CHAPTER 14
Clinical Pharmacology
Catherine H. Smith
St John's Institute of Dermatology, Guy's and St Thomas’ Hospitals NHS Foundation Trust, London, UK
Introduction
Drug therapy offers a real opportunity to dramatically improve the lives of people suffering from skin disease, and there has been a transformative change over the last 10 years in terms of treatment options available. An abiding principle in medicine is ‘to help, or at least to do no harm’ (Hippocrates) and it is a sobering fact that 3–6% of hospital admissions are due to adverse drug reactions [1, 2] and, of these, over half could have potentially been avoided [3, 4]. Clinical pharmacology deals with the actions, mechanisms of action, uses, adverse effects and fate of drugs in humans, and underpins all aspects of drug therapy from novel drug development through to safe, effective prescribing.
Types of drugs and terminology
Traditional pharmaceutical agents are typically small (<500 kDa), organic molecules (small-molecule drugs) and have, until recently, dominated medicine. With the advent of recombinant DNA technology and advances in biotechnology manufacturing, the design and large-scale production of proteins became possible and consequently brought in the era of biological therapy (large-molecule drugs). Biological therapy, biotherapeutics and biological medicinal products are all terms used interchangeably and can be used to describe therapeutic agents that are produced by or extracted from a biological source. They include recombinant proteins, monoclonal antibodies, fusion proteins, blood products, immunological medicinal products such as sera and vaccines, allergens and advanced technology products such as gene and cell therapy products [6]. These agents have revolutionized treatment approaches to disease in general, and skin disease is no exception. The principles of clinical pharmacology apply to both types of drug but due to the size and complexity of protein-based therapies, the pharmacokinetics and pharmacodynamics often differ, as does the ethical and regulatory framework around their development and licensing.
Pharmacokinetics
Pharmacokinetics describes the processes involved in drug absorption, distribution, metabolism (biotransformation) and elimination, and is often referred to by the acronym ADME (i.e. what the body does to the drug) (Table 14.1) [7]. Understanding these principles is fundamental to safe, effective prescribing as it dictates how, and when, a drug will be delivered to the main site(s) of action. Clinical pharmacokinetics aims to describe and quantify the relationship between a given dose of drug and the pharmacological effect, be it therapeutic, toxic or ‘off target’ (i.e. unrelated to the known therapeutic efficacy or toxicity), and assumes that the concentration of drug measured in the central compartment (usually blood or plasma) can be related to the concentration at the site of pharmacological action. In general, for most small-molecule drugs administered within the therapeutic dose range, this relationship is linear (first order kinetics) because the systems in place for drug elimination such as metabolizing enzymes and transporters are not saturated. In contrast, biological drugs are generally characterized by non-linear pharmacokinetics due to saturation of binding, distribution and/or elimination pathways as well as the development of antidrug antibodies [8, 9]. Target-mediated drug disposition, which refers to the situation where a significant proportion of a drug (relative to dose) is bound with high affinity to a pharmacological target, such that this interaction is reflected in the pharmacokinetic properties of the drug, is commonly reported with therapeutic monoclonal antibodies [10, 11]. Complex, multiple compartment pharmacokinetic modelling is frequently required to adequately describe biological drug disposition and is an area of ongoing development and challenge. Even so, for many biological drugs, the central premise that drug level concentration in the central compartment is related to the therapeutic effect remains true.
Table 14.1 Common terms used in clinical pharmacology
| Term | Definition |
| Drug disposition | The absorption, distribution, metabolism and elimination of a drug |
| Bioavailability | The fraction of a drug absorbed into the systemic circulation |
| Volume of distribution | A measure of the apparent space in the body available to contain a drug calculated according to the amount of drug given, and the concentration found in the systemic circulation |
| Clearance | A measure of the body's efficiency in eliminating a drug from the systemic circulation |
| Elimination half-life | A measure of the rate of removal of a drug from the systemic circulation |
| Equilibrium dissociation constant (KD) | A measure of drug/receptor binding affinity (a drug with high affinity binding will have a low KD) |
| Half maximally effective concentration (EC50) | A measure of drug potency |
| Median effective dose (MD50) | Dose of drug required to produce a specified effect in 50% of the population |
| Lethal effective dose (LD50) | Dose of drug required to cause death in 50% of experimental animals (pre-clinical) |
| Therapeutic window | Range of steady state drug concentration required to produce the desired clinical effect with minimal toxicity |
Adapted from Buxton and Beret 2011 [7].
Absorption
Absorption describes the movement of a drug from its site of administration to the central compartment, and is critically dependent on the route of administration used.
Topical
In skin disease, a topical approach to treatment (see Chapter 0) is often preferred as it allows direct application of the drug to the diseased area, whilst also limiting ‘off target’ effects and systemic exposure (and hence toxicity). Not all drugs can be effectively delivered in a topical form. The decision as to whether a topical or systemic approach is required will depend on the nature, extent, site and severity of disease, practicability and patient choice. In instances where topical therapy alone is insufficiently effective, a combined topical and systemic approach may minimize the dose requirement of potentially more toxic systemic drug therapy.
Oral
Oral drug administration is convenient for patients but bioavailability is highly variable and dependent on drug characteristics (e.g. lipophilicity, pH), concomitant food intake (e.g. chelation of tetracyclines by calcium in milk) and rate of gastric emptying. Drug formulation is also important [7]. Enteric-coated preparations are useful for drugs that cause significant gastric irritation, such as prednisolone, but may be incompletely dissolved and absorbed. Similarly, controlled release preparations have relevance where the half-life of the drug is short (<4 h) but are associated with wide interpatient variation and risk of ‘dose dumping’ when the dosage form fails.
Parenteral administration
Parenteral administration (intravenous, intramuscular, subcutaneous) circumvents the gastro-intestinal tract with consequent improved drug bioavailability. Therapeutic proteins have very limited oral bioavailability due to intestinal enzymes and poor permeability across the intestinal mucosal membrane barrier, and are almost exclusively administered via parenteral routes, either as an intravenous infusion (for example rituximab or infliximab) or, more conveniently, subcutaneously [9, 10]. Following subcutaneous administration, a drug is absorbed via the lymphatic and/or capillary networks into the systemic circulation at varying rates that are determined, at least in part, by molecular size. For example, the maximum systemic concentration (Tmax) occurs 3–7 h after subcutaneous administration of anakinra (molecular weight 17.3 kDa) as compared with 5 days for the monoclonal antibody adalimumab (molecular weight 148 kDa). Absorption, and consequent drug bioavailability following subcutaneous administration, is influenced by multiple factors, including patient characteristics (body weight, sex, age, activity level), local factors at the injection site (e.g. subcutaneous blood flow, adiposity) and injection technique, as well as drug-specific factors (e.g. formulation, volume, dose, concentration, presence of an Fc receptor, degree of glycosylation). Predictably, then, the range of bioavailabilities reported for different biological agents currently marketed ranges from 25% to 95%, and there is significant between-patient variation for any single agent.
Novel methods of drug delivery aim to limit systemic drug exposure (and therefore toxicity) and/or optimize bioavailability, particularly for large-molecule drugs (including DNA and proteins) either by facilitated access to the systemic circulation through traditional routes (skin, lung, gut) or by harnessing the specificity of the immune system to target drug action once inside the body. Advances in transdermal drug delivery systems [12] include the use of chemical enhancers, iontophoresis, electroporation (where short, high-voltage pulses disrupt the lipid bilayer of the skin), cavitational ultrasound (already approved for optimizing the delivery of topical lignocaine), microneedles (painless, spontaneous dissolution) and thermal ablation. Inhalation also offers potential for rapid systemic delivery of small-molecule drugs and can confer better bioavailability than oral ingestion given the much lower concentrations of drug-metabolizing enzymes in the lungs compared with the gastro-intestinal tract [13]. Oral drug delivery systems for large molecules capitalize on the multiple existing physiological transport processes within the gastro-intestinal tract, such as the neonatal Fc receptor (FcRn), the mechanism by which neonates absorb maternal immunoglobulins [14].
