3
The boy who wasn’t short
You will never make a crab walk straight.
ARISTOPHANES
Different people are prone to different types of mistake. I’m particularly vulnerable to the mistake that underlies much of magic. Magicians rely, in part, on misdirection — guiding your gaze over there so you don’t notice something important that’s happening right here. In medicine, misdirection can come from other doctors, from the patient, or just from unlucky happenstance, and it tends to lead to what look like simple mistakes — errors you would never make if you were paying attention to the right thing.
If the magician’s art is to misdirect us, the art of medicine often lies in finding ways not to be misdirected. We talk about ‘traps for young players’ — but the truth is that old players can also be ensnared.
A few years ago, a general practitioner, not a young player, referred a small boy to me for investigation of short stature. This was a bit unusual, because most of the time such referrals go to a paediatrician first. Then they might go to an endocrinologist, a specialist in hormones, including those that direct growth. But there are many genetic conditions that can make a child short, so, although unusual, it wasn’t an unreasonable thought to ask a geneticist’s opinion. In this case, the story was quite worrying, because there had been a rapid crossing of centile lines.
Paediatricians track children’s growth using centile charts. These are graphs that show normal growth patterns, with lines representing different percentiles. Three per cent of children are taller than the 97th percentile for height. A quarter of children are shorter than the 25th centile for height. Half of all children are lighter than the 50th centile for weight. And so on. Most children, most of the time, grow along a particular line. Start small, relative to other babies — you will probably continue to be small.
The neat thing about centile charts for growth is that you can very easily use them to track whether things are progressing normally or not. Does that baby have a big head purely because he’s from a family who take large hat sizes, and is destined to do the same? Then he should track along the same centile line over time. Is his head crossing lines upwards? That might be a problem. Cross enough lines and he will most likely score a brain scan. Similarly, when someone has been on a particular track for height and then drops — like the boy in this story — that’s a worry, and it makes us sit up and pay attention. Growing is one of the most important things that children do, and, when they stop doing it, it’s important to find out why. It’s not that this child had shrunk, of course — more that he had grown one centimetre over a period when we would have expected him to grow seven.
I went through the usual process we follow with any new patient. I asked about the boy’s family and their heights. I found out about the pregnancy, about his birth and early development. I examined him, looking particularly for abnormal limbs, for disproportion between limbs and body. I looked at the creases on the palms of his hands, because, if the bones in your hands are short, the creases can form differently.
I found nothing. Not a thing. He was a completely normal-looking child, who to all intents and purposes had been doing fine — until his growth went off a cliff.
So I went back to the growth chart. Fortunately, his mother had brought in his ‘Blue Book’, a book new parents are given for recording health information about their child. Even more fortunately, there were several previous height measurements in the book. I plotted them on the growth chart — and the answer jumped out at me.
Every measurement throughout his life had placed this child a little below the 25th centile for height — except for the one that someone had done nine months earlier, which had him above the 90th centile. In retrospect, it was obvious that that measurement had been a mismeasurement: the boy had not plummeted from the 90th to below the 25th centile, because he had never been on the 90th in the first place.
The boy wasn’t short, and certainly didn’t need to see a geneticist. But I didn’t count this appointment as a waste of time. And in the long run, neither did the boy’s mother, because that referral may have saved her life.
*
When I was a medical student, cancer was a mysterious thing. Not that we were completely ignorant — far from it. We knew that there were plenty of things that could give you cancer. Smoking, of course. Asbestos. Certain viruses, such as HIV. Exposure to mustard gas, fortunately not a common problem nowadays. In Australia, melanoma heartland of the world (Come visit! You’ll love it here!) … the sun.
We even knew that there were some inherited types of cancer, and there was evidence as far back as the late 1950s that there were genetic changes in cancer cells. In particular, in 1959, two researchers in Philadelphia (Peter Nowell and David Hungerford) noticed that, in some leukaemia cells, chromosome 22 was abnormally short; it was named the Philadelphia chromosome. In 1973, Janet Rowley17 discovered that the reason for the short chromosome 22 was that part of the chromosome had broken off, and was stuck onto chromosome 9. This turned out to be enormously important, because it was the first of a whole class of chromosome abnormalities uncovered in cancer.
