5
Non-alcoholic fatty liver disease (NAFLD) as cause of cryptogenic cirrhosis
Jay H. Lefkowitch
Department of Pathology and Cell Biology, College of Physicians and Surgeons, Columbia University, New York, NY, USA
Introduction
Most classifications of cirrhosis dating back 50 years or more have reserved the final entry on the list for the category “cryptogenic cirrhosis”: cirrhosis of unknown cause. With each passing era, this category has been winnowed because of major advances in diagnostic methods and in our understanding of the etiopathogenesis of hepatobiliary diseases. Two prime examples are the identification of the hepatitis C virus (HCV) in 1989 and the seminal description of non-alcoholic steatohepatitis (NASH), published by Ludwig and colleagues at the Mayo Clinic in 1980 [1]. The discovery and, then, availability of serologic tests for HCV lifted the veil from many cases of cirrhosis thought to be cryptogenic or attributed to the viral agent non-A, non-B hepatitis. The impact of such discoveries is evident when comparing prevalence rates of cryptogenic cirrhosis in publications from nearly 35 years ago to current data. A relatively high prevalence rate of cryptogenic cirrhosis (35–59.6% of cases of cirrhosis) was reported in a 1981 20-year prospective study of cirrhosis from Birmingham, United Kingdom [2] (published before the identification of HCV and only 1 year after the description of NASH by Ludwig et al.). More recent estimates show a considerably lower prevalence of cryptogenic cirrhosis of 5–30% of cases [3], predominantly because of wider recognition and understanding of the clinicopathological features of NAFLD and NASH in a period of alarming growth of worldwide obesity and diabetes. Thus, at present, patients with newly diagnosed cirrhosis of uncertain cause will very likely be scrupulously assessed for risk factors for non-alcoholic fatty liver disease (NAFLD), and many will have a liver biopsy to confirm such a diagnosis. Recent data suggest that NAFLD and NASH are actually responsible for 30–70% of cryptogenic cirrhosis [4]. Thus, when considering individuals with cirrhosis at the present time (Table 5.1), after exclusion of the well-established causes and any clinicopathological evidence supporting NAFLD/NASH as a possible cause, the percentage of cases that can truly be deemed “cryptogenic” is likely to be of the order of only 5–10%. Schuppan and Afdahl in a recent review of cirrhosis [5] also provide a similar overall assessment: “the diagnosis of cirrhosis without apparent cause (cryptogenic cirrhosis) is rarely made.” In the next decade or two, even this number will decline due to the advent of molecular and genomic diagnostics and application of the technologies that are currently referred to as “systems biology,” as will be discussed later.
TABLE 5.1 Causes of cirrhosis
| Well established |
| Chronic hepatitis |
| Hepatitis B virus (HBV) |
| Hepatitis C virus (HCV) |
| Autoimmune hepatitis (AIH) |
| Alpha-1-antitrypsin deficiency |
| Wilson’s disease |
| Fatty liver disease (macrovesicular) |
| Alcoholic (AFLD) |
| Non-alcoholic (NAFLD) |
| Other metabolic disorders |
| Hemochromatosis |
| Drug-induced liver injury (DILI) |
| Biliary tract disease |
| Chronic large bile duct obstruction |
| Primary biliary cirrhosis |
| Primary sclerosing cholangitis |
| Chronic ductopenic disorders |
| Hepatic venous outflow obstruction (e.g., Budd–Chiari syndrome) |
| Possible |
| Late, inactive autoimmune hepatitis |
| Occult viral hepatitis HBV or HCV |
| Antihepatitis viral therapy with sustained viral response (SVR) |
| “Posthepatitic” cirrhosis (following unknown viral infection or drug hepatitis) |
| Chronic hepatitis E virus (HEV) infection (immunosuppressed individuals) |
| Unknown/rare (“cryptogenic”) |
| Mitochondriopathies |
| Keratin mutations |
| Short telomere syndromes |
| Metabolic/enzyme mutations |
Cryptogenic cirrhosis: Definition and characteristics
The term “cryptogenic cirrhosis” implies that no recognizable cause of cirrhosis has been found by clinical, pathological, serological, biochemical, or historical investigations [3]. For the clinician, the prototypic patient with cryptogenic cirrhosis is likely to be 60 years or older, often female, with obesity and diabetes and is discovered to have cirrhosis while asymptomatic and being evaluated for another condition, or will have recently presented with new ascites, variceal hemorrhage, encephalopathy, or hepatocellular carcinoma (HCC) [3]. Serum liver tests are usually nondiscriminating, with normal or slightly raised aminotransferases. Serum autoantibody evaluation to exclude autoimmune hepatitis (AIH) is usually negative, unless there is positivity (typically low titer) for antinuclear or antismooth muscle antibodies (a nonspecific/generic immune response that is seen in many individuals with NAFLD) [6].
