CHAPTER 48
Adamantiades–Behçet disease
Christos C. Zouboulis
Departments of DermatologyVenereology, Allergology and Immunology, Dessau Medical Center, Germany
Definition and nomenclature
Adamantiades–Behçet disease (ABD), otherwise known as Behçet disease, is a multisystem inflammatory disease of unknown aetiology, classified as a systemic vasculitis and as a neutrophilic dermatosis involving all types and sizes of blood vessels. It is characterized clinically by recurrent courses of oral aphthous ulcers, genital ulcers, skin lesions (papulopustules, erythema nodosum) and iridocyclitis/posterior uveitis. It is occasionally accompanied by arthritis and vascular, neurological, gastrointestinal or other manifestations [1, 2].
Epidemiology
Incidence and prevalence
ABD has a worldwide occurrence with varying prevalence, being endemic in eastern and central Asian and the eastern Mediterranean countries (along the so-called Silk Road) and rare in northern European countries, central and southern Africa, the Americas and Australia [3]. A prevalence of 80–420 patients per 100 000 inhabitants has been reported in Turkey, 7–30 patients per 100 000 inhabitants in the rest of the Asian continent (Japan: 14–31/100 000; Korea: 7.3/100 000; northern China: 14/100 000; Saudi Arabia: 20/100 000; Iran: 17/100 000) and 1.5–7.5/100 000 in southern Europe [3]. In northern Europe (0.27–1.18/100 000) and the USA (0.12–0.33/100 000) [3], the disease is rare. The increasing prevalence of the disease is due to its chronic character. Its annual incidence is low; 0.75–1.0 new cases per 100 000 inhabitants were assessed in Japan (1990) and Germany (2005) [4].
Age
ABD most often affects patients in their twenties and thirties; however, early and late onsets (first year of life to 72 years of age) have been reported. Juvenile disease rates are 2–21% in different ethnic groups; its prevalence was estimated to be 0.17/100 000 in France.
Sex
In contrast to old Japanese and Turkish reports of male predominance, the male to female ratio has decreased drastically in the last 20 years. In the years 1983–2012 significant proportional declines occurred with respect to male sex, complete type of the disease and the major presenting features (genital ulcers, ocular involvement and skin lesions) in 3674 hospital-based patients of the Korean registry, whereas the mean patient age rose progressively, as did the frequencies of joint, gastrointestinal and central nervous system manifestations [5]. Currently, both genders are equally affected; a male predominance is still observed in Arab populations, whereas female predominance is evident in Korea, China, some northern European countries and the USA.
Ethnicity
In countries with several ethnic populations, including such of Turkmen and Mongol descents, the latter are mainly affected.
Associated diseases
Recurrent aphthous stomatitis (benign aphthosis) is associated.
Pathophysiology
The aetiology of the disease remains unknown, although genetic factors, infectious agents, environmental pollution, immunological mechanisms and endothelial and clotting factors have been implicated and studied intensively [6, 7]. The endemic occurrence along the historical Silk Road, the major involvement of certain ethnic groups (mostly of Turkish and Mongol descent), and associated immunogenetic data support the hypothesis that the disease followed the migration of these old nomadic tribes. On the other hand, the wide variation of the disease prevalence in the same ethnic group in association with different geographical areas of residence indicates an additional environmental triggering factor. Therefore, transfer of genetic material and/or of an unknown exogenous agent may have been responsible for the expansion of the disease.
Pathology
Characteristic histopathological features of ABD are vasculitis and thrombosis (Figure 48.1). Biopsies from early mucocutaneous lesions show a neutrophilic vascular reaction with endothelial swelling, extravasation of erythrocytes, and leukocytoclasia or a fully developed leukocytoclastic vasculitis with fibrinoid necrosis of the blood vessel walls [1, 4]. Although there are reports of lesions that consist primarily of a lymphocytic perivasculitis, most of these lesions are likely to be older. The neutrophilic vascular reaction should be considered the predominant histopathological finding [8]. Aneurysms can also develop in large arteries as a result of vasculitis of the vasa vasorum with penetration of the lamina elastica.
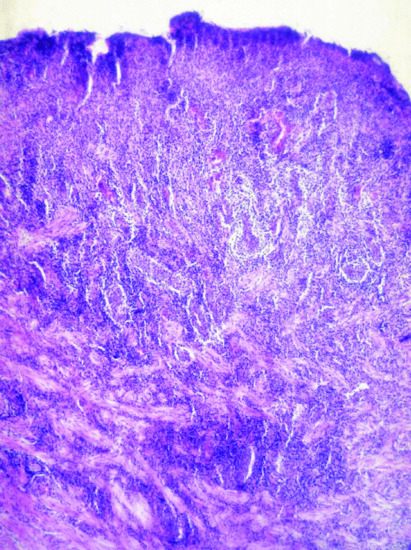
Figure 48.1 Abundant mixed inflammatory infiltrate dominated by neutrophils in an oral ulcer of Adamantiades–Behçet disease.
Causative organisms
ABD is not considered contagious as no horizontal transmission has ever been reported. However, viral and bacterial infections have been implicated in initiating immunopathological pathways, leading to the onset of the disease [6, 7].
Viral agents
Early theories of the pathogenesis of ABD proposed a viral or other infectious aetiology [3]. Partial transcription of herpes simplex virus type 1 (HSV-1) DNA in patients’ peripheral blood lymphocytes has been reported [6]. HSV-1 DNA was detected in patients’ saliva and oral and genital ulcers, and HSV-1 antibodies were found in patients’ sera.
Bacterial agents
Disease activity has been known to correlate with bacterial infection, particularly streptococci [6]. Streptococcus sanguinis dominates the flora of the oral mucosa in patients with the disease and appears to be the most relevant Streptococcus strain as a provoking factor for initiation of the disease [9]. Streptococcus antigens and antistreptococcal antibodies are frequently found in the oral mucosa and serum of patients. The involvement of immunoglobulin A protease-producing S. sanguinis is proposed as an explanation for a chronic infection leading to initiation of ABD. High titres of the immunogenic S. sanguinis antigen KTH-1 have been detected in patients. In addition, exposure of the patients to Streptococcus antigens may be a major provoking factor for disease activity [10]. The lipoprotein of Mycoplasma fermentas MALP-404 was found in the sera of patients with ABD but not in healthy controls [11]. Interestingly, MALP-404 contains a peptide motif, which can be presented by human leukocyte antigen B51 (HLA-B51). A possible role for bacterial stimulation of monocytes via Toll-like receptor 2 (TLR2) producing neutrophil-stimulating pro-inflammatory factors in ABD was detected. Moreover, markedly higher expression at the mRNA and protein level of TLR2, TLR3, TLR4 and TLR8 was observed in patients with active ABD as compared with controls [12].
