CHAPTER 19
Principles of Systemic Therapy
Michael J. Tidman1 and Catherine H. Smith2
1Department of Dermatology, Royal Infirmary of Edinburgh, Edinburgh, UK
2St John's Institute of Dermatology, Guy's and St Thomas' NHS Foundation Trust, London, UK
Introduction
General aspects
Drugs can target the skin by either topical or systemic routes, with intralesional administration an additional option for very localized conditions. Whilst topical application is often a very effective therapeutic modality for dermatological disease, drugs with physical characteristics that enable them to be absorbed through the skin may not always be sufficiently efficacious and will be ineffective beyond the limit of passive diffusion from the skin surface. Conversely, systemic agents distributed via the cutaneous vasculature have the potential to exert their pharmacological actions on all elements of the skin.
However, the systemic route also determines that not only the skin but also most of the other bodily organs are exposed to the drug, and many of the systemic therapies used in dermatology have the potential for significant adverse effects, sometimes life threatening. One of the principal precepts of medical ethics is embodied in the Latin aphorism primum non nocere, meaning ‘first, do no harm’, a reminder to the clinician always to consider the possibility of unwanted and perhaps dangerous consequences of a therapeutic intervention. The Yellow Card Scheme in the UK is a pharmacovigilance system that has involved collecting information from health professionals and the general public on suspected side effects of medications for more than four decades. It is worth noting that the Yellow Card data show that a number of the widely used dermatological therapies have been associated with fatalities: for instance, methotrexate (MTX) has been considered a significant contributing factor in six deaths per annum in the UK, on average, although not necessarily in a dermatological context, and the corresponding figures for azathioprine (AZA) and ciclosporin are three and five, respectively [1].
Whilst it behoves the clinician to use systemic medications as safely as possible [2], it is also important to appreciate the patient's perspective, and to assess the detrimental impact of a skin disorder on a patient's quality of life, when assessing the risk–benefit balance of a particular drug, and the clinician is well advised always to have the patient's best interests at heart.
Systemic dermatological medication is treatment and is rarely a cure. Furthermore, it is not always desirable to strive officiously for complete clearance of a dermatological condition as to do so may require excessive doses of a systemic drug. The ideal may be a combination of systemic and topical therapy. For these reasons, clinician and patient should try to establish mutually agreed achievable therapeutic goals.
Risk reduction
It is a fundamental truism of clinical practice that all clinicians treating medical disorders with potent systemic agents will inevitably, sooner or later, encounter significant adverse events in their patients. Drug side effects can be divided into those that are predictable due to known toxic properties of the drug, and those that are idiosyncratic and therefore unpredictable. It is largely possible for the clinician to take measures to minimize the likelihood of serious predictable complications, but even the most experienced and attentive clinician will still be subject to the capriciousness of drug reactions, and this should be clearly explained to the patient. Notwithstanding, any severe drug-related incident should prompt reflective analysis by the clinician, to the possible benefit of future patients.
Standards of care
There is no perfect medical risk management plan, but the astute clinician will consider following peer-determined standards of care, such as those contained in evidence-based guidelines produced by reputable organizations, including the British Association of Dermatologists, the American Academy of Dermatology and the National Institute for Health and Care Excellence (NICE), local policies and protocols on prescribing, as well as heeding the recommendations of individual pharmaceutical companies for individual drugs, an example being the pregnancy prevention plan and iPLEDGE for oral isotretinoin.
Patient selection
The question of whether systemic medication is justified for a particular individual should always be asked. The risks to a patient of active treatment should be balanced with the risks of no treatment in terms of disease progression (risk–risk assessment). Weighing the potential disadvantages against the potential advantages of a specific drug (risk–benefit assessment) can sometimes be very difficult: for instance, the person may have co-morbid conditions that might influence the effectiveness of the drug or its potential for causing side effects (e.g. postmenopausal women, smokers and those who are physically inactive being more at risk of osteoporosis from corticosteroid therapy, and alcohol dependency increasing the risks of liver damage by hepatotoxic drugs); there may be a paucity of evidence regarding the therapeutic benefits of systemic drug therapy for the particular skin disorder; and there may be doubts as to whether the severity of the condition warrants potent drug therapy. Properly validated quality-of-life assessment and clinical severity tools can be helpful in making therapeutic decisions.
Also to be factored in are assessments of whether the patient and their relatives fully understand the issues relating to systemic drug therapy and whether they will adhere to advice. Comprehension can be addressed by appropriate education, but the concept of compliance exposes the clinician to the vagaries of human nature.
In coming to a decision about systemic therapy, two or more heads may be better than one, and seeking the opinion of trusted colleagues can be very beneficial to patient care.
Patient education
Active participation of the patient and sometimes their relatives and the family doctor (the ‘therapeutic partnership’) in the decision-making process of treatment choice is to be encouraged as a means of enabling informed consent and reducing the risk of dissatisfaction with the outcome. This necessitates careful patient education. Verbal explanation, by physician or specialist nurse, may be entirely satisfactory, but most patients will welcome written information about a drug that should be in a form appropriate to a lay person. For instance, the patient information leaflets available from the British Association of Dermatologists represent a consensus view of clinicians, are produced by a process that has been certified for quality and accuracy, and, furthermore, are monitored by a lay panel for comprehensibility and readability. Time to consider the options, and the opportunity to ask questions, is often best resolved by offering a separate consultation. Do not assume patient knowledge.
Once a decision on treatment has been made, patients should be directed to read the package insert that comes with the drug, advised of the basics of when and how to take the medicine, the warning symptoms of severe side effects explained, and the importance of monitoring tests stressed. They should be instructed how to seek medical advice, and in this respect secretaries, nurses and ancillary staff, by facilitating access to the clinician, are included in the therapeutic partnership.
In the event of an emergency, patients on a systemic medication, despite being knowledgeable about their treatment, may not be able to impart that information to their medical attendants, exposing them to the risk of suboptimal care. For instance, the provision of a steroid card for someone on systemic corticosteroid therapy could reduce the risk of adrenal insufficiency caused by medication being stopped abruptly. Advising a patient to wear a MedicAlert® bracelet or necklace with appropriate medical information inscribed onto it may also be considered.
Pre-treatment screening and monitoring tests
In order to minimize the predictable risks of systemic drug therapy, it is prudent to undertake appropriate baseline investigations, haematological and otherwise, and to carefully monitor necessary parameters during the course of treatment. There are few systemic dermatological treatments that do not require some form of pre-treatment screening and subsequent monitoring.
Drug interactions
Clinicians are strongly advised, when they recommend a systemic treatment, to exclude potential interactions with the patient's existing medication, to provide the patient with a list of agents that might interact with the new drug and to ensure the patient knows to make the prescribers of any future medication aware of their current medicines. In this context, the hospital pharmacist, utilizing the local drug information service, also becomes part of the therapeutic partnership.
Record-keeping
Good clinical documentation is essential for risk management. The principle ‘What's not recorded didn't happen!’ may appear less than fair, but it is likely to be the tenet against which a clinician will be judged. Well-maintained records are the best defence against unwarranted criticism: failure to keep adequate documentation can be interpreted as falling short of expected standards of care. To avoid censure, it is recommended that records are made of discussions with patients regarding medications, paying particular attention to possible adverse effects and the provision of educational material. Hand-written notes in the patient record should be legible and attributable, but documentation can include tick boxes and formal letters to the primary care physician. Copy letters sent to the patient can be helpful in identifying possible misconceptions and also serve to keep them fully informed.
It is also advisable to keep a register of patients who are on high-risk therapies, so as to identify more readily clinic non-attenders who may therefore be receiving suboptimal monitoring of their therapy.
Basic pharmacology of systemic therapy
Dermatological drugs are usually administered by oral, subcutaneous, intramuscular or intravenous routes, and it is the pharmacokinetic properties of a drug that determine its absorption into the circulation, its distribution throughout the body, its bioavailability at the intended site of action, its metabolism and its excretion. It is the pharmacodynamic properties of a drug that determine its pharmacological activity in producing the desired (and unwanted) effects. Furthermore, differences in the genes encoding transportation mechanisms, targets and metabolizing enzymes may result in variations in the pharmacokinetic and pharmacodynamic aspects of a drug, and thus in its efficacy and toxicity: such pharmacogenetic factors may go some considerable way towards explaining individual differences in therapeutic response and adverse reactions to particular agents. For instance, screening for genetic polymorphisms that affect the activities of the enzymes thiopurine methyltransferase and glucose-6-phosphate dehydrogenase, predisposing to adverse haematological effects from AZA and dapsone respectively, is currently routine, and it is very likely that future testing for the wide variety of polymorphisms that have a clinically significant influence on drug efficacy and side effects will become widely available and desirable, raising the eventual prospect of a personalized dosing strategy being tailored to an individual's genotype.
Whilst every effort has been made in this chapter to provide the clinician with sufficient knowledge of the pharmacology, potential adverse effects, interactions and monitoring requirements of the most used systemic dermatological therapies, to enable the employment of these drugs to their maximum benefit and minimum detriment in the treatment of cutaneous conditions, the authors make no claim to complete comprehensiveness. Furthermore, in recognizing that practices may vary between individual dermatologists depending on personal experience, local factors and international differences, our suggestions for drug usage should not be considered prescriptive.
Immunomodulatory drugs
The majority of the systemic drugs utilized for dermatological conditions are immunomodulatory, and a number exert potent immunosuppressive effects. Such immunosuppressive drugs demand particular pre-treatment screening (summarized in Box 19.1) and subsequent monitoring.
Prior to initiating treatment with immunosuppressive agents, patients should be carefully counselled about the risk/benefit ratio, with particular regard to infection, systemic and cutaneous malignancy, bone marrow suppression and conception-related issues. Written information should be provided. When appropriate, women should be given contraceptive guidance, and pregnancy excluded before treatment is initiated. Their cervical cytology screening history should be established. A history of malignancy in any organ system should prompt the seeking of appropriate specialist advice. The entire skin should be examined to exclude the presence of dysplastic or neoplastic lesions, and sun protection measures should be discussed.
In order to minimize the risk of reactivation of dormant infections, which can be severe in the context of immunosuppression, consider screening for latent blood-borne viruses (e.g. hepatitis B and C, and HIV), latent tuberculosis, and varicella zoster virus immune status in the absence of a definite history of chickenpox infection. Seek specialist input in the event of positive findings. Patients should have annual influenza vaccination, and be vaccinated against varicella, in those who are seronegative, and Pneumococcus, and consideration given to hepatitis B vaccination. The varicella vaccine should be given at least several weeks prior to starting an immunosuppressive agent as it is a live vaccine.
Periodically, at follow-up appointments, the opportunity should be taken to reiterate the potential adverse effects of immunosuppressive agents, remind patients to adhere to sun protection measures and encourage the re-reading of the information leaflet.
Occasional, perhaps annual, general physical examination should be undertaken with a view to excluding lymphoma and cutaneous neoplasia.
Antihistamines
Histamine is synthesized by and stored in mast cells and basophils, together with a variety of other pro-inflammatory mediators, and its release results in inflammation, which contributes to a variety of cutaneous disorders, particularly urticaria. Histamine receptors are expressed in skin on endothelial cells, neurons and T lymphocytes. Specifically, histamine-induced itch, vasodilatation and increased vascular permeability are mediated by H1 receptors: H2 receptors do not participate in the aetiology of itch [1], but contribute to vasodilatation and vascular permeability. The role of H3 and H4 receptors in cutaneous inflammation remains to be fully defined. In theory, supressing the inflammatory actions of histamine may result from inhibiting its synthesis in mast cells, reducing its release (e.g. with ketotifen, a mast cell stabilizer) and accelerating its degradation in tissues, but in clinical practice the only effective way of minimizing histamine-mediated inflammation is by the use of antihistamines, primarily those that act on H1 receptors [1].
The first generation of antihistamines (represented by alimemazine, chlorphenamine, clemastine, cyproheptadine, hydroxyzine and promethazine) are able easily to cross the blood–brain barrier, interfering with the neurotransmitter function of histamine, thus causing sedation and impairing cognitive function. The second-generation antihistamines (such as acrivastine, bilastine, cetirizine, levocetirizine, loratadine, desloratadine, fexofenadine, mizolastine and rupatadine) are, in contrast, minimally sedating [2].
Dermatological uses (see Chapters 42, 44 and 46)
H1 antihistamines are the mainstay of treatment for chronic urticaria and angio-oedema, and may be symptomatically beneficial in the physical urticarias, urticarial vasculitis, cutaneous mastocytosis, insect bite reactions, anaphylaxis and allergic reactions to drugs. Their effectiveness in atopic eczema remains to be established, but the sedating H1 antihistamines may have a role in the management of nocturnal pruritus in atopic eczema [1]. The combination of H1 and H2 antihistamines (used off-licence) has been advocated for the treatment of urticaria, although the quality of evidence is low [3, 4]. H2 receptor-mediated down-regulation of T-lymphocyte activation is the rationale for the treatment of chronic mucocutaneous candidiasis and human papillomavirus infection with H2 antihistamines [1].
Pharmacological properties
Formula and structure
H1 antihistamines differ markedly in structure, with six structural classes: alkylamines, ethanolamines, ethylenediamines, phenothiazines, piperidines and piperazines [5]. The first generation of antihistamines have representatives in each structural group, but the majority of second-generation antihistamines are piperidines and piperazines. Doxepin is a tricyclic antidepressant with antihistamine activity.
Pharmacokinetics
Just as H1 antihistamines differ in structure so they differ in their pharmacokinetic characteristics. In general, they are readily absorbed from the gastrointestinal tract, reaching peak plasma levels within 2–3 h, and are substantially protein bound [1]. The lipophilicity of the first-generation antihistamines determines their ability to cross the blood–brain barrier, in contrast to the second-generation antihistamines. The first-generation drugs are metabolized by the hepatic cytochrome P450 system, usually followed by predominantly renal excretion. A number of the second-generation antihistamines, some of which are derived from a prodrug or are active metabolites of other antihistamines, may be excreted unchanged into the gut (fexofenadine) or urine (cetirizine and levocetirizine).
Pharmacodynamics
Traditionally, antihistamines have been thought of as reversible competitive inhibitors of histamine. However, molecular characterization has suggested that histamine receptors have an intrinsic level of activity, and that H1 and H2 antihistamines are now best regarded as inverse agonists, not just blocking the interaction of histamine with its receptors but inducing an opposite pharmacological response by decreasing the constitutive activity of the receptors [1, 6].
Additionally, it is thought possible that H1 antihistamines, especially the second-generation compounds, may have H1 receptor-independent anti-inflammatory effects, including inhibiting the release of histamine from mast cells and basophils, inhibiting the tissue activation and accumulation of inflammatory cells such as eosinophils, and directly inhibiting pro-inflammatory mediators such as bradykinin [6]. However, the experimental data are inconclusive and the potential clinical relevance is questionable [6].
The first-generation antihistamines, in contrast to the highly specific actions of the second generation, generally tend to have a high affinity for muscarinic cholinergic receptors, thereby inducing anticholinergic side effects [1]. Cyproheptadine is thought to have additional anti-serotonin activity and to interfere with hypothalamic function [1]. The phenothiazine H1 antihistamines have α-adrenergic blocking activity, which may cause hypotension [5].
Potential adverse effects
The first-generation antihistamines tend to cause sedation, impaired cognitive function, paradoxical excitability (especially in children and the elderly when given in high doses), anticholinergic effects (blurred vision, dry mouth, micturition difficulties and constipation) and weight gain, whereas the second-generation antihistamines are relatively free from such adverse effects and have a high therapeutic index. Rarely, antihistamines may cause headaches, hypotension, palpitations, arrhythmias, sleep disturbances, dizziness, confusion, extrapyramidal effects, tremor, convulsions, depression and hypersensitivity reactions [2]. The cardiotoxic antihistamines terfenadine and astemizole are no longer prescribable.
Contraindications
Antihistamines are contraindicated in those rare instances of hypersensitivity to them [7].
Cautions
Although the available data do not suggest that H1 antihistamines pose a significant risk to fetal well-being, it is generally recommended that they be avoided during pregnancy and lactation unless there is a clearly favourable benefit/risk ratio [2]. Chlorphenamine is generally acknowledged to be safe during pregnancy [1].
The first-generation antihistamines in particular, because of their sedating and anticholinergic actions, should be used with caution in patients with epilepsy, glaucoma and prostatic hypertrophy [8]: advice should be given regarding concurrent alcohol consumption, driving and operating dangerous machinery in view of the possibility of impaired judgement or dexterity [8].
In general, caution should be exercised if there is severe hepatic or renal impairment, severe hypertension, respiratory problems, porphyria or a predisposition to cardiac arrhythmias [2, 8].
Drug–drug interactions
- CYP3A4 inhibitors, such as macrolide antibiotics, azole antifungal agents, HIV-1 protease inhibitors and grapefruit juice, may inhibit the metabolism and increase the serum levels of those antihistamines that are metabolized in the liver and may cause an increase in adverse events.
- Alcohol, anxiolytic and hypnotic drugs may exacerbate the sedative effects of first-generation antihistamines.
- Antacids reduce the absorption of fexofenadine.
- Antidepressant drugs (tricyclics and monoamine oxidase inhibitors (MAOIs)) may increase the sedative and antimuscarinic effects of antihistamines.
Dose and regimens
If the recommended dose of individual antihistamines [2, 8] is not clinically effective, it is common clinical practice in the case of second-generation antihistamines to prescribe higher doses, although there is a limited evidence base for the efficacy and safety of this [1]. Furthermore, the combination of two or more antihistamines can be more effective than monotherapy [3].
Antimalarial agents
Quinine, a natural anti-inflammatory alkaloid derived from the bark of the South American Cinchona tree, has medicinal properties that have been utilized since the 17th century. It was the treatment of choice for malaria until synthetic derivatives with fewer side effects were developed in the 1930s and 1940s. Of these, mepacrine (known in the US as quinacrine), chloroquine and hydroxychloroquine have been employed as anti-inflammatory agents in various clinical settings, primarily dermatological and rheumatological. Mepacrine is now relatively little used, and in the UK is available only from ‘special-order’ manufacturers or specialist importing companies [1], but both chloroquine and the more commonly prescribed hydroxychloroquine have product licences in the UK for chronic cutaneous (discoid) and systemic lupus erythematosus as well as for rheumatoid arthritis. Additionally, the product licence for hydroxychloroquine extends to dermatological conditions caused or aggravated by sunlight [1]. Hydroxychloroquine appears less toxic but also less effective than chloroquine [2, 3].
Dermatological uses (see Chapters 51, 60, 97, 98, 99 and 127)
The dermatological licensed indications include cutaneous forms of lupus erythematosus and photodermatoses (including polymorphic light eruption, solar urticaria and porphyria cutanea tarda), but, off-label, antimalarials are used in granulomatous dermatoses (cutaneous sarcoidosis and granuloma annulare), panniculitides (lupus panniculitis, chronic erythema nodosum, idiopathic panniculitis), and a miscellany of other conditions (including Jessner lymphocytic infiltrate, oral lichen planus, urticarial vasculitis and reticular erythematous mucinosis) [3].
Pharmacological properties
Formula and structure
Hydroxychloroquine (empirical formula: C18H26ClN3O, systematic name: (RS)-2-[{4-[(7-chloroquinolin-4-yl)amino]pentyl}(ethyl)amino]ethanol) is a substituted 4-aminoquinoline, differing from chloroquine only by the presence of a hydroxyl group in a β position at the end of the ethyl side chain.
Mepacrine (empirical formula: C23H30ClN3O, systematic name: (RS)-N′-(6-chloro-2-methoxy-acridin-9-yl)- N,N-diethyl-pentane-1,4-diamine) differs from the 4-aminoquinolines in having a third benzene ring.
Administration
Hydroxychloroqine sulphate, chloroquine phosphate or sulphate and mepacrine hydrochloride are administered orally, in tablet form [1].
Pharmacokinetics
The pharmacokinetic characteristics of the antimalarials are complex, and there is great variability between individuals. Hydroxychloroquine and chloroquine are water soluble and readily absorbed from the gastrointestinal tract, with peak plasma concentration within 8–12 h and approximately 60% binding to plasma proteins [3]. They are widely distributed to the tissues, and accumulate in the liver, spleen, kidney, lung, leucocytes and within melanin-containing cells of the skin and retina [3]. The 4-aminoquinolines are metabolized in the liver by enzymes of the cytochrome P450 (CYP) group (CYP2D6, -2C8, -3A4 and -3A5) to active metabolites, hydroxychloroquine into N-desethylhydroxychloroquine, and chloroquine into desethylchloroquine and bisdesethylchloroquine [3]. Excretion is largely in the urine, but also in the bile, sweat and saliva. Elimination is slow, because of tissue accumulation, with a terminal half-life of 30–60 days [3], and maximum clinical efficacy may take up to 3–6 months.
The pharmacokinetics of mepacrine is similar to the 4-aminoquinolines [3].
Pharmacodynamics
Despite recent advances, the mechanisms underlying the immunomodulating and anti-inflammatory properties of the antimalarials are still incompletely understood. The traditional explanation centres on antimalarials, which are lipophilic weak bases, being ‘lysosomotropic’, penetrating cellular membranes and preferentially accumulating in lysosomes [3]. The resultant rise in lysosomal pH results in inhibition of proteases and consequent dysfunctional protein processing. In immunologically competent cells, this causes disruption of protein secretion, receptor recycling and reduced production of pro-inflammatory cytokines and other immunological mediators (such as tumour necrosis factor (TNF)-α, interleukin (IL)-6 and interferon (IFN)-γ). The effects of this include reduced antibody production and cell-mediated cytotoxicity by lymphocytes, reduced natural killer cell activity, impaired antigen presentation by monocytes, macrophages and dendritic cells to CD4 T cells, and decreased chemotaxis, phagocytosis and superoxide production by neutrophils [3, 4], thereby down-regulating immune responsiveness.
More recently, the 4-aminoquinolines have been demonstrated to have a potent and important inhibitory effect on intracellular toll-like receptor (TLR) signalling, which may be their principal mode of action [3]. The lysosomotropic nature of antimalarials appears to prevent the proper functioning of endosomal TLRs, particularly TLR9, primarily within antigen-presenting cells, thereby inhibiting the activation of the innate immune system that would otherwise result from the recognition by TLRs of self nucleic acid components, including immune complexes, found in connective tissue disorders [4].
Antimalarials also exert other complex immunological and anti-inflammatory effects, including inhibition of phospholipase A2, thereby reducing prostaglandin formation, and photoprotection by virtue of being able to absorb UV light [3].
Furthermore, antimalarials have anticoagulant, lipid-lowering and hypoglycaemic properties [3]. Hydroxychloroquine has been demonstrated to restore the binding of annexin A5 (a potent natural antithrombotic agent) to intravascular surfaces in the presence of antiphospholipid antibodies [5], and thus reduce thrombotic events in the antiphospholipid antibody syndrome. Antimalarials have a beneficial effect on cholesterol, triglyceride and low-density lipoprotein (LDL) levels [3, 6], and thus development of atherosclerosis, via effects on cholesterol synthesis and inhibition of a TLR9-mediated accumulation of lipids in macrophages [7]. Antimalarials also influence glucose metabolism, decreasing insulin degradation [3] and reducing the risk of diabetes [4], possibly also involving blockade of TLR9 [8]. The relevance, if any, of these interesting antimalarial properties to cutaneous disorders is as yet unclear.
Pharmacogenetics
The majority of patients respond to hydroxychloroquine and chloroquine, although a significant proportion do not or cannot tolerate them. Little is known about the effect of genetic factors on the pharmacokinetic and pharmacodynamic properties of the 4-aminoquinolines, and whether genotypic differences in the relevant cytochrome P450 enzymes influence their efficacy and toxicity. The available evidence has not demonstrated that CYP polymorphisms influence clinical outcome with hydroxychloroquine [9].
Potential adverse effects (Box 19.2)
Pharmacological
Myelotoxicity
Although very uncommon, all the antimalarials (but particularly chloroquine and mepacrine) can suppress bone marrow function [3] and leukopenia is thus a risk: agranulocytosis and aplastic anaemia have been recorded. Antimalarials may cause haemolysis in glucose-6-phosphate dehydrogenase (G6PD) deficient individuals [3].
