
1 Introduction
Natural products have long played a leading role in successful chemical biology and drug discovery, providing chemotypes sufficiently tailored to serve as chemical probes, drug leads or, at the very least, as sources of inspiration for molecular design [1–4]. While the development of innovative chemistry has facilitated the access to new and more diverse natural products in amounts suitable for bioactivity screening [5], prioritizing target-based assays remains not only a bottleneck in drug discovery but is also troublesome [6]. In fact, screening natural products of interest in target-based assays is often motivated by a prior phenotype change observation induced by the studied natural product in cell-based assays, e.g., cancer cell growth inhibition [6, 7]. Typically, the effective development of such bioactive natural products as useful drug leads relies on the deconvolution of the phenotypic readout and correlation of the said phenotype with the engagement of any given drug target or targets [7]. It is now widely accepted that natural products, like small molecules, rarely are selective but engage dozens of related or unrelated targets [8], resulting in intricate pharmacology networks that might be explored in a drug discovery context [9, 10]. Crucially, such knowledge may bring benefits to the design of leads with lower probability of attrition and ultimately afford efficacious disease modulators.
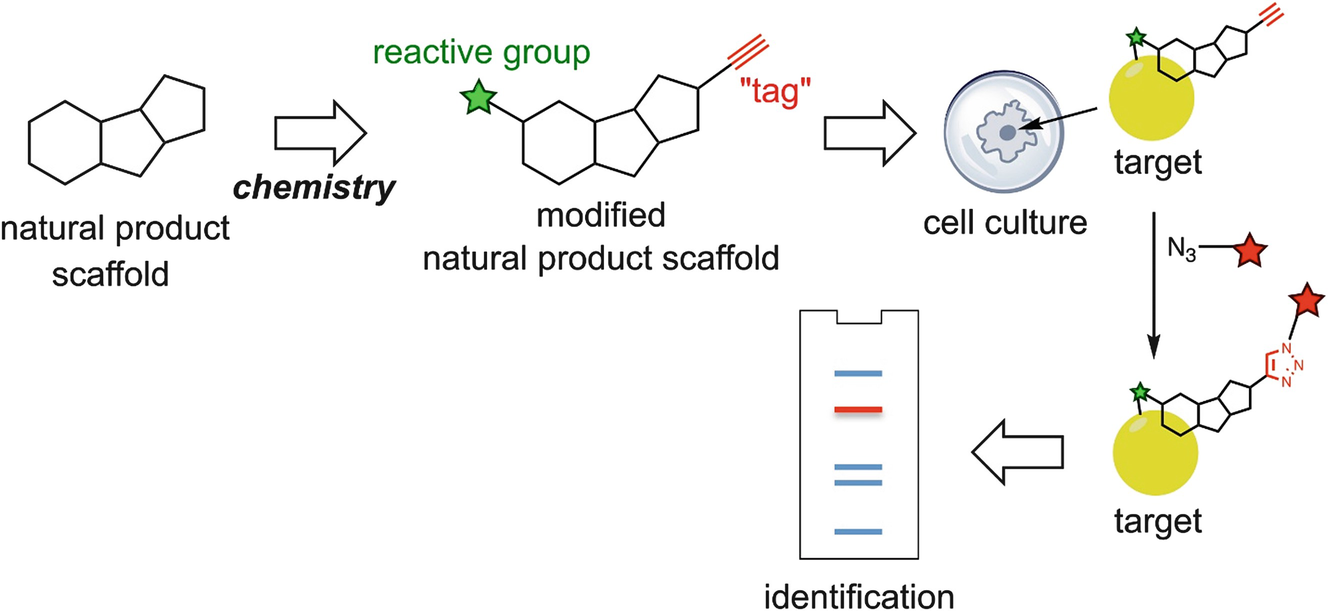
Typical workflow for identifying drug targets through chemical proteomics approaches
It is conceivable that in silico methods can provide viable alternatives to generate such motivated research hypotheses, yet within a fraction of time and resources spent. Virtual screening of an enumerated fraction of chemical space has been employed widely with vendor libraries as a means of accelerating hit discovery and prioritizing chemical matter for screening campaigns [14]. In contrast to chemical proteomics, where target identification is a step downstream from phenotypic assays, in silico screening often focuses on a drug target for which ligands are sought [3]; only in the event of successful experimental validation of the predicted ligand-target relationship is the engagement of the target correlated with modulation of disease or adverse drug reactions [13, 15].
