CHAPTER 28
Leprosy
Diana N. J. Lockwood
London School of Hygiene & Tropical Medicine, London, UK
Definition and nomenclature
A chronic granulomatous disease caused by Mycobacterium leprae, principally affecting peripheral nerves and skin.
Introduction and general description
Endemic in India and the Far East since ancient times, leprosy was imported into Europe in the 4th century BC, perhaps by the troops of Alexander the Great. The European epidemic peaked in the 13th century, then slowly died out [1]. French settlers took leprosy to Canada and African slaves took it to America. Even after the discovery of the agent, Mycobacterium leprae, by Armauer Hansen in Norway in 1873 (the first bacillus to be associated with a human disease), the infectious nature of leprosy was not readily accepted [2]. The slow spread of the disease and its familial association suggested that it was inherited. This belief, and the fear of the deformities that leprosy may cause, have contributed to the stigma and ostracization that still characterize attitudes towards leprosy. Stigma remains a major obstacle to leprosy control, despite advances in bacteriology, chemotherapy and epidemiology. New patients today can be reassured that their bacterial infection is readily treatable with antibiotics although associated nerve damage is more difficult to treat.
Epidemiology
Incidence and prevalence
About 4 million people have, or are disabled by, leprosy. The apparent fall in registered patients from 12 million in 1988 to 0.25 million on treatment in 2014 hides an intriguing picture. Prevalence has fallen due to a combination of effective antibiotic therapy and a change in case definition. Incidence, however, remains stable at around 250 000 new cases annually, with high rates of childhood cases [3].
India dominates the global picture with 60% of the world's leprosy cases; 86% of leprosy patients reside in six countries (India, Brazil, Indonesia, Nigeria, Ethiopia and Bangladesh). Leprosy has not always been a tropical disease; it was endemic in Norway until the early 20th century. Nearly all the new cases now seen in Europe and North America acquired their infection abroad [4].
Although leprosy is rarely a primary cause of death, patients have a standardized death rate at least twice that of the general population due to the indirect secondary effects of the disease [5]. It is estimated that 1 million disability-adjusted life-years (DALY) are lost globally each year due to leprosy, with 6.3 years of healthy life being lost per patient.
Age
Leprosy can occur at all ages but there is a peak in the teens and early twenties.
Sex
Leprosy occurs equally in both sexes until puberty then an excess of male cases has regularly been found, although this may be due to women being reluctant to present to health workers with skin lesions [6].
Pathophysiology
Predisposing factors
Living in a leprosy endemic area, age, sex and household contact predispose to acquiring leprosy [7]. Bacille Calmette–Guérin (BCG) vaccination protects against leprosy. Clustering of cases is well recognized [8]. Although poor nutritional status was thought to predispose to leprosy, no good evidence substantiates this. Improved socioeconomic conditions, extended schooling and good housing reduce the risk of leprosy. Various genes and regions in the human genome have been linked to or associated with susceptibility to leprosy per se or with a particular type of leprosy. Perhaps not surprisingly these are not all reproducible in different populations.
Mira et al. have identified that certain alleles in the PARK2 and PACRG region on chromosome 6 are associated with susceptibility to leprosy in Vietnamese and Brazilian cohorts [9]. PARK2 is expressed by both Schwann cells and macrophages. It is an ubiquination E3 ligase and is involved in the delivery of polyubiquinated proteins to the proteosome complex involved in protein degradation [10].
In an Indian cohort homozygotes for the different alleles of the vitamin D receptor (VDR) gene were associated with tuberculoid or lepromatous disease [11]. Upregulation of the VDR gene on macrophages is associated with increased intracellular killing of M. tuberculosis.
Polymorphisms of the tumour necrosis factor (TNF) α promoter region were shown to be associated with increased susceptibility to lepromatous leprosy (LL) [12], while in a cohort from southern Brazil the same allele was protective against leprosy per se [13]. A study of Malawians did not find any association of this TNF promoter with leprosy [14].
Brazilian and Indian groups have demonstrated that polymorphisms of the interleukin 10 (IL-10) promoter are associated with resistance to leprosy [15, 16].
The differing and sometimes conflicting results of genetic studies may be attributed to differences in study design and sample size. It is also possible that different populations have distinct genetic susceptibilities.
Early in the HIV epidemic it was predicted that HIV infection would worsen leprosy outcomes with more patients developing lepromatous disease, an impaired response to multidrug therapy (MDT) and fewer reactions. However, none of these have happened and instead HIV immune dysregulation rather than immune suppression dominates the clinical picture in patients with leprosy and HIV. Being HIV positive does not appear to confer increased susceptibility to leprosy. The full spectrum of standard clinical and histopathological leprosy features have been seen in co-infected persons [17]. These patients also have normal granuloma formation even with low circulating CD4 counts. Co-infected patients appear to be at increased risk of developing leprosy reactions and the borderline types of leprosy dominate the clinical picture. Leprosy may also present as an immune reconstitution syndrome (IRIS) phenomenon in HIV-positive patients who have recently started highly active antiretroviral therapy [18, 19]. A recent study on co-infection in Brazil found that patients had active skin lesions and active granulomas in their skin biopsies [20].
Subclinical infection with M. leprae is probably common, but the development of established disease is rare. There is no reliable test for determining whether a person has encountered M. leprae and mounted a protective immune response. In contacts of leprosy patients, there is frequently evidence of specific sensitization to M. leprae, using markers of infection such as serum antibody levels, in vitro lymphocyte transformation tests (LTTs) and skin test responses to soluble M. leprae antigen [20]. Contacts of an untreated elderly man with borderline lepromatous (BL) leprosy in a British residential home showed that 23/30 and 25/30 had positive Mitsuda skin test and positive LTT responses, respectively, to M. leprae sonicate, but only two contacts had positive antibody (immunoglobulin M phenolglycolipid antigen (IgM PGL)) responses. Self-healing often occurs in early monomacular tuberculoid cases. Leprosy is probably analogous to tuberculosis, in which only 10% of infections manifest as clinical disease [21]. In the 20th century, there were only two reported cases of secondary transmission within Britain, cases acquired from a known case of leprosy [22].
Nasal discharges from untreated LL patients, who are often undiagnosed for several years, are the main source of infection in the community [23]. In Indonesia and Ethiopia, M. leprae DNA has been detected in nasal swabs in up to 5% of the population. Infection probably also occurs through the nose. Mycobacterium leprae is inhaled, multiplies on the inferior turbinates and has a brief bacteraemic phase before binding to Schwann cells and macrophages. In patients with the tuberculoid types of leprosy, M. leprae remains within the skin and nerve compartments and these patients are probably never infectious.
The skin is unimportant in leprosy transmission. Bacilli are rarely found in the epidermis and are only excreted by untreated LL patients. The only evidence of bacilli entering via the skin comes from case reports of direct inoculation. Leprosy has been transmitted to nude mice through pricks from infected cactus thorns [24]. There is no evidence that biting arthropods transmit leprosy. It is surprising that, in contrast to tuberculosis, there are few documented cases of leprosy occurring in both the medical and non-medical attendants of leprosy patients.
Pathology
The pathogenesis [25], and thus the clinical features, reflect four principal causes of tissue damage (Figure 28.1):
- The degree to which cell-mediated immunity (CMI) is expressed [26]. LL represents a failure of CMI specifically towards M. leprae with resultant bacillary multiplication, spread and accumulation of antigen in infected tissues. The absence of activated lymphocytes and macrophages means that nerve damage is slow and gradual in onset. In tuberculoid leprosy, CMI is strongly expressed, so that the infection is restricted to one or a few skin sites and peripheral nerves. Lymphocytic infiltration rapidly causes nerve damage. Between those two polar forms lies the borderline forms of disease, with the extent of disease reflecting the balance between CMI and bacillary load.
- The extent of bacillary spread and multiplication. In LL, haematogenous spread of bacilli occurs [27] to cool, superficial sites, including eyes, upper respiratory mucosa, testes, small muscles and bones of hands, feet and face, as well as peripheral nerves and skin. In tuberculoid leprosy, bacillary multiplication is restricted to a few sites and bacilli are not readily found.
- The appearance of tissue-damaging immunological complications: lepra reactions [28]. Borderline patients (borderline tuberculoid (BT); borderline borderline (BB); BL) are immunologically unstable and at risk of developing immune-mediated reactions. Type 1 (reversal) reactions are delayed hypersensitivity reactions caused by increased recognition of M. leprae antigens in skin and nerve sites. Type 2 reactions, erythema nodosum leprosum (ENL), are due in part to immune complex deposition and occur in BL and LL patients who produce antibodies and have a large antigen load.
- The development of nerve damage and its complications. Nerve damage occurs in two settings, in skin lesions and in peripheral nerve trunks. In skin lesions, the small dermal sensory and autonomic nerve fibres supplying dermal and subcutaneous structures are damaged, causing local sensory loss and loss of sweating within the area of the skin lesion [29]. Peripheral nerve trunks are vulnerable at sites where they are superficial or are in fibro-osseous tunnels. At these points, a small increase in nerve diameter leads to raised intraneural pressure, with consequent neural compression and ischaemia. Damage to peripheral nerve trunks produces characteristic signs, with dermatomal sensory loss and dysfunction of muscles supplied by that peripheral nerve. Physiological evidence of central and peripheral autonomic nerve involvement has also been reported [30, 31].

Figure 28.1 Mechanisms of damage in leprosy, and tissues affected. Mechanisms under the broken line are characteristic of disease near the lepromatous end of the spectrum, those under the solid line of the tuberculoid end. They overlap in the centre where, in addition, instability predisposes to type 1 reactions. Abbreviations are given in the text.
Nerve damage leads to anaesthesia, muscular weakness and contracture, and autonomic dysfunction. These permit trauma, bruising, burns, cuts and, especially, tissue necrosis from prolonged, inappropriate or repetitive trauma, which in turn lead to ulceration, secondary cellulitis and osteomyelitis and loss of tissue, so that deformity is added to disability.
Histology [25]
Mycobacterium leprae has a predilection for neural tissue, and the first evidence of infection is often found in the peripheral nervous system. Patients with early leprosy of both tuberculoid [32, 33] and lepromatous types have abnormalities in nerve conduction studies and a histological picture of small fibre loss, with segmental demyelination and remyelination [34]. Bacilli probably enter nerves via endoneural blood vessels [35], the target cell being the Schwann cell. In the dermis, the type and degree of cellular infiltrate reflects the degree of CMI. The classification of Ridley and Jopling [36] describes five groups on the immunological spectrum designated tuberculoid tuberculoid (TT), BT, BB, BL and LL. In this classification, epithelioid cells and lymphocytes at the tuberculoid end of the spectrum give place to macrophages, which appear increasingly foamy as the lepromatous pole is reached.
Tuberculoid leprosy
Tuberculoid granulomas collect in foci surrounding neurovascular elements (Figure 28.2). The granuloma invades the papillary zone and may even erode the epidermis, but acid-fast bacilli (AFB) are not seen. Cutaneous nerves that are not completely destroyed appear greatly swollen by epithelioid cell granulomas and surrounded by a zone of lymphocytes; occasionally there may be caseation within the nerve.
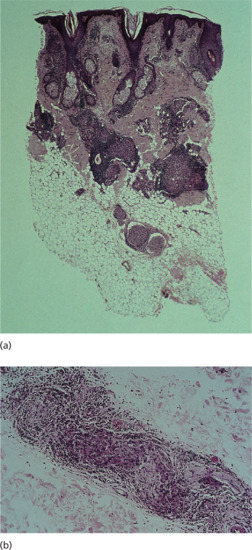
Figure 28.2 (a) Low-power view (H&E) showing tuberculoid granulomas around nerve and skin appendages in mid-dermis and a swollen, deep dermal nerve. There is no epidermal erosion. Around the granulomas there is a dense lymphocytic infiltrate. (b) A different case. Medium-power view (H&E) of a deep nerve in tuberculoid leprosy showing granulomatous disruption of the nerve with surrounding lymphocytic infiltrate.
(Courtesy of Professor S. Lucas, Kings College, London, UK.)
Lepromatous leprosy
Histological examination of skin lesions (H&E) shows thinning of the epidermis and flattening of rete ridges (Figure 28.3). The papillary layer of the dermis appears as a clear band, whilst deeper in the dermis lies the typical diffuse leproma consisting of foamy macrophages, with the addition of a few lymphocytes and plasma cells. The dermis contains enormous numbers of AFB, singly or in clumps (globi). With treatment, the leproma shows increased foamy change, vacuolates and breaks up into discrete foci with fibrocytes at the periphery. These foci shrink as treatment is continued and bacilli become fragmented and granular. In lepromatous neuropathy, there is quiet asymptomatic bacillation of Schwann cells leading to foamy degeneration of these cells. Demyelination, damage and destruction of the axis cylinder are prominent features and later Wallerian degeneration occurs [37]. Despite the large numbers of organisms in the nerve there is only a small inflammatory response; ultimately the nerve fibroses and is hyalinized [38].
