5
Meltdown of the Cellular Power Plants
![]()
The cellular components—“organelles”—known as mitochondria play a large role in aging, hand in hand with reactive chemicals known as free radicals. When I came into the field in the mid-1990s, however, this role was not clearly defined; evidence and interpretations were contradictory and awaiting synthesis. In response, I developed what is now a widely accepted mitochondrial free radical theory of aging. Read on: In order to understand aging, you must understand a little of the way in which our cells work.
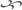
![]() In Chapter 4, I explained that there are seven major classes of lifelong, accumulating “damage” that we must address if are to uncouple their causes—the processes of life—from their eventual consequences, the pathology of aging, and thereby prevent those consequences. Six of those seven are the subject of one chapter each in this part of the book—Chapters 7 through 12. But the first one I’m going to address, mitochondrial mutations, is going to take two chapters. That’s because the question of whether mitochondrial mutations matter at all in aging is actually a really complicated one, and we must do our best to answer it in order to know whether we need to worry about them. Remember that in Chapter 4, I briefly noted that SENS does not incorporate a plan to address mutations in our cell nucleus at all unless they cause cancer, because non-cancer-causing mutations accumulate too slowly to matter in a normal lifetime. (I will explain this logic much more thoroughly in Chapter 12.) A lot of gerontologists feel that way about mitochondrial mutations. I disagree with them, so I need to tell you why. For each of the other six SENS damage categories, by contrast, there’s no argument: at least one of the major pathologies of aging is clearly caused or accelerated by that type of damage. So those six categories will only require one chapter each, focusing mainly on the solution and with a relatively brief description of why there’s a problem to solve.
In Chapter 4, I explained that there are seven major classes of lifelong, accumulating “damage” that we must address if are to uncouple their causes—the processes of life—from their eventual consequences, the pathology of aging, and thereby prevent those consequences. Six of those seven are the subject of one chapter each in this part of the book—Chapters 7 through 12. But the first one I’m going to address, mitochondrial mutations, is going to take two chapters. That’s because the question of whether mitochondrial mutations matter at all in aging is actually a really complicated one, and we must do our best to answer it in order to know whether we need to worry about them. Remember that in Chapter 4, I briefly noted that SENS does not incorporate a plan to address mutations in our cell nucleus at all unless they cause cancer, because non-cancer-causing mutations accumulate too slowly to matter in a normal lifetime. (I will explain this logic much more thoroughly in Chapter 12.) A lot of gerontologists feel that way about mitochondrial mutations. I disagree with them, so I need to tell you why. For each of the other six SENS damage categories, by contrast, there’s no argument: at least one of the major pathologies of aging is clearly caused or accelerated by that type of damage. So those six categories will only require one chapter each, focusing mainly on the solution and with a relatively brief description of why there’s a problem to solve.
![]() Free Radicals: A Brief Primer
Free Radicals: A Brief Primer
Almost everyone has heard of free radicals by now. Their involvement in aging is asserted so often and so confidently in popular press articles—especially articles trying to promote the latest “antioxidant” nutritional supplement—that you’d think the matter was done and dusted. As we’ll see, however, the exact roles played by free radicals in the aging process—and the best ways to deal with the problems they cause—are a lot more complicated, and more controversial, than these articles let on.
Free radicals in biology are, for the most part,1 oxygen-based molecules that are missing one of the electrons in their normal complement. Electrons are charged particles that surround the central nucleus of the atom, and they occupy well-defined locations (you can think of these as distances from the nucleus) termed orbitals. By their nature, molecules can only remain chemically stable when each of the electrons in the orbitals of their constituent atoms has a paired twin to complement it; an orbital with only one electron is unstable. So when a molecule loses one half of an electron twosome, it becomes chemically reactive until it gets that electron back. Usually, the free radical’s stability is restored when it tears an electron out of the nearest available normal, balanced molecule—but with this electron stolen from it, that second molecule generally loses its chemical stability, and will seek in turn to restore its balance by a similar theft. It’s a chain reaction.
Some unusual molecules—antioxidants—finesse this logic and are relatively stable even when they contain an unpaired electron. These molecules can “quench” free radical chain reactions. Until they do, however, free radicals will tear their way through your body like biochemical vandals, trashing whatever essential biomolecule they bump into: the structural proteins that make up your tissues, the fatty membranes that compartmentalize and facilitate your cells’ various specialized functions, the DNA code that holds the blueprints of the enzymes and proteins required by the cell, and so on. In biology, function follows structure, so the ability of these molecules to support metabolism and hold you together is impaired when they are chemically deformed by this process.
This is obviously not good for you—and unfortunately, it’s unavoidable. Free radicals are part of being alive.
While popular press articles on aging often give the impression that free radicals come mostly from environmental pollutants or toxins from an impure diet, the fact is that the overwhelming majority of the free radicals to which your body is exposed are generated in your very own cells—in the mitochondria, our cellular “power plants.” Mitochondria are one of several types of “organelle,” or self-contained cellular component that exists outside the nucleus. Each cell has hundreds to thousands of mitochondria. Man-made power plants take energy that is locked away in an inconvenient form of fuel—such as coal, natural gas, the strong nuclear force which holds atoms together, or wind—and convert it into a more convenient medium, electricity, which you can use to run your blender or computer. In just the same way, mitochondria convert a difficult-to-use energy source (the chemical energy locked up in the glucose and other molecules in your food) into a more convenient one: adenosine triphosphate, or ATP, the “universal energy currency” that your cells use to drive the essential biochemical reactions that keep you alive.
Mitochondria generate most of their cellular power using almost identical principles to the ones used by hydroelectric dams—right down to the turbines (see Figure 1). Using a series of preliminary biochemical reactions (each of which generates a small amount of energy), energy from food in the form of electrons is transferred to a carrier molecule called NAD+ (and a similar one called FAD). These electrons are used to run a series of “pumps” called the electron transport chain (ETC) that fill up a “reservoir” of protons held back by a mitochondrial “dam” (the mitochondrial inner membrane).
The buildup of protons behind the “dam” creates an electrochemical force that sends them “downhill” to the other side of the mitochondrial inner membrane, just as water behind a dam is drawn downward by gravity. And just as a hydroelectric dam exploits the flow of water to run a turbine, the inner membrane contains a quite literal turbine of its own called “Complex V” (or the “Fo/F1 ATP synthase”) that is driven by the flow of protons. The rushing of protons through the Complex V turbine causes it to spin, and this motion is harnessed to the addition of phosphate ions (“phosphorylation”) to a carrier molecule (adenosine diphosphate, or ADP), transforming it to ATP.
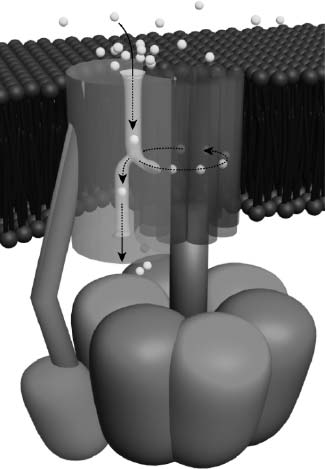
Figure 1. The Fo/F1 ATP synthase.
