CHAPTER 9
Photobiology
Antony R. Young
St John's Institute of Dermatology, Division of Genetics and Molecular Medicine, Faculty of Life Sciences and Medicine, King's College London, London, UK
Basic principles
Photobiology is the study of the effects of ultraviolet (UV) and visible radiation on life, and this chapter focuses on the photobiology of normal skin. This is important because the skin is the body's interface with the environment, and solar ultraviolet radiation (UVR) is probably our most important natural environmental hazard. Increasingly, people are also exposing themselves to artificially generated UVR for cosmetic tanning. Most of the effects of solar UVR on the skin are detrimental and should be minimized by photoprotection strategies. However, solar UVR is also necessary for vitamin D production, and may even be beneficial for cardiovascular disease (CVD). UV and visible radiation may be exploited in photo(chemo)therapy and photodynamic therapy for the treatment of skin disorders (see Chapters 21 and 22), and indeed for internal diseases. Furthermore, many people have disorders that render their skins abnormally sensitive to UV and visible radiation. These aspects of photodermatology are discussed in Chapter 127.
UVR and its production and sources
Ultraviolet and visible radiation, which comprises a very small part of the electromagnetic radiation spectrum (Figure 9.1), is energy released during the transition of a molecular electron from a higher energy outer molecular orbital to a less energetic inner one. Each such emission, known as a photon, is a discrete oscillating electromagnetic pulse of energy E (joules, J), wavelength λ (nanometres, nm, 10–9 m) and velocity through space c (3 × 108 m/s), such that E = hc/λ, where h = 6.63 × 10–34 J/s (Planck's constant) (Figure 9.2). Thus, a 300-nm photon has energy of 6.63 × 10–19 J, where 1 J is defined as the work required to accelerate 1 kg over 1 m in 1 s to a velocity of 1 m/s in a frictionless environment. Repeated molecular emissions from a point source lead to multiple spherical radiation wave fronts, each of total energy equal to the sum of the individual photon energies, but diverging with gradually diminishing intensity per unit surface area. UVR can be defined in terms of photons (i.e. particles) or in waveform.
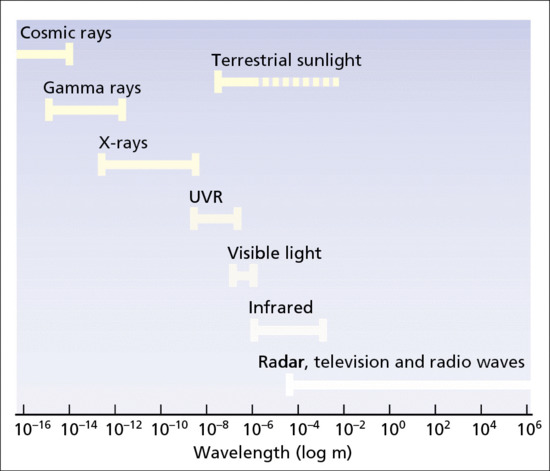
Figure 9.1 The electromagnetic spectrum.
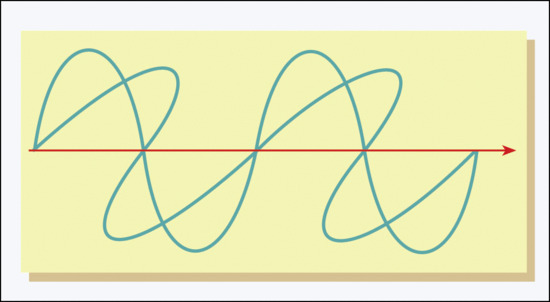
Figure 9.2 Schematic representation of electromagnetic wave.
Ultraviolet radiation is, by convention, divided into three subcategories officially designated by the Commission Internationale de l'Eclairage (CIE):
- UVC (200–280 nm).
- UVB (280–315 nm).
- UVA (315–400 nm).
However, a UVB/UVA boundary of 320 nm is very widely used in photodermatology. Furthermore, UVA is often subdivided into UVA1/I (340–400 nm) and UVA2/II (315/320–340 nm). This subdivision of UVA is based on different molecular mechanisms within the UVA spectral region. In general, UVA2 is thought to act more like UVB in the ways that it exerts its molecular and biological effects.
Measurement of UVR
Accurate measurement of UVR is important for all aspects of photobiology. Such measurement is called dosimetry, which can be approached physically [1] or biologically.
Physical dosimetry
Physical dosimeters contain sensors, typically photodiodes that respond to particular wavebands. The amount of such energy incident on a surface is known as the radiant exposure, exposure dose or fluence (J/m2 or mJ/cm2), and the rate of incidence as the irradiance, dose rate or intensity (W/m2 or mW/cm2), where 1 W = 1 Watt = 1 J/s.
In practice, exposure dose to the skin, measured as J/m2, is the product of W/m2 × time (s). Most modern phototherapy cabinets have built in dosimeters that can be programmed to calculate exposure time for a prescribed dose. However, it is wise to have these independently verified, and phototherapy and research departments will benefit considerably from close links to physicists with expertise in UVR measurement.
The spectral responsivities of different photodiodes in hand-held dosimeters vary considerably and typically have a bell shape. This means, for example, that a designated UVB sensor may also have some sensitivity in the UVA region or vice versa. This can result in false measurements when measuring UVB irradiance with a source that contains UVA or vice versa, and indeed there are many errors of UVR measurement in published literature because this has not been appreciated. Such errors can also give rise to false conclusions about the spectral dependence of biological outcomes. Thus, a given UVR dosimeter must be calibrated for a given UVR source and such a calibration is only valid for the given source. Calibration is widely done with a spectroradiometer that measures the irradiance of a given UVR spectrum on a wavelength-by-wavelength basis. Such measurement provides the emission spectrum of a given source. A good spectroradiometer will have a dynamic range of about six orders of magnitude. This is very important because even low levels (<1.0%) of a given waveband (usually UVB) may have very important biological effects. Spectroradiometers are themselves calibrated against national standard lamps.
Biological dosimetry
In biological dosimetry, the skin is the sensor and the most widely used measurement outcome is the minimal erythema dose (MED), which is a response that is typically assessed 24 h after UVR exposure. The MED is determined by exposing small areas of skin (e.g. 1 cm2) to a geometrically increasing series (e.g. × 1.25 or × √2) of exposures and determining the lowest UVR dose that results in either just perceptible redness or redness with definite borders. The MED is widely used as a measure of exposure in phototherapy and experimental photobiology. A threshold assessment by eye such as the MED is subjective but is reproducible and reliable if performed carefully with standardized lighting. It is also used in the diagnosis of the photodermatoses (see Chapter 127). By definition, the MED is a measure of individual sensitivity to UVR of a given spectrum. It should be noted that MED varies with body site [2], especially when comparing sites that are habitually sun exposed with those that are habitually sun protected. There is increasing use of the standard erythema dose (SED) [3], especially in research and epidemiology. One SED is equivalent to an erythemal effective radiant exposure of 100 J/m2. This is obtained by weighting a spectroradiometric measurement with the CIE action spectrum for erythema (see section later on action spectroscopy) [4]. One great advantage of the SED is that, in contrast to the MED, it is independent of personal UVR sensitivity and the emission spectrum of the UVR source.
Terrestrial UVR
The spectrum of terrestrial solar UVR is modified by several factors including the absorption of UVC and UVB by oxygen (O2) and stratospheric ozone (O3). Thus, the stratospheric ozone layer absorbs all UVC and attenuates UVB, but has virtually no effect on UVA. The UVR spectral profile of terrestrial sunlight is strongly influenced by the height of the sun that is dependent on latitude, season and time of day. Particulate and gaseous (e.g. O3, NO2, SO2) pollution in the troposphere (0–10 km above sea level) can also influence UVR spectrum. UVB is at its most intense when the sun is directly overhead, e.g. at solar noon on the equator. This is because the pathway through the stratosphere is shorter (i.e. less attenuation of UVB) than when the sun is low in the sky (Figure 9.3). This means that the ratio of UVB to UVA varies with the height of the sun (e.g. time of day year) [5], especially at higher latitudes, because there is more seasonal fluctuation of UVB irradiance compared with UVA. However, even when the sun is directly above (e.g. on the equator at noon at sea level) the vast majority of solar UVR is UVA (>90%). Examples of solar spectra are given in Figure 9.4, in which UVB content is about 5% when the sun is high in the sky. Thus, it can be said that the sun is primarily a UVA source, especially UVA1 (>70%). It should be noted that solar irradiance increases with altitude, e.g. on mountains. For example, in an Alpine region in summer, total UVR and UVA irradiance increases by about 8–9%/1000 m. When total irradiance is erythemally weighted (see section on action spectroscopy) this increases to 18%/1000 m [6].
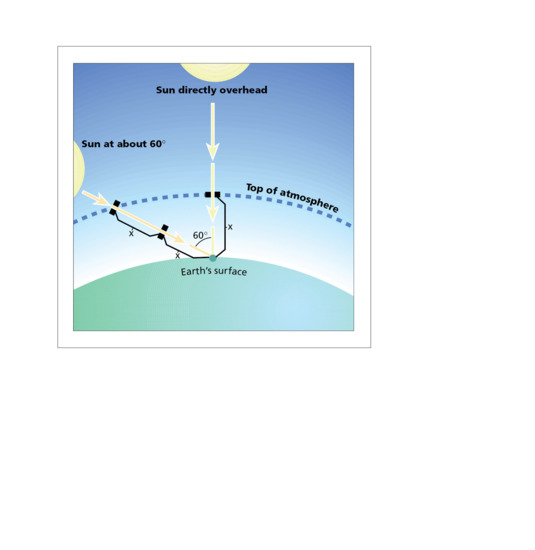
Figure 9.3 UVR path lengths for differing solar elevations.
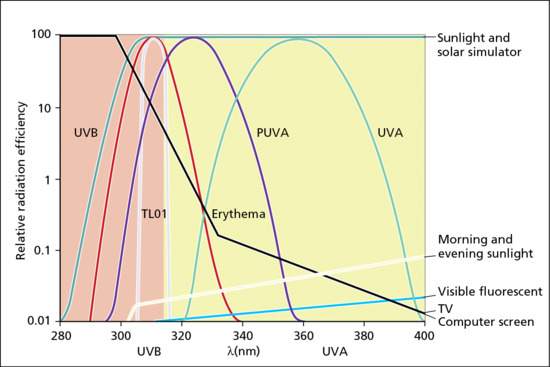
Figure 9.4 Approximate emission spectra of typical sources encountered in dermatological photobiology and elsewhere, with action spectrum for human cutaneous erythema for comparison.