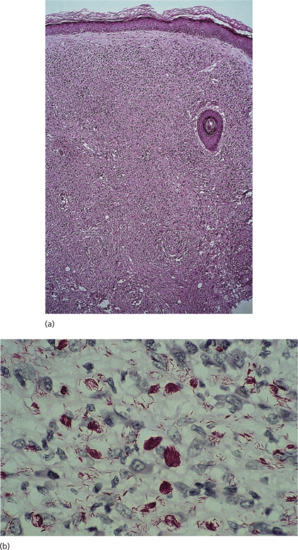
Figure 28.3 (a) Lepromatous leprosy. Medium-power view (H&E) showing thin epidermis, a clear subepidermal zone and a dense, uniform macrophage infiltrate in the dermis. Towards the bottom of the photograph there is an ‘onion skin’ perineurial lamination. (b) High-power view, Wade–Fite stain showing single and clustered acid-fast bacilli, part solid, part fragmented; bacillary index (BI) = 5.
(Courtesy of Professor S. Lucas, Kings College, London, UK.)
Borderline leprosy
In BT leprosy, the epithelioid cell granuloma is more diffuse than in TT with a free, but narrow, papillary zone. Giant cells tend to be foreign body rather than Langhans in type, and dermal nerves are moderately swollen by cellular infiltration, or may show only Schwann cell proliferation. AFB are usually absent or scanty. In mid-BB, there is diffuse epithelioid cell granuloma with very scanty lymphocytes and no giant cells; the papillary zone is clear, nerves are slightly swollen by cellular infiltrate, and AFB are present in moderate numbers. In BL leprosy, macrophages may show slight foamy change. Lymphocytes are present in dense clumps or are widely distributed in parts of the granuloma; a few epithelioid cells may be seen occasionally. The formation of small granulomas is characteristic of BL and granulomatous regions may abut strands of normal looking but heavily bacillated Schwann cells [39]. Nerve damage in BL results from a combination of lepromatous bacillation and a tuberculoid tissue damaging response producing widespread nerve damage. Acute neuritis damage occurs particularly during reversal reactions: oedema of the epithelioid cell granuloma compresses the remaining Schwann cells causing rapid functional loss in an already compromised nerve. In ENL nerve damage occurs more slowly and is probably due to inflammation associated with ENL nodule formation in nerve trunks.
Indeterminate leprosy
This early and transitory stage of leprosy occurs in those whose immunological state has not yet been determined, and histologically there is a scattered non-specific histiocytic and lymphocytic infiltration with some concentration around skin appendages (Figure 28.4). Rarely, a single bacillus will be found within a dermal nerve. The indeterminate phase may last for months or years before resolving or giving way to one of the determinate types of leprosy described above.
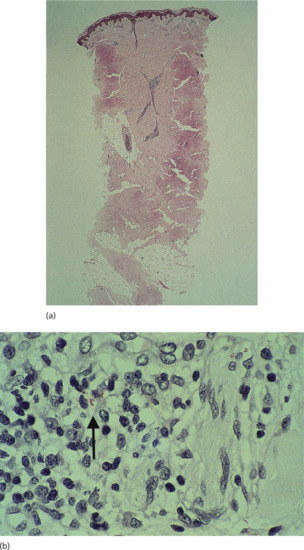
Figure 28.4 (a) Indeterminate leprosy. Low-power view (H&E) showing minimal perineurovascular inflammation in the mid-dermis. (b) High-power view, Wade–Fite stain showing one acid-fast bacillus in a Schwann cell.
(Courtesy of Professor S. Lucas, Kings College, London, UK.)
Reactions: type 1 and erythema nodosum leprosum
Type 1 reactions are characterized by an increase in lymphocytes within lesions, severe oedema with disruption of the granuloma, giant cell formation and HLA expression [40, 41]. In type 2 (ENL) reactions polymorphs infiltrate the granuloma and there is vasculitis and macrophage degeneration together with breakdown of foam cells (Figure 28.5).
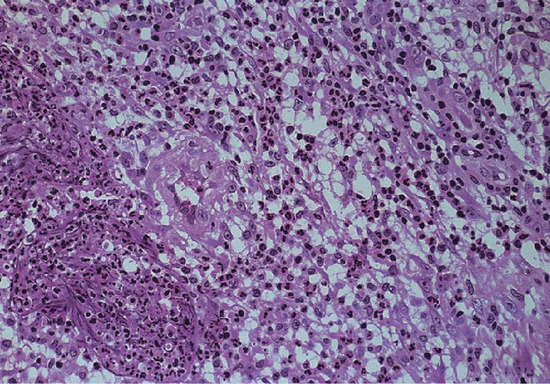
Figure 28.5 Erythema nodosum leprosum. Medium-power view (H&E) showing foamy macrophages with infiltrating polymorphs. There is also a swollen small artery in the centre of the photograph.
(Courtesy of Professor S. Lucas, Kings College, London, UK.)
Immunology [42]
The immune response to M. leprae determines not only whether disease will develop, but also which type of leprosy. The highly conserved Toll-like receptors on the surface of monocytes and macrophages recognize mycobacterial lipoproteins [43]. For M. leprae the TLR2/1 heterodimer is activated with monocyte differentiation into macrophages and dendritic cells [44]. The latter present antigen and activate naïve T cells by IL-12 secretion [45]. The IL-12βR2 portion of the IL-12 receptor is expressed more on T helper 1 (Th1) lymphocytes, preferentially shifting the immune response further towards a Th1 response.
In addition, IL-2 stimulates the expansion of α/β CD8 T cells and antigen non-specific natural killer cells in the lesion. All three types of cell can produce interferon-γ (IFN-γ), the major cytokine responsible for activating bactericidal mechanisms within the parasitized macrophage [46].
In tuberculoid leprosy, there is good evidence of a strong CMI response. Tests of T-cell function such as LTTs show that tuberculoid leprosy patients respond to M. leprae antigens as in whole M. leprae, separated M. leprae antigens and cloned antigens (18 kDa and 65 kDa) [47]. Skin tests with lepromin, a heat-killed M. leprae sonicate preparation, are strongly positive in these patients. Staining of skin biopsies from tuberculoid lesions with T-cell markers shows highly organized granulomas composed predominantly of CD4 cells and macrophages, with a peripheral mantle of suppressor/cytotoxic CD8 cells [48]. This strong CMI response has been misdirected at some stage, and the end result is a late, strong cell-mediated response that clears antigen at the expense of local tissue destruction.
Lepromatous leprosy patients are unable to mount a CMI response to M. leprae, with a failure of the T-cell response, and lymphocytes from LL patients respond poorly in LTT to whole M. leprae and cloned antigens. Similarly, LL patients fail to mount a skin test response to intradermal challenge with lepromin. The anergy of the lepromatous patient is striking because it is specific for the leprosy mycobacterium. Lepromatous patients can respond to antigens of other mycobacteria such as M. tuberculosis, both in in vitro and in skin tests [49, 50]. Identification of cell types in LL granulomas shows them to be a disorganized mixture of macrophages and T cells, mainly CD8 cells [51].
Both T-cell and macrophage dysfunction occur in lepromatous patients. The T-cell failure may be due to clonal anergy or active suppression. Defects in cytokine production have been demonstrated in lepromatous patients; addition of IL-2 to T-cell culture media restored the proliferative response to M. leprae in lepromatous patients [52], and intralesional injections of recombinant IL-2 reconstituted the local immune response with elimination of M. leprae from macrophages [53]. Macrophage defects described in LL disease include defective antigen presentation and recognition, defective IL-1 production, a failure of macrophages to kill M. leprae and a macrophage suppression of the T-cell response [43].
Studies of circulating cytokines in leprosy patients, and cytokine production in skin lesions, show that tuberculoid patients have a Th1 type response to M. leprae with predominant IL-2 and IFN-γ production, whilst lepromatous patients have low cytokine production and mainly of a Th2 type.
The inflammation seen in type 1 reactions is due to T-cell activity, with enhanced T-cell proliferation towards M. leprae antigens, increased numbers of CD4 and IL-2 producing cells in granulomas, and local production of cytokines such as IFN-γ and TNF-α [54]. Type 1 reactions are associated with an overproduction of Th1 type cytokines [55]. ENL has classically been regarded as an immune complex disorder. This is supported by the presence of immunoglobulin and complement in the lesions and circulating immune complexes [56, 57]. There is also evidence of enhanced T-cell activity during ENL episodes, with increased numbers of CD4 cells [58] in lesions, and production of the cytokines TNF-α and IL-6, increased circulating IL-2 receptors and high levels of circulating TNF-α [59] in acute episodes. Despite increased immune activity during ENL episodes, lepromatous patients revert to a state of immunological unresponsiveness after an episode.
Serology
Specific anti-M. leprae antibodies are produced against lipoarabinomannan (LAM), PGL and the protein antigens of M. leprae. No single antigen has been identified for use in detecting early subclinical infection. For all three types of antigen, multibacillary patients produce antibodies prolifically, while paucibacillary patients show a variable, often undetectable, response.
Lepromatous patients produce a range of autoantibodies, both organ specific (i.e. directed against thyroid, nerve, testis and gastric mucosa), and non-specific, such as rheumatoid factors, anti-DNA, cryoglobulins and cardiolipin.
Causative organisms
Mycobacterium leprae has never been grown in vitro. Limited growth has been achieved in the mouse footpad [60], and more widespread growth and disease in immunosuppressed and nude mice [61] and the nine-banded armadillo [62]. Leprosy has also been established as a zoonosis in the southern USA. The latter has provided mycobacteria for genetic and biochemical analysis and the production of trial vaccines. Mycobacterium leprae grows at 30–33°C, with a doubling time of 12 days. It is a remarkably hardy organism, remaining viable in the environment for up to 10 days [63]. The sequence of the M. leprae genome was published in 2001. Mycobacterium leprae has a 3.27-Mb genome that displays extreme reductive evolution [64]. Less than half the genome contains functional genes: many pseudogenes are present. One hundred and sixty-five genes are unique to M. leprae, and functions can be attributed to 29 of them. Comparison of biosynthetic pathways with M. tuberculosis shows that for lipolysis M. leprae has only two genes (M. tuberculosis has 22). Mycobacterium leprae has lost many genes for carbon catabolism, and many carbon sources (e.g. acetate and galactose) are unavailable to it. Mycobacterium leprae growth may be restricted to a few carbon sources on which it can maintain a balanced carbon metabolism. It has also lost anaerobic and microaerophilic electron transfer systems. Mycobacterium leprae has many genes for haem and iron-based proteins, but it is severely limited in its iron uptake capacity, since it has lost the ability to produce iron scavenging sideropores [65]. Recent genomic analysis of M. leprae shows that there are four main strains of the mycobacterium. One strain was probably taken across the Atlantic. Recent skeletal evidence shows that M. leprae has had a stable genome for at least 10 centuries [66, 67, 68].
Mycobacterium leprae has a complex antigenic cell wall composed of lipids, carbohydrates and proteins. The organism also synthesizes a species-specific lipid, phenolic glycolipid (PGL) [69]. Several polymerase chain reaction (PCR) probes (18 kDa, 36 kDa, 65 kDa and ribosomal RNA sequences) have been developed for the detection of M. leprae DNA in tissues from leprosy patients [70]. Although these are specific, they are not yet sensitive enough to be useful in diagnosing patients whose skin is bacteriologically negative on conventional staining. The molecular basis for rifampicin resistance has been elucidated and it is now possible, using a PCR–single strand polymorphism (PCR-SSC) technique, to identify rifampicin-resistant isolates within hours [71].
Clinical features
History
Leprosy is a chronic disease with a long incubation period. An average incubation time of 2–5 years has been calculated for tuberculoid cases, and 8–12 years for lepromatous cases. American servicemen who developed leprosy after serving in the tropics presented up to 20 years after their presumed exposure [72].