Genetics
There is no specific mode of Mendelian transmission in ABD [6, 7]. Familial occurrence with regional differences has been reported, being more frequent in Korea (15%) than in Japan or China (2–3%), and in Arab countries, Israel and Turkey (2–18%) than in Europe (0–5%). An earlier disease onset in children compared with their parents and a higher frequency of familial cases in juveniles than in adults has been observed.
A significant association exists between the disease and HLA-B51 in Japan, the Middle East and the Mediterranean countries; however, this relationship is not as strong in western countries [13]. The allele also seems to be associated with a more severe prognosis [14]. Its exact role in the disease mechanism is still unknown, although it may be involved in disease development through specific antigen presentation, molecular mimicry with microbial antigens, or participation in linkage disequilibrium with a presently unknown susceptibility gene [15, 16]. Among the 24 described alleles, HLA-B5101 and -B5108 have most frequently been associated with ABD [17]. Shared amino acid residues (defining the Bw4 epitope) are crucial for antigen binding and natural killer cell interactions [18], and Bw4 was also reported to contribute to the severity of the disease [19]. Genes possibly associated with the disease have been localized on chromosome 6 in the region between the tumour necrosis factor (TNF) gene and HLA-B or -C genes, including the major histocompatibility complex class I chain A gene (A6 allele) and genes for heat shock proteins (HSPs) [7, 15, 17, 20]. In addition, a novel susceptibility locus mapped to 6p22–23 was detected [17]. Lately, associations on chromosomes 1p31.3 (interleukin 23R (IL-23R)/IL-12RB2) and 1q32.1 (IL-10) were found by genome-wide association studies [18, 21]. A haplotype association of IL-8 gene with ABD was also detected [22]. Polymorphisms in genes encoding for host effector molecules may contribute to the disease susceptibility to and/or severity of the disease, such as in IL-23R reported in a Chinese Han population [23]. New gene associations with ERAP-1, CCR1–CCR3, KLRC4 and STAT4 genes have been reported [24].
Immunogenetic factors
Immunological mechanisms are considered to play a major role in the pathogenesis of ABD [6, 7, 17, 20]. The disease has currently been classified among the autoinflammatory disorders [25].
Autoimmune mechanisms
The major microscopic finding at most sites of active disease is an immune-mediated occlusive vasculitis. The pathergy reaction (see the clinical features section) is induced by the rapid accumulation of neutrophils (hyperchemotaxis) and later by T lymphocytes and monocytes/macrophages at the needle-prick sites. T cells in the peripheral blood and in the involved tissues are increased, and a predominant T-helper 1 immune response induced by IL-12 has been demonstrated [26]. Patients’ lymphocytes also express CD29 molecules and bind to endothelial cells in active disease. In addition to defective T-cell immunity, B-cell activation is impaired. Circulating immune complexes, together with enhanced neutrophil migration, may be involved; the diversity of T cells indicates that specific T-cell responses to several antigens may lead to the variety of symptoms [27]. Tropomyosins and the 160 kDa polypeptide kinectin have been detected as autoantigens in ABD [28, 29].
Heat shock proteins
Increased levels of HSP-specific antibodies in serum have been found in ABD [30, 31]. T cells respond to 60 kDa HSP, and four different peptide determinants within 60 kDa HSP identified by T-cell epitope mapping have been suggested to be involved in the pathogenesis of the disease.
Cytokine mediators
Various proinflammatory cytokines, such as IL-1, -8, -12, -17, -22, -23 and -33 and TNF-α are elevated in the sera of patients with active ABD [32–40]. In particular, IL-8 seems to play an important role, can also be released by endothelial cells, has a potent effect on the inflammatory response, and is a sensitive marker of disease activity [32–34, 38, 40]. Cytokine release may be dependent on the involved organ [30, 31, 33, 37].
Endothelial cells
The endothelium seems to be the primary target, or it may only be involved as an innocent bystander of pathological changes of the immune system [41]. An immunoglobulin M type, 47 kDa cell surface HSP against endothelial α-enolase has been identified in the serum of patients with ABD [10, 42]. Plasma endothelin 1 concentrations were found to be significantly increased, perhaps indicating vasoconstriction and being the direct result of elevated synthesis by injured vascular endothelial cells. Thrombomodulin, a cell surface glycoprotein of vascular endothelium, which is also increased in the plasma of patients with active disease, potentially damages the endothelial cells.
Clinical features
History
Hippocrates of Kos (460–377 BC) used the designation , (oral aphthous ulcers) in a probable first description of a patient with the disease (Epidemion book III, case 7). The disease is named after Benediktos Adamantiades, a Greek ophthalmologist, and Hulûsi Behçet, a Turkish dermatologist, who, in 1931 and 1937 respectively, described patients with the characteristic clinical complex and argued for this to be a single clinical entity [43]. The first international multidisciplinary conference was organized by two dermatologists, M. Monacelli and P. Nazarro, in 1964 in Rome, Italy.
Presentation
ABD is a chronic, recurrent, multisystem and, occasionally, life-threatening disorder [1, 4]. Recurrent oral aphthous ulcers, recurrent genital ulcers, skin manifestations, ocular lesions and arthritis/arthropathy are the most frequent clinical features. Vascular, neurological, gastrointestinal, psychiatric, pulmonary, renal and cardiac manifestations, epididymitis and other findings can also occur. The clinical picture usually develops within a few months after the presenting sign. Both an acute multisystem presentation and long-term development of the disease over years are possible.
Diagnosis of ABD is based on clinical signs as neither a pathognomonic laboratory test nor histological characteristics are available. There are several sets of diagnostic criteria, the most popular of them being the criteria of the International Study Group [44] and those of the Behçet's Disease Research Committee of Japan [45]. However, there have been several problems with these criteria, including their performance in selectivity and specificity, so that both of them have been revised. The currently published Revised International Criteria for Behçet's Disease provide the most accurate diagnosis (Table 48.1) [46].
Table 48.1 Revised international criteria for Behçet disease [46]
| Clinical feature | Pointsb |
| Ocular lesions (recurrent) | 2 |
| Oral aphthosis (recurrent) | 2 |
| Genital aphthosis (recurrent) | 2 |
| Skin lesions (recurrent) | 1 |
| Central nervous system lesions | 1 |
| Vascular manifestations | 1 |
| Positive pathergy testb | 1 |
aScoring: a score ≥4 indicates Adamantiades–Behçet disease.
bThough the main scoring system does not include pathergy test, where pathergy testing is conducted, a positive result may be included for one extra point.