Unlike hydroelectric dams, however, the use of chemical energy in food to generate ATP via this system is a chemical reaction. As with the burning of coal or wood to release energy, the powering-up of ADP into ATP consumes oxygen, which is why we have to breathe to keep the whole system running: oxygen is the final resting place of all those electrons that are released from food and channelled through the proton-pumping electron transport chain. Thus, the whole cycle is called oxidative phosphorylation (OXPHOS).
But while hydroelectric dams are (for the most part) environmentally benign, mitochondria are in one key aspect more like conventional power sources. Just like coal or nuclear power plants, mitochondria create toxic wastes during the conversion of energy from one form into another. As the proton-pumping complexes of the electron transport chain pass electrons from one to the next, they occasionally “fumble” an electron here or there. When this happens, the electron usually gets taken up by an oxygen molecule, which suddenly finds itself with an extra, unbalanced electron. (I just mentioned that oxygen is also the sink for the electrons that are not fumbled—that are properly processed by the mitochondria—but that process loads four electrons onto each oxygen molecule, not just one, so there’s no problem of electron imbalance.) Adding one electron, by contrast, transforms benevolent oxygen into a particularly important free radical, superoxide. With your mitochondria generating ATP day and night continually, the ongoing formation of superoxide is like having a constant stream of low-grade nuclear waste leaking out of your local reactor.
Once scientists established that mitochondria were the main source of free radicals in the body, it was quite quickly realized that these organelles would also be their main target. Free radicals are so rabidly reactive that they never travel far, attacking instead the first thing that they come across—and the mitochondria themselves are at ground zero. And there are plenty of potentially sensitive targets for these radicals in the mitochondrion. Free radicals produced in the mitochondria are right next to the very membranes and proteins on which ATP production depends, and also within spitting distance of the mitochondrial DNA. What’s that, you say? Well, whereas other components of the cell have all their proteins coded for them by the cell’s centralized genetic repository in the nucleus, mitochondria have their own DNA for thirteen of the proton-pumping, ATP-generating proteins in their membranes.2 If that DNA is significantly damaged, the mitochondrial machinery will go awry. Unfortunately, it’s clear that mitochondrial DNA does suffer a lot of self-inflicted damage, taking as much as a hundredfold more initial oxidative “hits” than the cell’s central, nuclear DNA, and suffering many times more actual, enduring mutations with age.
Starting with a classic paper put out in 1972 by chemist Denham Harman3 (who already had the distinction of being the father of the original “free radical theory of aging”), researchers put these facts together with a range of experimental findings and came up with several variations of a “mitochondrial free radical theory of aging.” Let’s briefly survey that experimental evidence.4
First of all, there was evidence from comparative biology. Slower-aging organisms, relative to faster-aging ones of similar size and body temperature, are always found to have slower mitochondrial free radical damage accumulation. They produce fewer free radicals in their mitochondria; they have mitochondrial membranes that are less susceptible to free radical damage; and sure enough, they accumulate less damage to their mitochondrial DNA. Calorie restriction (CR)—the only nongenetic intervention known to slow down aging in mammals—improves all of these parameters: it lowers the generation of mitochondrial free radicals, toughens their membranes against the free radical assault, and above all it reduces the age-related accumulation of mitochondrial DNA mutations—the irreparable removal or overwriting of “letters” in the genetic instruction book.
Leading toward the same conclusion from the opposite direction, CR does slow down aging, yet it has no consistent effect on the levels of most self-produced antioxidant enzymes. The enzymes that people examined in this regard in the 1980s were ones that are found predominantly in the rest of the cell, not in the mitochondria. This again suggests that free radical damage outside of the mitochondria is not a directly important cause of aging, since aging can in fact be slowed down (via CR) without doing the one thing that would most directly interdict that damage.
Fast-forward, for a moment, to 2005. In that year, the most direct evidence so far on this point came to light. It involved mice that had been given genes allowing them to produce extra amounts of an antioxidant enzyme (catalase), specifically targeted to different parts of their bodies.5,6 There was little to no benefit provided by giving these organisms catalase to protect their nuclear DNA—the genetic instructions that build the entire cell and determine its metabolic activity, except for those parts that the mitochondria code for themselves. And there was also no benefit observed from targeting catalase to organelles called peroxisomes, which are involved in processes that produce hydrogen peroxide (the molecule that catalase detoxifies) and accordingly are already stoked up with the enzyme. Yet, delivering catalase to the animals’ mitochondria, which significantly reduced the development of mitochondrial DNA deletions, extended their maximum lifespan by about 20 percent—the first unambiguous case of a genetic intervention with an effect on this key sign of aging in mammals.
These mice were no more than a twinkle in their creators’ eyes a decade ago, when I first addressed the question of mitochondrial oxidative damage. But even back then, it seemed unassailable that free radical damage to the mitochondria was a key driver of aging. The question was: what linked the one to the other?
That might sound like a stupid question, given that free radicals are obviously toxic, but it turned out to be decidedly tricky to come up with a coherent, detailed, mechanistic explanation for the connection. Scientists convinced that mitochondrial free radicals play a role in aging all begin with the undeniable observation that free radicals spewing out from the mitochondrion damage the membranes and proteins that it needs to generate ATP, and also cause mutations in the mitochondrial DNA that codes for some of those same proteins. But any such theory must explain how this self-inflicted damage contributes to the progressive, systemic decay that constitutes biological aging. Until recently, nearly all such theories postulated the existence of some form of mitochondrial “vicious cycle” of self-accelerating free radical production and bioenergetic decay.7,8
In these highly intuitive schemes, the mitochondrion is pictured to be like a hydroelectric dam whose turbines become rusted, worn, or broken by the forces to which they are subjected every day. Thanks to free radical damage to their membranes, proteins, and DNA, mitochondria in cells throughout the body would become progressively less able to pump protons and to keep a lid on the “hot potato” chain of ATP synthesis with age, leading to inefficient energy generation and increased production of free radicals as more and more electrons escape from increasingly banged-up transport complexes. Thus would begin the “vicious cycle,” as more and more renegade electrons tore into more and more mitochondrial constituents, leading to further damage to those same constituents, causing yet more inefficient, dirty energy generation, and so on and so forth, spiralling downward and ultimately starving the cell of energy and rendering it a hazardous waste site. See Figure 2.
Some version of this scenario is repeated in nearly all popular books and articles about the role of mitochondria in aging, as well as most scientific journal publications. Yet we’ve had evidence for nearly twenty years showing conclusively that it can’t be right.
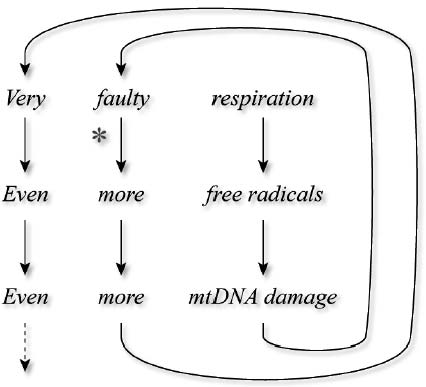
Figure 2. The “vicious cycle” theory of mitochondrial mutations accumulation. The key tenet of the theory is denoted by the asterisk: that typical mitochondrial mutations raise the rate of release of free radicals.
