6
Getting Off the Grid
![]()
There are good reasons to believe that most present attempts to modify metabolism to produce less damage to mitochondria (and thereby to the body at large) are a poor use of resources. Fortunately, a better path forward exists, promising far greater results for the same application of time and money. It seems possible and plausible to prevent damage to mitochondria from harming us as we age—and scientists are already working on many options for the first steps of this process.
![]() In the previous chapter, I explained in excruciating detail my views about the complex mechanisms whereby mitochondrial DNA deletions may act as a major engine of aging. I must now tell you that in a very real sense, it simply does not matter if that hypothesis is correct or not.
In the previous chapter, I explained in excruciating detail my views about the complex mechanisms whereby mitochondrial DNA deletions may act as a major engine of aging. I must now tell you that in a very real sense, it simply does not matter if that hypothesis is correct or not.
This point applies equally strongly to the other SENS interventions, and it is so central to the engineer’s approach to anti-aging medicine—and so enormously counterintuitive—that I must beg your indulgence if you find me to be repeating it too often. We must all keep it in the forefront of our minds when thinking about these problems. If our purpose were simply to understand aging, then teasing apart the specific pathways that lead to the accumulation of age damage would indeed be absolutely imperative. But that is not our purpose. Our purpose is to put an end to aging’s consequences: the daily descent into decrepitude, and subsequent deaths, of tens of thousands of people.
Aging is a deadly pandemic disease, and I believe that our understanding of its mechanisms, while still highly imperfect, is now good enough that we are in a position to intervene in it. We need only to identify the nature of the damage itself—the accumulating lesions that are the source of age-related loss of functionality in the organism—and then either to reverse that damage, or to eliminate its threat to our health and life expectancy. This goal should become the central focus of biogerontological work, and the major target of biomedical funding generally.
The problem of mutant mitochondria is a case in point. Mitochondrial DNA mutations are a form of molecular disorder that distinguishes the biologically young from the biologically old, and there is powerful evidence that they are deleterious.1 So, whether mutant mitochondria take over their host cells via “Survival of the Slowest” or through some other mechanism, and whether these cells exert their toxic effects on the rest of the body by way of the export of electrons through the PMRS or via a completely unrelated process, the nature of the task at hand is ultimately the same. Our therapeutic goal is clear: either to fix the mutations themselves, or to make them functionally irrelevant. How to achieve that goal is the subject of this chapter.
Before laying out my proposals to accomplish this goal, however, I must first spell out why the appealing-sounding solutions that many biogerontologists would propose are probably wastes of time and scarce resources.
![]() You Can’t Stop a Moving Train (Safely!)
You Can’t Stop a Moving Train (Safely!)
I termed the “over-preventative” approach to combating aging the “gerontology” approach because biogerontologists predominently favor it. By and large, when my colleagues think seriously about actually doing something about aging rather than just refining their understanding of it, their first instinct is to find some way to make metabolism run more cleanly. After all, aging is the result of the accumulation of the deleterious by-products of our metabolic processes; surely, the thinking goes, if we could just tweak or dampen down those processes a little bit, we could reduce the exposure of the organism to metabolism’s reactive by-products, reduce the rate at which our cells and tissues accumulate microscopic damage over time, and thus slow down the gradual decay of our bodies into age-related frailty and accelerating vulnerability to death.
This approach has a strong intuitive appeal, reinforced by the continuous stream of encouragement from supplement vendors and public health authorities alike. We are constantly urged to clean up our lifestyles and practice preventive medicine: surely, we think, it makes more sense to put one’s energies into attempting to interfere with the causes of aging and age-related diseases than to try to undo an established molecular mess. But, as we saw in Chapter 3, the causes of aging lie in the fundamental chemistry of life, and our capacity to interfere beneficially with that chemistry is limited by what the organism’s underlying biology will accommodate.
I gave examples of this general principle in Chapter 3, but let’s now look at the more concrete case of intervention into the problem of mitochondrial mutations. The obvious, old-school approach would be to try to reduce the formation of mutant mitochondrial DNA by cutting back on the bombardment of the mitochondrial DNA by free radicals. Just such a trick has been pulled off with some success in mice2 by giving them a copy of the gene for the antioxidant enzyme catalase, specially targeted to their mitochondria. Catalase breaks down the free radical-like molecule hydrogen peroxide, turning it into harmless water before it can become more vicious and do serious molecular damage. Animals that received the targeted catalase gene enjoyed a fifty-fold jump in the activity of the enzyme within their mitochondria, preventing a great deal of mitochondrial DNA damage—including some of the mutations that initiate the whole destructive process of decay that I described in the last chapter.
Compared to the repeated, abject failures of dietary antioxidants to extend lifespan, or the ambiguous3,4,5 or negative6 results of previous attempts to hold back the free radical onslaught in mice using genetic manipulations, these results are quite impressive. Mice given mitochondrially-targeted catalase gained a 20 percent extension not only of average, but of maximum lifespan—the gold standard for data relevant to aging. And while no detailed analysis of the level of age-related pathology in these animals has been published, we do know that they suffer less age-related heart degeneration and fewer cataracts.
This being so, why should we not vigorously pursue the development of a targeted boost in mitochondrial catalase for humans? Well, in part this goes back to the testability (and, thereafter, the possibility of full clinical development) of this intervention. It suffers the same sorts of weaknesses on these fronts that characterize all “preventive” anti-aging medicines, as outlined in Chapter 3. Firstly, because there is no short-term disease against which extra catalase could be tested as a cure, regulatory bodies won’t let clinical trials be performed using it, and will never approve it for human use; and secondly, the timescales required to prove its effectiveness against aging make it too expensive and risky for venture capital to touch it. That probably means that no amount of agitation by scientists or the public will actually put mitochondrially targeted catalase into the hands of clinicians to save people’s youth, health, and lives: the interests of those with the power to fund or halt development are aligned against moving forward.
