10
Of men and microbes: Role of the intestinal microbiome in non-alcoholic fatty liver disease
Mohammad Bilal Siddiqui1, Mohammad Shadab Siddiqui2, and Arun J. Sanyal2
1 Department of Internal Medicine, University of Texas Medical School, Houston, Texas, USA
2 Virginia Commonwealth University, Richmond, VA, USA
Introduction
NAFLD affects a third of the US population with increased prevalence among obese or diabetic individuals [1–4] and is closely associated with other metabolic disorders including obesity, insulin resistance (IR), hypertension, dyslipidemia, and obstructive sleep apnea [5, 6]. The higher incidence of cardiovascular disease, extrahepatic malignancies, and progression to cirrhosis is largely responsible for the decreased long-term survival seen in patients with NAFLD [5, 7, 8]. The disease burden from NAFLD is projected to mirror the rise in other metabolic disorders and will be the leading listing diagnosis for progression to cirrhosis and liver transplantation [9].
NAFLD is characterized by intrahepatic fat accumulation in the absence of excessive alcohol consumption (<20 g/day in women and <30 g/day in men) and exclusion of secondary causes of hepatic steatosis [10–13]. NAFLD represents a histopathologic spectrum ranging from simple hepatic steatosis to steatohepatitis characterized by hepatic steatosis with necroinflammatory changes. Non-alcoholic steatohepatitis (NASH) is associated with increased risk of progression to advanced fibrosis and cirrhosis [5, 14, 15].
Although NAFLD is commonly seen in conjunction with other features of the metabolic syndrome, an independent epidemiological association between IR and NAFLD has been confirmed in nondiabetic patients [16, 17]. Patients with steatohepatitis are more likely to have higher total insulin secretion and lower insulin sensitivity than controls even in the absence of overt diabetes. Fasting insulin levels are the best predictor of hepatic steatosis, and IR is an independent risk factor for development of NASH, irrespective of body mass index (BMI). In patients with NAFLD and normal alanine aminotransferase (ALT) values, IR has been independently associated with the presence of advanced fibrosis [18]. Emerging data implicates intestinal microbiome as an integral component in pathogenesis of metabolic disorders, particularly obesity and diabetes. The focus of the current review will be to explore the relationship between intestinal microbiome and NAFLD and its link to obesity and IR.
Intestinal microbiome
The human gut contains >1014 microorganisms and is collectively referred to as the intestinal or gut microbiota [19]. The intestinal microbiota is important in the maturation of the human immune system, strengthening of the host defenses, vitamin synthesis, and absorption and fermentation of polysaccharides [20–22]. Already implicated in the pathogenesis of obesity and diabetes, emerging data suggest a causal link between intestinal microbiome and NAFLD [23–28].
Obesity, IR, and the intestinal microbiome
Diet is an important determinant of the composition of the intestinal microbiome, and indigestible polysaccharides are the main substrate available to the intestinal microbiome. The metabolism of polysaccharides by intestinal microbiome results in short-chain fatty acids (SCFAs) and other metabolites that play a key role in bacterial–host interaction. The role of intestinal microbiome in weight regulation was initially demonstrated in animal studies [20, 29]. Compared to conventionally raised mice, germfree mice had reduced adiposity and weight gain despite increased food consumption [29]. Furthermore, transplantation of cecal-derived microbial species from conventionally raised mice into germfree mice, a process known as conventionalization, led to a dramatic increase in body weight and adiposity that was independent of chow diet consumption or energy expenditure [29, 30]. Using 16S RNA pyrosequencing, the obese phenotype was associated with a decrease in the Bacteroides species and a concurrent increase in Firmicutes species [30, 31]. These findings were corroborated in human studies that showed that weight loss proportionally correlated with the extent of restoration of the Bacteroidetes population [32]. Similarly, dietary intake of high-fat diets has been shown to alter the intestinal microbiome by promoting the relative concentrations of Firmicutes and Proteobacteria [33, 34]. Although the mechanism of weight gain due to changes in intestinal microbiome remains an area of ongoing investigation, several microbial processes have been identified that favor an obesogenic phenotype. For instance, in the presence of excess polysaccharides, Bacteroides thetaiotaomicron upregulates glycoside hydrolases thereby increasing fermentation of otherwise indigestible polysaccharides into short-chain fatty acids (SCFA) that are more readily absorbed by the host [21, 35]. Additionally, the microbiome induces host monosaccharide transporters to further enhance host utilization of liberated polysaccharides and allow more efficient caloric extraction from dietary intake. Butyrate, a SCFA, has been shown to stimulate leptin production and GLP-1 secretion thereby promoting an obesinogenic phenotype, while propionate is used for gluconeogenesis and lipogenesis by the hepatocytes [36].
The intestinal microbiome also regulates weight by affecting fasting-induced adipose factor (Fiaf), a circulating lipoprotein lipase (LPL) inhibitor that is produced in the small intestines, liver, and adipocytes. Elevated Fiaf levels protect against weight gain by inhibiting adipocyte LPL and thereby decreasing available free fatty acids necessary for adipose tissue expansion [20]. Conventionalization of germfree mice leads to significant reduction in Fiaf and increase in body weight and adiposity [20]. Fiaf-null germfree mice had the same amount of weight gain and increase in adiposity as the conventionally raised mice reaffirming the role of intestinal microbiome in Fiaf regulation.