In this contribution, an overview will be provided and a discussion of strengths and limitations of computational methods that have been successfully employed for unveiling targets in the natural product realm. In particular, molecular docking and pharmacophore model-based strategies will be described as a means of accounting for three-dimensionality in scrutinizing potential drug targets for either natural products or synthetic small molecules. Importantly, with the advent of big data in biological and chemical sciences [16, 17], molecular docking and pharmacophore screening have become suboptimal approaches to process large volumes of information. In fact, the increasing computer power, storage capacity, and improved algorithms to analyze unstructured and sparse data, are setting the tone for a new era of cheminformatics where artificial intelligence promises to tackle some of the long-standing problems in molecular informatics and chemistry in general [17, 18]. As such, a special focus is given to emerging machine learning tools that leverage topological descriptors as a workhorse to building predictive models, and how such approaches can drive future chemical biology and early drug discovery programs. By comparing different tools, some of them accessible through webservers [8, 19], this contribution aims at being a reference work for the motivated selection of any given tool according to the goal of the project.
2 Molecular Docking
Molecular docking has become a standard means of screening virtual libraries within the realm of receptor-based methods [20, 21]. In short, such methods sample the ligand conformation space in a user-defined “box”/binding site in an attempt to predict the so-called “docking pose” and rationalize, on