Cutaneous toxicity
It is generally considered that antimalarials, especially chloroquine and mepacrine, may result in exacerbation of psoriasis [3], and it is reasonable to warn patients regarding this, although a systematic review has concluded that the evidence for this is equivocal [10]. Mepacrine can give the skin a yellow colour and the 4-aminoquinolines may impart a blue-grey discoloration [11]. Antimalarials have also been associated with bleaching of hair and transverse bands affecting the nails [3]. A miscellany of adverse cutaneous reactions has been reported with antimalarials, including pruritus, erythroderma, urticaria, lichenoid eruptions, photosensitivity and alopecia [12].
Idiosyncratic
Oculotoxicity
Antimalarial-induced retinopathy is an important vision-threatening side effect [13], for which the mechanism is still poorly understood. It appears to be largely idiosyncratic, and individual differences in drug metabolism may eventually prove to be reason for this. The onset of central visual field loss may be insidious, and early detection at a reversible stage difficult [14], even with sophisticated ophthalmic techniques such as multifocal electroretinography and fundus autofluorescence imaging. Once developed, the maculopathy tends to be permanent and even progressive following cessation of antimalarial therapy [14, 15]; there is no effective treatment. If allowed to progress, the typical ‘bull's eye’ macular changes become visible on fundoscopy. The most common symptom is difficulty with reading [15].
The risk of retinal toxicity with hydroxychloroquine is small during the first 5 years of treatment but rises to approximately 1% after 5–7 years of use or a cumulative dose of 1000 g; this risk becomes greater with the continued use of the drug [16, 17]. Retinal damage is significantly more common with chloroquine [18, 19] and the safety parameters are less clear.
Hydroxychloroquine is also deposited in the cornea: it is usually asymptomatic, but may cause visual haloes [3, 19]. Corneal deposits are reversible on stopping the drug and are not a contraindication to continuing treatment [3].
The Royal College of Ophthalmologists in collaboration with the British Association of Dermatologists and the British Society for Rheumatology issued recommendations on screening for hydroxychloroquine-induced retinopathy in 2009 [19]. These suggest baseline and annual enquiry as to reading difficulties and reading performance with a near vision test type. The self-detection of perifoveal visual field defects using an Amsler grid is described but is only implicitly recommended.
The American Academy of Ophthalmology produced revised recommendations on screening for antimalarial-induced retinopathy in 2011 [16], which suggest using multifocal electroretinography, fundus autofluorescence or spectral domain optical coherence tomography in addition to Humphrey 10-2 static perimetry. Baseline assessment is advised, with annual screening commencing after 5 years unless there are risk factors, in which case annual assessment is recommended. Specifically, this guideline does not recommend Amsler grid testing.
Mepacrine is considered to be relatively non-oculotoxic [20].
Neuromuscular toxicity
Rarely, antimalarials can induce myalgia, fatigue and myopathy [21] and they have been associated with headaches, dizziness, tinnitus, hearing loss, nightmares, irritability, seizures and psychosis [12].
Cardiotoxicity
Antimalarial-induced cardiac side effects are rare, especially with hydroxychloroquine, but conduction abnormalities have been reported [22].
Gastrointestinal toxicity
Hydroxychloroquine is the least likely of the antimalarials to cause gastrointestinal symptoms [12], which include nausea, vomiting, diarrhoea, anorexia, heartburn, abdominal distension and elevated transaminases. They tend to be transient [3], although chloroquine and hydroxychloroquine should be used with caution in moderate to severe hepatic impairment [1].
Contraindications
Hypersensitivity reactions to antimalarials are a contraindication to their use. The 4-aminoquinolines and mepacrine do not cross-react, and so an adverse reaction to one does not preclude the use of the other [3].
Should a patient develop retinopathy on a 4-aminoquinoline, its continued use is contraindicated.
Cautions
The 4-aminoquinolines readily cross the placenta and are also excreted into breast milk. Data concerning 4-aminoquinolines during pregnancy and lactation are limited and, although the risk of fetal damage appears low [3], the use of chloroquine and hydroxychloroquine during pregnancy and lactation is not recommended [23].
Antimalarials should be used with caution in patients with a neuromuscular disease or a psychotic condition. Patients with G6PD deficiency should be monitored closely for haemolysis, although this is unlikely at the doses of antimalarial used for the treatment of cutaneous disorders [3]. Antimalarials should be given in low dosage when used to treat porphyria cutanea tarda (PCT) in view of the risk of causing hepatitis. G6PD deficiency and PCT are, however, both considered rare enough that routine testing to exclude them before antimalarial therapy is not necessary [3].
Pre-treatment screening
A baseline full blood count and liver and renal function tests should be undertaken, as should G6PD and porphyria screening in appropriate clinical settings [24].
Patients should be counselled about the risk of visual damage and provided with written information. Guidance produced by the Royal College of Ophthalmologists [19] recommends enquiring about any visual impairment not corrected with spectacles and recording the near visual acuity of each eye, with reading glasses if worn, using a near vision test type. If the patient can read a small print size (N6 or N8), treatment with hydroxychloroquine can proceed. If visual impairment is suspected, assessment by an optometrist should be advised: if any impairment is correctable by refraction, treatment can be started; non-correctable abnormalities will require ophthalmological assessment before proceeding with hydroxychloroquine [19].
Cigarette smoking has been associated with a poor response to hydroxychloroquine, although epidemiological evidence for this is lacking [9] and screening for smoking habit seems unnecessary.
Dose and regimens
The standard dose of hydroxychloroquine for adults is 200–400 mg daily, but not exceeding 6.5 mg/kg/day based on ideal body weight [1]. The dose of chloroquine for adults is up to 2.5 mg/kg/day, based on ideal body weight [1], and for mepacrine from as little as 50 mg three times each week up to 100 mg thrice daily [25].
Although there is only limited published evidence, combination therapy of hydroxychloroquine or chloroquine with mepacrine (quinacrine) may result in an enhanced therapeutic response [26, 27].
Monitoring
The full blood count and liver function tests should be checked monthly for 3 months, and then every 4–6 months whilst on treatment [3].
Visual symptoms should be enquired about, especially any problem with reading.
Reading performance should be checked annually with a near vision test type [19, 28].
Azathioprine
Azathioprine is a potent immunosuppressive, anti-inflammatory and antiproliferative drug that has been used over the last five decades to prevent graft rejection and to treat haematological malignancies and a variety of rheumatological, gastrointestinal, neurological and dermatological inflammatory disorders.
Dermatological uses (see Chapters 30, 41, 51, 53 and 127)
Azathioprine is licensed in the UK for the treatment of systemic lupus erythematosus and dermatomyositis and it has been used off-label for a range of cutaneous conditions, including immunobullous diseases (in particular pemphigus and pemphigoid), atopic eczema and chronic actinic dermatitis. It has also been advocated for use in lichen planus, contact dermatitis, polymorphic light eruption, leukocytoclastic vasculitis, pyoderma gangrenosum, Behçet disease and chronic cutaneous lupus erythematosus [1]. It is often used as an adjunct to other immunosuppressive agents such as prednisolone and may exert a steroid-sparing effect, thereby minimizing the adverse effects associated with the prolonged high-dose use of systemic corticosteroids.
Pharmacological properties
Formula and structure
Azathioprine (empirical formula: C9H7N7O2S, systematic name: 6-[(4-methyl-4-nitro-1H-imidazol-5-yl)sulfanyl]-7H-purine) is a thiopurine with a molecular structure consisting of imidazole and mercaptopurine moieties.
Administration
Azathioprine is available in the UK as oral (25 and 50 mg tablets) and intravenous preparations.
Pharmacokinetics
Azathioprine is readily absorbed from the gastrointestinal tract, the peak plasma concentration occurring 1–2 h after an oral dose, and it is distributed rapidly throughout the body. The plasma half-life is 3–5 h with up to 30% bound to plasma proteins [2].
Azathioprine is a prodrug that is rapidly transformed in vivo into 6-mercaptopurine (6-MP) by non-enzymatic cleavage of the imidazole ring, facilitated by glutathione. Further metabolism of 6-MP continues by three metabolic pathways: methylation of 6-MP to 6-methyl-mercaptopurine (6-MMP), which is biologically inactive, catalysed by thiopurine methyl transferase (TPMT); oxidation of 6-MP to thiouric acid, also biologically inactive, catalysed by xanthine oxidase (XO); and conversion of 6-MP, by a number of enzymatic steps, via 6-thioinosine monophosphate (6-TIMP) and 6-thioguanine monophosphate (6-TGMP) to a variety of 6-thioguanine nucleotides (6-TGNs), which are considered to be the active metabolites (Figure 19.1).
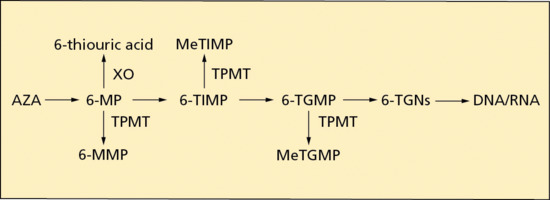
Figure 19.1 A simplified diagram of thiopurine metabolism (see text for abbreviations).
Azathioprine and its metabolites are excreted via the kidneys and gastrointestinal tract. There is no enterohepatic circulation. A lowered dose in cases of reduced renal function may be necessary [2].
Despite the rapid absorption, metabolism and excretion of AZA, its active metabolites only slowly accumulate in tissues, and thus the therapeutic immunosuppressive effects of AZA may take 6–12 weeks to develop.
Pharmacodynamics
Despite five decades of clinical use, the precise mechanism of action of AZA remains uncertain. AZA is a purine analogue and the accepted theory has been that its antiproliferative, anti-inflammatory and immunosuppressant properties are the result of the incorporation of 6-TGNs into DNA, thus interfering with its replication [3], and are perhaps also due to the suppression of purine synthesis, as methylthioinosine monophosphate (MeTIMP) is an in vitro inhibitor of de novo endogenous purine synthesis [4]. The relative specificity of its action on lymphocytes, with the consequence of altered T-cell and B-cell function, can be explained by the lack of a purine salvage pathway in lymphocytes [3]. However, it appears that AZA may additionally induce lymphocyte apoptosis and T-cell anergy, possibly by the modification of CD28 signalling [5], and thus the immunosuppressive effect of AZA may be due to a combination of antimetabolic and pro-apoptopic actions [6].
Pharmacogenetics
There are a number of allelic variants of TPMT and this polymorphism results in clinically important phenotypes. Approximately 90% of white individuals have normal TPMT activity (homozygous for the wild-type allele), 10% intermediate (heterozygous) and less than 1% low (homozygous or compound heterozygous for a mutant allele) TPMT activity [6]. TPMT is an important enzyme in the metabolic pathway of AZA. Not only does it catalyse the conversion of 6-MP to inactive 6-MMP, but it also inactivates 6-TIMP and 6-thioguanine by methylation. Therefore, in those individuals with inherited TPMT deficiency, the metabolism of AZA is shunted in the direction of excessive production and toxic accumulation of 6-TGNs (see Figure 19.1): standard dosages of AZA in such individuals risk causing severe bone marrow suppression and consequent pancytopenia. Conversely, TPMT deficiency results in reduced levels of 6-MMP, and, as 6-MMP is hepatotoxic [7], AZA-induced liver damage may be less likely in those with reduced TPMT activity. Pretreatment testing for TPMT polymorphism has become standard practice in the UK, either by measurement of TPMT activity in erythrocytes or undertaking genotyping, and the result is used to determine the dose of AZA [8].
Polymorphisms in the gene coding for inositol triphosphate pyrophosphatase may be associated with AZA hypersensitivity syndrome [9].
Potential adverse effects (Box 19.3)
Pharmacological
Myelosuppression
Bone marrow suppression is more likely with intermediate TPMT levels, and increasingly likely in individuals with low TPMT activity. However, a normal TPMT level with low 6-TGN production does not negate the possibility of myelotoxicity. Haematological monitoring is therefore required throughout the duration of treatment with AZA, independent of the TPMT status [1]. Leukopenia is the most common haematological side effect, but anaemia, thrombocytopenia and pancytopenia can occur. Patients should be warned to report infection, bruising, mouth ulcers and sore throat, which might be indicative of myelotoxicity.
Carcinogenesis
There is a paucity of data concerning immunosuppression-related carcinogenesis with AZA use in dermatological conditions, but by analogy with data obtained from transplantation medicine, it appears prudent to monitor dermatological usage of AZA for the development of lymphomas and squamous cell carcinomas (cutaneous, oral and female genital). Although the risk of lymphoma due to AZA appears to be low [8, 10, 11], courses of AZA should be restricted when possible to the short-to-medium term [8].
Infection
Azathioprine therapy is associated with an increased risk of infection, in particular by disseminated herpes simplex, varicella zoster virus and human papillomavirus [1]. However, true opportunistic infections are uncommon in patients treated with AZA for dermatological conditions [1].
Gastrointestinal side effects
Nausea, vomiting and diarrhoea are common side effects of AZA and can be minimized by dose reduction, dose division and taking AZA with food [1]. Pancreatitis has been reported with AZA therapy [1].
Hepatic side effects
Although generally well tolerated, mild derangement of liver function tests is not uncommon and may not require alteration of dosage [8]. However, AZA can rarely cause severe, occasionally life-threatening hepatitis. The mechanism is uncertain but may involve oxidative stress [12]. Furthermore, AZA, in the context of treating inflammatory bowel disease, has been associated with nodular regenerative hyperplasia of the liver [13] with resultant portal hypertension presenting with thrombocytopaenia and splenomegaly. Liver function should be monitored throughout treatment with AZA [1].
Idiosyncratic
Hypersensitivity syndrome
Azathioprine hypersensitivity syndrome is a rare but potentially fatal adverse effect. It occurs in approximately 2% of patients treated with AZA and typically develops early in the course of treatment, usually within the first month. It may be easily overlooked as it can mimic infection or disease exacerbation [14, 15, 16, 17]. The syndrome is characterized by fever, malaise, arthralgia, myalgia, nausea, vomiting and diarrhoea, with occasional renal and liver dysfunction, pancreatitis, hypotension, cardiogenic shock and leucocytosis [14, 15]. In about 50% of cases it has a cutaneous component, which may assist in its recognition [14, 15]. In the majority of cases in which rash is a feature, the clinical and histological features are of a neutrophilic dermatosis and, although the eruption can be non-specific in appearance, it may resemble Sweet syndrome, erythema nodosum, acute generalized exanthematous pustulosis or leukocytoclastic vasculitis [14]. The symptoms and signs of hypersensitivity syndrome settle within days of discontinuing AZA: rechallenge with AZA should not be undertaken for fear of causing a life-threatening shock reaction [14, 15].
Contraindications
Azathioprine is contraindicated if the TPMT activity is low; if there is a history of hypersensitivity to AZA; in the presence of severe infection, severely impaired hepatic or bone marrow function, or pancreatitis; with concomitant administration of live attenuated vaccines; and in women who are pregnant or considering conception (unless the benefits to the patient outweigh the potential risks) [8].
Cautions
Lactation and conception
Azathioprine is not recommended in breastfeeding women [1, 8]. Men taking AZA should receive counselling regarding the possibilities of reduced fertility and teratogenicity [18].
Pre-treatment screening [8]
As AZA has significant immunosuppressive activity, pre-treatment screening should incorporate all the aspects of Box 19.1.
A full blood count, including white cell differential, kidney function and liver function should be determined at baseline. Renal impairment may require a dose reduction [19], and hepatic dysfunction will necessitate close supervision of haematological and liver parameters.
The pre-treatment assessment of TPMT genotypic or phenotypic status is recommended in order to minimize the risk of potentially life-threatening myelosuppression [8]. If the patient has received a recent blood transfusion, genotyping is preferable to phenotyping.
Dose and regimens
For individuals where TPMT activity is in the normal range, the standard daily dose of AZA is 2–3 mg/kg [8]. If the TPMT activity is in the intermediate range, the recommended daily dose is 1–1.5 mg/kg [8]. The use of AZA should be avoided if the TPMT activity is low or absent [8].
Monitoring
Patients taking AZA should be monitored regularly for signs of toxicity, in particular hepatotoxicity and myelosuppression. Liver function tests and full blood counts should be checked weekly for 4–8 weeks after initiating therapy and after any dose increment, and, once the dose is stable, at least 2–3-monthly thereafter. Macrocytosis is a common unremarkable feature of AZA therapy.
It is not yet routine clinical practice to measure levels of 6-TGNs and other azathoprine metabolites (such as 6-MMP), but such monitoring may prove to assist in the determination of the dose required to optimize efficacy and minimize adverse effects.
Ciclosporin
Ciclosporin is a highly effective and rapidly acting potent inhibitor of T-cell function. It has remained of central importance in the management of severe inflammatory skin disease, particularly psoriasis, since the early 1990s.
Dermatological uses (see Chapters 35, 41, 42 and 47)
Cicloporin is licensed for use in psoriasis (plaque type) and atopic eczema. Common off-label uses include chronic urticaria, pyoderma gangrenosum, hand eczema and palmoplantar pustulosis. It is also used sporadically for a variety of other inflammatory conditions including Behçet disease, lichen planus, prurigo nodularis, chronic actinic dermatitis and toxic epidermal necrolysis.
Pharmacological properties
Formula and structure
Ciclosporin (originally named cyclosporine A) is a neutral, strongly hydrophobic, cyclic undecapeptide (hence the prefix ‘cyclo’ or ‘ciclo’) of 11 amino acids that was first detected in the early 1970s in the spores (hence the suffix ‘sporin’) of the fungus Tolypocladium inflatum Gams. A hydrophilic, microemulsion formulation was developed to circumvent problems with variable bioavailability, and there are a number of different generic formulations now available.
Administration
Ciclosporin is given orally, in the dose range 2.5–5 mg/kg daily, in two divided daily doses.
Pharmacokinetics [1, 2]
The absorption of ciclosporin from the gastrointestinal tract is incomplete and variable, and depends on the individual patient, the patient population and the formulation. The absolute bioavailability is poor (around 25% of the total administered dose) due to extensive first-pass metabolism as well as active transport of absorbed drug back into the intestinal lumen by the efflux transporter P-glycoprotein (encoded for by the ABCB1 gene), which is present at high concentrations in the villus tip of enterocytes of the small intestine. If taken with a high-fat meal, bioavailability may be further reduced. There may be differences in bioavailability between different formulations of ciclosporin (including generics which are licensed on the basis of bioequivalence) that may be clinically important in certain circumstances and so, in general, when switching from one formulation to another, additional monitoring and careful review is advised, specifying which particular brand of ciclosporin is to be dispensed.
Ciclosporin is highly lipophilic, readily absorbed through cell membranes and distributed widely throughout the body. The average apparent distribution volume is 3.5 L/kg. It is highly protein bound in circulating blood.
Ciclosporin undergoes extensive metabolism, mainly in the liver via cytochrome P450 3A4 (CYP3A4), which gives rise to a number of important drug interactions. The main pathways of metabolism consist of mono- and di-hydroxylation and N-demethylation at various positions in the molecule. All metabolites (up to 25) identified so far retain the intact peptide structure of the parent compound; some possess weak immunosuppressive activity (up to one-tenth that of the unchanged drug).
Excretion is primarily biliary; of the total oral dose, only around 6% is detected in the urine (a fraction of which is unchanged). Renal impairment thus has only minimal impact on the pharmacokinetics of ciclosporin. There is a high variability in the data reported on the terminal half-life of ciclosporin depending on the assay applied and on the target population, ranging from 6.3 h in healthy volunteers to 20.4 h in patients with severe liver disease.
Pharmacodynamics
Ciclosporin is a prodrug that becomes active only after forming a complex with an intracytoplasmic immunophilin (protein) known as cycliphilin. This ciclosporin–cycliphilin complex inhibits calcineurin phosphatase, an enzyme catalysing dephosphorylation of the cytoplasmic protein nuclear factor of activated T cells (NF-ATc). Ordinarily, dephosphorylation of NF-ATc allows it to translocate to the nucleus, where it enables transcription of a number of pro-inflammatory cytokines including IL-2, IL-4, IFN-γ, transforming growth factor-β and up-regulation of receptors such as IL-2R (CD25). Inhibition of calcineurin phosphatase, and possibly also JNK (MAPK8) and p38 (MAPK14) pathways, thus suppresses production of IL-2 and IL-2 receptor expression, key regulators of T-cell activation. Additional actions of therapeutic relevance include reduced histamine release and down-regulation of high-affinity immunoglobulin E (IgE) receptors on mast cells and basophils.
Pharmacogenetics
CYP3A4, CYP3A5 and ABCB1 genes code for enzymes and transporters that play a central role in ciclosporin disposition. All three genes are highly polymorphic, with marked differences in population prevalence depending on ethnicity. These genetic variants are likely to be major contributors to the wide interpatient variation in drug pharmacokinetics and have been subject to intensive investigation, predominantly in the transplant field [3]. For example, in one large renal cohort, variation in liver CYP3A4 and enterocyte P-glycoprotein activity explained up to 75% of the variation in ciclosporin clearance. Whilst promising, the clinical utility of these genetic variants to individualize ciclosporin dosing remains to be established.
Potential adverse effects
There are a large number of potential adverse effects reported with ciclosporin [1, 2], with hypertension, nephrotoxicity, hyperlipidaemia, myalgia and headache being the most common. Others include gingival hyperplasia, fatigue, gastrointestinal disturbances, tremor and paraesthesiae in the hands and feet, and a variety of metabolic abnormalities (hyperbilirubinaemia, hypercalcaemia, hypomagnesaemia, hyperuricaemia).
Most are dose related and respond rapidly to dose reduction or, if necessary, treatment cessation. In general though, ciclosporin is a very well-tolerated drug when used in the short term (6–12 months), whereas longer term use carries significant, predictable risk, particularly of nephrotoxicity, and is generally not recommended if other interventions can be used.
Nephrotoxicity
Acute nephrotoxicity can occur within weeks of treatment initiation, is reversible and arises due to dose-dependent vascular dysfunction involving afferent arteriolar constriction, which results in increased vascular resistance and a decrease in glomerular filtration rate. Tubular dysfunction may also occur and is characterized by decreased magnesium reabsorption, decreased uric acid excretion, decreased potassium and hydrogen ion secretion and distal tubular acidosis. Chronic nephrotoxicity [4, 5] is largely irreversible and is characterized by progressive arteriolar hyalinosis, interstitial fibrosis, tubular atrophy and glomerular sclerosis. Chronic nephrotoxicity is more likely to occur with higher daily doses, larger cumulative doses and long-term therapy (more than 1–2 years): particular risk factors include age over 50 years, pre-existing hypertension and/or renal impairment and concomitant treatment with non-steroidal anti-inflammatory drugs (NSAIDs) and other nephrotoxic drugs. Intermittent rather than continuous therapy is widely cited as a sensible, dose-minimizing strategy. However, with each treatment course, the time taken for creatinine levels to rise tends to become shorter. Change in serum creatinine from baseline is the single most important indicator of nephrotoxicity and thus is a critical component of monitoring.
Hyperlipidaemia
Triglycerides and, less commonly, cholesterol levels, may become elevated within 2 weeks of treatment initiation, usually returning to normal on withdrawal of therapy. If hyperlipidaemia does require active intervention, management requires care due to an increased risk of statin-induced myopathy; fibrates may be a preferred treatment option (see drug–drug interactions).
Malignancy
Long-term use of immunosuppressants, such as ciclosporin, is associated with a potential increase in the risk for developing certain types of malignancy, particularly cancers of the skin and lymphoid system. This risk has been identified in organ transplant populations where the burden of immunosuppression and duration of therapy is likely to be greater than in those being treated for skin disease. Data from psoriasis cohorts indicate that the incidence of non-melanoma skin cancer (principally squamous cell carcinoma) is significantly increased with use of ciclosporin [6, 7], predominantly in those who have previously received psoralens and ultraviolet A (PUVA), for whom the increased risk of squamous cell carcinoma following exposure to ciclosporin approaches that associated with 200 PUVA treatments [6]. The absolute risk of systemic malignancy is unclear, with the studies that do exist often being underpowered and/or not controlling for important confounders and any background disease-associated cancer risk, such as is the case in psoriasis [7].
Contraindications
Ciclosporin is contraindicated in uncontrolled hypertension, renal disease, serious infections and in those with a previous history of malignancy (excluding basal cell carcinoma) or a high cumulative dose of PUVA.