Clinical variants
Mucocutaneous lesions
Recurrent oral aphthous and genital ulcers are the most frequently observed mucosal manifestations. Oral aphthous ulcers are the presenting sign in more than 80% of patients (see Figure 48.1) [1, 4, 20]. Although recurrent aphthous stomatitis is a common disorder, only a few patients progress to ABD, and it is not possible to determine in whom or when the transition may occur [47]. Typically, lesions are multiple, painful, 1–3 cm in diameter and sharply margined with a fibrin-coated base and surrounding erythema (Figure 48.2). Oral aphthous ulcers usually heal without scarring (92%). Genital ulcers may not recur as often and usually heal with a characteristic scar (64–88%) (Figure 48.3). Spontaneous healing of aphthae occurs within 4 days to 1 month; genital ulcers may persist longer. Large oral ulcerations can also be associated with problems such as pharyngeal involvement, dysphagia, and dyspnoea or fistulae involving the pharynx, larynx, trachea or oesophagus. Genital ulcers can occur on the penis, scrotum, vagina, labia and urethra, and also in the anal, perineal and inguinal regions.
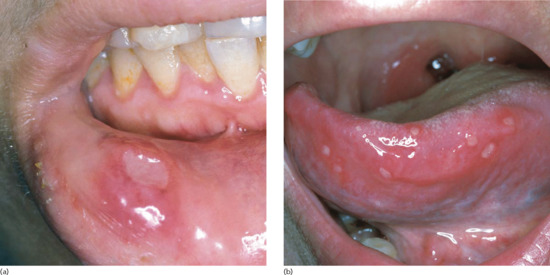
Figure 48.2 Single (a) and multiple (b) oral aphthous ulcers. (Part (a) from Altenburg et al. 2006 [4].
Reproduced with permission of John Wiley & Sons.)
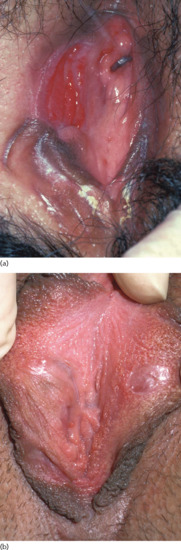
Figure 48.3 Genital ulcer (a) healing with a demarcated flat scar (b).
Skin lesions that should be accepted as diagnostically relevant in ABD should be confined to pustular vasculitic lesions (including pathergy lesions), erythema nodosum-like lesions, Sweet disease-like lesions, pyoderma gangrenosum-like lesions and palpable purpuric lesions of necrotizing venulitis. All lesions are characterized in their early stages by a neutrophilic vascular reaction. Acneform lesions or follicle-based pustules should not be considered relevant [8].
Systemic lesions
Ocular involvement is the major cause of morbidity in patients with ABD. The most diagnostically relevant lesion is posterior uveitis (also called retinal vasculitis), which can lead to blindness (Figure 48.4). Other ocular lesions include anterior uveitis, hypopyon (pus in the anterior chamber of the eye, which is now – due to early treatment – uncommon) and secondary complications such as cataract, glaucoma and neovascular lesions [48]. Retinal inflammation can lead to vascular occlusion and, ultimately, tractional retinal detachment. Severe vitreous involvement, chronic cystoid macular oedema and possible – presumably also vasculitic – involvement of the optic nerve can result in loss of vision. Recurrent vasculitic changes can ultimately lead to ischaemic optic nerve atrophy.
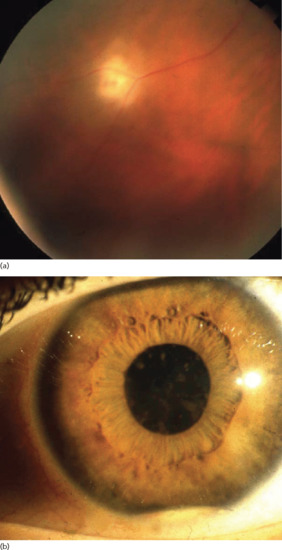
Figure 48.4 (a) Posterior uveitis. (b) Hypopyoniritis. (From Altenburg et al. 2006 [4].
Reproduced with permission of John Wiley & Sons.)
The characteristic arthritis is a non-erosive, asymmetrical, sterile, seronegative oligoarthritis; however, symmetrical polyarticular involvement is common. Joint manifestations frequently occur first in one knee or ankle and then the other as a migratory monoarthritis, then in both joints simultaneously, and finally affecting nearly all joints. An HLA-B27-positive erosive sacroiliitis has to be excluded.
Systemic vascular involvement can be significant and includes venous occlusions and varices, arterial occlusions and aneurysms, often being migratory. Cases of large-vein thrombosis (inferior vena cava, cranial venous sinuses) or large-artery aneurysms are potentially fatal [1, 4]. Arterial involvement is rather rare and usually presents in the form of thromboses and, less often, of aneurysms, resulting from multicentric arteritis. Pulmonary artery aneurysms are the principal feature of pulmonary involvement in ABD, occasionally resulting in coughing and haemoptysis. Cardiac involvement can include myocarditis, coronary arteritis, endocarditis and valvular disease. A wide spectrum of renal manifestations can occur, varying from minimal change disease to proliferative glomerulonephritis and rapidly progressive crescentic glomerulonephritis. Immune complex deposition is thought to be responsible for the underlying pathogenesis in some cases of glomerulonephritis. Gastrointestinal complaints can be a symptom for aphthae throughout the gastrointestinal tract and can rarely result in perforation and peritonitis (0.5%). Inflammatory bowel disease has to be excluded. Sterile prostatitis and epididymitis can be present in male patients without genital ulcers.
Significant neurological manifestations occur in approximately 10% of patients and may be delayed in onset. Meningoencephalitis, cerebral venous sinus thrombosis, benign intracranial hypertension, cranial nerve palsies, brainstem lesions and pyramidal or extrapyramidal lesions have been described. Poor prognosis is associated with a progressive course, relapses after treatment, repeated attacks and cerebellar symptoms or parenchymal disease. Neurological manifestations usually present with severe headache. Further symptoms include gait disturbance, dysarthria, vertigo and diplopia, as well as hyperreflexia, epileptic seizures, hemiplegia, ataxia or a positive Babinski reflex. Psychiatric symptoms, such as depression, insomnia or memory impairment, are also signs of neurological involvement.
Differential diagnosis [4, 49]
There are many possible differential diagnoses (Box 48.1).
Classification of severity
A severe course, including blindness, meningoencephalitis, haemoptysis, intestinal perforation and severe arthritis, occurs in approximately 10% of patients. Lethal outcome has been seen in 0–6% of affected patients in different ethnic groups. Central nervous system and pulmonary and large vessel involvement, as well as bowel perforation, are the major life-threatening complications; death may also result as a complication of immunosuppressive therapy.
Complications and co-morbidities
Ophthalmic and neurological sequelae are leading causes of morbidity, followed by severe vascular and gastrointestinal manifestations. Their effects on morbidity may be cumulative.