But even if these structural hurdles did not exist, I would still conclude that there are more fruitful soils into which to plow our limited resources for dealing with mitochondrial mutations. Boosting these mice’s catalase supply reduces the incidence of mitochondrial mutations—but it doesn’t eliminate or obviate them. Thus, it slows, but cannot treat, the progressive accumulation of this form of aging damage. I think we can do a lot better than this. My evaluation of the evidence indicates that it is not worth diverting our intellectual and financial capital into an intervention that might yield a 20 percent increase in lifespan (making the average person in a developed country live into his or her late nineties), because the same bricks, boards, and brains could be put to work in the development of an intervention that would not prevent this damage from happening, but would instead render it harmless. My analysis suggests that this intervention might, in turn, not only slash the rate of aging damage primarily caused by other mechanisms in half, but—when combined with a panel of similar therapies—would ultimately give us an indefinite youthful lifespan. More on that in Chapter 14.
In fact, if we were to eliminate, one by one, the other forms of molecular damage that cause us to wither and to die as we age, but were to “only” slow down the incidence of mitochondrial mutations by the degree seen in the mitochondrially targeted catalase mice, we might find ourselves stranded one step away from a future that stretches out further than our eyes can see. When advanced glycation endproduct (AGE)–breaking drugs reverse the gumming up of our structural proteins; when careful exploitation of the immune system clears out the extracellular junk that impedes cellular function; and when dormant cells that cause the decay of our immune system itself have been removed; at that time, with all the other key SENS platform interventions in our hands, we would still have mitochondria that are playing a game of Russian roulette with their DNA. However much more slowly they may be spinning the chamber, they could still end up being the weak link in a chain that could otherwise give us indefinite youthful lifespan.
We could apply these same objections to any approach to anti-aging biomedicine based on prevention of aging damage instead of genuine remediation. But there is a more specific objection to the “catalase solution.” Relying on an extra dose of catalase to deal with mitochondrial mutations could actually become harmful in important ways in a person whose body had already been cleared of—or rendered immune to—all of the other identified aging damage.
Catalase cleans up hydrogen peroxide, which can be damaging when it is the result of imperfections in oxidative phosphorylation—as most of it is. But evolution is, over the long term, an extremely clever engineer, and has learned ways of making the best of a bad job, harnessing hydrogen peroxide for its own purposes. While randomly spewing the stuff out of the cellular power plants does us no particular good, our cells also generate some hydrogen peroxide on purpose, for use as a chemical signal that regulates everything from glucose metabolism to cellular growth and proliferation.7 In this way, damping down the level of hydrogen peroxide in a cell—even using a technique designed to focus on the mitochondria—might well be expected to interfere with the functioning of our complex intracellular machinery.
One of the most dramatic examples of this potential problem is the need for hydrogen peroxide to reach the mitochondria themselves as part of the signaling cascade that triggers apoptosis, or “programmed cell death.” Apoptosis is important during embryonic development as part of a “remodelling” process that rids the nascent organism of excess cells that are only required during specific phases of its growth. But its main role in the body is similar to the self-destruct mechanisms built into James Bond’s Aston Martin or the Starship Enterprise: to give your cells a way to destroy themselves if they have been hijacked by “enemy forces” (viruses or cancer, for example) before they can threaten the integrity of the organism as a whole. When the cells of our immune system detect a hijacked cell, they bind to its surface, flipping a switch that signals its mitochondria to “blow their tops” and destroy it. Hydrogen peroxide is a player in that signaling system, and studies have shown that antioxidants—including catalase—can block the proper activation of this apoptotic program.
Thus, the massive boosting of mitochondrial catalase activity that is required to give these animals their partial protection from mitochondrial DNA damage has a dark side. And while it’s clear that the net effect of this boost is good for them—as evidenced by their extended lives and reduced age-related pathology—the overall balance of risks and benefits would in all probability be totally thrown off in an organism in which all other types of aging damage had been eliminated. In this otherwise rejuvenated body, a chronic dysregulation of cell signaling pathways would be a high price to pay for lower oxidative stress.
Finally, there is reason to think that the catalase boost given to these mice—which was performed in the mice while they were still early embryos, rather than in adult organisms—might not even work if done in adult organisms. Catalase genes only expressed the enzyme in some cell types, not in others, and the scientists who performed the study suggested that this might have been the result of a form of evolutionary selection during their development in the womb. The idea is that the extra catalase might be beneficial in some kinds of cells but harmful in others, and therefore those cells that were expressing a lot of the enzyme would tend to die off if they were of a type in which the extra catalase was deleterious. Meanwhile, other cells of the same type that did not express the new gene would survive, replicate, and come to dominate. But performing the same trick in mature organisms would short-circuit this internal evolutionary process, supplying the new gene to all cell types indiscriminately, so that the benefits of the catalase gene therapy in some cell types might be outweighed by negative effects elsewhere. This would mean that people unfortunate enough to have already been born would not be able to reap any benefit from such therapy—and no proposal to insert the gene into healthy developing infants, let alone embryos, is going to get past a medical ethics board.
None of this should dishearten us; it should simply remind us of the need to focus our efforts elsewhere. As I outlined in Chapter 4, I advocate a fundamentally different approach to dealing with age-related molecular damage. Instead of trying to “mess with metabolism” in ways that might prevent aging damage such as mitochondrial DNA deletions, it is my contention that we need to focus on developing anti-aging biomedicine that can repair or render harmless any mutations that may occur in mitochondrial DNA. While most people—whether laypersons or professional scientists—tend to assume that this must be far more difficult to achieve than a preventive strategy, there are in fact several promising techniques sitting on the drawing board that require no biotechnology more advanced than that which would already be required to put catalase into the mitochondria—namely, gene therapy. This suggests that we could actually have either type of intervention in the clinic at about the same time. In fact, it really tells us that the remedial technologies should be able to reach people suffering aging damage sooner than preventive ones, for the regulatory and pragmatic reasons I outlined earlier.
![]() The Best Bet: “Head for the Bomb Shelter!”
The Best Bet: “Head for the Bomb Shelter!”