a molecular structure level, the activity that any compound might present against a given protein [20]. Thus, molecular docking software tools do not aim at identifying ready-made and optimized ligands, but rather discriminate relevant chemical features responsible for a molecular recognition event. These compounds and spatial arrangement of features might then be further tuned through medicinal chemistry to enhance binding affinity and, ideally, improve functional activity. Despite the simplicity of the concept and the existence of several user-friendly tools to carry out molecular docking studies, the researcher must bear in mind several caveats for proper data interpretation [20, 22, 23]. For instance, docking solely provides motivated research hypotheses or can rationalize them prior to experimental observations. Given that docking models account for only a snapshot of the protein in a conformational ensemble [21], they ought to be validated in biochemical studies (e.g., site mutagenesis) and the accuracy of the output is tightly connected to the quality of the protein X-ray structure where docking is performed. Since X-ray structures represent electron density models, careful selection of the starting data is fundamental to avoid the exponential propagation of errors and inaccurate predicted poses. To this end, it is often advisable to select high-resolution structures (≤2.5 Å) and screen/correct amino acid residue rotamers, as assessed through Ramachandran plots [24–26].

2.1 Identification of Modes of Action with Docking
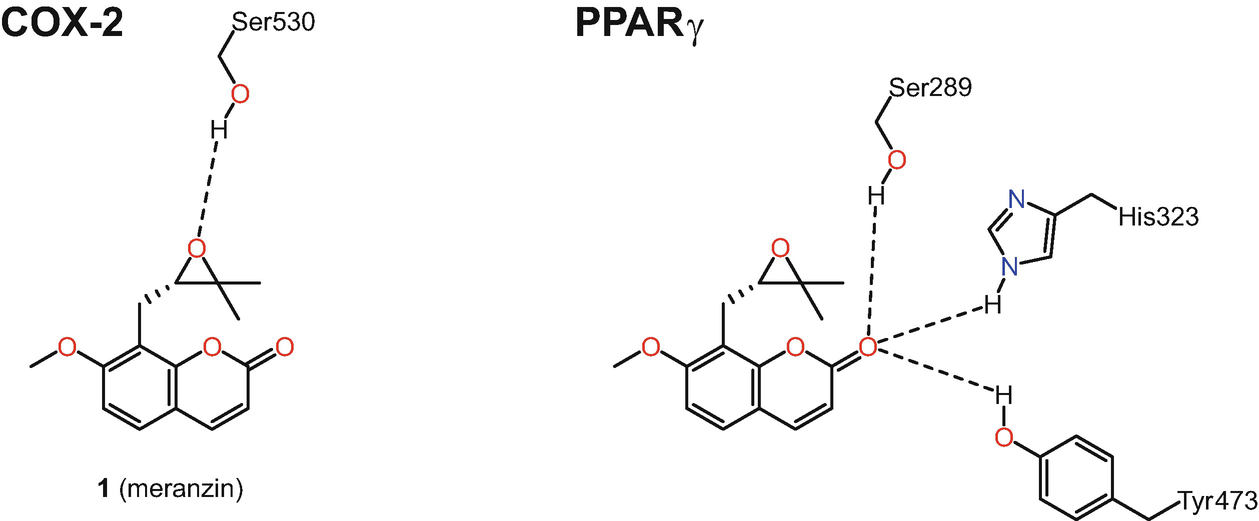
Predicted interactions between the natural product meranzin (1) and cyclooxygenase-2 (COX2) and the peroxisome proliferator-activated receptor gamma (PPAR γ)
