8
The Phylogenomic Origins and Definition of Homo sapiens
Peter J. Waddell
Introduction
In this chapter I examine Homo sapiens from a phylogenetic perspective and, using a variety of phylogenetic methods (Swofford et al. 1996, Felsenstein 2004), look at the definition and origins of this species. Phylogenetics is sometimes viewed as a subset of genetics, but the reverse also has validity. Every single nucleotide in every single organism follows a tree, and that is the basis of phylogenetics/phylogenomics. Genetic methods, in practice, often aim to avoid the difficulty of knowing each of these trees in order to yield tractable methods and interpretable results, which may or may not be trees or networks of descent (Felsenstein 2004).
Taking a phylogenetic view of Homo sapiens requires a definition of that species (Wilkins 2011). Darwin (1871) even once pondered the question of whether there was one or more species of living human. Although before World War II numerous species as well as genera of fossil hominids had been proposed, post-World War II there was a radical shift towards uniting nearly all these into the genus Homo, with many falling into one super-species, or a species swarm, designated with the suffix sensu lato. On the belief that one species graded imperceptibly into the other, Wolpoff et al. (1994) even went so far as to suggest lumping all specimens subsumed in H. erectus into H. sapiens sensu lato, and getting rid of the former altogether. Although extreme, this perspective derives from and characterizes the Multi-Regional (MR) hypothesis of human evolution (e.g., Wolpoff 1989, Wolpoff et al. 2001).
In contrast, the “Out of Africa” (OA) set of hypotheses (Cann et al. 1987, Vigilant et al. 1991), for the origin of living human population differences—which, according to the model, emerged as the descendants of an African ancestor migrated from that continent and spread throughout the Old World only ~100-60kya (~ means approximately) is neatly tailored to a precise phylogenetic species definition. Nevertheless, the boundaries of this taxically constrained species are often vaguely delineated, even by those who embrace some elements of the OA model. One example is the desire to call fossil forms such as LH-18 anatomically modern Homo sapiens (e.g., Rightmire 1998), while others will call it Homo heidelbergensis (e.g., Profico et al. 2016). Thus, the first task of this chapter is to define Homo sapiens using the same phylogenetic principals (Hennig 1966, Jefferies 1979) used to define anthropological groups all the way up to the mammalian super-order containing Primates, Euarchonta.
In this regard, I begin by reviewing how morphological data can objectively help us diagnose what may and may not be Homo sapiens, phylogenetically defined. Recent reevaluations of fossil skulls make it clear that the term “anatomically modern H. sapiens” must be refined (Schwartz 2016). Independently, quantitative phylogenetic analysis of skull shape is yielding statistically robust results supporting the OA model (Waddell 2015) and, perhaps, instances of hybridization (Waddell 2014). Importantly, these are broadly consistent with recent subjective views (e.g., Stringer 2016). Surprisingly, there are few “traditional” qualitative morphological character-based phylogenetic analyses (Swofford et al. 1996, Felsenstein 2004) of the origins of H. sapiens.
The ongoing efforts to sequence modern and ancient, or aDNA, genomes are making major impacts on the field (Der Sarkissian et al. 2015). These studies have a huge advantage over single nucleotide polymorphism (SNP) panels in not having ascertainment bias, which is due to only incorporating SNP’s already seen to be variable in previously sequenced genomes. Large amounts of genomic DNA have the advantage over mtDNA of having considerably more informative sites, at many unlinked loci, of which many are effectively near neutral and thus amenable to mathematical models of evolution. aDNA sequencing at the full genome level has progressed rapidly (Pääbo 2014), and now includes extensive sampling of individuals (e.g., Fu et al. 2016). Sequenced ancient Homo genomes now extend back to about 100+ kya (kya = one thousand years ago) for exceptionally preserved specimens (Prufer et al. 2014), and partial genomes have been analyzed from specimens as old as ~420 kya (Meyer et al. 2016).
The analysis of these genomes requires methods compatible with population genetics. They are able to detect introgression/hybridization, even when the DNA comes from an unknown form, a “ghost lineage,” as is the case in suggestions of an early diverging (Homo erectus-like?) lineage leaving genes in the Denisova genome (Waddell 2013, Prufer et al. 2014 Kuhlwilm 2016). A wide range of methods, including Buneman, P and D-statistics, plus well-established phylogenetic methods, are considered. A full spectrum of a site-patterns approach allows a detailed look at residuals (Waddell 1995, Waddell et al. 2011b). Computationally expensive Bayesian and Hidden Markov models are also providing a richer view of phylogenies (Gronau et al. 2011, Schiffels and Durbin 2014). An example of how unexpected splits in the autosomal genome data are still being discovered is the suggested introgression of a non-sapiens lineage into West Africans analyzed below. This contrasts with Chromosome X, which shows clear introgression between distinct populations of Homo sapiens, but much less evidence of non-sapiens introgression.
It should also be emphasized that the individuals/populations selected for these studies are often somewhat atypical of most recent modern populations, which often show high levels of mixing between lineages, regions, or continents due to major migratory events of the past ~10 kya. The hope of building trees and networks of populations that were relatively isolated 10 kya is to recover the major deep patterns of where H. sapiens came from and how it spread around the world. At the beginning and end of this chapter, I will examine how the OA model was conceived in the early 1990s, versus how it is supported and/or contradicted by the latest results.
Out-of-Africa Predictions: Early 1990s
For the last three decades the OA hypothesis (Cann et al. 1987, Vigilant et al. 1991) has been the favored explanation for the origin of Homo sapiens. Waddell and Penny (1996) reassessed and reanalyzed the key data available at that time using novel statistical analyses, to examine the When, Where, Who, and How set of hypotheses. To recap, these are:
When: Origin of Homo sapiens almost certainly less than 200 kya.
Where: Most consistently Africa, almost certainly sub-Saharan, with the very deepest lineages dominated by pygmies of Central Southern Africa.
Who: The actual people that left Africa seem to have been closer to the ancestors of East and West African, rather than Southern African populations.
How: OA involved a population bottleneck, followed by a relatively rapid expansion across Eurasia and then down into Sahul (Australia and New Guinea), replacing earlier inhabitants with no apparent evidence of interbreeding.
In terms of when, the occupation of Sahul at about 50–60 kya (Bowler et al. 2003, Roberts et al. 1990) remains a key calibration point for any genomic OA study, and is a valid test of the rather wide range of current rate estimates.
The dividing line between an OA-like scenario that emphasizes the basic genetic origins of a species or lineage in one region (with perhaps a co-adapted genome) versus a MR-like model that emphasizes essential genetic components/modules inherited from widely scattered independent lineages, is not easy to draw. Here, I propose that a rough line can be drawn if descendant lineages average more than 80% from a single parental population, and if genetic regions coming in from independent lineages also show clear depletion in most functional genes and few or no inherited genetic modules. In this case, evolution is likely reflected in a model of independent lineages with limited introgression (not necessarily adaptive overall). Templeton 2017 (this volume) argues that there is evidence suggesting over 10% of the genomes of non-Africans came from earlier Eurasian lineages, such as Neanderthals. However, for MR supporters, it is not clear how little gene flow or essential surviving genes there must be before they would reject the MR model. Thus, some researchers may never be able to accept that the OA model represented here was ever different from an MR model.
To contrast these two models another way, one may ask: If all hominids outside of Africa were exterminated immediately after the first lineage split among living humans, would H. sapiens still exist (the essence of OA)? If not, the answer might include the historic MR model. In this regard, my conclusion diverges from Templeton’s (2017, this volume).
A Phylogenetic Definition of Homo sapiens
Thanks to Hennig (1966), a rigorous approach to taxonomic classification—phylogenetic systematics—has increasingly dominated biology, and will be followed here. The desire to have a formal objective classification founded on monophyletic groups (a group or clade = ancestor and all its descendants) has, in turn, driven and been driven by a major effort to estimate the phylogeny (a tree or more complicated network of descent) of a set of homologous (same origin) and reliably aligned DNA sequences. Because a hypothesis of monophyly is precise and testable, it brings with it a level of rigor and objectivity to the understanding of “species” and “higher taxa” lacking in other approaches.
The most powerful phylogenetic definition of a species is that of an “extant crown group.” For living organisms, and in order to be fully testable, such a hypothesis should include a list of all members of that group. If it shows precise congruence with at least some of the properties of other popular concepts of “species” (Wilkins 2011), all the better, but equivalence is not essential. For H. sapiens, the most inclusive living crown group to be considered is the last ancestral population of all living humans and all descendants of that population.
It is increasingly being demonstrated that one ancestral population is the source of > 90% of all living human DNA. This is somewhat surprising since the genus Homo, being relatively recent (~2.5 million years old) and encompassing mobile, adaptive individuals, has been expected to show elements of a species complex, including a significant amount of introgression/hybridization. The MR hypothesis is based on such an expectation. In contrast, the OA model consists of a set of hypotheses that are consistent with H. sapiens being a phylogenetically recent and easily defined species. However, a consequence of H. sapiens being nested deep within the genus Homo, is that other proposed species, such as the purportedly widely distributed Homo erectus, are likely to be para- or polyphyletic, and themselves in need of taxonomic revision. Indeed, the disparate morphologies of specimens attributed to “erectus” suggest revision that goes well beyond allocating the East African specimens attributed to H. erectus to H. ergaster (Schwartz and Tattersall, 2002, 2003, 2005).
Why an Extant Crown-Based Definition of Homo sapiens?
In phylogenetic systematics, the basic principals should be the same across all taxonomic levels. For example, the clade and superorder Euarchonta (Waddell et al. 1999, 2001), to which humans belong, is defined as the common ancestor of the extant mammalian orders Primates, Dermoptera, and Scandentia, plus all descendants. As there are living species on both sides of the deepest split within Euarchonta, it is considered a “living” or “extant crown group,” which has attained special meaning in phylogenetics (stem and crown group terminology was developed by Jefferies [1979], based on the work of Hennig [1966]). By extension, it also has special meaning in computational biology, genomics, evolution, and other areas of biology that have been transformed or are emergent because of the methods of phylogenetics. For the last common ancestor of an extant crown group is, on balance, the ancestral population on which the best inferences of its biological properties can be made, which in turn, best predicts the properties of its descendants. Using extant crown groups, these ancestor-based hypotheses can be generated with a maximum amount of interpolation and a minimum amount of extrapolation. They are, therefore, expected to maximize the predictive power of the full range of biological hypotheses. In the case of the species H. sapiens, its crown encompasses the ancestral population of all living humans. As we shall see below, research over the past 30 years on sub-Saharan African populations and, with increasing focus on the Khoisan of Southern Africa, suggests they provide half of the deepest split amongst living humans.
Potential complication of this definition arises when a “species” is expected to present “special” properties: for example, there are at least two-dozen proposals along this line, many of which are contradictory (Wilkins 2011). Here I follow the view that species do not have any essential quality or typological essence, but that, given a sufficient amount of “evolutionary time,” a separate lineage will not readily mix parts of their genomes with other species and tends to express emergent properties. With regard to the Biological Species Concept, there are various forms, each with different emphases on pre- or post-mating mechanisms, different ecologies, prolonged geographical isolation and adaptation, and distinct mate recognition systems. Another process may, however, also be at work with distinct co-adapted genomes: There may be introgression, but over time, purifying selection will remove most of the functionally distinct introgressed elements, leaving mostly near neutral genomic regions.
Unfortunately, any emergent property can come into conflict with phylogenetic classification. For example, when a single descendant population does not mix with its relatives, the latter would constitute a non-monophyletic group. Ultimately, what phylogenetics offers biology is the encouragement to map as accurately as possible the pattern of evolution, as well as guidelines for how this “map” might most informatively be divided up. Such a phylogeny can certainly have properties related to “failure to introgress” mapped upon it. Phylogenetic classification is the natural extension of taking phylogenies as fundamental in understanding biology.
Finally, phylogenetic systematics permits some genetic introgression. If it is great enough, a hybrid species may need to be diagnosed when its properties are sufficiently distinct from either parent lineage. On the other hand, the sharing of relatively small genomic blocks, particularly if they are not fixed and account for less than 10 percent of the genome, constitutes introgression, which is expected to be a common pattern among closely related species that do not always have strongly developed pre- and post-zygotic barriers to gene flow.
Semantic Corollaries of an Extant Crown-based Definition of Homo sapiens
This phylogenetic definition of H. sapiens leads to certain semantic corollaries. Firstly, if it is accepted that human equals H. sapiens then, in the absence of a better equivalence, Neanderthals and other non-Homo sapiens lineages should not be referred to as human. Another is that “modern human” and “human” effectively become synonymous, with living humans remaining a distinct category. Such precision is particularly important in reference to specimens, such as those from Qafzeh (about 90–100 kya Israel). Specifically, Qafzeh 9 is the oldest fossil that appears to be consistent with the current understanding of morphological variability in living humans. It might even be shown to be morphologically indistinguishable from Homo sapiens and even nestled well inside that envelope of variability. Nevertheless, as argued here, Qafzeh 9 would not be identified as H. sapiens if it derived most of its genetic material from a population that predates the last common (ancestral) population of living humans. While such a strict use of the term “human” may seem pedantic, even discriminatory, I suggest it is appropriate if the goal is to come to solid scientific conclusions. Where there is the potential for ambiguity, H. sapiens with a phylogenetic definition should be used.
As mentioned earlier, the term “anatomically modern humans” (AMH) should be used with more caution. According to Schwartz (2016), only extant humans and a very small set of fossils can be accurately called anatomically modern, that is, morphologically indistinguishable from living humans. Much of the literature is full of imprecise uses of the term AMH, for specimens that, are likely “pre-Homo sapiens.” More precisely, and, for instance, with regard to Neanderthals, “pre-H. sapiens” would specify all lineages that originated along the lineage between the common ancestor of H. sapiens and the common ancestor of H. sapiens and H. neanderthalensis, but not including H. sapiens. Further, in recognition of the the need to refer to a specimen that is either H. sapiens or pre-H. sapiens, one might consider using the phrase “pro-H. sapiens vis-à-vis H. neanderthalensis,” to mean “of the clade that is closer to H. sapiens than to H. neanderthalensis.”
The currently popular terms “archaic human” or “archaic Homo” (AH) basically means any member of the genus Homo that is not H. sapiens according to some definition of that species (e.g., Schwartz 2016). The term “archaic” is nearly synonymous with “primitive” in general biology (which in turn typically equals “ancestral”). The term “primitive” in phylogenetics and evolutionary biology needs to be used very carefully, as it often means nonderived. As discussed below, there may have been many lineages (species in their own right with their own sets of derived features) during the last ~500 kya that were closely related to Homo sapiens, of which none were ancestral for H. sapiens. In this case, a more balanced term is simply “non-H. sapiens.”
