CHAPTER 2
Structure and Function of the Skin
John A. McGrath1 and Jouni Uitto2
1St John's Institute of Dermatology, Division of Genetics and Molecular Medicine, Faculty of Life Sciences and Medicine, King's College London, London, UK
2Department of Dermatology and Cutaneous Biology, Thomas Jefferson University, Philadelphia, PA, USA
Components of normal human skin
Skin is the largest organ in the body. In a 70 kg individual, the skin weighs over 5 kg and covers a surface area approaching 2 m2. Human skin consists of a stratified, cellular epidermis and an underlying dermis of connective tissue, separated by a dermal–epidermal basement membrane (Figure 2.1). Beneath the dermis is a layer of subcutaneous fat, which is separated from the rest of the body by a vestigial layer of striated muscle.
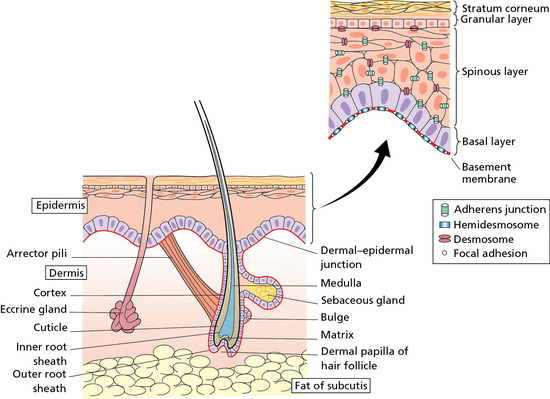
Figure 2.1 The skin and its appendages.
The epidermis is mainly composed of keratinocytes and, for the living cell layers, is typically 0.05–0.1 mm in thickness. It is formed by the division of cells in the basal layer, which give rise to the spinous layer. This layer contains cells that move outwards and progressively differentiate, forming the granular layer and the stratum corneum. The cellular progression from the basal layer to the skin surface takes about 30 days but is accelerated in diseases such as psoriasis. The ‘brick-like’ shape of keratinocytes is provided by a cytoskeleton made of keratin intermediate filaments. As the epidermis differentiates, the keratinocytes become flattened. This process involves the filament aggregating protein, filaggrin, a protein component of keratohyalin granules. Indeed, keratin and filaggrin comprise 80–90% of the mass of the epidermis.
The outermost layer of the epidermis is the stratum corneum, where cells (now called corneocytes) have lost the nuclei and cytoplasmic organelles. The corneocyte has a highly insoluble, cornified envelope within the plasma membrane, formed by cross-linking of soluble protein precursors, including involucrin and loricrin, the latter contributing 70–85% to the mass of the cornified cell envelope. It also contains several lipids (fatty acids, sterols and ceramides) released from lamellar bodies within the upper, living epidermis. The stratum corneum can be divided into three distinct biochemical and functional zones – an outer absorber of solutes, a middle absorber of water for hydration, and an inner mechanical defence barrier.
Other cells in the epidermis are the melanocytes, Langerhans cells and Merkel cells. Melanocytes are dendritic cells that distribute packages of melanin pigment in melanosomes to the surrounding keratinocytes to give skin its colour. The number of melanocytes does not differ much between skin types. Rather it is the nature of the melanin and the size of the melanosomes that account for the different appearances. The Langerhans cells are also dendritic in nature, although these are of mesenchymal origin and originate from bone marrow. Langerhans cells are antigen-presenting cells and process antigens encountered by the skin to local lymph nodes and thus have a key role in adaptive immune responses in the skin. Merkel cells are probably derived from keratinocytes. They have a role as mechanosensory receptors in response to touch.
Human skin contains pilosebaceous follicles and sweat glands. The hair follicles comprise pockets of epithelium that are continuous with the superficial epidermis but which also envelop a small papilla of dermis at their base. A bundle of smooth muscle, the arrector pili, extends at an angle between the surface of the dermis and a point in the follicle wall. Above the insertion, there are holocrine sebaceous glands which open into the pilary canal. In some sites, such as the axillae, the follicles may be associated with apocrine glands. Also derived from the epidermis and opening directly to the skin surface are the eccrine sweat glands.
The epidermis is attached to the dermis via a complex network of proteins and glycoproteins that extend from inside basal keratinocytes into the superficial dermis. Besides adhesion, the dermal–epidermal junction components also contribute to cell migration (for example during wound healing) as well as epithelial–mesenchymal signalling events. Over 30 different macromolecules (collagens, laminins and integrins) interact within a basement membrane zone that is less than 200 μm across.
The dermis consists of a supporting matrix in which polysaccharides and proteins are enmeshed to a network that provides resilience to the skin and has a remarkable capacity for retaining water. The thickness of the dermis varies from less than 0.5 mm to more than 5 mm depending on the skin site. There are two principal types of protein fibre: collagen and elastic tissue.
Collagen is the major extracellular matrix protein, comprising 80–85% of the dry weight of the dermis. Twenty-nine different collagens have been identified in vertebrate tissue (depicted by Roman numerals in the order of their discovery, from I to XXIX), of which at least 12 are expressed in skin. The main interstitial dermal collagens are types I and III, whereas the principal basement membrane collagen (at the dermal–epidermal junction and around dermal blood vessels, nerves and appendages) is type IV collagen. Triple-helical collagen monomers polymerize into fibrils and fibres, which then become stabilized by the complex formation of both intra- and intermolecular cross-links. Collagen fibres are extremely resilient and provide skin with its tensile strength.
In sun protected adult skin, elastic fibres account for no more than 2–4% of the extracellular matrix in the dermis and consist of two components, elastin and elastin-associated microfibrils, which together give skin its elasticity and resilience. Elastic microfibrils are composed of several proteins, including fibrillin, which surround the elastin and which can extend throughout the dermis in a web-like configuration to the junction between the dermis and the epidermis. The dermis also contains a number of non-collagenous glycoproteins, including fibronectins, fibulins and integrins. These extracellular matrix components facilitate cell adhesion and cell motility.
Between the dermal collagen and elastic tissue is the ground substance made up of glycosaminoglycan/proteoglycan macromolecules. These contribute only 0.1–0.3% of the total dry weight of the dermis but provide a vital role by maintaining hydration, mostly due to the high water-binding capacity of hyaluronic acid. About 60% of the total weight of the dermis is water.
The dermis has a very rich blood supply, although no vessels pass through the dermal–epidermal junction. There is a superficial and a deep vascular plexus. The motor innervation of the skin is autonomic, and includes a cholinergic component to the eccrine sweat glands and adrenergic components to both the eccrine and apocrine glands, to the smooth muscle and the arterioles and to the arrector pili muscle. The sensory nerve endings are of several kinds; some are free, some terminate in hair follicles and others have expanded tips.
Skin development
The skin arises by the juxtaposition of two major embryological elements: the prospective epidermis that originates from a surface area of the early gastrula, and the prospective mesoderm that comes into contact with the inner surface of the epidermis during gastrulation. The mesoderm not only provides the dermis, but is essential for inducing differentiation of the epidermal structures, such as the hair follicle. The melanocytes are derived from the neural crest.
After gastrulation, there is a single layer of neuroectoderm on the embryo surface; this layer will go on to form the nervous system or the skin epithelium depending on the molecular signals it receives (Figure 2.2). Activation of Wnt signalling will block the ability of the ectoderm to respond to fibroblast growth factors (FGFs). Without FGFs the cells express bone morphogenic proteins (BMPs) and progress to an epidermal lineage. Conversely, lack of Wnt signalling promotes a neural fate [1]. One key transcription factor in skin development is p63, which contributes to epidermal lineage commitment, epidermal differentiation, cell adhesion and basement membrane formation [2]. The embryonic epidermis consists of a single layer of multipotent epithelial cells, which is covered by a special layer known as periderm that is unique to mammals (Figures 2.3 and 2.4). Periderm provides some protection to the newly forming skin as well as exchange of material with the amniotic fluid.
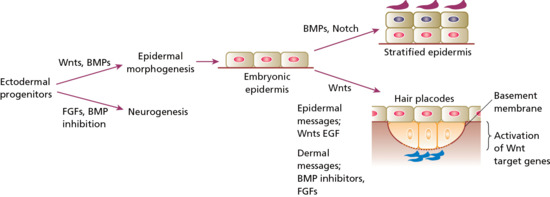
Figure 2.2 Embryonic development of the skin depends on specific signalling molecules. Relative stimulation or inhibition by these signalling molecules also determines whether embryonic epidermis progresses to a stratified epidermis or whether formation of skin appendages is induced. BMP, bone morphogenic protein; EGF, epidermal growth factor; FGF, fibroblast growth factor.
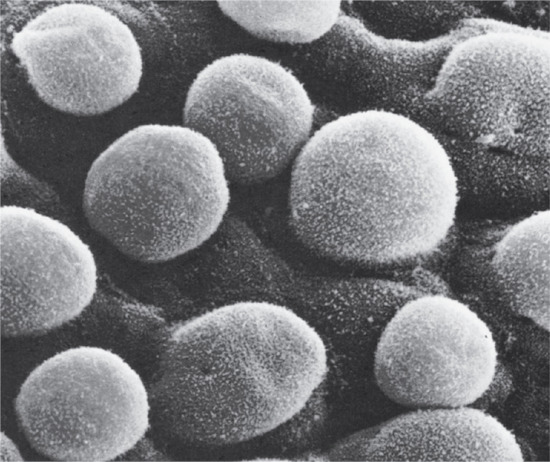
Figure 2.3 Scanning electron micrograph of an 85–110-day (estimated gestation age) human embryo. Single globular blebs project from the periderm cells.
(Courtesy of Professor K. A. Holbrook.)
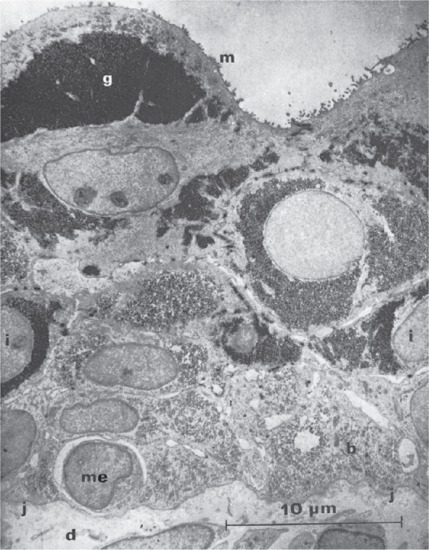
Figure 2.4 Electron micrograph of the full-thickness epidermis from the back of a 14-week human fetus. The periderm cells are full of glycogen (g) and have microvilli (m) at their amniotic border. Cells of the intermediate layer (i) also contain glycogen. Basal layer cells (b) have lost glycogen by this stage. Just above the dermal–epidermal junction (j) there is a melanocyte (me); the surrounding space indicates that it is a recent immigrant from the dermis (d). Osmium fixation and lead staining.
(Courtesy of Professor A. S. Breathnach.)
The embryonic dermis is at first very cellular, and at 6–14 weeks three types of cell are present: stellate cells, phagocytic macrophages and granule-secretory cells, either melanoblasts or mast cells. From weeks 14 to 21, fibroblasts are numerous and active, and perineural cells, pericytes, melanoblasts, Merkel cells and mast cells can be individually identified. Two distinct lineages of fibroblasts are present: one that gives rise to the upper dermis and hair follicle formation, and another that helps generate the deep dermis and subcutaneous fat [3].
The various structural components of the skin that can be recognized postnatally start to appear at different embryonic time points, for example hair follicles and nails (9 weeks), sweat glands (9 weeks for the palms and soles, 15 weeks for other sites) and sebaceous glands (15 weeks). Touch pads become recognizable on the fingers and toes by week 6 and reach their greatest development at week 15. After this, they flatten and become indistinct. It is these areas that determine the pattern of dermatoglyphics that take their place.
The mesoderm not only provides the dermis but is essential for inducing differentiation of the epidermal structures, such as the hair follicle in mammals [4]. The earliest development of the hair rudiments occurs at about 9 weeks in the regions of the eyebrow, upper lip and chin (Figure 2.5). Mesenchymal cells, derived from the dermomyotome, populate the skin and interact with the overlying epidermis to induce the formation of hair placodes [5]. Key components of the mesenchymal signals to produce hair follicles include FGFs and BMP-inhibitory factors such as Noggin; excessive BMP stimulation can reduce hair follicle density. The epidermal response to form the hair placode is generated by Wnt signals such as Wnt10b and sonic hedgehog (Shh), which also has a key role in the formation of the dermal papilla [6]. After it is formed, the dermal papilla sends further signals to transform the placode into a hair follicle. At the centre of the signalling cross-talk is the bipartite transcription factor composed of lymphoid enhancer-binding factor 1 (LEF1) and stabilized β-catenin, which is essential for hair follicle formation. Hair follicle development is also influenced by Smads, a group of signalling mediators and antagonists of the transforming growth factor β (TGF-β) superfamily. Smad-4 affects hair follicle differentiation by mediating BMP signalling; Smad-7 affects hair follicle development and differentiation by blocking TGF-β/Activin/BMP pathways [7]. Skin development is governed by complex, balanced waves of gene activation and silencing; cross-talk between small non-coding micro-RNAs and messenger RNAs is very important for the coordination of signal transduction and transcriptional activation [8].
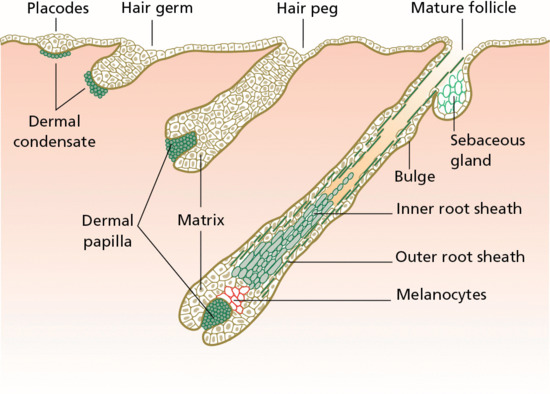
Figure 2.5 Embryonic stages of hair follicle morphogenesis.
Signalling responses differ between follicular and interfollicular epidermis: BMP signalling is active in the interfollicular epidermis and is both an epidermis-promoting signal as well as a follicle-inhibiting signal; epidermal growth factor receptor (EGFR) signalling may have a similar role in governing follicle density. As hair follicles mature to form inner and outer root sheaths, several signalling pathways are involved, including Wnt, Notch and BMP receptors. There are also marked changes in certain cell adhesion proteins, notably E-cadherin and P-cadherin. The hair follicles are arranged in patterns, usually in groups of three. It appears that the first follicles develop over the surface at fixed intervals of between 274 and 350 μm. As the skin grows, these first hair germs become separated, and new rudiments develop between them when a critical distance, dependent on the region of the body, has been reached. There is no large-scale destruction of follicles during postnatal development, only a decrease in actual density as the body surface increases; nor do any new follicles develop in adult skin. In interfollicular epidermis, the undersurface of the epidermis is smooth, but during the fourth month, at the same time as the hair follicle starts to develop, it becomes irregular.
Sebaceous glands first appear as hemispherical protuberances on the posterior surfaces of the hair pegs. The cells contain moderate amounts of glycogen, but soon the cells in the centre lose this, and become larger and accumulate droplets of lipid. The sebaceous glands become differentiated at 13–15 weeks, and are then large and functional. The sebum forms part of the vernix caseosa. At the end of fetal life, sebaceous glands are well developed and generally large. After birth, the size is rapidly reduced, and they enlarge to become functional again only after puberty. The molecular signals that induce sebaceous gland differentiation involve the c-Myc transcription factor as well as the adipogenic transcription factor peroxisome proliferator-activated receptor γ (PPAR-γ) [9].
Eccrine glands start to develop on the palms and soles at about 3 months, but not over the rest of the body until the fifth month [10]. In embryos of 12 weeks, the rudiments of eccrine sweat glands are first identifiable as regularly spaced undulations of the developing epidermis. Cells that go on to form the eccrine sweat glands are oblong, palisading and lie closely together, but otherwise they do not differ from the rest of the developing basal epidermis. By 14–15 weeks, the tips of the eccrine sweat gland rudiments have penetrated deeply into the dermis, and have begun to form the coils. In the overlying epidermis, columns of cells that are destined to form the intraepidermal sweat ducts are recognizable. Each column is composed of two distinct cylindrical layers, comprising two inner cells that are elongated and curved so that they embrace the inner cylinder. The intraepidermal duct appears to form by the coalescence of groups of intracytoplasmic cavities formed within two adjacent inner cells. In the intradermal segment, the lumen forms by dissolution of the desmosomal attachment plaques between the cells that compose the inner core of the eccrine duct germ.
Nails begin to develop in the third month. Key signalling events in nail development involve the R-spondin family of transcription factors [11]. In fetuses at 16–18 weeks (crown to rump length 120–150 mm), keratinizing cells from both dorsal and ventral matrices can be distinguished. Melanocytes take their origin from the neural crest. This can be identified in early human embryos, but the elements arising from it soon lose themselves in the mesenchyme, and pigmented melanocytes cannot be identified, even in darker skin fetuses, before 4–6 months of gestation. However, dopa-positive melanocytes can be demonstrated earlier. Langerhans cells are derived from the monocyte–macrophage–histiocyte lineage and enter the epidermis at about 12 weeks. Merkel cells appear in the glabrous skin of the fingertips, lip, gingiva and nail bed, and in several other regions, around 16 weeks.
Although some cells of the dermis may migrate from the dermatome (ventrolateral part of the somite) and take part in the formation of the skin, most of the dermis is formed by mesenchymal cells that migrate from other mesodermal areas [12]. These mesenchymal cells give rise to the whole range of blood and connective tissue cells, including the fibroblasts and mast cells of the dermis and the fat cells of the subcutis. In the second month, the dermis and subcutis are not distinguishable from each other but distinct collagen fibres are evident in the dermis by the end of the third month. Later, the papillary and reticular layers become distinct and, at the fifth month, the connective tissue sheaths are formed around the hair follicles. Elastic fibres are first detectable at 22 weeks.
Epidermal and adnexal structures
The normal epidermis is a terminally differentiated, stratified, squamous epithelium. The major cell type, making up 95% of the total, is the keratinocyte, which moves progressively from attachment to the epidermal basement membrane towards the skin surface, forming several well-defined layers during its transit [1]. Thus, on simple morphological grounds, the epidermis can be divided into four distinct layers: stratum basale or stratum germinativum, stratum spinosum, stratum granulosum and stratum corneum. The term Malpighian layer includes both the basal and spinous cells. Other constitutive cells within the epidermis include melanocytes, Langerhans cells and Merkel cells (Figure 2.6).
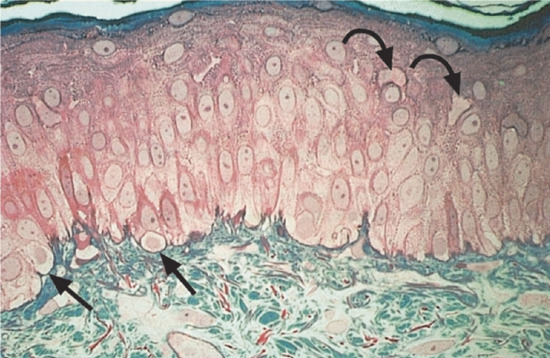
Figure 2.6 Photomicrograph of a 1 μm-thick plastic section of normal human skin. The tissue was fixed with half-strength Karnovsky medium and embedded in Epon. This technique allows the cellular components of the epidermis, including keratinocytes, melanocytes (straight arrows) and probable Langerhans cells (curved arrows) to be clearly resolved. Magnification 400× (basic fuchsin and methylene blue).
(Courtesy of Professor R. A. J. Eady, St John's Institute of Dermatology, King's College London, UK.)
The stratum basale is a continuous layer that is generally only one cell thick but it may be two to three cells thick in glabrous skin and in hyperproliferative epidermis. The basal cells are small and cuboidal (10–14 μm in diameter) and have large, dark-staining nuclei, and dense cytoplasm that contains many ribosomes and dense tonofilament bundles. Immediately above the basal cell layer, the epibasal keratinocytes enlarge to form the spinous/prickle cell layer or stratum spinosum (Figure 2.7).
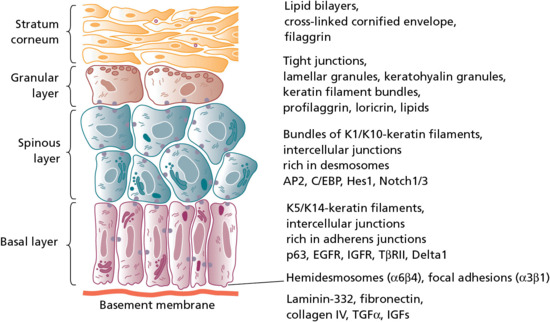
Figure 2.7 The process of epidermal differentiation is associated with the expression of different structures, macromolecules, transcription factors and other signalling molecules and their receptors in the different keratinocyte layers. EGFR, epidermal growth factor receptor; IGF, insulin-like growth factor; IGFR, IGF receptor; TGF, transforming growth factor.
The stratum spinosum is succeeded by the stratum granulosum or granular layer, which contains intracellular granules of keratohyalin. At high magnification, the dense mass of keratohyalin granules from human epidermis has a particulate substructure, with particles of irregular shape, on average 2 nm in length, and occurring randomly in rows or lattices. The cytoplasm of cells of the upper, spinous layer and granular cell layer also contains smaller lamellated granules averaging 100–300 nm in size. These are known as lamellar granules or bodies, membrane-coating granules or Odland bodies. They are numerous within the uppermost cells of the spinous layer and migrate towards the periphery of the cells as they enter the granular cell layer (Figure 2.8). They discharge their lipid components into the intercellular space, playing important roles in barrier function and intercellular cohesion within the stratum corneum (Figure 2.9) [2].
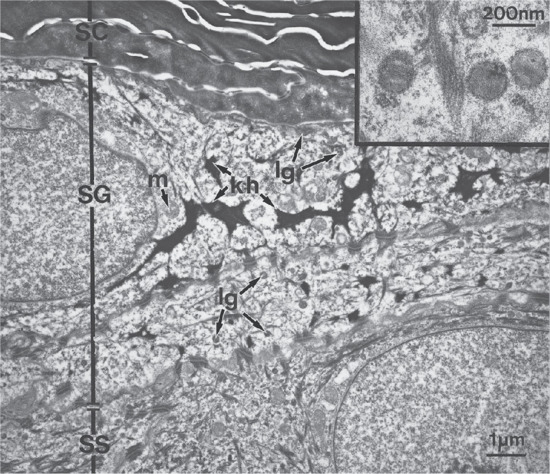
Figure 2.8 Electron micrograph showing details of the upper part of the epidermis including the stratum corneum (SC), stratum granulosum (SG) and most superficial cell layer of stratum spinosum (SS). Note the irregularly shaped keratohyalin granules (kh) and the small, round, lamellar granules (lg). The latter are present in both SS and SG and are smaller than mitochondria (m). The inset shows details of lamellar granules.
(Courtesy of Professor R. A. J. Eady, St John's Institute of Dermatology, King's College London, UK.)
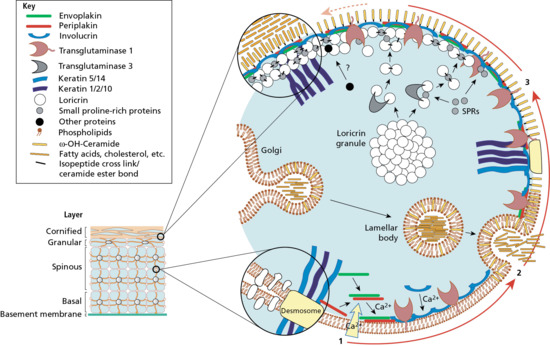
Figure 2.9 Assembly of the epidermal cornified cell envelope. In response to increasing intracellular calcium, an internal scaffold of desmosomal proteins is made along the plasma membrane. The contents of lamellar bodies (ceramides and other fatty acids, cholesterol and cholesterol esters) are released into the extracellular milieu to form a lipid membrane. The developing envelope is then added to and reinforced by the recruitment of various proteins, including loricrin, small proline-rich proteins (SPRs), other desmosomal remnants and attached keratin filaments. The resulting cornified cell envelope is durable and flexible and provides important mechanical and barrier functions.
The outermost layer of epidermis is the stratum corneum where cells, now known as corneocytes, have lost the nuclei and cytoplasmic organelles. The cells become flattened and the keratin filaments align into disulphide cross-linked macrofibres, under the influence of filaggrin, the protein component of the keratohyalin granule. Filaggrin is responsible for keratin filament aggregation, and is subsequently broken down into individual hygroscopic amino acids that form the basis of the natural moisturizing factor within corneocytes. The key role of filaggrin in skin biology has been demonstrated by the discovery of common loss-of-function mutations in the filaggrin gene as the cause of the genetic disorder ichthyosis vulgaris and as a major risk factor for the development of atopic eczema, atopic asthma and systemic allergies [3]. The corneocyte has a highly insoluble, cornified envelope within the plasma membrane, formed by cross-linking of the soluble protein precursor, involucrin, following the action of a specific epidermal transglutaminase also synthesized in the high stratum spinosum (Figure 2.10). Many of the proteins involved in terminal differentiation are derived from a cluster of about 25 genes located within a c. 2 Mb region on the long arm of chromosome 1. Termed the epidermal differentiation complex (EDC), these coding elements are derived from at least three families of structurally, functionally and evolutionarily related genes. Together, the EDC proteins have roles in structural integrity, signal transduction and cell cycle progression and may be primarily or secondarily disrupted in several inflammatory or neoplastic disorders.
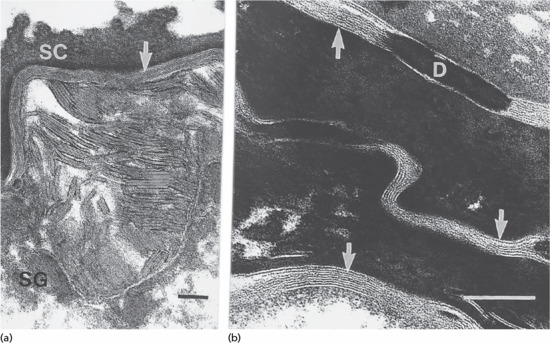
Figure 2.10 Electron micrograph showing the location of epidermal lipids by ruthenium oxide staining. (a) Extrusion of lamellar body lipids or sheets can be seen at the interface between the stratum granulosum (SG) and stratum corneum (SC). Scale bar 0.1 μm. (b) Sheets of lipid bilayers (arrowed) are present in the intercellular spaces of the SC. Some regions show a repetitive pattern of staining. D, desmosome. Scale bar 0.1 μm.
(Courtesy of Dr M. Fartasch, Department of Dermatology, University of Erlangen, Germany.)
The process of desquamation involves degradation of the lamellated lipid in the intercellular spaces and loss of the residual intercellular desmosomal interconnections [4]. In palmoplantar skin there is an additional zone, also electron-lucent, the stratum lucidum, between the granulosum and corneum. These cells are still nucleated, and may be referred to as ‘transitional’ cells.
Keratinocytes
The filamentous cytoskeleton of all mammalian cells, including epidermal keratinocytes, comprises: actin-containing microfilaments approximately 7 nm in diameter; tubulin-containing microtubules 20–25 nm in diameter; and filaments of intermediate size, 7–10 nm in diameter, known as intermediate filaments. There are six types of intermediate filaments: keratins in epithelial cells; vimentin within mesenchymal cells; glial filament acidic protein (GFAP) in glial cells; neurofilaments in neurons; desmin in muscle cells; and peripherin in peripheral nerves. The nuclear matrix proteins, nuclear lamins A, B and C, are also intermediate filaments. The polypeptide building blocks of all intermediate filaments have a similar backbone structure of a classic α-helical region with heptad repeats, having four separate helical zones with interhelical linker sequences, and non-helical carboxy- and amino-terminals. There are 70 intermediate filament genes (including those encoding keratins, desmins and lamins), which are now known to be associated with at least 72 distinct human diseases, including skin blistering, muscular dystrophy, cardiomyopathy, premature ageing syndromes, neurodegenerative disorders and cataract [1, 2].
The human genome possesses 54 functional keratin genes located in two compact gene clusters, as well as many non-functional pseudogenes scattered around the genome [3]. Keratin genes are very specific in their expression patterns. Each one of the many highly specialized epithelial tissues has its own profile of keratin gene expression. Hair and nails express modified keratins, containing large amounts of cysteine which forms numerous chemical cross-links to further strengthen the cytoskeleton. The genes encoding individual keratins fall into two gene families: type I (basic) and type II (acidic). Mapping the tissue distribution of keratins shows co-expression of particular acidic–basic pairs in a cell- and tissue-specific manner. Heterodimers are assembled into higher order protofibrils and protofilaments by an antiparallel stagger of some complexity.
Simple epithelia are characterized by the keratin pair K8/K18, and the stratified squamous epithelia by K5/K14 (Figure 2.11). In addition, stratified squamous epithelia express up to four other keratin pairs during epithelial differentiation. In skin, suprabasal keratins K1/K10 are characteristic of epidermal differentiation. In the stratum granulosum, release of filaggrin from the keratohyalin granules forms macrofibres. Retinoid levels, growth factors and hormones may regulate keratin gene expression. Mesenchymal signals may also direct or permit intrinsic patterns of keratinocyte differentiation. K15 is expressed in basal keratinocytes of the hair follicle bulge region at the site of pluripotential stem cells. K9 and K2 expression is site restricted in skin: K9 to the palmoplantar epidermis and K2 to the superficial interfollicular epidermis. Apart from their structural properties, keratins may also have direct roles in cell signalling, the stress response and apoptosis [4]. In epidermal hyperproliferation, as in wound healing and psoriasis, the expression of suprabasal keratins K6/K16/K17 is rapidly induced.