Expansion of adipose tissue depots (Figure 10.1) as a result of weight gain leads to infiltration of overloaded adipose tissue by activated macrophages leading to low-grade chronic inflammation, which is an important cause of IR [37]. Gut-derived endotoxins, dietary fats, relative hypoxia, and hormones have all been implicated in adipose tissue inflammation [38]. Macrophage infiltration of the adipose tissue promotes production and secretion of proinflammatory cytokines by adipocytes characterized by high levels of tumor necrosis factor alpha (TNF-α) and interleukin-6 (IL-6) and a decrease in adiponectin levels [39, 40]. This proinflammatory metabolic milieu leads to further deterioration of IR. Additionally, these adipocyte-secreted cytokines (adipokines) enter the portal circulation and are sensed by the liver, which releases additional cytokines (i.e., hs-CRP) as part of the acute phase reaction. These cytokines promote endothelial dysfunction, promote atherogenesis, promote thrombosis, impair fibrinolysis, impair the metabolic clearance of glucose, and lead to hepatic steatosis.
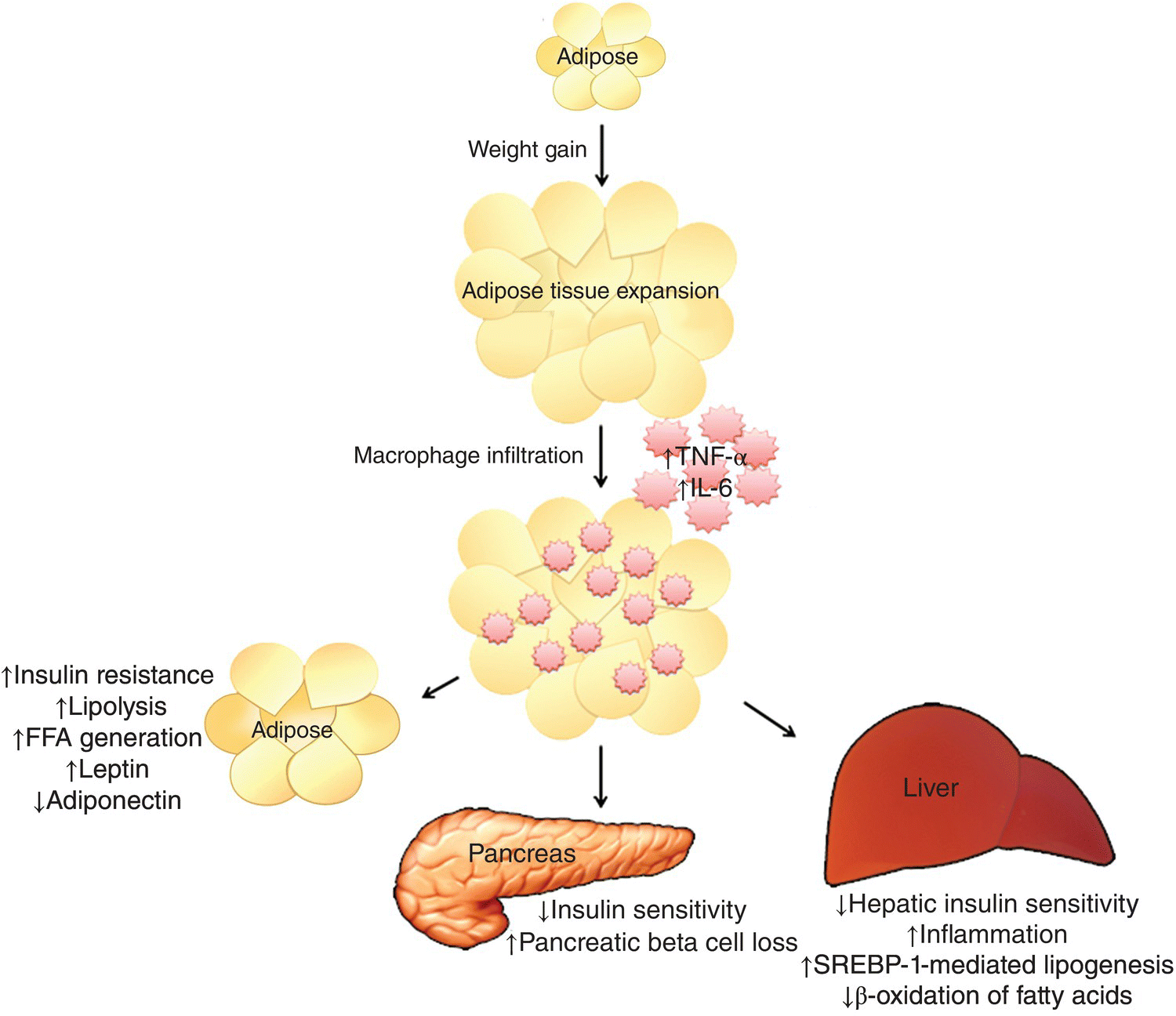
FIG 10.1 Weight gain leads to adipose tissue expansion and inflammation, which results in a proinflammatory state. These adipokines worsen adipose tissue and hepatic and systemic insulin resistance. The deterioration in insulin sensitivity leads to pancreatic β-cell loss, de novo lipogenesis, and hepatic steatosis.