As I discussed in the last chapter, the main reason for mitochondrial DNA’s unusual vulnerability to oxidative damage is its location: being next door to a leaking nuclear reactor (the mitochondrial electron transport chain) is a sure way to increase your risk of mutations, whether you’re a growing child or a tiny biological machine. Metabolism clean-up approaches try to make this reactor run more cleanly (so that it produces less damaging waste), or to install “pollution control” equipment that would catch more of its by-products before they do any harm (the cellular equivalent of the “smoke-stack scrubbers” on coal-fired power plants). My preferred approach is completely different: that we should put these mutations “beyond use” of harm. This could be accomplished by putting backup copies of the genes that are currently housed in the mitochondria in the safe haven of the cell’s nucleus, far from the constant bombardment of free radicals from the mitochondrion itself. This solution is called “allotopic expression” of the proteins these genes encode—i.e., expression from a different (Greek allo-) place (topos).
Let’s be clear about this. Allotopic expression would do absolutely nothing to prevent the native mitochondrial genes from suffering mutations: free radicals would hit the vulnerable mitochondria just as often, and mutations would occur at exactly the same rate, as they did before. But with a nuclear backup copy of these genes, any such mutations would be rendered functionally irrelevant, because the cell would be able to keep producing the proteins that the knocked-out genes in the mitochondrion had previously encoded. These mitochondria would thus enjoy functional proton-pumping, electron-transporting proteins, and would therefore behave exactly like mitochondria with intact DNA, just as if they had not suffered mutations in their “local” DNA. Electrons would continue to flow into the electron transport chain from NADH; protons would continue to be pumped; free radicals would continue to leak out of the system at random. The concept is illustrated in Figure 1.
Not only that: because such mitochondria would continue to damage their mitochondrial membranes, the cellular “incinerator system” (the lysosome) would be able to tell when they got old and haul them off for destruction. Therefore, the Survival of the Slowest mechanism that leads mutant mitochondria to take over their host cells would never take place, nor would cells be forced to shift into the abnormal metabolic state that cells with mutant mitochondrial DNA require in order to deal with an imbalance in their NADH-to-NAD+ ratio. Thus, despite the fact that these cells contain mutations in their mitochondrial DNA, they would not be unloading their excess electrons into LDL, would not spread oxidative stress to the rest of the body, and would make no more contribution to the aging of the organism as a whole than cells with perfectly intact mitochondrial DNA.
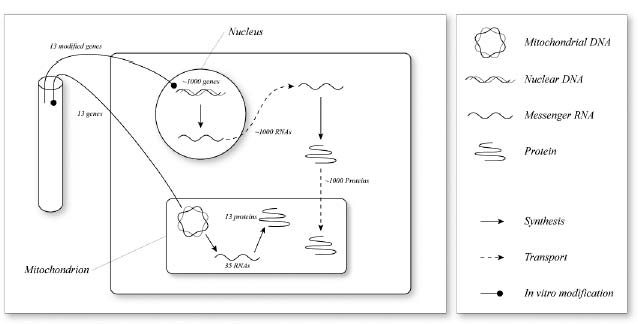
Figure 1. The concept of allotopic expression to obviate mitochondrial mutations.
“But,” I hear you ask, “what if one of these backup genes is itself mutated? Won’t we be facing the same catastrophe?” Fortunately, no! There is, in fact, no real risk of a functionally meaningful failure of this backup system occurring, even over the course of a lifespan that has been dramatically extended by a full panel of SENS anti-aging interventions.
To understand why this is so, let’s look at what would be required for such a failure to occur. First, in order for a cell with an allotopic copy of a mitochondrial gene to slide into Survival of the Slowest, it would have to have suffered mutations in both copies of the gene: the mitochondrial original and the duplicate copy that we would have placed in the nucleus.
This is less likely to happen than it may initially sound. It is already unusual for DNA located in the mitochondria to suffer permanent damage (remember that as things stand, less than 1 percent of cells are overtaken by mutant mitochondria), and the odds of a backup copy located in the nucleus being mutated are much lower. Aside from being better shielded from free radicals because of its location (DNA housed in the nucleus is many times less susceptible to mutations than its mitochondrial counterpart), there are many more proteins encoded by genes located in the nucleus: tens of thousands, versus only thirteen encoded by genes that are in the mitochondria themselves. So even when a free radical does get into the nuclear DNA, the odds of it damaging one of the allotopically expressed mitochondrial genes are many times lower than the odds that it will hit some other gene. Indeed, many such free radicals will not hit a gene (an instruction for building a protein) at all, but one of the many stretches of nonfunctional “junk” DNA. Therefore, the chances of both the mitochondrial copy and the nuclear backup of the same gene being mutated are vanishingly small.
Moreover, while the unusual design of the mitochondrial DNA ensures that large deletions in its structure wipe out its ability to synthesize any of its proteins, the same will not occur in the nuclear case: only the protein for the specific mutated gene will be affected. Of course, your mitochondria can’t function without all thirteen proteins, but we could help reduce the risk of any actual shutdown of oxidative phosphorylation—and the resulting clonal expansion of a mutant mitochondrion—by providing a double or even triple set of functioning backup copies.8
![]() The “Mitochondriopathies”
The “Mitochondriopathies”
One category of hurdle facing the clinical development of anti-aging biomedicines is structural: aging is not a recognized disease, so the FDA will not allow trials to be performed on interventions claimed to cure it. This is obviously a bucket of water thrown directly onto the heads of venture capitalists who might otherwise be interested in investing in startups working on a treatment for age-related mitochondrial DNA mutations. From the perspective of getting effective anti-aging interventions into the clinic as quickly as possible, allotopic expression has the advantage that it is already being pursued as a treatment for a recognized group of diseases: the mitochondriopathies.