Artificial sources of UVR
There are many man-made sources of UVR [7], but the most common is the fluorescent tube which is widely used in photo(chemo)therapy and by the tanning industry. This is a sealed glass tube that contains mercury and an inert gas, typically argon. The inner lining of the tube is coated with a phosphor powder. The mercury is excited when an electrical current is passed though the tube and releases UVR photons, which interact with the phosphor. UV and visible radiation are released by this interaction, the emission spectra of which can be tailored by the composition of the phosphor. The tubes widely used in phototherapy are typically narrow band, more or less monochromatic, UVB (∼313 nm), broadband UVB and UVA. Cosmetic tanning tubes generally contain primarily UVA1 with a small amount of UVA2 and UVB. See examples of phototherapy tube emission spectra in Figure 9.4.
Some UVR sources are based on the xenon (Xe) arc, which emits a very broad UV and visible spectrum that can be modified by suitable optics. Solar simulating radiation (SSR) (see Figure 9.4) can be generated with suitable glass filters, and is used in research, clinical phototesting and the assessment of sunscreens. The Xe arc can also be coupled with a monochromator to generate ‘monochromatic’ radiation that is required to determine action spectra (see section later). However, it should be noted that in reality this is narrow band polychromatic radiation with a bell shape. Such monochromatic spectra are characterized by their spectral bandwidth at full width at half maximum (FWHM), which is defined as the width of the spectral band at 50% of the peak maximum. For example, monochromatic radiation at 300 nm might have a FWHM of 5 nm.
Metal halide lamps are used to generate high output UVA1 that is used in high dose UVA1 phototherapy in which exposures are more than 60 J/cm2 [8]. A more recent development with promise in phototherapy is the light-emitting diode (LED) [9].
Interaction of UVR with the skin (physicochemical aspects)
Radiation incident on a surface is either reflected, or transmitted and scattered within the medium beneath, particularly at short wavelengths (Figure 9.5). It then eventually exits, unless it first collides with and is absorbed by an appropriately structured molecular moiety, known as a chromophore. In the skin these include DNA, trans-urocanic acid (tUCA), proteins with aromatic amino acids, melanins, metabolites of melanogenesis, flavins and porphyrins [10]. Any of a broad range of contiguous wavelengths may be absorbed, with slightly differing probabilities resulting in a highly characteristic absorption spectrum (Figure 9.6) for a given chromophore; for example naked DNA has an absorption maximum at 260 nm. This results in electronic excitation to an outer higher energy molecular orbital (Figure 9.7), enabling chemical reactions to occur which can lead to molecular, cellular and clinical outcomes. Thus the basis of all skin photobiology, whether acute or long term, is photochemistry.
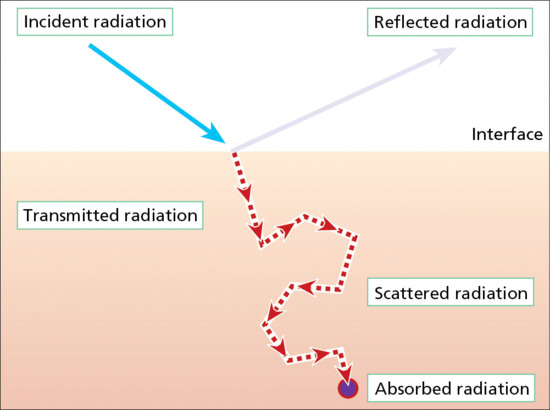
Figure 9.5 Interaction of UVR with physical matter.
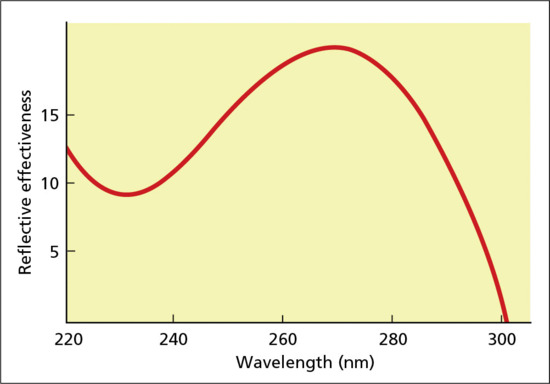
Figure 9.6 Typical appearance of wavelength probability curve for excitation of orbiting electron to outer electronic orbital (absorption spectrum).
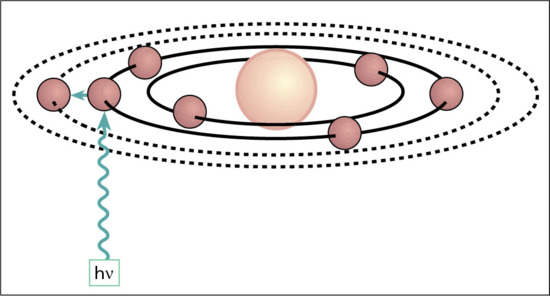
Figure 9.7 Schematic representation of photon absorption, leading to excitation of orbiting electron to outer electronic orbital (see text for abbreviations).
The penetration (transmission) of UVR through the skin is a major determinant of the location of photobiological outcome. Transmission studies of UVR have usually been done on isolated mouse and human epidermis and have shown much greater transmission of UVA than UVB. This is to be expected as some of the major chromophores in skin attenuate UVB as it passes to the basal layer and the dermis. For example, the stratum corneum is very rich in tUCA. Another approach to assess UVR penetration of the skin is to measure molecular markers of photodamage after in vivo irradiation. Such studies, using a marker of DNA damage, confirm that UVA penetrates better than UVB, but also suggest more scattering of UVA [11,12].
Chromophores may undergo structural change when they absorb UV or visible radiation energy. For example, di-pyrimidine lesions are formed in DNA and tUCA isomerizes to the cis form. The above examples are referred to as direct responses because the chromophore is also the target molecule that initiates the photobiological response. Indirect responses also occur, in which the chromophore is not the molecule that causes the response per se. For example, a chromophore may absorb UV or visible radiation energy and transfer this energy to oxygen that results in the generation of reactive oxygen species (ROS) that can damage cellular targets such as DNA and membranes. As a rule of thumb, UVB and UVA2 cause direct effects in normal skin whereas UVA1 causes indirect effects, but there are many exceptions. It should be noted that this rule of thumb is largely based on in vitro work.
Action spectroscopy
An action spectrum (wavelength dependence) determines the relationship between wavelength (λ) and photobiological outcome, usually expressed on a logarithmic scale because of the very large differences between different spectral regions for many biological outcomes. Figure 9.8 shows the CIE action spectrum for erythema in normal human skin and demonstrates that UVB is orders of magnitude more erythemogenic than UVA for a given UVR dose (J/m2). The relative difference between UVB and UVA for erythema will depend on the exact wavelengths being compared, but a rule of thumb for broad UVB and UVA spectra is that the former is about 1000 times more erythemogenic than the latter. Action spectroscopy has two main purposes. The first is the identification of the chromophore that is responsible for the biological response and this is critical for the understanding of a photobiological mechanism. Under ideal conditions, an action spectrum of a given biological response can be superimposed on the absorption spectrum of the chromophore that initiates the response. A good non-dermatological example is similarity of the action spectrum for photosynthesis and the absorption spectrum of chlorophyll. However, in more complex structures like human skin, such a match is much less likely because of competing chromophores and scattering that modify skin optics. The second and more important practical purpose of action spectra is their use as biological weighting functions for given UVR emission spectra. Biological weighting is the process whereby a given emission spectrum (e.g. solar UVR) is multiplied, wavelength by wavelength, by a given action spectrum to generate a third curve to define biologically effective energy. As described under Terrestrial UVR, the sun is primarily a UVA1 source; however, it is the small UVB (typically <5%) content that is primarily (>80%) responsible for erythema (sunburn). One practical consequence of this is that sunscreens would be ineffective without good UVB filtering. Biological weighting with action spectra can often give surprising results, such that <1% of a given emission spectrum may be responsible for the vast majority of a given biological or clinical response [13]. The determination of good action spectra for human skin is technically demanding and very time consuming. Reliable biological weighting requires very high quality spectroradiometric measurements. The action spectrum for normal erythema can be altered by administration of an exogenous chromophore. For example, the addition of 8-methoxypsoralen (8-MOP) to the skin, orally or topically, will result in an erythema action spectrum with a peak in the UVA region of about 340 nm [14].
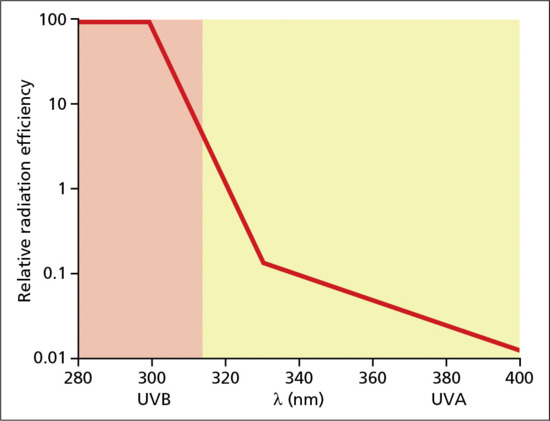
Figure 9.8 Action spectrum for induction of erythema in human skin.
Normal effects of UVR on skin
Molecular and cellular effects
The cellular epidermis is rich in UVR-absorbing chromophores and is therefore very vulnerable to photodamage. A summary of the main molecular and cellular effects is given in Table 9.1. The most important chromophore is probably nuclear DNA. Pyrimidine dimers (also known as dipyrimidine lesions) form after the absorption of photon energy that splits the C5=C6 double bonds of two adjacent pyrimidine bases, and forms new covalent bonds linking the two pyrimidines at the C5 and C6 positions. A 4-carbon cyclobutane ring is produced, giving the dimer its characteristic name; the cyclobutane pyrimidine dimer (CPD). Another lesion is the 6-4 pyrimidine-pyrimidone (6-4PP) in which the C5=C6 double bonds break and the surplus energy results in the rotation of one of the pyrimidine rings which offers its C4 to form a new bond with the C6 of the adjacent pyrimidine [15]. In this case, only one new covalent bond is formed. CPD and 6-4(PP) can be readily detected in skin in vivo after suberythemal exposure of UVB and SSR. The 6-4PP is also induced by UVB but not by UVA1 [11]. The induction of CPD and 6-4(PP) by UVB (300 nm) and UVA1 in human skin is shown in Figure 9.9.