Cautions
In women planning conception and during pregnancy, ciclosporin has a relatively favourable risk/benefit profile, especially when compared to agents such as MTX and retinoids since it is not teratogenic. However, data from use in the transplant population indicate an increased risk of pregnancy-associated complications such as pre-eclampsia and low birth weight and so it should only be used where benefits of use outweigh risk to the fetus. Small amounts of ciclosporin are excreted in breast milk, and while limited data suggest there are no side effects for breastfed children exposed to ciclosporin, recent European (psoriasis) guidelines suggest that breastfeeding whilst on ciclosporin should be avoided [8, 9].
Drug–drug interactions
Ciclosporin interacts with multiple drugs, many of which have important clinical implications. First, drugs may inhibit or induce enzymes involved in the metabolism of ciclosporin, in particular CYP3A4. For example, co-therapy with rifampicin or St John's wort, both potent CYP3A4 inducers, increases ciclosporin metabolism and so reduces efficacy; conversely, erythromycin inhibits CYP3A4 and thus increases ciclosporin levels by up to sevenfold with consequent risk of toxicity [2]. Second, ciclosporin itself is an inhibitor of CYP3A4, the multidrug efflux transporter P-glycoprotein and organic anion transporter proteins (OATP), and may therefore increase plasma levels of co-medications that are substrates of this enzyme and/or transporters. Statins, for example, are oxidized via CYP3A4, and so ciclosporin-mediated inhibition of statin metabolism is the likely mechanism underlying risk of rhabdomyolysis associated with concomitant ciclosporin therapy: certain statins (rosuvastatin and simvastatin) should be avoided completely whereas others may be used but at a reduced dose (e.g. a maximum dose of atorvastatin 10 mg or fluvastatin 20 mg) with monitoring of creatine kinase. Grapefruit juice increases ciclosporin bioavailability via inhibition of intestinal CYP3A4 [10].
Pre-treatment screening
Routine recommended clinical and investigational assessments aim to identify those at risk of the principal adverse effects (nephrotoxicity, hypertension), altered pharmacokinetics (liver disease, drug–drug interactions) and to minimize the risks of immunosuppression. Prior to commencing ciclosporin, screening as outlined in Box 19.1 should be undertaken.
A full history and examination should focus on past or current infection, malignancy (including a full skin check, counselling to ensure patients are up to date with national screening programmes), renal disease, liver disease, excess alcohol and previous phototherapy, especially PUVA.
Investigations include full blood count, fasting lipids, alanine aminotransferase (ALT), aspartate aminotransferase (AST), γ-glutamyl transferase (γ-GT), bilirubin; a comprehensive renal assessment (urinalysis, urine protein : creatinine ratio, baseline creatinine ideally calculated from the mean of two serum creatinine measurements, urea and electrolytes, including potassium and magnesium, and a urate level); in patients over 60, in those with suspected renal impairment or where therapy is likely to be required long term, an accurate assessment of renal capacity can be helpful (ethylenediaminetetraacetic acid (EDTA) clearance).
As with all immunosuppressive agents, ciclosporin may enhance the adverse and toxic effect of live attenuated vaccines (i.e. vaccinal infections), and in addition may diminish the therapeutic effects of vaccines, so that appropriate counselling about avoidance of live vaccination and a review of planned travel is important [11].
Dose and regimens
Generally, patients start treatment in the lower dose range (2.5 mg/kg/day), escalating to higher doses (up to 5 mg/kg/day) after a month of therapy in the event of a poor response. In circumstances where disease is acute, severe and/or unstable, treatment may be started at 5 mg/kg/day, but this carries an increased risk of side effects. The lowest possible therapeutic dose should be used to maintain remission and, ideally, a treatment course should last no more than 1 year. In obese patients, dosing per actual body weight may lead to toxicity as, although highly lipophilic, observations suggest that distribution of the drug is limited primarily to lean body mass.
Monitoring
In transplant medicine, in view of the narrow therapeutic index, requirement for long-term (lifelong) treatment and the critical importance of maintaining adequate immunosuppression to ensure organ viability, therapeutic drug monitoring with either serum trough and/or 2 h post dose measurement of ciclosporin is part of routine practice. In dermatology, routine measurement of drug levels offers no specific advantage in terms of optimizing efficacy, although measurement of trough levels to predict risk of nephrotoxicity may be helpful in patients who cannot avoid long-term treatment. Blood pressure, full blood count, liver function tests and renal function are generally checked every 2 weeks for 3 months after initiation and after any subsequent dose increase: once established on therapy, they should be checked at 8–12-weekly intervals, although fasting lipids may be monitored less frequently. If the creatinine level rises by more than 30% above baseline, the dose should be reduced: if it fails to normalize, the drug should be stopped.
Drug-induced hypertension should be managed as for ordinary hypertension; calcium-channel blockers are often the preferred first line agents due to their vasodilatory effect on the afferent renal arteriole, which is thought to protect against nephropathy. All drugs of this class are associated with gum hypertrophy (in common with ciclosporin). However, amlodipine has no impact on ciclosporin drug levels, in contrast to nifedipine and diltiazem, and so is often considered the preferred agent.
Angiotensin-converting enzyme inhibitors may be used although they lead to a rise in serum creatinine that, although not pathological, may be difficult to distinguish from changes attributable to ciclopsorin. Potassium-sparing diuretics should be avoided since ciclosporin tends to increase serum potassium levels, and, where psoriasis is the indication for use, β-blockers should also be avoided.
Summary
Ciclosporin remains an extremely useful, predictably effective and generally well-tolerated drug for short-term use. Long-term use is complicated by nephrotoxicity, as well as risks associated with ongoing potent immunosuppression.
Colchicine
Colchicine is an ancient drug, originally derived from the roots and seeds of plants of the genus Colchicum, particularly C. autumnale (the autumn crocus). In the UK, it is licensed only for the treatment of acute gout, but it has recognized benefit in familial Mediterranean fever, Behçet disease and recurrent pericarditis [1].
Dermatological uses (see Chapters 35, 49, 50, 53, 56, 102 and 110)
Colchicine has been used off-label for a wide variety of dermatological conditions, particularly those characterized by a neutrophilic inflammatory infiltrate, including neutrophilic dermatoses (Sweet syndrome, recurrent aphthous stomatitis), cutaneous vasculitis, autoimmune bullous disorders (dermatitis herpetiformis, epidermolysis bullosa acquisita, linear IgA disease), autoimmune connective tissue diseases (dermatomyositis, scleroderma) and papulosquamous disorders (psoriasis), with variable efficacy [2].
Pharmacological properties
Formula and structure
Colchicine (empirical formula: C22H25NO6, systematic name: N-[(7S)-1,2,3,10-tetramethoxy-9-oxo-5,6,7,9-tetrahydrobenzo[a]heptalen-7-yl] acetamide) is a tricyclic alkaloid consisting of two seven-member and one six-member carbon rings, with nitrogen in the side chain (a protoalkaloid).
Administration
Colchicine is administered orally; parenteral use has given rise to serious safety concerns [1].
Pharmacokinetics
Colchicine is lipophilic and is absorbed in the small intestine. There is a wide interindividual variation in bioavailability. The peak plasma concentration after oral administration is reached at 30–90 min, with a second peak at approximately 6 h [1]. Protein binding is between 10 and 30% and the terminal half-life is about 10 h. It is metabolized in the liver, with deacetylation via the CYP3A4 system, and excretion is predominantly into bile, with 10–20% eliminated unchanged in urine [2]. Colchicine is widely distributed in tissues but accumulates preferentially in neutrophils, where the concentration may exceed 16 times the peak plasma concentration [1].
Pharmacodynamics
Colchicine is both antimitotic and anti-inflammatory, but its precise mechanism of action is uncertain. By binding to β-tubulin it appears to interfere with the assembly of microtubules, thereby causing mitotic arrest in metaphase and inhibiting cellular chemotaxis. Its anti-inflammatory action results from the modulation of pro-inflammatory molecule production and the reduction of neutrophil degranulation, chemotaxis and phagocytosis [1, 3].
Pharmacogenetics
Transport of colchicine across cell membranes involves P-glycoprotein: polymorphisms of the ABCB1 gene may contribute to the interindividual bioavailability of colchicine and also to its drug–drug interactions. Furthermore, variant alleles of CYP3A4 may cause significant variability of enzyme activity. However, at present, these polymorphisms have limited clinical application [4].
Potential adverse effects
Gastrointestinal effects
Colchicine commonly causes watery diarrhoea, vomiting, abdominal pain, bloatedness and hyperperistalsis [1, 2].
Toxicity and poisoning
Acute overdosage with colchicine commences within hours with burning sensations in the mouth and throat, and severe gastroenteritis-like symptoms. After 24–72 h, signs of multiorgan dysfunction and sepsis may develop: bone marrow failure, renal and hepatic damage, respiratory distress, muscle weakness, central nervous system toxicity, myocardial damage, disseminated intravascular coagulation, metabolic acidosis, electrolyte disturbances and hypovolaemic shock may supervene, and are potentially fatal consequences [1, 5].
Contraindications
Colchicine is contraindicated if there is known hypersensitivity to it, and in the presence of blood dyscrasias [2].
Cautions
Colchicine should be used with caution if there is renal or hepatic dysfunction, and should be avoided during pregnancy [6].
Drug–drug interactions
Drugs that inhibit the CYP3A4 and P-glycoprotein systems may increase colchicine levels and toxicity: they include ciclosporin, erythromycin, clarithromycin, ketoconazole, itraconazole, antiviral drugs and verapamil, and grapefruit juice has a similar action [1, 5, 6]. Co-administration with statins may increase the risk of myopathy [1, 5, 6].
Pre-treatment screening
Full blood count, renal and hepatic biochemistry, urinalysis and, if appropriate, a pregnancy test should be undertaken.
Dose and regimens
A starting dose of 0.5 mg/day, increasing to 0.5 mg twice or thrice daily over several weeks, may enhance tolerability [2]. The dose can be subsequently tapered as disease activity allows [2].
Monitoring
Full blood count, renal and hepatic biochemistry and urinalysis should be checked monthly for several months, then 3-monthly thereafter [2].
Summary
Oral colchicine is a safe drug in the long term when used appropriately [1], but it has a narrow therapeutic range [1, 3], and care should be taken to avoid overdosage.
Dapsone
The synthesis of dapsone in 1908 developed out of research on azo dyes and it was subsequently discovered in the 1930s to have beneficial anti-infective properties like other sulphones [1, 2]. It still retains important roles in the treatment of leprosy and the prophylaxis of malaria and pneumocystis pneumonia, and has recently been shown to have anti-epileptic activity [3]; it was, however, the realization in the 1950s that dapsone is a potent anti-inflammatory agent that paved the way for its use in a wide variety of primarily dermatological inflammatory disorders. The related drugs, sulfapyridine and sulfamethoxypyridazine, have been used in the treatment of dermatological disorders, but are now only rarely prescribed.
Dermatological uses (see Chapters 49, 50 and 102)
In the UK, dapsone is licensed for the treatment of dermatitis herpetiformis and the rapidity of its action (usually between 1 and 3 days) has been used as a diagnostic test for this condition. Dapsone is also predictably beneficial for the treatment of linear IgA disease, chronic bullous disease of childhood, bullous lupus erythematosus, erythema elevatum diutinum, IgA pemphigus and subcorneal pustular dermatosis. It has been widely used in many other inflammatory dermatoses, although its efficacy tends to be unpredictable: diseases that may respond include autoimmune blistering disorders (bullous and cicatricial pemphigoid, pemphigus and epidermolysis bullosa acquisita), vasculitis (leukocytoclastic vasculitis, urticarial vasculitis, granuloma faciale and Behçet disease), neutrophilic dermatoses (Sweet syndrome and pyoderma gangrenosum) and a miscellany of other conditions (lupus erythematosus, panniculitis, acne vulgaris, pustular psoriasis, delayed pressure urticaria and relapsing polychondritis) [1, 4, 5].
Pharmacological properties
Formula and structure
Dapsone (4,4′-diaminodiphenylsulphone) is a sulphone with a simple structure consisting of an atom of sulphur linking two aromatic amine rings.
Administration
Dapsone is taken orally in tablet form: 50 mg and 100 mg tablets are available in the UK. A topical preparation, dapsone 5% gel is available in the US and Canada as a treatment for acne.
Pharmacokinetics [4, 5]
Dapsone is lipid soluble and water insoluble. Orally, it is absorbed very efficiently from the gastrointestinal tract and appears to have a significant enterohepatic circulation. The peak plasma concentration occurs 2–6 h after ingestion and in the circulation it is approximately 70% protein bound. It is widely distributed, crossing the placenta and passing into breast milk. Dapsone is metabolized in the liver along two pathways: acetylation (by an N-acetyltransferase) and hydroxylation (by an N-hydroxylase). Acetylation results in the non-toxic metabolites monoacetyl dapsone and diacetyl dapsone. Hydroxylation yields the potentially toxic dapsone hydroxylamine. Metabolites are subsequently glucuronidated and excreted in the urine, and a small percentage is excreted in bile. Dapsone has a relatively long elimination half-life of 1–2 days, with wide individual variation.
Pharmacodynamics
Dapsone affects the folic acid metabolic pathway, an important process in DNA synthesis. It is selectively toxic to bacterial cells as it inhibits bacterial synthesis of dihydrofolate (DHF) by competing with para-aminobenzoic acid for the catalytic activity of dihydropteroate synthetase [5]. Whilst this explains its antibiotic activity, the mechanisms underpinning the anti-inflammatory effects of dapsone are still poorly understood. The fact that dapsone seems to be particularly effective in inflammatory reactions characterized by a polymorph response has resulted in mechanistic theories centred on neutrophil function. Dapsone has inhibitory actions on neutrophil and eosinophil myeloperoxidase: the former, as part of the neutrophil ‘respiratory burst’, is an important element in tissue damage. It also inhibits neutrophil chemotaxis by inhibiting IL-8 release and function [6]. Further actions include stabilization of neutrophil lysosomes, inhibition of neutrophil lysosomal enzymes, and suppression of integrin-mediated neutrophil adhesion and neutrophil recruitment [1, 4, 5].
Pharmacogenetics
N-acetyl transferase has a number of polymorphisms, causing significant variation between individuals (slow, intermediate and fast acetylators) in the rate of acetylation of certain drugs. However, in the case of dapsone, such variability is not clinically relevant, and thus the acetylation phenotype does not need to be established before dapsone therapy is initiated [4].
The hydroxylation pathway involves the cytochrome P450 family of enzymes, and polymorphisms for these enzymes may contribute to the wide individual variation in the pharmacokinetics, clinical efficacy and toxicity of dapsone. Dapsone hydroxylamine has strong oxidizing properties, with the potential to induce a state of oxidative stress sufficient to cause severe haemolysis and methaemoglobinaemia in individuals with G6PD deficiency, the result of a number of polymorphisms of the G6PD gene on the X chromosome. G6PD is the rate-limiting enzyme in the pentose phosphate metabolic pathway, one of the main functions of which is the generation of nicotinamide adenine dinucleotide phosphate (NADPH), which is integral in maintaining the intracellular supply of reduced glutathione that prevents accumulation of free radicals that would otherwise cause oxidative damage to proteins. The pentose phosphate pathway is the only source of reduced glutathione in red blood cells, which are therefore at particular risk of damage to their cell membranes and haemoglobin by a variety of oxidants, including dapsone hydroxylamine, if G6PD is deficient. If this defence against oxidative stress is overwhelmed, consequent damage to the erythrocyte plasma membrane results in haemolysis or phagocytosis, and the ferrous ion of the haemoglobin molecule is oxidized to the ferric state (methaemoglobin), with a decreased oxygen-carrying capacity. Therefore, it is prudent to screen for functional G6PD deficiency prior to commencing dapsone therapy [4].
Potential adverse effects
Pharmacological
Haemolysis and methaemoglobinaemia
Haemolytic anaemia and methaemoglobinaemia are dose-dependent side effects, occurring to some degree in all dapsone-treated patients, but showing great individual variability [4]. Methaemoglobinaemia is manifest by lethargy and headache, and a cyanotic hue to the skin and mucous membranes. The decreased oxygen-carrying capacity of the blood consequent on haemolysis or methaemoglobinaemia may exacerbate pre-existing cardiac and pulmonary insufficiency. Mild or moderate degrees of methaemoglobinaemia may be treated with cimetidine (400 mg thrice daily), which reduces dapsone hydroxylamine formation by inhibiting the cytochrome P450 system of enzymes [8], although this effect declines after several months, possibly because of cytochrome P450 enzyme induction [9]. Vitamin E and ascorbic acid (vitamin C) have also been used to counter methaemoglobinaemia [4], and lipoic acid, as a dietary supplement, may prove to be a useful adjunct to cimetidine in improving patient tolerance of dapsone [10].
Idiosyncratic
Agranulocytosis
Agranulocytosis is a rare unpredictable idiosyncratic adverse effect of dapsone that is potentially life threatening and for which the mechanism is unknown. Dapsone-induced agranulocytosis is more common in older individuals (>60 years) and those of non-white descent, and represents a particular risk in the treatment of dermatitis herpetiformis (compared with a negligible risk when used in leprosy and as prophylaxis for malaria) [11]. Agranulocytosis may present with fever, sore throat and signs of infection and usually manifests within 3 weeks to 3 months of treatment being commenced. Recovery of the neutrophil count tends to occur within 7–14 days of withdrawing the drug, although there is a mortality rate of 14–33% [4].
Peripheral neuropathy
Rarely dapsone may cause peripheral neuropathy, which is more commonly motor than sensory [4]. The onset of distal neuropathy is often subtle and slowly progressive. Symptoms may persist long after dapsone therapy is terminated (sometimes as long as 1–3 years) [12, 13], although recovery usually occurs eventually. It typically presents with weakness of the hands or legs, loss of fine motor skills, gait disturbance, foot drop, glove and stocking loss of sensation, and wasting of the hand muscles [12]. Typically dapsone-induced peripheral neuropathy develops after several years of treatment, although it may occur as quickly as within 6 weeks[12, 13]. Electrophysiological studies have demonstrated axonal degeneration, although the mechanism is unknown [4, 12].
Ocular side effects
Dapsone therapy may also very rarely be associated with ophthalmic side effects including optic neuritis, optic atrophy and macular infarction, potentially resulting in severe visual impairment. Diabetes, hypertension, hypercholesterolaemia and coagulopathy are contributory risk factors [14].
Dapsone hypersensitivity syndrome
Dapsone hypersensitivity syndrome is an idiosyncratic adverse reaction of unknown mechanism, usually occurring in the first 3–5 weeks of commencement of dapsone. It comprises at least two of four signs: fever; lymphadenopathy; generalized rash; and hepatitis [15], and resembles DRESS syndrome (drug rash with eosinophilia and systemic symptoms) [16]. The prevalence is 1.4% and the fatality rate 9.9% [15], with liver failure the most frequent cause of death. Mucosal involvement, rash (which ranges from a maculopapular eruption to toxic epidermal necrolysis [4]) and delayed cessation of dapsone therapy are associated with an increased risk of a fatal outcome [15]. Nausea and vomiting are common, as are eosinophilia and leucocytosis. Other internal organs (kidneys, heart, lungs and pancreas) may be affected.
The presence of HLA-B*13:01 has been shown to be associated with the development of dapsone hypersensitivity syndrome in leprosy patients treated with dapsone [17].
Systemic glucocorticoid (GC) therapy appears to be beneficial when there is internal organ or mucosal involvement: it should be tapered over 1 month [15]. The cautious reintroduction of dapsone without recurrence of dapsone hypersensitivity syndrome has been reported [18, 19].
Other
Dapsone is generally well tolerated at the doses normally used for dermatological conditions. However, it may cause gastrointestinal upset and anorexia, hepatitis, hypoalbuminaemia, headache, insomnia, rashes (varying from a morbilliform eruption and exfoliation to erythroderma and toxic epidermal necrolysis) and, rarely, acute psychosis or photosensitivity [4].
Contraindications
Dapsone is contraindicated for patients with known hypersensitivity to it, and is relatively contraindicated in severe G6PD deficiency and in advanced cardiovascular or pulmonary disease that may be exacerbated by dapsone-induced haemolytic anaemia and methaemoglobinaemia [4].
Cautions
When its use during pregnancy is unavoidable, it is generally considered that dapsone is moderately safe [4, 20], although haemolysis and methaemoglobinaemia may develop in utero and breastfeeding infants, especially if the child is G6PD deficient [4, 7]. Folic acid (5 mg daily) for the mother is advised during pregnancy [7, 20].
Dapsone should be used with caution when there is G6PD deficiency, significant cardiopulmonary disease, severe hepatic and renal impairment and pre-existing peripheral neuropathy [4].
Drug–drug interactions
Drug interactions are relatively uncommon with dapsone [4], although the plasma concentration of dapsone may be reduced by rifamycins, carbamazepine, phenytoin, griseofulvin, proton pump inhibitors, calcium and H2 antihistamines, and be increased by probenecid. MTX, sulphonamides, trimethoprim and hydroxychloroquine may increase the risk from the haematological side effects of dapsone.
Pre-treatment screening
It is important to establish whether the patient has pre-existing conditions that may increase the risk of toxicity from dapsone, such as cardiopulmonary, renal, hepatic or neurological disease. There should be a baseline clinical examination of peripheral motor and sensory function. A full blood count, liver and renal function tests should be undertaken, and the G6PD level determined.
Dose and regimens
It is usual to commence dapsone in a single daily dose of 50–100 mg, depending on the pre-treatment screening, subsequently increasing to 100–200 mg/day. Once adequate disease control has been attained, the dose should be gradually tapered to the lowest effective level in order to minimize dapsone toxicity.
Monitoring
A full blood count with differential white cell count should be checked every week for the first 4 weeks, and then fortnightly for the next 8 weeks, to monitor for agranulocytosis. Patients should, of course, also be warned to discontinue the medication immediately in the event of fever, chills and sore throat occurring within 3 months of commencing dapsone.
A full blood count with reticulocytes should be checked 3–4 monthly: signs of haemolysis (such as raised reticulocytes and bilirubin) should prompt requests for a blood film (Heinz bodies within red blood cells in G6PD-induced haemolysis), lactate dehydrogenase (elevated in haemolysis) and a haptoglobin level (decreased in haemolysis). Methaemoglobin levels should be checked if there are signs or symptoms to suggest methaemoglobinaemia. It is prudent to check the liver function tests fortnightly for the first 3 months; thereafter liver and renal function tests should be performed with the full blood count every 3-4 months.
At each follow-up appointment, the peripheral motor and sensory nervous system should be assessed.
Fumaric acid esters
Common fumitory (Fumaria officinalis) is a plant rich in fumaric acid that is known to have been in use for the treatment of skin complaints, including leprosy, as early as the 17th century when Nicholas Culpeper claimed it to be ‘very effectual for . . . clarifying the blood from saltish, choleric humours which cause leprosy, scabs, tetters, and itches, and such like breakings-out of the skin’ [1]. Its use at this time may represent an early example of systemic therapy for psoriasis, as it was not until the 19th century that psoriasis was clearly differentiated from leprosy. The beneficial properties of fumaric acid in psoriasis were first reported by Schweckendiek in 1959: both a chemist and a psoriasis sufferer, he found through self-experimentation a combination of esters of fumaric acid (FAEs) that was both tolerable and effective in improving his psoriasis. Recently, there has been renewed interest in FAE with the development and licensing of dimethyl fumarate for the treatment of multiple sclerosis.
Dermatological uses (see Chapter 35)
FAEs are licensed in Germany (under the brand name Fumaderm®) for moderate to severe plaque psoriasis, but are widely used off-label for this indication elsewhere.
Pharmacological properties
Formula and structure
Fumaderm contains dimethylfumarate (DMF), an α,β-unsaturated carboxylic acid ester, and three monoethyl hydrogen fumarates (calcium, magnesium and zinc salts).
Administration
FAEs are taken orally in three divided doses each day, ideally before meals.
Pharmacokinetics
The pharmacokinetics of FAEs are still subject to investigation, but it seems likely that following oral ingestion DMF is absorbed from the small intestine in the preportal circulation, metabolized to mercapturic acids and then excreted in urine [2]; it is also rapidly hydrolysed by ubiquitous esterases to the main metabolite, monomethyl fumarate (MMF). Only MMF is detectable in serum, reaching maximum levels by 4 h [3]. Food intake may significantly delay or reduce absorption, and FAEs should therefore be taken before meals [3]. There is no evidence for a cytochrome P450-dependent metabolism in the liver. The half-life of FAE in vivo is approximately 90 min [4].