These diseases are caused by defects in the mitochondrial DNA that are inherited (or, more rarely, acquired through causes independent of the aging process). These mutations lead to a failure of energy production that causes a spectrum of dysfunctions in various organs, depending on the exact disorder: the brain, heart, and muscles tend to be the most vulnerable, but damage can also extend to the liver, kidneys, lungs, and certain glands. Because allotopic expression is a promising therapy for mitochondriopathies, government funding (albeit not nearly enough) is already available for work on its development; and once it is ready to move into the clinic, there will be an incentive for venture capital to invest in its development, giving a clear route forward for near-term testing in FDA-approved clinical trials.
In turn, once allotopic expression has been proven to be safe and effective as a treatment for inherited mutations of the mitochondrial DNA, we will be in an excellent position to make the small tweaks needed to adapt it for use as a treatment for mutations acquired during the aging process. This parallel applicability is a feature of most of the anti-aging interventions included in the SENS platform—and indeed, prototypical versions of several of the proposed interventions are already in clinical trials today.
![]() To Boldly Go Where Evolution Has Gone Before
To Boldly Go Where Evolution Has Gone Before
The other hurdles facing allotopic expression are the more purely scientific ones. Fortunately, as we will see, progress on these problems has been rapid in the last decade. But it’s better than that: evolution has been working on a similar job for untold millennia.
In ages long past, the ancestors of the mitochondria occupying our own cells were not just components of cells as they are today, but organisms in their own right, which formed an I’ll-scratch-your-back-you-scratch-mine relationship with our one-celled ancestors. Because they were independent organisms, these proto-mitochondria naturally had a full complement of their own DNA—at least one thousand genes.
But precisely because the hazardous environment of the mitochondrion put the genes housed there at extremely high risk of mutation, evolution has been performing allotopic expression on mitochondrial genes since long before humans appeared on the scene. Over the glacially slow timescales of evolution, organisms have copied mitochondrial genes that code for mitochondrial proteins into their cell nuclei, after which the original mitochondrial genes became redundant components and mutated into oblivion.
And to give Mother Nature her due, evolution has gone a long way in this direction. Out of more than one thousand original mitochondrial proteins, all but thirteen have had their genetic instructions moved into the nucleus.
Starting in the mid-1980s, scientists started showing that they, too, could perform allotopic expression of some mitochondrially coded proteins using biotechnology, albeit initially only in yeast—a crucial series of proofs-of-concept.
![]() Obstacles, Evolutionary and Otherwise
Obstacles, Evolutionary and Otherwise
But things get a lot trickier when we start trying to do the same thing with the thirteen protein-encoding genes that are still located within the mitochondria in human cells. The reason why evolution hasn’t finished the job for us already is a matter of some debate, but everyone agrees that there must be some kind of “forces” holding the process back. Whatever those forces are, this job is probably not going to be easy. What we have to do is figure out what forces are keeping those genes in the mitochondrion, and then devise ways of overcoming them. I have advanced the case that there are only two such forces that need concern us.9
One, which doesn’t apply to all organisms but does apply to us, is that the DNA “languages” of mitochondria and of the cell nucleus have evolved slightly different “dialects,” so that an exact copy of a given mitochondrial DNA sequence becomes indecipherable when it is dropped into the nucleus. This problem is called code disparity.
The case is rather like the changes that have occurred over time in the writing of the letter “s” in English. Up until the nineteenth century it was common for an “s” occurring in the middle of a word to be written in an elongated fashion that looks much more like a modern “f” than an “s.” Gradually, as writing became more widespread and irregularities in the written language more standardized, the elongated “s” came to be replaced by the shorter, more curved version of the letter that we use today. Thus a modern reader of an Enlightenment-era order to launch a naval attack (“Sail for the enemy”) might mistake it for a command to “throw” the battle (“Fail for the enemy”), and in other cases an instruction might be reduced to pure gibberish.
This disparity in the genetic codes of mitochondria and nucleus makes moving mitochondrial genes that contain such discrepant lettering into the nucleus a near impossibility for evolution. Indeed, all the genes still housed in the mitochondria contain such quirks, and this fact alone can explain why they haven’t made the jump to the nucleus. But code disparity doesn’t pose any serious problem for biotechnology: with our outsider’s understanding of the discrepancies in the two codes, we can simply create the new, allotopic gene using the nuclear version of the code (substitute “s” for “f”) and rest assured that it will be translated and turned into a protein just like any of the other genes for mitochondrial proteins that are already housed there.10
The second problem appears to have been a somewhat less imposing barrier to the evolutionary transmigration of mitochondrial genes to the nucleus, but is a much greater challenge for the anti-aging biotech engineer. It is the repulsion by water (“hydrophobicity”) of many of the mitochondrial proteins whose genes are still located within the mitochondria themselves. These proteins have sites in their chemical structures that have such a “fear” (phobia) of water (hydro) that, like a phobic human, they will literally curl themselves up into a ball in response to it.
Hydrophobicity is no problem for proteins when they are built from DNA that is already located within the mitochondrion, because the final three-dimensional shape of the protein is supposed to be twisted up, and there are special enzymes that guide such proteins into the proper conformation. But it becomes a showstopper when the same protein is constructed from DNA located in the nucleus. The genes for such proteins are translated into their products in the main body of the cell, and the proteins must then be moved from the fluid environment outside of the mitochondrion, through the outer and inner mitochondrial membranes, and into their final location in the mitochondrion’s core.
Of course, the membranes won’t just let proteins pass freely through them, or the integrity of the mitochondrion—and its ability to preserve its proton reservoir—would be compromised by the constant leak of material into and out of its chambers. But mitochondria do need to be able to import hundreds of proteins: for example, the dozens of the subunits of the electron transport chain whose genes have already been moved successfully into the bomb shelter of the nucleus by evolution. Mitochondria have therefore evolved elaborate machinery that specifically moves (translocates) proteins through these membranes. These are sensibly called the “Translocase of the Inner Mitochondrial” membrane (TIM) and the “Translocase of the Outer Mitochondrial” membrane (TOM), giving the whole system the glorious name TIM/TOM complex.