[1 Just a few weeks after Rowley’s discovery, Margaret Garson (a cytogeneticist in Melbourne) independently found the same thing. The two were friends, and the story told in Australia is that, when Rowley found out about Garson’s findings, she graciously offered to publish their findings together; but Garson declined, saying that Rowley had beaten her to it and deserved the credit.]
Many years later, the reason why this rearrangement of two chromosomes was a part of causing cancer was identified. The places where the two chromosomes break are in the middle of two different genes. Their fusion makes a new, hyperactive gene that drives abnormal cell growth. This discovery led in turn to the creation of a group of new treatments for some types of cancer (called tyrosine kinase inhibitors).
Over the past few decades, the biology of cancer has been worked out, in ever greater detail. It turns out that cancer is almost entirely a disease of the genome. The sickness that afflicts the genome of a cancer cell boils down to one thing: a mismatch between the accelerator and the brakes of the cell.
Growth is a fundamental part of life. At conception, you were a single cell. It was a huge cell, as cells go, about as wide as a strand of hair — but still a tiny, tiny thing. One of the most important tasks that cell had was to grow. As it divided and divided, signals were sent to the machinery of the new daughter cells, urging them to multiply and expand. These signals were obeyed, and, thanks to the rich bath of nutrients your mother provided, they were obeyed with gusto.
Which was fine — until it wasn’t. At some point, it’s not enough to be an ever-expanding ball of cells. You needed a shape. You needed some parts to grow, while others stopped. You even needed some cells to die. Six weeks after conception, you weighed about 500 times as much as when you were just a fertilised egg. If you’d continued growing at that rate, you’d have weighed more than the Earth before your first birthday.
This means that, to balance that first rush of acceleration, your cells also needed brakes. They needed a lot more than that, because all sorts of decisions had to be made. Which end is the top, and which is the bottom? Which side is the left, and which is the right? There are a pair of genes, named LEFTY1 and LEFTY2,18 that hold part of the answer to that one. Once you have a top and a bottom and a left and a right, you also have a front and a back. You’re still little more than a blob at this point, but you’re on your way.
[2 Yes, there should be a gene called RIGHTY. But there isn’t. The closest I can offer is a gene that’s important for the midline of your body, called MID1.]
There’s a beautiful complexity to what happens next. Proteins signal to each other in a kind of dance; instructions are sent to cells telling them their fate. You, and all your daughters, will be skin cells. You will be a nerve cell. You will be a liver cell. Grow along this line. Stop growing when you reach this point. Start doing your job, whatever it is: contracting, to make the heart beat; firing electrical signals, to make the brain work; filtering blood, to clean it and make urine.
But for some cells, the message reads: you will die. This is important in all sorts of ways — there is a process called programmed cell death, a pruning that takes out cells that aren’t needed. One of the places this is easiest to imagine is in the limbs. Your arm started out as a little nub, then grew to be a flipper. Your hand started as a lump on the end of the flipper. To make fingers, the cells in the gaps between the fingers had to go away — and so they did.
The message telling a cell that it needs to die is important later in life, too. Cells that are sick or damaged can trigger their own destruction. If this didn’t happen, a sick cell could cause problems by using up resources, by poisoning its neighbours, or by just getting in the way. Or by transforming into a cancer cell.
So there are three types of signal. Grow — the accelerators. Stop growing — the brakes. And the signal to die. All of these need to be in balance, and the balance is different for each cell type. In fact, for many individual cells, the brakes are locked fully on. Once a cell gets to be a mature white blood cell, for instance, by and large it is never supposed to divide again. Once it wears out, it has to be replaced by stem cells in your bone marrow. Stem cells hover around, not fully formed into a specialised cell but waiting until they are needed — at which point they divide, and one of the daughter cells matures into the needed blood cell (or liver cell or muscle cell, as the case may be) and the other steps back into the wings, waiting until it is needed again. Some other kinds of mature cell (such as skin cells) keep the ability to divide and replace themselves.