NAFLD and the intestinal microbiome
The portal vein receives 70% of its blood flow from the intestinal circulation and serves as a direct link between the intestinal microbiome and the liver by facilitating hepatic exposure to gut-derived microbial products. Patients with NAFLD have an increased prevalence of small intestinal bacterial overgrowth (SIBO), which augments the interaction between the microbiome and the liver by altering paracellular intestinal permeability [41, 42]. Normally, the integrity of the paracellular tight junctions is maintained by the interaction between transmembrane proteins occludins and claudins, which bind to membrane proteins zona occludens-1 and 2 (ZO-1 and ZO-2) [43–45]. SIBO disrupts the function of the tight junction protein ZO-1, increasing intestinal permeability and ultimately facilitating the increased absorption of microbial endotoxins into the portal circulation [42, 43] (Figure 10.2). Additionally, the association between intestinal permeability and the degree of steatosis is independent of other features of metabolic syndrome and SIBO, suggesting that the gut–liver axis is an important driver of hepatic steatosis [42].
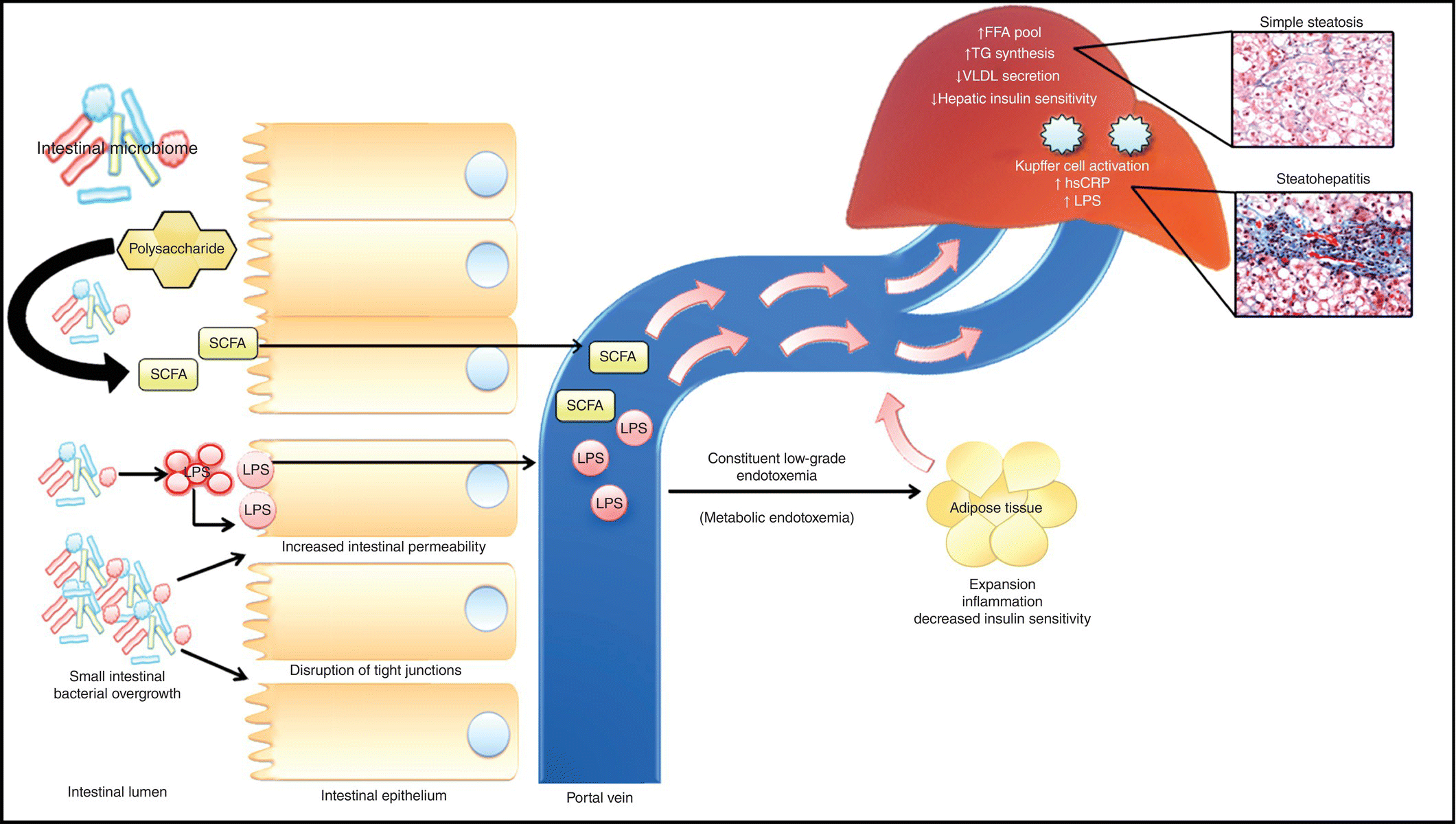
FIG 10.2 NAFLD is associated with an increase in small intestinal bacterial overgrowth (SIBO). The intestinal microbiome breaks down polysaccharides into short-chain fatty acids (SCFAs), and increased bacterial productions (i.e., LPS) are then readily absorbed into the portal circulation. These by-products of digestion and intestinal microbiome subsequently influence insulin resistance and hepatic triglyceride synthesis and impair hepatic triglyceride synthesis leading to hepatic steatosis.
