CHAPTER 50
Immunobullous Diseases
Enno Schmidt1 and Richard Groves2
1Department of Dermatology, Lübeck Institute of Experimental Dermatology, University of LLübeck, LLübeck, Germany
2Clinical Immunodermatology, St John's Institute of Dermatology, Guy's and St Thomas' NHS Foundation Trust, London, UK
Introduction
The immunobullous disorders represent a group of conditions characterized by antibody-mediated autoimmune responses against structural elements of the skin resulting in blistering of the skin and mucosae. Antibody targets include proteins in the hemidesmosomes and the basement membrane zone (pemphigoid group), desmosomes (pemphigus group) and epidermal and tissue-type transglutaminase (dermatitis herpetiformis). If left untreated, immunobullous disorders may be associated with significant morbidity and mortality and thus prompt accurate diagnosis and treatment are mandatory. Therapeutic options include corticosteroids, steroid-sparing immunosuppressants such as azathioprine and mycophenolates and immunomodulating agents, e.g. dapsone and tetracyclines. In severe and/or refractory patients, intravenous immunoglobulin, immunoadsorption, and the anti-B-cell drug rituximab are applied.
INTRAEPIDERMAL IMMUNOBULLOUS DISEASES
Pemphigus
Definition
Pemphigus is a group of chronic autoimmune blistering diseases characterized by the presence of antibodies against desmosomal adhesion proteins.
Introduction and general description
The term ‘pemphigus’ derives from the Greek pemphix meaning blister or bubble. A number of distinct subgroups of pemphigus have emerged and their relevant autoantigens have been characterized. The primary pathogenic antibodies are directed against desmogleins 1 and 3, members of the cadherin family of calcium-dependent cell–cell adhesion molecules that contribute to the structure of the desmosome. Characteristic clinical and immunopathological features of the various pemphigus disorders are summarized in Tables 50.1 and 50.2.
Table 50.1 The intraepidermal immunobullous diseases: characteristic clinical features
| Disease | Patients | Cutaneous distribution | Mucosal involvement | Lesions | Disease associations | Treatment | Prognosis |
| Pemphigus vulgaris | Middle age | Scalp, face, upper trunk, may be generalized | Oro-pharynx, conjunctiva, genital | Flaccid blisters, erosions, flexural vegetations | Autoimmune disease, thymoma | Steroids, cytotoxic immunosuppressants, rituximab | Variable, may remit |
| Pemphigus vegetans | Middle age | Flexural | Oro-pharynx, conjunctiva, genital | Vegetating plaques, vesicles, pustules, erosions | As pemphigus vulgaris | Steroids, cytotoxic immunosuppressants, acitretin, rituximab | Variable, may remit |
| Pemphigus foliaceus | Middle age | Scalp, face, chest, upper back, rarely generalized ‘seborrhoeic’ | None | Scaly papules, crusted erosions, erythroderma | As pemphigus vulgaris | Steroids, cytotoxic immunosuppressants, rituximab | Benign but chronic |
| Endemic pemphigus foliaceus | Children, young adults | Head, neck, generalized | None | Flaccid blisters, erosions, verrucous lesions, erythroderma | Steroids, cytotoxic immunosuppressants | Better when move from rural areas | |
| Intercelluar IgA dermatosis | Adults, children | Axillae, groins, face, scalp, proximal limbs | None | Flaccid pustules annular or circinate configuration | IgA monoclonal gammopathy | Dapsone, steroids, cytotoxic immunosuppressants | Chronic indolent |
| Paraneoplastic pemphigus | Adults, children | Upper body, palmoplantar | Severe mucositis | Polymorphous bullae, erosions, ‘target lesions’ | Lymphoproliferative disease, Castleman, other malignancies | Tumour resection, steroids, cytotoxic immunosuppressants, rituximab | Poor |
Table 50.2 The intraepidermal immunobullous diseases: immunopathology and immunogenetics
| Disease | Direct IMF | Isotype | Target antigens | Antigens (kDa) | Epitopes | Location | Immunogenetics |
| Pemphigus vulgaris/pemphigus vegetans | Intercellular | IgG (occasionally IgM, IgA), C3 | Desmoglein 3, sometimes desmoglein 1 desmocollins | 130 | Amino-terminal of extracellular domain | Desmosome | DRB1*0402 DRB1*1401 |
| Pemphigus foliaceus | Intercellular | IgG, C3 | Desmoglein 1, sometimes desmocollins | 160 | Amino-terminal of extracellular domain | Desmosome | HLA-DRB1*14 |
| Endemic pemphigus foliaceus | Intercellular | IgG, IgM | Desmoglein 1, sometimes desmocollins | 160 | Amino-terminal of extracellular domain | Desmosome | Several susceptibility alleles, all with same amino acid sequence in DRB-1 gene DRB1*0102 DRB1*0404, *1402 or *1406 |
| Paraneoplastic pemphigus | Intercellular and subepidermal | IgG | Multiple (desmogleins, desmoplakin, envoplakin, periplakin, BP230) | Various | Various | Desmosomes, BMZ; stratified, simple and transitional epithelia | Unknown |
BMZ, basement zone; IMF, immunofluorescence.
Epidemiology
Incidence and prevalence
The incidence of pemphigus is low but variable worldwide, ranging from 0.05 to 2.7/100 000/year [1, 2]. Pemphigus vulgaris (PV) is generally the commoner form, though there is some geographical variation in the incidence of the different subtypes; thus PV is more common in Europe, the US and India whereas pemphigus foliaceus (PF) is more common in Brazil and Africa. Recent data from the UK suggest that the incidence of PV may be increasing [3], though the reasons behind this are uncertain.
Age
PV can occur at any age but is usually seen between the fourth and sixth decades of life [4]. However, there is some variability in different countries. In Iran, North India and Pakistan, patients with PV have a relatively low age of onset of disease (mean c.40 years) [5–7]. In endemic PF in Tunisia, young women tend to be affected [10]. Although pemphigus is rare in childhood it has been reported in children as young as 3 years of age [8]. Children usually develop PV rather than PF [9] and the disease may be severe [4].
Sex
Pemphigus seems to affect men and women equally though some studies have suggested a slight female preponderance [7, 10, 11].
Ethnicity
PV has been reported in all ethnic groups, but is more common in Ashkenazi Jews, Mediterranean, Iranian and Indian populations [12]. Genetic variations are likely to play a major role.
Associated diseases
PV has been associated with many other autoimmune diseases, particularly thyroid disease and rheumatoid arthritis [13]. A novel disease cluster of PV, thyroid disease, rheumatoid arthritis and type 1 diabetes has recently been described [14]. Paraneoplastic pemphigus occurs in association with haematological malignancy and is described in more detail later.
Pathophysiology
Pemphigus target antigens and the desmoglein compensation hypothesis
The principal target antigens in pemphigus are desmogleins (Dsg) 1 and 3, which are expressed in the skin and mucosal tissue (reviewed in [15]; see Chapter 2). However, the distribution of the two proteins varies in different epithelia, such that in skin there is a high level expression of Dsg 1 throughout the epidermis whereas Dsg 3 is found only in the basal and immediate suprabasal layers. In oral epithelium, Dsg 3 is expressed through all layers whereas Dsg 1 is only present at low levels. Dsg 1 and 3 act cooperatively and consequently antibodies against Dsg 1 are unable to destabilize oral mucosa whereas they have a marked effect in the skin where there is little Dsg 3 to compensate [16]. In contrast, antibodies against Dsg 3 lead to severe oral mucosal blistering as there is little Dsg1. Consequently, patients with PF, who have anti-Dsg 1 antibodies alone, exhibit skin blistering without mucosal involvement. In PV, where anti-Dsg 3 antibodies predominate there is generally marked mucosal blistering.
Pemphigus antibodies
The predominant class of tissue-bound pemphigus antibody in pemphigus is immunoglobulin G (IgG), which can be demonstrated on direct and indirect immunofluorescence (IF) testing (Figure 50.1). Less commonly, IgM, IgA and IgE antibodies are present. There is now considerable evidence confirming the pathogenicity of the IgG fraction of pemphigus sera. Consistent with this, early observations suggested that there was a correlation between intercellular antibody titre and severity of disease [17]. Subsequently, murine passive transfer experiments with purified IgG autoantibodies derived from pemphigus sera were shown to induce blister development with histological and immunological features typical of pemphigus [18]. In vitro, IgG purified from pemphigus patient sera demonstrates a direct effect on dissociation of keratinocyte monolayer cultures [19].
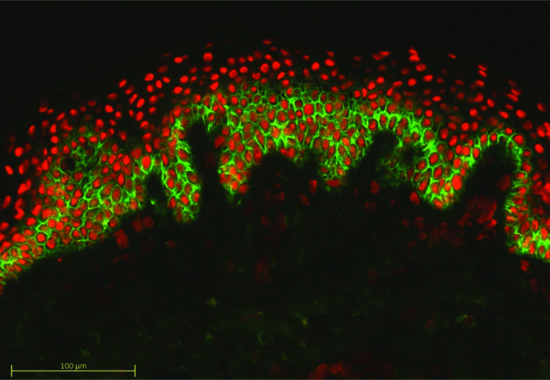
Figure 50.1 Direct immunofluorescence of pemphigus vulgaris. Antibody is deposited around the cell membrane of epidermal keratinocytes.
Although there is a large body of evidence that supports the role of Dsg 1 and 3 as targets for pathogenic antibodies in pemphigus, other antigenic targets have been implicated. Thus some patients have active pemphigus with intercellular antibodies detectable on direct or indirect IF but have negative Dsg antibodies as assessed by specific enzyme-linked immunosorbent assay (ELISA) assays. One explanation for this is that in some patients there may be significant non-Dsg targets and antibodies against desmocollins, plakoglobin, E-cadherin and acetylcholine receptors have been reported [20]. Indeed, acetylcholine receptor antibodies have been found in up to 85% of pemphigus patients [21]. These antibodies may weaken desmosomal junctions by inducing phosphorylation of adhesion molecules and prevent desmosomal reassembly.
In paraneoplastic pemphigus (reviewed in [22]) there is particularly good evidence that antibodies develop against multiple epidermal antigens in addition to Dsg 1 and 3. Not only are these antibodies directed against desmosomal proteins such as desmoplakins 1 and 2, envoplakin and periplakin but also constituents of the hemidesmosome including the 230 kDa bullous pemphigoid (BP) antigen 1. In consequence, direct IF of tissue from paraneoplastic pemphigus patients has features of pemphigus as well as basement membrane labelling (Figure 50.2).
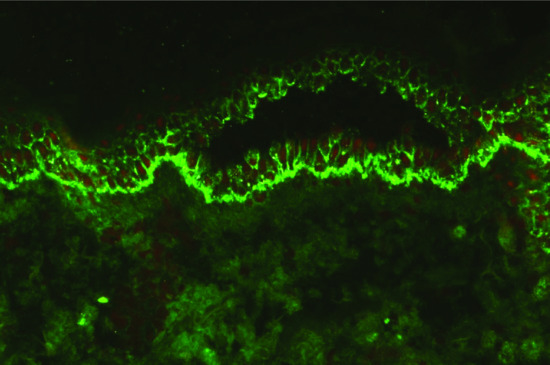
Figure 50.2 Direct immunofluorescence of paraneoplastic pemphigus. Intercellular antibody deposition is seen as in pemphigus vulgaris but in addition there is labelling of the basement membrane zone as a result of the broad spectrum antibody response.
Acantholysis
The key pathological process in PV is separation of keratinocytes from one another, a change known as acantholysis. Whilst acantholysis is not pathognomonic of pemphigus, all subtypes of the disease demonstrate it to some degree. There are a number of mechanisms that have been suggested to be involved including steric hindrance by anti-Dsg antibodies [23], protease activation and disruption of intracellular signalling pathways [24]. Apoptosis may also play a role and can be detected in pemphigus skin lesions [25] and is induced by PV-IgG in cultured keratinocytes [26]. Pemphigus antibodies can trigger secretion of factors from keratinocytes which are involved in apoptosis such as Fas ligand. However, evidence of apoptosis is seen late in the development of pemphigus lesions and after acantholysis. More recently, a new term ‘apoptolysis’ has been proposed to explain acantholysis and keratinocyte damage in pemphigus [27]. It is suggested that there are a series of events that follow binding of autoantibodies to pemphigus antigens resulting in epidermal growth factor receptor activation with initiation of cell death cascades, basal cell shrinkage, degradation of structural proteins and apoptosis of acantholytic cells.
The true mechanism for acantholysis is most likely a combination of multiple molecular and structural events and further work is needed to clarify this.
Genetics
The genetic basis of pemphigus is complex. Familial cases have been reported but are few [28, 29]. The presence of low titres of anti-Dsg antibodies in healthy relatives of patients with PV has been shown to be as high as 70% in some studies [30, 31] suggesting genetic control of the relevant immune response.
The primary genetic association in pemphigus in a number of populations appears to be with class II HLA alleles. Many population studies have shown strong associations between PV and HLA DRB1*04 and *14 alleles and with DQB1*0503 and *0302 [12] (see Table 50.2). Some class I HLA genes have been implicated in PV including HLA-A10 and HLA-B38 [32, 33].
Interestingly, Dsg 3 polymorphisms have been observed in association with HLA class II pemphigus susceptibility alleles and it has been suggested that this may contribute to development of PV [34]. Other non-HLA genes which have been implicated include the immunoglobulin heavy chain [35] and pemphigus-relevant cytokine genes such as interleukin 10 (IL-10) [36].
Although the major histocompatibility complex (MHC) clearly has a role in pemphigus susceptibility, this alone cannot explain disease development as (i) not all pemphigus patients have the disease-associated alleles; and (ii) healthy relatives of patients may have the relevant HLA allele(s) but do not develop disease. This supports the notion that pemphigus is most likely a multifactorial disease with a polygenic genetic component.
Environmental factors
A role for environmental factors in the pathogenesis of pemphigus is underscored by the recognition of an endemic form of the disease (fogo selvagem, reviewed in [37–39]) in several parts of the world, most notably rural Brazil and Tunisia. The disease is clinically and immunological very similar to PF, though it tends to affect children and young adults rather than the older population affected by sporadic PF. The majority of patients live near rivers and black flies (Simulium spp.) have been thought to be involved in disease pathogenesis. In endemic areas, as many as 50% of normal individuals have anti-Dsg type 1 antibodies.
A number of reports have suggested that smoking may have a protective or beneficial role in pemphigus [40–42]. Human keratinocytes have both nicotinic and muscarinic receptors for acetylcholine [43] and these receptors may play a role in regulating keratinocyte cell–cell adhesion.
Pesticides have also been postulated as possible triggers in disease development and an increased risk of pemphigus has been shown in exposed individuals [41]. Organophosphate pesticides block the acetylcholine breakdown pathway and so may lead to acetylcholine accumulation with resulting loss of cell–cell adhesion in the epidermis [44]. Pesticides implicated in contact pemphigus include glycophosate [45] and dihydrodiphenyltrichlorethane [46].
A link between diet and disease development in pemphigus has been suggested but difficult to prove. Although garlic has been proposed as a trigger for disease development [47] – and has been shown to induce acantholysis in vitro [48] – this area remains controversial.
Drug-induced pemphigus
Drug-induced pemphigus is rare. Most cases have been in association with drugs containing a thiol group such as penicillamine although non-thiol drugs including angiotensin-converting enzyme inhibitors and glibenclamide have also been implicated [49]. An additional group of drugs that may be responsible for the induction of pemphigus are those with a phenol group such as cephalosporins, rifampicin, pyritinol, phenobarbital and aspirin. Both PF and PV subtypes can occur.
Penicillamine is the most common culprit in drug-induced pemphigus and the disease may occur in 3–10% of patients on the drug, typically after around 1 year of exposure [50]. Penicillamine-induced pemphigus tends to occur in individuals with other autoimmune disorders such as rheumatoid arthritis suggesting that immune dysregulation may be an underlying factor [51]. Genetic factors may also play a role as an increase in frequency of HLA-B15 has been reported in penicillamine-induced pemphigus [52]. In some patients, simple withdrawal of the drug is sufficient to induce remission though in others treatment with corticosteroids and immunosuppressive medication may be required.
Clinical features
Pemphigus vulgaris
Nearly all patients have mucosal lesions, and PV presents with oral lesions in 50–70% of patients. These may precede cutaneous lesions by months or be the only manifestation of the disease. Intact bullae are rare in the mouth and more commonly patients have ill-defined irregularly shaped buccal or palatal erosions which are slow to heal (Figure 50.3). The erosions extend peripherally with shedding of the epithelium. Other mucosal surfaces may be involved, including the conjunctiva, nasopharynx, larynx, oesophagus [53], urethra, vulva and cervix [54].
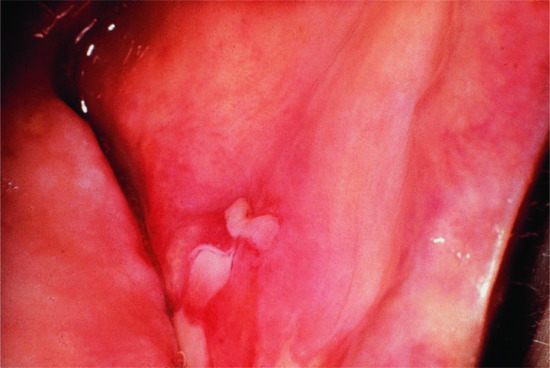
Figure 50.3 Pemphigus vulgaris. Mucosal erosions are an early sign in pemphigus vulgaris, often preceding the cutaneous changes.
(Courtesy of Dr R.J. Pye, Addenbrooke's Hospital, Cambridge, UK.)
Most patients develop cutaneous lesions. Involvement occasionally remains localized to one site but more commonly becomes widespread. The disease has a predilection for the scalp, face, neck, upper chest and back (Figure 50.4, 50.5). Flaccid blisters filled with clear fluid either arise on normal skin or an erythematous base. The contents may become turbid or the blisters rupture, producing painful erosions which extend at the edges as more epidermis is lost (Figure 50.5). At this stage, firm sliding pressure with a finger will separate normal-looking epidermis from dermis, producing an erosion – the Nikolsky sign – though this is not specific for pemphigus. Healing occurs without scarring but pigmentary change may occur in resolving lesions.
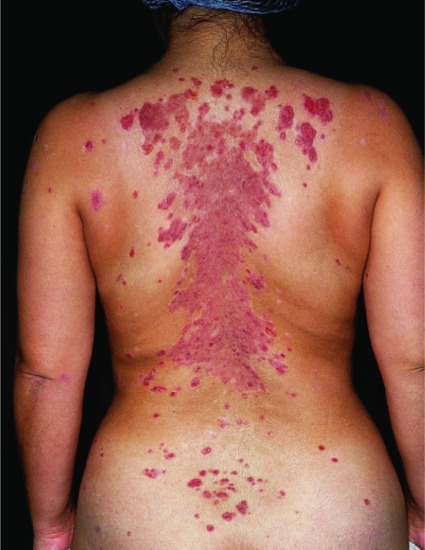
Figure 50.4 Pemphigus vulgaris. Cutaneous lesions typically affect the chest and back in addition to the scalp.
(Courtesy of Dr R.J. Pye, Addenbrooke's Hospital, Cambridge, UK.)
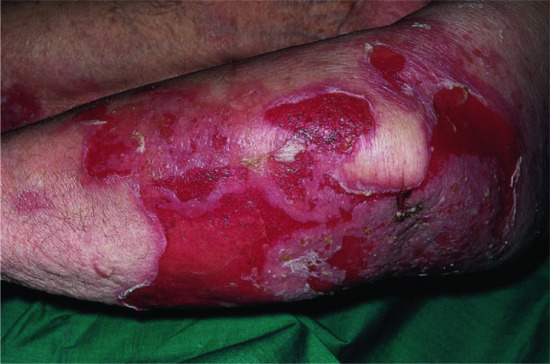
Figure 50.5 Pemphigus vulgaris. Because bullae occur within the epidermis they are fragile and frequently break down to leave widespread erosions.
(Courtesy of Dr R.J. Pye, Addenbrooke's Hospital, Cambridge, UK.)
Lesions in skin folds may form vegetating granulations (Figure 50.6), and flexural PV merges with its variant pemphigus vegetans. Nail dystrophies, acute paronychia and subungual haematomas have been observed in pemphigus [55, 56]. Occasional patients are described with typical PV type lesions confined to the skin and sparing the mucosae despite the presence of antibodies to Dsg 3 [57, 58]. Additionally, some patients will undergo phenotypic and immunological conversion from PV to PF or vice versa over the course of their disease [59–61].
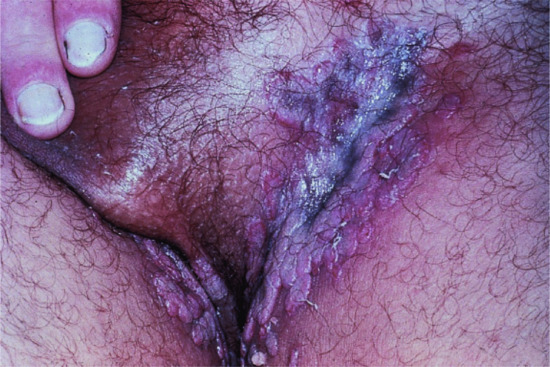
Figure 50.6 Pemphigus vegetans. Vegetating lesions typically occur in the flexures, often without evident blistering.
(Courtesy of Dr R.J. Pye, Addenbrooke's Hospital, Cambridge, UK.)
Pemphigus may deteriorate in pregnancy and the puerperium, and close observation is required at this time. In some patients, initial presentation is in pregnancy. Severe pemphigus in pregnancy may be associated with fetal prematurity and death, but it is difficult to separate the effects of treatment from those of the disease [62, 63]. Generally, the baby is healthy although neonatal pemphigus may occur with mucosal or mucocutaneous lesions which are generally short lived [62].
Pemphigus foliaceus
PF is often regarded as less severe than PV, though it can be as challenging to manage. The onset is usually insidious with scattered, scaly lesions involving the ‘seborrhoeic’ areas of the scalp, face, chest and upper back (Figures 50.7 and 50.8). Individual lesions typically have a fine collarete of scale. Localized disease slowly extends. Blistering may not be obvious because the cleavage is superficial and the small flaccid blisters rupture easily. Scales separate leaving well-demarcated crusted erosions surrounded by erythema, sometimes with small vesicles along the borders. In severe cases the patient may become erythrodermic with crusted oozing red skin (Figure 50.9).
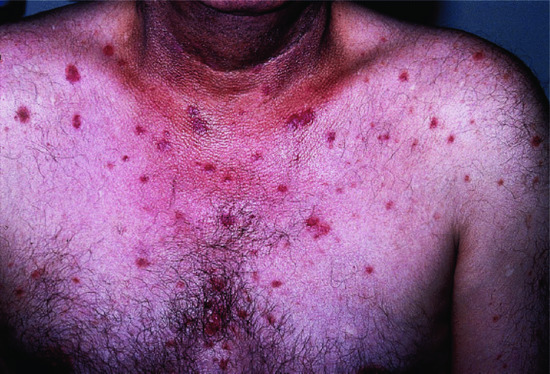
Figure 50.7 Pemphigus foliaceus. There are superficial erosions, frequently without obvious bullae.

Figure 50.8 Pemphigus foliaceus. Lesions frequently have a fine superficial scale, sometimes as a collarete.
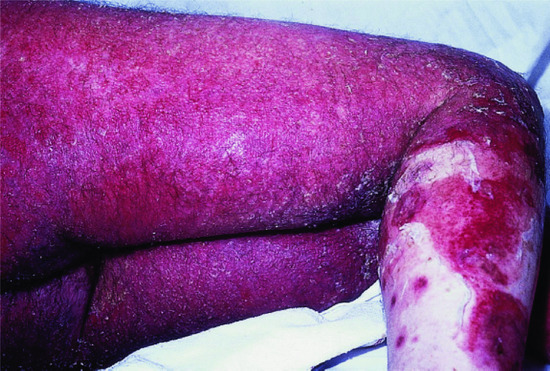
Figure 50.9 Pemphigus foliaceus. Occasionally, pemphigus foliaceus becomes widespread and can result in erythroderma.
Although the antibodies in PF can cross the placenta, the neonate is not usually affected. In two cases in which the neonate did develop PF, both mother and neonate had very high antibody titres.
Clinical variants
Pemphigus vegetans
Pemphigus vegetans is a rare variant of PV characterized by vegetating erosions, primarily in the flexures [64] (see Figure 50.6). Two subtypes are recognized that represent a clinical spectrum from the severe Neumann type to the milder Hallopeau type [65]. Patients have circulating antibodies against Dsg 3, as in PV. In some cases, antibodies in patients with pemphigus vegetans react with desmocollin molecules [66, 67].
The vegetating lesions are hyperkeratotic, papillomatous and acanthotic. Some suprabasal clefts may contain eosinophils but few acantholytic cells are present. Intraepidermal eosinophilic abscesses may be present in older lesions. Early lesions in the Neumann type show suprabasal acantholysis and intraepidermal vesicles without eosinophils. Eosinophilic spongiosis or eosinophilic microabscesses are common in the early pustular lesions of the Hallopeau type. The dermis contains a heavy infiltrate of lymphocytes and eosinophils with few neutrophils.
The disease chiefly affects middle-aged adults. Involvement of the oral mucosa is almost invariable, often with cerebriform changes on the tongue. Elsewhere, lesions are primarily flexural, although vegetations may occur at any site. In the Neumann type, vesicles and bullae rupture to form hypertrophic granulating erosions, which bleed easily. The lesions evolve into vegetating masses exuding serum and pus. The edges are studded with small pustules. Erosions at the edge of the lesions induce new vegetations, which eventually become dry, hyperkeratotic and fissured. In the Hallopeau type, pustules rather than vesicles characterize early lesions but these soon progress to vegetating plaques.
Pemphigus herpetiformis
This is a rare and atypical variant of pemphigus that clinically resembles dermatitis herpetiformis [68]. Widespread clusters of pruritic papules and vesicles develop on an erythematous background. Biopsies show subcorneal pustules, eosinophilic spongiosis or features of dermatitis herpetiformis often without acantholysis but IF studies reveal intercellular staining. Patients possess circulating IgG autoantibodies which recognize either Dsg 1 or 3 and some patients’ sera recognize desmocollins 1 or 3 [69, 70]. The condition may evolve into classical PF but has also been described preceding PV [71]. In general, the clinical course is benign.
Pemphigus erythematosus
Pemphigus erythematosus is a localized variant of PF, originally described by Senear and Usher [72]. Patients have immunological features of both lupus erythematosus and pemphigus, with granular IgG and C3 at the basement membrane zone, intercellular IgG and C3 in the epidermis and circulating antinuclear antibodies. The antibodies recognize Dsgs together with Ro, La and double-stranded DNA antigens [73]. Progression to systemic lupus erythematosus is rare. Pemphigus erythematosus may be associated with myasthenia gravis or thymoma [74].
Erythematous scaly lesions over the nose and cheeks in a butterfly distribution simulate cutaneous lupus erythematosus or seborrhoeic dermatitis. Sunlight may exacerbate the disease. Lesions on the trunk, either localized or generalized, are similar to those in PF.
Paraneoplastic pemphigus
Paraneoplastic pemphigus has been reported in association with a variety of neoplasms, almost exclusively of haematological origin [22]. The commonest association is with non-Hodgkin lymphoma but chronic lymphocytic leukaemia, Castleman disease, thymoma and Waldenström macroglobulinaemia have all been associated. IF changes are characteristic, with features of both pemphigus and pemphigoid reflecting the broad spectrum of circulating antiepithelial antibodies [75]. The early histopathology is frequently lichenoid [76].
Paraneoplastic pemphigus is characterized clinically by severe stomatitis, conjunctivitis which may be scarring and oesophageal, genital mucosal and flexural involvement together with bullous and erosive lesions elsewhere [77]. The respiratory tract may be severely affected in some patients. Cutaneous lesions may be erosive or blistering as in PV, though tense pemphigoid-like blisters may occur as well as erythema multiforme or lichenoid changes.
IgA pemphigus
Two histological forms of IgA pemphigus have been described: an intraepidermal neutrophilic type and a subcorneal pustular dermatosis type, both of which may be clinically indistinguishable from subcorneal pustular dermatosis (Sneddon–Wilkinson disease) [78]. Both are characterized by the deposition of IgA within the epidermis and, in some patients, by detectable circulating antiepidermal IgA antibodies. In the subcorneal pustular dermatosis type, antibodies appear to be directed against desmocollin 1 [79], though the target in the intraepidermal neutrophilic type is uncertain. Some patients also seem to have IgA anti-Dsg 1 or 3 antibodies [80, 81].
The disease chiefly affects adults, though childhood cases have been reported [82]. Patients with both histopathological types have flaccid vesicles or pustules arising on either erythematous or normal skin. The lesions may be intensely pruritic and show a circinate or annular configuration with central clearing, evolving to crusted or scaly erythematous macules. The sites of predilection are the axillae and groin though the trunk, face, scalp and proximal limbs may be affected. Whilst some patients are indistinguishable from Sneddon–Wilkinson disease, others may resemble PF or pemphigus herpetiformis. Flexural, oozing, verrucous plaques mimicking pemphigus vegetans have been described in a child [83]. Mucosal involvement is unusual. Most run a chronic indolent course.
The most frequently reported association is with monoclonal IgA gammopathy in the subcorneal type, a feature it has in common with classical subcorneal pustular dermatosis. Other cases have been linked with HIV infection [84], inflammatory bowel disease [85], rheumatoid arthritis and thiol drugs [86].
Differential diagnosis
Pemphigus vulgaris
Patients with mucosal lesions are likely to present to dental surgeons, oral surgeons or gynaecologists. Erosions may simulate acute herpetic stomatitis, erythema multiforme, aphthous ulcers, lichen planus or mucous membrane pemphigoid. Bullae are transient in the mouth and biopsies of erosions may not be diagnostic. The diagnosis is less difficult when the patients have cutaneous blisters or erosions, though pemphigoid and its variants, linear IgA disease and erythema multiforme may need to be ruled out. Blisters in pemphigoid are typically more tense than in pemphigus and may be haemorrhagic. Vegetating, pustular lesions in flexures must be differentiated from chronic infections or Hailey–Hailey disease (benign familial pemphigus). Vegetating plaques mimicking pemphigus vegetans may occur in IgA pemphigus and paraneoplastic pemphigus and the hyperkeratotic lesions of chronic pemphigus vegetans may simulate cutaneous tumours. The histological differential diagnosis of PV includes Darier disease, Hailey–Hailey disease (benign familial pemphigus) and transient acantholytic dermatosis (Grover disease). These conditions have distinctive clinical features in addition to negative IF studies. Eosinophilic spongiosis may be an early histological manifestation of either pemphigus or BP. IF studies are required to confirm the diagnosis.
Pemphigus foliaceus
PF may resemble seborrhoeic dermatitis or impetigo, but the histological and immunopathological features are diagnostic. Pemphigus erythematosus needs to be distinguished from both seborrhoeic dermatitis and chronic cutaneous lupus erythematosus. PF may occasionally simulate dermatitis herpetiformis both clinically and histologically, but immunopathological studies differentiate the two. Histological features may not be diagnostic in the early stages. Eosinophilic spongiosis may be an early manifestation. IF studies should always be performed if pemphigus is suspected.
Paraneoplastic pemphigus
In most patients, the diagnosis (and treatment) of the underlying malignancy precedes the development of pemphigus but this is not universally the case. The clinical differential diagnosis will include PV, erythema multiforme, graft-versus-host disease, lichen planus and viral infections including herpes simplex.
Classification of severity
A number of severity indices (including the pemphigus disease area index (PDAI, [87]) and autoimmune bullous skin disorder intensity score (ABSIS, [88]) have been proposed in order to standardize the assessment of patients with autoimmune blistering disorders but none has yet been universally adopted [89, 90]. However, as consensus develops, these tools will become useful both in the context of clinical trials and in the monitoring of the response to treatment in individual patients.
Disease course and prognosis
Pemphigus in its various forms typically has a chronic course with an average disease duration of 10 years [91]. However, there is great variability in disease length in patients. Various factors have been suggested to influence this including the site and severity of initial disease, with oral involvement an adverse prognostic factor [92–94]. Immunologically, the presence of both Dsg 1 and 3 antibodies tends to associate with more active disease [95]. Recent data from the UK suggest that early age of onset and Asian ancestry associate with more prolonged disease activity [96].
Investigations
Histopathology
The earliest histological changes consist of intercellular oedema with loss of intercellular attachments in the basal layer. Suprabasal epidermal cells separate from the basal cells to form clefts and blisters. Basal cells remain attached to the basement membrane but separate from one another and stand like a ‘row of tombstones’ on the floor of the blister. Blister cavities contain rounded-up acantholytic cells, which can be found in smears taken from the base of a blister or an oral erosion (Tzank preparation). Clefting may extend into the walls of adnexae. Blistering is preceded by eosinophilic spongiosis in some cases. The superficial dermis has a mild, superficial, mixed inflammatory infiltrate that may include eosinophils.
Direct immunofluorescence testing
Direct IF is the most accurate way to diagnose mucosal pemphigus [97, 98]. Specimens may be posted to a suitable laboratory in Michel's medium if facilities for snap-freezing and storage of the tissues are not available [99].
The diagnosis of pemphigus is confirmed by direct IF which shows IgG and often C3 deposited on the surface of keratinocytes throughout the epidermis in perilesional skin (see Figure 50.2). IgG1 and IgG4 are the most common subclasses; IgM and IgA are present less frequently than IgG.
Serological assays
Circulating pemphigus autoantibodies are detected by indirect IF in over 80% of patients. The use of more than one substrate improves sensitivity, oesophageal mucosal substrates being preferable for the detection of antibodies to Dsg 3 [100] whereas normal human skin shows higher sensitivity for the detection of antibodies against Dsg 1. Rat bladder, which is a transitional epithelium devoid of Dsg proteins, has proven a useful indirect IF substrate in the diagnosis of paraneoplastic pemphigus [101].
Recombinant Dsg 1 and 3 have been used recently to develop a sensitive and specific ELISA assays for the serological diagnosis of pemphigus [102]. Using ELISA, over 95% of PV patients have detectable Dsg 3 antibodies and around 50% have Dsg 1 antibodies. In appropriate dilutions, anti-Dsg ELISA assays can be used to monitor disease activity [103, 104]. Pemphigus-like circulating intercellular antibodies have been reported in conditions such as thermal burns [105], toxic epidermal necrolysis [106] and in first-degree relatives of relatives of pemphigus patients [107, 108].
Management
Because of the rarity of the pemphigus group of diseases, progress with informative randomized controlled trials has been slow. However, systemic corticosteroid therapy remains the mainstay of therapy, generally in combination with a steroid-sparing immunosuppressant. Treatment has been the subject of a number of reviews [109–111].
Topical therapy
Patients who present with oral disease and mild cutaneous involvement may remain in this localized phase for months. Potent topical or intralesional steroids may reduce the requirement for oral steroids. Good oral hygiene, including treatment of periodontal disease, is important.
In patients with widespread blistering, intensive nursing care is mandatory. Opportunist infection is the major cause of death in patients with widespread blistering who are also immunosuppressed and potassium permanganate and topical antiseptics may help reduce the risk of cutaneous infection. Liberal use of emollients will reduce frictional stress on affected skin.
Corticosteroids
Prednisolone together with a steroid-sparing adjuvant is the initial treatment for most patients with PV. Prednisolone 0.5–1 mg/kg/day in combination with adjuvant immunosuppression and appropriate topical therapy is sufficient to initiate disease control in many patients [112]. There seems to be little benefit in using higher doses [113]. The steroid dose should be titrated to the clinical response.
Patients with generalized disease may require aggressive immunosuppression to suppress blistering and a major difficulty in managing these patients is achieving a balance between the risks associated with high-dose steroid therapy and those of poorly controlled disease. In this situation IV pulses of either 1–g methylprednisolone or 100 mg dexamethasone are safer alternatives that may be used, often together with IVIG, immunoabsorption or plasmapheresis. Whilst steroid pulses have been shown to be useful in some studies [114] in others they did not show benefit [115, 116].
Azathioprine
Azathioprine has been widely used in the management of pemphigus at a dose of 2–3 mg/kg/day and multiple case series support its use. The combination of prednisolone and azathioprine is more effective than prednisolone alone both in terms of mortality and remission [117]. One randomized controlled trial of pemphigus treatment found azathioprine to be the most effective steroid-sparing agent, followed by cyclophosphamide (pulse therapy) and mycophenolate mofetil [118]. Another trial found azathioprine and mycophenolate mofetil to be equally effective [119]. Potential adverse effects of azathioprine include bone marrow suppression, nausea and liver dysfunction and careful blood monitoring is therefore mandatory, particularly in the early stages of treatment. Azathioprine toxicity is more frequent in patients with low levels of activity of the enzyme thiopurine methyl transferase (TPMT) and activity of the enzyme should be checked prior to treatment wherever possible [120]. In patients with low levels of TPMT, azathioprine dose should be adjusted accordingly.
Mycophenolate mofetil
Mycophenolate mofetil (1–3 g/day) has been found helpful as a steroid-sparing agent [119, 121, 122] in pemphigus. Mycophenolate mofetil is a pro-drug of mycophenolic acid and has a relatively selective effect on T and B lymphocytes through its effect on the inhibition of inosine monophosphate dehydrogenase. A double-blind randomized controlled study comparing azathioprine and mycophenolate showed no significant difference in efficacy between the two drugs though there was a trend towards fewer adverse effects and more rapid remission in the mycophenolate group [119]. Adverse effects of mycophenolate include bone marrow suppression and gastrointestinal symptoms and, as with azathioprine, patients require close monitoring in the early stages of treatment. Gastrointestinal symptoms may respond to the use of enteric-coated delayed-release mycophenolic acid in place of mycophenolate mofetil.
Cyclophosphamide
Cyclophosphamide is a potent anti-B-cell agent with significant activity in pemphigus and other antibody-mediated autoimmune diseases [123, 124]. Its use is limited by significant adverse effects, including gonadal failure, haemorrhagic cystitis, bone marrow suppression and an increased risk of bladder cancer. It is consequently generally reserved for patients who have failed to respond to conventional immunosuppression with azathioprine or mycophenolate mofetil. Monthly IV pulses of cyclophosphamide with dexamethasone combined with low-dose oral cyclophosphamide have been used, as has daily oral treatment [125].