![]() Everything You Know Is Wrong
Everything You Know Is Wrong
The “vicious cycle” theory of mitochondrial decay paints a picture that sounds seductively plausible, but it just isn’t compatible with the data. While many scientists remain oblivious to the glaring inconsistencies in these theories even today, a few mitochondrial specialists and biogerontologists have been pointing these problems out since the mid-1990s. So different were the predictions of “vicious cycle” theories from the experimental findings that many of these researchers went so far as to suggest that the findings simply ruled out a role for mitochondria free radicals in aging—forgetting that such a role might exist but not via a vicious-cycle mechanism.
The first problem with at least some versions of the vicious cycle theory had actually been pointed out decades earlier—just after Harman had originally put forward the first version of the mitochondrial free radical theory, in fact—by none other than Alex Comfort. (Yes, that’s the same Alex Comfort who wrote The Joy of Sex. He was a true polymath: he was also a controversial anarchist agitator, a poet, and a highly distinguished biogerontologist.)
In 1974, Comfort pointed out that while each mitochondrion could temporarily suffer progressively increasing damage to the proteins and membranes that make up its ATP synthesis machinery from the ongoing free radical barrage, no theory based on the idea that this damage would get worse and worse with age could fly, for the simple reason that the cell is constantly replacing and renewing those very components.
Periodically, old mitochondria are tagged for destruction in yet another type of organelle, the lysosome (the cellular “toxic waste incinerator,” about which I’ll be talking a great deal in Chapter 7). Then, to make up for the loss of energy factories, the cell puts out a signal for the remaining mitochondria to replicate themselves. During replication, each mitochondrion duplicates its DNA, and then that essential core “grows” itself a new “body,” including pristine new proton-pumping proteins and membranes. Whether you’re five years old or fifty, any given mitochondrion in your cells contains membranes and proteins that are on average only a few weeks old. Thus, the newest and the oldest of these mitochondrial components are present in the same proportions in the very old as in the very young. It just can’t be that aging is driven by a progressive process of degeneration in components that undergo a continuous process of renewal.
However, this objection wasn’t necessarily a problem for the more popular versions of the vicious cycle theory: those that assert that free radical damage to the mitochondrial DNA drives aging. Although mitochondrial membranes and proteins are periodically replaced, all mitochondria inherit their DNA directly from their “parent” power plants, which faithfully copy out their DNA and pass it on to their “children”—and just as whole organisms pass on any mutations that they may harbor in their DNA to their offspring, so errors in “parent” mitochondria appear in the next mitochondrial generation. If the mitochondria that have damaged DNA are preferentially destroyed by lysosomes, the effect will be as before—damage will be removed as fast as it spreads—and that’s what Comfort reasoned would be occurring. But if there’s no such bias in which mitochondria are and are not destroyed, DNA damage will accumulate.
These theories received a superficial plausibility boost from studies conducted in the 1990s, which showed that aging bodies do, indeed, accumulate cells populated with mutant mitochondria. However, the same studies shot entirely new—and even more deadly—holes into mitochondrial DNA-based “vicious cycle” theories.
For one thing, it was found that all of the mutant mitochondria in a given cell contain the same mutation. This is exactly the opposite of what the “vicious cycle” would predict. If each mitochondrion individually decayed as a result of a self-accelerating cycle of oxidative “hits” to its DNA, then each one would display an unique, random profile of mutations. In the same way, if one day—independently—two disgruntled librarians were each to snap, going on automatic rifle rampages through the collections in their care, you would naturally expect to see that the bullets would have hit different books, even if the collections themselves were the same: one would have put a bullet straight through the spine of a copy of Finnegans Wake, while the other would have punched an off-center dot atop the “i” in Bridget Jones’ Diary, and so on, at random, until each mad bibliophile ran out of bullets.
Instead, wherever cells that contain defective mitochondria are found, the mutants all harbor an identical DNA mutation. It’s as if librarians across the state had marched into work and each fired his or her guns exactly once, in every case into copies of The Catcher in the Rye. Random mutations happening continuously in each mitochondrion cannot reasonably result in each of the damaged mitochondria in a cell containing the same error in their DNA; a random mutation-creating process can’t explain the complete takeover of cells by such mitochondria, or the presence of other cells that contain nothing but healthy ones.
In fact, it’s even weirder than that, because the presence of mutant mitochondria turns out to be an all-or-nothing affair. That is: near enough, not only do all of the mutant mitochondria in a given cell turn out to contain the exact same mutation, but those cells that harbor any damaged mitochondria are found to contain nothing but mutants—while the other cells have nothing but pristine, youthful “power plants.”
But wait: the findings are even more bizarre yet. While each cell is full of mitochondria that all bear the same mutation, the mitochondria in different cells contain different mutations. It’s as if all of the librarians in Delaware had pumped their local branch’s copies of War and Peace full of lead, while their colleagues in California had simultaneously displayed an equally single-minded determination to purge their collections of Lady Chatterly’s Lover. It’s a case in which even the most skeptical conspiracy-theory debunker would be forced to admit that a “random act of violence” was not a credible explanation for the crime scene.
The nature of these mitochondrial mutations also proved to be inconsistent with vicious cycle theories of mitochondrial priority in aging. The assumption had been that damage to the DNA blueprints for mitochondrial proteins would most often result in minor defects in the instructions by which those proteins are coded. The ensuing proteins would be close enough to their proper structure to remain more-or-less capable of carrying on, but would be dysfunctional, “fumbling” more electrons into free radicals and generating less ATP. Instead, the mutations that accumulate in cells’ mitochondria were found to be overwhelmingly deletions of large blocks of DNA, which completely shut down the creation of all the mitochondrially-encoded proteins.
The vicious cycle theory proposed that mitochondria would make progressively less ATP and more free radicals as they accumulated more and more of these defects in their DNA. Instead, it was found that mutant mitochondria produce essentially no free radicals, and that the change in each mitochondrion could be attributed to a single, catastrophic event, rather than to a “death from a thousand cuts.”
![]() Forget the Quality, Feel the Quantity
Forget the Quality, Feel the Quantity
An additional finding seemed to leave no room for any way, vicious or otherwise, in which mitochondrial mutations could be involved in aging: very, very few cells actually contain mutant mitochondria at all. The vast majority of cells remain in perfect mitochondrial health well into old age. Yes, a tiny proportion of older people’s cells—about 1 percent—could be shown to be completely taken over by mitochondria that all suffer from an identical defect in their DNA; but 99 percent of cells were fine. How could 1 percent of cells matter?
Many biogerontologists concluded that these findings put the kibosh on any theory that asserted that mitochondrial decay was important in aging. If nearly every cell in the body still enjoys the same level of ATP output as it had in its youth, and suffers no more free radical damage than it did in its prime, how could a tiny proportion of cells that are low on power, but whose mitochondria produce no more free radicals than their neighbors—in fact, produce no free radicals at all—possibly have much negative effect on the function of the tissue in which they reside or the organism as a whole? To these scientists, the mitochondrial free radical theory of aging seemed dead in the water.
