7
Psychostimulant Treatment of ADHD
One might think that the whole nature of drug discovery is based on knowing what is wrong with the brain and then fixing it. Sadly, we are not at that point.
As is often the case in psychiatry, a lot of development in treatment has arisen out of luck and chance observation. For example, it was a serendipitous observation that amphetamine had a positive effect in hyperactive children [720]. The drugs that treat ADHD are not only useful for treatment but crucial for understanding the brain mechanisms involved in the disorder. The drugs can lead to theories about ADHD that can then inform treatment further.
The neuropharmacology of the most common drug treatments for ADHD is to correct a putative dopamine (DA) dysfunction. That we know there is a DA dysfunction, however, is due to the effects of the drugs – thus we enter a circular argument.
A Brief Review of the Neuropharmacology
In the previous chapter we looked at regions of the brain. These regions of the brain are made up of neural cells or neurons that carry out particular roles, both physiologically and, ultimately, behaviorally. The neurons communicate with each other, sending and receiving messages to one another. These messages are sent in two main ways: within a neuron, via electrical impulses; and between neurons, via chemicals.
There are three main components of a neuron: the cell body, the axon, and the dendrites (see Figure 7.1). The cell body (or soma) contains the nucleus with genetic information and the machinery for survival. The dendrites are the branches of the cell that receive incoming information from other neurons. The axon is the long(ish) projection from the cell body to the terminal region. At the terminal region, there is communication between neurons across a gap. The gap between neurons is called a synapse.
Figure 7.1 The neuron (see text for details)
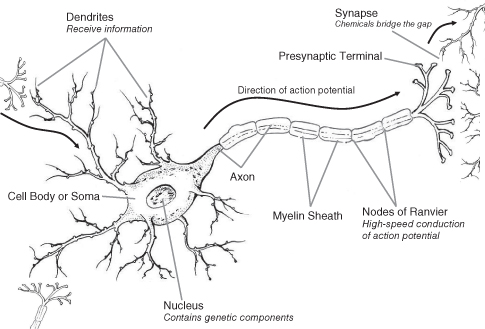
In order to comprehend what is happening in ADHD, we have to have a basic understanding of the two mechanisms of communication involved in brain activity: electrical communication and synaptic transmission.
Electrical communication
When we hear of a nerve firing, it is electrical communication that is being referred to. The basis of electrical communication is the difference in ion concentrations on either side of the neuron’s cell membrane: the inside and outside. Ions are molecules that have different electrical charges: positive or negative. There are different concentrations of ions in the intracellular fluid (inside the cell) compared with the extracellular fluid (outside of the cell). There are four main ions to consider: sodium (Na+), potassium (K+), Chloride (Cl-), and some negatively charged proteins. Calcium (Ca++) is also involved, but more so at the junctions between neurons called the synapse.
If we place one electrode inside the neuron (intracellular) and another outside (extracellular), we can measure the difference in the electrical charge between the two sides. The difference between the two sides of the cell wall is called the membrane potential. When there are no messages impinging on the neuron, it is said to be at rest, thus it is called the “resting potential.” Owing to the differences at rest, the neuron is polarized: that is, the two sides of the cell membrane are differentially charged.
When a neuron is activated (or fires), it produces what is called an action potential: a change form resting to being in action (see Figure 7.2). This is the process of conveying information within the cell. An action potential is a rapid reversal of the membrane potential from rest and occurs when the membrane allows an exchange of ions. An action potential reduces that polarity of the cell and is therefore depolarizing. The action potential is an all-or-none event: they either occur or they don’t. A big stimulus does not produce a bigger action potential. The action potential is always the same size.
Figure 7.2 The action potential (see text for details)
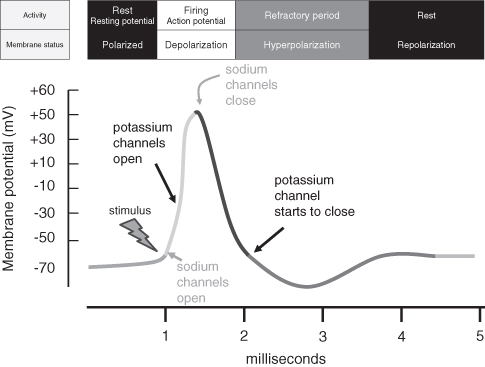
The action potential is a sequence of events in which the ions move across the cell membrane. Incoming messages change the cell membrane and the action potential starts with the opening of Na+ channels. When the channels open, Na+ enters the cell, causing a change in membrane potential. This upsets the balance between the ion concentrations on either side of the cell membrane and in response K+ channels open and K+ exits the cell. The next step is for Na+ channels to close, but, despite the closure, K+ ions still exit the cell, producing a repolarization of the cell: that is, heading back to the polarized state of the resting potential. The K+ channels close slowly and as a consequence K+ continues to exit, producing an overshoot of the resting potential which results in a hyperpolarization – more polarized then when at rest. Once all channels are closed, the membrane returns to the resting potential. All this activity happens in the space of 3–4 ms (see Figure 7.2). It is the action potential that EEG and ERP recordings aggregate to provide a measure of electrical activity in the brain.
The action potential travels from one end of the neuron to the other. The exchange of ions at any one point influences the neighboring section of the axon – it is like a domino effect in which one block knocks the other over, and so it continues. The speed of an action potential is increased by myelin, which acts as insulation (or a sheath) and prevents the ionic exchange. However, in the myelin sheath there are gaps for an action potential to occur. Thus the action potential appears to jump along the axon. Deficiencies in myelin have also been hypothesized to account for the symptoms of ADHD [721].
Synaptic transmission
It is at the synapse that a great deal of interest is focused in ADHD research. The synapse is the gap between two neurons. For a neuron to communicate with another, the synapse has to be crossed. The presynaptic neuron (the neuron sending the message) releases a chemical called a neurotransmitter, which crosses the synapse. Neurotransmitters interact with special receptors on another neuron (the postsynaptic neuron). The neuron that receives the message transforms the message into an action potential. The action potential travels down to the axon and releases a neurotransmitter into another synapse.
There are many neurotransmitters within the nervous system, which are classified into groups or families depending on chemical structure. For the purposes of ADHD, we will restrict ourselves to the main culprits: dopamine (DA) and noradrenaline (NA). Despite their central prominence in ADHD, they are not the most prevalent in the brain – that honor belongs to glutamate and gamma-aminobutyric acid (GABA). Glutamate has been hypothesized to have a role in ADHD [722]. For those of you who wish to research the role of neurotransmitters further, you should note that in the USA noradrenaline is referred to as norepinephrine.
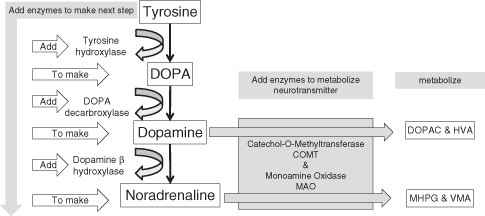
Neurotransmitters are made; they do not just appear. Synthesis of a neurotransmitter is like a recipe: the ingredients must be added before complete. The recipe for dopamine and noradrenaline is depicted in Figure 7.3. (It is noteworthy that NA shares much of the same synthetic pathway as DA.) The synthesis of a neurotransmitter takes place in the soma. The newly made neurotransmitter is put into packages called vesicles. Vesicles are spherical containers that are transported down microtubules of the axon to the presynaptic terminal. They remain in the presynaptic terminal until they are released into the synapse – the signal for their release is the action potential. This type of release is called phasic release.