At present, we do not have a name for the often-referred-to crown-group within Homo that comprises H. sapiens, H. neanderthalensis, and all other descendants of their common ancestor, such as the Denisovan individual. I propose that this clade of three genetically characterized hominids should be identified as “HND,” for human, Neanderthal, and Denisovan. This clade has the unusual property of being a “DNA-extant” crown group that includes nearly fully sequenced genomes on each descendant lineage, while lacking living individuals on all but one descendant lineage.
Finally, proposed subspecific names (for example, H. sapiens sapiens, H. s. idaltu) have no validity in this phylogenetic classification. H. s. sapiens is synonymous with H. sapiens, while H. s. idaltu is almost certainly neither H. sapiens (Schwartz 2016) nor a subspecies of H. sapiens, in which case H. idaltu is available.
Morphological Phylogenetic Information
Details of morphology can help diagnose, but not phylogenetically define (see previous sections) those specimens that may be H. sapiens and those that are not. These details should be analyzed in ways that are consistent with the major methodologies of phylogenetics. In this regard, a broad collaboration, representing a wide range of knowledge and disagreement, would seem to be desirable in order to arrive at a viable data matrix (e.g., O’Leary et al. 2013).
Derived features (evolutionary novelties) present in certain lineages (e.g., H. sapiens) that can be described succinctly and without need for measurement are qualitative or discrete characters. These can be encoded into a character data matrix for analysis by numerous valid phylogenetic methods, including parsimony (Swofford et al. 1996, Felsenstein 2004).
Properties of specimens that can be represented as measures or some other numerically continuous variable, such as shape or size, are quantitative. Various ways of divvying these up into discrete characters have been proposed, but they are problematic for reasons that include loss of information and bias (Swofford et al. 1996, Felsenstein 2004).
While there has been stagnation in the development of phylogenetic methods/models for quantitative geometric morphometric data (Adams et al. 2013), multiple new methods are now forthcoming. For example, extended distance-based phylogenetic techniques, which constitute a form of compound likelihood, have been shown to have considerable promise (Waddell 2014, 2015). Maximum likelihood and Bayesian analyses are also feasible, but methods for analyzing this specific type of data are still in need of refinement (e.g., Theobald and Wuttke 2008, Felsenstein 2015). Unlike gene frequencies, geometric morphometric data points are not independent characters, which makes problematic their analysis using methods designed for allele frequencies; however, analyses can rest on a simple additive genetic model wherein neutral drift gives rise to Brownian motion (Felsenstein 2002).
Qualitative Morphological Diagnosis of Homo sapiens
Schwartz and Tattersal (2000, 2002, 2003, 2005, 2010) have been particularly vigorous in trying to delineate derived qualitative features (apomorphies; Hennig 1966) found at high frequencies in living humans, that, because of their rarity or absence in fossils, should help in identifying H. sapiens. Among these, an inverted-T-shaped chin and a particular form of a bipartite brow seemed to be most promising. The major problem with the bipartite brow as a diagnostic character is that it has much less than 100 percent penetrance (e.g., young males and any female may lack it); it does not appear fully until later in life, and the H. sapiens unexpressed and the ancestral state appear to be the same (Schwartz 2016). Other potentially powerful diagnostic characters listed in (Schwartz 2016) need further investigation. A lacuna of all these studies, however, is that Khoisan, Pygmy, and Papuan/Australian populations have not been assessed.
The inverted T-shaped chin appears an excellent diagnostic character as its characteristic morphologies emerge early in fetal development and remain crisply defined in young children, after which, growth, differential bone deposition and resorption has the effect of accentuating or softening, but not obliterating, the basic features; it therefore seems to show 100 percent penetrance and persistence. It is interesting to note that Oase 1, with ~10% Neanderthal SNPs (Fu et al. 2015), presents a well-defined inverted-T-shaped chin (Schwartz 2016). Since chin shape emerges early in development, it might also be linked to fundamental developmental programs that do not easily admit to disruption with non-sapiens genes. Absence of this mandibular character, and/or supraorbital adornment that is not non-bipartite, exclude nearly all skulls and mandibles older than 45 kya from Homo sapiens (Schwartz 2016).
The oldest specimens clearly presenting a T-shaped chin are from Border Cave and Klasies River Mouth (~100 kya); penecontemporaneous Qafzeh 8, 9, and 10 may also show it. This seemingly removes many specimens widely proclaimed as H. sapiens from this species, including Chinese specimens older than Tianyuan (~45 kya) (e.g., Zhirendong, Liu et al. 2010). Further, and unexpectedly, the absence of this feature in various recent fossils from South Africa (Boskop perhaps 12–14 kya, Fish Hoek ~9 kya) and China (Curnoe et al. 2012, ~11–14.5 kya) suggests that multiple non-sapiens Homo survived until relatively recently (Schwartz 2016). The South African specimens lacking chins are particularly interesting because their basic skull shapes closely overlap those of living H. sapiens, which suggests their particularly close relationship. Their DNA would provide an excellent test of the utility of the inverted-T-shaped chin to diagnose H. sapiens.
Qualitative Morphological Phylogenetic Analysis
While it is useful to look for the best discrete characters to diagnose Homo sapiens, this endeavor does not replace a carefully constructed, and hopefully widely discussed, critiqued, and annotated character data matrix (Swofford et al. 1996, Felsenstein 2004). Matrices such as that of Mounier et al. (2011) have been analyzed using objective phylogenetic methods (e.g., Waddell 2013), but the characters are in serious need of scrutiny and repeatable description. For example, a key “discrete” character in Mounier et al.’s (2016) matrix is skull volume. However, the cut-off of 1200 cc, which is used to produce a binary character, has no objective justification. Indeed, this decision may derive from preconceived notions of which are the earlier- or later-diverging specimens. If so, this and similar “characters” are analogous to the old joke about Dwayne, the undisputed best shot in the county, whose bullet always hits the target dead center. The problem, as it turns out, is that Dwayne draws the target around his bullet hole. Thus, while the trees obtained to date are interesting, they contain relatively few specimens and are not highly resolved by unweighted parsimony (Mounier et al. 2016). The challenges of developing well-substantiated characters are discussed in Wagner (2001).
Quantitative Phylogenetic Morphological Analysis
Over relatively short periods of time, myriad genes can influence aspects of morphology, such as shape, without leaving easily agreed-upon discrete characters (Felsenstein 2002, Adams et al. 2013). The shape of the upper skull/calvaria is one example. Phylogenetic shape analysis (= geometric morphometric phylogenetics) aims to capture and quantify such change according to an explicit statistical model, one that often assumes Brownian motion.
In terms of the actual processing of 3D measurements, currently points in space are generally scaled, centered, and rotated to coincide with each other with a minimum sum of squared coordinante differences; this yields a Procrustes, or a minimum-scaled Euclidean, distance (Adams et al. 2013). A matrix of all such distances between specimens can serve as the starting point for distance-based phylogenetic analysis, for example, Caumul and Polly (2005).
Recent analyses employing improved methods indicate that there is a phylogenetic signal, with some apparently well-supported branching patterns, in the skullcap shape of specimens allocated to Homo (Waddell 2014, 2015). A reanalysis of that data (Harvati et al. 2011) with a refined set of constraints is shown in figure 8.1. The node where the two recent Khoisan skulls meet encapsulates approximately two-thirds of 58 Khoisan skulls and 62 percent of all 242 recent (last 10 kya) human skulls sampled (OLS+ tree not shown). Other Khoisan skulls fall around most of the sub-tree containing all recent H. sapiens skulls, and also near the particularly deep European female 012 (EurF012H).

The Ordinary Least Squares (OLS+) tree (Swofford 2000) of fossil Homo skulls based on a pairwise Procrustes distance (q = 0.5). Residual resampling support values of > 50% are shown (+2% increase in sampling variance to counter bias). Underlined skulls constrained a priori to fall together on the tree based upon the expectations expressed in Schwartz and Tattersal (2002, 2003, 2005), that is, no suggestion of more than one morph within that set (from top to bottom, Neanderthals, Jebel Irhoud site, Zhoukoudian Upper Cave) or timing and/or cultural artifacts (pre-Gravettian Europe and Gravettian Europe).
This data is consistent with a strong phylogenetic signal via multiple tests. These include an application of the statistics of Hillis and Huelsenbeck (1992), yielding a g1 statistic of −2.80 and a g2 of −15.94 (both larger than in the reduced dataset of Waddell [2014]), and the substantial levels of residual resampling support (Waddell et al. 2011a) on many internal edges. The tree is also in good agreement with the interpretations of some morphologists (e.g., Klein 2009, Stringer 2016), but suggests more specific detail. African specimens of the last ~300 kya fall closer to H. sapiens than to H. neanderthalensis in a phylogenetic, not simply a phenetic, sense. The results also suggest that H. heidelbergensis is not monophyletic. Even African specimens such as Kabwe and Saldanha appear well separated in the tree (Waddell 2015). Figure 8.1 also suggests that there may have been an accelerated evolutionary rate towards H. sapiens, as well as a branching pattern of multiple pre-sapiens lineages that may have emerged with some regularity in Africa after a split with Neanderthals ~500–800 kya.
A question of considerable debate is whether the specimens at ~100 kya Middle Eastern sites, such as Qafzeh and Skhul, are the result of hybridization (Trinkaus 2007) or if they represent different, even non-sapiens, forms (morphs) (Schwartz and Tattersal 2010, Schwartz 2016). In figure 8.1, Qafzeh 9 consistently falls in that part of the tree populated by H. sapiens skulls. If the positions of Chancelade and Ohalo II are ignored, Qafzeh 9 forms an exclusive group with the Khoisan skulls 85 percent of the time, while the pre-Gravettian skulls diverge more deeply than the Gravettian + Qafzeh 9 + Khoi skulls 99 percent of the time. Since Qafzeh 9 may be the oldest specimen of H. sapiens (Schwartz and Tattersal 2003), it might merit the accolade “the skull of Eve” (Waddell 2015) in similitude with mitochondrial Eve (Cann et al. 1987). Another specimen, Skhul 5, falls deeper in the tree than most H. sapiens, which is consistent with Schwartz and Tattersall’s (2005; also Schwartz 2016) suggestion that it represents a distinct morph.
In contrast, Qafzeh 6 diverges deeply (figure 8.1) and yields a result consistent with it being a hybrid when analyzed with Neighbor Net (figure 7 in Waddell 2014). Its ancestry seems to be approximately two-thirds close to H. sapiens, and approximately one-third close to H. neanderthalensis. These results would appear to make probable the existence of distinct morphs and of hybrids at Skhul and Qafzeh, respectively.
In figure 8.1, European pre-Gravettians (>32 kya) diverge significantly deeper than Gravettians (~22–32 kya), which are deeper than most living humans (the Khoisan pair), which is consistent with suggestions of a decrease in Neanderthal features (Trinkaus 2007) and DNA in Europe over time (Fu et al. 2016). Curiously, the pre-Gravettian skulls that show the most evidence of discrete features characteristic of H. neanderthalensis (see Trinkaus 2007) are not necessarily those that diverge deeper (Waddell 2015), thereby suggesting that overall shape and specific features are not tightly linked genetically. Another example of this effect might be seen amongst Neanderthals. Of the notably late Spy Neanderthals, in contrast to Spy 1, which has been suggested to show Homo sapiens-like features, the divergence of Spy 2 is closer to H. sapiens (Waddell 2015). Another example seems to be Irhoud 1, which in Figure 8.1 diverges more deeply than Irhoud 2 (Waddell 2015); the Mounier et al. (2016) analysis of discrete characters yields the reverse.
Specimens from Upper Paleolithic/Upper Cave Zhoukoudian, China (UC) (~13–30 kya) show a position in figure 8.1 that is consistent with even more inflow of non-sapiens genes than the European specimens. Indeed, UC1 is the most divergent skull yet (Waddell 2015), showing an inverted-T-shaped chin (Schwartz 2016). Inasmuch as the scattering of developmental outcomes is a common feature following hybridization (e.g., Ackermanna et al. 2006), early European, Qafzeh, and UC localities all yield skulls with very different positions on the tree, each including some skulls that are much closer than others to those typical of living humans. These results tend to contradict the common assertion that Upper Paleolithic skulls look different (more robust, for example) than living H. sapiens simply because of their lifestyle and, instead, suggest a genetic component that was introduced by introgression from non-sapiens individuals.
Similar to the classical study of morphology (Schwartz 2016), phylogenetic analysis of skull shape argues that non-Homo sapiens lineages persisted until relatively recently. The ~12 kya Iwo Eleru skull from Nigeria, West Africa, is located in the tree as a deep pre-sapiens lineage (Waddell 2014), which is consistent with expectations of Harvati et al. (2011) based on non-evolutionary models.
The edge lengths in figure 8.1 suggest that the most marked acceleration of change in skull shape, and thus also of brain shape and organization, occurred in the lineage leading to H. sapiens (Waddell 2014, 2015). Further, the long edges leading to Iwo Eleru or to Singa, also suggests an acceleration of change in these lineages as well. These results are consistent with the view that the brain of Neanderthals is essentially a scaled-up version of the brain of its ancestor, while in the lineage leading to H. sapiens its shape and developmental program were altered (Lieberman 2011). Figure 8.1 suggests that this renewed phase of evolutionary experimentation began soon after the divergence of Neanderthals and H. sapiens.
Overall, figure 8.1 shows a “caterpillar-like” pattern consisting of many fairly long terminal lineages, particularly amongst pre-H. sapiens specimens, coming off the main trunk. However, this picture changes somewhat when using the BME criterion (Gascuel and Steel 2006). While the likelihood of this model is not as high as OLS+, quite a few groups of two, three, or four specimens appear in this part of the tree. For example, a wide range of analyses yield the sub-tree ((Iwo Eleru, (LH18, Saldahna)); [Waddell and Swofford, unpublished]). This offers the tantalizing possibility that these three enigmatic skulls may belong to an ancient African clade that was widespread in both space (West, East, and Southern Africa) and time (from ~12 to >300 kya) and, therefore, a potential long-term competitor with the lineage leading to H. sapiens.