Disease course and prognosis
The clinical course of ABD is variable. There can be a delay of up to several years before the diagnosis is made, and this may influence the prognosis [50]. Mucocutaneous and joint manifestations usually occur first. Recurrent erythema nodosum and HLA-B51 positivity are risk factors for the development of superficial thrombophlebitis and vision loss; superficial thrombophlebitis, ocular lesions and male gender are risk factors for the development of systemic vessel involvement [14, 51, 52]. Blindness can often be prevented with early aggressive therapy of posterior uveitis. Markers of severe prognosis include HLA-B51 positivity, male gender and early development of systemic signs [14]. Onset in childhood does not necessarily predict a poor prognosis. Spontaneous remissions of certain or all manifestations of the disease have been observed. In a 3-year follow-up comparison of Turkish patients with ocular involvement during the years 1990–1994 and 2000–2004 [53], disease was milder at referral – as has been reported for Korean patients [5]. Posterior segment involvement was less common and visual acuity was better. The follow-up revealed no significant difference in number of uveitis attacks in the first 3 years, but fewer eyes lost useful vision, no patient became legally blind and fewer severe ocular complications occurred in the 2000s [53].
Investigations
Pathergy test
A positive pathergy test (hyperreactivity reaction) manifests within 48 h as an erythematous papule (>2 mm) or pustule at the site of a skin needle prick or after intracutaneous injection of 0.1 mL isotonic salt solution using a 20-gauge needle without prior disinfection of the injection site. The skin prick is generally placed at an angle of 45 degrees 3–5 mm intracutaneously on the volar forearm. Erythema without infiltration is considered a negative finding. Provoked oral aphthae and genital ulcers after injection or injury (such as chorioretinitis in the corneal region of the eye after photocoagulation of the ocular fundus region) can also be considered as positive pathergy phenomena. Broader pathergy phenomena also include the occurrence of aneurysms around vascular anastomoses as well as a local recurrence of ulcers after resection of affected bowel segments. Although a positive pathergy reaction is a sign of ABD, it is not pathognomonic, as it can also occur in patients with pyoderma gangrenosum, rheumatoid arthritis, Crohn disease and genital herpes infection.
Radiological findings
Scintigraphic evidence of arthritis is found in 50% of the patients [4]. Cranial magnetic resonance imaging allows documentation of hypodense or atrophic changes in the brain. Electroencephalographic detection of diffuse α-waves is considered a positive finding. Vascular lesions can be detected by angiography.
Management
The choice of treatment for patients with ABD depends on the site and severity of the clinical manifestations of the disease [54]. Recurrent aphthae are most often treated with palliative agents, such as mild diet, avoidance of irritating agents and potent topical glucocorticoids and local anaesthetics [55, 56, 57–61]. Lately, topical hyaluronic acid 0.2% gel applied twice a day over 30 days has been found to be effective (Box 48.2) [58]. For the topical treatment of genital ulcers and skin lesions, corticosteroid and antiseptic creams can be applied for up to 7 days. Painful genital ulcerations can be managed by topical anaesthetics in cream. Corticosteroid injections (triamcinolone acetonide 0.1–0.5 mL/lesion) can be helpful in recalcitrant ulcerations. They can also be beneficial on panuveitis and cystoid macular oedema as a single intravitreal injection (triamcinolone acetonide 4 mg) [59, 60].
Patients with mucocutaneous lesions resistant to topical treatment, those with systemic involvement, and patients with markers of poor prognosis are candidates for systemic treatment [54, 61–64]. Several compounds have been found to be effective in randomized, double-blind, placebo-controlled trials [56, 64, 65–89] (Table 48.2). Additional treatments have been successful in studies with a lower grade of evidence (Table 48.3) [2, 20, 54, 90–106]. Oral and intravenous prednisolone can be combined with other immunosuppressants, colchicine, dapsone, sulfasalazine or interferon-α (IFN-α). A synergistic effect with ciclosporin has been described in patients with ocular involvement. Prednisolone is one of the few medications that can be used during pregnancy. Colchicine can be combined with immunosuppressants and IFN-α. A rapid relapse often occurs after discontinuing dapsone, ciclosporin, IFN-α or infliximab [74, 91, 93, 98].
Table 48.2 Systemic treatment of Adamantiades–Behçet disease with grade A evidence (randomized, double-blind, placebo-controlled trial against placebo except otherwise mentioned)
| Drug and reference | Dose | Indications |
| Methylprednisolone [65] | 40 mg/every 3 weeks IM | Erythema nodosum (in females) but not oro-genital ulcers) |
| Rebamipide [66] | 300 mg/day PO (caveat: pregnancy, lactation) | Oral ulcers |
| Colchicine [67] | 1 mg/day PO (caveat: pregnancy, lactation – induces oligozoospermia) | Oral aphthous ulcers, genital ulcers, folliculitis, erythema nodosum |
| Colchicine [68] | 1.5 mg/day PO | Erythema nodosum and arthralgia |
| Colchicine [69] | 1-2 mg/day PO | Genital ulcers, erythema nodosum, arthritis (females),arthritis (males) |
| Colchicine [70] | Ineffective: does not reduce the use of immunosuppressives | |
| Colchicine versus colchicine + benzathine penicillin [71] | 1–2 mg/day PO; 1.2 mega units × 3/week IM | Combined treatment more effective in reducing frequency of arthritic episodes, duration and frequency of oral ulcers and erythema nodosum, and the frequency of genital ulcers |
| Colchicine versus colchicine + benzathine penicillin [72] | 1 mg/day PO;1.2 mega units/month IM | Combined treatment more effective than colchicine or penicillin alone |
| Zinc sulphate [73] | 300 mg/day PO | Mucocutaneous lesions |
| Dapsone [74] | 100 mg/day PO (caveat: pregnancy, lactation; methaemoglobin increase: ascorbic acid 500 mg/day) | Oral ulcers, genital ulcers, skin lesions, pathergy test, arthritis, epididymitis |
| Azathioprine [75] | 2.5 mg/kg/day PO (caveat: pregnancy, lactation, severe liver disease, bone marrow depression, severe infection, children) | Oral ulcers, genital ulcers, arthritis, recent-onset ocular disease. Prevents the development of new eye lesions |