Figure 7.3 Dopamine and noradrenaline synthesis and metabolism. These are the primary neurochemicals that are implicated in ADHD
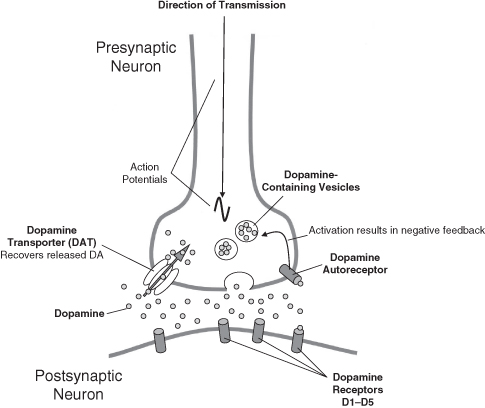
The vesicles are poised ready for communication. When the action potential reaches the presynaptic terminal, the cell membrane is depolarized. Depolarization of the membrane allows calcium (Ca++) ion channels to open and Ca++ to enter the presynaptic terminal, which makes the vesicles fuse with the presynaptic cell’s membrane. Once fused, the contents of the vesicle are released into the synapse. Particular neurons release particular neurotransmitters. A neuron that releases DA is referred to as a dopaminergic neuron. Neurons that release glutamate are glutamatergic neurons. This convention can be applied to many neurotransmitters.
The postsynaptic neuron requires a means of receiving the neurochemical message and does so by specialized receptors on its cell membrane. These receptors are proteins that are configured to receive certain neurotransmitters. When a neurotransmitter attaches itself to a receptor, it is said to bind. It is not just neurotransmitters that bind to receptors; drugs (sometimes called ligands) can also do so (see Figure 7.4).
Figure 7.4 A normal unaffected dopaminergic synapse (see text for details)
There are many types of receptors in the brain, but on the whole they fall into two categories: ionotropic and metabotropic. These two types of receptors respond to messages in different ways. The ionotropic receptor responds directly and quickly to a neurotransmitter. In contrast, the metabotropic receptor responds to ligands with an intermediate step and is slower.
There are numerous receptors in the brain for DA; they are imaginatively called D1, D2, D3, D4, etc. The differences between these receptors can be traced to the molecular structure. Basically, however, DA receptors fit into two families: the D1-like family (ionotropic) and the D2-like family (metabotropic) [723]. The differences are still the subject of research in order to understand their behavioral output. Receptors are not evenly distributed throughout the brain: there are clusters of D3 receptors in some areas and not others, whilst the D4 receptors may appear in completely different regions to that of the D3. This has clear implications for the treatment of disorders such as ADHD, as there is a potential for targeting specific receptors and therefore specific regions of the brain.
In the synapse, the released neurotransmitter is monitored by the presynaptic neuron. It does this by using receptors for the neurotransmitter that are located on the presynaptic membrane. These receptors are called autoreceptors. They act somewhat like a thermostat on a central heating system which monitors the temperature and adjusts according to your settings. We can apply this principle to DA. If there is too much DA in the synapse, the autoreceptor relays the message to the neuron and it shuts down production and release of DA. If there is too little DA, the neuron increases production and release. The neuron therefore uses feedback derived from the autoreceptor to regulate release of the neurotransmitter, but this can only work within certain limits. A failure may give rise to symptoms/behaviors.
Another type of presynaptic receptor that regulates neurotransmitter release is the heteroreceptors. The heteroreceptors modulate synaptic transmission via another neuron’s input. Heteroreceptors differ from autoreceptors as they receive messages from neurons from a different chemical family. In the case of ADHD, DA neurons can have heteroreceptors on them that receive glutamatergic input. The messages received via the heteroceptors are either excitatory or inhibitory. That is, they have the ability to increase or decrease the likelihood of a neurotransmitter being released. This type of neurotransmitter interaction is important in determining the background level of neurotransmitters released into the synapse and is called tonic release (in contrast to phasic release, which is in response to a stimulus).
Once a neurotransmitter is released, its active lifespan is very short. The lifespan of the neurotransmitter is brought to an end by either metabolism or reuptake. The process of metabolism involves the conversion of neurotransmitters into other chemicals called metabolites. These metabolites are often, but not always, inactive and can be measured in spinal fluid, urine, and blood, but this measure is for the whole body, not just the brain. With regard to DA, the metabolites are HVA and DOPAC and for NA they are MHPG and VMA. Many animal studies look at metabolism as an index of activity.
Reuptake also stops the action of a neurotransmitter by reclaiming it from the synapse. In reuptake, the presynaptic neuron has another set of receptors called transporters. The transporters are channels in the membrane that reclaim the neurotransmitter from the synapse. Once reclaimed, the neurotransmitter can be metabolized in the cell or repackaged into the vesicles for further use. This mechanism is critically involved in ADHD and is the basis of treatment strategies where a dopamine transporter (DAT) is blocked by methylphenidate, causing an increase in DA within the synapse. The fact the methylphenidate is effective and this is its modus operandi tentatively suggests a potential fault with DA. But of course it is not as clear-cut as that! Noradrenaline has a similar reuptake system that atomoxetine specifically targets and is the same mechanism by which many drugs operate such as antidepressants.
A brief note of caution is warranted before we continue with our explanations of ADHD in terms of neurochemistry. Often the textbooks portray disorders and diseases as being a result of one faulty transmitter, or being the consequence of a transmitter that is involved in a particular behavior. The truth is far more complex: DA, for example, is involved in many disease processes, such as schizophrenia, Parkinson’s disease, and addiction. It is not just confined to ADHD. The big questions relate to the exact nature of the DA problem in the different disorders. Furthermore, the use of drugs to determine what has gone wrong in ADHD is limited by the action of the drugs themselves. Many drugs do not have great pharmacological precision and act at many sites. The challenge is to determine the most important sites. Another caveat is that there can be knock-on effects of a neurotransmitter malfunction that can be found at sites distant from the primary point of the problem and drugs may work elsewhere. Looking at individual behaviors may not always give the full answer to where the problem is, and again the importance of diagnosis and the validity of those behaviors in the clinical picture of ADHD are crucial for sensible questions to be asked.
Pharmacotherapy in ADHD
Given that many consider ADHD to be a recent phenomenon, it may come as a surprise to learn that the treatment of behavioral problems with a psychostimulant dates back to 1937. Charles Bradley, a psychiatrist, was working with brain-injured children who required unpleasant diagnostic investigation – a pneumoencephalogram, in which the fluid from around the brain is drained off. Such procedures caused headaches and vomiting. To treat this, Bradley used the amphetamine Benzedrine, which was freely available at the time. By chance, Benzedrine improved the children’s behavior, with Bradley stating “there was a spectacular improvement in school performance in half of the children” [720]. He continued to use Benzedrine, publishing his findings in 1950 [724]. Whilst such studies were not rigorous, many clinical trials have now been conducted on amphetamine and methylphenidate (see chapter 1 for an overview of clinical trials). It is also worth noting that the treatment of adult ADHD lags somewhat behind childhood ADHD. The majority of drugs that are used in adult ADHD are not fully licensed for that use yet. They are used off-label, which means they are tested and known drugs, but used for a different purpose or under different circumstances to those for which they were originally intended.
What, then, is the evidence for these treatments being effective?
When evaluating the effectiveness of a drug, we have to decide what the outcome measures are going to be. This may appear a trivial point, but if the drugs are going to be considered effective, they need to be evaluated, and this can be done in as many different ways as there are studies. A large number of the rating scales have been designed to evaluate treatment effects (see chapter 2). However, these ratings scales may produce very different results from neuropsychological tests, or from those measuring academic performance or social well-being. Thus a number of outcome measures are preferable to a single simple rating scale.
The pharmacological treatment of ADHD is not a cure. It is a way of managing symptoms. If we could cure ADHD, we might have a better understanding of its cause. The more cynically minded might argue that the pharmaceutical industry has a financial interest in treatment rather than cure. If you cure someone, there is no more money to be made from that person; if you do not cure, but keep the symptoms under control, then you have a long-term income.
There is much more to ADHD treatment than drugs, but it is the drugs that cause most concern. All drugs have risks – even the aspirin or paracetamol you may take for a headache. An evaluation of the risks compared to the benefits is needed by parents and patients. It is a difficult decision for a parent to make with pressure from family members and friends, who often have their opinions based on the media portrayal of treatment and not on the balance of facts [725].
