3
Regulatory guidance – a quick tour
- The ICH E9 guideline issued in 1998 covers many of the important points to be considered with regard to missing data. The ICH guideline has the advantage that it is widely accepted by Japan and other countries outside the United States and Europe that use the results of clinical trials.
- Current positions of the European Union (EU) and US regulatory agencies regarding handling of missing data can be found in an official European Medicines Agency (EMA) Guideline and in a U.S. Food and Drug Administration (FDA) sponsored report by a U.S. National Research Council (NRC) panel.
- The EU guideline and the US NRC report (which hereafter may be referred to in this chapter as “the regulatory documents”) agree in most important areas.
- Regulators agree that methods of handling missing data are important in interpreting study results; that missing data should be prevented; that no single method will “cure” the missing data problem; that a primary analysis will need sensitivity analyses to assess its robustness to assumptions about missing data; that methods for handling missing data should be described and justified in the protocol; and that better use should be made of post-withdrawal data.
- There are differences in emphasis between the NRC report and the EU guidance document regarding last observation carried forward (LOCF); regarding the importance of a “conservative” approach; regarding post hoc analyses; and regarding intermediate or non-monotone missing data.
- The NRC report emphasizes that it is important that the assumptions made about missing data be described in terms that can be understood by clinicians.
- The regulatory documents have some useful comments on classes of methods for handling missing data, including the use of available cases; the assumption of missing at random (MAR); and methods where it is assumed that data are missing not at random (MNAR).
- In practice, the latest regulatory documents are not always adhered to by the regulator, but there is a growing tendency for regulators to treat the handling of missing data as important, and to apply the principles used in the regulatory documents.
3.1 International conference on harmonization guideline: Statistical principles for clinical trials: E9
Interestingly, the International Conference on Harmonization (ICH) guideline Statistical principles for clinical trials (1998) covers many of the important points that form the core of the two more recent NRC and EU regulatory documents on missing data (European Medicines Agency, 2010; National Research Council, 2010), 12 years before those documents were issued. The ICH guideline has the advantage that it is widely accepted by Japan and other countries outside the United States and Europe that use the results of clinical trials. Here are the points of overlap (page references are to the ICH guideline; overlaps with the NRC report and EU guidance):
- Missing data should be prevented where possible (pp. 8, 22, 24)
- “No universally applicable methods of handling missing data” (p. 22)
- Investigate “sensitivity of the results … to the method of handling missing data” (p. 24)
- “Methods of dealing with missing data … pre-defined in the protocol” (p. 24)
- Make a record of reasons for withdrawal: “frequency and type of … missing values should be documented” (p. 22)
Like the recent EU guidance, ICH E9 specifically recommends that the study report describe the extent of missing data and reasons for discontinuation (ICH E9, p. 30).
The ICH E9 guideline differs from the two more recent regulatory documents with regard to single imputation. In contrast to the more recent documents, it mentions the use of last observation carried forward (LOCF) without disapproval: “Imputation techniques, ranging from the carrying forward of the last observation to the use of complex mathematical models, may also be used in an attempt to compensate for missing data” (ICH E9, p. 23). Apart from this sentence, the E9 guideline does not comment on the acceptability of any particular assumptions about missing data. This contrasts with the more recent guidelines where, we will see, the strengths and weaknesses of common assumptions about missing data discussed in detail.
3.2 The US and EU regulatory documents
The two most recent regulatory documents on missing data for the United States and EU regions were issued almost simultaneously in July 2010. They agree on most important issues, but differ in character. The US document is a report commissioned by the FDA, while the EU document is an official regulatory guideline.
The FDA-commissioned report, “The Prevention and Treatment of Missing Data in Clinical Trials,” (National Research Council, 2010), as expected contains recommendations on the missing data problem, but also includes fairly detailed descriptions of the statistical basis of many approaches to the analysis of missing data. It thus acts partly as a tutorial in methods for missing data. This report was written by a panel of the NRC, and sometimes reads like an academic paper – it includes more than seven pages of references to the literature on missing data. The European Medicines Agency “Guideline on Missing Data in Confirmatory Clinical Trials” (2010) is compact (12 pages vs. the NRC report's 145 pages). In contrast to the NRC report, the EMA guideline tends to comment on the statistical basis of an approach to missing data only insofar as this might be needed to discuss regulatory acceptability of the approach; and the EMA guideline has just two references to other documents, both also guidance documents.
At present, there is no official general FDA guideline on missing data. Although the NRC report is not an official regulatory guidance, we have found that US regulators sometimes quote the report in their reviews and encourage industry practitioners to follow its recommendations during their presentations at public forums. The NRC report notes that some guidelines are available and we will describe these briefly later.
