Pharmacotherapy for Pain in a Family with Inherited Erythromelalgia Guided by Genomic Analysis and Functional Profiling*
Paul Geha, Yang Yang, Mark Estacion, Betsy R. Schulman, Hajime Tokuno, A. Vania Apkarian, Sulayman D. Dib-Hajj, and Stephen G. Waxman
Importance: There is a need for more effective pharmacotherapy for chronic pain, including pain in inherited erythromelalgia (IEM) in which gain-of-function mutations of sodium channel NaV1.7 make dorsal root ganglion (DRG) neurons hyperexcitable.
Objective: To determine whether pain in IEM can be attenuated via pharmacotherapy guided by genomic analysis and functional profiling.
Design, Setting, and Participants: Pain in 2 patients with IEM due to the NaV1.7 S241T mutation, predicted by structural modeling and functional analysis to be responsive to carbamazepine, was assessed in a double-blind, placebo-controlled study conducted from September 2014 to April 21, 2015. Functional magnetic resonance imaging assessed patterns of brain activity associated with pain during treatment with placebo or carbamazepine. Multielectrode array technology was used to assess the effect of carbamazepine on firing of DRG neurons carrying S241T mutant channels.
Main Outcomes and Measures: Behavioral assessment of pain; functional magnetic resonance imaging; and assessment of firing in DRG neurons carrying S241T mutant channels.
Results: This study included 2 patients from the same family with IEM and the S241T NaV1.7 mutation. We showed that, as predicted by molecular modeling, thermodynamic analysis, and functional profiling, carbamazepine attenuated pain in patients with IEM due to the S241T NaV1.7 mutation. Patient 1 reported a reduction in mean time in pain (TIP) per day during the 15-day maintenance period, from 424 minutes while taking placebo to 231.9 minutes while taking carbamazepine (400 mg/day), and a reduction in total TIP over the 15-day maintenance period, from 6360 minutes while taking placebo to 3015 minutes while taking carbamazepine. Patient 2 reported a reduction in mean TIP per day during the maintenance period, from 61 minutes while taking placebo to 9.1 minutes while taking carbamazepine (400 mg then 200 mg/day), and a reduction in total TIP, from 915 minutes while taking placebo over the 15-day maintenance period to 136 minutes while taking carbamazepine. Patient 1 reported a reduction of mean episode duration, from 615 minutes while taking placebo to 274.1 minutes while taking carbamazepine, while patient 2 reported a reduction of the mean episode duration from 91.5 minutes while taking placebo to 45.3 minutes while taking carbamazepine. Patient 1, who had a history of night awakenings from pain, reported 101 awakenings owing to pain while taking placebo during the maintenance period and 32 awakenings while taking carbamazepine. Attenuation of pain was paralleled by a shift in brain activity from valuation and pain areas to primary and secondary somatosensory, motor, and parietal attention areas. Firing of DRG neurons expressing the S241T NaV1.7 mutant channel in response to physiologically relevant thermal stimuli was reduced by carbamazepine.
Conclusions and Relevance: Our results demonstrate that pharmacotherapy guided by genomic analysis, molecular modeling, and functional profiling can attenuate neuropathic pain in patients carrying the S241T mutation.
Inherited erythromelalgia (IEM) is an autosomal dominant disorder characterized by severe burning pain in the distal extremities, triggered by warmth and relieved by cooling, caused by gain-of-function mutations of the NaV1.7 sodium channel, which is encoded by the SCN9A gene.1 NaV1.7 is preferentially expressed within peripheral sensory dorsal root ganglion (DRG) and sympathetic ganglion neurons,2–4 where it activates at relatively hyperpolarized potentials below the threshold for action potential generation. NaV1.7 amplifies small stimuli, thereby setting the gain for firing.2 In general, the NaV1.7 mutations that cause IEM shift channel activation in a hyperpolarizing direction, making it easier to open the channel; when expressed within DRG neurons, these mutations produce hyperexcitability.2,3 Most patients with IEM experience limited relief, if any, with available medications, and patients classically resort to cooling of the affected limbs, in some cases with prolonged ice baths that ultimately lead to tissue breakdown.1
While most patients with IEM do not respond to pharmacotherapy, a family with IEM, responsive to treatment with the sodium channel inhibitor carbamazepine, has been reported.5 The mutation in this family, V400M, hyperpolarizes activation, similar to other NaV1.7 mutations that cause IEM. Notably, carbamazepine at clinically relevant concentrations has a specific action on V400M mutant channels in which it normalizes activation.5 Yang et al.6 used this carbamazepine-responsive V400M mutation as a “seed” for atomic-level structural modeling and showed that another rare NaV1.7 mutation (S241T) identified in patients with IEM, 159 amino acids distant from V400M within the linear channel sequence, is located less than 2.8Å from V400M within the folded channel protein; they used thermodynamic analysis to demonstrate energetic coupling of the S241 and V400 amino acids during channel activation. As predicted by the atomic proximity and energetic coupling to a carbamazepine-responsive mutation, carbamazepine had a specific effect, not seen in other IEM mutant channels, on S241T where it normalizes activation.6
In this article, we translate our in silico and in vitro analyses of the S241T mutation to a family with IEM carrying this channel variant. We hypothesized that treatment with carbamazepine would attenuate pain in patients with IEM carrying the S241T mutation and that, compared with placebo, attenuation of pain would be paralleled by a decrease in brain activity, measured with functional brain imaging (fMRI), in valuation and pain areas previously implicated in chronic pain and modulated by treatment.7–14
Methods
Human Participants
The patients were 2 adults with IEM carrying the S241T mutation. Patient 1 reported onset in his teens with severe burning pain in his feet, triggered by mild warmth and relieved by cooling, followed by similar pain in his hands, knees, elbows, shoulders, and ears. He described up to 30 episodes per month, each lasting hours to days. He described his typical IEM pain episode as severe, at a 9 on the pain numerical rating scale (NRS). Venlafaxine and gabapentin did not provide relief, and lidocaine patches provided minimal relief. He reported that his IEM prevented him from sleeping through the night and that it limited physical activity. Patient 2 reported onset of burning pain in both feet, triggered by mild warmth and relieved by cooling, which began in her teens, subsequently involving her knees and ears. She rated her pain as severe, at 8 and 9 on the NRS pain scale. Aspirin did not provide relief.