3 Pharmacophore Model-Based Screening
Comparison of pharmacophore feature assignment schemes, by four popular software tools
Feature | LigandScout | MOE | Phase | Catalyst |
|---|---|---|---|---|
H-bond | Acceptor and donor located on heavy atom | Acceptor and donor located on heavy atom | Donor located on hydrogen and acceptor on heavy atom | Acceptor and donor located on heavy atom |
Lipophilic | Aromatic rings are recognized | Aromatic rings are not recognized | Aromatic rings are not recognized | Aromatic rings are recognized |
Aromatic | Represented with plane orientation | Depends on pharmacophore scheme | Represented with plane orientation | Represented with plane orientation |
Charge transfer | No explicit charges | Explicit charges | No explicit charges | No explicit charges |
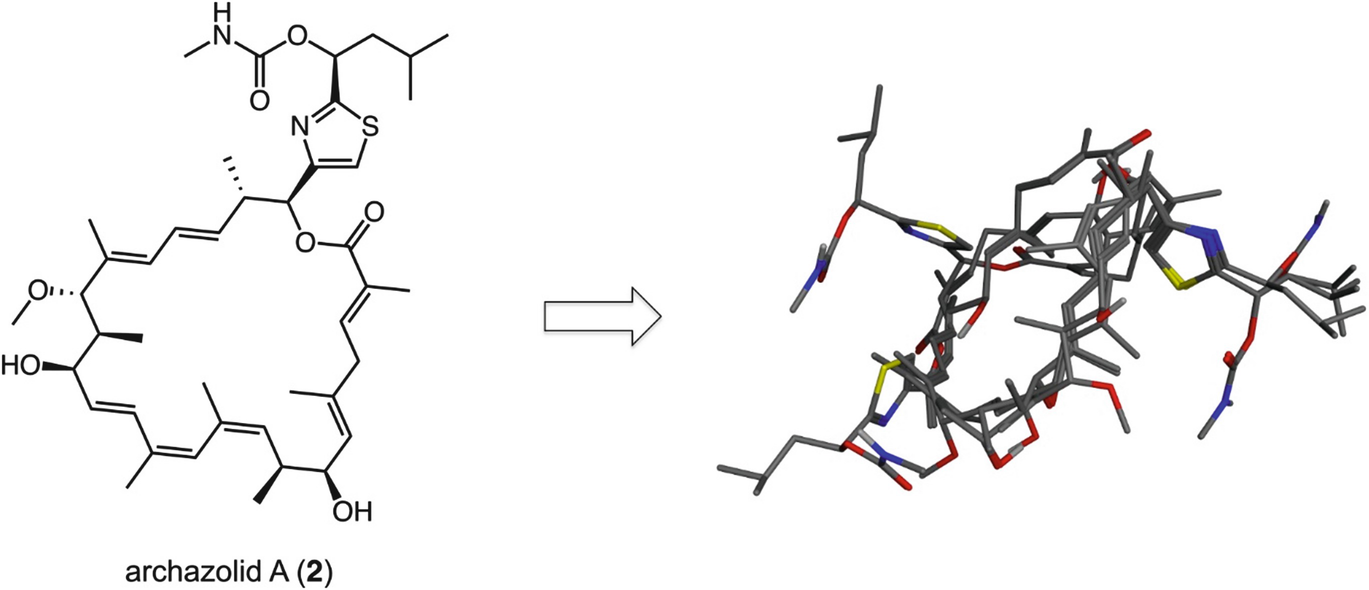
Structure of a natural product and the superimposition of energy-minimized conformers, as computed with MOE (Chemical Computing Group, Canada). Data show that several distinct conformers are generated from the same structure
3.1 Identification of Modes of Action with Pharmacophore Models

Ruta graveolens. Photograph: Jörg Hempel, Creative Commons 3.0
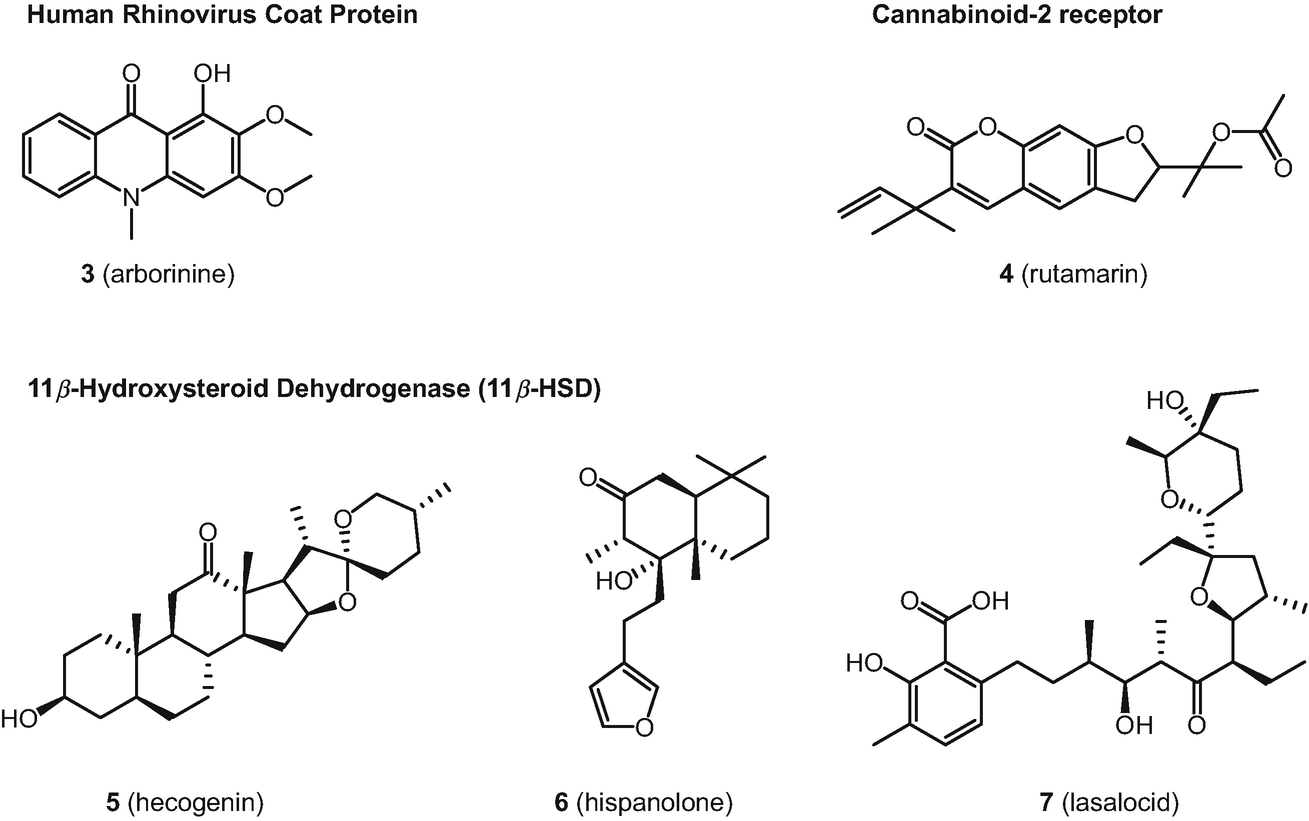

Structures of natural products deorphanized through pharmacophore model-based virtual screening
4 Molecular Similarity Searches
Both molecular docking and 3D pharmacophore screening have been applied with great effectiveness to unveil putative binding counterparts for natural products. However, they rely on the computation of meaningful conformations; as discussed above, which is a particularly challenging endeavor. Additionally, these methods persist in being computationally expensive and arguably are of limited throughput.