Intravenous immunoglobulin
Intravenous immunoglobulin (IVIG) is a potentially attractive treatment option because of its relative lack of immunosuppressive effects in comparison to most other pemphigus treatments. It is generally used at a dose of 2 g/kg split over 3–5 days and treatment may need to be continued monthly for prolonged periods [126]. Its mechanism of action is unclear though it may have a dilutional effect on pathogenic autoantibodies in addition to having anti-idioptypic effects [127]. A novel ‘time to escape from protocol’ multicentre double-blind controlled study in Japan has confirmed its effectiveness [128]. Whilst IVIG is generally well tolerated it can cause headaches, flu-like symptoms and hypotension. Anaphylaxis may occur in patients with complete IgA deficiency and this should be excluded prior to treatment.
Rituximab
Depletion of B lymphocytes using the anti-CD20 monoclonal antibody rituximab has been shown to be effective in the management of pemphigus in multiple case series. Several dose regimes have been used including 375 mg/m2 weekly for 4 weeks and, more recently, two infusions of 1 g, 2 weeks apart [129–131]. The mechanism of action of rituximab is likely through B-cell regulatory pathways as plasma cells, which are responsible for antibody production, do not express the CD20 molecule. Interestingly, T-cell responses to Dsgs are suppressed following rituximab therapy [132]. Onset of action of rituximab is typically 8–16 weeks following the first infusion, though it may take longer. Improvement may persist for 12–18 months. Rituximab may be combined with conventional steroid therapy and adjuvant immunosuppression, though care needs to be taken to avoid excessive immunosuppression and increased infection risk. In some centres, immunoabsorption has been combined with rituximab [133].
Adverse effects of rituximab in the treatment of pemphigus appear uncommon. Infusion reactions may occur, but are less frequent than when the drug is used in the treatment of B-cell malignancies. Infection has been reported [134], and particular care needs to be taken to avoid reactivation of viral hepatitis – patients should be rigorously screened prior to treatment [135].
Immunoabsorption and plasmapheresis
Removal of circulating antibodies by plasmapheresis was first used in 1978 [136] and is theoretically an attractive way of managing pemphigus in the acute stage, though other studies have not shown benefit [137]. More recently, selective immunoadsorption of IgG using columns containing protein A or other immunoglobulin binders [138–140] has been effective at inducing remission in pemphigus patients, but concomitant immunosuppression with steroids and steroid-sparing adjuvants is required to prevent rebound increase in the synthesis of antibody.
Other therapies
Although initial case reports were suggestive of benefit of ciclosporin in pemphigus [141, 142], more recent studies cast doubt on this [143]. Topical ciclosporin mouthwash may be helpful in severe oral pemphigus [144].
Gold may have modest effect in pemphigus [145, 146], though toxic effects limit its utility. Dapsone has been advocated as an adjunct in some patients with mild disease [147] and a recent prospective, placebo-controlled study showed a modest steroid-sparing effect [148]. Dapsone may be of particular utility in pemphigus herpetiformis. Acetretin has been used in conjunction with prednisolone in pemphigus vegetans [149].
Methotrexate is now seldom used, though there is some suggestion that it may have a steroid-sparing effect and moderate doses of methotrexate may permit withdrawal of prednisolone in steroid-dependent patients [150].
Other therapies that have been used in resistant cases of pemphigus include extracorporeal photophoresis [151, 152] and the tumour necrosis factor (TNF) inhibitors infliximab [153] and adalimumab [154] have also been reported as beneficial, though autoimmune bullous disease has been reported as a consequence of TNF blockade [155]. Tetracycline antibiotics with or without nicotinamide may be helpful as steroid-sparing agents in some patients [156, 157].
Resources
Management guidelines
- Harman KE, Albert S, Black MM; British Association of Dermatologists. Guidelines for the management of pemphigus vulgaris. Br J Dermatol 2003 Nov;149(5):926–37.
- Hertl M, Jedlickova H, Karpati S, et al. Pemphigus. S2 Guideline for diagnosis and treatment – guided by the European Dermatology Forum (EDF) in cooperation with the European Academy of Dermatology and Venereology (EADV). J Eur Acad Dermatol Venereol 2014 Oct 22.
- Joly P, Bernard P, Bedane C, Prost C, Ingen-Housz-Oro S. Centres de référence des maladies bulleuses auto-immunes. Société Française de Dermatologie. [Pemphigus. Guidelines for the diagnosis and treatment. Centres de référence des maladies bulleuses auto-immunes. Société Française de Dermatologie.] Ann Dermatol Venereol 2011 Mar;138(3):252–8.
Patient resources
- There are a number of patient support groups that provide useful information, including:
- The International Pemphigus and Pemphigoid Foundation http://www.pemphigus.org
- The Pemphigus Vulgaris Network http://www.pemphigus.org.uk All last accessed August 2015.
SUBEPIDERMAL IMMUNOBULLOUS DISEASES
Introduction
These bullous disorders include pemphigoid diseases and dermatitis herpetiformis. While pemphigoid diseases are characterized by autoantibodies against structural proteins of the dermal–epidermal junction (DEJ) in dermatitis herpetiformis, the autoantigen is epidermal transglutaminase [1, 2]. Via proteins of the DEJ, the cytoskeleton of the basal keratinocytes is linked to the extracellular matrix of the dermis (see Chapter 2). Binding of pemphigoid autoantibodies to their target antigens leads to the separation of epidermis and dermis. In bullous pemphigoid (BP) and epidermolysis bullosa acquisita (EBA), considerable progress has been made to understand this complex process.
Subepidermal bullous diseases share some clinical characteristics, such as tense blisters and erosions and, in contrast to pemphigus, a negative Nikolsky sign, i.e. friction of non-lesional skin does not lead to intraepidermal disruption and visible erosion. These disorders are, however, heterogeneous with regard to the clinical presentation, target antigen(s), autoantibody isotype and immunopathology. Importantly, prognosis and treatment may vary considerably, hence requiring exact diagnosis.
The target antigen(s) have been identified on the molecular level in most of the distinct entities amongst them BP, by far the most common pemphigoid disorder, mucous membrane pemphigoid, linear IgA disease, pemphigoid gestationis, lichen planus pemphigoides, anti-p200/ laminin γ1 pemphigoid, bullous systemic lupus erythematosus, EBA and cicatricial pemphigoid (Table 50.3). In addition, small numbers of patients with a pemphigoid disease characterized by renal insufficiency and autoantibodies against the a5 chain of type IV collagen or with autoantibodies against different not fully characterized antigens, amongst them 105 kDa, 168 kDa and 285 kDa proteins, have been reported [3–7]. New target antigens are still being described and it seems likely that many more yet only poorly characterized or undiscovered molecules within the DEJ may function as autoantigens in subepidermal blistering diseases.
Table 50.3 Pemphigoid diseases. Autoantibody specificities and diagnostically relevant clinical signs. Main target antigens are indicated in bold: for target antigens in italics, commercial detection systems are available
| Disease | Autoantibody | Clinical signs of diagnostic relevance |
| Bullous pemphigoid (BP) | BP180 NC16A, BP230 | Tense blisters, erosions, intense pruritus, old age (>75 years); no predominant mucosal involvement |
| Mucous membrane pemphigoid (MMP) | BP180, laminin 332, BP230, α6β4 integrin, laminin 311a | Predominant mucosal involvement |
| Linear IgA disease | LAD-1, BP230 (IgA reactivity) | Tense blisters, erosions; no predominant mucosal involvement |
| Pemphigoid gestationis | BP180 NC16A, BP230 | Erythema, papules, rarely vesicles, intense pruritus; pregnancy or postpartum period |
| Anti-p200/laminin γ1 pemphigoid | p200 antigen laminin γ1 | Tense blisters, erosions; <75 years of age; no predominant mucosal involvement |
| Epidermolysis bullosa acquisita | Type VII collagenb | Mechanobullous (like epidermolysis bullosa) and inflammatory variant (like BP or MMP) |
| Bullous systemic lupus erythematosus (SLE) | Type I: type VII collagenc Type II: BP180, BP230, laminin 332 |
SLE present; tense blisters, erosions; no predominant mucosal involvement; excellent response to dapsone |
| Lichen planus pemphigoides | BP180 NC16A, BP230 | Tense blisters independent of lichen planus lesions |
| Cicatricial pemphigoid | BP180, BP230, laminin 332 | Blisters and erosions that heal with scarring and/or milia formation. No predominant mucosal involvement |
Adapted from Schmidt and Zillikens 2013 [2].
aLaminin 311 shares the α3-chain with laminin 332 and is co-targeted.
bNot FDA cleared.
Bullous pemphigoid
Definition
BP is not only the most common disorder within the group of subepidermal immunobullous disorders but also represents the most frequent autoimmune blistering disease in general. It mainly affects elderly people although younger patients may also be affected and often starts with pruritus and urticated and erythematous lesions. Later, tense blisters are characteristic both on erythematous and on normal skin. Mucosal involvement only develops in a minority of patients and is not predominant. Histopathology of a lesional biopsy reveals subepidermal splitting. Autoantibodies, chiefly IgG, recognize two proteins of the DEJ, BP180 (type XVII collagen) in almost all patients and, in about half of them, BP230. The clinical features and immunopathology of BP are summarized in Table 50.3.
Introduction and general description
BP was first differentiated from pemphigus in 1953 by Walter Lever who described intraepidermal split formation and loss of cell adherence between keratinocytes (acantholysis) as histopathological hallmark of pemphigus, whereas he coined the term ‘pemphigoid’ for conditions in which a subepidermal split formation was typically present [8]. A decade later, Jordon et al. demonstrated that in BP, tissue-bound and serum autoantibodies against proteins of the DEJ were present [9]. Further milestones in the understanding of BP included the immunochemical characterization of the hemidesmosomal target proteins BP180 (type XVII collagen, also termed BPAG2) and BP230 (BPAG1-e), the cloning of their genes, and the demonstration that autoantibodies against BP180 are pathogenic [10–14].
Epidemiology
Incidence and prevalence
The reported incidence of BP ranges between 2.5 and 66 new cases per million per year depending on the studied population. The lowest incidences have been reported from Romania (2.5 per million per year) [15], Kuwait (2.6 per million per year) [16], Poland (4.5 per million per year) [17] and Singapore (7.6 per million per year) [18]. In Scotland, Italy, Germany and Switzerland, the incidence ranges from 10.0 to 14.0 per million per year [19–21], whereas a higher incidence of 22–24 new cases per million per year has been recently estimated in France, Guadeloupe and Olmsted County, Minnesota, USA [22–24]. An even higher incidence of 66 per million per year was reported in the UK based on a general practitioner database [25]. Interestingly, in Germany, France, Olmsted County and the UK, the incidence of BP appears to have increased considerably within the last 10 years (between 2- and 4.8-fold) [19, 22, 24–27]. This may be due to either the increasing age of the population, the increased prevalence of debilitating neurological disorders (shown to be major risk factors for BP) [28–30] and/or the increasing use of diuretics and psycholeptics, drugs that were associated with BP [28, 31, 32]. Furthermore, improved diagnostic assays and the better recognition of prodromal and atypical forms of BP are other explanations.
Age
BP is predominantly a disease of the elderly with a mean age at disease onset between 69 and 83 years [22, 30, 33–36]. A somewhat lower mean age of 64 years at disease onset was reported from China [37]. The incidence rises significantly with age to 190 to 312 per million per year in individuals older than 80 years of age [19–21, 25]. With the changing age structure of many populations worldwide even more people are expected to suffer from BP in the coming decades. In individuals younger than 50 years, BP rarely occurs (incidence of <0.5 per million per year) [19, 21, 25, 38]. Only about 80 cases of BP in children and adolescents have been described [39, 40]. In contrast, a retrospective study from Israel has indicated that BP in the first year of life occurs with an estimated incidence rate of 23.6 per million infants under the age of 1 year per year [41].
Sex
A slight female predominance was observed with 52–60% of BP patients being women [22, 25, 35, 36]. After age adjustment the incidence was, however, higher in men [22, 38] due to the generally higher life expectancy of women and the increasing incidence of BP with age.
Ethnicity
The incidence varies considerably between different countries (see earlier). No data are available about the incidence of BP in different ethnicities within a country. Thus, both genetic and environmental factors may contribute to the wide range of incidence. In addition, different diagnostic standards and healthcare systems may influence reported incidence.
Associated diseases
Neurological and psychiatric disorders including cognitive impairment, Parkinson disease, stroke, epilepsy, multiple sclerosis and uni- and bipolar disorders have been associated with BP [28, 29, 30, 33, 34, 42–44]. Between a third and half of all BP patients suffer from neurological diseases (odds ratios 2.4–10.6) [28–30, 33, 42] considerably increasing disease burden in this elderly patient population. These findings are intriguing since both target antigens, BP180 and BP230, are expressed in the central nervous system [45, 46] and mice with mutations in the dystonin gene encoding for various isoforms of BPAG1 including the epithelial isofom BP230 develop severe dystonia and sensory nerve degeneration [47]. In a nationwide study based on data from the national health insurance, an association with psoriasis (odds ratio 2.0) was reported in Taiwanese patients [43]. The co-occurrence of BP with autoimmune disorders and cancer has been described in several case reports [48, 49]. However, a case–control study did not find any increased risk for autoimmune disorders [50], while in two case–control studies with more than 1700 BP patients, only a low association with gastric cancer was observed in Japanese patients [51, 52]. Very recently, two studies based on patient registries in Germany and England including more than 1700 and 4700 BP patients identified a clear association with haematological malignancies (various lymphomas and myeloid leukaemia) and a weak association with lymphoid leukaemia, respectively [53, 54].
Pathophysiology
The pathogenic importance of humoral and cellular autoimmunity against BP180 has clearly been demonstrated. Both Fc receptor independent and, importantly, Fc receptor mediated effects were shown to be essential for blister formation in BP using in vitro, ex vivo and various animal models. More specifically, complement activation at the DEJ and the activation of mast cells appeared to be crucial to attract neutrophils and macrophages at the DEJ. Subsequent release of reactive oxygen species and various proteases then induced dermal–epidermal splitting (Figure 50.10). Targeting mast cells, neutrophils, complement activation and the cytokine network may open novel therapeutic avenues for this disease [55].
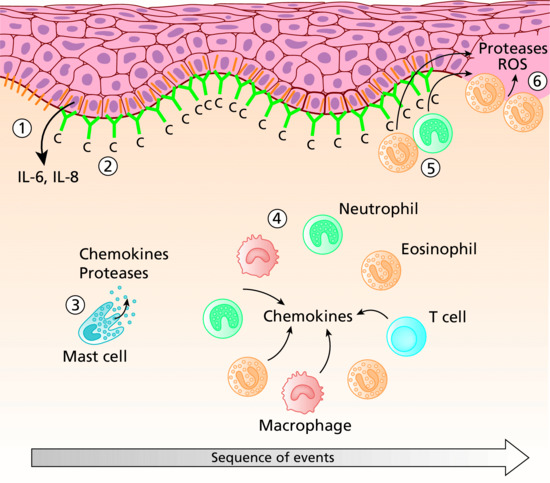
Figure 50.10 Sequence of events leading to blister formation in bullous pemphigoid. Binding of autoantibodies (green) against BP180 (orange) initiates Fc-independent events resulting in the secretion of interleukin (IL)-6 and IL-8 from basal keratinocytes as well as in internalization and decreased expression of BP180 (1). Complement is activated at the dermal–epidermal junction (DEJ) (2) and mast cells degranulate (3). Complement activation and chemokine gradients trigger the infiltration of inflammatory cells into the upper dermis (4). Their secretion of additional inflammatory mediators further increases the inflammatory reaction. Finally, granulocytes at the DEJ release reactive oxygen species and proteases (5) that ultimately induce dermal–epidermal separation (6).
(Adapted from Schmidt and Zillikens 2013 [2] © Elsevier.)
Autoantibodies
In nearly all BP patients, autoantibodies bind to BP180 (also termed type XVII collagen, and BPAG2) (see Chapter 2) [56]. The extracellular portion of the 16th non-collagenous domain (NC16A) located directly adjacent to the cellular membrane is the immunodominant region in BP and is recognized by autoantibodies in 75–90% of BP patients [57–63]. The importance of anti-BP180 NC16A reactivity is further highlighted by the observation that serum levels of BP180 NC16A-specific IgG antibodies correlate with the disease activity in BP patients [56, 60, 63–66]. IgG4 and IgG1 are the major IgG subclasses of anti-BP180 NC16A antibodies [67]. The majority of patients also raise IgG antibodies against epitopes outside the NC16A domain [56, 68, 69] while initial reactivity appeared to target the NC16A [70]. IgG reactivity with C-terminal epitopes appeared to be associated with mucosal involvement and more severe skin disease, whereas the intracellular domain was preferentially targeted at an early clinical stage [69, 71].
The majority of BP patients develop, beside IgG, IgA and IgE anti-BP180 reactivity [67, 72–75]. The presence of IgE anti-BP180 NC16A antibodies was associated with a severe form of BP, longer duration for remission and requirement for more intensive therapies [72].
BP230 (also known as BPAG1-e, and BPAG1) is recognized by 50–70% of BP sera [76–82]. As for BP180, B-cell epitopes are not equally distributed on the molecule but preferentially localize to the globular C-terminal domain of BP230 [56, 80, 83]. In addition to IgG reactivity, IgE antibodies against BP230 were detected in the majority of BP sera [84].
Cellular immune response
In contrast to the humoral immune response, the cellular immune response has been less widely studied in human BP [85]. T- and B-cell reactivity against the NH2-terminal portion of the BP180 ectodomain is associated with severe BP, while the central portion is more frequently recognized in patients with limited disease. In contrast, combined T- and B-cell response against the COOH- and NH2-terminal globular domains of BP230 were found in less than 50% [86]. The response to the BP180 ectodomain is restricted to the DQβ1*0301 allele [87, 88]. Autoreactive T cells in BP patients produced a Th1/Th2 mixed cytokine profile [86, 87]. While the number of circulating CD4+CD25+FOXP3+ regulatory T cells, natural killer T cells and natural killer cells are normal, γδT cell numbers are reduced in BP patients [89, 90]. The number of peripheral follicular helper T cells, a T-cell subset known to be pivotal for B-cell activation, was higher in patients with active disease compared to those in healthy volunteers and BP patients in remission and correlated with serum levels of anti-BP180 antibodies [91].
Cytokines and chemokines
Elevated levels of IL-1β, IL-2, IL-4, IL-5, IL-6, IL-8, IL-10, IL-15, IL-16, IL-17, IL-21, eotaxin, monocyte chemotactic protein 4 (MCP-4), TNF-α and CCL-18 occur in the sera and/or blister fluids of BP patients (reviewed in [92, 93]). Serum levels of TNF-α, IL-6, IL-8, IL-15, IL-21 and CCL18 correlated with the extent of BP skin lesions [91–93] pointing to a pathological relevance of these mediators. The assumption that Th2-type cytokines are important in human BP is supported by the increased frequency of cutaneous lymphocyte-associated antigen-positive IL-4- and IL-13-producing cells in the peripheral blood [94]. More recently, the potential role of Th17 cells in BP was highlighted [95].
Functionally relevant pathogenic mechanisms
Data about the functionally relevant pathogenic mechanisms in BP have been generated by in vitro studies using cultured human keratinocytes, ex vivo studies using cryosections of human skin and, importantly, various animal models.
When cultured normal human keratinocytes were treated with anti-BP180 IgG a signal-transducing event leading to a dose- and time-dependent release of IL-6 and IL-8 was observed [96]. In the same model, internalization and creased expression of BP180 and weakening of keratinocyte attachment in response to anti-BP180 IgG were seen [97, 98]. Recently, release of IL-6 and IL-8 and reduction of the number of hemidesmosomes was also seen after incubation with anti-BP180 IgE [99] (see Figure 50.10).
Using cryosections of normal human skin, BP180 NC16A-specific IgG induced a dermal–epidermal separation when co-incubated with leukocytes from healthy volunteers [100]. This effect was mediated by the Fc portion of autoantibodies and the Fcγ receptors IIA and IIIA on human neutrophils resulting in the release of matrix metalloproteinase-9 and neutrophil elastase [100–102]. Both enzymes were found in blister fluid and lesional biopsies from BP patients and were capable to degrade BP180 [103–105].
Passive transfer of rabbit IgG raised against the murine homologue of the human BP180 NC16A domain, into neonatal wild-type mice produced clinical, histopathological and immunopathological alterations similar to those seen in BP patients [13]. In this model, blister formation was dependent on the activation of complement [106, 107], degranulation of mast cells [108], recruitment of macrophages [108] and neutrophils [109], and the release of various proteases including the plasminogen/plasmin system [110], mast cell proteinase 4 [111], matrix metalloproteinase 9 [112], α1-proteinase inhibitor [113] and neutrophil elastase [65–68, 114, 115]. More specifically, in the early stages of blistering, matrix metalloproteinase 9 is mainly activated by plasmin, which is formed by activation of plasminogen by tissue plasminogen activator and/or urokinase plasminogen activator. Plasmin and the mast cell-specific serine protease 4 can activate matrix metalloproteinase 9 which then inactivates α1-proteinase inhibitor, the physiological inhibitor of neutrophil elastase. The unrestrained activity of neutrophil elastase is then responsible for the degradation of structural proteins of the DEJ including BP180 (see Figure 50.10). A recent passive transfer model in adult mice highlighted the importance of FcγR IIB, FcγR III and FcγR IV [116].
Subsequently, further mouse models were developed that allowed exploration of additional pathogenic mechanisms. Amongst these models, three distinct lines of transgenic mice that expressed human BP180 in murine skin elegantly replicated essential features of human BP [14, 117, 118]. In one of these models, the complement dependency of experimental BP was questioned when the passive transfer of F(ab')2 fragments of human BP led to skin frangibility in Col17-humanized mice [119]. Subsequently, two ‘active’ mouse models were developed that do not depend on the transfer of anti-BP180 antibodies: wild-type mice were immunized with recombinant murine NC15A and in the second model, Rag-2−/−/COL17m–/–,h+ mice (immunocompromised mice expressing human BP180) received splenocytes from wild-type mice that had been immunized by grafting of COL17m–/–,h+ mouse skin and subsequently developed anti-BP180 antibodies and a blistering phenotype [120, 121]. In the latter model, the importance of NC16A-reactive CD4+ T cells has corroborated previous in vitro studies with human cells that showed a restriction of NC16A-reactive CD4+ T cells to the HLA-DQB1*301 allele [87, 122].
In vivo evidence for the pathogenic role of IgE autoantibodies was provided by both clinical observations and two additional mouse models [123, 124]. IgE BP180 NC16A-specific antibodies were associated with more severe forms of human BP, correlated with the extent of skin lesions [67, 72, 73], and individual corticosteroid-resistant BP patients responded well to omalizumab, a humanized monoclonal antibody that inhibits IgE binding to its high-affinity receptor [125, 126].
In contrast to BP180, the pathogenic relevance of autoantibodies against BP230 remains elusive: two animal models investigating the pathogenic relevance of antibodies to BP230 have been reported; however, blisters were not [127] or were not consistently seen [128]. Studies on the correlation of serum anti-BP230 autoantibodies with disease have been contradictory, most of them finding no correlation between serum anti-BP230 levels and disease activity [56, 70, 78]. However, in the light of BP230−/− mice that, in addition to mild skin fragility, developed neurological defects with sensory neurone degeneration [47] and the recently highlighted association between BP and neurological disorders [28–30], autoimmunity to BP230 may contribute not only to the skin phenotype in patients with BP but also to the extracutaneous features.
Predisposing factors
Several triggers have been implicated in BP onset including trauma [129, 130], burns [131], skin grafting [132, 133], radiotherapy [134, 135] and UV radiation including sunlight [136], UVA1 [137], psoralen and UVA (PUVA) [138] and photodynamic therapy [139]. Furthermore, about 20 case reports have described the association of vaccination with the onset of BP, most frequently against influenza [130, 140]. Nevertheless, an epidemiological study over 9 years in Spain did not find a higher incidence of admissions for BP in the 10-week influenza vaccination period compared to the rest of the year [141].
Numerous case reports have described the triggering of BP by drugs, most frequently frusemide [142]. In three controlled studies, however, only a relatively weak association of BP with spironolactone (odds ratio 2.3), phenothiazines with aliphatic side-chains (odds ratio 3.7; 95% CI, 1.2–11.3) and loop diuretics (adjusted odds ratio 3.8; 95% CI, 1.5–9.7) was identified [31, 32, 143]. The use of these drugs should thus be carefully evaluated.
Pathology
Histopathology
Light microscopy of lesional skin typically shows a subepidermal blister with a dense eosinophil-rich infiltrate within the papillary dermis and along the DEJ that usually also includes neutrophils, macrophages, and T lymphocytes (Figure 50.11). The histopathological picture may vary considerably with the clinical picture and the age of the lesion. By electron microscopy, the split was shown to occur within the lamina lucida.
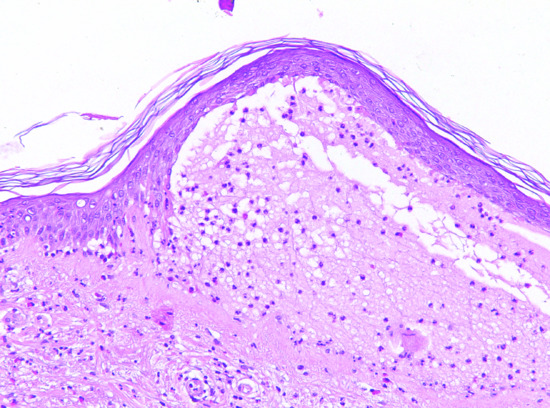
Figure 50.11 Histopathology of bullous pemphigoid. Lesional skin biopsy with subepidermal splitting and a dense inflammatory infiltrate of eosinophils and neutrophils in the upper dermis H&E, 400×.
Tissue-bound autoantibodies
Tissue-bound autoantibodies can be visualized by direct IF microscopy of a perilesional biopsy in almost all patients. Linear deposits of IgG (IgG4 and IgG1 are the predominant subclasses) and/or C3, and to a lesser extend of IgA and IgE, are seen along the DEJ (Figure 50.12) [144]. This binding pattern is observed in all pemphigoid disease. For further differentiation of the anti-DEJ staining, the n- versus u-pattern analysis can be performed [145] (see Figure 50.12, insert). At higher magnification and in thin sections (4 μm), IgG deposition appears serrated with arches closed at the top referred to as ‘n-serrated’ pattern. In all pemphigoid disease except EBA, an n-serrated pattern is seen. Alternatively, the biopsy is treated with 1 M NaCl solution to induce an artificial spit in the DEJ at the lamina lucida level [146]. Autoantibody specificities can then be detected similar to indirect IF microscopy on 1 M NaCl split skin (see later; Figure 50.13). Furthermore, although not routinely available computer-aided fluorescence overlay antigen mapping (FOAM) [147] and double IF labelling of tissue-bound and defined antigens of the DEJ analysed by laser scanning confocal microscopy [148] can be used to differentiate BP from for example antilaminin 332 pemphigoid, anti-p200 pemphigoid and EBA.

Figure 50.12 Direct immunofluorescence microscopy of bullous pemphigoid. In a perilesional biopsy, linear binding of immunoglobulin G (IgG) at the dermal–epidermal junction is seen (a). At higher magnification and in thin sections (4 μm), IgG deposition is no longer linear but appears curved with arches closed at the top referred to as ‘n-serrated’ pattern (b; inset).
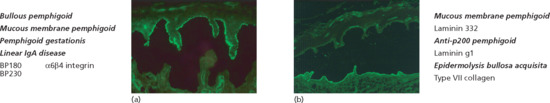
Figure 50.13 Serological screening for autoantibodies in pemphigoid diseases: indirect immunofluorescence microscopy on 1 M NaCl-split human skin. Autoantibodies against BP180, BP230 and α6β4 integrin show a linear binding at the roof of the artificial split (a). Antibodies to laminin 332, laminin γ1/p200 antigen and type VII collagen label the floor (b). The specific disorders and the corresponding target antigens are indicated to the left of (a) and to the right of (b).
Serum autoantibodies
Major characteristics of serum autoantibodies are described earlier (see Pathophysiology section). Circulating autoantibodies can be detected by: (i) indirect IF microscopy on monkey oesophagus, 1 M NaCl split human skin and a commercial IF BIOCHIP™ mosaic; (ii) immunoelectron microscopy [149] (not routinely available); (iii) commercial ELISA systems; and (iv) various in-house ELISA, immunoblotting and immunoprecipitation analyses using cell-derived or recombinant forms of BP180 and BP230 [144].
Indirect IF microscopy has traditionally been performed on monkey oesophagus with a sensitivity of 60–70% [9, 81, 150]. Higher sensitivities between 70% and 95% were obtained with normal human skin in which splitting of the DEJ had been induced by incubation in 1 M NaCl solution [82, 150–153]. Moreover, in this technique, BP autoantibodies bind to the roof of the artificial split and can therefore be differentiated from autoantibodies in antilaminin 332 pemphigoid, anti-p200 pemphigoid and EBA (see Figure 50.13). While IF binding signals by direct IF appear to be mainly due to anti-BP180 antibodies, signals by indirect IF on oesophagus and salt-split skin correlate with anti-BP230antibodies [154, 155]. More recently, a BIOCHIP mosaic has become available (Euroimmun, Luebeck, Germany). This indirect IF microscopy test allows the simultaneous analysis of several substrates including monkey oesophagus, salt-split skin, BP180 NC16A, a C-terminal stretch of BP230, Dsg 3 and Dsg 1 in asingle incubation field. This BIOCHIP mosaic has been shown to provide nearly the same diagnostic value compared to the routine multistep procedure (Figure 50.14) [156]. BP180 NC16A- and BP230-specific ELISA systems are also available [62, 63, 80, 157]. Both BP230 ELISA systems are based on recombinant C-terminal fragments that in one ELISA, is combined with a N-terminal fragment. By the combined use of the BP180 and BP230 ELISA, the diagnostic sensitivity can be increased by 4–8% to 87–91% compared to application of the BP180 ELISA alone [80–82]. Low serum levels of anti-BP180 or -BP230 antibodies may also be found in about 4% of dermatological patients not suffering from BP, especially in patients with pruritic dermatoses [158–160]. In a recent larger prospective study, patients with chronic pruritus did not reveal a higher frequency of anti-DEJ antibodies compared to healthy volunteers (unpublished data). In patients with low serum levels of anti-DEJ antibodies, direct IF microscopy is mandatory to exclude BP.
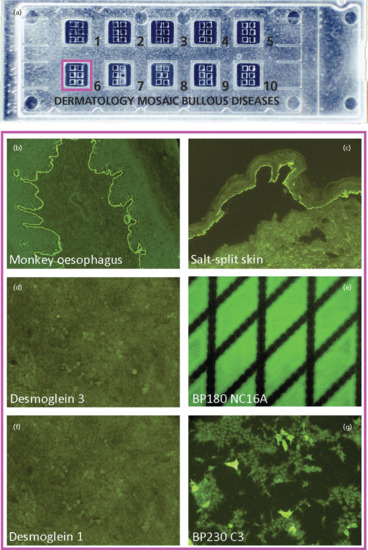
Figure 50.14 Serological screening for autoantibodies in pemphigoid diseases: indirect immunofluorescence microscopy using a BIOCHIP Mosaic™. Ten incubation fields each with six different BIOCHIPs are placed on a standard-sized slide (a). This technology allows the simultaneous testing for serum autoantibodies against various substrates including monkey oesophagus, salt-split human skin, recombinant BP180 NC16A and mammalian cells expressing the ectodomains of desmoglein 1 and 3, and the C terminus of BP230 (b–g) in patients with suspected autoimmune blistering diseases. Here, serum from a patient with bullous pemphigoid binds to monkey oesophagus, the roof of salt-split human skin, BP180 and BP230 but as expected not to desmoglein 1 or 3. This commercially available assay has a high agreement compared with the conventional multistep approach and similar specificities and sensitivities as the corresponding enzyme-linked immunosorbent assay (ELISA) assays [156].
(Adapted from van Beek et al. 2012 [156] with kind permission form BioMed Central © van Beek et al.)
Previously, detection of anti-BP180 and anti-BP230 serum reactivity relied on immunoblot and immunoprecipitation studies using cell-derived forms of BP180 and BP230 in extracts of cultured keratinocytes, human epidermis, placental amnion [10, 12, 161–163] and conditioned medium of cultured human keratinocytes as well as various recombinant forms [58, 164–167]. In fact, by combining different detection systems, autoantibodies against BP180 and/or BP230 can be found in virtually all BP sera [56, 166]. Currently, immunoblotting and immunoprecipitation studies are only required in exceptional cases and are mainly employed in investigative studies.
Genetics
Data about a genetic background in BP are sparse and partly inconsistent. In 21 white patients a significant association with DQB1*0301 was detected [168], an observation partly confirmed by the demonstration that the primary response of CD4-positive T cells to BP180 in vitro is restricted to DQB1*0301 [87]. Another study with 74 white patients identified an association with DQB1*0301 only in men [169]. In 25 patients from northern China, DRB1*08 and DRB1*08/DQB1*06 were detected with a lower frequency compared to controls [170]. In contrast, in 23 Japanese BP patients, HLA-DRB1*04/DQA1*0301/DQB1*0302, DRB1*1101/DQA1*0505/DQB1*0302, DRB1*1101 and DQB1*0302 were associated with BP [171]. Genome-wide association studies in different populations to further address the genetic susceptibility of BP are currently being performed.
Environmental factors
No environmental factors have been discovered so far. Trigger factors are described earlier (in Predisposing factors).
Clinical features
History
A prodromal non-bullous phase usually precedes the development of tense generalized blisters. This prodromal phase may last for several weeks or even months. In this stage, clinical diagnosis is difficult. Pruritus, from mild to intractable, is typical and may even occur without skin lesions. In the prodromal phase, excoriated papules, eczematous, or urticarial lesions, haemorrhagic crusts and excoriations prevail.
Presentation
The bullous stage is characterized by intense pruritus accompanied with widespread tense blisters and vesicles on apparently normal or erythematous skin (Figure 50.15). Frequently, partly haemorrhagic crusts and urticated and infiltrated erythematosus plaques with an occasionally annular or figurate pattern are present (Figure 50.16) [172]. The blisters may obtain a diameter of many centimetres and contain a clear sometimes haemorrhagic exudates; the Nikolsky sign is negative. Pruritus, which may be incapacitating, is almost constantly present [150]. Blisters are typically symmetrically distributed and may persist for several days. After mechanical irritation erosions and yellowish or haemorrhagic crusts develop. Predilection sites involve the flexural aspects of the limbs and abdomen [173]. In the intertriginal areas, vegetating plaques may occur and oral lesions develop in 10–20% of cases [173, 174]. The mucosae of the eyes, nose, pharynx, oesophagus and ano-genital areas are rarely affected (reviewed in [175, 176]). Without severe superinfection all lesions heal without scarring. Erythema may persist at the sites of previous blisters for many weeks or months. Milia formation only rarely occurs.
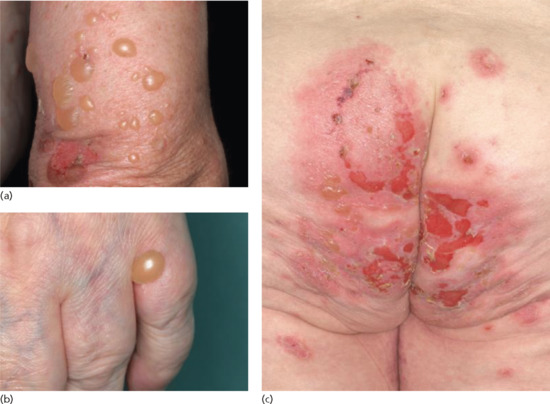
Figure 50.15 Classical bullous pemphigoid. Tense blisters and erosions on the arm (a), hand (b) and gluteal region (c). Blisters may arise on erythematous (a,c) or otherwise normal skin (b).
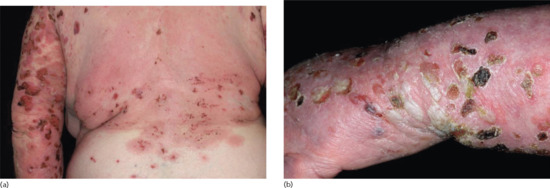
Figure 50.16 Classical bullous pemphigoid. Tense blisters, erosions, and partly haemorhagic crusts on the back and left arm (a) and left hand (b).
Clinical variants
Cutaneous manifestations of BP can be highly polymorphic. This notion has led to the description of several clinical variants (reviewed in [176]). In all of them, direct IF microscopy of a perilesional biopsy reveals linear deposits of IgG and/or C3 at the DEJ. At present, the fine specificities of serum autoantibodies were not shown to differ from the classical form.
Several clinical variants of BP have been described with a variety of different denominations, such as dyshidrosiform [177], prurigo nodularis-like [178, 179], prurigo-like [180], erythrodermic, ecthyma gangrenosum-like [181], intertrigo-like, papular, eczematous, lymphomatoid papulosis-like, vegetating [182–184], vesicular [185] and toxic epidermolysis-like pemphigoid [186, 187] (Figures 50.17 and 50.18). Some forms such as prurigo-like, papular, eczematous, vesicular and erythrodermic pemphigoid may later develop tense blisters and transform into the classical type. In a recent prospective study, about 20% of BP patients presented with the non-classical form at the time of diagnosis [174].
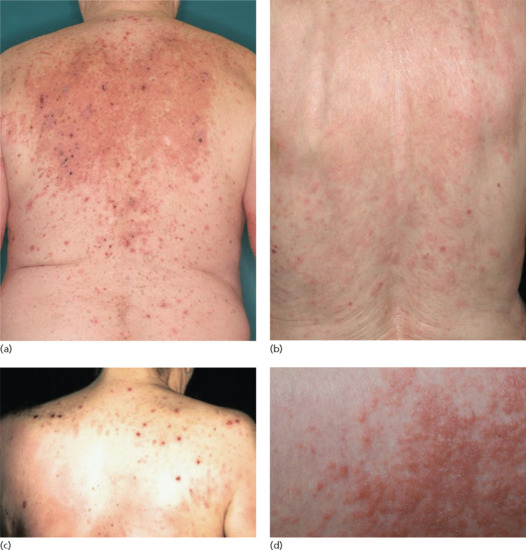
Figure 50.17 Clinical variants of bullous pemphigoid. Eczematous lesions with some erosions and crusts (a,b), prurigo-like variant with multiple excoriated papules (c) and papular variant (d).
