CHAPTER 37
Lichen Planus and Lichenoid Disorders
Vincent Piguet1, Stephen M. Breathnach2 and Laurence Le Cleach3
1Department of DermatologyCardiff University; University Hospital of Wales, UK
2St John's Institute of DermatologyGuy's and St Thomas' NHS Foundation Trust, UK
3Service de DermatologieSatellite Français du Cochrane Skin Group, Hôpital Henri Mondor, France
Definition
Lichenoid disorders are inflammatory dermatoses characterized clinically by flat-topped, papular lesions and histologically by a lymphocytic infiltrate with a band-like distribution in the papillary dermis [1]. Lichen planus is the prototype of lichenoid eruptions, but several skin diseases have a lichenoid tissue reaction and are included here [2, 3]. Graft-versus-host disease, which often presents as a lichenoid eruption, is addressed separately in Chapter 38.
LICHEN PLANUS
Introduction and general description
Lichen planus (LP), the most typical and best characterized lichenoid dematosis, is an idiopathic inflammatory skin disease affecting the skin and mucosal membranes, often with a chronic course with relapses and periods of remission. Its prevalence is approximately 0.5% of the population (reviewed in [1]).
Epidemiology
The incidence varies between 0.22% and 1% of the adult population worldwide. LP represented about 1.2% of all new dermatology patients in London and Turin [2], 0.9% in Copenhagen [3] and 0.38% in India [4]. Hypertrophic cases were reportedly to be common in Nigeria [5]. In contrast, oral LP seems to be more frequent with a reported incidence between 1% and 4% of the population [1]. LP is rare in children and commonly affects adults during their fourth to sixth decade. There was an overrepresentation of South Asians in a series of childhood cases in Birmingham, UK [6]. There is no obvious racial predisposition for LP, although a small overrepresentation with patients of Indian subcontinent origin has been suggested at least in oral disease [7]. Familial cases are unusual but have been reported in association with HLA-A3, -A5, -B7, -DR1 and -DRB*0101.
Pathophysiology
Lichen planus is thought to be a T-cell-mediated autoimmune disease, possibly targeting the basal keratinocytes, which can be triggered by a variety of situations, including viruses, drugs and contact allergens.
Immunopathology
The lymphocytic infiltrate of LP is composed of CD8+ T cells with a significant proportion of γ-δ T cells, which is unusual in the skin [1]. Polymerase chain reaction (PCR) studies analysing the T-cell receptor genes of the lymphocytes of the inflammatory infiltrate of LP are generally oligoclonal and potentially suggest the presence of autoantigens [2]. Electron microscopy studies and immunohistochemistry of LP demonstrate an increased number of epidermal Langerhans cells [3]. Although autoimmunity is clearly suggested to be central in LP, the exact pathomechanisms are unclear. Gene expression studies of LP have demonstrated an up-regulation of type I interferon-inducible genes and the presence of a specific chemokine signature; CXCL9, the ligand of the receptor CXCR3, was significantly increased in this study [4]. Global genetic studies of microRNAs (miRNA) in LP have revealed dysregulation in a number of miRNAs [5, 6]. Involvement of the Th17 pathway for LP has also been suggested [7]. That LP is thought to be an immunologically mediated disorder [8] is in part due to the fact that a lichenoid eruption may be seen as part of graft-versus-host disease [9–11]. Experimentally, cloned murine autoreactive T cells produce a lichenoid reaction in recipient animals following injection [12]. In LP lesions, T cells, both CD4+ and CD8+, accumulate in the dermis, whereas CD8+ T cells infiltrate the epidermis. It has been proposed that CD8+ cytotoxic T cells recognize an antigen (currently unknown) associated with major histocompatibility complex (MHC) class I on lesional keratinocytes, and lyse them [13–17]. T-cell receptor repertoires are skewed in tissue samples and in peripheral blood mononuclear cells of patients with oral LP [2].
The role of chemokines in the pathophysiology of lichenoid tissue reactions has been suggested with regard to recruitment and local activation of cytotoxic TH1 cells and plasmacytoid dendritic cells. Infiltrating CD8+ T cells, as well as keratinocytes, express a variety of different chemokines [18–20]. RANTES (regulated upon activation, normal T-cell expressed and secreted) secreted by T cells may trigger mast cell degranulation with a consequent release of tumour necrosis factor α (TNF-α), which in turn up-regulates lesional T-cell RANTES secretion. Such mechanisms may contribute to the chronicity of T-cell infiltration and clinical disease [21].
Finally, keratinocytes probably contribute to the disease and are also type I interferon producers in LP skin lesions [4]. Keratinocyte-derived cytokines may up-regulate the expression of cell surface adhesion molecules on, and migration by, T cells [22–24]. CD1a+ Langerhans cells and factor XIIIa+ cells are increased in LP [3, 15, 25] and may be involved in antigen presentation to T cells. Activin A – a transforming growth factor β (TGF-β) family member induced by pro-inflammatory cytokines and which promotes Langerhans cell differentiation and activation – is increased in the upper epidermis and dermis in LP [26]. Vascular endothelial expression of the adhesion molecules intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1) is also elevated [15]. Mast cell degranulation [27], and T-cell secretion of matrix metalloproteinase 9 (MMP-9) [28], may contribute to basement membrane disruption, enabling migration of CD8+ T cells into the epithelium. Granzyme B+ T cells have been observed close to apoptotic keratinocytes [29]. Overexpression of bone morphogenic protein 4 (BMP-4), another member of the TGF-β family, may contribute to apoptosis of epithelial cells in oral LP through up-regulation of p53, MMP-1 and MMP-3 [30]. Overexpression of MMP-2, membrane type 1 MMP and, especially, MMP-9 may be involved in malignant transformation in oral LP [31]. The role of c-Myc gains and overexpression in malignant transformation has also been established [32]. Myc is a transcription factor that is often dysregulated in cancers. The immune dysregulation and cytokine expression in LP has been reviewed [33].
Causative organisms
Several studies have suggested a role for hepatitis C virus (HCV) in LP, particularly in Japanese and Mediterranean populations [34–39]. This association has also been reported in other countries including the USA, Germany, Italy, Spain and Iran but this is not a consistent observation [40–43]. No association between LP and HCV was detected in France and England or India [44]. The role played by HCV in triggering LP remains unclear. Interestingly, tetramer analysis showed that HCV-specific CD8+ T cells with functional characteristics of terminally differentiated effector cells were identified in close vicinity to HCV (detected by PCR) in lesions of oral LP [45].
Other viruses have been implicated in the pathogenesis of LP, including hepatitis B virus (HBV) [46, 47], human herpesvirus 6 (HHV-6) and HHV-7 [48] and varicella zoster virus [49, 50]. Finally, a number of reports have described the presence of LP after various vaccines. Typically, hepatitis B vaccines have been shown in some patients to trigger LP, often after the second injection of vaccine [51, 52].
Genetics
There may be a genetic susceptibility to idiopathic LP. Familial cases are reported [53–58], and a familial incidence of 10.7% was quoted in one series [56]. LP has also been reported in monozygotic twins [59, 60]. An association with HLA-A3 and HLA-5 has been documented [61], although others found no such association [62, 63], as have associations with HLA-B7 [53], HLA-A28 in Jewish people with LP and carbohydrate intolerance [64], HLA-DR1 [65], HLA-DR10 [66] and HLA-DRB1*0101 [67]. Genetic heterogeneity in LP has led to the suggestion that idiopathic cutaneous, and purely mucosal, LP may each have a different pathogenesis [68]. An association between LP and polymorphisms in CIITA, the MHC class II transactivator, has been reported in a Chinese cohort of LP patients [69]. Another association has been reported between LP and TNF-α gene polymorphisms [70].
Environmental factors
Environmental factors including drugs, contact allergens and others have been potentially associated with LP.
- Drugs. A large variety of drugs has been linked with cutaneous eruptions similar or identical to LP. Often the terms LP-like eruption or lichenoid eruption are used in the context of an adverse drug reaction with features of LP. The list of drugs causing LP and LP-like eruptions has grown steadily and includes several classes, including antimicrobials, antihypertensives, antimalarials, antidepressants, anticonvulsants, diuretics, metals, non-steroidal anti-inflammatory drugs (NSAIDs) and more recently imatinib [71], intravenous immunoglobulin [72], etanercept [72] and adalimumab [73]. These reactions are described in more detail in Chapter 132.
- Dental amalgam. Another putative antigen in oral LP is mercury in dental amalgam [74], however more recent studies do not confirm a clear association between LP and dental amalgam [75].
- Betel nut. Social use of the betel nut is relatively common in India and South-East Asia. The product that is chewed, betel quid, is a mixture of substances, including the areca nut and betel leaf, and is associated with oral LP [76, 77].
- Lichen planus-like contact dermatitis. An LP-like eruption was reported in up to 25% of persons exposed to chemicals found in colour developer. Current automated equipment minimizes contact with these, and LP-like eruptions due to this source are now rare [78]. Two types of reaction were observed – acute (eczematous) and subacute (lichenoid). Patch-test reactions were usually positive to the substituted para-phenylenediamine and were eczematous in nature, but might become lichenoid [79]. LP-like lesions have developed on sites exposed to methacrylic acid esters used in the car industry [78], and more recently from dimethylfumarate, which can be found in sofas [81].
- Miscellaneous. LP has been induced by radiotherapy, and confined to a radiation field [80]. Anxiety, depression and stress are common in patients with LP [83] and may be risk factors for its development [84].
Clinical features
History and clinical presentation
The classic clinical presentation of LP includes primary lesions consisting of firm, shiny, polygonal, 1–3 mm diameter papules with a red to violet colour. More closely, a tracery of thin white lines can be seen on the surface of the lesions, known as Wickham's striae (Figure 37.1) [1]. Papules can be isolated or grouped, in a linear or annular distribution. Typically, a greyish brown pigmentation can be observed in lesions that have resolved due to deposition of melanin in the superficial dermis. Annular lesions are especially common on the penis (Figure 37.2) and rarely may be the predominant type of lesion present, later leading to atrophy [2]. A variant in which groups of ‘spiny’ lesions resembling keratosis pilaris develop around hair follicles (lichen planopilaris) is not uncommon (Figure 37.3). Lichen planopilaris more often forms only a minor feature of the disease, but occasionally this type of lesion may predominate. Co-localization of LP and vitiligo has been reported [3]. LP can affect any part of the body surface, but is most often seen on the volar aspect of the wrists (Figure 37.4), the lumbar region and around the ankles. The ankles and shins are the commonest sites for hypertrophic lesions. When the palms and soles are affected, the lesions tend to be firm and rough with a yellowish hue (Figure 37.5) [4]. A rare, erosive, flexural variant of LP has been described [5]. Linear LP occurring on Blashko's lines can also be observed [6]. Isolated lesions of LP have been reported to involve one or both eyelids [7, 8] or the lips [9–11]. Milia have been recorded in LP [12], and LP was reported as confined to the site of radiation therapy site in one patient [13].

Figure 37.1 Lichen planus. Close up to show Wickham's striae.
(Courtesy of the Welsh Institute of Dermatology, University Hospital of Wales, Cardiff, Wales, UK.)
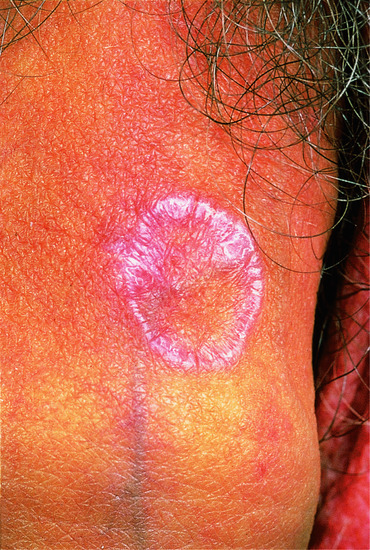
Figure 37.2 Lichen planus showing annular lesion on the shaft of the penis.
(Courtesy of St John's Institute of Dermatology, King's College London, UK.)

Figure 37.3 Lichen planopilaris showing hyperpigmented, follicular, ‘plugged’ lesions in the frontal scalp hairline.
(Courtesy of St John's Institute of Dermatology, King's College London, UK.)

Figure 37.4 Lichen planus. Classic eruption on the volar aspect of the wrist.
(Courtesy of the Welsh Institute of Dermatology, University Hospital of Wales, Cardiff, Wales, UK.)
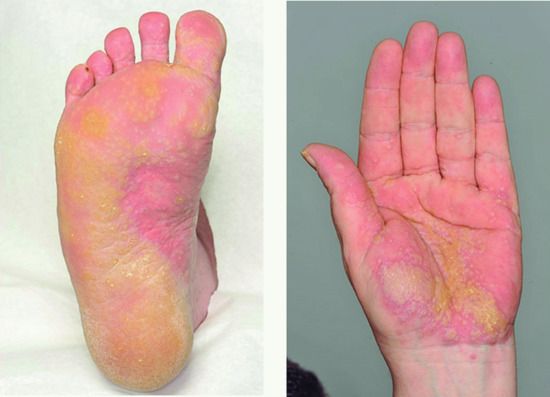
Figure 37.5 Lichen planus of the palms and feet showing hyperkeratosis and a yellow colour.
(Courtesy of the Welsh Institute of Dermatology, University Hospital of Wales, Cardiff, Wales, UK.)
Mucous membrane lesions are very common, occurring in 30–70% of cases, and may be present without evidence of skin lesions. They are, however, much less common in black people. The buccal mucosa and tongue are most often involved, but lesions may be found around the anus, on the genitalia, in the larynx and, very rarely, on the tympanic membranes or even in the oesophagus. White streaks, often forming a lacework, on the buccal mucosa are highly characteristic (Figure 37.6). They may be seen on the inner surface of the cheeks, on the gum margins or on the lips. On the tongue, the lesions are usually in the form of fixed, white plaques, often slightly depressed below the surrounding normal mucous membrane, especially on the upper surface and edges (Figure 37.7). Ulcerative lesions in the mouth are uncommon, but may be the site of epitheliomatous transformation (Figure 37.8). There may be striking pigmentation of oral LP in darkly pigmented races [14]. Diabetes is a possible associated disease of oral LP [15, 16], and candidiasis may coexist with LP in some patients.

Figure 37.6 Lichen planus on the buccal mucosa showing a lacework of white streaks.
(Courtesy of the Welsh Institute of Dermatology, University Hospital of Wales, Cardiff, Wales, UK.)

Figure 37.7 Lichen planus of the tongue showing irregular, fixed, white plaques.
(Courtesy of the Welsh Institute of Dermatology, University Hospital of Wales, Cardiff, Wales, UK.)

Figure 37.8 Erosive lichen planus of the buccal mucosa.
(Courtesy of the Welsh Institute of Dermatology, University Hospital of Wales, Cardiff, Wales, UK.)
Pruritus is a fairly consistent feature in LP and ranges from occasional mild irritation to more or less continuous, severe itching, which interferes with sleep and makes life almost intolerable; occasionally, pruritus is completely absent. Hypertrophic lesions usually itch severely. Paradoxically, there is seldom evidence of scratching, as the patient tends to rub to gain relief. Itching at sites without visible skin lesions can occur. Burning and stinging are less common complaints. In the mouth, the patient may complain of discomfort, stinging or pain; ulcerated lesions are especially painful. Great discomfort may be caused by hot foods and drinks.
Clinical variants
Lesions confined to the mouth, or with minimal accompanying skin involvement, are not uncommon [1–6], and account for about 15% of all cases. Mucosal lichen sclerosis/lichen planus overlap syndrome can also be observed [1]. The prevalence of oral LP was 1.5% among the villagers of Kerala in southern India [7], possibly related to chewing tobacco, and ranges between 0.5% and 2.2% in other epidemiological studies [2, 8]. The lesions do not differ from those found in connection with skin lesions, but, being confined to the mouth, may lead to great difficulty in diagnosis. They are often referred first to a dental surgeon. Distinct clinical subtypes such as reticular, atrophic, hypertrophic and erosive forms are well recognized, and more than one type may be present [2]. On the tongue and buccal mucosa, lesions are most likely to be mistaken for leukoplakia and on the gum margin for gingivitis or chronic candidiasis; the latter may coexist. Other conditions that must be excluded are ‘smoker's patches’, which characteristically involve the palate, and white-sponge naevi, in which the mucous membrane is thickened, irregularly folded and feels soft to the touch. These occur mainly on the floor of the mouth and histologically many of the prickle cells in the epidermis are vacuolated. It is important to bear in mind the possibility of a lichenoid drug reaction in patients with oral lichenoid changes (see Chapter 110). Oral lichenoid reactions may be asymmetrical on the buccal mucosa and occur adjacent to dental amalgam fillings. If patch testing reveals mercury allergy, changing to another type of filling may prove beneficial [9–14]. Very occasionally, LP lesions extend to the larynx or oesophagus [15–19]. Oesophageal LP may result in dysphagia and the formation of benign strictures.
In young men, the lesions of LP are sometimes restricted to the genitalia and/or mouth [20, 21]. Genital lesions, which are usually characteristic, may be present on the penile shaft (see Figure 37.2), glans penis, prepuce or scrotum. Ulceration is very unlikely, and syphilis can usually be excluded without difficulty. The presence of buccal mucosal lesions will usually confirm the diagnosis. Circumcision may be helpful in clearing up LP [20].
Lesions on the female genitalia are fairly common [22–29]; they may occur alone, be combined with lesions in the mouth only, or be part of widespread involvement. The clinical presentation of LP of the vulva spans a spectrum from subtle, fine, reticulate papules to severe erosive disease accompanied by dyspareunia, scarring and loss of the normal vulvar architecture. The condition should be distinguished from lichen sclerosus or leukoplakia, but this may be difficult when there is coexisting atrophy or vaginal stenosis. Diagnostic criteria have been proposed recently [30] and include well-demarcated erosions/erythematous areas at the vaginal introitus, the presence of a hyperkeratotic border to lesions and/or Wickham's striae in the surrounding skin, symptoms of pain/burning, scarring/loss of normal architecture, the presence of vaginal inflammation and the involvement of other mucosal surfaces. Histologically, a well-defined inflammatory band involving the dermal–epidermal junction and consisting predominantly of lymphocytes and signs of basal layer degeneration are seen. The association of erosive LP of the vulva and vagina with desquamative gingivitis has been termed the vulvo-vaginal–gingival syndrome (Figure 37.9) [21, 22]. Coexisting vulval lichen sclerosus and lichenoid oral lesions have been described [31].
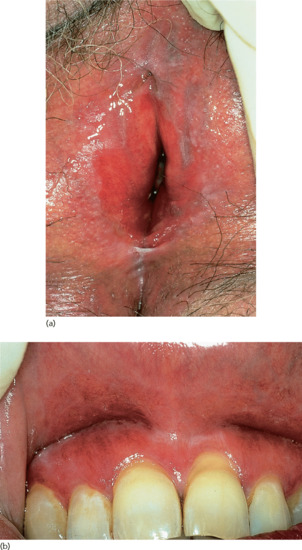
Figure 37.9 Vulvo-vaginal–gingival syndrome showing (a) vulvitis and (b) gingivitis in the same patient.
(Courtesy of Dr S. Neill, St John's Institute of Dermatology, King's College London, UK.)
Lichen planopilaris
Follicular lesions usually appear during the course of typical LP, but occasionally they predominate and diagnosis may then be difficult. An absence of arrector pili muscles and sebaceous glands, a perivascular and perifollicular lymphocytic infiltrate in the reticular dermis and mucinous perifollicular fibroplasia within the upper dermis with an absence of interfollicular mucin, and superficial perifollicular wedge-shaped scarring are all characteristic features of the histology [1]. Presentation with alopecia of the trunk is recorded [2]. Follicular lesions occurring in the scalp are accompanied by some scaling and are likely to lead to a scarring alopecia (Figure 37.10). Very rarely, the scalp alone is involved. The so-called Graham Little–Piccardi–Lassueur syndrome comprises the triad of multifocal scalp cicatricial alopecia, non-scarring alopecia of the axillae and/or groin, and keratotic lichenoid follicular papules [3–8]. The clinical, histological and immunofluorescence overlap between this syndrome and LP with follicular involvement (lichen planopilaris) suggest that both are variants of LP. Follicular LP must be distinguished by biopsy from keratosis pilaris, Darier disease, follicular mucinosis, lichen scrofulosorum and, in the scalp, from lupus erythematosus.