3.3 Key points in the regulatory documents on missing data
Both the NRC report and EU guideline have a straightforward logical line of reasoning about missing data. Its key points are as follows:
- Missing data are important and affect the validity, bias and interpretation of clinical trial results. Implied is that missing data have not in the past always been handled adequately. The EU document uses its status as an official guidance to indicate that regulatory review of missing data will in the future be more rigorous. It notes that “just ignoring missing data is not an acceptable option” and notifies the reader that “This guideline … provides an insight into regulatory standards that will be used to assess confirmatory clinical trials.”
- Missing data should be prevented as far as possible. The NRC report particularly emphasizes this, devoting 26 pages to it. Both documents cover how the study design and execution can help to reduce missing data. Chapter 2 of this book discusses this aspect of the guidance.
- Because missing data are missing, we cannot verify our assumptions about them. Therefore, no single method can be counted on to give a comprehensive treatment of missing data. A single primary analysis alone is not adequate when a study has missing data.
- Sensitivity analyses are needed to test the robustness of study results to the assumptions of the primary analysis.
- A study's approach to missing data should be pre-specified in the protocol and justified scientifically. The EU document is notable for recognizing that, since we cannot be sure about missing data, no planned approach to missing data will be perfect. The EMA criterion for the adequacy of an approach takes account of this: the handling of missing data should be “unlikely to be biased in favor of the experimental treatment to an important degree (under reasonable assumptions)” with “a confidence interval that does not underestimate …variability” (p. 3). This criterion may be regarded as achievable, even given a team's uncertainties about likely patterns of missing data in a planned study. The EMA guideline again recognizes the difficulty in specifying missing data analyses in advance by allowing for the possibility of post hoc analyses: “If unexpected missing data patterns are found in the data, it will be necessary to conduct some post hoc sensitivity analyses in addition to those pre-defined in the statistical analysis plan” (p. 7). The important proviso is that “It is not envisaged that these post hoc analyses can be used to rescue a trial which otherwise fails.”
- Most approaches to missing data have limitations and weaknesses which the guidance documents describe. As noted, the NRC report has more detail on the statistical basis of approaches, and covers a wider variety of statistical methods. We will summarize these regulatory comments on methods in the next section.
- Post-withdrawal data may be helpful in estimating treatment effect or in sensitivity analyses, but will need to be put in context. Sponsors are “strongly encouraged” to collect post-withdrawal data (EU guideline, p. 6, and see NRC Recommendation 3, p. 3). Such data could be used to provide a true intent-to-treat (ITT) analysis. However, both documents note that if “participants are offered an alternative treatment that is not part of the study following discontinuation … subsequent data collection may be considered to uninformative” (NRC report, p. 9). Therefore, such “retrieved dropout” information will need “to be put in context” (EU guideline, p. 6).
- Reasons for withdrawal are important in handling missing data and should be recorded carefully.
In discussions with planners, or when writing a protocol or statistical analysis plan (SAP), it may be useful to quote the guidance documents on particular aspects the study's approach to missing data. Table 3.1 can be used to find references to key points on which the NRC and EU documents agree. Despite the difference in character of the two documents, they have remarkably similar wording in their treatment of many key issues to do with missing data.
Table 3.1 Key points of agreement in the NRC and EU regulatory documents on missing data.
Key point in document |
|
| FDA commissioned NRC report | EMA guideline |
| Importance of missing data | |
| “crucial … should have higher priority” (p. 114) | “critical” (p. 3) |
| Prevent missing data | |
| “minimizing dropouts” (pp. 21–46) | “avoid … unobserved measurements” (p. 6 ) |
| Cannot verify assumptions about missing data | |
| “assumptions unverifiable” (p. 52) | “assumptions … cannot be verified” (p. 3) |
| There is no universal method for missing data | |
| “no universal method” (p. 48) | “no single method will provide a … solution” (p. 8) |
| Sensitivity analyses are needed | |
| “should (conduct) sensitivity analysis” (p. 49) | “sensitivity analyses should be presented” (p. 11) |
| Pre-specify the approach to missing data | |
| “should be specified … in study protocols” (p. 110) | “essential to pre-specify methods” (p. 6) |
| Pre-specify sensitivity analyses | |
| “prospective definition of sensitivity analyses” (p. 17) | “sensitivity analysis should be … in the protocol” (p. 12) |
| Not acceptable to use only observed data | |
| “generally inappropriate” (p. 55) | “a consequence … may be a bias” (p. 5) |
| Treat the MAR assumption with caution | |
| “not … valid estimator of intention-to-treat effect” (p. 55) | “(provides) estimate (had) patients continued on treatment” (p. 10) |
| Post-withdrawal data may give limited help | |
| “may be … uninformative” (p. 9) | “ ‘retrieved dropout’ information … put in context” (p. 6) |
| Important to record reasons for withdrawal | |
| “reasons for missing data must be documented” (p. 49) | “reasons for discontinuation should be given” (p. 7) |
3.4 Regulatory guidance on particular statistical approaches
3.4.1 Available cases
Both the EU guidance and NRC report discourage the use of available cases for a primary analysis. The NRC report notes (p. 55) that deleting incomplete cases provides valid inference only under the assumption that data are missing completely at random (MCAR) and adds that “this method is generally inappropriate for a regulatory setting.” The EMA guidance notes that “(if) patients are excluded from the analysis this may affect the comparability of the treatment groups … (and) the representativeness of the study sample in relation to the target population (external validity)” – in other words, the findings of the study may only apply to the type of subject who is likely to complete the study – a perhaps elite subset of the population.