Study Design
The Human Investigations Committees at Yale University and West Haven VAMC approved this study (NCT02214615), which was conducted from September 2014 to April 21, 2015, and written informed consent was obtained from both patients. In this double-blind crossover study, each of the 2 patients with IEM, carrying the S241T mutation, were assessed during a series of 7 hospital visits, which included 5 fMRI scans (efigure 1 in the Supplement). Details of visits, scans, drug ramp-up, maintenance, and taper-down periods are given in the eAppendix in the Supplement.
Carbamazepine Treatment and Monitoring
At each scanning visit, blood was obtained to monitor complete blood cell count and carbamazepine levels. Carbamazepine or placebo were started at 200 mg daily. Patients reported pain levels every 4 days using the NRS (0 = no pain to 10 = worst imaginable pain); if pain intensity had not improved by 2 NRS units and adverse effects were not experienced, the dose was increased by 200 mg until pain intensity improved. If pain intensity had improved, the carbamazepine dose was maintained.
Prescan Testing
Pain in IEM is triggered by warmth.1,2 We used a calibrated warming boot to reliably elicit pain as described in the eAppendix in the Supplement.
Continuous Pain Rating
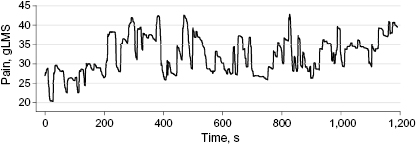
For continuous pain intensity ratings (figure 1),7,8,11,15 patients continuously indicated their level of pain during test sessions through a linear potentiometer device attached to the dominant thumb and index finger, with voltage output displayed by a computer that indicated the extent of their finger span, providing visual feedback. Maximum thumb-finger span was used to indicate “worst imaginable pain intensity” and thumb-and-finger touching to indicate “no pain” on the generalized labeled magnitude scale. Details are provided in the eAppendix in the Supplement.
Pain Rating and Visual Magnitude Rating Tasks during fMRI
Patients were scanned while (1) rating their pain in response to thermal stimuli, (2) rating ongoing pain (no stimulation) after an episode was elicited, and (3) rating the magnitude of a moving bar using the finger-span device. The first thermal stimulation run invariably elicited an IEM episode described at session debriefing to be similar to episodes experienced during daily life. Because we were assessing the response to treatment, we titrated the thermal stimulation until the pain intensity rating reached a predetermined level during all scans (visits 2, 3, 4, 6, and 7; efigure 1 in the Supplement). During the subsequent 2 pain runs, patients rated spontaneous fluctuations of their pain collected without thermal stimulation. A visual magnitude rating was performed last as a control for visuospatial and attention components inherent in our pain rating tasks (eAppendix in the Supplement).
fMRI Data Acquisition and Analysis
Imaging data were acquired with a Siemens 3T Trio scanner at Yale University Magnetic Resonance Research Center. Blood oxygen level–dependent images were acquired with parameters specified in the eAppendix in the Supplement. Image analysis was performed on each patient’s data using FMRIB Expert Analysis Tool (http://www.fmrib.ox.ac.uk/fsl) (eAppendix in the Supplement).
Assessment of DRG Neuron Excitability
We previously showed that carbamazepine attenuates firing induced by electrical stimuli in DRG neurons expressing S241T mutant channels in experiments carried out at room temperature.6 However, pain in IEM is triggered by warmth. To mimic the condition in human patients, we assessed the effect of carbamazepine on firing of DRG neurons expressing S241T at graded physiological temperatures (33°C, 37°C, and 40°C). Recording methods are described in the eAppendix in the Supplement.
Statistical Analysis
Multielectrode array data are expressed as mean (SEM). Statistical significance was determined by t test.
Results
Carbamazepine and Pain Attenuation in Patients Carrying the S241T Mutation
The effect of carbamazepine on S241T mutant channels6 suggested that carbamazepine might attenuate pain in patients with IEM carrying this mutation. Both patients in this study were blinded and asked to report duration and intensity of their IEM pain and the number of pain-induced awakenings from sleep on a daily basis (figure 2).