In some parts of the body (cartilage, brain), cells last a long time and seldom need replacement. Elsewhere, there is a great deal of wear and tear. Skin cells and the lining of your gut are constantly shearing off and being replaced. Rapid growth and replacement of cells creates opportunities for things to go wrong, which is one of the reasons skin cancer and bowel cancer are so common.
Every time a cell divides, its DNA has to be copied. Two sets of three billion pieces of information, copied out in the space of a few hours, in a tiny, tiny space. Mistakes get made. They get made all the time. Mostly, they are caught in time by the genome’s fact checkers, and are fixed. But there are plenty of chances for the fact checkers to get it wrong. A typical human body is made up of perhaps 30 trillion cells (30,000,000,000,000).19 In the course of an average lifetime, that might mean around ten quadrillion cell divisions (10,000,000,000,000,000). That’s a lot more than the number of cells, because of the many divisions needed to go from fertilised egg to adult, and because of all of the replacing of dead cells that has to happen.
[3 That’s just the human cells, of course — you are host to so many bacteria, protozoa, and fungi that there are about as many non-human as human cells in your body. Most of them are tiny, so at least the human part outweighs everything else. You probably woke up this morning thinking you were a human being, and you were only about 3 per cent wrong. Ninety-seven per cent human: not so bad.
It’s not just single-celled organisms, by the way. When I was at university, we had a lecturer who claimed that we have so many worms in our guts that, instead of saying ‘good morning’ to people, we should say, ‘How are your nematodes today?’
If that thought bothers you, I suggest you don’t look up face mites. No, really — forget I ever mentioned them.
Sorry.]
How many mistakes get missed? Well, if you’re typical, you have between 40 and 80 changes in your DNA, in every cell of your body, that you didn’t get from the genome of either parent, but will pass on to your children. Most of those had their origin in your father’s sperm. Making sperm is a high-speed, high-volume, and comparatively low-quality exercise compared with the bespoke tailoring that goes into making an egg.
Do these new changes in the DNA matter? It depends on exactly where the mistake happens. It can matter a great deal, as we shall discover. But mostly the changes happen in places where it doesn’t matter much: in between the genes, or in other places where it doesn’t make a difference if you change a C to a T or an A to a G.
What about your body’s allotted ten quadrillion cell divisions? How accurately is the DNA copied? Well, a recent study suggests that, typically, there is one new mistake per cell division. Yes, that’s right — almost every time a cell divides, something goes wrong, and the genome is imperfectly copied. Your genome is decaying, every minute of every day. In a very real sense, you don’t have ‘a’ genome — you have trillions of slightly different versions of the genome you started with. Almost no two cells share the exact same genetic make-up as each other.
If you have three billion bases of DNA, and there are ten quadrillion opportunities for it to be incorrectly copied, it follows that practically every single simple genetic change that is possible must happen many, many times over in a person’s lifetime. Plenty of chances for the brake genes to be damaged and to stop working, or for accelerator genes to be jammed in the ‘on’ position. Or for completely new accelerator genes to be created, as when the Philadelphia chromosome is formed.
You might ask how any of us are alive; how is it possible to survive from conception to birth without having cancer, let alone through childhood and into adult life?
It turns out that there are a couple of different answers to that. Firstly, and most importantly, the transformation from healthy cell to cancer cell is not something that happens overnight, or (usually) as the result of a single genetic mistake. There are backup systems in place, so that, for the most part, a single error isn’t enough to cause cancer — there need to be more piled on top of the first one, and of course they all have to happen in the very same cell. It’s a bit tricky working out exactly how many mistakes it takes, because one of the features of cancer cells is that their DNA copying often gets sloppy, leading to mistakes being made. Some of these aren’t part of why a cell is cancerous, but are a consequence of the chaos that goes along with being a cancer cell. This means that as well as ‘driver’ mutations, there are also ‘passengers’20 — not the cause of the problem, but along for the ride — and, when we study the DNA of cancer cells, it isn’t always obvious who’s doing what.