![$$ D=\sqrt[p]{\sum \limits_{i=1}^n{\left|{\mathbf{A}}_i-{\mathbf{B}}_i\right|}^p} $$](../images/480635_1_En_3_Chapter/480635_1_En_3_Chapter_TeX_Equ3.png)
In principle, any molecule with experimentally confirmed bioactivity against the target of interest can be used as starting point (reference) for similarity searches. However, taking into account that the goal of the method is to retrieve hits from a search database, a high-affinity ligand is a better motivated choice as reference molecule. Naturally, the selection of the descriptors employed and the metric used to assess similarity are cornerstones for the success of a screening campaign [49]. A wealth of screening techniques and software is available (some of which is implemented in open-source pipelining tools like KNIME), and their proper selection depends on the goal, suitability and availability, among others [50–52]. Irrespective of the screening strategy, similarity (or distance) values are calculated and stored in a database, which are then sorted in order of decreasing similarity (or increasing distance) to the query/reference molecules. The rank ordered list is provided as output for human inspection, wherein the molecule with the smallest distance or higher similarity is called the “nearest neighbor.”
4.1 Identification of Modes of Action Through Structural Similarity
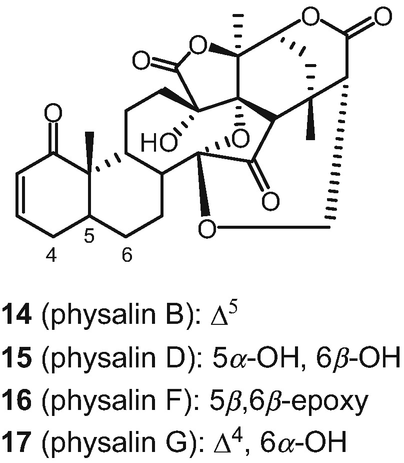
Structures of antiplasmodial physalins
5 Machine Learning Methods
- 1.
Regression (supervised learning) if the output is a numeric value
- 2.
Classification (supervised learning) if the output is a label
- 3.
Clustering (unsupervised learning) if the algorithm associates data solely based on its structure