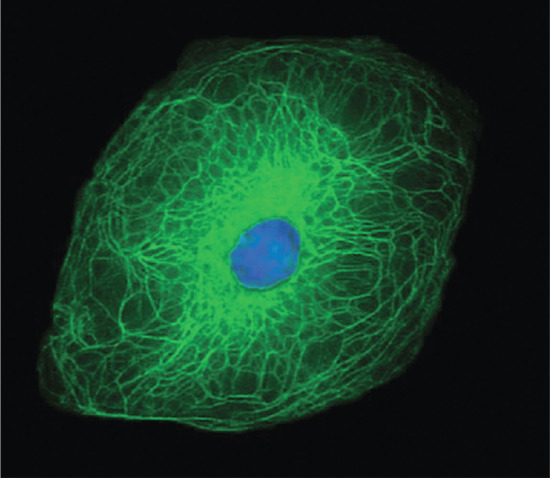
Figure 2.11 Structural organization of the keratin filament network within a keratinocyte.
(Courtesy of Professor W. H. I. McLean, University of Dundee, UK.)
Currently, 21 of the 54 known keratins (28 type I, and 26 type II) have been linked to monogenic genetic disorders, and some have been implicated in more complex traits, such as idiopathic liver disease or inflammatory bowel disease [5]. The first genetic disorder of keratin to be described was epidermolysis bullosa simplex, which involves mutations in the genes encoding K5 or K14. About half of the 54 keratin genes are expressed in the hair follicle (trichocyte ‘hard’ keratins), although only four of these have been linked to human genetic disorders (monilethrix, hair–nail ectodermal dysplasias, pseudofolliculitis barbae and woolly hair) [6].
Eccrine and apocrine glands
Human sweat glands are generally divided into two types: apocrine and eccrine [1]. The eccrine gland is the primary gland responsible for thermoregulatory sweating in humans. Eccrine sweat glands are distributed over nearly the entire body surface. Sweat glands become identifiable in the palms and soles in the 16th fetal week, and in the rest of the body from the 22nd week onwards. The number of sweat glands in humans varies greatly, ranging from 1.6 to 4.0 million. The structure of the eccrine sweat gland consists of a bulbous secretory coil leading to a duct. The secretory coil is located in the lower dermis, and the duct extends through the dermis and opens directly onto the skin surface. The active sweat glands are present most densely on the sole, forehead and palm, somewhat less on the back of the hand, still less on the lumbar region and the lateral and extensor surfaces of the extremities, and least on the trunk and the flexor and medial surfaces of the extremities. The uncoiled dimension of the secretory portion of the gland is approximately 30–50 μm in diameter and 2–5 mm in length. The size of the adult secretory coil ranges from 1 to 8 × 10−3 mm3.
Human perspiration is classified into two types: insensible perspiration and active sweating. Insensible perspiration involves water loss from the respiratory passages and the skin. In the skin, the epidermis is supplied with water originating from blood in the skin microcirculation and interstitial spaces so that water can evaporate from its dry surface. Thus, the evaporation of water from the skin may depend on several environmental factors, such as ambient temperature and ambient humidity. Heat, mental stimuli and muscular exercise can all induce active sweating in human beings. Active sweating may be classified into two types: thermal and mental/emotional. Thermal sweating plays an important role in keeping the body's temperature constant and involves the whole body surface [2]. Mental or emotional sweating usually appears on the palms and soles. The physiological features of mental sweating differ considerably from those of thermal sweating. Mental sweating has a shorter latent period for its onset and immediately attains a certain rate of secretion that corresponds to the intensity of stimulation, remaining for the duration of the stimulation and subsiding quickly after it ends [3]. Eccrine glands contribute to both types of sweating.
The secretory nerve fibres innervated in human sweat glands are sympathetic, and seem to be cholinergic in character as sweating is produced by pilocarpine and stopped by atropine [4]. Vasoactive intestinal peptide (VIP) coexisting in the cholinergic nerve fibres may act as a candidate neurotransmitter to control the blood circulation of the sweat glands. The sudorific nervous system is also separated into parts for thermal and emotional sweating, each being controlled by its own regulatory centre in the brain that is associated with the sweat glands in its respective region of the skin. The exact neurological pathways responsible for sweating are not entirely understood.
Sympathetic nerve terminals cluster mainly around the secretory coil of the sweat gland, but a few projections extend to the sweat duct. Acetylcholine is the primary neurotransmitter released from cholinergic sudomotor nerves and binds to muscarinic receptors on the eccrine sweat gland, although sweating can also occur via exogenous administration of α- or β-adrenergic agonists. Released acetylcholine is rapidly hydrolysed by acetylcholinesterase, and this response may be one of a number of mechanisms by which the rate of sweating rate is regulated.
When acetylcholine binds to muscarinic receptors on the sweat gland, intracellular Ca2+ concentrations increase. This results in an increase in the permeability of K+ and Cl− channels, which initiates the release of an isotonic precursor fluid from the secretory cells [5]. This precursor fluid is similar to plasma but is devoid of proteins. As the fluid travels up the duct toward the surface of the skin, sodium and chloride are reabsorbed, resulting in sweat on the surface being hypotonic relative to plasma. When the rate of sweat production increases, however, for example during exercise or heat stress, ion reabsorption mechanisms can be overwhelmed due to the large quantity of sweat secreted into the duct, resulting in higher ion losses. The sodium content in sweat on the skin's surface, therefore, is greatly influenced by sweat rate.
Apart from eccrine glands, the skin also contains apocrine sweat glands [6]. Eccrine glands do not show cytological changes during secretion, whereas apocrine glands are characterized by decapitation secretion, in which part of the cell is pinched off and released into the lumen. Apocrine glands are located only in genital, axillary and mammary areas, where they are always connected to a hair follicle. Apocrine glands have a low secretory output and hence no significant role in thermoregulation. They are composed of a coiled secretory portion and an excretory duct. The inner layer of the secretory portion contains a single columnar secretory cell type containing numerous, large, dense granules located at the apical aspect, which contribute to the lipid-rich secretion produced. The inner layer is also surrounded by a fenestrated layer of myoepithelial cells but the lumen is generally larger in diameter than that present in eccrine tissue. The apocrine excretory duct does not have any known reabsorptive function and consists of a double layer of cuboidal cells that merge distally with the epithelium of the hair follicle, resulting in emptying of the secretion into the hair follicle. The exact role of apocrine glands in humans is unknown. A third type of intermediate sweat glands, the apoeccrine glands, has also been described in axillary skin but their existence is not universally accepted.
Pilosebaceous unit
The pilosebaceous units develop from epidermal down-growths under the influence of specific mesenchymal cell condensations between the 10th and 14th week estimated gestational age. They have complex groups of specialized cell layers with distinctive pathways of differentiation. There are four classes of pilosebaceous unit: terminal on the scalp and beard; apopilosebaceous in the axilla and groin; vellus on the majority of skin; and sebaceous on the chest, back and face. The dermal papilla is located at the base of the hair follicle with a rich extracellular matrix. Around the papilla are germinative (matrix) cells that have a very high rate of division, and give rise to spindle-shaped central cortex cells of the hair fibre, and the single outer layer of flattened, overlapping cuticle cells. A central medulla is seen in some hairs, with regularly stacked, condensed cells interspersed with air spaces or low-density cores. The cortical cells are filled with keratin intermediate filaments orientated along the long axis of the cell, interspersed with a dense interfilamentous protein matrix.
Terminal differentiation of cortical cells is associated with the appearance of a contiguous, laminated, intercellular layer, which appears critical for filament integrity. The cuticular cells are morphologically distinct; these are flattened, outward-facing cells, with three layers inside the cuticle of condensed, flattened protein granules: endocuticle, exocuticle and ‘a’ layer [1]. Around the cuticle is the inner root sheath (IRS), which is composed of three distinct layers of cells that undergo keratinization: the IRS cuticle, the Huxley layer and the outermost Henle layer [2]. Differentiation in the IRS involves the development of trichohyalin granules, with 8–10 nm filaments orientated in the direction of hair growth. The IRS moves up the follicle, forming a support for the hair fibre, and degenerates above the sebaceous gland. The outermost layer is the outer root sheath (ORS), which is continuous with the epidermis and expresses epithelial keratins K5/K14, K1/K10 and K6/K16 in the upper ORS and K5/K14/K17 in the deeper ORS.
Normal growth of the hair fibre is 300–400 μm/day, generated by the high rate of proliferation of progenitor cells in the follicle bulb. Compartmentalization within the bulb gives rise to the different layers within the follicle, with the majority of bulb cells forming the IRS. There are three phases of cyclical hair growth: anagen, when growth occurs; catagen, a regressing phase; and telogen, a resting phase (Figure 2.12). The follicle re-enters anagen, and the old hair is replaced by a new one.

Figure 2.12 There are three components to the hair cycle: anagen (where new hair forms and grows), followed by catagen (regressing phase) and telogen (resting phase), and then loss of old hair. The hair cycle is associated with discrete changes in hair follicle anatomy, both in the shape of the follicle and in the subjacent dermal papilla. IRS, inner root sheath; ORS, outer root sheath.
Immediately above the basal layer in the hair bulb, cells undergo a secondary pathway of ‘trichocyte’ or hair differentiation, and express a further complex group of keratins, the hard keratins [3]. Two families of hair keratins, types I and II, are present in mammals, which have distinctive amino- and carboxy-terminals with high levels of cysteine residues, and lack the extended glycine residues of epidermal keratins. The proteins differ from epithelial keratins in their positions on two-dimensional gels but they can be grouped into acidic and basic families; there are four major proteins in each of these families and several minor proteins, Ha 1–4 and Hb 1–4. Recent cloning of the hair keratin genes, which cluster on chromosomes 12 and 17, has shown a greater number of hair keratin genes, HaKRT1–6 (including 3.1 and 3.2) and HbKRT1–6. Mutations in hair keratin genes have been found to be causative for autosomal dominant forms of the human disease monilethrix. In addition, keratin 17 null mice also demonstrate varying degrees of alopecia, depending on the age and strain of the mice.
Over the last two decades, several naturally occurring, inherited human disorders of hair have provided fascinating insight into hair development and growth. These include key signalling molecules such as ectodysplasin, as well as transcription factors, including hairless and the vitamin D receptor, structural hair keratins, desmosomal proteins, a G protein-coupled receptor, a serine protease and a copper transporter [4]. More common hair variants, such as curly hair, may be explained by dynamic changes during hair growth [5]. Curvature of curly hair is programmed from the very basal area of the follicle and the bending process is linked to a lack of axial symmetry in the lower part of the bulb, affecting the connective tissue sheath, ORS, IRS and hair shaft cuticle.
Nails
The main purpose of the nail apparatus is to provide protection to the digit tips, enhance sensory discrimination, help increase dexterity, facilitate scratching or grooming, and, in some individuals, to function as a cosmetic accessory [1]. The earliest signs of finer nail development occur at 8–9 weeks’ gestation: there is an invagination of the primitive epidermis to form an uninterrupted groove delineating a flattened surface at the end of each digit, known as the nail field. A key transcription factor in nail initiation is R-spondin 4, mutations in which underlie congenital anonychia [2]. A group of cells from the proximal part of the nail fold then grows proximally into the digit, stopping approximately 1 mm from the phalanx and giving rise to the matrix primordium. This site will eventually contribute to the epithelium of the proximal nail fold as well as the distal and intermediate matrix epithelium. From the distal part of the nail fold, a visible mound of cells emerges on the dorsum of the distal tip of each digit, which is known as the distal ridge [3]. At 13 weeks’ gestation, the proximal nail fold is formed and the first signs of nail plate growth are observed from the lunula. At this stage, the stratum corneum and the stratum granulosum start to materialize from the nail field epithelium, beginning distally and advancing toward the proximal nail fold. At 18 weeks’ gestation, the granular layer recedes, and the nail bed epithelium takes on a postnatal appearance. Likewise, at 20 weeks’ gestation, the process of cellular differentiation and maturation within the matrix is similar to that seen in adult nails. By 32 weeks’ gestation, virtually all the components of the nail can be recognized. Toe nail development is similar to that of finger nails but the stages occur about 4 weeks later.
The nail unit is composed of the nail plate and four epithelial structures: the proximal nail fold, the matrix, the nail bed and the hyponychium (Figure 2.13) [4]. The nail plate is a rectangular, translucent and relatively inflexible structure, and contains calcium, phosphate, iron, zinc, manganese and copper, but it is mainly the sulphur within the nail matrix that is responsible for the nail plate's physical qualities. The nail plate arises from beneath the proximal nail fold and is bordered on both sides by the lateral nail folds. The proximal aspect may contain white semicircular areas called lunulae, which are the visible portions of the distal matrix [5]. The dorsal surface of the nail unit appears pink in colour because of the enhanced vasculature of the underlying nail bed. The proximal nail fold has a dorsal and a ventral epithelial surface. It is a continuation of the skin of each digit (the dorsal surface) that folds underneath itself, resting above the nail matrix (the ventral surface). The dorsal proximal nail fold is devoid of hair follicles, sebaceous glands and dermatoglyphic markings and the ventral proximal nail fold also lacks rete ridges. At the junction between the dorsal and ventral surfaces is the eponychium (cuticle) which protects the matrix from damage. The lateral nail folds are extensions of the skin surface of the sides of the digits and join the nail bed medially.
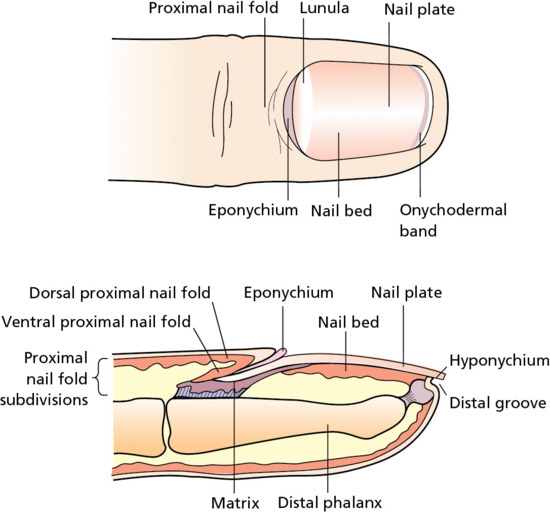
Figure 2.13 Anatomy and structure of the human nail.
The nail matrix forms the nail plate and is divided into three regions: the dorsal section of the matrix contributes to the most superficial layers of the nail plate, whereas the intermediate region of the matrix forms the deeper layers. The ventral subdivision is the most distal part of the nail matrix. The nail bed is the area underneath the nail plate (between the lunula and the hyponychium). It has a role in forming the deeper layers of the nail plate, as its thin epidermal layer represents the ventral portion of the nail matrix. The hyponychium is located underneath the free edge of the nail plate and denotes the transition of the nail bed to the normal epidermis of the fingers and toes. There is also part of the hyponychium, known as the onychodermal band, that reflects on to the ventral surface of the nail plate to protect the nail parenchyma from trauma.
The epithelium of the matrix is composed of at least two to three actively dividing, basal keratinocyte layers. These cuboidal cells have their vertical axes aligned in a diagonal manner, which allows the nail plate to develop in an upward and outward direction. As these cells differentiate and migrate they become flatter, losing their nuclei and becoming integrated into the developing nail plate as onychocytes, or nail plate cells. This process of cellular maturation is similar to stratum corneum formation within the epidermis but does not require keratohyalin. The matrix also contains melanocytes, which pigment the surrounding keratinocytes and manifest as longitudinal bands across the nail plate; this may be a common racial variant in darker skinned individuals. The nail bed is composed of a thin epidermal layer and a dermal layer, but there is no subcutaneous fat. As the epidermis is thin, the differentiation of keratinocytes to onychocytes occurs within one to two cell layers. The epidermis of the nail bed also contains parallel longitudinal ridges from the lunula to the hyponychium. These ridges interlock to provide strong binding between the nail bed and the nail plate. The dermal layer of the nail bed contains blood vessels to supply the nail unit, as well as lymphatics. Trauma to these vessels results in splinter haemorrhages.
Merkel cells
Merkel first gave the name tastzellen to certain cells that he found near the base of the rete peg in the snout skin of the mole. As there were intraepidermal neurites with expanded tips (Merkel discs) adjacent to them, he believed them to be transducers of physical stimuli. Merkel cells are post-mitotic cells scattered throughout the epidermis of vertebrates and constitute 0.2–5% of epidermal cells [1]. They are located amongst basal keratinocytes and are mainly found in hairy skin, tactile areas of glabrous skin, taste buds, anal canal, labial epithelium and eccrine sweat glands, all regions of high tactile acuity (Figure 2.14).
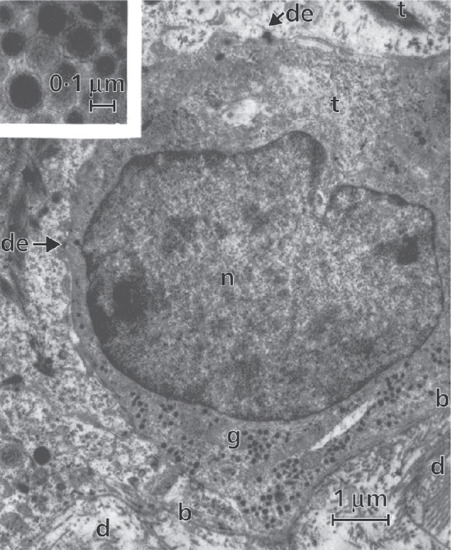
Figure 2.14 Merkel cell in human epidermis. The dermis (d) with collagen fibres is seen in the lower part of the picture; b, basement membrane; de, desmosomes making connections with adjacent basal keratinocyte; g, spherical granules (see inset); n, nucleus of Merkel cell; t, tonofilaments.
(Courtesy of Professor A. S. Breathnach.)
Sun-exposed skin may contain twice as many Merkel cells as non-sun-exposed skin. They form close connections with sensory nerve endings and secrete or express a number of peptides. Human Merkel cells express immunoreactivity for various neuropeptides including Met-enkephalin and vasoactive intestinal polypeptide, in addition to neuron-specific enolase and synaptophysin-like and pancreastatin-like material. They also contain chromogranin A [2].
Merkel cells are easily identifiable on transmission electron microscopy. They are oval with a long axis of approximately 15 μm, orientated parallel to the basement membrane. They also have a large bilobed nucleus and clear cytoplasm, which reflects a relative scarcity of intracellular organelles. Merkel cells contain numerous neurosecretory granules, each 50–160 nm across; these are found opposing the junctions with the sensory nerve endings. The close contact between Merkel cells and nerve fibres represents a Merkel cell–neurite complex. Indeed, Merkel cells actively participate in touch reception, displaying fast, touch-evoked mechanotransduction currents, and provide evidence for a direct, functional, excitatory connection between epidermal cells and sensory neurons [3].
Human skin contains an extensive neural network that consists of cholinergic and adrenergic nerves and myelinated and unmyelinated sensory fibres. The skin also contains several transducers involved in the perception of touch, pressure and vibration, including Ruffini organs surrounding hair follicles, Meissner's corpuscles, Vater–Pacini corpuscles located in the deep layer of the dermis, and nerve endings which pass through the epidermal basement membrane. Some of these contain Merkel cells, which form the Merkel cell–neurite complex, while others are free nerve endings. The cell bodies for all these neurons reside in the dorsal root ganglion. The Merkel cell–neurite complexes are thought to serve as mechanoreceptors and to be responsible for the sensation of touch.
In glabrous skin, the density of Merkel cells is approximately 50 per mm2. They are clustered near unmyelinated sensory nerve endings, where they group and form ‘touch spots’ at the bottom of rete ridges. These complexes are also known as hair discs, touch domes, touch corpuscles or Iggo discs. The complex is innervated by a single, slowly adapting type 1 nerve fibre. In hairy skin, Merkel cells also cluster in the rete ridges and in the outer root sheath of the hair follicle where the arrector pili muscles attach. The function of Merkel cells in hair follicles is unclear, although they may be involved in the induction of new anagen cycles.
There are two hypotheses for the origin of Merkel cells: one possibility is that they differentiate from epidermal keratinocyte-like cells and the other is that they arise from stem cells of neural crest origin that migrated during embryogenesis, in a similar fashion to melanocytes [4]. A unifying view, however, could be that there is very early migration of the Merkel cells from the neural crest and population of the epidermis during the sixth or seventh embryonic week in humans and that these cells subsequently only undergo further differentiation once in the epidermis.
Circulating autoantibodies against Merkel cells have been described in pemphigus and graft-versus-host disease. Merkel cells are absent in vitiligo lesions, in keeping with an autoimmune destruction or neural involvement. Merkel cell hyperplasia is a common histological finding and may accompany keratinocyte hyperproliferation as well as being frequently seen in adnexal tumours such as naevus sebaceus, trichoblastomas, trichoepitheliomas and nodular hidradenomas [5]. Merkel cell hyperplasia is associated with hyperplasia of nerve endings that occurs in neurofibromas, neurilemmomas, nodular prurigo or neurodermatitis. Merkel cell carcinoma is a highly aggressive neuroendocrine carcinoma, the incidence of which appears to be increasing; most cases are associated with the Merkel cell polyomavirus although the precise disease pathophysiology remains to be elucidated [6].
Innate immunity
The skin continuously encounters microbial pathogens, and to prevent infection, cells within the epidermis and dermis have evolved several innate strategies. One of the primary mechanisms used by the skin in the early stages of immune defence is the synthesis, expression and release of antimicrobial peptides [1]. There are more than 20 of them in the skin, including cathelicidins, β-defensins, substance P, RANTES, RNase 2,3,7 and S100A7 (Figure 2.15).
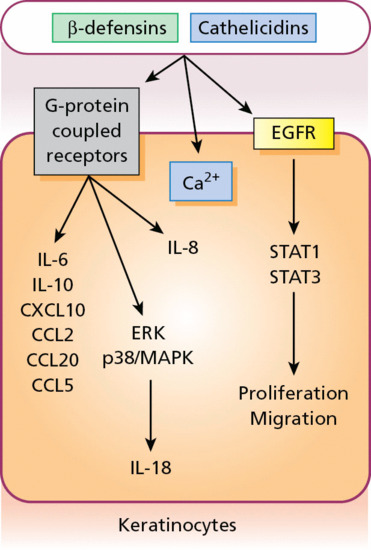
Figure 2.15 As part of the innate immune defence system, antimicrobial peptides can stimulate G-protein-coupled receptors to induce cytokine and chemokine release from keratinocytes as well as epidermal growth factor receptor (EGFR) signalling to influence cell proliferation and migration. CCL, chemokine (C–C motif) ligand; CXCL, C–X–C motif chemokine; IL, interleukin; MAPK, mitogen-activated protein kinase; STAT, signal transducer and activator of transcription.
Many peptides have antimicrobial action against bacteria, viruses and fungi. The antimicrobial activity of most peptides occurs as a result of unique structural characteristics that enable them to disrupt the microbial membrane while leaving human cell membranes intact. Some may play a specific role against certain microbes in normal skin, whereas others act only when the skin is injured and the physical barrier disrupted [2]. Other peptides may play a larger role, signalling host responses through chemotactic, angiogenic, growth factor and immunosuppressive activity; these peptides are known as alarmins [3]. For example, some alarmins not only kill bacteria but also stimulate expression of syndecan-1 and -4 in dermal fibroblasts, which is critical to the process of wound healing. Alarmins may also stimulate elements of the host defence system, such as barrier repair and recruitment of inflammatory cells [3].
The production by human skin of antimicrobial peptides such as defensins and cathelicidins occurs constitutively but also greatly increases after infection, inflammation or injury. Some skin diseases, including atopic eczema or rosacea, show altered expression of antimicrobial peptides, partially explaining the pathophysiology of these diseases [4]. In atopic eczema there is decreased expression of multiple antimicrobial peptides, which contributes to an increased susceptibility to infections, and in rosacea there are excessive and abnormally processed cathelicidin peptides, which can reproduce elements of the disease in mice. Certain antimicrobial peptides can influence host cell responses in specific ways. For example, the human cathelicidin peptide LL-37 can activate mitogen-activated protein kinase (MAPK) and extracellular signal-related kinase in epithelial cells, and blocking antibodies to LL-37 hinder wound repair in human skin equivalents. Defensins and cathelicidins have immunostimulatory and immunomodulatory capacities as catalysts for secondary host defence mechanisms. At nanomolar concentrations they are chemotactic for distinct subpopulations of leukocytes as well as some non-leukocytes. Human β-defensins (hBDs) 1–3 are chemotactic for memory T cells and immature dendritic cells. hBD2 attracts mast cells and activated neutrophils, whereas hBD3 and -4 are also chemotactic for monocytes/macrophages. Cathelicidins are chemotactic for neutrophils, monocytes/macrophages and CD4 T lymphocytes. Epidermal keratinocytes stimulated with either β-defensins or cathelicidins release an array of cytokines through the stimulation of G-protein-coupled receptors. In addition, antimicrobial peptides induce keratinocyte proliferation and migration, which involves EGFR signalling and STAT activation.
Skin microbiome
Present on the skin are thriving complex communities of bacteria, fungi and viruses, with approximately 1 million bacteria (involving hundreds of distinct species) inhabiting each square centimetre of skin [1]. The bacteria mostly comprise Actinobacteria, Firmicutes, Bacteroidetes and Proteobacteria, with numerous subspecies thereof. Actinobacteria represents the largest phylum and includes Propionibacteria and Corynebacteria; Firmicutes includes Clostridia and Bacilli, the latter including the class Staphylococcus. The precise composition of these organisms depends on sebaceous gland concentration, moisture content and temperature, as well as on host genetics and exogenous environmental factors, but may be very diverse (Figure 2.16). For example, a survey of the palm microbiome found 4742 distinct species in 51 healthy subjects, with an average of 158 species coexisting on a single palm [2]. It has also become clear that these organisms are not just commensals but play a much bigger role in immune modulation and epithelial health than previously expected. Understanding microbe–host interactions and discovering the factors that drive microbial colonization is likely to provide greater insight into the pathogenesis of skin diseases, such as the role of staphylococci in atopic eczema, and the development of new promicrobial and antimicrobial therapeutics [3].
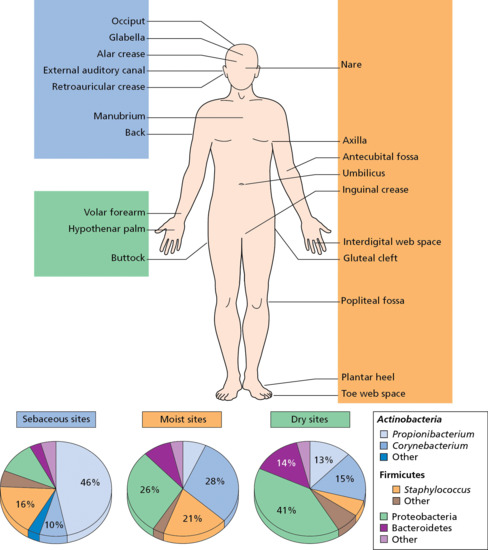
Figure 2.16 The skin microbiome contains numerous bacteria that are variably present in different body regions. (Adapted from Chen and Tsao 2013 [3].)
Langerhans cells
Dendritic cells of a form similar to melanocytes, but free from pigment and dopa negative, were first described by Langerhans, who demonstrated their existence in human epidermis by staining with gold chloride. More recently, the dynamic behaviour of epidermal Langerhans cells has been investigated by combining time-lapse, intravital, confocal imaging technology and I-Aβ-enhanced green fluorescent protein (EGFP) knock-in mice in which Langerhans cells can be identified by EGFP-associated fluorescence. Without stimulation, some Langerhans cells exhibit a unique motion, which has been termed dendrite surveillance extension and retraction cycling habitude (dSEARCH), and which is characterized by rhythmic extension and retraction of dendritic processes between intercellular spaces. The topical application of an antigen such as dinitrofluorobenzene leads to greater dSEARCH motion and also triggers direct cell-to-cell contact formation between adjacent Langerhans cells. It appears that, in vivo, dSEARCH motion allows for a more efficient antigen sampling through scanning of a wide area. It is also evident that, under pathological stimulation, adjacent Langerhans cells may exchange antigens between cells (Figure 2.17) [1].

Figure 2.17 Dendritic appearance of epidermal Langerhans cells. Exposure to antigen provokes an increased movement of Langerhans cells as well as direct cell–cell contact between Langerhans cells.
(Courtesy of Dr R. Mohr, University of Toledo, Ohio, USA.)
Langerhans cells, in combination with macrophages and dermal dendrocytes, represent the skin's mononuclear phagocyte system [2]. Langerhans cells are capable of phagocytosis, antigen processing, antigen presentation and interactions with lymphocytes (Figure 2.18). They can also release cytokines, such as interleukin-1, to promote lymphocyte chemotaxis and activation [3]. Langerhans cells are intraepidermal macrophages whose dendrites trap antigens among keratinocytes. The cells then leave the epidermis and migrate via lymphatics to a regional lymph node. In the paracortical region of lymph nodes, the Langerhans cell (or ‘interdigitating reticulum cell’ as it is then known) expresses protein on its surface to present to a T lymphocyte that can then undergo clonal proliferation. There may be some selectivity in whether certain antigens are presented to lymph nodes by Langerhans cells or by dermal dendrocytes. The timing of antigen presentation may also vary, with the possibility that sequential presentation of skin-acquired antigens may regulate cell-mediated immunity.
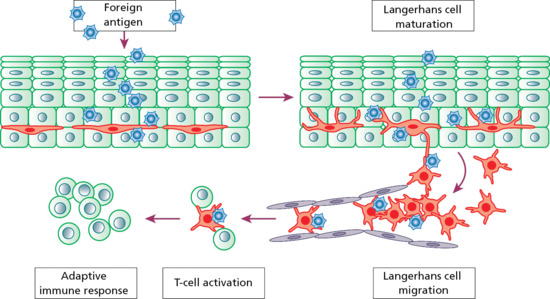
Figure 2.18 When exposed to foreign antigen, the activity of resting Langerhans cells increases and the cells mature. Antigen is then processed and transported to the lymph nodes. T cells are then activated and an immune response is triggered.