The problem is that once a protein has been gnarled into a balled-up Twister configuration by its repulsion to the water in the fluid compartment of the cell, the cell becomes unable to jam it through the pores of the TIM/TOM machinery—rather like trying to force a wildly bent-out-of-shape coat hanger down a drainpipe. This would be not just a failure, but actually counterproductive: not only would it prevent the newly allotopically expressed proteins from getting where they have to go, it would also “clog up” the TIM/TOM complex, disrupting the import of the many proteins that were previously being naturally, successfully imported.
In fact, all of the thirteen proteins that are still coded directly in our mitochondria are very hydrophobic. The instructions for several of those proteins have never been moved into the nucleus in any species, and those proteins are the most hydrophobic of all. This suggests that hydrophobicity is indeed the biggest hurdle to the allotopic expression project. There are several cases that seem to violate this rule in other species, but I have published detailed analyses9 that explain why all of these apparent counterexamples are misleading—why they would have happened in the course of evolution despite the fact that hydrophobicity really is the most important barrier to making the move.
So, if we are to obviate mitochondrial mutations via allotopic expression (my preferred solution), then in addition to the relatively simple task of editing the code in cases where the DNA “language” of mitochondria and nucleus are mismatched, we will have to find ways of tweaking the proteins that, in their present form, can’t be imported into the mitochondria.
When I first began contemplating this problem, I came up with a workable-sounding but technically challenging way of dealing with it, which I published in the journal Trends in Biotechnology in 2000.11 I’ll describe this approach later on. The reason I’m putting off discussion of this solution is that recent experiments suggest that we may not need to go to the lengths that I then proposed in order to overcome the hydrophobicity problem. There are at least two alternative ways to engineer these proteins to make them importable—ways that appear to be much easier.
![]() Pirating Mother Nature’s Intellectual Property
Pirating Mother Nature’s Intellectual Property
The first solution to the hydrophobicity problem is to look around for cases in which evolution has already done the yeoman’s work for us—in other species. We humans (and our evolutionary ancestors) have not yet enjoyed the simultaneous good fortune of the right random mutations, the right environment and the right selective pressures to pass nuclear versions of any of these genes along to us as their descendents. But that doesn’t mean that the same happy confluence of circumstances has never occurred in other species’ evolutionary history. Natural selection has been working on the hydrophobicity problem in many species independently of our own, and has in several cases come up with workable solutions—solutions that we did not inherit, simply because they occurred in a separate evolutionary lineage. By looking beyond our own mitochondrial DNA into the evolutionary inheritance of other species, we might find viable solutions that we only need to tweak slightly for use in our own cells.
Other species’ mitochondrially encoded proteins are not identical to our own, of course, but their structure is close enough to make it reasonable to believe that they could stand in for the originals if inserted into our mitochondria, or at least show us how to modify the sequences of the human counterparts to render them importable. If species could be found whose genes for their versions of some of the thirteen proteins had spontaneously moved into the nucleus, we would expect to be able to put those same genes into our own cells’ nuclei with only minimal modification. Those proteins would be constructed in our cell bodies, imported into our mitochondria, and take the place of the native version if and when mutations shut down the mitochondria’s ability to do the job themselves.
This idea is not just a fancy of mine: it’s been done in isolated human cells already. Work on such a project began in 1998, shortly after I started shouting about the importance of a discovery that had been made eight years earlier, when the mitochondrial genetic library of the green alga Chlamydomonas reinhardtii was sequenced. When the proteins for which these creatures’ mitochondrial genes code were identified by comparison with the equivalent genes in other species, it was found that they are missing versions of six of our thirteen hydrophobic electron transport protein genes.
In fact, of course, these genes are rather like your mysteriously “missing” car keys: you haven’t actually lost them, they just aren’t where you thought you’d left them. In these organisms, the changes needed to make these proteins less hydrophobic have taken place, because the greatest barrier to the change—code disparity—was never erected. These algae are so close to the “root” of the evolutionary “tree” that the disparity between the DNA coding systems of the nucleus and the mitochondrion never took place in them. Without that hurdle to leap, evolution has only had to address the much less challenging hydrophobicity problem—and it has done so with some success. The genetic instructions for these proteins are now housed in the algal cells’ nuclei. The cell’s machinery reads those instructions, manufactures the proteins in the main body of the cell, and then the mitochondria import them—the same thing that happens with most of our own mitochondrial proteins.
At this point Dr. Mike King at Thomas Jefferson University, Philadelphia, comes into the picture. King was not originally interested in aging, but in the inherited mitochondrial diseases (mitochondriopathies). Researchers had long dreamed of gene therapy for these disorders, but there are immense technical challenges to putting genes directly into the mitochondria. King thought that allotopic expression in the nucleus could provide a faster route to a cure.
But the hydrophobicity problem loomed over this potential solution to inherited mutations in the genes for the thirteen mitochondrially coded proteins, just as it does for the plan for mitochondrial genes mutated during the aging process via free radical damage. When he heard about the existence of nuclear-coded versions of some of these proteins in the algae in the late 1990s, Mike saw that they offered a potential blueprint for replicating the algae’s tricks in human patients with mitochondrial diseases.
What followed was an astonishing surge of progress. In 1998, King began a fruitful collaboration with Dr. Diego Gonzalez-Halphen of the Department of Molecular Genetics at the Autonomous National University of Mexico, to identify and clone the algae’s genes for the six analogous proteins. Within three years, these scientists had identified three of them. One of these (ATP6, a component of the mitochondrion’s Complex V “turbine”) was of particular interest because inherited mutations in it cause two rare but extremely serious disorders of the brain and muscular system in humans: NARP (Neuropathy, Ataxia, and Retinitis Pigmentosa) and maternally inherited subacute necrotising encephalomyopathy. In these diseases, ATP synthesis is reduced by 50 to 70 percent, leading to severe dysfunction of the neuromuscular system. Thus, the identification of an importable version of the mutated protein held forth therapeutic potential for people suffering from these diseases, as well as for all of us as we age.