Presentation [73]
Early lesions and presenting symptoms
The commonest early lesion is an area of numbness on the skin, or a visible skin lesion. The classic early skin lesion, especially in surveys, is that of indeterminate leprosy, which is most commonly found on the face, extensor surface of the limbs, buttocks or trunk (Figure 28.6) [74]. Scalp, axillae, groins and lumbar skin tend to be spared. Indeterminate lesions consist of one or more slightly hypopigmented or erythematous macules, a few centimetres in diameter, with poorly defined margins. Hair growth and nerve function are unimpaired. A biopsy may show the perineurovascular infiltrate, and only a prolonged search will reveal scanty acid-fast organisms. Alternatively, the initial skin lesion has features of one of the established forms of the disease.

Figure 28.6 Indeterminate leprosy. Face of a Nepali child showing vague hypopigmented patch with some central healing. Note the mark of a recent slit-skin smear.
Patients frequently present with signs of nerve damage: weakness or anaesthesia due to a peripheral nerve lesion, or a blister, burn or ulcer in an anaesthetic hand or foot. Borderline patients may present in reaction with nerve pain, sudden palsy, multiple new skin lesions, pain in the eye or a systemic febrile illness [75, 76].
Features of established leprosy
Careful attention to the eight different clinical aspects of leprosy listed in Table 28.1 will enable accurate recognition and classification, which are prerequisites to correct treatment and accurate prognosis.
Table 28.1 Characteristics of lesions of polar leprosy
| Tuberculoid | Lepromatous | |
| Number of lesions | 1–10 | Hundreds, confluent |
| Distribution | Asymmetrical, anywhere | Symmetrical, avoiding ‘spared’ areas |
| Definition and clarity | Defined edge, markedly hypopigmented | Vague edge, slight hypopigmentation |
| Anaesthesia | Early, marked, defined, localized to skin lesions or major peripheral nerve | Late, initially slight, ill-defined, but extensive, over ‘cool’ areas of body |
| Autonomic loss | Early in skin and nerve lesions | Late, extensive as for anaesthesia |
| Nerve enlargement | Marked, in a few nerves | Slight but widespread |
| Mucosal and systemic | Absent | Common, severe during type 2 reactions |
| Number of Mycobacterium leprae | Not detectable | Numerous in all affected tissues |
Clinical variants
Tuberculoid leprosy
Only nerves and skin show clinical evidence of disease; lesions are few, often solitary. The condition may be purely neural, with pain and swelling of the affected nerve followed by anaesthesia and/or muscle weakness and wasting. Alternatively, a skin lesion appears with or without evidence of nerve involvement. The typical lesion is a plaque that is conspicuous, erythematous, copper coloured or purple, with raised and clear-cut edges sloping towards a flattened and hypopigmented centre (Figures 28.7 and 28.8). Dark skins may not show the erythema. The surface is dry, hairless and insensitive, and sometimes scaly. Sensory impairment may be difficult to demonstrate on the face because of the generous supply of sensory nerve endings. If the examiner runs a finger around the lesion, just beyond the outer edge, a thickened sensory nerve may be palpated or a thickened nerve trunk may be felt in the vicinity, for example a thickened ulnar nerve if the lesion is on the arm. Less commonly the lesion is a macule, erythematous in light skins and hypopigmented (never depigmented) in dark skins (Figure 28.9). Such macules have a dry, hairless and insensitive surface.
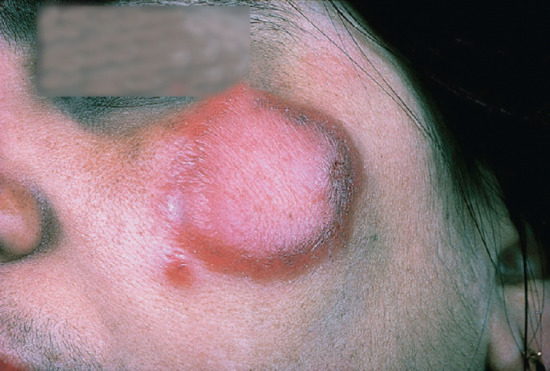
Figure 28.7 Tuberculoid leprosy. Face of Pakistani woman showing erythematous plaque with a well-defined active edge, and a small satellite lesion. On the face, such lesions may not be anaesthetic.
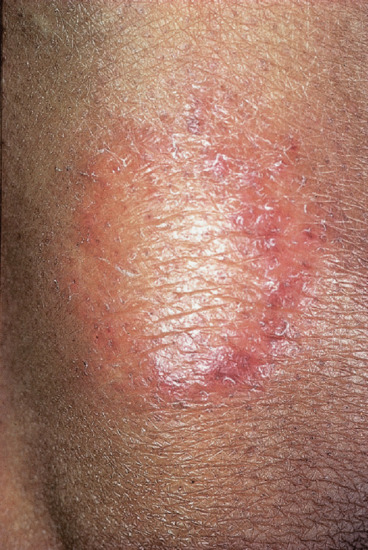
Figure 28.8 Tuberculoid or borderline tuberculoid leprosy. Upper arm of Indian man, showing typical dry, hairless, hypopigmented plaque with scaly, erythematous edge. Such lesions are usually anaesthetic.

Figure 28.9 Tuberculoid or borderline tuberculoid leprosy. Back of a Nigerian child showing well-defined hypopigmented macule with altered skin texture. The lesion was anaesthetic.
Lepromatous leprosy
The first clinical manifestations are usually dermal (because early nerve involvement is usually asymptomatic), but they may go unnoticed by the patient, who often complains of other early symptoms; these include nasal symptoms of stuffiness, discharge and epistaxis [77], and oedema of legs and ankles due to increased capillary stasis and permeability. Dermal signs comprise macules, diffuse papules, infiltration or nodules, or all four. Macules are small, multiple, erythematous or faintly hypopigmented, with vague edges and shiny surface (Figure 28.10). Papules and nodules usually have normal skin colour but sometimes are erythematous (Figure 28.11), with a bilaterally symmetrical distribution on face, arms, legs and buttocks, but may be anywhere apart from hairy scalp, axillae, groins and perineum (i.e. regions of skin with the highest temperature). Hair growth and sensation are not initially impaired over the lesions. In polar LL, diffuse infiltration and gradual thickening of the dermis may precede nodulation by months or years (Figure 28.12). Lesions of oral mucosa occur as papules on lips and nodules on palate (which may perforate), uvula, tongue and gums (Figure 28.13). The nasal mucosa is hyperaemic or ulcerated and bleeds easily; epistaxis is common (Figure 28.14).
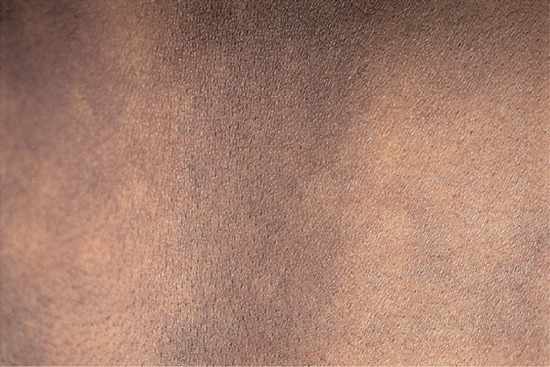
Figure 28.10 Lepromatous leprosy (borderline lepromatous/lepromatous. Back of a Bangladeshi boy showing numerous, often confluent hypopigmented macules, with relative sparing of the midline.
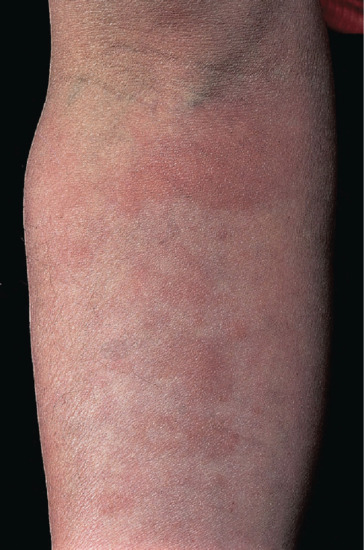
Figure 28.11 Lepromatous leprosy. Forearm of an English man showing erythematous macules and infiltration.
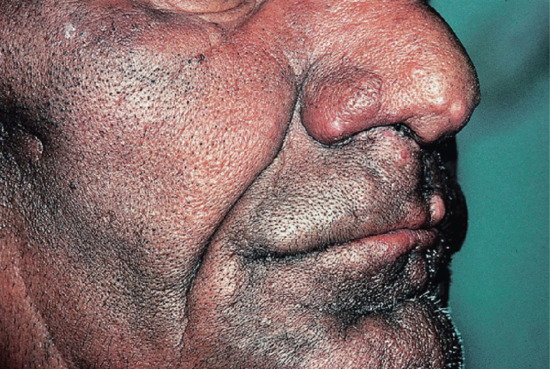
Figure 28.12 Lepromatous leprosy. Face of a man showing diffuse infiltration of the skin and appearance of nodules on the nose and lip.
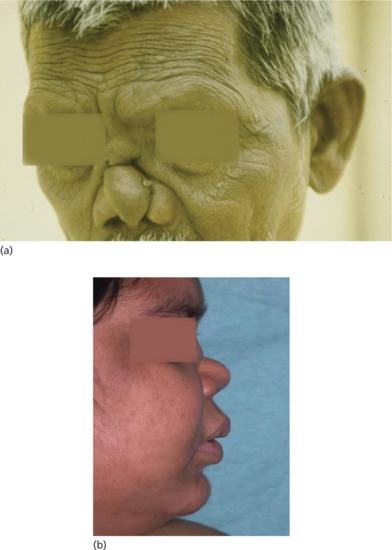
Figure 28.13 Lepromatous leprosy. (a) Man with nasal collapse and forehead wrinkling. (b) Woman with nasal collapse.
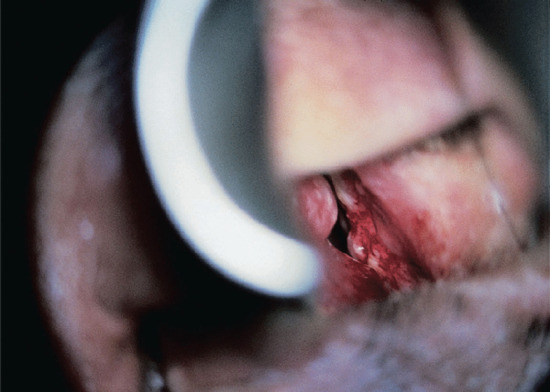
Figure 28.14 Lepromatous leprosy. Examination via a nasal speculum shows a pale septal nodule and bleeding. Such lesions in untreated lepromatous patients constitute a major source of infection.
The longest peripheral sensory nerve fibres are first affected, causing numbness and anaesthesia on the dorsal surfaces of hands and feet, and later on extensor surfaces of arms and legs, and finally over the trunk. Infiltration of corneal nerves causes anaesthesia, which predisposes to injury, infection and blindness if there is also lagophthalmos due to damage to the facial nerve. The hands and feet swell and may become oedematous. Radiographs may show osteoporosis in the phalanges, small osteolytic cysts and often hairline or compression fractures. The fingers may become crooked or short (Figure 28.15). Nails are thin and brittle.
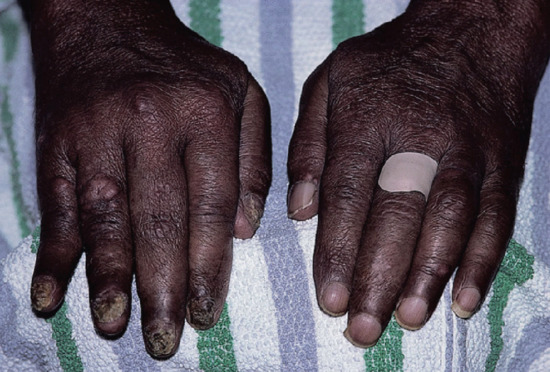
Figure 28.15 Lepromatous leprosy. Hands of an Indian man, showing swollen fingers due to leprous dactylitis and one crooked finger due to a pathological fracture. Note also the lepromatous nodules. The nail dystrophy is due to a dermatophyte infection.
If the patient remains untreated the lines of the forehead become deeper as the skin thickens (leonine facies), eyebrows and eyelashes become thinned or lost (madarosis), ear lobes are thickened, the nose becomes misshapen, and may collapse due to septal perforation and loss of the anterior nasal spine, the voice becomes hoarse and the upper incisor teeth loosen or fall out [78]. The skin of the legs becomes ichthyotic and thickened, ulcers may form on the legs when nodules break down, and a slow fibrosis of peripheral nerves results in nerve thickening and bilateral ‘glove and stocking’ anaesthesia. Sensation of palms and soles is retained until late in the disease. Leprous deposits in the eyes cause keratitis, iridocyclitis and iris atrophy. Testicular atrophy causes sterility, impotence and gynaecomastia.
