CHAPTER 13
Topical Drug Delivery
Richard H. Guy
Department of Pharmacy and Pharmacology, University of Bath, Bath, UK
Introduction: skin barrier function
The skin performs a variety of barrier functions, not the least of which is that to the passive, or insensible, loss of tissue water and that to the ingress of materials contacting its surface. The latter, of course, plays a crucial role in determining the effectiveness of any topical drug treatment designed to alleviate skin disease. It follows that the optimization of any dermatological therapy requires an understanding of the skin's physical barrier and of the physicochemical mechanisms involved in cutaneous drug uptake and transport.
At the macroscopic level, the skin consists of two principal components: the dermis and the epidermis. The former, in order of magnitude terms, is about 1 mm thick and represents a connective tissue, the main structural components of which are collagen and elastic fibres in an aqueous glycoprotein gel. The dermis also ‘houses’ mechanoreceptors of touch and heat, hair follicles, sweat and apocrine glands, sebaceous glands, lymphatic vessels and a rich microcirculation. These blood vessels deliver nutrients to the skin as well as providing a clearance mechanism by which penetrating xenobiotics are eliminated into the systemic circulation.
The epidermis is, on average, only 0.1 mm in thickness and is a stratified epithelial membrane comprising (primarily) proliferating basal and differentiated suprabasal keratinocytes. At the outermost surface of the epidermis, the cells are terminally differentiated creating the stratum corneum (SC) or horny layer, the thickness of which is only about 0.01 mm (except on the palms and soles where it is much more substantial). The epidermis also contains melanocytes, Langerhans cells and Merkel cells, but is avascular, such that transfer of material across this tissue layer occurs by passive diffusion only.
As the keratinocytes move up through the epidermis, they flatten and their nucleus begins to degenerate. A complex, lipid mixture of cholesterol, free fatty acids and ceramides is secreted into the intercellular spaces as the keratinocytes undergo their final transformation into corneocytes – anucleate flattened cells, filled with keratin – and create the SC. Microscopically, the SC is often compared, therefore, to a ‘brick wall’, with the corneocytes representing the bricks and the intercellular lipids the cement (Figure 13.1) [1, 2, 3]. Corneo-desmosomes act as bridges holding corneocytes together until their controlled enzymatic degradation towards the SC surface provokes (the roughly daily) desquamation of the outer cell layer.
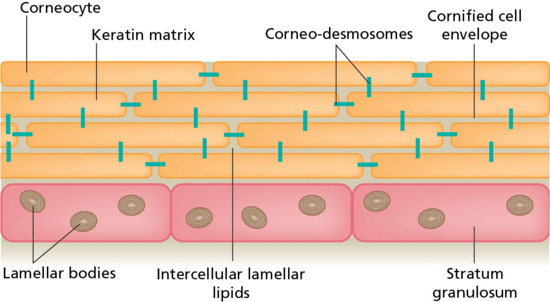
Figure 13.1 A schematic representation of the stratum corneum as a brick wall. The corneocytes act as the bricks, with the intercellular lipids providing the cement. Corneo-desmosomes are analogous to rivets holding the corneocytes together until their desquamation at the skin surface [1, 2, 3].
Remarkably, it is the SC that represents the skin's principal resistance to molecular diffusion either from the ‘inside–out’, as in the case of water, or from the ‘outside–in’ with respect to topically contacting drugs or other foreign substances. The effectiveness of the SC to limit transepidermal water loss (TEWL) is demonstrated easily by measuring the rate of water ‘escape’ across the skin as the outer layers of corneocytes are progressively removed by adhesive tape stripping (Figure 13.2), with TEWL increasing by about an order of magnitude once the barrier has been fully deranged [4].
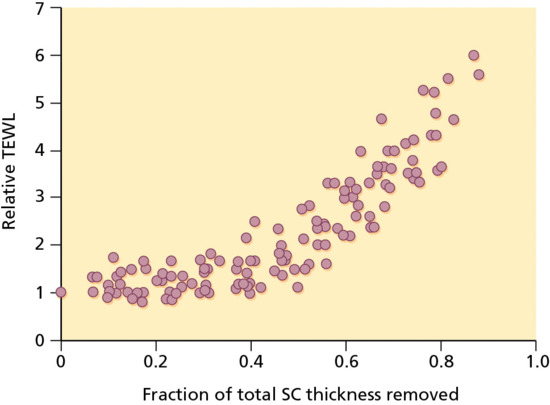
Figure 13.2 Transepidermal water loss (TEWL) across human skin in vivo as the stratum corneum (SC) is progressively removed by adhesive tape stripping [4]. The graph illustrates the relative change in TEWL versus the fraction of the total SC thickness removed.
The typical, normal value of TEWL across the skin of a human at rest is in the order of 0.5 mg/cm2/h. Given that the surface area of skin in an adult male will approach 2 m2 (or 20 000 cm2), this means that the amount of water lost passively across the skin – assuming no active sweating – is between 200 and 300 mL/day, a remarkably small volume given that the concentration of water inside the body is about 1 g/mL (i.e. c. 50 mol/L)!
Similar tape stripping experiments have also shown that the SC presents the principal barrier to drug penetration following topical application. Exceptions to this general rule are limited to very lipophilic compounds which, although taken up into the SC quite easily, are then limited in their entry into the underlying, viable epidermal tissue by their unfavourable partitioning and very low solubility in this predominantly aqueous environment.
Penetration pathways: mechanisms of percutaneous absorption
The ‘brick and mortar’ model of the SC immediately presents two potential pathways for penetration across the barrier (Figure 13.3) [5, 6]. The first is transcellularly, involving the most direct route and multiple transfers of the permeant between corneocytes and interstitial lipids. The other is intercellularly, a tortuous route constraining transport uniquely to the lipid ‘cement’ between the bricks. In addition, the opportunity for topically contacting chemicals to access apparent ‘shunt’ paths, involving the skin appendages (and, in particular, the follicles), represents a further alternative.