Figure 37.10 Lichen planus of the scalp leading to large areas of cicatricial alopecia.
(Courtesy of St John's Institute of Dermatology, King's College London, UK.)
Frontal fibrosing alopecia is a scalp condition that affects elderly women in particular and frequently involves the eyebrows. It has been regarded by some authors as a clinically distinct variant of lichen planopilaris [9] but is considered by most to be a variant [10], and is certainly associated with mucocutaneous LP [11, 12].
Hypertrophic lichen planus
This usually develops during the course of a subacute attack, but occasionally only hypertrophic or warty lesions are found. They most often occur on the lower limbs, especially around the ankles; venous stasis has been put forward as an explanation (Figure 37.11). The development of hypertrophic lesions greatly lengthens the course of the disease, as they may persist for many years. When such lesions eventually clear, an area of pigmentation and scarring may remain and there is often some degree of atrophy. They must be distinguished from lichen simplex chronicus and lichen amyloidosus (papular). Multiple cutaneous horns overlying hypertrophic LP are recorded [1], as are keratoacanthoma [2, 3] and malignant transformation [4–6] arising in hypertrophic LP, as well as metastatic squamous cell cancer [7].

Figure 37.11 Hypertrophic lichen planus of great chronicity occurring on the lower leg and ankle.
(Courtesy of St John's Institute of Dermatology, King's College London, UK.)
Lichen planus of the palms and soles
Although lesions on the volar aspect of the wrists or at the ankles occur in more than 50% of cases of LP, lesions on the adjacent palms and soles are less common, lack the characteristic shape and colour of lesions elsewhere, and are firm to the touch and yellow in hue (see Figure 37.5) [1]. They may be broadly sheeted or show up as punctate keratoses [2]. Vesicle-like papules are recorded [3]. Itching may be absent. When such changes occur in isolation, diagnosis is very difficult; syphilis, psoriasis, callosities and warts must be excluded.
A rare, very chronic form of LP involves large, painful, indolent ulcers on the sole of one or both feet [4], with gradual, permanent loss of the toenails; there may be secondary webbing of the toes [5]. The sole may resemble lichenified dermatitis or psoriasis rather than LP before the ulcers appear. The onset is insidious and there may be no other evidence of LP, although cicatricial alopecia has been associated in some cases.
Actinic lichen planus
‘Actinic’ or (sub)tropical LP generally occurs in children or young adults with dark skin living in tropical countries; virtually all cases originate from the Middle East, East Africa or India [6–10]. Lesions occur on exposed skin (usually the face) as well-defined annular or discoid patches, which have a deeply hyperpigmented centre surrounded by a striking hypopigmented zone (Figure 37.12). Erythematous actinic LP has been associated with oral erosive LP and chronic active hepatitis [11]. Sunlight exposure appears to be central to the pathogenesis of actinic LP [12], although evidence for photo-induction of lesions in actinic LP is still lacking [13]. Actinic LP may mimic melasma [14]. There is a histological spectrum comprising: a form with features of classic idiopathic LP; an intermediate form (lichenoid melanodermatitis) with foci of spongiosis and parakeratosis; and a more overtly eczematous type [15]. All share striking pigmentary incontinence. Some of these ‘hybrids’ of actinic LP may not be mere variants of LP.

Figure 37.12 Lichen planus actinicus showing well-defined, pigmented, nummular patches on the face.
(Courtesy of St John's Institute of Dermatology, King's College London, UK.)
Lichen planus pigmentosus
This is a pigmentary disorder seen in India or in the Middle East, which may or may not be associated with typical LP papules [16–18]. The macular hyperpigmentation involves chiefly the face, neck and upper limbs, although it can be more widespread, and varies from slate grey to brownish black. It is mostly diffuse, but reticular, blotchy and perifollicular forms are seen [3, 18]. The duration at presentation ranged from 2 months to 21 years in one series [18]. Occasionally, there is a striking predominance of lesions at intertriginous sites, especially the axillae [19, 20]. The mucous membranes, palms and soles are usually not involved, but involvement of mucous membranes has been observed [21]. LP pigmentosus has been anecdotally reported in association with acrokeratosis of Bazex [22].
Annular lichen planus
Although small annular lesions are common in LP, cases showing a few large annular lesions only are unusual. They may be widely scattered, and usually have a very narrow rim of activity and a depressed, slightly atrophic centre (Figure 37.13) [23]. Much less often, the margin is wide and the central area is quite small. Annular lesions are characteristically found on the penis (see Figure 37.2), sometimes associated with lesions on the buccal mucosa. The differential diagnosis includes granuloma annulare. A distinct entity termed annular lichenoid dermatitis of youth [2, 3, 24, 25] has been described, characterized by persistent, asymptomatic erythematous macules and round, annular patches with a red-brownish border and central hypopigmentation, mostly distributed on the groin and flanks, in children and adolescents. Histology reveals a lichenoid dermatitis with necrosis/apoptosis of the keratinocytes limited to the tips of rete ridges.

Figure 37.13 Annular lichen planus.
(Courtesy of the Welsh Institute of Dermatology, University Hospital of Wales, Cardiff, Wales, UK.)
Guttate lichen planus
Lesions are widely scattered and remain discrete, may be all small (1–2 mm across), or larger (up to 1 cm), and individual lesions seldom become chronic (Figure 37.14). Guttate psoriasis must be excluded as a differentiatial.

Figure 37.14 Guttate lichen planus.
(Courtesy of St John's Institute of Dermatology, King's College London, UK.)
Acute and subacute lichen planus with a confluence of lesions
In these forms, small lesions are widely distributed; where they become confluent, eczema may be simulated. Colour changes may be marked, with individual lesions initially red but progressing rapidly to black as they fade. Successive crops may occur, such that the total time for clearance may be little different from other forms. A small minority clear in under 3 months. Differential diagnosis includes pityriasis rosea in the earliest phase, and eczema later; drug-induced lichenoid eruptions may present in this fashion.
‘Mixed’ lichen planus/discoid lupus erythematosus disease patterns
Discoid lupus erythematosus (DLE) and LP are usually considered as distinct entities with characteristic clinical, histopathological and immunopathological features, with basement membrane deposition of IgG in DLE [1, 2]. However, similarities between LP and DLE have been noted. In addition, there have been several reports of patients showing overlapping features of both disorders [3–8]. Chronic atrophic DLE-like lesions on the head, neck and upper trunk may accompany reticular white lesions in the oral cavity, and combinations of lichenoid or verrucous lesions are seen. Eyelid involvement is recorded [9]. The association of extensive generalized LP with subacute cutaneous DLE has also been documented [10].
Bullous lichen planus and lichen planus pemphigoides
Lichen ruber pemphigoides was first described by Kaposi in 1892. Bullous LP and LP pemphigoides were in the past differentiated solely on clinical and histological criteria [1], but can now be differentiated using immunofluorescence (IMF) procedures and immunoelectron microscopy. In bullous LP, blisters arise only on or near the lesions of LP, as a result of severe liquefaction degeneration of the basal cell layer [3]. Histologically, there is subepidermal bulla formation with typical changes of LP, and direct and indirect IMF are negative [2, 3]. The eruption is usually only of short duration [2]. In LP pemphigoides the LP tends to be acute and generalized and is followed by the sudden appearance of large bullae on both involved and uninvolved skin (Figure 37.15) [4–7]. Occasionally, even in LP pemphigoides, blisters may arise only on the lesions of LP [8]. LP pemphigoides has been precipitated by psoralen and UVA (PUVA) [9] and has evolved into pemphigoid nodularis [10]. An LP pemphigoides-like eruption has been reported to overlap with paraneoplastic pemphigus [11, 12]. In LP pemphigoides, the histology shows a subepidermal bulla with no evidence of associated LP [2]. Direct IMF shows linear basement membrane zone deposition of IgG and C3 in perilesional skin [2, 7]. Immunoelectron microscopic studies reveal deposition of IgG and C3 in the base of the bulla and not in the roof as found in bullous pemphigoid [13].

Figure 37.15 Lichen planus pemphigoides showing large bulla arising on and around the vicinity of lichen planus around the ankle.
(Courtesy of St John's Institute of Dermatology, King's College London, UK.)
Immunoblotting data have revealed that circulating autoantibodies in LP pemphigoides react with an epitope within the C-terminal NC16A domain of bullous pemphigoid 180 kDa antigen, and also with a 200 kDa antigen detected in bullous pemphigoid [14–18]. It seems that epidermal damage from liquefaction degeneration in LP exposes basement membrane antigens, and a consequent stimulation of autoantibody production. The mean age of patients with LP pemphigoides is lower than that of those with classic bullous pemphigoid, and the course of the disease also tends to be less severe.
Lichen nitidus
Lichen nitidus is a rarer condition than idiopathic LP and is characterized clinically by the presence of pinpoint to pinhead-sized papules, which are usually asymptomatic and flesh-coloured, with a flat, shiny surface. The view that lichen nitidus represents a variant of LP tends to be supported by the fact that early, tiny LP papules may be clinically and histopathologically indistinguishable from lichen nitidus. Immunophenotypic studies also reinforce the association between LP and lichen nitidus [1]. However, some authors favour a separation into two dermatoses because of histopathological differences, or differences in cytokine expression in lichen nitidus [2].
Typical lichen nitidus papules are minute, pinpoint to pinhead sized, and have a flat or dome-shaped, shiny surface. They usually remain discrete, although they may be closely grouped (Figure 37.16). They are found on any part of the body but the sites of predilection are the forearms, penis (Figure 37.17), abdomen, chest and buttocks. The eruption is sometimes generalized [3, 4]. When the palms or soles are involved the changes can be those of a confluent hyperkeratosis resembling chronic fissured eczema, or there may be multiple, distinctive, minute papules [5, 6]. On the palms, the minute papules can become purpuric and may occasionally resemble pompholyx. Such cases may lack lesions of lichen nitidus elsewhere, so a biopsy is essential to confirm the diagnosis [5, 7].

Figure 37.16 Lichen nitidus showing a close-up of aggregated, pinhead-sized papules.
(Courtesy of St John's Institute of Dermatology, King's College London, UK.)

Figure 37.17 Lichen nitidus showing aggregates of pinhead-sized papules on the shaft of the penis.
(Courtesy of St John's Institute of Dermatology, King's College London, UK.)
Linear lichen nitidus has been described, but is exceptionally rare [8]. The development of lesions along scratch marks is not uncommon. The lesions are flesh coloured or reddish brown. Although intense pruritus can occur [4], the lesions are generally quite symptomless. Nail pitting may coexist with lichen nitidus [9], or the affected nails may appear rough due to increased linear striations and longitudinal ridging [5, 10]. An actinic variant has been documented [11]. Mucous membrane lesions occur occasionally and are much rarer than in LP. Lichen nitidus must be distinguished from lichen scrofulosorum [12], where there are follicular papules grouped in small patches on the trunk, and from keratosis pilaris, where there are horny follicular papules mainly on the extensor surface of the limbs. In cases of doubt, a biopsy usually clarifies the diagnosis. Lichen nitidus has been described in association with Crohn disease, trisomy 21, congenital megacolon [13] and Niemann–Pick disease type B [14].
Surprisingly, direct IMF studies in lichen nitidus have given negative results [15, 16]. However, ultrastructural studies have shown identical changes in lichen nitidus and LP [15]. The histology of a typical papule is characteristic. The papule is formed by an intense infiltrate situated immediately below the epidermis and is well circumscribed. The infiltrate consists of lymphocytes and histiocytes and there are often a few Langhans giant cells (Figure 37.18). Sometimes, plasma cells are numerous in the infiltrate [17]. The overlying epidermis is flattened and sometimes there is liquefaction degeneration of the basal cell layer. The rete ridges at the margin of the infiltrate are elongated and tend to encircle it. Although tuberculoid in appearance, true tubercle formation or caseation never occurs. The histology of a palmar lesion may show a deep parakeratotic plug, which distinguishes it from the palmar lesions of LP [7]. Perifollicular granulomas can occur in spinous and follicular lichen nitidus, which may simulate lichen scrofulosorum [12]. Perforating lichen nitidus has also been described [18]. In its characteristic monomorphic form, lichen nitidus is rare, but lesions of lichen nitidus occurring in association with LP are more common. The age incidence tends to be lower than that of LP. Most cases occur in children or young adults. Familial lichen nitidus has rarely been observed [19].
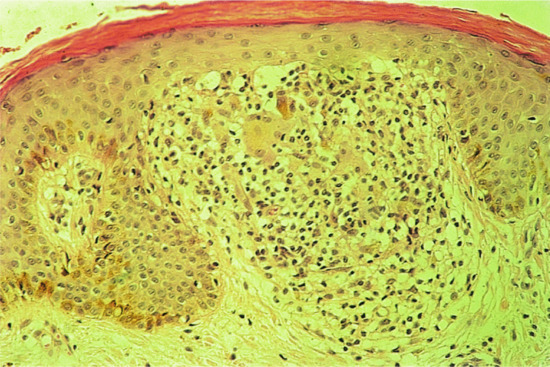
Figure 37.18 Lichen nitidus. Typical histology showing the focally dense infiltrate containing a few giant cells. Magnification 100× (H&E).
(Courtesy of St John's Institute of Dermatology, King's College London, UK.)
Nékam disease
The variety of synonyms used implies that there is no complete consensus of agreement about this rare disorder. The great majority of cases are adults between the ages of 20 and 40 years [1], although children are occasionally affected [2]. Nékam disease is characterized by violaceous, papular and nodular lesions typically arranged in a linear and reticulate pattern (Figure 37.19), most marked on the extremities and buttocks, and accompanied by a seborrhoeic dermatitis-like eruption on the face. Nékam's original case was also associated with palmoplantar keratoderma. The individual lesions are erythematous verrucous papules covered by a hyperkeratotic plug that can only be removed with difficulty, revealing irregular indentations and prominent capillary loops [1, 3]. In extensive Nékam disease, the lesions tend to be symmetrical bilaterally, mainly involving the antecubital fossae, extensor forearms, lumbosacral area and buttocks, posterior thighs, popliteal fossae and less commonly the oral cavity and genitalia. Oral manifestations occur in 50% of patients – recurrent aphthous ulcers, larger chronic ulcers or erythrokeratotic papules being the commonest oral features [4]. The nails can become thickened, longitudinally ridged and prone to paronychia [5]. Cases have followed trauma [6] and erythroderma [7]. A limited variant of Nékam disease presenting with erythematous hyperkeratotic papules and plaques on the face, clearing in the summer months, has been described in two young siblings [8]. Histologically, changes are often non-specific and consistent with a chronic dermatitis, but lichenoid features can be seen [1, 3]. Some authors believe that the condition is an unusual variant of LP [1], while others consider it to be a distinct entity [9]. A case of Nékam disease associated with porokeratotic histology and amyloid deposition may also point against the view that Nékam disease is a subset of LP [10]. A possible association of Nékam disease with glomerulonephritis and lymphoproliferative disorders has been commented on [4].