One particular variety of LL requires special mention, namely, the pure diffuse type described by Lucio and Alvarado in Mexico in 1852. The patients first notice impairment of sensation in the hands and feet, and this is followed by gradual loss of the eyebrows, eyelashes and body hair. At the same time, the skin of the whole body becomes diffusely thickened, rendering it stiff and smooth as in scleroderma. There may be alopecia, nasal and laryngeal involvement, and widespread small telangiectases, but cutaneous nodules and plaques do not develop. The eyes have a shining appearance [79].
Histoid lesions are distinctive round, regular, cutaneous nodules that stand out on normal skin [80].
Borderline leprosy
Skin lesions are intermediate in number between those of the two polar types already described, depending on the position of the patient on the borderline spectrum, and are distributed asymmetrically. They may take the form of macules, plaques, annular lesions or bizarre-shaped bands. Plaques with a ‘punched-out’ appearance are characteristic of the middle of the spectrum (Figure 28.16). Towards the tuberculoid end of the spectrum, lesions are fewer and drier (Figures 28.17 and 28.18), have more hair loss and anhidrosis, are more insensitive, and have fewer bacilli in smears and biopsies, and vice versa towards the lepromatous pole. One or more nerves are likely to be thickened and non-functioning (Figure 28.19). Neural symptoms may precede the appearance of skin lesions by as much as 8 years [81]. When BL downgrades to lepromatous, the resulting subpolar LL patients (designated LLs) can be differentiated from polar lepromatous (LLp) because, in addition to typical lepromatous skin lesions, there are several asymmetrical thickened nerves and one or more typical borderline skin lesions. Damage to structures other than skin and nerves will not be manifest clinically in BL, even though bacilli may be present in other tissue. BL is the commonest type of disease encountered and is unstable and ‘downgrades’ towards lepromatous, especially if untreated, or ‘upgrades’ towards tuberculoid. The clinical change lags behind the immunological and histological changes (Figure 28.20).
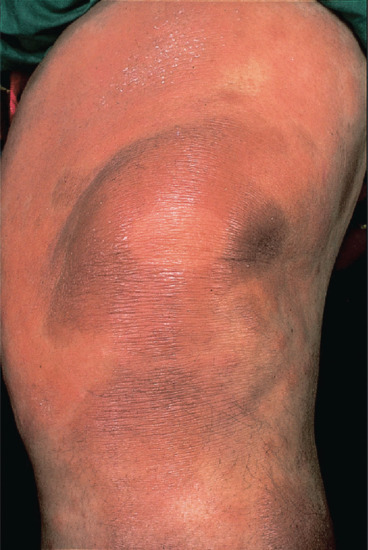
Figure 28.16 Borderline leprosy. Knee of Saudi-Arabian woman, showing classical annular lesions with well-defined centres. Slit-skin smears show acid-fast bacilli.
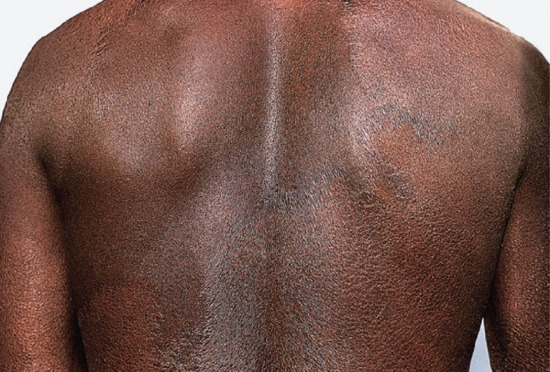
Figure 28.17 Borderline tuberculoid leprosy. Back of Nigerian man showing large well-defined scaly macules with some marginal elevation. Extensive disease, such as this, may seriously impair sweating and heat control.

Figure 28.18 Borderline tuberculoid leprosy. Arm of an Indian woman. A large scaly macule is developing secondary ichthyotic change.
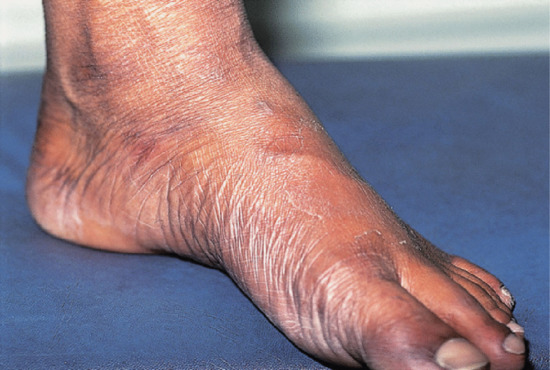
Figure 28.19 Borderline leprosy. Foot of a Bangladeshi child showing enlargement of posterior tibial and anterior tibial nerves.
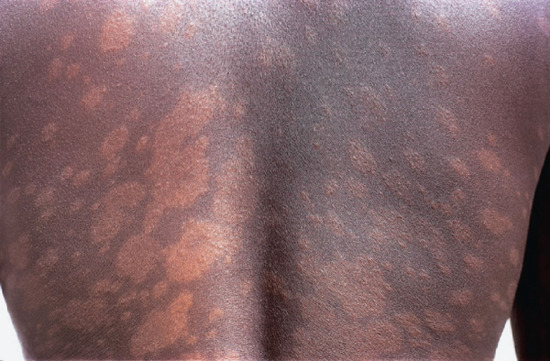
Figure 28.20 Borderline leprosy (BL). Borderline tuberculoid (BT) downgrading to BL. Back of a Nigerian woman, showing typical well-defined hypopigmented macules of BT leprosy and many small lesions, some of which are papular. Slit-skin smears showed acid-fast bacilli.
Pure neuritic leprosy
Pure neuritic leprosy presents with asymmetrical involvement of peripheral nerve trunks and no visible skin lesions; on histology of a cutaneous nerve biopsy, all types of leprosy are seen [82]. It is seen most frequently, but not exclusively, in India and Nepal where it accounts for 5–10% of patients.
Differential diagnosis
Leprosy tends to be overdiagnosed in endemic countries and underdiagnosed in non-endemic countries. Of new patients seen in the period 1995–99 at the Hospital for Tropical Diseases, London, diagnosis had been delayed in over 80% of cases [4]. Patients had been misdiagnosed by dermatologists, neurologists, orthopaedic surgeons and rheumatologists. A common problem was failure to consider leprosy as a cause of peripheral neuropathy in patients from leprosy endemic countries. These delays had serious consequences for patients, with over half of them having nerve damage and disability. In any population it is also important for the doctor to know the normal range of skin colour and texture, the common endemic skin diseases, such as onchocerciasis, that may coexist with leprosy and the common medical and artefactual practices that may cause lesions resembling those of leprosy.
Macular lesions
Birthmarks are abnormally pigmented but otherwise physiologically normal. Vitiligo lesions are depigmented; leprosy lesions are never completely depigmented. Hypopigmented lesions of eczema, especially of pityriasis alba in children, are difficult to distinguish from lepromatous macules, but their surface is often scaly and smears do not contain AFB. Pityriasis versicolor is not always scaly, but central distribution on the trunk, and the presence of minute distinct macules, are contrary to the characteristics of lepromatous macules. Lesions of tinea corporis itch and may have a vesicular edge, characteristically absent in tuberculoid patches, and scrapings usually show the fungus.
Plaques and annular lesions
In addition to ringworm, granuloma multiforme, sarcoidosis and cutaneous tuberculosis may resemble tuberculoid leprosy, having a similar immunological basis and often indistinguishable histological pattern. However, the lesions are not anaesthetic. Peripheral nerves may occasionally be enlarged in sarcoidosis [83].
Nodules
Cutaneous leishmaniasis causes nodules, but they usually crust and ulcerate after some weeks or months, and are seldom as numerous as those of LL. Slit-skin smears, appropriately stained, reveal Leishmania, and the leishmanin test is positive. Lesions of the rare diffuse cutaneous leishmaniasis may be confusing, until slit-skin smears have been examined [84]. Post-kala-azar dermal leishmaniasis in India and East Africa has a similar distribution and appearance to the skin lesions of LL [85].
Nerves
Peripheral nerve thickening is rarely seen except in leprosy. Hereditary sensory motor neuropathy type III is associated with palpable peripheral nerve hypertrophy. Amyloidosis, which can also complicate leprosy, causes thickening of peripheral nerves. Peroneal muscular atrophy (Charcot–Marie–Tooth disease) is an inherited neuropathy that causes distal atrophy and weakness. Nerve biopsy is characteristic. The causes of other polyneuropathies such as HIV infection, diabetes, alcoholism, vasculitides and heavy metal poisoning should all be considered where appropriate [86].
Likewise, there are many causes of eye disease in endemic countries which may cause signs that in isolation mimic leprosy, especially trachoma, in which trichiasis and entropion follow scarring of the lids, and onchocerciasis, which causes uveitis and its complications.
Classification of severity
All new leprosy patients need to be classified according to either the Ridley–Jopling classification or the World Health Organization (WHO) classification. Patients should be assessed to determine whether they have new nerve damage. Finally, they should be assessed for being in a reaction.
Complications and co-morbidities
Reactions: type 1, erythema nodosum leprosum and neuritis
Type 1 reactions occur in borderline disease and are characterized by acute neuritis and/or acutely inflamed skin lesions [87]. Nerves often become tender with loss of sensory and motor functions. Existing skin lesions become erythematous or oedematous and may desquamate or rarely ulcerate. New lesions may appear (Figure 28.21). Occasionally, oedema of the face, hands or feet is the presenting symptom, but constitutional symptoms are unusual. Patients often first present for medical advice when a previously quiescent BT lesion develops a spontaneous type 1 reaction. A recent cohort study in India found that most type 1 reactions occurred in the first 12 months after starting treatment [88]. Women are also at risk during the puerperium.
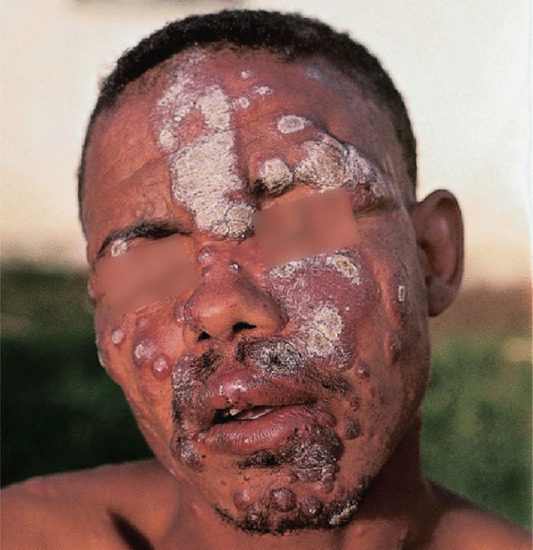
Figure 28.21 Type 1 reaction: borderline leprosy in an Ethiopian man. Existing lesions become acutely inflamed, scale and threaten ulceration. Many small new lesions have appeared. The histology shows a borderline tuberculoid pattern.
Type 2 (ENL) reactions occur in patients with multibacillary disease (LL and BL). They may occur before, during or after treatment. Up to 50% of LL and 15% of BL patients may experience ENL reactions [89]. Attacks are often acute at first, but may be prolonged or recurrent over several years and eventually quiet but insidious, especially in the eye. ENL manifests most commonly as painful red nodules on the face and extensor surfaces of limbs. The lesions may be superficial or deep, with suppuration (Figure 28.22), ulceration (Figure 28.23) or brawny induration when chronic. Acute lesions crop and desquamate, fading over several days (Figure 28.23). ENL is a systemic disorder producing fever and malaise and may be accompanied by uveitis, dactylitis, arthritis, neuritis, lymphadenitis, myositis and orchitis [90, 91]. Peripheral nerve neuritis and uveitis with its complications of synechiae, cataract and glaucoma are the most serious complication of ENL.

Figure 28.22 Type 2 reaction in lepromatous leprosy in a Nigerian man: erythema nodosum leprosum. Several of the reaction nodules have broken down, releasing pus.
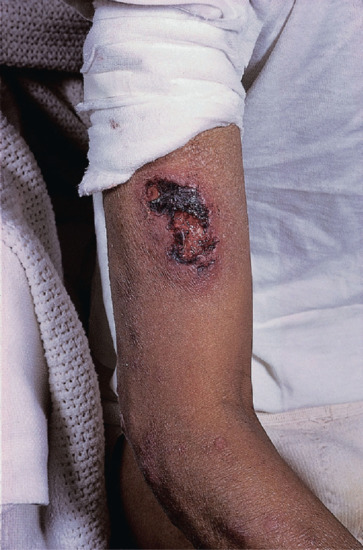
Figure 28.23 Type 2 reaction in lepromatous leprosy in a Bangladeshi man, showing severe necrosis and ulceration. Erythema nodosum leprosum tends to be more severe in Asian than in African people.