3.4.2 Single imputation methods
Methods such as LOCF and baseline observation carried forward (BOCF) replace a missing data point by a single value. Analyses are then carried out as if all the data were observed. The EU document points out that single imputation “risks biasing the standard error (of the estimate of treatment effect) downwards by ignoring the uncertainty of imputed values” (p. 9). The NRC report agrees: “statistical precision is overstated because the imputed values are assumed to be true” (p. 65). The EU document points out that, for conditions that are expected to deteriorate over time, LOCF can favor the treatment group with earlier withdrawals. We have seen a mild example of this in Chapter 1. However, the EMA guideline concedes that “where the condition is expected to improve … LOCF … might be conservative … where patients in the experimental group tend to withdraw earlier.” If LOCF can be shown to be conservative, then an LOCF approach, despite its “suboptimal statistical properties,” would provide “compelling evidence of efficacy from a regulatory perspective.” Similarly, BOCF “may be appropriate in, for example, a chronic pain trial” as a quantitative representation of the fact that “the patient does not … derive benefit from treatment.” In the final analysis, the EU guidance does not rule out the use of LOCF and BOCF. The NRC report simply provides the arguments against LOCF: LOCF “is not necessarily (conservative), since, for example, LOCF is anti-conservative in situations where participants off study treatment generally do worse over time” (p. 66).
3.4.3 Methods that generally assume MAR
Both the NRC and EU documents are blunt about a weakness of methods that assume MAR, that is, assume that missing values can be adequately predicted on the basis of observed values of patients with similar previous outcomes. The EMA declares that “the MAR assumption (provides) an unbiased estimate of the treatment effect that would have been observed if all patients had continued on treatment for the full study duration” and is thus “likely to overestimate the size of the treatment effect likely to be seen in practice” (EMA guidance, p. 10). The FDA-commissioned report agrees: “any method that relies on MAR is estimating the mean on the condition that everyone had remained on treatment. This generally will not provide a valid estimator of the intention-to-treat effect” (NRC report, p. 55). The NRC report also reminds us that where the mean of the response is non-linear in the explanatory variables (e.g., where the response is binary and logistic regression is used) the protocol or SAP must be clear as to whether the between-subject (population-averaged) or within-subject (subject-specific) treatment effect is being estimated. These are identical for some variables, for example, normally distributed, but may be different for others, for example, binary variables.
The EMA guideline is also cautious about the danger of data dredging when arriving at a model for the most frequent implementations of MAR – mixed models with repeated measures (MMRM), multiple imputation (MI) and weighted generalized estimating equations (GEE). The guideline emphasizes the need to pre-specify the model used and, in the case of MI, the random seed used. The NRC report (p. 64) notes the need to be cautious about MMRM because of its dependence on the correctness of its parametric model. This also applies to MI (NRC report, p. 69). The estimation of the variance–covariance matrix in MMRM may involve assumptions that are difficult to verify, such as the assumption of normality (NRC report, p. 64 again).
As noted in Chapter 1, an improved version of weighted GEE has been proposed recently, which has the attribute of being doubly robust. That is, its estimates will be valid if either the model for missingness or the model for the responses is correct. The NRC report opines that “With more published applications to real data and carefully designed simulation studies, the use of doubly robust estimators could become more (common) in the near future. However, at present, the operating characteristics of this method in applied settings with finite samples need to become more completely understood” (NRC report, p. 59). Chapter 8 of this book gives an account of doubly robust estimation.
The NRC report gives a list of possible estimands (pp. 22–29) formulated so as to be interpretable when a trial has missing data. (An “estimand” is that which is to be estimated in the trial analysis). We note that among these estimands, the only ITT-like one “Outcome Improvement for All Randomized Participants” requires that post-withdrawal efficacy be available and included in the analysis. The report itself notes that this particular estimand pertains to the effect of treatment policy, rather than the effect due to the experimental treatment. (See Section 4.2.1 on post-withdrawal data for a further discussion). Perhaps because of the weakness of the other approaches, the use of MAR is not ruled out, despite the reservations clearly expressed in both guidance documents. “In many cases, the primary assumption can be missing at random” (NRC report, p. 49). The EU guidance does not make this kind of explicit concession to the MAR approach. Nevertheless, its criterion that the analysis should be “unlikely to be biased in favor of the experimental treatment to an important degree (under reasonable assumptions)” may allow the justification of MAR for the primary analysis in some cases, and even perhaps in the majority of cases. An important proviso is that, given the weaknesses pointed out above, any MAR analysis will need to be supplemented by sensitivity analyses that test the robustness of results to the MAR assumption. Recommendation 11 of the NRC report states “random effects models in particular, should be used with caution, with all their assumptions clearly spelled out and justified” (NRC report, p. 77).