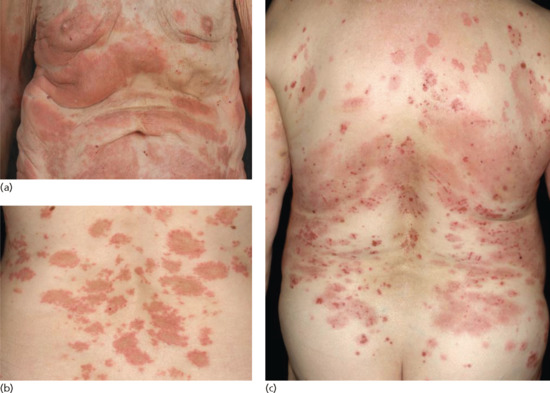
Figure 50.18 Clinical variants of bullous pemphigoid. Urticarial and erythematous plaques (a–c) accompanied by erosions and excoriations (c).
Localized bullous pemphigoid
In some patients, the disease is limited to certain body parts, most frequently the lower extremities notably the pretibial area [188–190]. Also other regions such as the flexures, palms, soles, genital area and umbilicus have been described [191, 192] as well as around stomata and haemodialysis fistulae (Figure 50.19) (see also Predisposing factors). Localized lesions may remain localized or develop into classical BP.
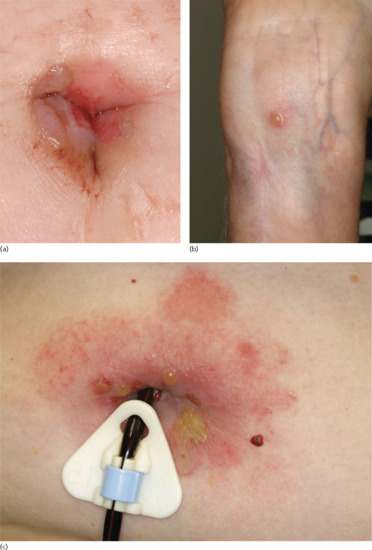
Figure 50.19 Localized bullous pemphigoid. Tense blisters and erosions limited to the umbilical area (a). Single tense blister at the site of major surgery (b). Eczema, erosions and tense blisters restricted to the site of percutaneous endoscopic gastrostomy (c).
Childhood bullous pemphigoid
Two peaks of incidences of BP in childhood were reported: in the first year of life (infantile BP), and around the age of 8 years [41]. Multiple cases with a close association with preceding vaccinations were reported, most of them in infants. Due to the high rate of vaccinations in this age group, a causative relation is difficult to confirm. In infants, the distribution of the lesions is often acral, in particular palmar and plantar [41]. In older children, involvement of the genital region occurs in almost half of the cases [41] (Figure 50.20). No immunopathological differences between BP in childhood and adults have been reported. Autoantibodies mainly target the NC16A domain of BP180 [193]. Generally, infants and children with BP have a good prognosis with remissions within weeks to a few months under therapy. For treatment, systemic corticosteroids are usually combined with dapsone or sulfapyri-dine [194].
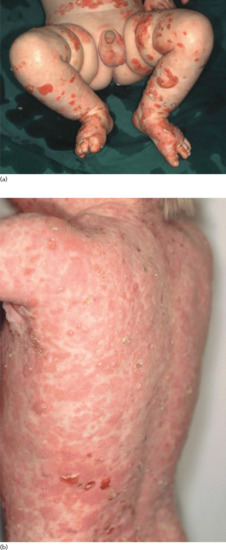
Figure 50.20 Childhood bullous pemphigoid. Disseminated tense blisters, erosions and crusts on the lower abdomen, genitalia and lower extremities in an infant (a). Generalized erythema, numerous tense vesicles and some erosions on the back of a 3-year-old boy (b).
Clinical features
Differential diagnosis
BP must be differentiated from other blistering autoimmune dermatoses (see Investigations; Figure 50.21). Diagnostic hallmarks are summarized in Table 50.3. In brief, diseases of the pemphigus group can be easily differentiated on the basis of distinctive clinical (positive Nikolsky sign) and immunopathological features. Mucous membrane pemphigoid is differentiated from BP by its predominant involvement of mucosal surfaces [195]. In contrast, the distinction of BP from linear IgA disease, EBA and anti-p200/laminin γ1 pemphigoid based simply on clinical and histopathological features is usually impossible and requires direct IF microscopy (for linear IgA disease) and serological analyses (for the latter two entities). In dermatitis herpetiformis, direct IF microscopy findings, and particularly the serological profile (presence of antitransglutaminase 1 and 2 as well as antigliadin IgA antibodies) are often required for diagnosis [172].
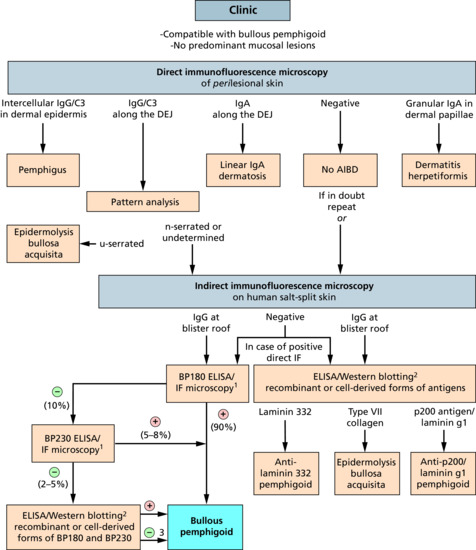
Figure 50.21 Diagnostic pathway for bullous pemphigoid. The diagnostic gold standard is still direct immunofluorescence (IF) microscopy of a perilesional biopsy. About 90% of patients can be diagnosed based on the clinical picture and serological tests. 1Commercially available assay; 2only available in specialized diagnostic centres; 3with positive direct IF microscopy and epidermal binding of immunoglobulin G (IgG) by indirect IF microscopy on salt-split skin or n-serrated/undetermined IgG binding by direct IF microscopy and no reactivity against laminin 332, p200 antigen, laminin γ1 and type VII collagen. AIBD, autoimmune bullous dermatoses; DEJ, dermal–epidermal junction; ELISA, enzyme-linked immunosorbent assay.
Furthermore, in the non-bullous prodromal stage or in atypical presentations, BP can closely resemble a variety of dermatoses including localized or generalized drug reactions, contact and allergic dermatitis, prurigo, urticaria, urticarial vasculitis, arthropod reactions, scabies, ecthyma or even pityriasis lichenoides. A detailed patient history, clinical evaluation, histopathological features and, above all, direct IF microscopy and serology are essential to distinguish these disorders from BP [172].
Classification of severity
An international panel of experts has recently proposed outcome measures for clinical studies and a clinical scoring system, the BP Disease Area Index [196]. The index records severity of: (i) skin lesions separately for classical lesions, i.e. erosions and blisters and atypicial lesions such as erythema, urticaria and others in 12 body regions; (ii) mucosal lesions; and (iii) pruritus.
Complications and co-morbidities
Patients with BP have a three times higher risk for pulmonary infection and embolism compared to controls [197, 198]. The association with neurological and psychiatric disorders as well as psoriasis is detailed earlier (see Associated diseases).
Disease course and prognosis
Untreated BP runs a chronic, self-limiting course over a number of months or years. Remission may occur within a few months or eruptions may continue for many years. Relapses are frequent and appear in about half of the patients [172]. Relapses appeared to be more frequent in patients with extensive disease, dementia and high serum levels of anti-BP180 NC16A antibodies at the time when treatment was omitted [199, 200]. The disease duration is usually 3–6 years, with most patients achieving complete remission off treatment.
The first-year mortality rates range from 10 to 40% which is about 2–6-fold higher compared to age- and sex-matched controls [22, 25, 33, 37, 174, 201]. Mortality varies considerably between different populations (out-patients versus in-patients, different countries, secondary or tertiary referral centres), and treatments. A 1-year mortality as high as 41% was observed in patients with extensive disease treated with prednisolone 1 mg/kg/day [202], while it was only 8% when class IV topical corticosteroids were combined with methylprednisolone 0.5 mg/kg/day, and dapsone [203].
Risk factors for lethal outcome are old age (greater than 80 years), extensive disease, high doses of prednisolone (>35 mg/day), serum albumin levels of less than 3.6 g/dL, a Karnofsky score of 40 or less, and the presence of heart disease, diabetes, or neurological diseases [33, 35, 36, 204, 205].
Investigations
The diagnosis is based on the combination of the clinical picture, direct IF microscopy and serology (Figure 50.22) [2, 144, 172, 206]. Guidelines for the diagnosis of BP have recently been published by the German Dermatological Society [207]. Direct IF microscopy and the serological assays are described above in more detail (see Pathology). About 90% of patients can be diagnosed based on the clinical picture and commercially available BP180- and B230-specific serological test such as ELISA and indirect IF microscopy [80–82, 144, 156].
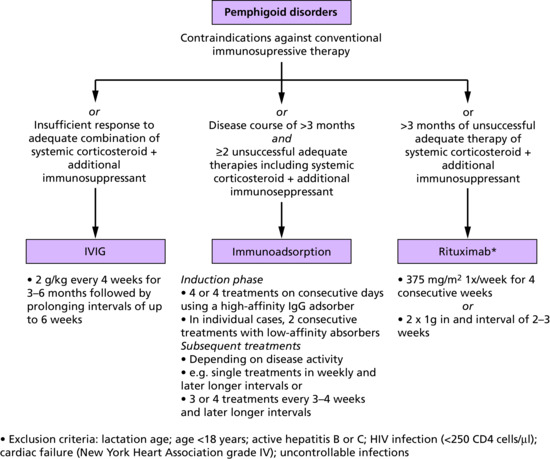
Figure 50.22 Indications and protocols for the treatment of refractory pemphigoid diseases with intravenous immunoglobulin (IVIG), immunoadsorption and rituximab. Summarized from four distinct consensus statements of international experts on autoimmune bullous disorders [233–235, 237].
Histopathology cannot differentiate BP from other pemphigoid diseases such as antilaminin 332 mucous membrane pemphigoid and anti-p200/laminin γ1 pemphigoid in all cases [208, 209]. The knowledge of the histopathological spectrum of BP, however, allows the histopathologist to arrange for direct IF microscopy and appropriate serological analyses to obtain a definite diagnosis. If these investigations are negative, other diagnoses may be considered (see Differential diagnosis). Linear deposits of C3d at the DEJ were detected in formalin-fixed biopsies of BP skin [210]. This method is, however, less sensitive than direct IF microscopy of frozen section and is hampered by its usually lesional site.
Management
The aim of treatment is to suppress disease activity with the minimum dose of drugs necessary. BP patients are: elderly; usually with several co-morbidities; commonly on many drugs; and susceptible to adverse reactions.
Topical and systemic steroids are the mainstay of treatment. Very potent topical steroids (0.05% clobetasol propionate ointment) should be considered in all patients. For localized BP, the lesional application of very potent topical steroids is often sufficient.
Controlled randomized trials
Only nine controlled prospective studies on treatment of BP have been published including oral corticosteroids (in several studies), the long-term whole-body use of very potent topical corticosteroids (in two), azathioprine (in two), plasmapheresis (in two) as well as mycophenolate mofetil and nicotinamide plus tetracycline (Table 50.4) [211].
Joly et al. showed in patients with moderate BP that clobetasol propionate 0.05% ointment (40 g/day, tapering over 12 months) was as effective and safe as oral prednisolone 0.5 mg/day [202]. In a subsequent study, lower doses of this high-potent topical corticosteroid (10–30 g daily, tapering over 4 months) did not differ in the time to achieve disease control compared to the more intensive topical regimen and was associated with less severe adverse events but more relapses in patients with moderate disease [212]. In patients with extensive disease (defined as >10 new bullae/day), treatment with oral prednisolone at an initial dose of 1.0 mg/kg/day resulted in significantly more severe adverse events compared to application of long-term very potent topical corticosteroids [202]. These two studies point to either oral prednisolone at a dose of 0.5 mg/kg/day or very potent topical corticosteroids as initial treatment of BP. Latter concept of long-term application of very potent topical corticosteroids on the entire body is, however, challenging in the elderly population that frequently suffers from neurological impairments. The controlled randomized clinical trials in BP are summarized in Table 50.4.
Controlled trials with dapsone, doxycycline, and IVIG are currently being analysed or recruiting.
Table 50.4 Controlled prospective trials for bullous pemphigoid
| Year [ref.] | Intervention | Number of patients | Outcome | Comment |
| 2009 [212] | Moderate disease: topical clobetasol propionate 0.05% 40 g/day for 1 year (high dose) versus topical clobetasol propionate 0.05% 30 g/day for 4 months (low dose) Extensive disease: low-dose versus high-dose regimen |
312 | Moderate disease: 2-fold lower risk for death and severe adverse events in the low-dose group. More relapses in the low-dose group (51% versus 32%) Extensive disease: no difference in efficacy and adverse events |
1-year mortality: 28–45% |
| 2007 [238] | Methylprednisolone 0.5 mg/kg/day plus azathioprine 2 mg/kg/day versus methylprednisolone 0.5 mg/kg/day plus mycophenolate mofetil 2 g/day | 73 | No difference in time to healing of all lesions and cumulative steroid dose (primary end points). More patients with elevated liver enzymes in the azathioprine arm | – |
| 2002 [202] | Moderate disease: topical clobetasol propionate 0.05% 40 g/day for 1 year versus prednisolone 0.5 mg/kg/dayExtensive disease: topical clobetasol propionate 0.05% 40 g/day for 1 year versus prednisolone 1.0 mg/kg/day | 341 | Moderate disease: no difference in efficacy and adverse eventsExtensive disease: lower 1-year mortality (primary end point) and less severe adverse events with topical steroids. More patients with no new blister after 3 weeks with topical steroids | Landmark study that showed the efficacy of topical steroids and the high risk of long-term higher dose (1 mg/day) prednisolone |
| 1994 [239] | Nicotinamide 3 × 500 mg/day plus oxytetrazycline 4 × 500 mg/day versus prednisolone 40–80 mg/day | 20 | Less adverse events in the tetracycline arm. No difference in efficacy | Low number of patients |
| 1993 [240] | Prednisolone 1 mg/kg/day versus prednisolone 1 mg/kg/day plus azathioprine 100–150 mg/day versus prednisolone 1 mg/kg/day plus plasmapheresis (4 times 1–5-fold plasma volume within 2 weeks) | 100 | No difference in efficacy (disease control after 4 weeks and 6 months). More severe adverse events in the azathioprine arm | Termination of the study following interim analysis after 6 months due to severe adverse events in 30% of patients, in 14% fatal |
| 1993 [241] | Prednisolone versus methylprednisolone, 1.0–1.5 mg/kg/day prednisolone equivalent | 57 | No difference in efficacy after 5 days. After 10 days less pruritus in the methylprednisolone group. 4 severe adverse events within the first 10 days | Double blinded, only 10 days follow-up |
| 1984 [242] | Prednisolone 0.75 mg/kg/day versus prednisolone 1.25 mg/kg/day for 3 weeks, then tapering | 46 | No difference in efficacy after 21 and 51 days (complete healing of lesions) and number of adverse events | – |
| 1984 [243] | Prednisolone 0.3 mg/kg/day versus prednisolone 0.3 mg/kg/day plus plasmapheresis (8 times 1.5-fold plasma volume within 2 weeks). When no response after 1 week prednisolone 1 mg/kg/day | 41 | Higher efficacy in the plasmapheresis group (more patients could be treated with the initial prednisolone dose, lower cumulative steroid dose) | Only mild adverse reactions of plasmapheresis |
| 1978 [244] | Prednisolone 30–80 mg/day versus prednisolone 30–80 mg/day plus azathioprine 2.5 mg/kg/day | 25 | Lower cumulative steroid dose with azathioprine after follow-up of 3 years | Long follow-up time |
Other clinical studies
Larger uncontrolled studies were performed with chlorambucil (prospective) as well as with dapsone, methotrexate and low-dose oral cyclophosphamide (all retrospective) [203, 213–216]. In addition, in small patient cohorts and individual cases, the successful use of erythromycin [217], ciclosporin [218], leflunomide [219], rituximab [220–222], immunoadsorption [223–225], high-dose IVIG [226–228] and omalizumab [126] has been reported.
Treatment guidelines
Guidelines, from the French reference centres for autoimmune bullous diseases, the British Association of Dermatologists, the German Dermatological Society and the European Academy of Dermatology and Venereology, for the treatment of BP have been proposed [229–232]. French centres recommend as first line treatment the long-term use of topical clobetasol propionate 0.05% ointment on the entire body (30 g/day for patients <10 new blisters/day; 40 g/day for patients >10 new blisters/day) [229]. British colleagues proposed the lesional application of topical clobetasol propionate 0.05% ointment with or without prednisolone 0.3 mg/kg/day or the combination of the topical steroid with anti-inflammatory antibiotics, e.g. doxycycline, tetracycline, minocycline and erythromycin as initial treatment for mild disease [230]. For moderate and severe disease, they recommend: (i) topical clobetasol propionate 0.05% (5–15 g twice daily to whole skin surface); (ii) prednisolone 0.5–1.0 mg/kg/day plus lesional topical steroid; or (iii) the combination of the topical steroid with anti-inflammatory antibiotics. The European guideline followed the French recommendations for the topical regimen. Alternatively, prednisolone at doses of 1.0 mg/kg/day for extensive disease and 0.5 mg/kg/day for mild BP is recommended. In patients with co-morbidities, tetracycline ± nicotinamide, azathioprine, mycophenolate, dapsone, chlorambucil and ciclosporin can be added or used alone [231].
Expert recommendations on indications and treatment protocols for rituximab [233], immunoadsorption [234] and IVIG [235–237] in BP are summarized in Figure 50.22.
Most clinicians would treat localized and mild BP with very potent topical corticosteroids alone (see Treatment ladder). For moderate and extensive BP, very potent topical corticosteroids on the whole body surface and/or oral prednisolone 0.5 mg/kg/day are recommended (see Treatment ladder). Depending on personal experience, this regimen can be combined with azathioprine, chlorambucil, dapsone, methotrexate, mycophenolic acid and tetracyclines (in alphabetical order). Due to the low 1-year mortality of only 8% in a large cohort of BP patients treated with methylprednisolone 0.5 mg/kg/day in combination with topical corticosteroids and dapsone [203], the author favours dapsone as combination therapy, and in case of contraindications, doxycycline. In refractory patients, immunoadsorption/plasmapheresis, rituximab or IVIG may be added (see Treatment ladder).
Mucous membrane pemphigoid
Definition and nomenclature
An international consensus conference defined mucous membrane pemphigoid (MMP) as an immunobullous disease with autoantibodies against components of the DEJ and predominant mucosal involvement [1] (see Table 50.3). Previously, the term cicatricial pemphigoid was used synonymously for MMP. Currently, cicatricial pemphigoid only refers to the rare clinical variant in which mucous membranes are not predominantly affected and skin lesions heal with scarring [1]. Due to the new definition, there is some overlap with linear IgA disease (in patients with predominant IgA anti-DEJ antibodies) and EBA (in patients with autoantibodies against type VII collagen) [2] (Figure 50.23).
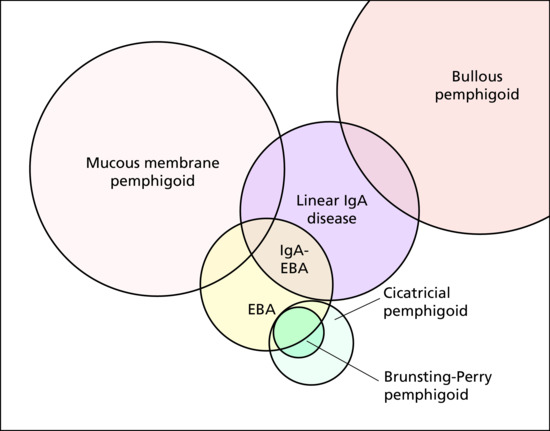
Figure 50.23 Diagnostic overlap between mucous membrane pemphigoid, linear IgA disease, epidermolysis bullosa acquisita (EBA), cicatricial pemphigoid and bullous pemphigoid. Brunsting–Perry pemphigoid can be regarded as variant of cicatricial pemphigoid or EBA. The size of the circles approximates the incidence of the diseases.
Introduction and general description
MMP is the second most frequent autoimmune blistering disease in central Europe. It predominantly affects the mucous membranes, most frequently the oral cavity followed by the conjunctivae, naso-pharynx, skin, ano-genital region, larynx and oesophagus [1, 3, 4]. Severe complications may arise with laryngeal and oesophageal involvement; conjunctival lesions result in scarring with a high risk of vision impairment and, ultimately, blindness.
The disease was first differentiated from pemphigus and BP in 1953 by Walter Lever as benign MMP or pemphigus conjunctivae [5]. Earlier reports of the disease are available by Wichmann (1798), Cooper (1858), Morris and Roberts (1889) and Thost (1911) [6–8].
The diagnosis is based on the clinic presentation and the detection of linear deposits of IgG and/or IgA and/or C3 at the DEJ by direct IF microscopy of a perilesional biopsy [1]. Serum autoantibodies are only present in low titres compared to BP. In about half of the patients, indirect IF microscopy on salt-split human skin, the standard screening test for serum autoantibodies in pemphigoid diseases, is negative [9, 10]. In MMP, six target antigens have been characterized at the molecular level: BP180; BP230; laminin 332; both subunits of α6β4 integrin; and type VII collagen [2]. Reactivity with laminin 332 was shown to be associated with a malignancy in 30% of patients [11]. The pathogenic relevance of autoantibodies against the α6 and the β4 subunits of α6β4 integrin has been demonstrated in vitro and against laminin 332 in vivo [12–16]. The clinical features and immunopathology are summarized in Table 50.3.
Epidemiology
Incidence and prevalence
In the early 1990s, the incidence was determined to be 0.9 and 1.3 per million per year in Germany and France, respectively. Ten years later, the incidence appeared to have increased to 2.0 per million per year in Germany [17–19]. The incidence of ocular MMP was calculated to be 0.8 per million per year in the UK [20].
Age
MMP is a disease of late middle to old age with a mean age of onset of 60–65 years [4, 21–24]. It can also affect children and teenagers [25].
Sex
In a compilation of large case series, a female predominance of 1.5- and 2.3-fold was reported [21, 24].
Ethnicity
No geographical or racial predilection was observed.
Associated diseases
In several patient cohorts, about 30% of patients with antilaminin 332 antibodies presented with a solid cancer [11, 26, 27]. No association with a malignancy was found in MMP patients overall irrespective of the target antigen as well as in patients with anti-α6β4 reactivity [28–30]. In some patients, an association with other autoimmune disorders such as rheumatoid arthritis, systemic lupus erythematosus and polyarthritis nodosa was observed [1].
Pathophysiology
Six different target antigens in patients with MMP have been identified at the molecular level: BP180 (in about 75% of patients [9, 31]); BP230 (in 25%); laminin 332 [32] (formerly termed laminin 5 and epiligrin; in 20–25% [9, 33]; α6 integrin; β4 integrin; and type VII collagen (see Figure 50.10 and Table 50.3). Reactivity against multiple antigens has frequently been reported [31, 34–36]. In particular, BP230 antibodies are often found in MMP patients with anti-BP180 and anti-β4 integrin reactivity [31, 34]. In contrast to BP, C-terminal epitopes on BP180 are predominantly recognized in MMP rather than the NC16A domain which is targeted by about 50% of BP180-reactive sera [9, 31, 34, 37–41]. In about a third of BP180-reactive sera, additional reactivity against epitopes on the intracellular portion of the molecule are described [9]. In almost all patients with antilaminin 332 reactivity, the α3 chain is targeted [42]. Autoantibodies to α6 integrin have been predominantly detected in patients with oral lesions and reactivity against β4 integrin in ocular involvement [43–45]. Interestingly, antibodies against β4 integrin predominantly react with the intracellular domain [46], thus a preceding event leading to a partially damage of the DEJ seems likely.
Autoantibodies in MMP may be IgG, IgA or of both isotypes [9, 10, 47–49]. In a series of 19 patients, a third exclusively had IgA antibodies against BP180 [9]. A dual IgG and IgA autoantibody reactivity and antibodies against laminin 332 were associated with a more severe clinical phenotype [33, 50]. Higher disease activity was also linked to reactivity with multiple BP180 epitopes and the HLA class II alleles DQB1*301, DRB1*04 und DRB1*11 [10, 31]. IgA and IgE antibodies against laminin 332 have also been reported in individual MMP patients [51, 52].
The pathogenic relevance of autoantibodies in MMP has been demonstrated both in vitro and in vivo. Passive transfer of antilaminin 332 IgG to neonatal or adult mice induced subepidermal blisters of skin and mucous membranes reproducing clinical, histological and immunopathological features of human MMP [14]. Of note and in contrast to BP, injection of Fab fragments against laminin 332 resulted in lesion formation and lesions could also be induced in mice lacking complement, mast cells or T cells, suggesting that such antilaminin 332 antibodies elicit epidermal detachment in an Fc-independent manner [12, 14]. Antibodies to the α6 and β4 subunits of α6β4 integrin were shown to induce separation along the DEJ in organ cultures of oral and conjunctival mucosa [15, 16, 44, 53]. Recently, injection of rabbit IgG against intracellular fragments of β4 integrin in neonatal mice induced subepidermal skin blisters [54]. In line with these data, a correlation of the ocular disease activity and serum autoantibody levels against β4 integrin was reported [55].
Limited data about the role of T cells are available in MMP. In a subgroup of patients, peripheral T cells released interferon (IFN)-γ after stimulation with NC16A peptides [56].
Since scarring is the major pathogenic process in conjunctival disease, fibrosis has been intensively studied in biopsies and cultured conjunctival fibroblasts. Various profibrotic factors were identified, including heat shock protein 47, connective tissue growth factor, transforming growth factor (TGF)-β, IL-4, IL-5, IL-13 and TNF-α [57–59].
Predisposing factors
No predisposing factors are known. It may be speculated that in the subgroup of antilaminin 332 MMP associated with solid cancers, pathological expression of laminin 332 in the tumour tissue may have triggered the autoimmune disease.
Pathology
Histopathology
Histological examination of a blister is helpful only if the blister is intact and recent. Biopsy of an erosion is not adequate. Blisters in the mouth and on the skin show subepithelial or subepidermal blister formation, but often lack distinctive and diagnostic features. There are usually fewer eosinophils present in the cutaneous lymphohistiocytic infiltrate than in BP; however, MMP cannot be differentiated from other pemphigoid diseases based on histopathology [7, 60]. At a later stage, fibrosis, the distinctive feature of MMP, may develop. The conjunctiva shows epithelial metaplasia, reduced numbers of goblet cells, a lymphocytic infiltrate with plasma cells and mast cells in the substantia propria, fibrosis of the lamina propria accompanied by inflammatory cells and an appearance of granulation tissue in the submucosa [61].
Tissue-bound autoantibodies
Tissue-bound autoantibodies can be visualized by direct IF microscopy of a perilesional biopsy. Linear deposits of IgG, C3 and/or IgA at the DEJ are diagnostic together with a compatible clinical phenotype (see Figure 50.13) [1]. For further differentiation of the anti-DEJ staining, the n- versus u-pattern analysis can be performed [62] (see Figure 50.13). In all pemphigoid disease except EBA, an n-serrated pattern is seen. Alternatively, the biopsy is treated with 1 M NaCl solution to induce an artificial split in the DEJ at the lamina lucida level [63]. Autoantibody specificities can then be detected by indirect IF microscopy on 1 M NaCl-split skin (see below; see Figure 50.14). A further, not routinely available, method is the double IF labelling of tissue-bound and defined antigens of the DEJ analysed by laser scanning confocal microscopy [64] that differentiates antilaminin 332 MMP from BP, anti-p200 pemphigoid and reactivity with type VII collagen.
Serum autoantibodies
Major characteristics of serum autoantibodies have been described above (see Pathophysiology section).
For routine analysis for circulating autoantibodies indirect IF microscopy on 1 M NaCl-split human skin is most appropriate (Figure 50.14). Its sensitivity of 50–70% which is considerably lower as in BP [9, 65–67]. Reactivity depends on the target antigen(s) and can be epidermal (most cases), dermal or both (Figure 50.14). Using skin from patients with epidermolysis bullosa that lack laminin 332 or type VII collagen expression by indirect IF in comparison with normal skin is an elegant method to detect antibodies against the two antigens [68].
Alternatively, monkey oesophagus, a BIOCHIP mosaic (Figure 50.15), and ELISA systems for anti-BP180 and anti-BP230 antibodies (all commercially available) can be applied. Furthermore, indirect immunoelectron microscopy [69] (rarely available) and various in-house ELISA, immunoblotting and immunoprecipitation analyses using cell-derived or recombinant forms of BP180, BP230, laminin 332, α6β4 integrin and type VII collagen may be used.
Genetics
Associations with HLA-DQB1*03(01) [70–75], DRB1*04 [74] and DRB1*11(01) [73, 74] have been reported. In contrast, a decreased frequency of the HLA-DRB1*02 allele was noted [73]. In MMP with ocular involvement, associations with HLA-DQB1*03(01) [70, 72, 76–78], DRB1*04 [72] and HLA-B12 [79] were found.
Environmental factors
No environmental factors have been discovered so far.
Clinical features
Presentation
MMP most frequently affects the oral cavity (in 85% of patients) followed by conjunctivae (65%), skin (25–30%), nasal cavity (20–40%), ano-genital area (20%), pharynx (20%), larynx (5–10%) and oesophagus (5–15%) [1, 3, 4]. At all affected body sites except the oral cavity, lesions tend to heal with scarring.
The extent of oral lesions may vary considerably from mild almost asymptomatic erosions and chronic gingivitis to extensive extremely painful ulcers (Figure 50.24). Nasal lesions may present as haemorrhagic crusts and epistaxis and can finally lead to disfiguring fibrosis and septum perforation. Pharyngeal lesions manifest with odynophagia, initial involvement of the larynx as hoarseness. Oesophageal disease becomes symptomatic with dysphagia, odynophagia and heartburn. Genital lesions usually present with erosions (Figure 50.25). Scarring may lead to labial fusion and introital shrinkage with end-stage scarring that may be indistinguishable from lichen sclerosus [80]. Skin lesions may either resemble BP or heal with scarring and milia formation (Figure 50.26).
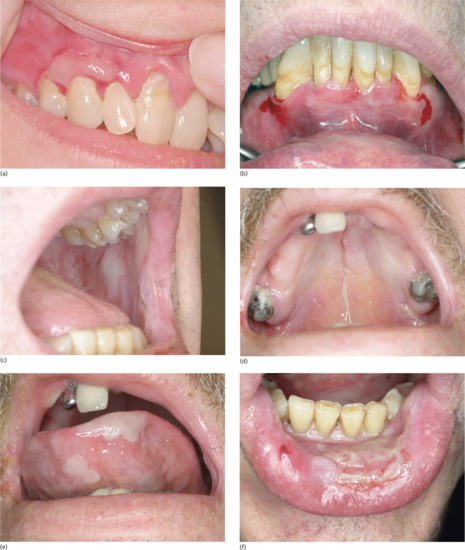
Figure 50.24 (a–f) Oral lesions in mucous membrane pemphigoid.
Figure 50.25 Genital involvement in mucous membrane pemphigoid: (a) male, and (b) female.
Figure 50.26 Skin lesions in mucous membrane pemphigoid. Brownish erythema, erosions, and some crusts on the right shin (a). Erosions, ulcerations and some atrophic scars on the right buttock (b). Multiple crusted erosions on the right hand (c). Atrophic alopecic scar on the right occiput (d). Of note, all patients had concurrently predominant mucous membrane lesions (not shown).
Ocular lesions, usually unilateral initially, with subtle symptoms such as burning, dryness and foreign-body sensation and may proceed to scar formation causing shortening of the inferior fornix, symblepharon, trichiasis, neovascularization and, finally, blindness (Figure 50.27) (reviewed in [57, 61, 81]). Within 2 years, the disease is usually bilateral [3]. All patients with MMP should be examined by an ophthalmologist to detect subtle changes by slit-lamp examination and measurement of the fornix depth (Figure 50.28). The latter is an objective clinical parameter for disease activity. In patients without initial ocular involvement, the annual risk for developing eye lesions was 5% over the first 5 years [4].

Figure 50.27 Ocular disease in mucous membrane pemphigoid. Conjunctival hyperaemia, inferior fornix shortening and loss of the plica in early disease (Foster II, Mondino II, Tauber IIb) (a). Conjunctival inflammation and limbal scarring (black arrows) in early disease (Foster II, Mondino I, Tauber IIa) (b). Loss of the temporal fornix and symblepharon with loss of lashes (Foster III, Mondino stage III, Tauber IId) (c). Complete loss of inferior fornix with some symblephara and loss of lashes (Foster III, Mondino IV, Tauber IId, IIIb) (d). (Parts b, e and f, courtesy of Dr J. K. D. Dart, Moorfields Eye Hospital, London, UK.). Diffuse conjunctival hyperaemia and lower lid entropion with lashes brushing the cornea (trichiasis). Since the lower lid is not everted no staging can be performed (e). End‐stage disease showing a ‘frozen globe' and keratin covering the dry surface (Foster IV, Mondino stage IV, Tauber IV) (f).
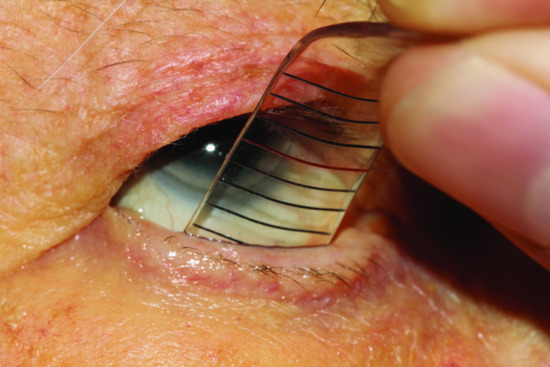
Figure 50.28 Fornix meter. Measurement of the fornix depth by an experienced ophthalmologist is important for the objective assessment of ocular disease activity.
(Courtesy of Dr G. Geerling, University of Düsseldorf, Germany.)
Of note, since in about 30% of patients with antilaminin 332 MMP a solid cancer is present, a thorough search for malignancy is required in patients with this subtype of MMP [11].
Clinical variants
Oral pemphigoid
The term oral pemphigoid is used when the disease is restricted to the oral cavity. In this entity, Bhol et al. have preferentially detected autoantibodies against the α6 chain of α6β4 integrin [44].
Ocular pemphigoid
When MMP is confined to the conjunctivae, which is found in about 20% of patients, the term ocular pemphigoid is applied [1, 4]. In this subgroup of MMP patients, Tygai et al. observed predominant autoantibody reactivity with the β4 chain of α6β4 integrin [45].
Vulvar pemphigoid
This rarely reported entity is characterized by recurrent blistering confined to the vulva of young girls, which does not result in scarring [82, 83]. This form usually responds well to topical corticosteroids.
Differential diagnosis
The oral lesions, which may appear first, must be differentiated from pemphigus vulgaris, paraneoplastic pemphigus, oral lichen planus, Behçet disease, Stevens–Johnson syndrome and bacterial gingivitis [4]. Pemphigus can be excluded by direct IF microscopy or serologically. Lichen planus and Stevens–Johnson syndrome may be diagnosed histopathologically or, more easily, when skin or nail involvement is present.
For ocular disease, in particular when the conjunctivae are the only site of manifestation (ocular pemphigoid) and direct IF microscopy is negative (in about 20% of ocular pemphigoid), diagnosis can be challenging. Ocular rosacea, chronic antiglaucoma therapy, conjunctival lichen planus, Stevens–Johnson syndrome, toxic epidermal necrolysis, Sjögren syndrome, graft-versus-host disease, chronic allergic conjunctivitis, severe atopic eczema, trauma, and viral and bacterial infections need to be excluded [4, 20, 57, 81]
.Classification of severity
Several classifications for ocular involvement of MMP have been proposed. The classification of Foster et al. is relatively simple and may also be applied by dermatologists [84]. The Mondino and Tauber systems require the measurement of fornix depth and better allows the documentation of disease progression (Table 50.5) [85–88]. An international panel of experts has recently proposed definitions for outcome measures and a clinical scoring system, the MMP Disease Area Index. The index records severity of skin lesions at 12 anatomical sites, scalp, mucosal lesions at 10 sites, and both eyes [89].
Table 50.5 Classification of ocular disease in mucous membrane pemphigoid
| Stage | Foster et al. [84] | Mondino et al. [85–87] | Tauber et al. [88] |
| I | Subconjunctival scarring/fibrosis | Loss of inferior fornix0–25% | Subconjunctival scarring/fibrosis |
| II | Shortening of fornix | 25–50% | Shortening of fornix a–d describes loss of inferior fornix deptha:
|
| III | Symblepharon | 50–75% | Symblepharon a–d describes horizontal involvement by symblepharaa:
|
| IV | Ankyloblepharon | 75–100% | Ankyloblepharon |
Complications and co-morbidities
Ocular inflammation and fibrosis can lead to the destruction of the tear ducts, corneal ulceration, corneal pannus and, ultimately, blindness. If laryngeal involvement is severe, life-threatening stenosis can occur requiring tracheotomy [90]. Deafness from involvement of the middle ear has been reported [22] as well as carcinoma arising from chronic oral and oesophageal lesions [91].
In about 30% of patients with antilaminin 332 MMP, a solid cancer develops [11].
Disease course and prognosis
MMP is typically a chronic and progressive disease. The disease often extends over many years with periods of activity and extension followed by quiescent phases. Unlike BP and linear IgA disease, MMP rarely goes into spontaneous remission except in localized oral disease [7, 92, 93]. Patients with dual IgG and IgA anti-DEJ autoantibodies were shown to have a more severe and persistent disease [10].
Investigations
Diagnosis is based on the combination of the clinical picture, direct IF microscopy and serology (Figure 50.29). In contrast to BP, diagnosis is complicated by the generally lower amounts of autoantibodies both tissue bound and circulating. In ocular involvement, close collaboration with an ophthalmologist experienced with the disease is mandatory for diagnosis, treatment decisions and follow-up.