Independently of the method, all machine-learning approaches have proven useful in early drug discovery by streamlining processes and facilitating the design of relevant experiments. On one hand, regression and classification models have been employed prospectively for de novo design of small-molecule effectors [59], prediction of pharmacokinetics [60], prediction of drug-likeness [61], prediction of synthesis routes [62], optimization of chemical reactions [63], and conformational sampling [39], among many others. On the other hand, clustering methods have proven useful in the analysis of bioactivity landscapes [64, 65].
Given its utility for a number of tasks and the increase of bioactivity data for small molecules, machine learning has found applicability in research programs aiming at identifying targets for bioactive molecules of synthetic and natural origin [17]. Indeed, the need for minimal computational effort to afford statistically motivated research hypotheses renders machine learning as an attractive alternative to molecular docking and pharmacophore-based virtual screening.
5.1 Identification of Modes of Action Using Learning Algorithms
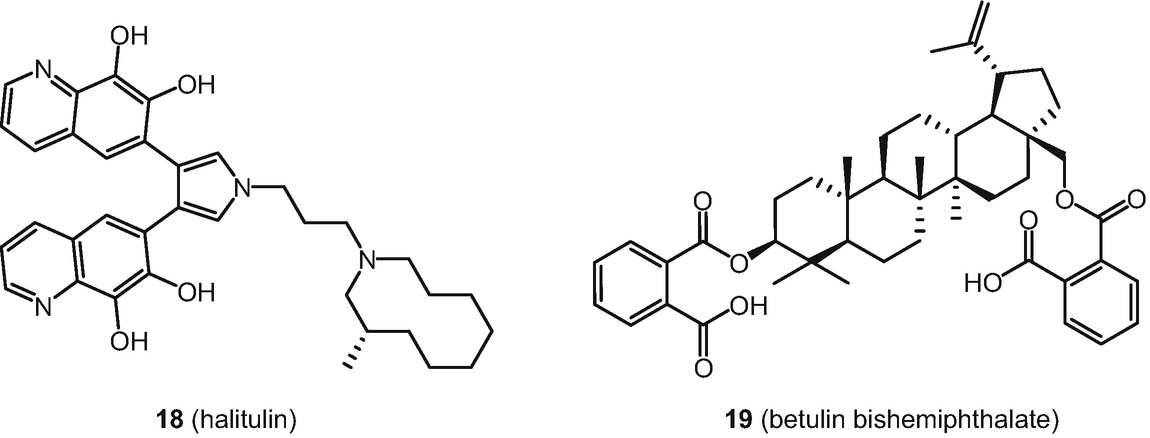
Examples of a natural product 18 and a natural product derivative 19 studied with PASS
Considering the intricate frameworks in natural products and their dissimilarity to those entailed in synthetic molecules in reference datasets, one may argue that fingerprints and substructural descriptors are suboptimal to leverage confident target predictions in natural product space. Indeed, SEA and PASS were designed for synthetic entities, and may afford less accurate predictions than software tools tailored for natural products. To mitigate this limitation, the Chemically Advanced Template Search (CATS) computes topological pairwise correlations of atom types in a given molecule, up to a distance of 10 bonds [70, 71]. This simple pharmacophore descriptor provides a fuzzy and size-independent molecular representation, which has proven well suited for scaffold hopping and correlation of structurally