This was where the field sat in the mid-1990s, when I first became aware of the unsatisfactory state of aging science and decided to try to do something to change the situation. When I saw the confused state of the mitochondrial free radical theory of aging, I felt that the field looked ripe for a new synthesis. On the one hand the evidence supporting the existence of a central role for mitochondrial free radicals in aging seemed strong; on the other hand, the standard vicious cycle theories simply could not be reconciled with the emerging results. It was into this fray that I stepped with my first formal scientific papers in 19979 and 1998,10 and where I have made my most widely acknowledged contributions to biogerontology. My key insights were, firstly, an explanation of how aging cells accumulate mitochondria that share one mutation in common instead of the random set predicted by the vicious circle theory, and secondly, an explanation of how only a small number of cells taken over by such mutant mitochondria could drive aging in the body as a whole. Let’s walk through these ideas one at a time.
![]() SOS: Survival of the Slowest
SOS: Survival of the Slowest
The vicious cycle theory had assumed that each mitochondrion would slowly accumulate minor, random mutations over the course of its lifetime. The fact that, in those cells where there were mutant mitochondria, all the mutants shared the same mutation—and that the mutants had completely replaced all healthy mitochondria in the cell—proved that assumption wrong.
The only reasonable alternative seemed to be “clonal expansion”: the idea that a single mitochondrion had originally gone bad, and that its progeny had slowly taken over the entire cell. Remember, mitochondria reproduce themselves by splitting themselves in half, much like amoebae: the original mitochondrion makes a copy of its DNA, and then forms two identical genetic “clones” of itself. This means that each clone will contain exact copies of any mutations present in the original organelle. So it seemed inescapable that the strange mitochondrial monocultures found in the all-mutant cells were the result of one mitochondrion initially acquiring a mutation, passing it on to its offspring, and then having its lineage somehow outcompete all of its neighbors until it eventually becomes the only game in town.
However, the idea that mitochondria with mutated DNA could somehow win a battle for dominance within the cell was itself a bit of a paradox. After all, these mitochondria are defective, with one or more enormous chunks blasted out of their DNA by free radicals or replication errors. While it’s true that once in a blue moon a mutation turns out to be beneficial—this is, after all, what allows evolution to happen—it’s supremely unlikely that it would happen over and over again, such that random mutations occurring in mitochondria in widely separated cells would turn out to be so beneficial to the mitochondrion as to give it a Darwinian “fitness advantage” over its fellows. And indeed, the mutations in question were known to be deleterious: they completely knock out the mitochondrion’s ability to perform oxidative phosphorylation, and thus turn off the great majority of their contribution to the cellular ATP supply.
The “clonal expansion” explanation was also hard to reconcile with the fact that many different mutations can cause a specific mitochondrial lineage to replace all other alternative lineages in the cell. That is: while the mutant mitochondria in a given cell all contain the same, specific mutation, a second such cell often contains mitochondria that all harbor a completely distinct mutation from the one that was found in the first. So it wasn’t that there is a single, specific mutation that gives the mutants their selective advantage over their neighbors: numerous mutations, independently arising in single mitochondria within widely separated cells, confer the same competitive edge. Was it really likely, I asked myself, that there were this many advantageous, yet unrelated, mutations to be had?
Yet, these various mutations do have one thing in common. They aren’t mild mutations, damaging just one protein: all of them are of a type that prevent the synthesis of all thirteen of the proteins that the mitochondrial DNA encodes. This shared property, I felt, might be the key to how they managed to take over the cell.
I set myself to thinking what would distinguish such mitochondria from their healthy counterparts. They would not generate nearly so much ATP, of course: only the small amount of cellular energy that gets produced in the initial stages of extracting chemical energy from food, which was a fraction of the total that a functioning oxidative phosphorylation system could churn out. This would be unhealthy for the cell, certainly, but I realized that it would have little negative impact on the mitochondrion itself, which normally exported nearly all of the ATP that it produced anyway. So, while I couldn’t see how this reduced energy output could explain the selective advantage enjoyed by the mutants, I saw that—contrary to what one might initially think—it really wasn’t a direct disadvantage relative to other mitochondria in the cell that might hinder them from rising to dominance in the host.
The other thing that would set mitochondria with no oxidative phosphorylation capacity apart from other mitochondria in a cell seemed more likely to be advantageous: such mitochondria would no longer be producing free radicals. Remember that mitochondrial produce free radicals when electrons leak out of the regulated channels through which they pump protons into the “reservoir” that drives the “turbines” of the mitochondrial inner membrane. If you aren’t feeding electrons into the pump system because the system itself is missing, then clearly there will be no leakage—and no free radicals.
Not having to deal with constant free radical vandalism sounded like it might be good for the mitochondrion—but it was not clear exactly how it might lead to an actual competitive advantage vis-à-vis the healthy mitochondria with which it was surrounded. True, its DNA would stop being bombarded—but of course, by this time it would already be suffering from a gaping hole in its DNA.
It was also obvious that the mitochondrion’s inner membrane would no longer suffer free radical damage—but again, it didn’t seem that this would matter in aging, since mitochondria are constantly having their membranes torn down and replaced anyway, either during replication or at the end of their brief individual lives, when mitochondria with defective membranes are sent off to the cellular “incinerator” in any case.
Now hang on a minute, I thought.
Like other researchers who had puzzled over this question, I’d been trying to imagine some improvement to the mitochondria’s function conferred by the mutation—the equivalent, in the microscopic evolutionary struggle, to sharper teeth, faster running, or greater fecundity. But what if, instead, the important thing about the mutation was not that it made its carriers “better,” but that it prevented them from being destroyed?
![]() Recycling Cellular Trash
Recycling Cellular Trash
There’s still a lot of work to be done to explain what exactly causes mitochondria to be sent to the cellular garbage disposal system. Still, even as early as Alex Comfort’s criticism of the original mitochondrial free radical theory of aging, it was widely believed that there was some selective process that specifically targeted old, damaged organelles for destruction. This could not be taken for granted, however. It was long believed that some components of the cell are turned over by an ongoing process of random recycling, in which the lysosome (strictly, a special sort of pre-lysosome called an autophagosome or autophagic vacuole) simply lumbers about the cell, swallowing a given number of various cellular constituents at random each day, so that everything is ultimately turned over sooner or later.
It is now widely accepted that this isn’t the way the lysosome works: the engulfment of proteins and other cellular components is known to be a highly directed process. In part, this is simply a matter of good use of scarce resources. Imagine if, in order to ensure that old, decaying vehicles were taken off the road (to improve the nation’s overall air quality and greenhouse gas emissions, eliminate the eyesore of old cars rusting on blocks, and bring down the price of recycled steel), the government were to send its agents wandering aimlessly through economically depressed neighborhoods to randomly select cars to be sent off to the scrap heap. Such a program would achieve some of its goals, but it would hit too many fully functional vehicles to make it viable, even setting aside the question of individual property rights.