Picking up the ball, Eric Schon and his coworkers from the Department of Neurology at Columbia took the next step, inserting a cloned copy of the algae’s ATP6 gene into the nucleus of human cells whose mitochondrial DNA harbored the same mutations that cause these neuromuscular diseases in humans. The cells decoded the genetic instructions, turned out the protein in the main chamber of the cell, imported it into the mitochondria, trimmed off its targeting sequence (a special string of amino acids that, when appended to the “nose” of a protein, directs it into the mitochondria), inserted it into the electron transport chain, and apparently took the place of the mutated protein, rescuing the cells from the destructive effects of the mutation.12
In other words, these researchers did exactly what I had been calling for someone to do. They found, in an alien species, a nuclear gene for a mitochondrial protein whose human counterpart is located in the mitochondrion itself; they inserted it into the nuclear DNA of human cells; and they showed that those cells could use it in just the way that the algae do, restoring near-normal functionality to cells with otherwise disabling mutations.
There is still a lot of work ahead, of course. To turn this into a viable therapy for people with inherited or age-related mitochondrial mutations, we will have to do two things. One is to identify in other organisms, or engineer ourselves, genes that can be put in the nucleus for the rest of the mitochondrially encoded proteins, and show that they restore function to mutant cells. And the other will be to do the trick in whole organisms burdened with these mutations: first in mice (a relatively simple task: genetic manipulation of mice is now relatively routine), and then in grown humans (a technology that we have not yet mastered, but upon which work is proceeding with an intensity that may well yield safe, viable therapies in the foreseeable future).
![]() Borrowed Ideas, Novel Solutions
Borrowed Ideas, Novel Solutions
But of course, there is no guarantee that we will find all of the necessary genes in other species. It would be smashing luck if we did, of course, but it’s entirely possible that many of the genes for electron transport chain proteins have never been transferred from the mitochondria to the nucleus in any species, or that the protein will have been so changed in the intricate branching of life’s evolutionary family tree that it will not work in human cells. In that case, we’ll just have to figure out for ourselves how to tweak our existing genes to make the proteins they encode importable.
Even here, however, we’ll be able to borrow tricks that we’ve learned from other species. A remarkable example was reported in 2002,13 when a group at the University of Western Australia discovered that several legume species—including the soybean and the common mung bean (Vigna radiata)—are actually in an intermediate evolutionary state, having evolved a nuclear copy of the mitochondrial gene for subunit two of the electron transport chain component cytochrome c oxidase, but without having yet discarded their original, mitochondrial copy.
The fact that these organisms survive while expressing the protein from both sites at once is itself good news, as it relieves (to some extent, anyway) a concern that some people have expressed about using allotopic expression as a solution to mutations. The worry is that in cells with healthy mitochondrial DNA and allotopic versions of the electron transport chain proteins, the existence of two independent, working copies of the same gene might lead to too many copies of the duplicate-coded proteins being produced, and that this might somehow imbalance or overload the capacity of the mitochondria to fit the various components together into working electron transport complexes. This would be like having two departments in the management of a factory, each using an independent system to order components for their product—a serious glitch in any “just-in-time” inventory system. The observation that no such problem occurs in these organisms suggests that we may not have much to worry about here—and that’s good news.
But when other scientists compared the two versions of the legume electron transport gene, they discovered something that makes me even more bullish about our ability to move the full complement of mitochondrial electron transport chain genes into the nucleus. The two versions of the protein differ in twenty-five amino acids (the building blocks of protein) out of hundreds—but only two of these differences are necessary to allow the nuclear version to be imported into the mitochondria! This suggests that we may only need to do some relatively minor fiddling with our thirteen proteins of interest in order to make feasible their import into the mitochondria.
Again, I’m no longer just extrapolating from what evolution has achieved in other organisms: progress in adapting these solutions to new problems is definitely under way. Around the same time Schon’s group expressed ATP6 in human cells allotopically using the algal version of the gene, they and another group also reported having developed different solutions to the challenge of engineering new, nuclear-coded versions of that protein. The difference was that these new proteins were not taken wholesale from another species, but modified from the mammalian original. As with the success using the algal gene, these human-generated versions were reported to rescue cells bearing inactive versions of ATP6 in their mitochondria.14,15
Not long after this, a third group engineered a nuclear-coded version of another mitochondrial gene named ND4, mutations in which cause one of the mitochondriopathies, Leber’s Hereditary Optic Neuropathy (LHON).16 To do this, they first had to find solutions to the problems that had previously been dealt with in the allotopic expression of ATP6. First, the DNA code for the protein had to be altered to make the “spelling” compatible with the nucleus, since ND4 suffers from the “code disparity” problem I discussed earlier. Then, the researchers had to tag on a “targeting sequence” copied from a completely different gene (aldehyde dehydrogenase) to guide it into the mitochondria. Next, they had to figure out a way to get the gene into the nucleus in the first place; this was accomplished using a trick borrowed from viruses that sneak their DNA into their infectees’ nuclei. And finally, they attached an additional sequence to the gene to allow it to be picked up by the gene-decoding machinery of the nucleus, so that it would be “read” and turned into a protein.
Given the need for so many alterations to the original gene, borrowed from so many different inspirations, you might reasonably be concerned that something would fail somewhere along the way. But no. The heavily modified protein, custom-built out of the original by reasoned analysis of what would be required to make it importable (rather than being copied in whole cloth from one of our distant relatives), was successfully incorporated into human cells bearing mutant ND4, which promptly began churning out functional, mitochondrially targeted ND4. The allotopically expressed protein then found its way into the mitochondria, took its place in the electron transport chain…and promptly tripled the cells’ ATP output, bringing it back to levels similar to those seen in normal, nonmutant cells. Not only that: when the cells were subjected to conditions under which they were forced to lean heavily on OXPHOS for energy, these cells with the new allotopically expressed mitochondrial electron chain subunits enjoyed three-fold better odds of survival than mutant cells lacking the engineered gene.