In addition the economic cost of treatment for adult ADHD has been shown to be less than that for depression and diabetes [726]. A Norwegian study indicated that childhood stimulant treatment has a positive effect on the economic burden of ADHD in adulthood [727], although this study only compared a small group of people treated in childhood against a large group of adults now seeking treatment. The total excess cost of ADHD in the US in 2000 was $31.6 billion. Of this total, $1.6 billion was for the treatment of ADHD; the remainder was for other health care and loss of work and the health care of the family members of those with ADHD [728].
Such costings need to be considered and compared to the economic cost of not treating ADHD, e.g. loss of production due to inability to work, health care consumption and criminality [729]. Forensic reports indicate that a large proportion of those in prison have some symptoms of ADHD, e.g. 45 percent with the DSM-IV criteria [730]. At an individual level, the cost of keeping one person in prison far exceeds health care costs. The average cost of a prison place in 2002 was £38,753 per year (Institute for the Study of Civil Society 2004).1 A year’s worth of methylphenidate is considerably less.
In the USA the cost of ADHD was estimated to be on average $14,576 per individual case, with the majority of the money spent not on health care but on education and crime [731], whereas an earlier review suggested that the cost of improvement was between $15,509 and $27,776 [732]. In the UK the cost of methylphenidate compared to placebo per child was £9,177 (with a range of £5,965–£14,233) [733]. The cost of the drug and its availability may be a reason why it is becoming a first line in treatment [734]. One of the concerns raised about cost effectiveness, and also clinical effectiveness, is that the long-term effects are relatively unknown. To quote Michael Schlander, who is a critic of the guidelines of the UK’s National Institute for Health and Clinical Excellence (NICE), “Limitations of currently available economic evaluations include their short time horizon, and future research should assess treatment effects on long-term sequelae associated with ADHD” [735] (p. 421). This might be critically important in light of the recent three-year follow-up of the MTA study in the USA, which was not indicative of a benefit above and beyond behavioral interventions with methylphenidate (see page 164).
The main treatment for ADHD is methylphenidate, and in a small number of cases amphetamine is used, and, more rarely, pemoline. Although it may look like there are a number of drugs for ADHD (e.g. Ritalin, Equasym, and Concerta), these are all brand names of methylphenidate. Methylphenidate is a psychostimulant and is a controlled substance because of its abuse liability (see chapter 9). The clinical effects of methylphenidate are well documented, and it is used as the main tool in trying to understand the psychopharmacology of ADHD.
Just because methylphenidate works in the majority of people with ADHD, it cannot be used to support or validate the diagnosis of the disorder. If methylphenidate works, it does not mean that the person has ADHD. Methylphenidate can have similar cognitive enhancing effects in non-ADHD groups [736].
Methylphenidate is a broad-spectrum drug with many effects, but it is important to establish its modus operandi so that we can hypothesize and explore etiology in ADHD. Unfortunately we do not have a large range of drugs to probe ADHD, and therefore our conclusions about the underlying neuropharmacology of the disorder are really limited to explanations of an individual drug, i.e. methylphenidate. The drugs are not precision tools that hit only one target; there is, to use a military term, collateral damage. That is, methylphenidate acts where it is therapeutic, but it also acts in other regions where the effects can be undesirable. More drugs are needed if this psychopharmacological approach is to succeed. With this in mind, we can investigate how these drugs work and what they can tell us about ADHD.
The Pharmacology and Efficacy of Psychostimulants Used in Treating ADHD
Both methylphenidate and amphetamine are dopamine-enhancing drugs or agonists. In reality they are more than just DA agonists; they act on different neurotransmitters, but it just happens that it is DA where the greatest effect is seen. They also act on noradrenaline, which may be critically important in ADHD [737]. There are two components to the pharmacological action of a drug: pharmacokinetics and pharmacodynamics. Pharmacokinetics is all about how the drug gets to and from its target. This involves the route of administration (in the case of ADHD it is the slow oral route and not the fast intravenous route), speed of release (slow is best), and metabolism (how it is deactivated). By contrast, pharmacodynamics is about what the drug does when it gets to the target. In ADHD the focus has been predominantly dopaminergic.
The Pharmacology of Methylphenidate
Methylphenidate comes in a number of different preparations. The nature of these different preparations is based upon their duration of action. They are either short-lasting or long-lasting. The drug is the same; it is the length and speed of delivery that is different. Immediate-release (IR) methylphenidate gets into the brain quickly and the behavioral effects are clear within approximately 20–30 minutes and come to an end after three to four hours, thereby requiring multiple doses. This is clearly problematic and results in oscillating symptom management and increased non-compliance. It has also been associated with an increased risk of abuse (see chapter 9).
Other versions of methylphenidate have a slow-release mechanism that allows the drug to be released gradually into the bloodstream (e.g. Ritalin SR). This provides an even concentration of the drug without the peaks and troughs associated with IR methylphenidate. One problem with the slow-release version is that it can take some time to take effect – approximately 40–60 minutes. An hour of ADHD symptoms is a long hour!
As the slow-release preparations take longer to become effective, they often come with an IR coating of methylphenidate to avoid a time delay in action. Essentially these preparations have a mixed release pattern: an IR that lasts for approximately four hours and a slow release that comes into effect after the initial release of methylphenidate comes to an end. The choice of drug preparation and the person’s response require close monitoring by health professionals in order to ensure adequate titration and optimal treatment response.
The mechanism by which slow-release methylphenidate works varies across brands. Concerta, made by the ALZA Corporation, uses an osmotic controlled-release system (OROS). Osmotic pressure delivers methylphenidate at a controlled rate. The system comprises of an osmotically active core surrounded by a semi-permeable membrane with an immediate-release drug outer layer. The core comprises two layers: one containing the drug, and another layer containing osmotically active components which push the drug out. There is a precision-laser-drilled hole on the drug layer at the end of the tablet. In the stomach, the drug overcoat dissolves quickly, providing an initial dose of methylphenidate. Slowly fluid crosses the semi-permeable membrane into the core of the tablet. This makes non-active parts of the formulation expand, like a sponge, pushing methylphenidate through the hole. The biologically inert components of the tablet remain intact and are eliminated in the stool as a tablet shell along with insoluble core components.
Equasym XR capsules, made by UCB, contain tiny beads of methylphenidate. The capsules are designed to release 30 percent of the methylphenidate immediately, giving an initial dose of medicine for the morning. The remaining 70 percent of the dose is released gradually through the afternoon. According to the manufacturers, this means the Equasym XR provides medication without the need to take a dose at lunchtime, thus covering the whole school day.
You do not need to stick with one brand of methylphenidate. Different versions can be used to make a bespoke treatment strategy for the individual’s life circumstances. The Rubina Chart in Table 7.1 shows how this can be achieved when looking at release patterns of the various formulations.
Table 7.1 Release rate of methylphenidate in different extended-release preparations (the Rubina Chart)
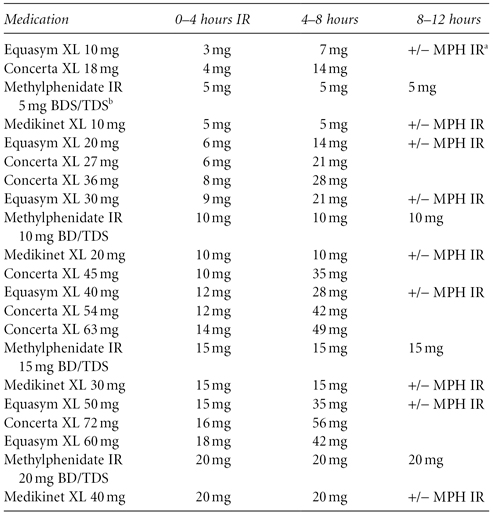
a MPH IR = methylphenidate immediate release
b BD/TDS = twice a day/three times a day
Source: Courtesy of Dr David Coghill, University of Dundee (personal communication). Copyright University of Dundee.
Other innovative methods of delivering methylphenidate throughout the day have taken the same way a nicotine patch works for smoking cessation and applied it to ADHD medication. Daytrana, made by Shire Pharmaceuticals, is a methylphenidate-containing skin patch. The methylphenidate is absorbed through the skin into the bloodstream. After the patch is applied, it takes about two hours before the medicine begins to take effect [738] – that’s a long time! Following removal of the patch, the effect slowly wears off over the 12 hours since application; thus after a day of wearing the patch, sleep may be difficult to come by and appetite remains suppressed [739]. The effects of transdermal delivery systems are also dependent upon the area on which the patch is placed, with the hip producing the best effect [740].