3.4.4 Methods that are used assuming MNAR
Methods used to handle missing data under the MNAR assumption include pattern-mixture models (PMMs), selection models (SEM), and shared parameter models. We present a non-technical overview here, with a more technical overview of PMMs in Chapter 7 as well as a technical summary of SEMs and shared parameter models in an appendix to that chapter. We note that most of the methods below can be used with binary as well as with continuous responses, although the theory on the validity of some of the methods is not as clear as in the binary case.
With PMMs one can specify a variety of assumptions, including MNAR assumptions, and the assumptions may differ by pattern of missingness. The subjects allotted to each pattern are often determined by treatment group and time of discontinuation, but can also be determined by reason for discontinuation, or in other clinically justifiable ways. The pattern-mixture approach requires clearly defined assumptions about clearly defined types or patterns of missingness. In other words, PMMs facilitate transparent pre-specification of assumptions tailored to each pattern of withdrawal, which is helpful to clinicians reviewing the study plan and eventually the clinical study report. As an example of an MNAR assumption, subjects who withdrew early because of lack of efficacy might be assumed to worsen over time until the protocol-defined end of the study (even if their previous efficacy gives no evidence for this); while subjects who withdrew for administrative reasons might be assumed to follow the MAR assumption. The EMA guideline sees merit in presenting results under such a mixture of assumptions: “It may be appropriate to treat data missing for different reasons in different ways” (p. 11). The NRC report states that “Many pattern mixture formulations are well suited to sensitivity analyses because they explicitly separate the observed data distribution from the predictive distribution of missing data given observed data.” It adds “The models are transparent with respect to how missing observations are being imputed because the within-pattern models specify the predictive distribution directly” (p. 74). The report's Recommendation 9 states that “assumptions (should be) stated in a way that can be understood by clinicians” (p. 76). As noted, PMMs suit this requirement.
SEMs assume a single distribution for the “full” data – both the observed and missing values – and model this jointly with the missingness indicator. The NRC report notes in favor of SEMs that “it may seem natural to assume the combined distribution … over observed and missing cases follows a single distribution” (p. 74). Sensitivity analyses can be implemented in SEMs by estimating the dependence of missingness on the unobserved values via a model parameter. However, the NRC report acknowledges that there is in fact no information about that very parameter, and that the model can only be fit “because of the parametric and structural assumptions being imposed on the full-data distribution.” While the full-data approach may be seen as an advantage, the authors of the report state that the dependence of SEMs on parametric assumptions is “a reason to exercise extreme caution (because) none of the assumptions underlying this parametric model can be checked from the observed data.” As Carpenter and Kenward (2007, p. 120) put it “the relationship between response and the unseen data can only be estimated subject to uncheckable modeling and distributional assumptions.” See Section 4.2.4.2 and appendix for a further discussion of limitations of SEMs as an approach for sensitivity analysis. Semi-parametric SEMs may offer a way of using the selection model approach that is more robust. However, semi-parametric SEMs are challenging to implement, and software is not readily available. Furthermore, there remains the difficulty of interpreting the results of a selection model in terms that clinician or a patient can understand. The NRC report notes that “it may not be intuitive to specify the relationship between non-response probability and the outcome of interest, which typically has to be done in the logit or probit scale.”
Shared parameter models attempt to model the relationship between missingness and outcome by positing a latent variable common to both. One may generate sensitivity analyses by estimating this variable and then artificially altering it to posit scenarios that are closer or farther away from MAR. As with SEMs, the NRC report notes the difficulty of interpreting the results: “Although these models can be enormously useful for complex data structures, they need to be used with extreme caution in a regulatory setting because of the many layers of assumptions needed to fit the models to data” (p. 76).
Given all the advantages and disadvantages outlined in the report (p. 103) for these three approaches, PMMs would seem to be most suitable for transparent interpretation.
We note that the above reasoning applies mostly to monotone missingness, and that the NRC report identifies dealing with non-monotone missingness as one of the key areas for future research, arguing that it is not always appropriate to treat it as MAR.
The EMA guideline in its Introduction states that “This document is not an extensive review of all the available methods” (p. 4), and later simply notes that “approaches that investigate different MNAR scenarios such as a PMM, SEM and a shared parameter model (SPM) may be useful” (p. 10).
