sodium-calcium exchanger. Scale bar: 20 μm. From Persson et al. (2010).
Figure 9.4 Patch-clamp recording from a small nerve fiber with a diameter of approximately 1 μm extending from a dorsal root ganglion neuron in tissue culture. The micrograph shows (upper right) the tip of the electrode where it contacts the nerve fiber. The top trace shows a nerve impulse (action potential) from this nerve fiber. The bottom trace shows the sodium currents, produced by NaV1.7 (blue trace) and then NaV1.8 channels (red), which underlie the action potential. The black trace indicates the summed NaV1.7 and NaV1.8 currents. From Vasylyev and Waxman (2012).
Another feature of small nerve fibers also drove my interest. Earlier studies in our laboratory and others (Donnelly 2008; Waxman et al. 1989) had shown us that small-diameter axons are especially sensitive to small changes in sodium channel activity. These studies had shown that small axons are poised to fire in response to very small stimuli, or to degenerate in response to small, subtle changes in the number or properties of the channels. Neuroscientists attribute this to a characteristic of small nerve fibers called “high input impedance.” The effect of input impedance can be appreciated by considering, for example, the impact of a small whistle on rooms of various sizes. Within a large auditorium, the whistle sounds may be lost or drowned out. In contrast, within the confines of a small room, the whistle can be heard and may even echo and disrupt conversation. Similar to a small room where the sound of a whistle can be impactful, we knew that small nerve fibers are highly sensitive to changes—even small changes—in the sodium channels within them. And based on this, it seemed logical to me that even minor degrees of hyperactivity of sodium channels might have a dramatic impact on small-diameter axons.
Now, working with Faber and Merkies, I wanted to find out whether NaV1.7 mutations contributed to development of peripheral neuropathy. Faber and Merkies described themselves as “just clinicians.” In my view this understated their contributions to our work together. They began with a series of 248 patients referred to them with a presumptive diagnosis of small fiber neuropathy. Pain was a common presenting symptom. In some of these patients, even the touch of a bedsheet was painful. To winnow this large group down to a smaller cohort of patients with small fiber neuropathy with a low likelihood of a nongenetic cause, they excluded cases in which there were underlying conditions such as diabetes, treatment with medications known to produce peripheral neuropathy as a side effect, inflammatory disorders, or other diseases that were known to be accompanied by neuropathy. This left 63 patients with small fiber neuropathy and no apparent underlying medical disease.
Forty-four of these patients agreed to participate in a research study; this was testimony to the close bond that Faber and Merkies established with their patients. But still another layer of filtering was needed before the ball moved to my court. Functional analysis, to determine whether any particular mutation was pathogenic, was going to require a substantial effort in my laboratory. Because of this, we decided to focus on patients in whom the diagnosis of small fiber neuropathy had been confirmed by the most stringent criteria. To accomplish this, after obtaining consent, Faber and Merkies studied all 44 patients by skin biopsy and by QST. These tests were both abnormal in 28 patients who, using these stringent criteria, gave us a “gold standard” cohort of subjects with well-documented, biopsy-confirmed small fiber neuropathy, pain, and no identifiable nongenetic cause. Remarkably, within this group of 28 patients, mutations of NaV1.7 were present in eight, or nearly 30%.
Some mutations are pathogenic and alter the function of ion channels in a way that contributes to disease, while others are functionally silent and clinically inconsequential. The challenge was to determine whether these mutations were functionally significant. Eight mutations may not sound like a large number, but functional assessment of eight sodium channel mutations represented an immense task. Each mutation required production of the gene for NaV1.7 containing the mutation and then, insertion of the DNA into cells in the laboratory. After the cells had grown in sterile tissue culture dishes and made the mutant channels, we could do a voltage-clamp analysis using patch-clamp recording to assess the effect of the mutation on the function of the ion channel. We also decided to assess each mutation by current clamp so that we could examine the effect of the mutant channels on the behavior of pain-signaling neurons. For each of the eight mutations, this analysis required extended study by a team that included an experienced electrophysiologist supported by a molecular biology technician to produce the mutant genes and a tissue culture technician who prepared tissue cultures containing DRG neurons in which the mutant channels could be expressed. We knew, at the outset, that analysis of each mutation could take up to three or four months if things went smoothly. Or even longer if they did not.