For the pathologist, the “cryptogenic cirrhosis” found in liver biopsy, explant, or postmortem specimens is a “faceless” cirrhosis characterized by bland fibrous septa surrounding regenerative nodules without steatosis, cholestasis, or hemosiderin (Figure 5.1). The specific histological features associated with known etiologies will largely be absent (or potentially overlooked), including interface hepatitis (chronic hepatitis B and C and AIH); ground-glass hepatocellular inclusions (chronic hepatitis B); portal tract lymphoid aggregates (chronic hepatitis C and other causes of chronic hepatitis); bile duct damage, loss, or absence (primary biliary cirrhosis, primary sclerosing cholangitis); periductal onion-skin fibrosis (primary sclerosing cholangitis); copper and copper-binding protein positivity (Wilson’s disease, chronic cholestatic diseases); and diastase-treated periodic acid–Schiff (DPAS) stain-positive globules within periportal hepatocytes (alpha-1-antitrypsin deficiency).
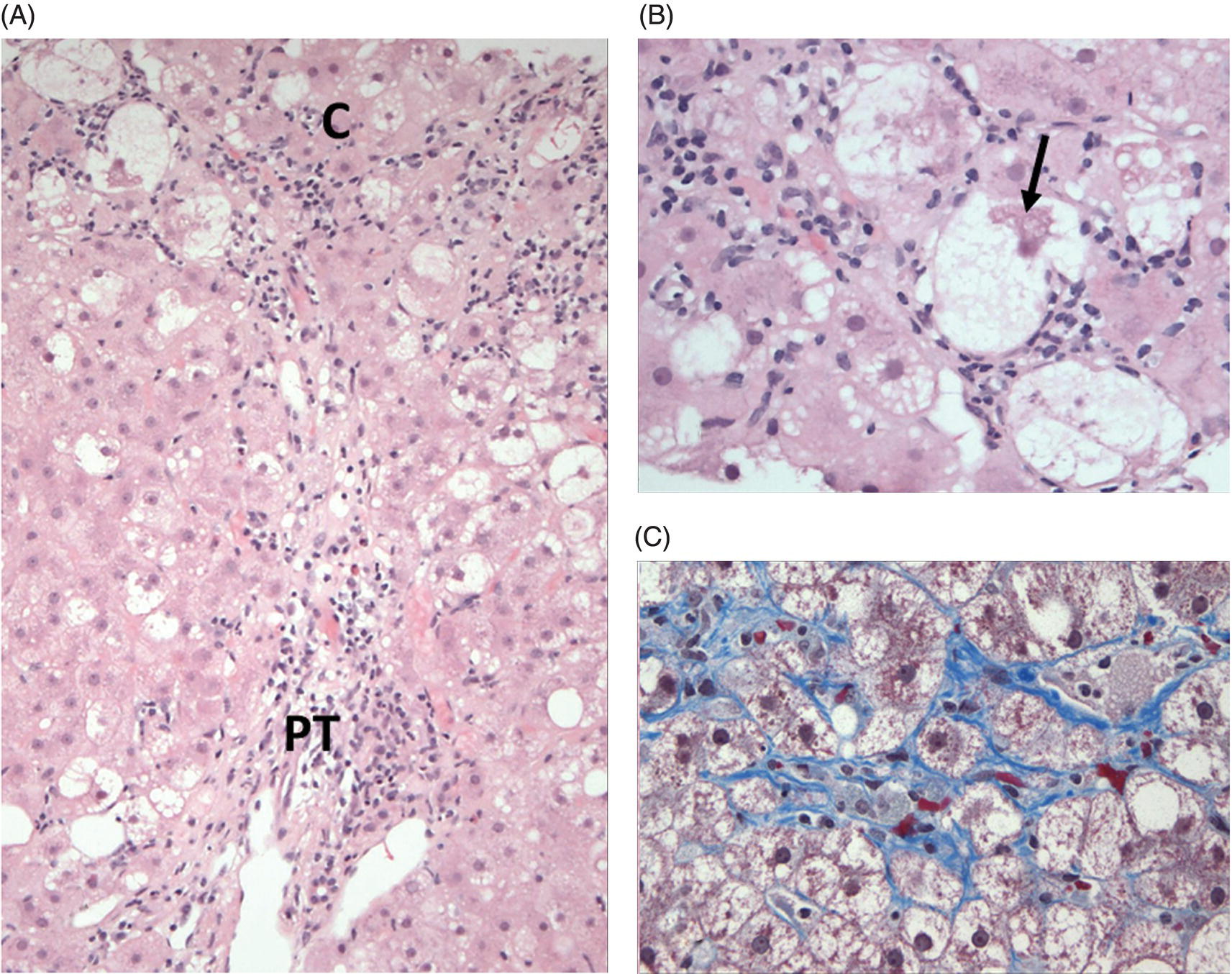
FIG 5.1 Classical non-alcoholic steatohepatitis (NASH). (A) Steatosis, hepatocyte ballooning, and inflammation (the trio of changes representing the minimal histological criteria of steatohepatitis) are evident in the centrilobular region (C). (B) Hepatocyte ballooning and intracellular Mallory–Denk bodies (arrow) are shown at high magnification. (C) The characteristic pericellular/perisinusoidal “chicken-wire” pattern of fibrosis surrounding centrilobular hepatocytes is seen on this trichrome stain (A and B, hematoxylin and eosin stain; C: Masson trichrome stain).
Pathological recognition of NAFLD/NASH in cryptogenic cirrhosis
The range of histological findings that constitute NAFLD includes the spectrum from macrovesicular steatosis to steatohepatitis, cirrhosis, and HCC [7–11]. Although the diagnostic histological features of NASH have been the subject of many studies since its first description by Ludwig and colleagues [1], many, if not all, of the hallmark changes may disappear once cirrhosis has developed, particularly steatosis (discussed later). NAFLD and NASH in adults chiefly affect centrilobular regions (acinar zone 3). In its fullest expression, the features of NASH include macrovesicular steatosis, hepatocyte ballooning, inflammation (lymphocytes and neutrophils), intrahepatocellular Mallory–Denk bodies [12], and perisinusoidal fibrosis surrounding hepatocytes in a characteristic “chicken-wire” pattern (Figure 5.1). The minimum criteria for a diagnosis of NASH include macrovesicular steatosis, hepatocyte ballooning, and inflammation [11], and these can be evaluated semiquantitatively to determine the NAFLD activity score [13] (NAS) and the likelihood that NASH is present. Additional helpful diagnostic features may be seen, such as giant mitochondria [14, 15] and, in diabetic subjects, “glycogen nuclei” in periportal hepatocytes (Figure 5.2).