But in some cases there is an even more powerful reason than mere efficiency to be sure that specific organelles get sent to the scrap heap. Some cellular components can become actively toxic to the cell if they aren’t quickly degraded once they’ve outlived their usefulness. Like the enchanted broom in Fantasia that continues to fill the vat in the sorcerer’s hall with water until it overflows and floods the room, many proteins and organelles are only useful for a limited period; when their job is done, they must be “put away,” and in the cell, this means torn apart for recycling. For instance, producing a pro-inflammatory enzyme can be critical to mobilizing an immune reaction against an invading pathogen, but leaving that enzyme to keep generating inflammation after the invader has been defeated would lead to a destructive, chronic inflammatory state with effects similar to those of autoimmune diseases like rheumatoid arthritis or lupus.
It then occurred to me that there were very good reasons for the cell to take care that its mitochondria were destroyed when their membranes had suffered free radical damage. Recall that the inner mitochondrial membrane acts as a “dam” to hold back the reservoir of protons that powers the energy-generating turbine of Complex V. Holes in that membrane would be “leaks” in the dam, depleting the reservoir as ions simply seeped through the holes without generating ATP. Evidence to confirm this basic scenario, in the form of leaks created from damaged membrane molecules, had actually been uncovered as early as the 1970s.11
This would make the “leaky” mitochondrion a serious drain on scarce resources, as the electron chain would continue to consume energy from food in a furious, futile attempt to refill the reservoir. Food-derived electrons would continue to be fed into the chain, which would use them to keep pumping protons across the membrane, but these ions would leak back across as fast as they were pumped “uphill,” without building up the electrochemical reservoir needed to create usable energy for the cell. This would drain the cell of energy, turning nutrients not into ATP but instead into nothing more useful than heat.
Moreover, the damage to the inner membrane might also allow many of the smaller proteins of the mitochondrial inner space to be released out of the mitochondrion and into the main body of the cell. If they continued to be active, these components could well be toxic to the cell when released from the controlled environment of the mitochondrion.
It would make sense, then, for the cell to have a system in place that would ensure that mitochondria are hauled off to the lysosome for destruction when their membranes become damaged by their own wastes. This prediction seems to have been fulfilled with the recent discovery of a specific targeting protein that “tags” yeast mitochondria for lysosomal pickup.12 We still don’t know for sure what makes the cell decide which mitochondria to “tag,” but it has now been shown that the formation of holes in the mitochondrial membrane does send a signal that increases the rate at which these organelles get sent to the scrapyard.13
I had no doubt that this was all to the good: I’m all for the removal of defective and potentially toxic components from the cell, and as usual nature has evolved an ingenious way to make sure that it happens. But I saw that, ironically, large deletions in the mitochondrial DNA would actually allow them to escape from the very mechanism that cells use to ensure that damaged mitochondria get slated for destruction. When mitochondria suffer the mutations that have been shown to accumulate with aging, they immediately cease performing oxidative phosphorylation (OXPHOS)—and with it, generating the resultant free radical waste. But reduced free radical production, in turn, should lead to less free radical damage to their membranes. Don’t forget that the prevailing vicious cycle theory proposed that mitochondrial mutations proliferate by causing their host mitochondria to make more free radicals than nonmutant mitochondria do. That, I saw, was where the proponents of the vicious cycle theory had gone wrong.
![]() Hiding Behind Clean Membranes
Hiding Behind Clean Membranes
Having advanced this far, I immediately saw how the mutants gained their advantage over their healthy counterparts. Even perfectly functional mitochondria constantly produce a steady, low-level stream of free radicals, leading to membrane damage. Every few weeks, this damage builds up to a level at which the mitochondria are consigned to the rubbish tip, and then the cell sends out a signal for a new round of mitochondrial reproduction to replace the decommissioned “power plants.”
But this process only weeds out mitochondria with damaged membranes—which will overwhelmingly be power plants whose DNA is still healthy enough to allow for the very electron transport that leads to the free radical damage to their membranes in the first place. Mitochondria with intact membranes, but damaged DNA, would not show outward signs of their internal injuries, and so would be passed over by the Angel of Death.
After a certain number of damaged mitochondria have been hauled off to the lysosome, the cell will send out the signal for mitochondria to replicate. Some or all of the remaining mitochondria—the genetically healthy and the mutants alike—will reproduce themselves, and because those mitochondria that bear large DNA deletions will almost always have survived the purge of outwardly damaged power plants, they will enjoy the opportunity to reproduce. But many of the membrane-damaged—but genetically intact!—mitochondria will already have been removed before replicating themselves. This will give the mutants a selective advantage over the nonmutants: every time replication happens, more and more of them will have survived a cull that has sent many of their genetically healthy competitors to the garbage disposal unit.
This is exactly how evolution works in organisms, of course. Animals that are slower runners, or less able to find food, or have poorer eyesight are more vulnerable to death from predators, exposure, or disease, which prevents them from successfully reproducing and passing on their genes. Meanwhile, a disproportionately high number of better-adapted organisms get the chance to breed, leaving behind progeny that carry their genetic legacy into the future. Over time, the genes that are best adapted to the specific threats in their environment come to dominate in the population.
In the cell, the threat to mitochondrial survival is the lysosome—a “predator,” which is supposed to ensure that only mitochondria fit to safely support cellular energy production survive. What the mutant mitochondria evolve (yes, evolve) is, in effect, camouflage that masks them from the eagle eye of this predator. Thanks to their undamaged membranes, these highly dysfunctional mitochondria appear healthy to the cellular surveillance system. Like the proverbial pharisees, their outsides are clean—but inwardly, they are full of ravening and wickedness.
I concluded that this “camouflaged mutant” concept provided the first consistent, detailed explanation for the takeover of cells by defective mitochondria. I named it “Survival of the Slowest” (SOS), because it postulates that the quiescent (“slow”) mitochondria enjoy a fitness advantage in the Darwinian fight for survival in the cellular jungle. See Figure 3.
But now, having explained how a small number of cells become a monoculture of defective mitochondria, there remained a question which might be thought to be more important—namely, how does that tiny fraction of the body’s cells drive aging across the entire body? It wasn’t long before I had a good explanation for that, too.
![]() The “Reductive Hotspot Hypothesis”
The “Reductive Hotspot Hypothesis”
The old vicious cycle models didn’t need to invoke any additional mechanism to explain how mitochondrial mutation could contribute to aging, because they assumed that there would be an accumulation of increasingly defective mitochondria in lots of cells with age. As more and more of their mitochondria randomly went on the blitz, the cells would suffer more and more oxidative hits, and be more and more starved for ATP, as the power plants’ efficiency sank with every new defect. It was a nice, common-sense explanation for the role of mutant mitochondria in the aging of the body.
But, as we’ve seen, it was also clearly wrong. Most cells in the body simply do not accumulate mutant mitochondria as we age: at most, about 1 percent of all of the trillions of cells in the body do so. Most cells and tissues suffer no decline in production or availability of ATP, and far from increasing free radical production, most of the mutant mitochondria that accumulate in the body produce no free radicals at all, because their radical-generating electron transport chains are simply absent.