Interaction between liver and intestine via bile acids
Bile acids, produced as glycine or taurine conjugates from cholesterol in the liver and secreted into the small intestine, facilitate the cross talk between the liver and intestine. Bile acids have a direct bacteriostatic effect, and in animal models of cirrhosis, admiration of conjugated bile acids reduced bacterial overgrowth and translocation [46]. In intestinal epithelial cells, conjugated bile acids bind to farnesoid X receptor (FXR), which increases production of antimicrobial proteins angiogenin 1 and ribonuclease family receptor member, thereby preventing bacterial overgrowth and promoting epithelial cell integrity [47]. Additionally, bile acid metabolism was disrupted in animal models of NASH potentially due to underlying hepatic inflammation and injury [48]. Corroborating the animal data, a recent phase 2 clinical trial of the FXR agonist obeticholic acid in patients with type 2 diabetes and NAFLD, showed significantly improved markers of inflammation and fibrosis [49].
Intestinal microbiome and chronic inflammation
In addition to the recruitment of adipokines, the microbiome also contributes to chronic inflammation through the production of endotoxins. Endotoxins are key constituents of the microbial cell wall that illicit an immune response upon their release into the host. Lipopolysaccharide (LPS) is an active component of endotoxins. Food intake impacts serum endotoxin levels, which normally increase during the fed state and decline with fasting. However, a high-fat diet disrupts these cyclic changes in serum endotoxin levels and leads to chronic endotoxemia, which can be seen in as little as 3 days after initiation of the high-fat feeding (Figure 10.2) [26, 31]. Additionally, the proportion of LPS-expressing bacteria also increases with a high-fat diet [28]. Serum endotoxin levels in this chronic low-grade endotoxemia or metabolic endotoxemia are usually 10–50-folds lower than is normally seen in septicemia or infections and have been implicated in the pathogenesis of metabolic disorders including hepatic steatosis [28, 50]. LPS binds to LPS binding protein (LBP), which subsequently binds to CD-14 and activates toll-like receptor-4 (TLR-4). TLRs are pattern recognition receptors that recognized highly conversed microbial molecules termed pathogen-associated molecular patterns (PAMPs). LPS-mediated activation of TLRs in Kupffer results in increased expression of proinflammatory cytokines and hepatic injury [28]. Importance of CD-14 and TLR-4 in propagating the host response to LPS-mediated hepatotoxicity was confirmed in CD-14 and TLR-4 null mice that had a blunted response to high-fat diets [28, 51, 52].
The inducible proinflammatory state seen with metabolic endotoxemia in the liver and visceral adipose tissue is characterized by increased levels of TNF-α, IL-1β, IL-6, and plasminogen activator inhibitor-1 (PAI-1) [27, 53–55]. These proinflammatory cytokines interfere with insulin signaling and lead to deterioration of both hepatic and systemic insulin sensitivities [40]. Metabolic endotoxemia also promotes hepatic triglyceride synthesis and liver fat accumulation. Interestingly, the presence of preexisting obesity exaggerates the host response to LPS-induced liver injury. Compared to lean littermates, obesity prone zucker (fa/fa) rats developed significant steatohepatitis and hepatic injury after i.p. administration of LPS. Although TNF-α production by Kupffer cells was similar, the phagocytic activity of Kupffer cells was significantly increased in obese animals. This data suggests that the liver in metabolic endotoxemia likely primes the Kupffer cells to LPS making them more sensitive to toxic effects of TNF-α [56].
Additional data confirming the importance of intestinal microbiome in NAFLD came from studies investigating probiotics in NAFLD. In animal studies, treatments with probiotics lead to significant improvement in serum endotoxemia, intestinal barrier function, IR, and liver fat content [57]. These observations were confirmed in human studies where treatment with VSL#3, a mix of probiotic strains, lead to significant improvement in serum TNF-α levels and hepatic inflammation [58]. More recently in a cross-sectional cohort, relative deficiency in intestinal microbial species of Bacteroides and Prevotella was associated with steatohepatitis compared to healthy subjects and those with simple hepatic steatosis [59]. Additionally, compared to those with simple hepatic steatosis, those with steatohepatitis had higher fecal content of Clostridium coccoides. This association was independent of dietary fat intake and BMI, suggesting a role of intestinal microbiome in the development of steatohepatitis [59]. In another cross-sectional study comparing NAFLD with healthy controls, there was overrepresentation of Firmicutes (Lactobacillaceae, Veillonellaceae, and Lachnospiraceae families) and Proteobacteria (Kiloniellaceae and Pasteurellaceae families) while a relative decrease in Bacteroides (Porphyromonadaceae) [60]. Volatile organic compounds (VOC) are microbial-derived compounds whose effects are unknown in the human gut. Interestingly, ester VOCs were identified more frequently in fecal samples of those with NAFLD, although the clinical implication of these findings is unclear at this point [60].
Furthermore, this chronic inflammatory state has been shown to promote hepatocarcinogenesis in mice [61]. Mice treated with constituently low levels of LPS had increased tumor size and number in carbon tetrachloride (CCl4)-induced hepatocarcinogenesis [61]. This increase in tumor burden is likely related to TLR-4-dependent pathways, as TLR-4-deficient mice had an 80–90% reduction in tumor size and number [61]. Additionally, reduced tumor burden was noted in germfree mice and in mice that underwent gut sterilization, implicating the intestinal microbiome as a key mediator of hepatocellular carcinoma progression [61].