Faced with eight mutations, I decided to deploy our entire electrophysiology team, including five highly skilled PhD electrophysiologists, each with substantial expertise in sodium channels, to work in parallel with the necessary support staff. This was a heavily leveraged bet. It was going to divert precious manpower from other projects. Indeed, if the bet did not work out, we would have invested several years of work by more than half a dozen team members, with no reward.
The ensuing months were very busy, as members of our team marched forward day by day, completing the intricate measurements needed for analysis of each of the eight mutations. We met nearly every day in the War Room to review progress. From our knowledge of the molecular architecture of the channel, and from the coordinates of the substituted amino acids which were located outside of the membrane-spanning segments of the channel protein molecule, we expected that the mutations would cause relatively subtle changes in channel function. This turned out to be the case, and it meant that we had to make an especially large number of measurements to detect small but functionally important changes in how the mutant channels worked. Our sense of urgency was amplified by frequent inquiries from Maastricht—“Have you found anything yet?” “What about D623N?” I did not regard this as a bother. Our Dutch collaborators were cheering us on.
As the months rolled by, the work progressed. The data from one, then another, then a third mutation arrived in the War Room, and a striking picture began to emerge. Some mutations impaired a process called slow inactivation, which essentially immobilizes channels in an inoperable state after prolonged depolarization. Other mutations produced a small impairment of slow inactivation together with a small impairment of fast inactivation, a process that temporarily locks channels in an inoperable state after brief periods of depolarization. And still another mutation enhanced resurgent current, which is triggered by repolarization at the end of a sustained depolarization so as to produce additional firing. The common thread was that every one of the mutations produced pro-excitatory changes that enhanced the activity of the channel.
When we put the mutant NaV1.7 channels into DRG neurons (the sensory neurons where they normally reside), each of the mutations produced hyperexcitability. The abnormal excitability was manifested by a decrease in current threshold (making it easier to excite the neuron) and a higher-than-normal frequency of firing in response to stimulation. These changes provided a basis for understanding the pain that could be evoked by even a light touch in our patients. Making the story even more complete, the mutations produced abnormal spontaneous firing in DRG neurons. This provided an explanation for the spontaneous pain, in the absence of stimulation, that brought most of our patients to the clinic. Here we had, in patients with a relatively common disorder, another example of the role of NaV1.7 in pain.
Our studies in this initial gold-standard cohort of 28 patients with painful small neuropathy had demonstrated gain-of-function mutations in about 28% (Faber, Hoeijmakers, et al. 2012). In follow-up studies on a larger cohort of more than 100 patients, in whom biopsy confirmation was not available and diagnosis rested on clinical criteria, NaV1.7 mutations were again found, although the percentage of patients carrying NaV1.7 mutations was about 15%. As I discussed this with our Maastricht collaborators, we speculated that the discrepancy might have reflected inclusion of some patients without neuropathy, or with mild neuropathy, in the larger group of patients, in which confirmation of the diagnosis by biopsy was not required. Irrespective of the precise percentage, we had demonstrated gain-of-function mutations in NaV1.7 in a substantial number of patients with painful neuropathy, a relatively common disorder. Subsequent to our 2012 report, we documented the gain-of-function changes in other NaV1.7 mutations linked to painful neuropathy (Estacion et al. 2011; Han, Hoeijmakers, Ahn, et al. 2012; Han, Hoeijmakers, Liu, et al. 2012; Hoeijmakers, Han, et al. 2012).