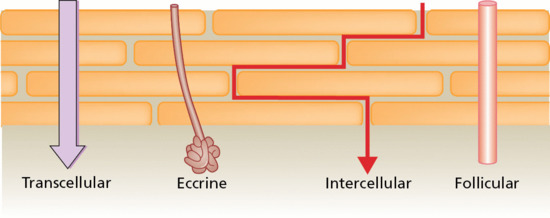
Figure 13.3 Potential penetration pathways across the stratum corneum [5, 6].
As no active transport mechanisms across the SC have been identified, and accepting therefore that percutaneous absorption involves passive diffusion, it follows that molecules will follow the path of least resistance across the barrier. At face value, the transcellular route appears most attractive: it has the largest surface area and volume available for transport and the path length is short (c. 0.01 mm). In contrast, the intercellular lipid domains comprise only about 15% of the SC volume and, given the flattened corneocyte dimensions, the path length around the cells is closer to 0.5 mm (i.e. 50 times longer than transcellularly).
However, a combination of elegant analytical chemistry, electron microscopy and biophysics research in the 1980s and early 1990s has led to a consensus that the intercellular route is, in fact, the dominant pathway. Unravelling the composition and arrangement of the SC intercellular lipids was a key first step in reaching this conclusion [1, 2, 3]. The absence of phospholipids and the presence of an array of ceramides were important and noteworthy features and their precise organization, with the approximately equimolar quantities of fatty acids and cholesterol, into two distinct lamellar phases immediately suggested a more important role for these domains than that of a simple ‘filler’ in between the bricks. It was then noted, in an attempt to visualize chemical penetration across the SC in situ, that the permeating molecules were indeed localized to the intercellular spaces, reinforcing the impression that transport might be constrained to these lipid-rich regions. This led to a defining and confirmatory study that demonstrated that water permeability through the SC was highly correlated with the conformational order of the intercellular lipid domains: as the lipids became progressively disordered by increasing temperature, the permeation of water increased concomitantly. In addition, even as the lipids went through a phase transition (and became markedly more disordered), the permeation of water was enhanced in a correspondingly discontinuous way (Figure 13.4) [7, 8]. The inescapable conclusion from these data was that the path of diffusion for water through the SC was via the intercellular lipids. This deduction was reinforced in a further series of experiments that independently assessed the path length of transport to be more than 50-fold greater than the SC thickness.
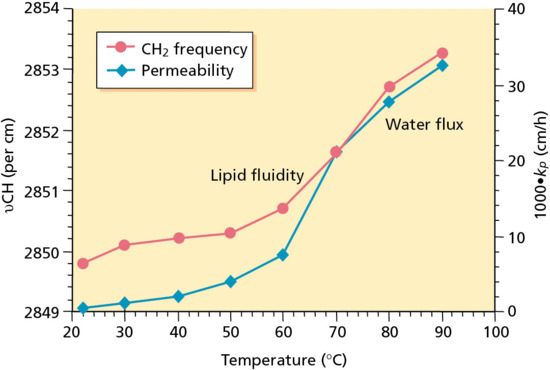
Figure 13.4 Correlation between the permeability coefficient (kp) of water across the stratum corneum (SC) with the conformational order of the SC intercellular lipid domains (as measured by the frequency of the asymmetrical methylene group vibration (υCH2)) as a function of increasing temperature [7, 8].
In terms of appendageal transport, a variety of experimental data point to two common features: (i) the hair follicles, in particular, appear to offer more rapid, ‘shunt’ pathways circumventing the SC barrier; and (ii) the capacity of the pathway is limited by the low density of follicles on most body sites. As a result, although the first molecules reaching the viable skin layers after the application of a topical drug formulation, for example, may well have diffused via the available hair follicles [1, 2, 3], the overall contribution to delivery at a ‘steady state’ (when transport across the SC will have caught up, so to speak) is small.
Factors determining drug permeation into the skin
The absorption of a chemical into the skin depends upon its physicochemical properties, its presentation to the skin (i.e. the ‘vehicle’ in which it is applied), the skin ‘environment’ (i.e. skin condition, disease state, etc.) and the duration of exposure.
The inherent permeability of a specific molecule may be expressed in terms of its maximum potential flux (Jmax) across the SC [9], defined as:
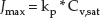
where Cv,sat is the solubility of the compound in the vehicle in which it is applied, and kp is its permeability coefficient across the skin when it is applied in that vehicle. The units of Jmax are amount per unit area per unit time (e.g. μg/cm2/h). kp has units of velocity (e.g. cm/h) and depends on the chemical's diffusivity (D) across the SC, its partition coefficient between the SC lipids and the vehicle in which it is applied (Ksc/v) and its path length of diffusion across the SC (h):
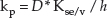
The permeability coefficients of a large number of drugs and other chemicals from water have been determined experimentally and this has permitted a simple algorithm to estimate kp (in units of cm/h) from water to be derived [9] and validated:
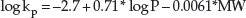
where P is the octanol–water partition coefficient of the chemical and MW is its molecular weight (in Daltons). The log P and MW are readily available physicochemical parameters of the permeant, the first defining its lipophilicity (a very commonly used metric in pharmaceutical development to assess a molecule's ability to be absorbed across lipid barriers), the second describing its size, and hence the rate at which it is able to diffuse.
Equation 13.3 assumes (as is typically the case) that the SC is the rate-determining barrier for percutaneous absorption. However, as noted above, for very lipophilic compounds, penetration is controlled by their ability to partition/dissolve into the viable skin layers. In this case, the value of kp predicted by equation 13.3 must be corrected [9]:
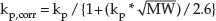
An estimate of Jmax for a chemical is now possible using equation 13.1, with kp,corr determined from equations 13.3 and 13.4 and its water solubility (i.e. Cwater,sat). The latter is frequently determined in the course of drug development, or (like log P, in fact) can be calculated using any one of a number of available algorithms.