Figure 37.19 Nékam disease. Reticulate keratotic erythematous papules on (a) the volar aspect of the wrist and (b) the dorsum of the hand.
(Courtesy of St John's Institute of Dermatology, King's College London, UK.)
Complications
Hair
Alopecia is uncommon in LP, but most often occurs in small areas on the scalp, producing patches of atrophic cicatricial alopecia (see Figure 37.10) [1–5]. It results from follicular destruction by the inflammatory infiltrate, with scarring. Areas of alopecia may continue to appear, or to extend, for months after the skin lesions have faded. The final result is one of pseudopelade [2, 5]; this is probably best considered as a distinct clinical entity due to a number of independent conditions, of which LP is only one. Lichen planopilaris is more common in women and about half will develop involvement of the glabrous skin, mucous membranes or nails [1]; it may occur in children [3]. Tumid forms of LP follicularis have been described in which clusters of milium cysts and comedones develop [6]. LP pilaris has been reported in association with dermatitis herpetiformis [7].
Nails
Nail involvement occurs in up to 10% of cases, but is usually a minor feature of the disease [8, 9]. The majority of cases present during the fifth or sixth decades. Long-term permanent damage to the nails is rare [10]. Fingernails are more frequently affected than toenails [10], with initial involvement of two or three fingernails before subsequent involvement of the remaining digits. The most common changes are exaggeration of the longitudinal lines and linear depressions due to slight thinning of the nail plate (Figure 37.20). These changes usually occur in the context of severe generalized LP, although skin lesions may not be seen in the vicinity of the affected nail. Elevated ridges may be seen on the nail [9]. Adhesion between the epidermis of the dorsal nail fold and the nail bed may cause partial destruction of the nail (pterygium unguis) (Figure 37.21a). Rarely, the nail is completely shed; there is usually clinical evidence of LP at the base of the nail before shedding. Nails may partially regrow or be permanently lost (Figure 37.21b); the nails of the great toes are the ones most often affected. LP has been shown to cause childhood idiopathic atrophy of the nails [11]. LP of the nails in childhood is rare [12] but may overlap with the condition of twenty-nail dystrophy of childhood (idiopathic trachyonychia); the exact relationship is unclear [13–18]. The rare variant of LP that causes ulceration of the soles is often accompanied by permanent destruction of several toenails. LP of the nail bed may give rise to longitudinal melanonychia [19], hyperpigmentation, subungual hyperkeratosis or onycholysis [9], or changes mimicking the yellow nail syndrome [20, 21].
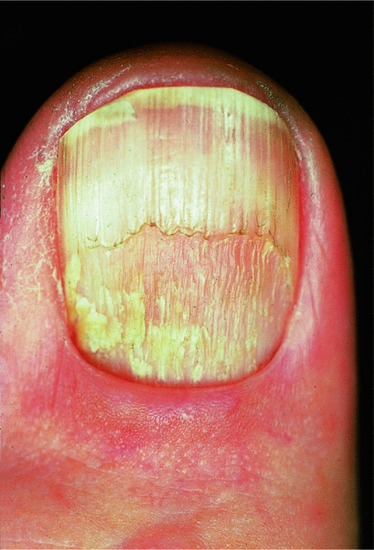
Figure 37.20 Lichen planus of the thumbnail showing thinning of the nail plate and longitudinal lines.
(Courtesy of St John's Institute of Dermatology, King's College London, UK.)
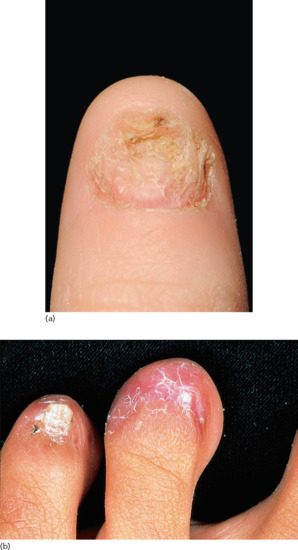
Figure 37.21 (a) Severe lichen planus of the fingernails showing involvement of the nail fold areas and early pterygium formation.
(Courtesy of the Welsh Institute of Dermatology, University Hospital of Wales, Cardiff, Wales, UK.) (b) Severe, destructive lichen planus of the toenails. (Courtesy of St John's Institute of Dermatology, King's College London, UK.)
Mucous membranes
Squamous cell carcinoma developing on oral lesions is uncommon but is a potential danger, especially with ulcerated lesions [22–30], although the incidence varies greatly in different series. Lesions may occur on the lip (Figure 37.22), the buccal mucosa or the gum margin. It has been postulated that the high expression of cyclo-oxygenase 2 reported in oral LP may be of aetiological significance in the development of squamous cell carcinoma [31]. Squamous cell carcinoma arising on cutaneous lesions of LP [32] and ano-genital lesions [33–35] is definitely a rare phenomenon. Cicatricial conjunctivitis [36] and lacrimal canalicular obstruction are recorded [37].

Figure 37.22 Squamous carcinoma on the lower lip developing at the site of lichen planus cheilitis.
(Courtesy of St John's Institute of Dermatology, King's College London, UK.)
Associated conditions
Idiopathic LP has been reported in association with diseases of altered or disturbed immunity, including ulcerative colitis [1–3], alopecia areata [3, 4], vitiligo [3], dermatomyositis [5], morphoea and lichen sclerosus [6], systemic lupus erythematosus [7, 8], pemphigus [7] and paraneoplastic pemphigus [9, 10]. In addition, LP has been observed in association with thymoma [4, 7, 11, 12], hypothyroidism [13], myasthenia gravis [2, 3, 12], hypogammaglobulinaemia [4, 5, 14], primary biliary cirrhosis [15–17], especially in those treated with penicillamine, and primary sclerosing cholangitis [18]. The literature with regard to LP and HCV infection is reviewed in the section on pathogenesis. In Italy, possibly because of a higher prevalence of HBV and HCV infection, patients with LP seem more prone to develop liver disease, including chronic active hepatitis [19, 20]. A high prevalence of anticardiolipin antibodies has been documented in patients with HCV-associated oral LP [21]. Elsewhere, the association between LP and chronic active hepatitis or primary biliary cirrhosis is unusual and probably coincidental [22–24]. Overall, the majority of patients with LP live to old age, despite an association with autoimmunity [25].
LP has also been associated with diabetes [26]. Anecdotally, LP has occurred in patients with Castleman tumours (giant lymph node hyperplasia) [27] or with generalized lichen amyloidosus [28]. LP has been described in certain tattoo reactions, particularly in those areas where there is coexisting mercury hypersensitivity to the injected dye [29, 30].
Disease course and prognosis
Although a few cases evolve rapidly and clear within a few weeks, the onset is usually insidious. In most cases, the papules eventually flatten after a few months, often to be replaced by an area of pigmentation that retains the shape of the papule and persists for months or years. There may be a gradual change in colour from pink to blue to black. The residual pigmentation may be intense, especially in dark-skinned races, or almost imperceptible in fair-skinned individuals. New papules may form while others are clearing. Some papules persist much longer, enlarge and thicken, and develop a roughened surface with a prominent violaceous hue – so-called hypertrophic LP. Such lesions may resolve with atrophy or scarring. More warty lesions are seen occasionally. Areas of pigment loss are described in black South Africans.
Investigations
Histology is the most useful investigation to confirm a diagnosis of LP. More recently, the use of dermoscopy has been assessed and found useful in some cases when Wickham's striae can be visualized better with the device (Figure 37.23) [1]. Typical features of dermoscopy images of LP include a network of whitish striae with red globules at the periphery [1, 2]. Histology will be routinely obtained to confirm the diagnosis of LP [3, 4]. The earliest finding is an increase in epidermal Langerhans cells [3], associated with a superficial perivascular infiltrate of lymphocytes and histiocytes, impinging on the dermal–epidermal junction (DEJ). Mild spongiosis is followed by vacuolar alteration and clefting along the DEJ, with accumulation of necrotic keratinocytes (colloid bodies) [5]. The characteristic histological changes are best seen in biopsies of fully developed LP papules (Figure 37.24) [6]. The centre of the papule shows irregular acanthosis of the epidermis, irregular thickening of the granular layer and compact hyperkeratosis. The mid-epidermal cells appear larger, flatter and paler than usual [6]. In oral LP, epithelial proliferation is increased [7]. Parakeratosis is rarely found in idiopathic LP, in contrast to some drug-induced lichenoid tissue reactions. A focal increase in thickness of the granular layer and infiltrate corresponds to the presence of Wickham's striae [8]. Degenerating basal epidermal cells are transformed into colloid bodies (15–20 μm diameter), which appear singly or in clumps [9–11]. The rete ridges may appear flattened or effaced (‘saw-tooth’ appearance), and focal separation from the dermis may lead to Max Joseph spaces (Figure 37.25). In older or hypertrophic lesions, the number of colloid bodies is considerably reduced. In ‘active’ LP, a band-like infiltrate of lymphocytes and histiocytes, rarely admixed with plasma cells [12, 13], obliterates the DEJ. Epidermal melanocytes are absent or considerably decreased in number [6], while pigmentary incontinence with dermal melanophages is characteristic. When the disease is becoming inactive, the infiltrate, with melanophages, becomes sparser and is arranged around papillary blood vessels, which may show ectasia and surrounding fibroplasia.

Figure 37.23 Dermoscopy image of lichen planus with typical Wickham's striae.
(Courtesy of Dr Alan Cameron, School of Medicine, University of Queensland, Brisbane, Australia.)
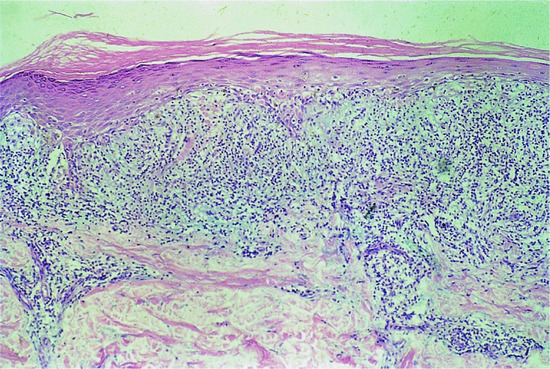
Figure 37.24 Typical histology of lichen planus. Magnification 40× (H&E).
(Courtesy of St John's Institute of Dermatology, King's College London, UK.)
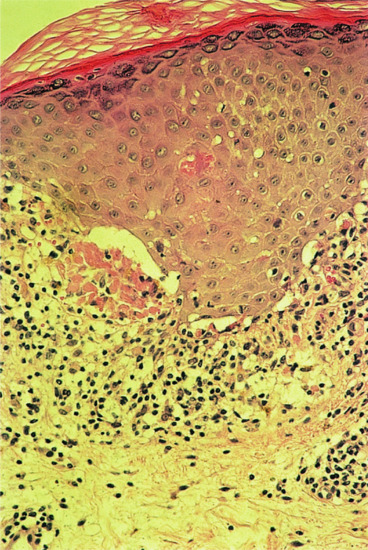
Figure 37.25 Lichen planus: photomicrograph to show clumps of cytoid bodies. Magnification 100× (H&E).
(Courtesy of St John's Institute of Dermatology, King's College London, UK.)
In hypertrophic LP, the epidermis may show a pseudoepitheliomatous appearance with extreme irregular acanthosis. Follicles may be expanded and at times have a cyst-like appearance. The infiltrate may not appear very band-like, but serial sections will usually show foci of basal cell liquefaction and colloid body formation, often around the follicular epithelium. Longstanding cases usually demonstrate coexistent dermal fibrosis adjacent to the inflammatory changes.
In atrophic LP, the epidermis may be greatly thinned almost to the level of the granular layer, although relatively compact hyperkeratosis remains. The rete ridges are usually completely effaced with relatively few colloid bodies. The papillary dermis shows fibrosis and loss of elastica.
In follicular lesions, an infiltrate extends around, and may permeate, the base of the hair follicle epithelium, with follicular keratin plugging (Figure 37.26) [14, 15].