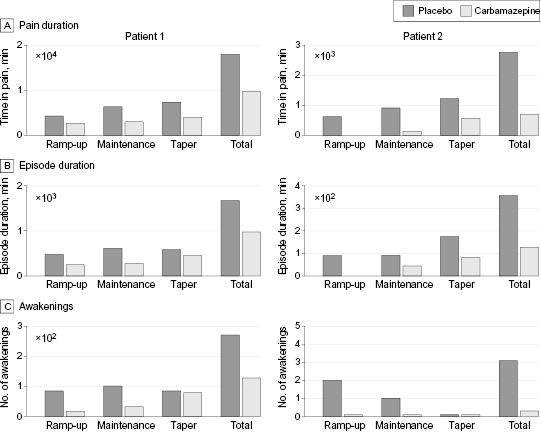
Patient 1 reported a reduction in mean time in pain (TIP) per day during the 15-day maintenance period of the study (at 400 mg/day of carbamazepine), from 424 minutes while taking placebo to 231.9 minutes while taking carbamazepine, and a reduction in total TIP over the 15-day maintenance period, from 6360 minutes while taking placebo to 3015 minutes while taking carbamazepine (figure 2A). Patient 2 reported a reduction in mean TIP per day during the maintenance period (at 400 and then 200 mg/day of carbamazepine), from 61 minutes while taking placebo to 9.1 minutes while taking carbamazepine, and a reduction in total TIP, from 915 minutes while taking placebo over the 15-day maintenance period to 136 minutes while taking carbamazepine (figure 2A). Patient 1 reported a reduction of mean episode duration from 615 minutes while taking placebo to 274.1 minutes while taking carbamazepine, while patient 2 reported a reduction of the mean episode duration from 91.5 minutes while taking placebo to 45.3 minutes while taking carbamazepine (figure 2B). Patient 1, who had a history of night awakenings from pain, reported 101 awakenings while taking placebo during the 15-day maintenance period and 32 awakenings while taking carbamazepine (figure 2C). Patient 2 reported 1 night awakening while taking placebo during the maintenance period and none while taking carbamazepine (figure 2C). Carbamazepine blood levels were in the therapeutic range (3.6–6.0 g/L) when patients were receiving carbamazepine (etable 1 in the Supplement).
Neither patient reported significant pain at arrival for hospital visits. Pain was provoked using a heating boot on the right foot with circulating water maintained at controlled temperatures. The empirically determined thermal stimulus in each patient invariably elicited a pain episode, which was described at session debriefing to be similar to episodes during daily life. Once pain was provoked, the thermal stimulus was terminated. Pain intensity ratings were continuously collected (figure 1; efigure 2 in the Supplement) as reported previously.7,11,13,15 To investigate long-term effects of carbamazepine vs placebo, we compared baseline fMRI scans of chronic (4-week) carbamazepine vs placebo treatment with a triple-paired t test implemented in FMRIB toolbox across the 2 patients. We previously demonstrated that brain activity maps obtained while patients with chronic pain rate intensity of their ongoing (stimulus-free) pain are more specific to the clinical condition under study compared with brain maps obtained during application of an external stimulus.7,11,13 This approach dissociates disease-specific ongoing fluctuations of pain, which in time shift away from sensory regions to engage valuation circuitry from acute thermal pain perception.7,11,13,14 The analysis reported here used scans where our patients rated their pain after the stimulus was terminated. Pain ratings collected during all scans and used to derive IEM pain maps are shown for both participants in efigure 2 in the Supplement.
Association between Carbamazepine and Brain Activity
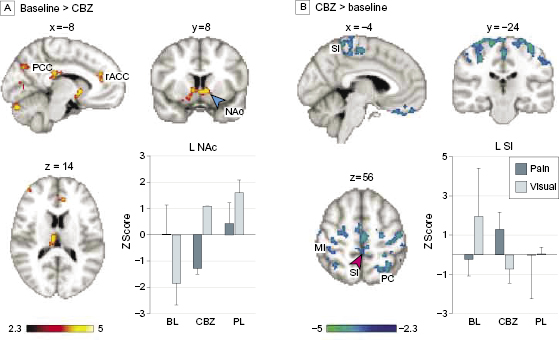
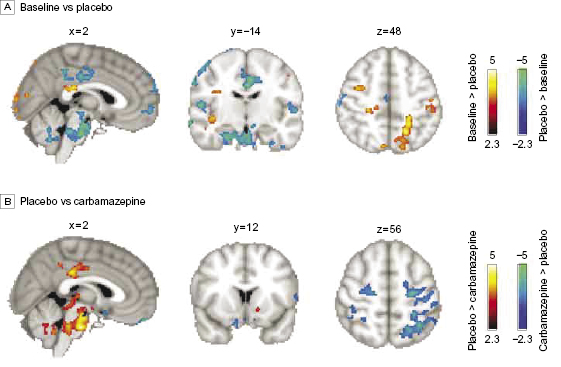
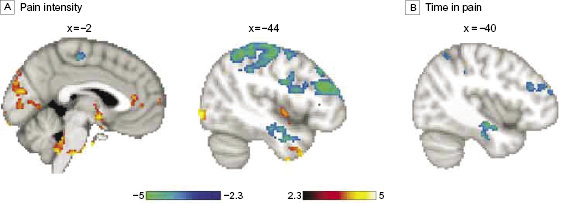
Carbamazepine treatment (carbamazepine scan < baseline; corrected for a visual control task) was associated with decreased activity in valuation areas14,16 including the ventral striatum (nucleus accumbens), ventral pallidum, rostral anterior cingulate (rACC; Brodmann Area [BA] 32), and posterior cingulate cortex (PCC), in addition to the ventral putamen, bilateral anterior insula, right thalamus, and hypothalamus (figure 3A; efigure 3 and etable 2 in the Supplement). By contrast, treatment was associated with increased activity (carbamazepine scan > baseline) in bilateral primary and secondary somatosensory-motor areas including the medial wall foot area according to the Jüelich Histological Atlas,17 bilateral parietal dorsal attention areas, supplementary motor area BA 6, and ventromedial prefrontal cortex (BA 11) compared with the baseline scan (figure 3B; efigure 3 and etable 2 in the Supplement; paired t test; fixed effects; n = 2, Z > 2.3; P < 0.05 corrected for multiple comparisons). However, treatment with placebo did not affect activity in the ventral striatum and had an effect opposite that of carbamazepine within the PCC, motor, and parietal areas. Placebo (baseline > placebo) decreased activity in the right dorsolateral prefrontal cortex (DLPFC) (BA 44/48), right posterior insula, and left parietal and bilateral visual areas. On the other hand, it increased activity (placebo > baseline) in the medial prefrontal cortex (BA 10), right inferior frontal gyrus (BA 45/47), bilateral central opercular areas (BA 48), PCC, bilateral hippocampi, midbrain, and pons (figure 4A; efigure 4 in the Supplement). Similar to the contrast of carbamazepine and baseline, the contrast of carbamazepine vs placebo showed a shift in activity from valuation areas to primary sensory motor and attention areas with carbamazepine (figure 4B; efigure 5 in the Supplement). To confirm the latter result, we collapsed placebo and baseline sessions together; paired t test with the carbamazepine treatment scans demonstrated an increase in sensory motor/attention areas with a concomitant decrease in valuation/reward areas after carbamazepine treatment (efigure 6 in the Supplement).
Next, we asked how pain intensity modulates brain activity to compare the effects of carbamazepine and placebo. We averaged pain intensity ratings within each scanning run and regressed them against brain activity across all visits. The regression results were similar to the effects of carbamazepine and opposite to placebo treatment. Activity within valuation areas, left nucleus accumbens and rACC, in addition to the left insula, right thalamus, hypothalamus, and midbrain covaried positively with pain intensity, whereas activity within primary and secondary sensory-motor cortices and dorsal parietal and DLPFC areas covaried negatively with pain intensity (figure 5A; efigure 7 and etable 3 in the Supplement). Additionally, activity in the left hippocampus was negatively correlated with pain intensity. Regression analysis that excluded visits when patients received carbamazepine confirmed that pain intensity covaries positively with valuation areas and negatively with primary somatosensory, motor, and parietal areas (efigure 8 in the Supplement). Hence, treatment with carbamazepine was associated with a shift of brain activity toward a pattern associated with decreased pain intensity. Using a similar analysis, we found that brain activity only in the left hippocampus, parietal cortex, and DLPFC were inversely correlated with TIP as reported in patients’ diaries (figure 5B), suggesting that there was increased activity in brain areas associated with less TIP during treatment with carbamazepine.
Carbamazepine and Warmth-Induced Firing of DRG Neurons Expressing S241T Mutant Channels
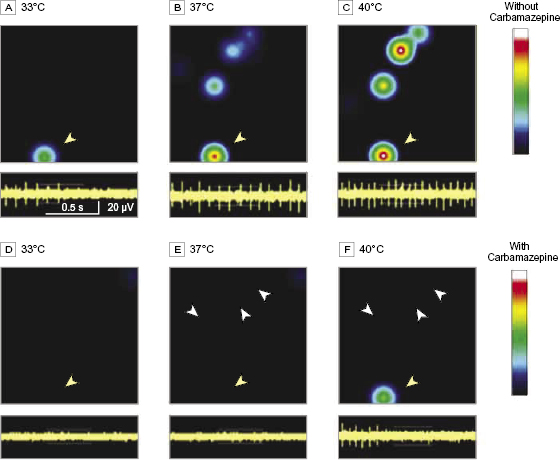
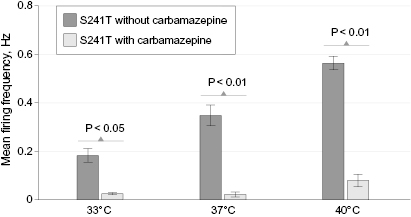
We showed previously that carbamazepine attenuates firing of DRG neurons expressing S241T mutant channels in response to graded electrical stimuli using current-clamp assays.6 However, pain in patients with IEM, including the patients we studied, is triggered by warmth. To determine whether carbamazepine had an effect on the firing of DRG neurons expressing NaV1.7 S241T channels in response to this naturally occurring stimulus, we assayed the firing of intact cultured DRG neurons using multielectrode arrays at normal skin temperature (33°C), core body temperature (37°C), and nonnoxious warmth (40°C). Firing of adult DRG neurons expressing NaV1.7 S241T was evoked in a temperature-dependent manner, as reflected by a heat map (figure 6A-C), increasing from a mean (SEM) frequency of 0.18 (0.03) Hz at 33°C (3 cultures using a total of 6 rats, 66 active electrodes/neurons) to 0.36 (0.04) Hz at 37°C (83 active electrodes/neurons) and to 0.56 (0.03) Hz at 40°C (98 active electrodes/neurons) (figure 7). Elevated temperature increased both the mean firing frequency and number of DRG neurons firing action potentials.