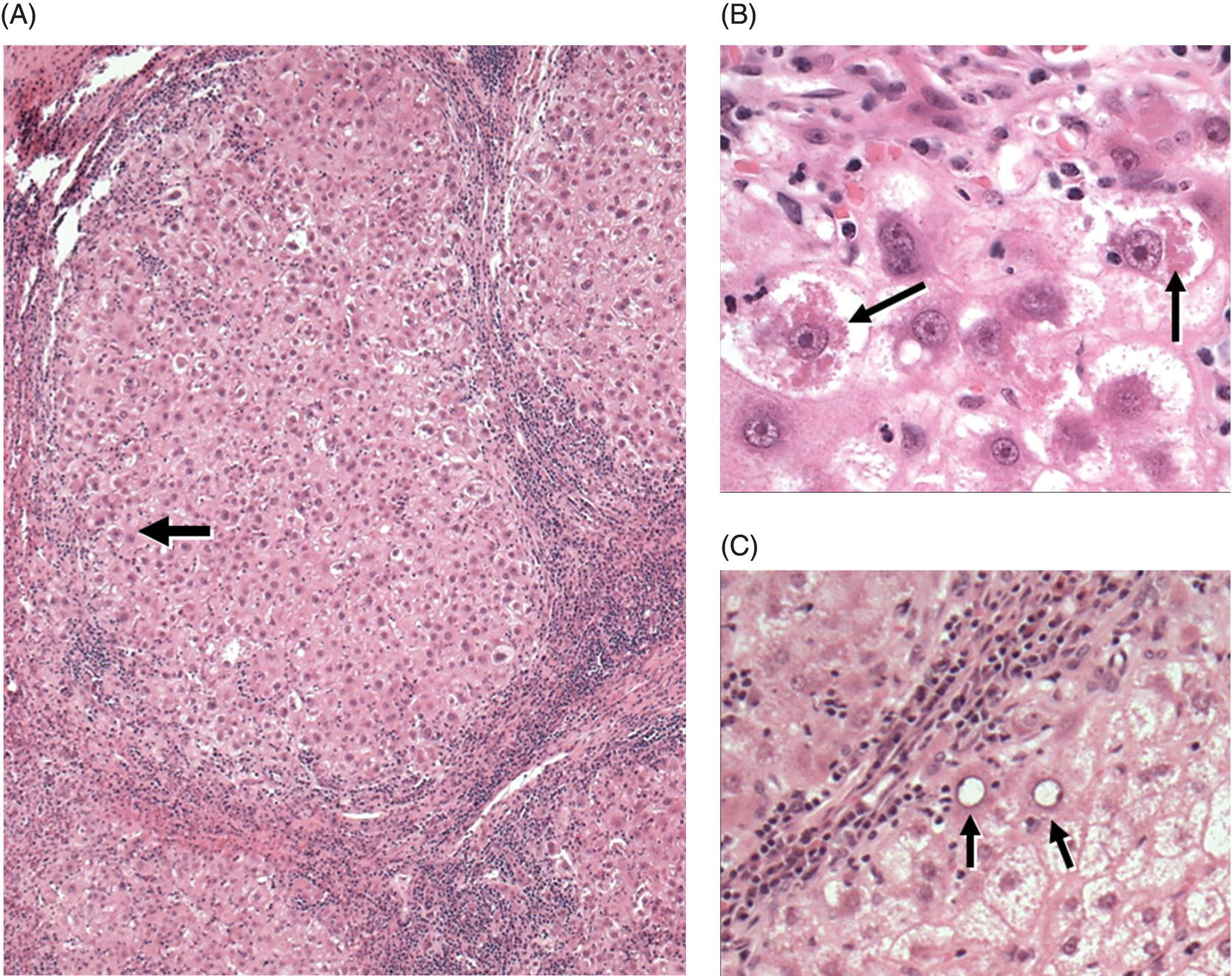
FIG 5.2 “Cryptogenic cirrhosis” due to NAFLD. (A) Cirrhosis without fat is seen at low magnification. Portal tracts and fibrous septa contain mild to moderate chronic inflammatory cell infiltrates, rendering a superficial resemblance to chronic hepatitis. However, careful examination of the hepatocytes located at the periphery of the nodules (arrow) allows recognition of remaining NASH features (seen at high power in panel B). (B) High magnification of the parenchyma near the arrow in panel A. Hepatocytes are variably ballooned and there is robust Mallory–Denk body formation (arrows). (C): Occasional periportal/periseptal hepatocytes show glycogenated nuclei (arrows) (A, B, and C: hematoxylin and eosin stain).