However, there is significant variability and even contradictory findings in human studies attempting to better characterize intestinal microbiome in NAFLD [59, 60, 62, 63]. These discrepant findings could potentially be due to differences in demographics (i.e., geographic locations, ethnicities, age, etc.), methodology, small sample size, and nonhistological characterization of NAFLD.
Choline deficiency and hepatic steatosis
Choline is an integral component of biological membranes and necessary for triglyceride secretion into very low-density lipoprotein (VLDL) from the liver [64–66]. Choline deficiency results in decreased hepatic triglyceride clearance and hepatic steatosis, which can be reversed with choline supplementation [65, 67]. Relative reduction in intestinal concentrations of Gammaproteobacteria and Erysipelotrichi is associated with choline deficiency and hepatic steatosis in humans [63]. Intestinal microbiome-mediated choline deficiency is likely multifactorial. Enzymes produced by the gut microbiota catalyze the first step in the conversion of dietary choline to methylamines, dimethylamine, and trimethylamine [68]. Methylamines are subsequently absorbed through the microvilli and reach the liver via portal vein where they activate inflammatory pathways leading to hepatic injury. Furthermore, conversion of choline into methylamines reduces the bioavailability of choline for production of phosphatidylcholine [69]. The relative choline and phosphatidylcholine deficiency subsequently interferes with VLDL assembly and triglyceride secretion from the liver leading to hepatic steatosis [69] (Figure 10.3).
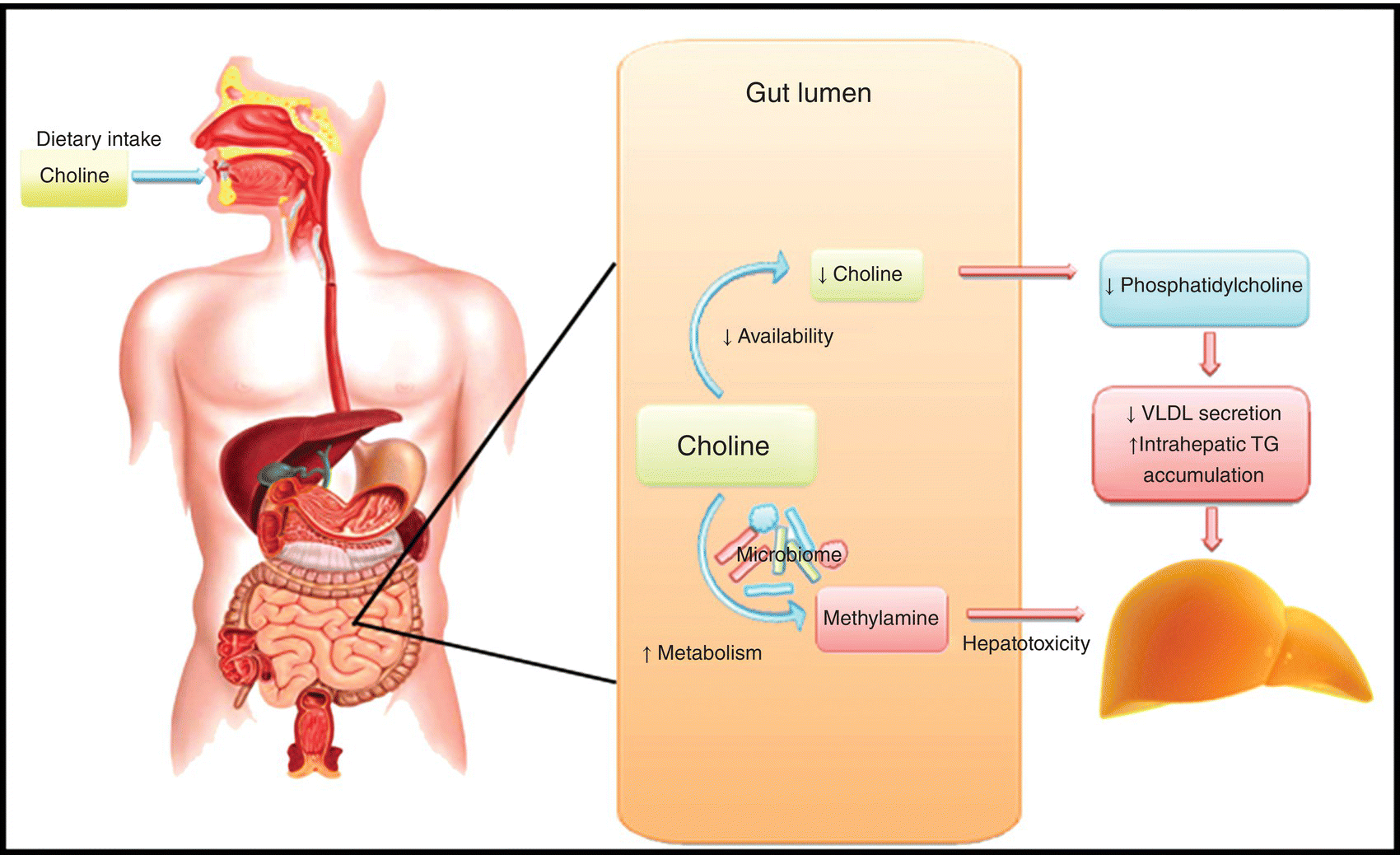
FIG 10.3 The gut microbiota catalyzes the conversion of dietary choline into methylamines, which enter the portal circulation and promote hepatocellular injury. The conversion of choline to methylamines also reduces the bioavailability of choline, leading to phosphatidylcholine deficiency, which impairs VLDL secretion and promotes steatosis.