The Lucio reaction only occurs in patients with Lucio leprosy [92]. It is due to infarction consequent upon deep cutaneous vasculitis, and causing the appearance of irregularly shaped erythematous patches which become dark and heal, or form bullae and necrose, leaving deep painful ulcers that are slow to heal (Figure 28.24). The systemic upset is severe and can be fatal. This form of leprosy has been associated with a putative new species of mycobacterium, M. lepromatosis.
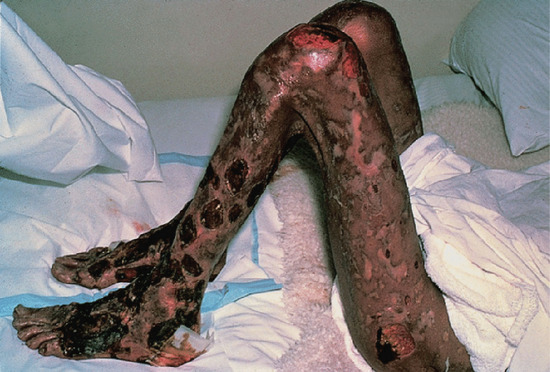
Figure 28.24 Lucio phenomenon in a Mexican man. The severe recurrent ulceration due to deep subcutaneous vasculitis may be fatal.
(Courtesy of Dr J. Keystone, the Toronto Hospital, Toronto, Canada.)
Nerve damage [93]
Of the three physiological functions of nerves, the sensory component is commonly the first and most severely affected, but occasionally there is a pure motor lesion. Autonomic dysfunction is always present in severe nerve damage [94]. In skin lesions this is associated with loss of hair growth, and of sebaceous and sweat secretion, and poor pigment formation. In a limb it causes capillary stasis, cyanosis and dryness, which predispose to fissuring. Two large cohort studies with systematic nerve examination at entry showed that the posterior tibial nerve is the most frequently affected nerve, followed by ulnar, median, lateral popliteal and facial [95, 96]. Ulnar and median nerve lesions are usually low, causing small muscle but not deep flexor weakness, and anaesthesia of the two halves of the hand. Isolated median nerve lesions are unusual. Common peroneal nerve lesions cause difficulty in dorsiflexion and eversion of the foot and anaesthesia of the outer border of the foot, a combination which predisposes to traumatic damage and plantar ulceration (Figure 28.25). Posterior tibial nerve damage is serious because it causes paralysis and contracture of the small muscles of the foot and anaesthesia of the sole.
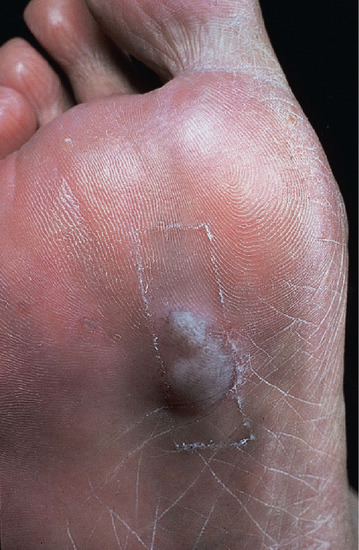
Figure 28.25 Necrosis blister in an anaesthetic foot. Necrotic material has tracked to soft skin from the site of trauma, beneath the metatarsal heads.
Eye involvement in leprosy
Blindness due to leprosy is a devastating complication, especially for a patient with anaesthetic hands and feet. Eye damage results from both nerve damage and bacillary invasion, and a recent cohort study found that 2.8% of multibacillary patients were blind at diagnosis, and a further 11% of patients had potentially blinding ocular pathology [97]. Lagophthalmos results from paresis of the orbicularis oculi due to involvement of the zygomatic and temporal branches of the facial (VIIth) nerve. Facial lesions cause a 10-fold increase in the risk of facial nerve damage [98]. In lepromatous disease, lagophthalmos occurs later and is usually bilateral. Damage to the ophthalmic branch of the trigeminal (Vth) nerve causes anaesthesia of the cornea and conjunctiva, which results in drying of the cornea, a reduction in blinking, and leaves the cornea at risk of minor trauma and ulceration. Bacillary invasion of the iris and ciliary body makes them extremely susceptible to reactions.
Investigations [99]
The diagnosis is usually made clinically on the basis of two out of three characteristic findings, or by the demonstration of AFB in slit-skin smears, or by histology typical of leprosy. The cardinal signs are:
- Anaesthesia of a skin lesion, or in the distribution of a peripheral nerve, or over dorsal surfaces of hands and feet.
- Thickened nerves, especially at the sites of predilection.
- Typical skin lesions.
The AFB load of a patient is determined by modified Ziehl–Neelsen staining of slit-skin smears. Suspect lesions, and sites commonly affected in LL, should be sampled (e.g. the forehead, earlobes, chin, extensor surface of the forearm, buttocks and trunk). The density of bacilli is expressed using a logarithmic scale, extending from very few AFB to many per high-power field. A mean score, the bacterial index (BI), is derived by adding the scores from each site and dividing by the number of sites sampled [100]. In untreated LL, the BI is 5 or 6. The BI falls to zero in TT disease. Slit-skin smears only detect bacilli present at a concentration greater than 104/g tissue, and so cannot be used as a test of microbiological cure. With treatment, bacilli disappear from BB lesions in a few months and from BL lesions in a year or two. It may take 6–10 years for the last bacillary remnants to disappear from the skin in LL.
Slit-skin smears [101]
The lesion is cleaned with ether or alcohol, and a fold is gripped firmly between thumb and forefinger to render it blood free. An incision 5 mm long and 3 mm deep is made with a small-bladed scalpel (size 15 Bard Parker blade); the blade is turned at right angles to the cut, and without relaxing finger pressure, the wound is scraped several times in one direction. Fluid and pulp from the dermis, collected on one side of the blade, are gently smeared on to a glass slide. A bloody smear is useless. The smear is then fixed over a flame and stained.
Skin biopsy [102]
The incision should be made down to subcutaneous fat, so that the whole depth of the dermis is included, otherwise leprous changes in the deeper layers of the dermis will be missed.
Nerve biopsy
In pure neural leprosy a nerve biopsy is necessary to establish the diagnosis. A purely sensory thickened peripheral nerve should be sampled, for example radial cutaneous at the wrist, superficial peroneal in front of the ankle or sural nerve at the ankle.
Serology
The evaluation of PGL serology in extensive field studies shows that more than 90% of untreated multibacillary patients have positive serology in comparison with 40–50% of paucibacillary patients and 5–10% of healthy controls [103]. PGL-1 antibodies can now be detected using a dipstick type test (so-called lateral flow). This has been evaluated in field settings and seropositivity correlates with bacterial load measured by slit-skin smears. The use of PGL-1 antibody testing may be useful in assessing paucibacillary patients who are smear negative but have a low bacterial load and are at risk of undertreatment [104].
Pregnancy
Women may present with all types of leprosy during pregnancy. This may be partly due to increased contact with health providers since they are relatively immunosuppressed during pregnancy. They are at high risk of T1R in the postpartum period and may present with leprosy then [105].
Management
There are five main principles of treatment:
- Stop the infection with chemotherapy.
- Treat reactions and reduce the risk of nerve damage.
- Educate the patient to cope with existing nerve damage, in particular anaesthesia.
- Treat the complications of nerve damage.
- Rehabilitate the patient socially and psychologically.
These objectives can only be achieved with the patient's cooperation and confidence. Patients will often need several consultations soon after diagnosis to address their fears and build up their confidence.
Expectations of treatment
Antibacterial treatment for leprosy is highly effective, with low relapse rates, but needs to be taken over 6, 12 or 24 months. However, new patients can be reassured that their infection is curable and that the aim of treatment is to minimize further nerve damage. Patients should not develop the mutilating sequelae seen previously.
Left untreated, borderline patients will downgrade towards the lepromatous end of the spectrum, and lepromatous patients will suffer the consequences of bacillary invasion. Borderline patients are at risk of developing type 1 reactions, which may result in devastating nerve damage. Many patients present with established nerve damage, which cannot be reversed. Treatment of the neuritis is currently unsatisfactory, and some patients with active neuritis will develop permanent nerve damage despite treatment with corticosteroids. It is not possible to predict which patients will develop reactions or nerve damage. Nerve damage and its complications may be severely disabling, especially when all four limbs and both eyes are affected.
First line
Chemotherapy
All leprosy patients should be given an appropriate multidrug combination. The effectiveness of dapsone against M. leprae was discovered in the late 1940s, and it was used widely as a single agent. This lead to the widespread development of dapsone resistance, initially presenting as disease relapse 15 years after treatment with dapsone monotherapy, but then also as primary dapsone resistance in untreated patients [106]. In response to the failure of dapsone monotherapy, WHO proposed an MDT for the treatment of leprosy [107]. In a multibacillary patient, there are three distinguishable types of bacilli: fully drug-sensitive bacteria, drug-resistant mutants and a small population of ‘persisters’, dormant non-multiplying bacilli. Treatment with an MDT should eliminate nearly all organisms. The first line antileprosy drugs are rifampicin, dapsone and clofazimine. Table 28.2 gives the details of drug dosages and duration of treatment.
Table 28.2 World Health Organization (WHO) multidrug therapy regimen
| Drug treatment | |||
| Type of leprosy | Monthly supervised | Daily self-administered | Duration of treatment |
| Paucibacillary | Rifampicin 600 mg | Dapsone 100 mg | 6 months |
| Multibacillary | Rifampicin 600 mg | Clofazimine 50 mg | 12 months |
| Clofazimine 300 mg | Dapsone 100 mg | ||
Rifampicin is a potent bactericidal for M. leprae. Four days after a single 600 mg dose, bacilli from a previously untreated multibacillary patient are no longer viable [108]. It acts by inhibiting DNA-dependent RNA polymerase, thereby interfering with bacterial RNA synthesis. Rifampicin is well absorbed orally. Hepatotoxicity may occur with a mild transient elevation of hepatic transaminases, but this is rare at the dosage and intervals recommended for leprosy and is not an indication for stopping treatment. Because M. leprae resistance to rifampicin can develop as a one-step process, rifampicin should always be given in combination with other antileprotics [109].
Dapsone (4,4-diaminodiphenylsulphone (DDS)) acts by blocking folic acid synthesis. It is only weakly bactericidal. Oral absorption is good and it has a long half-life, averaging 28 h. Dapsone, in the doses recommended for leprosy, commonly causes mild haemolysis, and rarely anaemia or psychosis. Glucose-6-phosphate dehydrogenase deficiency seldom causes a problem and is not routinely looked for. The ‘DDS syndrome’, which is occasionally seen in leprosy, starts 6 weeks after commencing DDS and manifests as exfoliative dermatitis associated with lymphadenopathy, hepatosplenomegaly, fever and hepatitis, and may be fatal [110]. Agranulocytosis, hepatitis and cholestatic jaundice occur rarely with DDS therapy.
Clofazimine is a brick red, fat-soluble crystalline dye. The mechanism of its weakly bactericidal action against M. leprae is not known. It has an anti-inflammatory effect, which is useful in the management of ENL reactions. High drug concentrations are found in the intestinal mucosa, mesenteric lymph nodes and body fat. The most noticeable side effect is skin discoloration, ranging from red to purple–black, the degree of discoloration depending on the dose and amount of leprous infiltration. The pigmentation usually fades within 6–12 months of stopping clofazimine, although traces of discoloration may remain for up to 4 years. Urine, sputum and sweat may become pink. Clofazimine also produces a characteristic ichthyosis on the shins and forearms. Gastrointestinal side effects, ranging from mild cramps to diarrhoea and weight loss, may occur as a result of clofazimine crystal deposition in the wall of the small bowel.
Relapse
Relapsed multibacillary patients are also retreated with triple therapy regardless of any change in classification [111].
The distinction between relapse and reaction may be difficult (Figure 28.26). Therefore, paucibacillary patients require 2 years, and multibacillary patients at least 5 years, of monitoring after treatment. Patients can be discharged if there is no evidence of activity or reaction, but should be advised to return if new symptoms develop, especially in the hands, feet or eyes. Patients with reactions or physical or psychological complications may need much longer care.