[4 In this case, a ‘driver’ can be either a faulty brake or an overactive accelerator. It’s anything that makes the cells grow faster.]
Thanks to our ability to sequence the whole genome of a tumour and compare it with healthy tissue, we are building up a picture of what it takes to go from being a well-behaved, functional cell to being a menace to society. It turns out that it’s probably not a very large number of mistakes — often as few as six or seven, and for some types of cancer it can be even fewer, maybe just one or two. But remember that they have to be exactly the right (or wrong) combination of mistakes. Our bodies are full of almost-cancers that will sit there and never bother us.
How frightening is the problem? Well, if you are of a nervous disposition, you may want to skip this next bit.
They probably didn’t think of it in quite this way, but, in 2015, a group of scientists from the United Kingdom set about working out exactly how scary the problem is. Four people who were having operations to tighten up sagging upper eyelids donated the excess skin for research. The skin samples looked completely normal under the microscope. The researchers in question took the leftover bits of skin, punched out 234 tiny biopsies, and read the genetic sequence in each of them. They looked only at a set of 74 genes that were known to be involved in the development of cancer, and within that they looked for changes in the genes that they could reasonably identify as potential drivers for cancer.
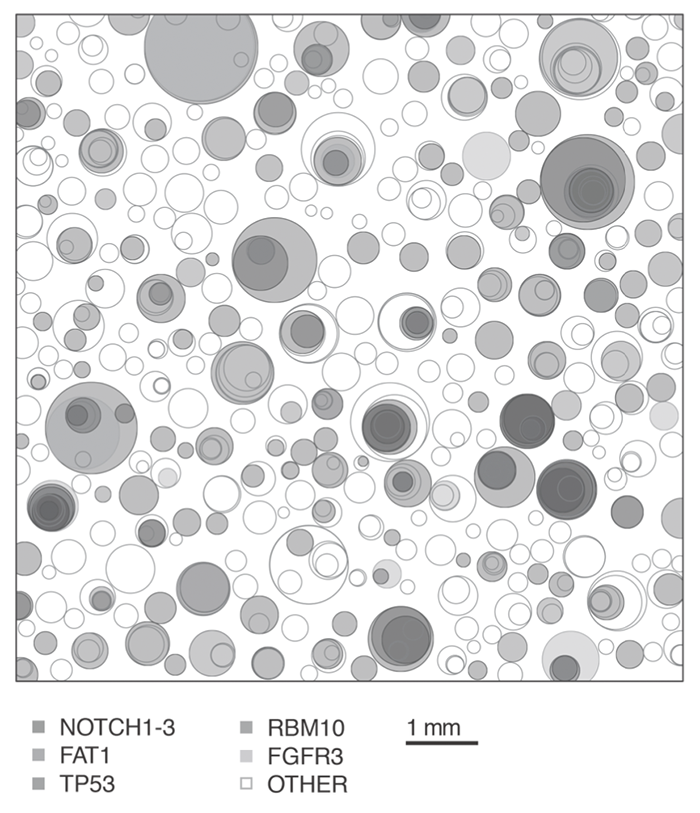
I don’t know what they expected to find, but what they actually found was … horrifying. Here’s an image from the paper:21
[5 From DOI: 10.1126/science.aaa6806. Reprinted with permission from AAAS.]
This picture represents one square centimetre of skin. Every circle is a region filled by cells that have a potential cancer-driver mutation. Each circle would have started as one cell that multiplied and expanded until its daughter cells filled the space. Where there are overlaps, there are cells that have more than one driver mutation. Five key genes or sets of genes get their own colours, and the other 67 genes are represented by the unfilled circles.