Distribution
The volume of distribution of a drug is mainly determined by its physicochemical properties (such as charge and lipophilicity), protein-binding capacity and the degree to which it is subject to active transport mechanisms. Small-molecule drugs are distributed into interstitial and intracellular fluids with well-perfused organs such as liver, kidney and brain receiving most of the drug initially, and skin and fat levels accumulating more slowly [7]. The degree to which a drug is bound to plasma proteins and tissues determines blood–tissue partitioning, which in turn can be affected by disease states. Preferential accumulation of drugs in certain tissues may also be of clinical relevance. For example, retinoids tend to preferentially accumulate in adipose tissue so that dosing may need alteration in obese patients. Albumin is a major carrier for acidic drugs, which when reduced by liver, renal or malnutritional disease can lead to elevated levels of unbound drug and increased clearance. On the other hand, α1-acid glycoprotein binds basic drugs and so if it is increased in acute inflammatory states, unbound drug levels may be correspondingly reduced.
For most therapeutic proteins, in contrast to small-molecule drugs, the measured volume of distribution is limited and close to the plasma volume, with tissue drug concentrations generally a fraction of those present in plasma. However, as a consequence of their specific, high-affinity binding properties these very low concentrations of drug are still able to effect marked pharmacological action within the target tissue. The distribution of a drug may also be influenced by the distribution and concentration of the target antigen, as exemplified by monoclonal antibodies, which in turn may be affected by disease activity. This phenomenon is also referred to as ‘antigen sink’, and can further contribute to non-linear pharmacokinetics [15].
Metabolism [7]
Drug metabolism is traditionally described as occurring in two phases. Phase I reactions are catalysed by cytochrome P450s (CYPs), flavin-containing mono-oxgenases and epoxide hydrolases, and lead to oxidation, reduction or hydrolysis of the drug. Usually this results in a loss of drug function, but for some drugs (so-called pro-drugs), it results in drug activation (e.g. mycophenolate mofetil is an ester pro-drug, which is hydrolysed to biologically active mycophenolic acid by plasma esterases). Of the large CYP superfamily, subfamilies CYP2C, CYP2D and CYP3A account for the majority of drug metabolizing activity, and CYP3A4, specifically, metabolizes over half of all small-molecule drugs in clinical use. These CYPs demonstrate significant overlapping substrate specificity and can also metabolize a single compound at different positions, which explains their significant role in the context of drug interactions (see later in this chapter).
Phase II reactions catalyse conjugation of the phase I product with a second molecule (sulphate, glucuronic acid, glutathione, acetyl group, methyl group). This inactivates potentially toxic phase I metabolites, and also facilitates drug elimination as a consequence of improved water solubility and increased molecular weight. Drug-metabolizing enzymes are found in most tissues in the body, including the skin, but are found in the greatest quantity in the gastro-intestinal tract (liver, small and large intestine). Thus when a drug is given orally, substantial metabolism and clearance may occur before it reaches the systemic circulation (so-called first pass effect), firstly by metabolizing enzymes within the gastro-intestinal epithelium, and subsequently following absorption into the portal vein, within the liver. Liver disease (as well as bowel disease or resection) may therefore have a clinically relevant impact on drug metabolism (and therefore drug concentration in the systemic circulation) but the degree to which this occurs is unpredictable and standard measures of liver function are not especially helpful.
Excretion
Drugs are eliminated from the body either unchanged or as drug metabolites, with the kidney being the principle site of drug and drug metabolite elimination. Renal function is therefore a critical factor in determining drug bioavailability and potential toxicity, and when reduced due to age, disease or concomitant therapy (e.g. non-steroidal anti-inflammatory drugs (NSAIDs), diuretics) becomes a very common source of adverse drug reactions.
Therapeutic proteins are removed from the circulation and interstitial tissue fluids in a variety of ways, including: (i) target-mediated clearance [15], where a drug is taken up intracellularly following antigen binding; (ii) non-specific endocytosis; and (iii) formation of circulating immune complexes [7, 16]. Once inside cells, whether in the circulation or target tissue, proteins are broken down into peptide fragments and/or amino acids. These protein degradation products, as well as biologicals of low molecular weight (<30 kDa, e.g. anakinra), are then renally excreted. Molecules with an Fc domain such as monoclonal antibodies and fusion proteins are actively protected from this intracellular proteolytic degradation via the FcRn present in endothelial cell endosomes; this FcRn/Fc complex is then transported back to the cell surface where it dissociates, releasing the active drug back into the circulation. This FcRn-mediated salvage pathway, which presumably evolved to maintain levels of endogenous IgG for health, results in a therapeutically helpful long half-life (around 4 weeks) for most monoclonal antibodies, and is also relevant when using these agents during pregnancy (see the section ‘Conception, pregnancy and lactation’).
Pharmacodynamics
Pharmacodynamics is the study of the biochemical and physiological effects of drugs and their mechanisms of actions (i.e. what the drug does to the body) [17]. The term encompasses desired therapeutic effects as well as unwanted toxic effects. Most drugs work by interacting with a specific cellular macromolecule (drug target or drug receptor). This drug–receptor interaction is the initiating event in a multistep process that ultimately alters tissue function and depends on two key principles: (i) the affinity and specificity of drug/receptor binding; and (ii) the intrinsic activity of receptor-bound drug to activate the receptor. The intrinsic activity of a drug describes whether binding to a receptor completely mimics the effect of the endogenous ligand (an agonist), prevents or blocks this response (an antagonist) or is somewhere in between (partial agonists). Many receptors exhibit constitutive activity in the absence of endogenous ligand binding; inverse agonists inhibit this activity by binding to and stabilizing the receptor in an inactive form. Drugs may bind to the same recognition site as the endogenous ligand (syntopic binding) or to a different region (allosteric or allotropic binding), and this may be reversible or irreversible, competitive or non-competitive. All of these variables ultimately determine the potency of the drug (Table 14.2).
Table 14.2 Major mechanisms underlying drug actions
| Structural family | Functional family | Physiological ligands | Effectors and transducers | Example drugs |
| Transmembrane transduction mechanisms | ||||
| G-protein-coupled receptors | Muscarinic cholinergic receptors | ACh | Gi and Gq; AC, ion channels, phospholipase | Botulinum toxin |
| Eicosanoid receptors | Prostaglandins, leukotrienes, thromboxanes | Gs, Gi and Gq proteins | Montelukast | |
| Histamine receptors (1–4) | Histamine | Gq/11, Gs, Gi/10 | Fexofenadine, ranitidine | |
| Ion channels | Ligand gated | ACh, GABA, 5-HT | Na+, Ca2+, K+, Cl− | Nicotine, gabapentin |
| Voltage gated | None (activated by membrane polarization) | Na+, Ca2+, K+, other ions | Lignocaine | |
| Transmembrane enzymes | Receptor tyrosine kinases | Insulin, PDGF, EGF, VEGF, growth factors | SH2 and phosphotyrosine-binding domain containing proteins | Herceptin, imatinib, vemurafenib |
| Transmembrane non-enzymes | Cytokine receptors | Interleukins and other cytokines | JAK/STAT, soluble tyrosine kinases | Janus kinase inhibitors |
| Toll-like receptors | LPS, bacterial products | NF-κB, MyD88, IRAKs | Imiquamod | |
| Intracellular transduction mechanisms | ||||
| Nuclear receptors | Steroid receptors (includes retinoic acid receptors, retinoid X receptors) | Oestrogen, testosterone | Co-activators | Corticosteroids, alitretinoin, bexarotene |
| PAR-ϒ | PAR-ϒ | Thiazolidinediones, clofibrate | ||
| Intracellular enzymes | Cyclic phosphodiesterases | Cyclic GMP, cAMP | Protein kinase A, exchange proteins activated by cAMP, cAMP responsive element binding protein | Vasodilators, anti-inflammatory agents (apremilast, rofluminast) |
Adapted from Blumenthal and Garrison 2011 [17].