Carbamazepine at a clinically relevant concentration (30 µM)5,6 markedly attenuated firing of DRG neurons expressing S241T mutant channels (figure 6D-F). In the presence of carbamazepine, the mean (SEM) firing frequency of neurons expressing S241T at 33°C was 0.024 (0.003) Hz (P < 0.05 compared with neurons before carbamazepine treatment) with 52 active electrodes/neurons, at 37°C was 0.026 (0.011) Hz (P < 0.01) with 48 active electrodes/neurons, and at 40°C was 0.089 (0.026) Hz (P < 0.01) with 44 active electrodes (figure 7). These data indicate that carbamazepine at a clinically relevant concentration inhibits warmth-evoked firing of DRG neurons expressing NaV1.7 S241T across a physiological temperature range.
Discussion
Inherited erythromelalgia is caused by gain-of-function mutations of NaV1.7 and is characterized clinically by severe pain, triggered by mild warmth.1,2 Most patients with IEM do not respond to pharmacotherapy and resort to cooling, in some cases with ice or iced water that causes gangrene, to alleviate pain.1,2 On the basis of atomic-level structural modeling and functional analysis, we predicted that carbamazepine would attenuate pain in patients with IEM due to the S241T mutation. We previously showed that within the folded channel protein, the S241 residue is located within 2.8Å of the carbamazepine-responsive V400M mutation.6 That study demonstrated that S241T and V400M are energetically coupled during activation, a finding that predicted that carbamazepine should have a specific effect on the abnormal activation of S241T mutant channels; voltage- and current-clamp analyses showed that, indeed, carbamazepine has a specific effect, not seen in other IEM mutant channels, on S241T where it restores essentially normal activation, thereby reducing electrically induced firing of DRG neurons expressing S241T channels.6 In the present study using double-blind, placebo-controlled assessment, we demonstrated that carbamazepine attenuated pain induced by warmth in patients carrying the S241T mutation and showed a shift in brain activity from valuation and pain areas toward primary and secondary somatosensory-motor and parietal attention areas, a pattern of brain activity that has been associated with a shift from chronic to acute pain states.13,18 We also showed that warmth within a physiological range triggers abnormal firing in DRG neurons carrying S241T mutant channels, recapitulating in vitro the clinical picture of sensitivity to mild increases in temperature displayed by patients with IEM, and we demonstrated that carbamazepine inhibits warmth-induced hyperactivity of DRG neurons carrying S241T mutant channels.
There were some limitations to this study. We stress that this study was based on a small number of patients and the long-term effects of carbamazepine were not assessed. Moreover, we emphasize that our results, based on study of patients with the S241T mutation, do not imply that patients with erythromelalgia due to other NaV1.7 mutations will experience pain relief from carbamazepine. Rather, our results provide proof of principle, based on the S241T mutation, that genomic analysis together with molecular modeling and functional profiling can guide pain pharmacotherapy.
Carbamazepine acts on multiple sodium-channel subtypes,19 including NaV1.7,20 and thus we cannot exclude a contribution of sodium-channel blockade within the central nervous system to the effects of carbamazepine that we observed with behavioral and fMRI measurements. While some evidence suggests an inverse association between carbamazepine levels and blood oxygen level–dependent brain activity,21 treatment with carbamazepine was associated with increased activity in sensory/motor/attention areas, together with decreased activity in valuation areas. This shift of activity cannot be accounted for by a generalized dampening of blood oxygen level–dependent signal by carbamazepine. Importantly, the in vitro recordings in the current study demonstrated a strong attenuation of physiologically relevant warmth-induced firing of DRG neurons expressing S241T mutant channels by a clinically relevant concentration of carbamazepine. Taken together with the specific action of carbamazepine on S241T mutant channels,6 our observations support the idea that pain in IEM reflects abnormal hyperactivity of DRG neurons carrying gain-of-function mutant NaV1.7 channels2,3,22 and suggest that carbamazepine relieves pain in human patients carrying the S241T mutation at least in part via an action on the mutant NaV1.7 channel in DRG neurons.
Functional MRI revealed that brain activity shifted during carbamazepine treatment from valuation (ventral striatum, rACC, and PCC)14,16 and pain (thalamus and insula) areas18,23 toward primary somatosensory-motor and parietal attention areas including the medial sensory-motor cortical wall with afferent and efferent fibers to the foot. This shift in brain activity was observed during carbamazepine treatment despite the decades-long history of severe pain in these patients. Previous fMRI and clinical studies showed that placebo can reduce pain in some patient populations while concurrently modulating activity in valuation and pain areas.10,24–28 However, in our study, carbamazepine treatment achieved this change, whereas placebo did not.
