CHAPTER THREE
FEED ME
There is a war going on for your ass. And no, I don’t mean “ass” in a metaphorical sense, like when the funkmaster George Clinton shouts, “Free your mind and your ass will follow.” Nor am I using “ass” like Kevin, the kid who had the locker next to mine in seventh-grade gym class, did when he said, “Linden, your mouth just wrote a check your ass can’t cash.” For the purposes of this discussion, “ass” does not refer to one’s corporeal self in a general sense.1 Rather, I’m speaking literally: There’s a war going on for the fat deposits in your tuchus, and your brain’s pleasure circuits are a major front in that campaign.
In 2008 I ate about 1.2 million calories and loved every one of them. They came in many forms, from fat-laden restaurant meals with pretentious lists of ingredients to sensible portions of vegetarian fare cooked at home to nasty little bags of Cheetos furtively wolfed down in my office with the door closed. I went for many weeks at a time in which I dutifully rode my bicycle for forty minutes every night and whole months where I was an utterly sedentary sofa slug. During that year my weight never fluctuated by more than five pounds and my weight on the first and the last days of that year was identical. Now, putting aside my frustration at this state of affairs, it’s remarkable that, with 1.2 million calories of food coming in, my body regulated my appetite and expended precisely the right amount of energy to break even.
My own experience with weight homeostasis is typical for others with unrestricted access to food. In studies where the food intake and energy expenditure of subjects are carefully monitored over a period of weeks to months (which tends to average out day-to-day fluctuations) a remarkable balance between calories consumed and calories burned was observed. When various mammals, from mice to monkeys, are either overfed or starved for a few weeks, their weight soon returns to normal levels when free access to food is resumed. Crucially, our mammalian bodies seem to be able to regulate feeding based on the amount of energy available in the food we consume, not just on the volume of that food. One example of many: When groups of rats were fed nutrient solutions of varying concentrations, they adjusted the volume consumed to achieve a constant inflow of calories. It’s a lot like the thermostat in your house: When its thermometer registers a drop in temperature, it sends a signal to the heater to warm the house until the desired set point is reached.
These observations suggest that the brain must receive signals from the body that indicate its weight and that the brain makes use of the signals to modulate appetite and energy expenditure in order to maintain an individual’s weight within a fairly narrow range. The signals are received in a structure at the base of the brain called the hypothalamus. The hypothalamus is involved in the control of many basic, subconscious drives and reflexes including sex, feeding, aggression, drinking, and regulation of body temperature.2 When rats received lesions in a particular subregion of the hypothalamus called the ventromedial area, they became obese. They behaved as if they were starving and compensated with an increase in food intake and a decrease in energy expenditure. Conversely, when a different part of the hypothalamus, called the lateral area, was destroyed, the rats behaved as if they had been overfed. They reduced food intake and increased energy use and thereby became dangerously lean. This is not just a rat trick: These experiments have been replicated in a wide variety of mammals, and humans who sustain damage to the ventromedial hypothalamus (usually from a tumor of the adjacent pituitary gland) will also increase their food intake and become obese.
This model raises one obvious question: How does your hypothalamus know how much you weigh? Let’s step back and play God for a moment. If you wanted to build this system, how would you do it? By measuring blood glucose? Fat deposits? Core body temperature? Pressure on the soles of the feet?
This all remained a mystery until 1994, when Jeffrey Friedman and his colleagues at Rockefeller University reported their observations of two strains of mutant mouse, one called obese and the other called db. (These mutations were not created by scientists using genetic tricks but arose spontaneously in a breeding colony.) Both strains of mice were extremely fat, a trait that was passed on to their offspring in a simple, dominant pattern of inheritance, like eye color. This suggested that obesity in both obese and db mouse strains resulted from a mutation in a single gene in each case. Friedman’s group was able to track down the mutation in the obese mice and found that it blocked production of a particular protein hormone, which they named leptin. The leptin protein is only secreted by fat cells. When similar analysis was performed on the db mice, it was found that the disrupted db gene was responsible for encoding a protein that functions as a leptin receptor: When it binds circulating leptin at the cell surface, it sets in motion a biochemical cascade inside the cell. Most provocatively, the leptin receptor is expressed strongly on neurons in those areas of the hypothalamus that cause obesity or leanness when destroyed.3
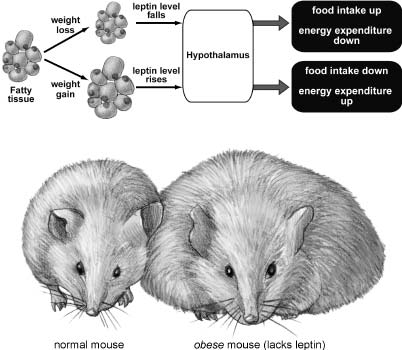
Figure 3.1 Leptin is a hormone produced by fat that acts in the brain to reduce appetite and increase energy expenditure, thus keeping weight constant. Top: When weight is lost, fatty tissue is reduced in mass, and so less leptin circulates throughout the body. This triggers increased food intake and reduced energy use, leading to compensatory weight gain. Conversely, when weight is gained, the increase in fat mass causes leptin levels to rise, suppressing feeding and increasing metabolism and activity to burn more energy, resulting in weight loss. Bottom: When the leptin gene is deleted in mice (either through random mutation of the DNA or by genetic engineering), no leptin is produced and severe obesity results. This drawing shows a leptin-lacking obese mouse on the right compared to a normal mouse on the left. The same effect is seen in mutant mice that lack the leptin receptor. Illustration by Joan M. K. Tycko.