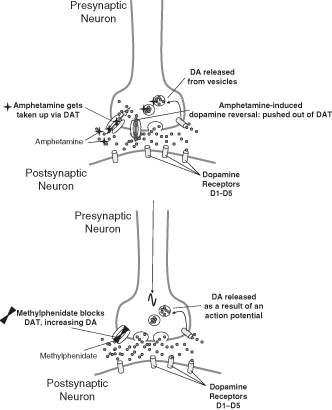
The technology of methylphenidate is all focused on release mechanisms. When methylphenidate is absorbed, it has exactly the same pharmacological action. Methylphenidate shares a great deal in common with cocaine (see chapter 9). Methylphenidate blocks the DAT. The DAT recovers released DA from the synapse, thereby deactivating it. When methylphenidate is given, the recovery of DA is prevented because the DAT is no longer working. The net effect of such a blockade is the accumulation of DA in the synapse; the DA has nowhere to escape and therefore it remains active in the synapse, stimulating DA receptors both pre- and postsynaptically. To use a domestic analogy, methylphenidate is the plug in the DA bath that stops DA going down the drain (see Figure 7.5).
Figure 7.5 The effects of amphetamine and methylphenidate on the neuron (see text for details)
Methylphenidate is made up of two stereoisomers, called D- methylphenidate and L-methylphenidate. These isomers are identical in chemical composition, with the exception that they are mirror images of each other – same drug, different view. In the majority of drugs a mix of the two is given. On the whole the D-isomer is more active in humans. This knowledge has been used in the treatment of ADHD with methylphenidate. Dexmethylphenidate (marketed as Focalin) is the D-isomer of methylphenidate, which has a higher affinity for binding to the DAT [741]. The evidence suggests that this is the effective part of the medication [742]. Why use this isomer? The manufacturers argue that the D-isomer is three times more potent than mixed methylphenidate; thus you need less of the drug to get an effect because you are only using the active ingredients.
The Pharmacology of Amphetamine
Amphetamine works by increasing the dopamine release from the presynaptic neuron. Amphetamine mimics DA and enters the presynspatic terminal via the DAT. This has two effects: (1) it competes with DA for reuptake and thus less DA can be removed from the synapse; and (2) it causes a release of DA that is independent of action potentials. This release of DA is via reversing the reuptake processes [743] (see Figure 7.5).
To use the domestic analogy again, amphetamine is like turning the taps full on in the bath and putting and undersized plug in.
Like methylphenidate, amphetamine comes in a D- and L-isomer. Dexedrine is D-amphetamine. Dexedrine was first shown to have a beneficial effect in 1950 [724] and has since been deployed for the treatment of ADHD.
Adderall is a combination of amphetamine isomers, in which the predominant isomer is D-amphetamine and L-amphetamine only makes up 19 percent of the drug. Adderall comes in an immediate- and extended-release formulation, the latter providing symptom relief for the day. The D-isomer of amphetamine is thought to be twice as potent as the L-isomer on DA reuptake and release [744]. The L-isomer was twice as potent at stimulating NA release in the cortex [745]. The L-isomer was also found to have a more widespread effect in the rat brain, which the authors link to different behavioral effects [746]. In the spontaneous hypertensive rat (SHR) model of ADHD, L-amphetamine was associated with improvements only in sustained attention, whereas the D-isomer was associated with improvements in sustained attention as well as reducing over-activity and impulsivity [747]. The duration of Adderall’s dopaminergic action has been seen to be increased above that of D-amphetamine and for a longer period of time [748]. Sometimes it is the mix of the drugs and not the precision that can lead to the desired clinical effects. Much of the work in psychopharmacology involves reduction and isolation of the elements involved. These elements may not be powerful on their own, but only when they are in combination. Numerous studies have found Adderall to be of benefit in children and adults [749–756].
An interesting development is lisdexamfetamine (Vyvanse), which is a prodrug (i.e. it is administered in an inactive form) marketed by Shire. This is a new concept in treating ADHD with amphetamine. Lisdexamfetamine is a combination of L-lysine and D-amphetamine. The L-lysine makes the D-amphetamine inactive until it is separated. Thus as a prodrug, lisdexamfetamine itself is inactive; in order to be active, it needs to converted via metabolic processes into D-amphetamine. As lisdexamfetamine needs to be metabolized before it can exert its pharmacological and clinical effects, this turns the drug into a slow-release formulation, allowing for D-amphetamine to be slowly released from the bond of L-lysine. The effect is similar to the slow-release formulations of methylphenidate; however, with lisdexamfetamine, the mechanism is based on the molecular characteristics of the drug rather than the delivery system seen in Concerta, for example. The studies to date looking at the efficacy of lisdexamfetamine support its use [738, 757–765], although these studies have varying methodological problems and some do not compare against other treatment but rather against placebo. Given that amphetamine has abuse potential, one of the advantages of lisdexamfetamine is that this is reduced [764, 766–767].
Clinical Effects of Methylphenidate and Amphetamine
Dexedrine was first shown to have a beneficial effect in 1937 (see [724]) and has since been deployed for the treatment of ADHD. Methylphenidate was first licensed for use in 1955 in the USA. In terms of safety and efficacy, more is known about methylphenidate and amphetamine in the pediatric population than any other drug.
The question of the psychostimulants’ effectiveness in treating ADHD could fill a whole book, and then other books would provide counter-arguments as to why they should not be used. Numerous studies have looked at the effectiveness of the drugs across many domains, ranging from cognitive functions to parental reports. It is beyond the scope of this chapter to review all the studies, some of which use questionable methodologies. However, taken together, the reports suggest that there is a considerable improvement upon commencement of drug treatment. Of course not everyone benefits from the drug. According to Mary Solanto [768], up to 25 percent of ADHD cases may not respond. This group represent an interesting group to treat, but they are also interesting from a scientific perspective given that the hypotheses generated about the pathological changes of ADHD stem from successful treatment strategies. Why are they different? This question remains unanswered, but some may benefit from a change of medication.
Methylphenidate is licensed for use “as part of a comprehensive treatment programme for attention-deficit hyperactivity disorder (ADHD) when remedial measures alone prove insufficient” (Summary of Product Characteristics for Ritalin and Equasym). Thus, drug treatment is to be used when other non-pharmacological treatments (remedial measures) have not been successful. However, an early review of the evidence for NICE “suggests that this treatment strategy is sub-optimal compared to first-line treatment with stimulant medication, followed up if necessary by behavioural intervention” [769]. The NICE guidelines of September 2008 continue to support the use of drug treatment in ADHD.2
Methylphenidate needs to be evaluated in each individual. There is a potential for adverse reaction and therefore it is better to start someone off with a low dose that acts for a short duration. From this position, the dose can be changed and different preparations of methylphenidate evaluated. The use of methylphenidate is on the increase, but this should not be surprising because so are the cases of ADHD, and from diagnosis comes treatment.
The short-term benefit of stimulants is well documented [770–772]. Children show improvement in the three core symptom domains of ADHD. Furthermore, there are indirect effects on other aspects of the child’s functioning such as their social interactions. The effects of methylphenidate have also been associated with positive effects on academic performance [773]. In addition, one must note that methylphenidate, whilst having addictive properties, is not used to get high, but rather to aid concentration and productivity (see chapter 9).
The effects of these drugs on the symptoms of ADHD can be looked at in a number of ways, and this has given rise to the large number of rating scales that are not necessarily diagnostic, but, rather, sensitive to changes in behavior (see chapter 2). Such rating scales have been used by the influential MTA study (see below). Other studies have looked at changes in cognition using laboratory-based tasks. In particular it would be considered important that methylphenidate and amphetamine were able to reverse the deficits in behavioral inhibition that are regarded to be prevalent in ADHD. A number of studies have founds a beneficial effect on such measures [774–780]. Similarly, working memory deficits were minimized by methylphenidate [781–787]. Thus the action of methylphenidate does not illuminate the debate on the relative importance of either of these processes in ADHD.