The ranges of water solubilities and log P values encompassed by 91 drugs, currently approved for topical and transdermal administration, either in Europe or the USA (or both) are shown in Figure 13.5 as a function of their molecular weight (MW). Immediately apparent is that the majority of compounds have a MW less than 500 Daltons, that their lipophilicities and aqueous solubilities span many orders of magnitude, and that, in broad terms, there is an inverse correlation between log P and Cwater,sat, the former generally increasing with MW and the latter decreasing.
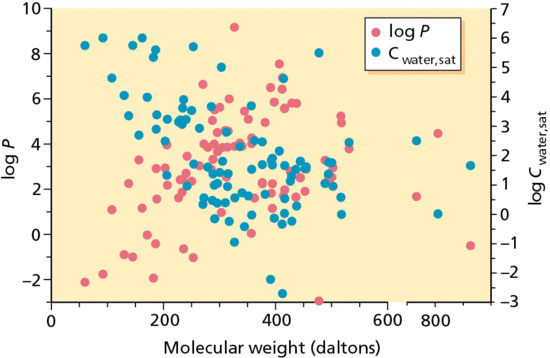
Figure 13.5 Water solubilities and log P values of 91 drugs approved for topical and transdermal administration in Europe and/or the USA as a function of their molecular weights.
Estimated Jmax values for these drugs are presented in Figure 13.6, as functions of MW, log P and Cwater,sat. As anticipated by equation 13.3, there are two clear patterns of behaviour: first, that drug flux is inversely dependent upon MW and, second, that within a group of compounds of approximately similar MW, Jmax increases with increasing lipophilicity (log P). Notably, the majority of topical and transdermal drugs manifest rather modest maximum flux rates across the skin (the dotted horizontal lines in Figure 13.6 signifying a Jmax of only 1 μg/cm2/h), and attest to the considerable pharmacological potency of these compounds.
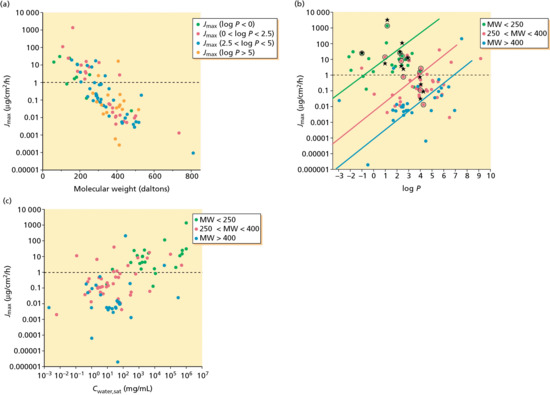
Figure 13.6 Estimated maximum flux (Jmax) values for approved topical and transdermal drugs as functions of: (a) their size (MW); (b) their lipophilicity (log P); and (c) their water solubility (Cwater,sat).
Validation of the conclusions drawn from this physicochemical analysis of the percutaneous absorption process is provided in Figure 13.6b, in which predicted values of Jmax are compared with published data in the literature (black circles and stars) for 12 different drugs. The mean ratio of the experimental to the predicted maximum flux for these compounds is 2.04, that is, very close to the ideal value of 1.
In sum, therefore, the solubility–diffusion approach to the prediction of skin penetration provides a decent ‘first estimate’ of the flux that might be expected. Key parameters that impact on percutaneous absorption are molecular size (or MW), lipophilicity (as expressed by log P, for example) and a measure of solubility (most accessibly provided by aqueous solubility). The criteria for drugs to have good oral bioavailability have been captured in the so-called Rule of Five: specifically, MW less than 500, log P less than 5, and that the compound is capable of donating no more than five hydrogen bonds or of accepting no more than 10 (the latter two conditions being clearly linked to water solubility). The behaviour illustrated in Figure 13.6 fully supports a similar set of criteria for drug delivery across the skin, perhaps with even more stringent limits required to confer adequate permeability.
Topical drug formulations used to treat dermatological disease
It is a logical objective to evolve a scientific rationale for the selection and optimization of a formulation for the topical delivery of specific drugs into and across the skin [10, 11]. Such an approach would recognize that the ‘ideal’ vehicle: (i) elicits no pharmacological effect; (ii) solubilizes the drug; (iii) releases the drug with appropriate kinetics; (iv) is chemically and physically stable; (v) is cosmetically appealing; and (vi) is non-allergenic and non-irritating.
In parallel, there are important issues to address that may significantly complicate the formulation development process. For example, the manner in which the vehicle interacts with the skin and, in particular, the stratum corneum, is very important. For example, there may be components of the vehicle that can act as penetration enhancers, or that may be irritating to the skin or (not atypically) both. Equally, the potential interactions between a drug and a vehicle are not necessarily easy to predict and there are no clear ‘rules’ for matching a formulation to a particular drug. This situation is complicated by a literature full of difficult to interpret data, often obtained from in vitro experiments using skin from animal models, which are known to exaggerate vehicle effects.
In short, there is no consistent strategy to maximize drug delivery via the use of specific formulations and little attention has been paid to the ‘metamorphosis’ of a vehicle that occurs when it is massaged into the skin. That is, the residual film of formulation left after inunction is clearly quite different from that which was originally applied (volatile excipients having evaporated, for example, and/or permeated rapidly into the skin, and so on).
Progressively, however, a more robust ‘decision tree’ strategy to define formulation options for specific drugs is emerging. As discussed already, this begins with an evaluation of the active's physicochemical properties (size, lipophilicity, solubility) and an estimation of the maximum delivery possible when applied as a saturated solution, assuming no effect of the vehicle on skin barrier function. From this point, a formulation can be assembled step by step, identifying constituents suitable to ensure sufficient loading of the drug (i.e. that the ultimate product contains enough of the active to provide an effective treatment over the dosing period), optimizing the drug release characteristics from the formulation and from the residual phase remaining after application, and creating a stable and cosmetically acceptable vehicle.