Langerhans cells may contribute to several skin pathologies including infections, inflammation and cancer, and they play a pivotal role in regulating the balance between immunity and peripheral tolerance [4]. Langerhans cells appear, however, to have characteristics distinct from other dendritic cells in that they are more likely to induce Th2 responses than the Th1 responses that are usually necessary for cellular immune responses against pathogens. It has also been shown that Langerhans cells are dispensable for contact hypersensitivity and that dermal dendrocytes can serve as antigen-presenting cells in the absence of Langerhans cells. Indeed, Langerhans cell-deficient mice appear to have enhanced contact hypersensitivity. With regard to a specific function, Langerhans cells, or a subset thereof, may have regulatory properties that counteract the proinflammatory activity of surrounding keratinocytes. It is plausible that under non-inflammatory steady state conditions, Langerhans cells carry skin-specific components to draining lymph nodes to prevent immunization and to induce peripheral tolerance against epidermal self-determinants [5]. It also appears that Langerhans cells may indeed consist of distinct subsets, since Langerhans cells that repopulate the skin after inflammation have been shown to derive from monocyte precursors.
Under the electron microscope, Langerhans cells share with melanocytes a lobulated nucleus, a relatively clear cytoplasm and well-developed endoplasmic reticulum, Golgi complex and lysosomes (Figure 2.19). They differ in lacking melanosomes or premelanosomes, and in possessing a characteristic granule that is either rod- or racquet-shaped. These ‘Birbeck’ granules have been shown to represent subdomains of the endosomal recycling compartment and form at sites where the protein Langerin accumulates. Using ultrastructural evidence of the presence of the characteristic granules, Langerhans cells have been identified in the outer root sheath of the human hair and the secretory duct of the sebaceous gland and in the epithelium of the crypts of the human tonsil. The discovery of similar granules in cells in the dermis in histiocytosis X resulted in the renaming of this condition as Langerhans cell histiocytosis.
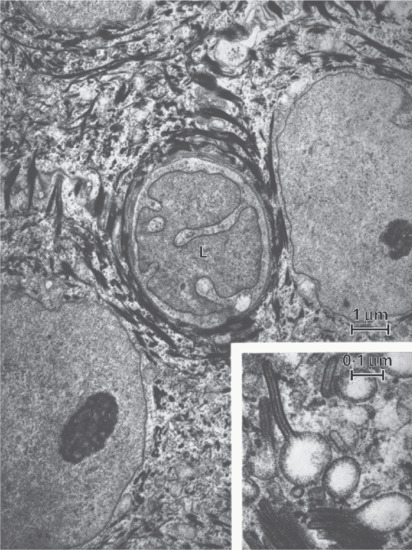
Figure 2.19 Langerhans cell (L) with its characteristically indented nucleus, situated between keratinocytes. The inset shows Langerhans cell granules with racquet-shaped profiles.
(Courtesy of Professor A. S. Breathnach.)
Immune surveillance
Besides the antigen detection and processing role of epidermal Langerhans cells, cutaneous immune surveillance is also carried out in the dermis by an array of tissue-resident T cells, macrophages and dendritic cells (Figure 2.20) [1]. These immune sentinel and effector cells are able to provide rapid and efficient immunological backup to restore tissue homeostasis should the epidermis be breached. The dermis contains a very large number of resident T cells; remarkably, there are approximately 2 × 1010 skin-resident T cells, which is twice the total number of T cells in the circulating blood [2, 3]. There are several distinct populations of dermal dendritic cells; some have potent antigen-presenting capacities, others have low antigen-presenting capacity but the potential to develop into CD1a+ and Langerin-positive Langerhans cells, while some are proinflammatory.
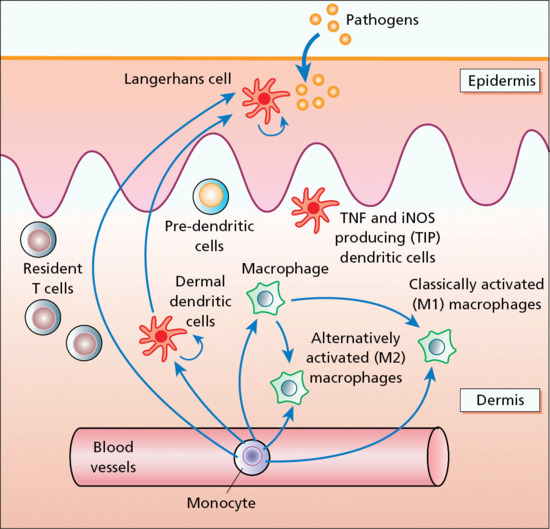
Figure 2.20 Immune surveillance in normal skin is carried out by an array of skin-based dendritic cells, macrophages and resident T cells. iNOS, inducible nitric oxide synthase; TNF, tumour necrosis factor.
Another recent addition to the family of skin immune sentinels is type I interferon-producing plasmacytoid predendritic cells, which are rare in normal skin but which can accumulate in inflamed skin [4]. A further component of the dermal immune system is the dermal macrophage. This cellular diversity of dermal immune sentinels is reflected in some flexibility or plasticity in function. For example, immature dendritic cells, including dermal dendritic cells, can be phagocytic, which is a cellular function usually attributed to macrophages [5]. Alternatively, macrophages, which normally are phagocytic cells, can also be potent antigen-presenting cells for CD8+ T cells. This means that tissue-resident mononuclear sentinels of the dermis are likely to exist in a pluripotent state. Depending on microenvironmental factors and cues, they may acquire an antigen-presenting mode, a migratory mode or a tissue-resident phagocytic mode.
Mast cells
Mast cells were first described by Ehrlich in 1877, who distinguished them from other connective tissue cells by their ability to stain metachromatically with basic aniline dyes. Mast cells are larger than eosinophils and basophils. They occur in most tissues, but are particularly numerous in the skin, bronchus, nasal mucosa and gut. In the skin, mast cells are distributed close to blood vessels, nerves and appendages, and are most numerous in the subpapillary dermis, in the region of the superficial dermal vascular plexus. There are about 7000 mast cells per mm3 in normal skin.
Dermal mast cells are ovoid or spindle-shaped, mononuclear or occasionally binuclear, and only rarely show signs of mitosis in normal skin. Their major distinguishing feature is the presence of numerous, round, cytoplasmic granules (Figures 2.21 and 2.22). Mast cells are heterogeneous and fall into two main types – connective tissue and mucosal – which can be differentiated by their morphology, tissue distribution, histochemical characteristics and responses to degranulating agents. Solubility of the granules in formaldehyde and the content of neutral proteinase, namely tryptase and chymase (chymotryptic proteinase), will vary according to the type of cell. For example, human foreskin mast cells contain both proteinases, whereas mast cells in intestinal mucosa and the lung contain mainly tryptase [1].
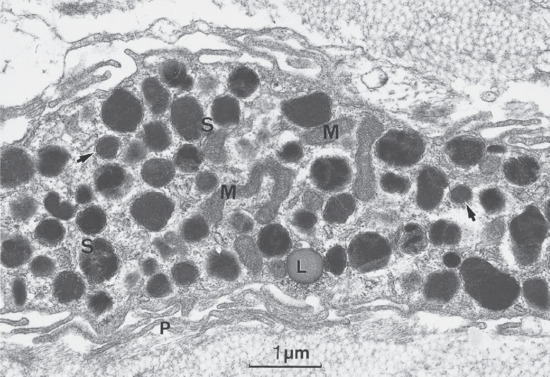
Figure 2.21 Part of a human skin mast cell showing characteristic granules, some with scroll-like profiles (S). Arrows indicate perigranular membrane; L, lipid droplet; M, mitochondria; P, peripheral processes.
(Courtesy of Professor R. A. J. Eady, St John's Institute of Dermatology, King's College London, UK.)
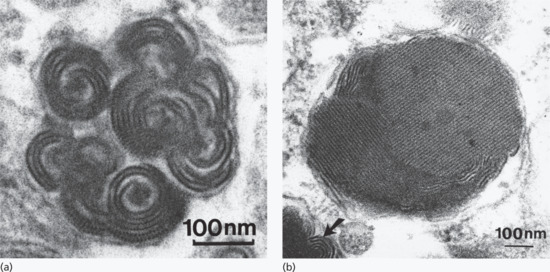
Figure 2.22 High-magnification views of dermal mast cell granules. (a) Typical scroll-like configuration of lamellae, some of which show a cross-banding of regular periodicity. (b) The substructure of this granule is a highly organized lattice (arrow).
(Courtesy of Professor R. A. J. Eady, St John's Institute of Dermatology, King's College London, UK.)
Human mast cells arise from CD34+ pluripotent stem cells in the bone marrow. They then circulate in the blood as precursors and home to tissues where they mature under the influence of stem cell factor (SCF) and local cytokines and other factors. Mast cell growth and differentiation are also influenced by several other cytokines, including interleukin 3 (IL-3), -4, -6, -9, -10 and nerve growth factor. Mast cells are long lived and may proliferate in association with IgE-dependent activation and in the presence of IL-4 [2].
Kit (CD117), expressed on haematopoietic stem cells and progenitor cells, is the tyrosine kinase transmembrane receptor for SCF that is involved in the differentiation of both myeloid and lymphoid lineages. While Kit is down-regulated on other bone marrow-derived cells during their differentiation, Kit remains highly expressed on mast cells and is critical for many mast cell functions such as survival, differentiation, chemotaxis and enhancement of signalling events during mast cell activation. The importance of Kit is shown by the finding of activating mutations in the KIT gene in patients with urticaria pigmentosa [3].
Upon activation of mast cells via cross-linking of the high affinity IgE receptor (FcεRI) or non-IgE-mediated activation through complement receptors or toll-like receptor (TLR) activation, mast cells can release histamine, serotonin and proteases as well as newly synthesized leukotrienes, prostaglandins, cytokines and chemokines. In addition to IgE-mediated activation, human mast cells exposed to interferon γ (IFN-γ) can be activated following IgG-mediated aggregation of FcγRI to release similar mediators. Additional IgE-independent mast cell triggers have been described, including SCF, complement (C3a and C5a), neuropeptides (substance P), adenosine, TLR and scavenger receptors.
Mast cell products may both induce an immediate reaction and contribute to a late phase reaction. The immediate phase reaction occurs within minutes of FcεRI cross-linking and its consequences are referred to as an immediate hypersensitivity reaction. Late phase reactions peak 6–12 h following antigen challenge and are associated with cytokine and chemokines from eosinophils, neutrophils and basophils that have been secondarily recruited. Mast cell activation results in increased vascular permeability and smooth muscle contraction, as well as fibroblast deposition of collagen, induction of B cells to class switch to synthesize IgE, basophil histamine release, recruitment of neutrophils and eosinophils, and promotion of T cells to a T helper 2 (Th2) phenotype.
Mast cells play an important role in both adaptive and innate immunity, and contribute to the skin pathology seen in contact dermatitis, atopic eczema (AE), immunobullous disease, scleroderma and chronic graft-versus-host disease [4]. In AE, there is an increase in mast cell numbers in lesional skin. Mast cells reside in the papillary dermis and undergo migration through the basal lamina into the epidermis. Although overall levels of histamine are not increased in AE, tryptase and activation of proteinase-activated receptor-2 (PAR-2) may contribute to the pruritus seen in AE, as tryptase is reported to be increased up to fourfold in AE patients and PAR-2 expression is markedly enhanced on primary afferent nerve fibres in skin biopsies from patients with AE. Chymase may play a role in eliciting and maintaining chronic inflammation in AE by increasing spongiosis and compromising the skin barrier. Mast cell–nerve interactions may also play a role in promoting inflammation in AE [5]. There is an increased number of contacts between mast cells and nerves in both lesional and non-lesional skin, which may lead to inflammation mediated by neuropeptides such as substance P, calcitonin gene-related peptide, vasoactive intestinal peptide and nerve growth factor.
Melanocytes
Melanocytes are pigment-producing cells located in the skin, inner ear, choroid and iris of the eye. In the skin and hair, two forms of melanin pigment are produced: brown/black eumelanin and yellow/red phaeomelanin (Figure 2.23). The melanin is subsequently transferred in melanosomes to neighbouring keratinocytes in the epidermis and into the growing shaft in hair follicles. Variations in the types of melanin pigment produced and their distribution within the skin and hair contribute to the vast diversity in colour. A key protein involved in melanosome assembly is NCKX5, encoded by the gene SLC24A5 [1]. Loss of expression of this gene in mice results in marked changes in skin colour with loss of pigment. Melanin production also provides skin protection by reducing damage from harmful ultraviolet radiation. In humans, alterations in melanocyte development and function can lead to various pigmentary disorders. These include disorders with reduced melanocytes in skin, such as piebaldism and Waardenburg syndrome, and disorders with defective pigment production or processing, such as albinism and Hermansky–Pudlak and Chediak–Higashi syndromes.
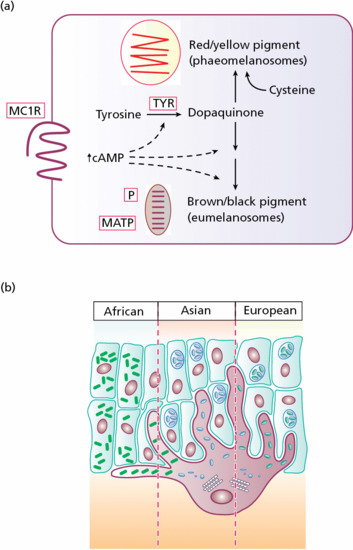
Figure 2.23 (a) Activation of the melanocortin 1 receptor (MC1R) promotes the synthesis of eumelanin at the expense of phaeomelanin. Oxidation of tyrosine by tyrosinase (TYR), however, is required for synthesis of both pigment types. Melanosomal membrane components, including the membrane-associated transport protein (MATP) and the pink-eyed dilution protein (P), play a role in determining the amount of pigment synthesis within melanosomes. (b) In African, Asian and European skin there is a gradient of melanosome size and number; in addition, melanosomes in African skin are more widely dispersed.
The melanin-producing melanocytes in adult skin and hair develop from embryonically derived melanocyte precursors called melanoblasts. During development, melanoblasts emerge from a subset of neural crest cells and migrate to the skin and developing hair follicles. In the hair follicle, melanocytes are divided into two distinct populations: differentiated melanocytes, located in the hair matrix region, and melanocyte stem cells, located at the lower permanent portion of the hair follicle. The life cycles of the follicular melanocytes and melanocyte stem cells are closely related to the cyclical nature of the hair follicle, and during anagen new melanocytes are generated from the pool of slow-proliferating melanocyte stem cells [2]. Differentiated melanocytes express Sox10, Kit, Mitf, Pax3 and Dct but melanocyte stem cells only express Pax3 and Dct [3]. Whether a subpopulation of melanocyte stem cells exists that may be relevant to the pathogenesis of melanoma is not yet known, although some of the melanocyte stem cells do express markers such as CD166, CD133 or Nestin, which are found in stem cells from other lineages and may be multipotent in culture, capable of differentiating into various cell types (e.g. adipocytes, chondrocytes).
Alterations in melanocyte signalling pathways also contribute to common hair abnormalities such as hair greying [4]. One particular pathway involves Notch signalling [5]. The Notch signalling pathway is an essential cell–cell interaction mechanism, which regulates processes such as cell proliferation, cell fate decisions, differentiation or stem cell maintenance. Notch signalling in melanocytes is essential for the maintenance of proper hair pigmentation, including regeneration of the melanocyte population during hair follicle cycling. Deletion of Notch1 and Notch2 or RBP-Jkappa in melanocyte lineages results in a gene dosage-dependent, precocious hair greying, due to the elimination of melanoblasts and melanocyte stem cells. Aberrant Notch signalling may also be relevant in the development or progression of melanoma.
Melanocytes possess melanocyte-specific receptors including melanocortin-1 (MC1R) and melatonin receptors [6]. The activation or the inhibition of melanocyte-specific receptors can augment normal melanocyte function, skin colour and photoprotection. Moreover, receptor polymorphisms are known to underlie red hair phenotypes. Receptor targeting may also be relevant to the treatment of melanoma. Notably, melanocytes also possess G-protein-coupled receptors, such as Frizzled5, and receptor tyrosine kinases, including c-Kit and hepatocyte growth factor receptor. These receptors activate two crucial cell signalling pathways, RAS/RAF/MEK/ERK and PI3K/AKT, integral to melanoma cell survival, and could serve as targets for future therapies of disseminated melanoma.
Desmosomes
Desmosomes are the major adhesion complex in the epidermis, anchoring keratin intermediate filaments to the cell membrane and bridging adjacent keratinocytes, and allowing cells to withstand trauma. Desmosomes are also found in the myocardium, meninges and cortex of lymph nodes. The desmosome has a characteristic ultrastructural appearance, in which the cell membrane of two adjacent cells forms a symmetrical junction with a central intercellular space of 30 nm containing a dense line (Figure 2.24). Plaques of electron-dense material run along the cytoplasm parallel to the junctional region, in which three ultrastructural bands can be distinguished: an electron-dense band next to the plasma membrane, a less dense band and then a fibrillar area [1].

Figure 2.24 Electron micrograph of desmosomes in the spinous layer. These intercellular junctions are closely associated with tonofilaments (tf), many of which, in this view, are cross-sectioned.
(Courtesy of Professor R. A. J. Eady, St John's Institute of Dermatology, King's College London, UK.)
The main components of desmosomes in the epidermis consist of the products of three gene superfamilies: the desmosomal cadherins, the armadillo family of nuclear and junctional proteins, and the plakins [2]. The transmembranous cadherins comprise mostly heterophilic associations of desmogleins and desmocollins. There are four main epidermis-specific desmogleins (Dsg1–4) and three desmocollins (Dsc1–3), all of which show differentiation-specific expression. For example, Dsg1 and Dsc1 are preferentially expressed in the superficial layers of the epidermis, whereas Dsg3 and Dsc3 show greater expression in basal keratinocytes. The intracellular parts of these glycoproteins are attached to the keratin filament network via desmoplakin, plakoglobin and other macromolecules, including the armadillo protein, plakophilin 1, an important stabilizer of keratinocyte adhesion in differentiated keratinocytes, as well as other site-specific plakin cell envelope proteins, such as envoplakin and periplakin [3]. The network of the major interactive desmosomal proteins is depicted in Figure 2.25.
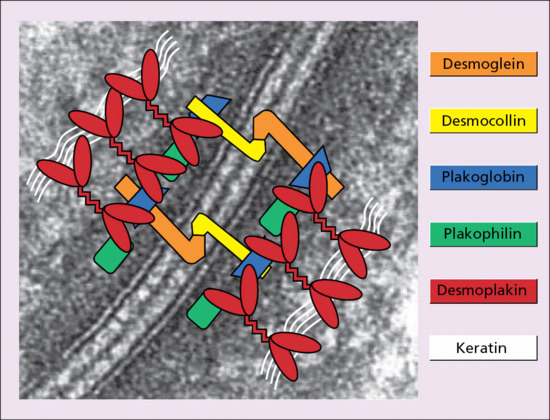
Figure 2.25 Macromolecular composition of desmosomes linking adjacent keratinocytes. Cells are connected via transmembranous cadherin glycoproteins (desmogleins and desmocollins). Attachment of these molecules to the keratin filament cytoskeleton occurs via a network of desmosomal plaque proteins (desmoplakin, plakoglobin and plakophilin). The background to this figure is a transmission electron micrograph of a desmosome to highlight how the molecules function as an adhesive complex.
Further clues to the biological function and in vivo contribution to keratinocyte adhesion of these desmosomal components have arisen from various mouse models and human diseases, both inherited and acquired [4, 5], and desmosome proteins may also serve as autoantigens in several immunobullous blistering skin diseases [6]. Antibodies to multiple desmosomal proteins may develop in diseases such as paraneoplastic pemphigus, possibly through the phenomenon of epitope spreading [7]. Disruption of the extracellular domain of Dsg1 has also been demonstrated as the basis of staphylococcal scalded skin syndrome and bullous impetigo in which this desmosomal cadherin is cleaved by the bacterial toxin [8].
Adherens junctions
Adherens junctions are electron-dense, transmembrane structures that engage with the actin skeleton [1]. They can associate with tight junctions and desmosomes or exist separately from these junction complexes. Adherens junctions contribute to epithelial assembly, adhesion, barrier formation, cell motility and changes in cell shape. They are characterized by two opposing membranes separated by approximately 20 nm and are 0.2–0.5 μm in diameter. Adherens junctions may also spatially coordinate signalling molecules and polarity cues as well as serving as docking sites for vesicle release. They comprise two basic adhesive units: the nectin–afadin complex and the classic cadherin complex (Figure 2.26) [2, 3].
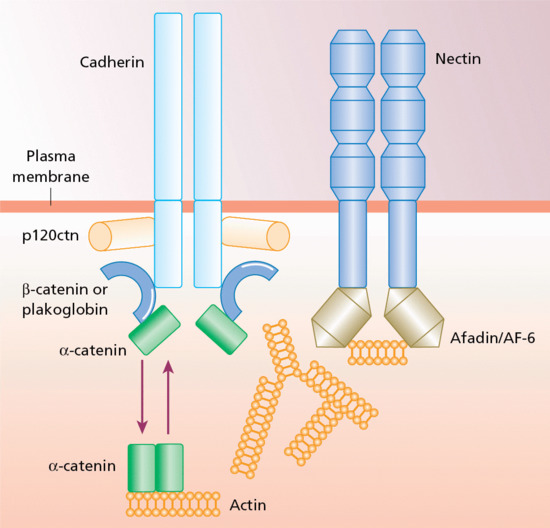
Figure 2.26 Macromolecular composition of an adherens junction in keratinocytes. There are two main components, nectin–afadin and the classic cadherin–catenin complex, which can both attach to the actin cytoskeleton.
There are several different nectins and cadherins and these may be variably incorporated into adherens junctions; the precise composition will impact on the adhesive specificity and other functions of the junction. The nectins form a structural link to the actin cytoskeleton via afadin (also known as AF-6) and may be important in the initial formation of adherens junctions. The cadherins form a complex with the catenins (α-, β-, and p120 catenin) and help mediate adhesion and signalling. Cell signalling via β-catenin can activate several Wnt pathways, which implicates adherens junctions in coordinating morphogenetic movements with cell fate determination. Adherens junctions are also associated with a variety of actin-binding molecules, suggesting multiple dynamic interactions with the cytoskeleton.
The first human gene mutation reported in a component of adherens junctions was in plakoglobin, also a component of desmosomes, in individuals with Naxos disease [4]. However, mutations have subsequently been reported in the CDH3 gene, which encodes P-cadherin; these mutations result in autosomal recessive hypotrichosis with juvenile macular dystrophy [5]. P-cadherin mutations are also found in a different disorder, ectodermal dysplasia–ectrodactyly–macular dystrophy (EEM) syndrome, in which there is hypotrichosis, macular degeneration, hypodontia and limb defects, including ectrodactyly, syndactyly and camptodactyly [6]. Mutations in nectin-1 and -4 have also been reported in a group of ectodermal dysplasia syndromes, sometimes refered to as nectinopathies [7].
Gap junctions
Gap junctions comprise clusters of intercellular channels, known as connexons, which directly form connections between the cytoplasm of adjacent keratinocytes (and other cells) [1]. Thirteen different human connexins have been described. Connexons originate following the assembly of six connexin subunits within the Golgi network that are then transported to the plasma membrane. The connexins are divided into three groups (α, β and γ) according to their gene structure, overall gene homology and specific sequence motifs [2]. At the plasma membrane, connexons associate with other connexons to form a gap junction (Figure 2.27).
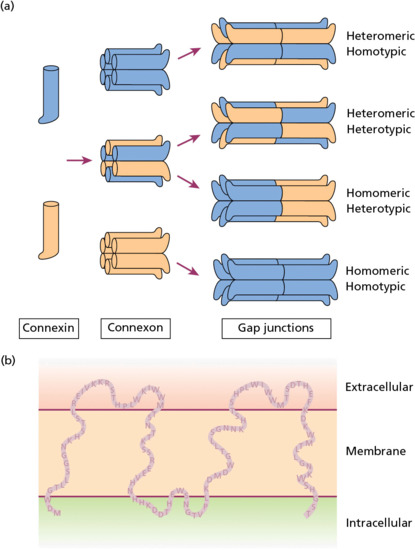
Figure 2.27 Formation and structure of gap junctions in human skin. (a) In the Golgi network six connexin subunits assemble to form a connexon. The connexon is then transported to the plasma membrane. Other connexons then co-aggregate to form homotypic or heterotypic gap junctions. (b) The gap junction protein is a transmembranous molecule with intracellular, transmembranous and extracellular domains (here illustrated for Cx26).
Homotypic or heterotypic connexins (formed from one or more than one type of connexin, respectively) can be identified, and the formation and stability of gap junctions can be regulated by protein kinase C, Src kinase, calcium concentration, calmodulin, adenosine 3′,5′-cyclic monophosphate (cAMP) and local pH [3]. Apart from the connexins, vertebrates also contain another class of gap junction proteins, the pannexins, which are related to the innexins found in non-chordate animals [4]. The function of gap junctions is to permit sharing of low-molecular-mass metabolites (<1000 Da) and ion exchange between neighbouring cells, thus allowing intercellular coordination and uniformity to maintain tissue/organ homeostasis in multicellular organisms [3]. Gap junction communication is essential for cell synchronization, cell differentiation, cell growth and metabolic coordination of avascular organs, including epidermis.
Inherited abnormalities in genes encoding four different connexins (Cx26, -30, -30.3 and -31) have been detected in several forms of keratoderma and/or hearing loss but non-dermatological disorders can also arise from mutations in the higher-molecular-weight connexins (Cx32, -40, -43, -46 and -50).
Tight junctions
Tight junctions are the major regulators of permeability in simple epithelia, but they are also present in the skin, with a key role in skin barrier integrity and maintaining cell polarity [1]. Tight junctions regulate the paracellular flux of water-soluble molecules between adjacent cells [2]. The principal structural proteins of tight junctions are the claudins, of which there are approximately 24 subtypes. The other component transmembranous proteins are the IgG-like family of junctional adhesion molecules (JAMs) and the occludin group of proteins. Seven claudins are expressed in human epidermis, although the main claudins are 1 and 4. These transmembranous proteins do not bind to one another, but the claudins and occludins can bind to the intracellular zonula occudens proteins ZO-1, ZO-2, ZO-3. These proteins can also interact with actin thus providing a direct link with the cytoskeleton [1, 3]. The structural organization of a tight junction is shown in Figure 2.28.

Figure 2.28 Structural composition of a tight junction in human skin. There are three transmembranous families of proteins, the junctional adhesion molecules (JAMs), the claudins and the occludins, of which the latter two bind to zonula occludens proteins (ZOs) and then directly to actin. MUPP1, multiple PDZ domain protein; MAGI, membrane-associated guanyl kinase inverted protein.
Mutations in claudin 16 (also known as paracellin-1) result in familial hypomagnesaemia with hypercalciuria and nephrocalcinosis; mutations in claudin 14 underlie the autosomal recessive deafness disorder DFNB29 leading to cochlear hair cell degeneration; mutations in claudin 19 result in renal and ocular disease [4]. In addition, the gene encoding the ZO-2 protein may be mutated in some cases of familial hypercholanaemia. With respect to skin, mutations in claudin 1 have been demonstrated in one pedigree with clinical features of diffuse ichthyosis with large scales, hypotrichosis, scarring alopecia and sclerosing cholangitis [5]. Collectively, these human genetic disorders, all of which are autosomal recessive in nature, demonstrate the key role tight junctions play in the skin, kidney, ear, eye and liver.
Dermal–epidermal basement membrane
The interface between the lower part of the epidermis and the top layer of the dermis comprises the dermal–epidermal basement membrane zone (BMZ), which consists of a number of extracellular matrix macromolecules (Box 2.1; Figure 2.29) [1, 2]. Many of these components are glycoproteins, and the BMZ can be recognized histologically by positive labelling with PAS stain. Ultrastructural examination of the BMZ by transmission electron microscopy shows the presence of two distinct layers with different optical densities (Figure 2.30).
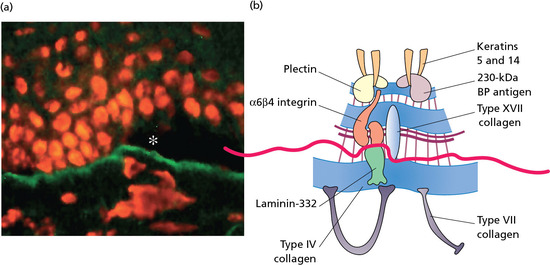
Figure 2.29 NaCl-induced separation between the epidermis and dermis and antigen mapping within the cutaneous basement membrane. (a) The dermal–epidermal basement membrane visualized by immunofluorescence staining with antibodies to type VII collagen. This maps to the base of the split because cleavage occurs through the lamina lucida, and type VII collagen is located below the lamina densa. (b) Molecular complexity at the dermal–epidermal junction and how various proteins map above or below the NaCl-induced split (red line and asterisk). BP, bullous pemphigoid.
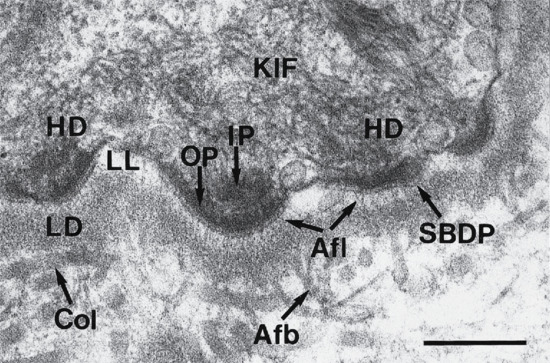
Figure 2.30 Transmission electron microscopy of the dermal–epidermal junction in human skin recognizing hemidesmosomes (HD), anchoring filaments (Afl) and anchoring fibrils (Afb). The HD consists of an intracellular inner plaque (IP) and outer plaque (OP) as well as a sub-basal dense plate (SBDP) in the upper lamina lucida (LL). The anchoring filaments traverse the LL, appearing as thread-like structures that concentrate under the HDs and merge with the lamina densa (LD). Anchoring fibrils extend from the lower part of the LD to the upper papillary dermis where they closely associate with interstitial collagen fibres (Col). Keratin intermediate filaments (KIFs) associate with intracellular components of the HDs. Scale bar 0.25 μm.
(Courtesy of Professor R. A. J. Eady, St John's Institute of Dermatology, King's College London, UK.)