The fibrosis surrounding central veins in NASH eventually involves many or most central veins. Once established perivenular fibrosis extends through the hepatic lobules toward other central veins and, importantly, also toward portal tracts, thereby subdividing the lobular parenchyma into small units of hepatocytes. The process of central-to-portal bridging fibrosis in progressive NASH is associated with activation of periportal hepatic progenitor cells that form bile ductular structures (ductular reaction), reflecting the impaired replication capacity of hepatocytes in NAFLD and NASH [16, 17]. In late NAFLD-related cirrhosis when fat is absent (or minimal and patchy), other features of NASH may be subtle and must be sought assiduously. Hepatocyte ballooning and Mallory–Denk bodies may be found microscopically in the peripheral regions of the cirrhotic nodules, near fibrous septa and portal tracts (Figure 5.2). Nonballooned hepatocytes may contain Mallory–Denk bodies. Trichrome connective tissue stain may disclose residual foci of perisinusoidal/pericellular “chicken-wire” fibrosis. The predilection of NASH for centrilobular regions and central-to-central bridging fibrosis may spare some portal tracts that come to be localized in the centers of regenerative nodules, a process referred to as “reversed lobulation.” The central-to-portal bridging fibrosis that often evolves from NASH may be remodeled to produce slender fibrous septa that are helpful regions for histological examination for residual hepatocyte ballooning and/or Mallory–Denk bodies. The hepatocytes near such slender septa may also show persistent immunostain positivity in adjacent hepatocytes for the urea cycle enzyme glutamine synthetase that is normally localized to centrilobular hepatocytes [18].
Evidence for NAFLD as the cause of cryptogenic cirrhosis
Two landmark papers from the 1990s provided critical evidence for NAFLD/NASH as the etiology of many cases of cryptogenic cirrhosis. In 1990, Powell and colleagues [19] described a cohort of patients with NASH who transitioned to cirrhosis over a 5-year period with loss of fat and inflammation, thereby highlighting the potential for so-called “burnt-out” NAFLD. This was followed in 1999 by the study by Caldwell and colleagues [20] in which diabetes and obesity were found significantly more often in a cohort of subjects with cryptogenic cirrhosis in comparison to patients with primary biliary cirrhosis or chronic hepatitis C-related cirrhosis. These studies initiated a line of evidence that has been assembled during the subsequent decade that lends further support for NAFLD as the etiology of many, if not most, cases of cryptogenic cirrhosis. This evidence was reviewed by Caldwell and Crespo [4] and included (i) studies of explant livers from patients transplanted for cryptogenic cirrhosis who later developed posttransplantation NASH, with patchy steatosis, Mallory–Denk bodies, and hepatocyte ballooning [21]; (ii) correlative clinicopathological studies of individuals with cryptogenic cirrhosis in whom a cause was found in 85% (33% of which had features of NASH) [22]; and (iii) patients with cryptogenic cirrhosis and HCC in whom obesity and diabetes were significantly more common in comparison to matched controls [23].
Loss of steatosis in late NAFLD/NASH with cirrhosis
NAFLD has eluded pathological detection as the cause of many cases of cryptogenic cirrhosis in large part due to the loss of large droplet steatosis from hepatocytes in the late cirrhotic stage (Figure 5.2). The pathogenesis of this fat loss has been the subject of several studies and hypotheses [24] (Table 5.2). Most hypotheses have addressed those physiological features of cirrhosis that would directly or indirectly affect acquisition of triglyceride by hepatocytes, chiefly the altered vascular inflow patterns [25, 26] (and delivery of factors such as insulin and fatty acids) due to portal hypertension and abnormal sinusoidal access of steatogenic factors to underlying hepatocytes because of changes in the sinusoidal endothelium (abnormal fenestrae) or increased collagenous barriers located under the endothelium in the space of Disse (“capillarization” of the sinusoids in cirrhosis, with gains of basement membrane collagen) [27]. More recent work has focused on the antisteatotic effects of adiponectin on hepatocytes and the mechanisms whereby serum adiponectin becomes elevated in late NAFLD cirrhosis and mediates loss of fat [24]. Elevation of serum bile acids develops in many late forms of cirrhosis, including late NAFLD-related cirrhosis, and bile acid “cross talk” between adipocytes and hepatocytes and binding to bile acid receptors on adipocytes are among the pathways considered to be important in mediating elevation of serum adiponectin in late cirrhosis due to NAFLD.