This analysis, and others (e.g., Harvati et al. 2011, Stojanowski 2014, Waddell 2014), along with Iwo Eleru having “no pronounced chin” (Brothwell and Shaw, 1971), and an accessible type specimen, arguably present a stronger case for this specimen being recognized a distinct non-sapiens lineage than for H. idaltu, consistent with it being dubbed H. iwoelerueensis (Waddell 2014). As yet there are few clues to the capabilities of this lineage, other than its Middle Stone age context and an apparent, but crude, burial.
Although a tree is presented here, a major challenge of phylogenetics is to accurately reconstruct a more general network when there has been reticulation. While this may have been achieved in the diagnosis of Qafzeh 6 as a hybrid (Waddell 2014), an ongoing methodological challenge is to achieve this reliably for larger data sets such as this.
Molecular Phylogenetic Information
Mitochondrial (mt) DNA
As mentioned above, mtDNA data was key to developing the OA set of hypotheses. The initial data (Cann et al. 1987) were from restriction sites, soon to be followed by sequences of approximately 1,000 bases across the control region (Vigilant et al. 1991). By the late 1990s, sequencing the mtDNA in its entirety led to finer resolution of trees and a better estimation of edge lengths.
Although overall tree structure is similar to that of earlier studies, extensive sampling continues to find the deepest branches of this tree in southern and central southern African populations (e.g., Tishkoff et al. 2007, Gonder et al. 2007, Tishkoff and Gonder 2006). Using models of sequence evolution and calibration points (like those introduced by Waddell and Penny [1996]), the divergence time of the root of the mtDNA tree for living humans also remains similar. The confidence intervals for the age of the ancestral population, however, have not tightened dramatically because other sources of error—calibration error and coalescent dynamics—that are incorporated in the estimates of Waddell and Penny (1996) have not improved much. Further, questions—for example, the impact of slightly deleterious alleles—remain concerning the adequacy of the models of substitution in estimating relative edge lengths (e.g., Peterson and Masel 2009). Uncertainties about the rate of mutation in the genomes of H. sapiens apply equally to mtDNA. Trio sequencing, for example, of the genomes of parents and offspring suggests that the mutation rate over the last million years or so might have been about twice as slow as was assumed based on the overall rate after the human-chimpanzee divergence (Scally 2016).
The ultimate problem with mtDNA is that it is a single genetic locus and, simply because of the random fluctuation of allele frequencies between populations, it may give the wrong impression of the most closely related populations. An example might be the frequency of haplogroup M in living populations leading to the hypothesis of an early coastal H. sapiens migration route to Australia. This hypothesis suggests a later domination of Eurasia by inland routes whereby the order of splitting of lineages would be (Australia (Europe, Asia)), as has been proposed in some full-genome studies (e.g., Rasmussen et al. 2011), but not in others (e.g., Reich et al. 2010, Prufer et al. 2014).
Single nucleotide polymorphisms (SNPs)
Counting allele frequencies, first with allozymic protein techniques and eventually with DNA alterations, particularly SNPs, has lent considerable support for the OA model (e.g., Li et al. 2008) as well as for a (Europe [Asia, Australia]) migration pattern (Li et al. 2008).
Nevertheless, counting SNP frequencies suffers from the weakness that the variants must be pre-selected on the basis of known populations, such that whichever populations are chosen creates an ascertainment bias (that is, one cannot discover what one doesn’t already know). This bias even exists when large data sets of ≥ 100,000 SNPs derived from across the genome are used. Other issues involve methodology, as is the case in interpreting the results of non-phylogenetic methods such as Principal Component Analysis. However, SNP-based studies continue to play an essential role, for example, to screen for the individuals that show the least evidence of admixture, but minimal inbreeding, as candidates for full genome sequencing (e.g., Kim et al. 2014).
Whole genome sequences
The advent of affordable, massively parallel sequencing, in conjunction with advances in sequencing “library” preparation, has made possible whole-genome sequencing of many individuals, whether recent or ancient (Der Sarkissian et al. 2015). These data are amenable to a diversity of phylogenetic methods, including detecting signals that contradict a single tree. Whole-genome sequences of a small number of specimens of Neanderthal, and a phalanx from Denisova Cave, Russia, present an increasingly detailed picture of how distinct lineages within Homo interacted (Green et al. 2010, Reich et al. 2010, Waddell et al. 2011b, Meyer et al. 2012, Prufer et al. 2014, Vernot and Akey 2015, Fu et al. 2016).
Each whole-genome sequence is ~3 billion bases long each; filtering and alignment from short reads often yield >1 billion well-aligned sites. The total number of discrete loci is unclear because recombination hot spots shift over time and recombination can occur anywhere in a sequence. Consequently, although analyses may produce estimates of the predominant tree for a given region of the genome, techniques that assume discrete loci can introduce bias and/or cause most of the data to be overlooked. Alternatively, one can use marginal statistics, for example, pairwise distances (Swofford et al. 1996), rooted triples, or “unrooted” quartets (Bandelt and Dress 1992, Waddell et al. 2001, Reich et al. 2010), or a full spectrum of site pattern frequencies (basically, all binary numbers, Penny and Hendy 1993). These approaches may provide estimates of the splits in the data, both tree and non-tree.
Trees
Simple edit, mismatch, or Hamming distances between genomes of closely related organisms are essentially unrooted two-taxon trees that converge at the average coalescence time (Felsenstein 2004). As such, they integrate over all gene trees to produce a set of estimates, which is the set of all pairwise distances that, in spite of the randomness of the coalescent process in a purely splitting or tree model, are expected to be additive on the “species” tree. In this tree, edge lengths represent the expected number of substitutions, which are usually normalized by dividing by the total number of analyzed sites. This in turn, and assuming a neutral model of evolution, is dependent on mutation rate as well as changes in population size, which determine the rate at which mutations are “fixed” to become substitutions or are lost. By ignoring possible multiple changes at a nucleotide, these requirements permit application of consistent distance-based phylogenetic methods (Swofford et al. 1996, Felsenstein 2004).
Another potentially fruitful aspect of working with whole genomes is that, as long as branching events occurred many generations before, a single human genome can effectively sample an entire ancestral population. Assuming about one recombination event per generation, and per chromosome arm, each genome will undergo about 50 “cuts” per generation, wherein each “chunk” of genome might have been inherited from a different ancestor. That is, because random mating and recombination have divvied the ancestral genomes into many smaller pieces, an individual descendant autosomal genome samples two pieces at each autosomal position. Consequently, and multiplying that by 100 generations (about 3,000 years for humans), one might be sampling up to about 5,000 ancestors, which would be close to many effective population sizes.
The full genome sequences used in Reich et al. (2010) yielded a highly resolved tree: ((Neanderthal, Denisovan), (Khoisan, (West African, (European, (Asian, Papuan))))). Reanalyzing their data with a different method (figure 8.2) depicts the same clear structure in Africa, with the Khoisan diverging markedly deeper than the Yoruba following an even longer internal edge leading to non-African genomes, in which Asians and the Papuan group together with 85 percent residual resampling support (Waddell 2011b).
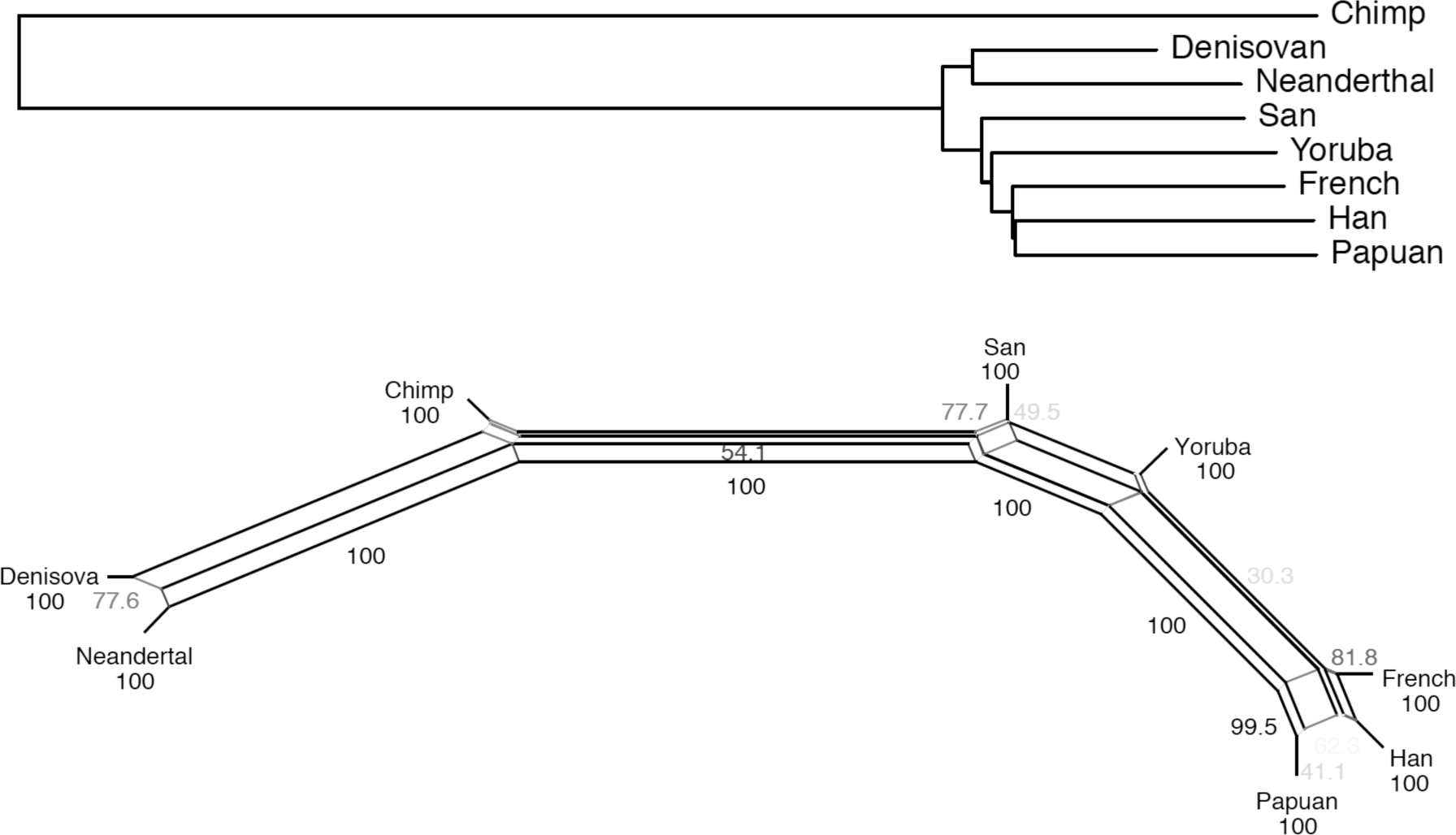
(a) The Weighted Least Squares (WLS+), power (P) = 1, tree estimated in Waddell et al. (2011) from the data presented in Reich et al. (2010). Shown on the edges are the degree of confidence in a split using the technique of residual resampling for trees or networks (Waddell et al. 2011), unless 100%.
(b) The Neighbor Net with the external edges trimmed. Edge weights estimated with weighted least squares (WLS+) power (P) = 1 (Felsenstein 2004). Note, the total residual for (b) (as measured by the ig%SD statistic, Waddell et al. 2011a), is less than 10 percent, suggesting that any or all “missing” signals will not be large enough to overwhelm the generally tree-like structure observed here.
A re-sequencing of these genomes, in conjunction with additional genomes, yielded a tree among African populations that presents the San diverging first, then Mbuti Pygmy, followed by Yoruba, Mandenka, and Dinka branches, and then non-African groups (Meyer et al. 2012, Prufer et al. 2014). Nevertheless, there are differences between these two analyses. For example, Meyer et al. (2012) provide strong support for an edge separating Papuan and Han (Chinese) from French of length 4.5 (× 10−6). This edge shrinks to less than half this length with the data and analyses in Prufer et al. (2014). When reanalyzed with the methods in Waddell et al. (2011b), the Papuan moves outside other non-Africans, separated by an edge of length 1.5 (× 10−6). Further, the scale used is ×10−6, which means that, for every million well-aligned/sequenced bases, one should expect only ≤ 5 substitutions underlying this split, that is, a coalescence on that edge of the tree. This is also the approximate size of many reported signals of non-sapiens-H. sapiens interbreeding (e.g., Waddell 2013). Clearly, good-quality data and robust analytical methods are essential.
Networks from pairwise distances
A powerful aspect of these simple distances is that they jointly allow the inference of a system of non-tree splits, including reticulation or gene swapping (hybridization, introgression, etc.) between lineages of the main tree. A popular method for doing this is Neighbor Net, which is a generalization of the popular Neighbor-Joining method of tree reconstruction (Huson and Bryant 2006). Neighbor Net can infer a circular split system via a heuristic process. A circular split system is one in which all genomes can be placed, in any order, around a circle, with any or all splits being depicted as chords across this circle. If, however, a pattern of interbreeding becomes more complicated, a circular split system cannot represent it. Thus, problems of not being able to capture all splits in the data will increase the degree of misfit: for example, the residual sum of squared errors will increase. This, in turn, will cause a method such as residual resampling to downgrade its confidence in the splits in the graph (Waddell et al. 2011b). Nevertheless, splits that are missed in such a network might be revealed by scrutiny of the residuals (Waddell 2013, 2014).
Application of circular-split systems to Reich and colleagues’ (2010) data yielded unexpected results, such as the largest non-tree split presented the Denisovan as splitting with the outgroup (Pan) rather than with other Homo genomes (Waddell et al. 2011b; figure 8.2). This split would be generated by the Denisovan interbreeding with a lineage that diverged somewhere along the edge of the chimpanzee outgroup (an unknown “ghost lineage,” such as a pre-H. sapiens/H. neanderthalensis lineage). An a priori possibility is that individuals of a Denisovan lineage interbred with a lineage that might loosely be called H. erectus-like, as possibly represented by specimens from Zhoukoudian, which fall roughly in the correct time interval of the last ~800 kya. (Neurocranial shape is often used to lump these with other specimens into a spatially and temporally very broad group identified as H. erectus sensu lato [e.g., Lordkipanidze et al. 2013]).