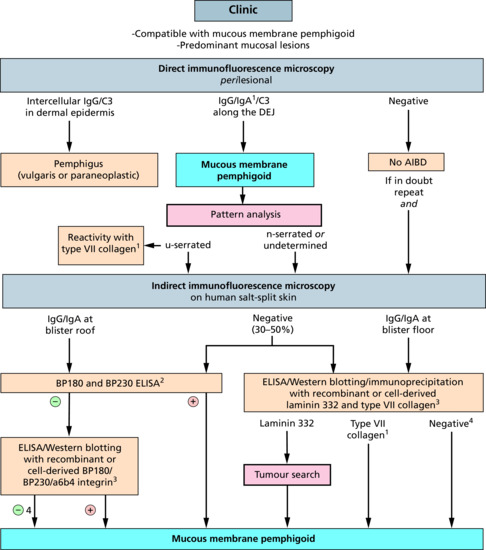
Figure 50.29 Diagnostic pathway for mucous membrane pemphigoid. (1) Overlap with linear IgA disease (with exclusive IgA reactivity) and EBA (with reactivity against type VII collagen) (see Figure 50.1). (2) Commercially available (for IgG antibodies). (3) Only available in specialized diagnostic centres. (4) With positive direct and/or indirect immunofluorescence microscopy. AIBD, autoimmune bullous dermatoses; DEJ, dermal–epidermal junction; ELISA, enzyme-linked immunosorbent assay.
Direct IF microscopy is indistinguishable from BP, deposition of IgG and/or C3 and, to a lesser degree, of IgA along the DEJ is detected. In ocular MMP, detection of autoantibodies by direct IF microscopy may fail in up to 20% [4]. The site for biopsy often presents a problem. We recommend the intact buccal mucosa for patients with oral lesions and also for ocular pemphigoid. If negative, a conjunctival biopsy is required in suspicion of ocular pemphigoid. Since conjunctival biopsies bear the risk of disease exacerbation infiltration anaesthesia may be avoided, biopsy size limited to 2 × 3 mm, the upper fornix or limbus chosen as biopsy site, and surgery may follow topical treatment with corticosteroids and antibiotic for 1 week.
By indirect IF microscopy on salt-split skin, epidermal and/or dermal staining of the artificial split is seen depending on the target antigen (Figure 50.13). In contrast to BP, MMP sera contain anti-DEJ reactivity at low titres (1 : 10–1 : 40) and in a lower percentage (50–80%) [3, 9, 10, 34]. In about 60% of MMP sera, IgA autoantibodies can be detected [9, 10, 31, 34, 94] and in a third of sera, autoantibody reactivity is restricted to the IgA isotype [9]. Due to the relatively low indirect IF microscopy reactivity it is recommended to further use antigen-specific test systems to detect serum autoantibodies even in indirect IF-negative patients.
Various recombinant fragments as well as the cell-derived forms of BP230 and BP180 including the soluble ectodomain of BP180 (LAD-1) and the commercially available ELISA systems were used to detect anti-BP230 and anti-BP180 autoantibodies [9, 31, 34, 37–41].
Serum antibodies against laminin 332 can be determined by various methods, none of them being commercially available yet. The most practical appeared to be immunoblotting and ELISA with the extracellular matrix of cultured human keratinocytes and purified laminin 332 from medium of cultured keratinocytes, respectively, the most sensitive is immunoprecipitation of radiolabelled keratinocytes [33, 95, 96]. Elegantly, indirect IF microscopy on laminin 332-deficient skin can be performed [68]. Immunoblotting with bovine gingiva lysate allows detection of α6β4 integrin-specific antibodies [43–45].
Management
Treatment of MMP is challenging for three reasons: (i) only three controlled therapeutic trials have been conducted; (ii) clinical response to immunosuppression in patients with severe disease, in particular with ocular lesions, is poor; and (iii) conjunctival fibrosis is irreversible and, in contrast to other pemphigoid diseases, causes permanent damage when treatment is delayed or ineffective [1, 61]. In fact, conjunctival scarring may even continue for some time after inflammation has been successfully treated.
Controlled randomized trials
In all three trials, only ocular involvement was studied. One trial with 24 patients showed a superior effect of oral cyclophosphamide 2 mg/day plus prednisolone 1.0 mg/kg/day versus prednisolone 1.0 mg/kg/day alone. The other trial included 40 patients and compared dapsone 2 mg/kg/day and cyclophosphamide 2 mg/day with response in 14 and 20 patients, respectively [61, 97]. In a more recent study with 30 MMP patients from Egypt, the IV application of the anti-TNF-α drug pentoxyfylline in combination with corticosteroid and cyclophosphamide pulses was more effective than corticosteroid and cyclophosphamide pulses alone [98].
Other clinical studies
Larger uncontrolled studies showed efficacy of the sulfa drugs dapsone, sulfapyridine and sulfamethoxypyridazine (in oral, ocular and generalized MMP) [97], cyclophosphamide (ocular, oral and generalized MMP) [97, 99], minocycline (oral MMP) [97], topical mitomycin (ocular MMP) [100, 101], methotrexate (ocular MMP) [102], mycophenolate mofetil (ocular MMP) [103] and IVIG (ocular and oral MMP) [104–106]. A comprehensive overview was given by the Cochrane Collaboration including studies until 2005 [107]. In addition, individual patients were successfully treated with adjuvant etanercept and infliximab [108–111]. These reports are in line with the high levels and profibrotic activity in ocular lesions and the clinical effect of pentoxyfylline described by Saw et al. and by El Darouti et al. [59, 98].
A retrospective study with 115 patients with ocular lesions (223 eyes) reported the highest efficacy for cyclophosphamide followed by mycophenolate, azathioprine and dapsone with the lowest number of adverse reactions for mycophenolate, the highest for azathioprine [112]. In a similarly designed study with 15 MMP patients, colchicine was identified as most effective drug in combination with prednisolone 40 mg/day compared to azathioprine, cyclophosphamide, tetracycline and dapsone [113].
Since the first use of rituximab in severe treatment refractory MMP that led to healing of all oral lesions but could not prevent progression of ocular disease [114], data from more than 40 patients, mostly with generalized MMP, are available [115–119], the largest study including a cohort of 25 cases [117]. In about 70% of patients, all lesions had healed, which is only about 10% less than in treatment-refractory pemphigus [118, 120]. However, relapses occurred in about half of the MMP patients [115, 119].
Treatment guidelines
Guidelines were by provided by the international consensus conference of MMP and the French reference centres for autoimmune bullous diseases [1, 121].
In ‘low-risk’ patients (lesions are limited to the mouth with or without skin involvement), potent topical corticosteroids, tetracycline plus nicotinamide, and dapsone have been proposed. If ineffective, oral prednisolone (0.5 mg/kg/day) with or without azathioprine 100–150 mg/day was recommended [1]. For ‘high-risk’ patients (any other mucosal site affected), prednisolone 1.0–1.5 mg/kg/day plus cyclophosphamide (1–2 mg/kg/day orally or 500–1000 mg IV in 3–4-week intervals) has been recommended in ocular, oesophageal or laryngeal involvement with rapid progression [1]. For patients with mild disease, dapsone 50–200 mg/day can be used [1].
In patients without laryngeal involvement and without severe ocular disease (Foster I and II), French colleagues recommended topical corticosteroids or dapsone and in case of inefficacy, sulfasalazine (alone or in combination with dapsone), mycophenolates, azathioprine, cyclophosphamide, IVIG or etanercept. For patients with severe ocular involvement, dapsone plus IV cyclophosphamide 750 mg/m2, etanercept, IVIG or rituximab plus prednisolone 0.5–1.0 mg/kg were suggested [1, 121].
Treatment of ocular disease, including local and surgical therapy, is reviewed elsewhere [57, 81].
Management algorithm
Treatment of MMP is largely tempered by its severity and sites of involvement. Local treatments are crucial and may be sufficient to control oral and genital disease to an acceptable level [122]. Secondary infection at these sites with candida is common and should be treated with antifungal therapy. Oral hygiene including patient instructions and professional periodontal therapy are recommended since they significantly reduced periodontal plaques and bleeding in oral MMP [123]. In ocular involvement, close cooperation with an ophthalmologist experienced in MMP is required to monitor ocular disease activity, adjust topical treatment, and surgical therapy including control of trichiasis [57, 81]. Suggested treatment regimens are detailed in the Treatment ladder. In refractory patients, rituximab [124], immunoadsorption [125, 126] and IVIG [127, 128] can be chosen (see Figure 50.22).
Linear IgA disease
Definition and nomenclature
This subepidermal blistering disease is defined by its main immunopathological feature, the exclusive or predominant binding of IgA along the DEJ (Table 50.3). Linear IgA disease (LAD) is the most frequent autoimmune blistering disease in infants and children. For this age group, several terms have been designed (see Synonyms and inclusions). It later became clear that immunopathologically, diseases in children and adults are identical although some clinical features may slightly differ. The use of the term LAD for both age groups is therefore recommended. Some overlap is seen with BP (in patients with dual IgG and IgA deposition along the DEJ), with MMP (in patients with predominant mucosal involvement) and EBA (in patients with IgA autoantibodies against type VII collagen; Figure 50.23) [1]50.23.
Introduction and general description
Tense blisters, vesicles and annular erythema are the clinical hallmarks. Frequently, blisters are arranged annularly, a formation referred to as ‘crown of jewels’, and mucous membrane lesions are present. Skin lesions have the same morphology in children and adults; however, they arise more abruptly in children and sites of predilection are different [2, 3].
LAD was first differentiated from dermatitis herpetiformis in 1975 by Chorzelski and Jablonska by the finding of linear deposits of IgA antibodies at the DEJ by direct IF microscopy [4–6]. The linear deposits of predominant or exclusive IgA antibodies at the DEJ are still the diagnostic hallmark of LAD [1]. As for the other pemphigoid diseases, indirect IF microscopy on human salt-split skin is an appropriate screening test for serum IgA antibodies in suspected LAD [7, 8]. Immunodominant epitopes were later localized on the ectodomain of BP180 [9, 10]. Reactivity with type VII collagen, BP230, laminin 332 and various not molecularly characterized antigens have also been described [11–18].
The clinical features and immunopathology are summarized in Table 50.3.
Epidemiology
Incidence and prevalence
An incidence of 0.25–1.0 patients with LAD per million per year was reported in central Europe, Singapore and Kuwait [19–22]. The vast majority of patients are adults. The incidence appeared to be higher in developing countries such as Malaysia, India, Thailand, Tunisia, Mali, South Africa and Uganda [23–29]. It may be speculated that this is related to the different age distribution of the populations with up to half of the population being minors in these countries [23].
Age
LAD is the most frequent immunobullous disorder in children, there being two peaks of onset, below the age of 5 and between the age of 60 and 65 years [2].
Sex
A slight female predominance was observed [2], however, larger studies are lacking.
Ethnicity
No geographical or racial predilection was observed.
Associated diseases
A slightly higher frequency of lymphoproliferative disorders and non-lymphoid malignancies as well as ulcerative colitis compared to the general population has been found [30–32]. Furthermore, numerous case reports with concomitant diseases associated with LAD have been published [3].
Pathophysiology
The major target antigen is the ectodomain of BP180. Initially, two target molecules were described. Zone et al. reported a 97 kDa protein in the extract of human epidermis and dermis termed the linear IgA bullous dermatosis antigen (LABD97) and Marinkovich et al. described a 120 kDa protein in the conditioned culture supernatant of human keratinocytes, referred to as linear IgA antigen 1 (LAD-1) [9, 10]. Later, it became clear that both antigens represent C-terminal portions of BP180 [33, 34] (Figure 50.30). The N termini of both antigens were localized within the NC16A domain of BP180 [35]. LAD-1 has different N termini since the shedding of this fragment from the cell surface is mediated by at least three different sheddases (ADAM 9, 10 and 17) [36] producing BP180 fragments with different cleavage sites between Asp514 and Ala531 [34, 35, 37] (Figure 50.30). Some of them may function as neoepitopes [37]. In contrast to BP and MMP, the BP180 NC16A domain is targeted in only 20% of LAD patients [38]. Exclusive IgA reactivity against the NC16A domain has only be described in individual patients [38, 39]. Most patients react with multiple epitopes on the BP180 ectodomain [38, 40]. In line with this, NC16A-specific T cells have been identified in LAD patients [41].
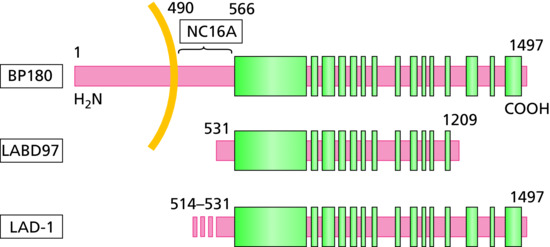
Figure 50.30 Schematic diagram of BP180 (type VII collagen) and its cell-derived fragments recognized by linear IgA disease (LAD) sera. Most LAD sera contain IgA reactivity against two fragments of the BP180 ectodomain, the 97 kDa linear IgA bullous dermatosis antigen (LABD97) and the 120 kDa linear IgA disease antigen 1 (LAD-1). While LABD97 is present in extract of human epidermis and dermis, LAD-1 is shed by ADAM 9, 10 and 17 from membrane-bound full-length BP180 of cultured human keratinocytes and, depending on the particular ADAM, has different N termini. In only about 20% of LAD sera, IgA antibodies against the extracellular portion of the 16th non-collagenous domain (NC16A), the immunodominant region in bullous pemphigoid and pemphigoid gestationis, are found. Green boxes, collagenous domains; yellow, cell membrane. Amino acid numbers are depicted above the molecules.
IgA antibodies were reported to be exclusively of the IgA1 subclass [42]. In most patients, in addition to IgA antibodies, anti-DEJ antibodies of the IgG isotype are present [43, 44]. In fact, the majority of LAD sera, in addition to IgA anti-BP180 antibodies, also contain IgG antibodies against BP180 [40, 45]. Interestingly, in most BP sera, IgA anti-BP180 antibodies can also be detected [45]. The two diseases may thus be regarded as different ends of a continuous spectrum with some overlap [45] (see Figure 50.23). This notion is supported by the finding that the isotype of anti-DEJ reactivity was associated with the age of the patients: in younger patients, IgA autoantibodies predominated, whereas in older patients, preferentially anti-DEJ antibodies of the IgG isotype were found [23]. For patients with equal IgG and IgA reactivity against the DEJ, the diagnosis ‘mixed immunobullous dermatosis’ or ‘linear IgA/IgG bullous dermatosis’ was proposed [43, 46] (see Clinical variants later).
The mechanism of blister formation in LAD is not fully understood, but is likely to involve IgA- and complement-mediated neutrophil chemotaxis. The pathogenic role of autoantibodies was first suggested by the adherence of stimulated human neutrophils along the DEJ after preincubation with LAD serum [47]. In line with this, in cultured human skin samples, the incubation with LAD sera resulted in dermal–epidermal separation [48]. In both approaches, the direct effect of serum proteases or IgG antibodies could not be excluded. The injection of monoclonal IgA anti-LAD-1 antibodies in human skin grafted onto SCID mice leading to microscopic subepidermal splitting in some of the mice, was the first direct proof of the pathogenic potential of IgA antibodies in LAD [49].
In conjunction with anti-BP180 antibodies, IgA reactivity against BP230 can be found in some LAD sera [15, 50]. In drug-induced LAD, IgA autoantibodies also target BP180 and BP230 [51]. Predominant or exclusive IgA antibodies against type VII collagen have been reported in a number of patients [12, 16, 52]. These patients may also be diagnosed as IgA EBA (see later) (see Figure 50.23). In two patients, IgA antibodies against laminin 332 were reported in conjunction with IgA and IgE reactivity against this protein, respectively [13, 14]. An individual patient with exclusive IgA reactivity against the p200 antigen was also described [53].
Predisposing factors
LAD can be triggered by various drugs, most frequently vancomycin, followed by non-steroidal anti-inflammatory drugs and penicillins [2, 30, 54–58]. Various other drugs were also associated with the onset of LAD [3, 59]. Controlled studies as reported in BP are, however, not available. Furthermore, infections, trauma, vaccination, UV radiation exposure and building work in the home, were also reported as precipitating factors [2, 30, 58, 60–62].
Pathology
Histopathology of a lesional biopsy typically shows subepidermal splitting and an infiltrate with neutrophils in the papillary dermis sometimes forming microabscesses as typically seen in dermatitis herpetiformis. Some CD4+ lymphocytes and CD30+ lymphocytes are present perivascularly in the upper dermis and eosinophils may be admixed [2, 63, 64] (Figure 50.31). Similar alterations may be seen in other pemphigoid diseases including BP, MMP and anti-p200 pemphigoid. Accumulation of IL-8 was observed in the epidermis and perivascularly and of IL-5 at the site of blistering [64].
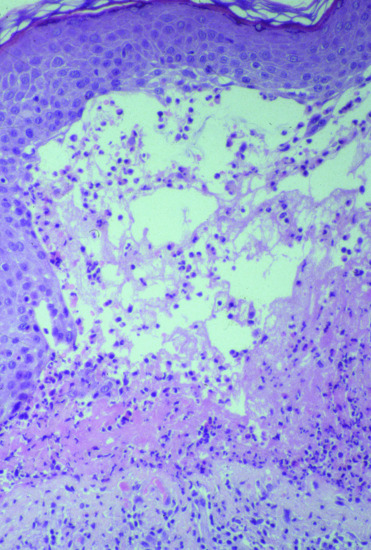
Figure 50.31 Lesional histopathology of linear IgA disease. H&E staining shows subepidermal splitting with a dense inflammatory infiltrate in the blister cavity and the underlying upper dermis composed of neutrophils, eosinophils and some lymphocytes. A similar histopathological pattern may also be seen in bullous pemphigoid, mucous membrane pemphigoid, and anti-p200 pemphigoid.
Tissue-bound autoantibodies can be visualized by direct IF microscopy of a perilesional biopsy. Linear deposits of IgA, frequently accompanied by weaker staining of IgG and/or C3, at the DEJ are diagnostic (Figure 50.32). By pattern analysis, a u-serrated pattern is seen with anti-type VII collagen autoantibodies (Figure 50.40). In all other autoantibody specificities, an n-serrated pattern is observed [52, 65, 66] (Figure 50.13).
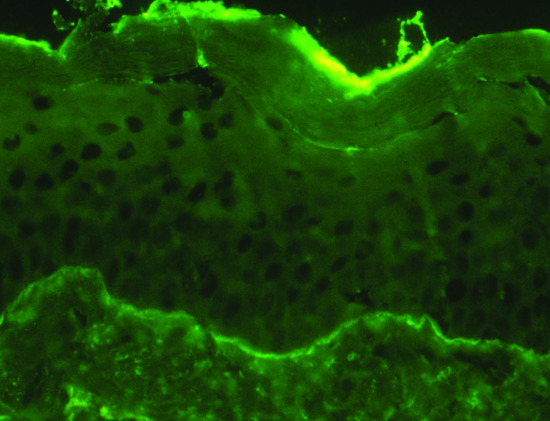
Figure 50.32 Direct immunofluorescence microscopy of a perilesional biopsy in linear IgA disease. Linear deposition of IgA at the dermal–epidermal junction.
Genetics
The only study on the genetic susceptibility of LAD identified an association with HLA-B8, HLA-Cw7, DR3, DR2 and the TNF2 allele as well as in children, with B8, DR3, DQ2 and also TNF2 [67]. In addition, five of 20 South African children with LAD (all black Africans) had the B8 allele which is uncommon in this population [25].
Environmental factors
No environmental factors have been discovered so far. Trigger factors were described in the section on Predisposing factors.
Clinical features
Presentation
In both children and adults, the individual lesions are similar including tense blisters and vesicles, urticated plaques, erosions and erythema (Figures 50.33 and 50.34) [2]. Blisters and vesicles frequently arise in an annular pattern with blistering along the edge of lesions forming the so-called ‘string-of-pearls’, ‘crown of jewels’ or ‘cluster of jewels’ sign (Figure 50.35a) [3]. Of note, this sign in not pathognomonic for LAD and may also be seen in BP (Figure 50.35b). Pruritus is variable from absent to severe. In children, lesions arise more abruptly compared to adults and tend to involve the perioral area and perineum in addition to the other predilection sites, trunk and limbs. Latter localizations are mainly involved in adult patients [2]. Mucosal involvement is common (in about 70% of patients) with mostly oral erosions and ulcers; nasal crusting and genital lesions may also occur [2]. When mucous membrane lesions are predominant there is a diagnostic overlap with MMP (see Figure 50.23). In patients with ocular scarring, the diagnosis of MMP is appropriate. As in BP, lesions tend to heal without scarring unless extensive superinfection occurs. Milia formation is uncommon.
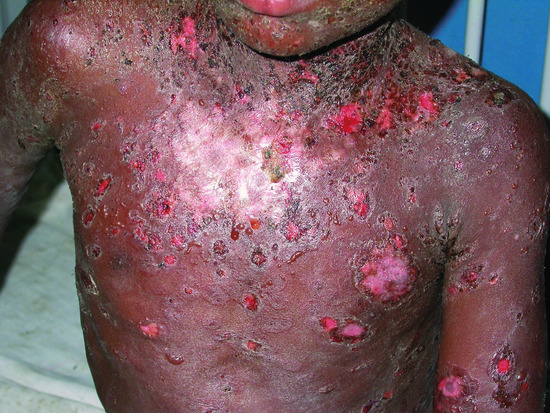
Figure 50.33 Linear IgA disease. Erosions and tense blisters on the trunk in a Ugandan child. Lesions were also present on the face.
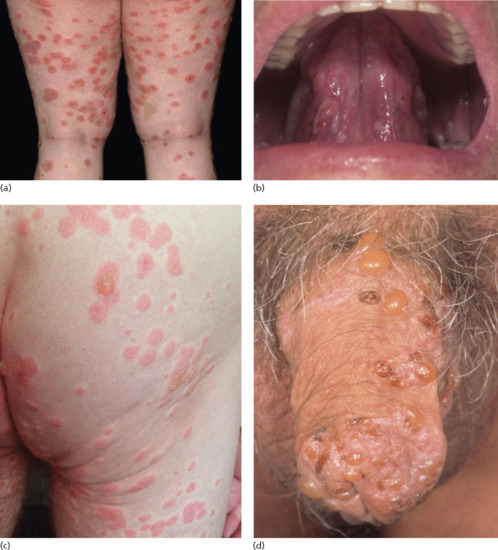
Figure 50.34 Linear IgA disease. Tense blisters in an annular pattern on the thighs (a), erosions on the tongue (b), erythema and blisters on the right gluteal region (c), and tense blisters and crusted erosions on the penis (d) in adult patients.
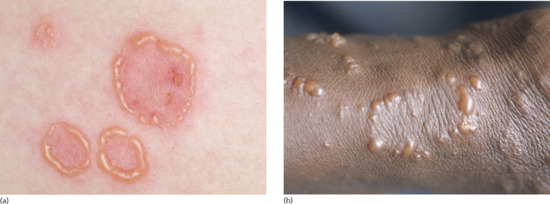
Figure 50.35 ‘Cluster of jewels’ or ‘ring of pearls’ sign. The peculiar appearance of vesicles in an annular pattern or along the edge of a lesion is frequently seen in linear IgA disease (a), however, it is not pathognomonic and may also be observed in bullous pemphigoid (b).
Clinical variants
Mixed immunobullous disease and linear IgA/IgG bullous dermatosis
For patients with equal IgA and IgG deposits at the DEJ by direct or indirect IF microscopy these two terms have been proposed [43, 46, 68]. No specific clinical phenotype was identified in these entities. Since in most LAD patients, IgG autoantibodies against proteins of the DEJ can also be found [40, 43–45, 68] and most BP sera also contain IgA autoantibodies, these cases may represent the centre of a LAD–BP spectrum.
Differential diagnosis
In young children, bullous impetigo may resemble initial lesions. Hereditary epidermolysis bullosa is often present at birth and the family history is helpful. The adult disease may be confused with atypical erythema multiforme, neurotic excoriations and prurigo.
In both age groups, dermatitis herpetiformis (antibodies against epidermal and or tissue transglutaminase with granular deposits of IgA in the dermal papillae by direct IF microscopy), BP (autoantibodies against the DEJ preferentially of the IgG isotype and reactivity against BP180 and/or BP230), MMP (predominant mucosal involvement) and EBA (reactivity against type VII collagen) need to be distinguished. Key clinical and immunopathological features of these diseases are summarized in Table 50.3.
Classification of severity
No system for the scoring of disease activity has been validated in LAD. The BP-Disease Activity Index, developed for BP, may be an alternative for individual patients or clinical studies.
Complications and co-morbidities
No specific complications and co-morbidities related to the pemphigoid disease have been described.
Disease course and prognosis
Patients with LAD almost always respond well to treatment. Relapses may occur over the next 2–4 years but are usually less severe than the initial disease episode. During pregnancy, the disease appears to improve with relapses within a few months postpartum [69]. Most children go into complete remission within 2 years of disease onset and only very rarely the disease persists after puberty [2, 70, 71]. Drug-induced LAD usually heals within 4–8 weeks after discontinuation of the drug. Some cases, however, have persisted for months [72].
Investigations
Diagnosis is based on the combination of the clinical picture and direct IF microscopy. Linear deposits of predominant or exclusive IgA deposits at the DEJ are diagnostic for LAD. With predominant mucosal involvement there is overlap with MMP. When scarring ocular disease or laryngeal and oesophageal lesions are present most clinicians would diagnose MMP (with predominant IgA reactivity).
For screening of serum autoantibodies indirect IF microscopy on monkey oesophagus or, even more sensitive and informative, on 1 M NaCl-split human skin can be used. The latter assay usually reveals IgA binding at low titres (1 : 10–1 : 40) with a sensitivity of 60–70% [8, 18, 38] and can differentiate anti-BP180/BP230 reactivity (epidermal binding) from anti-type VII collagen reactivity (dermal binding; Figure 50.14). Blister fluid can be applied as an alternative to serum and may be easier to obtain in a child [73].
To pinpoint the fine specificities of serum autoantibodies various assays, all of them not commercially available, can be performed. Antibodies against the ectodomain of BP180 can be detected by immunoblotting with epidermal and dermal extracts (97 kDa protein) [10], conditioned medium of cultured keratinocytes (LAD-1) [9, 74, 75], and various recombinant fragments [40, 74, 76], as well as by ELISA [77]. Assays for serum antibodies against type VII collagen are described in the EBA section.
Management
No prospective controlled clinical trial or larger case series have been reported. Patients usually well respond to treatment. In localized or limited disease very potent topical corticosteroids may suffice. Otherwise, dapsone 1.0–1.5 mg/kg/day is regarded as first line treatment which may be used in combination with the topical corticosteroids [1, 78]. Glucose-6-phosphate dehydrogenase deficiency should be excluded before dapsone is prescribed. The most frequent adverse events are anaemia (a reduction of haemoglobin of 1–2 g/dL can be expected), methaemoglobulinaemia and increased liver enzymes. Agranunlocytosis is rare but can be fatal and at least monthly blood counts are required and in case of fever, agranulocytosis needs to be excluded [79]. Sulfapyridine and, often better tolerated, sulfamethoxypyridazine are an alternative for dapsone. Some patients may require concomitant low-dose prednisolone (0.25–0.5 mg/kg/day) to suppress blister formation [1, 78]. In refractory patients, erythromycin, colchicine, flucloxacillin, methotrexate, ciclosporin, tetracycline and nicotinamide, IVIG, azathioprine, mycophenolates and immunoadsorption have been employed successfully [80].
Anti-p200 pemphigoi
Definition and nomenclature
Anti-p200 pemphigoid is a distinct subepidermal bullous skin disease characterized by autoantibodies against a 200 kDa protein (p200) of the DEJ detected by Western blotting with extract of human dermis. Clinically, patients resemble BP, but tend to be younger. Recently, the recombinant C terminus of laminin γ1 was shown to be recognized by 90% of anti-p200 pemphigoid sera [1].
Introduction and general description
Anti-p200 pemphigoid was first described in 1996 by Zillikens and Hashimoto [2, 3]. Subsequently, the p200 protein was shown to be an acidic non-collagenous N-linked glycoprotein that was localized within the lower lamina lucida outside of hemidesmosomes by electron microscopy [2–6]. The p200 protein is different from all major target antigens of the DEJ including BP180, BP230, α6β4 integrin, laminin 332, laminin 331, type VII collagen and nidogen [7–9]. It is synthesized by both keratinocytes and dermal fibroblasts [9]. In 2009, Dainichi et al. reported that 90% of anti-p200 pemphigoid sera contained antibodies against the C-terminal 245 amino acids of laminin γ1 and coined the term anti-laminin γ1 pemphigoid [8].
Laminins are cross- or T-shaped heterotrimers and composed of three non-identical protein chains, laminin α, β and γ . Laminins are extracellular matrix glycoproteins and major constituents of basement membranes. They interact for instance with nidogen (via the N terminus), perlecan and integrins (via the C terminus) [10]. Laminin γ1 is a component of various laminins including laminin 111, 211, 121, 213, 221, 212/222, 311, 321, 411, 421, 511 and 521 of which laminin 311, 321 and 511 are expressed in the DEJ [11].
Diagnosis is made by the detection of autoantibodies against the p200 protein by Western blotting against an extract of the upper portion of the human dermis [1, 2]. Detection depends on the quality of the dermal extract and was only possible in a handful of laboratories. With the discovery that reactivity against laminin γ1 can be found in 90% of anti-p200 sera, immunodetection by ELISA using the recombinant C terminus of laminin γ1 has greatly facilitated the diagnosis [8, 12]. This is of importance since serum autoantibodies in anti-p200 pemphigoid label the dermal side of the artificial blister by indirect IF microscopy on salt-split skin and thus EBA could be diagnosed mistakenly. In contrast to the latter disease, patients with anti-p200 pemphigoid usually respond well to topical or medium-dose oral corticosteroids [1].
The clinical features and immunopathology are summarized in Table 50.3.
Epidemiology
Incidence and prevalence
Anti-p200 pemphigoid is a rare chronic autoimmune disease with about 50 patients published in the English literature [7, 13]. The actual incidence is most likely considerably higher with many patients classified as BP or EBA. This assumption is supported by the, however highly biased, serological diagnosis of 41 patients with anti-p200 pemphigoid in our routine autoimmune laboratory in 2011 and 2012. In the same period, eight patients were diagnosed with EBA [1]
Age
The mean age of all published patients and our own was 69 years ranging from 50 to 91 years [1].
Sex
In all published patients and our own, 73% were male and 27% female [1].
Ethnicity
No reliable data are available.
Associated diseases
Psoriasis was associated in about 30% of the published cases, almost all of them were Japanese [7, 13].
Pathophysiology
The C terminus of laminin γ1 has been described as immunodominant region in anti-p200 pemphigoid [8, 12, 14]. About a third of sera additionally react with epitopes outside the 245 C-terminal amino acids [14] while in 10% of sera, no reactivity with laminin γ1 can be found [8]. Most autoantibodies belong to the IgG4 subclass, an individual patient with exclusive IgA autoantibodies against the p200 antigen but unreactive with laminin γ1 has been reported [15]. Intermolecular epitope spreading appears to be relatively frequent in anti-p200 pemphigoid with various reports of concomitant autoantibody reactivity against BP180, BP230, laminin 332 and type VII collagen, respectively [7, 16–20, 22].
To investigate the role of IL-8 in the neutrophil infiltration frequently seen in the patients’ upper dermis by histopathology, Iwata et al. determined the IL-8 release of cultured human keratinocytes in response to anti-p200 pemphigoid IgG. In contrast to BP-IgG, incubation with anti-p200 IgG did not result in the secretion of IL-8 [23]. These results point to different mechanisms of neutrophil accumulation in skin lesions in the two pemphigoid diseases.
Recently, as for BP and EBA sera, anti-p200 pemphigoid sera were shown to induce dermal–epidermal separation when employed in an ex vivo model using cryosections of human skin incubated with patients’ sera or IgG and subsequently, with leukocytes from healthy volunteers [14]. Of note, anti-p200 pemphigoid IgG affinity purified against various forms of the C terminus of laminin γ1 as well as the entire laminin γ1 molecule had no effect in this model [14]. Interestingly, patients’ sera depleted of laminin γ1 reactivity led to the same extent of dermal–epidermal separation as non-depleted patients’ sera [14]. In the same vein, investigators were unable to induce clinical disease in mice after transfer of antimurine laminin γ1 IgG or immunization of mice with recombinant murine laminin γ1 [14, 24]. Subsequently, antibodies against the N-terminus of laminin γ1, which are found in about a third of anti-p200 pemphigoid sera, were shown to induce dermal–epidermal separation in the cryosections model [14, 25].
Taken together, the ex vivo and in vivo data raise serious doubts about the pathogenic relevance of antibodies against laminin γ1 in anti-p200 pemphigoid. Another puzzling observation comes from direct IF microscopy of patients’ skin. While no laminin containing the γ1 chain is exclusively expressed in the DEJ [26], dermal blood vessels that express several γ1-containing laminins are almost never stained. These phenomena may be explained by a skin-specific splice variant or posttranslational modifications unique to the laminin γ1 of the DEJ. Alternatively, laminin γ1 is recognized by patients’ autoantibodies but the true autoantigen of anti-p200 pemphigoid remains yet to be discovered.
Predisposing factors
None described.
Pathology
As in all pemphigoid diseases, the histopathological hallmark of a lesional biopsy is the subepidermal split accompanied by the accumulation of neutrophils in the upper dermis [27] (Figure 50.36). In some patients a mixture of neutrophils and eosinophils was found and sometimes, microabscesses may develop at the tips of dermal papillae [7, 27]. Importantly, histopathology was shown to not allow the differentiation of anti-p200 pemphigoid from other pemphigoid diseases [27].
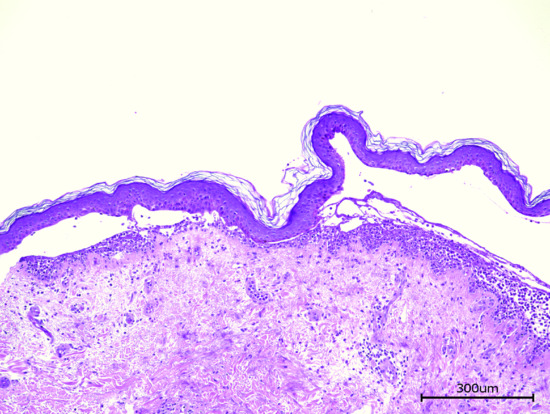
Figure 50.36 Lesional H&E histopathology of anti-p200 pemphigoid. Subepidermal splitting and a dense neutrophilic infiltration below the blister.
As in all pemphigoid disorders, tissue-bound autoantibodies can be visualized by direct IF microscopy of a perilesional biopsy as linear deposits of IgG at the DEJ. Pattern analysis reveals an n-serrated pattern (Figure 50.12). Exclusive IgA deposits were described in a single patient [15].
Genetics
No data available.
Environmental factors
None described.
Clinical features
Presentation
Most patients present with tense blisters on erythematosus or normal skin resembling BP [7, 18, 20] (Figure 50.37). As in BP, the clinical picture may vary between different patients and cases reminiscent of linear IgA dermatosis, dermatitis herpetiformis or pompholyx (Figure 50.38) have been described [7, 15, 29]. In some patients, oral and/or genital mucous membranes were involved [7, 16, 23]. Lesions usually heal without scarring; milia formation has rarely been observed [7].
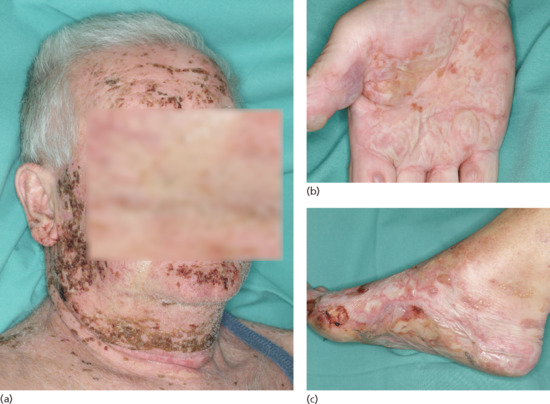
Figure 50.37 Anti-p200 pemphigoid. Erosions, haemorrhagic crusts and tense blisters on the face (a), palm (b) and foot (c).
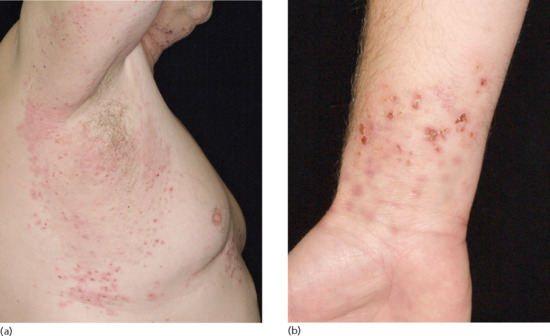
Figure 50.38 Anti-p200 pemphigoid. Erythematous partly excoriated papules and erythema on the right axilla (a) and vesicles and erythematosus papules on the right wrist (b) in a 52-year-old patient.
Clinical variants
None described.
Differential diagnosis
Clinically, BP and linear IgA disease may be differentiated. Since by indirect IF microscopy on salt-split skin IgG autoantibodies label the dermal side of the artificial blisters, EBA needs to be excluded by the detection of antibodies against the p200 antigen and/or laminin γ1.
Classification of severity
None described.
Complications and co-morbidities
No specific complications and co-morbidities related to the pemphigoid disease have been described.
Disease course and prognosis
The disease typically shows a prompt response to treatment. In mild cases, potent topical corticosteroids alone may be sufficient. While most patients remain in remission after tapering of the immunosuppressive medication, others relapse and require treatment over months and years 50.39 [7].
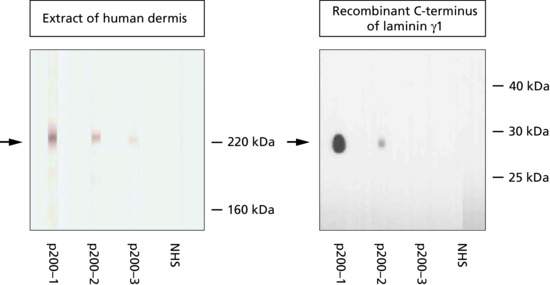
Figure 50.39 Anti-p200 pemphigoid: serum autoantibodies against the p200 antigen and laminin γ1. By Western blotting with extract of human dermis a 200 kDa protein is recognized in all three patients (p200-1-3). Reactivity with laminin γ1 is seen in two patients (p200-1, p200-2) but not in patient p200-3 with anti-p200 pemphigoid. The migration positions of molecular weight markers, the p200 antigen (a), and the C terminus of laminin γ1 (b) are indicated. NHS, normal human serum.