The upper layer, the lamina lucida, is a less electron-dense region and directly abuts the plasma membranes of the basal keratinocytes. Below the lamina lucida is the lamina densa, an electron-dense region that at the lower part interacts with the mesenchymal matrix of the upper dermis. The major biochemical components of the BMZ are type IV collagen and a number of non-collagenous glycoproteins, including laminin 322 [3]. Associated with the cutaneous BMZ are ultrastructurally recognizable attachment structures that form a contiguous network extending from the intracellular milieu of basal keratinocytes through the plasma membrane of basal cells, traversing the dermal–epidermal basement membrane and extending to the upper papillary dermis (Figure 2.29). The components of this network are the hemidesmosomes, anchoring filaments and anchoring fibrils. The biochemical components of the BMZ are synthesized by basal keratinocytes and dermal fibroblasts, which both contribute to the development and repair of the basement membrane [4].
The critical importance of this network structure in securing the adherence of the epidermis to the underlying dermis is reflected in the group of diseases, epidermolysis bullosa, in which components of the hemidesmosomes, anchoring filaments or anchoring fibrils are genetically altered or missing. As a result, fragility at the dermal–epidermal junction ensues, clinically manifesting as erosions and blisters following minor trauma (Figure 2.31) [5].
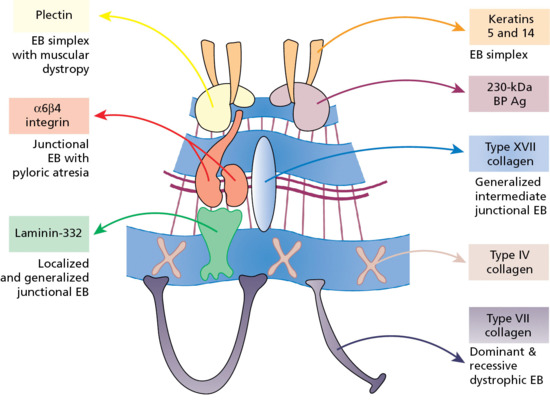
Figure 2.31 Gene/protein systems within the cutaneous basement membrane zone that can harbour mutations and result in blistering of the skin in different forms of epidermolysis bullosa (EB). BP, bullous pemphigoid.
Basement membrane collagen
The major component of the dermal–epidermal basement membrane is type IV collagen, a heterogeneous group of macromolecules that are present in diverse combinations in various basement membranes (Figure 2.32) [1]. Like all collagens, each type IV collagen molecule consists of three polypeptide subunits, known as α-chains. Some collagen molecules are homopolymers, that is the three α-chains are genetically identical, as in the case of type II, III and VII collagens. Others are heteropolymers so that there are two or even three different kinds of polypeptides, as for example in type I and type VI collagens. For type IV collagen, there are six genetically distinct but structurally related α-chains, and the precise composition of the α-chains varies with the tissue location of the basement membranes. In the case of the cutaneous BMZ, the major type IV collagen consists of α1 and α2 chains, with the chain composition [α1(IV)]2α2(IV), although other type IV collagen subunit polypeptides are also present in lower quantities. The α3 chain of type IV collagen has been shown to be the antigen recognized by circulating autoantibodies characteristic of Goodpasture syndrome, while structural aberrations in the α5 chain of type IV collagen are associated with Alport syndrome [2, 3]. Autoantibodies against α5 and α6 chain epitopes have also been reported in patients with glomerulonephritis and subepidermal blistering [4].
Figure 2.32 Immunofluorescence staining of the dermis and cutaneous basement membrane zone with an antibody for type IV collagen. Note positive staining at the dermal–epidermal basement membrane and around the dermal blood vessels. Original magnification 250×.
The characteristic fibre structure of interstitial collagens, as exemplified by type I collagen, results from the lateral aggregation of individual molecules in a quarter stagger array; this gives rise to a 64 nm cross-striation pattern when examined by transmission electron microscopy (Figure 2.33). In the case of type IV collagen, the non-collagenous globular domains both at the amino- and carboxyl-ends of the individual collagen molecules interact to form dimers and tetramers which then assemble into lattice-like structures and associate laterally in a complex hexagonal arrangement (Figure 2.34). This arrangement allows the basement membrane structure to be highly flexible and makes interactions with other collagenous and non-collagenous basement membrane components possible [1].
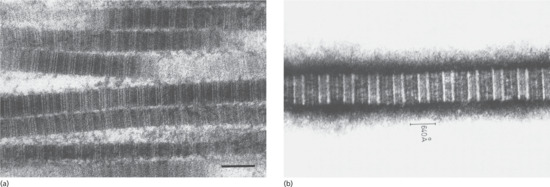
Figure 2.33 Demonstration of periodicity in collagen fibres of 640 Å. (a) Collagen fibrils in the reticular dermis show a characteristic banding pattern after standard processing and staining with uranyl acetate and lead citrate for transmission electron microscopy. Scale bar 0.1 μm. (b) Transmission electron micrograph of a shadowed replica of unfixed, freeze-frozen and surface-sublimated rat-tail tendon collagen showing the step-like banding of the fibres. Original magnification 40 500×.
(Courtesy of G. A. Meek.)
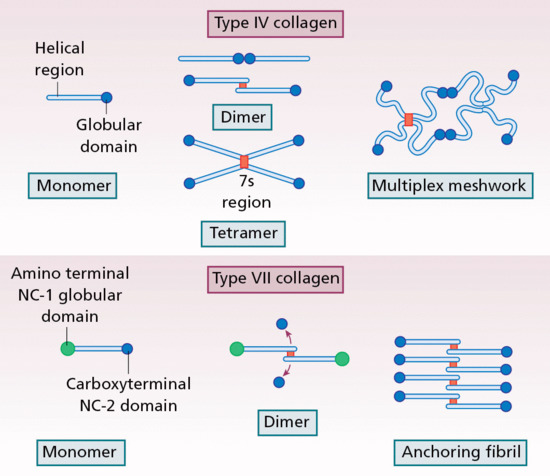
Figure 2.34 Assembly of type IV and type VII collagen molecules into supramolecular structures. The red boxes represent intermolecular disulphide bonds.
Laminins
Other components that contribute to the cutaneous BMZ include members of the laminin family of multidomain proteins. As many as 16 different laminins have been identified thus far and at least four of them are physiologically present in the skin in significant quantities (Table 2.1) [1, 2]. Each laminin molecule consists of three polypeptide subunits, α-, β- and γ-chains, which form a cruciform structure with three short arms and one long arm when visualized by rotary shadowing electron microscopy (Figure 2.35).
Table 2.1 Chain composition of the major laminins in the skin
| Type | Chain composition | Old designation | Distribution in basement membranes |
| 111 | α1β1γ1 | 1 | Blood vessels, LD |
| 332 | α3β3γ2 | 5 | LL/LD |
| 311 | α3β1γ1 | 6 | LL/LD |
| 511 | α5β1γ1 | 10 | LL/LD |
LD, lamina densa; LL, lamina lucida.
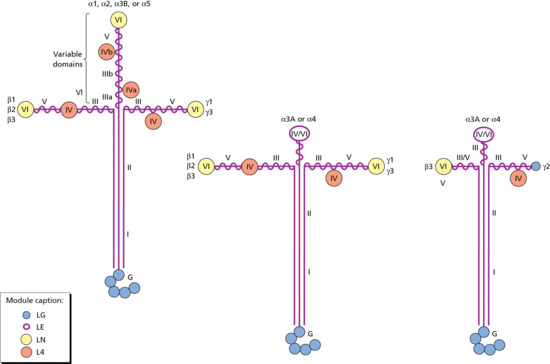
Figure 2.35 Different isoforms and domain organizations of laminin, each consisting of three distinct subunit polypeptides, α-, β- and γ-chains. The LE modules are formed by approximately 60 residues each and have homology to epidermal growth factor. The LN and L4 modules are folded into globular structures located between the LE modules (domain IV) or at the amino-terminus of each chain (domain VI). The coiled-coil central region, comprising all three polypeptides, is represented as vertical straight lines. Note the presence of a G domain consisting of five globular segments (LG) at the carboxy-terminus of the α-chains. (Adapted from Aumailley and Rouselle 1999 [8].
Reproduced with permission of Elsevier.)
The short arms represent the N-terminal segments of the α-, β- and γ-chains. The long arm consists of an extended rod-like structure of the triple-stranded, coiled-coil domain of all three chains. This domain serves as the site of chain assembly of the three subunit polypeptides. The α-chain contains an additional C-terminal segment that has five globular segments located at the tip of the long arm, known as the G domain. The major laminin within the cutaneous basement membrane zone is laminin 332, previously known as laminin 5 (Figure 2.36). In addition, laminin 311 and laminin 511 are integral components of the cutaneous BMZ, while laminin 111 is also present in basement membranes of the blood vessels in human dermis [3].
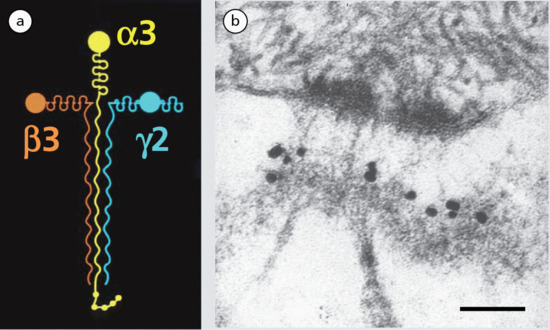
Figure 2.36 (a) Laminin 332 expressed in the cutaneous basement membrane zone. (b) Immunogold electron microscopy using an antibody to the γ2 chain of laminin 332 showing labelling at the lamina lucida–lamina densa interface below a hemidesmosome. Scale bar, 50 nm.
The cell binding of laminins is mediated by integrins, a family of cellular receptors, each consisting of two subunit polypeptides (Figure 2.37). Integrins also mediate outside-in signal transduction elicited by laminins and regulate cell migration, proliferation, differentiation and adhesion [4]. The principal integrin in the cutaneous BMZ is the α6β4 integrin, which is critical for the adhesion of basal cells to the underlying BMZ.
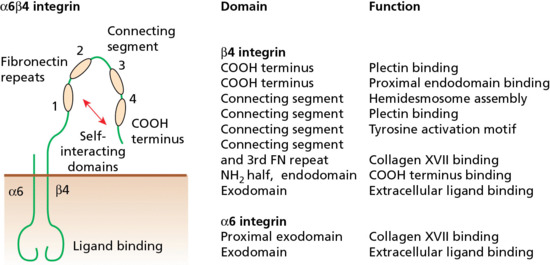
Figure 2.37 Molecular composition, domain organization and functions of the α6β4 integrin, the main keratinocyte integrin in hemidesmosomes. This integrin is important in hemidesmosome assembly and protein–protein interactions. FN, fibronectin.
The cruciform structure of laminins contains both globular and rod-like segments that have been individually implicated in various functions, including interactions with other extracellular matrix molecules, such as the hemidesmosomal components and type VII collagen, as well as in cell attachment and spreading, neurite outgrowth and cellular differentiation. Collectively, the laminins play vital roles in the development and maintenance of the supramolecular organization of the basement membrane [5]. The critical role of laminin 332 in providing integrity to the cutaneous BMZ is evident by observations that genetic mutations in any of the three polypeptide subunits – that is the LAMA3, LAMB3 or LAMC2 genes which encode the α3, β3, and γ2 chains, respectively – can result in junctional forms of epidermolysis bullosa (EB) with profound fragility of the skin. Furthermore, mutations in the ITGA6 and ITGB4 genes – encoding the α6 and β4 subunit polypeptides of integrin, respectively – cause a form of junctional EB frequently associated with pyloric atresia [6].
Other basement membrane zone components at the dermal–epidermal junction include a glycoprotein known as nidogen (previously called entactin) that interacts with type IV collagen either alone or as a laminin–nidogen complex (Figure 2.38) [7]. Nidogens are a family of highly conserved, sulphated glycoproteins within a spectrum of binding proteins. Although nidogens are an abundant component of various basement membranes, genetic analyses have shown that they are not required for the overall architecture of the basement membrane. Instead, nidogens appear to play a critical role in the development of basement membranes in tissues undergoing rapid growth or turnover.
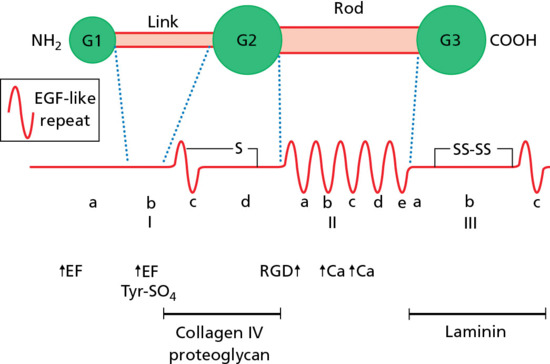
Figure 2.38 Model of nidogen, containing subdomains with predicted binding activities to type IV collagen, proteoglycans and laminin 1. EF and Ca refer to putative calcium-binding sites; RGD is a putative cell-binding sequence. EGF, epidermal growth factor. (Adapted from Niessen et al. 1994 [9].
Reproduced with permission of Elsevier.)
Another class of integral basement membrane constituents is that of the heparan sulphate proteoglycans (HSPGs). These molecules consist of a core protein with different numbers of covalently associated heparan and sulphate chains which make these molecules highly negatively charged and hydrophilic. These proteoglycans are capable of interacting with a number of basement membrane components and they are likely to contribute to the overall architecture of the basement membrane. Specific cell surface HSPGs are found also on the surface of epithelial cells and possibly mediate cell–matrix interactions.
Hemidesmosomes
The dermal–epidermal junction of skin is characterized by the presence of specific structures that are critical for the functional integrity of the skin. These ultrastructurally recognizable components include hemidesmosomes, anchoring filaments and anchoring fibrils. The molecular composition and the specific domain organizations of the component macromolecules have been largely characterized (Figure 2.39).

Figure 2.39 Structure and domain organization of the major protein components at the cutaneous basement membrane zone, with their molecular weights and chromosomal locations of the corresponding genes. BPAG, bullous pemphigoid antigen. (From Pulkkinen and Uitto 1998 [7].
Reproduced with permission of John Wiley & Sons.)
The hemidesmosomes are seen ultrastructurally as electron-dense attachment complexes, which extend from the intracellular compartment of the basal keratinocytes to the lamina lucida in the upper portion of the dermal–epidermal basement membrane (Figure 2.40). The intracellular domains of hemidesmosomes within the basal keratinocytes attach to the keratin intermediate filament network, while in the extracellular space within the lamina lucida the hemidesmosomes are contiguous with anchoring filaments; this unit is termed the hemidesmosome–anchoring filament complex.
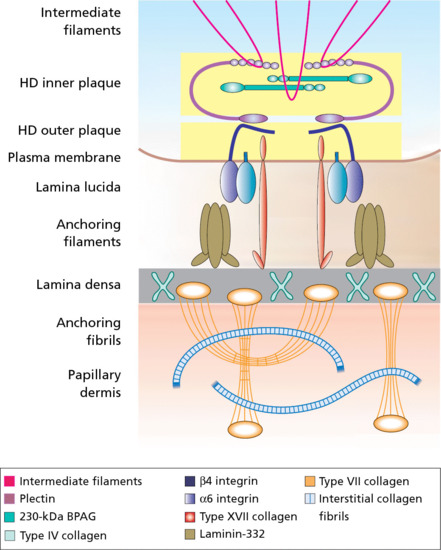
Figure 2.40 Molecular interactions of the major components of the cutaneous basement membrane zone. The individual components are identified in the colour key and their domain organizations are given in Figure 2.39. BPAG, bullous pemphigoid antigen; HD, hemidesmosome. (Adapted from Pulkkinen and Uitto 1998 [7].
Reproduced with permission of John Wiley & Sons.)
Early biochemical studies identified at least five major components of hemidesmosomes, originally designated as HD1 to HD5 with molecular masses of approximately 500, 230, 200, 180 and 120 kD, respectively. Various molecular and immunological approaches have subsequently identified HD2 and HD4 as the 230 and 180 kDa bullous pemphigoid antigens (BPAG1 and BPAG2), respectively [1]; HD3 and HD5 correspond to the β4 and α6 integrin subunit polypeptides, respectively; and HD1 corresponds to plectin, a large intracytoplasmic adhesion molecule. The intracellular hemidesmosomal plaque contains the 230 kDa bullous pemphigoid antigen, a non-collagenous protein of the plakin family that serves as an autoantigen in bullous pemphigoid. The 180 kDa bullous pemphigoid antigen, a transmembrane collagenous protein, also known as type XVII collagen, interacts with α6β4 integrin and extends from the intracellular compartment of basal cells to the extracellular space, thus stabilizing the association of basal keratinocytes to the underlying basement membrane.
Attesting to the critical importance of the hemidesmosomes in providing stability to the association of basal keratinocytes with the underlying BMZ, is the finding that mutations in the genes encoding hemidesmosomal proteins have been shown to result in different forms of EB (see Figure 2.31) [2, 3]. The causative nature of the mutations in these hemidesmosomal genes has been verified by the development of targeted mutant (‘knock-out’) mouse models, which frequently recapitulate the clinical, genetic, histological and ultrastructural features encountered in patients with EB [4, 5].
The hemidesmosomes are complexed with anchoring filaments, which are thread-like structures that tend to coalesce below the hemidesmosomal outer plaques (see Figure 2.36). Electron microscopy and immunohistochemical analyses have suggested that laminin 332 may be the major component of the anchoring filaments, although the presence of additional molecules has also been suggested. As indicated, laminin 332 is a disulphide-bonded complex of α3, β3 and γ2 chains that associate into a trimeric, cruciform structure. After the initial assembly, the α3 and γ2 chains are proteolytically processed, and many of the conformational epitopes are recognized by specific antibodies. Complete absence of any of the three subunit polypeptides of laminin 332, due to loss-of-function mutations in both alleles of the corresponding gene, results in severe generalized junctional EB [6]. Since laminin 332 binds to α6β4 integrin in the hemidesmosomes and to type VII collagen in the anchoring fibrils, the severity of skin fragility in this form of EB apparently reflects the loss of its ability to bridge the hemidesmosomes and the anchoring fibrils. This results in a separation of the epidermis from the dermis within the lamina lucida as a result of minor trauma to the skin.
Anchoring fibrils
Anchoring fibrils are ultrastructurally recognizable, U-shaped structures that extend from the lower part of the lamina densa to the upper reticular dermis (Figure 2.41). Type VII collagen is the major, if not the exclusive, component of anchoring fibrils [1]. Individual collagen molecules are approximately 450 nm long, consisting of a central, triple helical segment flanked by non-helical globular domains at each end of the triple helix: NC1 at the amino-terminus and NC2 at the carboxy-terminus of the molecule (see Figure 2.34). In addition, the triple helical segment of type VII collagen contains imperfections in the triple helix, including a central 39 amino acid non-collagenous segment. These interruptions in the glycine-X-Y sequence are thought to provide flexibility to the type VII collagen molecules.
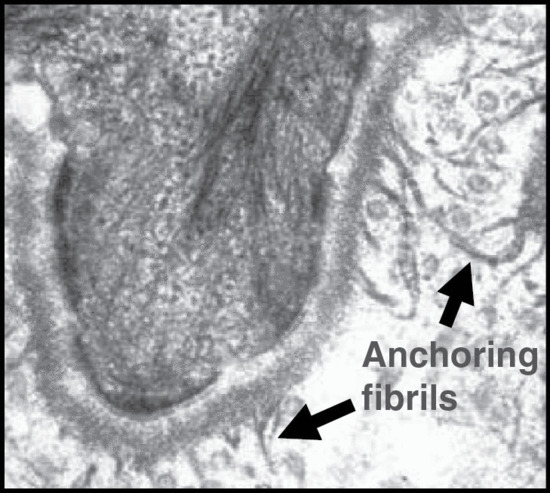
Figure 2.41 Transmission electron microscopy of the dermal–epidermal junction revealing wheatsheaf-shaped anchoring fibrils beneath the lamina densa. These fibrils help secure adhesion between the epidermal basement membrane and interstitial collagens within the dermis.
The gene encoding type VII collagen (COL7A1) is extremely complex, consisting of 118 exons on the short arm of human chromosome 3 [2]. Type VII collagen is synthesized by both dermal fibroblasts and epidermal keratinocytes, although the basal cell appears to be the major source of type VII collagen during the early prenatal development of skin. Upon secretion to the extracellular space, two type VII collagen molecules align into an antiparallel dimer with overlapping NC2 domains, and after partial proteolysis of the NC2 domain, the dimer is stabilized by intermolecular disulphide bonds. Subsequently, a large number of type VII molecules laterally aggregate to form anchoring fibrils in which the NC1 domains at both ends attach to the basement membrane. The U-shaped loops then entrap, and possibly interact with, large interstitial collagen fibres consisting of type I, III and V collagens [3].
The critical importance of the anchoring fibrils in securing the adhesion of the dermal–epidermal basement membrane to the underlying dermis as well as in wound healing is illustrated by the dystrophic forms of EB [4]. Specifically, a complete absence of type VII collagen results in severe, generalized, recessive dystrophic EB with fragility of the skin and mucous membranes, leading to mutilating scarring of the hands and feet. Missense mutations, particularly glycine substitution mutations, can result in somewhat milder, dominantly inherited dystrophic EB [5].
Extracellular matrix
The major component of human skin is the dermis, demarcated on the top by the lamina densa in the lower border of the dermal–epidermal basement membrane and at the bottom by the subcutis. In contrast to the epidermis, the dermis is largely acellular and consists primarily of the extracellular matrix of connective tissue, a complex meshwork of various macromolecules. There are four major classes of extracellular matrix components: (i) collagen fibres, which provide tensile strength to allow the skin to serve as a protective organ against external trauma; (ii) elastic structures, which provide elasticity and resilience to normal human skin; (iii) non-collagenous glycoproteins, such as fibrillins, fibulins and integrins, which often serve as organizers of the matrix and facilitate cell–matrix interactions; and (iv) proteoglycan/glycosaminoglycan macromolecules, which provide hydration to the skin. The maintenance of proper quantities and appropriate interactions between the extracellular matrix components is a prerequisite for the physiological homeostasis of the dermis.
The major extracellular matrix component in the dermis is collagen, which comprises a family of closely related yet genetically distinct proteins [1, 2, 3, 4]. The major collagen fibres in the dermis provide tensile strength to the skin to serve as a protective organ against external trauma (Figure 2.42). A characteristic feature of all collagens is the triple helical conformation, which is predicated upon the primary amino acid sequence of the subunit polypeptides, α-chains, depicting a repeating glycine-X-Y sequence. The collagens also demonstrate non-collagenous flanking segments at the ends of the individual molecules. Currently, 29 distinct collagens have been identified in vertebrate tissues, and have been characterized to the extent that they are referred to by Roman numeral designation (I–XXIX) in the order of their discovery, many of them being present in the skin (Table 2.2).
Table 2.2 Genetic heterogeneity of collagens
| Collagen type | Chain composition | Supramolecular assembly | Tissue distributiona |
| I | [α1(I)]2α2(I) | Fibrillar | Dermis, bone, tendons |
| III | [α1(III)]3 | Fibrillar | Fetal dermis, blood vessels, GI tract |
| IV | [α1(IV)]2α2(IV)b | Basement membrane | Ubiquitous |
| V | [α1(V)]2α2(V)c | Fibrillar | Ubiquitous |
| VI | [α1(VI)α2(VI)α3(VI)]d | Microfibrils | Ubiquitous |
| VII | [α1(VII)]3 | Anchoring fibrils | Epithelial basement membranes |
| VIII | [α1(VIII)]3 | Network forming | Endothelia |
| XIII | [α1(XIII)]3 | Transmembrane | Ubiquitous, including epidermis |
| XIV | [α1(XIV)]3 | FACIT | Skin, cornea |
| XV | [α1(XV)]3 | Basement membrane | Ubiquitous |
| XVII | [α1(XVII)]3 | Transmembrane | Hemidesmosomes |
| XXIX | Unknown | Unknown | Epidermis |
aDistribution in the skin and other major tissues is indicated; lesser amounts may be present in other tissues.
b Additional α-chains have been identified.
FACIT, fibril-associated collagens with interrupted triple helices; GI, gastro-intestinal.

Figure 2.42 Transmission electron micrograph of a section of dermis from the human forearm showing bundles of collagen fibres, both in transverse and longitudinal sections. Original magnification 4900×.
(Courtesy of Professor A. S. Breathnach.)
All collagen molecules consist of three subunit polypeptides, which can either be identical as homotrimers or can consist of two or even three genetically different polypeptides in heterotrimeric molecules. Since the different subunits are all distinct gene products, there are over 40 different genes in the human genome that encode the different subunit polypeptides, the α-chains, of these distinct collagens [5].
Collagens
On the basis of their fibre architecture in tissues, the genetically distinct collagens can be divided into different classes [1, 2]. Types I, II, III, V and IX align into large fibrils and are designated as fibril-forming collagens. Type IV is arranged in an interlacing network within the basement membranes, while type VI is a distinct microfibril-forming collagen, and type VII collagen forms anchoring fibrils. FACIT collagens (fibril-associated collagens with interrupted triple helices) include types IX, XII, XIV, XIX, XX and XXI. Many of the latter collagens associate with larger collagen fibres and serve as molecular bridges, stabilizing the organization of the extracellular matrices. The major collagens significantly contributing to skin physiology and pathology are presented in Table 2.2. Other collagen types, including types II, IX, X, XI, XIX, XX and XXI, are not discussed in detail because they are not present in the skin to a significant extent or their participation in maintaining skin physiology is unclear.
Type I collagen, the most abundant form, is the predominant collagen in human dermis, accounting for approximately 80% of the total collagen. Type I collagen contains two different kinds of α-chain with an [α(I)]2α2(I) stoichiometry. A collagen consisting of three identical α1(I) chains has also been identified (so-called α1(I) trimer), but it appears to be only a minor component of connective tissue in the skin. Type I collagen associates with type III collagen to form broad, extracellular fibres in the human dermis. Mutations in type I and III collagens, or in their processing enzymes, can result in connective tissue abnormalities in the different forms of Ehlers–Danlos syndrome, and mutations in the type I collagen gene are responsible for the fragility of bones in osteogenesis imperfecta.
Type III collagen accounts for about 10% of the total bulk of collagen found in adult human dermis. It was originally shown to predominate in human skin during embryonic development, but during the early postnatal period type I collagen synthesis accelerates, resulting in the ratio of type I to type III collagen in the adult human skin being approximately 8 : 1. Type III collagen is most prominent in vascular connective tissues, the gastro-intestinal tract and the uterus. It consists of three identical α-chains, α1(III), and mutations in the type III collagen gene can cause the vascular type of the Ehlers–Danlos syndrome [3].
Type IV collagen is a basement membrane collagen present within the dermal–epidermal junction as well as in the vascular basement membranes. The predominant form of type IV collagen in human skin is a heterotrimer of [α1[IV)]2α2(IV), although occasional homopolymers from these two chains may be assembled. The type IV collagen molecule is characterized by the presence of non-collagenous interruptions within the triple helical domains, thus conferring flexibility to the molecule. In addition to α1 and α2 chains, four other polypeptides of type IV collagen have been identified [4]. Type IV collagen molecules containing these polypeptides are present primarily in the glomerular basement membranes, and their importance for renal physiology is attested by the fact that mutations in the gene encoding the α5(IV) polypeptide results in Alport syndrome. The α3(IV) chain harbours the epitopes recognized by antibodies in Goodpasture syndrome, and the α5 chain of type IV collagen has been shown to be a target of circulating autoantibodies in a novel autoimmune disease with subepidermal blisters and renal insufficiency.
Type V collagen consists of interrelated collagens containing four different types of α-chains. Type V collagen is present in most connective tissues, including dermis, where it represents less than 5% of the total collagen. Type V collagen is located on the surface of large collagen fibres in the dermis, and its function is to regulate the lateral growth of these fibres. Thus, in the absence of type V collagen, the collagen fibre diameter is variable and the contour of the individual fibres can appear irregular, some of them having ‘flower-like’ morphology in cross-section. The importance of type V collagen in contributing to connective tissue stability is attested by the fact that mutations in the type V collagen gene underlie most patients with classic, autosomal dominant forms of Ehlers–Danlos syndrome [3].
Type VI collagen, as originally discovered, consists of three distinct α-chains, α1(VI), α2(VI) and α3(VI), which fold into a relatively short, triple helical domain and contain large globular domains at both ends. More recently, three additional α-chains have been suggested to belong to the type VI family of collagens [5]. Type VI collagen is a relatively minor collagen in human dermis, where it assembles into thin microfibrils independent of the broad collagen fibres, which consist primarily of type I and type III collagens. The microfibrillar network has an anchoring function, stabilizing the assembly of collagen fibres as well as basement membranes. Mutations in the three type VI collagen genes can lead to different forms of muscular dystrophy with little effect on the physiology of skin [6].
Type VII collagen, the major if not the exclusive component of anchoring fibrils, consists only of one type of α1 chain, α1(VII). This polypeptide has a characteristic modular structure, with the central collagenous domain being flanked by amino-terminal (NC1) and carboxy-terminal (NC2) collagenous domains with homology to known protein sequences.
Type VII collagen molecules become organized into anchoring fibrils through the formation of antiparallel dimers linked through their carboxy-terminal ends (Figure 2.34). The large amino-terminal, non-collagenous NC1 domains interact with type IV collagen and laminin 332 at the dermal–epidermal basement membrane, forming U-shaped loops that entrap larger fibres in a manner that stabilizes the association of the lower part of the lamina densa to the upper papillary dermis (Figure 2.41). Consequently, altered expression or changes in the molecular interactions of type VII collagen with other basement membrane components can result in skin fragility as exemplified by the dystrophic forms of epidermolysis bullosa [7]. In addition to the heritable forms of EB, type VII collagen serves as an autoantigen in the autoimmune blistering skin disease EB acquisita, the majority of antigenic epitopes residing within the NC1 domain [8].
Type XVII collagen was initially identified as the 180 kDa bullous pemphigoid antigen (BPAG2) recognized by circulating autoantibodies in the sera of patients with bullous pemphigoid or herpes gestationis. Subsequent characterization of the protein and the corresponding gene has indicated that BPAG2 is, in fact, a collagenous molecule consisting of 15 collagenous domains with characteristic Gly-X-Y repeat sequences which form triple helices (Figure 2.43) [9]. Type XVII collagen is a transmembrane protein in type 2 topography, that is the amino-terminal segment of the molecule is intracellular while the carboxy-terminal ectodomain, containing the collagenous segments, is in the extracellular space. The importance of type XVII collagen to the stability of the dermal–epidermal junction is attested to by the fact that mutations in the corresponding gene (COL17A1) result in a generalized intermediate variant of junctional EB, originally designated as non-Herlitz or generalized atrophic benign EB, and circulating autoantibodies to type XVII collagen (BPAG2) are associated with bullous pemphigoid [10].