Another difference generated by applying circular-split systems to Reich and colleagues’ (2010) data is that the Papuan split not with the Denisovan, but with a Neanderthal-Denisovan-Pan grouping. In theory, removal of the Denisovan should group the Papuan with only the Neanderthal lineage if genetic introgression came from that direction only. However, doing this, the Papuan still splits off with Neanderthals and the outgroup (Waddell et al. 2013); this split is also much larger than expected due to the Denisovan lineage interacting with an earlier ghost lineage. Although Prufer et al. (2014) re-sequenced most of Reich et al.’s (2010) genomes, with the distances presented, the Papuan still presents a non-tree split with Neanderthals, the Denisovan, and Pan (result not shown). Even if the Denisovan lineage that introgressed into the Papuan was a distant relative of the sequenced Denisovan and not particularly coalesced with the patterns of that individual, this split is difficult to understand (Waddell et al. 2011b). Indeed, although Prufer et al. (2014) prefer this explanation, Kuhlwilm et al. (2016) depict the source of introgression into the Papuan as close to the sequenced Denisovan.
A distance-based calculation that estimated the fraction of the Denisovan genome that came from the earlier “ghost” lineage gave a point estimate of about 2 percent introgression from an early “erectus-like” ancestor (Waddell et al. 2013). This point estimate, which resulted from different methods and dataset is in agreement with Kuhlwilm et al. (2016; ~0.2–1.2%); Prufer et al.’s (2014) estimate of ~2.7–5.8% seems rather high. This notwithstanding, Kuhlwilm (2016) suggested that the genes that introgressed into the Denisovan lineage came approximately 300 kya from a lineage that diverged over 2.5 mya (perhaps biased upwards by over a half million years, due to assuming a premature slowdown of the mutation rate). If over 2.5 mya is correct, since specimens identified as H. erectus significantly postdate that, a H. erectus-like Denisovan donor seems to be excluded. Thus, while there appears to be broad agreement on the reality of this introgression, there is disagreement over timing and percentages of contribution.
Buneman condition, P-statistics, D-statistics
Another way to learn about splits in the data that do not fit a single tree is via the analysis of all quartets, which in some cases are the same as rooted triplets. Under models with binary data, the differences between these quartets are equivalent to the Buneman four-point conditions (BFPC) of distances on a four-tipped tree (Buneman 1971, Bandelt and Dress 1992). This BFPC is used to build networks that are more complicated than trees, for example, Split Decomposition (Bandelt and Dress 1992), by adding a split only if that split is always one of the two strongest splits implied by distances for all possible quartets; when the network is drawn, the weight of that split is the minimum over all quartets.
Thus, if there is more disagreement, Split Decomposition collapses that split and, in this sense, is more conservative than Neighbor Net, which averages split differences (Huson and Bryant 2004). (Indeed, unless the data has a low incidence of rate of error or of conflict, and includes relatively few taxa, Split Decomposition often seems too conservative, while Neighbor Net, with residual resampling, seeks an appropriate balance [Waddell et al. 2011a.]) One should note that, with binary data, the BFPC is calculated from the frequencies of the site patterns 0011, 0101, and 0101, where 0 is ancestral and 1 the derived state. Here, pattern 0011 means that taxon 1 and taxon 2 have state 0, and taxon 3 and 4 the derived state 1. This is because no other patterns with four taxa change the basic relationships of the distances from which quartet structure is inferred, when multiple changes at a site are discounted
Rooted triplets test whether the data clearly favor one tree over others and are also consistent with a single tree (Waddell 1995, Waddell et al. 2001, Waddell et al. 2011b). They work with site patterns that take the form f(0011), f(0101), and f(0110), where f = frequency and the first taxon from the left is the outgroup, which is always assigned state 0. Rooted triplets can be applied to a variety of data, e.g. SINEs (short interspersed nucleotide sequences) and SNPs. Assuming that site patterns are unlinked, and f(011) is the greatest, one can use likelihood ratio tests (Waddell et al. 2001, 2005), with the P2 probability assessing f(101) = f(110).
When these tests are applied to Reich and colleagues’ (2010) genome data for order Pan (outgroup), the San, the Yoruba, and the Han, f(San+Yoruba) and f(San+Han) are not significantly different (Waddell et al. 2011b). If there are no parallelisms, convergences, or mistakes in the data, this result is consistent with the interpretation that the San population was long separated from the other populations with little introgression (Waddell et al. 2011b). Indeed, analysis of a larger sample of genomes has demonstrated that the long run ancestral Khoisan population was considerately larger than the others and, until recently (~5 kya), isolated from them for a vast amount of time (Kim et al. 2014). Based on trio-based mutation rates (Scally 2016), this period of isolation may have been ~140–200 kya, which predates considerably the fossil record of H. sapiens.
In their genomic analyses, Green et al. (2010) use what they call “D-statistics” (i.e., [f(0101) − f(0110)]/[f(0101) + f(0110)]). Reich et al. (2011) also employ “S-statistics” of the form f(0101) − f(0110) (a Buneman condition on binary data). If informative sites are assumed to be independent, the latter is the P2 statistic (above; Waddell et al. 2001). Green et al. (2010) used these and other statistics to infer a network of genetic interchange. Reich et al. (2010) then added to the data and analyses to estimate a Neanderthal introgression into non-Africans of 1−4%, and of Denisovan into the Papuan of 4–6%.
Challenges for all methods of building networks of descent include realizing that (1) finding the optimal network is NP Hard, which is a difficult search problem; (2) many different phylogenies may all look the same based on “distance” quartets; (3) systematic as well as sampling error must be accounted for when assessing robustness; and (4) detecting and dealing with convergence, parallelism, and even errors in the data are difficult indeed. With regard to the latter point, extension to true quartets, such as those obtained via a Hadamard conjugation on four sequences (Waddell et al. 2011b), may help. Further, true quartets have the potential of untangling more complex networks of descent than Buneman/D-statistics alone (Yang et al. 2013).
Full-Site Pattern Spectrum
In phylogenetic analysis, character-based methods tend to work with a full-site pattern spectrum (that is, the frequency of all possible assignments of 0 and 1 across all genomes [Hendy and Penny 1993]). However, these methods tend to discard sequence order, which is both an advantage and a disadvantage. If the method of analysis can integrate out the gene tree at each location in the genome, this can be a huge advantage since misidentification of a gene tree at each location can lead to major biases (Waddell 1995). Another advantage is that this approach does not need haplotype or phasing information.
A conceptually simple way to do this is to calculate, for a given species tree, what spectrum is predicted when all weighted gene trees and their frequencies under exact coalescent models of population change are taken into consideration. This can be greatly simplified using mathematical coalescent theory (Felsenstein 2004).
Waddell et al. (2011b) fitted such a model to the data of Reich et al. (2010), including a single flow of genes from Neanderthal to all non-Africans, and another from a Denisovan relative to the Papuan lineage. This significantly improved the fit of the model that allowed for these introgressions. A further advantage of these methods is that they identify the residual of every single site pattern with X2-like statistics tuned to either high sensitivity (which helps spot the worst fitting part of the data), or high robustness (which gives more robust parameter estimates in spite of misfits) (Waddell et al. 2011b, 2012). Upon doing so, it turned out that one of the residuals using sensitive fit measures showed that while the Neanderthal shared with present-day humans a significant number of fixed, derived alleles, the Denisovan did not. This would be expected if a fraction of the Denisovan genome came from a more deeply diverging “ghost” lineage.
In addition to the above, the site pattern spectrum unequivocally rejects a strong form of the MR hypothesis. The site pattern frequency spectrum in such a fully mixed population is given by Tajima (1983). It predicts the same frequency for each site pattern of two, three, or more derived SNPs. Testing this series of linear invariants leads to rejecting fully mixing genomes, a result expected to become stronger as errors in the data are removed. For example, testing that the frequency of the derived pattern FHP being equal to DFH yields a X2 statistic of ~4000. This alone is far larger than the misfit of all site patterns to an OA model that assumed limited introgression of genes from Neanderthals and the Denisovan (~2000 or less).
Sequencing-error patterns in this type of data can lead to biases in parameter estimates. When a complete likelihood-mixture model, produced by modeling and adding a spectrum of error, was added to the previous site pattern spectral model, optimal fitting indicated that sequence-error rates affecting more than one individual were much higher than expected based on singleton error (Waddell et al. 2011b). The predicted order of genome quality remained in agreement with Reich et al. (2010). However, with the best fitting model, the predicted proportion of “surviving” Neanderthal introgression into non-Africans drops from ~4.4% to ~1.9% (Waddell et al. 2011b). The latter figure is very close to a ~1.8% estimate based on effectively re-sequencing all these genomes (Prufer et al. 2014).
The X Chromosome
The human X chromosome (Reich et al. 2010, Meyer et al. 2012) is particularly deficient in alleles associated with non-sapiens interbreeding events (Waddell et al. 2011b, Meyer et al. 2012, Waddell 2013, Prufer et al. 2014, Sankararaman 2014). This could be due to mating with non-sapiens males (Waddell 2011b). However, the re-sequencing and remapping data of Meyer et al. (2012) reveals a near complete absence of the signal of non-sapiens alleles in the Papuan X chromosome (Waddell 2013). This suggests purifying-selection, because male-biased gene flow should depress non-sapiens allelic transfer on the X chromosome by one-third at most. Re-sequencing Neanderthal genomes further suggests negative (or purifying) selection against Neanderthal gene flow (Prufer et al. 2014). Indeed, when Sankararaman et al. (2014) studied the genomes of more than 1,000 Eurasian individuals, they discovered that Neanderthal alleles are less common in areas that are gene enriched: for example, genes showing high testes expression levels. On the X chromosome, which comprises numerous male hybrid sterility genes, overall Neanderthal ancestry is about one-fifth that of autosomal genes (Sankararaman et al. 2014). In contrast, non-tree splits found on the X chromosome involving only H, sapiens population (see Waddell 2013) far better match those of autosomal genes (figure 8.3 below).

Beyond Sequence Spectrums
The ongoing development of methods that analyze complete haplotypes and their phasing offer promise of inferring local genome-sequence history, including recombination, the timing of coalescence and population splitting, rates of migrants, and population size along each line of descent, for example, Multiple Sequentially Markovian Coalescent (MSMC; Schiffels and Durbin 2014). These methods tend to embody many “hidden” parameters, such as unknown recombination points, that are dealt with using methods such as a Hidden Markov Model or a Bayesian framework (Felsenstein 2004). Another method does not look for recombination points, but uses spaced blocks of sequences that are hopefully unlinked, “non-functional,” and individually single tree (i.e., they have not recombined; Gronau et al. 2011). Although these methods are presenting interesting insights, it remains unclear how they are related to methods reviewed above in terms of being unbiased, efficient, and robust. For example, while Kuhlwilm et al. (2016) corroborated the earlier-discussed ghost-lineage introgression hypotheses, there were unexpected twists. Using the methods of Gronau et al. and Schiffels and Durbin, Kuhlwilm et al. (2016) were able to place estimates of effective population size onto an expanded phylogeny, that included allele flow from another ghost population (sapiens or pre-sapiens) near the root of H. sapiens into the Altai Neanderthal genome.
Another Non-sapiens Introgression?
Figure 8.3 presents the Neighbor Network results for the autosomal data presented in Meyer et al. (2012) using analyses in Waddell (2013) for Chromosome X. The Denisovan is a particularly useful outgroup for examining what may have occurred with pre-sapiens in Africa. A large non-tree split between the Denisovan and Papuan is consistent with the earlier analyses of Reich et al. (2010),Waddell et al. (2011b) and Meyer et al. (2012). There is also evidence of a complex set of splits between Europeans, Native Americans, and East Asians that is consistent with, for example, the suggestion that Native American genomes consist of about one-third West Eurasian and about two-thirds East Asian genes (Fu et al. 2015).
Figure 8.4 shows a Q-Q plot of residuals; deviation away from the linear trend indicates significant outliers. A large negative residual between Mbuti and Dinka, and another between Mandenka and Denisova, suggests that these pairs are far too close; also the residual between Mbuti and Mandenka is too large. In the 3D plot of the residuals (figure 8.4b), the residuals of Denisova to Africans are negative leading up to Mandenka, where they become largest in absolute value and then abruptly switch sign and drop toward the Denisova to Papuan residual. The pattern is similar near the Mbuti-Dinka pair. These patterns may be reflective of a least squares fitted network (NeighborNet) attempting to accommodate unexpectedly small distance pairs; indeed, only a few hundred kilometers separate Northeastern Congolese Mbuti and South Sudanese Dinka. Interactions between these groups tend to be male Dinka with female Mbuti and may be the basis for their having the largest residual (figure 8.4). The large negative residual of the Mandenka/Denisova pair seems most consistent with genetic input from pre-sapiens individuals.

(a) A Q-Q plot of the standardized residuals from figure 8.3.
(b) A 3D plot of the standardized residuals. The large negative outliers are Denisova with Mandenka (–2.17) and Mbuti with Dinka (–2.41).
(c) As for (a), but the Q-Q plot after allowing for a Denisova + Mandenka and a Mbuti + Dinka split.
By reducing the expected distance from Denisova to Mandenka, the pre-sapiens-Mandingo hypothesis is implicitly modeled modeled with an “unseen” split of Denisova and Mandenka versus all other individuals. That is, minus twice the raw residual of Denisova to Mandenka (corresponding to a split of 8.63 × 10−6) was added to the observed Denisova to Mandenka distance to account for the fact that, in least-squares fitting, a misfit induced by pushing one residual down results in other residuals going up. The same adjustment was made for the Mbuti-Dinka pair (split size 12.49 × 10−6). In comparison, the split modeling the Denisova + Papuan-versus-all-others splits on the same data is 15.53 × 10−6.
Modeling these additional splits results in a considerable improvement in fit, with the residual weighted sum of squares going from ~0.12 to ~0.05. The lnL improves by over 29.5 units, with criteria such as AIC, BIC, and AICc favoring the revised model. Figure 8.4c shows there are now no obvious outliers in the Q-Q plot and figure 8.4d shows that not only the directly modeled residuals, but near neighbors (such as the two previously largest positive residuals) have approached zero. Further, similar to previously proposed interactions with pre-sapiens (Reich et al. 2010, Waddell et al. 2011b, Meyer et al. 2012, Waddell 2013, Prufer et al. 2014), there is much less evidence of gene transfer on the X chromosome for the Denisova/Mandenka signal (Waddell 2013). This contrasts with visible signals of introgression on the X chromosome amongst sapiens lineages, including Mbuti/Dinka (Waddell 2013).