ACh, acetyl choline; EGF, epidermal growth factor; GABA, γ-aminobutyric acid; 5-HT, 5-hydroxytryptamine; IRAKs, interleukin-associated kinases; JAK, Janus kinase; LPS, lipopolysaccharide; MyD88, myeloid differentiation primary response 88; NF-κB, nuclear factor κB; PAR-ϒ, proteinase activated receptor ϒ; PDGF, platelet-derived growth factor; SH2, Src homology 2; STAT, signal transducers and activators of transcription; VEGF, vascular endothelial growth factor.
Molecular mechanisms underlying drug action
Initial drug/receptor coupling may result in a direct effect on the cellular function, or convey a message to intermediary cellular signalling molecules (transducers). The receptor, its cellular target and any intermediary molecules are termed the receptor–effector system or signal transduction pathway. Often, the transducer proteins mediate the actual physiological effect via generating, moving or degrading small molecules known as second messengers (e.g. nitric oxide or cyclic adenosine monophosphate (cAMP)). This system allows the cell to coordinate, and to amplify, signals from multiple ligands, and explains how tiny amounts of drug at a particular receptor result in a significant biological effect. This complexity can also underpin unintended or unexpected drug effects. The site of drug/receptor coupling and associated underlying mechanism can be outside the cell, at the cell membrane or within the cell. In dermatology, as in other areas of medicine, for many of the drugs in clinical use the site of drug action is unknown, having been introduced either by serendipity, trial and error, or following traditional drug development where drugs were selected for development based on their effects at a whole organism, rather than molecular, level. However, this is changing as a result of our increased understanding of cellular biology, as well as the human genome project which has revealed a vast array of receptors – some of which have as yet unknown ligands (orphan receptors); the number of potentially ‘druggable’ targets has consequently increased substantially.
Extracellular mechanisms
A number of drugs act outside the cell to affect cellular function, typically in one of two ways. The first is to alter the activity of extracellular enzymes involved in the synthesis or degradation of endogenous signalling molecules (e.g. angiotensin-converting enzyme (ACE) inhibitors) and the second by directly interacting with the endogenous ligand to prevent binding to its site of action (e.g. monoclonal antibodies such as tumour necrosis factor (TNF) antagonists adalimumab or infliximab).
Transmembrane mechanisms
Hydrophilic drugs (and their physiological counterparts) cannot easily access the cell and so rely on membrane-bound receptors to exert their action (see Table 14.2 for further details and examples of drugs that exploit these receptors). These fall into five broad categories:
- G-protein-coupled receptors (GPCRs) couple to a family of heterotrimeric guanosine triphosphate (GTP) binding regulatory G proteins. Following ligand binding, G proteins signal to various effector proteins including enzymes such as adenyl cyclase phospholipase C and cyclic guanosine monophosphate (cGMP), which then leads to a cascade of intracellular events via second messenger systems and ultimately a drug effect. G proteins exist in as many as 23 isoforms coupled to different signalling paths and it is the type(s) of G protein coupled to the receptor that determines the response to receptor activation.
- Receptors linked to intracellular enzymes have an extracellular binding domain directly coupled in some way to enzymatic activity within the cell so that, on ligand binding, consequent enzymatic activity initiates and amplifies the intracellular signals and feedback responses by changing the phosphorylation status of the cellular proteins. The types of receptor described include: (i) receptor tyrosine kinases (RTKs), where the tyrosine kinase is part of the transmembrane receptor; (ii) transmembrane receptors that recruit cytoplasmic tyrosine Janus kinases (JAKs), which then phosphorylate STAT (signal transducers and activators of transcription) proteins that translocate to the nucleus and regulate transcription; and (iii) receptor serine/threonine kinases, analogous to the RTKs except they have a serine/threonine cytoplasmic domain.
- Transmembrane receptors without enzyme-linked activity include Toll-like receptors, fundamental components of innate immune signalling, which on ligand binding ultimately recruit interleukin-associated kinases and downstream pathway signalling via the nuclear factor κB (NF-κB) pathway. TNF-α receptor signalling operates in a very similar way to that of Toll-like receptors in that it is has a transmembrane domain with a non-enzymatic cytoplasmic domain (death domain) that on ligand binding triggers a cascade of intracellular events.
- Ion channels facilitate the flux of cations and anions across the impermeable plasma cell membrane to maintain electrochemical gradients critical to excitable cells such as nerves and muscles as well as non-excitable cells, to trigger biochemical and secretory cell function. These channels may be open, closed or inactive and drugs may affect their function by directly opening or closing the channel (ligand-gated channels), by influencing the voltage-dependent characteristics of the channels (voltage-gated channels) and by the amount of time the channel spends in a given state, or by generating second messengers that subsequently open or close the channel (second messenger gated).
- Membrane-bound transporters play a key role in drug pharmacokinetics as they determine drug entry to and elimination from cells, but are also drug targets, particularly those used in neuropsychiatric disorders (e.g. the transporter SERT (SLC6A4) is responsible for the uptake and clearance of serotonin in the brain, and the target for selective serotonin reuptake inhibitors).
Intracellular mechanisms
Some small-molecule drugs diffuse or are actively transported into the cell to access intracellular drug targets. There are two main types:
- Nuclear hormone receptors comprise a superfamily of 48 receptors that act as transcription factors able to directly regulate gene expression following ligand binding. Classic family members include sex hormones, cortisol, thyroid hormones and vitamin D receptors.
- Intracellular enzymes may be targeted directly by drugs, and with better understanding of the pathways involved, are an area of active drug development. Cyclic nucleotide phosphodiesterases (PDEs) are a major family of cytoplasmic enzymes important in cell signalling as they hydrolyse cAMP and cGMP. The specificity of PDEs varies, with some able to hydrolyse cAMP, cGMP or both. Inhibitors of PDE3 are drug targets for the treatment of asthma and cardiovascular disease, whilst the PDE4 inhibitor, apremilast, has been recently licensed for the treatment of psoriasis. Other examples include calcineurin inhibitors such as ciclosporin, tacrolimus and pimecrolimus, which act as antagonists to the cytoplasmic phosphatase enzyme, calcineurin, so preventing dephosphorylation of the cytoplasmic nuclear factor of activated T cells (NFAT) and its translocation to the nucleus.
Drug toxicity and adverse effects
An ideal drug would result only in the desired therapeutic effect. However, few drugs are sufficiently specific, so unwanted effects (side effects, adverse effects) are part of therapeutics. Drug toxicities can be classified in five, somewhat overlapping, broad categories based on the underlying mechanism.
- On target drug toxicity can be defined as drug toxicity following modulation of the primary, pharmacological target (e.g. receptor or enzyme). This may arise due to altered drug exposure at the target site, leading to an exaggerated pharmacological response. Sources of this type of event include deliberate or accidental dosing error, alterations in the pharmacokinetics of the drug (e.g. due to liver or kidney disease or to interactions with other drugs) or changes in the pharmacodynamics of the drug–receptor interaction that alter the pharmacologic response (e.g. changes in receptor number). Activation of the drug target in an unintended tissue is another example of on-target toxicity (e.g. osteoporosis secondary to corticosteroid use, myositis with statin therapy).
- Off target toxicity results from the interaction of a drug with targets other than the intended therapeutic targets, for example the H1-receptor antagonist terfenadine also inhibits a cardiac potassium channel (Kv11.1 encoded by the gene hERG) which led to fatal cardiac arrhythmias and ultimately drug withdrawal.
- Biological activation of drugs to toxic metabolites capable of binding to proteins, DNA and small molecules such as glutathione (GSH) is an increasingly recognized mechanism of drug toxicity. Paracetamol-induced hepatic necrosis is a classic example of this, due to GSH binding (and depletion) by the active intermediate metabolite N-acetyl-p-benzoquineimine.
- Allergic reactions to drugs extend from type I to type IV reactions, are not dose related and are generally unpredictable.
- Idiosyncratic drug reactions are very rare, significant adverse effects (e.g. toxic epidermal necrolysis), and historically describe events for which no obvious mechanism is apparent. Pharmacogenetics offers an opportunity to interrogate the molecular mechanisms underlying many of these reactions.