Figure 2.43 Type XVII collagen, a transmembrane protein in type 2 orientation. Note that the ectodomain traversing the lamina lucida contains 15 distinct triple helical collagenous segments (COL1–15). The non-collagenous segment between COL15 and the transmembrane domain, NC-16A, harbours the major epitopes recognized by autoantibodies in bullous pemphigoid. ADAMTS, a disintegrin and metalloproteinase with thrombospondin motifs, cleaves the protein at a sequence in the NC-16A, resulting in release of the ectodomain. (Adapted from Powell et al. 2005 [12].)
Type XXIX collagen is a putative epidermal collagen with a specific gene expression pattern; the highest level of expression is in the skin, lung and gastro-intestinal tract [11]. In the skin, expression is restricted to the epidermis with the highest level in suprabasal layers. This collagen was initially identified through genetic linkage of patients with AE to a locus on the short arm of chromosome 3q21. The locus contained a single gene encoding a collagenous segment flanked by multiple von Willebrand factor A-like domains, with a high degree of homology with the α3 chain of type VI collagen. Immunofluorescence staining of skin from patients with AE demonstrated a striking lack of collagen XXIX in the viable, outermost spinous and granular layers, suggesting a role in AE.
Collagen biosynthesis
The genetically distinct collagens demonstrate considerable tissue specificity, and, accordingly, they are synthesized by a number of different cell types, including dermal fibroblasts, epidermal keratinocytes, vascular endothelial cells and smooth muscle cells. The individual α-chains are initially synthesized as precursor molecules, pro-α-chains, with non-collagenous extensions at the ends of the collagenous domain (Figure 2.44).
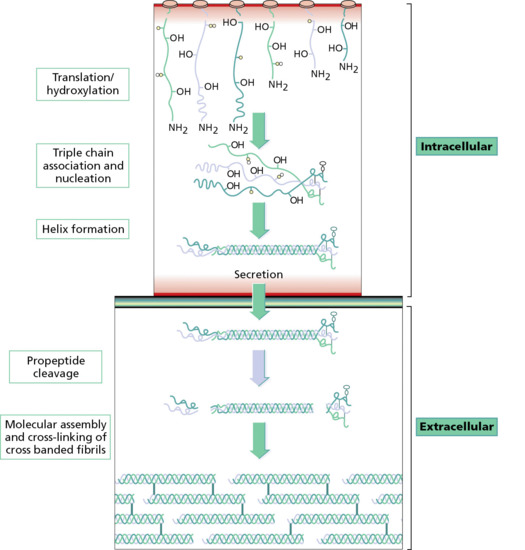
Figure 2.44 Steps in the intracellular biosynthesis of triple helical type I procollagen, its secretion into the extracellular space, and assembly and cross-linking of mature collagen fibres in the extracellular space.
While in the rough endoplasmic reticulum, three individual pro-α-chains assemble into a trimeric molecule through interactions of the non-collagenous sequences at the carboxy-terminal end. Upon completion of the prolyl hydroxylation reactions, the collagenous domains of the α-chains fold into a triple helical conformation, and the collagen molecules are then secreted through Golgi vesicles into the extracellular milieu. In the extracellular space, parts of the non-collagenous peptide extensions are cleaved by specific proteases, and the collagen molecules then assemble into their tissue-specific supramolecular organization. For example, the fibrillar collagens align into a characteristic quarter-stagger arrangement and form fibres, the growth occurring at the tip of the growing fibre. The coarse collagen fibres in the mid-dermis consist primarily of type I and III collagens, and type V collagen associates with them on the surface of the fibre so as to regulate the diameter of the growing fibre. Type VII collagen assembles into centrosymmetrical anchoring fibrils within the dermal–epidermal basement membrane zone, and type XVII collagen assumes a transmembrane type 2 orientation as a component of the hemidesmosomes. A characteristic feature of collagen is the presence of hydroxyproline and hydroxylysine residues, two amino acids that are post-translationally synthesized by hydroxylation of proline and lysine residues, respectively (Figure 2.45) [1].
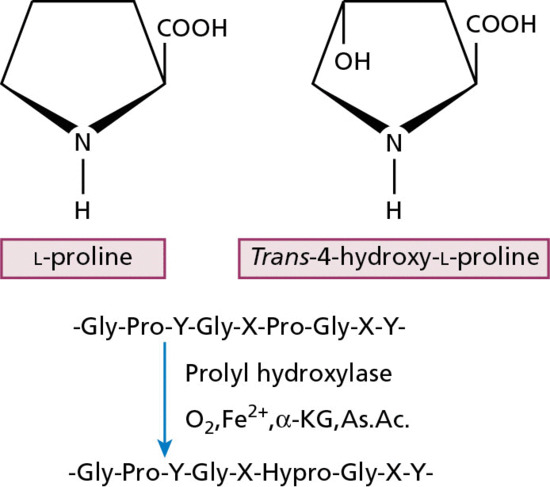
Figure 2.45 Enzymatic hydroxylation of prolyl residues in the Y-position of the repeating Gly-X-Y amino acid sequence to form hydroxyproline, an amino acid characteristic of collagen. Note that the reaction requires molecular oxygen, ferrous iron, α-ketoglutarate (α-KG) and ascorbic acid (As.Ac.) as co-factors.
These hydroxylation reactions are catalysed in the rough endoplasmic reticulum by prolyl and lysyl hydroxylases, respectively, enzymes that require ascorbic acid, molecular oxygen and ferrous iron as co-factors. The hydroxylation of prolyl residues is necessary for the stabilization of the triple helical conformation at physiological temperatures, and hydroxylysyl residues are required for the formation of stable covalent cross-links. Thus, for example, as a result of ascorbic acid deficiency in scurvy, the hydroxylation reactions are suboptimal, the newly synthesized collagen is poorly functional, and clinically scurvy manifests with connective tissue weakness. Similarly, low oxygen tension in chronic ulcers and wounds due to poor circulation may impair collagen production, resulting in poor healing.
Hydroxylation of the lysyl residues is followed by O-glycosylation, catalysed first by galactosyltransferase, which adds a galactosyl residue to the hydroxyl group of hydroxylysine, followed by a glucosyltransferase reaction to form glucosyl-galactosyl-hydroxylysine in O-glycosidic linkage. Additional glycosylation in N-glycosidic linkage will take place on the non-collagenous extensions at the end of the triple helical molecule, but the functional significance of these glycosylation reactions is currently unclear.
Collagen biology
The regulation of collagen gene expression has to be tightly controlled in order to maintain normal amounts and ratios of genetically distinct collagens under physiological conditions. At the same time, regulatory mechanisms have to be responsive to the needs of rapid collagen synthesis in repair processes, such as wound healing. On the other hand, uncontrolled collagen synthesis can lead to excessive accumulation of collagen in fibrotic diseases, as exemplified by systemic sclerosis, keloids and hypertrophic scars [1, 2].
An important control mechanism is at the level of collagen mRNA formation through regulation of the transcriptional activity of the corresponding genes. In general, there is a good correlation between the rate of collagen biosynthesis and the corresponding procollagen mRNA levels, as demonstrated in several in vitro models, including cultured fibroblasts. The transcriptional regulation of collagen gene expression involves a number of both cis-acting elements and trans-acting factors. The cis-acting elements, representing nucleotide sequences within the regulatory regions of the gene that serve as binding sites for trans-acting regulatory proteins, have been identified in most collagen gene regulatory regions. Such factors can either up-regulate or suppress the transcriptional promoter activity. An example of such trans-acting factors are the retinoic acid nuclear receptors (RAR and RXR) that form a complex with the ligand (a retinoid), which then binds to the retinoic acid-responsive elements (RARE) in the target gene. Retinoids, such as all-trans-retinoic acid, have been shown to modulate collagen gene expression both in vitro and in vivo, and quiescent non-proliferating cells can be stimulated by retinoic acid to activate type I collagen synthesis. These observations may have relevance to the elevated rate of collagen synthesis observed in photodamaged dermis treated by the topical application of retinoids [2]. All-trans-retinoic acid has also been demonstrated to increase the density of anchoring fibrils along the cutaneous BMZ in adult human skin, suggesting that retinoids are capable of up-regulating type VII collagen gene expression.
Collagen gene expression can also be modulated by a number of cytokines and growth factors, and one of the most powerful modulators of connective tissue gene expression is TGF-β [1, 3]. In general, TGF-β is pro-fibrotic and it has been shown to up-regulate the expression of a number of extracellular matrix protein genes, including those encoding collagen types I, III, IV, VI and VII. Elevated levels of TGF-β have also been demonstrated in various fibrotic lesions, including the skin in systemic sclerosis and keloids. The up-regulatory activity of TGF-β can be counteracted by other cytokines, including tumour necrosis factor α and interferon γ, which antagonize the TGF-β action [1]. These cytokines have been tested for their efficacy for the treatment of keloids and other fibrotic diseases, with variable results.
A number of hormones clearly regulate collagen gene expression, as certain endocrine disorders dramatically change the amount of collagen found in connective tissues, including the skin. Glucocorticosteroids also affect collagen biosynthesis; inhibition is much more pronounced with fluorinated steroids compared with hydrocortisone. The glucocorticosteroid inhibition of collagen biosynthesis occurs in lower concentrations at the transcriptional level through inhibition of promoter activity. In higher concentrations and with more potent glucocorticosteroids, inhibition of prolyl hydroxylase activity also is evident, leading to deficient hydroxylation of collagen polypeptides and subsequently to reduced amounts of newly synthesized collagen. These mechanisms would explain the connective tissue side effects, such as dermal atrophy, associated with intralesional or prolonged topical application of fluorinated glucocorticosteroids.
Collagen cross-linking
The alignment of collagen molecules into their specific supramolecular organization occurs spontaneously, but these fibre structures do not attain the necessary tensile strength until the molecules have been covalently linked together by specific intra- and intermolecular cross-links [1]. The commonest forms of cross-link in type I collagen are derived from lysine and hydroxylysine residues, and in some collagens there are also cysteine-derived disulphide bonds. The first step in the cross-linking process is enzymatic synthesis of aldehyde residues from lysyl and hydroxylysyl residues by removal of the ε-amino group of these amino acids (Figure 2.46).
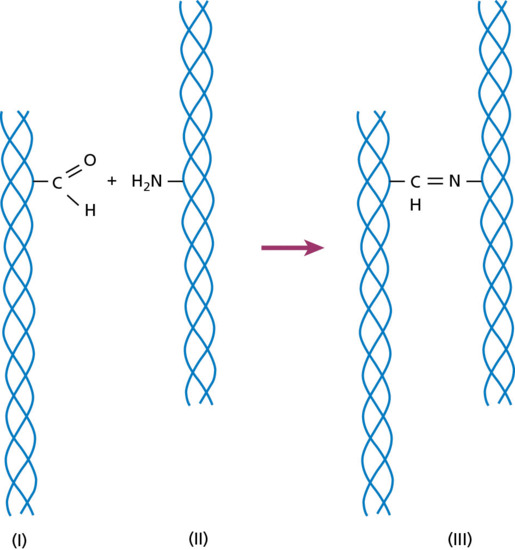
Figure 2.46 Formation of intermolecular cross-links between individual collagen molecules. The cross-linking is initiated by the conversion of lysine or a hydroxylysine residue that contains an ε-amino group to a corresponding aldehyde (I). The aldehyde then reacts with an unmodified ε-amino group in an adjacent collagen molecule (II) to form a Schiff base-type covalent cross-link (III).
This oxidative deamination reaction is catalysed by the lysyl oxidases, a group of enzymes that require copper as a co-factor. These enzymes act primarily upon native collagen fibrils and poorly, if at all, on denatured collagen (gelatin) or isolated α-chains. Similar deamination reaction catalysed by lysyl oxidase occurs also in elastin.
In addition to the classic human lysyl oxidase, four additional lysyl oxidase-like (LOXL1–4) genes/proteins have been identified. These lysyl oxidase-like enzymes have been postulated to play a role in a number of disease processes. For example, LOXL1 gene sequence variants confer susceptibility to exfoliation glaucoma and elastin was found to be the major component of the ocular lesions in this disease [2, 3]. The expression of LOXL2 is increased in a number of cancers, and LOXL2 has been shown to be a marker for poor prognosis with decreased overall and disease-free survival in squamous cell carcinomas [4]. LOXL3 and -4 demonstrate structural features similar to lysyl oxidase and have wide tissue distribution [5, 6]. Alterations in lysyl oxidase activities have also been described in a number of experimental systems involving age-related changes in the cardiovascular system [7].
Collagen degradation
Collagen fibres, once fully matured by the cross-linking processes, are relatively stable and can exist in tissues under normal physiological conditions for long periods. However, there is continuous, yet slow, degradation and turnover of collagen in normal situations, as attested by continuous urinary excretion of hydroxyproline as a marker of collagen degradation. In addition, in certain physiological situations, as exemplified by reabsorption of the postpartum uterus, and in pathological conditions, such as tissue invasion and tumour metastases, degradation of connective tissues and particularly collagen is accelerated. There are a number of enzymes that comprise the family of matrix metalloproteinases (MMPs), enzymes capable of degrading the extracellular matrix components. These proteinase families include the collagenases, gelatinases, stromelysins, matrilysins and membrane-type MMPs (Figure 2.47) [1, 2].
Figure 2.47 Structural organization of various matrix metalloproteinases (MMPs), divided into different subclasses. The signal, propeptide, active catalytic hinge and haemopexin regions are indicated. Note that MMP7 lacks the haemopexin region, while MMP14–17 harbour membrane-binding sequences at the carboxy-terminal end.
Native collagen is resistant to non-specific proteolytic degradation in physiological situations due to the fact that the triple helical conformation is not readily degradable by general proteases. However, collagenases have the ability to degrade collagen triple helix at physiological pH and temperature. The vertebrate collagenase was initially isolated from tadpole tails which, when cultured upon reconstituted type I native collagen substrate, exercise proteolytic activity. Similar techniques were subsequently employed to demonstrate the presence of collagenase in human skin. Interstitial collagenase (MMP1) was initially shown to be synthesized as a proenzyme by cultured fibroblasts, and later, different cell types, including epidermal keratinocytes, were shown to express a similar or identical enzyme. The ability of interstitial collagenase to digest the type I collagen triple helix is based on its ability to specifically cleave the α1(I) chain at a particular glycine–isoleucine peptide bond, or the α2(I) chain at a glycine–leucine peptide bond. This initial cleavage results in two degradation products, three-quarters and one-quarter of the size of the original collagen molecule. These shortened triple helical fragments have a lower helix-to-coil transition temperature (Tm) than the full-length molecule. Subsequently, at temperatures below 37°C the triple helix unravels, rendering the individual polypeptides susceptible to general proteolytic degradation. It should be noted that type I collagen has several additional glycine–isoleucine and glycine–leucine sequences, but these are not susceptible to collagenase degradation in this collagen when in the native triple helical conformation. A similar enzyme (MMP8) has been identified in human neutrophils with comparable degrading characteristics. The neutrophil collagenase is stored in neutrophil granules and released upon stimulation. MMP1 and MMP8 can, in addition to type I collagen, degrade a number of other collagens, including types III and VII.
Another group of extracellular proteolytic enzymes is that of the gelatinases, which are able to degrade denatured collagen (gelatin) but can also cleave certain native collagens, such as types IV, V and VII, with certain interruptions or imperfections in their collagenous triple helices, thus allowing the proteolytic cleavage at these sites. Basement membrane collagen IV can also be degraded by MMP3 (stromelysin-1) and MMP10 (stromelysin-2).
In general, MMPs are synthesized and secreted as inactive proenzymes, which become activated proteolytically by removal of the propeptide. The MMPs are zinc metalloenzymes and require calcium for their activity. Consequently, the enzymes can be inhibited by chelators of divalent cations, and, pharmacologically, tetracyclines have been suggested to inhibit MMP proteolytic activity due to their ability to bind calcium. The MMPs also have specific, small-molecular-weight peptide inhibitors, so-called tissue inhibitors of metalloproteinases (TIMPs). These proteins complex stoichiometrically with MMPs to prevent the degradative events.
In normal human skin, a number of MMPs are synthesized and secreted by fibroblasts and keratinocytes. The expression of these enzymes is activated in various pathological situations, including the invasion and metastasis of cutaneous malignancies, as well as during dermal wound healing and epidermal regeneration [2, 3, 4]. Finally, proteolytic enzymes play a pathophysiological role in tissue separation in a number of blistering diseases, such as bullous pemphigoid, dermatitis herpetiformis and epidermolysis bullosa acquisita [5].
Another metalloproteinase family has been designated as ADAMTS (a disintegrin and metalloproteinase with thrombospondin motifs). The prototype, ADAMTS-1 gene can be induced by interleukin 1 in vitro or by lipopolysaccharide injection in mice, and, thus, this metalloproteinase was initially associated with inflammatory processes. Subsequently, a number of ADAMTS genes have been identified with similar domain organizations, consisting of a signal sequence, a propeptide, a metalloproteinase domain, a disintegrin-like domain, a cysteine-rich region and a variable number of thrombospondin type 1 molecules (Figure 2.48). These molecules are zinc-dependent proteases with a high level of expression in fetal tissues. ADAMTS proteases also have high levels of expression in tumour cells and tissues, including melanoma and colon carcinoma. Of particular interest in the context of collagen processing is ADAMTS-2, which serves as a procollagen I/II amino-propeptide processing enzyme.
Figure 2.48 Main organization of various ADAMTS family metalloproteinases. The catalytic domain is shown in red, while other domains include the thrombospondin type I repeat sequences (green), disintegrin-like domain (blue) and cysteine domain (yellow). The signal peptidase is shown in black, the propeptide sequence is coloured pink and the spacer domain is in orange. ADAMTS, a disintegrin and metalloproteinase with thrombospondin motifs.
Regulation of extracellular matrix turnover and collagen degradation during postpartum involution of the uterus has been attributed to relaxin, a hormone initially implicated in pregnancy-related conditions. More recently, it has become clear that a number of tissues, including skin, can serve as targets of relaxin. These tissues contain a relaxin family peptide receptor 1 (RFPR1) that mediates the relaxin effects on connective tissue metabolism, contributing to the maintenance of tissue homeostasis. The critical role of relaxin and its receptor has been illustrated by targeted mutant mice in which the absence of relaxin leads to collagen accumulation in a number of tissues, similar to systemic sclerosis [6, 7].
Elastic fibres
An integral component of the dermal connective tissue is the elastic fibre network, which provides resilience and elasticity to the skin [1, 2]. Elastic fibres are a relatively minor component in normal sun-protected adult skin, being less than 2–4% of the total dry weight of the dermis. The elastic fibre system in the reticular dermis consists of horizontally orientated fibres that interconnect to provide a network structure. Extending from these horizontal fibres is a network of vertical extensions of relatively fine fibrils, which consist either of bundles of microfibrils (oxytalan fibres) or of small amounts of cross-linked elastin (elaunin fibres) (Figure 2.49).
Figure 2.49 Immunofluorescence staining of type I collagen (a,d) and the elastic fibre network (b,e) in the dermis of human skin visualized by confocal laser scanning microscopy. Merging of the images (c,f) reveals that the elastic fibres assume a horizontal orientation in the mid-dermis while vertical extensions (oxytalan and elaunin fibres) reach the upper dermis, terminating just below the dermal–epidermal junction. Note that in sun-damaged skin there is a dramatic decrease and disorganization of both collagen and elastic fibres in comparison with sun-protected skin. (Adapted from Uitto and Bernstein 1998 [3].)
Elastic fibres have two principal components: (i) elastin, a well-characterized connective tissue protein that forms the core of the mature fibres; and (ii) the elastin-associated microfibrils which consist of a family of proteins, some of them less defined. Examination of mature elastic fibres by transmission electron microscopy reveals an electron-lucent core that consists of elastin (Figure 2.50). Surrounding the elastin core are distinct microfibrillar structures, which appear electron dense under routine electron microscopy staining. While elastin is the major component and the microfibrils are less conspicuous in fully mature elastic fibres, the relative proportion of these two components varies during the embryonic development of elastic fibres and/or connective tissue repair. The first elements of elastic fibres that form consist of bundles of microfibrils, which can be visualized by electron microscopy during the first trimester of gestation. These microfibrils form a scaffold, allowing alignment of the elastin molecules in parallel array so as to guide the growth of fibres with relatively uniform diameters. During the second trimester of fetal development, the elastic fibres remain immature, but with increasing fetal age maturation of the fibres occurs and the elastin component becomes more prominent. In fully developed elastic fibres, well over 90% of the total content is elastin with relatively few microfibrillar components, mostly confined to the outer surface of the fibres.
Figure 2.50 Transmission electron microscopy of an elastic fibre in the reticular dermis. The central electron-pale core consists of elastin (E), while the electron-dense areas represent the elastin-associated microfibrillar proteins which are particularly evident at the periphery of the fibre (arrow). Scale bar 0.5 μm.
(Courtesy of Professor R. A. J. Eady, St John's Institute of Dermatology, King's College London, UK.)
Elastin
Elastin is initially synthesized as a precursor polypeptide, ‘tropoelastin’, which consists of approximately 700 amino acids with a molecular mass of approximately 70 kDa [1]. The amino acid composition of tropoelastin is similar to collagen, in that about one-third of the total amino residues consist of glycine. However, glycine is not evenly distributed in elastin in every third position as it is in a typical collagenous sequence. Instead, the tropoelastin primary sequence shows domains rich in glycine, valine and proline, alternating with lysine- and alanine-rich sequences. A characteristic sequence motif in the latter setting is the presence of two lysine residues separated by two or three alanine residues (Figure 2.51).
Figure 2.51 Assembly and cross-linking of elastic fibres. Newly synthesized elastin precursor polypeptides, tropoelastins, with alternating hydrophobic and cross-link regions are secreted into the extracellular milieu. Lysine residues in characteristic lys-ala-ala-lys or lys-ala-ala-ala-lys sequences in the cross-link region undergo oxidative deamination of the ε-amino groups catalysed by lysyl oxidase, an enzyme requiring copper as a co-factor. Three resulting allysine residues fuse with an unmodified lysine residue to form desmosines, elastin-specific cross-link molecules. (Adapted from Mahoney et al. 2009 [7].)
The lysine residues in tropoelastin are critical for the formation of covalent cross-links, desmosine and its isomer, isodesmosine, which are unique to elastin. The first step in the formation of these elastin-specific cross-links is oxidative deamination of three lysine residues to form corresponding aldehydes, so-called allysines. Three of these resulting aldehydes, together with a fourth lysine residue containing the intact unmodified ε-amino group, spontaneously fuse to form a stable desmosine compound which covalently links two of the tropoelastin polypeptides. The addition of desmosines to other parts of the molecule progressively converts tropoelastin molecules into an insoluble fibrous structure which can be stretched, but upon release the fibres recoil, providing resilience and elasticity to the skin (Figure 2.52). The content of desmosine in various elastin preparations is fairly constant, with approximately 1.5 residues per 1000 amino acids, and desmosine or isodesmosine can therefore provide a quantitative measure of the insoluble elastin content in tissues.
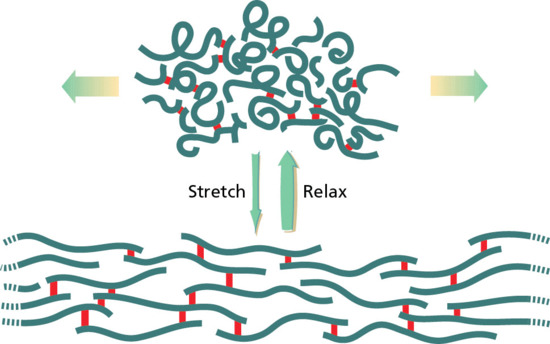
Figure 2.52 Elastic fibres cross-linked by desmosines (red). In the relaxed state, the fibres assume coiled-coil conformations. When the fibres are stretched and then released, they return to a relaxed state, the contraction providing elasticity and resilience to the skin.
The human elastin gene spans approximately 45 kb of genomic DNA on chromosome 7 and consists of 34 exons corresponding to 3.5 kb of human elastin mRNA sequences. Examination of the gene structure reveals that the alternating cross-link domains, characterized by the presence of lysyl residues separated by two or three alanines, and the hydrophobic domains, are encoded by individual exons. The elastin mRNAs are synthesized in a number of cell types present in elastin-rich tissues, as for example, the vascular smooth muscle cells in arterial connective tissues. In the skin, the primary cell type responsible for elastin production appears to be the fibroblast which, under tissue culture conditions, expresses the elastin gene. Keratinocytes have also been suggested to express the elastin gene, but the level of expression is very low in comparison with dermal fibroblasts and the potential significance of elastin in the epidermis remains unclear.
Primary mutations in the elastin gene have been demonstrated in cutis laxa, a group of diseases that manifest with loss or fragmentation of elastic fibres [2, 3]. It should be noted, however, that this group of heritable diseases is highly heterogeneous, and mutations in the fibulin-4 and fibulin-5 genes have also been observed. Williams syndrome is a contiguous gene deletion syndrome that also involves the elastin gene, with clinical manifestations predominantly in the cardiovascular system [4]. Finally, cutis laxa can develop as a post-inflammatory condition, probably mediated by proteolytic enzymes released from the inflammatory cells [5].
An interesting observation during the processing of elastin mRNA precursor molecules is that they undergo extensive alternative splicing, leading to the formation of elastin molecules of varying primary sequences. In fact, at least six exons in the human elastin gene have been reported to be subject to alternative splicing, and this mechanism can provide significant variation in the primary sequence composition of elastin polypeptides, leading to different types of elastic fibres in different tissues. However, the physiological significance of the alternative splicing has not been established.
The oxidative deamination of lysyl residues to corresponding aldehydes is catalysed by a group of enzymes, lysyl oxidases, which require copper for their activity. Thus, copper deficiency can lead to reduced lysyl oxidase activity and the synthesis of elastic fibres that are not stabilized by sufficient amounts of desmosines. In such a situation, the individual tropoelastin polypeptides remain soluble and susceptible to non-specific proteolysis, and the elastin-rich tissues are fragile. Clinical manifestations of copper deficiency can vary depending on the level of copper and its circulating transport protein, caeruloplasmin, as manifested by Menkes syndrome and the occipital horn syndrome, two allelic conditions due to mutations in the copper transporter protein gene, ATP7A [6]. Copper deficiency can also occur in patients undergoing long-term treatment with high doses of D-penicillamine, a copper chelating agent, which can result in abnormalities in the elastic structures in the skin and other tissues.
The metabolic turnover of elastin is slow, but a portion of elastin in the body is continuously degraded, as reflected by the continuous presence of desmosines in the urine. Thus there may be an ongoing turnover and repair of elastic fibres in normal tissues. In addition, there are a number of pathological conditions in which degradation of elastin is the histopathological hallmark, such as in some forms of cutis laxa and cutaneous ageing. Elastic fibres are degraded by elastases, a group of elastolytic enzymes in different tissues and with different cleavage specificities. The classic elastases, such as those originally isolated from the pancreas, are serine-proteases, and their activity can be inhibited by serum factors such as α1-antitrypsin and α2-macroglobulin. In addition to these classic serine-elastases, there are a number of metalloenzymes that are capable of degrading elastic structures, particularly the microfibrillar components. These metalloelastases are present in the skin and originate from fibroblasts and monocyte–macrophages.
Elastin-associated microfibrils
Elastin-associated microfibrils consist of tubular structures of approximately 10–12 nm in diameter. Both ultrastructural evidence and biochemical analyses have confirmed that the microfibrils differ from elastin, and they may also be found in a number of tissues as individual microfibrillar structures without direct association with elastin. It is now known that elastin-associated microfibrils consist of a number of proteins, which can be divided into several different categories based on their molecular characteristics. Many of them form gene families with closely related structure and function, but clearly different from other groups in their structural features.
One of the microfibrillar protein families is that of the fibrillins, which are a critical part of the microfibrillar structure [1]. Two distinct, yet closely homologous, human genes encode fibrillin 1 (FBN1) and fibrillin 2 (FBN2), proteins characterized by multiple repeats of sequence motifs previously observed in the epidermal growth factor (EGF) precursor molecule, with each motif having six conserved cysteine residues. Electron microscopy has established that monomeric fibrillin molecules synthesized by fibroblasts show an extended flexible molecule, which is approximately 148 nm long and 2.2 nm wide. Multiple fibrillin molecules can then align in a parallel, head-to-tail fashion to form microfibrils associated with elastin in tissues, such as skin and the arterial connective tissues. It should be noted that fibrillin is also a major component of microfibrils in tissues such as the ocular ciliary zonule and the periodontal ligament, without microscopic or immunoreactive evidence of elastin. The importance of fibrillin 1 is illustrated by the fact that the mutations in the corresponding gene (FBN1) underlie Marfan syndrome, manifesting with skeletal abnormalities, aortic dilatations, subluxation of the lens and cutaneous laxity [2]. Fibrillin 2 (FBN2) mutations cause congenital contractural arachnodactyly with some similarities, but also differences, to Marfan syndrome.
The latent TGF-β binding family of proteins (LTBP) has some structural similarities with the fibrillins, including repeating EGF-like domains [3]. TGF-β, a pro-fibrotic cytokine, is secreted as a latent complex bound to LTBP. There are at least four distinct proteins in the family, with a molecular weight ranging from 125 to 310 kD. One of the putative functions of LTBP is to facilitate the secretion of TGF-β or binding of the inactive complex to the cell surface where activation takes place. However, LTBPs have also been found as free proteins associated with components of the extracellular matrix. LTBP1, a prototype of this subfamily of elastin-associated microfibrillar proteins, is clearly a component of the elastic fibres in human skin, and its levels are altered in solar elastosis.