The drop in brain activity within valuation and pain areas with carbamazepine was consistent with previous work showing decreases in the ventral striatum, rACC, and insula activity, as well as changes in their functional connectivity with successful treatment of chronic pain.11,25 Baliki et al.8 suggested that nucleus accumbens activity tracks the value of pain relief in chronic pain, while the ventral striatum and rACC are activated by pain and pain predictive cues.8,29 It has also been reported that the valuation circuitry mediates reward-related decision making.14,16 We observed a concomitant increase in activity of areas mediating somatosensory, motor, and attention tasks.30 This observation suggests that attenuation of pain with carbamazepine may allow patients to shift brain resources from areas mediating emotional decision making and pain to sensory-motor, attention, and executive function areas mediating accurate movements and sensory perception while rating their pain experience, consistent with the suggestion that persistence of pain shifts brain activity from sensory-motor regions to emotional decision-making circuitry.13 Activity in the parietal and DLPFC areas were particularly inversely associated with TIP as reported in patients’ diaries, suggesting that the increase in activity in these areas with carbamazepine might have positive effects on attention and executive function.31 Whether this shift is associated with improved functioning on a daily basis remains to be determined.
Segerdahl et al.32 reported cerebral blood flow differences between states of acute thermal heating and cooling in a study that used arterial spin labeling to assess 1 patient with IEM. While some of the areas affected by carbamazepine treatment overlap with their report, mainly rACC, insula, and thalamus, methodological differences preclude direct comparisons between the 2 studies. Unlike in the study by Segerdahl et al.,32 our patients rated the intensity of pain after a provocatory thermal stimulus was terminated. Our approach allows the identification of brain maps specific to different clinical pain conditions without the added component of ongoing stimulation,13 which could mask differences between patients and healthy control individuals.7
Conclusions
In this study, genomic analysis, molecular modeling, and functional profiling provided a basis for reduction of neuropathic pain with carbamazepine in patients with IEM carrying the S241T mutation in sodium-channel NaV1.7. Functional brain imaging demonstrated a change in brain activity within the pain, valuation, and somatosensory/motor/attention circuitry in patients carrying this variant, providing a potential correlate within the brain for the report of an effect of carbamazepine on pain in their home environment. As the number of sodium-channel variants linked to pain grows,33 structural and functional analysis of other mutations may provide additional opportunities for genomically guided pain pharmacotherapy.
Acknowledgments
Author Contributions: Drs. Geha and Waxman had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Study concept and design: Geha, Tokuno, Dib-Hajj, Waxman. Acquisition, analysis, or interpretation of data: All authors. Drafting of the manuscript: Geha, Waxman. Critical revision of the manuscript for important intellectual content: All authors. Statistical analysis: Geha, Yang, Apkarian. Obtained funding: Waxman. Administrative, technical, or material support: Geha, Yang, Estacion, Schulman, Dib-Hajj. Study supervision: Waxman.
Conflict of Interest Disclosures: None reported.
Funding/Support: This work was supported in part by grants from the Rehabilitation Research Service and Medical Research Service, Department of Veterans Affairs, the Erythromelalgia Association, and the Kenneth Rainin Foundation (Dr Waxman). Dr Geha was supported by grant 1K08DA037525-01 from the National Institute on Drug Abuse and the Yale University Department of Psychiatry.
Role of the Funder/Sponsor: The funders had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
About the Authors
Paul Geha, MD, Department of Psychiatry, Yale University School of Medicine, New Haven, CT; The John B. Pierce Laboratory, New Haven, CT
Yang Yang, PhD, Department of Neurology, Yale University School of Medicine, New Haven, CT; Neurorehabilitation Research Center, Department of Neurology, Veterans Affairs Medical Center, West Haven, CT
Mark Estacion, PhD, Department of Neurology, Yale University School of Medicine, New Haven, CT; Neurorehabilitation Research Center, Department of Neurology, Veterans Affairs Medical Center, West Haven, CT
Betsy R. Schulman, PhD, Department of Neurology, Yale University School of Medicine, New Haven, CT; Neurorehabilitation Research Center, Department of Neurology, Veterans Affairs Medical Center, West Haven, CT
Hajime Tokuno, MD, Department of Neurology, Yale University School of Medicine, New Haven, CT; Neurorehabilitation Research Center, Department of Neurology, Veterans Affairs Medical Center, West Haven, CT
A. Vania Apkarian, PhD, Department of Physiology, Northwestern University, Chicago, IL
Sulayman D. Dib-Hajj, PhD, Department of Neurology, Yale University School of Medicine, New Haven, CT; Neurorehabilitation Research Center, Department of Neurology, Veterans Affairs Medical Center, West Haven, CT
Stephen G. Waxman, MD, PhD, Department of Neurology, Yale University School of Medicine, New Haven, CT; Neurorehabilitation Research Center, Department of Neurology, Veterans Affairs Medical Center, West Haven, CT
Box 1 Key Points
Question Is genomically guided pharmacotherapy feasible in a genetic model of pain?
Findings In this study of 2 patients with inherited erythromelalgia due to the NaV1.7 S241T mutation, a double-blind cross-over study showed that carbamazepine attenuated pain, as predicted by genomic/molecular analysis and functional profiling. Pain relief was paralleled by a shift in brain activity from valuation and pain areas to primary and secondary somatosensory, motor, and parietal attention areas.
Meaning Pharmacotherapy guided by genomic analysis, molecular modeling, and functional profiling reduces pain in patients with inherited erythromelalgia due to the S241T mutation. As more channel variants are linked to pain, structural and functional analyses may provide additional opportunities for genomically guided pharmacotherapy.
Figure 1 Inherited erythromelalgia pain rating. An example of rating of pain fluctuations after an episode is elicited with the thermal boot. The rating shown here was recorded after the thermal stimulus was switched off. gLMS indicates generalized Labeled Magnitude Scale.