Investigations
Diagnosis is based on the combination of the clinical picture, direct IF microscopy, and serology. By indirect IF microscopy on 1 M NaCl-split normal human skin autoantibodies label the floor of the artificial split (see Figure 50.13). Final diagnosis is then made by the demonstration of serum autoantibodies against the p200 antigen by immunoblotting with extract of normal human skin [1, 2] and/or against laminin γ1. In addition to immunoblotting with the recombinant C terminus of laminin γ1 (amino acids 1364 to 1609) that has a sensitivity of about 90%, an ELISA has been developed revealing a sensitivity and specificity of 69% and 98.7%, respectively [8, 12]. The release of commercial test systems is awaited. Since reactivity with laminin γ1 cannot be demonstrated in all patients, the term antilaminin γ1 pemphigoid may be reserved for those patients with autoantibody reactivity against laminin γ1.
Management
Most patients have been treated with tapering doses of prednisolone 0.5 mg/kg/day. Dapsone, tetracyclines, azathioprine and ciclosporin have been used in addition to prednisolone or alone [7]. Individuals succesfully treated with topical corticosteroids, IVIG, ustekinumab, plasmapheresis and immunoadsorption have also been reported [7, 30, 33].
Epidermolysis bullosa acquisita
Definition
EBA is a clinically heterogeneous subepidermal blistering disease defined by autoantibodies against type VII collagen [1]. In patients with predominant IgA anti-type VII collagen antibodies and patients with predominant mucosal involvement, there is diagnostic overlap with LAD and MMP, respectively (Figure 50.23).
Introduction and general description
EBA is a rare pemphigoid disorder with autoantibodies against type VII collagen. Two main clinical subtypes can be differentiated, the so-called classical or mechanobullous form initially described by Roenigk et al. and resembling hereditary dystrophic EB [2] and an inflammatory variant that mimics BP, LAD or MMP [3–5].
The first cases of what we now call EBA were most likely described by Elliot in 1895 when reporting two patients with adult onset of skin fragility, erosions and blisters healing with scarring and milia formation [6]. In 1970, Roenigk et al. proposed clinical criteria for EBA including: (i) negative family or personal history for skin blistering; (ii) adult disease onset; (iii) skin lesions that resemble hereditary dystrophic EB; and (iv) the exclusion of all other bullous diseases [2]. Three years later, linear deposits of IgG and C3 at the DEJ were noted in an EBA patient by direct IF microscopy [7]. In the early 1980s, direct immunoelectron microscopy localized Ig deposition in EBA in the sublamina densa area differentiating the disease from BP [8, 9]. In 1984, Woodley et al. described a 290 kDa protein of the DEJ as a target of serum autoantibodies in EBA [10] and, 4 years later, identified this target antigen as type VII collagen [1].
Diagnosis is made by the detection of autoantibodies against type VII collagen. An ELISA and IF microscopy-based assay for serum anti-type VII collagen antibodies are currently commercially available [11, 12]. Importantly, since in only half of the EBA patients circulating autoantibodies can be detected, the visualization of type VII collagen-specific antibodies that reveal a u-serrated pattern by direct IF microscopy is recommended [13]. The pathogenesis of the inflammatory variant of EBA is relatively well understood due to recent advances based on several in vitro and in vivo models [14]. The clinical features and immunopathology are summarized in Table 50.3.
Epidemiology
Incidence and prevalence
The estimated incidence of EBA lies between 0.2 and 0.5 new cases per million per year in central Europe, Kuwait and Singapore [15–19].
Age
The disease occurs at any age with reported mean ages of disease onset of 44 and 54 years [20, 21]. A considerable number of patients (estimated at 5%) are children and adolescents [20, 22–27].
Sex
No clear sex preference has been reported.
Ethnicity
In two studies, EBA occurred more frequently in black patients of African descent (66% and 54% of patients, respectively) [28, 29].
Associated diseases
While two case series from the US and France revealed an association with IBD, in particular Crohn disease, in 25% and 16% of patients, respectively; this association was not found in 30 Korean patients [21, 29, 30]. All reported cases of EBA and IBD were recently reviewed [31]. Two studies detected antibodies to type VII collagen in 6% and 60% of patients with IBD [30, 32]. In line with these findings, expression of type VII collagen was found in the oral cavity, oesophagus, small intestine and colon. In two murine models of EBA, intestinal inflammatory lesions were seen in about 20% of animals [33].
Associations with numerous systemic diseases, e.g. rheumatoid arthritis, diabetes, cryoglobulinaemia and psoriasis, were reported in individual patients [34–36]. Recently, an association with haematological malignancies, i.e. lymphoma, was observed in about 8% of EBA patients [37].
Autoantibodies against type VII collagen are also characteristic for bullous systemic lupus erythematosus (see Bullous systemic lupus erythematosus).
Pathophysiology
The autoantigen of EBA is homotrimeric type VII collagen, a constituent of the anchoring fibrils (see Chapter 2) [1, 10]. Its N terminal 145 kDa NC1 domain has been identified as the immunodominant region with epitopes spreading across all of this region [1, 38–41]. Only rare cases with reactivity against the C terminal NC2 or the central collagenous domain are reported [24, 42, 43]. Patients with intermolecular epitope spreading and reactivity with other DEJ antigens, i.e. BP180, BP230, laminin 332 and the p200 antigen in addition to type VII collagen-specific antibodies, may occasionally been found [20, 44–46].
The pathogenic relevance of autoantibodies in EBA has been shown unequivocally. Serum levels of anti-type VII collagen antibodies correlate with the disease activity in patients and transient neonatal disease was observed following placental transfer of autoantibodies from an affected mother [47, 48]. Furthermore, different in vitro and in vivo models have been established that have resulted in a relatively well-understood pathophysiology [40, 49–54]. In these models, patient or rabbit antimurine type VII collagen IgG was incubated with skin sections leading to dermal–epidermal splitting or injected into mice resulting in a disease phenotype clinically and immunopathologically mimicking human EBA. These models are suitable to explore the process of tissue destruction induced by anti-type VII collagen antibodies. In a further model, immunization of susceptible mouse strains with the recombinant murine NC1 domain triggered a longstanding autoimmune response resulting in a subepidermal blistering skin disease in 80% of mice [50]. This model allows studying the process of loss of tolerance which was shown to be T-cell dependent [55]. Furthermore, it is ideally suited to explore the effect of anti-inflammatory mediators when after the development of first skin lesions and subsequent randomization of mice these and control substances are applied. For example, in the immunization-induced EBA model inhibition of T-cell proliferation by heat shock protein 90 led to disease regression [56]. In human EBA, only limited data are available about the role of T cells that were shown to recognize identical regions on the NC1 domain as B lymphocytes [57]. Furthermore, neutrophils were identified as major effector cells that are recruited and activated via the murine Fcγ receptor IV following interaction with the Fc portion of skin-bound type VII collagen-specific antibodies [52, 58, 59]. The soluble Fcγ receptor IIB has recently been shown to suppress disease progression in experimental murine EBA [60]. Glycosylation of autoantibodies was highlighted to be essential for the interaction of autoantibodies with their Fcγ receptors as well as their interplay with complement activation [61, 62]. In addition, activation of the IL-1 system and the alternative complement cascade are important for the recruitment of neutrophils at the DEJ [63, 64]. Finally, their release of reactive oxygen species, elastase and matrix metalloproteinase 9 induced dermal–epidermal splitting [65, 66]. In contrast to BP, mast cells do not appear to be of relevance for lesion formation [67]. The therapeutic influence of neutrophil and complement activation, the cytokine network and autoantibody glycosylation may open novel therapeutic avenues for this difficult to treat disease [68].
Predisposing factors
Penicillin, vancomycin in conjunction with gentamycin, UV radiation and contact allergy to metals have been implicated as precipitating factors [69–72].
Pathology
As in all pemphigoid diseases the histopathological hallmark is subepidermal blistering. Depending on the clinical subtype, the inflammatory infiltrate in the dermis is variable, scarce in the mechanobullous variant and dense with predominant neutrophils, eosinophils, monocytes and lymphocytes reminiscent of BP in the inflammatory subtype [35]. The cleavage plane of the blister may be within the lamina densa corresponding to the autoantibody deposits as seen by direct electron microscopy or within the lamina lucida [8, 9, 73]. The latter finding may be explained by the lamina lucida as locus minoris resistentiae most fragile to the proteolytic enzymes secreted within the vicinity of the DEJ.
Tissue-bound autoantibodies can be detected by direct IF microscopy of a perilesional biopsy as linear deposits of IgG and/or C3 at the DEJ. An important contribution to the diagnosis of EBA is the pattern analysis introduced by Jonkman and co-workers. By a 600-fold magnification the linear binding shows an u-serrated pattern with a grass-like appearance of Ig deposits [13] (Figure 50.40). This pattern is unique to EBA and of particular importance since only in about half of patients can circulating autoantibodies be detected. In all other pemphigoid diseases, an n-serrated pattern is seen (Figure 50.13). In biopsies in which the serration pattern analysis remains inconclusive, more sophisticated alternatives comprise the computer-aided fluorescence overlay antigen mapping (FOAM) [74]; the double IF labelling of tissue-bound and defined antigens of the DEJ analysed by laser scanning confocal microscopy [75]; and the treatment of biopsies with 1 M NaCl solution to induce an artificial split at the lamina lucida level and subsequent autoantibody detection as by indirect IF microscopy on 1 M NaCl-split skin [76] (Figure 50.13).
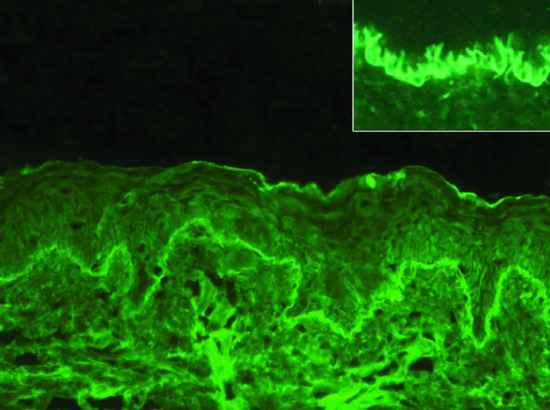
Figure 50.40 Direct immunofluorescence microscopy of a perilesional biopsy of a patient with epidermolysis bullosa acquisita. Linear deposition of IgG at the dermal–epidermal junction. A u-serrated pattern is seen (inset).
Exclusive or predominant IgA deposits by direct IF microscopy has been described in several patients [77–80]. In a recent larger study, about one third of EBA patients revealed exclusive IgA reactivity and in another third, a combined IgA and IgG autoantibody response was seen [20]. These patients may either be diagnosed as IgA-EBA or, due to overlapping diagnostic criteria, as LAD (Figure 50.23).
Genetics
EBA is associated with the HLA-DR2 (corresponding to HLA-DRB1*15) and DRB1*15:03, an allele found frequently in the general population [28, 29]. Korean patients more frequently carried DRB1*13 compared to controls [81]. Recently, the relevance of HLA (H2s) and non-HLA genes for disease susceptibility was described in the immunization-induced mouse model of EBA [82, 83].
Environmental factors
No environmental factors have been discovered so far. For trigger factors see earlier (Predisposing factors).
Clinical features
Presentation
Two main clinical forms can be differentiated, the classical mechanobullous variant, in about a third of patients, and the inflammatory subtype [2, 20, 21, 84, 85]. The classical mechanobullous phenotype mimics dystrophic hereditary EB when it is severe and porphyria cutanea tarda when it is mild [2]. Clinical characteristics are skin fragility, erosions, blisters, crusts and scars on trauma-prone areas such as the hands, knuckles, elbows, knees and toes (Figures 50.41 and 50.42). Scarring alopecia and nail loss may occur [2]. The inflammatory variant resembles other pemphigoid diseases such as BP (in about half of EBA patients), MMP (in 5–10%), and LAD (in 5–10%) [20, 21] (Figures 50.43 and 50.44). However, both the classic and the inflammatory forms may coexist in the same patient and the clinical presentation of a given EBA patient may change during the disease course [86–88]. Differentiation between the inflammatory subtypes remains somehow arbitrary. Both subtypes may occur in adults (Figures 50.41 and 50.43) and children (Figures 50.42, and 50.44). Mucous membranes are affected in about half of the patients [20] (Figures 50.42 and 50.43). Due to diagnostic overlap, patients with the MMP variant, i.e. with predominant mucosal lesions, may also be diagnosed as MMP [89] (Figure 50.23).
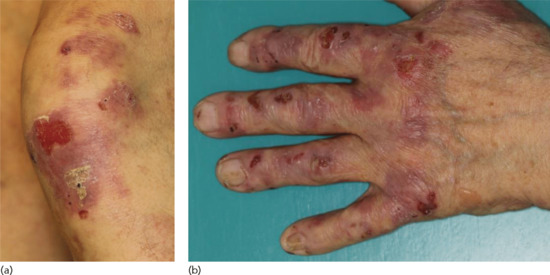
Figure 50.41 Epidermolysis bullosa acquisita, mechanobullous variant. Erythema, erosions and crusts on the left knee (a) and erythematous plaques, atrophic scars, milia, crusts and a tense blister on trauma-prone extensor surface of the left hand of a 75-year-old man (b).
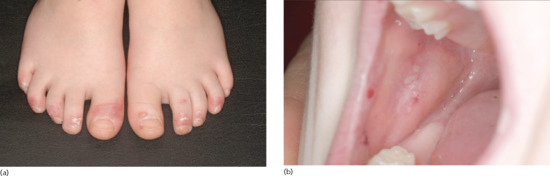
Figure 50.42 Childhood epidermolysis bullosa acquisita, mechanobullous variant. Erythema, erosions and tense blisters on the trauma-prone dorsal aspects of the toes (a) and erosions of the buccal mucosa (b) in a 5-year-old boy.

Figure 50.43 Epidermolysis bullosa acquisita, inflammatory variant. Erosions and tense blisters (insert) on the upper back (a), milia (b), and erosions of the buccal mucosa (c).
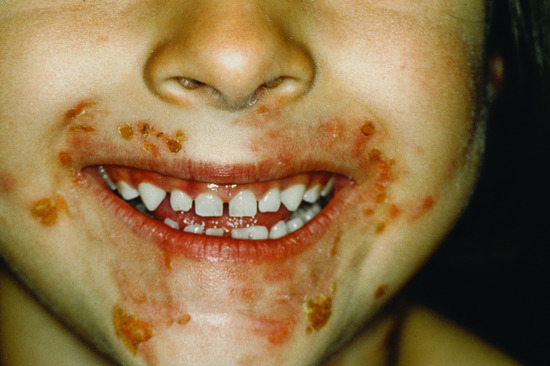
Figure 50.44 Childhood epidermolysis bullosa acquisita, inflammatory variant. Perioral yellowish crusts and erosions.
Clinical variants
Brunsting and Perry described patients with a subepidermal blistering autoimmune disease and skin lesions of the head and neck region that healed with scarring [90]. These patients are best classified as variants of cicatricial pemphigoid [89] (see Very rare pemphigoid disorders). In some patients with the Brunsting–Perry phenotype, autoantibodies against type VII collagen have been described [20, 21, 91–93]. These patients may also be classified as EBA [20, 21, 35] (Figure 50.23).
Differential diagnosis
Patients with the mechanobullous subtype need to be distinguished from dystrophic epidermolysis bullosa (family history, direct IF microscopy) and porphyria cutanea tarda (porphyrins in urine, direct IF microscopy). The inflammatory variants mimic BP (reactivity with BP180), MMP and LAD. With the latter two diseases there is a diagnostic overlap. We would recommend the diagnosis of EBA in all patients with antibodies to type VII collagen unless ocular scarring or laryngeal or oesophageal involvement is present. Those patients may best be classified as MMP.
Classification of severity
No scoring system for the disease activity has been validated in EBA.
Complications and co-morbidities
No specific complications and co-morbidities related to the pemphigoid disease have been described.
Disease course and prognosis
EBA is a chronic relapsing disease that usually is more difficult to treat compared to other pemphigoid disorders except MMP with ocular, oesophageal and laryngeal involvement [94]. A better overall prognosis was attributed to children with EBA compared to adults [23, 27]. No difference with respect to response to treatment was observed between the mechanobullous and the inflammatory variants [21].
Investigations
Diagnosis is based on the combination of the clinical picture, direct IF microscopy and serology (Figure 50.45). The diagnostic value of direct IF microscopy including pattern analysis are detailed earlier (see Pathology).

Figure 50.45 Diagnostic pathway for epidermolysis bullosa acquisita (EBA). (1) commercially available; (2) depending of availability; positivity in any of the four assays will allow diagnosis of EBA; (3) only availalble in specialized laboratories; and (4) from patients with dystrophic epidermolysis bullosa. AIBD, autoimmune bullous diseases; BP, bullous pemphigoid; DEJ, dermal–epidermal junction; ELISA, enzyme-linked immunosorbent assay; IF, immunofluorescence; LAD, linear IgA dermatosis; MMP, mucous membrane pemphigoid.
As in all pemphigoid diseases, indirect IF microscopy on 1 M NaCl-split normal human skin is a useful screening test for serum autoantibodies in EBA with a sensitivity of about 50% [20, 21]. In this assay, autoantibodies in EBA label the floor of the artificial split (see Figure 50.13). Importantly, testing for IgA antibodies is essential since the majority of EBA sera contain IgA reactivity [20, 95]. In about 15% of EBA patients, exclusive serum IgA reactivity was found. In this subgroup, patients are more likely to have the inflammatory phenotype [20]. For further differentiation and diagnosis of EBA, two commercially available assays for the detection of serum IgG autoantibodies against type VII collagen are available [11, 12]. One is based on indirect IF microscopy of NC1-expressing human cells (Euroimmun, Lübeck, Germany), the other is an ELISA based on both the NC1 and NC2 domains (MBL, Nagoya, Japan) revealing sensitivities of 92% and 94%, respectively. However, only sera reactive by indirect microscopy on human salt-split skin were included in these studies. Alternative methods for the detection of serum type VII collagen-specific antibodies are immunoblotting with dermal extract, human placental amnion or cultured A431 cells and, elegantly, indirect IF microscopy on hereditary EB skin deficient of type VII collagen [96–98]. If serology is negative and pattern analysis of the direct IF microscopy was inconclusive, the visualization of Ig deposits by direct immunoelectron microscopy is the diagnostic gold standard [8, 9, 99]. Alternatively, the perilesional biopsy can be subjected to FOAM using an overlay of red-stained anti-type VII collagen and green-stained IgG [74].
Management
Treatment of EBA is challenging: disease activity is more difficult to suppress compared to most other pemphigoid diseases and no controlled prospective trials are available [34, 94, 100–103]. The mainstay is systemic corticosteroids (prednisolone 0.5–2.0 mg/kg/day, depending on disease severity) in combination with colchicine [104–106] and dapsone [100]. For refractory patients or severe forms, ciclosporin, azathioprine, mycophenolate mofetil, gold, cyclophosphamide, extracorporal photophoresis, anti-CD25 therapy (daclizumab, basiliximab) [107, 108], plasmapheresis and immunoadsorption [109], IVIG [110] and rituximab [111, 112] have been successfully used in altogether about 80 individual patients (reviewed in [101–103, 113]).
Most children have been treated with a combination of systemic corticosteroids (prednisolone 1 mg/kg/day) and dapsone [23, 26, 27, 43].
Treatment recommendation
Japanese and Australian colleagues proposed prednisolone 0.5–1.5 mg/kg/day (depending on the disease activity) combined with colchicine 0.5–3.0 mg/day with or without dapsone 1–2 mg/kg/day as first line therapy [34, 102]. For refractory patients, ciclosporin, mycophenolate mofetil, IVIG, plasmapheresis/immunoadsorption, IVIG and rituximab were recommended [34, 102]. The US-American colleagues Gürcan and Ahmed emphasized that the benefit of systemic corticosteroids is low in EBA and proposed the following algorithm:
- In limited disease or paediatric patients: dapsone, colchicine, dapsone + colchicine + low-dose prednisolone, IVIG and/or rituximab.
- For moderate or mucocutaneous disease: dapsone, IVIG, rituximab, IVIG + rituximab (without financial restrictions); <6 weeks of prednisolone, ciclosporin, myophenolates, mycophenolates + immunoadsorption, IVIG and/or rituximab (with financial restrictions) [113].
Bullous systemic lupus erythematosus
Definition and nomenclature
Bullous systemic lupus erythematosus (BSLE) is an autoimmune subepidermal blistering disease that occurs in patients with SLE [1–3]. Blisters and erosions are not restricted to LE lesions, tend to arise in sun-exposed skin and heal without scarring. Most BSLE patients have antibodies against type VII collagen and were designated as type I BSLE. Type II BSLE is characterized by antibodies against other antigens of the DEJ [4–6]. As in SLE, BSLE typically affects women with an African background in the third decade. Bullous lesions usually rapidly respond to dapsone [7].
Introduction and general description
BSLE is a clinically and immunopathologically distinct subepidermal autoimmune bullous disease that arises in patients with SLE. Diagnostic criteria from the 1980s comprised: (i) a diagnosis of SLE based on the American Rheumatism Association criteria; (ii) blisters predominantly but not exclusively in sun-exposed areas; (iii) histopathology compatible with dermatitis herpetiformis; and (iv) linear or granular deposits of IgG, and/or IgA, and/or C3 at the DEJ by direct IF microscopy and immune reactants below the basal lamina by electron microscopy [1–3]. Gammon et al. recognized that the majority of BSLE patients have autoantibodies against the same antigen as EBA patients [4] later identified as type VII collagen [8]. In the mid-1990s, Gammon and Briggaman as well as Yell et al. proposed a broader definition and subdivided BSLE into type I (with reactivity against type VII collagen) and type II (with anti-DEJ reactivity without anti-type VII collagen antibodies) [5, 6]. Meanwhile, autoantibodies against other target antigens of the DEJ including BP180, BP230 and laminin 332 were found in BSLE [9, 10] and patients with concomitant SLE and BP, LAD or MMP were reported [5, 11–13].
The first historical case compatible with BSLE was described in 1889 and a patient with positive direct IF microscopy was reported in 1973 before in 1982 Hall et al. assembled the first case series of BSLE and recognized the high efficacy of dapsone [7, 14].
Diagnosis is still based on the criteria of Camisa and colleagues [1–3]. Serum autoantibodies against constituents of the DEJ may be detected using modern diagnostic assays [15]. Serration pattern analysis of direct IF microscopy has been established as practical tool to differentiate tissue-bound autoantibodies against type VII collagen (u-serrated pattern) from all other anti-DEJ antibodies (n-serrated pattern) [16, 17]. The clinical features and immunopathology of BSLE are summarized in Table 50.3.
Epidemiology
Incidence and prevalence
The incidence of BSLE was estimated to be 0.22 and 0.26 cases per million per year in France and Singapore, while no cases were found in three epidemiological studies in Germany and Kuwait [18–22]. The disease was found in about 1–2% of patients with SLE [23–25]. However, up to 10% of patients with SLE were reported to develop bullous skin eruptions [23, 26] and, thus, the incidence of BSLE may be higher than 1–2%. In large cohorts of sera with immunobullous disorders, 1–2% were identified as BSLE [16, 22, 27].
Age
Most patients are in the second, third or fourth decade of life reflecting the age distribution known for SLE [27]. BSLE may, however, also affect children and patients in their 60s and 70s and are described [27].
Sex
The female preponderance of BSLE reflects the gender distribution of SLE.
Ethnicity
Although BSLE manifests in all ethnicities, as in SLE, black patients of African descent have a higher risk of developing BSLE [11].
Associated diseases
No particular diseases were associated with BSLE.
Pathophysiology
The target antigen in type I BSLE is type VII collagen [4, 8]. As in EBA, autoantibodies in BSLE predominantly bind to the NC1 domain, in particular the fibronectin-like region [28]. The pathogenic relevance of anti-type VII collagen antibodies has been shown in various in vitro and in vivo models [29–35] and is detailed above (see Epidermolysis bullosa acquisita). In type II BSLE, either no serum autoantibodies or antibodies against structural proteins of the DEJ other than type VII collagen such as BP180, BP230 and laminin 332 were reported [5, 9–13, 36].
It may be speculated that, as discussed for lichen planus pemphigoides (see Very rare pemphigoid disorders), the interface dermatitis typically present in skin lesions of LE may have triggered an additional autoimmune response against proteins of the DEJ and transglutaminases resulting in a second autoimmune disease, BSLE. Alternatively, other factors that mediate the increased occurrence of several autoimmune diseases in individual patients may be responsible. Thus, the association of SLE and pemphigus or dermatitis herpetiformis can be explained [5].
Predisposing factors
Precipitating factors have only been described in individual patients and comprise drugs (hydralazine, penicillamine, methimazole) and UVB radiation [37–40].
Pathology
Histopathological findings are relatively uniform and resemble dermatitis herpetiformis with subepidermal splitting, a dense neutrophil-dominated infiltrate in the upper dermis sometimes accumulating in microabscesses of the papillary tips, and dermal oedema [3, 6, 7, 41]. In addition, mucin depositions are usually seen in the reticular dermis and sometimes signs of leukocytoclastic vasculitis [42]. Basal layer vacuolization characteristic for cutaneous LE is not present.
Direct IF microscopy of a perilesional skin biopsy shows linear (in about 40% of cases) and granular (in about 60%) staining of the DEJ by IgG. Additional linear or granular labelling of IgM, IgA, and/or C3 at the DE is found in about 70–80% of the biopsies [38]. In the major BSLE form (type I), an u-serrated staining pattern is seen [16] (Figure 50.40). By immunoelectron microscopy immunoreactants were localized beneath the lamina densa as in EBA [6]. The cleavage plane is usually in the area of Ig deposition, however, patients with splitting in the lamina lucida have been described [43]. These discrepancies are best explained by the different autoantibody specificities that have not always been explored in the initial reports.
Genetics
In a US-American cohort, presence of autoantibodies against type VII collagen (in BSLE and EBA) was associated with DRB1 1501 allele [25].
Environmental factors
No environmental factors have been discovered so far. For trigger factors see earlier (Predisposing factors).
Clinical features
History
The disease has an acute onset with usually generalized vesicles and blisters in patients with SLE [27].
Presentation
Primary lesions comprise tense vesicles and bullae with a clear or haemorrhagic content on otherwise normal or erythematosus skin (Figures 50.46 and 50.47). In addition, erythematous plaques in an annular or targetoid configuration may arise. Lesions are usually generalized with predilection for sun-exposed areas, neck, upper trunk and proximal extremities [27]. In contrast to other pemphigoid diseases, the face is relatively often affected and in some patients the only site of manifestation [43, 44]. Clinically, BSLE may mimic BP, LAD, dermatitis herpetiformis and EBA. When associated with autoantibodies against type VII collagen (type I BSLE), patients with the classical mechanobullous phenotype and the inflammatory variant known from EBA were described [16, 27]. Mucosal lesions and scar or milia formation, however, are unusual. Pruritus is absent or not intense and a burning sensation may occur. Interestingly, the onset of the bullous eruption does not necessarily correlate with the activity of the systemic involvement [6, 7, 27].
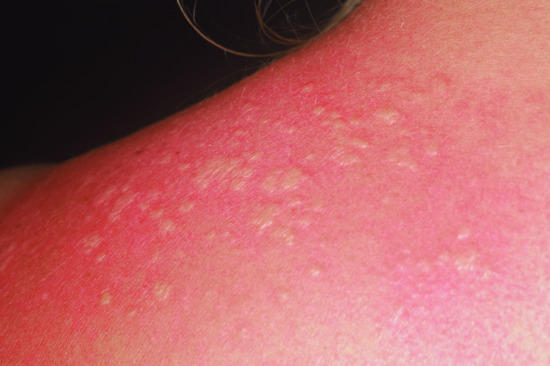
Figure 50.46 Bullous systemic lupus erythematosus. Tense blisters on erythematosus skin.

Figure 50.47 Bullous systemic lupus erythematosus. Erythematous macules and patches and a flaccid blister some days after the initiation of systemic corticosteroids.
(Courtesy of Dr K. Steinbrink, University of Mainz, Germany).
Clinical variants
None.
Differential diagnosis
Rarely, skin lesions of SLE may blister due to the high disease activity of the systemic disease. In these patients, extensive vacuolization of the DEJ with subepidermal cleavage and a mononuclear cell infiltrate in the upper dermis is seen by histopathology. Alternatively, blisters may arise due to photosensitivity or a drug eruption. By direct IF microscopy, band-like deposits of IgG, IgM, and IgA may be present, however, no linear or granular Ig depositions [5]. Other pemphigoid diseases, i.e. BP, LAD, EBA, anti-p200 pemphigoid as well as dermatitis herpetiformis, can be differentiated by the absence of SLE based on the American College of Rheumatology criteria for SLE, the generally good prognosis of BSLE and its rapid response to dapsone.
Classification of severity
No scoring system for the disease activity has been validated for BSLE.
Complications and co-morbidities
No specific complications and co-morbidities related to the pemphigoid disease have been described.
Disease course and prognosis
In most patients, the disease is transient. Unlike the other pemphigoid diseases (except pemphigoid gestationis), BSLE usually completely regresses with no further flares irrespective of the disease activity of the systemic disease [27].
Investigations
Diagnosis is still based on the criteria of Camisa with some modifications with respect to further observations in larger patient cohorts and the development of novel tools for the detection of serum autoantibodies [1–6]. In addition to the vesicobullous eruption, essential conditions for BSLE are a known SLE and linear or granular deposits of IgG, and/or IgA and/or C3 at the DEJ by direct IF microscopy. Histopathology is helpful and typically shows a neutrophil-rich infiltrate in the upper dermis and a subepidermal split formation. Serum autoantibodies are mainly directed against type VII collagen (type I BSLE) but reactivity against BP180, BP230 and laminin 332 were also found (in BSLE type II) [5, 9, 10, 12, 13, 36] (Figure 50.48). Serum anti-type VII collagen IgG levels were described to parallel the extent of bullous lesions in two patients with BSLE [45, 46]. In one patient, low autoantibody serum levels preceded the onset of the vesicobullous eruption by 3 months [45].
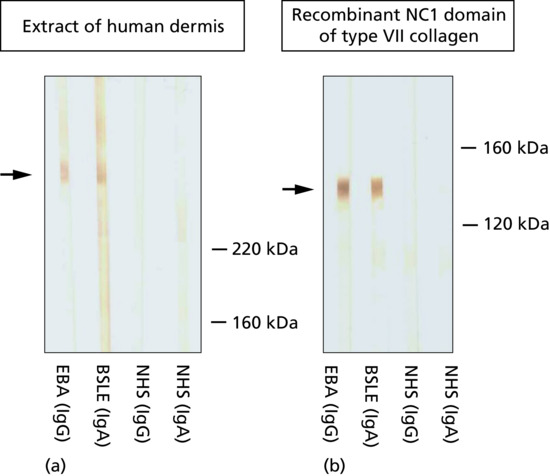
Figure 50.48 Serum autoantibodies in bullous systemic lupus erythematosus (BLSE) type I. IgA autoantibodies label the 290 kDa type VII collagen by immunoblotting with dermal extract (a) and the 140 kDa recombinant NC1 domain of type VII collagen by immunoblotting (b). The clinical picture of this patient is shown in Figure 50.47. EBA, epidermolysis bullosa acquisita; NHS, normal human serum.
Management
Dapsone is effective in about 90% of BSLE patients and leads to clinical improvement within days or a few weeks [27, 45]. In fact, the rapid response to dapsone is one of the characteristics that differentiate BSLE from SLE with concomitant pemphigoid disease. In patients unresponsive to dapsone, prednisolone and azathioprine have been advocated [6, 46, 47]. Individual patients effectively treated with methotrexate (10 mg/week) or rituximab have also been reported [48, 49].
Very rare pemphigoid disorders
Lichen planus pemphigoides
Lichen planus pemphigoides (ICD-10: L43.1) always arises in conjunction with lichen planus and can affect both adults and children [1–3]. At present, less than 50 patients have been described in the literature. In contrast to BP, the disease: (i) affects relatively young patients (average disease onset in the forties); (ii) mainly arises on the extremities; (iii) preferentially targets C-terminal epitopes within the immunodominant NC16A domain; and (iv) tends to be less severe [4]. For these reasons, lichen planus pemphigoides can also be regarded as a distinct entity within the pemphigoid disorders [5].
As in BSLE, it may be speculated that the chronic interface dermatitis typical for lichen planus may trigger the immune response against BP180. This hypothesis is supported by the notion that lichen planus pemphigoides only remits after lichen planus lesions are sufficiently controlled.
Diagnosis is made by the presence of tense blisters also outside of lichen planus lesions (Figure 50.49) and the detection of linear deposits of IgG and/or C3 at the DEJ by direct IF microscopy of a perilesional biopsy and circulating IgG antibodies against BP180 NC16A. Treatment requires therapy of lichen planus to avoid further stimulation of the anti-DEJ autoimmune process. Otherwise, treatment may follow the same algorithm as in BP.
Cicatricial pemphigoid
The term cicatricial pemphigoid has previously been applied to patients who are now classified as MMP [6]. Following an international expert conference, the term cicatricial pemphigoid is currently only used for the rare clinical variant of a pemphigoid disease in which mucous membranes are not predominantly affected and skin lesions heal with scarring [6]. In cases of a u-serrated pattern by direct IF microscopy or serum autoantibodies against type VII collagen, the patient would be classified as having EBA (Figures 50.50 and 50.51; and see Figure 50.23).

Figure 50.49 Lichen planus pemphigoides. Erosions, erythema, partly ruptured and subsequently desiccated blisters, and a tense vesicle on the left foot. In addition, a lichen planus lesion is seen. Of note, erosions and blisters are separate from the lichen planus lesion.
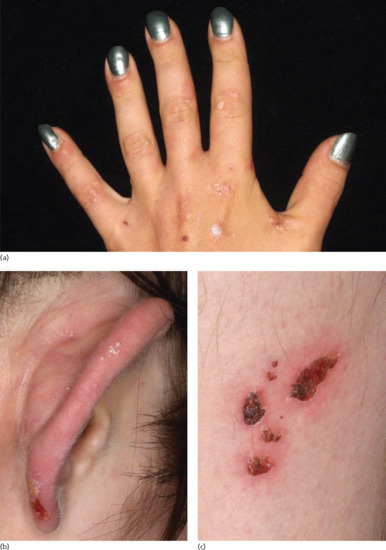
Figure 50.50 Cicatricial pemphigoid. A 36-year-old woman with tense vesicles between the thumb and ring finger and several atrophic papules and plaques on the dorsum of the left hand (a), milia, crust and erosion on the right ear (b), and crusts on the right thigh (c). Mucous membranes were not involved (not shown). Direct immunofluorescence (IF) microscopy showed linear deposits of immunoglobulin G (IgG) and C3 at the dermal–epidermal junction (DEJ). Serum autoantibodies labelled the epidermal side of human salt-split by indirect IF microscopy but no reactivity with BP180, BP230 and α6 integrin was detected by various enzyme-linked immunosorbent assay (ELISA) and immunoblotting analyses.
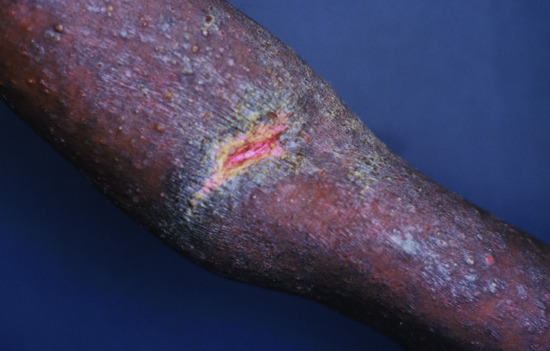
Figure 50.51 Cicatricial pemphigoid. Tense vesicles, erosions, milia and scarring on the right arm of a 24-year-old Ugandan man. Few oral lesions were present without conjunctival involvement. Serum autoantibodies reacted with BP180, BP230 and laminin 332 [32].
Brunsting–Perry pemphigoid is, at present, best regarded as a clinical subtype of cicatricial pemphigoid. Less than 100 patients with this phenotype have been reported so far. In 1957, Brunsting and Perry described a group of patients without mucous membrane involvement who developed subepidermal blisters, erosions, haemorrhagic crusts and atrophic scars on the head and neck [7]. The disease is common in middle-aged and elderly populations. Skin lesions are characteristically confined to the head, neck and upper trunk (Figures 50.52 and 50.53). Mucous membranes can also be involved, however not predominantly [8–10]. Direct IF microscopy shows linear deposits of IgG and/or C3 at the DEJ. Brunsting–Perry pemphigoid probably represents a heterogeneous disorder with several target antigens including type VII collagen, BP180, BP230, laminin 332 and desmoplakin [9–19]. Type VII collagen is the most frequent target antigen, therefore representing a localized form of EBA in these patients [8, 9, 17, 18]. In line with this, in most patients with Brunsting–Perry pemphigoid, electron microscopy revealed split formation below the lamina lucida [20]. In individual patients, disease onset was associated with trauma [21] and the combination of furosemide intake and sun exposure [22]. Differential diagnosis includes squamous cell carcinoma, basal cell carcinoma [23], pyoderma, erosive pustulosis of the scalp and dermatitis artefacta. Treatment usually comprises topical corticosteroids. Good responses to topical tacrolimus and the combination of topical corticosteroids with colchicine or dapsone have been reported [14, 16, 24].
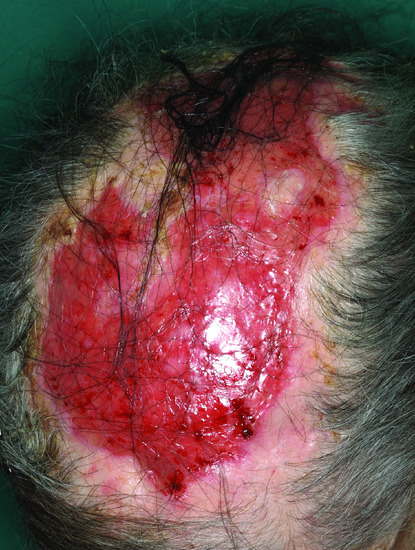
Figure 50.52 Brunsting–Perry pemphigoid. Erosions and some crusts on the scalp of a 76-year-old woman. By direct immunofluorescence (IF) microscopy linear deposits of immunoglobulin G (IgG) and C3 were seen at the dermal–epidermal junction (DEJ) and serum autoantibodies recognized multiple epitopes on BP180 (NC16A, LAD-1, C terminus) and BP230.