The researchers boldly concluded that “Restoration of respiration by allotopic expression opens the door for gene therapy of Leber Hereditary Optic Neuropathy.” I would add that it also puts a large foot into the same door for the solution of the reductive hotspot problem. Doubts have recently arisen concerning the methods used to demonstrate rescue of the treated cells in two of the above studies, but at least two others are still considered clear-cut.
![]() Inteins: Splitting the Difference
Inteins: Splitting the Difference
I am hopeful that these two approaches may be sufficient to allow us to deal with the hydrophobicity issue; however, it is possible that we may have to go further with some of the very hydrophobic proteins. One potential solution—which I originally put forward before the above successes suggested that it might not be necessary—is to further modify the proteins in question using inteins.
Inteins are sequences that are inserted temporarily into some proteins when they are first synthesized, possibly to help the protein mature into its final form properly, and are then snipped out once they’ve served their purpose. In some cases, the two halves of a final, functional protein are expressed separately, each with a complementary “semi-intein” at the end where the two protein segments must finally be joined. When the two halves of the final protein come together, the two semi-inteins are first bound together, a little like the male-to-female electrical connectors on strings of Christmas-tree lights. But then the united intein sequence is snipped out and the two precursors of the final protein are permanently, directly fused together into the final, completed structure.
We might be able to use inteins of this sort to help us deal with a hydrophobic protein. One approach is to split the protein up close to a site in its structure where it would otherwise curl up when exposed to water, and put semi-intein “caps” on either side of the break. As an analogy, imagine that you wanted to move a long piece of steel with a right angle in its center (the protein) down a straight drainpipe (the TIM/TOM complex). As it stands, the job is impossible. But if you could cut the piece of steel at or near the place where it bends, and then attach a short segment to each of the severed ends of the bar to show your coworker at the other end of the pipe where to weld the two halves together again, you could easily drop the two halves down the tube individually for reassembly at the other end.
Alternatively, whole inteins can actually be built right into the middle of the two halves of the protein, creating one long structure with the intein in its center. I’m afraid the way that this can be exploited is a bit harder to analogize, but I’ll try. Imagine if, instead of cutting your bent bar in half where the bend occurs and sending each half down the drainpipe separately, you were to cut the bar, rotate it through a half-twist, and put in a central joining segment, so that the final structure looks more like a stylized lightning bolt than a right angle. You could then drop the “straightened” bar (protein complete with intein) down the drainpipe (TIM/TOM complex) and then excise the joining segment (intein), allowing you to reassemble the bar into its proper configuration after intein removal.
This procedure is a good deal more complicated than switching a couple of amino acids, so I’d rather not have to resort to it if one of the former options will work. When I first came up with the idea of using inteins as a solution to the hydrophobicity problem, I foresaw a whole series of potentially crippling technical hurdles that might have to be overcome to get it to work, and I’m still not sure how easy that would be. Inteins would have to be placed in just the right place, and be of an appropriate length, and this would require a lot of fiddling. Also, natural inteins are designed to be snipped out as soon as the protein that contains them has been constructed, so mitochondrial inteins would need to be designed in ways that prevent them from being removed until the complete protein has been imported into the mitochondria.
Another problem we might face is that of ensuring that, in the “semi-intein” version of this approach, the protein segments are joined to their proper partners. If we have used inteins to break up several proteins that are imported simultaneously, or if one protein has been broken down into more than two pieces (as seems likely to be necessary in at least a couple of cases), the “exposed” ends of protein segments might be mismatched once the inteins are removed. Fortunately, it appears that some such multiple inteins do exist naturally and somehow “know” their proper partners, so this may not really be a problem—and if it is, the alternative solution of putting inteins into the hydrophobic stretches of these proteins is still available.
Yet another potential headache: segments of proteins could begin to fold too early, either before joining to their “other halves” in a way that obstructs access to the fusion site (so that the intended fusion with the mate is no longer possible), or even right in the midst of the TIM/TOM machinery, causing the exact problem that the use of inteins is supposed to prevent. And finally, a protein’s separate, newly inteinless segments might be altered by enzymes while they wait to be joined to their “mates” any such alterations could render them nonfunctional, or prevent them from “hooking up.”
The good news is that, despite all of these potential obstacles, at least one split intein job has already been pulled off in cell culture:17 Yoshio Umezawa and coworkers took an unimportable green fluorescent protein and introduced it into a cell’s mitochondria by first adding on a borrowed mitochondrial targeting sequence, and then adding inteins inserted into its structure using a protein splicing system taken from algae, before finally “infecting” the cell’s DNA with the composite structure’s code. The result was that the protein was indeed expressed and imported into the cell’s mitochondria, where it allowed the scientists to detect the presence of other proteins that they were trying to direct into the organelle.
![]() Not TINA but TATA
Not TINA but TATA
This string of successes indicates that we can expect rapid progress in expressing the remaining mitochondrially coded proteins from nuclear genes in reasonably short order, through some combination of the different techniques I’ve discussed here. Once we have figured out how to put all thirteen of these proteins’ genes into the nuclear “bomb shelter,” we will be very close to being able to negate the insidious effects of mitochondrial mutations in aging, turning existing “reductive hotspots” into normal, healthy cells and preventing the formation of new ones. When that is accomplished, we will be able not only to stop but to reverse the slow upward creep in oxidative stress—and the accelerating spiral of molecular damage and metabolic disruption—that we think is driven by these mutations today.
But, of course, the precedent of medical history tells us that some pitfalls may still lie in wait for this solution. One can make one’s most educated predictions, based on a sober consideration of the published science, and still fail to anticipate an intractable roadblock. My biggest worry is that, even after a bit of tweaking, the volume of TIM/TOM traffic required to import the new allotopically expressed proteins will be so high, and its rate of flow through the system so slow, that we will not be able to transfer the full complement of proteins via this route even if we succeed with each of them individually. I am especially concerned about this since some of the proteins that have been allotopically expressed move quite poorly through the TIM/TOM machinery; one of the examples we discussed above involves a protein that is only incorporated into the mitochondria at 40 percent of the efficiency of the native, mitochondrially-coded version.