Figure 2 Pain characteristics in patients 1 and 2. Pain characteristics and effects of carbamazepine treatment vs placebo for patients 1 and 2. (A) Time in pain as reported in patients’ diaries during the 3 phases of treatment ramp-up, maintenance, and taper. Histograms represent means. (B) Same as in panel A for the reported duration of inherited erythromelalgia episodes. (C) Number of awakenings due to pain during 3 phases of ramp-up, maintenance, and taper.
Figure 3 Brain activity modulation with carbamazepine (CBZ). Treatment effects of CBZ vs baseline. (A) Brain activity obtained when contrasting baseline to carbamazepine (baseline > CBZ) (paired t test; n = 2; fixed effects; P < .05, corrected for multiple comparisons). The bar plot shows mean (SEM) brain activity in z scores during pain rating (blue) and visual tracking (white) within the left (L) nucleus accumbens (NAc) (blue arrowhead) plotted for baseline (BL, left), chronic CBZ treatment (CBZ, middle), and chronic placebo (PL, right) treatment, respectively. (B) Brain activity when contrasting CBZ > baseline; the bar plot depicts mean activity within primary somatosensory area (SI) (red arrowhead). MI indicates primary motor cortex; PCC, posterior cingulate cortex; PC, parietal cortex; R, right; rACC, rostral anterior cingulate cortex; and SI, primary somatosensory cortex.
Figure 4 Brain activity modulation by placebo. (A) Treatment effects of placebo vs baseline. Areas shown in red to yellow represent the contrast (baseline > placebo) and areas shown in blue to green represent the contrast (placebo > baseline). Unlike carbamazepine, placebo decreases activity in somatosensory parietal areas and increases activity in the posterior cingulate cortex and medial prefrontal cortex, among others. (B) Contrast results between placebo scans (placebo > carbamazepine, red to yellow) and carbamazepine scans (carbamazepine > placebo, blue to green). Differences in activations are similar to those shown in figure 3 for carbamazepine and baseline.
Figure 5 Brain activity associated with decreased pain. (A) Regression of brain activity during pain rating scans across all visits against pain intensity reported during scanning. (B) Regression of brain activity against time in pain as reported in patients’ diaries after masking with results shown in panel A. Areas in red to yellow represent positive correlations, whereas areas in blue to green represent negative correlations.
Figure 6 Carbamazepine attenuation of warmth-evoked firing in dorsal root ganglion neurons expressing NaV1.7 S241T mutant channels. (A–C) Heat maps of a representative multielectrode array recording of dorsal root ganglion neurons expressing NaV1.7 S241T before carbamazepine treatment (upper panels). The firing frequency of each active electrode is color coded with white/red representing high firing frequency and blue/black representing low firing frequency. Each circle corresponds to an active electrode within an 8 × 8 electrode array. There is only 1 active electrode in the heat map at 33°C (A). The number of active electrodes and firing frequency increase at 37°C (B) and 40°C (C). (D–F) Heat maps of the same multielectrode array recording well after (30-µM) carbamazepine treatment (upper panels). The number of active electrodes and firing frequency of neurons are both markedly reduced at all 3 temperatures: 33°C (D), 37°C (E), and 40°C (F). White arrowheads indicate silent neurons after carbamazepine treatment. In the lower panels in A–F, recordings from a representative neuron in the heat map indicated by yellow arrowheads are shown. Note increased firing as temperature increased in the absence of carbamazepine (A–C) and attenuation of firing by carbamazepine (D–F).
Figure 7 Firing frequency. Mean firing frequency of neurons (n = 98) expressing NaV1.7 S241T before and after carbamazepine treatment at all 3 temperatures.
References
1 Drenth JP, Waxman SG. 2007. Mutations in sodium-channel gene SCN9A cause a spectrum of human genetic pain disorders. J Clin Invest 117(12): 3603–3609.
2 Dib-Hajj SD, Yang Y, Black JA, Waxman SG. 2013. The Na(V)1.7 sodium channel: From molecule to man. Nat Rev Neurosci 14(1): 49–62.
3 Rush AM, Dib-Hajj SD, Liu S, Cummins TR, Black JA, Waxman SG. 2006. A single sodium channel mutation produces hyper- or hypoexcitability in different types of neurons. Proc Natl Acad Sci USA 103(21): 8245–8250.
4 Toledo-Aral JJ, Moss BL, He ZJ, et al. 1997. Identification of PN1, a predominant voltage-dependent sodium channel expressed principally in peripheral neurons. Proc Natl Acad Sci USA 94(4): 1527–1532.
5 Fischer TZ, Gilmore ES, Estacion M, et al. 2009. A novel Nav1.7 mutation producing carbamazepine-responsive erythromelalgia. Ann Neurol 65(6): 733–741.
6 Yang Y, Dib-Hajj SD, Zhang J, et al. 2012. Structural modelling and mutant cycle analysis predict pharmacoresponsiveness of a Na(V)1.7 mutant channel. Nat Commun 3: 1186.
7 Baliki MN, Chialvo DR, Geha PY, et al. 2006. Chronic pain and the emotional brain: Specific brain activity associated with spontaneous fluctuations of intensity of chronic back pain. J Neurosci 26(47): 12165–12173.