Confounding factors, usually of biological origin, mean that the strategy is necessarily iterative. For instance, formulations optimized on normal skin may need to be ‘tweaked’ for optimal use on diseased skin; or the long-term effects of a vehicle on the skin must be particularly monitored if a penetration-enhancing excipient is present because such compounds can be irritating.
Broadly speaking, current topical formulations can be categorized as follows.
- Hydrocarbon-based formulations. For chronic skin disease, these formulations are preferred for their occlusive and protective properties. However, while useful as emollients, the value of these vehicles as topical release systems is limited by poor drug solubility. The latter can be enhanced by using a formulation with hydrocarbon-miscible solvents such as isopropyl myristate or propylene glycol. These anhydrous formulations are usually rather inelegant, sticky, greasy, white or off-white ointments, providing high occlusion of the skin. They are typically used to treat psoriasis, chronic eczema and mycosis, involving small areas of application to dry or very dry skin.
- Polar gel formulations. These are more elegant single-phase systems. They are usually water- and/or alcohol-based (hydrogel, hydro-alcoholic gel) with low or no lipid content. Physically, the gels are transparent to opaque semi-solid gels, rapidly absorbed, non-greasy, non-occlusive and may elicit a skin ‘cooling’ effect upon application. These gels are used to treat acne, acute eczema, rosacea and allergic skin conditions (but are not favoured for psoriasis and chronic eczema) and are applied to small areas of oily or inflamed skin (e.g. on the face). There is a variety of gelling agents available (such as cellulose derivatives, polysaccharide gums and acrylate polymers, such as carbomers) allowing the creation of diverse formulations. Variations of this vehicle class include hydrogels containing a dispersed lipid phase, a so-called emulsion gel or emulgel, and suspensions of water-insoluble drugs in hydrogels (suspension gel).
- Creams. Creams are disperse systems – that is, emulsions – and represent the majority of aqueous-based topical formulations. The readily adjustable properties of these vehicles have led to their wide application in dermatological therapy. Creams require ‘emulsifiers’ (i.e. surfactants) to stabilize the mixture of oil and water phases; the dispersion of oil droplets in water or water droplets in oil is complicated and the arrangement of emulsifier films at the oil–water interface determines the internal structure (crystalline or liquid–crystalline regions, hexagonal or lamellar structures, etc.) of these formulations. Water acts as either the outer, continuous phase (oil-in-water, o/w emulsion) or the inner, discontinuous phase (water-in-oil, w/o emulsion). The latter blend easily with SC lipids, improving the bioavailability of lipid-soluble drugs and moisturizing the skin via a modest occlusive effect. The continuous lipid phase typically comprises triglycerides or waxes. These white to off-white emulsions have a high lipid-replenishing effect and are applied to broad areas of normal to dry skin, treating diseases such as psoriasis, chronic eczema and mycosis. The former (o/w emulsions) are more cosmetically appealing (less sticky and greasy) as the lipids therein are finely dispersed, and contact of the outer aqueous phase with the skin increases water evaporation, producing a cooling effect after application (however, it should also be noted that the surfactant-like emulsifiers in o/w emulsions can withdraw moisture from the skin and facilitate both SC lipid extraction and perturbation). These formulations are hydrating, milky white, spreadable emulsions, suitable for application to large skin areas (both normal, slightly dry and inflamed) affected by acne and acute and subacute eczema.
- Other disperse systems for specific applications. These more complex formulations include: (i) those that are created with vesicular structures, such as liposomes or niosomes, based on phospholipids and non-ionic surfactants, respectively; (ii) multiple, nano- and microemulsions, which require relatively high levels of surfactant to maintain their stability; and (iii) sprays, foams and lacquers, the latter being most typically used in treating diseases of the nail.
Assessment of topical drug bioavailability and bioequivalence between formulations
Bioavailability may be defined as the ‘rate and extent to which the drug is absorbed from the formulation and becomes available at the site of action’. Bioequivalence is ‘the absence of a significant difference in the rate and extent to which the active ingredient or active moiety in pharmaceutical equivalents or pharmaceutical alternatives become available at the site of drug action when administered at the same molar dose under similar conditions in an appropriately designed study’. For new chemical entities, their approval as drug products requires the successful demonstration of safety and efficacy in a series of increasingly detailed clinical trials. With respect to the commercialization of bioequivalent, generic drug products for oral delivery, the accepted approach is relatively straightforward and is principally based on matching blood level profiles (rate and extent of absorption). For topical drug products, in contrast, other than the corticosteroids, a clinical trial is essentially the only route for the approval of a generic product or for the replacement of an already approved dermatological product that has appreciable compositional changes. Comparative clinical trials are relatively insensitive, time consuming and costly; to gain the adequate statistical power needed to clearly evaluate bioequivalence can require a very large number (i.e. hundreds or more) of subjects.
Methods for the determination of topical bioavailability (BA) and bioequivalence (BE) are summarized in Figure 13.7 and may be separated into in vitro and in vivo approaches. The methods in italics have not yet received official sanction as independent means with which to evaluate topical BA/BE; the others have each, to some extent, been employed to compare different topical drug products. With respect to in vitro approaches, the US Food and Drug Administration (FDA) has described a method to assure consistent release kinetics of the active component(s) from semi-solid formulations typical of those found in topical products. The technique measures the delivery of the active pharmaceutical ingredient (API) from the product across an artificial (e.g. polymeric), porous membrane and serves as a valuable quality control measurement of a number of important product characteristics, such as solubility and particle size of the API and its rheological properties. Thus, in many cases, the in vitro release rate is a useful test to assess product sameness when minor formulation changes are made. However, the in vitro release test for semi-solid dosage forms is not a surrogate test for in vivo BA or BE (as is often the case for the in vitro dissolution of solid oral dosage forms).