| Interferon-α 2a [76] | 6 × 106 IU × 3/week SC (caveat: pregnancy, lactation – induces psychotic signs, psoriasis, myopathy) | Oral ulcers, genital ulcers, papulopustular lesions, erythema nodosum, articular symptoms |
| Interferon-α [77] | 1000 and 2000 IU/day PO | Ineffective |
| Thalidomide [78] | 100 mg/day or 300 mg/day PO (caveat: pregnancy, lactation – induces polyneuropathy; minimized at 25 mg/day) | Oral ulcers, genital ulcers, papulopustular lesions |
| Ciclosporin versus colchicine [79] | 10 mg/kg/day PO; 1 mg/day PO (caveat: lactation, renal insufficiency – induces pathological central nervous system findings) | Ocular manifestations, oral ulcers, skin lesions, genital ulcers |
| Ciclosporin versus conventional treatments (prednisolone, chlorambucil) [80] | 10 mg/kg/day PO | Ocular attacks (conventional therapy superior in controlling oral ulcers, genital ulcers and arthritis) |
| Ciclosporin versus conventional treatments (prednisolone, chlorambucil) [81] | 10 mg/kg/day PO | Hearing loss (25%) |
| Ciclosporin versus conventional treatments (prednisolone, azathioprine) [81] | 5 mg/kg/day PO | Oral ulcers, genital ulcers, cutaneous lesions, thrombophlebitis, articular and neurological symptoms |
| Ciclosporin versus cyclophosphamide pulses [83] | 5 mg/kg/day PO | Visual acuity |
| Cyclophosphamide + corticosteroids versus corticosteroids [84] | 1 g/month IV; 2 g/month IV | Combined more effective in eye disease |
| Etanercept [85] | 25 mg × 2/week PO (caveat: pregnancy, lactation) | Oral ulcers, nodular skin lesions, papulopustular lesions (not pathergy test) |
| Rituximab versus cytotoxic combination therapy [86] | Two 1000 mg PO courses (15-day interval) | Significant improvement in total adjusted disease activity index |
| Aciclovir [87] | 800 mg × 5/day for 1 week + 400 mg × 2/day PO for 11 weeks | Ineffective |
| Azapropazone [88] | 900 mg/day over 3 weeks PO | Arthritis |
| Daclizumab (anti-CD25) [89] | 1 mg/kg/fortnight IV | Ineffective (eye disease) |
Table 48.3 Systemic treatment of Adamantiades–Behçet disease with grade B evidence (well-conducted open clinical trial) or grade C evidence (small open clinical triall)
| Drug | Dose | Indication |
| Corticosteroids [2, 20, 54] | 5–60 mg/day prednisolone equivalent PO100–1000 mg/day over 1–3 days IV (alone or in combinations) (caveat: diabetes – induces psychosis) | Active diseaseAcute exacerbation (particularly uveitis, neurological manifestations) |
| Indometacin [2, 20] | 100 mg/day PO | Mucocutaneous lesions, arthritis |
| Pentoxifylline [54] | 300–400 mg ×1–3/day PO | Oral ulcers (particularly in children) |
| Irsogladine [90] | 2–4 mg/day PO | Recurrent aphthous ulcers |
| Ciclosporin [54, 91] | 3–6 mg/kg/day PO (serum levels: 100–150 ng/mL) (caveat: lactation, renal insufficiency – induces pathologic CNS findings) | Uveitis, mucocutaneous signs, thrombophlebitis, acute hearing loss |
| Sulfasalazine [54] | 1.5–3 g/day PO | Gastrointestinal ulcers |
| Thalidomide [54, 92] | 2 mg/kg/day PO; increased to 3 mg/kg/day if necessary or decreased to 1–0.5 mg/kg/day PO according to response(caveat: neurotoxicity) | Intestinal involvement (in children) |
| Tacrolimus [54] | 0.05–0.2 mg/kg/day PO (serum levels: 15–25 ng/mL) | Refractory uveitis |
| Interferon-α [93, 95] | 9 × 106 IU × 3/week or 3–9 × 106 IU × 5/week SC (3 × 106 IU × 3/week SC maintenance dose) (caveat: pregnancy, lactation – induces psychotic signs, psoriasis, myopathy)1.5–3 × 106 IU × 3/week SC according to body weight | Ocular lesions, long-term visual prognosis, arthritis, vascular lesionsCortico-dependent uveitis in children |
| Cyclophosphamide [2, 20, 54] | 1 g/month IV bolus (caveat: haemorrhagic cystitis give mesna 200 mg) | Uveitis, neurological manifestations |
| Cyclophosphamide + azathioprine + prednisolone [96] | Cyclophosphamide 1 g/month IV (for 6 months); azathioprine 2–3 mg/kg/day IV (for 2–3 months); prednisolone 0.5 mg/kg/day IV (for 2–3 months) | Improvement of visual acuity (44%), active posterior uveitis (73%) and retinal vasculitis (70%) of eyes. Improvement of total adjusted disease activity index in 72% of patients |
| Chlorambucil [54] | 0.1 mg/day PO (2 mg/day maintenance dose) (caveat: cumulative toxicity) | Neurological manifestations, uveitis, thrombosis, mucocutaneous lesions |
| Methotrexate [2, 20] | 7.5–20 mg × 1/week PO (caveat: pregnancy, lactation, severe bone marrow depression, liver dysfunction, acute infections, gastrointestinal ulcers, kidney insufficiency) | Severe mucocutaneous lesions, arthritis, progressive psychosis or dementia |
| Methotraxate + prednisolone [97] | 7.5–15 mg × 1/week PO; 0.5 mg/kg/day PO | Improvement of visual acuity (46.5%), active posterior uveitis (75.4%) and retinal vasculitis (53.7%) of eyes. Improvement of total inflammatory activity index in 74% and total adjusted disease activity index in 69.4% of patients |
| Infliximab [98–104] | 5 mg/kg IV on days 1, 7, 14 and 28 and every 2 weeks/4 weeks/6 weeks subsequently (caveat: pregnancy, lactation) | Acute uveitis, refractory posterior uveitis, neurological manifestations, intestinal involvement |
| Adalimumab [105] | 40 mg/fortnight SC | Refractory ocular lesions |
| Gevokizumab (anti-IL1β) [106] | 0.3 mg/kg IV | Severe uveitis |
Prevention
Patients with severe or progressive recurrent aphthous stomatitis should be followed up for years as potential candidates for ABD, particularly those patients with familial occurrence of the disease.
Patients with suspected ABD should be referred early for specialist advice. Male patients with systemic involvement as a presenting sign and/or an early age of onset should be treated systemically because of the poor prognosis.
References
- McCarty MA, Garton RA, Jorizzo JL. Complex aphthosis and Behçet's disease. Dermatol Clin 2003;21:418.
- Suzuki Kurokawa M, Suzuki N. Behçet's disease. Clin Exp Med 2004;4:10–20.
- Zouboulis CC. Epidemiology of Adamantiades–Behçet's disease. In: Zierhut M, Ohno S, eds. Immunology of Behçet's Disease. Lisse: Swets and Zeitlinger, 2003:1–16.
- Altenburg A, Papoutsis N, Orawa H, et al. Epidemiology and clinical manifestations of Adamantiades–Behçet disease in Germany – current pathogenetic concepts and therapeutic possibilities. J Dtsch Dermatol Ges 2006;4:49–66.
- Kim DY, Choi MJ, Cho S, et al. Changing clinical expression of Behçet disease in Korea during three decades (1983–2012): chronological analysis of 3674 hospital-based patients. Br J Dermatol 2014;170:458–61.