Former prime minister Margaret Thatcher, the “Iron Lady,” was famous for intoning “There Is No Alternative” (TINA) to the neoliberal economic agenda. I have no intention of painting myself into an ideological corner over my preference for allotopic expression: lives are at stake, not just my ability to admit that I’m wrong. I will instead join the ranks of her anti-globalization critics, who spilled into Trafalgar Square in their thousands chanting “Not TINA but TATA!”—that is, “There Are Thousands of Alternatives.”
Having made that bold declaration, I hasten to add that I think we can safely trim down “thousands” to “a few, and even fewer that are viable.” In fact, quite a number of alternative fixes for mitochondriopathies have been advanced, and at first glance these solutions might also be expected to treat age-related mitochondrial DNA deletion accumulation. Unfortunately, I predict that most of those solutions will be of no help in preventing the development of “reductive hotspots,” because the mechanism whereby the underlying mutations accumulate (mitochondrial free radical generation and clonal expansion through the Survival of the Slowest) is quite distinct from the mostly inherited problems in mitochondriopathy victims. Research toward cures for mitochondriopathies using these alternatives will still doubtless yield techniques and insights that will in some way help us to develop a cure (via allotopic expression or some other therapy), but I do not believe that most of these will be adaptable as anti-aging biotechnology. I shot down the major flawed proposals down in the 2000 article in Trends in Biotechnology that I mentioned earlier. Two approaches exist that I feel have much more promise, however.
One, advanced by Drs. Rafal Smigrodzki and Shaharyar M. Khan of the Center for the Study of Neurodegenerative Diseases at the University of Virginia, starts with the ability to introduce into the mitochondria modified versions, not of individual genes (as in the classical gene therapy approach), but of complete copies of the entire mitochondrial DNA. They accomplished this maneuver—which they have termed protofection18—using boldly simple techniques that most scientists would have dismissed as unworkable. It’s too early to say how versatile and repliable their method is, though—it’s too new to have been explored by other scientists.
The other very promising alternative to allotopic expression is to introduce genes for alternative versions of the mitochondrially encoded enzymes, versions that don’t quite work the way that the native ones do. The enzymes in question already exist in—and could thus, in theory, be borrowed from—some lower organisms (yeasts and plants). They perform the exact same electron transport activities of the enzymes that we partly encode in our mitochondrial DNA, even though they themselves are not particularly hydrophobic and are encoded in their species’ nuclear DNA. The problem—if it is one—is that they only do the electron transport, not the proton pumping. But introducing them into our cells might be a fair trade-off, because while they would not restore the mitochondrially mutant cell to its full capacity, they would prevent these cells from impairing the ability of the rest of the body to get on with the business of life. This has actually been documented in isolated human cells: when Complex I is chemically inhibited, normal cells quickly die off, but cells given the relevant yeast gene remain viable.19
One problem with this proposal is that, if these proteins were to be expressed in cells that had not suffered deletions, they would deny the cell of much of its energy source. We would use one gene to bypass the first enzyme in the electron transport chain (Complex I) and another to bypass the rest. Neither of them pumps protons, so having both of them substituting for much of the native, proton-pumping complexes in a mitochondrion would seriously impair the buildup of a proton “reservoir” and, thus, ATP production. This could lead to anything from a mild functional impairment to a severe energy deficit—and it would extend through every cell in the body, not just the “evil 1 percent” of cells with a population of mutant mitochondria. This would leave us in a very bad way indeed, so we would need to find a way to ensure that the genes for these “bypass enzymes” are expressed only in cells whose mitochondria are no longer producing the native proteins. There is no obvious way to do this at the moment, unfortunately. But luckily, initial research indicates that these enzymes naturally possess a system that activates them only when their proton-pumping counterparts are failing. Even expressing one of the two “bypass enzymes” indiscriminately would make cells less efficient at making ATP, so this is actually no surprise.
![]() The Way Forward
The Way Forward
Overall, the picture is a rosy one. We have not only a good idea of how mutations in the mitochondrial DNA contribute to the age-related decay of our bodies, but a clear path forward to obviating the problem—even if our understanding of the exact mechanistic link between mutations and pathology turns out to be mistaken. Allotopic expression would allow our mitochondria to keep functioning normally even when their DNA acquired mutations; protofection, alternatively, could simply clear out the old mutant DNA periodically, replacing it with a fully functional new set of genetic blueprints; and the use of easier-to-handle enzymes that pump no protons but keep electron metabolism harmless would at least keep mutant cells from causing trouble outside of their own membranes.
Again, we will need to develop safe, effective, stable gene therapy that works in grown humans in order to turn any of these interventions into a real biomedical intervention against this aspect of the aging process, and that will certainly be a challenge. But, again again, it’s a challenge that scientists all over the world are already vigorously pursuing in order to treat genetic disorders—ailments ranging from Huntington’s disease, to inherited cancer risk, to familial Alzheimer’s disease, to sickle-cell anemia. And we can piggyback even more closely on research that is specific to the mitochondriopathies—a much smaller field, but one that, for the moment, still receives more serious funding and attention than work designed to tackle the slow-motion global plague that is aging.
With the resources already being thrown at advancing gene therapy, we can confidently predict that the clinical readiness of this enabling biotechnology is foreseeable. I am therefore convinced that the major hurdle facing speedy implementation of allotopic expression (or its alternatives) will not be our ability to develop safe gene therapy for patients, but the dearth of investment into the basic science of moving mitochondrial genes into the nucleus.
Remember the positive result using inteins to import proteins into mitochondria in culture? This achievement came about because scientists were seeking a way to get rapid feedback on the results of a completely unrelated project of interest to them. Imagine what could be accomplished with resources specifically dedicated to developing allotopic expression for the purposes of reversing aging damage!
How to bring about that change in research priorities is the subject of Chapter 15; but now, let me take you on a tour of the next of the “Seven Deadlies” of aging, and show you what we can do about it.