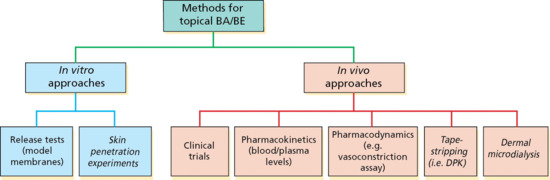
Figure 13.7 Methods, both accepted and under consideration by the regulatory authorities (see text for details), for the determination of topical bioavailability (BA) and bioequivalence (BE).
The use of ex vivo human skin to study the process of percutaneous absorption is widespread and has applications within multiple areas of the drug development process as well as in the field of toxicology. Though substantial literature has emerged utilizing this model system, the degree to which it mimics the living state and, therefore, the extent to which the data can be extrapolated with confidence must be critically examined. While not a perfect model, perhaps, the weight of evidence accumulated over more than 40 years suggests that, when a consistent and robust protocol is employed, the data are reliable and correlated with those obtained from living subjects. Despite having carefully examined the principles and practise of in vitro percutaneous absorption studies, and their relevance to BA/BE, the regulatory authorities have yet to formally embrace this approach. In the interim, however, it is clear that in vitro skin penetration studies are in routine use in manifold applications, including the screening and optimization of transdermal, topical, cosmetic and personal care formulations, and in the assessment of risk associated with dermal exposure to potentially harmful chemicals in the home, work place and environment.
A number of important issues have been identified with respect to in vivo approaches that rely on clinical end points for the assessment of topical BA/BE. These include: (i) when the reference listed drug (RLD) has low efficacy it is difficult to demonstrate that the test product and RLD are active, and that the study is sufficiently sensitive to detect differences between products; (ii) when the RLD is indicated for a relatively small patient population; (iii) the typically high variability of clinical end points means that BE can only be proven when the products are tested in very large patient populations – given the costs of clinical trials, this makes evaluation prohibitively expensive; and (iv) the creation of a potential barrier to post-approval changes to improve quality.
The use of a ‘classic’ pharmacokinetic approach (i.e. the measurement of blood or plasma levels of drug as a function of time post-application) for the evaluation of topical BA/BE has been largely ignored because, in general, the amount of API that can be found in the blood is small and difficult to quantify, and the levels present are typically considered (but rarely, if ever, proven) to be irrelevant to therapeutic activity at the site of action within the skin. Two key facts, however, are worthy of consideration. First, for a large fraction of all topically applied drugs (and this includes those used in dermatology, drugs to treat local, subcutaneous pain and inflammation, and transdermal drugs for systemic effect), the principal barrier to their absorption to the site of action – whether within the skin, beneath the skin at the application site, or in the central compartment – is the stratum corneum. Once the SC is breached, transport within the viable skin and uptake by the microcirculation proceeds relatively quickly, such that it is reasonable to hypothesize that drug levels in the sub-SC compartments (epidermis/dermis, subcutaneous tissue, blood/plasma) will be proportional to one another, albeit progressively smaller with increasing distance from the skin surface. If true, then a conventional plasma concentration (Cp) versus time profile may well reflect the fluctuation of drug levels at the target site in the skin, and a comparison of classic pharmacokinetic metrics for two topical drug products would then allow an assessment of local BE to be performed. Second, the recent and rapid development and evolution of coupled liquid chromatography and mass spectrometry (LC-MS) technology means that the very low levels of API in the blood post-administration of conventional topical dosage forms are now quantifiable with an accuracy and precision sufficient to allow pharmacokinetic studies to be performed. Of course, there are potential exceptions to the use of this approach, for which it makes little sense (e.g. for the evaluation of a topical antifungal, or for a drug with a specific action within a hair follicle). In contrast, for drugs acting in subcutaneous compartments, such as non-steroidal anti-inflammatory drugs in muscle, joints, etc., a sensitive comparison of Cp–time profiles may offer a far more sensitive approach to local BE assessment than currently available alternatives.
Corticosteroids, after permeation through the epidermal barrier, exert a quantifiable blanching effect on the skin, caused by vasoconstriction of the dermal microvasculature. This pharmacodynamic effect has been adopted as an acceptable method for assessing BA/BE of topical corticosteroids. While the approach is non-invasive and much less onerous than a clinical trial, refining the rather complicated protocol of subject selection, assessment of blanching (visual versus chromameter) and the determination of dose duration, has not reduced the significant variability associated with the assay. As a surrogate tool for comparing topical dosage forms, therefore, the vasoconstriction assay is of limited value, being applicable only to the steroids that elicit the requisite pharmacological response (which, it should be added, is not related to the actual mechanism of action of these drugs).
The so-called dermato-pharmacokinetic (DPK) method to assess the BE of topical products for application to the skin has been examined carefully over the past 15 years or so. A draft FDA Guidance, which was subsequently withdrawn, required quantification of the total amount of drug in the SC as a function of time, akin to the typical Cp–time profiles of traditional pharmacokinetic studies using blood sampling. However, the inconsistent results of a pivotal study, which compared tretinoin gel formulations, prompted concern about both the reproducibility and the adequacy of this method to assess drugs whose target site is beyond the SC. Considerable subsequent effort was directed at the identification of problems/limitations of the original DPK protocol and the development of an improved procedure to generate data of much higher quality and reproducibility. The end product of this research is a relatively straightforward method that examines only one uptake and one elimination time per formulation [12]. The total amount of drug in the SC is measured for each site, and the ratios of these quantities for an RLD and test formulations can then be compared to assess (in)equivalence. The method has been successively applied retrospectively to the comparison of retinoid gels and prospectively to the analysis of DPK data obtained for econazole creams consistent with clinical outcome.