So with Friedman’s key findings we now have a reasonable hypothesis for how the hypothalamus can sense body weight and use that information to maintain it within a narrow range (Figure 3.1). When weight is gained, the amount of body fat increases, and since fat cells secrete leptin in proportion to their mass, leptin levels will consequently rise. Leptin circulates in the blood and crosses into the brain, where it is sensed by leptin receptors expressed on neurons in the hypothalamus. Activation of those neurons by leptin suppresses appetite and increases energy expenditure. When weight is lost, the system works in the opposite direction: Less fat means reduced levels of circulating leptin, increased appetite, and reduced energy expenditure.
So far, the evidence that supports this hypothesis is quite promising. Leptin levels in the blood do indeed increase with weight gain and decrease with weight loss. Injections of leptin in obese mutant mice cause them to reduce food intake and lose weight (and these injections work even if tiny doses are delivered directly to the hypothalamus). Injections of leptin in db mutant mice have no effect, because there are no leptin receptors in the hypothalamus for the exogenous leptin to activate.
Of those people who are morbidly obese,4 less than 1 percent harbor DNA mutations that disrupt the function of the leptin gene—a low rate of incidence that is not surprising, as leptin-deficient humans and mice are both in fertile, so these mutations do not pass readily to subsequent generations. However, it’s encouraging that leptin-deficient patients can respond to exogenous leptin with a substantial reduction in food intake and subsequent weight loss. I. S. Farooqi and coworkers at Ad denbrooke’s Hospital in Cambridge, England, reported a case of a nine-year-old leptin-deficient girl with a nearly insatiable appetite. She ate enormous meals and constantly demanded between-meal snacks, as her leptin-deprived brain essentially made her feel as if she were starving. She weighed 208 pounds, which necessitated surgery on her legs to enable her to walk properly. After a year of leptin treatment, she had lost thirty-four pounds, almost all of which was fat. Her food consumption was reduced by 42 percent (which accounts entirely for her weight loss), and she reported that she no longer felt constantly hungry.5 The small number of patients who suffer from morbid obesity produced by mutation of the leptin receptor unfortunately cannot benefit from leptin therapy, much like db mutant mice.
While the leptin homeostatic system explains how the brain can receive information about long-term changes in weight as indicated by body fat, it doesn’t account for the short-term regulation of appetite. For example, what signals drive the initiation of eating? It used to be thought that reduced blood glucose levels were the primary trigger for the onset of a meal. However, more recent evidence indicates that eating is biochemically induced only in cases of severe starvation. More typically, in situations where food is abundant, meal onset is driven more by sociocultural and environmental factors.
How is the brain notified about the status of feeding during a meal? You don’t actually add fat mass during the course of a meal, so a different, rapid signal is required. In the short term, caloric intake is biochemically regulated by signals of satiety that influence the end of a meal. Sensors in the cells that line the stomach and the intestines can provide information to the brain about both the chemical and mechanical properties of the ingested food. (The chemical properties are things like sugar and protein levels, and the mechanical properties are mainly sensed by how much the gut is stretched.) Signals from the gut are conveyed through the secretion of protein hormones. There are several of these gut hormones that signal the brain in different ways: Some reach the brain directly through the bloodstream, while others activate neurons to send it electrical signals. Let’s consider one of these gut hormones as an example. When nutrients from a meal activate cells in the small intestine, a subset of cells lining this region of the gut secrete a hormone called CCK, which binds receptors on the endings of neurons in the nearby vagus nerve and activate it, causing electrical impulses to be passed up into the brain stem to a region called the nucleus tractus solitarius. When this region is activated, it in turn activates the mediobasal area of the hypothalamus, the same region that causes severe obesity if it is destroyed.
The mediobasal hypothalamus turns out to be a critical node in the feeding control circuit. In particular, an even smaller area within the mediobasal hypothalamus, the arcuate nucleus, receives both fast neural signals from the gut–vagus nerve–nucleus tractus solitarius pathway and slow body weight signals from circulating leptin secreted by fat cells (Figure 3.2). The arcuate nucleus contains a mixture of different types of neurons, which produce various effects on feeding when activated. A subset of neurons in the arcuate nucleus that contains the hormone POMC is activated by the nucleus tractus solitarius pathway from the gut and in turn inhibits neurons in the lateral hypothalamus. Activation of the lateral hypothalamus causes secretion of yet another hormone, called orexin, which produces feelings of hunger. At the same time, activation of the POMC neurons in the arcuate nucleus also activates a region called the paraventricular nucleus. Cells in the paraventricular nucleus secrete a hormone called CRH, which produces a feeling of satiety. So when you are eating that stack of pancakes, your gut is gradually sensing both the nutrients released from the food and the stretching of your stomach. These signals are conveyed via the complex pathway we just described to result in inhibition of orexin secretion and stimulation of CRH secretion, which work together to block your hunger and make you feel full.