- Zouboulis CC, May T. Pathogenesis of Adamantiades–Behçet's disease. Med Microbiol Immunol 2003;192:149–55.
- Hirohata S, Kikutchi H. Behçet's disease. Arthritis Res Ther 2003;5:139–46.
- Jorizzo JL, Abernethy JL, White WL, et al. Mucocutaneous criteria for the diagnosis of Behçet's disease: an analysis of clinicopathologic data from multiple international centers. J Am Acad Dermatol 1995;32:968–76.
- Kaneko, F, Tojo M, Sato, M, et al. The role of infectious agents in the pathogenesis of Behçet's disease. Adv Exp Med Biol 2003;528:181–3.
- Cho S, Zheng Z, Cho SB, et al. Streptococcus sanguinis and the sera of patients with Behçet's disease stimulate membrane expression of α-enolase in human dermal microvascular endothelial cells. Arch Dermatol Res 2013;305:223–32.
- Zouboulis CC, Turnbull JR, Mühlradt PF. High seroprevalence of anti-Mycoplasma fermentans antibodies in patients with malignant aphthosis. J Invest Dermatol 2003;121:211–12.
- Liu X, Wang C, Ye Z, et al. Higher expression of Toll-like receptors 2, 3, 4, and 8 in ocular Behcet's disease. Invest Ophthalmol Vis Sci 2013;54:6012–17.
- De Menthon M, Lavalley MP, Maldini C, et al. HLA-B51/B5 and the risk of Behçet's disease: a systematic review and meta-analysis of case-control genetic association studies. Arthritis Rheum 2009;61:1287–96.
- Zouboulis CC, Turnbull JR, Martus P. Univariate and multivariate analyses comparing demographic, genetic, clinical, and serological risk factors for severe Adamantiades–Behçet's disease. Adv Exp Med Biol 2003;528:123–6.
- Fietta P. Behçet's disease: familial clustering and immunogenetics. Clin Exp Rheumatol 2005;23(Suppl. 38):S96–105.
- Durrani K, Papaliodis GN. The genetics of Adamantiades–Behcet's disease. Semin Ophthalmol 2008;23:73–9.
- Zierhut M, Mizuki N, Ohno S, et al. Immunology and functional genomics of Behçet's disease. Cell Mol Life Sci 2003;60:1903–22.
- Remmers EF, Cosan F, Kirino Y, et al. Genome-wide association study identifies variants in the MHC class I, IL10, and IL23R-IL12RB2 regions associated with Behçet's disease. Nat Genet 2010;42:698–702.
- Papoutsis N, Bonitsis N, Altenburg A, et al. HLA-antigens and their importance as prognostic-marker in Adamantiades–Behçet's disease (ABD) – is HLA-Bw4 a new prognostic marker? Abstracts of the 14th International Conference on Behçet's disease, London, 2010:163.
- Escudier M, Bagan J, Scully C. Behçet's disease (Adamantiades syndrome). Oral Dis 2006;12:78–84.
- Mizuki N, Meguro A, Ota M, et al. Genome-wide association studies identify IL23R-IL12RB2 and IL10 as Behçet's disease susceptibility loci. Nat Genet 2010;42:703–6.
- Lee EB, Kim JY, Zhao J, et al. Haplotype association of IL-8 gene with Behcet's disease. Tissue Antigens 2007;69:128–32.
- Jiang Z, Yang P, Hou S, et al. IL-23R gene confers susceptibility to Behcet's disease in a Chinese Han population. Ann Rheum Dis 2010;69:1325–8.
- Hatemi G, Seyahi E, Fresko I, et al. Behçet's syndrome: a critical digest of the 2012–2013 literature. Clin Exp Rheumatol 2013;31(Suppl. 77):108–17.
- Gül A. Behçet's disease as an autoinflammatory disorder. Curr Drug Targets Inflamm Allergy 2005;4:81–3.
- Yanagihori H, Oyama N, Nakamura K, et al. Role of IL-12B promoter polymorphism in Adamantiades–Behçet's disease susceptibility: an involvement of Th1 immunoreactivity against streptococcus sangunis antigen. J Invest Dermatol 2006;126:1534–40.
- Freysdottir J, Hussain L, Farmer I, et al. Diversity of γδ T cells in patients with Behçet's disease is indicative of polyclonal activation. Oral Dis 2006;12:271–7.
- Mahesh SP, Li Z, Buggage R, et al. Alpha tropomyosin as a self-antigen in patients with Behçet's disease. Clin Exp Immunol 2005;140:368–75.
- Lu Y, Ye P, Chen S-L, et al. Identification of kinectin as a novel Behçet's disease autoantigen. Arthritis Res Ther 2005;7:R1133–9.
- Direskeneli H, Saruhan-Direskeneli G. The role of heat shock proteins in Behçet's disease. Clin Exp Rheumatol 2003;21:S44–8.
- Birtas-Atesoglu E, Inanc N, Yavuz S, et al. Serum levels of free heat shock protein 70 and anti-HSP70 are elevated in Behçet's disease. Clin Exp Rheumatol 2008;26(Suppl. 50):S96–8.
- Katsantonis J, Adler Y, Orfanos CE, et al. Adamantiades–Behçet's disease: serum IL-8 is a more reliable marker for disease activity than c-reactive protein and erythrocyte sedimentation rate. Dermatology 2000;201:37–9.
- Durmazlar SP, Ulkar GB, Eskioglu F, et al. Significance of serum interleukin-8 levels in patients with Behcet's disease: high levels may indicate vascular involvement. Int J Dermatol 2009;48:259–64.
- Polat M, Vahaboglu G, Onde U, et al. Classifying patients with Behçet's disease for disease severity, using a discriminating analysis method. Clin Exp Dermatol 2009;34:151–5.
- Chi W, Zhu X, Yang P, et al. Upregulated IL-23 and IL-17 in Behçet patients with active uveitis. Invest Ophthalmol Vis Sci 2008;49:3058–64.
- Habibagahi Z, Habibagahi M, Heidari M. Raised concentration of soluble form of vascular endothelial cadherin and IL-23 in sera of patients with Behçet's disease. Mod Rheumatol 2010;20:154–9.
- Na SY, Park MJ, Park S, et al. Up-regulation of Th17 and related cytokines in Behçet's disease corresponding to disease activity. Clin Exp Rheumatol 2013;31(Suppl. 77):32–40.
- Cai T, Wang Q, Zhou Q, et al. Increased expression of IL-22 is associated with disease activity in Behcet's disease. PLOS One 2013;8:e59009.
- Sugita S, Kawazoe Y, Imai A, et al. Role of IL-22- and TNF-α-producing Th22 cells in uveitis patients with Behçet's disease. J Immunol 2013;190:5799–808.
- Kim D-J, Baek S-Y, Park M-K, et al. Serum level of interleukin-33 and soluble ST2 and their association with disease activity in patients with Behcet's disease. J Korean Med Sci 2013;28:1145–53.