An interesting set of studies has shown that the parental interactions with a child with ADHD were more directive and critical when unmedicated [788], whereas upon treatment they were less critical and directive and expressed greater warmth [789–790]. Such data have been used to suggest that the parent’s interaction with the ADHD child is a response to the condition and not a cause of the condition.
According to the Royal College of Psychiatrists “The evidence for the long-term efficacy is much weaker than for short-term use. There are no truly long-term trials of stimulant treatment of ADHD.”3 In fact over a year is considered to be a long time! This is somewhat worrying as the efficacy and management of ADHD treatments is a long-term concern. To address this hole in our knowledge, a large multicentered study on treatment was conducted in the USA. This hugely influential study, which is still ongoing, is known at the MTA study.
The MTA Study
The Multimodal Treatment Study of Children with Attention Deficit Hyperactivity Disorder (MTA) is a trial spanning six different university medical centers and hospitals to evaluate treatments for ADHD. The study has included nearly 600 elementary school children, ages 7–9, randomly assigned to one of four treatment modes: (1) medication alone; (2) psychosocial/behavioral treatment alone; (3) a combination of both; or (4) routine community care [116]. The importance of the trial is highlighted in a paper by Pelham in which he states that design issues
make the MTA a landmark study for clinicians and researchers working with ADHD children. These include the large, heterogeneous sample, the state-of-the-art treatment, the lengthy treatment period, the extensive documentation of treatment manuals, and the attention paid to treatment fidelity and adherence.
[791] (p. 981)
At 14 months into the trial, medication was superior to behavioral treatment and community care. The combined treatment was no better than medication alone [792]. Negative parental interactions were also reduced in the behavioral, pharmacological, and combination groups [793]. Related to this, combination treatment produced the greatest reductions in Negative/Ineffective Discipline, which had the effect upon teacher-reported disruptive behavior that it was essentially normalized [794].
At 24 months follow-up, the MTA medication strategy showed a greater benefit over behavioral treatment and community care; however, the effect was not as large as at 14 months [795]. Additionally, at 24 months the effect of medication was in the modest to large category on neuropsychological tests and was consistent with such theories of ADHD as behavioral inhibition [796]. In another analysis the MTA group also found that growth was suppressed, but those who had not received medication showed growth rate beyond that of normal [797].
At 24 months the results of the MTA study were generally in favor of medication, with little benefit being conveyed with additional behavioral interventions. In 2007 four papers were published that reported on the 36-month outcome of the trial. The results of these papers were reported on widely by the media. In the BBC television program Panorama, aired on November 12, 2007, the makers provided an interpretation of the MTA 36-month follow-up study. Whilst they were able to correctly identify that the groups of children who were on either behavioral therapy or drug therapy did not differ at 36 months, the conclusion drawn by the program makers was that methylphenidate was ineffective. They used two case studies to highlight their cause: a male who was essentially a non-responder and potentially comorbid with ODD; and a female who appeared to have a poor reaction to the drug and was symptom-free after cessation. Both were adolescents, with all that that entails, and from difficult backgrounds (e.g. divorced parents etc.). The reporting was biased, favoring behavioral therapy, with a psychosocial cause of ADHD, and generally anti-methylphenidate. There was no mention of a biological basis of ADHD, and furthermore no mention of other treatments (which may be fair enough as the MTA study did not investigate these). However, the program makers did not read/understand the original document and the conclusions that were made were, in my opinion, based on a non-scientific understanding. Close scrutiny reveals some important features of the MTA study that make the media interpretation at best vulnerable and at worst misinformed.
So what did this research actually say? In the paper by Jensen et al. [798], the initial advantages of medication alone or in combination with behavioral treatment over behavioral or community care was no longer evident in the years after 14 months of controlled treatment ended. All groups in the study showed an improvement over the earlier baseline measures. This might be evidence that the symptoms of ADHD reduce with age. However, strong conclusions on the use of medication need to be tempered by understanding the methodological limitations of the three-year review. These limitations are acknowledged by the MTA group in their papers, but did not feature in the BBC program. The most notable limitation in terms of a clinical trial surrounds the random allocation of participants to treatment groups. After the initial 14 months of treatment, when the families could then choose their intervention (if at all), adherence to the groups dissipated. Even at 14 months, there was some movement within the groups. The behavioral-treatment-only group at 36 months was no longer that, with 45 percent taking medication. The medication group alone, and in combination with behavioral therapy, dropped to 71 percent taking medication. The community care group remained the same. This led the authors to suggest that there is a tendency for those who are doing well on medication to discontinue and those who are not doing well on behavioral interventions to start taking medications. A final note from the Jensen et al. paper on the limitations states that
the inclusion/exclusion criteria and the necessity for informed consent limit the generalizability of our findings to children with ADHD Combined type whose treatment started before the age of 10 and whose parents were willing to have them randomized to the study treatment possibilities and could commit to frequent treatment visits.
[798] (p. 999)
If that is you, then the MTA study should make interesting reading.
Unlike the BBC program, the MTA group have not relegated medication to a lower status. Jensen is quoted as saying “our results suggest that medication can make a long-term difference for some children if it’s continued with optimal intensity, and not started or added too late in a child’s clinical course.”4
In one of the companion papers, it was reported that there was substantial individual variability in responses to medication. Three groups of children with different patterns of response were identified: (1) 34 percent showed a gradual, moderate improvement; (2) 52 percent of the children showed larger initial improvement, which was sustained through the third year; and (3) 14 percent of the children responded well initially, but then deteriorated as symptoms returned during the second and third years [799]. Swanson et al. suggest “trial withdrawals” to determine if children still need to take medications.
As for the growth of children on stimulant medication, at 36 months slowed growth was confirmed and the case that they were bigger to start with was made. A group of 65 children with ADHD who had never taken medication grew somewhat larger than a group of 88 children who stayed on medication for three years. Growth rates (but not actual growth) normalized for the medicated children by the third year, but they had not made up for the earlier slowing in growth [800].
In the fourth paper of the three-year follow-up, Molina et al. reported that all the children with ADHD showed significantly higher-than-normal rates of delinquency (27.1 percent vs 7.4 percent) and substance use (17.4 percent vs 7.8 percent) after three years [801].
The question of maintaining consistent therapy in the MTA group was recently addressed and the importance of adherence was emphasized. In the study by Pappadopulos et al. [802], 24.5 percent of saliva samples indicated nonadherence, with only 53.5 percent of participants adherent at every time measure were taken, indicating some degree of nonadherence in nearly half of all the MTA children. Nonadherence is also argued to produce greater deleterious effects in children in the medication-only condition compared with those receiving both medication and behavioral treatment.
At eight years after enrollment on the MTA study it was noted that the type or intensity of the 14-month treatment plans for ADHD does not predict functioning six to eight years later, whereas early ADHD symptom trajectory is prognostic. Molina et al. continue to suggest that children with behavioral and socio-demographic advantage, with the best response to any treatment, will have the best long-term prognosis [803].
As a final note to the series of MTA reports, one has to note that medications for ADHD have changed somewhat since 1999. Whilst the pharmacology of the drugs remains the same, we now have several different ways of delivering drugs which may lead to long-term improvements and adherence [804]. Such are the differences that another MTA study would be extremely illuminating.
Side-Effects of Amphetamine and Methylphenidate
There are many side-effects associated with psychostimulants. The most notable are the effects on sleep and growth – these drugs are also used for their reversal of narcolepsy and as appetite suppressants.