Even where a trial is planned to have a primary efficacy parameter that is continuous, we briefly note here that a responder analysis can sometimes be useful as a potentially conservative sensitivity or MNAR analysis. In a typical responder analysis, a success/failure endpoint is calculated from a value or combination of values in a subject's data, while a subject with values missing for the endpoint is treated as a failure. For such an analysis to be potentially conservative, a greater proportion of missings must be expected in the experimental arm. The EMA guideline includes responder analysis as a possible supportive or sensitivity analysis, notably “where the … missing data is so substantial that no imputation or modeling strategies can be considered reliable.” The NRC report does not assess responder analysis.
3.5 Guidance about how to plan for missing data in a study
The NRC report gives six principles for handling missing data (p. 48–49):
The EU guideline emphasizes throughout the importance of pre-specification of analyses (the NRC report mentions the need for pre-specification but does no more than that). The EU guideline (p. 6) suggests using data from previous studies to help define assumptions with regard to missing data for future studies. The guideline suggests that the following factors could help the study team to decide a reasonable primary analysis and a set of sensitivity analyses (EU guidance, p. 8):
3.6 Differences in emphasis between the NRC report and EU guidance documents
We noted at the beginning of this chapter that the NRC report and the EU guidance document agree on most important issues. The previous section covered some comments from the NRC report on the detail of statistical methods, and we have noted that the EU guidance does not go into the same level of detail about statistical bases of approaches. Table 3.2 presents other key differences in emphasis, complete with page numbers so that the reader can view the context. However, we shall see that even here there are almost no real conflicts between the documents.
Table 3.2 Items where the NRC report and EU regulatory guidance document differ in emphasis.
Where emphasis differs between the documents |
|
| FDA commissioned NRC report | EMA guideline |
| Conservative approach | |
| “the need for conservative methods receives too much emphasis in … guidelines” (p. 19) | “important that … the method can be considered ‘conservative’ ” (p. 4) |
| LOCF | |
| “LOCF is anticonservative … where participants … do worse over time” (p. 66) | “LOCF … might be conservative … where the condition (improves) over time” (p. 9) |
| Post hoc analyses | |
| “secondary and sensitivity analyses … are certainly more valuable than post-hoc exploratory analyses” (p. 17) | “If unexpected missing data patterns are found in the data, it will be necessary to conduct some post hoc sensitivity analyses” (p. 7) |
| Non-monotone or interval missing data | |
| “non-monotone dropouts may require more specialized methods (than simple MAR-based ones” (p. 103) | “(MAR) could lead to biased results especially when data are missing due to withdrawal (rather than data for an interim visit being missing)” (p. 10) |
| Assumptions should be understood by clinicians | |
| “assumptions (should be) stated in a way that can be understood by clinicians” (p. 76) | Not mentioned in the document |
| Study report | |
| “sensitivity analyses should be part of the primary reporting” (p. 106) “more standardized documentation of missing data” (p. 112) | Section 5.3 (p. 7) describes items required for the final study report |
| Training | |
| “(FDA) and … companies that sponsor clinical trials should carry out continued training (in) missing data analysis” (p. 113) | Not mentioned in the document |
3.6.1 The term “conservative”
The NRC report has the academic's preference for a more technical term of an approach that aims to minimize bias as opposed to a general notion of a “conservative” approach. The frequent use of the term “conservative” in the EU guidance is tempered by frequent qualification of this word as meaning “unlikely to be biased in favor of the experimental treatment.” Such a qualified meaning for the term brings the EU guidance reasonably close to the NRC report in its recommendations for minimizing the bias of estimators.
3.6.2 Last observation carried forward
While we have seen that both NRC report and the EU guideline agree that single imputation methods like LOCF risk underestimating the variance of the estimate of treatment effect, the NRC report expresses only the down side of LOCF as an estimator. The EU guidance describes a limited potential use for LOCF. The NRC highlights important and widespread misconceptions about the alleged conservatism of the LOCF approach (pp. 65–66).
3.6.3 Post hoc analyses
As with LOCF, the NRC report describes only the disadvantages of post hoc analyses. In contrast, the EU guidance allows a place in the study report for post hoc sensitivity analyses, if missing data patterns are not as expected. This is consistent with the general awareness in the EU guidance that perfect planning for missing data is not always going to be possible.
3.6.4 Non-monotone or intermittently missing data
This rather technical item seems to be the only one on which the NRC and EU documents truly disagree. Recall from Chapter 1 that intermittently missing data typically occur when a subject who completes a clinical trial fails to turn up for some visits in the middle of the trial. The EU guidance only mentions intermittently missing data in passing, and implies (EU guidance, p. 7) that it is not as important a consideration as data missing due to early discontinuation (i.e., monotone missing data). It seems that the NRC report is aware of controversy raised by Robins and Gill (1997), regarding apparent difficulties in applying MAR assumptions to intermittently missing data. The arguments of Robins and Gill are quite complex, but in essence the reasoning seems to be that intermittently missing data are MAR only if their missingness is not dependent on the missingness of the previous observation – they argue that this assumption is not plausible. (See also Daniel and Kenward (2012), for a discussion of the Robins and Gill argument).