Figure 37.26 Lichen planus: photomicrograph of a follicular lesion. (a) Scanning view. Magnification 20× (H&E). (b) At higher power. Magnification 40× (H&E).
(Courtesy of St John's Institute of Dermatology, King's College London, UK.)
In mucosal LP, the epithelium is usually thinned with parakeratosis, although both types of keratinization may be seen [16, 17]. The epithelium may resemble epidermis from the skin, plasma cells may be prominent in the band-like infiltrate, and colloid bodies are fewer than in typical cutaneous papular LP. The erythematous subtype of oral LP is associated with increased cell proliferation compared with the reticular form [18]. Moderate or severe epithelial dysplasia on oral biopsy should probably be regarded as an increased risk for subsequent cancer development [19].
Bullous LP is rare. Blister formation takes place predominantly between the basal lamina and the cytomembranes of basal keratinocytes (i.e. intrabasal) [9], such that there is a wide separation between the epidermis and infiltrate.
Direct immunofluorescence shows globular deposits of immunoglobulin M (IgM) (Figure 37.27) and occasionally IgG and IgA, representing apoptotic keratinocytes, around the DEJ and lower epidermis, with fibrin deposition at the region of the DEJ [16, 20]. Direct IMF studies may be useful in differentiating LP from lupus erythematosus or LP pemphigoides [21, 22], and can be carried out on routine histological material (23).
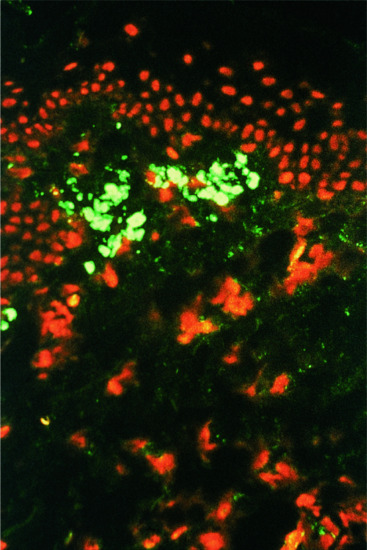
Figure 37.27 Lichen planus: photomicrograph of direct immunofluorescence showing the bright fluorescence of cytoid bodies with anti-IgM. Magnification 100×.
(Courtesy of St John's Institute of Dermatology, King's College London, UK.)
A clinicopathological study of lichenoid dermatitis concluded that it was usually possible to provide a specific diagnosis [24]. Occasionally, lichenoid dermatitis can become generalized and clinically mimic an exfoliative dermatitis; such eruptions are usually triggered by drugs.
Benign lichenoid keratoses are usually solitary but may be multiple [25–27], and show characteristic lichenoid infiltrates of lymphocytes, occasional parakeratosis, and apoptotic bodies in the epidermis without nuclear atypia of keratinocytes. Rarely, the histology may show features of mycosis fungoides [28]. Melanoma may be associated with a lichenoid tissue reaction [29], and melanoma in situ with lichenoid regression may mimic a benign lichenoid keratoses histologically [30].
Management
Treatment of LP can represent a challenge and depends on the localization, clinical form and severity [1]. For cutaneous LP, which can clear spontaneously within 1–2 years, the aim of treatment is to reduce pruritus and time to resolution [2]. Therapeutic abstention is recommended for asymptomatic oral LP. However, painful, erosive LP may need aggressive and long-term treatments [3]. Nail or scalp involvement [4, 5], which may induce scars, genital LP [6] and oesophageal and conjunctival involvement, which may induce strictures and fibrosis, may require rapid treatment to avoid scaring or avoid a fatal course.
The main therapy for LP is corticosteroids. Overall, the level of evidence for treatments used in LP is low [7]. Surprisingly, there is still little evidence for the efficacy of topical or systemic corticosteroids, commonly recommended as first line treatment for LP. Treatment is mainly based on clinical experience. In contrast, for oral LP, there are several randomized controlled trials (RCTs) and one Cochrane review [7–9]. However, the majority of these trials are small, have used various outcomes and are at high risk of bias. For cutaneous LP, there are only a few small RCTs. No RCT has assessed treatment for genital or nail LP, lichen planopilaris or other lichenoid disorders.
Cutaneous lichen planus
The goals of therapy are to improve itching and reduce time to resolution of the lesions.
First line
Topical corticosteroids are the first line treatment for limited cutaneous LP. The use of very potent corticosteroids is required (clobetasol propionate ointment 0.05%) once daily at night until remission. If no evolution is observed after 6 weeks, second line therapy has to be considered. After remission, the optimal length of maintenance therapy is unknown and will be adapted to each patient. For hypertrophic LP, very potent corticosteroids need to be applied under an occlusive bandage. Oral corticosteroids are the first line treatment for widespread cutaneous LP: prednisolone 0.5–1 mg/kg per day until improvement (usually 4–6 weeks). In a small RCT in which potent corticosteroid cream alone was compared with oral prednisolone (30 mg per day for 10 days) combined with topical corticosteroids, the time to clearing was significantly shorter in the oral prednisolone group (18 weeks versus 29 weeks) than in the topical steroid cream only group. Two patients in the corticosteroid group experienced a severe relapse after treatment withdrawal [10]. After remission, oral corticosteroids should be tapered down slowly.
Second line
An RCT showing a rate of regression or remission at 8 weeks significantly higher with acitretin (30 mg per day for 8 weeks) than with placebo has been published [11]. Considering the side effects of teratogenicity and mucocutaneous dryness, acitretin is used as second line therapy in cutaneous LP. Another second line option is photochemotherapy or phototherapy. In a small trial, PUVA used three times weekly on one side of the body was compared with no treatment on the other side of the body. Clearance was observed on the treated side only in 50% of the patients [12]. In a retrospective series, clearance within a mean of 3 months was observed in 70% of patients treated with narrow-band UVB therapy (13). PUVA phototherapy may also be combined with acitretin but evaluation of this association has not been assessed. Phototherapy can increase the risk of residual hyperpigmentation in dark-skinned patients. Oral corticosteroids in combination with phototherapy are also a possible second line therapy.
Third line
The best level of evidence for methotrexate in cutaneous LP is a prospective series. A complete remission was observed in 14/24 patients at 24 weeks [14]. However, by analogy with other cutaneous diseases, methotrexate can be used as third line treatment for generalized LP. There are four RCTs assessing griseofulvin, hydroxychloroquine or sulfasalazine [15–17]. Because of risk of bias in these trials, discrepancies of results between studies for griseofulvin and life-threatening adverse reaction for sulfasalazine, these treatments are not recommended in cutaneous LP.
Oral lichen planus
The aims of treatment are to heal erosive lesions in order to reduce pain and permit normal food intake. Education of the patient should emphasize that oral LP frequently has a chronic course marked by treatment-induced remission followed by relapse [4, 7]. Considering the potential higher risk of squamous cell carcinoma, the need for regular clinical surveillance on a long-term basis has also to be explained [18]. Alcohol and tobacco should be avoided as well as spicy or acidic foods and drinks. Good oral hygiene and professional dental care are recommended.
First line
Topical corticosteroids are the first line treatment for symptomatic oral LP. In two small RCTs comparing topical corticosteroids, higher cure rates were observed in the topical corticosteroids group (80% versus 30% with fluocinonide and 66% vsersus 18% with betamethasone) [19, 20]. In the absence of specific available formulation for oral lesions, super-potent corticosteroids (0.05% clobetasol propionate ointment) can be applied three times daily with a gloved finger on erosive lesions. It is recommended to not drink or eat for 1 h after the application. Alternatively, for widespread, less severe lesions or maintenance therapy, a patient can use a soluble prednisolone tablet 5 mg dissolved in 15 mL water for a mouthwash swish and rinse three times daily. Oral candidiasis is the most frequent complication of this treatment [21]. If no improvement is observed after 6 weeks, second line therapy has to be considered. After remission, the optimal length of maintenance therapy is unknown and should be adapted to each patient. In severe case of erosive LP associated with important eating difficulties and eventual weight loss, first line therapy is oral corticosteroids (1 mg/kg/day) until remission, generally 4–6 weeks. Slow reductions in dose are needed because of the risk of relapse. For popular plaque-like forms of oral LP, topical retinoids can be recommended as first line treatment alone or in association with topical corticosteroids. Three small RCTs compared topic retinoid (retinoic acid 0.05%, isotretinoin gel and tazarotene) with placebo. Improvement or cure rates were significantly higher in the treated group [22–24].
Second line
For erosive LP, if no or insufficient improvement is observed after topical corticosteroids, second line treatment is oral corticosteroids (1 mg/kg/day) until remission, generally 4–6 weeks [21].
Third line
No superiority over topical corticosteroids was demonstrated in the four RCTs comparing oral ciclosporin to topical corticosteroids [8]. No topical formulation is available. Two trials compared tacrolimus ointment 0.1% with clobetasol propionate 0.5%. One found a statistically significant difference favouring tacrolimus [25], while no difference between treatments was observed in the other [26]. An RCT comparing pimecrolimus cream 1% to triamcinolone acetonide paste 0.1% found no difference between the two treatments [27]. Topical calcineurin inhibitors (ciclosporin, pimecrolimus and tacrolimus) have thus not demonstrated their superiority over topical corticosteroids. Current US Food and Drug Administration (FDA) labelling states that topical pimecrolimus and tacrolimus should not be used to treat premalignant conditions. They have to be used with caution in oral LP due to the inherent risk of malignant transformation.
Several others treatments were assessed in RCTs (curcuminoids, hyaluronic acid, ignatia, purslane, oral lycopene, intralesional injection of the polysaccharide nucleic acid fraction of BCG, thalidomide, Aloe vera gel) [28–34]. A pooled estimate for the mean clinical score of two RCTs comparing topical Aloe vera with placeo was risk ratio 1.34 (95% confidence interval 1.10–1.62) [35, 36] and one RCT comparing Aloe vera gel with triamcinolone acetonide paste 0.1% found no difference between the groups [37]. These treatments are not recommended for the treatment of oral LP.
Ano-genital lichen planus
Early super-potent corticosteroid treatment (0.05% clobetasol propionate ointment once daily) aims to resolve symptoms and prevent synechiae and scarring of the genital area, although efficacy for the prevention of scars or relapses has not been demonstrated. Hydrocortisone suppositories, foam or cream every other day is used for vaginal localization. Potent corticosteroids can be used daily during the initial phase until symptom resolution [6]. The frequency of application during maintenance treatment and its duration should be adapted according to evolution. Foreskin retraction or removal surgery in uncircumcised men and vaginal dilators in women are used to prevent synechiae formation. Surgery is required after complete resolution of the active lesions if adhesion occurs.
Lichen planopilaris
In order to lessen pain and pruritus, and to avoid irreversible alopecia, first line treatment is potent corticosteroids (e.g. 0.05% clobetasol propionate ointment twice daily). In severe cases or in cases of insufficient improvement after corticosteroid ointment, second line treatment is monthly intralesional injection of triamcinolone acetonide (0.5–10 mg/mL) or systemic oral corticosteroids (prednisolone 1 mg/kg/day) [6, 37, 38].
Frontal fibrosing alopecia
Treatments can be of limited efficacy but several options have been proposed including topical and intralesional corticosteroids, antibiotics, hydroxychloroquine, topical and oral immunomodulators, tacrolimus and 5α-reductase inhibitors [38, 39].
Nail lichen planus
Evidence for nail LP is based only on retrospective case series and depends on the number of affected nails. The aims of treatment are to reduce pain and avoid irreversible nail scars. Two-thirds of 142 patients treated with intramuscular injection or oral systemic glucocorticoids and/or intralesional injection or topical glucocorticoids during 6 months were cured or had a major improvement [4, 40].
When less than four nails are involved, the recommended treatment is a twice daily application of super-potent corticosteroids (e.g. 0.05% clobetasol propionate ointment), or if lesions are more severe, monthly intralesional injection of triamcinolone acetonide (0.5–10 mg/mL) in the periungual sites.
When more than two or three nails are involved, systemic corticosteroids are required. Treatment with oral prednisolone (0.5–1 mg/kg/day) for 4–66 weeks or intramuscular triamcinolone acetonide injection have been reported. Etanercept has been suggested in a case report [41], although LP-like eruptions can be triggered by tumour necrosis factor antagonists [42].
Severe erosive lichen planus
Systemic corticosteroids are first line treatment [7]. Systemic immunosuppressive agents (e.g. azathioprine, mycophenolate mofetil and methotrexate) are used as corticosteroid-sparing agents for patients with erosive or potentially scarring LP requiring prolonged systemic corticosteroids or in disease unresponsive to corticosteroids. Complete remission was obtained following sessions of extracorporeal photochemotherapy in 16 of 19 patients in two prospective series of patients with resistant severe erosive LP [43, 44].
Lichen nitidus
As the disease is often asymptomatic and eventually self-limiting, no treatment is required in most cases. However, fluorinated topical corticosteroid preparations may be recommended if treatment is demanded, for example for lesions on the penis, and can be highly successful [45]. Clearance of generalized lichen nitidus has been described with sun exposure [46], PUVA [47], narrow-band UVB phototherapy [48, 49] and astemizole [50, 51]. Acitretin can lead to a gradual improvement in palmoplantar lichen nitidus [52].
Nékam disease/keratosis lichenoides chronica
The course of the dermatosis is chronic and progressive and very resistant to therapeutic approaches, but has shown a favourable response to photochemotherapy without [53, 54] or with [55] acitretin. Etretinate [56] and photodynamic therapy [57] have been reported to be helpful.
‘Mixed’ lichen planus/discoid lupus erythematosus disease patterns
Both ciclosporin [58] and acitretin [59] can be of benefit in treating LP/DLE overlap syndrome.
Actinic lichen planus
Actinic LP has been treated with acitretin and topical corticosteroids [60] and with ciclosporin [61].
Bullous lichen planus and lichen planus pemphigoides
Some cases require treatment with systemic steroids or azathioprine and fatalities can occur [60]. A combination of corticosteroids and acitretin has been used [61, 62].
LICHEN STRIATUS
Definition and nomenclature
Lichen striatus is a distinctive, usually self-limiting and asymptomatic inflammatory dermatosis characterized by pink or red papules in a linear distribution that develop in the lines of Blaschko. These usually occur as isolated lesions on the limbs in children aged 5–15 years and generally resolve over 6–12 months. Areas of post-inflammatory hypopigmentation may occur and persist for longer. The aetiology is incompletely understood but approximately 40% of cases have a background of atopy. Clustering of cases in families and in winter suggests an infectious agent; potentially, a virus may be involved.
Epidemiology
Incidence and prevalence
The incidence or prevalence is unknown, but, between January 1989 and January 2000, 115 cases were identified at the Paediatric Dermatology Unit, University of Bologna, Bologna, Italy [1].
Age
Over 50% of cases occur in children, usually between the ages of 5 and 15 years, but onset in early infancy and in adults has been reported.
Sex
Females are affected approximately two to three times as frequently as males [1].
Associated diseases
Lichen striatus occurs more often in individuals with atopy; atopy was reported in approximately 43% of cases in a recent series [2].
Pathophysiology
As the condition develops in the lines of Blaschko, which represent embryonic migratory patterns of ectoderm (see Chapter 7), it has been hypothesized that post-zygotic mutation or loss of heterozygosity may lead to cutaneous mosaicism and play a role in its pathophysiology, perhaps predisposing to the effects of an environmental trigger or an infectious agent. The presence of atopy in approximately 40% of cases is therefore intriguing. On the other hand, the resolution of lichen stratus over a period of months in the majority of cases suggests a temporary phenomenon, leading to the proposal of cutaneous antigenic mosaicism and a localized inflammatory T-cell response, again potentially related to viral infection or injury [3].
Pathology
The histological appearances are variable and depend on the stage of disease [3]. Usually, a band-like infiltrate composed of lymphocytes and histiocytes is observed with associated overlying epidermal change including parakeratosis and hyperkeratosis, resembling lichen planus. The earliest change is intercellular oedema, stretching the tonofilament–desmosome complexes and separating the epidermal cells. Like the spongiosis, acanthosis is variable in degree. Dyskeratotic keratinocytes, like the ‘corps ronds’ of Darier disease, are seen in about 50% of cases. There is often focal liquefactive degeneration of the basal layer. The dermis is oedematous, and the vessels and appendages are surrounded by an infiltrate of lymphocytes and histiocytes, which may be quite dense and extend deeply. Scattered lymphocyte cells often penetrate into the epidermis.
Immunohistochemical studies have revealed a predominance of CD8+ T cells compared with CD4+ T cells [3, 4].
Genetics
Familial cases are documented [1, 5, 6], although whether this relates to clustering related to contact with a common infectious agent remains to be determined.
Environmental factors
Possible triggering factors include infectious agents and trauma. For example, in a retrospective case review of 115 cases from Italy, the majority occurred in the cold season, five had prodromal symptoms of viral infection and two had a preceding history of skin trauma [1]. Other reported triggers include varicella infection [7] and measles, mumps and rubella vaccination [3].
Clinical features
History
The onset is usually sudden. Frequently, there are no symptoms, but pruritus may occasionally occur and is more common in adults. The course is variable. The area affected typically reaches its maximum extent within 2–3 weeks, but gradual extension can continue for several months. Spontaneous resolution can be expected within 6–12 months in most cases, but some lesions may persist for over a year [8]. Resolution may be followed by hypopigmentation or more rarely hyperpigmentation [1, 2].
Presentation
The initial presentation is characterized by the sudden appearance of small, discrete, pink, flat-topped, lichenoid papules in a typical linear distribution. In a recent series of 24 patients from Spain, 11 presented with erythematous papules [2]. Occasionally, vesicles are observed, but were present in only two out of 24 patients. The lesions extend over the course of a week or more and rapidly coalesce to form a dull-red to brown, slightly scaly, linear band, usually 2 mm to 2 cm in width, and often irregular. Occasionally, the bands broaden into plaques, especially on the buttocks. The lesion may be only a few centimetres in length or may extend the entire length of the limb, and may be continuous or interrupted (Figure 37.28).
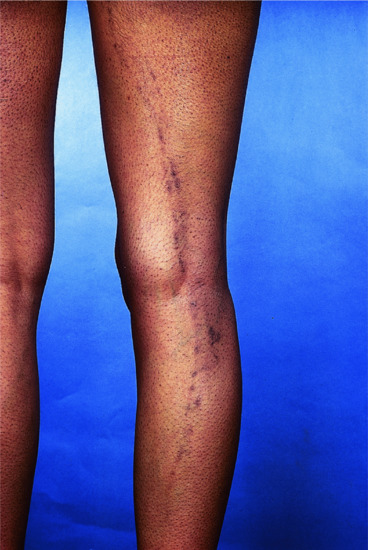
Figure 37.28 Lichen striatus of the inner thigh in a girl aged 16 years. The histological changes were those of chronic eczema.
(Courtesy of Dr R.A. Marsden, St George's Hospital, London, UK.)
The initial lichenoid papules are pink and not violaceous, and show no umbilication or Wickham's striae. The papules may be hypopigmented in dark-skinned people and hypopigmentation was noted at presentation in one-third of 24 cases reported from Spain [2]. Residual persistent hypopigmentation was also noted in approximately 30% of cases in a review of over 190 patients [2]. Post-inflammatory hyperpigmentation is reported less commonly (approximately 7% of 135 reported cases) [2].
The lesions occur most commonly on one arm or leg, or on the neck, but may develop on the trunk [1]. A review of more than 300 cases showed that over 75% occurred on the limbs [2]. The abdomen, buttocks and thighs may be involved in single extensive lesions, but multiple lesions are rare, and bilateral involvement is exceptional. Involvement of the nails may result in longitudinal ridging, splitting, onycholysis or nail loss [1, 9].
Clinical variants
Parallel linear bands or zosteriform patterns have been recorded (Figure 37.29) and extensive bilateral lesions were reported in two out of 24 patients in the series from Barcelona, Spain [2]. The majority of episodes are solitary, but occasionally repeated episodes can occur in different locations.