The Pre-sapiens Mandingo Hypothesis
There is increasing evidence of pre-sapiens gene flow into the ancestors of different African groups (Hammer et al. 2011). Based on deeply diverging haplotypes, Mbuti pygmies present the greatest evidence for pre-sapiens gene flow (Hammer et al. 2011). Evidence in West Africa of a specific event or set of events includes: (1) deeply divergent autosomal haplotypes in the Yoruba (Hammer et al. 2011); (2) a late transition from a Middle Stone Age culture across large areas; (3) a deeply diverging pre-sapiens lineage represented by the the Iwo Eleru skull (Harvati et al. 2011; figure 8.1); (4) a few copies of the most divergent H. sapiens Y chromosome found in Mbo (almost twice as deep as that of the Khoisan; Cruciani et al. 2011, Mendez et al. 2016); and (5) a particularly large and unexplained residual in the Mandenka toward the outgroup (Denisova) in the autosomal genome but not in the X chromosome (figure 8.4). Further, the locations of Yoruba, Iwo Eleru, Mandenka, and Mbo fall in a rough line in West Africa trending toward the southeast for ∼1000 km. This constitutes the “pre-sapiens Mandingo” hypothesis, whereby Mandenka refers to the language and Mandingo the people.
Using crania, it is possible to estimate when the Iwo Eleru lineage split from the Khoisan relative to the timing of the split between the latter and Neanderthals. First, is a simple “backbone” estimate (Waddell et al. 1999), which is the ratio of the path length from the Iwo Eleru/H. sapiens ancestor to Khoisan divided by the path length from the Neanderthal/sapiens ancestor to the same Khoisan (figure 8.1). The result is 79%; if the path lengths are traced to Iwo Eleru instead of Khoisan, it is 75%. In comparison, using the most filtered data from Mendez et al. (2016), the equivalent numbers of the edge length of Y chromosome A00 to a Neanderthal Y chromosome yield a ratio of 11/(11+12) (~ 48%). Since the tree suggests acceleration of skull-shape change toward H. sapiens and Iwo Eleru, these divergence-time estimates are likely to be too large. The evolutionary rate-change penalty of Kitazoe et al. (2007), which is specifically designed to accommodate this bias when making divergence-time estimates, should in future be applied.
The proportions of differences back to the H. sapiens/Neanderthal ancestor do not account for the “coalescence” time within populations, that is, here, the average difference in shape between two skulls. Using the difference between two Khoisan skulls as such an estimate, which, one should note, is similar to the difference between a pair of Neanderthal skulls, this fraction of the path length is removed, resulting in an estimate of 69% when traced to a Khoisan skull, or 57% if applied to the path to Iwo Eleru. If the divergence time of H. sapiens and Neanderthal is taken as 600 kya (e.g., Meyer et al. 2016, Kuhlwilm et al. 2016 based on slower H. sapiens mutation rates), it seems that the Iwo Eleru lineage diverged ~300–450 kya. A similar adjustment for Y chromosomes should produce less change, since their expected coalescence time is ~one-fourth as large as the autosomal genes assumed to generate skull shape. Based on these divergence times, and their uncertainty, it remains possible a Y A00 introgression was via a member of the Iwo Eleru lineage.
Out-of-Africa at Present
In this section I summarize how the latest data and analyses confirm, challenge, or enrich this hypothesis-set (Waddell and Penny 1996).
When?
The age of the split between Khoisan and other living humans seems to be ~100–200 kya, this time range being largely due to conflicting methods of rate calibration. As mentioned above, a recent estimate is ~250 kya, which is surprisingly old and will certainly need more validation. There is some suggestion that values may be trending toward ~150–200 kya (Scally 2016). However, values toward the lower end of this range tend to be favored by the archeological record, which does not show clear signs of a “package of modernity,” found abundantly among the Khoisan, for example, until < 100 kya (Klein 2009). This raises the issue of why it took so long for H. sapiens to populate Africa. Perhaps part of this puzzle lies in the co-existence until the Holocene of H. sapiens with closely related, but competing hominids (e.g., Iwo Eleru, Fish Hoek, and Boskop specimens). Earlier, at 100 to 200kya, Africa and the Middle East may have been full of competing lineages, suggested by multiple morphs/lineages likely present in that period, including, LH18, Jebel Irhoud, Singa, Omo 2, Herto, and Skhul 5.
Where?
Autosomal diversity gradient genetic maps among African populations suggest Southern Africa (south of Kenya; e.g., Henn et al. 2011). mtDNA analyses are consistent with the geographic range of the Khoisan (Gonder et al. 2007), as are those of the Y chromosome, if the very deep rare A00 type is attributed to introgression (see above). A Southern African origin is also suggested by tree and split diagrams of full genomes (figures 8.2 and 8.3), which depict the divergence first of the Khoisan, and then Mbuti, West Africans, Dinka, and, finally, all non-Africans. The deep coalescence time, greatest genetic diversity, and largest long-term effective population size of the Khoisan are consistent with this conclusion (Kim et al. 2014).
Nevertheless, three lines of evidence suggest East or Southeast (e.g., Kenya, Uganda and Tanzania) Africa might have been the birthplace of H. sapiens. First, archeologists see in that region more evidence of a continuous trend towards technological modernity (e.g., stone tool forms) and of larger, long-term population sizes (Blome, et al. 2012). Second, the Hadza and Sandawe of Tanzania are similar to the Khoisan in having a “click” language, and harboring some of the same types of deep mtDNA and Y lineages (Tishkoff et al. 2007). After removing apparent recent (< 5 kya) West African (Bantu) introgression, a Hadza genome branches deeply as the sister to the Khoisan, with the suggestion that the former may have had the larger ancestral population (Pickrell 2012). Third, late-occurring, apparently pre-sapiens fossils from South Africa (Boskop ~12–14 kya, and Fishhoek (~6.7 kya) might also be consistent with Khoisan moving into the region, and taking some time to fully populate it. If the Khoisan harbor a significant component of a very late pre-sapiens population’s genes, this could boost their genetic diversity relative to Hadza. It might also accentuate the “long fuse” issue of missing archeological evidence for H. sapiens, but would be difficult to detect with Khoisan at the root by themselves (accentuating the importance reconstructing the Hadza lineage genotypes pre-10 kya).
Who?
The tree/graph of figure 8.3 suggests the expansion, over a long period of time, of within-Africa H. sapiens populations that preceded the OA event, beginning with Mbuti, followed by West Africa, and then Dinka (Sudan). The timing of OA is linked to the age of the split with Khoisan, so OA was likely not older than ∼100 kya, and not younger than 60 kya. The introgresssion of possibly H. sapiens genes into the Altai Neanderthal, the individual represented by the morphologically H. sapiens Qafzeh 9 skull, and the archeological record together suggest a population that existed well before 60 kya, albeit one apparently leaving no genetic legacy. Specimens in China (∼100–150 kya) may indicate an earlier pre-sapiens wave. Although no genetic split is compatible with such earlier migrations (figure 8.3), this may be due to inadequacies of statistical power and sampling. In terms of who left Africa, and if heavily admixed genomes (e.g., from Egypt) are not considered, the South Sudanese Dinka currently appear to be the most representative of sampled living populations. A more detailed estimate will require reconstruction of ancient genomes from populations in areas such as Egypt, Ethiopia, Eritrea, and Somalia.
How?
It seems that human adaptive genetic/cultural advantage eventually overcame all pre-sapiens lineages. There is evidence of considerable interbreeding upon initial contact (Trinkaus 2007, Waddell 2014, figure 8.1) that may have been rapidly diluted by subsequent bands of H. sapiens, perhaps in conjunction with negative selection. In addition, male sterility may have been a factor, with purifying selection removing most, but not the adaptively advantageous (e.g., immune system, hair color [Sankararaman et al. 2014]), non-sapiens genes. Further, although Chromosome X is heavily depleted of non-sapiens-introgressed genes, there is evidence of abundant mixing of H. sapiens genes on the X chromosome between lineages, even when cultures were swamped. Surviving non-sapiens blocks in the genome probably globally average < 4% of the genomes of living people (perhaps less than 2.5%). The larger portions of these blocks have little or no function, and/or differ minimally from the common ancestral form in function.
It is now also becoming clear that the effective population size of H. sapiens and its ancestors was probably always of a healthy size (e.g., >20,000, and comparable to Pan subspecies [Prufer et al. 2014, Kuhlwilm et al. 2016]), which probably reflects the presence in Africa of more than one late Homo lineage, perhaps each with a geographically defined home territory (as seen with the 3 or 4 chimp subspecies). Further, there is no reliable evidence to support a “saltational,” genetic drift origin of H. sapiens, which, in turn, reinforces the role of natural selection. These points are elaborated later in this chapter.
Morphological Evidence for Homo sapiens Origins
In terms of a fossil-based geographic origin of H. sapiens, the tree in figure 8.1 is generally consistent with morphology (e.g., Klein 2009, Schwartz and Tattersall 2005, Stringer 2016). Estimating trees and networks based on morphological data should help to move paleoanthropology away from a still-entrenched “Great Chain of Being” or “Chronospecies” view of human evolution, into which virtually every newly discovered fossil is inserted as a direct ancestor of something else. Improved phylogenetic analyses may even support hypotheses of distinct clades (e.g., Iwo Eleru, LH-18, Saldahna), whose individuals would have competed with H. sapiens as well as with its ancestors.
Based on analysis of fossils, the archeological record, and DNA, the Middle East, from the Sinai well into Eurasia, and between ~120 and ~60 kya may have been a region of hybridization. For example, qualitative phylogenetic analysis suggests that Qafzeh 9 might have been H. sapiens, while Qafzeh 6 may be ~one-third hybrid with a lineage close to Neanderthals (Waddell 2014). Skhul 5 may represent a distinct pre-sapiens lineage (or at least not H. sapiens [Schwartz 2016]). Evidence of H. sapiens/pre-H. sapiens genes in the very roughly dated 100 kya Altai Neanderthal might reflect this phase (Kuhlwilm et al. 2016). Since these remnant genes cannot be attributed to potential ancestors of living OA descendants, this might be indicative of at least one “failed” major movement of pro-H. sapiens into Eurasia at ~100kya or older. Interestingly, the shape of the 35–55 kya Shanidar 1 Neanderthal skull that draws it towards sapiens (Waddell 2015) also suggests the existence of a large and dynamic zone of hybridization.
To the east, China has yielded ~80–150 kya specimens that appear to be close to H. sapiens, although at least one from this period, from Zhirendong, appears to be pre-sapiens (or at least not H. sapiens [Schwartz 2016]). China also seems to have harbored very unusual and potentially early diverging forms until the very late Pleistocene-early Holocene, for example, from Maludong and Longlin (Curnoe 2014, Schwartz 2016). Although having inverted-T-shaped chins (Schwartz 2016), the Zhoukoudian Upper Cave skulls (~20 kya) are placed unusually deep in figure 8.1, which suggests possible hybridization of H. sapiens with earlier forms. In spite of some enthusiasm for China in the scenario of H. sapiens origins, the large, long-term effective population of this species’ ancestors (e.g., Prufer et al. 2014, Kuhlwilm et al. 2016) suggests otherwise. That is, the hypothesized relatively large, ancestral population sizes, and their general continuity, do not seem consistent with a scenario of H. sapiens’ ancestors enduring an earlier OA bottleneck, migrating to China, and then enduring another bottleneck when returning to Africa.
Figure 8.1 suggests that quantitative morphological phylogenetics can detect a shift in the shape of early European H. sapiens skulls over the period ~40–25 kya, with the earlier being more deeply divergent. Although this appears to be consistent with Trinkaus’ (2007) proposal, specimens are highlighted differently in one versus the other analysis. The recent work of Fu et al. (2016) shows an ongoing decrease due to selection in the quantity of Neanderthal-derived genes during this period. A tree in Waddell (2015) also shows a late European Neanderthal skull, Spy 2, drawn in the direction of H. sapiens, which is consistent with suggestions of H. sapiens gene flow into Neanderthals in Europe just prior to the latter species’ extinction.
Given the frequency of potential hybrids in contact zones in Europe, the Middle East, and China, it seems possible that there was quite “leaky” pre-mating isolation of H. sapiens from its close relatives. Varying degrees of hybrid sterility, followed by predominately purifying selection within populations, together with possible group selection favoring populations with fewer introgressed genes, may finally have driven down non-sapiens genetic contributions. This certainly seems to be the case for Neanderthal introgression into living Europeans (Fu et al. 2016). Of course, since selective purification is hypothesized to act more quickly in populations with larger effective sizes, relatively smaller, often isolated, populations might tend to hold longer onto their genetic relics (perhaps the situation in Oceania), only to be swamped later by populations with fewer introgressed genes. Such a process might be accentuated, and causally confounded, if cultural/technological evolution was also more rapid in earlier occupied areas of denser population/larger population size.
Patterns of Introgression
It is worth emphasizing that while there is solid evidence of introgressed genes into H. sapiens, they are typically in small percentages, which validates the OA hypothesis of complete or nearly complete genetic replacement. There is now also evidence of numerous introgression events, including in Africa, but the fraction of retained non-sapiens genomes seems largely to have dropped to ∼0–3%.
Reported introgressions between major lineages of Homo include multiple Neanderthal introgressions into non-Africans of 1.5–2.5% (average ~2%), multiple Denisovan into Melanesians, East Asians and possibly South Asians (0–5%), Neanderthal into the Denisovan (0–2%), an early diverging ghost lineage into the Denisovan (0–2%), H. sapiens or close pre-sapiens into the Altai Neanderthal (0–1.5%), and pre-sapiens African into African H. sapiens (~0–5%?). Interestingly, the limited nDNA data extracted from a ~430 kya pre-Neanderthal from Sima de los Huesos, Sierra de la Atapuerca (Meyer et al. 2016), has no signs of introgression as yet, barring unresolved discrepancies of the mtDNA tree.
Since multiple Homo lineages appear to have a low-surviving introgressed fraction, they should be considered separate species. This pattern contrasts with that within H. sapiens, with often large percentages of genomes mixing and genes surviving, despite documented cases of strong cultural pre-zygotic isolation, and even culturally influenced post-zygotic effects. This general theme does suggest that selection did have significant impact in evolution within the genus Homo in favoring a fairly coordinated gene complex that operated in spite of small-to-medium long-term effective population sizes (e.g., Neanderthals and Denisovans <5,000 individuals, versus the ancestors of H. sapiens, ~15,000–30,000 individuals). The evidence for near-sapiens nuclear DNA introgression into the Altai Neanderthal suggests that the previously hypothesized Middle East hybrid zone had considerable long-range effects. However, a long-term hybrid zone need not equate with significant gene exchange if hybrids fail to successfully move into the ranges of parent species.