dissimilar chemical entities. According to the CATS descriptors, feature pairs are expressed as the number of bonds along the shortest path connecting two non-hydrogen nodes in the molecular graph. Atoms are typed as one of six possible features: hydrogen bond donor, hydrogen bond acceptor, positively charged, negatively charged, lipophilic, and aromatic, resulting in a 210-dimensional vector (21 feature combinations × 10 bonds) that can be employed to predict drug targets.

Schematics of the SPiDER method workflow
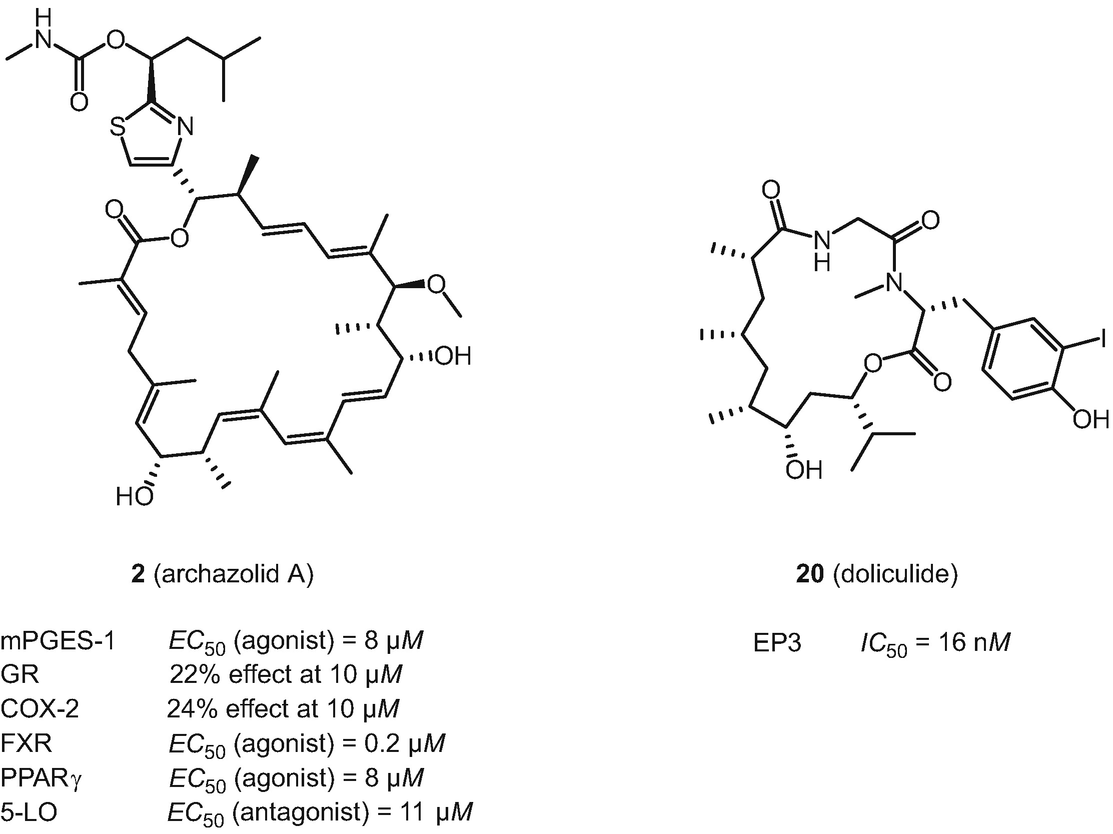
Structures of archazolid A (2) and doliculide (20) and bioactivities identified by SPiDER
Structures of fragment-like natural products deorphanized with SPiDER

(–)-Englerin A (24) and piperlongumine (25) display pharmacophore feature commonalities that allow cross-structure target inference. Cyan = hydrogen bond donor/acceptor; green = lipophilic; orange = aromatic and/or sp2 hybridized

Structures of natural products that have been studied by the TIGER method
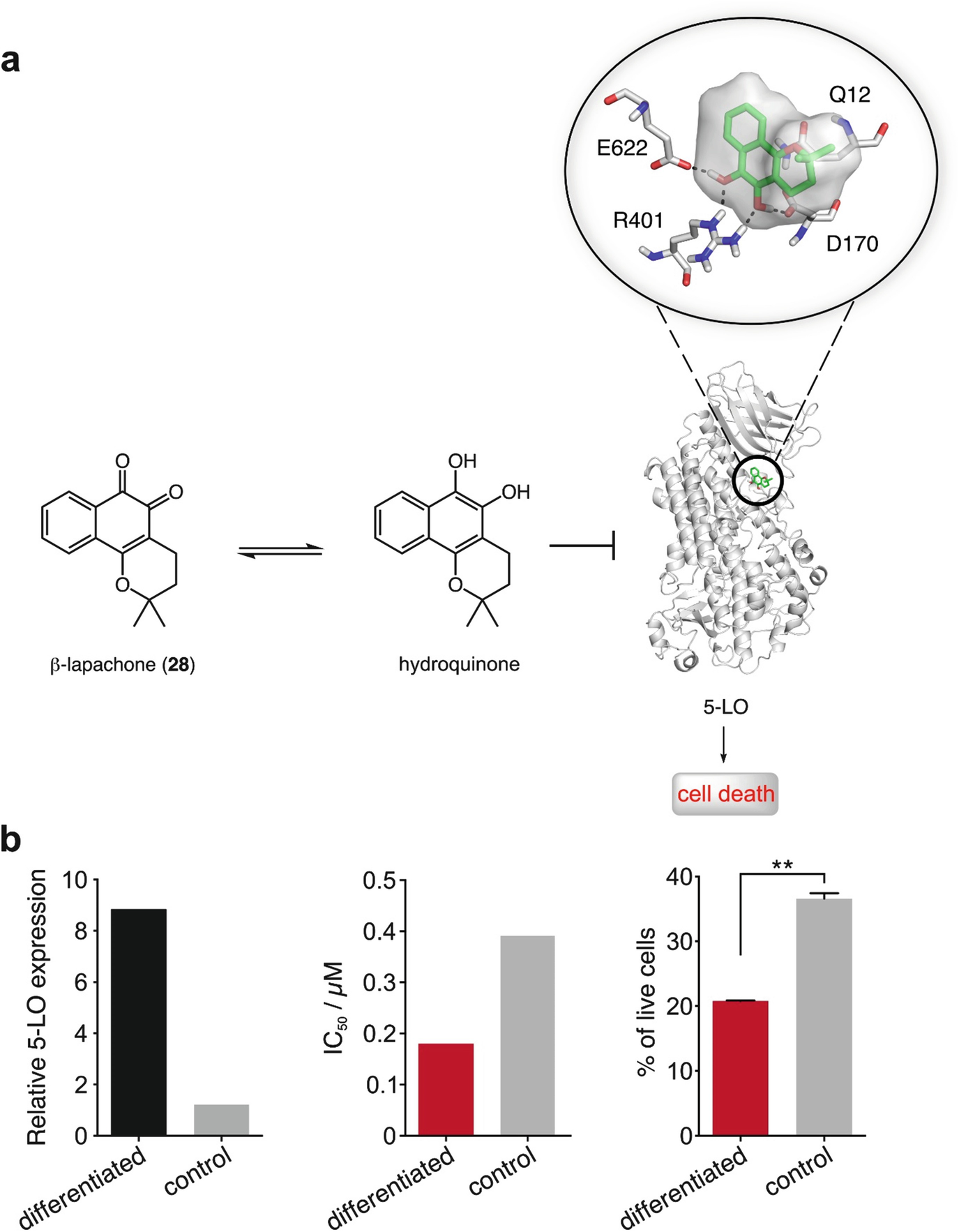
Mechanism of anticancer activity of β-lapachone (28). (a) Natural product 28 is converted in the intracellular compartment to the corresponding hydroquinone, which is a potent, reversible, allosteric inhibitor of 5-lipoxygenase (5-LO). (b) Differentiated HL-60 cell line overexpresses 5-LO (left) and are more sensitive to 28 (middle and right). IC 50 (differentiated) = 0.18 μM; IC 50 (control) = 0.39 μM (middle). Percentage of live HL-60 cells in the differentiated and control groups when treated with 0.5 μM of 28. ∗∗p < 0.005 (two-tailed t-Student test)
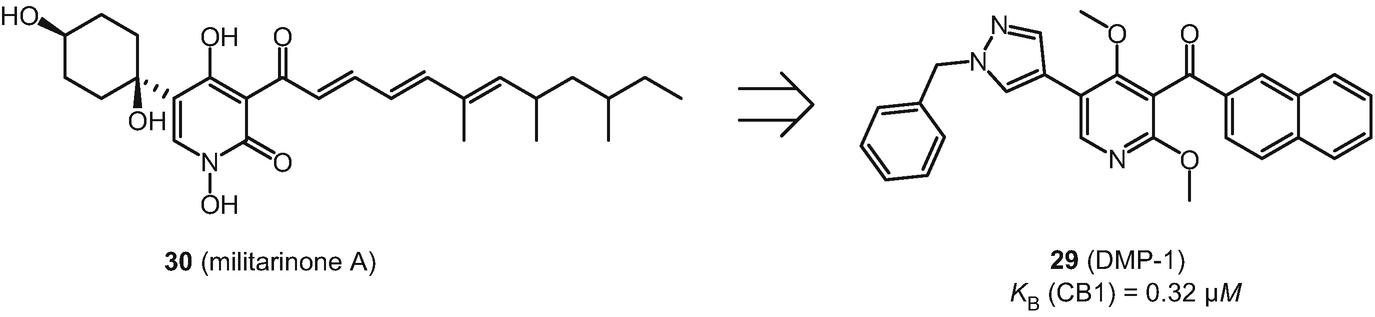
Structure of militarinone A (30) and its derivative DMP-1 (29)
6 Outlook