8 Baliki MN, Geha PY, Fields HL, Apkarian AV. 2010. Predicting value of pain and analgesia: Nucleus accumbens response to noxious stimuli changes in the presence of chronic pain. Neuron 66(1): 149–160.
9 Baliki MN, Petre B, Torbey S, et al. 2012. Corticostriatal functional connectivity predicts transition to chronic back pain. Nat Neurosci 15(8): 1117–1119.
10 Ellingsen DM, Wessberg J, Eikemo M, et al. 2013. Placebo improves pleasure and pain through opposite modulation of sensory processing. Proc Natl Acad Sci USA 110(44): 17993–17998.
11 Geha PY, Baliki MN, Chialvo DR, Harden RN, Paice JA, Apkarian AV. 2007. Brain activity for spontaneous pain of postherpetic neuralgia and its modulation by lidocaine patch therapy. Pain 128(1–2): 88–100.
12 Geha PY, Baliki MN, Harden RN, Bauer WR, Parrish TB, Apkarian AV. 2008. The brain in chronic CRPS pain: Abnormal gray-white matter interactions in emotional and autonomic regions. Neuron 60(4): 570–581.
13 Hashmi JA, Baliki MN, Huang L, et al. 2013. Shape shifting pain: Chronification of back pain shifts brain representation from nociceptive to emotional circuits. Brain 136(pt 9): 2751–2768.
14 Kable JW, Glimcher PW. 2007. The neural correlates of subjective value during intertemporal choice. Nat Neurosci 10(12): 1625–1633.
15 Foss JM, Apkarian AV, Chialvo DR. 2006. Dynamics of pain: Fractal dimension of temporal variability of spontaneous pain differentiates between pain states. J Neurophysiol 95(2): 730–736.
16 Levy DJ, Glimcher PW. 2012. The root of all value: A neural common currency for choice. Curr Opin Neurobiol 22(6): 1027–1038.
17 Eickhoff SB, Stephan KE, Mohlberg H, et al. 2005. A new SPM toolbox for combining probabilistic cytoarchitectonic maps and functional imaging data. Neuroimage 25(4): 1325–1335.
18 Apkarian AV, Bushnell MC, Treede RD, Zubieta JK. 2005. Human brain mechanisms of pain perception and regulation in health and disease. Eur J Pain 9(4): 463–484.
19 Qiao X, Sun G, Clare JJ, Werkman TR, Wadman WJ. 2014. Properties of human brain sodium channel α-subunits expressed in HEK293 cells and their modulation by carbamazepine, phenytoin and lamotrigine. Br J Pharmacol 171(4): 1054–1067.
20 Jo S, Bean BP. 2014. Sidedness of carbamazepine accessibility to voltage-gated sodium channels. Mol Pharmacol 85(2): 381–387.
21 Jokeit H, Okujava M, Woermann FG. 2001. Carbamazepine reduces memory induced activation of mesial temporal lobe structures: A pharmacological fMRI-study. BMC Neurol 1: 6.
22 Dib-Hajj SD, Rush AM, Cummins TR, et al. 2005. Gain-of-function mutation in Nav1.7 in familial erythromelalgia induces bursting of sensory neurons. Brain 128(pt 8): 1847–1854.
23 Lamm C, Decety J, Singer T. 2011. Meta-analytic evidence for common and distinct neural networks associated with directly experienced pain and empathy for pain. Neuroimage 54(3): 2492–2502.
24 Diederich NJ, Goetz CG. 2008. The placebo treatments in neurosciences: New insights from clinical and neuroimaging studies. Neurology 71(9): 677–684.
25 Hashmi JA, Baria AT, Baliki MN, Huang L, Schnitzer TJ, Apkarian AV. 2012. Brain networks predicting placebo analgesia in a clinical trial for chronic back pain. Pain 153(12): 2393–2402.
26 Tracey I. 2010. Getting the pain you expect: Mechanisms of placebo, nocebo and reappraisal effects in humans. Nat Med 16(11): 1277–1283.
27 Wager TD, Rilling JK, Smith EE, et al. 2004. Placebo-induced changes in FMRI in the anticipation and experience of pain. Science 303(5661): 1162–1167.
28 Zubieta JK, Stohler CS. 2009. Neurobiological mechanisms of placebo responses. Ann N Y Acad Sci 1156: 198–210.
29 Seymour B, O’Doherty JP, Koltzenburg M, et al. 2005. Opponent appetitive-aversive neural processes underlie predictive learning of pain relief. Nat Neurosci 8(9): 1234–1240.
30 Mesulam MM. 1998. From sensation to cognition. Brain 121(pt 6): 1013–1052.
31 Corbetta M, Shulman GL. 2002. Control of goal-directed and stimulus-driven attention in the brain. Nat Rev Neurosci 3(3): 201–215.
32 Segerdahl AR, Xie J, Paterson K, Ramirez JD, Tracey I, Bennett DL. 2012. Imaging the neural correlates of neuropathic pain and pleasurable relief associated with inherited erythromelalgia in a single subject with quantitative arterial spin labelling. Pain 153(5): 1122–1127.
33 Waxman SG, Merkies IS, Gerrits MM, et al. 2014. Sodium channel genes in pain-related disorders: Phenotype-genotype associations and recommendations for clinical use. Lancet Neurol 13(11): 1152–1160.