Finally, microdialysis is an in vivo technique for sampling free drug concentrations in the subcutaneous or intradermal extracellular fluid. A thin dialysis membrane tube is inserted into the skin and perfused with a site-compatible physiological solution. Molecules of interest diffuse across the membrane, allowing the percutaneously absorbed drug content of the extracellular fluid of the dermis to be continuously sampled and measured over a period of hours. The successful monitoring of the skin penetration of a number of drugs has been reported. However, the technically demanding procedure remains a research tool, with challenges related to tissue reactions as the probe is inserted, the need for highly sensitive analytical tools to detect very low amounts of the permeating drug (especially those that do not cross the SC rapidly) and the length of time (c. 6 h) that subjects can be comfortably immobilized with dialysis probes in place. Inter- and intraindividual variability is relatively large, with coefficients of variation as large as 100%.
In conclusion, it is evident that topical BA/BE can be assessed using appropriately selected in vitro and/or in vivo surrogate tests. All surrogate tests have some, but different, limitations, meaning that results from distinct approaches can be complementary and increase confidence in the outcome of (in)equivalence deduced. The adoption of alternative methodologies to facilitate the approval of topical drug products can be anticipated in the not too distant future.
Optimization of dermatological medicines
To optimize the use of dermatological medicines (i.e. drug plus delivery system) [10, 11], a useful starting point is to revisit the earlier discussion of a drug's maximum flux (Jmax) across the skin. Combining equations 13.1 and 13.2 yields:

Assuming for the moment that the vehicle (v) is incapable of changing either the drug's diffusivity (D), or its diffusion path length (h) through the SC, then Jmax is directly dependent upon the product of the compound's SC/vehicle partition coefficient (Ksc/v) and its saturation solubility in the vehicle (Cv,sat). The partition coefficient, of course, is a measure of the drug's relative affinity for the SC as compared with that for the vehicle and, as such, can be expressed as the ratio of its solubilities in these two phases, that is:

Two deductions are immediately apparent upon the examination of equations 13.5 and 13.6. First, substitution of the latter into the former gives:

and indicates that the maximum flux of a drug is the same from any vehicle in which it is applied at its saturation concentration, and depends only on the compound's solubility in the SC. While there are examples in the literature that confirm this prediction [9], the derivation of equation 13.7 assumes an ideal situation in which the vehicles (or components thereof) do not themselves partition into the SC, and/or alter the drug's solubility therein, and/or interact with the SC lipids, for example, and change the value of D.
Second, to achieve Jmax in accord with equation 13.5 involves an attempt to maximize two parameters, namely Ksc/v and Cv,sat, that are in ‘conflict’ with one another. That is, if the properties of the vehicle are altered to increase the drug's solubility then, in accord with equation 13.6, this leads to a reduction in the SC/vehicle partition coefficient. The logic is inescapable: the creation of a vehicle in which the drug has improved solubility means that the drug ‘likes’ the vehicle, and hence its affinity for the SC relative to the ‘improved’ vehicle must be diminished (again, following equation 13.6).
This point can be illustrated with the practical problem of formulating a lipophilic drug, such as a corticosteroid of low water solubility, in an aqueous gel [10, 11]. To improve the ‘loading’ of the formulation, a co-solvent – for example, propylene glycol (PG) – can be introduced to increase Cv,sat. While this works efficiently, as shown in Figure 13.8a, the opposite impact on the SC/vehicle partition coefficient is clear. In this ideal case, as the %PG in the vehicle is increased from 0% to 100%, the Cv,sat increases from 0.1. to 10 mg/mL, while the Ksc/v falls from 100 to 1.
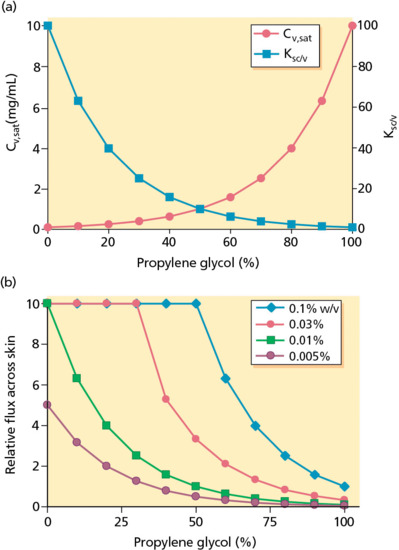
Figure 13.8 (a) Formulation of a lipophilic drug (e.g. a corticosteroid) of low water solubility, in an aqueous gel [9]. The profiles illustrate the manner in which the presence of a co-solvent, propylene glycol (PG), impacts on the saturation solubility of the drug (Cv,sat) and its SC/vehicle partition coefficient (Ksc/v). (b) The relative flux of the drug (which is directly proportional to Ksc/v*Cv) as a function of the %PG in the vehicle and the drug ‘loading’ therein [10, 11].
The selection of the optimum %PG for a formulation depends on the desired drug concentration to be incorporated. Figure 13.8b demonstrates how the relative flux of the active compound (which is directly proportional to Ksc/v*Cv) varies as a function of the %PG in the vehicle and the drug ‘loading’ therein [10, 11]. For example, at a target drug concentration of 0.1% w/v (i.e. 1 mg/mL), the flux from a gel consisting of 100% PG is rather small: at this level of co-solvent, the drug is completely solubilized and is far from its saturation solubility. As a result, the flux is well below Jmax and its ‘leaving tendency’ from the vehicle is low because the drug is quite ‘comfortable’ in that environment. Decreasing the %PG progressively to 50% causes the relative flux to increase to an obvious maximum; because the drug level in the vehicle is being held constant, it follows that, as the percentage co-solvent is reduced, the drug concentration becomes closer and closer to its saturation solubility. At 50% PG (where Jmax is attained), therefore, there is just enough co-solvent to solubilize 0.1% w/v of the drug. Further reduction of the %PG leads to the creation of a suspension of the drug, with a smaller and smaller fraction of the compound in solution as the co-solvent level falls. While the relative flux theoretically remains at Jmax, the period for which delivery into the skin can be maintained becomes more and more compromised. The solubilized molecules are quickly depleted but likely to be only very slowly replenished by dissolution of solid drug because of the diminishing solubility of the compound as the co-solvent level is reduced.