Assessing the potential for a drug to produce adverse effects begins at the earliest stage of drug development right through to post-marketing surveillance mechanisms (Table 14.3). In general, clinical trials are powered for efficacy, rather than safety, and are performed in patient populations that are not necessarily generalizable to those in whom the drug will be used in clinical practice. So although common adverse effects are likely to be picked up prior to drug marketing, rare (<1 : 10 000) idiosyncratic events (e.g. cases of progressive multifocal leukoencephalopathy observed with efalizumab), events with a long latency period (e.g. skin cancer secondary to psoralen and UVA) or problems using a drug in the context of other morbidities, may take many years to establish. Pharmacovigilance registries are critical to evaluate these longer term safety issues, especially those with a longer latency period.
Table 14.3 Methods used to identify drug-related adverse effects and their respective roles within the lifetime of a drug, from drug development through to post-licensing pharmacoviligance
| Method | Benefits | Problems |
| Clinical trials (phase I–IV) | Controlled Rigorous collection of high-quality data Will identify common, short-term side effects |
Short term Powered for efficacy not safety Data may not be relevant to clinical practice Will miss effects with long latency period |
| Spontaneous reporting | Identifies new/rare/unexpected adverse events and drug–drug interactions | May overestimate the risk (no denominator) May miss common/less severe morbidity |
| Registries | Reflects real-life clinical practice Population-based May allow relative risk/benefit analysis Allows collection of adverse effects with longer latency period |
Incomplete datasets Observational; multiple confounders May lack control group |
Factors that affect therapeutic outcome
Drug choice and medical decision making
Before initiating any therapeutic measures, a comprehensive assessment of the patient is essential, considering the disease that requires treatment in the context of the whole patient and shared treatment goals. Each therapeutic intervention in a patient can be considered a trial (of n = 1) with appropriately defined ‘outcome measures’ and timelines for judging success or failure. This is perhaps especially important in dermatological practice. The rarity of many skin diseases and historic lack of investment in drug development means that robust evidence to guide treatment choice and optimize drug use is often unavailable. Added to this, many drugs are unlicensed so that clear documentation of benefit (or harm) is also important. Continuing an ineffective or insufficiently effective treatment exposes patients to the ongoing risk of adverse events and expense, quite apart from suboptimal management of their skin disease.
Multiple factors determine the relationship between the dose of any particular drug and outcome (Figure 14.1) and thus therapy needs to be carefully and precisely tailored to the needs and clinical circumstance of each patient. This can be a highly complex, dynamic process that requires constant re-evaluation and refinement during the course of treatment.

Figure 14.1 An overview of the relationship between the dose of any particular drug and therapeutic outcome. Multiple, often interdependent, factors impact at critical points (dark blue circles) along the pharmacological pathway, some of which are relevant throughout (right hand side arrow).
Clinical factors that affect drug pharmacokinetics and pharmacodynamics
Age
Drug pharmacokinetics and pharmacodynamics are altered in the very young and in older people, although available drug-specific information is limited largely because these groups are often excluded from clinical trials. There are also age-specific considerations that are pertinent to medical errors, adherence to treatment and co-therapy.
Growth and development during childhood is associated with marked physiological change, especially during infancy and puberty, with consequent, non-linear changes in drug disposition [18]. Systemic exposure to topically applied therapies is more likely in infants and children compared with adults. During infancy, this relates in part to the presence of a thinner stratum corneum and, during childhood, to increased cutaneous perfusion and hydration of the epidermis compared with adults. The ratio of total body surface area to body mass in infants and young children also far exceeds that in adults. Orally administered drugs require normalization of drug dose for either body weight or body surface area to take into account liver and renal developmental change. Hepatic drug-metabolizing enzymes are expressed at very low levels at birth, and then undergo distinct patterns of isoform-specific developmental changes in the first year of life, with levels of activity in children exceeding those of adults. The glomerular filtration rate increases rapidly during the first 2 weeks of life and then rises steadily until adult values are reached at 8–12 months of age [7, 18].
Ageing is associated with a number of progressive physiological changes including cutaneous atrophy, reduction in lean body mass, 40% reduction in hepatic blood flow, reduced activity of hepatic enzyme activity (especially CYP phase I) and reduced renal function. The decline in glomerular filtration rate has been estimated to be around 1 mL/min/year (or less) after middle age and is a critical determinant of disposition for many drugs. The high prevalence of co-morbidities such as hypertension, diabetes and ischaemic heart disease, together with widespread use of nephrotoxic therapies such as diuretics and NSAIDs, contribute to this decline. In completely healthy older people, renal function may not necessarily be reduced significantly. Collectively, physiological and pathological change in older people tends to increase drug bioavailability and reduce drug metabolism and elimination (for comprehensive reviews see [19, 20]), so accounting for the general advice to start drug therapy at the lowest possible dose and increase the dose slowly. Pharmacodynamic responses to drugs may also change with age – the increased effect of drugs that depress the central nervous system is a good example – and the pathophysiology of the disease itself may be different from that in younger people, as in the case of psoriasis where genetic susceptibility loci are distinct in late-onset disease.
Polypharmacy is common with 20–40% of older people taking five or more medications, with consequent impact on medicine adherence and risk of drug interactions. All these factors lead to increased variability in drug response with age. Adverse drug reactions are also more common, and tend to be more severe and less likely to be recognized or reported by the patient [1, 21, 22].
Conception, pregnancy and lactation
Any drug exposure prior to, during or after conception can result in an adverse fetal outcome. This risk, which is closely related to drug dose and duration, should always be considered when prescribing and the patient fully informed of the potential hazard. The pre-implantation period (fertilization to implantation) is considered the ‘all or nothing’ period since injury will result either in embryonic loss, or, if only a small number of cells are affected, embryo survival, as the remaining cells are not yet committed to a particular path of development and can therefore compensate for the loss. Drug exposure during the embryonic period (week 2 to week 9) carries the greatest risk of fetal malformation as this is when organogenesis occurs. Nevertheless, maternal drug exposure at any period may be associated with longer term functional or developmental changes that are not immediately apparent. For example, in a cohort of 31 children born to mothers exposed to isotretinoin, nearly half had evidence of developmental delay, regardless of whether the children had structural malformations at birth [23]. Drugs given just before term or during labour can have adverse effects on labour or on the neonate (e.g. sedative antihistamines), whilst ongoing maternal drug use post-pregnancy may be secreted in breast milk.
Thalidomide, acitretin and isotretinoin are notable examples of drugs with specific, known risks of teratogenicity, and all of these are used in dermatology with rigorous pregnancy prevention plans attached. For the majority of drugs, there is insufficient evidence to properly categorize the level of risk. Very few drugs can be considered completely safe. In general, drug exposure should be avoided or minimized as far as possible in women planning conception or who are pregnant unless the risks and/or impact of the underlying disease warrant it; it is important to recognize that women with significant skin disease may well require active intervention. Topical therapy is generally considered the safest route of drug use for skin disease during pregnancy since systemic, and therefore fetal, exposure is minimized unless large volumes are applied to extensive, inflammatory skin disease. The same rules of minimizing unnecessary drug exposure do still apply, with recent studies suggesting an increased risk of low birth weights with the use of significant amounts of potent or very potent corticosteroids during pregnancy [24].
Drug disposition can alter significantly during pregnancy [25] with most of the changes beginning during the first trimester and peaking during the second trimester. Overall these physiological changes tend to increase the volume of drug distribution, reduce the amount of albumin-bound drug, and increase hepatic and renal clearance, with consequent variable effects on drug concentration depending on the drug. The use of therapeutic monoclonal antibodies and therapeutic peptides during pregnancy requires particular care since maternal antibodies are actively transported across the placenta from around the 14th week of pregnancy, via the Fc receptor expressed on trophoblasts, rapidly increasing over the second and third trimesters [26]. Data on the use of anti-TNF monoclonal antibodies during pregnancy suggest that therapeutic antibodies are likely to be handled in the same way as these naturally occurring antibodies, with drug levels of infliximab and adalimumab in infants at birth being at least equivalent to those of the mother. Interestingly, transplacental transfer of certolizumab pegol, a pegylated humanized antibody Fab′ fragment against TNF, appears to be low or absent, probably because it lacks an Fc receptor. Maternal immunoglobulins (predominantly IgA) are present in breast milk, so therapeutic antibodies might also be predicted in breast milk. Limited human data indicate that drug levels in breast milk are only a fraction of those in the mother, with infant drug levels falling over time despite continuation of breast feeding. This suggests that the infant intestinal mucosal barrier and enzymatic digestion limit the bioavailability of any drug present in breast milk.