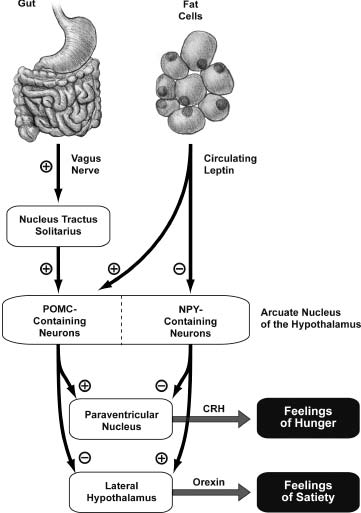
Figure 3.2 The feeding control circuit in the hypothalamus integrates slow body weight signals conveyed by circulating leptin and faster signals from the gut conveyed by the vagus nerve. In the end, the decision to start and stop eating is driven by the competition between two opposing signals: a hunger signal that uses orexin and a satiety signal that uses CRH. One analogy for this is an old-fashioned bathtub with separate hot and cold water taps: The temperature of the bath is determined by the relative flow of hot and cold water. Illustration by Joan M. K. Tycko.
A different group of neurons within the arcuate nucleus, those that use the neurotransmitter called NPY, are unaffected by the vagus nerve–nucleus tractus solitarius pathway, but are inhibited by circulating leptin. Like the POMC-containing neurons, these NPY cells also send axons to both the paraventricular nucleus and the lateral hypothalamus. But their actions are opposite to those of the POMC neurons: The NPY cells inhibit the paraventricular nucleus and excite the lateral hypothalamus. So if you are starving (literally so, not just as you might exclaim while waiting in line for a free table at the House of Pancakes), your fat mass is reduced, and you have less circulating leptin. This means less inhibition of the NPY cells in the arcuate nucleus, resulting in more orexin and less CRH. The end result: You’re ravenous.
Why does this circuit have to be so complicated?6 Why couldn’t it be built with a single center that both stimulates hunger and is turned off by a body fat signal or a gut-is-full-of-nutrients signal? Of course, we don’t really know the answer to these questions. However, we can speculate that the feeding control system was designed with redundancy and diverging signals to make it more robust and less susceptible to disruption of this crucial behavior. It is also worth noting that feeding is influenced by many factors (time of day, mood, exercise, odors, etc.), and all of these streams of information must be integrated somehow in the feeding control circuit. There are many aspects of this circuit we still don’t understand. For example, how and where does orexin act to stimulate and CRH act to suppress appetite?
The idea that eating is primarily a conscious and voluntary behavior is deeply rooted in our culture. We humans are invested in the notion that we have free will in all things. We want to believe that weight can be controlled by volition alone. Why can’t that fat guy just eat less and exercise more? He just lacks willpower, right? Not at all. Our homeostatic feeding control circuits make it very hard to lose a lot of weight and keep it off. As weight drops, fat mass decreases and leptin levels decline, triggering the biochemical cascade we just explored, producing signals that both reduce metabolic rate and produce a strong subconscious drive to eat. The more weight that is lost, the stronger the drive to eat will be and the greater the reduction in energy use. This is the sad but unavoidable truth that the multibillion-dollar-a-year diet industry doesn’t want you to know.
In the movies we typically see a time-lapse diet montage accompanied by upbeat music: Obese woman on a treadmill, obese woman eating salad, slightly less obese woman on an elliptical trainer, an obviously thinner woman running, ending with lean woman contentedly munching a celery stick and looking self-confident. Very inspirational! However, while moderate weight loss can be maintained through conscious monitoring of food intake and exercise, and dramatic weight loss can be achieved temporarily, it is extremely difficult for most people to maintain an extreme loss of weight over the long term. Even liposuction is only a temporary fix: As with dieting, removal of fat from the body reduces circulating levels of leptin, thereby reducing energy use and increasing appetite.
We share our homeostatic feeding control systems with other mammals. The subconscious drive to eat produced by leptin reduction that we experience is essentially the same as that felt by a mouse or a dog. Although humans are able to overlay these subconscious drives with slightly more cognitive control, at the root, we mammals are all the same. The main factor to appreciate in terms of humans and body weight is that for most of our evolutionary history we have not had access to unlimited calories. Also, for most of our evolutionary history, we belonged to hunter-gatherer societies and burned a lot of energy in everyday tasks. In that context it made sense to have a biological control system that set body weight (and appetite) at an optimum level: Too little of either and you’d be at risk of starvation during the next protracted famine. Too much and your mobility would be compromised. Today, when you try to lose large amounts of weight and keep it off, you are struggling against millions of years of evolutionary selective pressure. But wait. It gets worse. We haven’t even started talking about pleasure yet.
As we have seen, the pleasure circuit can be artificially activated, hijacked by drugs, or zapped by implanted electrodes. But is this circuit also activated by naturally pleasurable behaviors like eating? The answer is clearly yes. When a recording electrode is positioned in the VTA of a rat’s brain, it reveals a burst of neuronal activity when the rat begins to eat and some degree of continuing activity throughout the meal. Furthermore, when biochemical probes are implanted into the target regions of VTA neurons, eating is found to trigger a surge of released dopamine.