Table 9.1 Summary of the main normal effects of UVR on the skin
| Acute | Chronic | |
| Molecular/cellular | Clinical | Clinical |
| DNA photodamage (and its repair) and mutation | Erythema | Skin cancer |
| Melanogenesis | Delayed tanning | Photoageing |
| Inflammatory infiltrate, hyperplasia and stratum corneum thickening | Immediate pigment darkening | |
| Reactive oxygen species that damage DNA and cell membranes | Persistent pigment darkening | |
| Apoptosis (sunburn cells) | Suppression of acquired immunity | |
| Langerhans cell depletion | Enhancement of innate immunity | |
| Gene and protein expression | Reduction of blood pressure (by UVA) | |
| Vitamin D photosynthesis | ||
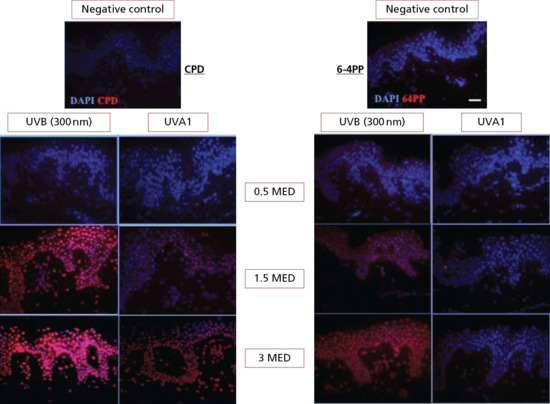
Figure 9.9 Cyclobutane pyrimidine dimer (CPD) and 6-4(PP) in skin immediately after UVB (300 nm) and UVA1 exposure. Note UVR dose response, damage to dermal cells and lack of 6-4(PP) with UVA1. This damage is potentially mutagenic and is removed by NER. DAPI identifies nuclei.
(DNA damage images courtesy of Dr Angela Tewari, St John's Institute of Dermatology, with permission.)
Oxidative stress (a state in which the cellular antioxidant system is overwhelmed by ROS) induces modification of cellular biomolecules such as lipids, proteins and over 30 types of nucleobase modifications. Guanine has a low threshold for oxidation, and commonly two types of oxidative DNA damage occur: 8-oxo-7,8-dihydroguanine (8oxoGua) and 8oxo7,8 dihydro2deoxguanosine (8oxodG) [15]. These can be induced by UVA and UVB, although it is widely reported that UVA produces relatively more oxidative damage than UVB.
Repair of DNA damage
DNA photodamage is ubiquitous in life and most organisms have elaborate DNA repair mechanisms. The basic principle of DNA repair in the skin is the recognition of the photolesion that is to be removed and replaced with a new base(s) followed by ligation to give intact DNA. Faithful DNA repair is critical for genomic integrity. The insertion of an incorrect base in proliferating epidermal cells may result in mutations that give rise to skin cancer.
Nucleotide excision repair
The most important repair process in human skin is nucleotide excision repair (NER). This process is dependent on a large group of enzymes with specific roles in the recognition and excision of DNA photolesions, and the repair of DNA using the opposite strand as a template. NER is triggered by the activation of p53 protein that occurs in response to cellular stress including DNA damage, oncogenic stimulation and hypoxia. This triggers a G1/S cell cycle arrest and transcriptional activation of NER genes. NER is also called unscheduled DNA synthesis (UDS), as opposed to scheduled DNA synthesis that is observed in cells undergoing mitosis. Failure to repair DNA by the lack of one or more repair proteins may have catastrophic consequences exemplified by patients with xeroderma pigmentosum (XP) who are extremely prone to skin cancer (see Chapter 78) [16]. Studies on cell cultures from XP patients have greatly increased our understanding of DNA repair and indeed many of the DNA repair proteins are described by the XP prefix.
Nucleotide excision repair has two subpathways: global genome repair (GGR) and transcription coupled repair (TCR) in which repair is selectively directed at the actively transcribing strand [16, 17]. In GGR damage is recognized by XPE and/or XPC, whilst in TCR the photoproducts cause RNA polymerase II to stall and damage recognition is done by CSA and CSB. In both processes, repair continues through a transcription factor replicosome complex consisting of 30 distinct proteins in human cells. Assessment of GGR in human epidermis in vivo shows that repair of 6-4(PP) is relatively rapid and more or less complete within 6 h. In contrast, the half-life for the removal of the CPD is about 30 h [18]. This difference may be because the 6-4(PP) causes a more significant distortion in the double helix than the CPD, which may result in more rapid recognition and repair.
Base excision repair
Base excision repair (BER) repairs damage to non-distorting single base modifications caused by oxidation [17]: 8-oxoguanine, 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapyG, FapyA), alkylation (3-methyladenine, 7-methylguanine), hydrolysis or deamination (hypoxanthine from deaminated adenine or xanthine from deaminated guanine). BER is initiated by DNA glycosylases that recognize and remove specific damaged or inappropriate bases. This leaves an apurinic/apyrimidic site or naked sugar phosphate backbone commonly referred to as an AP site. Once cleaved by an AP endonuclease, the resulting single-strand break can then be processed by either short-patch (where a single nucleotide is replaced) or long-patch BER (where between two and 10 new nucleotides are synthesized). One of the most well-described glycosylases is human 8-oxoguanine glycosylase (hOGG1) which repairs 8oxodG, and is poorly expressed in the basal layer. Consequently, UVA-induced 8-oxodG is repaired more slowly in the basal than in the upper layers of human epidermis in which lesions can be repaired within 2 h of UVA exposure [19].
In vitro studies have shown that UVR can upregulate a wide range of genes and this is also the case for skin in vivo. This includes nuclear transcription factors and genes associated with inflammation, immunoregulation and the remodelling of extra cellular proteins. The mechanisms by which this upregulation occurs, especially for specific genes, is not well understood but studies have shown a role for direct damage to DNA as well as ROS. Thus, some genes are more responsive to UVB and others to UVA.
Cellular effects
Ultraviolet radiation exposure of the skin results in a wide range of cellular effects. One of the most striking is the formation of epidermal sunburn cells (SBC) with pycnotic nuclei and eosinophilic cytoplasm. SBC are apoptotic keratinocytes that are readily induced by SSR and UVB but not by UVA1. Their formation is regulated by p53 that can be readily demonstrated by immunostaining. Its role in UVR-induced apoptosis is clear from mouse studies [20, 21] in which UVR irradiation of p53 wild-type mice induced the formation of SBC whilst p53-null mice were resistant to such apoptosis and accumulated CPD. p53 induces apoptosis through Bcl2 [22] family members (BAX, PUMA) and an upregulation of the death receptor FAS (also known as CD95 and APO-1) which eliminates DNA-damaged keratinocytes cells through a caspase-associated pathway. Thus, potentially carcinogenic cells are eliminated.
The loss of Langerhans cell (LC) dendricity and number is also seen. These changes may be accompanied by epidermal spongiosis along with dermal vasodilatation, neutrophil and CD3+ lymphocyte infiltration [23, 24]. Skin hyperplasia develops over hours to days after UVB or SSR (but generally not UVA) exposure, and results from a marked increase in cell mitosis, DNA, RNA and protein synthesis rates, after some hours of early inactivity. This is followed by a thickening of the epidermis, particularly the stratum corneum.
Clinical effects of UVR
The acute and chronic effects of UVR depend on skin type, which is usually a phenotypic assessment of skin colour coupled with self-reported acute responses to solar UVR. A summary of skin type and associated risks is shown in Table 9.1. Fair-skinned skin types I and II are at greatest risk from the acute and long-term effects of solar UVR. In general, MED on previously unexposed buttock skin increases with skin type [25], but there is considerable overlap and MED cannot be used to predict skin type or vice versa. The genetic basis of skin type is poorly understood.
Acute
The acute clinical effects of UVR are those that occur within seconds to weeks after a single UVR exposure.
Erythema
The most obvious acute clinical effect is erythema, which is UVR-induced inflammation. Typically with SSR/UVB, this is apparent after about 6 h and peaks at about 24 h and gradually resolves over a few days. Erythema may be associated with pain, warmth and oedema (blanching) in severe cases. Erythema is also associated with increased sensitivity to pain from mechanical and thermal stimuli, resulting in the skin feeling tender after sunburn [26]. Repeated daily suberythemal (0.65 MED) exposure in skin types II has a cumulative effect resulting in frank erythema after a few days, which is not the case in skin types IV [27], even though they received twice the physical dose because of their higher MED. This suggests that those with fair skin resolve erythema less well than those who tan readily.
Erythema action spectra have been studied for over 90 years and a library of such spectra has been compiled [28]. A standard CIE action spectrum for erythema [4] is shown in Figure 9.8. Typically, the MED is around 500 J/m2 for the UVC–UVB boundary (280 nm), 250 J/m2 for UVB (300 nm) and 320 kJ/m2 for UVA (360 nm). This action spectrum is similar, to that for CPD induction in human skin in vivo in the solar UVB and UVA range and provides indirect evidence that the CPD initiates the erythemal response [29]: wavelengths around 300 nm are the most DNA damaging and erythemogenic. A role for DNA in erythema is also supported by some animal studies. However, it is likely that other unknown non-DNA chromophores/mechanism contribute to erythema, especially in the UVA1 region. For example, it has been shown that UVA erythema is oxygen dependent which is not the case for UVB [30]. This is supported by studies that show a lack of correlation between UVB and UVA1 MED in the same individuals, suggesting different independent mechanisms [31]. The pharmacology of UVR-induced erythema is incompletely understood but there is evidence that it is mediated by prostaglandins and nitric oxide (NO) [23, 32].
Tanning
Three ‘tanning’ processes occur in response to UVR, are listed below and their action spectra have been compiled [28]:
- Immediate pigment darkening (IPD).
- Persistent pigment darkening (PPD).
- Melanogenesis or delayed tanning (DT).
Immediate and persistent pigment darkening
Immediate pigment darkening presents as a transient grey colour which is thought to be due to immediate photo-oxidation of existing colourless melanin precursors after exposure to UVA (action spectrum peak at 340 nm) and visible (400–500 nm) radiation. IPD, which fades within 15 min, is induced at low UVA doses (1–4 J/cm2). It is almost undetectable in fair-skinned individuals, but is easily observed in skin types IV (or darker). IPD is not protective against erythema and its biological significance is unclear, although it has been recently suggested that its oxidized products absorb in the visible wavebands and thus prevents photodamage by visible radiation to other important molecules such as folate (5-methyltetrahydrofolate (5MTHF)) in the blood [33]. 5MTHF deficiency may increase the risk of CVDs, colorectal carcinoma, megaloblastic anaemia, depression and dementia.
Persistent pigment darkening is a brown colour in response to higher UVA doses (>10 J/cm2) peaking 2 h post-irradiation and lasting for 1–5 days. PPD is not due to new melanin synthesis but rather to a more persistent oxidation of melanin precursors. PPD is an end point that is used to assess UVA protection of sunscreens.