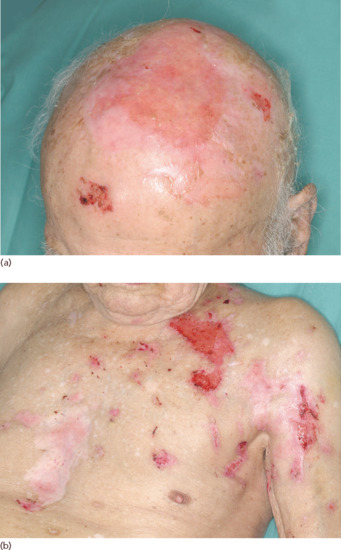
Figure 50.53 Brunsting–Perry pemphigoid. Erosions on the scalp (a), chest and upper left arm as well as atrophic scars (b) in a 95-year-old man. Linear staining of immunoglobulin G (IgG) and C3 were seen at the dermal–epidermal junction (DEJ) by direct immunofluorescence (IF) microscopy. Serum autoantibodies exclusively labelled the epidermal side by indirect IF microscopy on salt-split skin and reacted with BP180 NC16A.
Anti-type IV collagen pemphigoid
Ghohestani et al. reported three patients with generalized blistering and renal insufficiency [25, 26]. By lesional histopathology, subepidermal splitting and a dense eosinophilic infiltration of the upper dermis were seen. Direct IF microscopy revealed linear staining of IgG in two patients, of IgA (in one patient) and C3 (in all three patients) at the DEJ and, after splitting of the DEJ by 1 M NaCl solution, binding of IgG/IgA at the floor of the artificial blister. Serum autoantibodies reacted with the glomerular basement membrane and the a5 chain (in all three patients) and both the α5 and α6 chains (in one patient) of type IV collagen [25, 26].
Anti-105 kDa antigen pemphigoid
Chan and co-workers described two patients, 59 and 85 years old, with a generalized bullous eruption reminiscent of erythema multiforme and BP, respectively [27–29]. Both patients suffered from severe oral lesions and one from additional pharyngeal, oesophageal and conjunctival involvement. Neither scarring nor milia formation were observed. Lesional biopsies showed subepidermal splitting with dense neutrophilic infiltration within the dermal papilla compatible with dermatitis herpetiformis and direct IF microscopy revealed linear deposits of IgG and C3 at the DEJ [27–29]. Serum autoantibodies in both patients labelled the dermal side of the artificial split by indirect IF microscopy on human salt-split skin and reacted with a 105 kDa protein by immunoblotting with extracts of cultured keratinocytes and human skin. In one patient, additional reactivity with BP230 was detected [27–29]. Subsequent attempts to further characterize the target antigen did not lead to its molecular identification [30, 31].
Resources
Further information
- British Association of Dermatologists’ guidelines for the management of pemphigus and the management of bullous pemphigoid www.bad.org.uk/healthcare-professionals/clinical-standards/clinical-guidelines
- French guidelines (protocoles nationaux de diagnostic et de soins) for the management of pemphigus, management of bullous pemphigoid, management of mucous membrane pemphigoid, management of linear IgA disease, management of epidermolysis bullosa acquisita and management of dermatitis herpetiformis www.has-sante.fr
- Guidelines of the German Dermatological Society for the diagnosis of pemphigus and bullous pemphigoid and treatment of pemphigus and bullous pemphigoid www.awmf.org/leitlinien.
All last accessed August 2015.
Patient resources: support groups
- UK www.pemfriends.co.uk and www.pemphigus.org.uk
- Germany www.pemphigus-pemphigoid-selbsthilfe.de
- Italy www.assoc-apai.org and www.pemfigo.it
- France www.pemphigus.asso.fr
- Turkey www.turkdermatoloji.org.tr
- Australia www.blisters.org.au
- Canada www.pemphigus.ca
- International Pemphigus and Pemphigoid Foundation [33]
- All last accessed August 2015.
Dermatitis herpetiformis
Definition and nomenclature
Dermatitis herpetiformis is a chronic, intensely pruritic, skin condition associated with gluten-sensitive enteropathy. It results in deposition of IgA antitransglutaminase autoantibodies in the dermal papillae which lead to neutrophil infiltration and blister formation.
Introduction and general description
Dermatitis herpetiformis was first described by Duhring in 1884 [1]. It is a chronic autoimmune blistering disease that results in an intensely pruritic rash predominantly affecting the extensor surfaces [2, 3]. The characteristic vesicles are often not apparent as they are destroyed by excoriation. Dermatitis herpetiformis is closely associated with gluten-sensitive enteropathy (GSE [4, 5]): both conditions are characterized by the development of IgA autoantibodies against transglutaminases that, in the case of dermatitis herpetiformis, are deposited in the superficial papillary dermis. Therapy of dermatitis herpetiformis involves strict gluten avoidance and the use of sulphonamide drugs such as dapsone.
Epidemiology
Incidence and prevalence
It is most common in people of northern European descent, where prevalence ranges from 1.2 to 39.2 per 100 000 people and an incidence range of 0.4–2.6 per 100 000 people per year [6, 7]. A recent large population-based study from the UK suggests that, although the incidence of GSE increased over the period 1990–2011, the incidence of dermatitis herpetiformis fell from 1.8 per 100 000 to 0.8 per 100 000 person-years over the same period [8]. Interestingly, patients with dermatitis herpetiformis seem to have lower age-adjusted mortality than expected, possibly as a result of the modifications required to their diet as a result of the disease [9].
Age
Onset is most commonly in adult life, typically in the fourth decade, though cases have been reported from childhood to old age [10].
Sex
Several studies have suggested that dermatitis herpetiformis is commoner in men than women, with a male to female ratio of 1.5–2 : 1 [11].
Ethnicity
It is largely a condition of people of northern European descent and is most uncommon in Asian and African populations, though occasional cases have been reported [12]. Interestingly, the incidence and prevalence of the disease is similar in North American populations of European descent to those seen in northern Europe, suggesting that genetic factors are important in disease susceptibility [11]. Asian patients with dermatitis herpetiformis tend to have a distinct fibrillar pattern of IgA deposition in the skin and only very rarely associated with GSE [13, 14].
Associated diseases
The commonest association of dermatitis herpetiformis is with GSE. However, the severity of GSE in dermatitis herpetiformis patients varies hugely, and may be clinically silent or mild [15, 16]. Consequently, all patients with dermatitis herpetiformis should be reviewed by a gastroenterologist and investigated accordingly. An important consequence of a diagnosis of dermatitis herpetiformis is an increased risk of small bowel lymphoma [17].
Autoimmune diseases tend to associate with one another, presumably because of genetic susceptibility factors, and dermatitis herpetiformis is no different. The commonest association is with autoimmune thyroid disease [18, 19], and all patients with dermatitis herpetiformis should be screened for this. Additionally, patients with dermatitis herpetiformis have increased risk of type 1 diabetes [20], Addison disease [21] and vitiligo [18].
Pathophysiology
Pathology
The pathology in dermatitis herpetiformis and GSE results from an IgA dominant autoimmune response to transglutaminase molecules [2]. In GSE, the principal target is tissue transglutaminase (tTG) [22] whereas in dermatitis herpetiformis there is increasing evidence that epidermal transglutaminase 3 (TG3) may be the dominant antigen [23]. Both proteins have significant homologies in their enzymatic domains and are expressed in normal epidermis, where TG3 is important in cross-linking and maintenance of the cornified envelope [24].
The series of events that results in inflammation and itch in dermatitis herpetiformis remains to be fully elucidated [25, 26]. Hypotheses include release of TG3 from keratinocytes into the papillary dermis, where it is able to bind circulating IgA antibodies; or that circulating complexes of TG3 and IgA are deposited in the skin. Subsequently neutrophilic infiltration occurs, leading to inflammation and clefting within the lamina lucida.
Genetics
Multiple studies have revealed an association between dermatitis herpetiformis and certain HLA types, particularly HLA-DQ2 and, to a lesser extent, HLA DQ8 [27, 28]. The central role of the MHC in pathogenesis has been confirmed by studies using HLA DQ8 transgenic mice, which develop GSE following gluten challenge [29, 30]. Other non-MHC genes have been linked to GSE in genome-wide association studies including myosin IXB, IL-12, IL-23 and CCR3 [2], though the relevance of these to the pathogenesis of dermatitis herpetiformis (or for that matter GSE) remains unclear.
First-degree relatives of patients with GSE or dermatitis herpetiformis are significantly more likely to be affected by one or other disorder and thus family screening may be indicated [31, 32]. Monozygotic twins have a disease concordance rate of over 0.9 [33].
Environmental factors
The principal environmental factor involved in the pathogenesis of GSE and dermatitis herpetiformis is dietary gluten and its constituent gliadin [2]. Interestingly, tTG is responsible for the modification of gliadin in the gut mucosa into a more immunogenic form capable of better binding to HLA-DQ2 [34]. Other environmental factors relevant to dermatitis herpetiformis include iodine exposure (which can precipitate flares of the disease [35]) and tobacco smoking, which may ameliorate it [36, 37].
Clinical features
History
The principal symptom of patients with dermatitis herpetiformis is itch. Patients report a rash, most typically over the extensor surfaces of the elbows, knees, buttocks and scalp. There may also be symptoms of associated disorders such as GSE, with bloating, diarrhoea and other gastrointestinal complaints or of other autoimmune diseases including hypothyroidism.
Presentation
The characteristic lesions of dermatitis herpetiformis are grouped erythematous papules and vesicles located over extensor sites (Figure 50.54). Because the condition is so pruritic, intact vesicles are rarely seen and the patient may simply present with excoriations [38]. Lesions tend to be symmetrical and heal without scarring. Patients may develop punctate purpura on the palms and soles [39, 40]. Mucosal change may occur [41], and dental abnormalities have been reported, particularly enamel pits [42, 43]. Interestingly, first-degree relatives of patient with GSE may also show enamel defects [44].
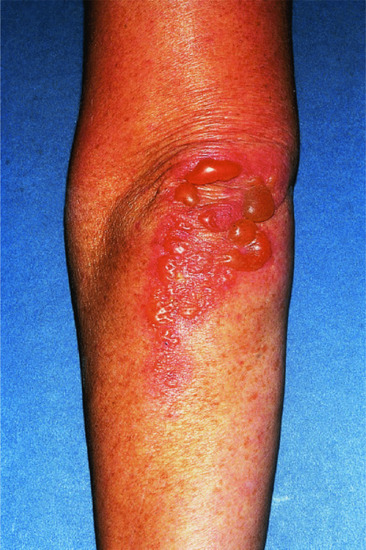
Figure 50.54 Dermatitis herpetiformis. Intact tense bullae on the elbow.
(Courtesy of Dr R.J. Pye, Addenbrooke's Hospital, Cambridge, UK.)
Differential diagnosis
The differential diagnosis of dermatitis herpetiformis includes many pruritic and vesiculobullous disorders including linear IgA disease, pemphigoid, eczema and scabies. Because of the often non-specific nature of dermatitis herpetiformis lesions, skin biopsy for direct IF should be considered in the investigation of patients with unexplained or treatment-refractory pruritic rashes.
Complications and co-morbidities
The principal complications and co-morbidities of dermatitis herpetiformis relate to GSE and the associated risk of small bowel lymphoma [45]. GSE may lead to malabsorption resulting in anaemia, weight loss and osteoporosis. In children, short stature is a consequence. Rarely, GSE (and consequently dermatitis herpetiformis) may be associated with neurological changes including ataxia and neuropathy [46, 47].
Disease course and prognosis
Dermatitis herpetiformis is a chronic disease that requires patients to adopt a long-term gluten-free diet. Those that are able to do this, and respond, seem to have excellent long-term survival [9] and are able to decrease or discontinue dapsone treatment. Recent data suggest that patients with GSE who do not respond to a gluten-free diet do poorly [48]. Whilst the disease is a chronic one, remission is recognized, and seems to be more common in adult patients over the age of 40 years [49].
Investigations
The diagnosis of dermatitis herpetiformis is made by the presence of characteristic clinical features, histopathology, direct IF testing and serology [3].
The histopathology of an intact vesicle in dermatitis herpetiformis demonstrates subepidermal blister formation with neutrophils located at the tips of the dermal papillae. There is frequently perivascular inflammatory cell infiltrate. Because vesicles may not survive the pruritus, clefting may not be seen.
Direct IF samples are best taken from perilesional skin as characteristic changes may be lost in lesional tissue [50, 51]. The diagnostic finding is that of deposition of IgA in the papillary dermis in a granular or fibrillar pattern (Figure 50.55). The latter has been particularly associated with dermatitis herpetiformis in Asian patients. Complement 3 may also be found in association with the IgA deposits.

Figure 50.55 Dermatitis herpetiformis. Direct immunofluorescence demonstrating granular IgA deposition in the dermal papillae.
Management
There are no randomized clinical trials in the management of dermatitis herpetiformis. Therapy is thus based on clinical experience and case series (see Treatment ladder).
The cornerstone of long-term dermatitis herpetiformis management is strict adherence to a gluten-free diet [52–54]. Not only does this improve skin changes over time but it is essential in the management of associated GSE. It should be remembered that the improvement in skin changes can be slow, taking months or years. Patients are best managed in consultation with an experienced dietitian who will be able to provide appropriate support as maintenance of a strict gluten-free diet can be challenging. Pure uncontaminated oats do not cause problems but patients need to avoid all exposure to wheat, barley and rye. Effective gluten avoidance, with resolution of manifest or subclinical GSE will lead to an increased sense of general well-being and may decrease the long-term risk of lymphoma [55].
Because of the slow response to gluten-free diet, most patients with dermatitis herpetiformis require pharmacological intervention to control their disease in the short to medium term. Dapsone and related sulphonamide drugs have proven highly effective, often suppressing pruritus within days of initiation of treatment [15]. It should be noted that dapsone does not impact on the gastrointestinal aspects of associated GSE.
The mechanism of action of dapsone in dermatitis herpetiformis is through its effects on neutrophil function and recruitment [56] and the drug is typically used at doses of 25–150 mg daily [57]. It is generally well tolerated though haematological side effects may occur including haemolysis [58], methaemoglobinaemia [59] and agranulocytosis [60]. Consequently, patients require regular blood monitoring, particularly in the early stages of treatment. Non-haematological adverse effects include a severe drug sensitivity reaction (‘dapsone hypersensitivity syndrome’, [61]) and a peripheral neuropathy [62]. Patients with glucose-6-phosphatase (G6PD) deficiency are more prone to adverse effects of dapsone [63] and G6PD activity should be checked before treatment wherever possible, particularly in at-risk populations. Concurrent administration of cimetidine, via inhibition of cytochrome p-450, has been shown to decrease the adverse effects of dapsone [64–66] and may be helpful in some patients. Dapsone should always be used in combination with a gluten-free diet. With time, it may be possible to decrease and potentially withdraw dapsone without relapse, as long as the patient is able to adhere to the diet.
Other sulphonamide drugs have utility in dermatitis herpetiformis including sulphamethoxypyridazine [67], sulphapyridine [68] and sulphasalazine [69]. Their mechanism of action is similar to that of dapsone, and they may be of utility in patients intolerant of dapsone. At the time of writing, however, sulphamethoxypyridazine is currently unavailable. Systemic corticosteroids are of little benefit, though potent topical corticosteroids may be of use in the short term to lessen pruritus.
Currently, there is just one guideline to the management of dermatitis herpetiformis in English [53], though others exist in Dutch [70], French [71] and Spanish [72].
Resources
Patient resources
- Patient information from Coeliac UK https://www.coeliac.org.uk/coeliac-disease/about-coeliac-disease-and-dermatitis-herpetiformis/dermatitis-herpetiformis/
- Patient information from the British Association of Dermatologists http://www.bad.org.uk/library-media/documents/Dermatitis%20Herpetiformis%20Update%20Jan%202013%20-%20lay%20reviewed%20Dec%202012.pdf
All last accessed August 2015.
References
Intraepidermal immunobullous diseases
- Bertram F, Bröcker E-B, Zillikens D, Schmidt E. Prospective analysis of the incidence of autoimmune bullous disorders in Lower Franconia, Germany. J Dtsch Dermatol Ges 2009;7:434–40.
- Pisanti S, Sharav Y, Kaufman E, Posner LN. Pemphigus vulgaris: incidence in Jews of different ethnic groups, according to age, sex, and initial lesion. Oral Surg Oral Med Oral Pathol 1974;38:382–7.
- Langan SM, Smeeth L, Hubbard R, Fleming KM, Smith CJP, West J. Bullous pemphigoid and pemphigus vulgaris – incidence and mortality in the UK: population based cohort study. BMJ 2008;337:a180.
- Korman N. Pemphigus. J Am Dermatol 1988;18:1219–38.
- Kanwar AJ, Ajith AC, Narang T. Pemphigus in North India. J Cutan Med Surg 2006;10:21–5.
- Hafeez ZH. Pemphigus in Pakistan, a study of 108 cases [corrected]. J Pakistan Med Assoc 1998;48:9–10.
- Salmanpour R, Shahkar H, Namazi MR, Rahman-Shenas MR. Epidemiology of pemphigus in south-western Iran: a 10-year retrospective study (1991–2000). Int J Dermatol 2006;45:103–5.
- Berger BW, Maier HS, Kantor I, Wexler DE. Pemphigus vulgaris in a 3-and-one-half-year old boy. Arch Dermatol 1973;107:433–4.
- Ahmed AR, Salm M. Juvenile pemphigus. J Am Dermatol 1983;8:799–807.
- Bastuji-Garin S, Souissi R, Blum L, et al. Comparative epidemiology of pemphigus in Tunisia and France: unusual incidence of pemphigus foliaceus in young Tunisian women. J Invest Dermatol 1995;104:302–5.
- Mahé A, Flageul B, Cissé I, Kéita S, Bobin P. Pemphigus in Mali: a study of 30 cases. Br J Dermatol 1996;134:114–19.
- Tron F, Gilbert D, Mouquet H, et al. Genetic factors in pemphigus. J Autoimmun 2005;24:319–28.
- Leshem YA, Katzenelson V, Yosipovitch G, David M, Mimouni D. Autoimmune diseases in patients with pemphigus and their first-degree relatives. Int J Dermatol 2011;50:827–31.
- Parameswaran A, Attwood K, Sato R, Seiffert-Sinha K, Sinha AA. Identification of a new disease cluster of pemphigus vulgaris with autoimmune thyroid disease, rheumatoid arthritis, and type I diabetes. Br J Dermatol 2014 Oct 1.
- Waschke J. The desmosome and pemphigus. Histochem Cell Biol 2008;130:21–54.
- Mahoney MG, Wang Z, Rothenberger K, Koch PJ, Amagai M, Stanley JR. Explanations for the clinical and microscopic localization of lesions in pemphigus foliaceus and vulgaris. J Clin Invest 1999;103:461–8.
- Judd KP, Lever WF. Correlation of antibodies in skin and serum with disease severity in pemphigus. Arch Dermatol 1979;115:428–32.
- Anhalt GJ, Labib RS, Voorhees JJ, Beals TF, Diaz LA. Induction of pemphigus in neonatal mice by passive transfer of IgG from patients with the disease. N Engl J Med 1982 May 20;306:1189–96.
- Ishii K, Harada R, Matsuo I, Shirakata Y, Hashimoto K, Amagai M. In vitro keratinocyte dissociation assay for evaluation of the pathogenicity of anti-desmoglein 3 IgG autoantibodies in pemphigus vulgaris. J Invest Dermatol 2005;124:939–46.
- Kurzen H, Brenner S. Significance of autoimmunity to non-desmoglein targets in pemphigus. Autoimmunity 2006;39:549–56.
- Vu TN, Lee TX, Ndoye A, et al. The pathophysiological significance of nondesmoglein targets of pemphigus autoimmunity. Development of antibodies against keratinocyte cholinergic receptors in patients with pemphigus vulgaris and pemphigus foliaceus. Arch Dermatol 1998;134:971–80.
- Anhalt GJ. Paraneoplastic pemphigus. J Investig Dermatol Symp Proc 2004;9:29–33.
- Sekiguchi M, Futei Y, Fujii Y, Iwasaki T, Nishikawa T, Amagai M. Dominant autoimmune epitopes recognized by pemphigus antibodies map to the N-terminal adhesive region of desmogleins. J Immunol 2001 Nov 1;167:5439–48.
- Sharma P, Mao X, Payne AS. Beyond steric hindrance: the role of adhesion signaling pathways in the pathogenesis of pemphigus. J Dermatol Sci 2007;48:1–14.
- Schmidt E, Waschke J. Apoptosis in pemphigus. Autoimmun Rev 2009;8:533–7.
- Puviani M, Marconi A, Cozzani E, Pincelli C. Fas ligand in pemphigus sera induces keratinocyte apoptosis through the activation of caspase-8. J Invest Dermatol 2003;120:164–7.
- Grando SA, Bystryn J-C, Chernyavsky AI, et al. Apoptolysis: a novel mechanism of skin blistering in pemphigus vulgaris linking the apoptotic pathways to basal cell shrinkage and suprabasal acantholysis. Exp Dermatol 2009;18:764–70.
- Brenner S, Dorfman B, Himelfarb M. Familial pemphigus vulgaris. Dermatologica 1985;171:38–40.
- Ahmed AR, Sofen H. Familial occurrence of pemphigus vulgaris. Arch Dermatol 1982;118:423–4.
- Ahmed AR, Mohimen A, Yunis EJ, et al. Linkage of pemphigus vulgaris antibody to the major histocompatibility complex in healthy relatives of patients. J Exp Med 1993 Feb 1;177:419–24.
- Brandsen R, Frusic-Zlotkin M, Lyubimov H, et al. Circulating pemphigus IgG in families of patients with pemphigus: comparison of indirect immunofluorescence, direct immunofluorescence, and immunoblotting. J Am Dermatol 1997;36:44–52.
- Slomov E, Loewenthal R, Goldberg I, Korostishevsky M, Brenner S, Gazit E. Pemphigus vulgaris in Jewish patients is associated with HLA-A region genes: mapping by microsatellite markers. Hum Immunol 2003;64:771–9.
- Hashimoto K, Miki Y, Nakata S, Matsuyama M. HLA-A10 in pemphigus among Japanese. Arch Dermatol 1977;113:1518–19.
- Capon F, Bharkhada J, Cochrane NE, et al. Evidence of an association between desmoglein 3 haplotypes and pemphigus vulgaris. Br J Dermatol 2006;154:67–71.
- Gibson WT, Walter MA, Ahmed AR, Alper CA, Cox DW. The immunoglobulin heavy chain and disease association: application to pemphigus vulgaris. Hum Genet 1994;94:675–83.
- Eberhard Y, Burgos E, Gagliardi J, et al. Cytokine polymorphisms in patients with pemphigus. Arch Dermatol Res 2005;296:309–13.
- Diaz LA, Sampaio SA, Rivitti EA, et al. Endemic pemphigus foliaceus (fogo selvagem). I. Clinical features and immunopathology. J Am Dermatol 1989;20:657–69.
- Diaz LA, Sampaio SA, Rivitti EA, et al. Endemic pemphigus foliaceus (fogo selvagem): II. Current and historic epidemiologic studies. J Invest Dermatol 1989;92:4–12.
- Aoki V, Sousa JX, Diaz LA, Cooperative Group on Fogo Selvagem Research. Pathogenesis of endemic pemphigus foliaceus. Dermatol Clin 2011;29:413–18, viii.
- Mehta JN, Martin AG. A case of pemphigus vulgaris improved by cigarette smoking. Arch Dermatol 2000;136:15–17.
- Valikhani M, Kavusi S, Chams-Davatchi C, et al. Pemphigus and associated environmental factors: a case-control study. Clin Exp Dermatol 2007;32:256–60.
- Valikhani M, Kavusi S, Chams-Davatchi C, et al. Impact of smoking on pemphigus. Int J Dermatol 2008;47:567–70.
- Grando SA. Cholinergic control of epidermal cohesion. Exp Dermatol 2006;15:265–82.
- Brenner S, Tur E, Shapiro J, et al. Pemphigus vulgaris: environmental factors. Occupational, behavioral, medical, and qualitative food frequency questionnaire. Int J Dermatol 2001;40:562–9.
- Fisher KR, Higginbotham R, Frey J, Granese J, Pillow J, Skinner RB. Pesticide-associated pemphigus vulgaris. Cutis 2008;82:51–4.
- Tsankov N, Kazandjieva J, Gantcheva M. Contact pemphigus induced by dihydrodiphenyltrichlorethane. Eur J Dermatol 1998;8:442–3.
- Ruocco V, Ruocco E, Schiavo Lo A, Brunetti G, Guerrera LP, Wolf R. Pemphigus: etiology, pathogenesis, and inducing or triggering factors: facts and controversies. Clin Dermatol 2013;31:374–81.
- Brenner S, Ruocco V, Wolf R, de Angelis E, Lombardi ML. Pemphigus and dietary factors. In vitro acantholysis by allyl compounds of the genus Allium. Dermatology 1995;190:197–202.
- Brenner S, Goldberg I. Drug-induced pemphigus. Clin Dermatol 2011;29:455–7.
- Pisani M, Ruocco V. Drug-induced pemphigus. Clin Dermatol 1986;4:118–32.
- Bialy-Golan A, Brenner S. Penicillamine-induced bullous dermatoses. J Am Dermatol 1996;35(5 Pt 1):732–42.
- Zone J, Ward J, Boyce E, Schupbach C. Penicillamine-induced pemphigus. JAMA 1982 May 21;247:2705–7.
- Gomi H, Akiyama M, Yakabi K, Nakamura T, Matsuo I. Oesophageal involvement in pemphigus vulgaris. Lancet 1999 Nov 20;354(9192):1794.
- Akhyani M, Chams-Davatchi C, Naraghi Z, et al. Cervicovaginal involvement in pemphigus vulgaris: a clinical study of 77 cases. Br J Dermatol 2008;158:478–82.
- Schlesinger N, Katz M, Ingber A. Nail involvement in pemphigus vulgaris. Br J Dermatol 2002;146:836–9.
- Habibi M, Mortazavi H, Shadianloo S, et al. Nail changes in pemphigus vulgaris. Int J Dermatol 2008;47:1141–4.
- Yoshida K, Takae Y, Saito H, et al. Cutaneous type pemphigus vulgaris: a rare clinical phenotype of pemphigus. J Am Acad Dermatol 2005;52:839–45.
- Carew B, Wagner G. Cutaneous pemphigus vulgaris with absence of desmoglein 1 autoantibodies. An example of the extended desmoglein compensation theory. Australas J Dermatol 2014;55:292–5.
- Awazawa R, Yamamoto Y-I, Gushi M, et al. Case of pemphigus foliaceus that shifted into pemphigus vulgaris after adrenal tumor resection. J Dermatol 2007;34:549–55.
- Harman KE, Gratian MJ, Shirlaw PJ, Bhogal BS, Challacombe SJ, Black MM. The transition of pemphigus vulgaris into pemphigus foliaceus: a reflection of changing desmoglein 1 and 3 autoantibody levels in pemphigus vulgaris. Br J Dermatol 2002;146:684–7.
- Lévy-Sitbon C, Reguiaï Z, Durlach A, Goeldel A-L, Grange F, Bernard P. [Transition from pemphigus vulgaris to pemphigus foliaceus: a case report.] Ann Dermatol Venereol 2013;140:788–92.
- Kardos M, Levine D, Gürcan HM, Ahmed RA. Pemphigus vulgaris in pregnancy: analysis of current data on the management and outcomes. Obstet Gynecol Surv 2009;64:739–49.
- Daneshpazhooh M, Chams-Davatchi C, Valikhani M, et al. Pemphigus and pregnancy: a 23-year experience. Indian J Dermatol Venereol Leprol 2011;77:534.
- Zaraa I, Sellami A, Bouguerra C, et al. Pemphigus vegetans: a clinical, histological, immunopathological and prognostic study. J Eur Acad Dermatol Venereol 2011;25:1160–7.
- Ahmed AR, Blose DA. Pemphigus vegetans. Neumann type and Hallopeau type. Int J Dermatol 1984;23:135–41.
- Hashimoto K, Hashimoto T, Higashiyama M, Nishikawa T, Garrod DR, Yoshikawa K. Detection of anti-desmocollins I and II autoantibodies in two cases of Hallopeau type pemphigus vegetans by immunoblot analysis. J Dermatol Sci 1994;7:100–6.
- Saruta H, Ishii N, Teye K, et al. Two cases of pemphigus vegetans with IgG anti-desmocollin 3 antibodies. JAMA Dermatol 2013;149:1209–13.
- Kasperkiewicz M, Kowalewski C, Jabłońska S. Pemphigus herpetiformis: from first description until now. J Am Acad Dermatol 2014;70:780–7.
- Kozlowska A, Hashimoto T, Jarzabek-Chorzelska M, et al. Pemphigus herpetiformis with IgA and IgG antibodies to desmoglein 1 and IgG antibodies to desmocollin 3. J Am Dermatol 2003;48:117–22.
- On HR, Hashimoto T, Kim S-C. Pemphigus herpetiformis with IgG autoantibodies to desmoglein 1 and desmocollin 1. Br J Dermatol 2014 Sep 21.
- Santi CG, Maruta CW, Aoki V, Sotto MN, Rivitti EA, Diaz LA. Pemphigus herpetiformis is a rare clinical expression of nonendemic pemphigus foliaceus, fogo selvagem, and pemphigus vulgaris. Cooperative Group on Fogo Selvagem Research. J Am Dermatol 1996;34:40–6.
- Steffen C, Thomas D. The men behind the eponym: Francis E. Senear, Barney Usher, and the Senear-Usher syndrome. Am J Dermatopathol 2003;25:432–6.
- Pérez-Pérez ME, Avalos-Díaz E, Herrera-Esparza R. Autoantibodies in senear-usher syndrome: cross-reactivity or multiple autoimmunity? Autoimmune Dis 2012;2012:296214–17.
- Jablonska S, Chorzelski T, Blaszczyk M, Maciejewski W. Pathogenesis of pemphigus erythematosus. Arch Dermatol Res 1977 Apr 27;258:135–40.
- Yong AA, Tey HL. Paraneoplastic pemphigus. Australas J Dermatol 2013;54:241–50.
- Horn TD, Anhalt GJ. Histologic features of paraneoplastic pemphigus. Arch Dermatol 1992;128:1091–5.
- Frew JW, Murrell DF. Paraneoplastic pemphigus (paraneoplastic autoimmune multiorgan syndrome): clinical presentations and pathogenesis. Dermatol Clin 2011;29:419–25, viii.
- Tsuruta D, Ishii N, Hamada T, et al. IgA pemphigus. Clin Dermatol 2011;29:437–42.
- Hashimoto T, Kiyokawa C, Mori O, et al. Human desmocollin 1 (Dsc1) is an autoantigen for the subcorneal pustular dermatosis type of IgA pemphigus. J Invest Dermatol 1997;109:127–31.
- Zaraa I, Kerkeni N, Sellami M, et al. IgG/IgA pemphigus with IgG and IgA antidesmoglein 3 antibodies and IgA antidesmoglein 1 antibodies detected by enzyme-linked immunosorbent assay: a case report and review of the literature. Int J Dermatol 2010;49:298–302.
- Tajima M, Mitsuhashi Y, Irisawa R, Amagai M, Hashimoto T, Tsuboi R. IgA pemphigus reacting exclusively to desmoglein 3. Eur J Dermatol 2010;20:626–9.
- Caputo R, Pistritto G, Gianni E, et al. IgA pemphigus in a child. J Am Dermatol 1991;25(2 Pt 2):383–6.
- Weston WL, Friednash M, Hashimoto T, Seline P, Huff JC, Morelli JG. A novel childhood pemphigus vegetans variant of intraepidermal neutrophilic IgA dermatosis. J Am Dermatol 1998;38:635–8.
- Myers SA, Rico MJ. Intraepidermal neutrophilic IgA dermatosis in an HIV-infected patient. J Am Dermatol 1994;31(3 Pt 1):502–4.
- Borradori L, Saada V, Rybojad M, et al. Oral intraepidermal IgA pustulosis and Crohn's disease. Br J Dermatol 1992;126:383–6.
- Kishimoto K, Iwatsuki K, Akiba H, Motoki Y, Kaneko F. Subcorneal pustular dermatosis-type IgA pemphigus induced by thiol drugs. Eur J Dermatol 2001;11:41–4.
- Shimizu T, Takebayashi T, Sato Y, et al. Grading criteria for disease severity by pemphigus disease area index. J Dermatol 2014;41:969–73.
- Pfütze M, Niedermeier A, Hertl M, Eming R. Introducing a novel Autoimmune Bullous Skin Disorder Intensity Score (ABSIS) in pemphigus. Eur J Dermatol 2007;17:4–11.
- Daniel BS, Hertl M, Werth VP, Eming R, Murrell DF. Severity score indexes for blistering diseases. Clin Dermatol 2012;30:108–13.
- Rahbar Z, Daneshpazhooh M, Mirshams-Shahshahani M, et al. Pemphigus disease activity measurements: pemphigus disease area index, autoimmune bullous skin disorder intensity score, and pemphigus vulgaris activity score. JAMA Dermatol 2014;150:266–72.
- Herbst A, Bystryn JC. Patterns of remission in pemphigus vulgaris. J Am Dermatol 2000;42:422–7.
- Seidenbaum M, David M, Sandbank M. The course and prognosis of pemphigus. A review of 115 patients. Int J Dermatol 1988;27:580–4.
- Chams-Davatchi C, Valikhani M, Daneshpazhooh M, et al. Pemphigus: analysis of 1209 cases. Int J Dermatol 2005;44:470–6.
- Kavusi S, Daneshpazhooh M, Farahani F, Abedini R, Lajevardi V, Chams-Davatchi C. Outcome of pemphigus vulgaris. J Eur Acad Dermatol Venereol 2008;22:580–4.
- Harman KE, Gratian MJ, Bhogal BS, Challacombe SJ, Black MM. A study of desmoglein 1 autoantibodies in pemphigus vulgaris: racial differences in frequency and the association with a more severe phenotype. Br J Dermatol 2000;143:343–8.
- Saha M, Bhogal B, Black MM, Cooper D, Vaughan RW, Groves RW. Prognostic factors in pemphigus vulgaris and pemphigus foliaceus. Br J Dermatol 2014;170:116–22.
- Black MM, Thomas RH, Bhogal B. The value of immunofluorescence techniques in the diagnosis of bullous disorders: a review. Clin Exp Dermatol 1983;8:337–53.
- Beutner EH, Chorzelski TP, Jablonska S. Immunofluorescence tests. Clinical significance of sera and skin in bullous diseases. Int J Dermatol 1985;24:405–21.
- Vaughan Jones SA, Salas J, McGrath JA, Palmer I, Bhogal GS, Black MM. A retrospective analysis of tissue-fixed immunoreactants from skin biopsies maintained in Michel's medium. Dermatology 1994;189 Suppl. 1:131–2.
- Feibelman C, Stolzner G, Provost TT. Pemphigus vulgaris. Superior sensitivity of monkey esophagus in the determination of pemphigus antibody. Arch Dermatol 1981;117:561–2.
- Kelly S, Culican S, Silvestrini RA, et al. Comparative study of five serological assays for the diagnosis of paraneoplastic pemphigus. Pathology 2014 Dec 3;47:58–61.
- Schmidt E, Zillikens D. Modern diagnosis of autoimmune blistering skin diseases. Autoimmun Rev 2010;10:84–9.
- Cheng SW, Kobayashi M, Kinoshita-Kuroda K, Tanikawa A, Amagai M, Nishikawa T. Monitoring disease activity in pemphigus with enzyme-linked immunosorbent assay using recombinant desmogleins 1 and 3. Br J Dermatol 2002;147:261–5.
- Schmidt E, Dähnrich C, Rosemann A, et al. Novel ELISA systems for antibodies to desmoglein 1 and 3: correlation of disease activity with serum autoantibody levels in individual pemphigus patients. Exp Dermatol 2010;19:458–63.
- Ablin RJ, Milgrom F, Kano K, Rapaport FT, Beutner EH. Pemphigus-like antibodies in patients with skin burns. Vox Sang 1969;16:73–5.
- Park GT, Quan G, Lee JB. Sera from patients with toxic epidermal necrolysis contain autoantibodies to periplakin. Br J Dermatol 2006;155:337–43.
- Torzecka JD, Woźniak K, Kowalewski C, et al. Circulating pemphigus autoantibodies in healthy relatives of pemphigus patients: coincidental phenomenon with a risk of disease development? Arch Dermatol Res 2007;299(5–6):239–43.
- Kricheli D, David M, Frusic-Zlotkin M, et al. The distribution of pemphigus vulgaris-IgG subclasses and their reactivity with desmoglein 3 and 1 in pemphigus patients and their first-degree relatives. Br J Dermatol 2000;143:337–42.
- Kasperkiewicz M, Schmidt E, Zillikens D. Current therapy of the pemphigus group. Clin Dermatol 2012;30:84–94.
- Ruocco E, Wolf R, Ruocco V, Brunetti G, Romano F, Schiavo Lo A. Pemphigus: associations and management guidelines: facts and controversies. Clin Dermatol 2013;31:382–90.
- Harman KE, Albert S, Black MM, British Association of Dermatologists. Guidelines for the management of pemphigus vulgaris. Br J Dermatol 2003;149:926–37.
- Chams-Davatchi C, Esmaili N, Daneshpazhooh M, et al. Randomized controlled open-label trial of four treatment regimens for pemphigus vulgaris. J Am Acad Dermatol 2007;57:622–8.
- Ratnam KV, Phay KL, Tan CK. Pemphigus therapy with oral prednisolone regimens. A 5-year study. Int J Dermatol 1990;29:363–7.
- Femiano F, Gombos F, Scully C. Pemphigus vulgaris with oral involvement: evaluation of two different systemic corticosteroid therapeutic protocols. J Eur Acad Dermatol Venereol 2002;16:353–6.