In men, drugs may affect fertility and/or spermatogenesis, although reliable information on absolute risk is scarce. Drugs that directly interact with DNA (e.g. methotrexate, azathioprine, cytotoxics) theoretically carry particular risk and although studies have suggested an increased incidence of congenital abnormalities [27], data are conflicting [28]. This may reflect inadequate study power and/or difficulties with controlling for the confounding effect of disease indication.
Drug interactions
A drug's effect may be significantly altered by the co-administration of another drug with a consequent impact on efficacy and/or induction of toxicity. The principal mechanisms underlying most drug interactions relate to drug-metabolizing enzymes and transporters. Whilst all major drug-metabolizing enzymes have been associated with drug–drug interactions, cytochrome P40 enzymes are the major contributors to clinically relevant drug interactions, with CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 and CYP3A most commonly implicated [29]. For example, ciclosporin is extensively metabolized by CYP3A isoforms. Co-therapy with erythromycin or itraconazole, both potent inhibitors of CYP3A, can therefore lead to significantly increased levels of ciclosporin, whereas phenytoin, a potent inducer of CYP3A, may reduce ciclosporin levels. Alternative remedies are also relevant here. St John's wort contains a potent inducer of CYP3A (hyperforin), for example, and has been reported in association with reduced levels of ciclosporin, whilst echinacea variously inhibits intestinal CYP1A2 and CYP3A and induces hepatic CYP3A activities [30].
Drug–transporter interactions arise either due to a change in activity (increased or decreased) or competition at the transporter site. For example, the activity of the efflux transporter P-glycoprotein, which normally acts to reduce gastro-intestinal absorption and increase biliary excretion of drugs, is inhibited by intraconazole and ciclosporin, with a resultant increase in relevant drug substrate levels (e.g. fexofenadine) [31].
Aside from these pharmacokinetic mechanisms, drugs may interact at a pharmacodynamic level. This may be used to therapeutic advantage – prednisolone is often combined with azathioprine or mycophenolate mofetil in order to achieve the same level of immunosuppressant effect, whilst reducing the toxicity of steroid monotherapy, for example – but may equally give rise to adverse events such as the synergistic nephrotoxicity seen when ciclosporin is combined with NSAIDs.
Relatively few clinically relevant drug interactions have been reported with biological therapies but they do occur. For example, interferons have been shown to both inhibit and induce specific CYP isoenzymes [32], and concomitant administration of weekly methotrexate appears to decrease the clearance of adalimumab, an interaction that is used to therapeutic advantage.
Patient adherence to treatment
The extent to which patients follow the instructions they are given for a prescribed medication (i.e. adherence) varies enormously, from missing an occasional dose to completely stopping treatment, and therefore may or may not be relevant to treatment outcome. Non-adherence to treatment is very common but few patients will declare it to health care professionals voluntarily and assessment is best approached in a non-judgemental way. It arises for multiple and complex reasons that fall into two main overlapping categories: unintentional and intentional non-adherence. Examples of unintentional non-adherence include poor comprehension or recall of the details of the prescribed therapeutic regimen, difficulties applying or taking a medicine, and lack of access or ability to pay for treatment. Intentional non-adherence, when a patient decides not to take the prescribed medicine, arises due to patients’ beliefs, concerns or problems associated with their condition and/or prescribed medicine [33]. In general, patients wish to minimize the number of medicines they take, and where there are multiple treatments given they may prioritize one over the other. They will evaluate for themselves the side effects and efficacy of any given treatment – and, of course, the effectiveness of treatment in skin disease is manifestly obvious – and may stop therapy to see the outcome. Overall, adherence to prescribed therapy, especially for chronic conditions, is poor, with, for example, the NHS spend on unused or unwanted medicines estimated at £100 million annually [33].
Factors that may improve adherence include providing clear written and verbal communication about how, why and when the treatment needs to be taken, using the simplest dosing schedule possible, and employing various techniques to remind and support patients depending on the particular situation (e.g. telephone reminders, dosette boxes) [34]. Dermatology nurse specialists can play a key role in improving adherence to therapy through patient education and support, practical demonstration of where and how to apply topical treatments and, in some instances, application of treatments in formal out-patient settings. Unlicensed ‘special manufacture’ medicines (‘specials’) are widely used in topical therapy but can be associated with erratic supplies and hence poor adherence. In the UK, the use of ‘specials’ has been rationalized and manufacture is limited to fewer, approved centres in order to address this issue.
Medication errors
Some side effects and adverse reactions to drugs are accepted risks of treatment and, whilst they can be minimized with careful prescribing and medicine use, they are unavoidable. In contrast, those arising as a consequence of errors in prescribing, dispensing and/or administration of medicines are, by definition, avoidable. The steps involved between writing a prescription and the patient receiving the medicine are multiple, and each is subject to potential error. Medication errors that lead to serious or fatal adverse events invariably occur due to a series of events, and reflect human error (active failure) arising in the context of certain predisposing environmental or system factors (latent failures), the so-called ‘Swiss cheese model’ (Figure 14.2). Reasons for errors include inadequate knowledge of the patient, their clinical condition, drug history and known allergies, inadequate knowledge of the drug being prescribed, drug dose calculation errors, illegible handwriting, and confusing drug names and product labelling [21, 35, 36].
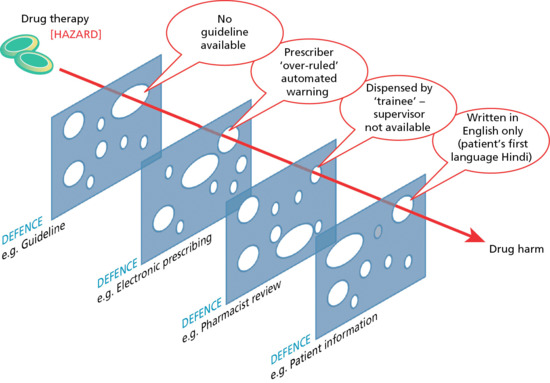
Figure 14.2 Swiss cheese model to illustrate the cumulative effect of multiple failures (holes) in defence mechanisms that ultimately translate drug hazard into actual, drug-related patient harm. The ‘holes’ in the defence may be latent such as organizational flaws (e.g. no supervisor being present) or active (e.g. the prescriber actively overruling an automated alert). Understanding the origin of near misses and/or actual drug-related adverse events is a crucial step towards safer prescribing and the avoidance of preventable drug harm.
One of the commonest sources of error is in the prescription itself [35, 37]. In hospital, often the most junior member of the team with the least knowledge and experience is tasked with prescribing; this in itself is a risk, but also fails to create a culture where prescribing is perceived to be important [36]. Additional factors that predispose to errors include repeated interruptions when writing a prescription and depression in the prescriber.
Poor communication amongst clinicians, particularly where care crosses or is transferred from one clinical setting (e.g. hospital) to another (e.g. primary care), is also a significant source of error. This is both in relation to the dose and type of medication to be used, and also, as importantly, review and monitoring of the treatment. In this situation, a disconnect can arise between the prescriber who initiated the treatment and the clinician responsible for ongoing care, so the necessary adjustments in drug therapy and monitoring fail to occur and/or drug errors remain unchecked for prolonged periods of time, with sometimes catastrophic results. This was specifically identified as a major contributory factor in a review of 137 adverse events arising in relation to methotrexate, which included 25 deaths and 26 serious incidents requiring hospital admission in the UK. It was also the commonest reason in ambulatory care for adverse drug reactions leading to hospital admission [10].