Melanogenesis
Delayed tanning (facultative pigmentation or neomelanogenesis) is due to stimulation of new melanin synthesis by basal epidermal melanocytes, which is then transported via dendrites to adjacent keratinocytes and redistributed towards the surface of the skin. The ability to tan depends on skin type as described in Table 9.2. The density of melanin varies with body site and declines with age. Melanin is synthesized either as dark-coloured brown-black insoluble eumelanin or light-coloured red-yellow, alkali soluble, sulphur-containing phaeomelanin. Eumelanin is thought to be the major factor in the photoprotective properties of melanin, which when induced in white skin types, results in a protection factor of about 2–3 against DNA photodamage and erythema as well as a visible tan [27]. There is in vitro and animal evidence that products relating to melanogenesis particularly phaeomelanin may have photosensitizing properties contributing to the skin cancer susceptibility of people with red hair. New melanin is redistributed with the transit of keratinocytes through the epidermis and is typically evident ∼3 days post-irradiation. The tan fades when the stratum corneum is shed over several weeks.
Table 9.2 Summary of skin types based on Fitzpatrick classification. There is a relationship between the acute and carcinogenic effects of UVR, the basis of which is poorly understood
| Skin type | Skin colour on sun-protected site | Sunburn risk | Approximate SED for 1 MED on sun-protected site | Tanning ability | Skin cancer risk |
| I | White | ++++ | 2–3 | ± | High |
| II | White | +++ | 2–3 | + | High |
| III | White | ++ | 4–6 | ++ | Moderate |
| IV | Olive | + | 4–6 | +++ | Moderate |
| V | Brown | ± | 6–20a | ++++ | Low |
| VI | Black/dark brown | ± | 6–20b | ++++ | Low |
aData on skin types V/VI less reliable because melanin masks erythema.
UVR-induced DT results from an increase in tyrosinase activity stimulating the conversion of tyrosine to levodopa (L-DOPA), which is the rate-limiting step. Other precursors include dopaquinone, 5,6-dihydroxyindole (DHI) and 5,6-dihydroxyindole-2-carboxylic acid (DHICA). DHICA can be methylated to produce 6-hydroxy-5-methoxyindole-2-carboxylic acid (6H5MICA) and 5-hydroxy-6-methoxyindole-2-carboxylic acid (5H6MICA) in melanocyte cytoplasm to yield melanin.
Tyrosinase activity can be stimulated directly by UVR on melanocytes and by indirect activation by keratinocytes, whereby UVR induces release of membrane-associated diacylglycerol (DAG) from plasma membranes, which activates PKC-β and in turn activates tyrosinase. A small degree of tyrosinase stimulation also occurs through fibroblasts, neurons and mast cells. A recent study of gene expression after multiple UVR doses in vivo in humans showed that UVB is a strong stimulator of various pigment-related genes, such as the tyrosinase (TYR) gene, tyrosinase-related protein 1 (TRP1) and dopachrome tautomerase (DCT), as well as the transcription factor microphthalmia-associated transcription factor (MITF) [34] and SSR is more effective in eliciting these effects than UVB suggesting a synergistic effect on melanogenesis when UVA and UVB are combined. UVA alone did not induce such upregulation of pigment cell-specific genes, suggesting UVA-induced pigmentation likely occurs via a distinct mechanism [35]. Melanogenesis is also mediated via DNA photodamage, which is indicated by its action spectrum. In vitro experiments show that the topical DNA repair enzyme (T4N5) treatment of murine S91 melanoma cells and human melanocytes exposed to SSR demonstrated greater melanogenesis than when treated with heat-inactivated enzyme [36] and that accelerated and/or more extensive excision of CPD enhances tanning. The release of single-stranded DNA fragments during CPD repair is thought to stimulate melanogenesis by increasing tyrosinase activity leading to an increase in tyrosinase protein and downstream new melanin formation [37].
UVR-induced DNA damage activates p53 protein [38, 39], which is also a mediator of melanogenesis. It causes an upregulation of pro-opiomelanocortin (POMC), which is then processed to adrenocorticotrophic hormone (ACTH) and α-melanocyte stimulating hormone (α-MSH). Secreted α-MSH binds to the melanocortin 1 receptor (MC1R) on melanocytes and via an increase in cyclic adenosine monophosphate (cAMP), increases the activity of tyrosinase.
Immunomodulation
The skin is an important arm of the innate and acquired immune system (see Chapter 8) and the epidermis is rich in antigen-presenting LC. UVR has profound effects on skin immunity [40,41, 42] and, for example, readily depletes the epidermis of LC. Classic experiments in the 1970s showed that UVR-induced immunosuppression in the mouse played a major role in the development of UVR-induced squamous cell carcinoma (SCC). A similar role is likely to occur for humans, though this is impossible to test experimentally. It is clear that an intact immune system is important for skin cancer surveillance and tumour rejection because organ transplant patients, maintained on immunosuppressive drugs, have a high incidence of all types of skin cancer, especially on sun-exposed sites. A major difference between drug-induced immunosuppression and photoimmunosuppression is that the latter is antigen specific. This can be demonstrated using the contact hypersensitivity (CHS) model of acquired immunity in mice and humans. This model is widely used to represent the photoimmunological events in skin cancer. Skin can be sensitized to a universal sensitizer such as dinitrochlorobenzene (DNCB) and shows a typical CHS response when subsequently challenged on a distal site with DNCB. However, the CHS response to DNCB is reduced or inhibited if the sensitization site is pre-exposed to UVR. Such animals could not be subsequently resensitized with DNCB, but could be sensitized with another antigen. Mechanistic studies in mouse skin show that suppression of the CHS response is mediated by the generation of antigen-specific T-regs which results in ‘immunotolerance’.
Suppression of the CHS response can readily occur in humans with suberythemal exposure [43]. There is good evidence that DNA [44] and tUCA [45] are chromophores for the suppression of the CHS response. It is likely that antigen-specific photoimmunosuppression evolved to prevent chronic allergic reactions to new antigens that arise from UVR-induced alterations to skin molecules that result in the generation of photoantigens. There is some evidence to suggest that polymorphic light eruption (PLE) is a consequence of the failure to suppress the CHS response to unspecified photoantigens [46, 47]. The downside of the suppression of photoimmunosuppression is the failure of the immune system to reject potentially carcinogenic clones of keratinocytes and melanocytes. Selection pressure is likely to have had a preference for the prevention of allergic reactions, given that skin cancer typically occurs well past the age of reproduction and child rearing. Interestingly, there is evidence to suggest that PLE patients are less prone to skin cancer [48].
Mouse studies have shown that UVR can suppress acquired immunity to a range of pathogens and this has raised concerns about a possible role of UVR in vaccination programmes. However, to date, there is no substantial evidence support these concerns [41].
The above discussion has focused on the effects of UVR on the sensitization (primary) arm of the CHS response. However, UVR can also suppress the elicitation (memory) arm of the CHS response, which can be demonstrated by irradiating skin prior to challenge with recall antigens. The significance of this is not known.
UVR suppresses acquired and recall immunity but recent studies have also shown that it enhances innate immunity, by the induction of antimicrobial peptides such as β-defensin, especially in the upper layers of the epidermis [49].
In summary, UVR inhibits acquired immunity with antigen specificity. This has probably evolved to prevent CHS reactions to photochemically induced antigens in the skin. However, its long-term consequences are increased susceptibility to skin cancer. In contrast, it appears that innate immunity is enhanced by UVR, which may give better protection against microbial infection.
Vitamin D
The production of vitamin D is the only established benefit of solar UVR exposure [50]. Vitamin D has long been known to be important for skeletal health, but there is increasing and often controversial evidence that vitamin D is important for a wide range of health outcomes. The skin chromophore for vitamin D synthesis is 7-dehydrocholesterol (7DHC), also known a pro-vitamin D. It is converted to pre-vitamin D by solar UVR, with an action spectrum that is similar to that for erythema in the UVB region. There is no evidence that UVA is involved in vitamin D production, but there is evidence that it may induce photodegradation. Pre-vitamin D is converted to the final biologically active vitamin D (in fact a hormone) by thermal and enzymatic steps. Vitamin D status is usually assessed by measuring serum 25-hydroxy vitamin D. Several studies have estimated that the sun is the most important source of vitamin D for most people, mainly because relatively few components of typical diets are rich in vitamin D, for example oily fish. In temperate climates, vitamin D status is seasonal.
Epidemiological studies have shown that people with darker skin types have poorer vitamin D status at a given latitude, and such observations have been attributed to photoprotection by melanin. Indeed, it has been argued that the loss of melanin as humans moved out of Africa was due to selection pressure to maintain vitamin D status [51]. The role of melanin in vitamin D synthesis has been addressed in a few laboratory studies with conflicting results. A recent unpublished comprehensive study by the author has demonstrated that skin pigment has a very modest effect on vitamin D status. This may be because 7DHC is in sufficient quantity above the heavily melanized basal layer.
Cardiovascular disease
Several studies have postulated a relationship between blood pressure and CVD and solar UVR exposure. This has been attributed to vitamin D (i.e. a UVB effect) but has not been supported by intervention studies. Recent studies have provided evidence that a single suberythemal dose (2 SED) of UVA (320–400 nm) can lower blood pressure via the release of NO from the skin into the systemic circulation [52]. This is an important finding with implications for other diseases that have latitude gradients. It will be important to confirm these data because they have important public health implications [53] and may also impact on photoprotection strategies.
Chronic
Subject to ethical approval, it is relatively easy to study the acute effects of UVR on human skin in the laboratory. However, this cannot be done for chronic effects for which an epidemiological approach is necessary. There has been extensive epidemiological research on skin cancer that has been supported by animal and in vitro studies. In contrast, there has been very little epidemiological work on photoageing, almost certainly because it has been traditionally viewed as a cosmetic rather than a health problem. Most of our understanding of photoageing has come from chronic animal studies and the use of acute molecular surrogates in human skin.
Skin cancer
Epidemiological data and body site studies have long supported an association between solar radiation exposure and skin cancer, including basal cell carcinoma (BCC) and SCC, which are keratinocyte cancers, and malignant melanoma (MM), which is a cancer of the melanocyte. Solar UVR exposure is also associated with actinic keratoses (AK) that are indicative of SCC risk. The risk of skin cancer is very dependent on skin type (see Table 9.2). For example, the risk factors for MM include self-reported higher levels of sunburn, and genetically determined phenotypic characteristics associated with greater sensitivity to UVR, including fair skin, light hair and eye colour, poor ability to tan, freckling and multiple naevi. See Lucas et al. [50] for recent review of UVR and skin cancer.