- Kalayciyan A, Zouboulis CC. An update on Behçet's disease. J Eur Acad Dermatol Venereol 2007;21:1–10.
- Lee KH, Chung HS, Kim HS, et al. Human alpha-enolase from endothelial cells as a target antigen of anti-endothelial cell antibody in Behçet's disease. Arthritis Rheum 2003;48:2025–35.
- Zouboulis CC, Keitel W. A historical review of early descriptions of Adamantiades–Behçet's disease. J Invest Dermatol 2002;119:201–5.
- International Study Group for Behçet's Disease. Criteria for diagnosis of Behçet's disease. Lancet 1990;335:1078–80.
- Kaneko F, Oyama T, Sato M, et al. Behçet's Disease and Diseases for its Differential Diagnosis in Dermatology. Annual Report of the Behçet's Disease Research Committee of Japan. Tokyo: Behçet's Disease Research Committee of Japan, 1999.
- International Team for the Revision of the International Criteria for Behçet's Disease (ITR-ICBD). The International Criteria for Behçet's Disease (ICBD): a collaborative study of 27 countries on the sensitivity and specificity of the new criteria. J Eur Acad Dermatol Venereol 2013;28:338–47.
- Oh SH, Han EC, Lee JH, et al. Comparison of the clinical features of recurrent aphthous stomatitis and Behçet's disease. Clin Exp Dermatol 2009;34:e208–12.
- Krause L. Morbus Adamantiades–Behcet. Ophthalmologe 2005;102:329–34.
- Rogers RS, 3rd. Pseudo-Behçet's disease. Dermatol Clin 2003;21:49–61.
- Zouboulis CC, Kötter I, Djawari D, et al. Current epidemiological data from the German Registry of Adamantiades–Behçet's disease. Adv Exp Med Biol 2003;528:43–8.
- Sakamoto M, Akazawa K, Nisioka Y, et al. Prognostic factors of vision in patients with Behçet disease. Ophthalmology 1995;102:317–21.
- Coskun B, Öztürk P, Saral Y. Are erythema nodosum-like lesions and superficial thrombophlebitis prodromal in terms of visceral involvement in Behçet's disease? Int J Clin Pract 2005;59:69–71.
- Cingu AK, Onal S, Urgancioglu M, et al. Comparison of presenting features and three-year disease course in Turkish patients with Behçet uveitis who presented in the early 1990s and the early 2000s. Ocul Immunol Inflamm 2012;20:423–8.
- Bonitsis NG, Altenburg A, Krause L, et al. Current concepts in the treatment of Adamantiades–Behçet's disease. Drugs Fut 2009;34:749–63.
- Zouboulis CC. Adamantiades–Behçet's disease. In: Katsambas AD, Lotti TM, eds. European Handbook of Dermatological Treatments, 2nd edn. Berlin: Springer, 2003:16–26.
- Alpsoy E. New evidence-based treatment approach in Behçet's disease. Pathol Res Intern 2012;2012:871019.
- Altenburg A, Abdel-Naser B, Abdallah M, et al. Practical aspects of management of recurrent aphthous stomatitis. J Eur Acad Dermatol Venereol 2007;21:1019–26.
- Lee JH, Jung JY, Bang D. The efficacy of topical 0.2% hyaluronic acid gel on recurrent oral ulcers: comparison between recurrent aphthous ulcers and the oral ulcers of Behçet's disease. J Eur Acad Dermatol Venereol 2008;22:590–5.
- Atmaca LS, Yalçindağ FN, Ozdemir O. Intravitreal triamcinolone acetonide in the management of cystoid macular edema in Behçet's disease. Graefes Arch Clin Exp Ophthalmol 2007;245:451–6.
- Tuncer S, Yilmaz S, Urgancioglu M, et al. Results of intravitreal triamcinolone acetonide (IVTA) injection for the treatment of panuveitis attacks in patients with Behçet disease. J Ocul Pharmacol Ther 2007;23:395–401.
- Altenburg A, Micheli CK, Maldini C, et al. Klinik und Therapie chronisch rezidivierender Aphthen. Hautarzt 2012;63:693–703.
- Pipitone N, Olivieri I, Cantini F, et al. New approaches in the treatment of Adamantiades–Behçet's disease. Curr Opin Rheumatol 2006;18:3–9.
- Hatemi G, Silman A, Bang D, et al. Management of Behçet disease: a systematic literature review for the European League Against Rheumatism evidence-based recommendations for the management of Behçet disease. Ann Rheum Dis 2009;68:1528–34.
- Comarmond C, Wechsler B, Cacoub P, et al. Traitement de la maladie de Behçet. Rev méd interne 2014;35126–38.
- Mat C, Yurdakul S, Uysal S, et al. A double-blind trial of depot corticosteroids in Behçet's syndrome. Rheumatology (Oxford) 2006;45:348–52.
- Matsuda T, Ohno S, Hirohata S, et al. Efficacy of rebamipide as adjunctive therapy in the treatment of recurrent oral aphthous ulcers in patients with Behçet's disease: a randomised, double-blind, placebo-controlled study. Drugs R D 2003;4:19–28.
- Davatchi F, Sadeghi Abdollahi B, Tehrani Banihashemi A, et al. Colchicine versus placebo in Behçet's disease: randomized, double-blind, controlled crossover trial. Mod Rheumatol 2009;19:542–9.
- Aktulga E, Altac M, Muftuoglu A, et al. A double blind study of colchicine in Behcet's disease. Haematologica 1980;65:399–402.
- Yurdakul S, Mat C, Tüzün Y, et al. A double-blind trial of colchicine in Behçet's syndrome. Arthritis Rheum 2001;44:2686–92.
- Hamuryudan V, Hatemi G, Tascilar K, et al. Colchicine in Behcet syndrome: a longterm survey of patients in a controlled trial. J Rheumatol 2014;41:735–8.
- Çalgüneri M, Ertenli I, Kiraz S, et al. Effect of prophylactic benzathine penicillin on mucocutaneous symptoms of Behçet's disease. Dermatology 1996;192:125–8.
- Al-Waiz MM, Sharquie KE, A-Qaissi MH, et al. Colchicine and benzathine penicillin in the treatment of Behçet disease: a case comparative study. Dermatology Online J 2005;11(3):3.
- Sharquie KE, Najim RA, Al-Dori WS, et al. Oral zinc sulfate in the treatment of Behçet's disease: a double blind cross-over study. J Dermatol 2006;33:541–6.
- Sharquie KE, Najim RA, Abu-Raghif AR. Dapsone in Behçet's disease: a double-blind, placebo-controlled, cross-over study. J Dermatol 2002;29:267–79.
- Yazici H, Pazarli H, Barnes CG, et al. A controlled trial of azathioprine in Behçet's disease. N Engl J Med 1990;322:281–5.