TABLE 5.2 Possible causes of fat loss in late NAFLD-related cirrhosis
Source: See Ref. [24] for further discussion and study citations.
|
Other possible causes of cryptogenic cirrhosis and future directions
Conditions other than NAFLD can result in cryptogenic cirrhosis and should be considered in the differential diagnosis (Table 5.1). Late AIH without residual interface hepatitis and with only minimal or mild portal and septal lymphocytes and plasma cells is one such disorder. Occult or inactive HBV or HCV infection can also be included in this category. Antiviral treatment of an individual with HBV or HCV cirrhosis may result in a sustained viral response (SVR) status and a paradoxically bland-appearing cirrhosis with little or no inflammation. In immunosuppressed individuals, particularly liver transplant recipients, the possibility of developing posttransplantation cirrhosis due to chronic hepatitis E virus (HEV) infection also requires serologic exclusion [28–30].
Rarer conditions that may underlie cryptogenic cirrhosis include mitochondriopathies, keratin mutations, and short telomere syndromes. Mitochondriopathies comprise a variety of point mutations and/or deletions of mitochondrial DNA (mtDNA) resulting in defects of the respiratory chain [31]. Such defects can affect hepatocytes, muscles, and tissues of the nervous system, thereby causing multiorgan syndromes (e.g., neurohepatopathies) with cirrhosis in neonates, children, and adults. Loss of function mutations affecting the telomerase complex (which normally allows multiple generations of cell division and regeneration by adding successive TTAGGG sequences to the 3′ end of chromosomes) may cause an otherwise unexplained cirrhosis associated with idiopathic pulmonary fibrosis [32] or with dyskeratosis congenita [33]. Some cases of cryptogenic cirrhosis may be due to keratin 8 gene mutations [34].
In the coming decade, for the relatively small numbers of individuals for whom a cause of cirrhosis cannot be determined, it is likely that one or more systems biology techniques [35] will elucidate the etiology. These techniques, collectively referred to as “omics” (genomics, transcriptomics, proteomics, and metabolomics), allow identification of distinct moieties such as genomic sequences, proteins, and peptides within cells and tissues, which define or provide a “signature” of that individual’s disease. NAFLD is an example where such techniques have already been broadly applied and have generated exceptionally useful data. Genomic studies assessing single nucleotide polymorphisms (SNPs) have demonstrated an important and consistent mutation in the PNPLA3 gene in subjects with NAFLD that results in a nonsynonymous substitution of a methionine for an isoleucine at position 148 in the enzyme PNPLA3 and the accumulation of fat in hepatocytes [36]. PNPLA3 (patatin-like phospholipase domain-containing protein 3 or adiponutrin) is a triacylglycerol lipase that mediates triacylglycerol hydrolysis in adipocytes. Metabolomics (detecting small-sized molecules, <1500 Da in size, by high-throughput nuclear magnetic resonance spectroscopy and/or mass spectrometry) in NAFLD has demonstrated a specific lipidomic signature that is associated with progressive disease [37, 38]. These are only several examples of the potential future uses of systems biology in clarifying the etiology of remaining cases of “cryptogenic” cirrhosis.
Summary
Cryptogenic cirrhosis has, by definition, always been a diagnosis of uncertainty as far as its etiology is concerned. The past 30 years have shown an alarming growth in the prevalence of obesity, diabetes, metabolic syndrome, and NAFLD in developed countries and elsewhere. Outcomes data on the natural history of NAFLD have emerged and indicate that ~9–20% of individuals with NASH will later develop cirrhosis [39]. Many of these individuals with cirrhosis will retain sufficient clinical and pathological features of NAFLD and NASH to make the etiology clear, but there yet remain individuals with NAFLD who are diagnosed as having “cryptogenic cirrhosis,” especially when major histological markers of NAFLD and NASH (particularly large droplet fat) have disappeared. Some 30–70% of cryptogenic cirrhosis cases today are believed to be due to NAFLD/NASH. Future investigations hold the promise of further winnowing the category of cryptogenic cirrhosis by identifying specific NAFLD/NASH genomic and/or metabolomic signatures and determining the presence of alternative unusual disorders by applying systems biology techniques (e.g., transcriptomics, genome-wide sequencing) to individual cases.