Here’s some consolation if that’s freaking you out a bit (it certainly gave me the willies when I first read the paper). This was sun exposed skin, and the changes were mainly the sort of changes caused by ultraviolet radiation.22 Also, the volunteers were aged 55–73. You wouldn’t find anything like this picture in, say, a piece of muscle sampled from a 20-year-old. On the other hand, these were all Brits. You call that sun exposure?
[6 Nowadays, I don’t even turn on the lights without putting on sunscreen first. You can’t be too careful.]
So why aren’t we all dead already? Firstly, you do need that series of unlucky breaks to happen to the same cell. Secondly, your immune system is pretty good at mopping up cancer cells — before any cancer can establish itself and get growing, it has to evade the cellular police. For this reason, people with immune deficiencies are at increased risk of getting cancer.
There’s another group of people who are at increased risk of getting cancer. Some people are unlucky enough to inherit a faulty brake or accelerator gene from one of their parents. If the change is there in every single cell from the word go, potential cancers have a head start. And so we see bowel cancer families, breast and ovarian cancer families, and so on. In families like this, cancers tend to be diagnosed at much younger ages than usual — because the cancer cells have been brewing since much earlier. So when we see a family in which there are multiple people affected by cancers that we know can be associated with a particular gene (like breast and ovarian cancer are), and many of them are being diagnosed at a young age — we suspect there may be one of these conditions in the family.
There are a surprising number of different conditions like this, but most of them are very rare. By far the most common involve a high risk of (mainly) bowel cancer and a high risk of (mainly) breast or ovarian cancer. It’s not always as easy as you might think to spot families with such a condition. Cancer is common, and you can get what looks like a familial cancer syndrome just by bad luck. On the other hand, it’s an increased risk, not a certainty, and there are many people who inherit one of these faulty genes but get away with it, living their whole lives unaffected by cancer. On top of that, men who inherit a mutation in one of the breast cancer genes, BRCA1 or BRCA2, are much less likely than the women to get cancer. They can get breast cancer — far more frequently than the general population, but it’s still uncommon. They have an increased risk of some other types of cancer, too, but the pattern is much less distinctive. All of this means that, sometimes, the family trees can be confusing and hard to sort out.
This was also one of the challenges faced by the people who were trying to find these genes in the first place. Nowadays, it’s possible to make a link between a genetic condition and a specific gene by studying a remarkably small number of affected people. This is because we have access to the sequence of the human genome, and because of the power of the new genetic sequencing technologies. For example, my recent PhD student Emma Palmer is working on the epileptic encephalopathies, a group of severe neurological conditions that affect small babies. In the course of her PhD studies, she played a major role in discovering that faults in no fewer than four different genes can cause epileptic encephalopathy. Terrific researcher though Emma is, this would have been an inconceivable feat for one PhD student until the last few years.
In the late 1980s and through to the mid-1990s, when BRCA1 and BRCA2 were found, it was a long, hard slog making such a discovery. BRCA1 and BRCA2 are brake genes, and mutations in the genes lift the cells’ feet (as it were) off the brake a little. It had been recognised for decades that some families have an inherited form of breast and ovarian cancer, with younger age at diagnosis on average than is usual for affected people in the general population. Over a number of years, researchers looked for genetic markers that could lead them to the genes. Think of the fly maps from the previous chapter, but, instead of finding that visible features like yellow body and white eyes were linked, the researchers were trying to find variable stretches of DNA that were connected to the occurrence of cancer. The closer they got to their targets, the tighter that linkage would be.