Those receiving treatment tend to show reduced growth in height in the first couple of years [805–806], which becomes a minimal concern with continued treatment [807–808]. In the MTA study, growth was reported as being suppressed after treatment [797]. The impact of treatment on growth has been addressed by Swanson et al. [809], in which children who remained on medication had annual growth rates that were reduced by 20.3 percent for height and 55.2 percent for weight, but despite the risks this should be balanced against the benefits. To complicate matters, Spencer has noted that reduced growth was more to do with ADHD in the early years rather than its treatment [810]. However, as Jackson [811] points out in a critique of the growth studies, often the design used is cross-sectional and therefore does not indicate changes in growth trajectories from baseline, and those studies that do show an increased size prior to treatment. In a meta-analysis of the growth effect studies, Poulton [812] found that 11 of 29 studies indicated growth suppression and those that did not were poorly executed. The deficit in height was on average 1 cm per year for the first three years of treatment. To date, the reason for the growth suppression is not understood. It could be due to disruption of the endocrine system or due to the suppression of appetite; however, these are yet to be verified.
Sleep disruption is a common side-effect to stimulant medication. This should not come as a surprise as one of the uses of methylphenidate is for narcolepsy, which is sleep disorder characterized by excessive daytime sleepiness.
As usual there are numerous papers that provide contradictory points of view regarding sleep. Some have found a detrimental effect [813–816] whilst others have found a beneficial effect of methylphenidate in adults [817].
It is not only the length of sleep or the increased time it takes to fall asleep that is affected by methylphenidate. REM sleep (rapid eye movement sleep) is reduced in ADHD groups [818–819] but is not affected by methylphenidate [820–821] or only has a small effect [819].
The sleep disturbances are clearly evident when objective measures are taken; however, parental ratings of sleep disturbances have been shown to be exaggerated [822]. This is perhaps understandable after a day on the frontline with ADHD!
Why is the sleep disruption important? Sleep has been associated with many processes. For example, hormones that regulate growth are released during different periods of the circadian cycle, which has been considered to be a possible mechanism of growth suppression [823]. Sleep is considered to be most important for the brain and effects cognition, especially memory [824]. Sleep restriction is associated with poor attention and slowed working memory [825].
A recent study by Faraone et al. did not find that sustained-release methylphenidate was a factor predicting sleep problems in treated children who were “carefully titrated to an optimal dose” [826] (p. 308). Perhaps that quote reveals that the prescribing and monitoring of the drug is crucial not only for its effectiveness but also for minimizing the side-effect profile, thus enhancing tolerability and compliance. Therefore, using the different formulations of methylphenidate that are available could minimize the sleep disturbances. That is, don’t give a long-acting drug such as Concerta three hours before bedtime; use an IR drug that only has an effect lasting three to four hours.
Other side-effects included in the BNF (British National Formulary – the prescribing bible for medics) are restlessness, irritability and excitability, nervousness, night terrors, euphoria, tremor, dizziness, headache, rash, urticaria, fever, arthralgia, alopecia, exfoliative dermatitis, erythema multiforme, thrombocytopenic purpura, thrombocytopenia, leucopenia and abnormal liver function, convulsions, dependence and tolerance, sometimes psychosis, anorexia, gastro-intestinal symptoms, growth retardation in children, dry mouth, sweating, tachycardia (and anginal pain), palpitations, increased blood pressure, and visual disturbances, Cardiomyopathy has been reported with chronic use, and central stimulants have provoked choreoathetoid movements, tics, and Tourette’s syndrome in predisposed individuals.
As there are few studies on the long-term effects of psychostimulants, there is a degree of concern that these drugs, particularly in the developing brain, have structural and functional consequences [see 330, 827].
Psychostimulants have been shown to have an up-regulating effect on the DAT, especially in the striatum [828]. Such changes may be of concern and the duration of such action is unknown. However, Feron et al. [829] found a reduction of the DAT in children who had received methylphenidate for three months. Upon cessation of treatment, ADHD patients four to six weeks later had increased DAT levels, thus there was not a long-term effect of methylphenidate. Some features of the studies need to be considered:
1 The developmental age of the child may have a critical bearing on the adaptations to methylphenidate – the effects of drugs on dopamine systems have been demonstrated to react differently depending on the age of the organism [830].
2 Studies do not necessarily look at people with ADHD – they may look at drug users or normal controls. Clearly the brain is different in these groups.
3 The type of delivery of methylphenidate may be critical to neural adaptations: slow vs sustained release may have differences.
A worrying study was published in 2007 which linked methylphenidate with DNA damage in the rat striatum, although this damage could be repaired [641]. Following this report and a similar one by El-Zein [831], a number of studies were conducted. Human studies have produced contradictory results with some finding an effect [831], but the majority do not find an adverse effect [642–643, 832–836]. Whilst this is good news it also demonstrates that one can influence DNA.
Pemoline
Pharmacology
Once marketed as Cylert, pemoline has been used to a lesser extent in the treatment of ADHD since 1975. Unlike methylphenidate and amphetamine, pemoline has no effect on noradrenergic systems (see [837]). Pemoline action is to increase DA release and block its reuptake [838–839]. The half-life of pemoline is 8.6 hours [840], with some finding effects still after 7 hours [841]. Therefore the advantage of pemoline is that a single dose can last a long time, thereby avoiding multiple dosing [837].
Clinical effects
There are comparatively few studies that have looked at pemoline in ADHD. Back in the days of minimal brain dysfunction, pemoline was seen to have a beneficial effect especially because of its duration of action [842–843]. In a study of ADHD children using a number of outcome measures, pemoline was seen as an effective treatment [844]. It was associated with an improvement in college students [845], in adolescents [846], and in adults it was better than placebo [847], but there appears to be a reduced effect of pemoline in adults because of adverse effects [848]. Whilst a benefit of pemoline over methylphenidate was seen in one study, adverse reactions to pemoline were greater and sufficient to prevent further use [849]. Another benefit with pemoline is its lack of abuse potential [850]. However, one paper does report a case study of pemoline abuse, but this was induced by prior amphetamine abuse [851].
Side-effects
One of the recurrent side-effects of pemoline that is reported is its hepatotoxicity. This can range from increases in liver enzymes through to full liver failure [852–854]. Such was the concern that the manufacturers stated it should not be a first-line medication for ADHD [837]. In contrast the effects of pemoline on growth appear to be transient [855]. Like methylphenidate, pemoline induces insomnia [841, 844] and is associated with abdominal pain and headaches [837].
Nicotine
The use of nicotine might appear at odds with all the health promotion surrounding smoking. However, the logic is supportive of the use – it is the smoking that is problematic to health. Nicotine is addictive and is therefore problematic, but, as you will see in chapter 9, drugs that are addictive can be delivered in such a way as to avoid dependence. The main way of delivering nicotine in these studies is via a patch. This provides a slow steady release of nicotine into the bloodstream and ultimately to the brain. Unlike methylphenidate, amphetamine, and pemoline, nicotine is not a direct stimulant of DA. Nicotine does not interact with the DAT or DA receptors. Instead nicotine acts on acetylcholine receptors that have been identified and called nicotine receptors. Nicotine is considered to be a cognitive enhancer (see [856]); it is also thought possibly to be a drug with which people with ADHD might self-medicate (see chapter 9). Thus a number of studies have looked at nicotine in ADHD, and they have found a beneficial effect [857–860]. The mechanism by which nicotine brings about improvements is still being unraveled. There are many types of nicotine receptors, some of which modulate DA in the prefrontal cortex [861]. Of course one should not rush out and buy a packet of cigarettes or patches as the data are limited and there are adverse effects associated with nicotine – addiction being only one. To avoid the complex issues of using nicotine, specific agonists that target receptors have been used in trials for ADHD with some success [862–864]. As yet, no nicotinic agents are on the market for the treatment of ADHD.
Tolerance and Withdrawal
A well-known phenomenon in drug use is that of tolerance. Tolerance is the gradual decline in the effectiveness of a drug. If this happens, then you need more of the drug to get a clinical response. It is akin to the drug addict needing more drugs to get high. As the psychostimulants are not cures but rather long-term interventions for symptom management, the chance of tolerance should be increased. A number of issues arise when looking at tolerance in a pediatric population:
1 There are developmental changes that are occurring. The effectiveness of drugs may wax and wane depending on the behavior being measured.
2 Despite the adverse effects of psychostimulants on growth, children still grow, and by virtue of their increasing size they may need more of the drug – that is not to say they have become tolerant.