The incidence of MM has steadily increased since the 1960s in fair-skinned populations, with an approximate doubling every 10–20 years. This has been attributed to changes in recreational behaviour leading to increased sun exposure. This trend is projected to continue for at least another 20 years. In Australia and the USA, there is evidence of a recent reduction in the rate of new cases in both sexes, and even a stabilization. This observation has occurred mainly in the younger age group (25–44 years); while large increases in people aged over 60 years, particularly in men, persist. It is likely that public health sun protection campaigns, beginning in the 1980s, have contributed to the decrease in new cases in younger age groups.
Both BCC and SCC are among the most frequently occurring cancers in white-skinned populations and are often referred to collectively as non-melanoma skin cancers (NMSC). There is considerable evidence that the incidence of NMSC is increasing, even though it is very difficult to obtain accurate incidence rates for NMSC for a variety of reasons including the widespread lack of mandatory reporting of NMSCs to cancer registries, and population-based studies are rare. Furthermore, most reports do record lesion multiplicity and there is no consistent approach to the reference population being used for age standardization.
In contrast, it is estimated that MM represents only about 4% of cases of skin cancer. However, it accounts for about 80% of skin cancer deaths, and a global burden of estimated 46 000 deaths in 2008. Mortality due to MM has increased in recent years, for example more than doubling in Europe between the early 1970s and the early 2000s.
Experimental evidence for UVR as a carcinogen
The association between skin cancer and solar range UVR (∼295–400 nm) component has been extensively confirmed with animal studies, especially with hairless mice that readily develop SCC with chronic UVR exposure, including SSR. Mouse models have also shown a relationship between UVR and MM [54]. Mouse studies have generated an action spectrum for SCC, which has been used to produce an action spectrum for human SCC after correcting for differences in the transmission optics in human and mouse skin [55]. This shows that UVB is the main cause of SCC. The human SCC action spectrum is fairly similar to those for human erythema and epidermal CPD, which implicates DNA as a chromophore. This has been supported by a direct molecular link with UVR in AK, BCC and SCC. UVR results in specific ‘fingerprint’ mutations (T → C and TT → CC transitions) that have been found at high frequencies in these lesions. For example, the p53 gene is mutated in more than 90% of SCC and about 50% of BCC [56, 57]. UVR induced C → T mutations have been found in 80% of p53 mutations in AK [57] and 63% of p53 mutations in Bowen disease [58]. An analysis of mutations in SCC and BCC combined in XP patients, showed that 90% were G:C to A:T [59, 60]. Mutations in p53 inhibit cellular arrest and DNA repair, which results in their incorporation into daughter cells. Similarly, UVR-specific mutations have also been found in the tumour suppressor PTEN gene in MM [61,62]. The action spectrum for MM has not yet been established though it has been argued by some that UVA may more important than for NMSC.
Effect of different patterns of UVR exposure on skin cancer
Patterns of UVR exposure seem to be important in the genesis of skin cancer [63]. SCC is associated with low-dose chronic exposure whereas MM and probably BCC, are associated with intermittent high dose sunburning exposure. Studies show that childhood sun exposure may be particularly important, with studies showing that migration from countries of low insolation (e.g. Scotland) to those of high insolation (e.g. Australia) after the age of 10 years is associated with a markedly lower risk of MM development in later life compared with migration before the age of 10 years. Interestingly, a mouse model for MM will only develop lesions if exposure is done within a few days of birth [54].
Photoageing
Skin ageing occurs as two distinct phenomena: intrinsic chronological ageing in which changes are attributable to the passage of time, and photoageing that is the superposition of structural and functional changes caused by chronic UVR exposure. This may be seen as a deterministic (i.e. inevitable) process in contrast to skin cancer that is stochastic. The main clinical features of photoageing are fine and course wrinkling, dryness, coarseness, telangiectasia, yellowness and irregular patchy pigmentation. Skin also loses its mechanical properties. Table 9.3 summarizes the main histological differences between photoageing and chronological skin ageing. The mechanical and elastic properties of the skin derive from the extracellular matrix of the dermis of which the major proteins are collagen, and elastin to a lesser extent. The degradation of elastin, known as elastosis, is a typical histological feature of skin that has been chronically exposed to solar UVR. Elastosis is often observed close to SSC, which is associated with chronic low-level UVR exposure.
Table 9.3 Summary of main histological features of chronological ageing and photoageing
| Location | Chronological ageing | Photoageing |
| Epidermis | Flattened dermal–epidermal junction Variable keratinocyte size and epidermal thickness Loss of melanocytes |
Flattened dermal–epidermal junction Epidermal thickening and atrophy Variable keratinocyte size, with increase in melanocytes (often atypical) and melanin content |
| Dermis | Atrophy with reduction of collagen, elastic fibres and proteoglycans Loss of vascularity |
Elastosis and collagen degeneration Increased proteoglycans Low-grade perivascular inflammation Thickening of vascular walls |
Modified from Herschenfeld & Gilchrest 1999 [110].
Collagen degradation
Collagens are subject to degradation by matrix metalloproteinases (MMP) that have low levels of expression in sun-protected skin. MMP are a large family of proteases that share common structural and functional elements. They contain a zinc-containing binding site that is found in their catalytic domain. MMP are released as proenzymes where a blocking cysteine residue must be removed to activate enzyme activity. This is largely in order to ensure MMP activity is focused at target sites. Their main physiological function is ascribed to the modulation and regulation of ECM turnover by direct proteolytic degradation of dermal ECM proteins and non-matrix proteins, as well as liberation of biologically active cytokine growth factors and chemokines from their membrane anchored preforms. MMP upregulation forms part of normal physiological processes such as wound healing and angiogenesis. However, MMP also play an important role in cancer invasion.
Both UVB and UVA1 have been shown to induce a range of MMP at the gene, protein and protein activity level [64, 65]. Most studies have focused on MMP1, which is a collagenase. Tissue inhibitors of MMP (TIMP), the expression of which is much less responsive to UVR exposure, regulate MMP activity [66]. Thus, photoageing appears to be the consequence of an imbalance between MMP and TIMP activity and possibly the consequence of imperfect repair when new extracellular proteins are synthesized in response to MMP-induced degradation [67]. Most laboratory studies on UVR induction of MMP protein and its activity have shown much greater expression/activity of MMP in the epidermis [64, 68] than the dermis. This conundrum has yet to be explained, but it has been suggested that MMP may migrate from the epidermis to the dermis. It should also be noted that laboratory studies are typically acute and that photoageing is a long-term process.
Several studies have indicated that ROS initiate MMP production. However, it has also been shown that the CPD is likely to be important, at least for the induction of MMP1 [69].
Role of damage to mitochondria
Genetic damage and instability outside the nuclear genome have been suggested to contribute to photoageing [70, 71, 72]. Cellular mitochondria generate energy (ATP) via a series of redox reactions mediated through the electron transport chain and antioxidants (nicotinamide adenine dinucleotide (NADH), flavin adenine dinucleotide 2 (FADH2), coenzyme Q). Despite these constitutive enzymes and mitochondrial BER, mitochondria DNA (mtDNA) are sensitive to ROS-induced damage and the mutation incidence for mtDNA is about 20-fold increased compared to nuclear DNA. Photodamaged skin has a higher mtDNA mutation frequency when compared with sun-protected skin, which also correlates with higher MMP1 levels [73].
Spectral dependence
It is often stated that UVA is the main cause of photoageing on the basis that it penetrates deeper into the dermis. Furthermore, there have been a few case reports of photoageing on one side of the face that has been habitually exposed to solar UVR through glass (which attenuates UVB). However, an action spectrum for elastosis in the hairless mouse is very similar to that for human erythema [74], which supports UVB as the major cause. Other mouse studies for different photoageing end points also reported UVB as the major cause apart from skin sagging [75], which was primarily a UVA effect. Erythemal responses to different spectra, including UVA1, have been shown to predict the expression of MMP1 mRNA in human skin [31]. Overall, the data suggest that in a solar environmental context, UVB is the major cause of photoageing, but UVA certainly plays a role.
Relationship between photoageing and skin cancer
Finally, it should be noted that photocarcinogenesis and photoageing have usually been treated as separate phenomena, when in reality it is very likely that they are closely related. This has been demonstrated in hairless mouse studies in which a neutrophil elastase deficient strain was resistant to UVR-induced SCC [76] and photoageing [77].
Photoprotection
The aim of photoprotection is to reduce the molecular and clinical damage caused by solar UVR. The prevention of erythema is a good starting point. A good rule of thumb is that photoprotection is necessary when your shadow is shorter than you, because this is indicative of high levels of UVB. This is especially important for skin types I and II but, depending on circumstances, it may also be advisable for more sun-resistant skin types. Tanning offers some protection against erythema and DNA damage in skin types II–IV, but the level of protection is modest [27, 78].
Sunscreens
Sunscreens are topical formulations that contain agents that filter and/or scatter UVR. In effect, UVR-absorbing molecules are exogenous chromophores applied to the skin [79]. A typical product contains a mix of organic filters with different absorption spectra as well as micropigments that may be physical (e.g. titanium dioxide) or organic. In Europe, sunscreens are regulated as cosmetics but are regarded as drugs in the USA. Sunscreens are designed and tested to prevent erythema and their main index of protection is the sun protection factor (SPF). The SPF is calculated by the following formula: SPF = [24 h MED on protected skin]/[24 h MED on unprotected skin] according to strict protocols prescribed by regulatory bodies. Included in these protocols is a sunscreen application density of 2 mg/cm2 on skin and the use of well-defined SSR. In theory, an SPF of 15 if used correctly will reduce erythemal exposure to 1/15th of that which would otherwise have been received for a given time in the sun.
The action spectrum for erythema in Figure 9.8 shows that no sunscreen could be effective without good UVB protection, but there has been an increasing trend for better UVA protection in recent years resulting in broad spectrum protection, and this is now required by regulatory bodies [80] though the indices of UVA protection may vary with regulatory body. These indices may be based on the ability to prevent PPD (of unknown biological significance) or the UVR transmission properties of the sunscreen. Recent changes to the testing and labelling of sunscreens within the EU have been agreed: the categories are divided into low (SPF 6–10), medium (SPF 15–25), high (SPF 30–50) and very high (SPF 50+) protection, with the level of UVA protection at least one-third of the SPF. One de facto consequence of better UVA protection for a given SPF is reduced UVB protection. However, the UVA protection index must be regarded as secondary to the prevention of erythema as indicated by the SPF. The long-term benefits of improved UVA protection have yet to be determined.