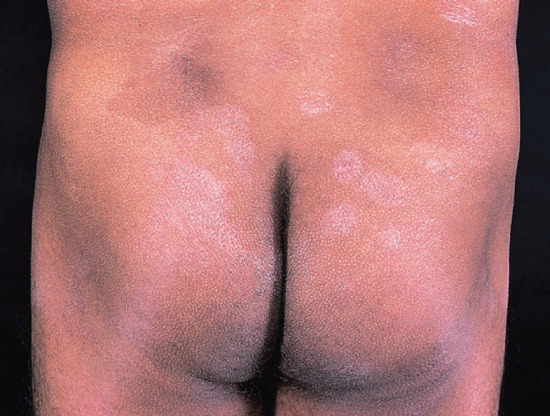
Figure 28.26 Borderline leprosy (BL). Borderline leprosy upgrading to borderline tuberculoid (BT). Buttocks of an Indian man who ‘relapsed’ 3 years after completing multidrug therapy for BL leprosy. The lesions here are typical hypopigmented, erythematous, scaly plaques of BT leprosy in reaction. No acid-fast bacilli were seen on slit-skin smears. In this situation, the distinction between (bacterial) relapse and simple reversal reaction may be impossible.
Published clinical outcomes for patients treated with the paucibacillary regimen show that 2–44% of patients had clinically active skin lesions at the end of treatment [112]. Nerve impairment occurred de novo in 2.5% of patients, and visible disabilities increased from 4% at enrolment to 7% at 8–10 years of follow-up. Relapse rates are low, ranging from 0% in Ethiopia [113] to 2.5% over 4 years in Malawi [114]. For patients treated with the multibacillary regime for 24 months, one study in Thailand found that 29% of lesions were still active after 3 years and that visible disabilities increased from 5% at enrolment to 13% at 8–10 years of follow-up [115]. Relapse rates have been reported from six observational studies varying from zero in China and Ethiopia to 2.04/100 person years in India. Data from West Africa [116] and India [117] shows that patients with a high initial bacterial load (BI >4+) treated with 2 years of rifampicin, clofazimine and dapsone had a relapse rate of 8/100 person years, whereas patients treated to smear negativity had a relapse rate of 2/100 person years. These patients may form a subgroup who need treating to skin-smear negativity [118]. Susceptibility testing of M. leprae strains from relapsed multibacillary patients has shown them to remain drug sensitive.
The recommended length of treatment for multibacillary patients has dropped from 24 months to 12 months. There was no controlled trial data to guide this decision, but the classification of multibacillary patients had been widened, so some patients who would previously have received paucibacillary treatment for 6 months are now receiving multibacillary treatment for 12 months. New proposals include testing a common 6-month regimen of dapsone, clofazimine and rifampicin for all patients [119]. This would simplify leprosy treatment but would give 60% of patients a third drug that they do not need, and undertreat patients with a high bacterial load [120].
Second line
Several new drugs bactericidal for M. leprae have been identified: fluoroquinolones, minocycline and clarithromycin. The fluoroquinolones pefloxacin and ofloxacin have a remarkable degree of bactericidal activity, with 22 daily doses killing 99.99% of viable M. leprae present in multibacillary cases at the start of treatment [121]. Daily minocycline (100 mg) treatment of multibacillary patients for 3 months resulted in killing of all viable M. leprae organisms [122]. Clarithromycin, given in 500 mg daily doses to multibacillary patients, has a similar bactericidal effect [123]. Antagonism between these new drugs has not been demonstrated [124]. Ofloxacin, minocycline and clarithromycin are established second line drugs, and may replace dapsone and clofazimine. Minocycline may also cause hyperpigmentation of skin lesions, and so may not be an appropriate substitute for clofazimine if pigmentation is to be avoided [125].
A triple-drug combination (rifampicin, ofloxacin and minocycline) which can be given as a single monthly dose for 6 or 12 months can be used to treat patients with adverse effects to one of the components of MDT [126].
Complications of treatment
Reactions and neuritis [127]
Nerve damage occurs before diagnosis, during and after multidrug treatment. It may occur during a reaction or without overt signs of nerve inflammation (silent neuropathy). In field cohort studies 16–56% of newly diagnosed patients have nerve damage [128]. In a Bangladeshi study, 25% of multibacillary patients developed nerve damage during treatment [129]. This study also showed that 65% of patients with multibacillary leprosy who had nerve damage at diagnosis developed new nerve damage over a 5-year follow-up; therefore this group should be closely monitored. Analysis from a large cohort study in Ethiopia showed that standardized nerve function testing was needed monthly to detect new nerve damage early [130].
Patients frequently seek medical advice for their leprosy only when a reversal reaction develops in a previously quiescent skin lesion, or when they develop pain, weakness or numbness. Awareness of the early symptoms of reversal reactions by both patient and physician is important, because if left untreated severe nerve damage may occur. The peak time for reversal reactions is in the first 6 months of treatment [131], so it is important to warn patients about reactions. The sudden development of reactional lesions soon after starting treatment is distressing and undermines confidence.
The treatment of reactions is aimed at controlling acute inflammation, easing pain, reversing nerve and eye damage and reassuring the patient. MDT should be continued. Neuritis (e.g. nerve tenderness, new anaesthesia and/or motor loss) or moderately inflamed skin lesions should be treated with corticosteroids. Standardized courses of prednisolone have been used, starting at 40 mg daily, reducing by 5 mg every 2–4 weeks [132]. Patients with borderline reactions commonly need 20 weeks of steroids. A recent Indian study compared different starting doses (60 versus 30 mg) and durations (12 versus 20 weeks) and showed that the longer durations gave the best outcomes. A study looking at cytokine profiles in reactional patients showed that, even after 6 months of steroid treatment, some patients still had high levels of pro-inflammmatory cytokines in their skin lesions [133]. In another approach, the prevention of reactions has been explored. In one study, multibacillary patients were randomized to either prednisolone 20 mg daily or placebo for the first 3 months of their MDT. Reactional episodes were significantly lower in the prednisolone-treated group at 4 months, but the protective effect was lost by 12 months. These studies all demonstrate that reactions are difficult to prevent and to switch off once established. Other immunosuppressants have been studied to assess their effect in reactions, 3 days of 1 g methylprednisolone did not reduce the recurrence rate [134]. Adding azathioprine to prednisolone did not improve nerve function or reduce recurrence rate [135].
Erythema nodosum leprosum is a difficult condition to treat, and frequently requires therapy with high-dose steroids (80 mg daily, tapered down rapidly) or thalidomide. Since ENL frequently recurs, steroid dependency can easily develop. Thalidomide (400 mg daily) is superior to steroids in controlling ENL, and is the drug of choice for young men with severe ENL [136]. Women with severe ENL may benefit from thalidomide treatment. This is a difficult decision for the woman and her physician, and needs careful discussion of the benefits and risks (phocomelia when thalidomide is taken in the first trimester). Women should use double contraception and report immediately if menstruation is delayed. Unfortunately, the problems with thalidomide mean that it is unavailable in several leprosy endemic countries despite its undoubted value. Clofazimine has a useful anti-inflammatory effect in ENL and can be used at 300 mg/day for several months. Low-grade chronic ENL, with iritis or neuritis, will require long-term suppression, preferably with thalidomide or clofazimine. Acute iridocyclitis is treated with 4-hourly instillation of 1% hydrocortisone eye drops and 1% atropine drops twice daily.
Additional aspects of management
Education of the patient
Educating a leprosy patient about their disease is the key to successful management. The patient needs to be reassured that within a few days of chemotherapy they will not be infectious and can lead a normal social life. A clear explanation of the disease and refutation of myths about leprosy will help the patient come to terms with their diagnosis and may well improve compliance. It is important to emphasize that gross deformities are not the inevitable end point of disease, and that care and awareness of their limbs is as important as chemotherapy. Anxieties about transmission and reactions, as well as issues about compliance, should be addressed.
Complications of nerve damage [137, 138]
The complications of nerve damage, which are the major causes of disability and deformity in leprosy, are preventable by early diagnosis, correct treatment and education of the patient. Monitoring sensation and muscle power in patient hands, feet and eyes should be part of the routine follow-up, so that new nerve damage is detected early.
Dry skin should be treated by soaking in water, followed by rubbing with emulsifying ointment or petrolatum. Callus should be rubbed down with pumice or an abrasive nylon pad, and fissures need to be covered to allow them to heal.
The morbidity and disability associated with leprosy is secondary to nerve damage. A major goal in the prevention of disability is to create patient self-awareness, so that damage is minimized. The patient with an anaesthetic hand or foot needs to understand the importance of daily self-care, especially protection when doing potentially dangerous tasks, and inspection for trauma. It is helpful to identify for each patient potentially dangerous situations, such as cooking, car repairs or smoking. Soaking dry hands and feet followed by rubbing with oil keeps the skin moist and supple.
An anaesthetic foot needs the protection of an appropriate shoe. For anaesthesia alone, a well-fitting ‘trainer’ with firm soles and shock-absorbing inners will provide adequate protection. Once there is deformity, such as clawing, shoes must be tailored specially to ensure protection of pressure points and even weight distribution.
The patient should be taught to question the cause of an injury so that the risk can in the future be avoided. Plantar ulceration occurs secondary to increased pressure over bony prominences. Ulceration is treated by rest. Unlike ulcers in diabetic or ischaemic feet, ulcers in leprosy heal if they are protected from weight-bearing. No weight-bearing is permitted until the ulcer has healed. Appropriate footwear should be provided to prevent recurrence.
Weakness or paralysis
These patients require physiotherapy, with the objective of permitting the return of function while preventing the formation of contracture. Patients with contractures are taught exercises to prevent fixation. Contractures of hands and feet, foot drop, lagophthalmos, entropion and ectropion are amenable to surgery.
Social, psychological and vocational rehabilitation
The social and cultural background of the patient determines the nature of many of the problems that may be encountered. The patient may have difficulty in coming to terms with leprosy. The community may reject the patient. Education, gainful employment, confidence from family, friends and doctor, and plastic surgery to correct stigmatizing deformity, all have a role to play.
Prevention and control
Dermatologists now play a key role in diagnosing and treating leprosy patients. For much of the 20th century, leprosy patients were detected and treated within vertical programmes dedicated to leprosy. Although these were effective they also became inefficient as the numbers of leprosy patients declined. The management of leprosy patients is now being integrated into a range of health services including combined leprosy and tuberculosis programmes, dermatology programmes and full integration with general health services. The current WHO strategy emphasizes the importance of quality services that are accessible, patient centred, provide free treatment with MDT, undertake appropriate prevention of disability, and refer patients on for the management of complications. There are now huge needs to train people to recognize leprosy. The development of referral services is also a critical need if an integrated approach is to work well.
Vaccines against leprosy
The substantial cross-reactivity between BCG and M. leprae has been exploited in attempts to develop a vaccine against leprosy. Trials of BCG as a vaccine against leprosy in Uganda, New Guinea, Burma and South India showed it to confer statistically significant but variable protection, ranging from 80% in Uganda to 20% in Burma, and a meta-analysis has estimated that BCG vaccination confers 50% protection against leprosy [139]. In Northern Malawi, BCG gives 50% protection against leprosy but no significant protection against tuberculosis [140]. A case–control study in Venezuela showed BCG vaccination to give 56% protection to the household contacts of leprosy patients [141]. The variability and unpredictability of BCG vaccination has led to various attempts to improve its protective efficacy. Combining BCG and killed M. leprae is one approach, but in both a large population-based trial in Malawi [142] and in an immunoprophylactic trial in Venezuela [143] there was no advantage with BCG plus M. leprae over BCG alone. Possibly the variable protection induced by BCG is due to early contact with environmental mycobacteria priming the immune system and conferring protective immunity against M. leprae. Vaccination with BCG after contact with environmental mycobacteria will then contribute little towards inducing improved immunity against M. leprae [144].
There was a WHO-led campaign to eliminate leprosy as a public health problem. Although this focused resources and energy on leprosy, the effect of a target-driven approach was eventually counterproductive [145]. Leprosy patients continue to present in many countries and will need diagnosis and treatment. Leprosy is unlikely to be eradicated until there is considerable improvement in general health, wealth, living conditions and education.
Resources
Further information
- World Health Organization: http://www.who.int/topics/leprosy/en/
- Public Health England: http://www.hpa.org.uk/Topics/InfectiousDiseases/InfectionsAZ/Leprosy/ (Both last accessed August 2014.)
References
- Irgens LM, Bjerkedal T. Epidemiology of leprosy in Norway. The history of the National Leprosy Registry of Norway from 1856 until today. Int J Epidemiol 1973;2:81–9.