Another family of the elastin-associated microfibrillar proteins consists of fibulins, extracellular matrix glycoproteins with characteristic calcium binding EGF-like domains. Five distinct fibulins have been characterized (fibulins 1–5; FBLN-1–5), and at least four of them have been located within the elastic fibres in different tissues [4]. In addition, FBLN-5 has been shown to bind both muscle cells and elastin, thus apparently facilitating cell–matrix interactions. The importance of FBLN-4 and FBLN-5 in skin physiology is attested by the demonstration of mutations in the corresponding genes in patients with cutis laxa, manifesting with loose and sagging skin and loss of recoil [5].
In addition to fibrillins, LTBPs and fibulins, a number of other proteins have been shown to be associated with elastic fibres in the microfibrillar network. Several of these proteins belong to the families of microfibril-associated glycoproteins (MAGPs) or microfibril-associated proteins (MFAPs), highly acidic, relatively small molecules, some of which have been characterized in detail. Finally, interface proteins, so-called emilins, as well as lysyl oxidases critical for the cross-linking and stabilization of elastic fibre structures, have been shown to be associated with elastic fibres [6].
Proteoglycan/glycosaminoglycans
Proteoglycans form a number of subfamilies defined by a core protein to which polymers of unbranched disaccharide units, glycosaminoglycans (GAGs), are linked by an O-linkage to serine residues (Figure 2.53). There are a number of distinct core proteins, the number of attached disaccharides varies, and the molecular mass of GAGs is highly variable. Commensurate with variability in structure, different proteoglycans are of different functional importance as critical components of cell membranes and the extracellular matrix of the skin during development, homeostasis and disease [1].
Figure 2.53 Prototypic proteoglycan in which the central core (green) is hyaluronic and the link proteins are represented by S-shapes, joining the protein side chains and carbohydrate polymers. (Adapted from Stryer 1995 [6].)
GAGs are highly charged polyanionic molecules that attach to the core protein. The characteristic feature of GAGs is their primary structure, consisting of alternating pairs of different monosaccharides, glucose or galactose, joined in 1–3 or 1–4 linkage (Figure 2.54). After the initial synthesis of GAGs, the polymers undergo complex post-assembly modifications catalysed by specific enzymes. Sulphatases catalyse replacement of N-acetyl by N-sulphate and epimerases convert d-glucuronic to l-iduronic acid. The linear GAG chains, consisting of linked disaccharide units, are highly variable in size, ranging from just a few to several thousands. Consequently, the molecular mass of naturally occurring GAGs can range from 5 × 103 to 5 × 107 Da.
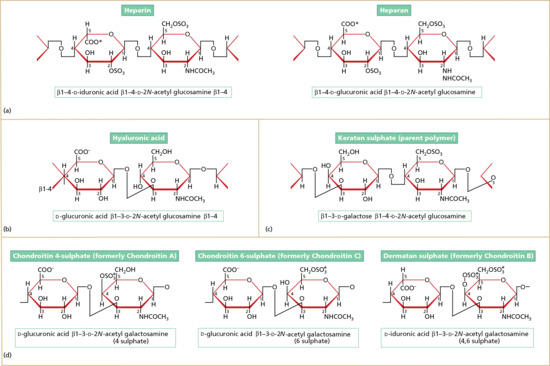
Figure 2.54 Glycosaminoglycan molecules that comprise the carbohydrate polymer side chains of proteoglycan molecules, including (a) heparin and heparan, (b) hyaluronic acid, (c) keratan sulphate and (d) various chrondroitin sulphates. Note the variants that include O-sulphation at the 6 position of both glucosamine and galactose.
The degree of post-assembly modifications is highly variable, and the control of this reaction depends on the specific characteristics of the GAGs, the associated core protein as well as the cell type and tissue environment (Figure 2.55). The simplest GAG, hyaluronic acid, is not sulphated, while other GAGs show sulphation of varying degrees. Within the individual GAG chains, there are regions that show either a low or high degree of sulphation, a feature that may facilitate interactions of proteoglycans and GAGs with their numerous binding partners.
Figure 2.55 Core protein aggrecan is joined by link proteins to hyluronate, with keratan sulphate (KS, blue) and chondroitin sulphate (CS, purple) side chains. aa, amino acids. (Adapted from Heinegard and Oldberg 1993 [7].)
The core proteins of proteoglycans have been increasingly characterized through cloning and sequencing of the corresponding genes. These genes are expressed in a number of different types of cells, including dermal fibroblasts which are the principal cell type for proteoglycan synthesis in the dermis. Newly synthesized core protein polypeptides are transferred to Golgi vesicles where the attachment of GAG chains occurs. The final product, consisting of a core protein with attached GAG chains, allows classification of the proteoglycans. It should be noted that hyaluronic acid is a GAG produced without synthesis of a core protein; instead, this macromolecule is synthesized by a complex of enzymes at the plasma membrane, with subsequent extrusion into the extracellular space.
Known core proteins with their predominant tissue distribution and associated GAG components are listed in Table 2.3. The core proteins can be intracellular, reside on the cell surface or be part of the extracellular matrix. For example, serglycin shows an intracellular core protein present in the secretory granules of haematopoietic cells, such as mast cells and eosinophils, associated with either heparan sulphate or chondroitin sulphate GAGs. This proteoglycan is found in the skin in areas infiltrated by mast cells or eosinophils, and, on subsequent release, serglycin is a major form of highly sulphated heparan sulphate GAG in the skin.
Table 2.3 Molecular characteristics and tissue distribution of selected proteoglycans (PG)
| PG | Protein (kDa) | Glycosaminoglycan(s) | Gene location | Tissue distribution |
| Decorin | 36 | CS/DS | 12q21–23 | Connective tissue |
| Biglycan | 38 | CS/DS | Xq28 | Cell surface |
| Fibromodulin | 42 | KS | 1q32 | Collagen matrix |
| Lumican | 38 | KS | 12q21–22 | Cornea, bowel, cartilage, muscle |
| Epiglycan | 36 | CS/DS | Epiphyseal cartilage | |
| Versican | 260–370 | CS/DS (10–30) | 5q13 | Skin, blood vessel, cartilage, brain |
| Aggrecan | 220 | CS (100) | 15q26 | Cartilage, blood vessel, brain |
| Perlecan | 400–470 | HS/CS | 1p36 | Cartilage, bone, marrow |
| Agrin | 210 | HS (3–6) | 1q32 | Cell membranes, kidneys, neuromuscular |
| Neurican | 136 | CS (3–7) | Brain | |
| Brevican | 100 | CS (1–3) | Brain | |
| Testican | 44 | HS/CS | 21 | Testis |
CS, chondroitin sulphate; DS, dermatan sulphate; HS, heparan sulphate; KS, keratan sulphate.
There are a number of cell surface proteoglycans that function at the interface between the plasma membranes and the extracellular matrix. The mode of attachment to the cell surface is variable. For example, the glypican family of proteoglycans is attached to the cell surface by a phospholipid anchor, while the syndecans have membrane-spanning core proteins. Syndecans and glypicans are present in a number of cells and tissues, including abundant expression in the skin. Syndecan expression varies during the development and maturation of tissues, and, for example, syndecan-1 is particularly abundant in keratinocytes. The nature of the attached GAG chains, however, changes as keratinocytes differentiate. Syndecans-1 and -4 are also induced in the dermis and granulation tissue, and it has been shown that deletion of syndecan-4 from mice greatly decreases the rate of wound repair. Furthermore, there are alterations in syndecan-1 expression as a result of malignant transformation.
The extracellular matrix contains a number of different proteoglycans as an integral component of the connective tissue meshwork. In the dermis, fibroblasts produce large proteoglycans, as exemplified by versican, consisting of a core protein with attachment sites for 12–15 GAG side chains (Figure 2.56). The GAGs in versican are primarily chondroitin sulphate or dermatan sulphate, but versican can also bind hyaluronic acid, resulting in the formation of large aggregates. In the skin, versican has been identified in the dermis and epidermis as a product of fibroblasts and keratinocytes, respectively.

Figure 2.56 Human versican gene: intron–exon organization (top) and deduced functional domains of the encoded protein. CRP, complement regulatory protein; EGF, epidermal growth factor; GAG, glycosaminoglycan; HBR, hyaluronan-binding region; SP, signal peptide; UTR, untranslated region. (Adapted from Dours-Zimmerman and Zimmerman 1994 [8].)
Extracellular matrix contains a number of small proteoglycans, exemplified by the family of leucine-rich repeat motifs. The prototype of this family is decorin, abundantly present in the skin. The decorin core protein is relatively small in size and has a single dermatan sulphate side chain covalently bound to a serine residue at the amino acid position 4 of the core protein. This proteoglycan was designated ‘decorin’ due to the observation that it associates with collagen and ‘decorates’ the fibres in vivo. This binding is attributed to the availability of decorin core protein to bind type I collagen, but the single GAG chain of decorin also binds to tenascin X, another extracellular component with affinity for collagen fibrils. Consequently, these interactions contribute to the connective tissue organization and architecture with functional consequences for normal skin physiology [2].
Proteoglycan–GAG complexes have a multitude of functions. For example, the proteoglycans containing heparan sulphate and dermatan sulphate have the ability to bind extracellular matrix components, including various collagens. In addition, these proteoglycans bind several growth factors, cytokines, cell adhesion molecules and growth factor-binding proteins and they can serve as antiproteases. In addition to binding to a number of extracellular molecules, proteoglycans also play a role in the adhesion of cells to the extracellular matrix. For example, syndecan-4, which is selectively enriched in dermal fibroblasts, facilitates the adherence of the cells in conjunction with other extracellular matrix-binding molecules, such as the integrins. Furthermore, the formation of focal adhesions requires heparan sulphate and subsequent activation of protein kinase C by a domain in the syndecan-4 core protein cytoplasmic tail [3].
Proteoglycans also interact with other extracellular matrix molecules besides collagen. In addition to decorin, which is known to associate primarily with type I collagen, chondroitin sulphate and dermatan sulphate bind fibronectin and laminin. The largest extracellular GAG, hyaluronic acid, plays an important role in providing physicochemical properties to the skin, mediated at least in part by its hydrophilicity and viscosity in dilute solutions. Most notably, hyaluronic acid has an expansive water-binding capacity, providing hydration to normal skin. The expression of hyaluronan is developmentally regulated in the skin, and the gene required for its synthesis, hyaluronan synthase, has been characterized. During wound healing, the physicochemical properties of hyaluronan may serve to expand the matrix and thus aid cell movement. The relatively high content of hyaluronan may also explain the finding that wounds in fetal skin heal without scarring. Other properties attributed to large proteoglycan complexes, such as those formed with versican or basement membrane proteoglycans, include their ability to serve as ionic filters, to regulate salt and water balance and to provide an elastic cushion [4].
Quantitative changes in the deposition of tissue proteoglycans have been encountered in a number of pathological processes. These include elevated hyaluronic acid synthesis in keloids and other fibrotic processes, as well as in pretibial myxoedema. In other skin conditions, including lichen myxoedematosus, systemic scleroderma and pseudoxanthoma elasticum, the lesional areas of skin have been reported to display abnormal amounts of proteoglycans. In most of these cases, the changes in proteoglycan/GAG content are secondary to an unrelated primary event. Finally, during innate cutaneous ageing in sun-protected areas of skin, the content of hyaluronic acid diminishes, possibly explaining the reduced turgor in aged skin [5].
Fibroblasts
The principal cell type responsible for the synthesis of connective tissue in the dermis is the fibroblast, which is of mesenchymal origin. The term fibroblast refers to a fully differentiated, biosynthetically active cell, while the term fibrocyte refers to an inactive cell. Biosynthetically active fibroblasts, as detected in developing or regenerating tissues, have an abundant cytoplasm, well-developed rough endoplasmic reticulum and prominent ribosomes attached to the membrane surfaces – features characteristic of cells engaged in active synthesis and secretion of extracellular matrix macromolecules (Figure 2.57). As indicated in the case of collagen, the newly synthesized polypeptides are first assembled in the cisternae of the rough endoplasmic reticulum, and the precursor polypeptides subsequently undergo extensive post-translational modifications. The polypeptides are then transferred to the Golgi vesicles and secreted to the extracellular milieu.

Figure 2.57 Transmission electron microscopy of an activated dermal fibroblast (F) in a healing wound. Note the prominent rough endoplasmic reticulum in the cytoplasm of this cell. There is an adjacent macrophage (M) with characteristic phagolysosomes, some of which contain ingested melanosomes.
(Courtesy of Professor R. A. J. Eady, St John's Institute of Dermatology, King's College London, UK.)
Human skin fibroblasts are the principal cell synthesizing collagen in the dermis. While the source of elastic fibres in the skin is less clear, fibroblasts clearly have the capacity to synthesize elastic tissues in vitro, and they probably are the primary source of elastin within the dermis as well. Finally, fibroblasts are the primary, if not the exclusive, cellular source of proteoglycan/glycosaminoglycan macromolecules. There is, however, considerable heterogeneity within fibroblast populations, and it has been demonstrated that the ratio of type I : III collagen synthesis or fibronectin expression in any given fibroblast population can be variable. For example, fibroblasts isolated from the papillary versus reticular dermis of skin have a higher rate of synthesis of type III collagen, and there can be as many as 30-fold differences in the level of fibronectin expression within individual cells [1].
The multiple functions of the stroma of vertebrate animals is dependent on the architecture of the extracellular matrix, which contains mesenchymal cells and provides a structural scaffold for blood and lymphatic vessels and nerves. Reciprocal interactions between the mesenchymal and epithelial cells are known to play a critical role in the development and morphogenesis of tissues, such as skin. More recently, the specific gene expression patterns in cultured fibroblasts derived from fetal and adult human skin at different anatomical sites have been explored [2, 3]. Fibroblasts from different sites were shown to display distinct and characteristic transcriptional patterns, and groups of differentially expressed genes include some involved in extracellular matrix synthesis, lipid metabolism and cell signalling pathways that control proliferation, cell migration and fate determination. Large differences in the gene expression programmes were also related to anterior–posterior, proximal–distal and dermal versus non-dermal anatomical divisions.
Remarkably, adult fibroblasts maintain key features of HOX gene expression patterns established during embryogenesis, suggesting that the HOX genes direct topographical differentiation and retain a detailed positional memory in fibroblasts. In that sense, fibroblasts from different parts of the skin should be considered distinct, differentiated cell types. Collectively, these findings suggest that site-specific variations in fibroblast gene expression programmes are systematically related to their positional identities relative to the major anatomical axes [2, 3].
While fibroblasts demonstrate certain variability in their gene expression profile, they are considered fully differentiated cells with relatively little plasticity. Recent, remarkable observations suggest, however, that fibroblasts can be induced to become pluripotent stem cells, essentially indistinguishable from embryonic stem cells [4, 5]. Specifically, the transduction of cultured fibroblasts with four transcription factors, Oct4, Sox2, Klf4 and cMyc, generated pluripotent stem cells, and similar protocols have been developed without the participation of the Myc retrovirus [6]. Furthermore, the transduction of human skin cancer cell lines with mir-302, a member of the micro-RNA family, which is highly expressed in slow-growing human embryonic cells and which quickly decreases after differentiation and proliferation, rapidly converted the cancer cells into a pluripotent state with the expression of key embryonic cell markers [7]. Collectively, development of these technologies holds promise for the reprogramming of fibroblasts in a manner that allows the development of patient- and disease-specific pluripotent stem cells for the treatment of diseases without a significant risk of immune rejection.
Micro-RNAs (miRNAs) are short, non-coding RNAs involved in the post-transcriptional regulation of gene expression, and over 500 miRNAs have been identified so far in humans [8]. Their biological importance was initially demonstrated for a role in cancer and, subsequently, they have been suggested to play a role in a number of clinical conditions. Certain miRNAs have also been suggested to contribute to skin development, for example by invoking differentiation through suppression of ‘stemness’ of the stem cells [9, 10]. Recent results have also suggested that miRNA deregulation may be involved in the pathogenesis of psoriasis by contributing to the dysfunction of the cross-talk between resident and infiltrating cells [11].
Blood vessels and lymphatics
The arteries entering the skin form a deep plexus, the ‘fascial’ network, from which individual vessels rise to the border between the subcutaneous adipose tissue and the dermis to form a ‘cutaneous’ vessel network. These vessels then branch out towards various cutaneous appendages and provide ascending arterioles to generate a subpapillary plexus, which forms capillary loops entering the papillary dermis between the rete ridges. From these capillaries the blood is drained by venules which form intermediate plexuses. Thus, the cutaneous vasculature is rather elaborate and limited to the dermis, while the epidermis has no blood vessels (Figure 2.58) [1, 2, 3].
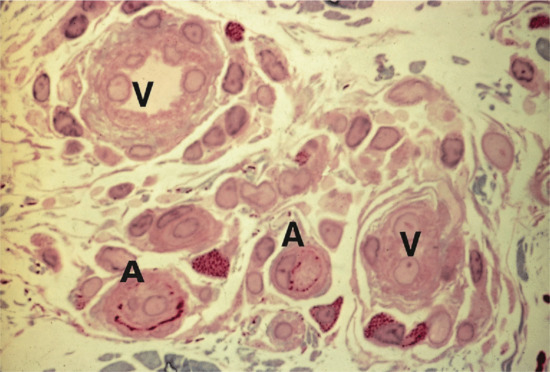
Figure 2.58 Histology of microvessels in the reticular dermis. Arterioles (A) can be distinguished from venules (V) by the presence of elastic lamina, which stains red. Surrounding mast cells can be distinguished by their prominent red/blue cytoplasmic granules. Original magnification 400× (basic fuchsin and methylene blue stain).
(Courtesy of Professor R. A. J. Eady, St John's Institute of Dermatology, King's College London, UK.)
In addition to providing nutrients and oxygen to the skin, the vasculature plays a major role in regulating the body temperature. This is accomplished by controlling the blood flow through the capillaries in the upper dermis so that opening blood vessels allows dissipation of excess heat while constriction of blood vessels slows the blood flow to the skin and conserves the core energy. The amount of blood flowing through the superficial layers of the dermis can also be controlled by arterial–venous anastomoses, which act as shunts to short-circuit the flow.
The innermost component of the blood vessels is the endothelium, consisting of adjoining endothelial cells that surround the lumen. Arterioles are characterized by a subendothelial layer of elastic tissue (Figure 2.59), while venules generally do not have elastic tissue in their walls (Figure 2.60). The endothelium of capillaries and small arterioles and venules is surrounded by pericytes, which appear to share certain characteristics with both endothelial and smooth muscle cells. Capillaries contain a single, discontinuous layer of pericytes, whereas venules may include more than one pericyte layer in their periendothelial investment. Smooth muscle cells are found chiefly in the walls of ascending arterioles but also within the arterioles of the superficial and deep plexus and in collecting venules. Smooth muscle cells and pericytes are surrounded by a basement membrane, which also encompasses the outer surface of endothelial cells. Veil cells are long, thin cells with an attenuated cytoplasm, and they more closely resemble fibroblasts than pericytes. They do not have a basement membrane investment and are located outside the vessel wall [4].
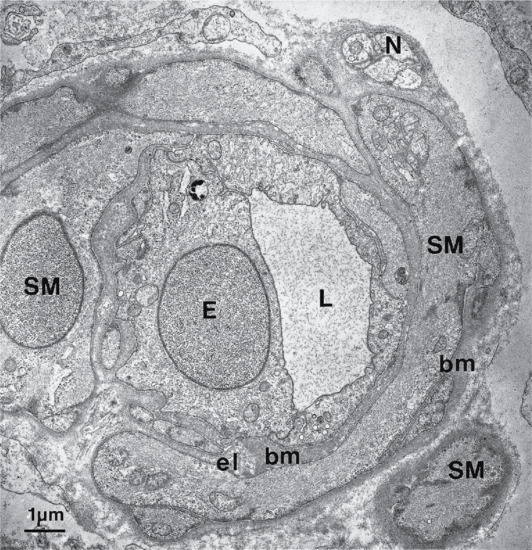
Figure 2.59 Transmission electron microscopy of a cross-section through a small arteriole in the skin. Note the relatively smooth surface of the endothelial cell (E) surrounding the lumen (L) and the presence of smooth muscle (SM) with an associated nerve (N). There is a small amount of elastic tissue (el) adjacent to the endothelial basement membrane (bm).
(Courtesy of Professor R. A. J. Eady, St John's Institute of Dermatology, King's College London, UK.)
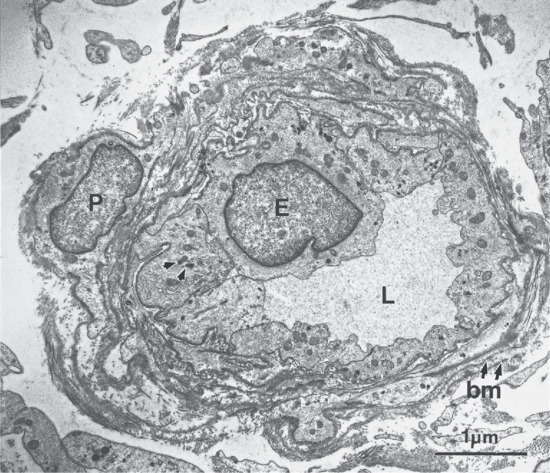
Figure 2.60 Transmission electron microscopy of a transverse section through a venule in the skin. The surface of the endothelial cells (E) in the lumen (L) is more convoluted than in its arteriolar counterpart (see Figure 2.59). The endothelial cells are surrounded by pericytes (P), and not smooth muscle cells, and the basement membrane (bm) contains dense strands (small arrows). The arrowheads indicate Weibel–Palade bodies.
(Courtesy of Professor R. A. J. Eady, St John's Institute of Dermatology, King's College London, UK.)
At the ultrastructural level, endothelial cells possess many of the common cellular organelles, including rough and smooth endoplasmic reticula, mitochondria and lysosomes; micropinocytotic vesicles are also evident. Intermediate filaments containing vimentin are present and have been reported to be more abundant on the venous than on the arterial site. Dense bodies associated with actin-like filaments of 5–6 nm diameter are found in the endothelial cells of the larger arterioles, and they may have a role in endothelial contraction.
Weibel–Palade bodies are endothelium-specific inclusions that occur more frequently in the venous side of the microvasculature (Figure 2.61). They are not found in dermal lymphatics but have been reported in larger lymph vessels. Weibel–Palade bodies contain factor XIII-related antigen, von Willebrand factor and GMP-140, a protein that was first described in platelet α-granules.
Figure 2.61 High magnification view of Weibel–Palade bodies revealing tubular profiles in cross-section.
(Courtesy of Professor R. A. J. Eady, St John's Institute of Dermatology, King's College London, UK.)
A major feature distinguishing arterial from venous microvessels is the ultrastructural appearance of the basement membrane. Venules and venous capillaries have a multilaminated basement membrane, whereas arterioles possess a more homogeneous matrix, lacking the electron-dense strands. Vascular basement membrane contains laminin 111, type IV collagen, fibronectin and heparan sulphate proteoglycans. It does not contain, however, bullous pemphigoid antigens, type VII collagen or laminin 332, components of the epidermal basement membrane zone.
A number of endothelium-specific antigens have been recognized, and they may have a special value in studies of cutaneous pathology. Endothelial cells are the major source of angiotensin-converting enzyme as well as various cytokines and adhesion molecules. The microvasculature is also a rich source of enzymes that may be involved in cellular processes, such as endocytosis and vesicular transport. Acid phosphatase has been localized to lysosome-like structures in the endothelium, and alkaline phosphatase reactivity has been used extensively to map the distribution and arborization of the arterial network in the upper dermis (Figure 2.62).
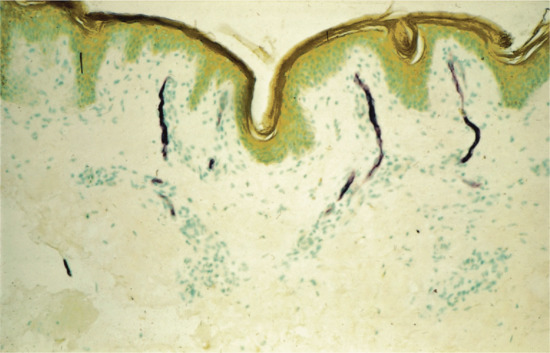
Figure 2.62 Histochemical detection of alkaline phosphatase activity indicating the presence of arterial microvessels in the superficial dermis. Original magnification 160×.
(Courtesy of Professor R. A. J. Eady, St John's Institute of Dermatology, King's College London, UK.)
The lymphatic network in the skin serves to transport particulate and liquid materials, such as proteins, from the extravascular compartment of the dermis. Interconnecting lymphatic spaces arise from terminal bulbs in the papillary layer and ultimately form the system that drains into the lymph nodes. The vessels have a broad lumen surrounded by a single endothelial layer, which is discontinuous in the terminal components and rests on an often discontinuous basal lamina. These processes are critical for the normal function of skin, as altered function and development of lymphatics can lead to diseases, including primary and secondary lymphoedemas [5].
Subcutaneous fat
Fat is a major component of the human body and approximately 80% of fat is in the subcutis; the rest surrounds internal organs. In non-obese males, 10–12% of body weight is fat, while in females the figure is 15–20%. Fat comprises white and brown adipose tissue. Brown fat is more common in infants and children and is characterized by different mitochondrial properties and increased heat production [1]. The function of fat is to provide insulation, mechanical cushioning and an energy store. In addition, fat may have an endocrine function, communicating with the hypothalamus via secreted molecules such as leptin to alter energy turnover in the body and to regulate appetite [2]. Adipocytes also have important signalling roles in osteogenesis and angiogenesis, and additional physical functions such as phagocytosis. Multipotent stem cells have been identified in human fat, which are capable of developing into adipocytes, osteoblasts, myoblasts and chondroblasts. Molecular biological insight into genes, proteins, hormones and other molecules that influence fat deposition and distribution are gradually being realized, both from research on rare inherited disorders (such as the lipodystrophies or obesity syndromes) as well as population studies on more common forms of obesity [3, 4].
Physiological functions of skin
A key role of skin is to provide a mechanical barrier against the external environment [1]. The cornified cell envelope and the stratum corneum restrict water loss from the skin, while keratinocyte-derived endogenous antibiotics (defensins and cathelicidins) provide an innate immune defence against bacteria, viruses and fungi [2]. The epidermis also contains a network of about 2 × 109 Langerhans cells, which serve as sentinel cells whose prime function is to survey the epidermal environment and to initiate an immune response against microbial threats, although they may also contribute to immune tolerance in the skin. Melanin, which is mostly found in basal keratinocytes, also provides some protection against DNA damage from ultraviolet radiation.
An important function of skin is thermoregulation. Vasodilatation or vasoconstriction of the blood vessels in the deep or superficial plexuses helps regulate heat loss. Eccrine sweat glands are found at all skin sites and are present in densities of 100–600/cm2; they play a role in heat control and produce approximately 1 litre of sweat per hour during moderate exercise [3]. Secretions from apocrine sweat glands contribute to body odour (pheromones). Skin lubrication and waterproofing is provided by sebum secreted from sebaceous glands.
Subcutaneous fat has important roles in cushioning trauma as well as providing insulation and a calorie reserve. In non-obese subjects, about 80% of the body's total fat is found in subcutaneous tissue. Fat also has an endocrine function, releasing the hormone leptin, which acts on the hypothalamus to regulate hunger and energy metabolism. Other functions of fat cells include tissue remodelling and phagocytosis [4].
Nails provide protection to the ends of the fingers and toes as well as being important in pinching and prising objects. Hair may have important social and psychological value, reflecting the notion that the appearances of human skin and its associated structures have a major impact on interpersonal relationships and personal well-being. Skin also has a key function in synthesizing various metabolic products, such as vitamin D.
There are two main kinds of human skin: glabrous skin (non-hairy skin) and hair-bearing skin. Glabrous skin is found on the palms and soles and has a grooved surface with alternating ridges and sulci giving rise to the dermatoglyphics (fingerprints). Glabrous skin has a compact stratum corneum which may be up to 10 times thicker compared with other body sites such as the flexures, where the epidermis is at its thinnest. Glabrous skin also contains encapsulated sense organs within the dermis, as well as a lack of hair follicles and sebaceous glands. In contrast, hair-bearing skin has both hair follicles and sebaceous glands but lacks encapsulated sense organs [5]. Hair follicle size, structure and density can vary between different body sites; the scalp has large hair follicles that may extend into the subcutaneous fat whereas the forehead has only small, vellus hair-producing follicles although sebaceous glands are large. The number of hair follicles remains unchanged until middle life but there is a changing balance between vellus and terminal hairs throughout life. In certain hair-bearing sites, such as the axilla, there are apocrine glands in addition to the eccrine sweat glands. Sebaceous glands are actively functioning in the newborn, and from puberty onwards, and the relative activity modifies the composition of the skin surface lipids. The structure of the dermal–epidermal junction also shows regional variations in the number of hemidesmosomal-anchoring filament complexes that exist (more in the leg than the arm). In the dermis, the arrangement and size of elastic fibres varies from very large fibres in perianal skin to almost no fibres in the scrotum. Marked variation in the cutaneous blood supply is found between areas of distensible skin such as the eyelid and more rigid areas such as the fingertips. There are also regional differences in biomechanical properties which can affect percutaneous absorption of creams and ointments.
Skin homeostasis
To maintain skin homeostasis, regenerate skin appendages and repair itself after injury, the skin contains stem cells that reside in the bulge area of hair follicles, the basal layer of interfollicular epidermis and the base of sebaceous glands (Figure 2.63) [1]. These stem cells generate a proliferative progeny that can undergo differentiation. The molecular signals involved in regulating epidermal stem cell proliferation and differentiation are illustrated in Figure 2.64.
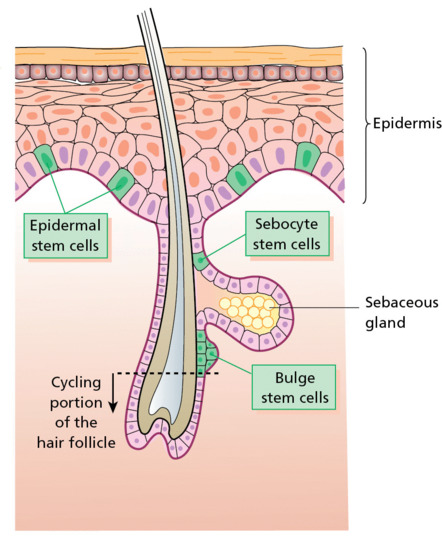
Figure 2.63 Epithelial stem cells are found within the interfollicular epidermis, the base of sebaceous glands and in the bulge area of hair follicles.