As stated, the SPF is an index of protection from erythema after a single exposure to SSR. Assuming beach use, a typical person should use about 100 mL/day, assuming three whole-body applications, to achieve the labelled SPF. In practice, people apply sunscreen much less thickly than SPF test conditions [81, 82] which results in much less protection than indicated by the labelled SPF. This means that people may overestimate their degree of protection. Thus, the typical use of a labelled SPF 15 product may result in an actual SPF of 3–5. Overall, people are probably getting much less photoprotection than they think from sunscreens, especially with intentional solar exposure. This is a public health issue that has to be addressed either by encouraging people to use more sunscreen or to use a higher SPF to compensate for inadequate application.
Effects of sunscreens on non-erythema end points
Protection against erythema does not mean protection against other types of photodamage that may occur with suberythemal exposure. For example, suppression of the induction phase of the CHS reaction in skin types I/II occurs with doses of <0.5 MED [43]. Little is known about the ability of sunscreens to prevent molecular damage to the skin that may result in skin cancer or photoageing. However, epidemiological data suggest that regular sunscreen use may be beneficial in the inhibition of AK [83, 84] and SSC [85]. The data for a beneficial effect against MM are more controversial [86, 87] but there are some recent indications of benefit [88]. There are also recent data that indicate that regular sunscreen use may inhibit photoageing [89].
Concerns about the possible adverse effect of sunscreen use on vitamin D synthesis have largely not been borne out in field studies [90, 91]. Although there is a theoretical risk [92], the overall conclusion is that vitamin D status has not been adversely affected because people do not apply sunscreens at sufficient thickness to achieve the stated SPF. This could change in the future with the use of higher SPF products or better application practice.
Traditional topical sunscreens depend on the filtering or scattering of UVR, and may be described as ‘passive’ photoprotection. There has however been a trend for ‘active’ photoprotection at a topical and systemic level. These include topical products with natural antioxidants and liposome-containing DNA repair enzymes. Another approach is the subcutaneous delivery of α-MSH analogues that stimulate melanogenesis [50].
Clothing and shade
It should be remembered that sunscreen use is not the only option for photoprotection. Clothing reduces the skin surface area exposed to solar UVR. It offers good protection against sunburn that is dependent on fabric properties such as colour, weave, wetness, etc. Some clothing is designed and tested for photoprotection and the level of protection is expressed by the UV protection factor (UVP) [93] that can be seen as equivalent to the SPF of a sunscreen. Clothing can be very effective as witnessed by the poor vitamin D status that is common in the Middle East and North Africa, especially in women [94]. Vitamin D supplementation is necessary when the body is typically fully clothed, even in sunny countries.
Shade seeking is a well-recognized and very effective way of reducing solar UVR exposure, especially when the sun is high. The construction of shaded areas and judicious tree planting is one way to reduce solar exposure in public areas [95], for example in children's playgrounds and parks.
Population exposure to UVR
The sun
The sun is the most important source of UVR exposure for most people and the degree of exposure is primarily dependent on behaviour. In people with indoor jobs, exposure may be adventitious (e.g. taking a lunch break) or from deliberate sun seeking, as with sunbathing and beach holidays. Occupational exposure will occur in those with outdoor work such as farmers. There are different ways to assess personal exposure, but the most reliable is with personal dosimetry, in which people wear the dosimeter on a specified part of the body. There are two basic types of personal dosimeter. The first is an integrating device, such as the polysulphone film badge, which records total exposure over a fixed period of time. The second is a time-stamped electronic device that records the time of the exposure. The latter are much more expensive and technically demanding, but provide important information on behaviour. Such data are very important because behavioural knowledge provides a better basis for informed public health interventions. For example, one recent study with time-stamped dosimeters showed that Danes on holiday in Tenerife received most of their UVR round about noon (88% of time outdoors between 12.00 and 15.00) [96], despite widespread advice to an educated population to avoid this period. Time-stamped dosimeters can also show how solar UVR exposure varies on different parts of the body with orientation and movement [97].
Several studies have been done in which personal UVR exposure has been measured over extended periods in different populations in spring/summer in Copenhagen, Denmark (56oN), using time-stamped personal electronic dosimeters. In people working indoors without ‘risk, or sun-seeking, behaviour’ the median daily dose was 0.3 SED (range 0–3.9) on working days and 0.6 SED (range 0.1–3.5 SED) on days off [98]. It has been estimated that indoor workers in northern Europe receive about 150–200 SED/year, which is approximately 5% of the total available ambient UVR. Outdoor workers are exposed to more UVR, perhaps up to 400–600 SED/year. Children spend more time outdoors and it has been previously estimated that those in the UK receive an annual dose of about 300 SED [7]; however, this may be less now because of increasing levels of indoor activity. A very high proportion of annual exposure may be received during holidays. It has been estimated that about a third of annual UVR exposure to the face in Northern Europe is received during a 2-week summer vacation at home [99]. A study of Danish holidaymakers showed that they obtained 43% of their annual erythemal UVR exposure in just 6 days in Tenerife in March [96]. This was associated with high levels of sunburn, despite the use of sunscreens.
Unsurprisingly, higher daily exposures have been measured using polysulphone badges in Queensland, Australia, with for example Brisbane (27.4oS) home workers having weekday shoulder exposure medians of 2.0–8.00 SED depending on the time of year [100]. Outdoor workers at the same latitude showed values of 3.0–10.0 SED. It has been estimated that an average American is exposed to about 250 SED per working year; mostly in the spring and summer [101]. This can be increased by 78 SED (i.e. about 30%) with a 3-week vacation (i.e. by 3.7 SED/day) when sunscreens are more likely to be worn. Behaviour is a major determinant of UVR exposure, such that it is possible for children in the UK to have higher solar exposure levels than in Queensland, Australia [102].
UV index
The UV index (UVI) is increasingly used as a means of informing the public about the levels of ambient erythemal UVR at a given location. Such information can help inform sun exposure behaviour and the need for photoprotection. The information on UVI is readily available and is often presented in weather forecasts, and can be easily found online for a given geographical location. In reporting the UVI, most emphasis is placed on the maximum UVR level on a given day, which is during the 4-h period around solar noon. The UVI ranges from 1 to 11+, the latter being classed as extreme. Photoprotection for fair-skinned people is recommended once the UVI reaches 3.
The UVI is a measure of ambient erythemal UVR. It could be argued that there is a need for cheap reliable personal user-friendly dosimeters so that people can measure their own UVR exposure that would allow them to modify their UVR exposure. In addition, there are smartphone applications for this purpose. One possible disadvantage of these devices is that they would encourage maximal exposure, especially in cases where, typically there is inadequate sunscreen application.
Non-solar sources
People may be exposed to UVR from a range of artificial sources: some indoor workers are significantly irradiated by, for example, arc welding devices and, possibly, from hospital phototherapy equipment if due care is not taken. Office workers and home dwellers may be at risk from low-intensity UVR from fluorescent or, more importantly, quartz halogen lamps used for indoor lighting.
Sunbeds
The use of tanning devices is increasingly popular and one study on sunbeds and melanoma reported that more than 50% of the study control population, based in five Northern European populations, had used such devices [103]. This is supported by other studies [104]. Considerable population UVR exposure can occur from such use. Sunbeds are designed to induce tanning (melanogenesis), the action spectrum of which is similar to that for erythema, with a peak in the UVB region at about 300 nm in the solar UVR range [28]. People also use sunbeds to ‘feel good’ and ‘look good’ [105]. A recent study in Germany showed that median annual exposure was 3 h/year and that the age of first use was declining [104].
Several studies have investigated the relationship between sunbed use and skin cancer. One systematic review and meta-analysis of 27 studies [106] showed that sunbed use significantly increases the risk of MM (relative risk (RR) of 1.20 with 95% confidence interval (CI) of 1.08–1.34) especially when first used by those under 35 years of age; the latter group having a RR of 1.87 (95% CI of 1.41–2.48). Many national and international organizations have issued guidelines/laws to restrict sunbed use, especially in those under 18 years of age. Some countries have banned sunbed use.
Generally, the emission spectra of tanning devices are primarily in the UVA1 region (∼80%), with a small amount of UVB (2–3%). This UVB will be responsible for most of the tanning based on the action spectrum for melanogenesis. The European Union (EU) standard is that the erythemally weighted irradiance of sunbeds should not exceed 0.3 W/m2, and that levels above that are unsafe. Surveys in the UK have shown that more than 80% of more than 500 sunbeds studied exceeded this limit [105, 107]. In England, the average erythemally weighted irradiance of 402 tanning devices was about twice the EU limit. When the emission spectra of these sources were weighted with an action spectrum for non-melanoma skin cancer, the average irradiance was over twice that of Mediterranean noon sun [107]. That is, the average carcinogenic risk per minute of exposure was 2.3 times greater than intense solar exposure. These data show that small errors in sunbed dosimetry/timing of exposure can easily result in erythema and give cause for concern, especially as sunbed operators are unlikely to have much technical knowledge. They also show that regulations are not being enforced.
Risks versus benefits of population UVR exposure
The acute and long-term risks of UVR exposure are well established with damage to DNA leading to mutation and skin cancer. Chronic UVR exposure also results in photoageing. In the context of terrestrial UVR (with no UVC), the vast majority of action spectrum studies, whether in vitro, in animal or human skin in vivo, have shown that these effects are primarily caused by UVB. The only established benefit of solar UVR exposure is vitamin D production, which is also caused by UVB. Field studies have shown that vitamin D production and DNA damage are related to the product of skin area exposed and solar UVB dose over a wide dose range [108]. Thus, benefit is always associated with some risk, which will be influenced by skin type. There are those who argue that the population benefits of maintaining optimal vitamin D status are more important than the burden of skin cancer [109], but such views remain highly controversial, especially in the dermatology community. More recently, it has been argued that exposure to solar UVA is beneficial because it reduces blood pressure and a reduction in blood pressure would have major health benefits at a population level. Any proposal to increase UVA exposure would be contrary to recent global trends for ever better UVA protection. Until recently, most emphasis on the immunological effects of UVR on the skin has been focused on its suppression of acquired immunity, but it is now recognized that UVR can enhance innate immunity. At present, we lack the information to prescribe solar UVR exposure to obtain the best risk–benefit outcome for health. As such, it is probably best to advise that daily exposure be restricted to suberythemal doses, e.g. about 2 SED whether through sunscreen or not, which are sufficient for vitamin D synthesis. There is, however, no case for additional UVR exposure from tanning beds which significantly adds to MM risk. Sunbed use, popular with the young, is often unregulated and the non-cosmetic beneficial effects can be readily obtained from the sun or vitamin D supplementation.
References
- Diffey BL. Sources and measurement of ultraviolet radiation. Methods 2002;28(1):4–13.