When drugs that flood the brain with dopamine, like cocaine or amphetamines, are given chronically, rats eat less and gain less weight. Loss of appetite is also produced when drugs that mimic dopamine (dopamine receptor agonists) are administered. Conversely, drugs that block dopamine receptors increase appetite and energy intake (total calories consumed) and cause weight gain. Cannabis is another drug that is a well-known appetite stimulant. In fact, it is so effective that it can counteract the dangerous appetite-suppressing effects seen in patients undergoing chemotherapy or who are suffering from AIDS. The brain’s own cannabislike molecules, the endocannabinoids, appear to have a role in feeding behavior. Drugs that block the brain’s endocannabinoid receptors suppress appetite and reduce body weight. Similarly, mutant mice that lack cannabis receptors in the brain have low appetites and are lean.
The pleasure circuit is also modified by body weight signals. There are leptin receptors on the dopamine neurons of the VTA, and when circulating leptin binds these receptors, a biochemical cascade is set in motion that inhibits their firing and consequent dopamine release in the VTA target areas. When genetic tricks are used in rats to delete leptin receptors only in the cells of the VTA (but not in other brain regions), the rats eat more and gain weight.7 This effect was further explored in a study in which leptin-deficient patients were placed in a brain scanner and shown images of food. In these patients the activation of certain VTA target regions (the nucleus accumbens and the caudate nucleus) by food images was similar to that seen in genetically normal patients when they are in the starved state. However, after chronic leptin therapy, the leptin-deficient patients showed normal pleasure circuit activation by food images, coincident with a decrease in appetite. When asked to rate how much they liked each of the food images, the leptin-deficient patients gave lower ratings after leptin treatment.8 Taken together, these results suggest that when you’re trying to sustain a substantial weight loss, the reduction in circulating leptin levels will modulate the pleasure circuit to make food actually seem more appealing.
Because food and addictive drugs activate overlapping pleasure circuits in the brain, there are well-known behavioral interactions between the two that are likely to result from this shared brain wiring. For example, starved rats show enhanced motivation for either addictive drugs like cocaine or amphetamines or direct electrical stimulation of the medial forebrain bundle. (They will press a lever faster and longer to receive these pleasures.)
Does this mean that obesity can be considered as a kind of food addiction? Emmanuel Pothos and his colleagues at Tufts University School of Medicine approached this question by breeding rats for several generations, crossing high weight-gainers to other high weight-gainers and low weight-gainers to other low weight-gainers in order to create obesity-prone and obesity-resistant strains. When allowed free access to standard laboratory rat chow for fifteen weeks, the obesity-prone rats ate significantly more food and gained an average of 22 percent more weight compared with their obesity-resistant cousins. Pothos and others have hypothesized that obesity-prone rats have blunted dopamine signaling in their midbrain pleasure circuits, a condition that causes them to eat more in an attempt to achieve a certain target level of dopamine signaling shared by all rats. Indeed, when dopamine levels in the nucleus accumbens were measured, the obesity-prone rats showed a significant reduction in both baseline dopamine levels and the dopamine surge evoked by electrical stimulation of the VTA. Is this reduced dopamine function something the obesity-prone rats are born with, or do they acquire it as they mature? Studies on obesity-prone rat pups found attenuation of dopamine signaling similar to that found in adults, suggesting the former. These findings support the hypothesis that obesity-prone rats must eat more to achieve the same set point of pleasure achieved by smaller meals in obesity-resistant rats.9
While obese rats are interesting for their own sake, how well does their model of heritable obesity carry over to humans? Is there evidence for a genetic component to obesity in humans, or is it all the result of environmental factors? Now, obviously, for a significant fraction of the world’s population, environmental concerns are overriding: If you don’t have access to sufficient nutrition, you can’t become obese. Likewise, many sociocultural factors as well as aspects of an individual’s life history also come into play (and we’ll get into these in more detail later in this chapter). But why, when given access to unlimited calories, do only some people become obese? Most of our cultural influences (and the diet industry) insist that overeating and obesity result from a failure of willpower. However, the evidence from genetics argues strongly against that idea: Data from adoptions and twin and family lineage studies indicates that about 80 percent of the variation in body weight is determined by genes. That’s about the same degree of heritability as a characteristic like height, and much greater than that for other conditions that we now clearly regard as running in families, including breast cancer, schizophrenia, and heart disease.
In a small fraction of cases, obesity results from mutation in a single gene like that encoding leptin or the leptin receptor. It’s not surprising that mutations in a number of the other molecules we’ve discussed in the feeding control circuits, including POMC and the receptors for CRH and MCH, can also cause obesity. However, our best estimates to date are that only about 8 percent of the cases of morbid obesity result from mutations in a single gene. In the vast remainder of the population, interactions of multiple genes with the environment appear to be involved.