What about the patients with neuropathy who did not carry mutations of NaV1.7? To examine that question, we first searched in the DNA of these patients for mutations of NaV1.8, another “peripheral” sodium channel. For this study (Faber, Lauria, et al. 2012) we assessed a series of 104 patients with painful neuropathy in which a NaV1.7 mutation had not been found. Seven mutations in NaV1.8 were identified in nine of these 104 patients. Computer algorithms that assessed the potential functional effects of these mutations, on the basis of their effects on channel structure, suggested that three of these mutations were likely to be pathogenic. We assessed these mutations by voltage-clamp and current-clamp. Two of these mutations were found to enhance the channel’s response to depolarization and produced hyperexcitability in DRG neurons (Faber, Lauria, et al. 2012). We subsequently studied two additional NaV1.8 mutations from patients with painful neuropathy and found that these, too, conferred gain-of-function changes on the channel and produced DRG neuron hyperexcitability (Han et al. 2014; Huang et al. 2013).
In other studies together with our European collaborators, we asked whether mutations of the third peripheral sodium channel, NaV1.9, might be present in patients with painful peripheral neuropathy in whom NaV1.7 or NaV1.8 mutations were not present. For this study, we assessed a cohort of 344 patients with painful neuropathy without mutations in NaV1.7 or NaV1.8 and found four mutations of NaV1.9 in conserved, membrane-spanning regions of the channel. The biophysical effects of these mutations were especially interesting. The normal NaV1.9 channel is unusual in displaying a large overlap between activation and inactivation. As a result of this overlap, NaV1.9 channels produce a “window current,” which is a small, sustained inward sodium current that depolarizes neurons. Functional analysis of the NaV1.9 mutations from patients with painful neuropathy is ongoing as this book is being written. We have thus far found three NaV1.9 mutations from patients with painful neuropathy that shift channel gating so as to increase the window current, to a degree that depolarizes DRG neurons to a level that makes them hyperexcitable, providing an explanation for the pain that these patients’ experience (Han et al. 2015; Huang et al. 2014). We have also studied a NaV1.9 mutation from a patient with a loss of pain sensation. Interestingly, in this case a massive change in channel gating produces a larger increase in the window current that profoundly depolarizes DRG neurons, inactivating the sodium channels within these cells so that they lose the ability to generate nerve impulses, thus explaining the loss of pain sensibility (Huang et al., 2017). In the aggregate, these studies are showing us that, while less common than NaV1.7 mutations, NaV1.8 and NaV1.9 mutations also contribute to disorders of pain signaling.
These findings may have therapeutic implications. New agents designed to selectively block each of the peripheral sodium channels—NaV1.7, NaV1.8, and NaV1.9—are under development. The basic idea is that, because NaV1.7, NaV1.8, and NaV1.9 do not play substantial roles within the brain, selective blockade of these peripheral sodium channels would not be expected to affect the brain; consequently, subtype-specific NaV1.7, NaV1.8, or NaV1.9 blockers would be expected to provide pain relief without “central” side effects such as double vision, loss of balance, confusion, or sleepiness. And, because they do not act on the brain, these peripheral sodium channel blockers would be expected to be devoid of addictive potential. Development of NaV1.7 blockers is the most advanced. Particularly promising, as described in chapter 11, are the results of early-phase clinical trials on NaV1.7 blockers as a treatment for pain.
There is another dimension to these observations. These studies may help us to understand the events that lead to degeneration of nerve fibers in peripheral neuropathy. Identification of specific molecules that play key roles in axonal injury might provide a basis for therapies that would prevent, or slow, the degeneration of axons, thus halting or slowing the progression of peripheral neuropathy. In thinking about injury to axons in peripheral neuropathy, we built upon lessons learned in our earlier studies (Stys, Waxman, and Ransom 1992) on degeneration of axons within the optic nerve. Since small pain-signaling axons were known to express the sodium-calcium exchanger in proximity to NaV1.7 (Persson et al. 2010), we hypothesized that increased sodium influx into axons expressing mutant NaV1.7 channels might trigger calcium influx via reverse (calcium-importing) sodium-calcium exchange, as we had shown in 1992 in the optic nerve. To begin to assess this hypothesis, we developed a tissue culture model in which we could study the effect of mutant NaV1.7 sodium channels on the axons of DRG neurons. These studies showed that the length of axons of DRG transfected with mutant NaV1.7 channels was reduced compared to similar neurons transfected with normal, wild-type channels (Persson, Liu, et al. 2013). This suggested that the mutant NaV1.7 channels might impair the integrity of these axons. To examine this hypothesis, we asked whether a sodium channel blocker would protect axons transfected with the NaV1.7 mutant channels, preventing the decrease in length. For a variety of reasons we started with the sodium channel blocker carbamazepine, and we found that it does, in fact, have a protective effect. We also assessed the effect of a pharmacological blocker of reverse sodium-calcium exchange and found that it, too, was protective, preventing injury to the axons of DRG neurons expressing the mutant channels (Persson, Liu, et al. 2013).