Endogenous ethanol production in NAFLD
Some of the microbial species in the intestinal microbiome produce potentially hepatotoxic agents like ethanol even in the absence of exogenous alcohol consumption. Endogenous ethanol is metabolized into acetate and acetaldehyde, which are subsequently transported to the liver via portal circulation [62]. In the liver, acetate can be used as a substrate for de novo lipogenesis, while acetaldehyde can activate Kupffer cells to produce reactive oxygen species (ROS) leading to hepatic injury. Compared to obese controls, the microbiome of patients with steatohepatitis was found to be enriched with Proteobacteria, a major source of endogenous ethanol production in humans [70]. The increase in relative concentration of Proteobacteria was associated with a parallel increase in blood ethanol concentration.
Conclusion
The role of intestinal microbiome in pathogenesis of NALFD is being continually defined. Although some of these mechanisms are likely linked to the development of other metabolic disorders like IR and obesity, other mechanisms are more specific to the liver like Kupffer cell activation, increase in de novo lipogenesis, and endogenous ethanol production.
References
- 1. Browning JD, Szczepaniak LS, Dobbins R, Nuremberg P, Horton JD, Cohen JC, Grundy SM, et al. Prevalence of hepatic steatosis in an urban population in the united states: Impact of ethnicity. Hepatology 2004; 40(6): 1387–1395.
- 2. Bhala N, Angulo P, van der Poorten D, Lee E, Hui JM, Saracco G, Adams LA, et al. The natural history of nonalcoholic fatty liver disease with advanced fibrosis or cirrhosis: An international collaborative study. Hepatology 2011; 54(4): 1208–1216.
- 3. Bellentani S, Tiribelli C, Saccoccio G, Sodde M, Fratti N, De Martin C, Cristianini G. Prevalence of chronic liver disease in the general population of northern Italy: The dionysos study. Hepatology 1994; 20(6): 1442–1449.
- 4. Kallwitz ER, Kumar M, Aggarwal R, Berger R, Layden-Almer J, Gupta N, Cotler SJ. Ethnicity and nonalcoholic fatty liver disease in an obesity clinic: The impact of triglycerides. Dig Dis Sci 2008; 53(5): 1358–1363.
- 5. Adams LA, Lymp JF, St Sauver J, Sanderson SO, Lindor KD, Feldstein A, Angulo P. The natural history of nonalcoholic fatty liver disease: A population-based cohort study. Gastroenterology 2005; 129(1): 113–121.
- 6. Vernon G, Baranova A, Younossi ZM. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Hepatology 2011; 34(3): 274–285.
- 7. Söderberg C, Stål P, Askling J, Glaumann H, Lindberg G, Marmur J, Hultcrantz R. Decreased survival of subjects with elevated liver function tests during a 28-year follow-up. Hepatology 2009; 51: 595–602.
- 8. Ekstedt M, Franzen LE, Mathiesen UL, Thorelius L, Holmqvist M, Bodemar G, Kechagias S. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology 2006; 44(4): 865–873.
- 9. Charlton MR, Burns JM, Pedersen RA, Watt KD, Heimbach JK, Dierkhsing RA. Frequency and outcomes of liver transplantation for nonalcoholic steatohepatitis in the united states. Gastroenterology 2011; 141: 1249–1253.
- 10. Ludwig J, Viggiano TR, McGill DB, Oh BJ. Nonalcoholic steatohepatitis: Mayo clinic experiences with a hitherto unnamed disease. Mayo Clin Proc 1980; 55(7): 434–438.
- 11. Chalasani N, Younossi Z, Lavine JE, Diehl AM, Brunt EM, Cusi K, Charlton M, et al. The diagnosis and management of non-alcoholic fatty liver disease: Practice guideline by the American association for the study of liver diseases, American college of gastroenterology, and the American gastroenterological association. Hepatology 2012; 55(6): 2005–2023.
- 12. Bacon BR, Farahvash MJ, Janney CG, Neuschwander-Tetri BA. Nonalcoholic steatohepatitis: An expanded clinical entity. Gastroenterology 1994; 107(4): 1103–1109.
- 13. Matteoni CA, Younossi ZM, Gramlich T, Boparai N, Liu YC, McCullough AJ. Nonalcoholic fatty liver disease: A spectrum of clinical and pathological severity. Gastroenterology 1999; 116(6): 1413–1419.
- 14. Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005; 41(6): 1313–1321.
- 15. Brunt EM, Janney CG, Di Bisceglie AM, Neuschwander-Tetri BA, Bacon BR. Nonalcoholic steatohepatitis: A proposal for grading and staging the histological lesions. Am J Gastroenterol 1999; 94(9): 2467–2474.
- 16. Pagano G, Pacini G, Musso G, Gambino R, Mecca F, Depetris N, Cassader M, et al. Nonalcoholic steatohepatitis, insulin resistance, and metabolic syndrome: Further evidence for an etiologic association. Hepatology 2002; 35(2): 367–372.