Does the genetic component of obesity result in overeating, in normal eating coupled with a slow metabolism, or in both? In most studies, when food intake is monitored closely, it appears as if overeating is by far the dominant factor, with reduced energy expenditure contributing, but to a lesser degree. Yes, obese people eat more and exercise less. But why? A large part of the answer will be found in molecular and cellular analysis of the brain’s feeding control and pleasure circuits.10
Can we reasonably extrapolate from the aforementioned rat studies and conclude that a blunted dopamine pleasure circuit might drive compensatory overeating in humans? As is the case with rats, eating in humans is associated with dopamine release in VTA target zones, including the dorsal striatum. The benefit of human subjects is that we can talk to them after they’ve been in the brain scanner and ask them how they felt. These studies revealed not only that eating was associated with dopamine release, but also that the degree of dopamine release could be used to predict how pleasurable the subject rated the experience of eating. Different foods produced different levels of dopamine release, a finding that correlated with the reported pleasure of eating. (For me, Louisiana hot sausages would peg the dopa-meter.) Also, as hungry subjects continued to eat and became sated, the amount of dopamine released in the dorsal striatum was reduced. Not surprising: The first bites of a meal give the most pleasure when you’re hungry.
Again, like rats, humans who take drugs that increase basal dopamine signaling show reduced appetite, as well as reduced caloric intake and weight gain, whereas drugs that reduce dopamine signaling produce the opposite effects. So far, so good. Another important observation is that, on average, dopamine receptor density is reduced in the VTA target regions of obese subjects as compared with those lean subjects (a characteristic that can be measured in a brain scanner). But the key question remains: Do obese individuals show reduced dopaminergic activation of VTA target areas in response to food? Is a blunted pleasure response to food involved in obesity?
A recent study by Eric Stice and coworkers at the University of Oregon placed obese and lean subjects, all young women, in a brain scanner while giving them sips of chocolate milkshake through a plastic tube.11 Not only is chocolate an unusually good activator of brain pleasure centers, but it’s a lot easier to run a flexible straw into the mouth of a head-fixed subject in the brain scanner than, say, proffering a pastrami sandwich or a plate of risotto. The main finding was that the obese subjects showed significantly less activation of the dorsal striatum in response to milkshake sips than did the lean subjects, supporting the blunted pleasure hypothesis.
These milkshake-sipping young women also agreed to DNA testing for a common genetic variant called the TaqIA A1 allele, which results in a reduction in the density of D2-type dopamine receptors in the pleasure circuit. Carriers of the A1 allele showed the greatest reduction in milkshake-evoked dorsal striatum activation. When follow-up examinations were performed a year later, A1 carriers also showed significantly greater weight gain than noncarriers. So do some obese people overeat to try to compensate for low-functioning pleasure circuitry? That’s likely to be part of the explanation, but there may be another twist. “If you look at the brain response when people are about to get the milkshake, obese individuals show greater activation of the reward circuitry, not less,” Stice observes. “So, ironically, they expect more reward but seem to experience less.”12 It’s a cruel double-edged sword: increased craving coupled with decreased pleasure. In fact, this pattern may be a general problem for many forms of compulsive and addictive behavior, not just overeating. Carriers of the TaqIA A1 allele of the D2 dopamine receptor are not only more likely to be obese, but they are more likely to struggle with drug and alcohol abuse as well as compulsive gambling.
Another front in the war for your body fat is located in the test kitchens and offices of restaurant chains, commercial bakeries, and other “food service” corporations. While body mass index is indeed about 80 percent heritable, it’s clear that environment and gene/environment interactions also play a major role in determining an individual’s weight. One telling statistic is that the average weight of an adult in the United States has increased by about twenty-six pounds between 1960 and the present.13 Clearly, this is not due to genetic changes in the population. Rather, it’s mostly a result of the concerted efforts of these corporations to produce food and drink, served in large portions, that maximally activate the pleasure circuit and thereby contribute to overeating.
Imagine that we’re test kitchen chefs for the Ruby Tuesday restaurant chain or the Nabisco Corporation or PepsiCo (which also owns KFC and Taco Bell). Our goal is to create delicious food that people will crave and will return to buy in large quantities. How do we go about this? How do we create foods that activate the pleasure circuit so strongly that they override the satiety and body weight signals that would normally prevent overeating? Basically, we do this by exploiting the mismatch between the food landscape we humans have evolved to navigate (that of our ancestors) and that of the present.