Figure 37.29 Lichen striatus showing parallel linear bands in a zigzag distribution on the thigh of a 15-year-old girl. Histology showed epidermal hyperkeratosis, focal liquefactive degeneration at the dermo-epidermal junction; upper dermal oedema with chronic inflammatory infiltrate in the upper dermis. The eruption resolved spontaneously.
Differential diagnosis
Linear inflammatory eruptions that are distributed along Blaschko's lines or show a zosteriform distribution, including linear psoriasis, linear Darier disease, linear lichen planus, linear porokeratosis and inflammatory linear verrucous epidermal naevus (ILVEN), should be considered in the differential diagnosis. Linear forms of lichen planus and psoriasis can usually be differentiated clinically, even in the absence of typical lesions in other sites, which should always be sought. ILVEN (Chapter 75), in particular, has many clinical and histological features in common with lichen striatus. However, ILVEN is always pruritic and persists despite periods of relative improvement. The differential diagnosis of hypopigmented lesions includes linear vitiligo or nevoid hypomelanosis [2].
Disease course and prognosis
The disease course is variable. The majority of lesions last for at least 6 months and resolve within 1 year, but lesions may last for just a few weeks or persist for several years, both in children and in adults [1, 8]. It may follow a prolonged and/or relapsing course, particularly in adults [1, 10]. Post-inflammatory hypopigmentation may last for years, particularly in darker skin types2. Hypopigmentation may also be the presenting feature in some patients [2]. Lichen striatus presenting in adults tends to be more extensive and symptomatic and may require treatment (see below) [10].
Investigations
Skin biopsy may be helpful.
Management
General principles of management
Usually no treatment is necessary in childhood cases which are largely asymptomatic and typically spontaneously resolve. In patients with troublesome itch (usually adults), topical corticosteroids are the first line of management. However, in resistant cases or when there are concerns over topical corticosteroid-induced skin atrophy, calcineurin inhibitors may be considered. Because of the rarity of the condition, no trials have been performed, but individual case reports and small case series report positive and prompt symptomatic response and rapid resolution of lesions in response to both tacrolimus and pimecrolimus [4, 10, 11]. A single case of a response to photodynamic therapy is reported [12]. Nail involvement may respond to potent steroid cream under occlusion.
References
Definition
- Darier J. Précis de Dermatologie, p. 118. Paris: Masson, 1909.
- Weedon D. The lichenoid tissue reaction. Int J Dermatol 1982;21(4):203–6.
- Boyd AS, Neldner KH. Lichen planus. J Am Acad Dermatol 1991;25(4):593–619.
Lichen planus
Introduction and general description
- Gorouhi F, Davari P, Fazel N. Cutaneous and mucosal lichen planus: a comprehensive review of clinical subtypes, risk factors, diagnosis, and prognosis. Sci World J 2014;2014:742826.
- Depaoli M. [Clinico-statistical data on lichen ruber planus.] Minerva Dermatol 1964;39:166–71.
- Schmidt H. Frequency, duration and localization of lichen planus. A study based on 181 patients. Acta Derm Venereol 1961;41:164–7.
- Bhattacharya M, Kaur I, Kumar B. Lichen planus: a clinical and epidemiological study. J Dermatol 2000;27(9):576–82.
- Daramola OO, Ogunbiyi AO, George AO. Evaluation of clinical types of cutaneous lichen planus in anti-hepatitis C virus seronegative and seropositive Nigerian patients. Int J Dermatol 2003;42(12):933–5.
- Balasubramaniam P, Ogboli M, Moss C. Lichen planus in children: review of 26 cases. Clin Exp Dermatol 2008;33(4):457–9.
- Kumar RR, Hay KD. Demographic analysis of oral lichen planus presentations to Auckland Oral Medicine Clinic from 1999 to 2006. NZ Dent J 2010;106(3):113–14.
Pathophysiology
- Bramanti TE, Dekker NP, Lozada-Nur F, Sauk JJ, Regezi JA. Heat shock (stress) proteins and gamma delta T lymphocytes in oral lichen planus. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1995;80(6):698–704.
- Gotoh A, Hamada Y, Shiobara N, et al. Skew in T cell receptor usage with polyclonal expansion in lesions of oral lichen planus without hepatitis C virus infection. Clin Exp Immunol 2008;154(2):192–201.
- Hasseus B, Jontell M, Brune M, Johansson P, Dahlgren UI. Langerhans cells and T cells in oral graft versus host disease and oral lichen planus. Scan J Immunol 2001;54(5):516–24.
- Wenzel J, Peters B, Zahn S, et al. Gene expression profiling of lichen planus reflects CXCL9+-mediated inflammation and distinguishes this disease from atopic dermatitis and psoriasis. J Invest Dermatol 2008;128(1):67–78.
- Gassling V, Hampe J, Acil Y, Braesen JH, Wiltfang J, Hasler R. Disease-associated miRNA-mRNA networks in oral lichen planus. PLOS One 2013;8(5):e63015.
- Terlou A, Santegoets LA, van der Meijden WI, et al. An autoimmune phenotype in vulvar lichen sclerosus and lichen planus: a Th1 response and high levels of microRNA-155. J Invest Dermatol 2012;132(3 Pt 1):658–66.
- Shen Z, Gao X, Ma L, Zhou Z, Shen X, Liu W. Expression of Foxp3 and interleukin-17 in lichen planus lesions with emphasis on difference in oral and cutaneous variants. Arch Dermatol Res 2014;306(5):441–6.
- Dutz JP. T-cell-mediated injury to keratinocytes: insights from animal models of the lichenoid tissue reaction. J Invest Dermatol 2009;129(2):309–14.
- Breathnach SM, Katz SI. Immunopathology of cutaneous graft-versus-host disease. Am J Dermatopathol 1987;9(4):343–8.
- Liddle BJ, Cowan MA. Lichen planus-like eruption and nail changes in a patient with graft-versus-host disease. Br J Dermatol 1990;122(6):841–3.
- Garcia FVMJ, Pascual-Lopez M, Elices M, Dauden E, Garcia-Diez A, Fraga J. Superficial mucoceles and lichenoid graft versus host disease: report of three cases. Acta Derm Venereol 2002;82(6):453–5.
- Shiohara T. The lichenoid tissue reaction. An immunological perspective. Am J Dermatopathol 1988;10(3):252–6.
- Fayyazi A, Schweyer S, Soruri A, et al. T lymphocytes and altered keratinocytes express interferon-gamma and interleukin 6 in lichen planus. Arch Dermatol Res 1999;291(9):485–90.
- Sugerman PB, Satterwhite K, Bigby M. Autocytotoxic T-cell clones in lichen planus. Br J Dermatol 2000;142(3):449–56.
- Villarroel Dorrego M, Correnti M, Delgado R, Tapia FJ. Oral lichen planus: immunohistology of mucosal lesions. J Oral Pathol Med 2002;31(7):410–14.
- Kawamura E, Nakamura S, Sasaki M, et al. Accumulation of oligoclonal T cells in the infiltrating lymphocytes in oral lichen planus. J Oral Pathol Med 2003;32(5):282–9.
- Sugerman PB, Savage NW, Walsh LJ, et al. The pathogenesis of oral lichen planus. Crit Rev Oral Biol Med 2002;13(4):350–65.
- Zhao ZZ, Sugerman PB, Walsh LJ, Savage NW. Expression of RANTES and CCR1 in oral lichen planus and association with mast cell migration. J Oral Pathol Med 2002;31(3):158–62.
- Iijima W, Ohtani H, Nakayama T, et al. Infiltrating CD8+ T cells in oral lichen planus predominantly express CCR5 and CXCR3 and carry respective chemokine ligands RANTES/CCL5 and IP-10/CXCL10 in their cytolytic granules: a potential self-recruiting mechanism. Am J Pathol 2003;163(1):261–8.
- Little MC, Griffiths CE, Watson RE, Pemberton MN, Thornhill MH. Oral mucosal keratinocytes express RANTES and ICAM-1, but not interleukin-8, in oral lichen planus and oral lichenoid reactions induced by amalgam fillings. Clin Exp Dermatol 2003;28(1):64–9.
- Zhao ZZ, Sugerman PB, Zhou XJ, Walsh LJ, Savage NW. Mast cell degranulation and the role of T cell RANTES in oral lichen planus. Oral Dis 2001;7(4):246–51.
- Shiohara T, Moriya N, Nagashima M. The lichenoid tissue reaction. A new concept of pathogenesis. Int J Dermatol 1988;27(6):365–74.
- Yamamoto T, Osaki T. Characteristic cytokines generated by keratinocytes and mononuclear infiltrates in oral lichen planus. J Invest Dermatol 1995;104(5):784–8.
- Yamamoto T, Nakane T, Osaki T. The mechanism of mononuclear cell infiltration in oral lichen planus: the role of cytokines released from keratinocytes. J Clin Immunol 2000;20(4):294–305.
- Deguchi M, Aiba S, Ohtani H, Nagura H, Tagami H. Comparison of the distribution and numbers of antigen-presenting cells among T-lymphocyte-mediated dermatoses: CD1a+, factor XIIIa+, and CD68+ cells in eczematous dermatitis, psoriasis, lichen planus and graft-versus-host disease. Arch Dermatol Res 2002;294(7):297–302.
- Musso T, Scutera S, Vermi W, et al. Activin A induces Langerhans cell differentiation in vitro and in human skin explants. PLOS One 2008;3(9):e3271.
- Zhou XJ, Sugerman PB, Savage NW, Walsh LJ, Seymour GJ. Intra-epithelial CD8+ T cells and basement membrane disruption in oral lichen planus. J Oral Pathol Med 2002;31(1):23–7.
- Zhou XJ, Sugerman PB, Savage NW, Walsh LJ. Matrix metalloproteinases and their inhibitors in oral lichen planus. J Cutan Pathol 2001;28(2):72–82.
- Ammar M, Mokni M, Boubaker S, El Gaied A, Ben Osman A, Louzir H. Involvement of granzyme B and granulysin in the cytotoxic response in lichen planus. J Cutan Pathol 2008;35(7):630–4.
- Kim SG, Chae CH, Cho BO, et al. Apoptosis of oral epithelial cells in oral lichen planus caused by upregulation of BMP-4. J Oral Pathol Med 2006;35(1):37–45.
- Chen Y, Zhang W, Geng N, Tian K, Jack Windsor L. MMPs, TIMP-2, and TGF-beta1 in the cancerization of oral lichen planus. Head Neck 2008;30(9):1237–45.
- Segura S, Rozas-Munoz E, Toll A, et al. Evaluation of MYC status in oral lichen planus in patients with progression to oral squamous cell carcinoma. Br J Dermatol 2013;169(1):106–14.
- Lu R, Zhang J, Sun W, Du G, Zhou G. Inflammation-related cytokines in oral lichen planus: an overview. J Oral Pathol Med 2013.
- Abell E, Presbury DG, Marks R, Ramnarain D. The diagnostic significance of immunoglobulin and fibrin deposition in lichen planus. Br J Dermatol 1975;93(1):17–24.
- Kulthanan K, Jiamton S, Varothai S, Pinkaew S, Sutthipinittharm P. Direct immunofluorescence study in patients with lichen planus. Int J Dermatol 2007;46(12):1237–41.
- Camisa C, Neff JC, Olsen RG. Use of indirect immunofluorescence in the lupus erythematosus/lichen planus overlap syndrome: an additional diagnostic clue. J Am Acad Dermatol 1984;11(6):1050–9.
- Mora RG, Nesbitt LT, Jr, Brantley JB. Lichen planus pemphigoides: clinical and immunofluorescent findings in four cases. J Am Acad Dermatol 1983;8(3):331–6.
- Kolde G, Wesendahl C, Stein H, Reichart PA. Oral lichen planus: diagnostic immunofluorescence testing on routine histological material. Br J Dermatol 2003;148(2):374–6.
- Oliver GF, Winkelmann RK, Muller SA. Lichenoid dermatitis: a clinicopathologic and immunopathologic review of sixty-two cases. J Am Acad Dermatol 1989;21(2 Pt 1):284–92.
- Toll A, Gilaberte M, Gallardo F, Iglesias M, Barranco C, Pujol RM. Multiple and extensive lichen planus-like keratoses: an underestimated cutaneous eruption observed in patients with intense sun damage. J Eur Acad Dermatol Venereol 2006;20(4):472–3.
- Al-Hoqail IA, Crawford RI. Benign lichenoid keratoses with histologic features of mycosis fungoides: clinicopathologic description of a clinically significant histologic pattern. J Cutan Pathol 2002;29(5):291–4.
- Dalton SR, Baptista MA, Libow LF, Elston DM. Lichenoid tissue reaction in malignant melanoma: a potential diagnostic pitfall. Am J Clin Pathol 2002;117(5):766–70.
- Dalton SR, Fillman EP, Altman CE, et al. Atypical junctional melanocytic proliferations in benign lichenoid keratosis. Hum Pathol 2003;34(7):706–9.
- Patil S, Khandelwal S, Rahman F, Kaswan S, Tipu S. Epidemiological relationship of oral lichen planus to hepatitis C virus in an Indian population. Oral Health Dent Manage 2012;11(4):199–205.
- Pilli M, Penna A, Zerbini A, et al. Oral lichen planus pathogenesis: a role for the HCV-specific cellular immune response. Hepatology 2002;36(6):1446–52.
- Daramola OO, George AO, Ogunbiyi AO, Otegbayo JA. Hepatitis B virus in Nigerians with lichen planus. West Afr J Med 2004;23(2):104–6.
- Rubsam K, Schroll A, Weisenseel P, Multhaup S, Ruzicka T, Prinz JC. Lichen planus and hepatitis virus infections: causal association? J Deut Dermatol Gesellschaft 2011;9(6):464–8.
- De Vries HJ, van Marle J, Teunissen MB, et al. Lichen planus is associated with human herpesvirus type 7 replication and infiltration of plasmacytoid dendritic cells. Br J Dermatol 2006;154(2):361–4.
- Mizukawa Y, Horie C, Yamazaki Y, Shiohara T. Detection of varicella-zoster virus antigens in lesional skin of zosteriform lichen planus but not in that of linear lichen planus. Dermatology 2012;225(1):22–6.
- Pourshahidi S, Fakhri F, Ebrahimi H, et al. Lack of association between Helicobacter pylori infection and oral lichen planus. Asian Pac J Cancer Prev 2012;13(5):1745–7.
- Rebora A, Rongioletti F, Drago F, Parodi. Lichen planus as a side effect of HBV vaccination. Dermatology 1999;198(1):1–2.
- Limas C, Limas CJ. Lichen planus in children: a possible complication of hepatitis B vaccines. Pediatr Dermatol 2002;19(3):204–9.
- Ragaz A, Ackerman AB. Evolution, maturation, and regression of lesions of lichen planus. New observations and correlations of clinical and histologic findings. Am J Dermatopathol 1981;3(1):5–25.
- Patterson JW. The spectrum of lichenoid dermatitis. J Cutan Pathol 1991;18(2):67–74.
- Neppelberg E, Johannessen AC, Jonsson R. Apoptosis in oral lichen planus. Eur J Oral Sci 2001;109(5):361–4.
- Black MM, Wilson-Jones E. The role of the epidermis in the histopathogenesis of lichen planus. Histochemical correlations. Arch Dermatol 1972;105(1):81–6.
- Da Silva Fonseca LM, do Carmo MA. Identification of the AgNORs, PCNA and ck16 proteins in oral lichen planus lesions. Oral Dis 2001;7(6):344–8.
- Summerly R, Jones EW. The microarchitecture of Wickham's dtriae. Trans St Johns Hosp Dermatol Soc 1964;50:157–61.
- Ebner H, Erlach E, Gebbart W. [Blister formation in lichen planus: electronmicroscopical observations.] Arch Dermatol Forsch 1973;247(2):193–205.
- Ebner H, Gebhart W. Light and electron microscopic differentiation of amyloid and colloid or hyaline bodies. Br J Dermatol 1975;92(6):637–45.
- Ebner H, Gebhart W. Light and electron microscopic studies on colloid and other cytoid bodies. Clin Exp Dermatol 1977;2(4):311–22.
- Lupton GP, Goette DK. Lichen planus with plasma cell infiltrate. Arch Dermatol 1981;117(2):124–5.
- Roustan G, Hospital M, Villegas C, Sanchez Yus E, Robledo A. Lichen planus with predominant plasma cell infiltrate. Am J Dermatopathol 1994;16(3):311–14.
- Horn RT, Jr, Goette DK, Odom RB, Olson EG, Guill MA. Immunofluorescent findings and clinical overlap in two cases of follicular lichen planus. J Am Acad Dermatol 1982;7(2):203–7.
- Matta M, Kibbi AG, Khattar J, Salman SM, Zaynoun ST. Lichen planopilaris: a clinicopathologic study. J Am Acad Dermatol 1990;22(4):594–8.
- Shklar G. Erosive and bullous oral lesions of lichen planus: histologic studies. Arch Dermatol 1968;97(4):411–16.
- Karatsaidis A, Schreurs O, Helgeland K, Axell T, Schenck K. Erythematous and reticular forms of oral lichen planus and oral lichenoid reactions differ in pathological features related to disease activity. J Oral Pathol Med 2003;32(5):275–81.
- Odukoya O, Gallagher G, Shklar G. A histologic study of epithelial dysplasia in oral lichen planus. Arch Dermatol 1985;121(9):1132–6.
- Wu D, Wang L, Sun M, et al. CIITA rs4774 and rs6498122 polymorphisms are associated with oral lichen planus in Chinese people: a case–control study. Eur J Oral Sci 2013;121(2):69–75.
- Jin X, Wang J, Zhu L, et al. Association between –308 G/A polymorphism in TNF-alpha gene and lichen planus: a meta-analysis. J Dermatol Sci 2012;68(3):127–34.
- Kuraishi N, Nagai Y, Hasegawa M, Ishikawa O. Lichenoid drug eruption with palmoplantar hyperkeratosis due to imatinib mesylate: a case report and a review of the literature. Acta Derm Venereol 2010;90(1):73–6.
- Tessari G, Barba A, Schena D. Lichen ruber planus following the administration of human anti-hepatitis B virus immunoglobulins. Acta Derm Venereol 1996;76(2):154.
- Asarch A, Gottlieb AB, Lee J, et al. Lichen planus-like eruptions: an emerging side effect of tumor necrosis factor-alpha antagonists. J Am Acad Dermatol 2009;61(1):104–11.
- Martin MD, Broughton S, Drangsholt M. Oral lichen planus and dental materials: a case–control study. Contact Dermatitis 2003;48(6):331–6.
- Lopez-Jornet P, Camacho-Alonso F. Do metal restorations in mouth alter clinical and histological appearance of oral lichen planus? NY State Dent J 2008;74(6):40–3.