In summary, morphology and genetics taken together suggest first frequent introgression of H. sapiens genes into near relatives in contact zones, followed by strong selection against the majority of introgressed genes, with positive selection acting on only a few (Abi-Rached et al. 2011, Fu et al. 2016). Although this pattern has been referred to as “adaptive introgression” (e.g., Kuhlwilm et al. 2016), the term should be used with caution since the overall effect may be maladaptive in terms of the genetic load of all genes subject to purifying selection. Genomic data so far suggests that the pattern and outcome described above may have been a common characteristic of many lineages within Homo, and not just during the last OA event.
Limitations
The reviewers of an earlier version of this manuscript raised important points that need clarification. One is the validity of the method used here—OLS+ with Procrustes distances—to infer a phylogeny based on skull shapes (Waddell 2014). This concern can be partly addressed by comparing this method to an emerging method of Maximum Likelihood (ML) estimation (Theobald and Wuttke 2008, Felsenstein 2015). An ML method being developed by Felsenstein and Bookstein (2016) aims to be a partial solution to the complexities of shape analysis of living forms that Bookstein (2009, 2015, 2016) discusses. Weber and Bookstein (2011) and Bookstein (2016) also address additional complications in current geometric morphometric analyses, pointing out the desirability of implicitly basing estimates of variance and covariance on mathematical equations that describe growth processes. This is a worthy goal in the long term, although phylogenetic systematics continues apace without this being achieved, for example, in virtually all DNA-based phylogenies. The danger, of course, is that many phylogenies will be misleading, at least in part, until the process is consistently well approximated (Swofford et al. 1996, Felsenstein 2004).
A Procrustes-based estimate of a phylogenetic pattern is effectively a sub-model of the ML method of Theobald and Wuttke (2008), or Felsenstein (2015), obtained by setting its variance-covariance matrix to the identity matrix. This simplification should run much faster, and may be a useful model for identifying “good” parts of tree-space. Other sub-models, such as a diagonal matrix only, may also offer advantages in practical analyses since they vastly reduce the need to estimate the values of a huge number of covariance parameters.
With regard to tree estimation, OLS+ and a Procrustes distance is expected to be less statistically efficient, and also perhaps less robust, than the equivalent ML method asymptotically, but it is computationally much faster (Swofford et al. 1996, Felsenstein 2004). As mentioned, a standard Procrustes distance assumes that the variance-covariance matrix between all landmarks is the identity matrix (zero variances and covariances). Felsenstein (2015) gives an elegant example in which some fish lineages have two different fins that grow either significantly more or less than other fins (larger variance) and are also closely correlated in size (non-zero covariances).
In Felsenstein’s example, Procrustes distances may give a useful estimate of the relative distance between short-finned fish, but it may also slightly overestimate the distances between long-finned fish and more strongly overestimate the distance between short-finned and long-finned-fish. With four taxa, this problem translates to the inverse-Felsenstein effect, where taxa on long terminal edges may be repelled from each other (Waddell 1995). This problem is broadly analogous to using a Jukes Cantor distance to build at tree for a set of nucleotides that evolved by a fully general Markov (GM) model with divergent base compositions. Yet, a Jukes Cantor tree may be a useful estimate, and the GM tree may yield a more erratic estimate, if there is insufficient information to estimate the greater number of parameters and/or if the complications are not so pronounced. As noted earlier, since there is an interesting pattern of long edges in the tree of figure 8.1, it would be valuable, as ML software becomes available, to reexamine this. Another potentially robust approach is the use of median-based geometric morphometric distances (Rohlf and Slice 1990) with OLS+ .
Clearly, phylogenetics and geometric morphometrics continue to wed in a variety of ways. However, as with marriage, some will be happier than others. There is a certainly a need to press on beyond Procrustes distances, but it is not yet clear what might work best in a given instance, or how the emphases of authors such as Gould (2002) and Hull (1988), who desire more process modeled in evolution, will be realized. Friston et al. (2012) provide one critique of least squares (e.g., Procrustes) when they are poorly equipped to handle unexpected changes, for example, in skull shape. Waddell et al. (2012) explore altering the fit function to reduce “surprise” or misfit human genome site patterns and, in a sense, this is what Theobald and Wuttke (2008) and Felsenstein and Bookstein (2016) are doing to move beyond Procrustes distances. A widespread transformation in this field will require development of different data/tools and a willingness to think of the problem in different ways (Fleck 1979). Bayesian platforms, such as Beast2, certainly offer still untapped tools for incorporating into a phylogenetic method not only expanded models of the evolution of shape, but also the ages, locations, associated technology, paleogeography, and so on of extinct lineages.
Concern has also been expressed regarding the conflicting conclusions of this and Templeton’s (2017, this volume) chapter. In terms of the amount of non-sapiens introgression, Nested Clade Phylogeographic Analysis (NCPA, Templeton et al. 1995) estimated an ~10% contribution of non-H. sapiens to non-African forms (about four to five times greater than estimates herein based on the latest data). This method, however, has been shown to yield false positives in up to 75% of cases (e.g., Knowles and Maddison 2002, Knowles 2008, Petit 2008). A second issue is that, similar to the MR model, NCPA is biased towards inferring isolation by distance, even when the data lack structure (Beaumont and Panchal 2008) or there is a tree-like divergence pattern (Knowles and Maddison 2002, Knowles 2008). Amongst most modern populations, isolation-by-distance effects overlay some early H. sapiens migrations. In attempting to explain why some modern populations show more Neanderthal genes than others, these effects have been mixed with a structured ancestral African population (Eriksson and Manica 2012). However, these processes to explain the data are falsified by specific tests (Yang et al. 2012).
A second difference between these chapters comes from different definitions of the MR and OA models. Waddell and Penny (1996) defined the OA model and contrasted it with the MR model, as it was conceived prior to the work of Cann et al. (1987). Whole genome data has since shown that current parameter values are much closer to those of OA than to MR. Technically, if the MR model is treated as a general graph and OA a tree, the former is a superset and the latter a subset (Templeton 2017). However, this does not reflect the history of the debate, wherein non-sapiens components are rare and non-essential to the genetics of living H. sapiens. Further, considering the OA model as strictly a tree was never an essential component of the OA hypothesis. Not only have surviving non-sapiens elements been shown to be rare, they are now known to be biased towards less functional regions of the genome (e.g., Sankararaman et al. 2014). Further, some of the introgressed gene variants in H. sapiens that have medical implications show evidence of being selected against.
A final concern is going beyond trees to networks for understanding different kinds of information, morphology included. This is a difficult methodological problem that affects phylogenetics as a whole, and many current implementations towards networks have limitations; for example, Neighbor Net is limited to circular split systems. On the other hand, just as trees infer false edges when the data do not fit model expectations, it seems that networks are far more likely to infer false edges because they allow freer choice of splits/edges.
Even though no current method can extract all historical information from genomes, there seem to be real caps on the amount of introgression of all types between H. sapiens and non-African lineages of Homo that survived. If there are still-unidentified introgressions/migrants into H. sapiens, the best way to find them may be through careful examination of all regions of the thousands of available genomes that do not fit well (e.g., low posterior probability), in the highly parameterized models that are now being developed (e.g., Gronau et al. 2011, Schiffels and Durbin 2014).
A Struggling Genus?
It is now commonplace for articles to assert how tenuously lineages within Homo persisted, as though our lineage and others were at an unusually high risk of extinction. Given current trends, it may be necessary to sharply adjust “near-catastrophe” thinking (such as giant volcanic eruptions, ice ages, etc.) to explain the low-to-moderate effective population sizes of 5,000 to 30,000 individuals characteristic of H. sapiens and inferred for H. neanderthalensis and Denisovans. Rather than being disastrous, these population sizes apparently permitted the emergence of a diversity of novel adaptations and behaviors/cultures. During the last few hundred thousand years, there was a rich patchwork of distinct populations with many characteristics typical of species that overran each other at different times, apparently partly powered by selective advantage. Given the morphological (e.g., Trinkaus 2007, Waddell 2014) and DNA (Fu et al. 2016) evidence for increased numbers of introgressed hybrids where and when lineages meet, failure to swap large amounts of the genome was likely not due to pre-mating isolation, with hybrids likely being less fit than at least one source population.
A corollary of OA is that the greatest risk of extinction any ≤100 kya lineage within Homo faced was H. sapiens. Given the apparently low rates of genetic introgression across Homo, this risk of extinction by a co-generic was probably also a factor earlier in the history of the genus. This view of evolution in the genus Homo, from OA on, may be moving towards the antithesis of the MR model. This in turn may be evidence of co-adapted gene complexes related to development and behavior/culture that did not tolerate disruption.
The ongoing acquisition of genomic data of species of Homo may lead to testing one of the most interesting species concepts of all: The co-adapted genome. In the case of Homo, a tightly constrained co-adaptation might by crucial to the evolution of a socially networked thinking machine. Hints of this possibility come from the lack of introgressed genes on chromosome X between long-separated lineages: X is overrepresented in determinants of intelligence (Skuse 2005) and social networking (Startin et al. 2015). Additional support for this scenario comes from the observation that genes associated with specific brain regions, including the parietal lobes, that are greatly expanded in H. sapiens, are seriously lacking in the remnants of Neanderthal/Denisovan to H. sapiens introgression (Vernot and Akey 2014, 2015).
Phylogenetic Definitions of Homo sapiens and Species with Which They Interacted
Here I have argued, and only argued, for an extant-clade or crown-based definition of H. sapiens. A similar crown-based definition of H. neanderthalensis that includes specimens at or after an arbitrary but accessible date (e.g. ∼100 kya, perhaps just prior to contact with H. sapiens) may also be possible. As for other fossils, phylogenetic systematics cautions against basing species on grades (non-monophyletic groups; Hennig 1966). It is also unwise to assume that all members of a lineage that is closer to H. sapiens than to H. neanderthalensis, but which is not H. sapiens, is an ancestor (or sister taxon) of the extant species. There may well be considerable structure amongst these extinct hominids, as is suggested by the here-hypothesized Saldahna/LH18/Iwo Eleru clade. In order to pursue this, it will be essential to include more detail, including sample identity, and conceive a narrow definition of “species” or “morph” (Schwartz and Tattersal 2002), in order to have a better chance of understanding how these morphs or species interrelate and therefore also how their evolution may be traced. This also applies to estimating trees as well as more general networks.
Based on current genomic data for different specimens of Homo, it seems unwise, and obfuscating, to continue using the systematically meaningless, grab bag term sensu lato (see also discussion in Schwartz, this volume). Among other things, and in the context of this contribution, this modifier implies a kind of hybrid swarm. In contrast, the latest genomic results suggest that later lineages of Homo were pretty frugal about persistent gene flow between themselves. Further, sensu lato has outworn its utility (if it ever had one), inasmuch as it adds further to the taxonomic confusion that already exists: for example, currently in the literature one finds Homo heidelbergensis, Homo heidelbergensis sensu stricto, Homo heidelbergensis sensu lato, Homo heidelbergensis sensu Stringer, and so on, each with a different and often idiosyncratic definition. If one insists on being vague, it seems more useful to simply write “Homo heidelbergensis-like” as a way of letting the reader know that one is referring broadly to specimens that are grossly similar to one another, and not making a phylogenetic statement.
Future Directions
Africa is the key area for a better understanding of the origin, both in time and specific location, of H. sapiens. Robust reconstructions of pre-admixture genomes such as exist for the Hadza, Sandawe, and other exceptional African populations that are based on “pieces” of them that can be identified in living descendants represent an essential first step in furthering this field of inquiry (Pickrell et al. 2012). Genomes and morphology both suggest that the last ancestors of H. sapiens kept pretty much to themselves (e.g., witness the abrupt appearance of the inverted-T-shaped chin), and likely had a fairly long history in one part of Africa before expanding to overrun multiple neighboring species. aDNA from not very ancient specimens (e.g., Fish Hoek and Boskop) could provide currently missing information on major demographic shifts.
Other important contributions to the progress of studies of human origins include:
1. Examination of morphological characters useful to diagnosing H. sapiens in key populations including Khoisan, Pygmies, and Oceanians.
2. A well-characterized morphological matrix of discrete characters within Homo.
3. A freely accessible database of shape measures, such as landmarks, curves, and internal and external 3D scans of key fossils and recently living humans, as well as improved analyses that include networks of descent.
4. Critical reexamination of articles on non-sapiens genomes with regard to changing data (errors?), correspondence between established phylogenetic methods, and tracking changing parameter estimates via the communication of key data sets in compact forms.
5. Careful residual analysis of all genomes with a focus on regions that do not fit parameterized models; this may provide the best clues toward identifying currently undetected instances of introgressions and other avenues of gene flow.
In summary, the OA replacement model has continually been reinforced by new data and analyses. It also appears that near-complete (>90%) genetic replacement of independent lineages may have been a common and repeating feature of evolution within the genus Homo—which certainly contradicts some of the basic assumptions of the MR model and the chronospecies concept. Further, similar studies of other genera of mammals will help us to understand the fit of the general history of genus Homo with the rest of nature—a long-time but still unattained goal of paleoanthropology.
Acknowledgments
I am firstly very grateful to workshop organizer Jeff Schwartz for his invitation to this invigorating conference. For comment and critique of the manuscript I thank Fred Bookstein, Alan Templeton, and Joe Felsenstein. I also thank David Bryant, Mike Hendy, Hiro Kishino, David Penny, and David Swofford for years of intellectual stimulation. This manuscript was accepted November 8, 2016, and the research behind it partly supported by NIH grant 5R01LM008626.
References
Abi-Rached, L., M. J. Jobin, S. Kulkarni, A. McWhinnie, K. Dalva, L. Gragert, F. Babrzadeh, B. Gharizadeh, M. Luo, F. A. Plummer, J. Kimani, M. Carrington, D. Middleton, R. Rajalingam, M. Beksac, S. G. E. Marsh, M. Maiers, L. A. Guethlein, S. Tavoularis, A.-M. Little, R. E. Green, P. J. Norman, and P. Parham. 2011. “The Shaping of Modern Human Immune Systems by Multiregional Admixture with Archaic Humans.” Science 334: 89–94.
Ackermanna, R. R., J. Rogers, and J. M. Cheverudc. 2006. “Identifying the Morphological Signatures of Hybridization in Primate and Human Evolution.” Journal of Human Evolution 51:632–645.
Adams, D. C., F. J. Rohlf, and D. E. Slice. 2013. “A Field Comes of Age: Geometric Morphometrics in the 21st Century.” Hystrix 24:7–14.
Bandelt, H.-J., and A. W. M. Dress. 1992. “Split Decomposition: A New and Useful Approach to Phylogenetic Analysis of Distance Data.” Molecular Phylogenetics and Evolution 1:242–252.