- Rose E, Wever S, Zilliken D, Linse R, Haustein U-F, Bröcker E-B. Intravenous dexamethasone-cyclophosphamide pulse therapy in comparison with oral methylprednisolone-azathioprine therapy in patients with pemphigus: results of a multicenter prospectively randomized study. J Dtsch Dermatol Ges 2005;3:200–6.
- Mentink LF, Mackenzie MW, Tóth GG, et al. Randomized controlled trial of adjuvant oral dexamethasone pulse therapy in pemphigus vulgaris: PEMPULS trial. Arch Dermatol 2006;142:570–6.
- Aberer W, Wolff-Schreiner EC, Stingl G, Wolff K. Azathioprine in the treatment of pemphigus vulgaris. A long-term follow-up. J Am Dermatol 1987;16(3 Pt 1):527–33.
- Chams-Davatchi C, Esmaili N, Daneshpazhooh M, et al. Randomized controlled open-label trial of four treatment regimens for pemphigus vulgaris. J Am Acad Dermatol 2007;57:622–8.
- Beissert S, Werfel T, Frieling U, et al. A comparison of oral methylprednisolone plus azathioprine or mycophenolate mofetil for the treatment of pemphigus. Arch Dermatol 2006;142:1447–54.
- Anstey AV, Wakelin S, Reynolds NJ, British Association of Dermatologists Therapy, Guidelines and Audit Subcommittee. Guidelines for prescribing azathioprine in dermatology. Br J Dermatol 2004;151:1123–32.
- Beissert S, Mimouni D, Kanwar AJ, Solomons N, Kalia V, Anhalt GJ. Treating pemphigus vulgaris with prednisone and mycophenolate mofetil: a multicenter, randomized, placebo-controlled trial. J Invest Dermatol 2010 Apr 22;130:2041–8.
- Powell AM, Albert S, Fares al S, et al. An evaluation of the usefulness of mycophenolate mofetil in pemphigus. Br J Dermatol 2003;149:138–45.
- Pasricha JS, Sood VD, Minocha Y. Treatment of pemphigus with cyclophosphamide. Br J Dermatol 1975;93:573–6.
- Saha M, Bhogal B, Black MM, Groves RW. Pulsed intravenous cyclophosphamide and methylprednisolone therapy in refractory pemphigus. Br J Dermatol 2010;162:790–7.
- Olszewska M, Kolacinska-Strasz Z, Sulej J, et al. Efficacy and safety of cyclophosphamide, azathioprine, and cyclosporine (ciclosporin) as adjuvant drugs in pemphigus vulgaris. Am J Clin Dermatol 2007;8:85–92.
- Ahmed AR. Intravenous immunoglobulin therapy in the treatment of patients with pemphigus vulgaris unresponsive to conventional immunosuppressive treatment. J Am Dermatol 2001;45:679–90.
- Aoyama Y. What's new in i.v. immunoglobulin therapy and pemphigus: high-dose i.v. immunoglobulin therapy and its mode of action for treatment of pemphigus. J Dermatol 2010;37:239–45.
- Amagai M, Ikeda S, Shimizu H, et al. A randomized double-blind trial of intravenous immunoglobulin for pemphigus. J Am Acad Dermatol 2009;60:595–603.
- Joly P, D'Incan M, Musette P. Rituximab for pemphigus vulgaris. N Engl J Med 2007 Feb 1;356:521, author reply 521–2.
- Ahmed AR, Spigelman Z, Cavacini LA, Posner MR. Treatment of pemphigus vulgaris with rituximab and intravenous immune globulin. N Engl J Med 2006 Oct 26;355:1772–9.
- Kanwar AJ, Vinay K, Sawatkar GU, et al. Clinical and immunological outcomes of high- and low-dose rituximab treatments in patients with pemphigus: a randomized, comparative, observer-blinded study. Br J Dermatol 2014;170:1341–9.
- Eming R, Nagel A, Wolff-Franke S, Podstawa E, Debus D, Hertl M. Rituximab exerts a dual effect in pemphigus vulgaris. J Invest Dermatol 2008;128:2850–8.
- Kasperkiewicz M, Shimanovich I, Meier M, et al. Treatment of severe pemphigus with a combination of immunoadsorption, rituximab, pulsed dexamethasone and azathioprine/mycophenolate mofetil: a pilot study of 23 patients. Br J Dermatol 2011 Oct 17;166:154–60.
- Kasperkiewicz M, Eming R, Behzad M, et al. Efficacy and safety of rituximab in pemphigus: experience of the German Registry of Autoimmune Diseases. J Dtsch Dermatol Ges 2012;10:727–32.
- Riedell P, Carson KR. A drug safety evaluation of rituximab and risk of hepatitis B. Exp Opin Drug Saf 2014;13:977–87.
- Cotterill JA, Barker DJ, Millard LG. Plasma exchange in the treatment of pemphigus vulgaris. Br J Dermatol 1978;98:243.
- Guillaume JC, Roujeau JC, Morel P, et al. Controlled study of plasma exchange in pemphigus. Arch Dermatol 1988;124:1659–63.
- Schmidt E, Zillikens D. Immunoadsorption in dermatology. Arch Dermatol Res 2010;302:241–53.
- Eming R, Hertl M. Immunoadsorption in pemphigus. Autoimmunity 2006;39:609–16.
- Kolesnik M, Becker E, Reinhold D, et al. Treatment of severe autoimmune blistering skin diseases with combination of protein A immunoadsorption and rituximab: a protocol without initial high dose or pulse steroid medication. J Eur Acad Dermatol Venereol 2014;28:771–80.
- Vardy DA, Cohen AD. Cyclosporine therapy should be considered for maintenance of remission in patients with pemphigus. Arch Dermatol 2001;137:505–6.
- Mobini N, Padilla T, Ahmed AR. Long-term remission in selected patients with pemphigus vulgaris treated with cyclosporine. J Am Dermatol 1997;36(2 Pt 1):264–6.
- Ioannides D, Chrysomallis F, Bystryn JC. Ineffectiveness of cyclosporine as an adjuvant to corticosteroids in the treatment of pemphigus. Arch Dermatol 2000;136:868–72.
- Gooptu C, Staughton RC. Use of topical cyclosporin in oral pemphigus. J Am Dermatol 1998;38(5 Pt 2):860–1.
- Pandya AG, Dyke C. Treatment of pemphigus with gold. Arch Dermatol 1998;134:1104–7.
- Iranzo P, Alsina MM, Martínez-De Pablo I, Segura S, Mascaro JM, Herrero C. Gold: an old drug still working in refractory pemphigus. J Eur Acad Dermatol Venereol 2007;21:902–7.
- Gürcan HM, Ahmed AR. Efficacy of dapsone in the treatment of pemphigus and pemphigoid: analysis of current data. Am J Clin Dermatol 2009;10:383–96.
- Werth VP, Fivenson D, Pandya AG, et al. Multicenter randomized, double-blind, placebo-controlled, clinical trial of dapsone as a glucocorticoid-sparing agent in maintenance-phase pemphigus vulgaris. Arch Dermatol 2008;144:25–32.
- Ichimiya M, Yamamoto K, Muto M. Successful treatment of pemphigus vegetans by addition of etretinate to systemic steroids. Clin Exp Dermatol 1998;23:178–80.
- Smith TJ, Bystryn JC. Methotrexate as an adjuvant treatment for pemphigus vulgaris. Arch Dermatol 1999;135:1275–6.
- Liang G, Nahass G, Kerdel FA. Pemphigus vulgaris treated with photopheresis. J Am Dermatol 1992;26(5 Pt 1):779–80.
- Gollnick HP, Owsianowski M, Taube KM, Orfanos CE. Unresponsive severe generalized pemphigus vulgaris successfully controlled by extracorporeal photopheresis. J Am Dermatol 1993;28:122–4.
- Pardo J, Mercader P, Mahiques L, Sánchez-Carazo JL, Oliver V, Fortea JM. Infliximab in the management of severe pemphigus vulgaris. Br J Dermatol 2005;153:222–3.
- Vojáčková N, Fialová J, Vaňousová D, Hercogová J. Pemphigus vulgaris treated with adalimumab: case study. Dermatol Ther 2012;25:95–7.
- Boussemart L, Jacobelli S, Batteux F, et al. Autoimmune bullous skin diseases occurring under anti-tumor necrosis factor therapy: two case reports. Dermatology 2010;221:201–5.
- Chaffins ML, Collison D, Fivenson DP. Treatment of pemphigus and linear IgA dermatosis with nicotinamide and tetracycline: a review of 13 cases. J Am Dermatol 1993;28:998–1000.
- McCarty M, Fivenson D. Two decades of using the combination of tetracycline derivatives and niacinamide as steroid-sparing agents in the management of pemphigus: defining a niche for these low toxicity agents. J Am Acad Dermatol 2014;71:475–9.
Subepidermal immunobullous diseases
Bullous pemphigoid
- Sardy M, Karpati S, Merkl B, Paulsson M, Smyth N. Epidermal transglutaminase (TGase 3) is the autoantigen of dermatitis herpetiformis. J Exp Med 2002;195:747–57.
- Schmidt E, Zillikens D. Pemphigoid diseases. Lancet 2013;381:320–32.
- Chan LS, Fine JD, Briggaman RA, et al. Identification and partial characterization of a novel 105-kDalton lower lamina lucida autoantigen associated with a novel immune-mediated subepidermal blistering disease. J Invest Dermatol 1993;101:262–7.
- Ghohestani RF, Hudson BG, Claudy A, Uitto J. The alpha 5 chain of type IV collagen is the target of IgG autoantibodies in a novel autoimmune disease with subepidermal blisters and renal insufficiency. J Biol Chem 2000;275:16002–6.
- Venning VA. Linear IgA disease: clinical presentation, diagnosis, and pathogenesis. Dermatol Clin 2011;29:453–58, ix.
- Zhu XJ, Niimi Y, Bystryn JC. Identification of a 160-kD molecule as a component of the basement membrane zone and as a minor bullous pemphigoid antigen. J Invest Dermatol 1990;94:817–21.
- Allen J, Wojnarowska F. Linear IgA disease: the IgA and IgG response to dermal antigens demonstrates a chiefly IgA response to LAD285 and a dermal 180-kDa protein. Br J Dermatol 2003;149:1055–8.
- Lever WF. Pemphigus. Medicine 1953;32:1–123.
- Jordon RE, Beutner EH, Witebsky E, Blumental G, Hale WL, Lever WF. Basement zone antibodies in bullous pemphigoid. JAMA 1967;200:751–6.
- Labib RS, Anhalt GJ, Patel HP, Mutasim DF, Diaz LA. Molecular heterogeneity of the bullous pemphigoid antigens as detected by immunoblotting. J Immunol 1986;136:1231–5.
- Diaz LA, Ratrie H, 3rd, Saunders WS, et al. Isolation of a human epidermal cDNA corresponding to the 180-kD autoantigen recognized by bullous pemphigoid and herpes gestationis sera. Immunolocalization of this protein to the hemidesmosome. J Clin Invest 1990;86:1088–94.
- Stanley JR, Hawley-Nelson P, Yuspa SH, Shevach EM, Katz SI. Characterization of bullous pemphigoid antigen: a unique basement membrane protein of stratified squamous epithelia. Cell 1981;24:897–903.
- Liu Z, Diaz LA, Troy JL, et al. A passive transfer model of the organ-specific autoimmune disease, bullous pemphigoid, using antibodies generated against the hemidesmosomal antigen, BP180. J Clin Invest 1993;92:2480–8.
- Nishie W, Sawamura D, Goto M, et al. Humanization of autoantigen. Nat Med 2007;13:378–83.
- Baican A, Baican C, Chiriac G, et al. Pemphigus vulgaris is the most common autoimmune bullous disease in Northwestern Romania. Int J Dermatol 2010;49:768–74.
- Nanda A, Dvorak R, Al-Saeed K, Al-Sabah H, Alsaleh QA. Spectrum of autoimmune bullous diseases in Kuwait. Int J Dermatol 2004;43:876–81.
- Serwin AB, Bokiniec E, Piascik M, Masny D, Chodynicka B. Epidemiological and clinical analysis of pemphigoid patients in northeastern Poland in 2000–2005. Med Sci Monit 2007;13:CR360–4.
- Wong SN, Chua SH. Spectrum of subepidermal immunobullous disorders seen at the National Skin Centre, Singapore: a 2-year review. Br J Dermatol 2002;147:476–80.
- Bertram F, Brocker EB, Zillikens D, Schmidt E. Prospective analysis of the incidence of autoimmune bullous disorders in Lower Franconia, Germany. J Dtsch Dermatol Ges 2009;7:434–40.
- Gudi VS, White MI, Cruickshank N, et al. Annual incidence and mortality of bullous pemphigoid in the Grampian Region of North-east Scotland. Br J Dermatol 2005;153:424–7.
- Marazza G, Pham HC, Scharer L, et al. Incidence of bullous pemphigoid and pemphigus in Switzerland: a 2-year prospective study. Br J Dermatol 2009;161:861–8.
- Joly P, Baricault S, Sparsa A, et al. Incidence and mortality of bullous pemphigoid in France. J Invest Dermatol 2012;132:1998–2004.
- Cordel N, Renier M, Samyn A, Fauvel C, Hope-Rapp E, Gilbert D. [Epidemiology of bullous pemphigoid in Guadeloupe (French West Indies).] Ann Dermatol Venereol 2009;136:907–9.
- Brick KE, Weaver CH, Lohse CM, et al. Incidence of bullous pemphigoid and mortality of patients with bullous pemphigoid in Olmsted County, Minnesota, 1960 through 2009. J Am Acad Dermatol 2014;71:92–9.
- Langan SM, Smeeth L, Hubbard R, Fleming KM, Smith CJ, West J. Bullous pemphigoid and pemphigus vulgaris – incidence and mortality in the UK: population based cohort study. BMJ 2008;337:a180.
- Zillikens D, Wever S, Roth A, Weidenthaler-Barth B, Hashimoto T, Brocker EB. Incidence of autoimmune subepidermal blistering dermatoses in a region of central Germany. Arch Dermatol 1995;131:957–8.
- Bernard P, Vaillant L, Labeille B, et al. Incidence and distribution of subepidermal autoimmune bullous skin diseases in three French regions. Bullous Diseases French Study Group. Arch Dermatol 1995;131:48–52.
- Bastuji-Garin S, Joly P, Lemordant P, et al. Risk factors for bullous pemphigoid in the elderly: a prospective case-control study. J Invest Dermatol 2011;131:637–43.
- Langan SM, Groves RW, West J. The relationship between neurological disease and bullous pemphigoid: a population-based case-control study. J Invest Dermatol 2010;131:631–6.
- Taghipour K, Chi CC, Vincent A, Groves RW, Venning V, Wojnarowska F. The association of bullous pemphigoid with cerebrovascular disease and dementia: a case-control study. Arch Dermatol 2010;146:1251–4.
- Bastuji-Garin S, Joly P, Picard-Dahan C, et al. Drugs associated with bullous pemphigoid. A case-control study. Arch Dermatol 1996;132:272–6.
- Lloyd-Lavery A, Chi CC, Wojnarowska F, Taghipour K. The associations between bullous pemphigoid and drug use: a UK case-control study. JAMA Dermatol 2013;149:58–62.
- Cortes B, Marazza G, Naldi L, Combescure C, Borradori L. Mortality of bullous pemphigoid in Switzerland: a prospective study. Br J Dermatol 2011;165:368–74.
- Kulthanan K, Chularojanamontri L, Tuchinda P, Sirikudta W, Pinkaew S. Prevalence and clinical features of Thai patients with bullous pemphigoid. Asian Pac J Allergy Immunol 2011;29:66–72.
- Parker SR, Dyson S, Brisman S, et al. Mortality of bullous pemphigoid: an evaluation of 223 patients and comparison with the mortality in the general population in the United States. J Am Acad Dermatol 2008;59:582–8.
- Rzany B, Partscht K, Jung M, et al. Risk factors for lethal outcome in patients with bullous pemphigoid: low serum albumin level, high dosage of glucocorticosteroids, and old age. Arch Dermatol 2002;138:903–8.
- Li J, Zuo YG, Zheng HY. Mortality of bullous pemphigoid in China. JAMA Dermatol 2013;149:106–8.
- Jung M, Kippes W, Messer G, Zillikens D, Rzany B. Increased risk of bullous pemphigoid in male and very old patients: A population-based study on incidence. J Am Acad Dermatol 1999;41:266–8.
- Amos B, Deng JS, Flynn K, Suarez S. Bullous pemphigoid in infancy: case report and literature review. Pediatr Dermatol 1998;15:108–11.
- Nemeth AJ, Klein AD, Gould EW, Schachner LA. Childhood bullous pemphigoid. Clinical and immunologic features, treatment, and prognosis. Arch Dermatol 1991;127:378–86.
- Waisbourd-Zinman O, Ben-Amitai D, Cohen AD, et al. Bullous pemphigoid in infancy: Clinical and epidemiologic characteristics. J Am Acad Dermatol 2008;58:41–8.
- Cordel N, Chosidow O, Hellot MF, et al. Neurological disorders in patients with bullous pemphigoid. Dermatology 2007;215:187–91.
- Chen YJ, Wu CY, Lin MW, et al. Comorbidity profiles among patients with bullous pemphigoid: a nationwide population-based study. Br J Dermatol 2011;165:593–9.
- Jedlickova H, Hlubinka M, Pavlik T, Semradova V, Budinska E, Vlasin Z. Bullous pemphigoid and internal diseases – A case-control study. Eur J Dermatol 2010;20:96–101.
- Seppanen A, Suuronen T, Hofmann SC, Majamaa K, Alafuzoff I. Distribution of collagen XVII in the human brain. Brain Res 2007;1158:50–6.
- Leung CL, Zheng M, Prater SM, Liem RK. The BPAG1 locus: Alternative splicing produces multiple isoforms with distinct cytoskeletal linker domains, including predominant isoforms in neurons and muscles. J Cell Biol 2001;154:691–7.
- Guo L, Degenstein L, Dowling J, et al. Gene targeting of BPAG1: abnormalities in mechanical strength and cell migration in stratified epithelia and neurologic degeneration. Cell 1995;81:233–43.
- Sakanoue M, Kawai K, Kanekura T. Bullous pemphigoid associated with type 1 diabetes mellitus responsive to mycophenolate mofetil. J Dermatol 2012;39:884–5.
- Ameri P, Cinotti E, Mussap M, Murialdo G, Parodi A, Cozzani E. Association of pemphigus and bullous pemphigoid with thyroid autoimmunity in Caucasian patients. J Am Acad Dermatol 2013;68:687–9.
- Taylor G, Venning V, Wojnarowska F, Welch K. Bullous pemphigoid and autoimmunity. J Am Acad Dermatol 1993;29:181–4.
- Lindelof B, Islam N, Eklund G, Arfors L. Pemphigoid and cancer. Arch Dermatol 1990;126:66–8.
- Ogawa H, Sakuma M, Morioka S, et al. The incidence of internal malignancies in pemphigus and bullous pemphigoid in Japan. J Dermatol Sci 1995;9:136–41.
- Ong E, Goldacre R, Hoang U, Sinclair R, Goldacre M. Associations between bullous pemphigoid and primary malignant cancers: an English national record linkage study, 1999-2011. Arch Dermatol Res 2014;306:75–80.
- Schulze F, Neumann K, Recke A, Zillikens D, Linder R, Schmidt E. Malignancies in pemphigus and pemphigoid diseases. J Invest Dermatol 2015;135:1445–7.
- Ludwig RJ, Kalies K, Kohl J, Zillikens D, Schmidt E. Emerging treatments for pemphigoid diseases. Trends Mol Med 2013;19:501–12.
- Di Zenzo G, Thoma-Uszynski S, Fontao L, et al. Multicenter prospective study of the humoral autoimmune response in bullous pemphigoid. Clin Immunol 2008;128:415–26.
- Zillikens D, Rose PA, Balding SD, et al. Tight clustering of extracellular BP180 epitopes recognized by bullous pemphigoid autoantibodies. J Invest Dermatol 1997;109:573–9.
- Giudice GJ, Emery DJ, Zelickson BD, Anhalt GJ, Liu Z, Diaz LA. Bullous pemphigoid and herpes gestationis autoantibodies recognize a common non-collagenous site on the BP180 ectodomain. J Immunol 1993;151:5742–50.
- Sakuma-Oyama Y, Powell AM, Oyama N, Albert S, Bhogal BS, Black MM. Evaluation of a BP180-NC16a enzyme-linked immunosorbent assay in the initial diagnosis of bullous pemphigoid. Br J Dermatol 2004;151:126–31.
- Tsuji-Abe Y, Akiyama M, Yamanaka Y, Kikuchi T, Sato-Matsumura KC, Shimizu H. Correlation of clinical severity and ELISA indices for the NC16A domain of BP180 measured using BP180 ELISA kit in bullous pemphigoid. J Dermatol Sci 2005;37:145–9.
- Mariotti F, Grosso F, Terracina M, et al. Development of a novel ELISA system for detection of anti-BP180 IgG and characterization of autoantibody profile in bullous pemphigoid patients. Br J Dermatol 2004;151:1004–10.
- Sitaru C, Dahnrich C, Probst C, et al. Enzyme-linked immunosorbent assay using multimers of the 16th non-collagenous domain of the BP180 antigen for sensitive and specific detection of pemphigoid autoantibodies. Exp Dermatol 2007;16:770–7.
- Kobayashi M, Amagai M, Kuroda-Kinoshita K, et al. BP180 ELISA using bacterial recombinant NC16a protein as a diagnostic and monitoring tool for bullous pemphigoid. J Dermatol Sci 2002;30:224–32.
- Schmidt E, Obe K, Brocker EB, Zillikens D. Serum levels of autoantibodies to BP180 correlate with disease activity in patients with bullous pemphigoid. Arch Dermatol 2000;136:174–8.
- Feng S, Wu Q, Jin P, Lin L, Zhou W, Sang H, Shao C. Serum levels of autoantibodies to BP180 correlate with disease activity in patients with bullous pemphigoid. Int J Dermatol 2008;47:225–8.
- Amo Y, Ohkawa T, Tatsuta M, et al. Clinical significance of enzyme-linked immunosorbent assay for the detection of circulating anti-BP180 autoantibodies in patients with bullous pemphigoid. J Dermatol Sci 2001;26:14–18.
- Dopp R, Schmidt E, Chimanovitch I, Leverkus M, Brocker EB, Zillikens D. IgG4 and IgE are the major immunoglobulins targeting the NC16A domain of BP180 in Bullous pemphigoid: serum levels of these immunoglobulins reflect disease activity. J Am Acad Dermatol 2000;42:577–83.
- Perriard J, Jaunin F, Favre B, et al. IgG autoantibodies from bullous pemphigoid (BP) patients bind antigenic sites on both the extracellular and the intracellular domains of the BP antigen 180. J Invest Dermatol 1999;112:141–7.
- Hofmann S, Thoma-Uszynski S, Hunziker T, et al. Severity and phenotype of bullous pemphigoid relate to autoantibody profile against the NH2- and COOH-terminal regions of the BP180 ectodomain. J Invest Dermatol 2002;119:1065–73.
- Di Zenzo G, Thoma-Uszynski S, Calabresi V, et al. Demonstration of epitope-spreading phenomena in bullous pemphigoid: results of a prospective multicenter study. J Invest Dermatol 2011;131:2271–80.
- Di Zenzo G, Grosso F, Terracina M, et al. Characterization of the anti-BP180 autoantibody reactivity profile and epitope mapping in bullous pemphigoid patients. J Invest Dermatol 2004;122:103–10.
- Iwata Y, Komura K, Kodera M, et al. Correlation of IgE autoantibody to BP180 with a severe form of bullous pemphigoid. Arch Dermatol 2008;144:41–8.
- Messingham KA, Noe MH, Chapman MA, Giudice GJ, Fairley JA. A novel ELISA reveals high frequencies of BP180-specific IgE production in bullous pemphigoid. J Immunol Methods 2009;346:18–25.
- Kromminga A, Scheckenbach C, Georgi M, et al. Patients with bullous pemphigoid and linear IgA disease show a dual IgA and IgG autoimmune response to BP180. J Autoimmun 2000;15:293–300.
- Christophoridis S, Budinger L, Borradori L, Hunziker T, Merk HF, Hertl M. IgG, IgA and IgE autoantibodies against the ectodomain of BP180 in patients with bullous and cicatricial pemphigoid and linear IgA bullous dermatosis. Br J Dermatol 2000;143:349–55.
- Kromminga A, Sitaru C, Hagel C, Herzog S, Zillikens D. Development of an ELISA for the detection of autoantibodies to BP230. Clin Immunol 2004;111:146–52.
- Thoma-Uszynski S, Uter W, Schwietzke S, et al. BP230- and BP180-specific auto-antibodies in bullous pemphigoid. J Invest Dermatol 2004;122:1413–22.
- Yoshida M, Hamada T, Amagai M, et al. Enzyme-linked immunosorbent assay using bacterial recombinant proteins of human BP230 as a diagnostic tool for bullous pemphigoid. J Dermatol Sci 2006;41:21–30.
- Tampoia M, Lattanzi V, Zucano A, et al. Evaluation of a new ELISA assay for detection of BP230 autoantibodies in bullous pemphigoid. Ann N Y Acad Sci 2009;1173:15–20.
- Blocker IM, Dahnrich C, Probst C, et al. Epitope mapping of BP230 leading to a novel enzyme-linked immunosorbent assay for autoantibodies in bullous pemphigoid. Br J Dermatol 2012;166:964–70.
- Charneux J, Lorin J, Vitry F, et al. Usefulness of BP230 and BP180-NC16a enzyme-linked immunosorbent assays in the initial diagnosis of bullous pemphigoid: a retrospective study of 138 patients. Arch Dermatol 2011;147:286–91.
- Roussel A, Benichou J, Randriamanantany ZA, et al. Enzyme-linked immunosorbent assay for the combination of bullous pemphigoid antigens 1 and 2 in the diagnosis of bullous pemphigoid. Arch Dermatol 2011;147:293–8.
- Skaria M, Jaunin F, Hunziker T, et al. IgG autoantibodies from bullous pemphigoid patients recognize multiple antigenic reactive sites located predominantly within the B and C subdomains of the COOH-terminus of BP230. J Invest Dermatol 2000;114:998–1004.
- Delaporte E, Dubost-Brama A, Ghohestani R, et al. IgE autoantibodies directed against the major bullous pemphigoid antigen in patients with a severe form of pemphigoid. J Immunol 1996;157:3642–7.
- Hertl M, Eming R, Veldman C. T cell control in autoimmune bullous skin disorders. J Clin Invest 2006;116:1159–66.
- Thoma-Uszynski S, Uter W, Schwietzke S, Schuler G, Borradori L, Hertl M. Autoreactive T and B cells from bullous pemphigoid (BP) patients recognize epitopes clustered in distinct regions of BP180 and BP230. J Immunol 2006;176:2015–23.
- Budinger L, Borradori L, Yee C, et al. Identification and characterization of autoreactive T cell responses to bullous pemphigoid antigen 2 in patients and healthy controls. J Clin Invest 1998;102:2082–9.
- Lin MS, Fu CL, Giudice GJ, et al. Epitopes targeted by bullous pemphigoid T lymphocytes and autoantibodies map to the same sites on the bullous pemphigoid 180 ectodomain. J Invest Dermatol 2000;115:955–61.
- Rensing-Ehl A, Gaus B, Bruckner-Tuderman L, Martin SF. Frequency, function and CLA expression of CD4+CD25+FOXP3+ regulatory T cells in bullous pemphigoid. Exp Dermatol 2007;16:13–21.
- Oswald E, Fisch P, Jakob T, Bruckner-Tuderman L, Martin SF, Rensing-Ehl A. Reduced numbers of circulating gammadelta T cells in patients with bullous pemphigoid. Exp Dermatol 2009;18:991–3.
- Li Q, Liu Z, Dang E, et al. Follicular helper T cells (Tfh) and IL-21 involvement in the pathogenesis of bullous pemphigoid. PLOS One 2013;8:e68145.
- Kasperkiewicz M, Zillikens D, Schmidt E. Pemphigoid diseases: pathogenesis, diagnosis, and treatment. Autoimmunity 2011;45:55–70.
- Ludwig RJ, Schmidt E. Cytokines in autoimmune bullous skin diseases. Epiphenomena or contribution to pathogenesis? G Ital Dermatol Venereol 2009;144:339–49.
- Teraki Y, Hotta T, Shiohara T. Skin-homing interleukin-4 and -13-producing cells contribute to bullous pemphigoid: remission of disease is associated with increased frequency of interleukin-10-producing cells. J Invest Dermatol 2001;117:1097–102.
- Arakawa M, Dainichi T, Ishii N, et al. Lesional Th17 cells and regulatory T cells in bullous pemphigoid. Exp Dermatol 2011;20:1022–4.
- Schmidt E, Reimer S, Kruse N, et al. Autoantibodies to BP180 associated with bullous pemphigoid release interleukin-6 and interleukin-8 from cultured human keratinocytes. J Invest Dermatol 2000;115:842–8.
- Iwata H, Kamio N, Aoyama Y, et al. IgG from patients with bullous pemphigoid depletes cultured keratinocytes of the 180-kDa bullous pemphigoid antigen (type XVII collagen) and weakens cell attachment. J Invest Dermatol 2009;129:919–26.
- Hiroyasu S, Ozawa T, Kobayashi H, et al. Bullous pemphigoid IgG induces BP180 internalization via a macropinocytic pathway. Am J Pathol 2013;182:828–40.
- Messingham KN, Srikantha R, DeGueme AM, Fairley JA. FcR-independent effects of IgE and IgG autoantibodies in bullous pemphigoid. J Immunol 2011;187:553–60.
- Sitaru C, Schmidt E, Petermann S, Munteanu LS, Brocker EB, Zillikens D. Autoantibodies to bullous pemphigoid antigen 180 induce dermal-epidermal separation in cryosections of human skin. J Invest Dermatol 2002;118:664–71.
- Shimanovich I, Mihai S, Oostingh GJ, et al. Granulocyte-derived elastase and gelatinase B are required for dermal-epidermal separation induced by autoantibodies from patients with epidermolysis bullosa acquisita and bullous pemphigoid. J Pathol 2004;204:519–27.
- Yu X, Holdorf K, Kasper B, Zillikens D, Ludwig RJ, Petersen F. FcgammaRIIA and FcgammaRIIIB are required for autoantibody-induced tissue damage in experimental human models of bullous pemphigoid. J Invest Dermatol 130:2841–4.
- Stahle-Backdahl M, Inoue M, Guidice GJ, Parks WC. 92-kD gelatinase is produced by eosinophils at the site of blister formation in bullous pemphigoid and cleaves the extracellular domain of recombinant 180-kD bullous pemphigoid autoantigen. J Clin Invest 1994;93:2022–30.
- Niimi Y, Pawankar R, Kawana S. Increased expression of matrix metalloproteinase-2, matrix metalloproteinase-9 and matrix metalloproteinase-13 in lesional skin of bullous pemphigoid. Int Arch Allergy Immunol 2006;139:104–13.
- Verraes S, Hornebeck W, Polette M, Borradori L, Bernard P. Respective contribution of neutrophil elastase and matrix metalloproteinase 9 in the degradation of BP180 (type XVII collagen) in human bullous pemphigoid. J Invest Dermatol 2001;117:1091–6.
- Liu Z, Giudice GJ, Swartz SJ, et al. The role of complement in experimental bullous pemphigoid. J Clin Invest 1995;95:1539–44.
- Nelson KC, Zhao M, Schroeder PR, et al. Role of different pathways of the complement cascade in experimental bullous pemphigoid. J Clin Invest 2006;116:2892–900.
- Chen R, Fairley JA, Zhao ML, et al. Macrophages, but not T and B lymphocytes, are critical for subepidermal blister formation in experimental bullous pemphigoid: macrophage-mediated neutrophil infiltration depends on mast cell activation. J Immunol 2002;169:3987–992.
- Liu Z, Giudice GJ, Zhou X, et al. A major role for neutrophils in experimental bullous pemphigoid. J Clin Invest 1997;100:1256–63.
- Liu Z, Li N, Diaz LA, Shipley M, Senior RM, Werb Z. Synergy between a plasminogen cascade and MMP-9 in autoimmune disease. J Clin Invest 2005;115:879–87.
- Lin L, Bankaitis E, Heimbach L, et al. Dual targets for mouse mast cell protease-4 in mediating tissue damage in experimental bullous pemphigoid. J Biol Chem 2011;286:37358–67.
- Liu Z, Shipley JM, Vu TH, et al. Gelatinase B-deficient mice are resistant to experimental bullous pemphigoid. J Exp Med 1998;188:475–82.
- Liu Z, Zhou X, Shapiro SD, et al. The serpin alpha1-proteinase inhibitor is a critical substrate for gelatinase B/MMP-9 in vivo. Cell 2000;102:647–55.
- Liu Z, Shapiro SD, Zhou X, et al. A critical role for neutrophil elastase in experimental bullous pemphigoid. J Clin Invest 2000;105:113–23.
- Lin L, Betsuyaku T, Heimbach L, et al. Neutrophil elastase cleaves the murine hemidesmosomal protein BP180/type XVII collagen and generates degradation products that modulate experimental bullous pemphigoid. Matrix Biol 2012;31:38–44.
- Schulze FS, Beckmann T, Nimmerjahn F, et al. Fc gamma receptors III and IV mediate tissue destruction in a novel adult mouse model of bullous pemphigoid. Am J Pathol 2014;184:2185–96.
- Olasz EB, Roh J, Yee CL, et al. Human bullous pemphigoid antigen 2 transgenic skin elicits specific IgG in wild-type mice. J Invest Dermatol 2007;127:2807–17.
- Liu Z, Sui W, Zhao M, et al. Subepidermal blistering induced by human autoantibodies to BP180 requires innate immune players in a humanized bullous pemphigoid mouse model. J Autoimmun 2008;31:331–8.
- Natsuga K, Nishie W, Shinkuma S, et al. Antibodies to pathogenic epitopes on type XVII collagen cause skin fragility in a complement-dependent and -independent manner. J Immunol 2012;188:5792–9.
- Ujiie H, Shibaki A, Nishie W, et al. A novel active mouse model for bullous pemphigoid targeting humanized pathogenic antigen. J Immunol 2010;184:2166–74.
- Hirose M, Recke A, Beckmann T, et al. Repetitive immunization breaks tolerance to type XVII collagen and leads to bullous pemphigoid in mice. J Immunol 2011;187:1176–83.
- Ujiie H, Shibaki A, Nishie W, et al. Noncollagenous 16A domain of type XVII collagen-reactive CD4+ T cells play a pivotal role in the development of active disease in experimental bullous pemphigoid model. Clin Immunol 2012;142:167–75.
- Fairley JA, Burnett CT, Fu CL, Larson DL, Fleming MG, Giudice GJ. A pathogenic role for IgE in autoimmunity: bullous pemphigoid IgE reproduces the early phase of lesion development in human skin grafted to nu/nu mice. J Invest Dermatol 2007;127:2605–11.
- Zone JJ, Taylor T, Hull C, Schmidt L, Meyer L. IgE basement membrane zone antibodies induce eosinophil infiltration and histological blisters in engrafted human skin on SCID mice. J Invest Dermatol 2007;127:1167–74.
- Fairley JA, Baum CL, Brandt DS, Messingham KA. Pathogenicity of IgE in autoimmunity: successful treatment of bullous pemphigoid with omalizumab. J Allergy Clin Immunol 2009;123:704–5.
- Yu KK, Crew AB, Messingham KA, Fairley JA, Woodley DT. Omalizumab therapy for bullous pemphigoid. J Am Acad Dermatol 2014;71:468–74.
- Hall RP 3rd, Murray JC, McCord MM, Rico MJ, Streilein RD. Rabbits immunized with a peptide encoded for by the 230-kD bullous pemphigoid antigen cDNA develop an enhanced inflammatory response to UVB irradiation: a potential animal model for bullous pemphigoid. J Invest Dermatol 1993;101:9–14.
- Kiss M, Husz S, Janossy T, et al. Experimental bullous pemphigoid generated in mice with an antigenic epitope of the human hemidesmosomal protein BP230. J Autoimmun 2005;24:1–10.
- Macfarlane AW, Verbov JL. Trauma-induced bullous pemphigoid. Clin Exp Dermatol 1989;14:245–9.
- Venning VA, Wojnarowska F. Induced bullous pemphigoid. Br J Dermatol 1995;132:831–2.
- Vassileva S, Mateev G, Balabanova M, Tsankov N. Burn-induced bullous pemphigoid. J Am Acad Dermatol 1994;30:1027–8.
- McGrath J, Black M. Split skin grafting and bullous pemphigoid. Clin Exp Dermatol 1991;16:72–3.
- Neri I, Antonucci VA, Balestri R, Tengattini V, Iozzo I, Bardazzi F. Bullous pemphigoid appearing both on thermal burn scars and split-thickness skin graft donor sites. J Dtsch Dermatol Ges 2013;11:675–6.
- Emery EW, Hare PJ, Abadir R. Pemphigoid, bronchial neoplasm and radiotherapy. Proc R Soc Med 1967;60:1271–2.
- Mul VE, van Geest AJ, Pijls-Johannesma MC, et al. Radiation-induced bullous pemphigoid: a systematic review of an unusual radiation side effect. Radiother Oncol 2007;82:5–9.
- Lee CW, Ro YS. Sun-induced localized bullous pemphigoid. Br J Dermatol 1992;126:91–2.
- Sacher C, Konig C, Scharffetter-Kochanek K, Krieg T, Hunzelmann N. Bullous pemphigoid in a patient treated with UVA-1 phototherapy for disseminated morphea. Dermatology 2001;202:54–7.
- Perl S, Rappersberger K, Fodinger D, Anegg B, Honigsmann H, Ortel B. Bullous pemphigoid induced by PUVA therapy. Dermatology 1996;193:245–7.
- Rakvit P, Kerr AC, Ibbotson SH. Localized bullous pemphigoid induced by photodynamic therapy. Photodermatol Photoimmunol Photomed 2011;27:251–3.
- Walmsley N, Hampton P. Bullous pemphigoid triggered by swine flu vaccination: case report and review of vaccine triggered pemphigoid. J Dermatol Case Rep 2011;5:74–6.
- Garcia-Doval I, Mayo E, Nogueira Farina J, Cruces MJ. Bullous pemphigoid triggered by influenza vaccination? Ecological study in Galicia, Spain. Br J Dermatol 2006;155:820–3.