Of course there is more to learn. Several aspects of peripheral neuropathy remain especially enigmatic. For example, we still have not solved the question “why does peripheral neuropathy become manifest in most patients relatively late in life?” Given that patients with NaV1.7-associated neuropathy carry their mutations throughout their entire lives, it is possible that axonal injury in peripheral neuropathy may build up via a multihit process: The sodium channel mutations may drive axonal degeneration in concert with other factors—genetic, or epigenetic, or environmental—that accumulate over time. Our recent observations suggest, for example, that dysfunction of mitochondria—intracellular organelles which act as energy generators for the cell—occurs with aging and accumulates as ever-increasing numbers of mitochondria are injured, finally combining with the gain-of-function in NaV1.7 channels to trigger axonal degeneration. As a postdoctoral fellow in 1976 I had demonstrated, using the computational methods available at that time, that multiple, cumulative insults to a nerve fiber, occurring at different times, could account for length-dependent injury (Waxman et al. 1976). We recently carried out experiments that show sodium channels contribute to degeneration of DRG axons induced by mitochondrial injury in tissue culture (Persson, Kim, et al. 2013), providing support for a multihit mechanism of axonal injury that involves mitochondrial dysfunction together with gain-of-function of NaV1.7. This work is ongoing (Estacion et al. 2015; Rolyan et al. 2016). These observations raise the possibility that in the future, it may be possible to devise neuroprotective strategies that will slow or prevent axonal degeneration in peripheral neuropathy.
Men on fire are very rare. In the parlance of medical rounds, they are “zebras.” But their genes pointed to NaV1.7 as a major player in pain. And helped us to discover that NaV1.7 is a participant in a more common disorder, peripheral neuropathy. People with peripheral neuropathy are “horses” and are seen every day in clinics around the world. The DNA of men on fire has helped to explain a disease in “the rest of us.”
References
Bakkers M, Merkies IS, Lauria G, Devigili G, Penza P, Lombardi R, Hermans MC, van Nes SI, De Baets M, Faber CG. 2009. Intraepidermal nerve fiber density and its application in sarcoidosis. Neurology 73(14): 1142–1148.
Black JA, Frezel N, Dib-Hajj SD, Waxman SG. 2012. Expression of Nav1.7 in DRG neurons extends from peripheral terminals in the skin to central preterminal branches and terminals in the dorsal horn. Mol Pain 8: 82.
Cummins TR, Dib-Hajj SD, Waxman SG. 2004. Electrophysiological properties of mutant Nav1.7 sodium channels in a painful inherited neuropathy. J Neurosci 24(38): 8232–8236.
Donnelly DF. 2008. Spontaneous action potential generation due to persistent sodium channel currents in simulated carotid body afferent fibers. J Appl Physiol 104(5): 1394–1401.
Estacion M, Han C, Choi JS, Hoeijmakers JG, Lauria G, Drenth JP, Gerrits MM, Dib-Hajj SD, Faber CG, Merkies IS, Waxman SG. 2011. Intra- and interfamily phenotypic diversity in pain syndromes associated with a gain-of-function variant of NaV1.7. Mol Pain 7: 92.