- 17. Sanyal AJ, Campbell-Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, Luketic VA, et al. Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001; 120(5): 1183–1192.
- 18. Fracanzani AL, Valenti L, Bugianesi E, Andreoletti M, Colli A, Vanni E, Bertelli C, et al. Risk of severe liver disease in nonalcoholic fatty liver disease with normal aminotransferase levels: A role for insulin resistance and diabetes. Hepatology 2008; 48(3): 792–798.
- 19. Savage DC. Microbial ecology of the gastrointestinal tract. Annu Rev Microbiol 1977; 31: 107–133.
- 20. Backhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, Semenkovich CF, et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A 2004; 101(44): 15718–15723.
- 21. Backhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science 2005; 307(5717): 1915–1920.
- 22. Xu J, Bjursell MK, Himrod J, Deng S, Carmichael LK, Chiang HC, Hooper LV, et al. A genomic view of the human-bacteroides thetaiotaomicron symbiosis. Science 2003; 299(5615): 2074–2076.
- 23. Tilg H, Moschen AR, Kaser A. Obesity and the microbiota. Gastroenterology 2009; 136(5): 1476–1483.
- 24. Tilg H, Kaser A. Gut microbiome, obesity, and metabolic dysfunction. J Clin Invest 2011; 121(6): 2126–2132.
- 25. Tilg H. Obesity, metabolic syndrome, and microbiota: Multiple interactions. J Clin Gastroenterol 2010; 44(Suppl 1): S16–S18.
- 26. Amar J, Burcelin R, Ruidavets JB, Cani PD, Fauvel J, Alessi MC, Chamontin B, et al. Energy intake is associated with endotoxemia in apparently healthy men. Am J Clin Nutr 2008; 87(5): 1219–1223.
- 27. Brun P, Castagliuolo I, Di Leo V, Buda A, Pinzani M, Palu G, Martines D. Increased intestinal permeability in obese mice: New evidence in the pathogenesis of nonalcoholic steatohepatitis. Am J Physiol Gastrointest Liver Physiol 2007; 292(2): G518–G525.
- 28. Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007; 56(7): 1761–1772.
- 29. Backhed F, Manchester JK, Semenkovich CF, Gordon JI. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc Natl Acad Sci U S A 2007; 104(3): 979–984.
- 30. Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006; 444(7122): 1027–1031.
- 31. Turnbaugh PJ, Backhed F, Fulton L, Gordon JI. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe 2008; 3(4): 213–223.
- 32. Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: Human gut microbes associated with obesity. Nature 2006; 444(7122): 1022–1023.
- 33. Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, Almeida M, et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013; 500(7464): 541–546.
- 34. Cotillard A, Kennedy SP, Kong LC, Prifti E, Pons N, Le Chatelier E, Almeida M, et al. Dietary intervention impact on gut microbial gene richness. Nature 2013; 500(7464): 585–588.
- 35. Sonnenburg JL, Xu J, Leip DD, Chen CH, Westover BP, Weatherford J, Buhler JD, et al. Glycan foraging in vivo by an intestine-adapted bacterial symbiont. Science 2005; 307(5717): 1955–1959.
- 36. Samuel BS, Shaito A, Motoike T, Rey FE, Backhed F, Manchester JK, Hammer RE, et al. Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proc Natl Acad Sci U S A 2008; 105(43): 16767–16772.
- 37. Chawla A, Nguyen KD, Goh YP. Macrophage-mediated inflammation in metabolic disease. Nat Rev Immunol 2011; 11(11): 738–749.
- 38. Lolmede K, Durand de Saint Front V, Galitzky J, Lafontan M, Bouloumie A. Effects of hypoxia on the expression of proangiogenic factors in differentiated 3T3-F442A adipocytes. Int J Obes Relat Metab Disord 2003; 27(10): 1187–1195.
- 39. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW, Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 2003; 112(12): 1796–1808.
- 40. Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 2003; 112(12): 1821–1830.
- 41. Sabate JM, Jouet P, Harnois F, Mechler C, Msika S, Grossin M, Coffin B. High prevalence of small intestinal bacterial overgrowth in patients with morbid obesity: A contributor to severe hepatic steatosis. Obes Surg 2008; 18(4): 371–377.
- 42. Miele L, Valenza V, La Torre G, Montalto M, Cammarota G, Ricci R, Masciana R, et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 2009; 49(6): 1877–1887.
- 43. Anderson JM, Van Itallie CM. Tight junctions and the molecular basis for regulation of paracellular permeability. Am J Physiol 1995; 269(4 Pt 1): G467–G475.
- 44. Furuse M, Hirase T, Itoh M, Nagafuchi A, Yonemura S, Tsukita S, Tsukita S. Occludin: A novel integral membrane protein localizing at tight junctions. J Cell Biol 1993; 123(6 Pt 2): 1777–1788.
- 45. Furuse M, Fujita K, Hiiragi T, Fujimoto K, Tsukita S. Claudin-1 and -2: Novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J Cell Biol 1998; 141(7): 1539–1550.
- 46. Kakiyama G, Pandak WM, Gillevet PM, Hylemon PB, Heuman DM, Daita K, Takei H, et al. Modulation of the fecal bile acid profile by gut microbiota in cirrhosis. J Hepatol 2013; 58(5): 949–955.
- 47. Inagaki T, Moschetta A, Lee YK, Peng L, Zhao G, Downes M, Yu RT, et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci U S A 2006; 103(10): 3920–3925.