3 The studies that have looked at the effectiveness over time will have used different release formulations. The speed and magnitude of the drug effect may have important effects on tolerance and therefore comparisons across studies may be limited.
Tolerance to methylphenidate has been shown in animal studies (e.g. [865, 866–867]), but animal studies often use high doses delivered quickly. Tolerance in humans is not as obvious. The majority of studies fail to find a diminished effect over the time course of treatment [868–871]. However, there are some reports that tolerance might develop in subgroups of individuals [872–873]. Swanson et al. looked at acute tolerance to methylphenidate within the day. Tolerance was observed to the clinical effects of a second dose if it was given closer to the initial dose compared to if it was given later [874].
Sometimes when a drug effect wears off, or the drug is removed suddenly, there can be a withdrawal effect called rebound. Rebound is when the symptoms become worse than before. This can appear at the end of the day, when the dose is no longer effective [772]. In children given D-amphetamine, activity was decreased for about eight hours with a following period “of slight but significant increases in activity” [875] (p. 688). Johnston et al. [876] found that parent ratings on one of two measures of behavior were higher following two daytime doses of methylphenidate rather than placebo, suggesting that rebound was minimal. In a study looking at inpatients on immediate-release psychostimulants, 30 percent were seen to have rebound effects [877]. One report suggests that stimulant rebound to immediate-release methylphenidate was similar to the symptoms of bipolar disorder, an effect that was lost when long-acting preparations were used [878]. A study looking at Concerta and Adderall found that, on a driving task, participants in the experiment were more inattentive after the drug had finished working than if they had been given a placebo [879]. These problems can be managed using short-lasting preparations towards the end of the day, with a reduced impact of sleep.
Psychopharmacology: From Treatment to Theory
Neurochemical theories of ADHD predominantly, but not exclusively, address a DA dysfunction. Such theories have evolved primarily from the use of methylphenidate in treatment. As methylphenidate works, then the secrets of ADHD should be uncovered by the study of the drug. However, to limit the application of methylphenidate and amphetamine to this quest, we must also note that it enhances cognition in healthy controls as well as ADHD [880–882]. From this standpoint we can see how and where methylphenidate operates. Methylphenidate is therefore the search engine (the Google) for looking at the brain in ADHD. However, this approach is only as good as the drug itself. To be able to make strong account of ADHD’s underlying chemical pathology using drugs, we need to have several drugs that are clinically effective. If we have many drugs to use, we can look for their common features which may pinpoint the underlying problems. Unfortunately we are limited to only three main drugs: methylphenidate, amphetamine, and atomoxetine.
A further problem with using drugs as a search engine for causality is that they do not always target the site of origin, but rather operate at a connected, yet distant and downstream, location. For example, the problems with ADHD may arise in the cortex, but this has a knock-on effect further along a pathway in distant regions such as the striatum, where methylphenidate can be seen to act.
Such limitations need to be noted and considered, but one does not accept a hypothesis on one small piece of evidence. More evidence can be obtained from the different areas of the medical sciences.
Hypo/Hyperfunctioning DA in ADHD
The logic is simple: methylphenidate increases DA; therefore ADHD is a result of too little DA [883]. In fact much of the support comes from animal studies, where lesions of the DA system are used as a model of ADHD [884] and genetically modified animals that are hypoactive dopaminergically [191, 885]. Given that methylphenidate acts on the DAT, perhaps this is also the faulty mechanisms in ADHD. Early studies indicated that the DAT was increased by as much as 70 percent in the human striatum [886]. However, the human data obtained from PET scans is not consistent (see [768]). One of the main problems is that stimulation of the DAT with a drug can have an up-regulating effect in animals and humans [887–888] increasing the number of DATs. Of course such studies are limited as they use radioactive tracers (which are not ethical in children) and small numbers of adults, and sadly magnetic resonance scanning is not able to detect DA.
Recent well-controlled studies of medication-naïve adults provide further conflict, with one study stating that there was little difference in the DAT between controls and ADHD, leading the authors to suggest that ADHD may be more about a reduction of DA release [889], and another supporting a specific reduction in the striatal DAT in ADHD [890].
Flying in the face of the hypoactive argument are the metabolite studies looking at homovanillic acid (HVA), the main breakdown product of DA. Increased levels of HVA in children were associated with increased severity and a positive response to treatment [891–892]. Increased HVA is an index of increased DA activity. However, the data on Monoamine Oxidase B, the enzyme that metabolizes DA, point to either an increase in activity [893] or a decrease in activity [894–895].
Mary Solanto [896] dared to speculate that ADHD was a consequence of hyper-dopaminergic activity and that methylphenidate was working not to increase DA levels postsynaptically, but rather to stimulate autoreceptors to provide feedback stopping DA activity (via various mechanisms such as synthesis inhibition and release inhibition). Autoreceptors are approximately 10 times more sensitive than their postsysnaptic counterparts, and therefore small doses can preferentially activate them [897]. Thus low doses stimulate the autoreceptor, turning off DA activity and associated behavior in animals [898], and reducing activity in children with a subclinical dose of methylphenidate [899]. In a subsequent elaboration of the hyperdopaminergic hypothesis, the effects of methylphenidate were thought to operate differently in the various DA neurocircuits [900]. Here the nigrostriatal pathway responds to methylphenidate-induced increases in DA by activating autoreceptors which are overactive. However, in the mesocortical DA circuit where there are no autoreceptors, the DA accumulation in response to methylphenidate treatment activates postsynaptic receptors.
According to Seeman and Madras [901–902], during normal nerve activity, tonic DA levels transiently rise. At the low therapeutic doses used to treat ADHD, psychostimulants reduce motor activity. These drugs raise tonic levels of DA several-fold, but reduce the extent to which dopamine is released with action potentials (phasic). Thus the signals from stimuli are smaller and result in less activation of post-synaptic DA receptors which underlie motor activity. In addition, the elevated tonic DA reduces the number of DA receptors. At high doses the stimulants activate the nervous system, owing to the very high concentrations of tonic DA and the increased release of phasic DA. These high levels of DA stimulate postsynaptic DA receptors, thereby negating presynaptic inhibition.
Thus a division of whether ADHD is a result of too little or too much DA is overly simplistic. The devil is in the detail of DA activity, and one must remember these are theories, not conclusive answers.
Grace’s Tonic and the Phasic Account of ADHD
The importance of DA in ADHD can be seen in Grace’s theory of ADHD. Professor Anthony Grace provides an elaborate account of methylphenidate’s action and ADHD itself [722]. Like Seeman and Madras, Grace’s account focuses on the autoreceptors which are presynaptic and regulate the presence of DA in the synapse. According to Grace, three types of dopaminergic autoreceptors are involved: (1) firing-rate-inhibiting autoreceptors (stopping the action potentials and the signal); (2) synthesis-inhibiting autoreceptors (stopping the DA being made); and (3) release-inhibiting autoreceptors (stopping the DA being released). All have the net effect of reducing dopaminergic transmission using negative feedback loops when activated. Furthermore, Grace describes two types of DA release: tonic and phasic release. Tonic DA release is the low constant background level of DA in the synapse, which is modulated by glutamatergic projections from the cortex. In contrast, phasic release consists of a large and transient discharge of DA from the synapse as a result of an action potential [903].
Grace [722] proposes that methylphenidate has a two-stage action to its therapeutic efficacy. Firstly, there is a short-term effect due to the immediate blockade of the DAT, with the resultant accumulation DA after phasic release. This effect is considered to be responsible for the reward and addiction (see chapter 9). Secondly, the methylphenidate-induced increases in DA are unable to escape via the DAT, which then goes on to elevate tonic levels of DA. This activates autoreceptors, leading to feedback inhibition and a decrease in phasic DA release. This is the synapse responding to maintain an equilibrium or homeostasis.
Grace states that the two stages of methylphenidate’s action explain why psychostimulants result in an increase in symptoms’ severity immediately after administration [749, 841]. The good news is that oral administration of methylphenidate minimizes the early accumulation of DA, but goes on to alter autoreceptor activity, bringing about a therapeutic change.