It was hard to see how so few cells, containing mitochondrial mutants that were not harming nearby cells in any obvious way, could possibly be driving aging in the body. In fact, these findings were enough to make plenty of biogerontologists talk about the death of the mitochondrial free radical theory of aging. Yet, as we briefly discussed earlier in this chapter, the circumstantial evidence that mitochondrial mutations somehow contribute to aging is too strong to dismiss. To reconcile the two sets of data would require a truly novel solution to the puzzle.
Figure 3. The “Survival of the Slowest” model for mitochondrial mutation accumulation. (a) The proposed normal mode of turnover and renewal of non-mutant mitochondria; spots denote membrane damage. (b) The clonal expansion of mutations (denoted by X) resulting from low free radical damage to membranes and slow lysosomal destruction.
I saw that any refined version of the mitochondrial free radical theory of aging would have to do two closely related things. First, since so few cells are taken over by these burned-out power plants, it would have to show that cells harboring mutant mitochondria somehow spread toxicity beyond their own borders. And second, it would have to explain the nature of that toxicity, since the usual suspect—free radicals—appeared to have been ruled out by the fact that the mitochondria in these cells would have their normal free radical production turned off at the source.
I began by trying to work out just what cells that had been taken over by mutant mitochondria were doing to survive in the first place. What were they using as an energy source? Not only were these mitochondria unable to perform the oxidative phosphorylation that provides their host cells with the great majority of their ATP, but it was not at all obvious how they could produce any cellular energy at all.
![]() Upstream of a Blocked Dam
Upstream of a Blocked Dam
In normal cells, the initial metabolism of glucose from food is performed in the main body of the cell through a chemical process called glycolysis. Glycolysis generates a small amount of ATP, a breakdown product called pyruvate, and some electrons which can drive oxidative phosphorylation in the mitochondria. To shuttle electrons into the mitochondria for this purpose, they are loaded onto a carrier molecule called NAD+. The charged-up form of NAD+ is called NADH.
The pyruvate formed during glycolysis is also delivered into the mitochondria, where it is further broken down into another intermediate called acetyl CoA. This process releases some more electrons, which are again harvested for use in electron transport by “charging” NAD+ into NADH. Acetyl CoA is then used as the raw material for a complex series of chemical reactions called the tricarboxylic acid (TCA) cycle (also called the Krebs cycle or citric acid cycle), which liberates many times more electrons (again leading to creation of NADH) than has been created in previous steps.
Finally, all of the NADH charged up via all these processes—glycolysis, the breakdown of pyruvate into acetyl CoA, and the TCA cycle—is delivered to the electron transport chain, which uses this electron payload to generate the proton “reservoir” that drives the generation of nearly all the cell’s energy.
This was well-understood biochemistry, taught in its simple form to students in middle-school science classes. But it’s all predicated on being able to feed these electrons into the electron transport chain machinery. So, I asked myself, what would happen when that machinery was shut down, as it is in mitochondrially mutant cells?
It seemed to me that the whole process might grind to a halt. Every step along the way—from glycolysis to the TCA cycle—loads electrons onto waiting NAD+ “fuel tankers” for delivery to the electron transport chain. There is, of course, only a limited supply of NAD+ “carriers” available to be charged up into NADH, but normally that isn’t a big deal: there are always plenty of these carriers available, because NADH is recycled back into NAD+ when it releases its electron cargo to the electron transport machinery in the mitochondria.
But with that natural destination shut off, there is no obvious way for NADH to relieve itself of its burden of electrons. (Similarly, you can imagine how, if all the refineries on Earth were suddenly decommissioned, the taps on the world’s oil wells would quickly need to be turned off. With nowhere to deliver their oil for processing, fuel tankers could only be filled once before their capacity would be taken out of circulation, and continuing to pump oil would lead to a logistical nightmare.) And since every step of the process—glycolysis, intermediate metabolism of pyruvate into acetyl CoA, and the TCA cycle—needs NAD+ to proceed, a lack of NAD+ would be expected, at first glance, to lead to the deactivation of the entire process, leaving no mechanism for the cell to produce even the small quantities of ATP energy that result from these early processing steps.
In fact, I could see how mitochondrially mutant cells might be even worse off than this. NAD+ is required for a wide range of cellular functions unrelated to energy production—and each time these functions utilize NAD+, they not only reduce the pool of available NAD+, they also convert it to yet more NADH, further upsetting the cell’s metabolic balance. In fact, some researchers believe that many of the complications of diabetes are caused by an excess of NADH and a lack of NAD+, leading to disruption of these various metabolic processes (although the imbalance of NAD+ and NADH in diabetics has different causes than the loss of OXPHOS capacity).
Despite all this, however, cells that have been taken over by mutant mitochondria do survive, as is shown by their gradual accumulation with aging. So they have to be getting ATP from somewhere. At the time I was exploring this matter, the general presumption in the field was that these cells could survive by shutting down the TCA cycle and relying entirely on glycolysis for energy production. This is what happens in muscle cells as a brief stopgap during intense anaerobic exercise, when the cell is working so hard that it uses up all of the available oxygen and can’t keep oxidative phosphorylation going. Glycolysis would provide the cell with a small but just adequate amount of ATP, and this school of thought suggested that the cell could deal with the resulting small excess of NADH through a biochemical process that converts pyruvate into lactic acid—the biochemical origin of the famous “burn” that weightlifters suffer during the very last possible rep in their set.
But I realized that this theory didn’t match the evidence. For one thing, the expected rise in lactic acid didn’t seem to happen. And even more bizarrely, rather than a shutdown of TCA activity (as one would expect because of the lack of the required free NAD+), enzyme studies had strongly suggested that mitochondrially mutant cells have hyperactive TCA cycles. So, I wondered, how do they keep this seemingly unsustainable process going?
![]() Learning from the Great Mr. Nobody
Learning from the Great Mr. Nobody
A big step toward understanding this phenomenon was achieved with the creation of so-called ![]() 0 (“rho-zero”) cells, whose mitochondria are completely lacking in DNA. This condition renders cells functionally very similar to cells that have been overtaken by mutant mitochondria, because the deletion mutations found in these mitochondria actually shut down the ability to turn any DNA instructions into functioning proteins. If your DNA can’t be decoded into usable blueprints, it might as well not be there, like instructions on how to build a bridge that are written in a language you don’t understand. So having these DNA deletions puts mitochondria in just the same condition as having no mitochondrial DNA at all.
0 (“rho-zero”) cells, whose mitochondria are completely lacking in DNA. This condition renders cells functionally very similar to cells that have been overtaken by mutant mitochondria, because the deletion mutations found in these mitochondria actually shut down the ability to turn any DNA instructions into functioning proteins. If your DNA can’t be decoded into usable blueprints, it might as well not be there, like instructions on how to build a bridge that are written in a language you don’t understand. So having these DNA deletions puts mitochondria in just the same condition as having no mitochondrial DNA at all.