- Hansen GA. [Undersolgelser angraaende spedalskhedens aasger.] Norsk Magazin Laegervidens Kaben 1874;4 (Suppl.):1–88.
- Anonymous. Leprosy: global situation 2012. Wkly Epidemiol Rec 2012;87:317–28.
- Lockwood DN, Reid AJ. The diagnosis of leprosy is delayed in the United Kingdom. QJM 2001;94:207–12.
- Noordeen SK. Mortality in leprosy. Indian J Med Res 1972;60:439–45.
- Noordeen SK. The epidemiology of leprosy. In: Hastings RC, ed. Leprosy. Edinburgh: Churchill Livingstone, 1994:15–30.
- Fine PE. Leprosy: the epidemiology of a slow bacterium. Epidemiol Rev 1982;4:161–88.
- Feldman RA, Sturdivant M. Leprosy in Louisiana, 1855–1970. An epidemiologic study of long-term trends. Am J Epidemiol 1975;102:303–10.
- Mira MT, Alcaïs A, Thuc NV, et al. Susceptibility to leprosy is associated with PARK2 and PACRG. Nature 2004;427:636–40.
- Ciechanover A. The ubiquitin proteolytic system. Neurology 2006;66(Suppl. 1):1–13.
- Roy S, Frodsham A, Saha B, et al. Association of vitamin D receptor genotype with leprosy type. J Infect Dis 1999;179:187–91.
- Roy S, McGuire W, Mascie-Taylor CG, et al. Tumor necrosis factor promoter polymorphism and susceptibility to lepromatous leprosy. J Infect Dis 1997;176:530–2.
- Santos AR, Suffys PN, Vanderborght PR, et al. Role of tumor necrosis factor-alpha and interleukin-10 promoter gene polymorphisms in leprosy. J Infect Dis 2002;186:1687–91.
- Fitness J, Floyd S, Warndorff DK, et al. Large-scale candidate gene study of leprosy susceptibility in the Karonga district of northern Malawi. Am J Trop Med Hyg 2004;71:330–40.
- Moraes MO, Pacheco AG, Schonkeren JJ, et al. Interleukin-10 promoter single-nucleotide polymorphisms as markers for disease susceptibility and disease severity in leprosy. Genes Immun 2004;5:592–5.
- Malhotra D, Darvishi K, Sood S, et al. IL-10 promoter single nucleotide polymorphisms are significantly associated with resistance to leprosy. Hum Genet 2005;118:295–300.
- Sampaio EP, Caneshi JR, Nery JA, et al. Cellular immune response to Mycobacterium leprae infection in human immunodeficiency virus-infected individuals. Infect Immun 1995;63:1848–54.
- Lawn SD, Wood C, Lockwood DNJ. Borderline tuberculoid leprosy: an immune reconstitution phenomenon in a human immuno-deficiency virus-infected person. Clin Infect Dis 2003;36:e5–6.
- Ustianowski AP, Lawn SD, Lockwood DNJ. Interactions between HIV infection and leprosy: a paradox. Lancet Infect Dis 2006;6:350–60.
- Deps P, Lucas S, Porro AM. Clinical and histological features of leprosy and HIV co-infection in Brazil. Br J Dermatol 2013;38:470–7.
- Rees RJ, Meade TW. Comparison of the modes of spread and the incidence of tuberculosis and leprosy. Lancet 1974;1:47–8.
- Gill AL, Bell DR, Gill GV, et al. Leprosy in Britain: 50 years experience in Liverpool. QJM 2005;98:505–11.
- Beyene D, Aseffa A, Harboe M, et al. Nasal carriage of Mycobacterium leprae DNA in healthy individuals in Lega Robi village, Ethiopia. Epidemiol Infect 2003;131:841–8.
- Chehl S, Job CK, Hastings RC. Transmission of leprosy in nude mice. Am J Trop Med Hyg 1985;34:1161–6.
- Ridley DS. Pathogenesis of Leprosy and Related Diseases. London: Wright, 1988.
- Myrvang B, Godal T, Feek CM, et al. Immune response to Mycobacterium leprae in indeterminate leprosy patients. Acta Pathol Microbiol Scand [B] Microbiol Immunol 1973;81:615–20.
- Drutz DJ, Chen TS, Lu WH. The continuous bacteremia of lepromatous leprosy. N Engl J Med 1972;287:159–64.
- Jopling WH. Reactions in leprosy. Lepr Rev 1970;41:62–3.
- Dastur DK. Cutaneous nerves in leprosy: the relationship between histopathology and cutaneous sensibility. Brain 1955;78:615–33.
- Shah PK, Malhotra YK, Lakhotia M, et al. Cardiovascular dysautonomia in patients with lepromatous leprosy. Indian J Lepr 1990;62:91–7.
- Beck JS, Abbot NC, Samson PD, et al. Impairment of vasomotor reflexes in the fingertips of leprosy patients. J Neurol Neurosurg Psychiatry 1991;54:965–71.
- Antia NH, Mehta L, Shetty V, Irani PF. Clinical, electrophysiological, quantitative, histologic and ultrastructural studies of the index branch of the radial cutaneous nerve in leprosy. I. Preliminary report. Int J Lepr Other Mycobact Dis 1975;43:106–13.
- Mehta LN, Shetty VP, Antia NH, Irani PF. Quantitative, histologic and ultrastructural studies of the index branch of the radial cutaneous nerve in leprosy and its correlation with electrophysiologic study. Int J Lepr Other Mycobact Dis 1975;43:256–64.
- Shetty VP, Mehta LN, Irani PF, Antia NH. Study of the evolution of nerve damage in leprosy. Part I – lesions of the index branch of the radial cutaneous nerve in early leprosy. Lepr India 1980;52:5–18.
- Weddell GM, Pearson JM. Leprosy – histopathologic aspects of nerve involvement. Contemp Neurol Ser 1975;12:17–28.
- Ridley DS, Jopling WH. Classification of leprosy according to immunity. A five-group system. Int J Lepr Other Mycobact Dis 1966;34:255–73.
- Job CK, Desikan KV. Pathologic changes and their distribution in peripheral nerves in lepromatous leprosy. Int J Lepr Other Mycobact Dis 1968;36:257–70.
- Job CK. Pathology of peripheral nerve lesions in lepromatous leprosy – a light and electron microscopic study. Int J Lepr Other Mycobact Dis 1971;39:251–68.
- Job CK. Mechanism of nerve destruction in tuberculoid-borderline leprosy. An electron-microscopic study. J Neurol Sci 1973;20:25–38.
- Ridley DS, Radia KB. The histological course of reactions in borderline leprosy and their outcome. Int J Lepr Other Mycobact Dis 1981;49:383–92.
- Lockwood DN, Lucas SB, Desikan KV, et al. The histological diagnosis of leprosy type 1 reactions: identification of key variables and an analysis of the process of histological diagnosis. J Clin Pathol 2008;61:595–600.
- Britton WJ, Lockwood DNJ. Leprosy. Lancet 2004;363:1209–19.
- Brightbill HD, Libraty DH, Krutzik SR, et al. Host defense mechanisms triggered by microbial lipoproteins through Toll-like receptors. Science 1999;285:732–3.
- Krutzik SR, Ochoa MT, Sieling PA, et al. Activation and regulation of Toll-like receptors 2 and 1 in human leprosy. Nature Med 2003;9:525–32.
- Demangel C, Britton WJ. Interaction of dendritic cells with mycobacteria: where the action starts. Immunol Cell Biol 2000;78:318–24.
- Birdi TJ, Antia NH. The macrophage in leprosy: a review on the current status. Int J Lepr Other Mycobact Dis 1989;57:511–25.
- Lee SP, Stoker NG, Grant KA, et al. Cellular immune responses of leprosy contacts to fractionated Mycobacterium leprae antigens. Infect Immun 1989;57:2475–80.
- Modlin RL, Hofman FM, Horwitz DA, et al. In situ identification of cells in human leprosy granulomas with monoclonal antibodies to interleukin 2 and its receptor. J Immunol 1984;132:3085–90.
- Ilangumaran S, Shankernarayan N, Ramu G, Muthukkaruppan V. Antibody response to recombinant 65-kDa, 70-kDa and 18-kDa mycobacterial antigens in leprosy patients and healthy contacts in a leprosy-endemic population. Int J Lepr Other Mycobact Dis 1984;62:245–55.
- Paul RC, Stanford JL, Carswell JW. Multiple skin testing in leprosy. J Hyg (Lond) 1975;75:57–68.
- Modlin RL, Melancon-Kaplan J, Young SM, et al. Learning from lesions: patterns of tissue inflammation in leprosy. Proc Natl Acad Sci USA 1988;85:1213–17.
- Haregewoin A, Godal T, Mustafa AS, et al. T-cell conditioned media reverse T-cell unresponsiveness in lepromatous leprosy. Nature 1983;303:342–4.
- Kaplan G, Kiessling R, Teklemariam S, et al. The reconstitution of cell-mediated immunity in the cutaneous lesions of lepromatous leprosy by recombinant interleukin 2. J Exp Med 1989;169:893–907.
- Khanolkar-Young S, Rayment N, Brickell PM, et al. Tumour necrosis factor-α (TNF-α) synthesis is associated with the skin and peripheral nerve pathology of leprosy reversal reactions. Clin Exp Immunol 1995;99:196–202.
- Yamamura M, Wang X-H, Ohmen JD, et al. Cytokine patterns of immunologically mediated tissue damage. J Immunol 1992;149:1470–5.
- Wemambu SN, Turk JL, Waters MF, Rees RJ. Erythema nodosum leprosum: a clinical manifestation of the arthus phenomenon. Lancet 1969;2:933–5.
- Kahawita IP, Lockwood DN. Towards understanding the pathology of erythema nodosum leprosum. Trans R Soc Trop Med Hyg 2008;102:329–37.
- Dharma Rao T, Ramchander Rao P. Enhanced cell-mediated immune response in erythema nodosum leprosum reactions of leprosy. Int J Lepr Other Mycobact Dis 1987;55:36–42.
- Sarno EN, Grau GE, Vieira LM, Nery JA. Serum levels of tumour necrosis factor-α and interleukin-1β during leprosy reactional states. Clin Exp Immunol 1991;84:103–8.
- Levy L, Ji B. The mouse foot-pad technique for cultivation of Mycobacterium leprae. Lepr Rev 2006;77:5–24.
- Rees RJW. Animal models in leprosy. In: Rees RJW, ed. Tuberculosis and Leprosy. British Medical Bulletin, Vol. 44. Edinburgh: Churchill Livingstone, 1988:650–64.
- Storrs EE. The nine-banded armadillo: a model for leprosy and other biomedical research. Int J Lepr Other Mycobact Dis 1971;39:703–14.
- Truman R, Singh P, Sharma S. Probable zoonotic for leprosy in the Southern United States. N Engl J Med 2011;364:1626–33
- Cole ST, Eiglmeier K, Parkhill J, et al. Massive gene decay in the leprosy bacillus. Nature 2001;409:1007–11.
- Eiglmeier K, Cole ST. The integrated genome map of Mycobacterium leprae. Lepr Rev 2001;72:462–9.
- Monot M, Honore N, Garnier N, et al. On the origin of leprosy. Science 2005;308(5724):1040–2.
- Schuenemann VJ, Singh P, Mendum T. A genome-wide comparison of medieval and modern Mycobacterium leprae. Science 2013;341:179–83.
- Han XY, Silva O. On the age of leprosy. PLOS Negl Trop Dis 2014;Feb 8(2):e2544.
- Rivoire B, Pessolani MC, Bozic CM, et al. Chemical definition, cloning, and expression of the major protein of the leprosy bacillus. Infect Immun 1994;62:2417–25.
- Jamil S, Keer JT, Lucas SB, et al. Use of polymerase chain reaction to assess efficacy of leprosy chemotherapy. Lancet 1993;342:264–8.
- Honore N, Cole ST. Molecular basis of rifampin resistance in Mycobacterium leprae. Antimicrob Agents Chemother 1993;37:414–18.
- Brubaker ML, Binford CH, Trautman JR. Occurrence of leprosy in US veterans after service in endemic areas abroad. Public Health Rep 1969;84:1051–8.
- Bryceson AD, Pfalzgraff RE. Leprosy, 3rd edn. Edinburgh: Churchill Livingstone, 1990.
- Sehgal VN, Srivastava G. Indeterminate leprosy. A passing phase in the evolution of leprosy. Lepr Rev 1987;58:291–9.