Beaumont, M. A., and M. Panchal. 2008. “On the Validity of Nested Clade Phylogeographical Analysis.” Molecular Ecology 17:2563–2565.
Blome, M.W., A.S. Cohen, C.A. Tryon, A.S. Brooks, and J. Russell. 2012. “The Environmental Context for the Origins of Modern Human Diversity: A Synthesis of Regional Variability in African Climate 150,000–30,000 Years Ago.” Journal of Human Evolution 62:563–92.
Bookstein, F. L. 2009. “Measurement, Explanation, and Biology: Lessons from a Long Century.” Biological Theory 4:6–20.
Bookstein, F. L. 2015. “Integration, Disintegration, and Self-Similarity: Characterizing the Scales of Shape Variation in Landmark Data.” Evolutionary Biology 42:395–426.
Bookstein, F. L. 2016. “The Inappropriate Symmetries of Multi-variate Statistical Analysis in Geometric Morphometrics.” Evolutionary Biology 43:277–313.
Bowler, J. M., H. Johnston, J. M. Olley, J. R. Prescott, R. G. Roberts, W. Shawcross, N. A. Spooner, O. Johnston, R. Prescott, and S. Shawcross. 2003. “New Ages for Human Occupation and Climatic Change at Lake Mungo, Australia.” Nature 421:837–840.
Brothwell, D., and T. Shaw. 1971. “A late Upper Pleistocene Proto-West African Negro from Nigeria.” Man 6:221–227
Buneman, P. 1971. “The Recovery of Trees from Measures of Dissimilarity.” In Mathematics in the Archaeological and Historical Sciences, edited by F. R. Hodson, D. G. Kendall, and P. Tautu, 387–395. Edinburgh: Edinburgh University Press.
Cann, R. L., M. Stoneking, and A. C. Wilson. 1987. “Mitochondrial DNA and Human Evolution.” Nature 325:31–36.
Caumul, R., and P. D. Polly. 2005. “Phylogenetic and Environmental Components of Morphological Variation: Skull, Mandible, and Molar Shape in Marmots Marmota, Rodentia.” Evolution 59:2460–2472.
Cruciani, F., B. Trombetta, A. Massaia, G. Destro-Bisol, D. Sellitto, and R. Scozzari. 2011. “A Revised Root for the Human Y Chromosomal Phylogenetic Tree: The Origin of Patrilineal Diversity in Africa.” American Journal of Human Genetics 88:814–818.
Curnoe, D., J. Xueping, A. I. R. Herries, B. Kanning, P. S. C. Taçon, B. Zhende, D. Fink, Z. Yunsheng, J. Hellstrom, L. Yun, G. Cassis, S. Bing, S. Wroe, H. Shi, W.C. Parr, H. Shengmin, and N. Rogers. 2012. “Human Remains from the Pleistocene-Holocene Transition of Southwest China Suggest a Complex Evolutionary History for East Asians.” PLoS ONE 7(3):e31918.
Darwin, C. 1871. The Descent of Man, and Selection in Relation to Sex. London: John Murray.
Der Sarkissian, C., M. E. Allentoft, M. C. Avila-Arcos, R. Barnett, P. F. Campos, E. Cappellini, L. Ermini, R. Fernandez, R. da Fonseca, A. Ginolhac, A. J. Hansen, H. Jonsson, T. Korneliussen, A. Margaryan, M. D. Martin, J. V. Moreno-Mayar, M. Raghavan, M. Rasmussen, M. S. Velasco, H. Schroeder, M. Schubert, A. Seguin-Orlando, N. Wales, M. T. P. Gilbert, E. Willerslev, and L. Orlando. 2015. “Ancient Genomics.” Philosophical Transactions of the Royal Society B 370: 20130387.
Eriksson, A., and A. Manica. 2012. “Effect of Ancient Population Structure on the Degree of Polymorphism Shared Between Modern Human Populations and Ancient Hominins.” Proceedings of the National Academy of Sciences USA 109:13956–13960.
Felsenstein, J. 2002. “Quantitative Characters, Phylogenies, and Morphometrics.” In Morphology, Shape, and Phylogenetics, edited by N. MacLeod, 27–44. Systematics Association Special Volume Series 64. London: Taylor and Francis.
Felsenstein, J. 2004. Inferring Phylogenies. Sunderland, MA: Sinauer Associates.
Felsenstein, J. 2015. “Morphometric Methods that Use Phylogenies, NIMBioS, 15 August 2015: Tutorial on Evolutionary Quantitative Genetics.” Accessed May 28, 2016. http://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.717.7187&rep=rep1&type=pdf.
Felsenstein, J., and Bookstein, F. L. n.d. “Morphometrics on Phylogenies,” unpublished manuscript.
Fleck, L. 1979. The Genesis and Development of a Scientific Fact (ed. T.J. Trenn and R.K. Merton, foreword T. Kuhn). Chicago: Chicago University Press.
Friston, K., C. Thornton, and A. Clark. 2012. “Free Energy Minimization and the Dark-Room Problem.” Frontiers in Psychology 3(130):1–7.
Fu, Q., M. Hajdinjak, O. T. Moldovan, S. Constantin, S. Mallick, P. Skoglund, N. Patterson, N. Rohland, I. Lazaridis, B. Nickel, B. Viola, K. Prüfer, M. Meyer, J. Kelso, D. Reich, and S. Pääbo. 2015. “An Early Modern Human from Romania with a Recent Neanderthal Ancestor.” Nature 524:216–219.
Fu, Q., C. Posth, M. Hajdinjak, M. Petr, S. Mallick, D. Fernandes, A. Furtwängler, W. Haak, M. Meyer, A. Mittnik, B. Nickel, A. Peltzer, N. Rohland, V. Slon, S. Talamo, I. Lazaridis, M. Lipson, I. Mathieson, S. Schiffels, P. Skoglund, A. P. Derevianko, N. Drozdov, V. Slavinsky, A. Tsybankov, R. G. Cremonesi, F. Mallegni, B. Gély, E. Vacca, M. R. G. Morales, L. G. Straus, C. Neugebauer-Maresch, M. Teschler-Nicola, S. Constantin, O. T. Moldovan, S. Benazzi, M. Peresani, D. Coppola, M. Lari, S. Ricci, A. Ronchitelli, F. Valentin, C. Thevenet, K. Wehrberger, D. Grigorescu, H. Rougier, I. Crevecoeur, D. Flas, P. Semal, M. A. Mannino, C. Cupillard, H. Bocherens, N. J. Conard, K. Harvati, V. Moiseyev, D. G. Drucker, J. Svoboda, M. P. Richards, D. Caramelli, R. Pinhasi, J. Kelso, N. Patterson, J. Krause, S. Pääbo, and D. Reich. 2016. “The Genetic History of Ice Age Europe.” Nature 534:200–205.
Gascuel, O., and M. Steel. 2006. “Neighbor-Joining Revealed.” Molecular Biology and Evolution 23:1997–2000.
Green, R. E., J. Krause, A. W. Briggs, T. Maricic, U. Stenzel, M. Kircher, N. Patterson, H. Li, W. Zhai, M. H. Y. Fritz, N. F. Hansen, E. Y. Durand, A. S. Malaspinas, J. D. Jensen, T. Marques-Bonet, C. Alkan, K. Prufer, M. Meyer, H. A. Burbano, J. M. Good, R. Schultz, A. Aximu-Petri, A. Butthof, B. Hober, B. Hoffner, M. Siegemund, A. Weihmann, C. Nusbaum, E. S. Lander, C. Russ, N. Novod, J. Affourtit, M. Egholm, C. Verna, P. Rudan, D. Brajkovic, Z. Kucan, I. Gusic, V. B. Doronichev, L. V. Golovanova, C. Lalueza-Fox, M. de la Rasilla, J. Fortea, A. Rosas, R. W. Schmitz, P. L. F. Johnson, E. E. Eichler, D. Falush, E. Birney, J. C. Mullikin, M. Slatkin, R. Nielsen, J. Kelso, M. Lachmann, D. Reich, and S. Paabo. 2010. “A Draft Sequence of the Neanderthal Genome.” Science 328:710–715.
Gonder, M. K., H. M. Mortensen, F. A. Reed, A. de Sousa, and S. A. Tishkoff. 2007. “Whole-mtDNA Genome Sequence Analysis of Ancient African Lineages.” Molecular Biology and Evolution 24:757–768.
Gould, S. J. 2002. The Structure of Evolutionary Theory. Cambridge, MA: Harvard University Press.
Gronau, I., M. J. Hubisz, B. Gulko, C. G. Danko, and A. Siepel. 2011. “Bayesian Inference of Ancient Human Demography from Individual Genome Sequences.” Nature Genetics 43:1031–1034.
Hammer, M. F., A. E. Woerner, F. L. Mendez, J. C. Watkins, and J. D. Wall. 2011. “Genetic Evidence for Archaic Admixture in Africa.” Proceedings of the National Academy of Sciences USA 108:15123–15128
Harvati, K., C. Stringer, R. Grün, M. Aubert, P. Allsworth-Jones, and C. A. Folorunso. 2011. “The Later Stone Age Calvaria from Iwo Eleru, Nigeria: Morphology and Chronology.” PLoS ONE 69:e24024.
Hendy, M. D., and D. Penny. 1993. “Spectral Analysis of Phylogenetic Data.” Journal of Classification 10:5–24.
Henn, B., C. R. Gignoux, and M. Jobin. 2011. “Hunter-Gatherer Genomic Diversity Suggests a Southern African Origin for Modern Humans.” Proceedings of the National Academy of Sciences USA 108:5154–5162.
Hennig, W. 1966. Phylogenetic Systematics, translated by D. Dwight Davis and Rainer Znagerl. Urbana: University of Illinois Press.
Hillis, D. M., and J. J. Bull. 1993. “An Empirical Test of Bootstrapping as a Method for Assessing Confidence in Phylogenetic Analysis.” Systematic Biology 42:182–192.
Hillis, D. M., and J. P. Huelsenbeck. 1992. “Signal, Noise, and Reliability in Molecular Phylogenetic Analyses.” Journal of Heredity 83:189–195.
Hull, D. L. 1988. An Evolutionary Account of the Social and Conceptual Development of Science. Chicago, IL: University of Chicago Press.
Huson, D. H., and D. Bryant. 2006. “Application of Phylogenetic Networks in Evolutionary Studies.” Molecular Biology and Evolution 23:254–267.
Jefferies, R. P. S. 1979. “The Origin of ChordatesA Methodological Essay.” In The Origin of Major Invertebrate Groups, edited by M. R. House, 443–447. London; New York: Academic Press for The Systematics Association.
Kitazoe, Y., H. Kishino, P. J. Waddell, N. Nakajima, T. Okabayashi, T. Watabe, and Y. Okuhara. 2007. “Robust Time Estimation Reconciles Views of the Antiquity of Placental Mammals.” PLoS ONE. 2007 2;e384:1–11.
Kim, H. L., A. Ratan, G. H. Perry, A. Montenegro, W. Miller, and S. C. Schuster. 2014. “Khoisan Hunter-Gatherers Have Been the Largest Population Throughout Most of Modern-Human Demographic History.” Nature Communications 5, Article number: 5692.
Klein, R. G. 2009. The Human Career: Human Biological and Cultural Origins. 3rd edition. Chicago IL: University of Chicago Press.
Knowles, L. L. 2008. “Why Does a Method that Fails Continue to Be Used?” Evolution 62:2713–2717.
Knowles, L. L., and W. P. Maddison. 2002. “Statistical Phylogeography.” Molecular Ecology 11:2623–2635.
Kuhlwilm, M., I. Gronau, M. J. Hubisz, C. de Filippo, J. Prado-Martinez, M. Kircher, Q. Fu, H. A. Burbano, C. Lalueza-Fox, M. de la Rasilla, A. Rosas, P. Rudan, D. Brajkovic, Ž. Kucan, I. Gušic, T. Marques-Bonet, A. M. Andrés, B. Viola, S. Pääbo, M. Meyer, A. Siepel, and S. Castellano. 2016. “Ancient Gene Flow from Early Modern Humans into Eastern Neanderthals.” Nature 530:429–435.
Li, J. Z., D. M. Absher, H. Tang, A. M. Southwick, A. M. Casto, S. Ramachandran, H. M. Cann, G. S. Barsh, M. Feldman, L. L. Cavalli-Sforza, and R. M. Myers. 2008. “Worldwide Human Relationships Inferred from Genome-wide Patterns of Variation.” Science 319:1100e1104.
Lieberman, D. E. 2011. The Evolution of the Human Head. Cambridge, MA: Belknap Press/Harvard University Press.
Liu, W., C.-Z. Jin, Y.-Q. Zhang, Y.-J. Cai, S. Xing, X.-J. Wu, H. Cheng, R. L. Edwards, W.-S. Pan, D.-G. Qin, Z.-S. An, E. Trinkaus, and X.-Z. Wu. 2010. “Human Remains from Zhirendong, South China, and Modern Human Emergence in East Asia.” Proceedings of the National Academy of Sciences USA 107:19201–19206.
Lordkipanidze, D., M. S. Ponce de León, A. Margvelashvili, Y. Rak, G. P. Rightmire, A. Vekua, and C. P. E. Zollikofer. 2013. “A Complete Skull from Dmanisi, Georgia, and the Evolutionary Biology of Early Homo.” Science 342:326–331.
Mendez, F. L., G. D. Poznik, S. Castellano, and C. D. Bustamante. 2016. “The Divergence of Neandertal and Modern Human Y Chromosomes.” American Journal of Human Genetics 98:728–734.
Meyer, M., M. Kircher, M.-T. Gansauge, H. Li, F. Racimo, S. Mallick, J. G. Schraiber, F. Jay, K. Prufer, C. de Filippo, P. H. Sudmant, C. Alkan, Q. Fu, R. Do, N. Rohland, A. Tandon, M. Siebauer, R. E. Green, K. Bryc, A. W. Briggs, U. Stenzel, J. Dabney, J. Shendure, J. Kitzman, M. F. Hammer, M. V. Shunkov, A. P. Derevianko, N. Patterson, A. M. Andres, E. E. Eichler, M. Slatkin, D. Reich, J. Kelso, and S. Paabo. 2012. “A High-Coverage Genome Sequence from an Archaic Denisovan Individual.” Science 338:222–226.
Meyer, M., J.-L. Arsuaga, C. de Filippo, S. Nagel, A. Aximu-Petri, B. Nickel, I. Martínez, A. Gracia, J. M. B. de Castro, E. Carbonell, B. Viola, J. Kelso, K. Prüfer, and S. Pääbo. 2016. “Nuclear DNA Sequences from the Middle Pleistocene Sima de los Huesos Hominins.” Nature 531:504–507.