Pharmacodynamics
FAEs appear to modulate T-helper cell differentiation away from Th1/Th17 development in favour of Th2, which may explain therapeutic efficacy in Th1-mediated disease and the eosinophilia and elevated IgE that can occur with therapy. The molecular mechanisms underlying this are incompletely understood, but data from studies on FAEs in the dermatology literature and recent data on experimental animal models of multiple sclerosis indicate that both DMF and MMF modulate oxidative stress response pathways by binding both to glutathione [2, 5] and to KEAP-1 (kelch-like ECH-associated protein 1) [7]. Glutathione is the most important intracellular scavenger of reactive oxygen species and leads to impaired IL-12 and IL-23 production with consequent induction of type II dentritic cells [6]. KEAP-1 is an inhibitor of NFE2L2 (nuclear factor erythroid 2-like 2 or Nrf2), which promotes the synthesis of antioxidant proteins protective against oxidative tissue injury [7].
Potential adverse effects
Two thirds of patients experience gastrointestinal symptoms, such as nausea, cramps and diarrhoea, and one third report episodic flushing lasting minutes to hours, with or without headache. These symptoms may settle with time and/or dose reduction but often lead to treatment discontinuation. Transient eosinophilia occurs between weeks 4 and 10, and lasts 1–2 months. A fall in lymphocyte count seen in nearly all patients is not usually of any clinical significance and may correlate with treatment response; in about 10% of patients, the fall is greater than a 50% reduction below baseline values. Four cases of confirmed progressive multifocal leucoencephalopathy [8, 9, 10] have been reported in patients taking FAEs, all of which were associated with profound lengthy lymphopenia, as well as with other potentially relevant factors (cancer and other immunosuppressive therapies): this highlights the importance of monitoring for and avoiding lymphocyte counts below 0.5 × 109/L. With long-term use, proteinuria with associated renal impairment may rarely develop, with a risk of acute renal failure [4].
Contraindications
Toxicology studies have shown that FAEs have neither teratogenic nor mutagenic potential; nonetheless, conception (for men and women), pregnancy and breastfeeding are contraindicated. Severe hepatic or renal disease, severe gastrointestinal disease (such as untreated peptic ulceration) and significant leucopenia are also contraindications.
Cautions
Recent guidelines suggest caution when FAEs are used with MTX, retinoids, psoralens, ciclosporin, immunosuppressants, cytostatics and any drugs with known nephrotoxicity [11]. Live vaccines or live attenuated vaccines should be avoided during FAE therapy.
Drug–drug interactions
There are no reported interactions of FAEs with other drugs; limited data are available so a high index of suspicion should remain for potential drug interactions.
Pre-treatment screening
Routine baseline blood tests (full blood count, liver function tests, renal function), lipids and urinalysis for protein are needed [11].
Dose and regimens
FAEs are available in white low-strength (Fumaderm initial®; 30 mg DMF, 67 mg monoethylfumarate calcium salt, 5 mg monoethylfumarate magnesium salt, 3 mg monoethylfumarate zinc salt) and blue high-strength (Fumaderm®; 120 mg DMF, 87 mg monoethylfumarate calcium salt, 5 mg monoethylfumarate magnesium salt, 3 mg monoethylfumarate zinc salt) preparations. Typically, treatment is initiated with one tablet (30 mg DMF) of low-strength Fumaderm, increasing each week, as tolerated, over 9 weeks (Table 19.1) up to the maximum dose of six tablets of high-strength Fumaderm (equivalent to a daily dose of 720 mg of DMF). Treatment at this dose can be continued as long as there is a clinical need and provided monitoring is satisfactory.
Table 19.1 Dosage (number of tablets).
| Week | Morning | Noon | Evening | Fumaric acid ester formulation |
| 1 | 1 | – | – | Low strength (30 mg) |
| 2 | 1 | – | 1 | Low strength (30 mg) |
| 3 | 1 | 1 | 1 | Low strength (30 mg) |
| 4 | 1 | – | – | High strength (120 mg) |
| 5 | 1 | – | 1 | High strength (120 mg) |
| 6 | 1 | 1 | 1 | High strength (120 mg) |
| 7 | 2 | 1 | 1 | High strength (120 mg) |
| 8 | 2 | 1 | 2 | High strength (120 mg) |
| 9 | 2 | 2 | 2 | High strength (120 mg) |
Monitoring
Full blood counts, renal and liver function tests and urinalysis for protein should be checked at monthly intervals during dose escalation and then bi-monthly once a therapeutic dose is established. Significant lymphopenia (<0.7 × 109/L) should prompt halving of the dose; in the absence of lymphocyte count recovery by 4 weeks or a further fall to <0.5 × 109/L the drug should be stopped [11].
Summary
For those patients who tolerate FAE, the therapy is straightforward to manage. Licensing of DMF for multiple sclerosis may improve availability and provide important additional safety data, particularly in relation to rare serious adverse effects.
Systemic glucocorticoids
The GCs are a family of steroid hormones that have vital immunomodulatory and metabolic functions and are the most frequently used and consistently effective anti-inflammatory agents for dermatological conditions. The term glucocorticoid is often used synonymously with corticosteroid, although, strictly, the latter includes both GCs and mineralocorticoids, both produced within the adrenal cortex. The principal naturally occurring GC is cortisol (hydrocortisone), possessing modest GC potency; there are also a number of synthetic systemic GCs with intermediate (prednisolone, methylprednisolone and triamcinolone) and high (betamethasone and dexamethasone) GC potency.
Dermatological uses – licensed indications and off-label usage (see Chapters 35, 36, 37, 39, 40, 41, 42, 49, 50, 51, 52, 53, 54, 55, 56, 57, 98 and 102)
GCs are used in a very wide variety of dermatological inflammatory conditions [1], including bullous dermatoses, connective tissue diseases, vasculitides, neutrophilic dermatoses, eczematous and papulosquamous disorders and miscellaneous conditions such as severe urticaria and sarcoidosis. Low-dose dexamethasone has also been used in androgen excess syndromes [1].
Pharmacological properties
Formula and structure
The basic ring structure of all GCs is the cyclopentanoperhydrophenanthrene nucleus, consisting of three hexane rings and one pentane ring. GCs with increased anti-inflammatory potency are formed by modifications of the hydrocortisone molecule: introducing a 1,2 double bond (prednisolone); a 1,2 double bond and a 6-methyl group (methylprednisolone); a 1,2 double bond and fluorine at the 9α position, with a 16α hydroxyl group (triamcinolone), a 16α methyl group (dexamethasone); or a 16β methyl group (betamethasone).
Administration
Although GCs are usually administered orally, there are occasional indications for intramuscular, intravenous and intralesional administration.
Oral GCs are ideally given in the early morning, to conform to the natural circadian rhythm of endogenous glucocorticoid production. Traditionally, prednisolone (the biologically active metabolite of prednisone) is the favoured oral GC for dermatological use in the UK, although betamethasone, deflazacort, dexamethasone, hydrocortisone and methylprednisolone can be given orally [2].
Intramuscular administration of GCs has the advantages of guaranteeing that a possibly unreliable patient receives treatment and also of avoiding the potential gastrointestinal side effects of oral GCs; however, intramuscular GCs may cause fat atrophy and occasional abscess formation at the injection site, as well as being more likely than oral GCs to induce menstrual irregularities in women [1], possibly because the intramuscular route results in constant levels of circulating GCs without diurnal variation, thereby suppressing gonadotropin release. Traditionally triamcinolone acetonide has been favoured for intramuscular use [3].
Intravenous administration of GCs, usually given in ‘pulses’ on an in-patient basis, is generally only used in situations where it is desirable to bring very serious steroid-responsive conditions under rapid control. Methylprednisolone is the usual intravenous GC of choice. The risks particularly associated with intravenous pulsed GCs include sudden death of presumed cardiac origin secondary to acute electrolyte shifts; cardiac arrhythmias; potentially life-threatening anaphylaxis; gastric erosions; and sepsis [1]. Serious cardiovascular adverse effects are rare, tending to occur in patients with heart or kidney disease [4], and pulsed intravenous GC therapy is considered to have an acceptable risk–benefit profile, in the appropriate clinical context [5].
Formerly, corticotropin (adrenocorticotropic hormone) and its analogue, tetracosactide, were used as an alternative to GCs but had the disadvantages of variable and unpredictable therapeutic responses and a gradual waning of effect [2].
Pharmacokinetics
The synthetic GCs are readily absorbed from the small bowel after oral administration and peak plasma concentrations occur after 1–2 h. They are significantly protein bound in the circulation to cortisol-binding globulin (transcortin) and corticosteroid-binding albumin, become widely distributed in body tissues and also cross the placenta.
They are metabolized predominantly in the liver and the metabolites are then conjugated with sulphate or glucuronic acid to make them water soluble, before being excreted in the urine. Inactivation is mainly by reduction of both the 3-keto group (by 3α-hydroxysteroid dehydrogenase) and the 4,5 double bond in the steroid A ring (by 5α-reductase and 5β-reductase).
The biological half-life of GCs varies from 8 to 12 h for the short-acting GCs (hydrocortisone and cortisone), 24–36 h for the intermediate-acting GC (prednisolone, methylprednisolone and triamcinolone), and 36–54 h for the long-acting GCs (betamethasone and dexamethasone) [1].
Pharmacodynamics
Endogenous GC (cortisol) plays vital physiological roles in anti-inflammatory homeostasis and in certain metabolic processes, including gluconeogenesis and fluid/electrolyte regulation. The synthetic GCs have been developed primarily as anti-inflammatory agents for the treatment of conditions unresponsive to natural anti-inflammatory processes.
The intracellular mechanisms of action of GCs are complex and still incompletely understood [6]. They are known to act within the nucleus at a genomic level but also to have non-genomic effects [7]. Being lipophilic, GCs cross plasma membranes into the cytoplasm of cells with ease, where they bind to cytosolic GC receptor (cGCR) encoded by the NR3C1 (nuclear receptor subfamily 3, group C, member 1) gene.
Unactivated cGCR, which is ubiquitous in vertebrate cells, is a 94 kDa protein present in the cytoplasm as a complex with various other proteins (chaperones), including heat-shock proteins and several kinases of the mitogen-activated protein kinase (MAPK) signalling system that are thought to be important in maintaining the conformational state of the cGCR to enable high-affinity binding with GCs. It has a complex structure that includes specific domains for binding GCs and DNA and for undertaking transcription functions. On binding to GC (endogenous or synthetic), the cGCR rapidly dissociates from its protein chaperones, enabling the GC/cGCR complex to pass into the nucleus. There, the GC/cGCR complex binds to specific DNA binding sites (GC response elements, GREs) within the promoter regions of GC-inducible genes, from where it modifies the transcription of those genes. Depending on the target gene, transcription is either activated (‘transactivation’) or inhibited [7, 8]. Numerous genes are targeted by the GC/cGCR complex, many of which produce proteins that regulate the inflammatory process and apoptosis and others which regulate metabolic functions: it is estimated that GCs influence the expression of approximately 1% of the entire genome [7].
In addition to its DNA binding effects on transcription, GC/cGCR complexes also prevent the binding to their target genes of a variety of natural transcription factors (such as activator protein 1 (AP1), nuclear factor of activated T cells (NF-AT) and NF-κB), resulting in the reduced expression of many immunoregulatory and pro-inflammatory proteins (‘transrepression’) [7, 8]. Furthermore, GCs also seem to have post-transcriptional and post-translational effects [7].
GCs have several non-genomically mediated actions still to be precisely defined. These include the release of the protein chaperones when the GC/cGCR complex is formed, which may have important signalling effects, such as those involving the T-cell receptor (TCR). Via this mechanism, GCs impair TCR signalling and consequently T-cell cytokine production, proliferation and differentiation [9]. There may also be an important non-specific non-genomic mechanism of GC action involving intercalation of GC molecules in cell plasma membranes, resulting in altered cation transport, which in the case of immune-competent cells is considered to contribute to immunosuppressive and anti-inflammatory actions [7]. Finally, GCs may also cause specific non-genomic actions, mediated through a cell surface membrane bound GCR known to be present on the cell surfaces of human monocytes and B lymphocytes [7].
This is a very simplified summary of the complex mechanisms that in combination are thought to regulate GC-induced immune modulation and apoptosis of immune-competent cells. In addition to these desirable anti-inflammatory properties in the context of treating disease states, the enhanced effect compared with that of endogenous cortisol of the synthetic GCs on gene transcription results in a number of undesirable metabolic consequences. These include important effects on gluconeogenesis, causing increased carbohydrate production by the enhanced metabolism of endogenous protein (from bone, muscle, skin and blood vessels) and on fat metabolism with associated peripheral insulin resistance; the less potent GCs also have important mineralocorticoid actions. It has long been assumed that the unwanted effects of prolonged treatment with GCs are largely the result of transactivation, whilst the beneficial anti-inflammatory effects are the result of transrepression [7, 8], but doubt has recently been cast on this hypothesis [10, 11].
Pharmacogenetics
The pharmacogenetic factors of clinical relevance to dermatologists are still to be established. However, GCR polymorphisms may explain in part the wide individual variation in response to GCs [12, 13, 14] and may influence GC-induced bone mineral density [13] and metabolic [15] changes. GCR polymorphisms have also been associated with depression and bipolar disorder [16, 17, 18] and may therefore possibly be relevant to the mood disturbances that may complicate GC therapy.
Potential adverse effects (Boxes 19.4, 19.5, 19.6, 19.7)
GCs have the potential for a wide variety of side effects, particularly when used in high (supra-physiological) doses and in long-term regimens. Short courses (2–3 weeks) of GCs are generally relatively safe [1].
Hydrocortisone has a significant mineralocorticoid action, but with increasing potency the mineralocorticoid effect of GCs diminishes: GCs of intermediate potency (prednisolone, methylprednisolone and triamcinolone) have some and the high-potency GCs (betamethasone and dexamethasone) negligible mineralocorticoid action [2].
Malignancy
The development of immunosuppression-related malignancies, such as lymphoma, squamous cell carcinoma and Kaposi sarcoma, is very uncommon when GCs are used in isolation [1].
Steroid withdrawal syndrome
The withdrawal of GCs may occasionally and apparently idiosyncratically be associated with signs and symptoms of variable degree resembling adrenal insufficiency [20]. Steroid withdrawal syndrome may occur when the dose of GC is still supra-physiological or even after complete withdrawal without biochemical evidence of subnormal hypothalamopituitary–adrenal (HPA) axis integrity. Symptoms include fever, anorexia, nausea, vomiting, lethargy, fatigue, weakness, malaise, emotional lability, depression, myalgia, arthralgia, headache, abdominal pain, skin peeling, influenza-like symptoms and weight loss. Signs include postural hypotension, hyponatraemia and hyperkalaemia [20].
The precise mechanism underlying the steroid withdrawal syndrome is unknown, although it is possible that the symptoms are mediated by increased circulatory levels of pro-inflammatory cytokines such as IL-6, TNF-α and IL-1β consequent on the reduction of GCs [21]. Its treatment is to reinstitute or increase GC therapy, followed by more gradual tapering of the dose [21].
Cautions
Adrenal suppression
Endogenous cortisol production in the physiological state is regulated by a negative feedback homeostatic mechanism on corticotropin-releasing factor secretion from the hypothalamus and adrenocorticotropic hormone release from the pituitary – the HPA axis. Synthetic GCs also suppress the hypothalamopituitary drive of the adrenal glands and will abolish endogenous cortisol production by the adrenals if the dose and duration of treatment is sufficient, resulting in the potential, albeit rare, for acute adrenal insufficiency (Addisonian crisis) if the GC is stopped abruptly. It can take very many months for the normal adrenocortical response to fully recover, and vulnerability to stress may last for a year or more [1].
There is significant individual variation in susceptibility to GC-induced adrenal suppression [1]. GCs given in divided doses or at times other than the morning are more likely to induce HPA axis suppression. Alternate day GC therapy reduces the risk of HPA axis suppression [1].
The early morning cortisol level and 24 h urinary cortisol level both provide a measure of basal HPA axis function for a patient on GCs, and the short synacthen stimulation test is a measure of the stress responsiveness of the adrenal: these are an option for assessing adrenal reserve towards the end of long-term GC therapy.
GCs taken in a daily dose of more than 40 mg for more than 1 week (or a lesser dose for more than 3 weeks) should be tapered gradually [2]. Intercurrent illness, infection, surgical procedure or trauma requires a temporary increase in GC dose or, if a prolonged GC course has recently been completed, a temporary reintroduction of GC to compensate for a diminished adrenal response. Anaesthetists must know if their patient is taking or has been taking a GC within 3 months of surgery [2]. Patients on long-term GC therapy should carry a steroid treatment card with them at all times.
Acquired resistance
The therapeutic response to GCs in chronic inflammatory disorders may be compromised by the development of an acquired resistance or insensitivity to GCs [22, 23]. The precise mechanisms for this phenomenon are not fully elucidated but are thought to include down-regulation of GCR expression and inhibition of GCR translocation by pro-inflammatory cytokines, oxidative stress and hypoxia in the cellular microenvironment occurring over time [23]. The consequent higher doses of GCs that are necessary to maintain disease control increase the risk of adverse effects.
Infections
Prolonged courses of GCs increase susceptibility to infection and may modify and perhaps mask signs of infection, especially septicaemia and tuberculosis [2]. GCs predispose to severe chickenpox and measles if the patient is not already immune to these infections: exposure to them requires passive immunization. Fungal or viral ocular infections may be exacerbated, as may amoebiasis and strongyloidiasis [2].
Bone protection
GC-induced osteoporotic fractures are a real risk with long-term therapy, even in low dosage, and monitoring with bone mineral density measurements (DEXA scans) is advisable in patients on long-term GC therapy. Preventative measures, such as calcium/vitamin D supplements and biphosphonate therapy, together with lifestyle advice including weight-bearing exercise, smoking cessation and avoiding excessive alcohol intake, should be considered early when initiating long-term GC therapy to limit the likelihood of osteoporosis.
Gastrointestinal protection
Although controversial, the prophylactic administration of a proton pump inhibitor or an H2 antagonist should be considered in high-dose GC therapy, particularly for high-risk groups (e.g. anticoagulation therapy or a history of peptic ulcer disease) [1].
Psychiatric reactions
GCs should be prescribed with caution for patients with a personal or family history of psychiatric disorders.
Pregnancy
GCs may increase the risks of spontaneous abortion and stillbirth, and the possibility of fetal HPA axis suppression should be considered when GCs are given to the mother close to delivery [1].
General cautions
Care should be taken when prescribing GCs for children, the elderly, pregnant women or patients with diabetes, hypertension, osteoporosis, raised intraocular pressure, a history of peptic ulceration, diverticulitis, recent bowel anastomosis or tuberculosis, or if there is hepatic or renal impairment.
Contraindications
GCs are relatively contraindicated if there is systemic infection unless specific therapy is being given [2]. Vaccination with live virus should be avoided in patients taking GCs.
Drug–drug interactions
There are a large number of potential interactions between systemic GCs and other drugs (see British National Formulary for complete list), although only a small number are potentially serious, with only very few of these involving prednisolone (19.08) [1].
Pre-treatment screening
If a short course (up to 3 weeks) of GC therapy is envisaged, it is reasonable to establish whether there are coexisting conditions that might be influenced by GCs, such as diabetes, psychosis, glaucoma, infections, peptic ulcer disease, active diverticulitis and recent surgery, to enquire about medications that might influence or be influenced by GCs, and to monitor for rising blood pressure and gycosuria.
If extended treatment is anticipated, baseline assessment of blood pressure, weight, height (in children), serum electrolytes, fasting glucose and fasting lipids should be undertaken. Consideration should be given to screening for active and latent tuberculosis, especially in those considered to be at particular risk, by means of a Mantoux test followed if positive by an IFN-γ test and chest X-ray [24]. Baseline ophthalmic examination for cataracts and ocular hypertension should also be considered.
Prior to initiating GC therapy, the patient and family members should receive appropriate education, in particular about the potential adverse effects and the monitoring details. A steroid treatment card should be provided and the information on it kept up to date.
Dose and regimens
Oral administration
Depending on the clinical diagnosis, its severity and the presence or otherwise of cautionary factors, it is reasonable to consider commencing prednisolone at a starting dose of up to 1 mg/kg body weight daily, ideally given as a single dose in the morning. A single daily dose is less likely than a divided dose to cause adverse effects, and a morning dose less likely to result in HPA axis suppression than when given at other times of day [1]. In some circumstances, higher doses may be considered [1].
Intramuscular administration
A typical regimen for intramuscular triamcinolone is 80 mg two or three times a year [1].
Pulsed intravenous administration
A typical regimen for inpatient-based pulsed intravenous methylprednisolone is 500–1000 mg (approximately10–15 mg/kg) daily, given over at least 60 min, for 3–5 consecutive days, with continuous cardiac monitoring and daily measurement of electrolytes and glucose levels recommended [1]. Thereafter, oral prednisolone and/or a non-steroidal immunosuppressive may be required to maintain the therapeutic effect of pulsed intravenous therapy.
Tapering steroid dose
Generally, as soon as there is adequate disease control, consideration should be given to tapering long-term GCs to the minimal effective dose in order to minimize the risk of side effects, although, despite their use over many decades, the optimal regimen has yet to be determined. The rate of dose reduction is determined by disease activity, assessed by clinical features and laboratory parameters. When withdrawing GCs, it is reasonable to attempt to reduce the dose rapidly to a physiological level (7.5 mg prednisolone daily or equivalent [2]), followed thereafter by a more gradual reduction. If supra-physiological doses of GCs are required in the long term, adjusting to an alternating day regimen may ultimately allow recovery of the HPA axis and cause less metabolic disruption than continuing to give GCs in a daily dose [1].
Monitoring
A reasonable follow-up frequency for patients on oral GC treatment is at 1 month and then every 2–3 months [1]. At each visit specific enquiries should be made for side effects, blood pressure and weight (and height in children) should be recorded, and serum electrolytes, fasting glucose and lipids measured [1]. An ophthalmic examination every 6–12 months is recommended [1].
A neutrophilia frequently occurs with GC therapy. This should not be assumed to be necessarily the result of infection as it is often innocent, resulting from a shift of neutrophils from the marginated to the circulating pool, with a minor contribution from an increased release from bone marrow [25].
Depending on therapeutic response and the likely duration of therapy with GCs, active consideration should be given to steroid-sparing strategies.
Summary
GCs have a very important place in the dermatological therapeutic armamentarium and their prudent use and monitoring will minimize, although not obviate, adverse effects. For the future, enhancement of the risk–benefit ratio of GCs may be possible by the targeted delivery of GCs (long-circulating liposomal GCs), by combining GCs with synergistic anti-inflammatory moieties (such as nitric oxide) or by the modified release of GC to conform to circadian rhythms (chronotherapy) [26]. Novel non-steroidal drugs such as SEGRAs (selective GCR agonists) and SGRMs (selective GCR modulators) may have the anti-inflammatory properties of conventional GCs with fewer side effects [26].
Hydroxycarbamide
Hydroxycarbamide (formerly known as hydroxyurea) is an antimetabolite cytotoxic drug that is used primarily in the treatment of chronic myeloid leukaemia and certain solid malignancies, for conditions with a high risk of thromboembolic complications (including polycythaemia rubra vera and essential thrombocythaemia), and for reducing the crises of sickle cell disease [1, 2].
Dermatological uses (see Chapter 35)
Off-label, the primary dermatological indication for hydroxycarbamide has been for the treatment of recalcitrant chronic plaque psoriasis [3, 4], although, with the advent of the biological era, it is now rarely used [5]. It has also been used for the treatment of Sweet syndrome, erythromelalgia and hypereosinophilic syndrome [5].
Pharmacological properties
Formula and structure
Hydroxycarbamide (empirical formula: CH4N2O2, systematic name: hydroxyurea) is a simple organic molecule consisting of a carbonyl functional group attached to single amine and hydroxylamine functional groups. It was first synthesized in 1869.
Administration
Hydroxycarbamide is administered orally and is available in the UK as 100 mg, 500 mg and 1000 mg tablets [1].