- Iveson JM, McDougall AC, Leathem AJ, Harris HJ. Lepromatous leprosy presenting with polyarthritis, myositis, and immune-complex glomerulonephritis. BMJ 1975;3:619–21.
- Malin AS, Waters MF, Shehade SA, Roberts MM. Leprosy in reaction: a medical emergency. BMJ 1991;302:1324–6.
- Barton RP. A clinical study of the nose in lepromatous leprosy. Lepr Rev 1974;45:135–44.
- Moller-Christensen V. Changes in the anterior nasal spine and the alveolar process of the macillae in leprosy: a clinical examination. Int J Lepr Other Mycobact Dis 1974;42:431–5.
- Latapi L, Chevez Zamora A. The ‘spotted’ leprosy of Lucio (la lepra ‘manchada’ de Lucio). An introduction to its clinical and histological study. Int J Lepr Other Mycobact Dis 1948;16:421–9.
- Wade HW. The histoid variety of lepromatous leprosy. Int J Lepr Other Mycobact Dis 1963;31:129–42.
- Suneetha S, Arunthathi S, Chandi S, Kurian N, Chacko CJ. Histological studies in primary neuritic leprosy: changes in the apparently normal skin. Lepr Rev 1998;69:351–7.
- Mahajan PM, Jogaikar DG, Mehta JM. A study of pure neuritic leprosy: clinical experience. Indian J Lepr 1996;68:137–41.
- Matthews LVB. Sarcoidosis of the nervous system. J Neurol Neurosurg Psychiatry 1965;28:23.
- Bryceson AD. Diffuse cutaneous leishmaniasis in Ethiopia. I. The clinical and histological features of the disease. Trans R Soc Trop Med Hyg 1969;63:708–37.
- Munro DD, Du Vivier A, Jopling WH. Post kala-azar dermal leishmaniasis. Br J Dermatol 1972;87:374–8.
- Nunzi E, Fiallo P. Differential diagnosis. In: Hastings RC, ed. Leprosy. Edinburgh: Churchill Livingstone, 1994:291–313.
- Lockwood DN, Vinayakumar S, Stanley JN, et al. Clinical features and outcome of reversal (type 1) reactions in Hyderabad, India. Int J Lepr Other Mycobact Dis 1993;61:8–15.
- Van Brakel WH, Nicholls PG, Das L, et al. The INFIR Cohort Study: assessment of sensory and motor neuropathy in leprosy at baseline. Lepr Rev 2005;76:277–95.
- Pocaterra L, Jain S, Reddy R, et al. Clinical course of erythema nodosum leprosum: an 11-year cohort study in Hyderabad, India. Am J Trop Med Hyg 2006;74:868–79.
- Kahawita IP, Lockwood DN. Towards understanding the pathology of erythema nodosum leprosum. Trans R Soc Trop Med Hyg 2008;102:329–37.
- Walker SL, Waters M, Lockwood DNJ. The role of thalidomide in the management of erythema nodosum leprosum. Lepr Rev 2007;78:197–215.
- Rea TH, Ridley DS. Lucio's phenomenon: a comparative histological study. Int J Lepr Other Mycobact Dis 1979;47:161–6.
- Pearson JM, Ross WF. Nerve involvement in leprosy – pathology, differential diagnosis and principles of management. Lepr Rev 1975;46:199–212.
- Dabholkar VR, Gaitonde BB. A study of autonomic functions in leprosy. Lepr India 1982;54:303–17.
- Croft RP, Richardus JH, Nicholls PG, Smith WC. Nerve function impairment in leprosy. Design, methodology, and intake status of a prospective cohort study of 2664 new leprosy cases in Bangladesh (the Bangladesh Acute Nerve Damage Study). Lepr Rev 1999;70:140–59.
- Saunderson P, Gebre S, Desta K, et al. The pattern of leprosy-related neuropathy in the AMFES patients in Ethiopia: definitions, incidence, risk factors and outcome. Lepr Rev 2000;71:285–308.
- Courtright P, Daniel E, Sundarrao Ravenes J, et al. Eye disease in multibacillary leprosy patients at the time of their leprosy diagnosis: findings from the Longitudinal Study of Ocular Leprosy (LOSOL) in India, the Philippines and Ethiopia. Lepr Rev 2002;73:225–38.
- Hogeweg M, Faber WR. Progression of eye lesions in leprosy. Ten-year follow-up study in the Netherlands. Int J Lepr Other Mycobact Dis 1991;59:392–7.
- Guinto RS, Abalos RM, Cellona RV. An Atlas of Leprosy. Tokyo: Sasakawa Memorial Health Foundation, 1983.
- Ridley DS. Therapeutic trials in leprosy using serial biopsies. Lepr Rev 1958;29:45–52.
- World Health Organization. Guidelines for slit skin smears. Int J Lepr Other Mycobact Dis 1987;55:421–2.
- Ridley DS. Skin Biopsy in Leprosy. Basel: Documenta Geigy, 1977.
- Smith PG. The serodiagnosis of leprosy. Lepr Rev 1992;63:97–100.
- Bührer-Sékula S, Visschedijk J, Grossi MA, et al. The ML flow test as a point of care test for leprosy control programmes: potential effects on classification of leprosy patients. Lepr Rev 2007;78:70–9.
- Lockwood DN, Sinha HH. Pregnancy and leprosy: a comprehensive literature review. Int J Lepr Other Mycobact Dis 1999;67:6–12.
- Ji BH. Drug resistance in leprosy – a review. Lepr Rev 1985;56:265–78.
- World Health Organization. Chemotherapy of Leprosy for Control Programmes. Technical Report Series 675. Geneva: World Health Organization, 1982.
- Levy L, Shepard CC, Fasal P. The bactericidal effect of rifampicin on M. leprae in man: (a) single doses of 600, 900 and 1200 mg; and (b) daily doses of 300 mg. Int J Lepr Other Mycobact Dis 1976;44:183–7.
- Jacobson RR, Hastings RC. Rifampin-resistant leprosy. Lancet 1976;2:1304–5.
- Frey HM, Gershon AA, Borkowsky W, Bullock WE. Fatal reaction to dapsone during treatment of leprosy. Ann Intern Med 1981;94:777–9.
- World Health Organization. Chemotherapy of Leprosy. Technical Report Series 847. Geneva: World Health Organization, 1994.
- Lockwood DN. Leprosy. Clin Evid 2002;8:709–20.
- Gebre S, Saunderson P, Byass P. Relapses after fixed duration multiple drug therapy: the AMFES cohort. Lepr Rev 2000;71:325–31.
- Boerrigter G, Ponnighaus JM, Fine PE, Wilson RJ. Four-year follow-up results of a WHO-recommended multiple-drug regimen in paucibacillary leprosy patients in Malawi. Int J Lepr Other Mycobact Dis 1991;59:255–61.
- Dasananjali K, Schreuder PA, Pirayavaraporn C. A study on the effectiveness and safety of the WHO/MDT regimen in the northeast of Thailand: a prospective study, 1984–96. Int J Lepr Other Mycobact Dis 1997;65:28–36.
- Jamet P, Ji B. Relapse after long-term follow up of multibacillary patients treated by WHO multidrug regimen. Marchoux Chemotherapy Study Group. Int J Lepr Other Mycobact Dis 1995;63:195–201.
- Girdhar BK, Girdhar A, Kumar A. Relapses in multibacillary leprosy patients: effect of length of therapy. Lepr Rev 2000;71:144–53.
- Ji B. Rifampicin resistant leprosy: a review and a research proposal of a pilot study. Lepr Rev 2002;73:2–8.
- World Health Organization. Report of the Third Meeting of the WHO Technical Advisory Group on the Elimination of Leprosy. Geneva: WHO/CDS/CPE/CEE, 2002.
- Lockwood DN. Leprosy elimination – a virtual phenomenon or a reality? BMJ 2002;324:1516–18.
- N'Deli L, Guelpa-Lauras CC, Perani EG, Grosset JH. Effectiveness of pefloxacin in the treatment of lepromatous leprosy. Int J Lepr Other Mycobact Dis 1990;58:12–18.
- Gelber RH, Fukuda K, Byrd S, et al. A clinical trial of minocycline in lepromatous leprosy. BMJ 1992;304:91–2.
- Ji B, Jamet P, Perani EG, Bobin P, Grosset JH. Powerful bactericidal activities of clarithromycin and minocycline against Mycobacterium leprae in lepromatous leprosy. J Infect Dis 1993;168:188–90.
- Xiong JH, Ji B, Perani EG, Petinon C, Grosset JH. Further study of the effectiveness of single doses of clarithromycin and minocycline against Mycobacterium leprae in mice. Int J Lepr Other Mycobact Dis 1994;62:37–42.
- Fleming CJ, Hunt MJ, Salisbury EL, et al. Minocycline-induced hyperpigmentation in leprosy. Br J Dermatol 1996;134:784–7.
- Lockwood DN. Cunha G Developing new MDT regimens for MB patients; time to test ROM 12 month regimens globally. Lepr Rev 2012;83(3):241–4.
- Rose P, Waters MF. Reversal reactions in leprosy and their management. Lepr Rev 1991;62:113–21.
- Van Brakel WH. Peripheral neuropathy in leprosy and its consequences. Lepr Rev 2000;71(Suppl.):S146–53.
- Croft RP, Richardus JH, Nicholls PG, Smith WC. Nerve function impairment in leprosy. Design, methodology, and intake status of a prospective cohort study of 2664 new leprosy cases in Bangladesh (the Bangladesh Acute Nerve Damage Study). Lepr Rev 1999;70:140–59.
- Saunderson P, Gebre S, Desta K, et al. The pattern of leprosy-related neuropathy in the AMFES patients in Ethiopia: definitions, incidence, risk factors and outcome. Lepr Rev 2000;71:285–308.
- Lienhardt C, Fine PE. Type 1 reaction, neuritis and disability in leprosy. What is the current epidemiological situation? Lepr Rev 1994;65:9–33.
- Kiran KU, Stanley JN, Pearson JM. The outpatient treatment of nerve damage in patients with borderline leprosy using a semi-standardized steroid regimen. Lepr Rev 1985;56:127–34.
- Little D, Khanolkar-Young S, Coulthart A, et al. Immunohistochemical analysis of cellular infiltrate and γ interferon, interleukin-12, and inducible nitric oxide synthase expression in leprosy type 1 (reversal) reactions before and during prednisolone treatment. Infect Immun 2001;69:3413–17.
- Walker SL, Nicholls PG, Dhakal S. A phase two randomised controlled double blind trial of high dose intravenous methylprednisolone and oral prednisolone versus intravenous Normal saline and oral prednisolone in individuals with leprosy Type 1 reactions and /or nerve function impairment. PLOS Negl Trop Dis 2011;Apr 12;5(4):e1041.
- Marlowe SNS, Leekassa R, Bizuneh E, et al. Clinical response to cyclosporin A treatment in Ethiopian and Nepali patients with severe leprosy type 1 reactions (T1R). Trans R Soc Trop Med Hyg 2007;101:1004–12.
- Walker SL, Waters M, Lockwood DNJ. The role of thalidomide in the management of erythema nodosum leprosum. Lep Rev 2007;78:197–215.
- Watson JM. Preventing Disability in Leprosy Patients. London: the Leprosy Mission International, 1986.
- Srinivasan H. Prevention of Disabilities in Patients with Leprosy: a Practical Guide. Geneva: World Health Organization, 1986.
- Setia MS, Steinmaus C, Ho CS, Rutherford GW. The role of BCG in prevention of leprosy: a meta-analysis. Lancet Infect Dis 2006;6:162–70.
- Ponnighaus JM, Fine PE, Sterne JA, et al. Efficacy of BCG vaccine against leprosy and tuberculosis in northern Malawi. Lancet 1992;339:636–9.
- Convit J, Smith PG, Zuniga M, et al. BCG vaccination protects against leprosy in Venezuela: a case-control study. Int J Lepr Other Mycobact Dis 1993;61:185–91.
- Karonga Prevention Trial Group. Randomised controlled trial of single BCG, repeated BCG, or combined BCG and killed Mycobacterium leprae vaccine for prevention of leprosy and tberculosis in Malawi. Lancet 1996;348:17–24.
- Convit J, Sampson C, Zuniga M, et al. Immunoprophylactic trial with combined Mycobacterium leprae/BCG vaccine against leprosy: preliminary results. Lancet 1992;339:446–50.
- Anonymous. Bettering BCG. Lancet 1992;339:462–3.
- Lockwood DN, Shetty V, Penna GO. Hazards of targets to eliminate disease: lessons from the leprosy campaign. BMJ 2014;15–21.