Mounier, A., A. Balzeau, M. Caparros, and D. Grimaud-Herve. 2016. “Brain, Calvarium, Cladistics: A New Approach to an Old Question, Who Are Modern Humans and Neandertals?” Journal of Human Evolution 92:22–36.
Mounier, A., S. Condemi, and G. Manzi. 2011. “The Stem Species of Our Species: A Place for the Archaic Human Cranium from Ceprano, Italy.” PLoS ONE 6:e18821.
O’Leary, M. A., J. I. Bloch, J. J. Flynn, T. J. Gaudin, A. Giallombardo, and N. P. Giannini. 2013. “The Placental Mammal Ancestor and the Post–K-Pg Radiation of Placentals.” Science 339:662–667.
Paabo, S. 2014. Neanderthal Man: In Search of Lost Genomes. New York, NY: Basic Books.
Peterson, G.I., and J. Masel. 2009. “Quantitative Prediction of Molecular Clock and Ka/Ks at Short Timescales.” Molecular Biology and Evolution 26:2595–2603.
Petit, R. J. 2008. “The Coup de Grace for Nested Clade Phylogeographic Analysis?” Molecular Ecology 17:516–518.
Pickrell, J. K., N. Patterson, C. Barbieri, F. Berthold, L. Gerlach, T. Güldemann, B. Kure, S. W. Mpoloka, H. Nakagawa, C. Naumann, M. Lipson, P.-R. Loh, J. Lachance, J. Mountain, C. Bustamante, B. Berger, S. Tishkoff, B. Henn, M. Stoneking, D. Reich, and B. Pakendorf. 2012. “The Genetic Prehistory of Southern Africa.” Nature Communications 2012;3:1143.
Profico, A., F. Di Vincenzo, L. Gagliardi1, M. Piperno, and G. Manzi. 2016. “Filling the Gap. Human Cranial Remains from Gombore II (Melka Kunture, Ethiopia; ca. 850 ka) and the Origin of Homo heidelbergensis.” Journal of Anthropological Sciences 94:1–24.
Prüfer, K., F. Racimo, N. Patterson, F. Jay, S. Sankararaman, S. Sawyer, A. Heinze, G. Renaud, P. H. Sudmant, C. de Filippo, H. Li, S. Mallick, M. Dannemann, Q. Fu, M. Kircher, M. Kuhlwilm, M. Lachmann, M. Meyer, M. Ongyerth, M. Siebauer, C. Theunert, A. Tandon, P. Moorjani, J. Pickrell, J. C. Mullikin, S. H. Vohr, R. E. Green, I. Hellmann, P. L. F. Johnson, H. Blanche, H. Cann, J. O. Kitzman, J. Shendure, E. E. Eichler, E. S. Lein, T. E. Bakken, L. V. Golovanova, V. B. Doronichev, M. V. Shunkov, A. P. Derevianko, B. Viola, M. Slatkin, D. Reich, J. Kelso, and S. Pääbo. 2014. “The Complete Genome Sequence of a Neanderthal from the Altai Mountains.” Nature 505:43–49.
Rasmussen, M., X. Guo, Y. Wang, K. E. Lohmueller, S. Rasmussen, A. Albrechtsen, L. Skotte, S. Lindgreen, M. Metspalu, T. Jombart, T. Kivisild, W. Zhai, A. Eriksson, A. Manica, L. Orlando, F. M. De La Vega, S. Tridico, E. Metspalu, K. Nielsen, M. C. Avila-Arcos, J. V. Moreno-Mayar, C. Muller, J. Dortch, M. T. P. Gilbert, O. Lund, A. Wesolowska, M. Karmin, L. A. Weinert, B. Wang, J. Li, S. Tai, F. Xiao, T. Hanihara, G. van Driem, A. R. Jha, F.-X. Ricaut, P. de Knijff, A. B. Migliano, I. Gallego Romero, K. Kristiansen, D. M. Lambert, S. Brunak, P. Forster, B. Brinkmann, O. Nehlich, M. Bunce, M. Richards, R. Gupta, C. D. Bustamante, A. Krogh, R. A. Foley, M. M. Lahr, F. Balloux, T. Sicheritz-Ponten, R. Villems, R. Nielsen, J. Wang, and E. Willerslev. 2011. “An Aboriginal Australian Genome Reveals Separate Human Dispersals into Asia.” Science 334:94–98.
Reich, D., R. E. Green, M. Kircher, J. Krause, N. Patterson, E. Y. Durand, B. Viola, A. W. Briggs, U. Stenzel, P. L. F. Johnson, T. Maricic, J. M. Good, T. Marques-Bonet, C. Alkan, Q. Fu, S. Mallick, H. Li, M. Meyer, E. E. Eichler, M. Stoneking, M. Richards, S. Talamo, M. V. Shunkov, A. P. Derevianko, J.-J. Hublin, J. Kelso, M. Slatkin, and S. Pääbo. 2010. “Genetic History of an Archaic Hominin Group from Denisova Cave in Siberia.” Nature 468:1053–1060.
Rohlf, F. J. and D. E. Slice. 1990. “Extensions of the Procrustes Method for the Optimal Superimposition of Landmarks.” Systematic Zoology 39:40–59.
Rightmire, G. P. 1998. “Human Evolution in the Middle Pleistocene: The Role of Homo heidelbergensis.” Evolutionary Anthropology 6:218–227.
Roberts, R. G., R. Jones, and M. A. Smith. 1990. “Thermoluminescence Dating of a 50,000-Year-Old Human Occupation Site in Northern Australia.” Nature 345:153–156.
Sankararaman, S., S. Mallick, M. Dannemann, K. Prüfer, J. Kelso, S. Pääbo, N. Patterson and D. Reich. 2014. “The Genomic Landscape of Neanderthal Ancestry in Present-Day Humans.” Nature 507:354–357.
Scally, A. 2016. “Mutation Rates and the Evolution of Germline Structure.” Philosophical Transactions of the Royal Society 371:20150137.
Schiffels, S., and R. Durbin. 2014. “Inferring Human Population Size and Separation History from Multiple Genome Sequences.” Nature Genetics 46:919–925.
Schwartz, J. H. 2016. “What Constitutes Homo sapiens? Morphology Versus Received Wisdom.” Journal of Anthropological Sciences 94:1–16.
Schwartz, J. H., and I. Tattersall. 2000. “The Human Chin Revisited: What Is It and Who Has It?” Journal of Human Evolution 38:367–409.
Schwartz, J. H., and I. Tattersall. 2002. The Human Fossil Record: Terminology and Craniodental Morphology of Genus Homo Europe, Volume 1. Hoboken, NJ: Wiley-Liss.
Schwartz, J. H., and I. Tattersall. 2003. The Human Fossil Record: Craniodental Morphology of Genus Homo Africa and Asia, Volume 2. Hoboken, NJ: Wiley-Liss.
Schwartz, J. H., and I. Tattersall. 2005. The Human Fossil Record: Craniodental Morphology of Early Hominids Genera Australopithecus, Paranthropus, Orrrorin and Overview, Volume 4. Hoboken, NJ: Wiley-Liss.
Schwartz, J. H., and I. Tattersall. 2010. “Fossil Evidence for the Origin of Homo Sapiens.” American Journal of Physical Anthropology 143 (Suppl. 51):94–121.
Skuse, D. H. 2005. “X-linked Genes and Mental Functioning.” Human Molecular Genetics 149 (Spec No 1):R27–32.
Startin, C. M., C. Fiorentini, M. de Haan, and D. H. Skuse. 2015. “Variation in the X-linked EFHC2 Gene Is Associated with Social Cognitive Abilities in Males.” PLoS ONE 10(6):e0131604.
Stojanowski, C.M. 2014. “Iwo Eleru’s Place Among Late Pleistocene and Early Holocene Populations of North and East Africa.” Journal of Human Evolution 75:80–89.
Stringer, C. 2016. “The Origin and Evolution of Homo sapiens.” Philosophical Transactions of the Royal Society B 371:20150237.
Swofford, D. L. 2000. Phylogenetic Analysis Using Parsimony *and Other Methods, Version 4.0b10. Sunderland, MA: Sinauer Associates.
Swofford, D. L., G. J. Olsen, P. J. Waddell, and D. M. Hillis. 1996. “Phylogenetic Inference.” In Molecular Systematics, 2nd edition, edited by D. M. Hillis and C. Moritz, 450–572. Sunderland, MA: Sinauer Associates.
Tajima, F. 1983. “Evolutionary Relationship of DNA Sequences in Finite Populations.” Genetics 105:437–460.
Templeton, A. R. 1998. “Nested Clade Analyses of Phylogeographic Data: Testing Hypotheses about Gene Flow and Population History.” Molecular Ecology 7:381–397.
Templeton, A. R., E. Routman, and C. A. Phillips. 1995. “Separating Population Structure from Population History: A Cladistic Analysis of the Geographical Distribution of Mitochondrial DNA Haplotypes in the Tiger Salamander, Ambystoma tigrinum.” Genetics 140:767–782.
Tishkoff, S. A., M. K. Gonder, B. M. Henn, H. Mortensen, A. Knight, C. Gignoux, N. Fernandopulle, G. Lema, T. B. Nyambo, U. Ramakrishnan, F. A. Reed, and J. L. Mountain. 2007. “History of Click-Speaking Populations of Africa Inferred from mtDNA and Y Chromosome Genetic Variation.” Molecular Biology and Evolution 24:2180–2195.
Tishkoff, S. A., and M. K. Gonder. 2006. “Human Origins within and OA.” In Anthropological Genetics, M. Crawford, ed. Cambridge, UK: Cambridge University Press.
Theobald, D. L., and D. S. Wuttke. 2008. “Accurate Structural Correlations from Maximum Likelihood Superposition.” PLoS Computational Biology 42(2):e43.
Trinkaus, E. 2007. “European Early Modern Humans and the Fate of the Neandertals.” Proceedings of the National Academy of Sciences USA 104:7367–7372.
Vernot, B., and J. M. Akey. 2014. “Resurrecting Surviving Neandertal Lineages from Modern Human Genomes.” Science 343:1017–1021.
Vernot, B., and J. M. Akey. 2015. “Complex History of Admixture between Modern Humans and Neandertals.” American Journal of Human Genetics 96:448–453.
Vigilant, L., M. Stoneking, H. Harpending, K. Hawkes, and A. C. Wilson. 1991. “African Populations and the Evolution of Human Mitochondrial DNA.” Science 253:1503–1507.
Waddell, P. J. 1995. “Statistical Methods of Phylogenetic Analysis, Including Hadamard Conjugations, Logdet Transforms, and Maximum Likelihood.” PhD thesis. Palmerston North, New Zealand: Massey University.
Waddell, P. J. 2013. “Happy New Year Homo erectus? More Evidence for Interbreeding with Archaics Predating the Modern Human/Neanderthal Split.” arXiv Quantitative Biology 1312.7749:1–29.
Waddell, P. J. 2014. “Extended Distance-based Phylogenetic Analyses Applied to 3D Homo Fossil Skull Evolution.” arXiv Quantitative Biology 1501.0019:1–42.
Waddell, P. J. 2015. “Expanded Distance-based Phylogenetic Analyses of Fossil Homo Skull Shape Evolution.” arXiv Quantitative Biology 1512.09115:1–20.
Waddell, P. J., A. Azad, and I. Khan. 2011a. “Resampling Residuals on Phylogenetic Trees: Extended Results.” arXiv Quantitative Biology 1101.0020:1–9.
Waddell, P. J., H. Kishino, and R. Ota. 2001. “A Phylogenetic Foundation for Comparative Mammalian Genomics.” Genome Informatics Series 12:141–154.
Waddell, P. J., H. Kishino, and R. Ota. 2005. “Statistical Tests for SINE Data and Resolution of the Species Tree.” Technical Report 216:1–21. Columbia: Department of Statistics, University of South Carolina.
Waddell, P. J., N. Okada, and M. Hasegawa. 1999. “Towards Resolving the Interordinal Relationships of Placental Mammals.” Systematic Biology 48:1–5.
Waddell, P. J., and D. Penny. 1996. “Evolutionary Trees of Apes and Humans from DNA Sequences.” In Handbook of Symbolic Evolution, edited by A.J. Lock and C.R. Peters, 53–73. Oxford: Clarendon Press.
Waddell, P. J., J. Ramos, and X. Tan. 2011b. “Homo denisova, Correspondence Spectral Analysis, and Finite Sites Reticulate Hierarchical Coalescent Models.” arXiv Quantitative Biology 1112.6424:1–43.
Waddell, P. J., and X. Tan. 2012. “New g%AIC, g%AICc, g%BIC, and Power Divergence Fit Statistics Expose Mating between Modern Humans, Neanderthals and Other Archaics.” arXiv Quantitative Biology 1212.6820:1–23.
Wagner, G. P. 2001. The Character Concept in Evolutionary Biology. Lieden: Elsevier.
Weber, G. W., and F. Bookstein. 2011. Virtual Anthropology. A Guide to a New Interdisiplinary Field. Vienna: Springer-Verlag.
Wilkins, J. S. 2011. Species: A History of the Idea. Berkeley: University of California Press.
Wolpoff, M. H. 1989. “Multiregional Evolution: the Fossil Alternative to Eden.” In The Human Revolution: Behavioral and Biological Perspectives on the Origins of Modern Humans, edited by P. Mellars and C. B. Stringer, 62–108. Edinburgh: Edinburgh University Press.
Wolpoff, M. H., J. Hawks, D. W. Frayer, and K. Hunley. 2001. “Modern Human Ancestry at the Peripheries: a Test of the Replacement Theory.” Science 291:293–297.
Wolpoff, M., A. G. Thorne, J. Jelinek, and Z. Yinyun. 1994. “The Case for Sinking Homo erectus: 100 Years of Pithecanthropus Is Enough!” Courier Forschungs Senckenburg 171:341–361.
Yang, J., S. Grünewald, and X.-F. Wan. 2013. “Quartet-Net: A Quartet-Based Method to Reconstruct Phylogenetic Networks.” Molecular Biology and Evolution 30:1206–1217.
Yang, M. A., A. S. Malaspinas, E. Y. Durand, and M. Slatkin. 2012. “Ancient Structure in Africa Unlikely to Explain Neanderthal and Non-African Genetic Similarity.” Molecular Biology and Evolution 29:2987–2995.