The NRC report states that uncertainty about how to handle non-monotone missing data “raises concern among members of the panel that non-monotone dropouts may require more specialized methods for modeling the missing data mechanism, and accounting for departures from MAR.” In practice, the amount of non-monotone data is not usually large in clinical trials, so this conflict between the two regions in regulatory tone is may not cause difficulties in implementing future development plans.
3.6.5 Assumptions should be readily interpretable
The NRC report emphasizes how important it is that the assumptions made in a clinical trial about missing data should be understood by non-statisticians and specifically, by clinicians. This is a criterion that is not covered by the EU guidance, and is a useful point to bear in mind when planning a study, especially when choosing sensitivity analyses. We saw above that the NRC report applied this criterion of interpretability to two methods for sensitivity analysis (SEMs and PMMs) and gave a more favorable “review” to PMMs as a result.
3.6.6 Study report
In line with its concern with the everyday practice of clinical trials, the EU guidance describes items it would expect in a final study report. In particular, the EU guidance recommends a “critical discussion of the number, timing, pattern, reason for and possible implications of missing values … in the clinical report” (p. 3). The NRC notes that “Systematic investigations of factors related to treatment dropout and withdrawal and to missing data more generally are needed,” recommends better standard reporting of information about missing data, and discusses options for deciding whether a trial result overturns the null hypothesis. However, it does not give detailed recommendations about the contents of an individual study report. See Section 4.6 for a more detailed discussion of the NRC suggestions for methods whereby it might be decided whether a trial result overturns the null hypothesis.
3.6.7 Training
The NRC report in its Recommendation 17 recommends “continued training … to keep abreast of up-to-date techniques for missing data analysis” for analysts both in industry and in FDA, and recommends also that FDA clinical reviewers should receive training.
3.7 Other technical points from the NRC report
3.7.1 Time-to-event analyses
On time-to-event analyses with missing data, the NRC report notes that ideally the statistical model should distinguish between administrative censoring (due to the subject reaching the scheduled end of the trial) and informative censoring. The key reference is to Scharfstein and Robins (2002). However, the Scharfstein and Robins paper is quite complex and no implementation of their method is publicly available at the moment. See Section 3.8.1 below for recommendations about missing data in time-to-event analyses from the EU.
3.7.2 Tipping point sensitivity analyses
It is noteworthy that in the chapter on sensitivity analyses in the NRC report, all the suggested sensitivity analyses involve adding an amount 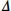 to some parameter so as to shift the assumption of the primary analysis and make the result less favorable to the experimental treatment. The report suggests that inference about the primary result can be tested as follows: “one can proceed by generating an estimate of m (the treatment effect) for each value of that is thought to be plausible … = 0 (might correspond) to MAR … .examining inferences about m over a set or range for that includes = 0 will summarize the effects of departures from MAR on inferences” (NRC report, p. 89). The NRC report shows how such an approach could be implemented using the selection model or PMM method. This kind of stress-test of a primary analysis is sometimes referred to as a “tipping point” test. The idea is described in a paper by Yan et al. (2009) from FDA: “Tipping points are outcomes that result in a change of study conclusion. Such outcomes can be conveyed to clinical reviewers to determine if they are implausibly unfavorable. The analysis aids clinical reviewers in making judgment regarding treatment effect in the study.” We shall describe straightforward methods to implement such tipping point analyses in SAS in Chapter 7.
to some parameter so as to shift the assumption of the primary analysis and make the result less favorable to the experimental treatment. The report suggests that inference about the primary result can be tested as follows: “one can proceed by generating an estimate of m (the treatment effect) for each value of that is thought to be plausible … = 0 (might correspond) to MAR … .examining inferences about m over a set or range for that includes = 0 will summarize the effects of departures from MAR on inferences” (NRC report, p. 89). The NRC report shows how such an approach could be implemented using the selection model or PMM method. This kind of stress-test of a primary analysis is sometimes referred to as a “tipping point” test. The idea is described in a paper by Yan et al. (2009) from FDA: “Tipping points are outcomes that result in a change of study conclusion. Such outcomes can be conveyed to clinical reviewers to determine if they are implausibly unfavorable. The analysis aids clinical reviewers in making judgment regarding treatment effect in the study.” We shall describe straightforward methods to implement such tipping point analyses in SAS in Chapter 7.