Certain types of medication are subject to frequent medication errors [1, 3]: (i) drugs with a narrow therapeutic index and/or where the therapeutic dose is close to the toxic dose (e.g. chemotherapeutic agents, anticoagulants, narcotics); (ii) drugs with an unusual dosing schedule that patients may easily confuse (e.g. once-weekly methotrexate); or (iii) drugs requiring intravenous administration where the speed of infusion or extravasation of drug into tissue can lead to problems (e.g. rapid immunoglobulin infusion and risk of thromboembolic events). The drug groups most frequently associated with preventable, drug-related hospital admissions are anticoagulants, antiplatelet agents, diuretics and NSAIDs, with around a third due to problems with prescribing, a third due to adherence issues and a third due to inadequate monitoring [3, 4, 35]. Patient groups vulnerable to medication error include infants and children, where complex dose calculations are required using drugs that may not be licensed for use in children, and older patients due to communication difficulties, polypharmacy and the presence of co-morbidities.
Whilst something is known about the cause and prevalence of medication errors, there is relatively little good evidence to indicate which interventions effectively reduce error. Prescribing is no longer the sole remit of doctors and extends to nurses, pharmacists and physiotherapists, all of whom need to take professional responsibility for their prescribing practice. Prescribers need to be supported by appropriate training in clinical pharmacology and the practicalities of prescribing with ready access to clear guidelines and protocols on the use of drugs. Clearly defined ‘care pathways’ can also provide clarity over where responsibility lies for prescribing and monitoring, as has been implemented as part of the National Patient Safety Agency alert on methotrexate in the UK. However, all humans are subject to errors and lapses and so additional, multiple checks need to be in place. Pharmacists play a critical role in cross-checking scripts (although knowing that this safeguard is in place has been identified as a source of inaccurate or lazy prescription writing in hospital practice). Numerous and varied information technology initiatives focusing on all aspects of the prescribing pathway have been developed, although not all are necessarily associated with a reduction in error risk. For example, electronic prescribing has been shown to reduce the incidence of illegible scripts and adverse drug reactions [38, 39] but automated alerts about potential drug interactions and dose errors may overwhelm the prescriber so that the excessively frequent alerts are ignored. It seems likely, though, that further refinement of existing systems and new developments in this area will yield substantial benefit.
New legislation from the UK Medicines and Healthcare Products Regulatory Agency on standards for product labelling should help to reduce confusion and thus minimize dispensing errors. Perhaps most important of all, ensuring the patient is fully briefed on the treatment plan can allow patients themselves to correct mistakes.
Pharmacogenetics and personalized medicine
Personalized medicine (reviewed in [40]) can be defined as giving the right drug to the right patient at the right time, and offers an opportunity to optimize efficacy, minimize adverse effects and save money. This is an area of highly significant, ongoing research investment given the enormous potential benefits, but to successfully achieve it is challenging. First, our current diagnostic paradigm is largely based on a variety of clinical and/or investigational ‘phenotypic’ markers and reveals little about the underlying disease mechanism, and therefore how best to ‘target’ therapy. Within any one particular phenotype, there will be significant underlying genetic and/or immunological heterogeneity and hence variable responses to drug therapy. Second, the pharmacokinetic and pharmacodynamic profile of the drug itself is highly variable, and reflects the sum of multiple processes, each of which is potentially subject to genetic variation. Significant duplication within biological systems also means that the deletion of one metabolic pathway, for example, may not necessarily result in a clinically relevant change. Third, many potentially promising genetic or other biomarkers of treatment outcome fail to move into the clinic because when evaluated in real life [41], they fail to demonstrate added value over and above more traditional ‘clinical’ indicators of treatment response. This may be for a variety of reasons, including ethnic diversity, and the fact that other influences are in play (such as adherence). Fourth, moving genetic or other biomarkers of response or toxicity from a research environment into clinical practice requires capacity: capacity for clinicians to absorb new investigative tools and understand their meaning in the context of the patient in front of them, capacity of laboratories to process new tests, and financial capacity within health care systems.
Pharmacogenetics
Genes encoding drug-metabolizing enzymes, transporters and drug targets may all be subject to functionally relevant polymorphisms and are estimated to account for 15–30% of interindividual variation in drug response.
Interindividual variation in drug-metabolizing enzyme activity has historically been described in terms of phenotypic variants (e.g. fast and slow ‘acetylators’, slow and fast ‘metabolizers’). These are now known to originate from specific genetic variants (NAT2 and CYP2D6 polymorphisms, respectively, in the following example) and more than half the drugs cited in adverse drug reaction studies are metabolized by at least one enzyme with a variant allele known to be associated with poor metabolism [42]. The polymorphic enzyme thiopurine methyltransferase (TPMT), which catalyses a key step in azathioprine metabolism, illustrates this concept well. Three common single nucleotide polymorphisms (SNPs) [42, 43] associated with the loss of enzyme function have been identified and linked to the risk of bone marrow suppression due to excess accumulation of biologically active thioguanine nucleotides. The determination of TPMT status (either by measuring enzyme activity directly, or less commonly by genotyping) is now accepted best practice in dermatology and meets several of the criteria necessary for a pharmacogenetic test to be clinically valuable. First, it allows the identification of a small subset of people in whom a drug is absolutely contraindicated due to the risk of developing a severe adverse reaction – one in 300 individuals are homozygous for loss-of-function TPMT gene variants and show complete absence of TPMT enzyme activity with consequent predictable, potentially fatal, bone marrow aplasia on exposure to azathioprine. Second, it identifies a further subset of patients who may benefit from a drug but who might, without prior dose adjustment, have had to discontinue it – 10% of the population carry one loss-of-function TPMT gene variant and although at risk of bone marrow suppression with standard dosing, tolerate azathioprine if the dose is reduced. Third, TPMT testing is widely available and results are easy to interpret [44]. It therefore provides additional, clinically relevant information that directly impacts on prescribing and ultimately patient outcome. Even so, since TPMT loss-of-function allelic variants account for less than 30% of all adverse reactions associated with azathioprine, additional mechanisms involving other polymorphic drug-metabolizing enzymes are likely to operate.
Polymorphisms in genes encoding drug transporters also influence drug response. Methotrexate toxicity, for example, is associated with specific polymorphisms in SLC19A1 (a member of the solute carrier transporter superfamily) [43] that controls the influx of methotrexate into cells, whilst variants in efflux transporters ABCC1 and ABCG2 (members of the second major drug transporter superfamily, ATP-binding cassette transporters) associate with methotrexate efficacy [45]. Variants in the ABCB1 gene (also known as the multidrug resistance 1 gene, MDR-1, since it was originally identified as a factor underlying anticancer-drug-resistant tumour cell lines) can influence the pharmacokinetics of many drugs, including calcineurin inhibitors. This is because it encodes P-glycoprotein, a transporter that limits intestinal absorption and facilitates biliary excretion of lipophilic drugs [46].
Much pharmacogenetic research has focused on genetic factors underlying differences in drug pharmacokinetics, and this has been largely driven by prior detailed knowledge about drug metabolic pathways. Genetic variation in drug targets also plays a major role in determining drug response; for example, vitamin K epoxide reductase (VKORC1), the target for warfarin, where common SNPs are associated with a lower dose requirement and shorter time to optimal international normalized ratio (INR).
Genes may also influence drug response through involvement in the underlying disease process. Certain HLA class I alleles appear to confer drug (and ethnic) specific risk of severe cutaneous reactions, possibly by functioning to present specific drugs or drug metabolites for HLA-restricted T-cell activation. HLA-B*5701 is strongly associated with hypersensitivity reactions to abacavir, which ordinarily occur in up to 7% of patients treated. Avoidance of abacavir in (white) individuals who carry the HLA-B*5701 allele reduces the risk of hypersensitivity by 50% and pre-testing is now part of routine practice [47].
Drug development and licensing procedures
The process of developing a drug that meets statutory efficacy and safety requirements for use in patients is long, complex and extremely costly (Figure 14.3). Very substantial investment in drug research and development over the past few decades, together with the genomics era and technological advances, have brought many new and ‘game changing’ interventions.