While our ancestral human diet varied for different groups of our forebears who lived in different habitats, it did have certain common features. It was a diet that was mostly vegetarian, with very little fat (probably about 10 percent of total calories) and very little sugar. Sweet flavors were rarely encountered—they typically occurred in ripe fruit or wild honey—and meat was a rare luxury and was usually quite lean when it could be obtained. For inland peoples, salty flavors were almost unknown. There were few foods with high moisture and oil content that would enable them to be chewed and swallowed quickly. Most important, in many locations intermittent famines were regular occurrences, so when energy-dense foods containing fat and sugar were available, it made sense to gorge on them to establish a body fat reserve for anticipated hard times.14
The result of this ancestral diet is that we are hardwired from birth to like certain tastes and smells, most notably those of sugar and fat, but also salt. Both humans and rats show much greater activation of the VTA and dopamine release in VTA target regions when eating energy-dense fatty and sugary foods. Like the difference between “chewed” and injected cocaine, this may reflect the different concentration profiles of glucose (or some other food-related signal) as conveyed to the brain: Large, fast-rising pleasure signals are the most rewarding and most addictive. Interestingly, the combination of fat and sugar is superaddictive, producing a significantly larger jolt to the pleasure circuit than either one given alone. Not only will rats in a Skinner box work hard and perform many lever presses for a sweet, fatty food reward, but those that have already eaten their fill of lab chow will readily consume more food if it is sweet or fatty. (Froot Loops cereal works particularly well.) Then again, we didn’t really need an experiment with rats to tell us this: Everyone has had an experience of feeling full at the end of a meal but still having “room for dessert.” Our love of salt, meanwhile, remains a bit of a puzzle: Rats won’t work for it, but humans crave it, possibly as an adaptation to compensate for salt loss from perspiration.
In our role as corporate test kitchen chefs, we don’t need rats, Skinner boxes, brain scanners, or even a knowledge of the scientific literature to figure out how to make foods people crave and overeat; all that is necessary is a collection of different recipes to try out on willing subjects.15 Even so, developing those recipes is actually a very complex business, for to make craveable foods you can’t simply add more salt, fat, and sugar to your existing ones. There is, for example, no single ideal salt concentration for food. We tend to like a lot more salt on our chips and crackers than we do on our meat or in our soup. We like intensely sweet foods better if they are combined with fat. We are also more likely to overeat if foods have a combination of contrasting tastes: Ice cream with chocolate and fruit bits mixed in is more compelling than a smooth, single-flavor ice cream. More Buffalo wings are eaten if they are paired with a contrasting dipping sauce (like cool, fat-laden ranch dressing). Sweet and spicy, fatty and salty, spicy and salty are all combinations that work. Contrasting textures are also highly rewarding: A crispy fried exterior with a soft filling will often be the basis for a craveable food. Flavors and odors from cooking fats also provoke a strong response, perhaps because we have an inordinate number of olfactory receptors devoted to fatty odors.
Another thing that test kitchen chefs have discovered is that people will eat more food if they don’t have to work too hard to chew and swallow it. Hence much of the meat served in chain restaurants has been mechanically tenderized and injected with marinade. It dissolves in your mouth quickly and is lubricated for swallowing by high water content. In essence, the factory has done half of your chewing and swallowing for you so you can eat more. And finally, one of the simplest strategies is to take advantage of the fact that people tend to finish what’s on their plate (or drink what’s in their bottle of soda). Bigger portion sizes are a simple and effective way to promote overeating, defeat the body’s appetite control system, and sell more food.
The increase in body weight that’s swept the United States in the last forty years, and that is also being seen in other affluent countries, is an enormous public health problem. Increases in weight put people at an elevated risk for a range of health problems, including diabetes, cancer, sleep disturbances, heart disease, and hypertension. The good news is that even modest weight loss—from five to twenty pounds, a level that can be reliably sustained through good eating habits and exercise—has significant health benefits. But what about the morbidly obese, who would need to achieve and sustain a dramatic reduction in weight to experience those benefits? As we have discussed, the brain’s homeostatic systems will work against such an effort, as they increase appetite and slow metabolism, making a dramatic weight loss very hard to maintain. Bariatric surgery, in which a portion of the gut is resected, is one option, but because it carries significant risk and expense it is elected in a small fraction of cases. Consequently, there has been a concerted effort by pharmaceutical companies to develop safe and effective anti-obesity drugs to augment diet and exercise.
It’s worth noting that there are already unsafe drugs available that will reduce appetite and cause weight loss in a broad spectrum of patients. Drugs like amphetamines, which artificially stimulate the midbrain dopaminergic reward circuit, are very effective but are also highly addictive and have disastrous side effects. Likewise, for many years the drug fenfluramine was prescribed for weight loss, typically combined with a weak amphetamine called phentermine in a formulation sold as “Fen-Phen.” Fenfluramine acts by blocking transporters that package the neurotransmitter serotonin into vesicles for later release and by reversing serotonin reuptake across the membrane of the presynaptic terminal to secrete serotonin into the synapse. Tragically, fenfluramine produced heart valve disease in about 20 percent of women and 12 percent of men—a condition that progressed long after these individuals stopped taking the drug. It was withdrawn from the market in 1997 and has been the subject of one of the largest product liability lawsuits of all time, with more than fifty thousand claims and a potential liability for the Wyeth drug company estimated at $14 billion.