References
- 1. Ludwig J, Viggiano TR, McGill DB, Oh BJ. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin Proc 1980; 55: 434–438.
- 2. Saunders JB, Walters JRF, Davies P, Paton A. A 20-year prospective study of cirrhosis. Br Med J 1981; 282: 263–266.
- 3. Caldwell S. Cryptogenic cirrhosis: what are we missing? Curr Gastroenterol Rep 2010; 12: 40–48.
- 4. Caldwell SH, Crespo DM. The spectrum expanded: cryptogenic cirrhosis and the natural history of non-alcoholic fatty liver disease. J Hepatol 2004; 40: 578–584.
- 5. Schuppan D, Afdhal NH. Liver cirrhosis. Lancet 2008; 371: 838–851.
- 6. Loria P, Lonardo A, Leonardi F, et al. Non-organ-specific autoantibodies in nonalcoholic fatty liver disease: prevalence and correlates. Dig Dis Sci 2003; 48: 2173–2181.
- 7. Yeh MM, Brunt EM. Pathology of nonalcoholic fatty liver disease. Am J Clin Pathol 2007; 128: 837–847.
- 8. Hübscher SG. Histological assessment of non-alcoholic fatty liver disease. Histopathology 2006; 49: 450–465.
- 9. Tannapfel A, Denk H, Dienes H-P, et al. Histopathological diagnosis of non-alcoholic and alcoholic fatty liver disease. Virchows Arch 2011; 458: 511–523.
- 10. Brunt EM. Non-alcoholic fatty liver disease: what’s new under the microscope? Gut 2011; 60: 1152–1158.
- 11. Yeh MM, Brunt EM. Pathological features of fatty liver disease. Gastroenterology 2014; 147: 754–764.
- 12. Zatloukal K, French SW, Stumptner C, et al. From Mallory to Mallory–Denk bodies: what, how and why? Exp Cell Res 2007; 313: 2033–2049.
- 13. Kleiner DE, Brunt EM, Van Natta M, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005; 41: 1313–1321.
- 14. Le TH, Caldwell SH, Redick JA, et al. The zonal distribution of megamitochondria with crystalline inclusions in nonalcoholic steatohepatitis. Hepatology 2004; 39: 1423–1429.
- 15. Lotowska JM, Sobaniec-Lotowska ME, Backowska SB, Lebensztejn DM. Pediatric non-alcoholic steatohepatitis: the first report on the ultrastructure of hepatocyte mitochondria. World J Gastroenterol 2014; 20: 4335–4340.
- 16. Richardson MM, Jonsson JR, Powell EE, et al. Progressive fibrosis in nonalcoholic steatohepatitis: association with altered regeneration and a ductular reaction. Gastroenterology 2007; 133: 80–90.
- 17. Gadd VL, Skoien R, Powell EE, et al. The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology 2014; 59: 1393–1405.
- 18. Lefkowitch JH, Morawski JL. Late nonalcoholic fatty liver disease with cirrhosis: a pathologic case of lost or mistaken identity. Semin Liver Dis 2012; 32: 92–98.
- 19. Powell EE, Cooksley WGE, Hanson R, Searle J, Halliday JW, Powell LW. The natural history of nonalcoholic steatohepatitis: a follow-up study of forty-two patients for up to 21 years. Hepatology 1990; 11: 74–80.
- 20. Caldwell SH, Oelsner DH, Iezzoni JC, Hespenheide EE, Battle EH, Driscoll CJ. Cryptogenic cirrhosis: clinical characterization and risk factors for underlying disease. Hepatology 1999; 29: 664–669.