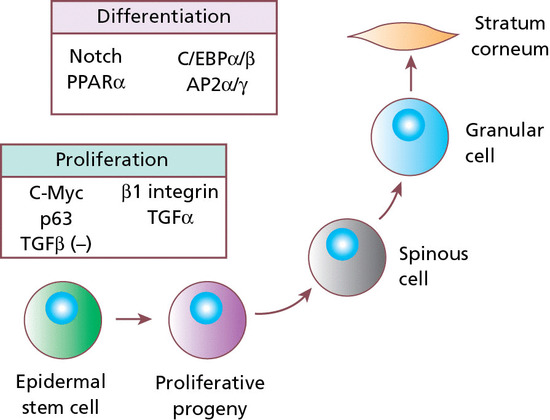
Figure 2.64 Epidermal stem cell proliferation is regulated positively by β1 integrin and transforming growth factor α (TGF-α) and negatively (–) by TGF-β signalling. The transcription factors c-Myc and p63 also promote epidermal proliferation. Notch signalling and the transcription factors peroxisome proliferator-activated receptor α (PPARα), AP2α/γ and C/EBPα/β control the differentiation of epidermal cells.
Stem cells are able to self-renew as well as give rise to differentiating cells [2]. In the epidermis, some basal cells can periodically withdraw from the cell cycle and commit to terminal differentiation. It is still not clear, however, what proportion of cells in the basal layer can function as a stem cell. One long-established theory divides basal keratinocytes into epidermal proliferation units, which comprise one self-renewing stem cell and about 10 tightly packed, transient, amplifying cells (each capable of dividing several times and then exiting the basal layer to undergo terminal differentiation) [3]. This unit gives rise to a column of larger and flatter cells that culminates in a single hexagonal surface. Stem cells within epidermal proliferation units are associated with a profile of particular chemical, molecular and biological characteristics. For example, stem cells retain labelling with injected 3H-thymidine or 5-bromo-2-deoxyuridine after repeated cell division. In culture, actively growing clones present after serial passaging are considered to indicate an origin from stem cells. Potential markers of interfollicular epidermal stem cells are α6 and β1 integrin as well as p63, whereas sebocyte stem cells express Blimp1. Markers of hair follicle bulge stem cells include CD34, NFATc1, vitamin D receptor, TCF3, Sox9 and Lhx2, although considerably more markers exist in this and other parts of the hair follicle, highlighting the protean nature of the stem cell population in being able to respond to the requirements of tissue homeostasis, injury or growth spurts [4].
In the epidermal proliferation unit concept of stem cell behaviour, the division of basal cells has been viewed as a symmetrical process in which equal daughter cells are generated; the basal cells progressively reduce their adhesiveness to the underlying epidermal basement membrane, delaminate and commit to terminal differentiation (Figure 2.65). However, data also suggest that basal cells can also undergo asymmetrical cell division, shifting their spindle orientation from lateral to perpendicular [5]. Asymmetrical cell divisions provide a natural means of maintaining a proliferative daughter cell that retains the cell markers associated with stem cells, while the other daughter cell has reduced markers such as β1 integrin, increased expression of Notch signals, and is committed to terminal differentiation. Asymmetrical cell divisions, therefore, can bypass the need for transient amplifying cells.
Figure 2.65 Possible mechanisms for the proliferative potential of stem cells (SC) in the basal keratinocyte layer. In the symmetrical division model, two stem cells are produced. Some of these cells in contact with the epidermal basement membrane are transient-amplifying cells (TA). These cells are capable of dividing four to five times before leaving the basal layer (delamination, black arrows) to become a spinous layer cell (SP) and entering terminal differentiation. In the asymmetrical division model, there is preferential partitioning of proliferation-associated factors into the stem cell daughter cell and, conversely, preferential partitioning of differentiation-inducing components into the daughter cell that is destined to become an SP. Depending on the orientation of the cell spindle, the daughter cell destined for differentiation can either become an SP directly or delaminate from the basal layer to enter terminal differentiation. In vivo, both mechanisms may exist.
The structural and biological composition of the dermal–epidermal junction also influences the proliferative properties of basal keratinocytes. Laminin 332 promotes anchorage as a ligand via α6β4 integrins in hemidesmosomes and signalling/migration via its association with α3β1 in focal adhesions. Signalling via α3β1 integrin stimulates the MAPK pathway, turnover of focal adhesions and epidermal migration. The basement membrane is also a reservoir for growth factors that can promote epidermal proliferation (e.g. TGF-α, EGFs, insulin growth factors) or restrict it (e.g. TGF-β). EGFR signalling also enhances proliferation and migration in the epidermis, possibly by phosphorylating β4 integrin and promoting hemidesmosome disassembly. Thus, the control of basal keratinocyte stem cell activity in maintaining homeostasis and responding to injury is through the regulation of at least two opposing tyrosine kinase pathways and two integrin structures.
A key transcription factor in regulating the self-renewal and long-term proliferative capacity of the stem cell is p63, a member of the p53 family of proto-oncogenes [6]. However, the precise role of p63 is not clear; it may have a direct effect on stem cell renewal, or lineage commitment, and/or an effect on switching from proliferation to terminal differentiation [6]. Notch signalling also appears to have an important gatekeeper function in the transition from basal to suprabasal cells; there is basal expression of Notch ligands such as Delta1 and suprabasal expression of Notch receptors and Notch downstream targets such as Hes1.
Stem cells in hair follicles are located in the lowest permanent part of the follicle, within the outer root sheath. These cells cycle more slowly than other cells and have the capacity to migrate (e.g. to the base of the hair follicle in follicular regeneration), as well as to differentiate into diverse lineages (e.g. outer root sheath, inner root sheath, hair shaft, sebocytes, interfollicular epidermis). Despite this multipotency, however, the follicle stem cells only function in pilosebaceous unit homeostasis and do not contribute to interfolliclular epidermis unless the skin is wounded [7].
Hair follicles undergo cycles of degeneration and regeneration throughout life. During the growth phase (anagen), which requires activation of hair follicle stem cells, matrix cells proliferate rapidly but then undergo sudden apoptosis (catagen). The hair bulb and root shrivel to form an epithelial strand which forces the dermal papilla to rest at the base of the non-cycling part of the hair follicle [8]. The hairs then enter a resting phase (telogen). At a molecular level, inhibition of BMP signalling and activation of Wnt signalling converge to regulate stem cell activation. From microarray studies, Sox9, Tcf3 and Lhx2 appear to be markers of follicular stem cells whether they are quiescent or proliferative.
Apart from stem cells in the hair follicles and interfollicular epidermis, other cells in the dermis and subcutis may have stem cell properties. These include cells that have been termed skin-derived precursors, which can differentiate into both neural and mesodermal progeny [9]. In addition, a subset of dermal fibroblasts can have adipogenic, osteogenic, chondrogenic, neurogenic and hepatogenic differentiation potential [10]. Moreover, dermal fibroblasts can be reprogrammed into cells bearing an embryonic stem cell (pluripotent) phenotype by the insertion of just four key transcription factors, Oct4, Sox2, Klf4 and Myc [11].
Skin ageing
Skin ageing represents an inevitable physiological consequence of getting older but the impact on personal health and well-being can be significant. Considerable efforts have therefore been made to understand the biology and pathophysiology of skin ageing to try to identify new targets that might offer therapeutic intervention and prevention.
For many years, attempts have been made to define and characterize the physiological and pathological changes that occur in skin ageing [1], which is often subdivided into intrinsic (chronological) skin ageing and photoageing (sun-exposed sites). However, it is clear that this distinction is somewhat arbitrary, with light microscopy typically showing overlapping features of loss of collagen fibres, elastic fibre disruption, irregular pigmentation, a reduced number of thin hair follicles with grey hairs and a reduced number of inflammatory cells in the dermis, as well as shared age-associated physical changes such as reduced epidermal water-retaining capacity and reduced skin surface acidification (Figure 2.66) [2, 3]. The often lax appearance to aged skin, however, does not reflect changes in skin water content. Indeed, the amount of water may not alter in intrinsically aged skin and may even increase in photoaged skin [4]. In skin ageing, there is an increase in the number of mast cells, mononuclear cells and neutrophils. In photoageing, the fibroblasts show a stellate phenotype and, ultrastructurally, a highly activated endoplasmic reticulum, reflecting increased biosynthetic activity [5].
Figure 2.66 Structural and functional changes associated with skin ageing.
Age often decreases sensory perception and increases the threshold for pain [6]. Aged skin displays a progressive disorganization or loss of some sense organs; for example, the density of Meissner corpuscles in terminal digits skin falls from over 30/mm2 in young adults to approximately 12/mm2 by the age of 70 years [7]. Langerhans cells become considerably reduced in number in elderly people, even in sun-protected areas [8]. There is a reduction in the number of epidermal Langerhans cells with age, coupled with a reduced ability to migrate from the epidermis in response to tumour necrosis factor α [9], although exogenous IL-1β can improve the impaired Langerhans cell migration in aged skin [10].
Peripheral immune function is altered in the elderly. The responses of both T and B cells to specific mitogens change in elderly skin, despite the fact that the absolute numbers of T and B cells do not alter significantly [11]. Nevertheless, a decreased intensity in delayed hypersensitivity reactions [11, 12], an increased risk of photocarcinogenesis, and a greater susceptibility to chronic skin infections are all consequences of the ageing of the (skin) immune system [11, 12].
Additional contributors to ageing may include endocrine factors, nutritional or calorie intake [13], mechanical stress/tissue tension or inflammation [14] and electromagnetic radiation [15].
Understanding why and precisely how these changes in the skin occur is a major challenge and one that is pivotal in trying to develop new and effective therapeutic agents that can delay or prevent the ageing skin phenotype. One approach in trying to address this has involved the comparison of young and old skin, using various chemical, physical, biological and molecular techniques, and over the last 60 years several theories have been proposed to explain ageing (in general and in the skin). In the 1950s, oxidative damage, with an accumulation of free oxygen radicals capable of damaging cell proteins, lipids and nucleic acids, was considered to be an aetiological factor [16]. Further cellular changes that promoted ageing were suggested to include the accumulation of nuclear DNA damage, misfolded proteins and increased frequency of mitochondrial DNA deletions. These are not mutually exclusive contributors and indeed chronic inflammation, both immune and non-immune mediated, has been identified as another factor that can promote ageing, in a manner linked to oxidative damage [17].
Apart from cellular damage, other factors may include telomere shortening, as ageing cells fail to express sufficient telomerase to maintain the telomere ends that prevent replicative senescence [18]. Overall, it is plausible that different types of damage occur at different rates in a population of individuals, with a variable impact (timing and extent of ageing) as thresholds for toxicity are reached. Indeed, studies on fibroblast cell senescence in culture have identified the contribution of cell stress, the accumulation of intracellular damage and the secretion of factors that can affect the behaviour of other cells in their vicinity [19]. Thus the surrounding microenvironment that bathes cells may have a direct impact on cellular ageing, tissue integrity and the ageing phenotype.
One other investigative approach in skin ageing has been to focus on differences in the severity and time course of skin ageing between different individuals. This has been done predominantly using genomics platforms facilitated by improvements in the annotation of the human genome as well as new technical advances for functional studies. The concept has been that key events in ageing might be gleaned from the analysis of cohorts of individuals who demonstrate traits such as shorter or longer than average lifespans, as well as those with phenotypically accelerated forms of ageing such as the progeria syndromes [20]. Such studies do not relate exclusively to skin ageing but, as an expression of the ageing process, skin provides a useful model to observe the downstream functional consequences of specific variations in the genome that might be implicated in ageing.
With regard to longevity, gene association studies using whole genome screening and targeted approaches have revealed certain genes or genetic variants that may be associated with an increased lifespan [21]. For example, polymorphisms in the p16/p15 locus (INK4a/INK4b, CDKN2a/b) have been identified in several age-associated disease studies, potentially with relevance to stem cell function [22]. In addition, other studies have identified several genes associated with the insulin or insulin-like growth factor 1 signalling pathways [23]. The development of suitable animal models to study these pathways in more detail is likely to generate further insight into longevity and their relevance (or not) to the biology of tissue ageing, including skin health and either resistance or susceptibility to skin ageing.
From a reductionist point of view, rare genetic diseases, in which the pathology associated with normal ageing seems to accumulate at an accelerated rate, also have the potential to improve our understanding of the pathophysiology of normal skin ageing. These conditions include diseases such as the Werner syndrome, Hutchinson–Gilford progeria syndrome, Cockayne syndrome and trichothiodystrophy [8]. Collectively, molecular characterization of these diseases has identified particular abnormalities in enhanced DNA damage, defective DNA repair, genomic instability, susceptibility to genotoxic stress and impaired epigenetic homeostasis – processes which, when studied in specific individual disease models, demonstrate some overlap with ageing-related events [24]. For example, fibroblasts from individuals with Hutchinson–Gilford progeria syndrome show similar changes to normally aged fibroblasts with regard to increased reactive oxygen species accumulation, increased basal DNA damage, reduced proliferation and early senescence [25]. It is plausible, although not yet proven, that more common coding or non-coding variants in the genes that cause these rare monogenic syndromes (i.e. less disruptive changes than the disease-associated mutations) may confer some increased or decreased susceptibility to ageing via similar processes that occur on a grander scale in the rare genetic diseases.
With regard to photoageing, studies on the pathological changes following UV exposure have provided other insights into skin ageing mechanisms. For example, it is known that UV radiation can induce the synthesis of various destructive enzymes, specifically the MMPs [26]. Notably, there is increased expression of MMP-1, MMP-3 and MMP-9 [27]. The MMP-1 enzyme can cleave the major interstitial collagens type I and type III and these fragments can be further degraded by MMP-3 and MMP-9. It has also been shown that different individuals can have variable activity in the MMP-1 enzyme and that some of this variability may be due to functional polymorphisms within the promoter of the MMP-1 gene [28]. Of note, at the −1607 position, there is a common polymorphic variant that either gives a single G nucleotide or a GG sequence. This GG sequence creates a binding site for ETS (E26 transformation-specific) transcription factors that leads to increased activity of MMP-1 and a greater breakdown in dermal collagen.
It is possible that this single nucleotide polymorphism in MMP-1 (or others with similar functionality) might provide some insight into differential rates of skin ageing: those with the GG genotype might be prone to accelerated or more marked visible changes of skin ageing. For now, however, this possibility is merely speculative. Nevertheless, the GG genotype in the MMP-1 promoter has been shown to have a marked effect on contrasting skin phenotypes, for example, in the severity of scarring in the inherited blistering skin disorder, recessive dystrophic epidermolysis bullosa [28]. MMP-1 activity has also been shown to be greater in the skin of smokers (sun-exposed and sun-protected), a finding that may partially explain why many smokers display greater signs of skin ageing compared with age-matched non-smokers [29, 30]. Nevertheless, MMP-1 is just one of c. 20 000 genes and it is clear that the analysis of single genes in isolation is not the optimal approach in trying to dissect out what contributes to a skin ageing phenotype in the general population.
An alternative approach to the study of ageing is to examine comparative gene expression, taking a more global assessment of different patterns of gene activation or repression. Transcriptome analysis has been performed in lymphocyte RNA derived from young and old subjects. This revealed that intrinsic ageing was associated with the modification of RNA processing genes and disruption of alternative splicing [31], but thus far only limited data on skin ageing have been published. Moreover, by comparing global gene expression in young skin versus old skin or UV-exposed skin versus UV-protected skin, there is the possibility of generating hypothesis-free data that also provide a distinct insight into both intrinsic ageing and photoageing.
Indeed, the analysis of gene expression in intrinsically and photoaged skin has revealed new insights into skin ageing processes – with some common changes in transcript levels and others that differ [32]. Notably, both types of ageing are associated with the reduced expression of genes involved in lipid biosynthesis and epidermal differentiation and display an increased expression of certain genes associated with inflammation and wound healing; changes in extracellular matrix are variable. Looking at processes rather than individual genes highlights pathways that are connected to proteases, matrix proteins and inflammation.
With regard to particular changes noted on transcriptome analysis, all aged skin shows alterations in lipid synthesis with significant reductions in many enzymes that are necessary for the synthesis of cholesterol, fatty acids and sphingolipids, consistent with a reduced capacity for skin barrier maintenance and repair. Aged skin also shows a decreased expression of several genes expressed in the epidermis, or hair follicles, a finding that indicates that skin ageing is not exclusively a dermal problem. Notably, a decreased expression of epidermal differentiation genes indicates that skin ageing affects epidermal integrity, keratinocyte structure, tight junctions, keratinocyte adhesion and stem cells. Although lipid metabolism and keratinocyte-associated gene expression changes may be common to intrinsic ageing and photoageing, the changes in epidermal differentiation-related genes tend to be greater in intrinsic ageing.
With regard to the extracellular matrix gene expression patterns, there is typically a markedly increased expression of elastic fibre components in photoaged skin, with reduced expression of some dermal collagens during skin ageing (particularly in intrinsically aged skin), although a paradoxical increase in some collagens can occur in both photoageing and intrinsic ageing. Aside from different changes in extracellular matrix patterns of gene expression, some similarities occur for markers of stress in aged skin that are indicative of oxidative stress. Of note, all aged skin shows a decreased capacity to detoxify hydrogen peroxide and other free radicals along with reduced antioxidant defences.
Overall, it is evident that intrinsic ageing and photoageing clearly affect many of the same biological processes. Themes common to both types of skin ageing include abnormalities in lipid biosynthesis, epidermal differentiation and oxidative stress. Contrasting differences between photoaged and intrinsically aged skin mainly involve extracellular matrix composition. Transcriptomic analysis also provides clues to biomarkers of aged skin, particularly involving abnormalities of proteases, matrix proteins and inflammation as well as structural protein pathology in both the epidermis and dermis. The challenge will be to see whether these types of data will generate new targets for intervention and a rationale for developing new agents that delay or prevent some of the pathophysiological changes noted at both an individual gene/protein level and as well as in the analysis of integrated or interactive biological pathways. An -omics approach to dissecting the pathophysiology of skin ageing is insightful at a basic science level, and at least provides a platform for further anti-ageing research and therapies, particularly to counter the visible effects of skin ageing.
References
Skin development
- Fuchs E. Scratching the surface of skin development. Nature 2007;445:834–42.
- Koster MI. p63 in skin development and ectodermal dysplasias. J Invest Dermatol 2010;130:2352–8.
- Driskell RR, Lichtenberger BM, Hoste E, et al. Distinct fibroblast lineages determine dermal architecture in skin development. Nature 2013;504:277–81.
- Duverger O, Morasso MI. To grow or not to grow: hair morphogenesis and human genetic hair disorders. Semin Cell Dev Biol 2014;25//ϖ26C:22–33.
- Sennett R, Rendl M. Mesenchymal–epithelial interactions during hair follicle morphogenesis and cycling. Semin Cell Dev Biol 2012;23:917–27.
- Lim X, Nusse R. Wnt signaling in skin development, homeostasis and disease. Cold Spring Harb Perspect Biol 2013;5:piia008029.
- Owens P, Han G, Li AG, Wang XJ. The role of Smads in skin development. J Invest Dermatol 2008;128:783–90.
- Botchkareva NV. MicroRNA/mRNA regulatory networks in the control of skin development and regeneration. Cell Cycle 2012;11:468–74.
- Honeycutt KA, Roop DR. c-Myc and epithelial stem cell fate determination. J Dermatol 2004;31:368–75.
- Loomis CA. Development and morphogenesis of the skin. Adv Dermatol 2001;17:183–210.
- Bergmann C, Senderek J, Anhuf D, et al. Mutations in the gene encoding Wnt-signaling component R-spondin 4 (RSPO4) cause autosomal recessive anonychia. Am J Hum Genet 2006;79:1105–9.
- Olivera-Martinez I, Thelu J, Dhouailly D. Molecular mechanisms controlling dorsal dermis generation from the somite dermomyotome. Int J Dev Biol 2004;48:93–101.
Epidermal and adnexal structures
- Houben E, De Paepe K, Rogiers V. A keratinocyte's course of life. Skin Pharmacol Physiol 2007;20:122–32.
- Feingold KR, Elias PM. Role of lipids in the formation and maintenance of the cutaneous permeability barrier. Biochim Biophys Acta 2014;1841:280–94.
- Brown SJ, McLean WH. One remarkable molecule: filaggrin. J Invest Dermatol 2012;132:751–62.
- Eckhart L, Lippens S, Tschachler E, Declerq W. Cell death by cornification. Biochim Biophys Acta 2013;1833:3471–80.
Keratinocytes
- Szeverenyi I, Cassidy AJ, Chung CW, et al. The human intermediate filament database: comprehensive information on a gene family involved in many human diseases. Hum Mutat 2008;29:351–60.
- Pan X, Hobbs RP, Coulombe PA. The expanding significance of keratin intermediate filaments in normal and diseased epithelia. Curr Opin Cell Biol 2013;25:47–56.
- Schweizer J, Bowden PE, Coulombe PA, et al. New consensus nomenclature for mammalian keratins. J Cell Biol 2006;174:169–74.
- Magin TM, Vijayaraj P, Leube RE. Structural and regulatory functions of keratins. Exp Cell Res 2007;313:2031–2.
- Uitto J, Richard G, McGrath JA. Diseases of epidermal keratins and their linker proteins. Exp Cell Res 2007;313:1995–2009.
- McLean WH, Moore CB. Keratin disorders:from gene to therapy. Hum Mol Genet 2011;20:R189–97.
Eccrine and apocrine glands
- Wollina U, Abdel-Naser MB, Ganceviciene R, Zouboulis CC. Receptors of eccrine, apocrine, and holocrine skin glands. Dermatol Clin 2007;25:577–88.
- Morimoto T, Itoh T. Thermoregulation and body fluid osmolality. J Basic Clin Physiol Pharmacol 1998;9:51–72.
- Harker M. Psychological sweating: a systematic review focused on aetiology and cutaneous responses. Skin Pharmacol Physiol 2013;26:92–100.
- Shibasaki M, Wilson TE, Crandall CG. Neural control and mechanisms of eccrine sweating during heat stress and exercise. J Appl Physiol 2006;100:1692–701.
- Quinton PM. Cystic fibrosis: lessons from the sweat gland. Physiology (Bethesda) 2007;22:212–25.
- Lonsdale-Eccles A, Leonard N, Lawrence C. Axillary hyperhidrosis: eccrine or apocrine. Clin Exp Dermatol 2003;28:2–7.
Pilosebaceous unit
- Schlake T. Determination of hair structure and shape. Semin Cell Dev Biol 2007;18:267–73.
- Fuchs E. Scratching the surface of skin development. Nature 2007;445:834–42.
- Schweizer J, Langbein L, Rogers MA, Winter H. Hair follicle-specific keratins and their diseases. Exp Cell Res 2007;313:2010–20.
- Harel S, Christiano AM. Genetics of structural hair disorders. J Invest Dermatol 2012;132:E22–6.
- Westgate GE, Botchkareva NV, Tobin DJ. The biology of hair diversity. Int J Cosmet Sci 2013;35:329–36.
Nails
- De Berker D. Nail anatomy. Clin Dermatol 2013;31:509–15.
- Blaydon DC, Ishii Y, O'Toole EA, et al. The gene encoding R-spondin 4 (RSPO4), a secreted protein implicated in Wnt signaling, is mutated in inherited anonychia. Nat Genet 2006;38:1245–7.
- De Berker DA, Andre J, Baran R. Nail biology and nail science. Int J Cosmet Sci 2007;29:241–75.
- Achten G, Parent D. The normal and pathologic nail. Int J Dermatol 1983;22:556–65.
- Cohen PR. The lunula. J Am Acad Dermatol 1996;34:943–53.
Merkel cells
- Boulais N, Misery L. Merkel cells. J Am Acad Dermatol 2007;57:147–65.
- Moll I, Roessler M, Brandner JM, et al. Human Merkel cells – aspects of cell biology, distribution and functions. Eur J Cell Biol 2005;84:259–71.
- Maksimovic S, Nakatani M, Baba Y, et al. Epidermal Merkel cells are mechanosensory cells that tune mammalian touch receptors. Nature 2014;509:617–21.
- Szrder V, Grim M, Halata Z, Sieber-Blum M. Neural crest origin of mammalian Merkel cells. Dev Biol 2003;253:258–63.
- Sidhu GS, Chandra P, Cassai ND. Merkel cells, normal and neoplastic. Ultrastruct Pathol 2005;29:287–94.
- Schrama D, Ugurel S, Becker JC. Merkel cell carcinoma: recent insights and new treatment options. Curr Opin Oncol 2012;24:141–9.
Innate immunity
- Nakatsuji T, Gallo RL. Antimicrobial peptides: old molecules with new ideas. J Invest Dermatol 2012;132:887–95.
- Afshar M, Gallo RL. Innate immune defense system of the skin. Vet Dermatol 2013;24:32–8.
- Chan JK, Roth j, Oppenheim JJ, et al. Alarmins: awaiting a clinical response. J Clin Invest 2012;122:2711–19.
- Steinhoff M, Schauber J, Leyden JJ. New insights into rosacea pathophysiology: a review of recent findings. J Am Acad Dermatol 2013;69:515–26.
Skin microbiome
- Grice EA, Kong HH, Renaud G, et al. A diversity profile of the human skin microbiota. Genome Res 2008;18:1043–50.
- Fierer N, Hamady M, Lauber CL, Knight R. The influence of sex, handedness, and washing on the diversity of hand surface bacteria. Proc Natl Acad Sci USA 2008;105:17994–9.
- Chen YE, Tsao H. The skin microbiome: current perspectives and future challenges. J Am Acad Dermatol 2013;69:143–55.
Langerhans cells
- Mohr RE, Takashima A. Epidermal Langerhans' cell movement in situ: a model for understanding immunologic function in the skin. Arch Dermatol 2007;143:1438.
- Tay SS, Roediger B, Tong PL, Tikoo S, Weninger W. The skin-resident immune network. Curr Dermatol Rep 2013;3:13–22.
- Bennett CL, Noordegraaf M, Martina CA, Clausen BE. Langerhans' cells are required for efficient presentation of topically applied hapten to cells. J Immunol 2007;179:6830–5.
- Chopin M, Nutt SL. Establishing and maintaining the Langerhans cell network. Semin Cell Dev Biol 2014;in press.
- Igyarto BZ, Kaplan DH. Antigen presentation by Langerhans cells. Curr Opin Immunol 2013;25:115–19.
Immune surveillance
- Pasparakis M, Haase I, Nestle FO. Mechanisms regulating skin immunity and inflammation. Nat Rev Immunol 2014;14:289–301.
- Loser K, Beissert S. Dendritic cells and T cells in the regulation of cutaneous immunity. Adv Dermatol 2007;23:307–33.
- Seneschal J, Clark RA, Gehad A, Baccher-Allan CM, Kupper TS. Human epidermal Langerhans cells maintain immune homeostasis in skin by activating skin resident regulatory T cells. Immunity 2012;36:873–84.
- Magyarics Z, Csillag A, Pazmandi K, et al. Identification of plasmacytoid pre-dendritic cells by one-color flow cytometry for phenotype screening. Cytometry A 2008;73:254–8.
- Chu CC, Di Meglio P, Nestle FO. Harnessing dendritic cells in inflammatory skin diseases. Semin Immunol 2011;23:28–41.
Mast cells
- Hide M, Yanase Y, Greaves MW. Cutaneous mast cell receptors. Dermatol Clin 2007;25:563–75.
- Galli SJ, Tsai M. Mast cells: versatile regulators of inflammation, tissue remodeling, host defense and homeostasis. J Dermatol Sci 2008;49:7–19.
- Hungness SI, Akin C. Mastocytosis:advances in diagnosis and treatment. Curr Allergy Asthma Rep 2007;7:248–54.
- Tete S, Tripodi D, Rosati M, et al. Role of mast cells in innate and adaptive immunity. J Biol Reg Homeost Agents 2012;26:193–201.
- Harvima IT, Nilsson G. Mast cells as regulators of skin inflammation and immunity. Acta Derm Venereol 2011;91:644–50.
Melanocytes
- Wilson S, Ginger RS, Dadd T, et al. NCKX5, a natural regulator of human skin colour variation, regulates the expression of key pigment genes MC1R and alpha-MSH and alters cholesterol homeostasis in normal human melanocytes. Adv Exp Med Biol 2013;961:95–107.
- Lang D, Macarenhas JB, Shea CR. Melanocytes, melanocyte stem cells, and melanoma stem cells. Clin Dermatol 2013;31:166–78.
- Murisier F, Beermann F. Genetics of pigment cells: lessons from the tyrosinase gene family. Histol Histopathol 2006;21:567–78.
- Nishimura EK. Melanocyte stem cells: a melanocyte reservoir in hair follicles for hair skin skin repigmentation. Pigment Cell Melanoma Res 2011;24:401–10.
- Schouwey K, Beermann F. The Notch pathway: hair graying and pigment cell homeostasis. Histol Histopathol 2008;23:609–19.
- Sturm RA. Molecular genetics of human pigmentation diversity. Hum Mol Genet 2009;18:R9–17.
Desmosomes
- Kowalczyk AP, Green KJ. Structure, function, and regulation of desmosomes. Prog Mol Biol Transl Sci 2013;116:95–118.
- Al-Jassar C, Bikker H, Overduin M, Chidget M. Mechanistic basis of desmosome-targeted diseases. J Mol Biol 2013;425:4006–22.
- Harmon RM, Green KJ. Structural and functional diversity of desmosomes. Cell Commun Adhes 2013;20:171–87.
- Petrof G, Mellerio JE, McGrath JA. Desmosomal genodermatoses Br J Dermatol 2012;166:36–45.
- Ishida-Yamamoto A, Igawa S. Genetic skin diseases related to desmosomes and corneodesmosomes. J Dermatol Sci 2014;74:99–105.
- Amagai M, Stanley J. Desmoglein as a target in skin disease and beyond. J Invest Dermatol 2012;132:776–84.