Pharmacokinetics
Hydroxycarbamide has excellent oral bioavailability, with maximum plasma concentrations reached between 0.5 and 2 h [1, 6]. Despite therapeutic use over many years, there is still only a limited understanding of hydroxycarbamide absorption, distribution, metabolism and clearance [6]. The metabolic pathways include saturable hepatic metabolism and also degradation by urease-producing intestinal bacteria [5]. It is cleared relatively rapidly, mainly via the kidneys, with a plasma half life of 2–4 h [6].
Pharmacodynamics
Hydroxycarbamide is thought to act by inhibiting the ribonucleotide reductase system, thus blocking the formation of deoxyribonucleotides and thereby inhibiting DNA synthesis [1].
Pharmacogenetics
Analysis of polymorphisms in candidate genes for aspects of hydroxycarbamide pharmacokinetics and pharmacodynamics, including ribonucleotide reductase and specific hydroxyurea drug transporters, did not reveal any clinically significant associations [6].
Potential adverse effects
Hydroxycarbamide appears to have an acceptable safety profile [7], and in particular it has little hepatic or renal toxicity [3, 4].
Myelosuppression
Dose-related bone marrow toxicity, causing anaemia, leucopenia or thrombocytopenia, is the most common adverse effect of hydroxycarbamide, although clinically significant complications are rare provided that there is close haematological monitoring [3]. Mild megaloblastic changes, which are common and of little significance [5], are unrelated to vitamin B12 or folate deficiency [1] and are a good indicator of compliance [3].
Gastrointestinal adverse effects
Diarrhoea, constipation, nausea, vomiting and stomatitis may occur [1]; upper gastrointestinal symptoms may be reduced by administration of hydroxycarbamide with food, milk or antacids [5].
Cutaneous adverse effects
Hydroxycarbamide is associated with a number of cutaneous side effects including leg ulceration [8], a dermatomyositis-like eruption [9, 10, 11], a lichen planus-like rash [12], alopecia [5], lupus erythematosus [13], photosensitivity [5], radiation recall [5] and hyperpigmentation of the skin and nails [14, 15, 16].
The mutagenic potential of hydroxycarbamide and its impairment of DNA repair mechanisms may increase the long-term risk of non-melanoma cutaneous malignancy [17, 18, 19, 20], particularly in patients being treated for myeloproliferative disorders. Although it is prudent to offer standard advice regarding sun protection, the risk to patients being treated for dermatological conditions appears to be low [3, 5, 7]. Similarly, although hydroxycarbamide has been associated with the development of leukaemic change when used in myeloproliferative disorders, myelodysplasia has not been established as a long-term concern in dermatological usage [3, 8].
Miscellaneous
Rarely, hydroxycarbamide has been associated with acute pulmonary inflammation, abnormal liver function, neurological symptoms, pyrexia, hypersensitivity and hallucinations.
Contraindications
Hydroxycarbamide is contraindicated if there is significant pre-existing bone marrow dysfunction, or if there is known sensitivity to the drug.
Cautions
Hydroxycarbamide should be avoided during pregnancy and breastfeeding, and men should be advised to avoid conception during treatment and for 3 months after its discontinuation [1].
Drug–drug interactions
Hydroxycarbamide is associated with few drug interactions likely to be of clinical relevance when used for dermatological indications [3].
- Other myelosuppressive and antineoplastic drugs should be avoided.
- Hydroxycarbamide may enhance the potential side effects of nucleoside reverse transcriptase inhibitors such as didanosine and stavudine.
Pre-treatment screening
Prior to commencing hydroxycarbamide, screening as outlined in Box 19.1 should be undertaken.
Dose and regimens
When used for cutaneous disorders, an initial dose of 1 g once daily is recommended [3]. Depending on clinical response and haematological parameters, this can be gradually increased to 2 g daily, in a divided dose. In the elderly and those with renal impairment, a starting dose of 500 mg daily should be considered [3].
Monitoring
It is reasonable for the full blood count to be checked weekly for the first month, then fortnightly for the next 3 months, monthly for a further 3 months, then quarterly thereafter [5]. Dose adjustment or temporary cessation of therapy should be considered if significant myelosuppression occurs [3]. Monthly electrolyte, creatinine and liver function tests, reducing to every 3–6 months if stable, is also reasonable [5].
Methotrexate
MTX is an antimetabolite that has been used as a chemotherapeutic agent since the early 1950s. It also has immunosuppressive and anti-inflammatory effects and is used therapeutically in a variety of rheumatological, gastrointestinal, neurological and dermatological inflammatory disorders. It has been used as an abortifacient.
Dermatological uses (see Chapters 35, 41, 42, 49, 50, 53, 54, 55, 56, 57, 97, 98, 102 and 140)
In the UK, MTX is licensed for and highly effective in the treatment of severe psoriasis. Off-label it is used (often as a steroid-sparing measure) in immunobullous disorders (pemphigus, bullous pemphigoid, cicatricial pemphigoid and epidermolysis bullosa acquisita), connective tissue diseases (dermatomyositis, lupus erythematosus and scleroderma), vasculitides, neutrophilic dermatoses (pyoderma gangrenosum and Sweet syndrome) and in a variety of other inflammatory (such as atopic eczema, sarcoidosis, cutaneous Crohn disease and chronic idiopathic urticaria) and proliferative (mycosis fungoides, Sézary syndrome, pityriasis lichenoides and pityriasis rubra pilaris) disorders [1].
Pharmacological properties
Formula and structure
MTX (empirical formula: C20H22N8O5, systematic name: (2S)-2-[(4-{[(2,4-diaminopteridin-6-yl)methyl](methyl)amino}benzoyl)amino]pentanedioic acid), abbreviated to 4-amino-N10-methyl folic acid, is a weak organic acid with a very similar structure to folic acid (pteroylglutamic acid).
Administration
In dermatological usage, MTX is usually taken orally in weekly (rather than daily) doses, although it can also be administered by intramuscular, intravenous, subcutaneous or intralesional routes. In the UK it is available in 2.5 mg and 10 mg tablets.
Pharmacokinetics
MTX is absorbed from the gastrointestinal tract rapidly and efficiently, actively transported by a carrier-mediated uptake system, reduced folate carrier 1 (RFC1). RFC1 is a saturable protein transporter and therefore the bioavailability of oral MTX (which demonstrates considerable interindividual variation) declines with higher doses [2]. Bioavailability can be improved with parenteral administration.
MTX is widely distributed in body tissues, although penetration of the blood–brain barrier is poor. In the circulation it is approximately 50% protein bound and, if displaced from protein by other drugs (such as aspirin, NSAIDs or sulphonamides), the increase in the level of active (non-bound) MTX will increase the risk of significant side effects, such as pancytopenia. Furthermore, alterations in circulating albumin levels (e.g. as the result of severe liver or kidney disease) may necessitate an adjustment of dosage.
MTX is actively transported into cells by RFC1. Contrary to the historical perception that it is not metabolized to a significant degree, approximately10% of MTX is converted to 7-hydroxymethotrexate in the liver and in all cells a proportion of MTX and 7-hydroxymethotrexate is transformed by γ-glutamyl hydrolase (GGH) to pharmacologically active polyglutamate derivatives [2]: this process may delay cellular clearance and contribute to MTX toxicity [3].
MTX and 7-hydroxymethotrexate are excreted mainly through the kidneys via glomerular filtration and active transport, although a small proportion is excreted into the bile. If renal function is compromised, a reduced dosage may be necessary to prevent accumulation and minimize adverse effects. Other weak acids, such as salicylates, probenecid and sulphonamides, may interact with MTX at this level.
The serum half-life of MTX is approximately 6–8 h and the drug is undetectable in the serum after 24 h, although intracellular accumulation is of long duration.
Pharmacodynamics
The mechanisms of action of MTX are complex. It inhibits a number of key enzyme systems. By virtue of it being a structural analogue of folic acid, MTX blocks the metabolism of folic acid through competitive inhibition of dihydrofolate reductase (DHFR), which catalyses the conversion of DHF to tetrahydrofolate (THF), a single-carbon transfer source essential to the generation of purine and pyrimidine nucleotides and therefore for nucleic acid and protein synthesis (Figure 19.2). It also inhibits thymidylate synthase (TS), which converts deoxyuridine monophosphate by methylation to the nucleotide thymidine monophosphate (dTMP), which is essential for normal DNA replication. Additionally, MTX inhibits the enzyme methylenetetrahydrofolate reductase (MTHFR), reducing the conversion of methylenetetrahydrofolate (methylene-THF) to methyltetrahydrofolate (methyl-THF), which is the methyl donor for the recycling of homocysteine to methionine: by inhibiting this recycling, MTX may prevent the accumulation of polyamines that contribute to inflammatory injury [4]. MTX also inhibits the enzyme 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) transformylase (ATIC), which plays important roles in de novo purine metabolism (by catalysing the reversible conversion of 10-formyl THF to THF) and inflammatory regulation.
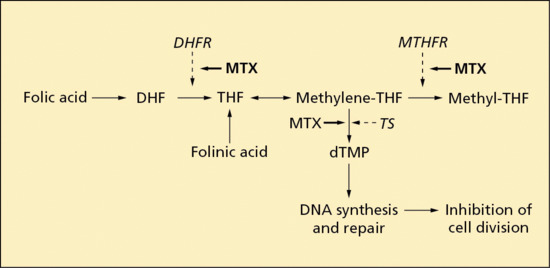
Figure 19.2 Methotrexate interaction with the folate metabolic pathway (see text for abbreviations).
Whilst the suppression of transmethylation reactions and the reduction of purine and pyrimidine synthesis by the inhibition of folate-dependent enzymes may play important roles in the antiproliferative and immunosuppressive properties of MTX, particularly in relation to lymphocytes which are dependent on de novo synthesis of nucleotides, there is a growing body of evidence supporting adenosine as a key mediator of the anti-inflammatory action of MTX through the potent inhibition of ATIC [2]. Inhibition of ATIC by MTX and hydroxymethotrexate results in an intracellular accumulation of AICAR which inhibits adenosine monophosphate (AMP) deaminase. This in turn causes the release of intracellular adenine nucleotides into the extracellular space, where they are dephosphorylated to adenosine, an endogenous purine nucleoside and an important signalling molecule mediating cytokine secretion and expression of adhesion molecules in immunologically competent cells. In the peripheral circulation and in inflammatory exudates, adenosine binds to specific cell surface receptors (belonging to the family of G-protein-coupled receptors), where it modulates a wide variety of physiological functions: these include the regulation of acute and chronic inflammatory processes, although the precise mechanisms for this regulation are still uncertain [2].
Pharmacogenetics
There are functionally significant polymorphisms in the genes encoding RFC1, DHFR, TS, MTHFR, ATIC and GGH, and very probably other elements of the folate metabolic pathway, although their use in a particular individual as predictors of therapeutic response to and toxicity of MTX remains to be defined. Single-nucleotide polymorphisms in the gene (SLC19A1) encoding RFC1 have been shown to influence intracellular folate levels [5]. Polymorphisms of DHFR, MTHFR, TS, ATIC and GGH have been associated with MTX toxicity and poor therapeutic response, although there are many conflicting data [6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16].
Potential adverse effects
Myelotoxicity
Most cases of dose-dependent MTX-induced bone marrow suppression occur during the first 2 months of treatment, although this can occur at any time during therapy and may result in pancytopenia or any combination of anaemia, leucopenia or thrombocytopenia, with potentially lethal consequences. Folate supplementation provides some protection. Particular risk factors include poor kidney function, old age, displacement of MTX from protein-binding sites (particularly by NSAIDs or sulphonamides) and hypoalbuminaemia. Inadvertent daily (rather than weekly) MTX dosing greatly increases the risk of pancytopenia. Should significant myelosuppression occur during MTX therapy, prompt folinic acid rescue should be considered [1].
Hepatotoxicity
MTX may be associated with an acute transaminitis or, in the long term, liver fibrosis [17]. The underlying mechanism of MTX-induced hepatic fibrosis appears to be associated with the adenosine A2A G-protein-coupled receptor [18]. Co-morbidities, such as obesity, high alcohol intake, previous hepatic problems and diabetes, may be significant aggravating or confounding factors. MTX-induced cirrhosis often has a fairly non-aggressive clinical course [19] and advanced hepatic fibrosis is uncommon [20].
Gastrointestinal toxicity
MTX commonly causes nausea and anorexia: vomiting, diarrhoea and stomatitis are occasionally encountered. Folate supplementation and/or administration of MTX parenterally can reduce gastrointestinal side effects [1].
Nephrotoxicity
Although high-dose MTX as used in the treatment of malignant conditions may cause renal damage from precipitation in the renal tubules, this is an unlikely consequence of low-dose therapy [1].
Reproductive toxicity
MTX is teratogenic and an abortifacient and therefore should be avoided in pregnancy. It is excreted in breast milk and should thus be avoided by breastfeeding women. MTX can impair fertility by adversely affecting oogenesis and spermatogenesis, and by causing menstrual dysfunction: these effects are considered to be reversible on discontinuing treatment [21]. It also has a mutagenic potential and thus it would seem prudent to advise both men and women to avoid conception whilst taking MTX and for up to 6 months thereafter [21].
Pulmonary toxicity
MTX-induced pulmonary toxicity, although well recognized in rheumatoid arthritis, is uncommon in psoriatic patients but pneumonitis or pulmonary fibrosis can occur with low doses of MTX and is a serious, unpredictable and potentially fatal adverse reaction. It may develop shortly after initiating MTX [22, 23] or after many years [24]. The development of cough and dyspnoea should prompt immediate cessation of MTX and appropriate investigation and treatment.
Malignancy
There appears to be an increased risk of systemic lymphoma with long-term MTX therapy, although this has been reported only infrequently in patients with dermatological disorders [25]. The available data do not allow any firm conclusions as to whether MTX predisposes to cutaneous malignancy.
Drug–drug interactions
See Box 19.9.
Miscellaneous
Other adverse effects of MTX include fatigue, headaches, dizziness, alopecia, phototoxicity, ‘recall’ reactions at sites of sunburn or radiotherapy, anaphylaxis, acral erythema, vasculitis and cutaneous ulceration [1].
Folate supplementation
Low-dose MTX is generally well tolerated. However, a number of toxic adverse reactions that appear to result from the inhibitory effect of MTX on purine and pyrimidine synthesis, such as myelotoxicity, hepatotoxicity and stomatitis, may limit its long-term use. Folate supplementation, which has as its rationale that folic acid competes with MTX for DHFR, has been shown in a number of principally rheumatological studies to reduce the adverse reactions to MTX, particularly those related to the gastrointestinal tract, liver and bone marrow, thus enabling MTX to be better tolerated without impairing its clinical efficacy [4, 26]. Two recent dermatological studies, however, have suggested a detrimental effect of folate supplementation on MTX efficacy [27, 28] and so the issue remains contentious. However, the balance of evidence favours supplementation [26], although the optimal dosage regimen remains uncertain.
Folinic acid (leucovorin, N5-formyl-THF) is as effective as folic acid in reducing MTX toxicity [26]. By direct conversion to THF it bypasses the step catalysed by DHFR, thus allowing some purine and pyrimidine synthesis despite DHFR inhibition by MTX. Additionally, folinic acid competes with MTX for RFC1 intracellular transportation. Folinic acid is particularly useful in the ‘rescue’ of bone marrow and gastrointestinal cells from MTX toxicity and acute MTX overdose. To avoid impairing intracellular uptake of MTX, folinic acid should not be taken within 12 h of the weekly MTX dose.
Furthermore, both folic acid and folinic acid reduce plasma homocysteine levels during MTX therapy and therefore theoretically may benefit those patients at risk of cardiovascular disease [26].
Contraindications
MTX is contraindicated in the treatment of pregnant or breastfeeding women and if there is known MTX hypersensitivity. Significant impairment of hepatic and renal function, pre-existing blood dyscrasias, immunodeficiency and latent infection (such as tuberculosis, viral hepatitis B and C) are relative contraindications, as are excess alcohol consumption and concern regarding the reliability of the patient to conform to necessary monitoring procedures [1].
Cautions
Where, as in the UK, there are two strengths of tablet (2.5 and 10 mg) available, confusion regarding dosage can easily occur. It is recommended that only one strength, usually 2.5 mg, is prescribed.
Pre-treatment screening
Potential recipients of MTX should be counselled about the risks of immunosuppressive therapy, provided with written details of the medication and warned about drug–drug interactions. Prior to commencing MTX, screening as outlined in Box 19.1 should be undertaken.
Pregnancy should be excluded before initiating treatment and contraception (ideally employing two methods) carefully discussed. A pre-treatment chest X-ray is probably not necessary unless the patient is at increased risk of tuberculosis, as in some ethnic communities or immigrants from countries with a high prevalence of tuberculosis.
Dose and regimens
For the treatment of dermatological conditions, the administration of MTX once weekly is standard practice (although the weekly dose may be divided into three doses given at 12 h intervals over a 24 h period: this may be particularly helpful for patients who suffer nausea with MTX). If oral MTX causes unacceptable nausea or if a patient is non-compliant with oral therapy, it can be given as a weekly intramuscular or self-administered subcutaneous (or, very occasionally, intravenous) injection.
It is common dermatological practice to give a test dose of 2.5–5 mg, followed by a full blood count and liver function tests after 1 week to detect idiosyncratic reactions to MTX, and thereafter for the dose to be gradually increased by 2.5–5 mg/week until there is a satisfactory response without significant toxicity. Ordinarily, the dose should not exceed 25 mg weekly and, in view of the reduced bioavailability of oral MTX at higher doses, consideration should be given to switching to a subcutaneous route of administration if response remains poor. Once an optimal response has been achieved, the dose of MTX can be tapered by 2.5 mg each week to determine the lowest dose necessary to maintain clinical benefit.
It is also standard practice to administer oral folic acid, an example regimen being 5 mg once weekly, usually on the day following MTX, although the timing is not crucial [29].
Oral folinic acid, three doses of 5 mg at 12 h intervals commencing 24 h after the weekly MTX dose, should be considered if folic acid is not improving gastrointestinal symptoms or correcting abnormalities of the liver enzymes or macrocytosis.
Monitoring
Close monitoring is required until MTX dosage has been stabilized, particularly for elderly patients and those with renal impairment. Initially, the full blood count, liver function tests and creatinine should be checked every 1–2 weeks for 4–8 weeks following the last dose escalation; thereafter the frequency of blood tests can be gradually reduced to 3-monthly. Deterioration in blood parameters or an intercurrent illness requires resumption of closer monitoring and consideration of dose reduction. Macrocytic red blood cell indices are common during MTX therapy.
During the early months of treatment, the opportunity should be taken at clinic attendances to reinforce warnings regarding symptoms of bone marrow suppression (bruising, sore throats, mouth ulcers), the importance of contraception and the avoidance of drugs that might interact with MTX.
The development of abnormal liver function tests can be a sign of MTX damage, but liver enzyme levels may be normal despite the presence of significant hepatic fibrosis. Elevation of liver transaminases to double the upper limit of the normal range necessitates discontinuation or dose reduction of MTX. There is continuing uncertainty over the optimal method for identifying and monitoring liver fibrosis [30]. Routine liver biopsies for the histological assessment of hepatic structure, for many years considered an integral part of MTX usage, are no longer justified in view of the attendant risks of the procedure and the low absolute risk of significant fibrosis [20, 31]. There are non-invasive tests of liver function that can assist in predicting significant hepatic toxicity, including a variety of serum fibrosis markers, including procollagen-III aminoterminal-propeptide [32, 33], hyaluronic acid [33, 34] and tissue inhibitor of matrix metalloproteinase-1 [35]. Any deterioration in these more sensitive tests of hepatic function should prompt referral to a hepatologist for consideration of measurement of liver elasticity [36] and possible liver biopsy.
Mycophenolate mofetil
Mycophenolate mofetil (MMF) is a potent immunosuppressant prodrug of mycophenolic acid (MPA), used primarily to prevent solid-organ graft rejection, but it is increasingly being employed off-label in the treatment of a variety of immunologically mediated dermatological conditions as a single agent or as a steroid-sparing drug.
Dermatological uses (see Chapter 50)
MMF has a predictably beneficial effect in the treatment of immunobullous disorders (in particular pemphigus and pemphigoid), and a less consistent effect in psoriasis, atopic eczema, connective tissue disorders and vasculitides [1, 2, 3, 4].
Pharmacological properties
Formula and structure
MMF (empirical formula: C23H31NO7, systematic name: 2-morpholinoethyl (E)-6-(1,3-dihydro-4-hydroxy-6-methoxy-7-methyl-3-oxo-5-isobenzofuranyl)-4-methyl-4-hexenoate) is the 2-morpholinoethyl ester of MPA.
Administration
When given for dermatological reasons, MMF is usually administered orally, although there is an intravenous preparation. An enteric-coated form of MPA is also available (720 mg of enteric-coated MPA is therapeutically equivalent to 1000 mg of MMF).
Pharmacokinetics
MMF is absorbed efficiently and completely from the gastrointestinal tract and is rapidly converted to its active metabolite, MPA, which is 97% bound to plasma albumin. The half-life of MPA is approximately 16 h. The peak plasma level occurs at about 1 h, followed by a second peak at 6–12 h that is the result of enterohepatic recirculation.
MPA is metabolized predominantly in the liver by uridine diphosphate glucuronosyl transferase to its phenolic glucuronide (MPAG), which is pharmacologically inactive but which can be converted back to MPA by β-glucuronidase, especially in the epidermis and gastrointestinal tract.
Biliary excretion of MPA/MPAG involves several transporters including organic anion transporting polypeptides and multidrug-resistant protein 2 (MRP-2). More than 90% of the administered dose of MMF is excreted in the urine, mainly as MPAG (and involving, at least in part, MRP-2) with the remainder eliminated in the faeces.
The pharmacokinetics of MPA are complex and there is substantial intersubject variation [6].
Pharmacodynamics
Like AZA, the mechanism underlying the immunosuppressive action of MMF involves purine biosynthesis. Purine nucleotides are formed by either a complex de novo pathway or are recycled by a salvage pathway (in which bases and nucleosides released in the breakdown of nucleic acids are recovered and converted back into nucleotides).
Adenine and guanine, the purine nucleobases, are both derived from the nucleotide inosine-5′-monophosphate (IMP), which is the first compound in the de novo pathway of purine synthesis to have a completely formed purine ring. MPA interferes with this process by inhibiting the action of IMP dehydrogenase (IMPDH) in catalysing the conversion of IMP to xanthine monophosphate, the rate-limiting step to the de novo formation of guanine nucleotides (guanosine mono-, di- and triphosphates), which have important roles in a variety of biological processes including the synthesis of DNA and RNA.
This effect of MMF is specifically targeted towards T and B lymphocytes as, in contrast to other nucleated cells, they lack the ability to produce purines via a salvage mechanism and are thus critically dependent for their DNA/RNA synthesis, growth and proliferation on the de novo synthesis of purines. Interruption of the de novo pathway by MMF therefore results in a relatively selective immunosuppressive effect on lymphocytes which, amongst other immunological effects, includes the suppression of cell-mediated immune responses and the inhibition of antibody production by activated lymphocytes.
Additionally, humans have two IMPDH genes, encoding hIMPDH1 and hIMPDH2 isoenzymes, which have similar structural and kinetic properties [7]. MPA has a fivefold greater inhibitory effect on the type 2 isoform of IMPDH, which is expressed in activated lymphocytes, than the type 1 isoform that is expressed preferentially in other cell types, thereby exerting a more potent cytostatic effect on lymphocytes than other cell types [8].
Furthermore, several other factors may contribute to the anti-inflammatory effects of MPA. MPA can induce apoptosis of activated T lymphocytes; it can inhibit the presentation of antigen to T lymphocytes by dendritic cells; by depleting guanosine nucleotides, it can suppress glycosylation and the expression of certain adhesion molecules, thereby decreasing lymphocyte and monocyte recruitment in sites of inflammation; and also, by depleting guanosine nucleotides, MPA depletes tetrahydrobiopterin, thus suppressing the production of nitric oxide by the inducible form of nitric oxide synthase and consequent tissue damage mediated by peroxynitrite [8, 9, 10, 11].
Clinical response to MMF is slow and typically takes 6–8 weeks [1].
Pharmacogenetics
A number of polymorphisms have been identified in the genes encoding IMPDH, uridine diphosphate glucuronosyltransferase, organic anion transporting polypeptides and MRP-2: although these do not yet have a known clinical relevance, they are likely to have a significant influence on the pharmacokinetics and pharmacodynamics of mycophenolate [12].