- Alpsoy E, Durusoy C, Yilmaz E, et al. Interferon alfa-2a in the treatment of Behçet's disease: a randomized placebo-controlled and double-blind study. Arch Dermatol 2002;138:467–71.
- Kiliç H, Zeytin HE, Korkmaz C, et al. Low-dose natural human interferon-alpha lozenges in the treatment of Behçet's syndrome. Rheumatology (Oxford) 2009;48:1388–91.
- Hamuryudan V, Mat C, Saip S, et al. Thalidomide in the treatment of the mucocutaneous lesions of the Behçet syndrome. A randomized, double-blind, placebo-controlled trial. Ann Intern Med 1998;128:443–50.
- Masuda K, Nakajima A, Urayama A, et al. Double-masked trial of cyclosporin versus colchicine and long-term open study of cyclosporin in Behçet's disease. Lancet 1989;1(8647):1093–6.
- BenEzra D, Cohen E, Chajek T, et al. Evaluation of conventional therapy versus cyclosporine A in Behcet's syndrome. Transplant Proc 1988;20(Suppl. 4):136–43.
- Elidan J, Cohen E, Levi H, et al. Effect of cyclosporine A on the hearing loss in Behçet's disease. Ann Otol Rhinol Laryngol 1991;100464–8.
- Assaad-Khalil SH. Low-dose cyclosporin in Behçet's disease: follow-up controlled study with emphasis on extraocular manifestations and neuro-Behçet's disease. In: O'Duffy JD, Kokmen E, eds. Behçet's Disease: Basic and Clinical Aspects. New York: Marcel Dekker, 1991:603–12.
- Ozyazgan Y, Yurdakul S, Yazici H, et al. Low dose cyclosporin A versus pulsed cyclophosphamide in Behcet's syndrome: a single masked trial. Br J Ophthalmol 1992;76:241–3.
- Davatchi F, Shahram F, Chams H, et al. Pulse cyclophosphamide (PCP) for ocular lesions of Behçet's disease: double blind crossover study. Arthritis Rheum 1999;42:S320.
- Melikoglu M, Fresko I, Mat C, et al. Short-term trial of etanercept in Behçet's disease: a double blind, placebo controlled study. J Rheumatol 2005;32:98–105.
- Davatchi F, Shams H, Rezaipoor M, et al. Rituximab in intractable ocular lesions of Behçet's disease; randomized single-blind control study (pilot study). Int J Rheum Dis 2010;13:246–52.
- Davies UM, Palmer RG, Denman AM. Treatment with acyclovir does not affect orogenital ulcers in Behcet's syndrome: a randomized double-blind trial. Br J Rheumatol 1998;27:300–2.
- Moral F, Hamuryudan V, Yurdakul S, et al. Inefficacy of azapropazone in the acute arthritis of Behcet's syndrome: a randomized, double blind, placebo controlled study. Clin Exp Rheumatol 1995;13:493–5.
- Buggage RR, Levy-Clarke G, Sen HN, et al. A double-masked, randomized study to investigate the safety and efficacy of daclizumab to treat the ocular complications related to Behçet's disease. Ocul Immunol Inflamm 2007;15:63–70.
- Nanke Y, Kamatani N, Okamoto T, et al. Irsogladine is effective for recurrent oral ulcers in patients with Behçet's disease: an open-label, single-centre study. Drugs R D 2008;9:455–9.
- Zouboulis, CC. Morbus Adamantiades–Behçet. In: Mrowietz U, ed. Ciclosporin in der Dermatologie. Stuttgart: Thieme, 2003:38–51.
- Yasui K, Uchida N, Akazawa Y, et al. Thalidomide for treatment of intestinal involvement of juvenile-onset Behçet disease. Inflamm Bowel Dis 2008;14:396–400.
- Zouboulis CC, Orfanos CE. Treatment of Adamantiades–Behçet's disease with systemic interferon alfa. Arch Dermatol 1998;134:1010–16.
- Krause L, Altenburg A, Pleyer U, et al. Longterm visual prognosis of patients with ocular Adamantiades–Behçet's disease treated with interferon-alpha-2a. J Rheumatol 2008;35:896–903.
- Guillaume-Czitrom S, Berger C, Pajot C, et al. Efficacy and safety of interferon-alpha in the treatment of corticodependent uveitis of paediatric Behcet's disease. Rheumatology (Oxford) 2007;46:1570–3.
- Davatchi F, Sadeghi Abdollahi B, Shams H, et al. Combination of pulse cyclophosphamide and azathioprine in ocular manifestations of Behcet's disease: longitudinal study of up to 10 years. Int J Rheum Dis 2014;17:444–52.
- Davatchi F, Shams H, Shahram F, et al. Methotrexate in ocular manifestations of Behcet's disease: a longitudinal study up to 15 years. Int J Rheum Dis 2013;16:568–77.
- Sfikakis PP, Markomichelakis N, Alpsoy E, et al. Anti-TNF therapy in the management of Behçet's disease: review and basis for recommendations. Rheumatology (Oxford) 2007;46:736–41.
- Niccoli L, Nannini C, Benucci M, et al. Long-term efficacy of infliximab in refractory posterior uveitis of Behcet's disease: a 24-month follow-up study. Rheumatology (Oxford) 2007;46:1161–4.
- Tognon S, Graziani G, Marcolongo R. Anti-TNF-alpha therapy in seven patients with Behcet's uveitis: advantages and controversial aspects. Ann NY Acad Sci 2007;1110:474–84.
- Pipitone N, Olivieri I, Padula A, et al. Infliximab for the treatment of neuro-Behçet's disease: a case series and review of the literature. Arthritis Rheum 2008;59:285–90.
- Naganuma M, Sakuraba A, Hisamatsu T, et al. Efficacy of infliximab for induction and maintenance of remission in intestinal Behçet's disease. Inflamm Bowel Dis 2008;14:1259–64.
- Sakai T, Watanabe H, Kuroyanagi K, et al. Health- and vision-related quality of life in patients receiving infliximab therapy for Behcet uveitis. Br J Ophthalmol 2013;97:338–42.
- Ha LJ, Hee CJ, Woo JS, et al. Efficacy of infliximab in intestinal Behcet's disease: a Korean multicenter retrospective study. Inflamm Bowel Dis 2013;19:1833–8.
- Perra D, Alba MA, Callejas JL, et al. Adalimumab for the treatment of Behçet's disease: experience in 19 patients. Rheumatology 2012;51:1825–31.
- Gül A, Tugal-Tutkun I, Dinarello CA, et al. Interleukin-1-regulating antibody XOMA 052 (gevokizumab) in the treatment of acute exacerbations of resistant uveitis of Behcet's disease: an open-label pilot study. Ann Rheum Dis 2012;71:563–6.