From the actions of methylphenidate on DA, Grace accounts for the actual symptoms of ADHD. Essentially the symptoms of ADHD are directly a result of an abnormally low level of tonic DA within the striatum and nucleus accumbens, which leads to increased phasic responses. The variability in tonic release and consequential changes in phasic release are arguably linked to symptom severity, so much so that methylphenidate works best when the severity of the symptoms is more pronounced [904–905]. Furthermore, Grace argues that this may explain the rate-dependency hypothesis of amphetamine’s and methylphenidate’s paradoxical effect in ADHD, in which high levels of activity are depressed and low levels of activity are increased (see later and also [904, 906–907].
Such a role of tonic and phasic DA has been supported by mathematical models, which have gone on to further emphasize a modulation of stimulus input in the amelioration of ADHD [908].
It is of note that Grace has used the notions of tonic and phasic DA release to address disorders such as schizophrenia and addiction [909–910]. This is not to say that schizophrenia is the same as ADHD – clinically and psychopharmacologically it is different – but the mechanisms of synaptic modulation that can vary in different regions of the brain for each disorder can thus be used to hypothesize on the development of the disorders without recourse to weaken the other. In fact this gives greater strength to the theory that differences within the dopamine framework can yield different disorders.
A Dynamic Developmental Theory of ADHD
This theory of ADHD stems from rat models and is pioneered by Sagvolden and colleagues [911]. The theory centers on DA circuits, because that is where the evidence points. The view is of a general under-activity or hypofunctioning of DA circuits and a failure to modulate other neurotransmitters such as GABA and glutamate. These circuits are the mesolimbic, mesocortical, and nigrostriatal DA pathways. They have all had particular behaviors associated with them and can be differentially affected in ADHD. A dysfunctioning mesolimbic dopamine circuit will alter reinforcement of behavior and produce deficient extinction of previously reinforced behavior. This will manifest as delay aversion, hyperactivity, impulsiveness, inattention, and a failure in behavioral inhibition – all cardinal symptoms of ADHD. Dysfunction in the mesocortical dopamine circuit will cause attentional problems such as poor orienting responses, impaired saccadic eye movements, and poor executive functions. A dysfunctioning nigrostriatal dopamine circuit will cause impaired modulation of motor functions and deficient habit learning and memory, which will be clinically manifest as clumsiness and a failure to inhibit responses when quick reactions are required. All of these circuits are discussed by other in the context of the cortical regions that they connect to (see chapter 6).
According to the theory, ADHD is a manifestation of differential dysfunction in these circuits. Central to the theory is the disruption of reinforcement in ADHD; from this the other symptoms stem. It also argues that ADHD is not a distinct disorder with qualitatively distinct behavior patterns, but rather is a case where the function of DA circuits deviates from normal variation. Psychostimulants would be seen to redress the balance of DA. From the developmental perspective, Sagvolden et al. suggest that early in development over-activity of mesolimbic dopamine neurons could activate DA receptors and have the knock-on effects of increasing glutamate receptors in the mesolimbic circuit. Increased glutamatergic activity could result in compensatory changes that would result in deactivation of dopamine neurons and hypoactivity of the mesolimbic dopaminergic system. This shares some similarities with Grace’s theory above.
The theory has received some criticism. Not all agree with the central position of a dysfunctional mesolimbic circuit and the lack of acknowledgment of the contribution of hyopfunctioning frontal cortical areas. Perhaps because the authors come from a perspective based on animal models of behavior, they have provided a reductionistic view of ADHD without evaluation of the heterogeneity and variability of the disorder. One must note that the theory addresses only the ADHD-H and -C subtypes and not the ADHD-I subtype.
The Rate-Dependent Hypothesis and the Effects of Psychostimulants
It is bizarre that a drug with a street name of “speed” should be effective in treating ADHD. One would expect methylphenidate and amphetamine to exacerbate the symptoms of the disorder. However, these drugs have been argued to work differently in those with ADHD. What is the difference? The difference in ADHD is the baseline of activity prior to treatment: amphetamine when given to someone with high levels of activity will show a reduction, whereas someone with a low level of activity may show activation. This is called the rate-dependent hypothesis.
The rate-dependent hypothesis of drug effects changed our preconceptions of how drugs work. We are familiar with the concept that drugs bring about an effect and that the drug’s pharmacology is central to the behavior. However, the effects of a drug are not exclusively determined by pharmacological factors, but also by environmental and behavioral factors independent of the drug.
When there are low rates of activity, psychostimulants were shown to increase behaviors (in the early studies pigeons were used), whereas when the rates of activity were high, they reduced the behavior [912]. Similar effects were seen in humans [913]. Numerous studies have built on this work and refined the rate-dependent hypothesis. It has been proposed that rate-dependency may account for the effects of methylphenidate and amphetamine in ADHD [904, 914]. There is some evidence to support a rate-dependent effect of ADHD drugs, but these have been in laboratory tasks [915–917]. However, comparisons between groups of people with and without ADHD have not provided support for rate-dependency, where one might expect to see a suppressant effect in ADHD and an activating effect in those without the disorder [880, 918–921].
A critic of the rate-dependent hypothesis, Professor Jim Swanson [922], argues that looking at behavior between groups of people was not a suitable test of the rate-dependent hypothesis. He suggested that the same people should be measured on both high and low baselines of behavior. A second criticism of rate-dependent studies when looking at differences between groups is they are in fact showing a regression-to-the-mean. This is when people are subjected to more than one test and show a lower level of responding on the second test. Given that this happens, then the rate-dependent hypothesis needs to show an even stronger effect for it to be considered. In a set of experiments by Teicher et al., these two criticisms were addressed. The rate-dependent effect was seen in response to methylphenidate on certain, but not all, measures of attention [923].
An interesting study would be to look at the rate-dependent effect of methylphenidate across the different severities and subtypes of ADHD. One might be able to predict that those with a hyperactive component would benefit most. Although such a study has yet to be conducted, differential effects of methylphenidate have been shown in subgroups with ADHD-C, the most common showing that increasing doses of stimulant medication were associated with increased improvement of inattention and hyperactivity symptoms, whereas in those with ADHD-I, symptoms improved with lower doses, with less benefit seen at higher doses [924].
Finally, methylphenidate is used for sleeping disorders such as narcolepsy. One might consider this to be at the opposite end to ADHD along a hyperactivity continuum. Thus the effects of these drugs may well be dependent upon the activity levels at baseline. But the question remains, “what is the neurophysiology of these baseline states?” The neurophysiological changes may be key to this effect and be accounted for by theories such as those proposed by Grace [722] and Carlsson [925].
Noradrenaline
Whilst dopamine remains center-stage in theories of ADHD, noradrenaline also has a role. However, that role has not been as extensively studied as dopamine. That may change with the introduction of atomoxetine in the treatment of ADHD. Some scientists have placed a greater emphasis on noradrenaline than dopamine (e.g. [737, 926]) and this will be discussed in chapter 8.
Summary
There are surprisingly few treatments for ADHD. Those that are available result in moderate improvements, but also come with a range of side-effects that can be problematic, and the long-term (more than three years) effects of these drugs still need to be established. The value of ADHD medication goes beyond treatment. As drugs’ pharmacological mechanism of action is known, they have provided a great deal of direction in attempts to understand ADHD. The predominant hypothesis is that of a dysfunctional dopamine system in the disorder. The exact nature of the deficit is yet to be determined, with some diametrically opposite hypotheses in existence. The view from psychopharmacology is follow the drug and the pathology of ADHD will emerge. Herein lies a problem with such research: the hypothesis is only as good as the drug(s), and with few available, this type of inquiry is all the more difficult. New drugs with different mechanisms of action will be crucial in elucidating the whole area of the neurochemistry of ADHD.
Notes
1 http://www.civitas.org.uk/pubs/prisonValue.php.
2 http://www.nice.org.uk/guidance/CG72.
3 http://www.rcpsych.ac.uk/training/cpdandrevalidation/adhd/therapy/methylphenidate/clinicaleffectiveness/child.aspx.
4 http://www.nimh.nih.gov/science-news/2007/improvement-following-adhd-treatment-sustained-in-most-children.shtml.