3.8 Other US/EU/international guidance documents that refer to missing data
3.8.1 Committee for medicinal products for human use guideline on anti-cancer products, recommendations on survival analysis
The Committee for Medicinal Products for Human Use (CHMP) guideline on the evaluation of anti-cancer medicinal products in man (European Medicines Agency, 2008), provides some recommendations for handling missing data in survival analysis. Key points in its line of reasoning are consistent with those in the recent US and EU regulatory documents on missing data: “There is no way to handle this problem that is optimal for all anti-cancer studies” (p. 2 of the Appendix), so any primary analysis should be accompanied by sensitivity analyses. “Outcome data should be collected … for all randomized patients,” so it is not generally acceptable to include only available cases. For survival data in particular, the Appendix suggests predefining and justifying rules of censoring. Censoring rules might, for example, “consider withdrawal or change of therapy prior to adjudicated progression/recurrence as events in an analysis of (progression free survival or disease free survival).” Regarding sensitivity analyses, the Appendix states only that they “should be sufficient to demonstrate that the trial results are robust and will depend on the clinical situation and nature of the trial data observed (e.g., patterns of patient withdrawals).”
3.8.2 US guidance on considerations when research supported by office of human research protections is discontinued
The US Office of Human Research Protections (OHRP, 2008) in the Department of Health and Human Services (DHHS) has issued a draft “Guidance on Important Considerations for when Participation of Human Subjects in Research is Discontinued.” This guidance is aimed at studies funded by or supported by the OHRP, and covers the basics (i.e., ensuring that the reason for discontinuation is documented and allowing for the use of data from discontinued subjects in a final study report.)
3.8.3 FDA guidance on data retention
In 2008, the FDA issued a “Guidance for Sponsors, Clinical Investigators, and IRBs: Data Retention when Subjects Withdraw from FDA-regulated Clinical Trials” (U.S. Food and Drug Administration, 2008). As with the OHRP guidance, the objective of this short guidance is to ensure that data from subjects who discontinued are retained. It also provides for continued collection of data after a subject withdraws, if permission is given by the subject (p. 6 of the data retention guidance).
3.9 And in practice?
The following anecdotal evidence gives a picture of regulators in the US and EU coming gradually to treat missing data as more important in their reviews, and beginning to apply the principles described in the US NRC report and the EU guidance.
Some regulators still recommend or approve LOCF, even for diseases whose symptoms worsen over time. We have seen a regulator approve LOCF as a primary analysis for Parkinson's disease, whose symptoms do worsen with time.
BOCF is regarded by some regulators as justifiable or required as a way of penalizing treatments that are not tolerated, particularly in studies of pain.
During a visit to regulators in EU countries recently, the potential inadequacy of single imputation methods was raised by the regulatory groups at three of the five meetings. The three groups that raised question of valid methods for missing data had a statistician among the regulators.
At a discussion on missing data at a meeting of the European Federation of Statisticians in the Pharmaceutical Industry (EFSPI) on Advances in the Treatment of Missing Data in 2011, a number of speakers noted that they had encountered regulators who required LOCF for the primary analysis. However, other speakers stated that they had encountered regulators who referred to the new regulatory documents and took into account those documents' deprecation of single imputation methods such as LOCF.
In a review of a SAP in 2011, FDA quoted the NRC report and, after discussion, approved a sensitivity analysis that used a PMM approach to implement control-based imputation, using the method described by Ratitch and O'Kelly (2011) and Ratitch et al. (2013). Control-based imputation models post-withdrawal data from the experimental treatment arm as if they were from the control arm (see Chapter 7 for a description of how to implement this method in SAS, with example code.) The regulator required a justification that the control-based imputation did in fact constitute a true and “conservative” stress-test of the primary study result. To do this, it was required to show from historic data evidence that in fact subjects who discontinued early from the experimental arm usually tended to continue to have symptoms that compared favorably to the control group, and that therefore the scenario with control-based imputation was rather less favorable to the experimental arm than would be likely to happen in clinical practice. The cautious nature of FDA's acceptance of control-based approaches is evident in a briefing document for a meeting of the Pulmonary Allergy Drugs advisory committee (U.S. Food and Drug Administration, 2013). The briefing document carefully notes the shortcomings of MAR and LOCF approaches (as might be expected); but the document also critiques the control-based analysis. The control-based analysis in this case was called copy difference from control (CDC), and is now better known as Copy Increment from Reference (see Section 7.3.5 for a full description). The critique in the briefing document echoes a criticism of control-based assumptions made during at least one conference by an FDA statistician, that the control subjects who remain in the study are themselves an “elite” group whose symptoms may be unrepresentatively good; and that therefore control-based assumptions are not as conservative as they appear to be, and perhaps not conservative enough. This argument is noted later in this book, in Section 4.2.2.3. Despite the critique, though, the conclusion in this case was “Nonetheless, it is reassuring that the results of the LOCF, MAR and the CDC MI analyses (applying various missing data assumptions) conducted by the applicant were all consistent in magnitude and direction to the primary analysis (MMRM).” Thus evidence from sensitivity analyses may be accepted, even when their assumptions are critiqued by the regulator.