- Dawe RS. Knowledge of body site variability in ultraviolet-induced erythemal responses guides choice of site for pre-therapy minimal erythema dose testing. J Invest Dermatol 2005;124(3):662; author reply 663.
- Diffey BL, Jansen CT, Urbach F, Wulf HC. The standard erythema dose: a new photobiological concept. Photodermatol Photoimmunol Photomed 1997;13(1–2):64–6.
- Webb AR, Slaper H, Koepke P, Schmalwieser AW. Know your standard: clarifying the CIE erythema action spectrum. Photochem Photobiol 2011;87(2):483–6.
- Hooke RJ, Pearson AJ, O'Hagan JB. Temporal variation of erythemally effective UVB/UVA ratio at Chilton, UK. Radiat Prot Dosimetr 2012;149(2):185–90.
- Blumthaler M, Ambach W, Ellinger R. Increase in solar UV radiation with altitude. J Photochem Photobiol B Biol 1997;39(2):130–4.
- Diffey BL. Human exposure to solar ultraviolet radiation. J Cosmet Dermatol 2002;1(3):124–30.
- Kerr AC, Ferguson J, Attili SK, et al. Ultraviolet A1 phototherapy: a British Photodermatology Group workshop report. Clin Exp Dermatol 2012;37(3):219–26.
- Takahashi H, Nakajima S, Ogasawara K, et al. Photodynamic therapy using a novel irradiation source, LED lamp, is similarly effective to photodynamic therapy using diode laser or metal-halide lamp on DMBA- and TPA-induced mouse skin papillomas. J Dermatol 2014;41(8):729–31.
- Young AR. Chromophores in human skin. Phys Med Biol 1997;42(5):789–802.
- Tewari A, Sarkany RP, Young AR. UVA1 induces cyclobutane pyrimidine dimers but not 6-4 photoproducts in human skin in vivo. J Invest Dermatol 2012;132(2):394–400.
- Tewari A, Grage MM, Harrison GI, Sarkany R, Young AR. UVA1 is skin deep: molecular and clinical implications. Photochem Photobiol Sci 2013;12(1):95–103.
- Woollons A, Kipp C, Young AR, et al. The 0.8% ultraviolet B content of an ultraviolet A sunlamp induces 75% of cyclobutane pyrimidine dimers in human keratinocytes in vitro. Br J Dermatol 1999;140(6):1023–30.
- Bech-Thomsen N, Wulf HC. A polychromatic action spectrum for photosensitivity to orally administered 8-methoxypsoralen in humans. Clin Exp Dermatol 1994;19(1):12–15.
- Cadet J, Mouret S, Ravanat JL, Douki T. Photoinduced damage to cellular DNA: direct and photosensitized reactions. Photochem Photobiol 2012;88(5):1048–65.
- DiGiovanna JJ, Kraemer KH. Shining a light on xeroderma pigmentosum. J Invest Dermatol 2012;132(3 Pt 2):785–96.
- Melis JP, van Steeg H, Luijten M. Oxidative DNA damage and nucleotide excision repair. Antioxid Redox Signal 2013;18(18):2409–19.
- Young AR, Chadwick CA, Harrison GI, Hawk JL, Nikaido O, Potten CS. The in situ repair kinetics of epidermal thymine dimers and 6-4 photoproducts in human skin types I and II. J Invest Dermatol 1996;106(6):1307–13.
- Halliday GM, Cadet J. It's all about position: the basal layer of human epidermis is particularly susceptible to different types of sunlight-induced DNA damage. J Invest Dermatol 2012;132(2):265–7.
- Ziegler A, Jonason AS, Leffell DJ, et al. Sunburn and p53 in the onset of skin cancer. Nature 1994;372(6508):773–6.
- Jiang, W, Ananthaswamy HN, Muller HK, Kripke ML. p53 protects against skin cancer induction by UV-B radiation. Oncogene 1999;18(29):4247–53.
- Burns AS, Jaros E, Cole M, Perry R, Pearson AJ, Lunec J. The molecular pathology of p53 in primitive neuroectodermal tumours of the central nervous system. Br J Cancer 2002;86(7):1117–23.
- Rhodes LE, Gledhill K, Masoodi M, et al. The sunburn response in human skin is characterized by sequential eicosanoid profiles that may mediate its early and late phases. FASEB J 2009;23(11):3947–56.
- Nicolaou A, Masoodi M, Gledhill K, et al. The eicosanoid response to high dose UVR exposure of individuals prone and resistant to sunburn. Photochem Photobiol Sci 2012;11(2):371–80.
- Harrison GI, Young AR. Ultraviolet radiation-induced erythema in human skin. Methods 2002;28(1):14–19.
- Harrison GI, Young AR, McMahon SB. Ultraviolet radiation-induced inflammation as a model for cutaneous hyperalgesia. J Invest Dermatol 2004;122(1):183–9.
- Sheehan JM, Cragg N, Chadwick CA, Potten CS, Young AR. Repeated ultraviolet exposure affords the same protection against DNA photodamage and erythema in human skin types II and IV but is associated with faster DNA repair in skin type IV. J Invest Dermatol 2002;118(5):825–9.
- Schmalwieser AW, Wallisch S, Diffey B. A library of action spectra for erythema and pigmentation. Photochem Photobiol Sci 2012;11(2):251–68.
- Young AR, Chadwick CA, Harrison GI, Nikaido O, Ramsden J, Potten CS. The similarity of action spectra for thymine dimers in human epidermis and erythema suggests that DNA is the chromophore for erythema. J Invest Dermatol 1998;111(6):982–8.
- Auletta M, Gange RW, Tan OT, Matzinger E. Effect of cutaneous hypoxia upon erythema and pigment responses to UVA, UVB, and PUVA (8-MOP + UVA) in human skin. J Invest Dermatol 1986;86(6):649–52.
- Tewari A, Lahmann C, Sarkany R, Bergemann J, Young AR. Human erythema and matrix metalloproteinase-1 mRNA induction, in vivo, share an action spectrum which suggests common chromophores. Photochem Photobiol Sci 2012;11(1):216–23.
- Rhodes LE, Belgi G, Parslew R, McLoughlin L, Clough GF, Friedmann PS. Ultraviolet-B-induced erythema is mediated by nitric oxide and prostaglandin E2 in combination. J Invest Dermatol 2001;117(4):880–5.
- Moan J, Nielsen KP, Juzeniene A. Immediate pigment darkening: its evolutionary roles may include protection against folate photosensitization. FASEB J 2012;26(3):971–5.
- Tadokoro T, Yamaguchi Y, Batzer J, et al. Mechanisms of skin tanning in different racial/ethnic groups in response to ultraviolet radiation. J Invest Dermatol 2005;124(6):1326–32.
- Choi W, Miyamura Y, Wolber R, et al. Regulation of human skin pigmentation in situ by repetitive UV exposure: molecular characterization of responses to UVA and/or UVB. J Invest Dermatol 2010;130(6):1685–96.
- Gilchrest BA, Zhai S, Eller MS, Yarosh DB, Yaar M. Treatment of human melanocytes and S91 melanoma cells with the DNA repair enzyme T4 endonuclease V enhances melanogenesis after ultraviolet irradiation. J Invest Dermatol 1993;101(5):666–72.
- Gilchrest BA, Eller MS. DNA photodamage stimulates melanogenesis and other photoprotective responses. J Investig Dermatol Symp Proc 1999;4(1):35–40.
- Eller MS, Ostrom K, Gilchrest BA. DNA damage enhances melanogenesis. Proc Natl Acad Sci USA 1996;93(3):1087–92.
- Chen H, Weng QY, Fisher DE. UV signaling pathways within the skin. J Invest Dermatol 2014;134(8):2080–5.
- Schwarz T. The dark and the sunny sides of UVR-induced immunosuppression: photoimmunology revisited. J Invest Dermatol 2010;130(1):49–54.
- Norval M, Halliday GM. The consequences of UV-induced immunosuppression for human health. Photochem Photobiol 2011;87(5):965–77.
- Gibbs NK, Norval M. Photoimmunosuppression: a brief overview. Photodermatol Photoimmunol Photomed 2013;29(2):57–64.
- Kelly DA, Young AR, McGregor JM, Seed PT, Potten CS, Walker SL. Sensitivity to sunburn is associated with susceptibility to ultraviolet radiation-induced suppression of cutaneous cell-mediated immunity. J Exp Med 2000;191(3):561–6.
- Vink AA, Yarosh DB, Kripke ML. Chromophore for UV-induced immunosuppression: DNA. Photochem Photobiol 1996;63(4):383–6.
- Norval M. Chromophore for UV-induced immunosuppression: urocanic acid. Photochem Photobiol 1996;63(4):386–90.
- Palmer RA, Friedmann PS. Ultraviolet radiation causes less immunosuppression in patients with polymorphic light eruption than in controls. J Invest Dermatol 2004;122(2):291–4.
- van de Pas CB, Kelly DA, Seed PT, Young AR, Hawk JL, Walker SL. Ultraviolet-radiation-induced erythema and suppression of contact hypersensitivity responses in patients with polymorphic light eruption. J Invest Dermatol 2004;122(2):295–9.
- Lembo S, Fallon J, O'Kelly P, Murphy GM. Polymorphic light eruption and skin cancer prevalence: is one protective against the other? Br J Dermatol 2008;159(6):1342–7.
- Felton S, Navid F, Schwarz A, Schwarz T, Glaser R, Rhodes LE. Ultraviolet radiation-induced upregulation of antimicrobial proteins in health and disease. Photochem Photobiol Sci 2013;12(1):29–36.
- Lucas RM, Norval M, Neale RE, et al. The consequences for humans of stratospheric ozone depletion in association with other environmental factors. Photochem Photobiol Sci 2015;14(1):53–87.
- Jablonski NG. The evolution of human skin colouration and its relevance to health in the modern world. J R Coll Phys Edinb 2012;42(1):58–63.
- Liu D, Fernandez BO, Hamilton A, et al. UVA irradiation of human skin vasodilates arterial vasculature and lowers blood pressure independently of nitric oxide synthase. J Invest Dermatol 2014;134(7):1839–46.
- Halliday GM, Byrne SN. An unexpected role: UVA-induced release of nitric oxide from skin may have unexpected health benefits. J Invest Dermatol 2014;134(7):1791–4.
- Wolnicka-Glubisz A, Noonan FP. Neonatal susceptibility to UV induced cutaneous malignant melanoma in a mouse model. Photochem Photobiol Sci 2006;5(2):254–60.
- de Gruijl FR, Van der Leun JC. Estimate of the wavelength dependency of ultraviolet carcinogenesis in humans and its relevance to the risk assessment of a stratospheric ozone depletion. Health Phys 1994;67(4):319–25.