At a desired drug concentration of 0.03% (0.3 mg/mL), a similar pattern of behaviour is observed to that described above. In this case, however, a lower %PG (c. 30%) is required to just solubilize this level of drug loading. The profile for the target drug loading of 0.01% w/v indicates that this concentration is equal to the aqueous solubility of the compound (Jmax being attained without the need for co-solvent); addition of PG to the formulation only leads to the flux falling off as the co-solvent progressively increases drug solubility. Finally, at 0.005% w/v of drug, the highest flux possible (again, in the absence of any co-solvent) is only one-half of Jmax, given that this concentration is 50% of the compound's water solubility.
Further practical lessons can be learned from this analysis of formulation optimization. Specifically, a ‘one size fits all’ approach to vehicle selection is clearly inappropriate: (i) optimal drug delivery from formulations containing different levels of the compound may require different vehicle compositions, as shown above; (ii) the dilution of formulations in which drugs are present as suspensions may not always lead to a concomitant reduction in delivery; and (iii) the optimal formulation of a structurally related, but more lipophilic analogue, of an existing drug, for example, will require a higher level of co-solvent in the formulation to maximize its flux into the skin.
In reality, the ‘art’ of topical formulation is even more complicated than that outlined above and the ‘ideal’ conditions assumed so far are rarely applicable except, perhaps, during the initial moments of formulation administration. A common aqueous-based vehicle or cream will probably contain [13] – in addition to the drug, water, one or more co-solvents and a polymeric gelling agent –a volatile solvent (such as ethanol or isopropyl alcohol), emulsifiers or surfactants to stabilize a disperse system and/or facilitate drug uptake into, or passage through, the SC. It will also contatin other ingredients to render the formulation cosmetically and pharmaceutically acceptable (e.g. fragrance, preservative, etc.). Upon application of the drug product and its massage into the skin, multiple, parallel events can and do take place [13] that influence the ultimate disposition and bioavailability of the active compound, including (but not limited to) the following.
- The incorporation of a volatile solvent is a useful means by which to solubilize a poorly soluble drug in the vehicle (i.e. the volatile excipient is also acting as a co-solvent). Upon administration, a solvent, such as ethanol, will itself rapidly partition into the SC and assist in transferring the drug quickly from the formulation into the barrier. In parallel, the solvent will be evaporating, causing the drug, which has been up to this point solubilized by the solvent's presence, to concentrate in the vehicle and hence increase the driving force for its diffusion across the SC. From a thermodynamic point of view, the loss (by evaporation and SC uptake) of the volatile will eventually reach the point at which the amount of drug remaining at the skin surface can no longer be fully solubilized and its precipitation must therefore occur – a step which will significantly impede any further transfer into the SC. However, because this change in the formulation occurs rather rapidly, it is possible that there will be a transient, so-called supersaturation [13] of drug in the vehicle allowing for a short-lived period during which the active compound may be delivered into the skin at a rate greater than Jmax. The kinetics of precipitation may be retarded by (for example) certain polymers, such as those used to gel the vehicle or to adjust its viscosity.
- A co-solvent like PG is frequently present in topical formulations at quite high levels. When the vehicle is applied, the co-solvent not only influences the drug's solubility and its partitioning into the SC, it also transfers itself from the administered delivery system into the skin. The physicochemical properties of co-solvents are such that they are typically well absorbed into the SC. Having done so, they are then capable of changing the drug's solubility in this outer layer of the skin [14]; that is, the principle, upon which equation 13.7 was derived, is violated and drug delivery can therefore exceed Jmax. Figure 13.9 illustrates this point for the delivery of ibuprofen into human SC in vivo from three formulations in which the drug was saturated: 50 : 50 v/v PG : water, 75 : 25 v/v PG : water and PG alone [14]. The ideal analysis developed mathematically above predicts that drug flux from the three vehicles should have been identical. In fact, with increasing PG, ibuprofen availability increased, relative to the 50 : 50 PG : water formulation, by about 2- and 6.5-fold, respectively, for 75 : 25 PG : water and PG alone.
- Clearly, the transfer of co-solvent(s) into the SC will also facilitate the uptake of surfactants or emulsifiers from the vehicle. These compounds have been shown capable of structurally perturbing and/or extracting SC intercellular lipids and thereby compromising barrier function to some extent. Such penetration enhancers may therefore impact upon both the SC/vehicle partition coefficient of the drug and its diffusivity across the skin.
- Finally, once the formulation has been completely massaged into the skin, it is evident, as mentioned previously, that a complete ‘metamorphosis’ of the vehicle has taken place [13], leaving only a residual (and not easily visualized) film of material on the SC surface. The loss of co-solvents by volatilization and/or uptake into the SC means that it is highly probable that the residual film contains solid drug particles or crystals that precipitated out of solution during the dramatic changes in the composition of the formulation (Figure 13.10) [15, 16]. It seems reasonable to speculate that this solid drug material is unlikely to be absorbed to its target site in the skin, given the absence of available solubilizing agents. Indeed, the events described leading to the residual layer of vehicle may well explain why only a few per cent of most topically applied drug doses are ever absorbed into the skin – the remainder are presumably ‘stranded’ in the solid state at the surface and ultimately removed by physical abrasion or washing prior to a subsequent dose.