Reviewing a protocol for a study to test for equivalence, the EMA pointed out that counting subjects with missing data as failures in a responder analysis would tend to bias towards a conclusion of equivalence – the more early discontinuations, the more equivalent the two treatment groups would appear. Instead, the regulator accepted an MAR approach, but also required sensitivity analyses to test the study result for robustness to the MAR assumptions.
Awareness among regulators of the importance of missing data is also applied in areas other than the planning and conduct of clinical trials. At an EMA-International Federation of Pharmaceutical Manufacturers Associations (IFPIA) workshop on Modeling and Simulation in London in November, 2011, regulators pointed out the importance of including a variety of assumptions about missingness in a specification of a simulation exercise, so that the robustness of a new method to plausible patterns of missingness could be assessed.
Finally, Section 2.2.2 includes an example showing that FDA may refuse to accept the results of a study with unwarranted proportions of missing data, when there is not an adequate plan in the protocol to prevent missing data and to deal with that missing data, when it occurs.
References
Carpenter JR, Kenward MG (2007) Missing Data in Randomised Controlled Trials – A Practical Guide. National Health Service Co-ordinating Center for Research Methodology, Birmingham, UK, http://www.hta.nhs.uk/nihrmethodology/reports/1589.pdf, accessed 18 June 2013.
Daniel R, Kenward MG (2012) A method for increasing the robustness of multiple imputation. Computational Statistics and Data Analysis 56: 1624–1643.
European Medicines Agency (2008) Appendix to the guideline on the evaluation of anticancer medicinal products in man (CHMP/EWP/205/95 REV. 3): methodological considerations for using progression-free survival (PFS) as primary endpoint in confirmatory trials for registration, EMEA/CHMP/EWP/27994/2008. http://www.ema.europa.eu/docs/en_GB/document_library/Other/2009/12/WC500017749.pdf, accessed 23 June 2013.
European Medicines Agency (2010) Guideline on Missing Data in Confirmatory Clinical Trials. EMA/CPMP/EWP/1776/99 Rev.1. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/09/WC500096793.pdf, accessed 23 June 2013.
International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (1998) Statistical principles for clinical trials: E9. http://www.emea.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002928.pdf, accessed 23 June 2013.
National Research Council. Panel on Handling Missing Data in Clinical Trials. Committee on National Statistics, Division of Behavioral and Social Sciences and Education (2010) The Prevention and Treatment of Missing Data in Clinical Trials. The National Academies Press, Washington, DC, available at http://www.nap.edu/catalog.php?record_id=12955, accessed 16 July 2013.
Office of Human Research Protections (2008) Guidance on important considerations for when participation of human subjects in research is discontinued. Washington DC. http://www.hhs.gov/ohrp/documents/200811guidance.pdf, accessed 23 June 2013.
Ratitch B, O'Kelly M (2011) Implementation of Pattern-mixture models using standard SAS/STAT procedures. In Proceedings of Pharmaceutical Industry SAS User Group, Nashville. http://pharmasug.org/proceedings/2011/SP/PharmaSUG-2011-SP04.pdf, acces-sed 23 June 2013.
Ratitch B, O'Kelly M, Tosiello R (2013) Missing data in clinical trials: from clinical assumptions to statistical analysis using pattern mixture models. Pharmaceutical Statistics, 12: 337-347, available at http://onlinelibrary.wiley.com/doi/10.1002/pst.1549/pdf, accessed 20 June 2013.
Robins J, Gill, R (1997) Non-response models for the analysis of non-monotone ignorable missing data. Statistics in Medicine 16: 39–56.
Scharfstein D, Robins J (2002) Estimation of the failure time distribution in the presence of informative censoring. Biometrika 89: 617–634, available at http://www.jstor.org/stable/4140606, accessed 23 June 2013.
U.S. Food and Drug Administration (2008) Guidance for sponsors, clinical investigators, and IRBs: data retention when subjects withdraw from FDA-regulated clinical trials. Rockville. www.fda.gov/downloads/RegulatoryInformation/Guidances/UCM126489.pdf, accessed 23 June 2013.
U.S. Food and Drug Administration (2013) Briefing document, Pulmonary Allergy Drugs Advisory Committee Meeting: NDA 204-275: fluticasone furoate and vilanterol inhalation powder for the long-term, maintenance treatment of airflow obstruction and for reducing exacerbations in patients with chronic obstructive pulmonary disease (COPD). http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Pulmonary-AllergyDrugsAdvisoryCommittee/UCM347929.pdf, accessed 23 June 2013.
Yan X, Lee S, Li N (2009) Missing data handling methods in medical device clinical trials. Journal of Biopharmaceutical Statistics 19: 1085–1098, available at http://www.tandfonline.com/doi/pdf/10.1080/10543400903243009, accessed 23 June 2013.