Potential adverse effects
Gastrointestinal toxicity
The most common side effects of MMF are gastrointestinal and dose dependent [2]. Nausea, vomiting, abdominal pain, diarrhoea and constipation occur in up to 20% of patients [2]. These are usually mild and tend to improve with time. Administration with food and the use of enteric-coated MPA may help to ameliorate these adverse effects [1]. MMF is not considered to be hepatotoxic [1]. Gastrointestinal ulceration and cytomegalovirus-associated colitis may rarely occur [1].
Haematological toxicity
Anaemia, neutropenia and thrombocytopaenia are not uncommon, and are usually mild, dose related and reversible with discontinuation of therapy or dose reduction [2]. MMF has also been associated with pseudo-Pelger–Huët anomaly, a form of neutrophil dysplasia in which the nuclei tend to be bilobed rather than hypersegmented [13].
Infections
MMF, by dampening the normal immune response, appears to increase susceptibility to opportunistic infections, particularly in organ transplant recipients and specifically to cytomegalovirus and BK virus [14], although other herpesvirus infections (varicella zoster and herpes simplex), bacterial sepsis, atypical mycobacterial and fungal infections have been reported in the dermatological literature [1]. Systemic immunosuppression carries with it the risk of reactivation of other latent virus infections, such as hepatitis B, hepatitis C and JC virus (associated with progressive multifocal leukoencephalopathy), as well as reactivation of dormant tuberculosis [1]. Conversely, MMF has antimicrobial activity against certain pathogens, including hepatitis C, HIV and Pneumocystis jirovecii [14]. Mucocutaneous candidosis, urinary tract infections and pneumonia are commonly associated with MMF therapy [5].
Carcinogenicity
The magnitude of the potential for MMF specifically to cause lymphoproliferative malignancy is uncertain, and much of the available evidence relates to the transplant population receiving combinations of immunosuppressive agents [5]. However, the development of Epstein–Barr virus related B-cell lymphoma involving the central nervous system has been recorded with MMF used to treat autoimmune conditions [1, 15].
There is no clear evidence base for an association between MMF and non-melanoma cutaneous malignancy [1], but it would appear prudent to offer sun-protection advice [5].
Teratogenicity
Despite limited data, MMF appears to be teratogenic when given in early pregnancy, being particularly associated with early miscarriage and with craniofacial and cardiac malformations [16, 17, 18, 19]. Appropriate measures should be taken to minimize the risk of pregnancy in women of childbearing potential [20].
Drug–drug interactions
See 19.10.
Miscellaneous
Genitourinary symptoms, including frequency, dysuria, urgency, haematuria and sterile pyuria, may occur during the first year of treatment [2]. Tremor, dizziness, anxiety, depression, confusional states, dysgeusia, headache, electrolyte abnormalities, hypercholesterolaemia, rashes (urticaria, hand dermatitis, alopecia), hypo- and hypertension, dyspnoea, cough, interstitial lung disease, pulmonary fibrosis, arthralgia and pyrexia are also recorded [5].
Contraindications
MMF is contraindicated in pregnancy, breastfeeding women and in those with a history of hypersensitivity to it [5].
Cautions
MMF should be used with caution in patients with active gastrointestinal disease, and live attenuated vaccines should be avoided.
Pre-treatment screening
Before commencing treatment with MMF, the items listed in Box 19.1 should be addressed. When appropriate, contraceptive counselling should be undertaken, and two forms of contraception, commencing 4 weeks prior to initiating treatment and continuing for 6 weeks after discontinuation of treatment, should be instituted. A pregnancy test within 1 week of starting treatment should be performed.
Dose and regimens
Twice-daily dosing is recommended, and a reasonable starting dose for dermatological indications is 250 mg twice daily for the first week, then 500 mg twice daily, increasing thereafter if tolerated by 500 mg daily or twice daily every 4 weeks until a maximum of 1.5 g twice daily is reached. Dosage should be tailored to individual tolerance and response. Patents should be reminded that the clinical response is slow.
Monitoring
The full blood count should be checked weekly for the first month, then fortnightly for 2 months, then monthly for the first year [21]. Thereafter, if stable, the full blood count should be checked every 2–3 months.
Electrolytes, creatinine and liver function tests should be monitored every 2–4 weeks following dose escalation, then every 2–3 months [1].
Potassium iodide
Medicinally, potassium iodide is used principally as an emergency treatment for hyperthyroidism, and for thyroid protection during treatment with radiopharmaceuticals and following exposure to nuclear radiation.
Dermatological uses (see Chapters 32, 47, 48 and 99)
Potassium iodide is also a potentially useful, traditional and inexpensive off-label option in a variety of cutaneous disorders, including cutaneous sporotrichosis, erythema nodosum, subacute nodular migratory panniculitis, nodular vasculitis, Sweet syndrome, Behçet syndrome, pyoderma gangrenosum and erythema multiforme [1, 2, 3].
Pharmacological properties
Formula and structure
Potassium iodide is an inorganic ionic compound, with chemical formula K+I−.
Administration
Potassium iodide can be given as drops of a saturated solution of potassium iodide (SSKI), but in the management of dermatological conditions it is most conveniently given in tablet form.
Pharmacokinetics
Potassium iodide is well absorbed orally, and is distributed to the thyroid and, to a lesser extent, to the salivary glands, breasts, choroid plexus and gastric mucosa [2]. It crosses the placenta and is found in breast milk [2]. It is excreted predominantly in urine, with small amounts excreted in sweat and faeces [2].
Pharmacodynamics
The mechanism by which potassium iodide exerts its anti-inflammatory effects is unknown, although there is evidence that it has a detrimental effect on neutrophil chemotaxis and the generation by neutrophils of pro-inflammatory oxygen intermediates [1, 2, 3].
Potential adverse effects
Wolff–Chaikoff effect
Potassium iodide can induce the Wolff–Chaikoff effect, which is the inhibition of organic binding of iodide in the normal thyroid gland by excess iodide. This is a protective mechanism, blocking thyroid hormone synthesis, and predisposing to temporary hypothyroidism [4, 5]. Resolution usually occurs within several weeks, although thyroid replacement therapy may need to be considered [1].
Jod–Basedow phenomenon
There is a risk that the therapeutic administration of potassium iodide to a person with thyroid dysfunction lacking pituitary control, such as Graves disease or a multinodular goitre, may cause a significant exacerbation of hyperthyroidism [2, 5].
Potassium toxicity
Rarely, potassium iodide may cause symptoms of hyperkalaemia, including fatigue, confusion, palpitations, muscle weakness and numbness and tingling of the extremities.
Other adverse effects (iodism)
- Sialadenitis (in particular parotid and submaxillary glands).
- Hypersalivation.
- Metallic taste, halitosis, burning sensation in the mouth.
- Coryzal symptoms, headache.
- Gastric irritation, diarrhoea, nausea.
- Urticaria/angio-oedema, acneform pustulation, iododerma.
Contraindications
Potassium iodide is contraindicated in pregnancy and if there is known hypersensitivity to iodine. It has been associated with fetal malformations [6] and may cause hypothyroidism in the fetus [1].
Cautions
Potassium iodide should be used with caution in patients with thyroid and cardiac disorders, with conditions associated with hyperkalaemia, or with active tuberculosis [3]. It may exacerbate dermatitis herpetiformis, pustular psoriasis [1] and hypocomplementaemic vasculitis [3].
Drug–drug interactions
Potassium iodide may interact with antithyroid drugs, lithium, amiodarone and sulphonamides to cause hypothyroidism, and with potassium-sparing diuretics and angiotensin-converting enzyme inhibitors to cause hyperkalaemia [2].
Pre-treatment screening
Specific enquiries should be made regarding a history of thyroid disease and the use of medications that might interfere with thyroid function. Suspicion of thyroid disease should prompt the checking of thyroid function (T4, thyroid-stimulating hormone (TSH) and circulating thyroid antibody). A baseline check of electrolyte and urea levels should be undertaken.
Dose and regimens
For dermatological conditions, a reasonable initial dose is 300 mg daily, increasing to 900 mg daily depending on disease activity and tolerability.
Monitoring
The TSH level should be checked 1 month after initiation of therapy to exclude iodide-induced hypothyroidism [2] and, if abnormal, it would be prudent to monitor thyroid function thereafter. It is reasonable to monitor potassium levels.
Biological therapies (protein therapeutics)
Biological therapy (also known as protein therapeutics) is a term that encompasses, in its broadest sense, a group of pharmacologically active protein (or peptide)-based molecules produced by living organisms that are designed to alleviate disease by inhibiting or imitating the actions of naturally occurring proteins in the body. Insulin could be considered the very first ‘biological’ therapy, purified from bovine and porcine pancreas in 1922 to treat diabetes, while pooled immunoglobulins (IVIg), introduced in the 1980s, remain an important intervention for a number of dermatological diseases. However, the advent of recombinant DNA technology, which allows the large-scale production of recombinant proteins with highly specific structure and function, together with a better understanding of the complex role of endogenous proteins in health and disease (including enzymes, receptors, membrane channels and molecule ‘transporters’), has led to an exponential increase in drug development in the field of biological therapy. Biological therapies include: those that are identical to an endogenous protein and function either to replenish or to enhance an endogenous supply (e.g. interferons, pooled immunoglobulins; and those proteins that target a specific pathway), using the specificity of the antigen-recognition site of immunoglobulins (monoclonal antibodies) or the receptor-binding domains of native protein ligands (immunoadhesins) to guide the immune system to destroy the relevant molecule or cell. Their structure can be inferred in part by nomenclature (Figure 19.3).
Figure 19.3 The International Nonproprietary Names (INN) of biological substances infer their function. For monoclonal antibodies (mAbs), the INNs are composed of: (i) a prefix, which is random, only required to give the molecule a distinctive name: a substem A indicates the target molecule, e.g. -k(i) interleukin; -l(i) immunomodulating; and -t(u) tumour; (ii) a substem B indicates the species on which the immunoglobulin sequence of the mAb is based; and (iii) a suffix – the common stem for all mAbs is -mab.
This therapeutic field confers many potential advantages over small molecule pharmacology. A high degree of functional specificity and extracellular binding should result in fewer ‘off target’ actions compared to small molecules, which often have direct intracellular effects with consequent hepatoxicity and other toxic effects. However, this does not necessarily translate to safer drugs, since the complexities of biological systems are such that interruption of a particular pathway may nevertheless result in undesired pharmacodynamic effects. Reactivation of latent tuberculosis with TNF-antagonist therapy for psoriasis is one such example, as was the catastrophic trial of the CD28 agonist which invoked a ‘cytokine storm’ in the six volunteers involved [1]. Further disadvantages include the fact that, since these drugs are proteins, all those currently available for dermatological conditions (and, at present, also for the majority of other indications) have to be given parenterally, and also that immunogenicity remains a significant problem, even in the context of ‘fully human’ monoclonal antibodies such as adalimumab.
The number of biological therapies available for use is rapidly increasing and the disease indications constantly changing; drug acquisition costs are high, and so use is generally limited to severe recalcitrant disease, although access may improve with the advent of ‘biosimilars’ (a biological medicine that is similar to another biological medicine that has already been authorised for use, without any meaningful differences in terms of quality, safety or efficacy). Treatment with biological therapies should always be supervised by clinicians who are experienced in their use, and, because these are (generally) novel treatments, participation in long-term pharmacovigilance registries actively encouraged. Brief detail is provided below on a number of biological therapies with a focus on those agents that are licensed for use in skin disease; however, the list is not comprehensive and additional information is available in the relevant disease chapters.
Biological therapies directed against cytokines
Cytokines are potent regulators of inflammation, and their neutralization by antibodies or receptor antagonists can be an extremely effective therapeutic strategy as well as a major contributor to understanding the underlying disease pathogenesis.
Tumour necrosis factor antagonists
TNF is a pro-inflammatory cytokine produced by a wide variety of cell types including keratinocytes. It plays a complex role in innate immunity and host defence, particularly against mycobacterial infection, and can both enhance and suppress adaptive immunity. Over the last 15 years, it has been shown to play a central role in the pathogenesis of a number of chronic inflammatory disease states, evidenced in large part by the striking clinical efficacy of TNF antagonists in disorders such as psoriasis, rheumatoid arthritis and Crohn disease. There are currently two types of biological agents that target TNF: the first binds directly to TNF and is represented by the monoclonal antibodies adalimumab, certolizumab, golimumab and infliximab; the second, represented by the fusion protein etanercept, a soluble TNF receptor, which competitively inhibits the binding of TNF to TNF receptors so inhibiting the biological activity of TNF.
Dermatological uses
Licensed indications (see Chapter 35)
Adalimumab, etanercept and infliximab have all been shown to be very effective in psoriasis in large-scale randomized controlled trials (RCTs). These interventions now form a standard part of the treatment approach for severe psoriasis (and also psoriatic arthritis). Golimumab and certolizumab are currently licensed only for use in psoriatic arthritis although they have also proved efficacious in psoriasis [2, 3].
Off-label uses (see Chapters 47, 48, 92 and 98)
TNF antagonists are used in a wide spectrum of inflammatory skin conditions, largely based on observational data and/or small trials. These include skin diseases associated with inflammatory bowel disease and rheumatoid arthritis where TNF antagonists are of proven efficacy: examples include pyoderma gangrenosum, Sweet syndrome, hidradenitis suppurativa and Behçet disease. Severe treatment-resistant cutaneous sarcoidosis has also been reported to benefit from infliximab and adalimumab [4, 5], whereas etanercept is of no benefit (and possibly harmful). Case reports of positive treatment responses to TNF antagonists (mainly infliximab) exist for an even wider spectrum of diseases [6], but require further study. Proper assessment of the risk/benefit ratio in all these ‘off label’ indications is hampered by a lack of controlled trial data, poorly defined treatment outcomes and limited follow-up.
Pharmacological properties
Formula and structure (see Figure 19.3)
Infliximab is a chimeric human–murine monoclonal antibody (~25% mouse-derived protein), whereas golimumab and adalimumab are fully human IgG1 antibodies. Etanercept is a fusion protein composed of a dimer of the extracellular portions of human TNFR2 (p75) fused to the Fc domain of human IgG1. Certolizumab pegol is a humanized anti-TNF Fab′ fragment conjugated to a polyethylene glycol that increases the half-life of the attached molecule and may also reduce immunogenicity.
Administration
All TNF antagonists are given parenterally and, with the exception of infliximab, can be self-administered subcutaneously (Table 19.2).
Table 19.2 Dose and regimens.
| Infliximab | Adalimumab | Etanercept | |
| Route | IV, over 2 h | s/c | s/c |
| Dosea | 5 mg/kg week 0, 2, 6 and then 8 weekly | 80 mg (week 0), 40 mg week 1 and then 40 mg every other week | 50 mg weekly |
| Bioavailability | [100%] | 64% | 76% |
| Terminal half-life (days) | 8–9.5 | 14 | 3–4 |
aFor psoriasis, licensed indication.
Pharmacokinetics
Following subcutaneous administration the drugs are slowly absorbed, probably predominantly via lymphatics, to reach peak plasma concentrations after 2–4 days. The disposition of TNF antagonists, as with other monoclonal antibodies and protein-based molecules, is complex and incompletely understood (see Chapter 14) [7]. It is affected by multiple interrelated factors including body weight (which is associated with more rapid clearance of adalimumab), the degree of drug binding to TNF within target tissues such as skin and synovium (so-called target-mediated drug disposition) and drug immunogenicity (see later). Trough plasma drug levels do, however, correlate with treatment efficacy and explain some of the variability in treatment response. In contrast to small molecules, renal filtration and biliary secretion are not relevant to drug elimination, so dose adjustment is not required in the event of impaired renal or hepatic function.
Drug immunogenicity
In common with biological agents in general, all TNF antagonists can generate an immune response with consequent development of antidrug antibodies [8, 9]. These are increasingly recognized as an important mechanism underlying treatment failure either by competing with endogenous ligand (TNF) and preventing the neutralizing effect of the TNF antagonist or through formation of drug/antidrug antibody complexes and accelerated drug clearance. These mechanisms may coexist and may also contribute to the adverse event profile. Whilst the majority of patients develop antidrug antibodies, typically within the first 6 months of therapy, not all lose treatment efficacy, as antibodies may be transient, or at a low level. This interindividual variability in development and amplification (or tolerance) of immune response appears to relate to multiple factors, including route and dosing schedule (e.g. an initial high dose and continuous dosing may confer a reduced risk), route of administration, patient factors (e.g. genetic factors, level of disease activity) as well as drug-specific considerations. Notably, methotrexate [9] (and possibly other immunosuppressants) significantly reduces the risk of development of antidrug antibodies and is often co-prescribed with TNF antagonists. The clinical utility of drug and antidrug antibody level evaluation to optimize drug dosing and strategies to minimize or avoid antibody formation are subject to ongoing research.
Pharmacodynamics
TNF is released from a wide variety of cells including keratinocytes as a soluble cytokine (sTNF) following cleavage from its cell surface bound precursor (tmTNF). Both sTNF and tmTNF are biologically active and bind to either of two distinct receptors: TNF receptor 1 (TNFR1, p55) and TNF receptor 2 (TNFR2, p75). This leads to NF-κB activation (which promotes inflammation) and/or cell apoptosis. In addition, tmTNF can itself act as a ligand via a process of reverse signalling to induce cell activation, cytokine suppression or apoptosis of the tmTNF-bearing cell.
All TNF antagonist biological agents specifically bind both soluble and transmembrane forms of TNF and act by: (i) blocking TNFR-mediated mechanisms; and (ii) inducing tmTNF (reverse-signalling) events. Etanercept also binds members of the lymphotoxin family (LTα3 (also known as TNF-β) and LTα2β1), whereas certolizumab, because it lacks an Fc′ portion, cannot bind complement or cause antibody-dependent cell-mediated cytotoxicity (in vitro). Whether these differences in function account for differences in overall efficacy and/or safety in clinical practice is unclear.
Adverse effects
Principal side effects include injection site reactions, infusion reactions (infliximab) and infection. Reactivation of tuberculosis is a particular risk, given the role of TNF in host defence against mycobacterial infection and granuloma formation, and frequently presents with extrapulmonary or other atypical site involvement. Heart failure (both new onset and worsening of pre-existing heart disease) and demyelinating disorders have also been reported. Paradoxically, new-onset psoriasis (including palmoplantar pustulosis) and sarcoidosis have developed in patients on TNF antagonists, particularly when given for rheumatological indications or Crohn disease, illustrating that the impact of TNF blockade on what is a highly complex cytokine network is not always predictable. Idiosyncratic hepatitic reactions, autoimmune hepatitis, cytopenias, pancytopenias and serious opportunistic infections are also rarely reported. Ongoing concern exists about whether or not TNF antagonists confer an increased risk of malignancy, particularly in relation to lymphoma and skin cancer. So far, the data are reassuring but largely relate to relatively short-term use in psoriasis (i.e. less than 5 years) and data from non-dermatological indications such as rheumatoid arthritis. Risks in patients with psoriasis and other inflammatory skin diseases may be different and data currently being collected in a number of registries in the UK [10], Europe [11] and the US [12] will be important in establishing the risk of malignancy in the pertinent populations.
Contraindications
Contraindications include the presence of severe cardiac failure (New York Heart Association class III/IV), a personal history of demyelinating disease, active infection or untreated latent tuberculosis, and a current or recent past history of malignancy (unless the malignancy has been diagnosed and treated more than 5 years previously, and /or where the likelihood of cure is high, e.g. adequately treated non-melanoma skin cancer). Live and live attenuated vaccinations may potentially lead to severe or fatal infections.
Cautions
Patients with multiple co-morbidities, who are older or have a history of cancer may be at increased risk of serious adverse events and should be carefully reviewed and monitored [13]. Caution is also required when considering use in patients with chronic viral infections including hepatitis B, C and HIV due to risks of reactivation and/or progression and lack of information on safety. Use during pregnancy and whilst breastfeeding should be avoided whenever possible. IgG is actively transported from mother to fetus via the neonatal Fc receptor (FcRN) from around 14 weeks, so that all the TNF antagonists with the exception of certolizumab, which lacks an Fc portion, would be expected to cross the placenta during the second and third trimester, and measureable drug levels in infants born to mothers on infliximab and etanercept have been reported [14]. TNF antagonists are also excreted in breast milk. Surgery whilst on TNF therapy is associated with an increased risk of postoperative infection. In all these instances, specialist advice and consideration on a case-by-case basis with reference to relevant clinical guidelines are strongly recommended.
Drug–drug interactions
There are no clinically important interactions between TNF antagonists and small-molecule drugs. With the exception of methotrexate, co-administration with other immunomodulatory agents, including biological therapies, should generally be avoided due to the potential for increased risk of infection.
Pre-treatment screening
All patients should have a comprehensive history taken and a thorough clinical examination and appropiate investigations performed to identify contraindications to or cautions for therapy, with particular focus on the presence or risks of infection, including active tuberculosis, demyelination or cancer. Planned conception, pregnancy and travel, given the risk of tuberculosis and possible vaccination requirements, should be discussed. Screening for latent tuberculosis should include a chest X-ray and a tuberculin skin test. In instances where this is unreliable, however, as in bacille Calmette–Guérin-vaccinated individuals or those on immunosuppressants, an in vitro IFN-γ release assay (IGRA) tests (QuantiFERON®-TB Gold In-Tube, Cellestis, Australia, and T-Spot®.TB, Oxford Immunotec, UK) should be performed [15]. In patients with investigations supportive of a diagnosis of latent tuberculosis, treatment with antituberculous therapy may be indicated, although it is worth noting that this only reduces the risk of reactivation of latent tuberculosis during subsequent TNF antagonist therapy by 50%, so a high index of suspicion should remain during therapy. Relevant national guidelines [16] are helpful since specific details on recommended pre-treatment screening and treatment for latent tuberculosis [15] will vary depending on population and individual patient risk profiles. Where available, patients should be encouraged to participate in national long-term safety registries.
Monitoring
Careful clinical review and advice to patients to seek early medical review in the event of new symptomatology, as well as routine blood investigations (full blood count, renal and liver function tests) are part of the routine monitoring of patients on TNF therapy. Annual IGRA testing (for those who are negative at treatment outset) may be useful in those at especially high risk of tuberculosis (e.g. people who work or live in high-risk settings, or who travel to high prevalence countries).
Other cytokine antagonists
IL-23/IL-17 antagonists
Drugs that target IL-23 and IL-17 include the anti-p40 monoclonal antibody ustekinumab and agents currently in the late stages of clinical development such as the anti IL-23 mAb guselkumab [17] and the IL-17 antagonists secukinumab, ixekizumab and brodalumab [18]. The striking efficacy of all of these agents in psoriasis underpins the critical importance of the IL-23/Th17 axis in the disease pathogenesis; in time, their clinical utility may well extend to other immune-mediated skin diseases.
Ustekinumab (see Chapter 35)
Ustekinumab is licensed for use in plaque psoriasis (and also psoriatic arthritis [19]). A small series suggested efficacy in palmoplantar pustulosis [20] but this has not been confirmed in a subsequent RCT [21]. There have also been recent reports of its value for treating pityriasis rubra pilaris but formal studies have yet to be undertaken (see Chapter 36).
- Pharmacological properties
- Formula and structure. Ustekinumab is a fully human IgG1κ monoclonal antibody that binds with specificity to the shared p40 protein subunit of human cytokines IL-12 and IL-23.
- Administration. It is given by self-administered subcutaneous injection.
- Pharmacokinetics. The estimated bioavailability of ustekinumab is 57% following a single subcutaneous injection, with a median half-life of approximately 3 weeks (range 15–32 days). Enhanced drug clearance occurs with increasing body weight and the presence of ustekinumab antidrug antibodies [22].
- Pharmacodynamics. IL-12 and IL-23 are heterodimeric cytokines secreted by activated antigen-presenting cells and share a common protein subunit, p40. IL-12 activates CD4 and NK cells to induce expression of type 1 cytokines (TNF and IFN-γ), whilst IL-23 stimulates survival and proliferation of a subset of T cells that produce IL-17 (Th17 cells). Ustekinumab binds specifically to the p40 protein subunit thereby preventing IL-12 and IL-23 from binding to the IL-12Rβ1 receptor protein expressed on the surface of T cells, NK cells and antigen-presenting cells. The marked efficacy of ustekinumab in psoriasis is likely to be mainly attributable to the inhibition of IL-23, given the central role for the IL-23/IL-17 pathway in the pathogenesis of psoriasis.