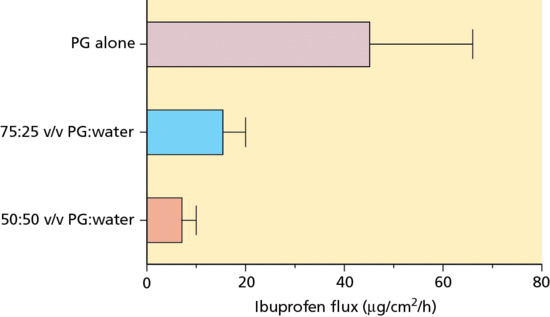
Figure 13.9 Flux of ibuprofen into human stratum corneum in vivo from three formulations [14] in which the drug was present at its saturation concentration. PG, propylene glycol.

Figure 13.10 Stimulated Raman spectroscopic images of (a) solid ibuprofen and (b) betamethasone valerate particles crystallized on and within the outer stratum corneum 30 min after application of the drugs as solutions in propylene glycol. Each image is approximately 150 × 150 μm2.
(Courtesy of N. Belsey.)
Conclusions
Skin barrier function is principally determined by the SC, a remarkable feat of bioengineering that provides an extremely efficient means to retard water loss from the ‘inside–out’ and to the penetration of xenobiotics from the ‘outside–in’. While there are multiple potential pathways for drug diffusion across the SC, it appears that most compounds are constrained to transport via the lipid-filled intercellular domain of the barrier, significantly extending the distance that must be travelled from one side of the barrier to the other (and hence substantially lowering the permeability of the membrane).
Analysis of the substantial percutaneous absorption literature reveals unequivocally that molecular penetration of the barrier depends upon the drug's lipophilicity (consistent with the principal permeation pathway), its size and its solubility properties. Key physicochemical parameters that ultimately determine a drug's maximum flux across the SC are hence its octanol–water partition coefficient (P), molecular weight (MW) and aqueous solubility (Cwater,sat); compounds with extreme values of lipophilicity (e.g. log P >5), MW of >400–500 and low Cwater,sat (less than c. 1 mg/mL) are unlikely to penetrate the skin well.
A variety of topical drug formulations have been developed to treat dermatological disease and many potent drugs are successfully used to resolve diverse skin problems. The efficiency of the majority of drug products approved for therapeutic use is, however, rather poor. Typically, only a few per cent of the applied drug dose actually becomes available at the site of action. Because of this disappointing performance, it is not surprising that drug treatment of skin disease is associated with large variability and that the demonstration of safety and efficacy (and indeed the bioequivalence between formulations of the same drug) currently require clinical trials with very large patient numbers.
Two substantial challenges remain, therefore, to improve the quality of topical dermatological therapy. First, it is essential to create better formulations that contain lower drug loads but which deliver a greater proportion of that load. At present, a substantial fraction of the applied drug is probably ‘stranded’ on the skin surface in the solid state following the loss of co-solvent excipients (by volatilization and skin permeation) during the application process (i.e. massage of the formulation into the skin). Greater attention needs to be paid to the nature of the residual formulation after this ‘metamorphosis’ of the vehicle to ensure that the drug remains in a molecularly absorbable form from the residual film. Second, additional research is required to evolve validated, surrogate tests with which to objectively, rapidly and economically evaluate dermato-pharmacokinetics – that is, the bioavailability of topically applied drugs and the bioequivalence between different formulations containing the same drug that are designed to achieve the same clinical end point.
References
- Menon GK, Cleary GW, Lane ME. The structure and function of the stratum corneum. Int J Pharmaceut 2012;435:3–9.
- Wertz PW, Downing DT. Stratum corneum: biological and biochemical considerations. In: HadgraftJ, GuyRH, eds. Transdermal Drug Delivery – Developmental Issues and Research Initiatives. New York: Dekker, 1989:1–22.
- Elias PM. Epidermal barrier function: intercellular lamellar lipid structures, origin, composition and metabolism. J Control Release 1991;15:199–208.
- Kalia YN, Alberti I, Sekkat N, et al. Normalization of stratum corneum barrier function and transepidermal water loss in vivo. Pharmaceut Res 2000;17:1148–50.
- Scheuplein RJ. Mechanism of percutaneous adsorption. I. Routes of penetration and the influence of solubility. J Invest Dermatol 1965;45:334–46.
- Scheuplein RJ. Mechanism of percutaneous absorption. II. Transient diffusion and the relative importance of various routes of skin penetration. J Invest Dermatol 1967;48:79–88.
- Potts RO, Francoeur ML. Lipid biophysics of water loss through the skin. Proc Natl Acad Sci USA 1990;87:3871–3.
- Potts RO, Francoeur ML. The influence of stratum corneum morphology on water permeability. J Invest Dermatol 1991;96:495–9.
- Mitragotri S, Anissimov YG, Bunge AL, et al. Mathematical models of skin permeability: an overview. Int J Pharmaceut 2011;418:115–29.
- Barry BW. Dermatological Formulations: Percutaneous Absorption. New York: Dekker, 1983.
- Williams AC. Transdermal and Topical Drug Delivery. London: Pharmaceutical Press, 2003.
- N'Dri-Stempfer B, Navidi WC, Guy RH, et al. Improved bioequivalence assessment of topical dermatological drug products using dermatopharmacokinetics. Pharmaceut Res 2009;26:316–28.
- Surber C, Davis AF. Bioavailability and bioequivalence of dermatological formulations. In: WaltersKA, ed. Dermatological and Transdermal Formulations. New York: Informa Healthcare, 2002:401–98.
- Herkenne C, Naik A, Kalia YN, et al. Effect of propylene glycol on ibuprofen absorption into human skin in vivo. J Pharmaceut Sci 2008;97:185–97.
- Saar BG, Contreras-Rojas LR, Xie XS, et al. Imaging drug delivery to skin with stimulated Raman scattering microscopy. Mol Pharmaceut 2011;8:969–75.
- Belsey NA, Garrett NL, Contreras-Rojas LR, et al. Evaluation of drug delivery to intact and porated skin by coherent Raman scattering and fluorescence microscopies. J Control Release 2014;174:37–42.