The process involved hundreds of researchers and many, many family members who volunteered as research subjects. In 1990, a group of researchers led by Mary-Claire King at the University of California, Berkeley announced that they had narrowed down the location of BRCA1 to chromosome 17. Gradually, her group and others whittled away at the chromosome. In May 1994, a group from the University of Utah, in collaboration with a group from Cambridge University in the UK, published a detailed map of the region, which by then contained only a little over 20 genes, one of which must be BRCA1. By October 1994, the race to find the gene was won … but along with the announcement, and publication of the sequence of the gene, by Mark Skolnick and colleagues from the University of Utah, came a shock for those in the field. A company called Myriad Genetics, formed by Skolnick’s group, had applied for a patent on the gene. This was one of a series of patents that essentially tied up commercial testing for BRCA1. And then they did it again. In a similar international effort, the location of BRCA2 had been narrowed down to chromosome 13. In December 1995, another British-led group, headed by Michael Stratton, published the sequence of BRCA2. But the day before the paper was published, Myriad announced that it, too, had found the gene, and had filed for a patent.
To most people in the field, the idea of patenting a gene seemed, and seems, absurd. Myriad didn’t invent either gene, after all — they discovered it. Moreover, Myriad’s discoveries would not have been possible without the work done to map the genes by many other, publicly funded groups, and all of that work rested on patients who volunteered to take part in research. It seemed deeply wrong to most of us that a company could profit from this. But profit they did, and for a long time.
This played out differently in different parts of the world. In the US, Myriad held a stranglehold on diagnostic testing for BRCA1 and BRCA2 for nearly 20 years, until finally the US Supreme Court struck down their key patents, which previously had been repeatedly upheld by lower courts. In Australia, a company called GTG had a patent on all of the genome that did not code for genes — i.e. most of it. Spoiler alert: GTG didn’t invent that either — but they had still been granted a patent for it, and they had argued that Myriad had infringed this patent. The end result was a deal that left GTG holding the BRCA patents in Australia. This was highly controversial and never really enforced, but it was a big relief to the field when the High Court of Australia also struck down the patents, in 2015.
*
It has been said that women have double the number of relatives that men do. When we take a family history from a woman, we generally learn twice as much about the family as we would if we had spoken to her brother instead. Sometimes, this really matters. When I saw the boy who wasn’t short, I took a family history from his mother, because this is something we always do. She knew that her first cousin, her uncle’s daughter, had recently been diagnosed with breast cancer at the age of 50. She also knew about two others in the family who had died of breast cancer at a young age, and another who had died of ovarian cancer. She didn’t know that breast and ovarian cancer could be inherited, so it didn’t occur to her to seek advice, and, in any case, we usually start by testing someone who is affected by cancer. Her cousin’s doctors could have put the pieces together — if her cousin’s father had known and passed on the information.
When I learned of the family history, I arranged for the cousin to be seen by a cancer genetics service. They tested her, and found that she carried a mutation in BRCA1. Later, the woman I had seen also decided to be tested, and found that she, too, had inherited the faulty copy of the gene. In one sense, of course, this was bad news for her — far better not to learn that you are at high risk of cancer. But this gave her options, which included having more extensive screening than she would otherwise have had, with a chance of detecting cancers early, meaning a better chance of cure. It also gave her the option of having risk-reducing surgery — removal of her ovaries and breasts before cancer had a chance to develop. A tough choice, but one that we know can reduce the chance that BRCA1 mutation carriers will die from cancer. Several other women in the family had the opportunity to be tested as well. It’s likely that sooner or later, someone in the family will have their life saved by this information.
Although I’m a doctor, Angelina Jolie has saved far more lives than I ever have or could hope to. When she told the story of learning that she, too, carried a BRCA1 mutation, and of her choice to have surgery, our cancer genetics services were swamped with people who had considered their family history in a new light and sought testing. We called this the Jolie effect. As a result, our labs were flooded with samples, many of which tested positive. The rate of people having risk-reducing surgery, as Jolie had done, doubled in Australia and in the US, and probably in many other countries. There are people who are alive today only because of Jolie’s decision to speak out.
The risks for people with BRCA1 and BRCA2 mutations are greatly increased compared with the general population, but there are no certainties. Some carriers live their lives unaffected by cancer, while others with the same genetic make-up die young. This means that making a choice to have surgery represents a decision in the face of uncertainty.
Offering people choices with uncertain outcomes is standard operating practice in genetics, as we shall see.