One of the first things that scientists working with ![]() 0 cells discovered was that they did, in fact, quickly die—unless their surrounding bath of culture medium contained one of a few compounds which are not normally present in the fluid that surrounds cells in the body. Intriguingly, however, some of these compounds are unable to enter cells, which meant that whatever it was that these compounds do to keep cultured
0 cells discovered was that they did, in fact, quickly die—unless their surrounding bath of culture medium contained one of a few compounds which are not normally present in the fluid that surrounds cells in the body. Intriguingly, however, some of these compounds are unable to enter cells, which meant that whatever it was that these compounds do to keep cultured ![]() 0 cells alive, it must be something that can be accomplished from outside the cell. This fact made little sparks go off in my brain, because I was looking for a way to explain how cells that had been overtaken by a clonal brigade of mutant mitochondria could export some kind of toxicity outside themselves to the body at large. Might these compounds rescue
0 cells alive, it must be something that can be accomplished from outside the cell. This fact made little sparks go off in my brain, because I was looking for a way to explain how cells that had been overtaken by a clonal brigade of mutant mitochondria could export some kind of toxicity outside themselves to the body at large. Might these compounds rescue ![]() 0 cells by unburdening them of this same toxic material?
0 cells by unburdening them of this same toxic material?
And might this toxic material be none other than…electrons?
I immediately drew a connection between the predicted excess of NADH in cells that could not perform oxidative phosphorylation, and the dependence of ![]() 0 cells on the presence of the “detoxifying” compounds in their medium. What the mitochondrially mutant cell needed to do was to dump electrons, so as to recover some NAD+—and the “rescue” compounds for
0 cells on the presence of the “detoxifying” compounds in their medium. What the mitochondrially mutant cell needed to do was to dump electrons, so as to recover some NAD+—and the “rescue” compounds for ![]() 0 cells were all electron acceptors, and they worked even if they were kept outside the cell’s boundaries. My hypothesis: Mitochondrially mutant cells prevent a crippling backlog of unused electrons by exporting them out of the cell, via a mechanism similar to that which is essential to the survival of
0 cells were all electron acceptors, and they worked even if they were kept outside the cell’s boundaries. My hypothesis: Mitochondrially mutant cells prevent a crippling backlog of unused electrons by exporting them out of the cell, via a mechanism similar to that which is essential to the survival of ![]() 0 cells in culture—and this export somehow spreads toxicity to the rest of the organism.
0 cells in culture—and this export somehow spreads toxicity to the rest of the organism.
![]() The Safety Valve
The Safety Valve
To turn this idea into a reformulation of the mitochondrial free radical aging theory, I needed clear answers to three questions. First, how were these cells delivering electrons to acceptors that were located outside their own membranes? Second, since the electron acceptors used in the ![]() 0 culture studies are normally not found in bodily fluids (or not at adequate concentrations), what electron acceptors are available to do the same job for mitochondrially mutant cells in the body? And third, could these processes provide an explanation for these cells’ systematic spread of toxicity throughout the body, as seemed required in order to accept that they might play a significant role in aging?
0 culture studies are normally not found in bodily fluids (or not at adequate concentrations), what electron acceptors are available to do the same job for mitochondrially mutant cells in the body? And third, could these processes provide an explanation for these cells’ systematic spread of toxicity throughout the body, as seemed required in order to accept that they might play a significant role in aging?
The first question turned out to have been answered already. For decades, scientists had known of the existence of an electron-exporting feature located at the cell membrane that we today call the Plasma Membrane Redox System (PMRS). While little was understood about its actual purpose in the body, its basic function was well established: it was known to accept electrons from NADH inside cells and to transport them out of the cell, thereby recycling the NADH to NAD+. This export allows even normal, healthy cells to have better control over the balance of chemically oxidizing and reducing factors within their boundaries, and to keep tighter control over the availability of NAD+ and NADH for essential cellular biochemistry. In other words, it does exactly what mitochondrially mutant cells would need to be able to do in order survive.
And the PMRS turned out to be an almost impeccable candidate for the job. PMRS researcher Dr. Alfons Lawen, of Monash University in Australia, had by this time already shown (with no thought of its application to aging, mind you) not only that the PMRS is able to deliver electrons to the same membrane-impermeant electron acceptors that allow ![]() 0 cells to survive, but that PMRS activity is required for the survival of these cells. This proved both that the export of electrons is a requirement for cell survival, and that the PMRS is the dock that sets them loose into the ocean of surrounding bodily fluids.
0 cells to survive, but that PMRS activity is required for the survival of these cells. This proved both that the export of electrons is a requirement for cell survival, and that the PMRS is the dock that sets them loose into the ocean of surrounding bodily fluids.
The ability of mitochondrially mutant cells to recycle NADH back to NAD+ would allow them to carry out their normal cellular processes, and without becoming so burdened with extra electrons as to create an internal environment in which other critical cellular chemistry becomes impossible. Moreover, I realized, this is a plausible explanation for the fact that these cells have an unusually active TCA cycle. With oxidative phosphorylation shut down, putting the TCA cycle into overdrive would allow the cell to double its production of precious ATP from sugars (and to do many other metabolic jobs in which the TCA cycle participates). The PMRS would make this possible, by recycling the greatly increased amounts of NADH that would be created and thereby providing the cell with the extra NAD+ required to keep the process going.
But the drastic increase in PMRS activity required to make the increased TCA activity sustainable would make the surfaces of mitochondrially mutant cells positively bristle with electrons undergoing export, forming a hotspot of electrically “reducing” pressure (i.e., an unstable excess of electrons). The next question, therefore, was onto what molecules the PMRS was unloading the electron surfeit. None of the electron acceptors being used to keep ![]() 0 cells alive in culture existed in adequate concentrations in the body to accomplish the task, so something else had to be performing this essential role. For example, some of the burden might be taken up by dehydroascorbate, the waste product of vitamin C that is created after it is used in quenching free radicals, but there wasn’t enough of that to deal with the powerful “reductive hotspot” created by these cells.
0 cells alive in culture existed in adequate concentrations in the body to accomplish the task, so something else had to be performing this essential role. For example, some of the burden might be taken up by dehydroascorbate, the waste product of vitamin C that is created after it is used in quenching free radicals, but there wasn’t enough of that to deal with the powerful “reductive hotspot” created by these cells.
At this point, an attractive candidate that could tie the story together flashed into my mind: our old two-faced friend, oxygen.
![]() Don’t Throw Your Junk in My Backyard, My Backyard…
Don’t Throw Your Junk in My Backyard, My Backyard…
Oxygen, of course, can and does absorb surplus electrons in its environment. As we discussed above, this is exactly how mitochondria generate free radicals during oxidative phosphorylation, as “fumbled” electrons slipping out of the electron transport chain are taken up by the oxygen dissolved in the surrounding fluid. And oxygen is the only such molecule in the body’s bathing fluids that is present in sufficient quantity to be able to sponge up the huge electron leak that mitochondrially mutant cells would be predicted to generate.
This uptake of electrons by oxygen could, in an ideal world, be safe: the PMRS might load four electrons onto each oxygen molecule, turning it into water, just as the electron transport chain does with electrons that it doesn’t “fumble.” But it was eminently possible that the PMRS would fumble some electrons—maybe quite a lot of them. If so, it would generate large amounts of superoxide radicals at the surface of mitochondrially mutant cells. The consequences of this would clearly be bad. But actually, maybe not so bad: one might think that the negative effects would mostly be confined to the immediate locality. Like nearly all free radicals, superoxide is highly reactive, and therefore short-lived. Either it would be dealt with by local antioxidants, or else it would attack the first thing with which it came into contact (a neighboring cell’s membrane, for instance)—but its aggressiveness would be quenched in the process. Superoxide certainly couldn’t remain a free radical for long enough to reach the far corners of the body, as the implications of the mitochondrial free radical theory required.