Target identification and deconvolution of phenotypic readouts is an important step in early discovery programs. While this is a challenging task for synthetic small molecules, the difficulty is typically magnified for natural products, given the poorer synthetic accessibility and troublesome derivatization tendencies. However, with such knowledge in hand, developing bioactive natural products and designing analogues may be facilitated and assisted by state-of-the-art computational technologies. In this contribution, different in silico methods that can be of utility to unveil pharmacology of natural products have been discussed, and in a broader sense any small molecule of interest, by generating motivated research hypotheses for confirmation in biochemistry laboratories.
There is no universal best method and both 3D and 2D approaches can be deployed efficiently by keeping in mind their caveats and limitations. Still, any computational method is certain to fail occasionally even when properly employed, but more often when applied outside its domain of applicability. However, there is compelling evidence that the accuracy and scope of computational methods are improving considerably. This offers great prospects for more successful case studies in the deconvolution of modes of action and biochemical liabilities of natural products. Much of the current enthusiasm is spearheaded by the emergence of big data, faster computers, and more efficient algorithms for pattern recognition, which parallels the need for sustainable drug discovery. Machine learning is primed to analyze large volumes of data; these algorithms will equally benefit from high-quality negative data, which historically tends to be neglected. With the rise of digital chemistry, it is expected that laborious tasks such as target identification will be increasingly automated, thus opening new avenues for probabilistic drug discovery.
Acknowledgments
Tiago Rodrigues is a Marie Skłodowska-Curie Fellow (Grant 743640) and acknowledges the FCT/FEDER (02/SAICT/2017, Grant 28333) for funding.