- Chan LS. Epitope spreading in paraneoplastic pemphigus: autoimmune induction in antibody-mediated blistering diseases. Arch Dermatol 2000;136:663–4.
- Stanley JR, Amagai M. Pemphigus, bullous impetigo and the staphylococcal scalded-skin syndrome. N Eng J Med 2006;355:1800–10.
Adherens junctions
- Niessen CM. Tight/adherens junctions: basic structure and function. J Invest Dermatol 2007;127:2525–32.
- Irie K, Shimizu K, Sasikawa T, Ikeda W, Takai Y. Roles and modes of action of nectins in cell–cell adhesion. Semin Cell Dev Biol 2004;15:643–56.
- Niessen CM, Gumbiner BM. Cadherin-mediated cell sorting not determined by binding or adhesion specificity. J Cell Biol 2002;156:389–99.
- Protonotarius NI, Tsatspoulou AA, Gatzoulis KA. Arrhythmogenic right ventricular cardiomyopathy caused by a deletion in plakoglobin (Naxos disease). Card Electrophysiol Rev 2002;6:72–80.
- Sprecher E, Bergman R, Richard G, et al. Hypotrichosis with juvenile macular dystrophy is caused by a mutation in CDH3, encoding P-cadherin. Nat Genet 2001;29:134–6.
- Kjaer KW, Hansen L, Schwabe GC, et al. Distinct CDH3 mutations cause ectodermal dysplasia, ectrodacytly, macular dystrophy (EEM) syndrome. J Med Genet 2005;42:292–8.
- Brancati F, Agolini E, Fortugno P. Nectinopathies: an emerging group of ectodermal dysplasia syndromes. G Ital Dermatol Venereol 2013;148:59–64.
Gap junctions
- Mese G, Richard G, White TW. Gap junctions:basic structure and function. J Invest Dermatol 2007;127:2516–24.
- Richard G. Connexin disorders of the skin. Clin Dermatol 2005;23:23–32.
- Scott CA, Kelsell DP. Key functions for gap junctions in skin and hearing Biochem J 2011;438:245–54.
- Velasquez S, Eugenin EA. Role of pannexin-1 hemichannels and purinergic receptors in the pathogenesis of human diseases. Front Physiol 2014;5:96.
Tight junctions
- Niessen CM. Tight/adherens junctions: basic structure and function. J Invest Dermatol 2007;127:2525–32.
- Morita K, Miyachi Y. Tight junctions in the skin. J Dermatol Sci 2003;31:81–9.
- Gupta IR, Ryan AK. Claudins: unlocking the code to tight junction formation during embryogenesis and in disease. Clin Genet 2010;77:314–25.
- Konrad M, Schaller A, Seelow D, et al. Mutations in the tight junction gene claudin 19 (CLDN19) are associated with renal magnesium wasting, renal failure, and severe ocular involvement. Am J Hum Genet 2006;79:949–57.
- Hadj-Rabia S, Baala L, Vabres P, et al. Claudin-1 mutations in neonatal sclerosing cholangitis associated with ichthyosis: a tight junction disease. Gastroenterology 2004;127:1386–90.
Dermal–epidermal basement membrane
- Bruckner-Tuderman L, Has C. Disorders of the cutaneous basement membrane zone – the paradigm of epidermolysis bullosa. Matrix Biol 2014;33:29–34.
- Uitto J, Pulkkinen L. Molecular complexity of the cutaneous basement membrane zone. Mol Biol Rep 1996;23:35–46.
- Tzu J, Marinkovich MP. Bridging structure with function: structural, regulatory, and developmental role of laminins. Int J Biochem Cell Biol 2008;40:199–214.
- El Ghalbzouri A, Jonkman MF, Dijkman R, et al. Basement membrane reconstruction in human skin equivalents is regulated by fibroblasts and/or exogenously activated keratinocytes. J Invest Dermatol 2005;124:79–86.
- Salam A, Proudfoot LE, McGrath JA. Inherited blistering skin diseases: underlying molecular mechanisms and emerging therapies. Ann Med 2014;46:49–61.
Basement membrane collagen
- Khoshnoodi J, Pedchenko V, Hudson BG. Mammalian collagen IV. Microsc Res Tech 2008;71:357–70.
- Hudson BG, Tryggvason K, Sundaramoorthy M, et al. Alport's syndrome, Goodpasture's syndrome, and type IV collagen. N Eng J Med 2003;25:2543–56.
- Kruegel J, Rubel D, Gross O. Alport syndrome – insights from basic and clinical research. Nat Rev Nephrol 2013;9:170–8.
- Ghohestani RF, Rotunda SL, Hudson B, et al. Crescentic glomerulonephritis and subepidermal blisters with autoantibodies to α5 and α6 chains of type IV collagen. Lab Invest 2003;83:605–11.
Laminins
- Miner JH. Laminins and their roles in mammals. Microsc Res Tech 2008;71:349–56.
- Tzu J, Marinkovich MP. Bridging structure with function: structural, regulatory, and developmental role of laminins. Int J Biochem Cell Biol 2008;40:199–214.
- Sugawara K, Tsuruta D, Ishii M, et al. Laminin-332 and -511 in skin. Exp Dermatol 2008;17:473–80.
- Belkin AM, Stepp MA. Integrins as receptors for laminins. Microsc Res Tech 2000;51:280–301.
- Rouselle P, Beck K. Laminin 332 processing impacts cellular behavior. Cell Adhes Migr 2013;7:122–34.
- Chung HJ, Uitto J. Epidermolysis bullosa with pyloric atresia. Dermatol Clin 2010;28:43–54.
- Breitkreutz D, Koxholt I, Thieman K, Nischt R. Skin basement membrane: the foundation of epidermal integrity – BM functions and diverse roles of bridging molecules nidogen and perlecan. Biomed Res Int 2013;2013:179784.
- Aumailley M, Rouselle P. Laminins of the dermo-epidermal junction. Matrix Biol 1999;18:19–28.
- Niessen CM, Hogervorst F, Jaspars LH, et al. The α6β4 integrin is a receptor for both laminin and kalinin. Exp Cell Res 1994;211:360–7.
Hemidesmosomes
- De Pereda JM, Ortega E, Alonso-Garcia N, et al. Advances and perspectives of the architecture of hemidesmosomes: lessons from structural biology. Cell Adhes Migr 2009;3:361–4.
- McGrath JA, Gatalica B, Christiano AM, et al. Mutations in the 180-kD bullous pemphigoid antigen (BPAG2), a hemidesmosomal transmembrane collagen (COL17A1), in generalized atrophic benign epidermolysis bullosa. Nat Genet 1995;11:83–6.
- Groves RW, Liu L, Dopping-Hepenstal PJ, et al. A homozygous nonsense mutation within the dystonin gene coding for the coiled-coil domain of the epithelial isofroms of BPAG1 underlies a new subtype of autosomal recessive epidermolysis bullosa simplex. J Invest Dermatol 2010;130:1551–7.
- Bruckner-Tuderman L, McGrath JA, Robinson EC, Uitto J. Animal models of epidermolysis bullosa: update 2010. J Invest Dermatol 2010;130:1485–8.
- Natsuga K, Shinkuma S, Nishie W, et al. Animal models of epidermolysis bullosa. Dermatol Clin 2010;28:137–42.
- Laimer M, Lanscheutzer CM, Diem A, Bauer JW. Herlitz junctional epidermolysis bullosa. Dermatol Clin 2010;28:55–60.
- Pulkkinen L, Uitto J. Hemidesmosomal variants of epidermolysis bullosa. Mutations in the α6β4 integrin and the 180-kD bullous pemphigoid antigen/type XVII collagen genes. Exp Dermatol 1998;7:46–64.
Anchoring fibrils
- Chung HJ, Uitto J. Type VII collagen: the anchoring fibril protein at fault in dystrophic epidermolysis bullosa. Dermatol Clin 2010;28:93–105.
- Christiano AM, Hoffman GG, Chung-Honet LC, et al. Structural organization of the human type VII collagen gene (COL7A1), composed of more exons than any previously characterized gene. Genomics 1994;21:169–79.
- Villone D, Fritsch A, Koch M. Supramolecular interactions in the dermo-epidermal junction zone: anchoring fibril-collagen VII tightly binds to banded collagen fibrils. J Biol Chem 2008;238:24506–13.
- Nystrom, Velati D, Mittapalli VR, et al. Collagen VII plays a dual role in wound healing. J Clin Invest 2013;123:3498–509.
- Varki R, Sadowski S, Uitto J, et al. Epidermolysis bullosa. II. Type VII collagen mutations and phenotype–genotype correlations in the dystrophic subtypes. J Med Genet 2007;44:181–92.
Extracellular matrix
- Myllyharju J, Kivirikko KI. Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet 2004;20:33–43.
- Shaw LM, Olsen BR. FACIT collagens: diverse molecular bridges in extracellular matrices. Trends Biochem Sci 1991;16:191.
- Franzke CW, Bruckner P, Bruckner-Tuderman L. Collagenous transmembrane proteins: recent insights into biology and pathology. J Biol Chem 2005;280:4005–8.
- Kuivaniemi H, Tromp G, Prockop DJ, et al. Mutations in fibrillar collagens (types I, II, III, and XI), fibril-associated collagen (type IX), and network-forming collagen (type X) cause a spectrum of diseases of bone, cartilage, and blood vessels. Hum Mutat 1997;9:300–15.
- Chu M-L, Prockop DJ. Collagen:gene structure. In: Royce PM, Steinmann B, eds. Connective Tissue and its Heritable Disorders: Molecular, Genetic and Medical Aspects, 2nd edn. New York: Wiley-Liss, 2002:223–43.
Collagens
- Myllyharju J, Kivirikko KI. Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet 2004;20:33–43.
- Kuivaniemi H, Tromp G, Prockop DJ, et al. Mutations in fibrillar collagens (types I, II, III, and XI), fibril-associated collagen (type IX), and network-forming collagen (type X) cause a spectrum of diseases of bone, cartilage, and blood vessels. Hum Mutat 1997;9:300–15.
- Symoens S, Syx D, Malfait F, et al. Comprehensive molecular analysis demonstrates type V collagen mutations in over 90% of patients with classic EDS and allows to refine diagnostic criteria. Hum Mut 2012;33:1485–93.
- Khoshnoodi J, Pedchenko V, Hudson BG. Mammalian collagen IV. Microsc Res Tech 2008;71:357–70.
- Fitzgerald J, Rich C, Zhou FH, et al. Three novel collagen VI chains, α4(VI), α5(VI) and α6(VI). J Biol Chem 2008;283:20170–80.
- Bonnemann CG. The collagen VI-related myopathies: muscle meets its matrix. Nat Rev Neurol 2011;7:379–90.
- Chung HJ, Uitto J. Type VII collagen: the anchoring fibril protein at fault in dystrophic epidermolysis bullosa. Dermatol Clin 2010;28:93–105.
- Remington J, Chen M, Burnett J, et al. Autoimmunity to type VII collagen: epidermolysis bullosa acquisita. Curr Dir Autoimmun 2008;10:195–205.
- Gatalica B, Pulkkinen L, Li K, et al. Cloning of the human type XVII collagen gene (COL17A1), and detection of novel mutations in generalized atrophic benign epidermolysis bullosa. Am J Hum Genet 1997;60:352–65.
- Schmidt E, della Torre R, Borradori L. Clinical features and practical diagnosis of bullous pemphigoid. Dermatol Clin 2011;29:427–38.
- Söderhäll C, Marenholz I, Kerscher T, et al. Variants in a novel epidermal collagen gene (COL29A1) are associated with atopic dermatitis. PLoS Biol 2007;5:e242.
- Powell AM, Sakuma-Oyama Y, Oyama M, et al. Collagen XVII/BP180: a collagenous transmembrane protein and component of the dermoepidermal anchoring complex. Clin Exp Derm 2005;30:682–7.
Collagen biosynthesis
- Myllyharju J. Prolyl 4-hydroxylases, key enzymes in the synthesis of collagens and regulation of the response to hypoxia, and their roles as treatment targets. Ann Med 2008;23:1–16.
Collagen biology
- Uitto J, Kouba DJ. Cytokine modulation of extracellular matrix gene expression: relevance to fibrotic skin diseases. J Dermatol Sci 2000;24:S60.
- Griffiths CE, Russman AN, Majmudar G, et al. Restoration of collagen formation in photodamaged human skin by tretinoin (retinoic acid). N Engl J Med 1993;329:530–5.
- Samarakoon R, Overstreet JM, Higgins PJ. TGF-β signaling in tissue fibrosis: redox controls, target genes and therapeutic opportunities. Cell Signal 2013;25:264–8.
Collagen cross-linking
- Yamauchi M, Shiiba M. Lysine hydroxylation and cross-linking of collagen. Methods Mol Biol 2008;446:95–108.
- Thorleifsson G, Magnusson KP, Sulem P, et al. Common sequence variants in the LOXL1 gene confer susceptibility to exfoliation glaucoma. Science 2007;317:1397–400.
- Challa P, Schmidt S, Liu Y, et al. Analysis of LOXL1 polymorphisms in a United States population with pseudoexfoliation glaucoma. Mol Vis 2008;14:146–9.
- Peinado H, Moreno-Bueno G, Haridsson D, et al. Lysyl oxidase-like 2 as a new poor prognosis marker of squamous cell carcinomas. Cancer Res 2008;68:4541–50.
- Mäki JM, Tikkanen H, Kivirikko KI. Cloning and characterization of a fifth human lysyl oxidase isoenzyme: the third member of the lysyl oxidase-related subfamily with four scavenger receptor cysteine-rich domains. Matrix Biol 2001;20:493–6.
- Kim DJ, Lee DC, Yang SJ, et al. Lysyl oxidase like 4, a novel target gene of TGF-β1 signaling, can negatively regulate TGF-β1 induced cell motility in PLC/PRF/5 hepatoma cells. Biochem Biophys Res Commun 2008;373:521–7.
- Pascual G, Mendieta C, Mecham RP, et al. Down-regulation of lysyl oxidase-like in aging and venous insufficiency. Histol Histopathol 2008;23:179–86.
Collagen degradation
- Hernandez-Perez M, Mahalingam M. Matrix metalloproteinases in health and disease: insights from dermatopathology. Am J Dermatopathol 2012;34:565–79.
- Peng WJ, Yan JW, Wan YN, et al. Matrix metalloproteinases: a review of their structure and role in systemic sclerosis. J Clin Immunol 2012;32:1409–14.
- Flannery CR. MMPs and ADAMTSs: functional studies. Front Biosci 2006;11:544–69.
- Pirilä E, Korpi JT, Korkiamäki T, et al. Collagenase-2 (MMP-8) and matrilysin-2 (MMP-26) expression in human wounds of different etiologies. Wound Repair Regen 2007;15:47–57.
- Shimanovich I, Mihai S, Oostingh GJ, et al. Granulocyte-derived elastase and gelatinase B are required for dermal-epidermal separation induced by autoantibodies from patients with epidermolysis bullosa acquisita and bullous pemphigoid. J Pathol 2004;204:519–27.
- Samuel CS, Lekgabe ED, Mookerjee I. The effects of relaxin on extracellular matrix remodeling in health and fibrotic diseases. Adv Exp Med Biol 2007;612:88–103.
- Samuel CS, Zhao C, Bathgate RA, et al. The relaxin gene-knockout mouse: a model of progressive fibrosis. Ann NY Acad Sci 2005;1041:173–81.
Elastic fibres
- Midwood KS, Schwarzbauer JE. Elastic fibers: building bridges between cells and their matrix. Curr Biol 2002;12:R279.
- Uitto J, Li Q, Urban Z. The complexity of elastic fibre biogenesis in the skin – a perspective to the clinical heterogeneity of cutis laxa. Exp Dermatol 2013;22:88–92.
- Uitto J, Bernstein EF. Molecular mechanisms of cutaneous aging: connective tissue alterations in the dermis. J Invest Dermatol Symp Prog 1998;1:41–4.
Elastin
- Wise SG, Weiss AS. Tropoelastin. Int J Biochem Cell Biol 2009;41:494–7.
- Uitto J, Li Q, Urban Z. The complexity of elastic fibre biogenesis in the skin – a perspective to the clinical heterogeneity of cutis laxa. Exp Dermatol 2013;22:88–92.
- Berk DR, Bentley DD, Bayliss SJ, et al. Cutis laxa: a review. J Am Acad Dermatol 2012;66:842.
- Pober BR, Johnson M, Urban Z. Mechanisms and treatment of cardiovascular disease in Williams–Beuren syndrome. J Clin Invest 2008;118:1606–15.
- Antonicellil F, Bellon G, Debelle L, et al. Elastin-elastases and inflamm-aging. Curr Top Dev Biol 2007;79:99–155.
- Kaler SG, Holmes CS, Goldstein DS, et al. Neonatal diagnosis and treatment of Menkes disease. N Eng J Med 2008;358:605–14.
- Mahoney MG, Brennan D, Starcher B, et al. Extracellular matrix in cutaneous aging: the effects of 0.1% copper-zinc malonate-containing cream on elastin biosynthesis. Exp Dermatol 2009;18:205–11.
Elastin-associated microfibrils
- Ramirez F, Sakai LY, Dietz HC, Rifkin DB. Fibrillin microfibrils: multipurpose extracellular networks in organismal physiology. Physiol Genomics 2004;19:151–4.
- Robinson PN, Arteaga-Solis E, Baldock C, et al. The molecular genetics of Marfan syndrome and related disorders. J Med Genet 2006;43:769–87.
- Hirai M, Horiguchi M, Ohbayashi T, et al. Latent TGF-β-binding protein 2 binds to DANCE/fibulin-5 and regulates elastic fiber assembly. EMBO J 2007;26:3283–95.
- Chu ML, Tsuda T. Fibulins in development and heritable disease. Birth Defects Res C Embryo Today 2004;72:25–36.
- Uitto J, Li Q, Urban Z. The complexity of elastic fibre biogenesis in the skin – a perspective to the clinical heterogeneity of cutis laxa. Exp Dermatol 2013;22:88–92.
- Verdone G, Doliana R, Corazza A, et al. The solution structure of EMILIN1 globular C1q domain reveals a disordered insertion necessary for interaction with the α4β1 integrin. J Biol Chem 2008;283:18947–56.
Proteoglycan/glycosaminoglycans
- Iozzo RV. Basement membrane proteoglycans: from cellar to ceiling. Nat Rev Mol Cell Biol 2005;6:646–56.
- Danielson KG, Baribault H, Holmes DF, et al. Targeted disruption of decorin leads to abnormal collagen fibril morphology and skin fragility. J Cell Biol 1997;136:729–43.
- Esko JD, Lindahl U. Molecular diversity of heparan sulfate. J Clin Invest 2001;108:169–74.
- Agren UM, Tammi M, Ryynänen M, Tammi R. Developmentally programmed expression of hyaluronan in human skin and its appendages. J Invest Dermatol 1997;109:219–24.
- Ghersetich I, Lotti T, Campanile G, et al. Hyaluronic acid in cutaneous intrinsic aging. Int J Dermatol 1994;33:119–22.
- Stryer L. Carbohydrates. In: Berg JM, Tymoczko JL, Stryer L, eds. Biochemistry, 4th edn. New York: WH Freeman, 1995:461–82.
- Heinegard D, Oldberg A. Glycosylated matrix proteins. In: Royce PM, Steinmann B, eds. Connective Tissue and its Heritable Diseases. New York: Wiley Liss, 1993:189–209.
- Dours-Zimmerman MT, Zimmerman DR. A novel glycosaminoglycan attachment domain, identified in two alternative splice variant of versican. J Biol Chem 1994;269:32992–8.
Fibroblasts
- Sorrell JM, Baber MA, Caplan AI. Clonal characterization of fibroblasts in the superficial layer of the adult human dermis. Cell Tissue Res 2007;327:499–510.
- Chang HY, Chi JT, Dudoit S, et al. Diversity, topographic differentiation, and positional memory in human fibroblasts. Proc Natl Acad Sci USA 2002;99:12877–82.
- Rinn JL, Bondre C, Gladstone HB, et al. Anatomic demarcation by positional variation in fibroblast gene expression programs. PLoS Genet 2006;2:e119.
- Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007;131:861–72.
- Park IH, Zhao R, West JA, et al. Reprogramming of human somatic cells to pluripotency with defined factors. Nature 2008;451:141–6.
- Nakagawa M, Koyanagi M, Tanabe K, et al. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol 2008;26:101–6.
- Lin SL, Chang DC, Chang-Lin S, et al. Mir-302 reprograms human skin cancer cells into a pluripotent ES-cell-like state. RNA 2008;14:2115–24.
- Fabbri M, Croce CM, Calin GA. MicroRNAs. Cancer J 2008;14:1–6.
- Yi R, O'Carroll D, Pasolli HA, et al. Morphogenesis in skin is governed by discrete sets of differentially expressed microRNAs. Nat Genet 2006;38:356–62.
- Yi R, Poy MN, Stoffel M, et al. A skin microRNA promotes differentiation by repressing ‘stemness.’ Nature 2008;452:225–9.
- Sonkoly E, Wei T, Janson PC. MicroRNAs: novel regulators involved in the pathogenesis of psoriasis? PLoS ONE 2007;2:e610.
Blood vessels and lymphatics
- Ryan TJ. Cutaneous circulation. In: Goldsmith LA, ed. Biochemistry and Physiology of the Skin. New York: Oxford University Press, 1983: 817–77.
- Braverman IM, Yen A. Ultrastructure of the human dermal microcirculation. II. The capillary loops of the dermal papillae. J Invest Dermatol 1977;68:44–52.
- Braverman IM, Yen A. Ultrastructure of the human dermal microcirculation. III. The vessels in the mid- and lower dermis and subcutaneous fat. J Invest Dermatol 1982;78:297–304.
- Pober JS. Cytokine-mediated activation of vascular endothelium: physiology and pathology. Am J Pathol 1988;133:426–33.
- Jurisic G, Detmar M. Lymphatic endothelium in health and disease. Cell Tissue Res 2009;335:97–108.
Subcutaneous fat
- Wehrli NE, Bural G, Houseni M, et al. Determination of age-related changes in structure and function of skin, adipose tissue, and skeletal muscle with computed tomography, magnetic resonance imaging, and positron emission tomography. Semin Nucl Med 2007;37:195–205.
- Farooqi IS, O'Rahilly S. Mutations in ligands and receptors of the leptin–melanocortin pathway that lead to obesity. Nat Clin Pract Endocrinol Metab 2008;4:569–77.
- Bessesen DH. Update on obesity. J Clin Endocrinol Metab 2008;93:2027–34.
- Algire C, Medrikova D, Herzig S. White and brown adipose stem cells: from signaling to clinical implications. Biochim Biophys Acta 2013;1831:896–904.
Physiological functions of skin
- Kubo A, Nagao K, Amagai M. Epidermal barrier dysfunction and cutaneous sensitization in atopic diseases. J Clin Invest 2012;122:440–7.
- Elias PM, Eichenfield LF, Fowler JF, Jr, et al. Update on the structure and function of the skin barrier: atopic dermatitis as an exemplar of clinical implications. Semin Cutan Med Surg 2013;32:S21–4.
- Noël F, Piérard-Franchimont C, Piérard GE, Quatresooz P. Sweaty skin, background and assessments. Int J Dermatol 2012;51:647–55.
- Ali AT, Hochfeld WE, Myburgh R, Pepper MS. Adipocyte and adipogenesis. Eur J Cell Biol 2013;92:229–36.
- Biggs LC, Mikkola ML. Early inductive events in ectodermal appendage morphogenesis. Semin Cell Dev Biol 2014;25//ϖ26C:11–21.
Skin homeostasis
- Blanpain C, Fuchs E. Epidermal homeostasis: a balancing act of stem cells in the skin. Nat Rev Mol Cell Biol 2009;10:207–17.
- Fuchs E. The tortoise and the hair: slow-cycling cells in the stem cell race. Cell 2009;137:811–19.
- Potten CS. The epidermal proliferative unit: the possible role of the central basal cell. Cell Tissue Kinet 1974;7:77–88.
- Rompolas P, Greco V. Stem cell dynamics in the hair follicle niche. Semin Cell Dev Biol 2014;25//ϖ26C:34–42.
- Lechler T, Fuchs E. Asymmetric cell divisions promote stratification and differentiation of mammalian skin. Nature 2005;437:275–80.
- Koster MI. p63 in skin development and ectodermal dysplasias. J Invest Dermatol 2010;130:2352–8.
- Ito M, Yang Z, Andl T, et al. Wnt-dependent de novo hair follicle regeneration in adult mouse skin after wounding. Nature 2007;447:316–20.
- Cotsarelis G. Epithelial stem cells: a folliculocentric view. J Invest Dermatol 2006;126:1459–68.
- Toma JG, McKenzie IA, Bagli D, Miller FD. Isolation and characterization of multipotent skin derived precursors from human skin. Stem Cells 2005;23:727–37.
- Chen FG, Zhang WJ, Bi D, et al. Clonal analysis of nestin (–) vimentin (+) multipotent fibroblasts isolated from human dermis. J Cell Sci 2007;120:2875–83.
- Park IH, Zhao R, West JA, et al. Reprogramming of human somatic cells to pluripotancy with defined factors. Nature 2008;451:141–6.
Skin ageing
- Zouboulis CC, Makrantonaki E. Clinical aspects and molecular diagnostics of skin aging. Clin Dermatol 2011;29:3–14.
- Tagami H. Functional characteristics of the stratum corneum in photoaged skin in comparison with those found in intrinsic aging. Arch Dermatol Res 2008;300:S1–6.
- Robert L, Labat-Robert J, Robert AM. Physiology of skin aging. Pathol Biol (Paris) 2009;57:336–41.
- Waller JM, Maibach HI. Age and skin structure and function, a quantitative approach (II): protein, glycosaminoglycan, water, and lipid content and structure. Skin Res Technol 2006;12:145–54.
- Ma W, Wlaschek M, Tantcheva-Poor I, et al. Chronological ageing and photoageing of the fibroblasts and the dermal connective tissue. Clin Exp Dermatol 2001;26:592–9.
- Schludermann E, Zubeck JP. Effect of age on pain sensibility. Percept Motor Skills 1962;14:295–301.
- Winkelmann RK. Nerve changes in aging skin. In: Montagna W, ed. Advances in Biology of Skin, Vol. 6. Aging. Oxford: Pergamon, 1965:51–61.
- Grewe M. Chronological ageing and photoageing of dendritic cells. Clin Exp Dermatol 2001;26:608–12.
- Bhushan M, Cumberbatch M, Dearman RJ, et al. Tumour necrosis factor-α induced migration of human Langerhans' cells:the influence of ageing. Br J Dermatol 2002;146:32–40.
- Bhushan M, Cumberbatch M, Dearman RJ, et al. Exogenous interleukin-1β restores impaired Langerhans cell migration in aged skin. Br J Dermatol 2004;150:1217–18.
- Rabe JH, Mamelak AJ, McElgunn PJ, et al. Photoaging: mechanisms and repair. J Am Acad Dermatol 2006;55:1–19.
- Thivolet J, Nicolas JF. Skin ageing and immune competence. Br J Dermatol 1990;122(Suppl. 35):77–81.
- Simpson SJ, Raubenheimer D. Caloric restriction and aging revisited: the need for a geometric analysis of the nutritional bases of aging. J Gerontol A Biol Sci Med Sci 2007;62:707–13.
- Giacomoni PU, Rein G. A mechanistic model for the aging of human skin. Micron 2004;35:179–84.
- Millington GWM. Epigenetics and dermatological disease. Pharmacogenomics 2008;9:1835–50.
- Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol 1956;11:298–300.
- De la Fuente M, Miquel J. An update of the oxidation-inflammation theory of aging: the involvement of the immune system in oxi-inflamm-aging. Curr Pharm Des 2009;15:3003–26.
- Blasco MA. Telomere length, stem cells and aging. Nat Chem Biol 2007;3:640–9.
- Adams PD. Healing and hurting: molecular mechanisms, functions, and pathologies of cellular senescence. Mol Cell 2009;36:2–14.
- Burtner CR, Kennedy BK. Progeria syndromes and ageing; what is the connection? Nat Rev Mol Cell Biol 2010;11:567–8.
- Melzer D. Genetic polymorphisms and human aging: association studies deliver. Rejuvenation Res 2008;11:523–6.
- Kim WY, Sharpless NE. The regulation of INK4/ARF in cancer and aging. Cell 2006;127:265–75.
- Bonafè M, Olivieri F. Genetic polymorphism in long-lived people: cues for the presence of an insulin/IGF-pathway-dependent network affecting human longevity. Mol Cell Endocrinol 2009;299:118–23.
- Mounkes L, Kozlov S, Burke B, et al. The laminopathies: nuclear structure meets disease. Curr Opin Genet Dev 2003;13:223–30.
- Viteri G, Chung YW, Stadtman ER. Effect of progerin on the accumulation of oxidized proteins in fibroblasts from Hutchinson Gilford progeria patients. Mech Ageing Dev 2010;131:2–8.
- Lahmann C, Young AR, Wittern K-P, et al. Induction of mRNA for matrix metalloproteinase 1 and tissue inhibitor of metalloproteinases 1 in human skin in vivo by solar simulated radiation. Photochem Photobiol 2001;73:657–63.
- Quan T, Qin Z, Xia W, et al. Matrix-degrading metalloproteinases in photoaging. J Investig Dermatol Symp Proc 2009;14:20–4.
- Titeux M, Pendaries V, Tonasso L, et al. A frequent functional SNP in the MMP1 promoter is associated with higher disease severity in recessive dystrophic epidermolysis bullosa. Hum Mutat 2008;29:267–76.
- Lahmann C, Bergemann J, Harrison G, et al. Matrix metalloproteinase-1 and skin ageing in smokers. Lancet 2001;357:935–6.
- Morita A. Tobacco smoke causes premature skin aging. J Dermatol Sci 2007;48:169–75.
- Harries LW, Hernandez D, Henley W, et al. Human aging is characterized by focused changes in gene expression and deregulation of alternative splicing. Aging Cell 2011;10:868–78.
- McGrath JA, Robinson MK, Binder RL. Skin differences based on age and chronicity of ultraviolet exposure: results from a gene expression profiling study. Br J Dermatol 2012;166(Suppl. 2):9–15.