At present large numbers of weight loss drugs are in various stages of development, from initial testing in rats and mice to late-stage human clinical trials. Not surprisingly, the potential market for a safe and effective weight loss drug is huge, so considerable commercial impetus is driving research in this area. Candidates include a set of drugs designed to enhance satiety signals from the gut. For example, the drug SR146131 targets the activation of specific receptors for the gut hormone CCK, thereby promoting a feeling of fullness. Another set of drugs is designed to target feeding control and pleasure circuits in the hypothalamus and medial forebrain. Recall that NPY released from a subset of neurons in the arcuate nucleus of the hypothalamus stimulates appetite (Figure 3.2). Animal studies have shown that drugs that bind and inactivate NPY receptors could block this action and promote weight loss.16
Another potentially useful class of weight loss drug is compounds that target receptors for endocannabinoids, the brain’s own THC-like molecules. This is a clever strategy inspired by the well-known stoner phenomenon of “the munchies”: Since smoking cannabis stimulates appetite, perhaps conversely drugs that block the action of endocannabinoids (at the major neuronal cannabinoid receptor called CB1) would suppress appetite. Indeed, the CB1-blocking drug rimonabant (also known by trade names including Acomplia and Slimona), a compound developed by the drug company Sanofi-Aventis, has been approved for treatment of obesity in over fifty countries. There’s no question that rimonabant can produce moderate weight loss. In clinical trials, patients receiving 20 mg/day of the drug for a year lost an average of about sixteen pounds, compared to about four pounds for a group that received a placebo. Other measures of body fat, such as waist circumference and triglyceride levels in the blood, were also reduced.
Unfortunately, its use has also raised some very serious concerns about side effects. Rimonabant has been linked to a significant increase in nausea, major depression, and even suicide in obese patients, a finding that has caused the European Medicines Agency to issue a recommendation to doctors to discontinue its use in European Union countries. In a unanimous vote the fourteen members of the Endocrinologic and Metabolic Drugs Advisory Committee of the Food and Drug Administration turned down a 2007 application from Sanofi-Aventis to allow the use of rimonabant for obesity treatment in the United States. It may ultimately be possible to create CB1-blocking drugs that will decrease appetite without involving such serious side effects, and indeed CB1 drugs being developed by Sanofi-Aventis and other companies have subtle differences in their mode of CB1-receptor blocking that could reduce these symptoms.17
Now, at this point you may logically be wondering, “What about leptin? You already told us how it can reduce appetite and cause weight loss in people with an inborn leptin deficiency. Why not just use it on all obese people?” Indeed, the biotechnology company Amgen had this idea in 1995 when it paid $20 million to license the hormone. Unfortunately, when Amgen sponsored a large clinical trial of leptin, few obese participants lost weight.18 In hindsight perhaps this is not surprising: The vast majority of obese people have high circulating leptin levels because they have a lot of body fat; adding extra, exogenous leptin doesn’t help much. The conclusion from these findings: Most obese people are not leptin-deficient, but rather are leptin-resistant. They lack some of the molecular machinery necessary to transduce circulating leptin into reduced appetite and increased energy expenditure. Little is known about the molecular basis for leptin resistance. It could involve changes in downstream feeding control molecules (like NPY or MCH or CRH or orexins) or their receptors. There are even some indications that leptin resistance can result from a failure of leptin to pass from the blood into the brain through a structure of tightly woven cells called the blood-brain barrier. Understanding the molecular and cellular basis of leptin resistance will certainly yield new insights for developing anti-obesity drugs.
Jane has been feeling totally stressed out. She is eighteen years old and lives with three other girls in a small apartment. She and her roommates bicker a lot, and Jane is clearly at the bottom of the social rank. The others push her around, and as a result she tends to avoid them. When she lived by herself, Jane was slim and ate a balanced diet, but since she has been in this pressure cooker of an apartment, she’s taken to snacking all day and all night and choosing high-fat foods over more healthy fare. Her weight and waistline have increased significantly. When she went for a checkup, tests revealed high levels of stress hormones in her blood. These days, when one of her domineering roommates approaches, Jane will grimace, emit a submissive squeal, and retreat to the corner.
“Jane” is a macaque monkey living at the Yerkes National Primate Research Center, and I’ve adapted her story (with some literary license) from a recent report by Mark Wilson and coworkers that appeared in the scientific journal Physiology & Behavior.19 Their findings are remarkable in their parallels with the well-known human phenomenon of stress-induced eating. Not only did Jane and the other low-social-status monkeys in the experiment eat more, but they began to eat “between meals” and they changed the composition of their diet to include more high-fat comfort foods.
Moderate stress will, in fact, stimulate appetite in a wide variety of mammals, from rodents to humans. In several rodent species, chronic restraint stress, forced swimming in cold water, or social stress produced by introduction of a dominant intruder animal triggers increased feeding and the choice of calorically dense foods with high levels of fat and sugar. This causes weight gain and especially the addition of abdominal body fat.
These results suggest that there is some stress-evoked biochemical signal that modifies the feeding and/or pleasure circuits to trigger overeating of comfort foods. Stress triggers a signaling cascade in which neurons of the hypothalamus secrete corticotropin-releasing hormone (CRH), which travels a very short distance through the bloodstream to the adjacent pituitary gland, where it activates pituitary cells to secrete corticotropin itself (also called adreno-corticotropic hormone, or ACTH) into the bloodstream, which then disperses it throughout the body. One important target is the adrenal gland, which when stimulated by corticotropin releases yet another hormone, corticosterone. Corticosterone and its metabolites can pass into the brain, where they contribute to brain responses to stress. The role of corticosterone in feeding is supported by studies showing that corticosterone injections can substitute for stressful experience in triggering overeating. One interesting implication of this work for us humans is that behavioral strategies for stress reduction (like meditation or exercise) can reduce the amplitude of stress hormone surges and are thereby effective in reducing stress-triggered overeating.