Estacion M, Vohra BP, Liu S, Hoeijmakers JG, Faber CG, Merkies IS, Lauria G, Black JA, Waxman SG. 2015. Ca2+ toxicity due to reverse Na+-Ca2+ exchange contributes to degeneration of neurites of DRG neurons induced by a neuropathy-associated Nav1.7 mutation. J Neurophysiol 114(3): 1554–1564.
Faber CG, Hoeijmakers JG, Ahn HS, Cheng X, Han C, Choi JS, Estacion M, et al. 2012. Gain of function NaV1.7 mutations in idiopathic small fiber neuropathy. Ann Neurol 71(1): 26–39.
Faber CG, Lauria G, Merkies IS, Cheng X, Han C, Ahn HS, Persson AK, et al. 2012. Gain-of-function Nav1.8 mutations in painful neuropathy. Proc Natl Acad Sci USA 109(47): 19444–19449.
Fertleman CR, Baker MD, Parker KA, Moffatt S, Elmslie FV, Abrahamsen B, Ostman J, Klugbauer N, Wood JN, Gardiner RM, Rees M. 2006. SCN9A mutations in paroxysmal extreme pain disorder: Allelic variants underlie distinct channel defects and phenotypes. Neuron 52(5): 767–774.
Han C, Hoeijmakers JG, Ahn HS, Zhao P, Shah P, Lauria G, Gerrits MM, et al. 2012. Nav1.7-related small fiber neuropathy: Impaired slow-inactivation and DRG neuron hyperexcitability. Neurology 78(21): 1635–1643.
Han C, Hoeijmakers JG, Liu S, Gerrits MM, te Morsche RH, Lauria G, Dib-Hajj SD, Drenth JP, Faber CG, Merkies IS, Waxman SG. 2012. Functional profiles of SCN9A variants in dorsal root ganglion neurons and superior cervical ganglion neurons correlate with autonomic symptoms in small fibre neuropathy. Brain 135(Pt 9): 2613–2628.
Han C, Vasylyev D, Macala LJ, Gerrits MM, Hoeijmakers JG, Bekelaar KJ, Dib-Hajj SD, Faber CG, Merkies IS, Waxman SG. 2014. The G1662S NaV1.8 mutation in small fibre neuropathy: Impaired inactivation underlying DRG neuron hyperexcitability. J Neurol Neurosurg Psychiatry 85(5): 499–505.
Han C, Yang Y, de Greef BT, Hoeijmakers JG, Gerrits MM, Verhamme C, Qu J, et al. 2015. The Domain II S4-S5 linker in Nav1.9: A missense mutation enhances activation, impairs fast inactivation, and produces human painful neuropathy. Neuromolecular Med 17(2): 158–169.
Herrmann DN, Griffin JW, Hauer P, Cornblath DR, McArthur JC. 1999. Epidermal nerve fiber density and sural nerve morphometry in peripheral neuropathies. Neurology 53(8): 1634–1640.
Hoeijmakers JG, Faber CG, Lauria G, Merkies IS, Waxman SG. 2012. Small-fibre neuropathies—advances in diagnosis, pathophysiology and management. Nat Rev Neurol 8(7): 369–379.
Hoeijmakers JG, Han C, Merkies IS, Macala LJ, Lauria G, Gerrits MM, Dib-Hajj SD, Faber CG, Waxman SG. 2012. Small nerve fibres, small hands and small feet: A new syndrome of pain, dysautonomia and acromesomelia in a kindred with a novel NaV1.7 mutation. Brain 135(Pt 2): 345–358.
Holland NR, Crawford TO, Hauer P, Cornblath DR, Griffin JW, McArthur JC. 1998. Small-fiber sensory neuropathies: Clinical course and neuropathology of idiopathic cases. Ann Neurol 44(1): 47–59.
Huang J, Han C, Estacion M, Vasylyev D, Hoeijmakers JG, Gerrits MM, Tyrrell L, et al, and the Propane Study Group. 2014. Gain-of-function mutations in sodium channel Na(v)1.9 in painful neuropathy. Brain 137(Pt 6): 1627–1642.