- Reichart PA, Schmidtberg W, Samaranayake LP, Scheifele C. Betel quid-associated oral lesions and oral Candida species in a female Cambodian cohort. J Oral Pathol Med 2002;31(8):468–72.
- Stoopler ET, Parisi E, Sollecito TP. Betel quid-induced oral lichen planus: a case report. Cutis 2003;71(4):307–11.
- Brancaccio RR, Cockerell CJ, Belsito D, Ostreicher R. Allergic contact dermatitis from color film developers: clinical and histologic features. J Am Acad Dermatol 1993;28(5 Pt 2):827–30.
- Liden C. Lichen planus in relation to occupational and non-occupational exposure to chemicals. Br J Dermatol 1986;115(1):23–31.
- Kawamura T, Fukuda S, Ohtake N, Furue M, Tamaki K. Lichen planus-like contact dermatitis due to methacrylic acid esters. Br J Dermatol 1996;134(2):358–60.
- Guillet G, Coindre M, Levillain P, Guillet MH. [Lichenoid dermatitis resulting from sensitization to dimethylfumarate: atypical presentation of “Chinese sofa dermatitis”.] Ann Dermatol Venereol 2009;136(3):279–81.
- Vergilis-Kalner IJ, Sharma V, Sethi A. Lichen planus arising in radiation therapy treatment sites. Cutis 2008;82(5):353–5.
- Lundqvist EN, Wahlin YB, Bergdahl M, Bergdahl J. Psychological health in patients with genital and oral erosive lichen planus. J Eur Acad Dermatol Venereol 2006;20(6):661–6.
- Gavic L, Cigic L, Biocina Lukenda D, Gruden V, Gruden Pokupec JS. The role of anxiety, depression, and psychological stress on the clinical status of recurrent aphthous stomatitis and oral lichen planus. J Oral Pathol Med 2014;43(6):410–17.
History and clinical presentation
- Rivers JK, Jackson R, Orizaga M. Who was Wickham and what are his striae? Int J Dermatol 1986;25(9):611–13.
- Friedman DB, Hashimoto K. Annular atrophic lichen planus. J Am Acad Dermatol 1991;25(2 Pt 2):392–4.
- Anstey A, Marks R. Colocalization of lichen planus and vitiligo. Br J Dermatol 1993;128(1):103–4.
- Sanchez-Perez J, Rios Buceta L, Fraga J, Garcia-Diez A. Lichen planus with lesions on the palms and/or soles: prevalence and clinicopathological study of 36 patients. Br J Dermatol 2000;142(2):310–14.
- Higgins CR, Handfield-Jones S, Black MM. Erosive, flexural lichen planus – an uncommon variant. Clin Exp Dermatol 1993;18(2):169–70.
- Long CC, Finlay AY. Multiple linear lichen planus in the lines of Blaschko. Br J Dermatol 1996;135(2):275–6.
- Vogel PS, James WD. Lichen planus of the eyelid: an unusual clinical presentation. J Am Acad Dermatol 1992;27(4):638–9.
- Sharma R, Singhal N. Lichen planus of the eyelids. A report of 5 cases. Dermatol Online J 2001;7(1):5.
- Itin PH, Schiller P, Gilli L, Buechner SA. Isolated lichen planus of the lip. Br J Dermatol 1995;132(6):1000–2.
- Cecchi R, Giomi A. Isolated lichen planus of the lip. Australas J Dermatol 2002;43(4):309–10.
- Yu TC, Kelly SC, Weinberg JM, Scheinfeld NS. Isolated lichen planus of the lower lip. Cutis 2003;71(3):210–12.
- Lucke T, Fallowfield M, Burden D. Lichen planus associated with milia. Clin Exp Dermatol 1999;24(4):266–9.
- Kim JH, Krivda SJ. Lichen planus confined to a radiation therapy site. J Am Acad Dermatol 2002;46(4):604–5.
- Kanwar AJ, Ghosh S, Dhar S, Kaur S. Oral lesions of lichen planus. Int J Dermatol 1993;32(1):76.
- Hornstein OP, Stuhler C, Schirner E, Simon M, Jr. [Lichen ruber and diabetes mellitus – pathogenetic relations?] Hautarzt 1984;35(6):287–91.
- Smith MJ. Oral lichen planus and diabetes mellitus: a possible association. J Oral Med 1977;32(4):110–12.
Lichen planus principally involving mucous membranes
- Warin RP, Crabb HS, Darling AI. Lichen planus of the mouth. BMJ 1958;1(5077):983–4.
- Setterfield JF, Black MM, Challacombe SJ. The management of oral lichen planus. Clin Exp Dermatol 2000;25(3):176–82.
- Alam F, Hamburger J. Oral mucosal lichen planus in children. Int J Paediatr Dent 2001;11(3):209–14.
- Agarwal R, Saraswat A. Oral lichen planus: an update. Drugs Today (Barc) 2002;38(8):533–47.
- Eisen D. The clinical manifestations and treatment of oral lichen planus. Dermatol Clin 2003;21(1):79–89.
- Ismail SB, Kumar SK, Zain RB. Oral lichen planus and lichenoid reactions: etiopathogenesis, diagnosis, management and malignant transformation. J Oral Sci 2007;49(2):89–106.
- Pindborg JJ, Mehta FS, Daftary DK, Gupta PC, Bhonsle RB. Prevalence of oral lichen planus among 7639 Indian villagers in Kerala, South India. Acta Derm Venereol 1972;52(3):216–20.
- Salonen L, Axell T, Hellden L. Occurrence of oral mucosal lesions, the influence of tobacco habits and an estimate of treatment time in an adult Swedish population. J Oral Pathol Med 1990;19(4):170–6.
- Laine J, Kalimo K, Forssell H, Happonen RP. Resolution of oral lichenoid lesions after replacement of amalgam restorations in patients allergic to mercury compounds. Br J Dermatol 1992;126(1):10–15.
- Ibbotson SH, Speight EL, Macleod RI, Smart ER, Lawrence CM. The relevance and effect of amalgam replacement in subjects with oral lichenoid reactions. Br J Dermatol 1996;134(3):420–3.
- Dunsche A, Kastel I, Terheyden H, Springer IN, Christophers E, Brasch J. Oral lichenoid reactions associated with amalgam: improvement after amalgam removal. Br J Dermatol 2003;148(1):70–6.
- Dunsche A, Frank MP, Luttges J, et al. Lichenoid reactions of murine mucosa associated with amalgam. Br J Dermatol 2003;148(4):741–8.
- Wong L, Freeman S. Oral lichenoid lesions (OLL) and mercury in amalgam fillings. Contact Dermatitis 2003;48(2):74–9.
- Thornhill MH, Pemberton MN, Simmons RK, Theaker ED. Amalgam-contact hypersensitivity lesions and oral lichen planus. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2003;95(3):291–9.
- Menges M, Hohloch K, Pueschel W, Stallmach A. Lichen planus with oesophageal involvement. A case report and review of the literature. Digestion 2002;65(3):184–9.
- Keate RF, Williams JW, Connolly SM. Lichen planus esophagitis: report of three patients treated with oral tacrolimus or intraesophageal corticosteroid injections or both. Dis Esophagus 2003;16(1):47–53.
- Chryssostalis A, Gaudric M, Terris B, Coriat R, Prat F, Chaussade S. Esophageal lichen planus: a series of eight cases including a patient with esophageal verrucous carcinoma. A case series. Endoscopy 2008;40(9):764–8.
- Chandan VS, Murray JA, Abraham SC. Esophageal lichen planus. Arch Pathol Lab Med 2008;132(6):1026–9.
- Westbrook R, Riley S. Esophageal lichen planus: case report and literature review. Dysphagia 2008;23(3):331–4.
- Porter WM, Dinneen M, Hawkins DA, Bunker CB. Erosive penile lichen planus responding to circumcision. J Eur Acad Dermatol Venereol 2001;15(3):266–8.
- Rogers RS, 3rd, Eisen D. Erosive oral lichen planus with genital lesions: the vulvovaginal-gingival syndrome and the peno-gingival syndrome. Dermatol Clin 2003;21(1):91–8, vi–vii.
- Edwards L. Vulvar lichen planus. Arch Dermatol 1989;125(12):1677–80.
- Eisen D. The vulvovaginal-gingival syndrome of lichen planus. The clinical characteristics of 22 patients. Arch Dermatol 1994;130(11):1379–82.
- Lewis FM, Shah M, Harrington CI. Vulval involvement in lichen planus: a study of 37 women. Br J Dermatol 1996;135(1):89–91.
- Lewis FM. Vulval lichen planus. Br J Dermatol 1998;138(4):569–75.
- Kirtschig G, Van Der Meulen AJ, Ion Lipan JW, Stoof TJ. Successful treatment of erosive vulvovaginal lichen planus with topical tacrolimus. Br J Dermatol 2002;147(3):625–6.
- Lotery HE, Galask RP. Erosive lichen planus of the vulva and vagina. Obstet Gynecol 2003;101(5 Pt 2):1121–5.
- Watsky KL. Erosive perianal lichen planus responsive to tacrolimus. Int J Dermatol 2003;42(3):217–18.
- Kennedy CM, Galask RP. Erosive vulvar lichen planus: retrospective review of characteristics and outcomes in 113 patients seen in a vulvar specialty clinic. J Reprod Med 2007;52(1):43–7.
- Simpson RC, Thomas KS, Leighton P, Murphy R. Diagnostic criteria for erosive lichen planus affecting the vulva: an international electronic-Delphi consensus exercise. Br J Dermatol 2013;169(2):337–43.
- Marren P, Millard P, Chia Y, Wojnarowska F. Mucosal lichen sclerosus/lichen planus overlap syndromes. Br J Dermatol 1994;131(1):118–23.
Lichen planopilaris
- Tandon YK, Somani N, Cevasco NC, Bergfeld WF. A histologic review of 27 patients with lichen planopilaris. J Am Acad Dermatol 2008;59(1):91–8.
- Gupta SN, Palceski D. Lichen planopilaris presenting as truncal alopecia: a case presentation and review of the literature. Cutis 2003;72(1):63–6.
- Graham Little EG. Folliculitis decalvans et atrophicans. Proc R Soc Med 1915;8(Dermatol Sect):139–41.
- Matta M, Kibbi AG, Khattar J, Salman SM, Zaynoun ST. Lichen planopilaris: a clinicopathologic study. J Am Acad Dermatol 1990;22(4):594–8.
- Horn RT, Jr, Goette DK, Odom RB, Olson EG, Guill MA. Immunofluorescent findings and clinical overlap in two cases of follicular lichen planus. J Am Acad Dermatol 1982;7(2):203–7.
- Ghislain PD, Van Eeckhout P, Ghislain E. Lassueur–Graham Little–Piccardi syndrome: a 20-year follow-up. Dermatology 2003;206(4):391–2.
- Viglizzo G, Verrini A, Rongioletti F. Familial Lassueur–Graham-Little–Piccardi syndrome. Dermatology 2004;208(2):142–4.
- Abbas O, Chedraoui A, Ghosn S. Frontal fibrosing alopecia presenting with components of Piccardi–Lassueur–Graham-Little syndrome. J Am Acad Dermatol 2007;57(Suppl. 2):S15–18.
- Kossard S, Lee MS, Wilkinson B. Postmenopausal frontal fibrosing alopecia: a frontal variant of lichen planopilaris. J Am Acad Dermatol 1997;36(1):59–66.
- MacDonald A, Clark C, Holmes S. Frontal fibrosing alopecia: a review of 60 cases. J Am Acad Dermatol 2012;67(5):955–61.
- Tan KT, Messenger AG. Frontal fibrosing alopecia: clinical presentations and prognosis. Br J Dermatol 2009;160(1):75–9.
- Vano-Galvan S, Molina-Ruiz AM, Serrano-Falcon C, et al. Frontal fibrosing alopecia: a multicenter review of 355 patients. J Am Acad Dermatol 2014;70(4):670–8.
Hypertrophic lichen planus
- Sharma VK, Achar A, Ramam M, Singh MK. Multiple cutaneous horns overlying lichen planus hypertrophicus. Br J Dermatol 2001;144(2):424–5.
- Badell A, Marcoval J, Gallego I, Moreno A, Peyri J. Keratoacanthoma arising in hypertrophic lichen planus. Br J Dermatol 2000;142(2):380–2.
- Chave TA, Graham-Brown RA. Keratoacanthoma developing in hypertrophic lichen planus. Br J Dermatol 2003;148(3):592.
- Yesudian P, Rao R. Malignant transformation of hypertrophic lichen planus. Int J Dermatol 1985;24(3):177–8.
- Singh SK, Saikia UN, Ajith C, Kumar B. Squamous cell carcinoma arising from hypertrophic lichen planus. J Eur Acad Dermatol Venereol 2006;20(6):745–6.
- Krasowska D, Bogaczewicz J, Chodorowska G. Development of squamous cell carcinoma within lesions of cutaneous lichen planus. Eur J Dermatol 2007;17(5):447–8.
- Ardabili M, Gambichler T, Rotterdam S, Altmeyer P, Hoffmann K, Stucker M. Metastatic cutaneous squamous cell carcinoma arising from a previous area of chronic hypertrophic lichen planus. Dermatol Online J 2003;9(1):10.
Lichen planus of the palms and soles, Actinic lichen planus, Lichen planus pigmentosus, Annular lichen planus
- Sanchez-Perez J, Rios Buceta L, Fraga J, Garcia-Diez A. Lichen planus with lesions on the palms and/or soles: prevalence and clinicopathological study of 36 patients. Br J Dermatol 2000;142(2):310–14.
- Sait MA, Garg BR. Punctate keratoses of palms in lichen planus. Int J Dermatol 1986;25(9):592–3.
- Gunduz K, Inanir I, Turkdogan P, Sacar H. Palmoplantar lichen planus presenting with vesicle-like papules. J Dermatol 2003;30(4):337–40.
- Cram DL, Kierland RR, Winkelmann RK. Ulcerative lichen planus of the feet. Bullous variant with hair and nail lesions. Arch Dermatol 1966;93(6):692–701.
- Sonnex TS, Eady RA, Sparrow GP, Mayou B. Ulcerative lichen planus associated with webbing of the toes. J R Soc Med 1986;79(6):363–5.
- Kilaimy M. Lichen planus subtropicus. Arch Dermatol 1976;112(9):1251–3.
- Singh OP, Kanwar AJ. Lichen planus in India: an appraisal of 441 cases. Int J Dermatol 1976;15:752–6.
- Peretz E, Grunwald MH, Halevy S. Annular plaque on the face. Actinic lichen planus (ALP). Arch Dermatol 1999;135(12):1543, 1646.
- Bouassida S, Boudaya S, Turki H, Gueriani H, Zahaf A. [Actinic lichen planus: 32 cases.] Ann Dermatol Venereol 1998;125(6–7):408–13.
- Meads SB, Kunishige J, Ramos-Caro FA, Hassanein AM. Lichen planus actinicus. Cutis 2003;72(5):377–81.
- Skowron F, Grezard P, Merle P, Balme B, Perrot H. Erythematosus actinic lichen planus: a new clinical form associated with oral erosive lichen planus and chronic active hepatitis B. Br J Dermatol 2002;147(5):1032–4.
- Isaacson D, Turner ML, Elgart ML. Summertime actinic lichenoid eruption (lichen planus actinicus). J Am Acad Dermatol 1981;4(4):404–11.
- Salman SM, Kibbi AG, Zaynoun S. Actinic lichen planus. A clinicopathologic study of 16 patients. J Am Acad Dermatol 1989;20(2 Pt 1):226–31.
- Salman SM, Khallouf R, Zaynoun S. Actinic lichen planus mimicking melasma. A clinical and histopathologic study of three cases. J Am Acad Dermatol 1988;18(2 Pt 1):275–8.
- Verhagen AR, Koten JW. Lichenoid melanodermatitis. A clinicopathological study of fifty-one Kenyan patients with so-called tropical lichen planus. Br J Dermatol 1979;101(6):651–8.
- Bhutani LK, Bedi TR, Pandhi RK, Nayak NC. Lichen planus pigmentosus. Dermatologica 1974;149(1):43–50.
- Kanwar AJ, Kaur S. Lichen planus pigmentosus. J Am Acad Dermatol 1989;21(4 Pt 1):815.
- Kanwar AJ, Dogra S, Handa S, Parsad D, Radotra BD. A study of 124 Indian patients with lichen planus pigmentosus. Clin Exp Dermatol 2003;28(5):481–5.
- Pock L, Jelinkova L, Drlik L, et al. Lichen planus pigmentosus-inversus. J Eur Acad Dermatol Venereol 2001;15(5):452–4.
- Kashima A, Tajiri A, Yamashita A, Asada Y, Setoyama M. Two Japanese cases of lichen planus pigmentosus-inversus. Int J Dermatol 2007;46(7):740–2.
- Laskaris GC, Papavasiliou SS, Bovopoulou OD, Nicolis GD. Lichen planus pigmentosus of the oral mucosa: a rare clinical variety. Dermatologica 1981;162(1):61–3.
- Sassolas B, Zagnoli A, Leroy JP, Guillet G. Lichen planus pigmentosus associated with acrokeratosis of Bazex. Clin Exp Dermatol 1994;19(1):70–3.
- Mehregan DA, Van Hale HM, Muller SA. Lichen planopilaris: clinical and pathologic study of forty-five patients. J Am Acad Dermatol 1992;27(6 Pt 1):935–42.
- Annessi G, Lombardo G, Gobello T, Puddu P. A clinicopathologic study of scarring alopecia due to lichen planus: comparison with scarring alopecia in discoid lupus erythematosus and pseudopelade. Am J Dermatopathol 1999;21(4):324–31.
- Sehgal VN, Bajaj P, Srivastva G. Lichen planopilaris [cicatricial (scarring) alopecia] in a child. Int J Dermatol 2001;40(7):461–3.
'Mixed' lichen planus/discoid lupus erythematosus disease patterns
- Potts ED, Rowell NR. Lichen planus: a distinct entity from lupus erythematosus. Acta Derm Venereol 1981;61(5):413–16.
- Nieboer C. The reliability of immunofluorescence and histopathology in the diagnosis of discoid lupus erythematosus and lichen planus. Br J Dermatol 1987;116(2):189–98.
- Copeman PW, Schroeter AL, Kierland RR. An unusual variant of lupus erythematosus or lichen planus. Br J Dermatol 1970;83(2):269–72.
- Davies MG, Gorkiewicz A, Knight A, Marks R. Is there a relationship between lupus erythematosus and lichen planus? Br J Dermatol 1977;96(2):145–54.
- Nagy E, Szakaly I. [Discoid lupus erythematosus or lichen planus?] Z Hautkr 1978;53(17):599–603.
- Piamphongsant T, Sawannapreecha S, Arangson PG, Sawchome Y, Kullavanijaya P. Mixed lichen planus–lupus erythematosus disease. J Cutan Pathol 1978;5(4):209–15.
- Romero RW, Nesbitt LT, Jr, Reed RJ. Unusual variant of lupus erythematosus or lichen planus. Clinical, histopathologic, and immunofluorescent studies. Arch Dermatol 1977;113(6):741–8.
- Uitto J, Santa-Cruz DJ, Eisen AZ, Leone P. Verrucous lesion in patients with discoid lupus erythematosus. Clinical, histopathological and immunofluorescence studies. Br J Dermatol 1978;98(5):507–20.
- Tursen U, Oz O, Ikizoglu G, Kaya TI, Dusmez D. A case of lichen planus-lupus erythematosus overlap syndrome with eyelid involvement. Eur J Ophthalmol 2002;12(3):244–6.
- Grabbe S, Kolde G. Coexisting lichen planus and subacute cutaneous lupus erythematosus. Clin Exp Dermatol 1995;20(3):249–54.
Bullous lichen planus and lichen planus pemphigoides
- Sarkany I, Caron GA, Jones HH. Lichen planus pemphigoides. Trans St Johns Hosp Dermatol Soc 1964;50:50–5.
- Gawkrodger DJ, Stavropoulos PG, McLaren KM, Buxton PK. Bullous lichen planus and lichen planus pemphigoides – clinico-pathological comparisons. Clin Exp Dermatol 1989;14(2):150–3.
- Ebner H, Erlach E, Gebbart W. [Blister formation in lichen planus: electronmicroscopical observations.] Arch Dermatol Forsch 1973;247(2):193–205.