But what if, instead of attacking the components of a cell’s immediate neighbors, superoxide generated by the PMRS were to damage some other molecule that was then stable enough to be carried throughout the body? There was an obvious suspect: serum cholesterol, especially the LDL (“bad”) particles—low-density lipoproteins, to give them their full name—that deliver their cholesterol payload to cells all over the body.
Oxidized (and otherwise modified) cholesterol was already known to exist in the body, and everyone now accepts that it’s the main culprit behind atherosclerosis (a subject to which we’ll return in Chapter 7). It was quite plausible, I realized, that superoxide originating at the surface of mitochondrially mutant cells could be oxidizing LDL as it passed by, not only because LDL is ubiquitous and therefore an easy target, but also because the presence of loosely bound reactive metals such as iron ions would multiply superoxide’s potential virulence. One might think that the presence of antioxidants—such as the vitamin E that’s dissolved in LDL—would prevent this from happening, but researchers had already discovered that it didn’t. Not only is oxidized LDL found routinely in the body, but studies using the most accurate available tests of lipid peroxidation had shown that vitamin E supplements were unable to reduce the oxidation of fats in healthy people’s bodies.14 In fact, the lack of antioxidant partners in the inaccessible core of the LDL particle means that when it is subjected to anything more intense than the most trivial free radical challenge, the particle’s vitamin E can actually accelerate free radicals’ spread to its center through a phenomenon called “tocopherol-mediated peroxidation.”15 (Tocopherol is the technical name for vitamin E.)
I saw the light at the end of the logical tunnel now. The oxidation of LDL would provide a very plausible mechanism to explain the ability of mitochondrially mutant cells to spread oxidative stress throughout the aging organism. Despite its ability to promote atherosclerosis when present in excessive amounts in the blood, LDL cholesterol serves an essential function in the body. Cells need cholesterol for the manufacture of their membranes, and LDL is the body’s cholesterol delivery service, taking it from the liver and gut (where it is either manufactured or absorbed from the diet) out to the cells that need it.
But if its cholesterol consignment became oxidized by mitochondrially mutant cells along the way, LDL would become a deadly Trojan horse, delivering a toxic payload to whichever cells absorbs its cargo of damaged cholesterol. This would spread free radical damage into the incorporating cell, as the radicalized fats propagated their toxicity through the well-established chemical reactions that underlie the rancidity of fats. As more and more cells were taken over by mutant mitochondria with age, more and more cells would accidentally swallow oxidized LDL, and oxidative stress would gradually rise systemically across the entire body. See Figure 4.
![]() The New Mitochondrial Free Radical Theory of Aging
The New Mitochondrial Free Radical Theory of Aging
I walked myself through the entire scenario again and again, and gradually satisfied myself that I had indeed developed a complete, detailed, and consistent scenario to explain the link between mitochondrial free radicals and the increase in oxidative stress throughout the body with aging. This model answered all of my key questions and resolved all of the apparent paradoxes that had led so many of my colleagues to abandon the mitochondrial free radical theory of aging entirely. Mitochondrial free radicals cause deletions in the DNA. These mutant mitochondria are unable to perform oxidative phosphorylation, drastically reducing their production of both ATP and free radicals. Because they are not constantly bombarding their own membranes with free radicals, the cell’s lysosomal apparatus will not recognize them as defective and they will gradually drive out their healthy neighbors. (This was the maladaptive evolutionary process that I had already, a year previously, termed “Survival of the Slowest.”)
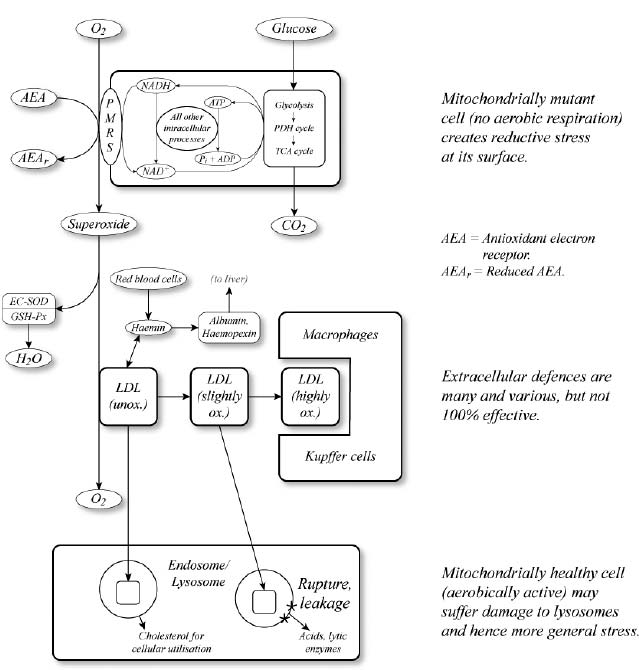
Figure 4. The “reductive hotspot hypothesis” for amplification of the toxicity of rare mitochondrially mutant cells.
To continue producing the ATP and other metabolites their host cells need for survival, these mitochondria must maintain the activity of their TCA cycle—but this, combined with other cellular processes in the absence of oxidative phosphorylation, will quickly deplete the cell of needed NAD+ carriers unless a way is found to relieve them of their electron burden. This is accomplished via the “safety valve” for excess electrons located at the cell membrane—the Plasma Membrane Redox System (PMRS).
The snarling buzz of electrons congregating at these cells’ outer surfaces turns them into “reductive hotspots,” generating a steady stream of superoxide free radicals. These radicals contaminate passing LDL particles, which then spread to cells far away in the body, driving a systemic rise in oxidative stress throughout the body with age. With oxidative stress comes damaged proteins, lipids, and DNA, as well as inflammation, disrupted cellular metabolism, and maladaptive gene expression. This could certainly be a central driver of biological aging.
The whole theory is unattractively elaborate, as you’ll have gathered, and as I immediately appreciated—but as far as I can tell, it’s the only hypothesis that can accommodate all of the data. I published the twin arms of the theory in rapid succession in the late 1990s, and a more detailed presentation of the integrated theory was accepted as my Ph.D. thesis for Cambridge and published in the Landes Bioscience “Molecular Biology Intelligence Unit” series.16 In the ensuing years the theory has enjoyed widespread appreciation and citation in the scientific literature. Unfortunately, despite this fact, both the popular press and many biogerontologists17 continue to cite the long-disproven vicious cycle theories either to support or to refute the role of mitochondria in aging, instead of seriously grappling with this detailed mechanistic account.
So now you know, in minute detail, my interpretation of why mitochondrial mutations are probably a major contributor to mammalian aging, and therefore why they are included as a category of “damage” in the definition of SENS. The question, then, is what to do about the toxic effects of mitochondrially mutant cells. The solution to this problem is the subject of the next chapter.