- Brash DE, Rudolph JA, Simon JA, et al. A role for sunlight in skin cancer: UV-induced p53 mutations in squamous cell carcinoma. Proc Natl Acad Sci USA 1991;88(22):10124–8.
- Ziegler AD, Leffell J, Kunala S, et al. Mutation hotspots due to sunlight in the p53 gene of nonmelanoma skin cancers. Proc Natl Acad Sci USA 1993;90(9):4216–20.
- Campbell C, Quinn AG, Ro YS, Angus B, Rees JL. p53. mutations are common and early events that precede tumor invasion in squamous cell neoplasia of the skin. J Invest Dermatol 1993;100(6):746–8.
- Dumaz N, Drougard C, Sarasin A, Daya-Grosjean L. Specific UV-induced mutation spectrum in the p53 gene of skin tumors from DNA-repair-deficient xeroderma pigmentosum patients. Proc Natl Acad Sci USA 1993;90(22):10529–33.
- Kraemer KH. Sunlight and skin cancer: another link revealed. Proc Natl Acad Sci USA 1997;94(1):11–14.
- Wang Y, Digiovanna JJ, Stern JB, et al. Evidence of ultraviolet type mutations in xeroderma pigmentosum melanomas. Proc Natl Acad Sci USA 2009;106(15):6279–84.
- Runger TM. Is UV-induced mutation formation in melanocytes different from other skin cells? Pigment Cell Melanoma Res 2011;24(1):10–12.
- Armstrong BK, Kricker A. The epidemiology of UV induced skin cancer. J Photochem Photobiol B 2001;63(1–3):8–18.
- Tewari A, Grys K, Kollet J, Sarkany R, Young AR. Upregulation of MMP12 and its activity by UVA1 in human skin: potential implications for photoaging. J Invest Dermatol 2014;134(10)2598–609.
- Wang FN, Smith R, Tran BA, Kang S, Voorhees JJ, Fisher GJ. Dermal damage promoted by repeated low-level UV-A1 exposure despite tanning response in human skin. JAMA Dermatol 2014;150(4):401–6.
- Lahmann C, Young AR, Wittern KP, Bergemann J. Induction of mRNA for matrix metalloproteinase 1 and tissue inhibitor of metalloproteinases 1 in human skin in vivo by solar simulated radiation. Photochem Photobiol 2001;73(6):657–63.
- Fisher GJ, Wang ZQ, Datta SC, Varani J, Kang S, Voorhees JJ. Pathophysiology of premature skin aging induced by ultraviolet light. N Engl J Med 1997;337(20):1419–28.
- Quan T, Qin Z, Xia W, Shao Y, Voorhees JJ, Fisher GJ. Matrix-degrading metalloproteinases in photoaging. J Investig Dermatol Symp Proc 2009;14(1):20–4.
- Yarosh D, Dong K, Smiles K. UV-induced degradation of collagen I is mediated by soluble factors released from keratinocytes. Photochem Photobiol 2008;84(1):67–8.
- Birket MJ, Birch-Machin MA. Ultraviolet radiation exposure accelerates the accumulation of the aging-dependent T414G mitochondrial DNA mutation in human skin. Aging Cell 2007;6(4):557–64.
- Krutmann J, Schroeder P. Role of mitochondria in photoaging of human skin: the defective powerhouse model. J Investig Dermatol Symp Proc 2009;14(1):44–9.
- Tulah AS, Birch-Machin MA. Stressed out mitochondria: the role of mitochondria in ageing and cancer focussing on strategies and opportunities in human skin. Mitochondrion 2013;13(5):444–53.
- Berneburg M, Plettenberg H, Krutmann J. Photoaging of human skin. Photodermatol Photoimmunol Photomed 2000;16(6):239–44.
- Kligman LH, Sayre RM. An action spectrum for ultraviolet induced elastosis in hairless mice: quantification of elastosis by image analysis. Photochem Photobiol 1991;53(2):237–42.
- Bissett DL, Hannon DP, Orr TV. Wavelength dependence of histological, physical, and visible changes in chronically UV-irradiated hairless mouse skin. Photochem Photobiol 1989;50(6):763–9.
- Starcher B, O'Neal P, Granstein RD, Beissert S. Inhibition of neutrophil elastase suppresses the development of skin tumors in hairless mice. J Invest Dermatol 1996;107(2):159–63.
- Starcher B, Conrad M. A role for neutrophil elastase in solar elastosis. Ciba Found Symp 1995;192:338–46; discussion 346–37.
- Sheehan JM, Potten CS, Young AR. Tanning in human skin types II and III offers modest photoprotection against erythema. Photochem Photobiol 1998;68(4):588–92.
- Jansen R, Osterwalder U, Wang SQ, Burnett M, Lim HW. Photoprotection: part II. Sunscreen: development, efficacy, and controversies. J Am Acad Dermatol 2013;69(6):867:e1–14;
- Fourtanier A, Moyal D, Seite S. UVA filters in sun-protection products: regulatory and biological aspects. Photochem Photobiol Sci 2012;11(1):81–9.
- Petersen B, Datta P, Philipsen PA, Wulf HC. Sunscreen use and failures – on site observations on a sun-holiday. Photochem Photobiol Sci 2013;12(1):190–6.
- Petersen B, Wulf HC. Application of sunscreen – theory and reality. Photodermatol Photoimmunol Photomed 2014;30(2–3):96–101.
- Thompson SC, Jolley D, Marks R. Reduction of solar keratoses by regular sunscreen use. N Engl J Med 1993;329(16):1147–51.
- Naylor MF, Boyd A, Smith DW, Cameron GS, Hubbard D, Neldner KH. High sun protection factor sunscreens in the suppression of actinic neoplasia. Arch Dermatol 1995;131(2):170–5.
- van der Pols JC, Williams GM, Pandeya N, Logan V, Green AC. Prolonged prevention of squamous cell carcinoma of the skin by regular sunscreen use. Cancer Epidemiol Biomarkers Prev 2006;15(12):2546–8.
- Huncharek M, Kupelnick B. Use of topical sunscreens and the risk of malignant melanoma: a meta-analysis of 9067 patients from 11 case–control studies. Am J Public Health 2002;92(7):1173–7.
- Gorham ED, Mohr SB, Garland CF, Chaplin G, Garland FC.Do sunscreens increase risk of melanoma in populations residing at higher latitudes? Ann Epidemiol 2007;17(12):956–63.
- Green AC, Williams GM, Logan V, Strutton GM. Reduced melanoma after regular sunscreen use: randomized trial follow-up. J Clin Oncol 2011;29(3):257–63.
- Hughes MC, Williams GM, Baker P, Green AC. Sunscreen and prevention of skin aging: a randomized trial. Ann Intern Med 2013;158(11):781–90.
- Norval M, Wulf HC. Does chronic sunscreen use reduce vitamin D production to insufficient levels? Br J Dermatol 2009;161(4):732–6.
- Springbett P, Buglass S, Young AR. Photoprotection and vitamin D status. J Photochem Photobiol B 2010;101(2):160–8.
- Sayre RM, Dowdy JC. Darkness at noon: sunscreens and vitamin D3. Photochem Photobiol 2007;83(2):459–63.
- Gambichler T, Laperre J, Hoffmann K. The European standard for sun-protective clothing: EN 13758. J Eur Acad Dermatol Venereol 2006;20(2):125–30.
- Bassil D, Rahme M, Hoteit M, Fuleihan H. Gel hypovitaminosis D in the Middle East and North Africa: Prevalence, risk factors and impact on outcomes. Dermatoendocrinology 2013;5(2):274–98.
- Diffey BL, Diffey JL. Sun protection with trees. Br J Dermatol 2002;147(2):397–9.
- Petersen B, Thieden E, Philipsen PA, Heydenreich J, Wulf HC, Young AR. Determinants of personal ultraviolet-radiation exposure doses on a sun holiday. Br J Dermatol 2013;168(5):1073–9.
- Weihs P, Schmalwieser A, Reinisch C, Meraner E, Walisch S, Harald M. Measurements of personal UV exposure on different parts of the body during various activities. Photochem Photobiol 2013;89(4):1004–7.
- Thieden E, Philipsen PA, Heydenreich J, Wulf HC. UV radiation exposure related to age, sex, occupation, and sun behavior based on time-stamped personal dosimeter readings. Arch Dermatol 2004;140(2):197–203.
- Diffey B. A behavioral model for estimating population exposure to solar ultraviolet radiation. Photochem Photobiol 2008;84(2):371–5.
- Parisi AV, Meldrum LR, Kimlin MG, Wong JC, Aitken J, Mainstone JS. Evaluation of differences in ultraviolet exposure during weekend and weekday activities. Phys Med Biol 2000;45(8):2253–62.
- Godar DE, Wengraitis SP, Shreffler J, Sliney DH. UV doses of Americans. Photochem Photobiol 2001;73(6):621–9.
- Diffey BL, Gies HP. The confounding influence of sun exposure in melanoma. Lancet 1998;351(9109):1101–2.
- Bataille V, Boniol M, De Vries E, et al. A multicentre epidemiological study on sunbed use and cutaneous melanoma in Europe. Eur J Cancer 2005;41(14):2141–9.
- Bock C, Diehl K, Litaker D, Breitbart EW, Greinert R, Schneider S. Sunbed use in Germany: trends, user histories and factors associated with cessation and readiness to change. Br J Dermatol 2013;169(2):441–9.
- Oliver H, Ferguson J, Moseley H. Quantitative risk assessment of sunbeds: impact of new high power lamps. Br J Dermatol 2007;157(2):350–6.
- Boniol M, Autier P, Boyle P, Gandini S. Cutaneous melanoma attributable to sunbed use: systematic review and meta-analysis. BMJ 2012;345:e4757.
- Tierney P, Ferguson J, Ibbotson S, Dawe R, Eadie E, Moseley H. Nine out of 10 sunbeds in England emit ultraviolet radiation levels that exceed current safety limits. Br J Dermatol 2013;168(3):602–8.
- Petersen B, Wulf HC, Triguero-Mas M, et al. Sun and ski holidays improve vitamin D status, but are associated with high levels of DNA damage. J Invest Dermatol 2014;134(11):2806–13.
- Grant WB, Cross HS, Garland CF, et al. Estimated benefit of increased vitamin D status in reducing the economic burden of disease in western Europe. Prog Biophys Mol Biol 2009;99(2–3):104–13.
- Herschenfeld RE, Gilchrest BA. The cumulative effects of ultraviolet radiation on skin: photoageing. In: Hawk JLM, ed. Photodermatology. London: Arnold, 1999.