- 21. Conto MJ, Cales W, Sterling RK, et al. Development of nonalcoholic fatty liver disease after orthotopic liver transplantation for cryptogenic cirrhosis. Liver Transpl 2001; 7: 363–373.
- 22. Ayata G, Gordon FD, Lewis WD, et al. Cryptogenic cirrhosis: clinicopathologic findings at and after liver transplantation. Hum Pathol 2002; 33: 1098–1104.
- 23. Bugianasi E, Leone N, Vanni E, et al. Expanding the natural history of nonalcoholic steatohepatitis from cryptogenic cirrhosis to hepatocellular carcinoma. Gastroenterology 2012; 123: 134–140.
- 24. van der Poorten D, Samer CF, Ramezani-Moghadam M, et al. Hepatic fat loss in advanced nonalcoholic steatohepatitis: are alterations in serum adiponectin the cause? Hepatology 2013; 57: 2180–2188.
- 25. Matsui O, Kadoya M, Takahashi S, et al. Focal sparing of segment IV in fatty livers shown by sonography and CT: correlation with aberrant gastric venous drainage. Am J Roentgenol 1995; 164: 1137–1140.
- 26. Nosadini R, vogaro A, Mollo F, et al. Carbohydrate and lipid metabolism in cirrhosis. Evidence that hepatic uptake of gluconeogenic precursors and of free fatty acid depends on effective hepatic flow. J Clin Endocrinol Metab 1984; 58: 1125–1132.
- 27. Schaffner H, Popper H. Capillarization of the sinusoids. Gastroenterology 1963; 44: 339–342.
- 28. Kamar N, Selver J, Mansuy J-M, et al. Hepatitis E virus and chronic hepatitis in organ-transplant recipients. N Engl J Med 2008; 358: 811–817.
- 29. Kamar N, Mansuy J-M, Cointault O, et al. Hepatitis E virus-related cirrhosis in kidney-and kidney-pancreas-transplant recipients. Am J Transplant 2008; 8: 1744–1748.
- 30. Singh GKJ, Ijaz S, Rockwood N, et al. Chronic hepatitis E as a cause for cryptogenic cirrhosis in HIV. J Infect 2012; 66: 103–106.
- 31. Pesce V, Cormio A, Marangi LC, et al. Depletion of mitochondrial DNA in the skeletal muscle of two cirrhotic patients with severe asthenia. Gene 2012; 286: 143–148.
- 32. Carulli L, Dei Cas A, Nascimbeni F. Synchronous cryptogenic liver cirrhosis and idiopathic pulmonary fibrosis: a clue to telomere involvement. Hepatology 2012; 56: 2001–2003.
- 33. Calado RT, Brudno J, Mehta P, et al. Constitutional telomerase mutations are genetic risk factors for cirrhosis. Hepatology 2011; 53: 1600–1607.
- 34. Ku N-O, Gish R, Wright TL, Omary MB. Keratin 8 mutations in patients with cryptogenic liver disease. N Engl J Med 2001; 344: 1580–1587.
- 35. Mato JM, Martínez-Chantar ML, Lu SC. Systems biology for hepatologists. Hepatology 2014; 60: 735–743.
- 36. Daly AK, Ballestri S, Carulli L, Loria P, Day CP. Genetic determinants of susceptibility and severity in nonalcoholic fatty liver disease. Expert Rev Gastroenterol Hepatol 2011; 5: 253–263.
- 37. Barr J, Caballeria J, Martinez-Arranz I, et al. Obesity-dependent metabolic signatures associated with nonalcoholic fatty liver disease progression. J Proteome Res 2012; 1: 2521–2532.
- 38. Barr J, Vázquez-Chantada M, Alonso C, et al. Liquid chromatography-mass spectrometry-based parallel metabolic profiling of human and mouse model serum reveals putative biomarkers associated with the progression of nonalcoholic fatty liver disease. J Proteome Res 2010; 9: 4501–4512.
- 39. Starley BQ, Calcagno CJ, Harrison SA. Nonalcoholic fatty liver disease and hepatocellular carcinoma: a weighty connection. Hepatology 2010; 51: 1820–1832.