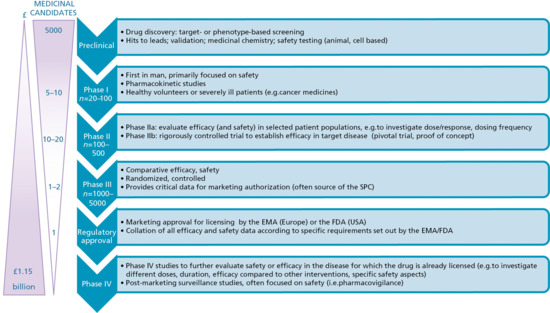
Figure 14.3 An overview of the phases of drug development from discovery through to licensing and post-marketing surveillance (phase IV). Toxicity, failed efficacy and commercial reasons contribute to the slow attrition in numbers of promising candidate drugs that successfully progress along this pathway. The parallel, almost exponential, increase in the cost of delivering each phase of drug development means that for each drug that reaches the market, the investment is substantial. EMA, European Medicines Agency; FDA, US Food and Drug Administration; SPC, summary of product characteristics.
Pre-clinical drug identification
The two principal approaches used to identify potential candidate medicines are phenotypic screening and target-based screening [48]. Phenotypic screening investigates the effects, or phenotypes, that a compound induces in cells, tissues or whole organisms and used to be the mainstay of new drug identification. With advances in molecular biology and genomics, phenotypic screens have been largely replaced with a target-based approach. This requires a detailed understanding of the molecular basis of a particular disease or pathogenic pathway. Potential candidate medicines are then screened for their ability to alter a specific biologically critical ‘target’ within that pathway (usually a protein). Structure-based techniques such as X-ray crystallography and computational modelling (virtual screening) enable precise delineation of the target. Libraries of ‘small-molecule’ compounds that interact with these targets can be rapidly screened using high throughput technology, and subsequently tailored to optimize target binding. The development of biological therapy similarly employs a ‘target-based’ approach, modulating endogenous pathways known to be pathogenically relevant. The two approaches can be very effectively combined, capitalizing on the benefit of knowing that candidate molecules identified in a phenotypic screen show pharmacological activity in a complex, biologically relevant system, and then using all the -omics technology to identify the underlying molecular mechanism of action and tailor the drug accordingly.
Drug development
Once candidate drugs are identified, further optimization and testing is completed in a variety of in vitro, cell, organotypic and animal models. Animal testing is kept to an absolute minimum (as mandated by the recent European ‘3 Rs’ directive: reduce, replace and refine [49]) but remains an essential component of drug development prior to first human trials in order to establish drug pharmacokinetics and safety (e.g. genotoxicity, mutatagenicity, unexpected toxicity). Phase I trials investigate the use of the drug in humans for the first time (Figure 14.3). These studies involve a small number of healthy volunteers or, particularly in cancer medicine, severely ill patients with poor prognosis. Because the drug effects can be unpredictable and occasionally catastrophic [50], these studies are always completed in a highly supervised, clinical research environment. Phase I studies intensively assess safety and pharmacokinetics, often in a staggered programme of drug exposure that starts with minimal drug exposure, up-titrating to a modelled or predicted therapeutic dose range, with the trial only progressing through each stage of escalated dosing if all safety parameters are met. If the phase I studies reveal no untoward findings and are consistent with the known pharmacology of the drug, phase II trials proceed in the target disease with the aim of establishing drug efficacy. Phase III trials follow, with at least two, large, randomized, comparative trials generally required for regulatory approval.
Application for regulatory approval is made to the European Medicines Agency (EMA) and/or the US Food and Drug Administration (FDA) on submission of a detailed dossier of requisite information. This includes all pre-clinical testing, data from the phase I–III trials, technical information on drug manufacturing processes and plans for post-marketing pharmacoviligance (see also the section ‘Drug toxicity and adverse effects’). Drugs assigned ‘orphan’ status are those indicated for rare diseases (‘orphan’ diseases) where the feasibility of large-scale trials as well as cost (in terms of likely market return) is likely to be prohibitive. Here regulatory requirements are adjusted to encourage industry investment (e.g. early access and marketing of drugs prior to the completion of formal approval) in order to maintain focus and investment on what are often very high-need groups.
Ethics and trial reporting
An internationally agreed ethical and scientific quality standard has been developed for the design, conduct, recording and reporting of drug intervention trials involving human subjects – the International Conference on Harmonisation Good Clinical Practice Guideline (ICH-GCP). This aims to ensure that the rights, safety and well-being of trial subjects are protected, consistent with the principles that have their origin in the Declaration of Helsinki, that the clinical trial data are credible, and to facilitate mutual acceptance of clinical data by the different regulatory authorities. All trials conducted on human subjects are mandated to adhere to ICH-GCP standards. The reporting of trials and trial results has been subject to much debate, with concern over lack of transparency and significant reporting bias [51, 52] given what is published and in the public domain when compared with the large tracts of trial data that remain either with regulators or unpublished within industry [53]. Collectively, this all contributes to clinicians having an incomplete picture of a drug's efficacy and safety, and thus limits their ability to achieve optimal outcomes. A number of strategies are being put in place to improve this situation. First, investigators are strongly encouraged to register trials on publically available databases such as ClinicalTrials.gov with protocol details, recruitment progress and eventually results. This strategy has been strongly endorsed by the International Committee of Medical Journal Editors who agreed to make this a requirement for journal publication. Compliance remains poor though with, for example, only 46% of 677 trials listed as completed by 2007 on ClinicalTrials.gov published in a peer-reviewed Medline-listed biomedical journal within 30 months of trial completion [54]. Second, standards have been developed to improve the quality of trial reporting by the EQUATOR Network and many journals subscribe to adherence to these standards for trial reports submitted for publication. Third, the EMA, as well as a number of large pharmaceutical companies, have agreed to the release of previously unpublished data.
Resources
Further information
EQUATOR (Enhancing the Quality and Transparency of Health Research) Network: http://www.equator-network.org/ (last accessed January 2015).
References
- Pirmohamed M, James S, Meakin S, et al. Adverse drug reactions as cause of admission to hospital: prospective analysis of 18 820 patients. BMJ 2004;329(7456):15–9.
- Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA 1998;279(15):1200–5.
- Howard RL, Avery AJ, Slavenburg S, et al. Which drugs cause preventable admissions to hospital? A systematic review. Br J Clin Pharmacol 2007;63(2):136–47.
- Leendertse AJ, Egberts AG, Stoker LJ, van den Bemt PA, Group HS. Frequency of and risk factors for preventable medication-related hospital admissions in the netherlands. Arch Intern Med 2008;168(17):1890–6.
- Co-ordination Group for Mutual Recognition and Decentralised Procedures – Human. CMDh Questions and Answers: Biologicals. 2012. http://www.hma.eu/fileadmin/dateien/Human_Medicines/CMD_h_/Questions_Answers/CMDh_269_2012_Rev0_2012_10.pdf (last accessed January 2015).
- Buxton ILO, Beret LZ. Pharmacokinetics: the dynamics of drug absorption, distribution, metabolism and elimination. In: Brunton LL, Chabner BA, Knollman BC, eds. Goodman and Gilman's The Pharmacological Basis of Therapeutics, 12th edn. New York: McGraw Hill Medical, 2011:17–40.
- Tang L, Persky AM, Hochhaus G, Meibohm B. Pharmacokinetic aspects of biotechnology products. J Pharm Sci 2004;93(9):2184–204.
- Vugmeyster Y, Xu X, Theil FP, Khawli LA, Leach MW. Pharmacokinetics and toxicology of therapeutic proteins: advances and challenges. World J Biol Chem 2012;3(4):73–92.
- Dostalek M, Gardner I, Gurbaxani B, Rose R, Chetty M. Pharmacokinetics, pharmacodynamics and physiologically-based pharmacokinetic modelling of monoclonal antibodies. Clin Pharmacokinet 2013;52(2):83–124.
- Wang W, Wang EQ, Balthasar JP. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther 2008;84(5):548–58.
- Prausnitz MR, Langer R. Transdermal drug delivery. Nature Biotech 2008;26:1261–9.
- Vlieghe P, Lisowski V, Martinez J, Khrestchatisky M. Synthetic therapeutic peptides: science and market. Drug Discov Today 2010;15(1–2):40–56.