- Potential adverse effects. Ustekinumab is generally well tolerated, with nasopharyngitis, headache, upper respiratory tract infection and injection site reactions being the most commonly reported adverse effects in clinical trials; rarely, serious infections may also occur. IL-12 and IL-23 have been implicated in atherogenesis, and there has been a reported possible excess of major adverse cardiovascular events in RCTs involving use of ustekinumab and another monoclonal antibody, briakinumab, with specificity for the p40 subunit: the latter was subsequently withdrawn from clinical development [23]. Long-term safety data for ustekinumab are limited to 5 years [24].
- Contraindications. These include active infection or untreated latent tuberculosis, a current or recent past history of malignancy (unless the malignancy has been diagnosed and treated more than 5 years previously, and /or where the likelihood of cure is high, e.g. adequately treated non-melanoma skin cancer) and live vaccination within 2 weeks preceding the first injection or within 15 weeks after the last injection.
- Drug–drug interactions. There are no known specific drug interactions. Co-therapy with other immunosuppressant drugs should generally be avoided due to risk of excess immunosuppression and limited data on the safety of such combinations.
- Pre-treatment screening. The recommended pre-treatment assessment for ustekinumab is comparable to that for TNF antagonists [16, 25].
- Dose and regimens. The standard licensed dosing regimen is as a fixed dose according to body weight (<100 kg, 45 mg; >100 kg, 90 mg) at week 0, 4 and then at 12 weekly intervals thereafter. Off-label use of escalated dosing schedules is occasionally used in poor responders, for example, increasing the dose from 45 mg to 90 mg (in those who are <100 kg) or increasing the frequency of injections (45 or 90 mg) to 8 weekly [26, 27].
- Monitoring. As for TNF antagonists [16, 25].
IL-1 antagonists
IL-1 plays a central role in a wide range of inflammatory conditions, and there are a number of novel biological agents targeting IL-1, both approved and in development, that may be of potential therapeutic benefit in pustular skin diseases such as autoinflammatory disorders, Behçet syndrome and SAPHO syndrome (synovitis, acne, pustulosis, hyperostosis and osteitis) [28, 29, 30]. All three agents detailed below are licensed for use in the ‘orphan indication’ cryopyrin-associated autoinflammatory syndromes that include familial cold autoinflammatory syndrome (FCAS) and Muckle–Wells syndrome (see Chapter 45).
Anakinra is a recombinant IL-1 receptor antagonist and acts to competitively inhibit both IL-1α and IL-ß binding to the IL-1 receptor. It is administered as a single daily self-administered subcutaneous injection (100 mg/day in adults or 1–2 mg/kg/day). Bioavailability is 95%, with rapid renal clearance (median half-life 4–6 h). Adverse effects include injection site reactions (very common), allergic reactions, anaphylaxis (rare), infection including serious infections, neutropenia and raised liver enzymes [31, 32].
Canakinumab is a human IgG1κ anti-IL-1β monoclonal antibody that acts by binding to endogenous IL-1β and thus preventing interaction with IL-1 receptors. It is administered as a single dose (initially 150 mg or 2 mg/kg escalating as indicated by clinical response) with maintenance therapy at 8 weekly intervals [33]. The bioavailability is 66% with a mean half-life of 26 days (comparable to other therapeutic monoclonal antibodies). The adverse event profile is similar to anakinra [34].
Rilonacept (IL-1 trap) is a dimeric fusion protein that contains in a single chain the extracellular domains of IL-receptor type 1 and IL-1 receptor accessory protein fused to the human Fc portion of IgG. It acts as a soluble decoy receptor to block primarily IL-1β signalling and, to a lesser extent, IL-1α and endogenous IL-1 receptor antagonist (IL-1ra) signalling. It is available only in the USA.
B-cell directed biological therapies
Available B-cell directed therapy has until recently been limited to rituximab, a potent B-cell depleting anti-CD20 monoclonal antibody that was originally developed for the treatment of a wide range of B-cell malignancies including cutaneous B-cell lymphoma. However, belimumab, now licensed for use in systemic lupus erythematosus, is one of a number of new recombinant antibodies targeting surface-bound proteins on B cells [35, 36] that may have clinical utility in autoimmune/inflammatory skin disease (Figure 19.4).
Figure 19.4 B-cell directed biological therapies. In common with rituximab, ofatumumab and ocrelizumab both target CD20 but with enhanced complement- and antibody-dependent cell-mediated cytoxicity [36]. Belimumab binds to B-cell activating factor of the tumour necrosis factor (TNF) family (BAFF; also known as B-lymphocyte stimulator (BLyS), a crucial soluble B-cell survival factor, to prevent activation of TNF receptor superfamily member 13C (BAFF receptor (BAFF-R) or BLyS receptor 3 (BR3)), TNF receptor superfamily member 17 (B-cell maturation antigen (BCMA)) and TNF receptor superfamily member 13B (transmembrane activator and cyclophilin ligand interactor (TACI)). Atacicept is a fusion protein containing the extracellular, ligand-binding portion of TACI and the modified Fc portion of human immunoglobulin G (IgG): it binds and neutralizes both BAFF and a second soluble B-cell activating factor known as a proliferation-inducing ligand (APRIL). Both belimumab and atacicept show efficacy in systemic lupus erythematosus and have potential applications in autoimmune skin disease [36, 42, 43]. Drugs given in italics are in clinical development. Adapted from Faurschou and Jayne 2014 [36] and Vincent et al. 2014 [42].
Rituximab
Dermatological uses (see Chapters 50, 51, 52, 53, 54, 55 and 102)
Rituximab is licensed for use, in combination with GCs, for the induction of remission in adults with antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis (granulomatosis with polyangiitis (GPA, previously known as Wegener granulomatosis) and microscopic polyangiitis) [36, 37]. It is used off-label in a wide spectrum of severe autoimmune and chronic inflammatory diseases including vasculitis, pemphigus, systemic lupus erythematosus, dermatomyositis and primary Sjögren syndrome, usually in combination with other immunomodulatory agents [36, 38].
Pharmacological properties
Formula and structure
Rituximab is an IgG1, chimaeric mouse/human anti-CD20 monoclonal antibody.
Administration
Rituximab is given as a slow intravenous infusion. Premedication with an analgesic/antipyretic (e.g. paracetamol), an antihistaminic drug (e.g. chlorphenamine) and a GC (e.g. methylprednisolone) is given to reduce or avoid infusion reactions.
Pharmacokinetics
In non-malignant conditions, the mean terminal half-life following IV infusion is around 20 days (range 9–35 days depending also to some extent on the dose), with metabolism and elimination as for IgG in general (see Chapter 14); population pharmacokinetic modelling indicates that body surface area and gender (men have a larger volume of distribution and clearance) are the most significant co-variates to explain interindividual variability in pharmacokinetic parameters, although gender differences are not considered clinically relevant.
Pharmacodynamics
CD20 is a four transmembrane phosphoprotein that is specifically expressed on the cell surface of B cells and is regulated by differentiation. It is expressed during the transition from pre-B to immature cell in the bone marrow, and on naïve, activated and memory B cells in the circulation and tissue, but is lost on differentiation into plasma cells. Following binding of the rituximab Fab fragment to CD20, B-cell lysis occurs, primarily via antibody-dependent cellular cytotoxicity mediated by one or more of the Fcγ receptors on the surface of granulocytes, macrophages and NK cells, although other mechanisms have also been implicated. Naïve B cells disappear rapidly from peripheral blood circulation and also, to a more variable extent, from tissues, depending on the pathology and setting. Circulating B-cell populations recover to normal over the ensuing 3–6 months. Since the CD20 antigen is not expressed by pro- or pre-B cells or by terminally differentiated plasma cells, rituximab does not prevent regeneration of CD20-positive B cells from precursor cells and does not directly interfere with the production of immunoglobulins. Demonstration of efficacy in diseases where autoantibodies per se have not historically been considered to be of direct pathogenic significance (e.g. rheumatoid arthritis, dermatomyositis and systemic sclerosis) reflects the increasingly recognized, highly diverse nature of B-cell function including antigen presentation, cytokine production, lymphoid organ remodelling and immune cell regulation (by regulatory B cells) [39].
Adverse effects
Infusion reactions are common and occur in up to 25% of patients following the first infusion; most are mild to moderate in degree and decrease in severity with subsequent infusions. There is a predisposition to infection, including serious and opportunistic infections, herpes zoster and candidosis (rate of clinically significant infection in rheumatoid arthritis trials 0.05 per patient-year); progressive multifocal leukoencephalopathy has been reported in patients treated for lymphoproliferative disease and, very rarely, for systemic lupus erythematosus [42, 43]. Cytopenias including neutropenia may occur months after treatment. The cohort of patients receiving rituximab tend to have severe recalcitrant disease and have therefore necessarily received significant, often very long-term immunosuppression prior to treatment, which probably further increases the risks of infection. Human antichimaeric antibodies develop in about 25% of patients treated and can be associated with worsening of infusion or allergic reactions and failure to deplete B cells, although not predictably so.
Contraindications
These include those with active infection; severely immunocompromised patients (e.g. with hypogammaglobulinaemia or where levels of CD4 or CD8 are very low); uncontrolled heart disease or heart failure (New York Heart Association grade IV); and live vaccination within 4 weeks of infusion and/or whilst B-cell depleted.
Cautions
Patients with pre-existing heart disease should be closely monitored as a variety of cardiac events have been reported during infusions (e.g. angina, cardiac arrhythmias) and infusion reactions may be poorly tolerated. Caution should be exercised when considering the use of rituximab in patients with a history of recurring or chronic infections or with underlying conditions that may further predispose patients to serious infection [40].
Drug–drug interactions
There are no known specific drug interactions.
Pre-treatment screening
To minimize risk, rituximab should be instigated only by clinicians experienced in its use, in a multidisciplinary environment and where a comprehensive pre-treatment history, clinical and investigation protocol is in place. Screening prior to rituximab should aim to identify in particular those with cardiac disease and any current or past infection at risk of progression or reactivation. This includes a past history of tuberculosis, risk factors for or presence of active infection together with relevant screening tests for latent tuberculosis and blood-borne viral infections (HIV, hepatitis B and C) [40, 41]. Fulminant hepatic failure following hepatitis B reactivation is well documented in the oncology literature, although rituximab may not be absolutely contraindicated in the presence of positive hepatitis B or C serology, given appropriate antiviral prophylaxis (for hepatitis B) and relevant expert hepatology advice [40]. Vaccination status against Pneumococcus and influenza as well as any travel plans likely to require live vaccination should be checked. The importance of avoidance of pregnancy during and for 12 months post-infusion should be emphasized. Commonly recommended additional investigations include full blood count, renal and liver function, serum immunoglobulins and lymphocyte subsets.
Dose and regimens
Dosing schedules have varied in different clinical trials and according to the disease being treated. For most dermatological conditions, a single cycle of treatment is given (usually 375 mg/m2 or 1 g total dose, weekly for 2–4 weeks) in the context of concomitant immunosuppressant agents such as corticosteroids. Subsequent cycles may be given on disease relapse if necessary, but not usually until 6 months have elapsed.
Monitoring
Ongoing monitoring is necessary for signs of infection, neurological disturbance. Routine blood investigations (full blood count, renal and liver function tests) should be carried out at regular intervals (e.g. monthly) or more frequently in the event of abnormalities. Immunoglobulins and lymphocyte subsets should be checked prior to any subsequent infusions.
Miscellaneous biological therapies
Omalizumab
Omalizumab was first licensed for use in severe allergic (IgE-mediated) asthma. More recently, it has been shown to be effective in chronic urticaria.
Dermatological uses (see Chapter 42)
It is licensed for use as an add-on therapy for the treatment of chronic spontaneous urticaria in adults and adolescents (12 years and above) with inadequate response to H1 antihistamine treatment. Off label, it is used in other forms of urticaria [44] and treatment-resistant atopic eczema, although with variable outcomes [45, 46].
Pharmacological properties
Formula and structure
Omalizumab is a recombinant humanized monoclonal antibody (IgG1κ) against the Cε3 domain of IgE.
Administration
It is given by subcutaneous injection.
Pharmacokinetics
Following injection, the absolute bioavailability is 62%, reaching peak serum concentrations after an average of 6–8 days; metabolism and elimination is as for IgG in general (see Chapter 14) via the reticuloendothelial system and by targeted binding (IgE/omalizumab complex formation), with a mean elimination half-life of 24 days.
Pharmacodynamics
Omalizumab binds specifically to free IgE only, since Cε3, the IgE antigenic determinant recognized by omalizumab, constitutes part of the high-affinity IgE receptor (FcεR) binding site. This results in lower serum levels of free IgE, prevention of IgE binding to FcεRI and subsequent down-regulation of FcεRI expression on basophils, mast cells and dendritic cells. Omalizumab–IgE immune complexes may also sequester allergens/autoantigens, and, through down-regulation of IgE-expressing B lymphoblasts and memory B cells, reduce the number of IgE-secreting plasma cells. These mechanisms clearly play a key role in the treatment effect where urticaria is caused by autoreactive IgG antibodies against FcεRI and/or IgE or autoreactive IgE antibodies against autoallergens. In other forms of urticaria the precise mechanism of action is unclear, although its effectiveness points to the IgE/mast cell axis being central to all forms of urticaria.
Potential adverse effects
The most common adverse effects include headache, sinusitis, joint pain, upper respiratory tract infection and injection site reactions. Reported adverse events that will require longer term, larger scale pharmacovigilance studies that properly control for confounders to evaluate true risk include arterial thromboembolic events (such as stroke, transient ischaemic attack, myocardial infarction), anaphylaxis, theoretical concerns around immune complex mediated pathology and abnormal immune responses to parasitic infections [47].
Contraindications
These include hypersensivity reactions to omalizumab or injection excipients.
Cautions
Type 1 allergic reactions including anaphylaxis have been reported following omalizumab, usually within 2 h of injection but occasionally up to 24 h post-injection. Patients should be warned about this possibility and injections always given under medical supervision (see monitoring). Omalizumab may present particular risk to patients with a susceptibility to helminthic/other parasitic infections in endemic areas.
Drug–drug interactions
No specific drug interactions have been reported.
Pre-treatment screening
This should include routine history, clinical assessment and blood tests (full blood count, liver function tests, renal function). Dosing for asthma (only) is based on serum IgE.
Dose and regimens
The licensed dosing for chronic urticaria is 300 mg subcutaneously every 4 weeks. Injections should be given under supervision with post-treatment monitoring for allergic reactions for 1–2 h.
Monitoring
Routine bloods investigations should be undertaken, in particular to exclude thrombocytopenia [45].
C1-esterase inhibitor replacement therapy
Three forms of C1-esterase inhibitor (C1INH) replacement therapy are approved for use in hereditary angio-oedema (type 1 and 2): Cinryze® and Berinert®, both of which are derived from human plasma, pasteurized and nano-filtered to reduce the risk of infection transmission; and Rhucin®, which is a recombinant human C1INH concentrate purified from rabbit breast milk [48, 49]. CINH replacement therapy is given as an intravenous infusion, either as short-term prophylaxis prior to procedures at high risk of triggering an attack (e.g. surgery, extensive dental work) or as an emergency intervention during an acute attack. Cinryze is also licensed for the ‘routine’ prevention of recurrent hereditary angio-oedema (e.g. 1000 IU every 3–4 days). Acute anaphylaxis (rarely) and the theoretical transmission of as yet unidentified infectious agents are the principal adverse effects.
Intravenous immunoglobulin therapy
IVIg refers to the intravenous infusion of high doses of human IgG pooled from the plasma of thousands of healthy donors. As well as its use as replacement therapy in primary and secondary immunodeficiency syndromes with impaired antibody production, IVIg has immunomodulatory actions and is licensed in the UK for use in primary immune thrombocytopenia, Guillain–Barré syndrome and Kawasaki disease [1].
Dermatological uses (see Chapters 38, 42, 50, 51, 52, 53, 54, 55, 56 and 59)
Off-label, IVIg, either as monotherapy or in combination with other immunomodulating drugs, has been utilized in a variety of autoimmune and inflammatory dermatoses including autoimmune bullous disorders (pemphigus, pemphigoid, epidermolysis bullosa acquisita and linear IgA disease), autoimmune connective tissue disorders (dermatomyositis, systemic sclerosis and systemic lupus erythematosus) and miscellaneous other conditions including chronic autoimmune urticaria, graft-versus-host disease and scleromyxoedema [2].
Pharmacological properties
Formula and structure
The IgG antibodies present in IVIg preparations reflect those present in the normal population, with the proportions of its four subclasses approximating to the in vivo state [1].
Administration
IVIg is given intravenously and should be administered in accord with the infusion rate recommended for the particular brand being used. Adequate hydration of the patient should be established prior to use [1].
Pharmacokinetics
Following infusion, IVIg is distributed relatively rapidly between intra- and extravascular compartments, with equilibrium after 3–5 days [1]. IVIg crosses the placenta and may be excreted into breast milk [2]. The half-life of the immunoglobulins, which are gradually degraded by cells of the reticuloendothelial system, is approximately 4 weeks [1].
Pharmacodynamics
The mechanisms underlying the immunomodulatory actions of IVIg remain poorly understood, and in particular the apparent contradiction between the well-established pro-inflammatory properties of IgG antibodies and the anti-inflammatory activity of high-dose pooled IgG has yet to be reconciled. Suffice to say that IVIg has complex effects on both the innate and adaptive components of the immune system that act in the direction of immune homeostasis. These include the blockade of Fc receptors on B lymphocytes by ‘natural’ antibodies, resulting in a reduction of pathogenic autoantibody and cytokine production, and B-cell apoptosis; blocking of Fc receptors on macrophages (particularly by IgG in IVIg with sialylated Fc, thereby inhibiting the secretion of pro-inflammatory cytokines) and on granulocytes, thus inhibiting degranulation; saturation of Fc receptors leading to increased clearance of pathogenic antibodies; anti-idiotypic antibodies in IVIg neutralizing the effect of pathogenic antibodies; inhibition by natural antibodies of complement-mediated cellular damage, including interference with the formation of the membrane attack complex; inhibition by natural antibodies of certain cytokines (including TNF-α and IFN-γ); inhibition of the maturation and function of dendritic cells, imposing on them a tolerogenic phenotype; and expansion of the population of regulatory T cells (T-regs) and enhancement of their suppressive functions [3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13]. However, the precise mechanistic process for the anti-inflammatory action of IVIg remains to be definitively established.
Pharmacogenetics
Little is known about the pharmacogenetic aspects of IVIg therapy, although there is some evidence that genetic factors may influence clinical response to IVIg in Kawasaki syndrome [14].
Potential adverse effects
In general IVIg is considered to have a good safety profile, with side effects tending to be mild and transient and with a low incidence of serious adverse effects [15, 16, 17, 18]. However, older patients tend to be at greater risk of acute renal failure and venous and arterial thrombosis [18].
General infusion-related effects
Symptoms, usually mild, may occur during the course of the infusion of IVIg and include fatigue, malaise, shivering, raised temperature, flushing, headache, myalgia, arthralgia, back pain, chest tightness, dyspnoea, nausea, vomiting, diarrhoea, rashes, blood pressure variation and tachycardia [1, 15]. These side effects usually settle if the infusion rate is slowed or temporarily discontinued and may be pre-empted in susceptible individuals by pretreatment with analgesia, antihistamines, NSAIDs or low-dose intravenous corticosteroid [2].
Acute kidney injury
Rarely IVIg may cause renal dysfunction by inducing osmotic nephrosis, in which cells of the proximal renal tubules are damaged [19, 20, 21, 22]. Risk factors for osmotic nephrosis include age (>65 years), diabetes, pre-existing renal impairment, dehydration, concomitant administration of other nephrotoxic drugs [21] and the use of a sucrose-stabilized brand of IVIg [20, 21]. Circulating rheumatoid factor and cryoglobulins may also be risk factors [2]. Gradual recovery of renal function is the rule [21], although there is a risk of fatal outcome [19]. If any risk factors are present, diuretics should be avoided for the period around the transfusion, hydration of the patient should be monitored and the infusion rate should be reduced [21].
Haemolysis
Haemolytic transfusion reactions with IVIg are uncommon, although non-O blood group recipients with an underlying inflammatory disorder appear at particular risk [23]. They have been linked to the presence of anti-A and anti-B haemagglutinins in the IVIg preparation and may be both IgG and complement mediated [23]. Haemolysis is manifest by a drop in haemoglobin following IVIg transfusion, elevated unconjugated bilirubin, elevated lactate dehydrogenase, a positive direct antiglobulin (Coombs’) test and evidence of spherocytes and polychromasia on a peripheral blood film [23].
The immediate management of IVIg-related haemolysis is to stop the transfusion and, if red cell replacement is required, group O cells should be given [23]. To minimize the risk of further haemolytic transfusion reaction for a particular individual, a different IVIg product containing a lower titre of ABO haemagglutinins may be considered, together with cross-matching between the IVIg preparation and the recipient [23].
Thrombotic events
By increasing blood viscosity and thereby altering the rheological properties of the blood and decreasing its flow, IVIg is associated with an increased risk of venous and arterial thrombosis and subsequent embolic complications, including deep-vein thrombosis, pulmonary embolism, myocardial infarction and cerebrovascular accidents [24, 25, 26, 27, 28]. In 50% of cases such thrombotic events may be manifest during the IVIg infusion, although detection may not occur until later [25], with arterial thrombosis tending to be evident before venous thrombosis [24]. IVIg-related thrombosis, especially arterial, is associated with significant mortality, with the risk factors being older age, arterial hypertension, atherosclerosis, obesity, immobility, dehydration, diabetes, history of thrombosis, hyperviscosity syndromes and hypercholesterolaemia [1, 24, 25]. The risk of thrombotic events may be reduced in those considered to be particularly vulnerable by administering IVIg at a slow rate, giving a lower dose, and using prophylactic aspirin or low-molecular-weight heparin [26].
IgA-mediated anaphylaxis and hypersensitivity reactions
The presence of trace amounts of IgA in IVIg preparations may induce allergic reactions and even anaphylaxis in otherwise symptomless IgA-deficient recipients with circulating anti-IgA antibodies of IgG or IgE class [29]. However, such IgA-dependent transfusion reactions are rare. Strategies have been developed to prevent IgA-related anaphylaxis in patients with a history of hypersensitivity [29].
Aseptic meningitis
Aseptic meningitis, presenting with headache, fever, photophobia, neck stiffness and vomiting, is a rare adverse reaction to IVIg, tending to occur within 48 h of the infusion [30]. Patients with pre-existing migraine may be more susceptible [30]. Examination and culture of cerebrospinal fluid is usually necessary to exclude bacterial meningitis. The prognosis for aseptic meningitis is good, and usually symptomatic treatment is all that is required. Ensuring adequate pre-treatment hydration, premedication analgesia or antihistamine and using a slow rate of infusion may enable continuation of IVIg therapy in patients who have experienced aseptic meningitis [30].
Infection risk
As a biological product derived from pooled human plasma, IVIg carries the potential risk of transmission of pathogens, which is minimized by the use of donor selection, screening of donations and plasma pools for specific markers of infection, scrupulous preparation hygiene and validated techniques for the removal or inactivation of infectious agents [1, 31]. Nonetheless, the possibility of transmitting infection with known and unknown organisms cannot be totally excluded [1] and for medicolegal reasons it has been suggested that a pre-treatment serum sample from the recipient be stored [2], although this does not appear to be a general view.
Contraindications
Severe anaphylaxis resulting from a previous infusion is a contraindication to further use; the presence of risk factors for acute kidney injury and thrombosis are relative contraindications.
Cautions
The recommended infusion rate should not be exceeded. IVIg administration may impair the efficacy of live attenuated virus vaccines for up to 3 months (1 year in the case of measles) [1].
Drug–drug interactions
For the period immediately before and after IVIg infusions, the administration of loop diuretics should be avoided [1].
Pre-treatment screening
When assessing the risk–benefit ratio of a course of IVIg, factors predisposing that individual to acute kidney injury and thrombosis should be assessed; it is reasonable to consider measuring serum IgA and undertaking a thrombophilia screen.
Before each cycle of IVIg, a full blood count, creatinine level and liver function tests should be checked. The hydration of the patient should be optimized, especially for the older person [18].