Huang J, Vanoye CG, Cutts A, Goldberg YP, Dib-Hajj SD, Cohen CJ, Waxman SG, George AL Jr. 2017. Sodium channel NaV1.9 mutations associated with insensitivity to pain dampen neuronal excitability. J Clin Invest 127(7): 2805–2814.
Huang J, Yang Y, Zhao P, Gerrits MM, Hoeijmakers JG, Bekelaar K, Merkies IS, Faber CG, Dib-Hajj SD, Waxman SG. 2013. Small-fiber neuropathy Nav1.8 mutation shifts activation to hyperpolarized potentials and increases excitability of dorsal root ganglion neurons. J Neurosci 33(35): 14087–14097.
Kennedy WR, Wendelschafer-Crabb G. 1999. Utility of the skin biopsy method in studies of diabetic neuropathy. Electroencephalogr Clin Neurophysiol 50(Supplement): 553–559.
Lacomis D. 2002. Small-fiber neuropathy. Muscle Nerve 26(2): 173–188.
McCarthy BG, Hsieh ST, Stocks A, Hauer P, Macko C, Cornblath DR, Griffin JW, McArthur JC. 1995. Cutaneous innervation in sensory neuropathies: Evaluation by skin biopsy. Neurology 45(10): 1848–1855.
Persson AK, Black JA, Gasser A, Cheng X, Fischer TZ, Waxman SG. 2010. Sodium-calcium exchanger and multiple sodium channel isoforms in intra-epidermal nerve terminals. Mol Pain 6: 84.
Persson AK, Kim I, Zhao P, Estacion M, Black JA, Waxman SG. 2013. Sodium channels contribute to degeneration of dorsal root ganglion neurites induced by mitochondrial dysfunction in an in vitro model of axonal injury. J Neurosci 33(49): 19250–19261.
Persson AK, Liu S, Faber CG, Merkies IS, Black JA, Waxman SG. 2013. Neuropathy-associated Nav1.7 variant I228M impairs integrity of dorsal root ganglion neuron axons. Ann Neurol 73(1): 140–145.
Peters MJ, Bakkers M, Merkies IS, Hoeijmakers JG, van Raak EP, Faber CG. 2013. Incidence and prevalence of small-fiber neuropathy: A survey in the Netherlands. Neurology 81(15): 1356–1360.
Rolyan H, Liu S, Hoeijmakers JG, Faber CG, Merkies IS, Lauria G, Black JA, Waxman SG. 2016. A painful neuropathy-associated Nav1.7 mutant leads to time-dependent degeneration of small-diameter axons associated with intracellular Ca2+ dysregulation and decrease in ATP levels. Mol Pain 12.
Stys PK, Ransom BR, Waxman SG. 1992. Tertiary and quaternary local anesthetics protect CNS white matter from anoxic injury at concentrations that do not block excitability. J Neurophysiol 67(1): 236–240.
Stys PK, Waxman SG, Ransom BR. 1992. Ionic mechanisms of anoxic injury in mammalian CNS white matter: Role of Na+ channels and Na(+)-Ca2+ exchanger. J Neurosci 12(2): 430–439.
Vasylyev DV, Waxman SG. 2012. Membrane properties and electrogenesis in the distal axons of small dorsal root ganglion neurons in vitro. J Neurophysiol 108(3): 729–740.
Walk D, Wendelschafer-Crabb G, Davey C, Kennedy WR. 2007. Concordance between epidermal nerve fiber density and sensory examination in patients with symptoms of idiopathic small fiber neuropathy. J Neurol Sci 255(1–2): 23–26.
Waxman SG, Black JA, Kocsis JD, Ritchie JM. 1989. Low density of sodium channels supports action potential conduction in axons of neonatal rat optic nerve. Proc Natl Acad Sci USA 86(4): 1406–1410.
Waxman SG, Brill MH, Geschwind N, Sabin TD, Lettvin JY. 1976. Probability of conduction deficit as related to fiber length in random-distribution models of peripheral neuropathies. J Neurol Sci 29(1): 39–53.