- 48. Tanaka N, Matsubara T, Krausz KW, Patterson AD, Gonzalez FJ. Disruption of phospholipid and bile acid homeostasis in mice with nonalcoholic steatohepatitis. Hepatology 2012; 56(1): 118–129.
- 49. Mudaliar S, Henry RR, Sanyal AJ, Morrow L, Marschall HU, Kipnes M, Adorini L, et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology 2013; 145(3): 574–582.e1.
- 50. Mydel P, Takahashi Y, Yumoto H, Sztukowska M, Kubica M, Gibson FC, III, Kurtz DM, Jr, et al. Roles of the host oxidative immune response and bacterial antioxidant rubrerythrin during porphyromonas gingivalis infection. PLoS Pathog 2006; 2(7): e76.
- 51. Roncon-Albuquerque R, Jr, Moreira-Rodrigues M, Faria B, Ferreira AP, Cerqueira C, Lourenco AP, Pestana M, et al. Attenuation of the cardiovascular and metabolic complications of obesity in CD14 knockout mice. Life Sci 2008; 83(13–14): 502–510.
- 52. Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest 2006; 116(11): 3015–3025.
- 53. Laugerette F, Vors C, Peretti N, Michalski MC. Complex links between dietary lipids, endogenous endotoxins and metabolic inflammation. Biochimie 2011; 93: 39–45.
- 54. Ruiz AG, Casafont F, Crespo J, Cayon A, Mayorga M, Estebanez A, Fernadez-Escalante JC, et al. Lipopolysaccharide-binding protein plasma levels and liver TNF-alpha gene expression in obese patients: Evidence for the potential role of endotoxin in the pathogenesis of non-alcoholic steatohepatitis. Obes Surg 2007; 17(10): 1374–1380.
- 55. Nolan JP. The role of intestinal endotoxin in liver injury: A long and evolving history. Hepatology 2010; 52(5): 1829–1835.
- 56. Yang SQ, Lin HZ, Lane MD, Clemens M, Diehl AM. Obesity increases sensitivity to endotoxin liver injury: Implications for the pathogenesis of steatohepatitis. Proc Natl Acad Sci U S A 1997; 94(6): 2557–2562.
- 57. Li Z, Yang S, Lin H, Huang J, Watkins PA, Moser AB, Desimone C, et al. Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatology 2003; 37(2): 343–350.
- 58. Loguercio C, Federico A, Tuccillo C, Terracciano F, D’Auria MV, De Simone C, Del Vecchio Blanco C. Beneficial effects of a probiotic VSL#3 on parameters of liver dysfunction in chronic liver diseases. J Clin Gastroenterol 2005; 39(6): 540–543.
- 59. Mouzaki M, Comelli EM, Arendt BM, Bonengel J, Fung SK, Fischer SE, McGilvray ID, et al. Intestinal microbiota in patients with nonalcoholic fatty liver disease. Hepatology 2013; 58(1): 120–127.
- 60. Raman M, Ahmed I, Gillevet PM, Probert CS, Ratcliffe NM, Smith S, Greenwood R, et al. Fecal microbiome and volatile organic compound metabolome in obese humans with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol 2013; 11(7): 868–875.e1-3.
- 61. Dapito DH, Mencin A, Gwak GY, Pradere JP, Jang MK, Mederacke I, Caviglia JM, et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell 2012; 21(4): 504–516.
- 62. Zhu L, Baker SS, Gill C, Liu W, Alkhouri R, Baker RD, Gill SR. Characterization of the gut microbiome in non-alcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013; 57(2): 609–609.
- 63. Spencer MD, Hamp TJ, Reid RW, Fischer LM, Zeisel SH, Fodor AA. Association between composition of the human gastrointestinal microbiome and development of fatty liver with choline deficiency. Gastroenterology 2011; 140(3): 976–986.
- 64. Zeisel SH, da Costa KA. Choline: An essential nutrient for public health. Nutr Rev 2009; 67(11): 615–623.
- 65. Rinella ME, Elias MS, Smolak RR, Fu T, Borensztajn J, Green RM. Mechanisms of hepatic steatosis in mice fed a lipogenic methionine choline-deficient diet. J Lipid Res 2008; 49(5): 1068–1076.
- 66. Yao ZM, Vance DE. The active synthesis of phosphatidylcholine is required for very low density lipoprotein secretion from rat hepatocytes. J Biol Chem 1988; 263(6): 2998–3004.
- 67. Buchman AL, Dubin MD, Moukarzel AA, Jenden DJ, Roch M, Rice KM, Gornbein J, et al. Choline deficiency: A cause of hepatic steatosis during parenteral nutrition that can be reversed with intravenous choline supplementation. Hepatology 1995; 22(5): 1399–1403.
- 68. Zeisel SH, Wishnok JS, Blusztajn JK. Formation of methylamines from ingested choline and lecithin. J Pharmacol Exp Ther 1983; 225(2): 320–324.
- 69. Dumas ME, Barton RH, Toye A, Cloarec O, Blancher C, Rothwell A, Fearnside J, et al. Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc Natl Acad Sci U S A 2006; 103(33): 12511–12516.
- 70. Clark DP. The fermentation pathways of Escherichia coli. FEMS Microbiol Rev 1989; 5(3): 223–234.