While moderate stress can trigger overeating, severe stress can have the opposite effect, suppressing appetite. In humans, for example, it is typical to have a short-term decrease in appetite while grieving the loss of a loved one. This effect of severe stress appears to be conserved across mammalian species as well. When the intensity of either restraint or social stress is increased, rodents will also decrease their food intake.
Overeating is not the only compulsive behavior that can be triggered by stress. As discussed in chapter 2, stress is often a trigger for the use of certain drugs—such as alcohol, heroin, nicotine, cocaine, and amphetamines—that activate the pleasure circuit. Stress plays a particularly critical role in triggering relapse following a period of abstention in addicts: More than 70 percent of relapsing addicts report a particularly stressful event as precipitating their return to drugs. As with overeating, this finding emphasizes the importance of behavioral stress-reduction techniques in programs to help addicts stay clean. It also suggests that drugs that interfere with stress hormone responses, such as blockers of the CRH receptors, hold promise for the treatment of both stress-induced drug relapse and stress-induced overeating.
How does stress influence the midbrain pleasure circuit (or the feeding control circuits)? The short answer is that we don’t really know. However, there are some tantalizing initial clues. Recall that twenty-four hours after a single exposure to cocaine, the excitatory glutamate-using synapses received by VTA dopamine neurons express LTP. This change, which will result in greater dopamine release in VTA target areas, could also be produced by nicotine, morphine, amphetamines, or alcohol. Amazingly, even brief exposure to stress (a rat’s five-minute-long forced swim in cold water) also produced LTP of the VTA synapses that was indistinguishable from that evoked by drugs. What’s more, the stress-induced LTP could be prevented by pretreatment with a corticosterone receptor blocker. This suggests that drugs and stress rewire the pleasure circuit in overlapping ways and that the stress response to trigger LTP in the VTA requires a stress hormone signaling loop from the brain to the body and back.20
In addition to stress actions mediated by corticosterone, there is also evidence that CRH can act directly on synapses in the VTA, as there are CRH-releasing axons that run from the hypothalamus to the VTA. In one recent study by Antonello Bonci and coworkers from the University of California at San Francisco, living brain slices containing the VTA were prepared from both cocaine-treated and saline-treated control mice. (These are kept alive for a few hours in a bath of oxygenated salt solution designed to mimic the milieu of the brain.) When CRH was added to the brain slices of control mice, no change in synaptic strength was produced. But when CRH was added to the brain slices derived from cocaine-treated mice, LTP of the glutamate-using synapses received by the VTA was observed. This is an exciting finding because it suggests a biological framework for how stress could trigger drug relapse.21
So we’ve seen that both certain foods and certain drugs can activate the pleasure circuit. We’ve also seen that, in many cases, obesity results from food addiction and that food addiction shares many properties and biological substrates with drug addiction, including a strong heritable component and triggering by stress. We know some of the ways in which chronic exposure to some drugs can rewire the pleasure circuit, by changing synaptic structure and function. These findings beg the question: Does chronic exposure to craveable foods—those high in combinations of fat, sugar, and salt—also rewire the pleasure circuit to promote and amplify continued craving?
A recent report by Paul Johnson and Paul Kenny of the Scripps Research Institute suggests that this is indeed the case.22 They began by allowing a group of rats near-constant access to both standard lab chow pellets and an energy-dense “cafeteria diet” including bacon, chocolate, sausage, cheesecake, and frosting. Forty days later, when compared to a control group of rats fed only lab chow, the cafeteria diet rats showed reduction in the levels of D2 dopamine receptors in the striatum, a central node of the reward circuit. Furthermore, when these groups of rats were implanted with electrodes to directly activate pleasure centers and were allowed to self-stimulate their brains, it was found that stronger electrical pulses delivered to the brain were required to maintain self-stimulation in cafeteria diet rats. Thus it appeared as if many days of the cafeteria diet left the reward circuit partially numbed, an effect that is also seen with chronic cocaine or heroin treatment in both rats and humans.
These findings are interesting and provocative correlations, but they do not speak to the question of whether reduced levels of striatal D2 receptors will actually contribute to continued food craving. To address this issue, the authors took a group of lab chow–fed rats and injected a genetically engineered virus into a subregion of the striatum (the dorsal striatum). This virus was designed to reduce levels of D2 dopamine receptors, and its efficacy for doing so was confirmed with biochemical measurements. Like rats that ate the cafeteria diet for forty days, these rats with artificial reduction of their striatal D2 receptors also showed increased thresholds for brain stimulation reward—they were also partially numbed to pleasure.
These are early days in our understanding of both food and drug addiction, and there’s a temptation to overreach. Do these addictions share some common biological and genetic substrates in the brain? Almost certainly. Are those substrates identical? Almost certainly not. Moving forward, it’s likely that some of the treatments for drug addiction (behavioral strategies leading to stress reduction and relearning as well as emerging biological treatments for drug addiction that act on the brain’s reward circuitry or the stress hormones that modify it) will also be useful in treating food addiction.