Dose and regimens
The standard regimen for high-dose IVIg for the treatment of dermatological conditions is 2 g/kg/cycle, based on the ideal weight of the patient, in divided doses over 2–5 days [1]. The rate of infusion should not exceed that recommended by the supplier. Cycles are repeated at approximately monthly intervals until effective disease control is obtained, after which the interval between cycles is gradually increased empirically up to 16 weeks, when IVIg can be discontinued if remission continues [2].
Fluid intake should be encouraged during and after the infusion. Consider paracetamol, with or without codeine, an NSAID or an antihistamine as a premedication if infusion-related symptoms have previously occurred. In those patients considered susceptible to thrombosis, aspirin or low-molecular-weight heparin are prophylactic options [26].
Monitoring
During and immediately after IVIg infusions, the vital signs, hydration status (to exclude both fluid overload and dehydration) and urine output should be monitored. Post-transfusion, a full blood count with film, and bilirubin level will screen for the possibility of haemolysis. Mobility should be encouraged in all patients to minimize the risk of thrombosis.
Systemic retinoids
The synthetic retinoids are a class of organic molecules derived from and with similar biological activity to the naturally occurring vitamin A group of retinoids, which includes retinol, retinal and retinoic acid.
Whilst the naturally occurring retinoids are involved in the regulation of diverse and important biological processes, not least cell signalling in embryogenesis, the synthetic retinoids exert their medicinal properties principally via their specific effects at the genome level on epidermal cell proliferation, differentiation and apoptosis, tumour suppressor gene expression and immune function [1, 2, 3, 4, 5]. Their development over the last six decades was initially prompted by the observed cutaneous effects of vitamin A deficiency, although their medical significance has subsequently extended far beyond the dermatological sphere. They are showing great potential in a number of different fields, including chemotherapeutic and chemoprotective use in a variety of haematological and solid malignancies; in metabolic diseases, as regulators of adipogenesis and as antidiabetic agents; in the prevention or treatment of neurodegenerative conditions; in certain renal disorders; and in stem cell-based regenerative medicine [6].
This section concerns those synthetic retinoids administered systemically for dermatological conditions, rather than the topical retinoid preparations. There are three generations of dermatologically useful systemic retinoids: first, the non-aromatic retinoids, represented by isotretinoin (13-cis retinoic acid) and alitretinoin (9-cis retinoic acid); second, the monoaromatic retinoids, including acitretin, a metabolite of etretinate, which it has now replaced; and third, the polyaromatic group, which includes bexarotene.
Dermatological uses (see Chapters 90, 91 and 92)
Isotretinoin is licensed for the treatment of severe acne resistant to adequate courses of standard therapy, although off-label it has been used in rosacea, hidradenitis suppurativa and dissecting cellulitis of the scalp.
Alitretinoin has a product license to treat severe chronic hand eczema (and has also been approved by the US Food and Drug Administration for the topical treatment of the cutaneous lesions of Kaposi sarcoma).
Acitretin is licensed in the UK for the treatment of severe psoriasis resistant to standard therapies, palmoplantar pustulosis, inherited ichthyoses and Darier disease. Off-license, it has been used as chemoprophylaxis to reduce the risk of actinically induced non-melanoma cutaneous malignancy [5, 7].
Bexarotene is indicated for the treatment of the cutaneous manifestations of advanced cutaneous T-cell lymphoma [8].
Retinoids have also been used off-license to treat pityriasis rubra pilaris, lupus erythematosus and lichen planus.
Pharmacological properties
Formula and structure
The basic structure of a retinoid has three parts: a hydrophobic trimethylated cyclohexene ring; a conjugated polyene linker side chain; and a hydrophilic polar moiety [6].
Isotretinoin (systematic name: 13-cis retinoic acid), acitretin (systematic name: (2E,4E,6E,8E)-9-(4-methoxy-2,3,6-trimethylphenyl)-3,7-dimethylnona-2,4,6,8-tetraenoic acid), alitretinoin (systematic name: (2E,4E,6Z,8E)-3,7-dimethyl-9-(2,6,6-trimethyl-1-cyclohexenyl)nona-2,4,6,8-tetraenoic acid) and bexarotene (systematic name: 4-[1-(3,5,5,8,8-pentamethyltetralin-2-yl)ethenyl]benzoic acid) are the four principal retinoids currently used in dermatological therapy.
Administration
All the systemic retinoids are administered orally.
Pharmacokinetics
The pharmacokinetics of retinoids are complex. The broad principles are absorption from the gut into intestinal mucosal cells, chylomicron-borne passage to the liver, intracellular processing by hepatocytes, secretion into blood (where they are bound to albumin) and finally uptake by and transport to the nucleus of the target cells [6]. This involves a sequence of intracellular transporter proteins that include cellular retinol-binding proteins, plasma retinol-binding proteins and cellular retinoic acid-binding proteins [6].
Being taken with food or milk enhances their bioavailability. In the circulation, they are bound to albumin, and are widely distributed: they cross the placenta and are secreted into breast milk.
Retinoids are metabolized via isomerization and oxidation by enzymes of the cytochrome P450 system, followed by glucuronidation into inactive water-soluble forms [5, 8], and biliary or renal elimination.
The elimination half-lives of the retinoids vary, with that of alitretinoin up to 10 h, isotretinoin approximately 20 h and acitretin 50–60 h. Isotretinoin and alitretinoin are endogenous retinoids and after cessation of treatment normal physiological levels are reached within 2 weeks for isotretinoin and a few days for alitretinoin [8]. Bexarotene has a clearance profile similar to isotretinoin [5].
Acitretin is cleared from the body within 1 month of stopping therapy [5]. However, because alcohol ingestion by patients on acitretin results in re-esterification to etretinate, which has a comparatively long half-life of 80–160 days, adequate contraceptive precautions are necessary for 2 years [5, 8].
Pharmacodynamics
In the target cells, retinoids enter the nucleus to regulate the transcription of a variety of target genes. The complex process of retinoid signalling involves two families of retinoid nuclear receptor, the retinoic acid receptors (RAR) and the retinoid X receptors (RXR) [9]. Each family has three isotypes (α, β and γ), and each isotype several isoforms [4, 6]. Each receptor has a DNA-binding domain and a ligand-binding domain. In order to regulate the expression of target genes, the receptors form dimers, each containing an obligatory RXR and either a second RXR (RXR-RXR) or an RAR (RAR-RXR). The receptor dimers bind to specific sequences of DNA (retinoic acid response elements) within the promoter regions of the target genes, and it is the conformational change in the receptor consequent to binding of the ligand that promotes the transcription process, resulting in mRNA and then protein formation. Isotretinoin and acitretin act as ligands for RAR, bexarotene is a ligand for RXR, and alitretinoin is a panagonist, binding to both RAR and RXR [4].
Details regarding the genes targeted by retinoids and the mechanistic processes by which their protein products influence epidermal cell and immune cell function remain to be fully elucidated.
In addition to this genomic action, retinoids also have non-genomic effects, in particular retinoylation, a post-translational modification of certain proteins, including cytokeratins [6].
Pharmacogenetics
Currently, there are no known pharmacogenetic factors of relevance to the clincal use of synthetic retinoids, but it seems very likely that their future use (in terms of optimizing clinical effectiveness and minimizing adverse effects) will be guided at an individual level by polymorphism profiling of a variety of genes involved in their pharmacokinetic and pharmacodynamic properties [10, 11, 12, 13].
Potential adverse effects
Teratogenicity
Natural retinoids, in particular retinoic acid, play a fundamental role in embryonic patterning, growth and organogenesis, and perplexingly excess retinoid activity results in many of the same embryonic developmental defects as vitamin A deficiency [6]. Fetal exposure to retinoids in early pregnancy puts at risk normal neurological, ocular, cardiovascular and renal development, as well as that of the pulmonary system, skeleton, pancreas and limbs [6]. Administration of retinoids to women during the first trimester of pregnancy may cause cranio-facial deformities (including cleft palate and external ear malformations), central nervous system abnormalities (such as hydrocephalus, microcephaly and cerebellar malformation), heart defects (tetralogy of Fallot, transposition of the great vessels and septal defects) and abnormalities of the thymus and parathyroid glands in up to 50% of pregnancies, with increased numbers of early and late stillbirths [5, 6].
There appears to be negligible risk of retinoid-induced embryopathy in fetuses fathered by men taking systemic retinoids [5].
Psychiatric
The relationship between retinoid therapy and psychiatric adverse effects, including depression, suicidal ideation and personality disorders, remains ill defined and controversial [14, 15]. The majority of the conflicting literature on this subject relates to isotretinoin for the treatment of acne vulgaris, although psychiatric adverse effects are documented for alitretinoin and bexarotene [8]. Acne per se is associated with anxiety, low self-esteem, social-phobic disorders, depression and even suicidal ideation, and there seems little doubt that isotretinoin may frequently be beneficial in alleviating the psychological effects of acne [16, 17, 18, 19, 20]. However, although the majority of retrospective and prospective clinical trials have failed to prove an association between oral isotretinoin therapy and psychiatric symptoms, some studies suggest that a proportion of patients treated with isotretinoin may develop depression [15]. Meta-analyses have either shown no relationship between oral isotretinoin usage and depression [21, 22] or have indicated a possible association [23, 24]. The mechanisms underlying such a putative effect may involve isotretinoin crossing the blood–brain barrier and modifying the expression of a variety of genes in the limbic system, the hippocampus in particular, thereby affecting the dopaminergic, serotonergic and noradrenergic regulation of mood and emotion [15].
In summary, depression may be a rare, idiosyncratic and unpredictable adverse effect of isotretinoin therapy [15, 19], and it would seem prudent to advise patients and members of their families to be watchful for signs and symptoms of depression and to make specific enquiries relating to mood and suicidal thoughts [25] at each review consultation. Severe or frequent headaches may be a warning of retinoid-induced depression [26]. It is not clear whether patients with a pre-existing history of psychiatric problems are at increased risk [5], but oral isotretinoin therapy should not automatically be refused to such individuals [15, 25, 27]; rather joint management with a psychiatric colleague should be undertaken, with measures in place to ensure that any deterioration in mental state is recognized at an early stage (including the use of a validated rating scale for anxiety and depression): introducing isotretinoin at a low dose (0.5 mg/kg body weight per day) before gradually increasing the dose of isotretinoin after 2 months if required may lessen the risk of this [15, 25].
Ocular
Dry irritated eyes caused by blepharoconjunctivitis, occasionally complicated by bacterial infection, results from retinoid-induced alterations in conjunctival epithelium [28] and decreased meibomium gland secretion [29]. These problems can be alleviated by artificial tears, and affected individuals should be advised to temporarily discontinue contact lens use.
Asymptomatic corneal opacities may occur with retinoid therapy but these do not adversely affect vision [5].
Retinal toxicity causing impaired night vision is a potential adverse effect of retinoids which may persist for a number of years [30, 31]. The possibility of this may justify electrophysiological screening in those individuals who have occupations that are dependent on satisfactory night vision.
Gastrointestinal
Of the retinoids, isotretinoin in particular has been implicated as a cause of inflammatory bowel disease, both ulcerative colitis and Crohn disease [5]. However, the evidence is conflicting [19], and, if indeed isotretinoin does predispose to inflammatory bowel disease, the overall risk is likely to be very low [19]. Patients with inflammatory bowel disease that require isotretinoin should be managed jointly with their gastroenterologist. Mild gastrointestinal upset is relatively common.
Musculoskeletal
Vitamin A toxicity may result in hyperostosis (in particular diffuse interstitial skeletal hyperostosis, DISH), premature epiphyseal closure, calcification of tendons and ligaments, and lowered bone mineral density [5]. However, under the clinical conditions that retinoids are normally used in the treatment of acne and keratinization disorders, there appears to be only a very low risk of these complications [5, 32, 33, 34]. The possibility, albeit rare, of premature epiphyseal closure should be considered in pre-adolescent children.
Myalgia is a common side effect, particularly in physically active patients on isotretinoin, and may be accompanied by elevated creatine phosphokinase levels [5].
Mucocutaneous
Dryness of the skin (particularly in atopic patients) and mucous membranes (especially the nasal lining, vermilion of the lips and conjunctivae) is very common and dose related. It may be associated with pruritus, facial erythema, impetiginization, asteatotic eczema and skin fragility. Epistaxis can be troublesome. Patients on acitretin occasionally complain of ‘sticky skin’. Emollient therapy is usually effective: a non-comedogenic preparation is recommended for acne-prone areas.
A temporary deterioration in acne frequently occurs when isotretinoin therapy is initiated but this does not normally require dose adjustment; very rarely, acne fulminans may develop [35]. Telogen effluvium, nail fragility, onycholysis and photosensitivity are occasional complications [5]. Isotretinoin-induced pyogenic granuloma-like lesions can arise in association with acne lesions and over nail folds: they tend to resolve on discontinuation of treatment [36]. There are rare post-marketing reports of severe skin reactions (Stevens–Johnson syndrome and toxic epidermal necrolysis) associated with isotretinoin therapy [37].
Neurological
Transient headaches are a common side effect, but very occasionally oral retinoids may cause benign intracranial hypertension (pseudotumor cerebri) with associated nausea, vomiting and visual disturbances, potentially with loss of vision [38], especially when isotretinoin is used concomitantly with an oral tetracycline.
Hyperlipidaemia
Dyslipidaemia, especially hypertriglyceridaemia but also hypercholesterolaemia, is a common consequence of retinoid therapy, particularly with bexarotene, and requires monitoring during retinoid therapy. The management of modest dyslipidaemia involves dietary modification, increased physical activity and weight control to reduce cardiovascular risk, with more severe impairment of lipid levels necessitating pharmacological intervention to minimize the risk of pancreatitis [39]. Retinoid-induced hyperlipidaemia is reversible on discontinuation of treatment [5].
Hepatotoxicity
Retinoid-induced elevations of liver transaminases are common, and, although usually of little clinical significance, require monitoring [5]. Severe hepatitis is rare and probably idiosyncratic.
Haematological
Neutropenia is a common laboratory observation with isotretinoin and bexarotene, and isotretinoin may result in platelet abnormalities and anaemia [8].
Endocrinological
Bexarotene [40] and alitretinoin [8] may both cause central hypothyroidism, and monitoring for this adverse effect is necessary.
Drug–drug interactions
See Box 19.11.
Contraindications
The oral retinoids are contraindicated in women who are pregnant or breastfeeding; women of childbearing potential unless all the conditions of a retinoid pregnancy prevention plan are met; patients who are receiving concomitant treatment with a tetracycline; and patients who are hypersensitive to the retinoid or an excipient (in particular soya and peanuts). Relative contraindications include renal and hepatic insufficiency and severe hyperlipidaemia.
Cautions
Patients should not donate blood during treatment or for at least 1 month thereafter in the case of isotretinoin and alitretinoin, and 6 months thereafter for acitretin, because of the potential risk to the fetus of a pregnant transfusion recipient [8]. Exposure to intense sunlight should be avoided and sun-protection measures used. Wax depilation should be discouraged for 6 months following retinoid treatment for fear of epidermal stripping, and dermabrasion or laser resurfacing should be postponed for at least 6 months because of the risk of hypertrophic scarring and dyspigmentation [8]. Retinoids may affect blood glucose control in diabetics.
Pre-treatment screening
Women of childbearing potential should receive explicit counselling on the teratogenicity of retinoids and either adopt birth control measures or abstain from coitus. Pregnancy should be excluded prior to commencing retinoid therapy, and pregnancy should be avoided for the duration of therapy and for an appropriate time thereafter (1 month for isotretinoin, alitretinoin and bexarotene, and 2 years for acitretin). The recommendations regarding isotretinoin, which can be adapted of the other retinoids, suggest that women deemed at risk of conceiving should be recruited into the isotretinoin pregnancy prevention plan [42] (iPLEDGE in the USA [43]), which involves patient education about retinoid adverse effects and contraception, written informed consent, the employment of ideally two adequate forms of contraception established at least 1 month prior to initiation of therapy and a negative urinary or serum pregnancy test (with a minimum sensitivity of 25 mIU/mL of human chorionic gonadotrophin) immediately before commencement of treatment, which should start on the second or third day of a menstrual cycle. Amenorrhoeic women (including those receiving the progestogen-only contraceptive) should have a pregnancy test 14 days after the last act of coitus. Women can be exempted from the pregnancy prevention plan if they are sexually abstinent or have had a hysterectomy.
Pre-treatment laboratory tests should include a full blood count, liver function tests, renal function tests and a full fasting lipid profile. Alitretinoin and bexarotene treatments necessitate monitoring of thyroid function.
Dose and regimens
There are standard dosage regimens for each of the retinoids [8, 41, 42, 44, 45].
Monitoring
A reasonable follow-up regimen is monthly clinical evaluation and blood tests (liver function tests and fasting lipid profile, with occasional renal function tests and full blood count) for 3–6 months, then 3-monthly reviews with blood tests [5]. Thyroid function should be monitored in patients receiving alitretinoin and bexarotene.
Serum or urinary pregnancy tests are required monthly for women of childbearing potential, and again 5 weeks after the drug is discontinued.
Specifically enquire about adverse effects, particularly those relating to mood and vision. Asymptomatic patients do not require routine monitoring for skeletal toxicity or osteoporosis [5] but if a patient reports restricted mobility and bone pain consideration should be given to relevant radiological examination.
Thalidomide
Thalidomide acquired pharmacological notoriety in the early 1960s when it became clear that its use as a sedative and antiemetic for pregnant women could cause limb defects (phocomelia) and internal deformities in their children. Its subsequent reinvention as a useful therapeutic agent with potent immunomodulatory, anti-inflammatory, antiangiogenic and antineoplastic properties has seen it licensed as a component of first line treatment for myeloma and, in the USA, for erythema nodosum leprosum.
Dermatological uses (see Chapters 31, 48, 51, 83, 127 and 136)
Thalidomide has also been used off-license in a wide variety of dermatoses, including nodular prurigo, actinic prurigo, cutaneous lupus erythematosus, aphthous stomatitis, Behçet disease, sarcoidosis, graft-versus-host disease, Langerhans cell histiocytosis, cutaneous manifestations of advanced HIV infection and Kaposi sarcoma [1, 2, 3, 4, 5, 6, 7, 8, 9, 10].
Pharmacological properties
Formula and structure
Thalidomide (empirical formula C13H10N2O4, systematic name (RS)-2-(2,6-dioxopiperidin-3-yl)-1H-isoindole-1,3(2H)-dione) is a piperidinyl isoindole, consisting of a phthaloyl ring and a glutarimide ring. It has a chiral centre and exists as a racemic mixture of left- and right-handed enantiomers [2].
Administration
Thalidomide Celgene® is available in the UK as capsules containing 50 mg of thalidomide [11].
Pharmacokinetics
Only limited pharmacokinetic data are available for thalidomide. It is lipid soluble and peak plasma levels are reached within 2–6 h [1]. It appears to be degraded by non-enzymatic hydrolysis, although there also seems to be hepatic metabolism involving the cytochrome P450 system [1]. Its half-life is about 9 h, but the precise details of its excretion are unknown, although it is predominantly non-renal. It readily crosses the placenta.
Pharmacodynamics
The mechanisms underlying the many biological actions of thalidomide remain obscure. There is evidence to support its teratogenic effect being caused by the reduced expression of fibroblast growth factor 8 (an essential regulator of limb development) consequent to the binding of thalidomide to cereblon [12], a ubiquitously expressed E3 ligase protein which inhibits its ubiquitin ligase activity. Other theories for its teratogenic action include oxidative stress and damage, DNA intercalation and inhibition of angiogenesis [13]. Cereblon may also prove to be the protein target for the immunomodulatory and antiproliferative properties of thalidomide [14]. Thalidomide influences both cellular and humoral immune responses, inhibits neutrophil and monocyte phagocytosis and neutrophil chemotaxis, and inhibits the production of a variety of inflammatory mediators including TNF-α and IL-12 [1].
Pharmacogenetics
Although not of relevance for current clinical use, genetic polymorphisms in genes encoding CYP450 and non-CYP450 metabolizing enzymes and transporters may explain the wide interindividual variation in the pharmacokinetic and toxicity profile of thalidomide [15, 16]. There is recent evidence that the risk of developing peripheral neuropathy is determined by polymorphisms in genes governing repair mechanisms and inflammation in the peripheral nervous system [17].
Potential adverse effects
Teratogenicity
In addition to phocomelia, thalidomide has been associated with congenital heart disease, ocular and aural malformations, urological abnormalities, autism and mental retardation [18]. The critical exposure period during which it appears to exert its devastating embryopathic effects is between 20 and 36 days after conception [13, 18, 19].
Peripheral neuropathy
Peripheral neuropathy is a very significant adverse effect of thalidomide, and the predominant factor limiting its use. It occurs in 20–55% of patients, with women and the elderly at greatest risk. The peak incidence is in the first year of treatment [20, 21]. The precise neurotoxic mechanism is not known but it seems likely that affected individuals may have a genetic susceptibility [17] which is dependent on the daily dose rather than the cumulative dose [21, 22]. This results in a symmetrical, mainly sensory, length-dependent axonal polyneuropathy, with reduced sensory nerve action potential (SNAP) amplitudes and relative conservation of nerve conduction velocities [23, 24, 25]. It presents clinically as symmetrical painful parasthesiae of the hands and feet, sensory loss and, occasionally, muscle weakness or cramps, signs of pyramidal tract involvement and carpal tunnel syndrome [2]. Recovery tends to be slow and often incomplete, and neurotoxicity may even be progressive after thalidomide therapy is withdrawn [2].
Other adverse effects
See Box 19.12.
Contraindications
Thalidomide is absolutely contraindicated during pregnancy and for those individuals with a known sensitivity to it [1]; it should be avoided in breastfeeding mothers [11].
Cautions
Women of childbearing potential receiving thalidomide must practice strict contraception. Men whose sexual partners are women of childbearing potential should use condoms as thalidomide is secreted into seminal fluid.
Thalidomide is relatively contraindicated in patients with an existing peripheral neuropathy or other neurological disorder, or if there is significant renal or hepatic impairment, congestive heart failure, hypertension, hypothyroidism, gastrointestinal disease or increased risk of thrombosis [1, 11].
When used in the treatment of myeloma, anticoagulation as prophylaxis for thrombosis is recommended for at least the first 5 months of treatment, particularly if there are specific risk factors such as smoking, hypertension or hyperlipidaemia [11].
Drug–drug interactions
Thalidomide may potentiate the effects of drugs which cause sedation, bradycardia or peripheral neuropathy. In women of childbearing potential requiring oral contraception because of thalidomide therapy, the possibility of drug interactions involving the contraceptive preparation should be considered [1]. Furthermore, the combined oral contraceptive is not recommended because of the possible increased risk of thrombosis with thalidomide.
Pre-treatment screening
Thalidomide is licensed by the European Medicines Agency [26], and it is mandatory to prescribe and dispense the drug according to the Thalidomide CelgeneTM Pregnancy Prevention Programme [27], based on the THALOMID Risk Evaluation and Mitigation Strategy (REMS)TM programme (formerly the System for Thalidomide Education and Prescribing Safety (STEPS) programme) in the USA. Both of these require formal registration of physician, pharmacist and patient, and informed consent from the patient.
Careful counselling and education of patients is necessary, and Celgene have produced an information booklet to facilitate this [28].
Patients should be assessed regarding risk of thrombosis, and, if considered necessary, anticoagulation commenced.
A clinical neurological examination should be undertaken and baseline electrophysiological testing (measurement of SNAP amplitudes) should be considered.
In women of childbearing potential, two reliable methods of contraception must be employed, commencing at least 1 month before therapy is started. Pregnancy must be excluded before initiating treatment.
A full blood count and renal and liver function should be checked.
Dose and regimens
The dose range for dermatological conditions is 50–300 mg/day, taken as a single dose at bedtime to reduce the impact of sedation.
Monitoring
Peripheral neuropathy
It is recommended that patients are examined at monthly intervals for the first 3 months of thalidomide therapy and periodically (1–6 monthly as indicated [1]) thereafter for signs or symptoms of peripheral neuropathy. Electrophysiological testing should be considered every 6 months to detect subclinical neuropathy.
Laboratory tests
Pregnancy testing should be performed monthly for women of childbearing age, and again 4 weeks following cessation of treatment. A full blood count and liver function tests should be performed monthly until the dose of thalidomide is stable, then every 2–3 months.