- Bouloc A, Vignon-Pennamen MD, Caux F, et al. Lichen planus pemphigoides is a heterogeneous disease: a report of five cases studied by immunoelectron microscopy. Br J Dermatol 1998;138(6):972–80.
- Swale VJ, Black MM, Bhogal BS. Lichen planus pemphigoides: two case reports. Clin Exp Dermatol 1998;23(3):132–5.
- Demircay Z, Baykal C, Demirkesen C. Lichen planus pemphigoides: report of two cases. Int J Dermatol 2001;40(12):757–9.
- Mora RG, Nesbitt LT, Jr, Brantley JB. Lichen planus pemphigoides: clinical and immunofluorescent findings in four cases. J Am Acad Dermatol 1983;8(3):331–6.
- Archer CB, Cronin E, Smith NP. Diagnosis of lichen planus pemphigoides in the absence of bullae on normal-appearing skin. Clin Exp Dermatol 1992;17(6):433–6.
- Kuramoto N, Kishimoto S, Shibagaki R, Yasuno H. PUVA-induced lichen planus pemphigoides. Br J Dermatol 2000;142(3):509–12.
- Sakuma-Oyama Y, Powell AM, Albert S, Oyama N, Bhogal BS, Black MM. Lichen planus pemphigoides evolving into pemphigoid nodularis. Clin Exp Dermatol 2003;28(6):613–16.
- Stevens SR, Griffiths CE, Anhalt GJ, Cooper KD. Paraneoplastic pemphigus presenting as a lichen planus pemphigoides-like eruption. Arch Dermatol 1993;129(7):866–9.
- Hsiao CJ, Hsu MM, Lee JY, Chen WC, Hsieh WC. Paraneoplastic pemphigus in association with a retroperitoneal Castleman's disease presenting with a lichen planus pemphigoides-like eruption. A case report and review of literature. Br J Dermatol 2001;144(2):372–6.
- Prost C, Tesserand F, Laroche L, et al. Lichen planus pemphigoides: an immuno-electron microscopic study. Br J Dermatol 1985;113(1):31–6.
- Tamada Y, Yokochi K, Nitta Y, Ikeya T, Hara K, Owaribe K. Lichen planus pemphigoides: identification of 180 kd hemidesmosome antigen. J Am Acad Dermatol 1995;32(5 Pt 2):883–7.
- Zillikens D, Caux F, Mascaro JM, et al. Autoantibodies in lichen planus pemphigoides react with a novel epitope within the C-terminal NC16A domain of BP180. J Invest Dermatol 1999;113(1):117–21.
- Skaria M, Salomon D, Jaunin F, Friedli A, Saurat JH, Borradori L. IgG autoantibodies from a lichen planus pemphigoides patient recognize the NC16A domain of the bullous pemphigoid antigen 180. Dermatology 1999;199(3):253–5.
- Hsu S, Ghohestani RF, Uitto J. Lichen planus pemphigoides with IgG autoantibodies to the 180 kd bullous pemphigoid antigen (type XVII collagen). J Am Acad Dermatol 2000;42(1 Pt 1):136–41.
- Yoon KH, Kim SC, Kang DS, Lee IJ. Lichen planus pemphigoides with circulating autoantibodies against 200 and 180 kDa epidermal antigens. Eur J Dermatol 2000;10(3):212–14.
Lichen nitidus
- Wright AL, McVittie E, Hunter JA. An immunophenotypic study of lichen nitidus. Clin Exp Dermatol 1990;15(4):273–6.
- Smoller BR, Flynn TC. Immunohistochemical examination of lichen nitidus suggests that it is not a localized papular variant of lichen planus. J Am Acad Dermatol 1992;27(2 Pt 1):232–6.
- Ocampo J, Torne R. Generalized lichen nitidus. Report of two cases treated with astemizol. Int J Dermatol 1989;28(1):49–51.
- Wall LM, Heenan PJ, Papadimitriou JM. Generalized lichen nitidus: a case report. Australas J Dermatol 1985;26(1):36–40.
- Munro CS, Cox NH, Marks JM, Natarajan S. Lichen nitidus presenting as palmoplantar hyperkeratosis and nail dystrophy. Clin Exp Dermatol 1993;18(4):381–3.
- Weiss RM, Cohen AD. Lichen nitidus of the palms and soles. Arch Dermatol 1971;104(5):538–40.
- Coulsown IH, Marsden RA, Cook MG. Purpuric palmar lichen nitidus – an unusual though distinctive eruption. Clin Exp Dermatol 1988;13(5):347–9.
- Prigent F, Cavelier-Balloy B, Lemarchand-Venencie F, Civatte J. [Linear lichen nitidus.] Ann Dermatol Venereol 1989;116(11):814–15.
- Kellett JK, Beck MH. Lichen nitidus associated with distinctive nail changes. Clin Exp Dermatol 1984;9(2):201–4.
- Natarajan S, Dick DC. Lichen nitidus associated with nail changes. Int J Dermatol 1986;25(7):461–2.
- Summey BT, Cusack CA. Actinic lichen nitidus. Cutis 2008;81(3):266–8.
- Madhok R, Winkelmann RK. Spinous, follicular lichen nitidus associated with perifollicular granulomas. J Cutan Pathol 1988;15(4):245–8.
- Patrizi A, Di Lernia V, Pauluzzi P. [Generalized lichen niditus, trisomy 21 and congenital megacolon.] Ann Dermatol Venereol 1991;118(10):725.
- Teixeira VB, Coutinho I, Cardoso JC, Tellhechea O. Generalized lichen nitidus in a boy with Niemann-Pick disease type B. An Bras Dermatol 2013;88(6):977–8.
- Clausen J, Jacobsen FK, Brandrup F. Lichen nitidus: electron microscopic and immunofluorescence studies. Acta Derm Venereol 1982;62(1):15–19.
- Waisman M, Dundon BC, Michel B. Immunofluorescent studies in lichen nitidus. Arch Dermatol 1973;107(2):200–3.
- Eisen RF, Stenn J, Kahn SM, Bhawan J. Lichen nitidus with plasma cell infiltrate. Arch Dermatol 1985;121(9):1193–4.
- Banse-Kupin L, Morales A, Kleinsmith DA. Perforating lichen nitidus. J Am Acad Dermatol 1983;9(3):452–6.
- Kato N. Familial lichen nitidus. Clin Exp Dermatol 1995;20(4):336–8.
Nékam disease
- Kersey P, Ive FA. Keratosis lichenoides chronica is synonymous with lichen planus. Clin Exp Dermatol 1982;7(1):49–54.
- Patrizi A, Neri I, Passarini B, Varotti C. Keratosis lichenoides chronica: a pediatric case. Dermatology 1995;191(3):264–7.
- Margolis MH, Cooper GA, Johnson SA. Keratosis lichenoides chronica. Arch Dermatol 1972;105(5):739–43.
- Masouye I, Saurat JH. Keratosis lichenoides chronica: the centenary of another Kaposi's disease. Dermatology 1995;191(3):188–92.
- Baran R, Panizzon R, Goldberg L. The nails in keratosis lichenoides chronica. Characteristics and response to treatment. Arch Dermatol 1984;120(11):1471–4.
- Haas N, Czaika V, Sterry W. [Keratosis lichenoides chronica following trauma. A case report and update of the last literature review.] Hautarzt2001;52(7):629–33.
- Criado PR, Valente NY, Sittart JA, Juang JM, Vasconcellos C. Keratosis lichenoides chronica: report of a case developing after erythroderma. Australas J Dermatol 2000;41(4):247–9.
- Arata J, Seno A, Tada J, Wada E, Tamaki H, Tamaki M. Peculiar facial erythematosquamous lesions in two siblings with cyclical summer improvement and winter relapse: a variant of keratosis lichenoides chronica? J Am Acad Dermatol 1993;28(5 Pt 2):870–3.
- Braun-Falco O, Bieber T, Heider L. [Chronic lichenoid keratosis: disease variant or disease entity?] Hautarzt1989;40(10):614–22.
- Stefanato CM, Youssef EA, Cerio R, Kobza-Black A, Greaves MW. Atypical Nekam's disease – keratosis lichenoides chronica associated with porokeratotic histology and amyloidosis. Clin Exp Dermatol 1993;18(3):274–6.
Complications
- Mehregan DA, Van Hale HM, Muller SA. Lichen planopilaris: clinical and pathologic study of forty-five patients. J Am Acad Dermatol 1992;27(6 Pt 1):935–42.
- Annessi G, Lombardo G, Gobello T, Puddu P. A clinicopathologic study of scarring alopecia due to lichen planus: comparison with scarring alopecia in discoid lupus erythematosus and pseudopelade. Am J Dermatopathol 1999;21(4):324–31.
- Sehgal VN, Bajaj P, Srivastva G. Lichen planopilaris [cicatricial (scarring) alopecia] in a child. Int J Dermatol 2001;40(7):461–3.
- Faulkner CF, Wilson NJ, Jones SK. Frontal fibrosing alopecia associated with cutaneous lichen planus in a premenopausal woman. Australas J Dermatol 2002;43(1):65–7.
- Amato L, Mei S, Massi D, Gallerani I, Fabbri P. Cicatricial alopecia; a dermatopathologic and immunopathologic study of 33 patients (pseudopelade of Brocq is not a specific clinico-pathologic entity). Int J Dermatol 2002;41(1):8–15.
- Vazquez Garcia J, Perez Oliva N, Peireio Ferreiros MM, Toribio J. Lichen planus follicularis tumidus with cysts and comedones. Clin Exp Dermatol 1992;17(5):346–8.
- Isaac M, McNeely MC. Dermatitis herpetiformis associated with lichen planopilaris. J Am Acad Dermatol 1995;33(6):1050–1.
- Samman PD. The nails in lichen planus. Br J Dermatol 1961;73:288–92.
- Zaias N. The nail in lichen planus. Arch Dermatol 1970;101(3):264–71.
- Tosti A, Peluso AM, Fanti PA, Piraccini BM. Nail lichen planus: clinical and pathologic study of twenty-four patients. J Am Acad Dermatol 1993;28(5 Pt 1):724–30.
- Colver GB, Dawber RP. Is childhood idiopathic atrophy of the nails due to lichen planus? Br J Dermatol 1987;116(5):709–12.
- Tosti A, Piraccini BM, Cambiaghi S, Jorizzo M. Nail lichen planus in children: clinical features, response to treatment, and long-term follow-up. Arch Dermatol 2001;137(8):1027–32.
- Arias AM, Yung CW, Rendler S, Soltani K, Lorincz AL. Familial severe twenty-nail dystrophy. J Am Acad Dermatol 1982;7(3):349–52.
- Menni S, Piccinno R, Sala F, Crosti C, Dalpozzo V. Twenty-nail dystrophy of childhood – two cases in one family. Clin Exp Dermatol 1984;9(6):604–7.
- Tosti A, Bardazzi F, Piraccini BM, Fanti PA. Idiopathic trachyonychia (twenty-nail dystrophy): a pathological study of 23 patients. Br J Dermatol 1994;131(6):866–72.
- Taniguchi S, Kutsuna H, Tani Y, Kawahira K, Hamada T. Twenty-nail dystrophy (trachyonychia) caused by lichen planus in a patient with alopecia universalis and ichthyosis vulgaris. J Am Acad Dermatol 1995;33(5 Pt 2):903–5.
- Scheinfeld NS. Trachyonychia: a case report and review of manifestations, associations, and treatments. Cutis 2003;71(4):299–302.
- Sehgal VN. Twenty nail dystrophy trachyonychia: an overview. J Dermatol 2007;34(6):361–6.
- Baran R, Jancovici E, Sayag J, Dawber RP. Longitudinal melanonychia in lichen planus. Br J Dermatol 1985;113(3):369–70.
- Tosti A, Piraccini BM, Cameli N. Nail changes in lichen planus may resemble those of yellow nail syndrome. Br J Dermatol 2000;142(4):848–9.
- Baran R. Lichen planus of the nails mimicking the yellow nail syndrome. Br J Dermatol 2000;143(5):1117–18.
- Rode M, Kogoj-Rode M. Malignant potential of the reticular form of oral lichen planus over a 25-year observation period in 55 patients from Slovenia. J Oral Sci 2002;44(2):109–11.
- Reichart PA. [Oral precancerous conditions – an overview.] Mund Kiefer Gesichtschir 2003;7(4):201–7.
- Epstein JB, Wan LS, Gorsky M, Zhang L. Oral lichen planus: progress in understanding its malignant potential and the implications for clinical management. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2003;96(1):32–7.
- Van der Meij EH, Schepman KP, van der Waal I. The possible premalignant character of oral lichen planus and oral lichenoid lesions: a prospective study. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2003;96(2):164–71.
- Larsson A, Warfvinge G. Malignant transformation of oral lichen planus. Oral Oncol 2003;39(6):630–1.
- Tomb R, El-Hajj H, Nehme E, Haddad A. [Verrucous carcinoma of the tongue occurring on lesions of lichen planus.] Ann Dermatol Venereol 2003;130(1 Pt 1):55–7.
- Warshaw EM, Templeton SF, Washington CV. Verrucous carcinoma occurring in a lesion of oral lichen planus. Cutis 2000;65(4):219–22.
- Lanfranchi-Tizeira HE, Aguas SC, Sano SM. Malignant transformation of atypical oral lichen planus: a review of 32 cases. Med Oral 2003;8(1):2–9.
- Mignogna MD, Lo Russo L, Fedele S, Ruoppo E, Califano L, Lo Muzio L. Clinical behaviour of malignant transforming oral lichen planus. Eur J Surg Oncol 2002;28(8):838–43.
- Lysitsa S, Samson J, Gerber-Wicht C, Lang U, Lombardi T. COX-2 expression in oral lichen planus. Dermatology 2008;217(2):150–5.
- Sigurgeirsson B, Lindelof B. Lichen planus and malignancy. An epidemiologic study of 2071 patients and a review of the literature. Arch Dermatol 1991;127(11):1684–8.
- Dwyer CM, Kerr RE, Millan DW. Squamous carcinoma following lichen planus of the vulva. Clin Exp Dermatol 1995;20(2):171–2.
- Jones RW, Rowan DM, Kirker J, Wilkinson EJ. Vulval lichen planus: progression of pseudoepitheliomatous hyperplasia to invasive vulval carcinomas. Br J Obstet Gynaecol 2001;108(6):665–6.
- Hoshi A, Usui Y, Terachi T. Penile carcinoma originating from lichen planus on glans penis. Urology 2008;71(5):816–17.
- Thorne JE, Jabs DA, Nikolskaia OV, Mimouni D, Anhalt GJ, Nousari HC. Lichen planus and cicatrizing conjunctivitis: characterization of five cases. Am J Ophthalmol 2003;136(2):239–43.
- McNab AA. Lacrimal canalicular obstruction in lichen planus. Orbit 1998;17(3):201–2.
Associated conditions
- Cox NH, Finlay AY, Watkinson G. Atypical lichen planus associated with ulcerative colitis. Dermatologica 1986;173(6):294–6.
- Miller TN. Myasthenia gravis, ulcerative colitis and lichen planus. Proc R Soc Med 1971;64(8):807–8.
- Tan RS. Ulcerative colitis, myasthenia gravis, atypical lichen planus, alopecia areata, vitiligo. Proc R Soc Med 1974;67(3):195–6.
- Tan RS. Thymoma, acquired hypogammaglobulinaemia, lichen planus, alopecia areata. Proc R Soc Med 1974;67(3):196–8.
- Al-Najjar A, Reilly GD, Harrington C. Dermatomyositis and lichen planus – an association or manifestation? Clin Exp Dermatol 1985;10(2):174–8.
- Connelly MG, Winkelmann RK. Coexistence of lichen sclerosus, morphea, and lichen planus. Report of four cases and review of the literature. J Am Acad Dermatol 1985;12(5 Pt 1):844–51.
- Ng PP, Ng SK, Chng HH. Pemphigus foliaceus and oral lichen planus in a patient with systemic lupus erythematosus and thymoma. Clin Exp Dermatol 1998;23(4):181–4.
- Koga T, Kubota Y, Kiryu H, Nakayama J, Takeda S, Kono K. Late onset systemic lupus erythematosus with lichen planus-like eruption and cardiac tamponade. Eur J Dermatol 2000;10(8):620–2.
- Passeron T, Bahadoran P, Lacour JP, et al. Paraneoplastic pemphigus presenting as erosive lichen planus. Br J Dermatol 1999;140(3):552–3.
- Bowen GM, Peters NT, Fivenson DP, et al. Lichenoid dermatitis in paraneoplastic pemphigus: a pathogenic trigger of epitope spreading? Arch Dermatol 2000;136(5):652–6.
- Calista D. Oral erosive lichen planus associated with thymoma. Int J Dermatol 2001;40(12):762–4.
- Aronson IK, Soltani K, Paik KI, Rubenstein D, Lorincz AL. Triad of lichen planus, myasthenia gravis, and thymoma. Arch Dermatol 1978;114(2):255–8.
- Siponen M, Huuskonen L, Laara E, Salo T. Association of oral lichen planus with thyroid disease in a Finnish population: a retrospective case–control study. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2010;110(3):319–24.
- Mann RJ, Wallington TB, Warin RP. Lichen planus with late onset hypogammaglobulinaemia: a casual relationship? Br J Dermatol 1982;106(3):357–60.
- Powell FC, Rogers RS, 3rd, Dickson ER. Primary biliary cirrhosis and lichen planus. J Am Acad Dermatol 1983;9(4):540–5.
- Sarkany I. The skin–liver connection. Clin Exp Dermatol 1988;13(3):151–9.
- Chu CY, Yang CY, Huang SF, Lu SC, Wang LF. Lichen planus with xanthomatous change in a patient with primary biliary cirrhosis. Br J Dermatol 2000;142(2):377–8.
- Tong DC, Ferguson MM. Concurrent oral lichen planus and primary sclerosing cholangitis. Br J Dermatol 2002;147(2):356–8.
- Rebora A, Rongioletti F. Lichen planus and chronic active hepatitis. A retrospective survey. Acta Derm Venereol 1984;64(1):52–6.
- Rebora A. Hepatitis viruses and lichen planus. Arch Dermatol 1994;130(10):1328–9.
- Nagao Y, Tsubone K, Kimura R, et al. High prevalence of anticardiolipin antibodies in patients with HCV-associated oral lichen planus. Int J Mol Med 2002;9(3):293–7.
- Epstein O. Lichen planus and liver disease. Br J Dermatol 1984;111(4):473–5.
- Mobacken H, Nilsson LA, Olsson R, Sloberg K. Incidence of liver disease in chronic lichen planus of the mouth. Acta Derm Venereol 1984;64(1):70–3.
- El-Kabir M, Scully C, Porter S, Porter K, Macnamara E. Liver function in UK patients with oral lichen planus. Clin Exp Dermatol 1993;18(1):12–16.
- Anonide A, Rebora A. What lichen planus patients die of. A retrospective study. Int J Dermatol 1989;28(8):524–6.
- Romero MA, Seoane J, Varela-Centelles P, Diz-Dios P, Garcia-Pola MJ. Prevalence of diabetes mellitus amongst oral lichen planus patients. Clinical and pathological characteristics. Med Oral 2002;7(2):121–9.
- Ashinoff R, Cohen R, Lipkin G. Castleman's tumor and erosive lichen planus: coincidence or association? Report of a case. J Am Acad Dermatol 1989;21(5 Pt 2):1076–80.
- Hongcharu W, Baldassano M, Gonzalez E. Generalized lichen amyloidosis associated with chronic lichen planus. J Am Acad Dermatol 2000;43(2 Pt 2):346–8.
- Clarke J, Black MM. Lichenoid tattoo reactions. Br J Dermatol 1979;100(4):451–4.
- Winkelmann RK, Harris RB. Lichenoid delayed hypersensitivity reactions in tattoos. J Cutan Pathol 1979;6(1):59–65.
Investigations
- Lallas A, Kyrgidis A, Tzellos TG, et al. Accuracy of dermoscopic criteria for the diagnosis of psoriasis, dermatitis, lichen planus and pityriasis rosea. Br J Dermatol 2012;166(6):1198–205.
- Zalaudek I, Argenziano G. Dermoscopy subpatterns of inflammatory skin disorders. Arch Dermatol 2006;142(6):808.