- Li X, Yu M, Fan W, Gan Y, Hovgaard L, Yang M. Orally active-targeted drug delivery systems for proteins and peptides. Expert Opin Drug Deliv 11(9):1435–47.
- Mager DE. Target-mediated drug disposition and dynamics. Biochem Pharmacol 2006;72(1):1–10.
- Tabrizi MA, Tseng C-ML, Roskos LK. Elimination mechanisms of therapeutic monoclonal antibodies. Drug Discov Today 2006;11(1–2):81–8.
- Blumenthal DK, Garrison JC. Pharmacodynamics: molecular mechanisms of drug action. In: Brunton LL, Chabner BA, Knollman BC, eds. Goodman and Gilman's The Pharmacological Basis of Therapeutics, 12th edn. New York: McGraw Hill Medicine, 2011:41–72.
- Kearns GL, Abdel-Rahman SM, Alander SW, Blowey DL, Leeder JS, Kauffman RE. Developmental pharmacology – drug disposition, action, and therapy in infants and children. N Eng J Med 2003;349(12):1157–67.
- Klotz U. Pharmacokinetics and drug metabolism in the elderly. Drug Metab Rev 2009;41(2):67–76.
- McLean AJ, Le Couteur DG. Aging biology and geriatric clinical pharmacology. Pharmacol Rev 2004;56(2):163–84.
- Gurwitz JH, Field TS, Harrold LR, et al. Incidence and preventability of adverse drug events among older persons in the ambulatory setting. JAMA 2003;289(9):1107–16.
- Zhang M, Holman CDAJ, Price SD, Sanfilippo FM, Preen DB, Bulsara MK. Comorbidity and repeat admission to hospital for adverse drug reactions in older adults: retrospective cohort study. BMJ 2009;338:a2752.
- Adams J, Lammer EJ. Neurobehavioral teratology of isotretinoin. Reprod Toxicol 1993;7(2):175–7.
- Chi C, Wang S, Mayon-White R, Wojnarowska F. Pregnancy outcomes after maternal exposure to topical corticosteroids: a UK population-based cohort study. JAMA Dermatol 2013;149(11):1274–80.
- Ke AB, Rostami-Hodjegan A, Zhao P, Unadkat JD. Pharmacometrics in pregnancy: an unmet need. Ann Rev Pharmacol Toxicol 2014;54(1):53–69.
- Hyrich KL, Verstappen SMM. Biologic therapies and pregnancy: the story so far. Rheumatology 2014;53(8):1377–85.
- Nørgård B, Pedersen L, Jacobsen J, Rasmussen SN, Sørensen HT. The risk of congenital abnormalities in children fathered by men treated with azathioprine or mercaptopurine before conception. Aliment Pharmacol Ther 2004;19(6):679–85.
- Weber-Schoendorfer C, Hoeltzenbein M, Wacker E, Meister R, Schaefer C. No evidence for an increased risk of adverse pregnancy outcome after paternal low-dose methotrexate: an observational cohort study. Rheumatology 2014;53(4):757–63.
- Huang S-M, Strong JM, Zhang L, Reynolds KS, Nallani S, Temple R, et al. New era in drug interaction evaluation: us food and drug administration update on cyp enzymes, transporters, and the guidance process. J Clin Pharmacol 2008;48(6):662–70.
- Gorski JC, Huang S-M, Pinto A, et al. The effect of echinacea (Echinacea purpurea root) on cytochrome p450 activity in vivo. Clin Pharmacol Ther 2004;75(1):89–100.
- Tateishi T, Miura M, Suzuki T, Uno T. The different effects of itraconazole on the pharmacokinetics of fexofenadine enantiomers. Br J Clin Pharmacol 2008;65(5):693–700.
- Huang SM, Zhao H, Lee JI, et al. Therapeutic protein–drug interactions and implications for drug development. Clin Pharmacol Ther 2010;87(4):497–503.
- National Institute for Health and Care Excellence. Medicines Adherence: Involving Patients in Decisions about Prescribed Medicines and Supporting Adherence. 2009. http://guidance.nice.org.uk/CG76/NiceGuidance/pdf/English (last accessed January 2015).
- Haynes RB, Ackloo E, Sahota N, McDonald HP, Yao, X. Interventions for enhancing medication adherence. Cochrane Database Syst Rev 2008;Issue 2:CD000011.
- Thomsen LA, Winterstein AG, Sondergaard B, Haugbolle LS, Melander A. Systematic review of the incidence and characteristics of preventable adverse drug events in ambulatory care. Ann Pharmacother 2007;41(9):15.
- Dean B, Schachter M, Vincent C, Barber N. Causes of prescribing errors in hospital inpatients: a prospective study. Lancet 2002;359(9315):1373–8.
- Van Doormaal JE, van den Bemt PMLA, Mol PGM, et al. Medication errors: the impact of prescribing and transcribing errors on preventable harm in hospitalised patients. Qual Safety Health Care 2009;18(1):22–7.
- Rinke ML, Bundy DG, Velasquez CA, et al. Interventions to reduce pediatric medication errors: a systematic review. Pediatrics 2014;134(2):338–60.
- Nuckols TK, Smith-Spangler C, Morton SC, et al. The effectiveness of computerized order entry at reducing preventable adverse drug events and medication errors in hospital settings: a systematic review and meta-analysis. Syst Rev 2014;3(56):12.
- Pirmohamed M. Personalized pharmacogenomics: predicting efficacy and adverse drug reactions. Annu Rev Genomics Hum Genet 2014;15(1):349–70.
- Cavallari LH, Nutescu EA. Warfarin pharmacogenetics: to genotype or not to genotype, that is the question. Clin Pharmacol Ther 2014;96(1):22–4.
- Tai HL, Krynetski EY, Yates CR, et al. Thiopurine S-methyltransferase deficiency: two nucleotide transitions define the most prevalent mutant allele associated with loss of catalytic activity in Caucasians. Am J Hum Genet 1996;58(4):8.
- Yates CR, Krynetski EY, Loennechen T, et al. Molecular diagnosis of thiopurine S-methyltransferase deficiency: genetic basis for azathioprine and mercaptopurine intolerance. Ann Intern Med 1997;126(8):608–14.
- Fargher EA, Tricker K, Newman W, et al. Current use of pharmacogenetic testing: a national survey of thiopurine methyltransferase testing prior to azathioprine prescription. J Clin Pharm Ther 2007;32(2):187–95.
- Warren RB, Smith RL, Campalani E, et al. Genetic variation in efflux transporters influences outcome to methotrexate therapy in patients with psoriasis. J Invest Dermatol 2008;128(8):1925–9.
- Barbarino JM, Staatz CE, Venkataramanan R, Klein TE, Altman RB. PharmGKB summary: cyclosporine and tacrolimus pathways. Pharmacogenet Genom 2013;23(10):563–85.
- Mallal S, Phillips E, Carosi G, et al. HLA-B*5701 screening for hypersensitivity to abacavir. N Eng J Med 2008;358(6):568–79.
- Swinney DC, Anthony J. How were new medicines discovered? Nat Rev Drug Discov 2011;10(7):507–19.
- Directive 2010/63/EU of the European Parliament and of the Council. On the Protection of Animals used for Scientific Purposes, 22 September 2010. http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2010:276:0033:0079:en:PDF (last accessed January 2015).
- Suntharalingam G, Perry MR, Ward S, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Eng J Med 2006;355(10): 1018–28.
- Bourgeois FT, Murthy S, Mandl KD. Outcome reporting among drug trials registered in ClinicalTrials.gov. Ann Intern Med 2010;153(3):158–66.
- Goldacre B, Heneghan C. Improving, and auditing, access to clinical trial results. BMJ 2014;348:g213.
- Saito H, Gill CJ. How frequently do the results from completed US clinical trials enter the public domain? – A statistical analysis of the ClinicalTrials.gov database. PLOS One 2014;9(7):e101826.
- Ross JS, Tse T, Zarin DA, Xu H, Zhou L, Krumholz HM. Publication of NIH funded trials registered in ClinicalTrials.gov: cross sectional analysis. BMJ 2012;344:d7292.