- Ragaz A, Ackerman AB. Evolution, maturation, and regression of lesions of lichen planus. New observations and correlations of clinical and histologic findings. Am J Dermatopathol 1981;3(1):5–25.
- Patterson JW. The spectrum of lichenoid dermatitis. J Cutan Pathol 1991;18(2):67–74.
- Neppelberg E, Johannessen AC, Jonsson R. Apoptosis in oral lichen planus. Eur J Oral Sci 2001;109(5):361–4.
- Black MM, Wilson-Jones E. The role of the epidermis in the histopathogenesis of lichen planus. Histochemical correlations. Arch Dermatol 1972;105(1):81–6.
- Da Silva Fonseca LM, do Carmo MA. Identification of the AgNORs, PCNA and ck16 proteins in oral lichen planus lesions. Oral Dis 2001;7(6):344–8.
- Summerly R, Jones EW. The microarchitecture of Wickham's striae. Trans St Johns Hosp Dermatol Soc 1964;50:157–61.
- Ebner H, Erlach E, Gebbart W. [Blister formation in lichen planus: electronmicroscopical observations.] Arch Dermatol Forsch 1973;247(2):193–205.
- Ebner H, Gebhart W. Light and electron microscopic differentiation of amyloid and colloid or hyaline bodies. Br J Dermatol 1975;92(6):637–45.
- Ebner H, Gebhart W. Light and electron microscopic studies on colloid and other cytoid bodies. Clin Exp Dermatol 1977;2(4):311–22.
- Lupton GP, Goette DK. Lichen planus with plasma cell infiltrate. Arch Dermatol 1981;117(2):124–5.
- Roustan G, Hospital M, Villegas C, Sanchez Yus E, Robledo A. Lichen planus with predominant plasma cell infiltrate. Am J Dermatopathol 1994;16(3):311–14.
- Horn RT, Jr, Goette DK, Odom RB, Olson EG, Guill MA. Immunofluorescent findings and clinical overlap in two cases of follicular lichen planus. J Am Acad Dermatol 1982;7(2):203–7.
- Matta M, Kibbi AG, Khattar J, Salman SM, Zaynoun ST. Lichen planopilaris: a clinicopathologic study. J Am Acad Dermatol 1990;22(4):594–8.
- Abell E, Presbury DG, Marks R, Ramnarain D. The diagnostic significance of immunoglobulin and fibrin deposition in lichen planus. Br J Dermatol 1975;93(1):17–24.
- Shklar G. Erosive and bullous oral lesions of lichen planus: histologic studies. Arch Dermatol 1968;97(4):411–16.
- Karatsaidis A, Schreurs O, Helgeland K, Axell T, Schenck K. Erythematous and reticular forms of oral lichen planus and oral lichenoid reactions differ in pathological features related to disease activity. J Oral Pathol Med 2003;32(5):275–81.
- Odukoya O, Gallagher G, Shklar G. A histologic study of epithelial dysplasia in oral lichen planus. Arch Dermatol 1985;121(9):1132–6.
- Kulthanan K, Jiamton S, Varothai S, Pinkaew S, Sutthipinittharm P. Direct immunofluorescence study in patients with lichen planus. Int J Dermatol 2007;46(12):1237–41.
- Camisa C, Neff JC, Olsen RG. Use of indirect immunofluorescence in the lupus erythematosus/lichen planus overlap syndrome: an additional diagnostic clue. J Am Acad Dermatol 1984;11(6):1050–9.
- Mora RG, Nesbitt LT, Jr, Brantley JB. Lichen planus pemphigoides: clinical and immunofluorescent findings in four cases. J Am Acad Dermatol 1983;8(3):331–6.
- Kolde G, Wesendahl C, Stein H, Reichart PA. Oral lichen planus: diagnostic immunofluorescence testing on routine histological material. Br J Dermatol 2003;148(2):374–6.
- Oliver GF, Winkelmann RK, Muller SA. Lichenoid dermatitis: a clinicopathologic and immunopathologic review of sixty-two cases. J Am Acad Dermatol 1989;21(2 Pt 1):284–92.
- Jang KA, Kim SH, Choi JH, Sung KJ, Moon KC, Koh JK. Lichenoid keratosis: a clinicopathologic study of 17 patients. J Am Acad Dermatol 2000;43(3):511–16.
- Morgan MB, Stevens GL, Switlyk S. Benign lichenoid keratosis: a clinical and pathologic reappraisal of 1040 cases. Am J Dermatopathol 2005;27(5):387–92.
- Toll A, Gilaberte M, Gallardo F, Iglesias M, Barranco C, Pujol RM. Multiple and extensive lichen planus-like keratoses: an underestimated cutaneous eruption observed in patients with intense sun damage. J Eur Acad Dermatol Venereol 2006;20(4):472–3.
- Al-Hoqail IA, Crawford RI. Benign lichenoid keratoses with histologic features of mycosis fungoides: clinicopathologic description of a clinically significant histologic pattern. J Cutan Pathol 2002;29(5):291–4.
- Dalton SR, Baptista MA, Libow LF, Elston DM. Lichenoid tissue reaction in malignant melanoma: a potential diagnostic pitfall. Am J Clin Pathol 2002;117(5):766–70.
- Dalton SR, Fillman EP, Altman CE, et al. Atypical junctional melanocytic proliferations in benign lichenoid keratosis. Hum Pathol 2003;34(7):706–9.
Management
- Le Cleach L, Chosidow O. Clinical practice. Lichen planus. N Engl J Med 2012;366(8):723–32.
- Irvine C, Irvine F, Champion RH. Long-term follow-up of lichen planus. Acta Derm Venereol 1991;71(3):242–4.
- Carbone M, Arduino PG, Carrozzo M, et al. Course of oral lichen planus: a retrospective study of 808 northern Italian patients. Oral Dis 2009;15(3):235–43.
- Goettmann S, Zaraa I, Moulonguet I. Nail lichen planus: epidemiological, clinical, pathological, therapeutic and prognosis study of 67 cases. J Eur Acad Dermatol Venereol 2012;26(10):1304–9.
- Assouly P, Reygagne P. Lichen planopilaris: update on diagnosis and treatment. Semin Cutan Med Surg 2009;28(1):3–10.
- Cooper SM, Wojnarowska F. Influence of treatment of erosive lichen planus of the vulva on its prognosis. Arch Dermatol 2006;142(3):289–94.
- Cheng S, Kirtschig G, Cooper S, Thornhill M, Leonardi-Bee J, Murphy R. Interventions for erosive lichen planus affecting mucosal sites. Cochrane Database Syst Rev 2012;Issue 2:CD008092.
- Kellett JK, Ead RD. Treatment of lichen planus with a short course of oral prednisolone. Br J Dermatol 1990;123(4):550–1.
- Lodi G, Carrozzo M, Furness S, Thongprasom K. Interventions for treating oral lichen planus: a systematic review. Br J Dermatol 2012;166(5):938–47.
- Laurberg G, Geiger JM, Hjorth N, et al. Treatment of lichen planus with acitretin. A double-blind, placebo-controlled study in 65 patients. J Am Acad Dermatol 1991;24(3):434–7.
- Gonzalez E, Momtaz TK, Freedman S. Bilateral comparison of generalized lichen planus treated with psoralens and ultraviolet A. J Am Acad Dermatol 1984;10(6):958–61.
- Pavlotsky F, Nathansohn N, Kriger G, Shpiro D, Trau H. Ultraviolet-B treatment for cutaneous lichen planus: our experience with 50 patients. Photodermatol Photoimmunol Photomed 2008;24(2):83–6.
- Kanwar AJ, De D. Methotrexate for treatment of lichen planus: old drug, new indication. J Eur Acad Dermatol Venereol 2013;27(3):e410–13.
- Sehgal VN, Abraham GJ, Malik GB. Griseofulvin therapy in lichen planus. A double-blind controlled trial. Br J Dermatol 1972;87(4):383–5.
- Sehgal VN, Bikhchandani R, Koranne RV, Nayar M, Saxena HM. Histopathological evaluation of griseofulvin therapy in lichen planus. A double-blind controlled study. Dermatologica 1980;161(1):22–7.
- Omidian M, Ayoobi A, Mapar MA, Feily A, Cheraghian B. Efficacy of sulfasalazine in the treatment of generalized lichen planus: randomized double-blinded clinical trial on 52 patients. J Eur Acad Dermatol Venereol 2010;24(9):1051–4.
- Warnakulasuriya S, Kovacevic T, Madden P, et al. Factors predicting malignant transformation in oral potentially malignant disorders among patients accrued over a 10-year period in South East England. J Oral Pathol Med 2011;40(9):677–83.
- Voute AB, Schulten EA, Langendijk PN, Kostense PJ, van der Waal I. Fluocinonide in an adhesive base for treatment of oral lichen planus. A double-blind, placebo-controlled clinical study. Oral Surg Oral Med Oral Pathol 1993;75(2):181–5.
- Tyldesley WR, Harding SM. Betamethasone valerate aerosol in the treatment of oral lichen planus. Br J Dermatol 1977;96(6):659–62.
- Malhotra AK, Khaitan BK, Sethuraman G, Sharma VK. Betamethasone oral mini-pulse therapy compared with topical triamcinolone acetonide (0.1%) paste in oral lichen planus: a randomized comparative study. J Am Acad Dermatol 2008;58(4):596–602.
- Boisnic S, Branchet MC, Pascal F, Ben Slama L, Rostin M, Szpirglas H. [Topical tretinoin in the treatment of lichen planus and leukoplakia of the mouth mucosa. A clinical evaluation.] Ann Dermatol Venereol 1994;121(6–7):459–63.
- Giustina TA, Stewart JC, Ellis CN, et al. Topical application of isotretinoin gel improves oral lichen planus. A double-blind study. Arch Dermatol 1986;122(5):534–6.
- Petruzzi M, De Benedittis M, Grassi R, Cassano N, Vena G, Serpico R. Oral lichen planus: a preliminary clinical study on treatment with tazarotene. Oral Dis 2002;8(6):291–5.
- Buajeeb W, Kraivaphan P, Pobrurksa C. Efficacy of topical retinoic acid compared with topical fluocinolone acetonide in the treatment of oral lichen planus. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1997;83(1):21–5.
- Radfar L, Wild RC, Suresh L. A comparative treatment study of topical tacrolimus and clobetasol in oral lichen planus. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2008;105(2):187–93.
- Gorouhi F, Solhpour A, Beitollahi JM, et al. Randomized trial of pimecrolimus cream versus triamcinolone acetonide paste in the treatment of oral lichen planus. J Am Acad Dermatol 2007;57(5):806–13.
- Chainani-Wu N, Madden E, Lozada-Nur F, Silverman S, Jr. High-dose curcuminoids are efficacious in the reduction in symptoms and signs of oral lichen planus. J Am Acad Dermatol 2012;66(5):752–60.
- Nolan A, Badminton J, Maguire J, Seymour RA. The efficacy of topical hyaluronic acid in the management of oral lichen planus. J Oral Pathol Med 2009;38(3):299–303.
- Mousavi F, Sherafati S, Mojaver YN. Ignatia in the treatment of oral lichen planus. Homeopathy 2009;98(1):40–4.
- Agha-Hosseini F, Borhan-Mojabi K, Monsef-Esfahani HR, Mirzaii-Dizgah I, Etemad-Moghadam S, Karagah A. Efficacy of purslane in the treatment of oral lichen planus. Phytother Res 2010;24(2):240–4.
- Saawarn N, Shashikanth MC, Saawarn S, Jirge V, Chaitanya NC, Pinakapani R. Lycopene in the management of oral lichen planus: a placebo-controlled study. Indian J Dent Res 2011;22(5):639–43.
- Xiong C, Li Q, Lin M, et al. The efficacy of topical intralesional BCG-PSN injection in the treatment of erosive oral lichen planus: a randomized controlled trial. J Oral Pathol Med 2009;38(7):551–8.
- Wu Y, Zhou G, Zeng H, Xiong CR, Lin M, Zhou HM. A randomized double-blind, positive-control trial of topical thalidomide in erosive oral lichen planus. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2010;110(2):188–95.
- Choonhakarn C, Busaracome P, Sripanidkulchai B, Sarakarn P. The efficacy of aloe vera gel in the treatment of oral lichen planus: a randomized controlled trial. Br J Dermatol 2008;158(3):573–7.
- Salazar-Sanchez N, Lopez-Jornet P, Camacho-Alonso F, Sanchez-Siles M. Efficacy of topical aloe vera in patients with oral lichen planus: a randomized double-blind study. J Oral Pathol Med 2010;39(10):735–40.
- Mansourian A, Momen-Heravi F, Saheb-Jamee M, Esfehani M, Khalilzadeh O, Momen-Beitollahi J. Comparison of aloe vera mouthwash with triamcinolone acetonide 0.1% on oral lichen planus: a randomized double-blinded clinical trial. Am J Med Sci 2011;342(6):447–51.
- Sperling LC, Nguyen JV. Commentary: treatment of lichen planopilaris: some progress, but a long way to go. J Am Acad Dermatol 2010;62(3):398–401.
- Racz E, Gho C, Moorman PW, Noordhoek Hegt V, Neumann HA. Treatment of frontal fibrosing alopecia and lichen planopilaris: a systematic review. J Eur Acad Dermatol Venereol 2013;27(12):1461–70.
- Vano-Galvan S, Molina-Ruiz AM, Serrano-Falcon C, et al. Frontal fibrosing alopecia: a multicenter review of 355 patients. J Am Acad Dermatol 2014;70(4):670–8.
- Piraccini BM, Saccani E, Starace M, Balestri R, Tosti A. Nail lichen planus: response to treatment and long term follow-up. Eur J Dermatol 2010;20(4):489–96.
- Irla N, Schneiter T, Haneke E, Yawalkar N. Nail lichen planus: successful treatment with etanercept. Case Reports Dermatol 2010;2(3):173–6.
- Asarch A, Gottlieb AB, Lee J, et al. Lichen planus-like eruptions: an emerging side effect of tumor necrosis factor-alpha antagonists. J Am Acad Dermatol 2009;61(1):104–11.
- Becherel PA, Bussel A, Chosidow O, Rabian C, Piette JC, Frances C. Extracorporeal photochemotherapy for chronic erosive lichen planus. Lancet 1998;351(9105):805.
- Guyot AD, Farhi D, Ingen-Housz-Oro S, et al. Treatment of refractory erosive oral lichen planus with extracorporeal photochemotherapy: 12 cases. Br J Dermatol 2007;156(3):553–6.
- Wright AL, McVittie E, Hunter JA. An immunophenotypic study of lichen nitidus. Clin Exp Dermatol 1990;15(4):273–6.
- Arizaga AT, Gaughan MD, Bang RH. Generalized lichen nitidus. Clin Exp Dermatol 2002;27(2):115–17.
- Randle HW, Sander HM. Treatment of generalized lichen nitidus with PUVA. Int J Dermatol 1986;25(5):330–1.
- Kim YC, Shim SD. Two cases of generalized lichen nitidus treated successfully with narrow-band UV-B phototherapy. Int J Dermatol 2006;45(5):615–17.
- Do MO, Kim MJ, Kim SH, Myung KB, Choi YW. Generalized lichen nitidus successfully treated with narrow-band UVB phototherapy: two cases report. J Korean Med Sci 2007;22(1):163–6.
- Ocampo J, Torne R. Generalized lichen nitidus. Report of two cases treated with astemizol. Int J Dermatol 1989;28(1):49–51.
- Vaughn RY, Smith JG, Jr. The treatment of lichen nitidus with astemizole. J Am Acad Dermatol 1990;23(4 Pt 1):757–8.
- Lucker GP, Koopman RJ, Steijlen PM, van der Valk PG. Treatment of palmoplantar lichen nitidus with acitretin. Br J Dermatol 1994;130(6):791–3.
- Lang PG, Jr. Keratosis lichenoides chronica. Successful treatment with psoralen-ultraviolet-A therapy. Arch Dermatol 1981;117(2):105–8.
- Kunte C, Kerschenlohr K, Rocken M, Schirren C. Keratosis lichenoides chronica: treatment with bath-PUVA. Acta Derm Venereol 2007;87(2):182–3.
- Koseoglu RD, Sezer E, Yuksek J. Keratosis lichenoides chronica treated with acitretin plus narrowband ultraviolet B phototherapy. J Dermatol 2008;35(3):172–4.
- David M, Filhaber A, Rotem A, Katzenelson-Weissman V, Sandbank M. Keratosis lichenoides chronica with prominent telangiectasia: response to etretinate. J Am Acad Dermatol 1989;21(5 Pt 2):1112–14.
- Lopez-Navarro N, Alcaraz I, Bosch RJ, Tejera A, Herrera E. Keratosis lichenoides chronica: response to photodynamic therapy. J Dermatol Treat 2008;19(2):124–5.
- Grabbe S, Kolde G. Coexisting lichen planus and subacute cutaneous lupus erythematosus. Clin Exp Dermatol 1995;20(3):249–54.
- De Jong EM, Van De Kerkhof PC. Coexistence of palmoplantar lichen planus and lupus erythematosus with response to treatment using acitretin. Br J Dermatol 1996;134(3):538–41.
- Mora RG, Nesbitt LT, Jr, Brantley JB. Lichen planus pemphigoides: clinical and immunofluorescent findings in four cases. J Am Acad Dermatol 1983;8(3):331–6.
- Yoon KH, Kim SC, Kang DS, Lee IJ. Lichen planus pemphigoides with circulating autoantibodies against 200 and 180 kDa epidermal antigens. Eur J Dermatol 2000;10(3):212–14.
- Kolb-Maurer A, Sitaru C, Rose C, Brocker EB, Goebeler M, Zillikens D. [Treatment of lichen planus pemphigoides with acitretin and pulsed corticosteroids.] Hautarzt 2003;54(3):268–73.
Lichen striatus
- Patrizi A, Neri I, Fiorentini C, et al. Lichen striatus: clinical and laboratory features of 115 children. Pediatr Dermatol 2004;21:197–204.
- Peramiquel L, Baselga E, Dalmau J, et al. Lichen striatus: clinical and epidemiological review of 23 cases. Eur J Pediatr 2006;165:267–9.
- Muller CS, Schmaltz R, Vogt T, Pfohler C. Lichen striatus and blaschkitis: reappraisal of the concept of blaschkolinear dermatoses. Br J Dermatol 2011;164:257–62.
- Jo JH, Jang HS, Park HJ, et al. Early treatment of multiple and spreading lichen striatus with topical tacrolimus. J Am Acad Dermatol 2007;57:904–5.
- Yaosaka M, Sawamura D, Iitoyo M, et al. Lichen striatus affecting a mother and her son. J Am Acad Dermatol 2005;53:352–3.
- Racette AJ, Adams AD, Kessler SE. Simultaneous lichen striatus in siblings along the same Blaschko line. Pediatr Dermatol 2009;26:50–4.
- Hafner C, Landthaler M, Vogt T. Lichen striatus (blaschkitis) following varicella infection. J Eur Acad Dermatol Venereol 2006;20:1345–7.
- Feely MA, Silverberg NB. Two cases of lichen striatus with prolonged active phase. Pediatr Dermatol 2014;31:e67–8.
- Baran R, Dupre A, Lauret P, Puissant A. [Lichen striatus with nail involvement. Report of 4 cases and review of the 4 cases in the literature (author's transl)]. Ann Dermatol Venereol 1979;106:885–91.
- Campanati A, Brandozzi G, Giangiacomi M, et al. Lichen striatus in adults and pimecrolimus: open, off-label clinical study. Int J Dermatol 2008;47:732–6.
- Sorgentini C, Allevato MA, Dahbar M, Cabrera H. Lichen striatus in an adult: successful treatment with tacrolimus. Br J Dermatol 2004;150:776–7.
- Park JY, Kim YC. Lichen striatus successfully treated with photodynamic therapy. Clin Exp Dermatol 2012;37:570–2.