- Lee JJ, Downham TF 2nd. Furosemide-induced bullous pemphigoid: case report and review of literature. J Drugs Dermatol 2006;5:562–4.
- Bastuji-Garin S, Joly P, Lemordant P, et al. Risk factors for bullous pemphigoid in the elderly: a prospective case-control study. J Invest Dermatol 2011;131:637–43.
- Schmidt E, Zillikens D. Modern diagnosis of autoimmune blistering skin diseases. Autoimmunity Rev 2010;10:84–9.
- Vodegel RM, Jonkman MF, Pas HH, de Jong MC. U-serrated immunodeposition pattern differentiates type VII collagen targeting bullous diseases from other subepidermal bullous autoimmune diseases. Br J Dermatol 2004;151:112–18.
- Gammon WR, Kowalewski C, Chorzelski TP, Kumar V, Briggaman RA, Beutner EH. Direct immunofluorescence studies of sodium chloride-separated skin in the differential diagnosis of bullous pemphigoid and epidermolysis bullosa acquisita. J Am Acad Dermatol 1990;22:664–70.
- De Jong MC, Bruins S, Heeres K, et al. Bullous pemphigoid and epidermolysis bullosa acquisita. Differentiation by fluorescence overlay antigen mapping. Arch Dermatol 1996;132:151–7.
- Wozniak K, Kazama T, Kowalewski C. A practical technique for differentiation of subepidermal bullous diseases: localization of in vivo-bound IgG by laser scanning confocal microscopy. Arch Dermatol 2003;139:1007–11.
- Ishiko A, Shimizu H, Kikuchi A, Ebihara T, Hashimoto T, Nishikawa T. Human autoantibodies against the 230-kD bullous pemphigoid antigen (BPAG1) bind only to the intracellular domain of the hemidesmosome, whereas those against the 180-kD bullous pemphigoid antigen (BPAG2) bind along the plasma membrane of the hemidesmosome in normal human and swine skin. J Clin Invest 1993;91:1608–15.
- Kippes W, Schmidt E, Roth A, Rzany B, Brocker EB, Zillikens D. [Immunopathologic changes in 115 patients with bullous pemphigoid.] Hautarzt 1999;50:866–72.
- Gammon WR, Briggaman RA, Inman AO 3rd, Queen LL, Wheeler CE. Differentiating anti-lamina lucida and anti-sublamina densa anti-BMZ antibodies by indirect immunofluorescence on 1.0 M sodium chloride-separated skin. J Invest Dermatol 1984;82:139–44.
- Ghohestani RF, Nicolas JF, Rousselle P, Claudy AL. Diagnostic value of indirect immunofluorescence on sodium chloride-split skin in differential diagnosis of subepidermal autoimmune bullous dermatoses. Arch Dermatol 1997;133:1102–7.
- Machado P, Michalaki H, Roche P, Gaucherand M, Thivolet J, Nicolas JF. Serological diagnosis of bullous pemphigoid (BP): comparison of the sensitivity of indirect immunofluorescence on salt-split skin to immunoblotting. Br J Dermatol 1992;126:236–41.
- Pas HH, de Jong MC, Jonkman MF, Heeres K, Slijper-Pal IJ, van der Meer JB. Bullous pemphigoid: serum antibody titre and antigen specificity. Exp Dermatol 1995;4:372–6.
- Sardy M, Kostaki D, Varga R, Peris K, Ruzicka T. Comparative study of direct and indirect immunofluorescence and of bullous pemphigoid 180 and 230 enzyme-linked immunosorbent assays for diagnosis of bullous pemphigoid. J Am Acad Dermatol 2013;69:748–53.
- van Beek N, Rentzsch K, Probst C, et al. Serological diagnosis of autoimmune bullous skin diseases: Prospective comparison of the BIOCHIP mosaic-based indirect immunofluorescence technique with the conventional multi-step single test strategy. Orphanet J Rare Dis 2012;7:49.
- Youinou P, Pers JO, Gershwin ME, Shoenfeld Y. Geo-epidemiology and autoimmunity. J Autoimmun 2010;34:J163–7.
- Feliciani C, Caldarola G, Kneisel A, et al. IgG autoantibody reactivity against bullous pemphigoid (BP) 180 and BP230 in elderly patients with pruritic dermatoses. Br J Dermatol 2009;161:306–12.
- Hofmann SC, Tamm K, Hertl M, Borradori L. Diagnostic value of an enzyme-linked immunosorbent assay using BP180 recombinant proteins in elderly patients with pruritic skin disorders. Br J Dermatol 2003;149:910–12.
- Wieland CN, Comfere NI, Gibson LE, Weaver AL, Krause PK, Murray JA. Anti-bullous pemphigoid 180 and 230 antibodies in a sample of unaffected subjects. Arch Dermatol 2010;146:21–5.
- Mueller S, Klaus-Kovtun V, Stanley JR. A 230-kD basic protein is the major bullous pemphigoid antigen. J Invest Dermatol 1989;92:33–8.
- Bernard P, Didierjean L, Denis F, Saurat JH, Bonnetblanc JM. Heterogeneous bullous pemphigoid antibodies: detection and characterization by immunoblotting when absent by indirect immunofluorescence. J Invest Dermatol 1989;92:171–4.
- Oyama N, Bhogal BS, Carrington P, Gratian MJ, Black MM. Human placental amnion is a novel substrate for detecting autoantibodies in autoimmune bullous diseases by immunoblotting. Br J Dermatol 2003;148:939–44.
- Marinkovich MP, Taylor TB, Keene DR, Burgeson RE, Zone JJ. LAD-1, the linear IgA bullous dermatosis autoantigen, is a novel 120-kDa anchoring filament protein synthesized by epidermal cells. J Invest Dermatol 1996;106:734–8.
- Haase C, Budinger L, Borradori L, et al. Detection of IgG autoantibodies in the sera of patients with bullous and gestational pemphigoid: ELISA studies utilizing a baculovirus-encoded form of bullous pemphigoid antigen 2. J Invest Dermatol 1998;110:282–6.
- Schmidt E, Kromminga A, Mimietz S, et al. A highly sensitive and simple assay for the detection of circulating autoantibodies against full-length bullous pemphigoid antigen 180. J Autoimmun 2002;18:299–309.
- Tanaka M, Hashimoto T, Amagai M, et al. Characterization of bullous pemphigoid antibodies by use of recombinant bullous pemphigoid antigen proteins. J Invest Dermatol 1991;97:725–8.
- Delgado JC, Turbay D, Yunis EJ, et al. A common major histocompatibility complex class II allele HLA-DQB1* 0301 is present in clinical variants of pemphigoid. Proc Natl Acad Sci U S A 1996;93:8569–71.
- Banfield CC, Wojnarowska F, Allen J, George S, Venning VA, Welsh KI. The association of HLA-DQ7 with bullous pemphigoid is restricted to men. Br J Dermatol 1998;138:1085–90.
- Gao XH, Winsey S, Li G, et al. HLA-DR and DQ polymorphisms in bullous pemphigoid from northern China. Clin Exp Dermatol 2002;27:319–21.
- Okazaki A, Miyagawa S, Yamashina Y, Kitamura W, Shirai T. Polymorphisms of HLA-DR and -DQ genes in Japanese patients with bullous pemphigoid. J Dermatol 2000;27:149–56.
- Schmidt E, della Torre R, Borradori L. Clinical features and practical diagnosis of bullous pemphigoid. Dermatol Clin 2011;29:427–38, viii–ix.
- Di Zenzo G, Marazza G, Borradori L. Bullous pemphigoid: physiopathology, clinical features and management. Adv Dermatol 2007;23:257–88.
- della Torre R, Combescure C, Cortes B, et al. Clinical presentation and diagnostic delay in bullous pemphigoid: a prospective nationwide cohort. Br J Dermatol 2012;167:1111–17.
- Liu HN, Su WP, Rogers RS, 3rd. Clinical variants of pemphigoid. Int J Dermatol 1986;25:17–27.
- Korman N. Bullous pemphigoid. J Am Acad Dermatol 1987;16:907–24.
- Yasuda M, Miyachi Y, Utani A. Two cases of dyshidrosiform pemphigoid with different presentations. Clin Exp Dermatol 2009;34:e151–3.
- Powell AM, Albert S, Gratian MJ, Bittencourt R, Bhogal BS, Black MM. Pemphigoid nodularis (non-bullous): a clinicopathological study of five cases. Br J Dermatol 2002;147:343–9.
- Cliff S, Holden CA. Pemphigoid nodularis: a report of three cases and review of the literature. Br J Dermatol 1997;136:398–401.
- Schmidt E, Sitaru C, Schubert B, et al. Subacute prurigo variant of bullous pemphigoid: autoantibodies show the same specificity compared with classic bullous pemphigoid. J Am Acad Dermatol 2002;47:133–6.
- Geiss Steiner J, Trueb RM, Kerl K, Muhleisen B, French LE, Hofbauer GF. Ecthyma-gangrenosum-like bullous pemphigoid. Dermatology 2010;221:142–8.
- Chan LS, Dorman MA, Agha A, Suzuki T, Cooper KD, Hashimoto K. Pemphigoid vegetans represents a bullous pemphigoid variant. Patient's IgG autoantibodies identify the major bullous pemphigoid antigen. J Am Acad Dermatol 1993;28:331–5.
- Ueda Y, Nashiro K, Seki Y, Otsuka F, Tamaki K, Ishibashi Y. Pemphigoid vegetans. Br J Dermatol 1989;120:449–53.
- Ogasawara M, Matsuda S, Nishioka K, Asagami C. Pemphigoid vegetans. J Am Acad Dermatol 1994;30:649–50.
- Lai FJ, Sheu HM, Lee JY, Cheng CL, Chen W. Vesicular pemphigoid with circulating autoantibodies against 230-kDa and 180-kDa proteins, and additional autoantibodies against 97-kDa and 45-kDa proteins. Int J Dermatol 2007;46:206–9.
- Alonso-Llamazares J, Dietrich SM, Gibson LE. Bullous pemphigoid presenting as exfoliative erythroderma. J Am Acad Dermatol 1998;39:827–30.
- Redondo P, Espana A, Idoate M, Sanchez-Ibarrola A, Quintanilla E. Unusual bullous disorder with features of toxic epidermal necrolysis, bullous pemphigoid and cicatricial pemphigoid. Clin Exp Dermatol 1995;20:65–9.
- Kakurai M, Demitsu T, Azuma R, et al. Localized pemphigoid (pretibial type) with IgG antibody to BP180 NC16a domain successfully treated with minocycline and topical corticosteroid. Clin Exp Dermatol 2007;32:759–61.
- Kohroh K, Suga Y, Mizuno Y, Ishii N, Hashimoto T, Ikeda S. Case of localized bullous pemphigoid with unique clinical manifestations in the lower legs. J Dermatol 2007;34:482–5.
- Tran JT, Mutasim DF. Localized bullous pemphigoid: a commonly delayed diagnosis. Int J Dermatol 2005;44:942–5.
- Mehta V, Balachandran C. Localized flexural bullous pemphigoid. Indian J Dermatol 2008;53:157–8.
- Schmidt E, Benoit S, Brocker EB. Bullous pemphigoid with localized umbilical involvement. Acta Derm Venereol 2009;89:419–20.
- Toyama T, Nakamura K, Kuramochi A, Ohyama B, Hashimoto T, Tsuchida T. Two cases of childhood bullous pemphigoid. Eur J Dermatol 2009;19:368–71.
- Mirza M, Zamilpa I, Wilson JM. Localized penile bullous pemphigoid of childhood. J Pediatr Urol 2008;4:395–7.
- Chan LS, Ahmed AR, Anhalt GJ, et al. The first international consensus on mucous membrane pemphigoid: definition, diagnostic criteria, pathogenic factors, medical treatment, and prognostic indicators. Arch Dermatol 2002;138:370–9.
- Murrell DF, Daniel BS, Joly P, et al. Definitions and outcome measures for bullous pemphigoid: Recommendations by an international panel of experts. J Am Acad Dermatol 2012;66:479–85.
- Savin JA. The events leading to the death of patients with pemphigus and pemphigoid. Br J Dermatol 1979;101:521–34.
- Langan SM, Hubbard R, Fleming K, West J. A population-based study of acute medical conditions associated with bullous pemphigoid. Br J Dermatol 2009;161:1149–52.
- Bernard P, Reguiai Z, Tancrede-Bohin E, et al. Risk factors for relapse in patients with bullous pemphigoid in clinical remission: a multicenter, prospective, cohort study. Arch Dermatol 2009;145:537–42.
- Fichel F, Barbe C, Joly P, et al. Clinical and immunologic factors associated with bullous pemphigoid relapse during the first year of treatment: a multicenter, prospective study. JAMA Dermatol 2014;150:25–33.
- Cai SC, Allen JC, Lim YL, Chua SH, Tan SH, Tang MB. Mortality of bullous pemphigoid in Singapore: risk factors and causes of death in 359 patients seen at the National Skin Centre. Br J Dermatol 2014;170:1319–26.
- Joly P, Roujeau JC, Benichou J, et al. A comparison of oral and topical corticosteroids in patients with bullous pemphigoid. N Engl J Med 2002;346:321–7.
- Schmidt E, Kraensel R, Goebeler M, et al. Treatment of bullous pemphigoid with dapsone, methylprednisolone, and topical clobetasol propionate: a retrospective study of 62 cases. Cutis 2005;76:205–9.
- Joly P, Benichou J, Lok C, et al. Prediction of survival for patients with bullous pemphigoid: a prospective study. Arch Dermatol 2005;141:691–8.
- Roujeau JC, Lok C, Bastuji-Garin S, Mhalla S, Enginger V, Bernard P. High risk of death in elderly patients with extensive bullous pemphigoid. Arch Dermatol 1998;134:465–9.
- Vaillant L, Bernard P, Joly P, et al. Evaluation of clinical criteria for diagnosis of bullous pemphigoid. French Bullous Study Group. Arch Dermatol 1998;134:1075–80.
- Schmidt E, Goebeler M, Hertl M, et al. S2k Guidelines for diagnosis of pemphigus vulgaris/foliaceus and bullous pemphigoid. J Dtsch Dermatol Ges 2015;13:713–27.
- Rose C, Schmidt E, Kerstan A, et al. Histopathology of anti-laminin 5 mucous membrane pemphigoid. J Am Acad Dermatol 2009;61:433–40.
- Rose C, Weyers W, Denisjuk N, Hillen U, Zillikens D, Shimanovich I. Histopathology of anti-p200 pemphigoid. Am J Dermatopathol 2007;29:119–24.
- Pfaltz K, Mertz K, Rose C, Scheidegger P, Pfaltz M, Kempf W. C3d immunohistochemistry on formalin-fixed tissue is a valuable tool in the diagnosis of bullous pemphigoid of the skin. J Cutan Pathol 2010;37:654–8.
- Kirtschig G, Middleton P, Bennett C, Murrell DF, Wojnarowska F, Khumalo NP. Interventions for bullous pemphigoid. Cochrane Database Syst Rev 2010:CD002292.
- Joly P, Roujeau JC, Benichou J, et al. A comparison of two regimens of topical corticosteroids in the treatment of patients with bullous pemphigoid: a multicenter randomized study. J Invest Dermatol 2009;129:1681–7.
- Kjellman P, Eriksson H, Berg P. A retrospective analysis of patients with bullous pemphigoid treated with methotrexate. Arch Dermatol 2008;144:612–16.
- Chave TA, Mortimer NJ, Shah DS, Hutchinson PE. Chlorambucil as a steroid-sparing agent in bullous pemphigoid. Br J Dermatol 2004;151:1107–8.
- Du-Thanh A, Merlet S, Maillard H, et al. Combined treatment with low-dose methotrexate and initial short-term superpotent topical steroids in bullous pemphigoid: an open, multicentre, retrospective study. Br J Dermatol 2011;165:1337–43.
- Gual A, Iranzo P, Mascaro Jr JM. Treatment of bullous pemphigoid with low-dose oral cyclophosphamide: a case series of 20 patients. J Eur Acad Dermatol Venereol 2014;28:814–18.
- Fox BJ, Odom RB, Findlay RF. Erythromycin therapy in bullous pemphigoid: possible anti-inflammatory effects. J Am Acad Dermatol 1982;7:504–10.
- Barthelemy H, Thivolet J, Cambazard F, et al. [Cyclosporin in the treatment of bullous pemphigoid: preliminary study.] Ann Dermatol Venereol 1986;113:309–13.
- Nousari HC, Anhalt GJ. Bullous pemphigoid treated with leflunomide: a novel immunomodulatory agent. Arch Dermatol 2000;136:1204–5.
- Schmidt E, Goebeler M. CD20-directed therapy in autoimmune diseases involving the skin: role of rituximab. Exp Rev Dermatol 2008;3:259–78.
- Kasperkiewicz M, Shimanovich I, Ludwig RJ, Rose C, Zillikens D, Schmidt E. Rituximab for treatment-refractory pemphigus and pemphigoid: a case series of 17 patients. J Am Acad Dermatol 2011;65:552–8.
- Shetty S, Ahmed AR. Treatment of bullous pemphigoid with rituximab: critical analysis of the current literature. J Drugs Dermatol 2013;12:672–7.
- Herrero-Gonzalez JE, Sitaru C, Klinker E, Brocker EB, Zillikens D. Successful adjuvant treatment of severe bullous pemphigoid by tryptophan immunoadsorption. Clin Exp Dermatol 2005;30:519–22.
- Ino N, Kamata N, Matsuura C, Shinkai H, Odaka M. Immunoadsorption for the treatment of bullous pemphigoid. Ther Apher 1997;1:372–6.
- Schmidt E, Zillikens D. Immunoadsorption in dermatology. Arch Dermatol Res 2010;302:241–53.
- Ahmed AR. Intravenous immunoglobulin therapy for patients with bullous pemphigoid unresponsive to conventional immunosuppressive treatment. J Am Acad Dermatol 2001;45:825–35.
- Engineer L, Ahmed AR. Role of intravenous immunoglobulin in the treatment of bullous pemphigoid: analysis of current data. J Am Acad Dermatol 2001;44:83–8.
- Gaitanis G, Alexis I, Pelidou SH, et al. High-dose intravenous immunoglobulin in the treatment of adult patients with bullous pemphigoid. Eur J Dermatol 2012;22:363–9.
- Bernard P, Bedane C, Prost C, Ingen-Housz-Oro S, Joly P. [Bullous pemphigoid. Guidelines for the diagnosis and treatment. Centres de reference des maladies bulleuses auto-immunes. Societe Francaise de Dermatologie.] Ann Dermatol Venereol 2011;138:247–51.
- Venning VA, Taghipour K, Mohd Mustapa MF, Highet AS, Kirtschig G. British Association of Dermatologists' guidelines for the management of bullous pemphigoid 2012. Br J Dermatol 2012;167:1200–14.
- Feciliani C, Borradori L, Ioannides D, et al. Pemphigoid. Guidelines for diagnosis and treatment.
- Eming R, Sticherling M, Hofmann S, et al. S2k guidelines for the treatment of pemphigus vulgaris/foliaceus and bullous pemphigoid. J Dtsch Dermatol Ges 2015;13:833–44.
- Hertl M, Zillikens D, Borradori L, et al. Recommendations for the use of rituximab (anti-CD20 antibody) in the treatment of autoimmune bullous skin diseases. J Dtsch Dermatol Ges 2008;6:366–73.
- Zillikens D, Derfler K, Eming R, et al. Recommendations for the use of immunoapheresis in the treatment of autoimmune bullous diseases. J Dtsch Dermatol Ges 2007;5:881–7.
- Enk A, Fierlbeck G, French L, et al. Use of high-dose immunoglobulins in dermatology. J Dtsch Dermatol Ges 2009;7:806–12.
- Ahmed AR, Dahl MV. Consensus statement on the use of intravenous immunoglobulin therapy in the treatment of autoimmune mucocutaneous blistering diseases. Arch Dermatol 2003;139:1051–9.
- Enk A. Guidelines on the use of high-dose intravenous immunoglobulin in dermatology. Eur J Dermatol 2009;19:90–8.
- Beissert S, Werfel T, Frieling U, et al. A comparison of oral methylprednisolone plus azathioprine or mycophenolate mofetil for the treatment of pemphigus. Arch Dermatol 2006;142:1447–54.
- Fivenson DP, Breneman DL, Rosen GB, Hersh CS, Cardone S, Mutasim D. Nicotinamide and tetracycline therapy of bullous pemphigoid. Arch Dermatol 1994;130:753–8.
- Guillaume JC, Vaillant L, Bernard P, et al. Controlled trial of azathioprine and plasma exchange in addition to prednisolone in the treatment of bullous pemphigoid. Arch Dermatol 1993;129:49–53.
- Dreno B, Sassolas B, Lacour P, et al. [Methylprednisolone versus prednisolone methylsulfobenzoate in pemphigoid: a comparative multicenter study.] Ann Dermatol Venereol 1993;120:518–21.
- Morel P, Guillaume JC. [Treatment of bullous pemphigoid with prednisolone only: 0.75 mg/kg/day versus 1.25 mg/kg/day. A multicenter randomized study.] Ann Dermatol Venereol 1984;111:925–8.
- Roujeau JC, Guillaume JC, Morel P, et al. Plasma exchange in bullous pemphigoid. Lancet 1984;2:486–8.
- Burton JL, Harman RR, Peachey RD, Warin RP. Azathioprine plus prednisone in treatment of pemphigoid. BMJ 1978;2:1190–1.
Mucous membrane pemphigoid
- Chan LS, Ahmed AR, Anhalt GJ, et al. The first international consensus on mucous membrane pemphigoid: definition, diagnostic criteria, pathogenic factors, medical treatment, and prognostic indicators. Arch Dermatol 2002;138:370–9.
- Schmidt E, Zillikens D. Pemphigoid diseases. Lancet 2013;381:320–32.
- Ahmed AR, Kurgis BS, Rogers RS 3rd. Cicatricial pemphigoid. J Am Acad Dermatol 1991;24:987–1001.
- Thorne JE, Anhalt GJ, Jabs DA. Mucous membrane pemphigoid and pseudopemphigoid. Ophthalmology 2004;111:45–52.
- Lever WF. Pemphigus. Medicine 1953;32:1–123.
- Holubar K. Historical background. In: Wojnarowska F, Briggaman RA, eds. Management of Blistering Diseases. London: Chapman and Hall Medical, 1990:1–12.
- Fleming TE, Korman NJ. Cicatricial pemphigoid. J Am Acad Dermatol 2000;43:571–91; quiz 591–74.
- Morris M, Roberts HK. Pemphigus of the skin and mucous membranes. Br J Dermatol 1889;1:175–81.
- Schmidt E, Skrobek C, Kromminga A, et al. Cicatricial pemphigoid: IgA and IgG autoantibodies target epitopes on both intra- and extracellular domains of bullous pemphigoid antigen 180. Br J Dermatol 2001;145:778–83.
- Setterfield J, Shirlaw PJ, Kerr-Muir M, et al. Mucous membrane pemphigoid: a dual circulating antibody response with IgG and IgA signifies a more severe and persistent disease. Br J Dermatol 1998;138:602–10.
- Egan CA, Lazarova Z, Darling TN, Yee C, Cote T, Yancey KB. Anti-epiligrin cicatricial pemphigoid and relative risk for cancer. Lancet 2001;357:1850–1.
- Lazarova Z, Hsu R, Briggaman RA, Yancey KB. Fab fragments directed against laminin 5 induce subepidermal blisters in neonatal mice. Clin Immunol 2000;95:26–32.
- Lazarova Z, Hsu R, Yee C, Yancey KB. Human anti-laminin 5 autoantibodies induce subepidermal blisters in an experimental human skin graft model. J Invest Dermatol 2000;114:178–84.
- Lazarova Z, Yee C, Darling T, Briggaman RA, Yancey KB. Passive transfer of anti-laminin 5 antibodies induces subepidermal blisters in neonatal mice. J Clin Invest 1996;98:1509–18.
- Chan RY, Bhol K, Tesavibul N, et al. The role of antibody to human beta4 integrin in conjunctival basement membrane separation: possible in vitro model for ocular cicatricial pemphigoid. Invest Ophthalmol Vis Sci 1999;40:2283–90.
- Rashid KA, Stern JN, Ahmed AR. Identification of an epitope within human integrin alpha 6 subunit for the binding of autoantibody and its role in basement membrane separation in oral pemphigoid. J Immunol 2006;176:1968–77.
- Bernard P, Vaillant L, Labeille B, et al. Incidence and distribution of subepidermal autoimmune bullous skin diseases in three French regions. Bullous Diseases French Study Group. Arch Dermatol 1995;131:48–52.
- Bertram F, Brocker EB, Zillikens D, Schmidt E. Prospective analysis of the incidence of autoimmune bullous disorders in Lower Franconia, Germany. J Dtsch Dermatol Ges 2009;7:434–40.
- Zillikens D, Wever S, Roth A, et al. Incidence of autoimmune subepidermal blistering dermatoses in a region of central Germany. Arch Dermatol 1995;131:957–8.
- Radford CF, Rauz S, Williams GP, Saw VP, Dart JK. Incidence, presenting features, and diagnosis of cicatrising conjunctivitis in the United Kingdom. Eye (London) 2012;26:1199–208.
- Ahmed AR, Hombal SM. Cicatricial pemphigoid. Int J Dermatol 1986;25:90–6.
- Alexandre M, Brette MD, Pascal F, et al. A prospective study of upper aerodigestive tract manifestations of mucous membrane pemphigoid. Medicine (Baltimore) 2006;85:239–52.
- Higgins GT, Allan RB, Hall R, Field EA, Kaye SB. Development of ocular disease in patients with mucous membrane pemphigoid involving the oral mucosa. Br J Ophthalmol 2006;90:964–7.
- Laskaris G, Sklavounou A, Stratigos J. Bullous pemphigoid, cicatricial pemphigoid, and pemphigus vulgaris. A comparative clinical survey of 278 cases. Oral Surg Oral Med Oral Pathol 1982;54:656–62.
- Wojnarowska F, Marsden RA, Bhogal B, Black MM. Chronic bullous disease of childhood, childhood cicatricial pemphigoid, and linear IgA disease of adults. A comparative study demonstrating clinical and immunopathologic overlap. J Am Acad Dermatol 1988;19:792–805.
- Matsushima S, Horiguchi Y, Honda T, et al. A case of anti-epiligrin cicatricial pemphigoid associated with lung carcinoma and severe laryngeal stenosis: review of Japanese cases and evaluation of risk for internal malignancy. J Dermatol 2004;31:10–15.
- Leverkus M, Schmidt E, Lazarova Z, Brocker EB, Yancey KB, Zillikens D. Antiepiligrin cicatricial pemphigoid: an underdiagnosed entity within the spectrum of scarring autoimmune subepidermal bullous diseases? Arch Dermatol 1999;135:1091–8.
- Nayar M, Wojnarowska F. No association between cicatricial pemphigoid and malignant disease. Br J Dermatol 1991;125:193–4.
- Letko E, Gurcan HM, Papaliodis GN, Christen W, Foster CS, Ahmed AR. Relative risk for cancer in mucous membrane pemphigoid associated with antibodies to the beta4 integrin subunit. Clin Exp Dermatol 2007;32:637–41.
- Malik M, Gurcan HM, Christen W, Ahmed AR. Relationship between cancer and oral pemphigoid patients with antibodies to alpha6-integrin. J Oral Pathol Med 2007;36:1–5.
- Oyama N, Setterfield JF, Powell AM, et al. Bullous pemphigoid antigen II (BP180) and its soluble extracellular domains are major autoantigens in mucous membrane pemphigoid: the pathogenic relevance to HLA class II alleles and disease severity. Br J Dermatol 2006;154:90–8.
- Domloge-Hultsch N, Gammon WR, Briggaman RA, et al. Epiligrin, the major human keratinocyte integrin ligand, is a target in both an acquired autoimmune and an inherited subepidermal blistering skin disease. J Clin Invest 1992;90:1628–33.
- Bernard P, Antonicelli F, Bedane C, et al. Prevalence and clinical significance of anti-laminin 332 autoantibodies detected by a novel enzyme-linked immunosorbent assay in mucous membrane pemphigoid. JAMA Dermatol 2013;149:533–40.
- Murakami H, Nishioka S, Setterfield J, et al. Analysis of antigens targeted by circulating IgG and IgA autoantibodies in 50 patients with cicatricial pemphigoid. J Dermatol Sci 1998;17:39–44.
- Kawahara Y, Amagai M, Ohata Y, et al. A case of cicatricial pemphigoid with simultaneous IgG autoantibodies against the 180 kd bullous pemphigoid antigen and laminin 5. J Am Acad Dermatol 1998;38:624–7.
- Shimanovich I, Petersen EE, Weyers W, Sitaru C, Zillikens D. Subepidermal blistering disease with autoantibodies to both the p200 autoantigen and the alpha3 chain of laminin 5. J Am Acad Dermatol 2005;52:S90–2.
- Balding SD, Prost C, Diaz LA, et al. Cicatricial pemphigoid autoantibodies react with multiple sites on the BP180 extracellular domain. J Invest Dermatol 1996;106:141–6.
- Bedane C, McMillan JR, Balding SD, et al. Bullous pemphigoid and cicatricial pemphigoid autoantibodies react with ultrastructurally separable epitopes on the BP180 ectodomain: evidence that BP180 spans the lamina lucida. J Invest Dermatol 1997;108:901–7.
- Kromminga A, Sitaru C, Meyer J, et al. Cicatricial pemphigoid differs from bullous pemphigoid and pemphigoid gestationis regarding the fine specificity of autoantibodies to the BP180 NC16A domain. J Dermatol Sci 2002;28:68–75.
- Lee JB, Liu Y, Hashimoto T. Cicatricial pemphigoid sera specifically react with the most C-terminal portion of BP180. J Dermatol Sci 2003;32:59–64.
- Roh JY, Yee C, Lazarova Z, Hall RP, Yancey KB. The 120-kDa soluble ectodomain of type XVII collagen is recognized by autoantibodies in patients with pemphigoid and linear IgA dermatosis. Br J Dermatol 2000;143:104–11.
- Kirtschig G, Marinkovich MP, Burgeson RE, Yancey KB. Anti-basement membrane autoantibodies in patients with anti-epiligrin cicatricial pemphigoid bind the alpha subunit of laminin 5. J Invest Dermatol 1995;105:543–8.
- Rashid KA, Gurcan HM, Ahmed AR. Antigen specificity in subsets of mucous membrane pemphigoid. J Invest Dermatol 2006;126:2631–6.
- Bhol KC, Goss L, Kumari S, Colon JE, Ahmed AR. Autoantibodies to human alpha6 integrin in patients with oral pemphigoid. J Dent Res 2001;80:1711–15.
- Tyagi S, Bhol K, Natarajan K, Livir-Rallatos C, Foster CS, Ahmed AR. Ocular cicatricial pemphigoid antigen: partial sequence and biochemical characterization. Proc Natl Acad Sci U S A 1996;93:14714–19.
- Bhol KC, Dans MJ, Simmons RK, Foster CS, Giancotti FG, Ahmed AR. The autoantibodies to alpha 6 beta 4 integrin of patients affected by ocular cicatricial pemphigoid recognize predominantly epitopes within the large cytoplasmic domain of human beta 4. J Immunol 2000;165:2824–9.
- Bean SF, Waisman M, Michel B, Thomas CI, Knox JM, Levine M. Cicatricial pemphigoid. Immunofluorescent studies. Arch Dermatol 1972;106:195–9.
- Sarret Y, Hall R, Cobo LM, Thivolet J, Patton DL, Woodley DT. Salt-split human skin substrate for the immunofluorescent screening of serum from patients with cicatricial pemphigoid and a new method of immunoprecipitation with IgA antibodies. J Am Acad Dermatol 1991;24:952–8.
- Bernard P, Prost C, Aucouturier P, Durepaire N, Denis F, Bonnetblanc JM. The subclass distribution of IgG autoantibodies in cicatricial pemphigoid and epidermolysis bullosa acquisita. J Invest Dermatol 1991;97:259–63.
- Setterfield J, Shirlaw PJ, Bhogal BS, Tilling K, Challacombe SJ, Black MM. Cicatricial pemphigoid: serial titres of circulating IgG and IgA antibasement membrane antibodies correlate with disease activity. Br J Dermatol 1999;140:645–50.
- Hayashi I, Shinkuma S, Shimizu S, et al. Mucous membrane pemphigoid with generalized blisters: IgA and IgG autoantibodies target both laminin-332 and type XVII collagen. Br J Dermatol 2012;166:1116–20.
- Natsuga K, Nishie W, Shinkuma S, et al. Circulating IgA and IgE autoantibodies in antilaminin-332 mucous membrane pemphigoid. Br J Dermatol 2010;162:513–17.
- Colon JE, Bhol KC, Razzaque MS, Ahmed AR. In vitro organ culture model for mucous membrane pemphigoid. Clin Immunol 2001;98:229–34.
- Rashid KA, Foster CS, Ahmed AR. Identification of epitopes within integrin beta4 for binding of autoantibodies in ocular cicatricial and mucous membrane pemphigoid-preliminary report. Invest Ophthalmol Vis Sci 2013;54:7707–16.
- Letko E, Bhol K, Foster SC, Ahmed RA. Influence of intravenous immunoglobulin therapy on serum levels of anti-beta 4 antibodies in ocular cicatricial pemphigoid. A correlation with disease activity. A preliminary study. Curr Eye Res 2000;21:646–54.
- Black AP, Seneviratne SL, Jones L, et al. Rapid effector function of circulating NC16A-specific T cells in individuals with mucous membrane pemphigoid. Br J Dermatol 2004;151:1160–4.
- Foster CS, Sainz De La Maza M. Ocular cicatricial pemphigoid review. Curr Opin Allergy Clin Immunol 2004;4:435–9.
- Saw VP, Offiah I, Dart RJ, et al. Conjunctival interleukin-13 expression in mucous membrane pemphigoid and functional effects of interleukin-13 on conjunctival fibroblasts in vitro. Am J Pathol 2009;175:2406–15.
- Saw VP, Dart RJ, Galatowicz G, Daniels JT, Dart JK, Calder VL. Tumor necrosis factor-alpha in ocular mucous membrane pemphigoid and its effect on conjunctival fibroblasts. Invest Ophthalmol Vis Sci 2009;50:5310–17.
- Rose C, Schmidt E, Kerstan A, et al. Histopathology of anti-laminin 5 mucous membrane pemphigoid. J Am Acad Dermatol 2009;61:433–40.
- Foster CS. Cicatricial pemphigoid. Trans Am Ophthalmol Soc 1986;84:527–663.
- Terra JB, Pas HH, Hertl M, Dikkers FG, Kamminga N, Jonkman MF. Immunofluorescence serration pattern analysis as a diagnostic criterion in antilaminin-332 mucous membrane pemphigoid: immunopathological findings and clinical experience in 10 Dutch patients. Br J Dermatol 2011;165:815–22.
- Gammon WR, Kowalewski C, Chorzelski TP, Kumar V, Briggaman RA, Beutner EH. Direct immunofluorescence studies of sodium chloride-separated skin in the differential diagnosis of bullous pemphigoid and epidermolysis bullosa acquisita. J Am Acad Dermatol 1990;22:664–70.
- Wozniak K, Kazama T, Kowalewski C. A practical technique for differentiation of subepidermal bullous diseases: localization of in vivo-bound IgG by laser scanning confocal microscopy. Arch Dermatol 2003;139:1007–11.
- Ghohestani RF, Nicolas JF, Rousselle P, Claudy AL. Diagnostic value of indirect immunofluorescence on sodium chloride-split skin in differential diagnosis of subepidermal autoimmune bullous dermatoses. Arch Dermatol 1997;133:1102–7.
- Kelly SE, Wojnarowska F. The use of chemically split tissue in the detection of circulating anti-basement membrane zone antibodies in bullous pemphigoid and cicatricial pemphigoid. Br J Dermatol 1988;118:31–40.
- Bernard P, Prost C, Lecerf V, et al. Studies of cicatricial pemphigoid autoantibodies using direct immunoelectron microscopy and immunoblot analysis. J Invest Dermatol 1990;94:630–5.
- Vodegel RM, de Jong MC, Pas HH, Yancey KB, Jonkman MF. Anti-epiligrin cicatricial pemphigoid and epidermolysis bullosa acquisita: differentiation by use of indirect immunofluorescence microscopy. J Am Acad Dermatol 2003;48:542–7.
- Bedane C, Prost C, Bernard P, Catanzano G, Bonnetblanc JM, Dubertret L. Cicatricial pemphigoid antigen differs from bullous pemphigoid antigen by its exclusive extracellular localization: a study by indirect immunoelectronmicroscopy. J Invest Dermatol 1991;97:3–9.
- Delgado JC, Turbay D, Yunis EJ, et al. A common major histocompatibility complex class II allele HLA-DQB1* 0301 is present in clinical variants of pemphigoid. Proc Natl Acad Sci U S A 1996;93:8569–71.
- Nayar M, Wojnarowska F, Venning V, Taylor CJ. Association of autoimmunity and cicatricial pemphigoid: is there an immunogenetic basis? J Am Acad Dermatol 1991;25:1011–15.
- Yunis JJ, Mobini N, Yunis EJ, et al. Common major histocompatibility complex class II markers in clinical variants of cicatricial pemphigoid. Proc Natl Acad Sci U S A 1994;91:7747–51.
- Drouet M, Delpuget-Bertin N, Vaillant L, Chauchaix S, Boulanger MD, Bonnetblanc JM, Bernard P. HLA-DRB1 and HLA-DQB1 genes in susceptibility and resistance to cicatricial pemphigoid in French Caucasians. Eur J Dermatol 1998;8:330–3.
- Setterfield J, Theron J, Vaughan RW, et al. Mucous membrane pemphigoid: HLA-DQB1*0301 is associated with all clinical sites of involvement and may be linked to antibasement membrane IgG production. Br J Dermatol 2001;145:406–14.
- Carrozzo M, Fasano ME, Broccoletti R, et al. HLA-DQB1 alleles in Italian patients with mucous membrane pemphigoid predominantly affecting the oral cavity. Br J Dermatol 2001;145:805–8.
- Chan LS, Hammerberg C, Cooper KD. Significantly increased occurrence of HLA-DQB1*0301 allele in patients with ocular cicatricial pemphigoid. J Invest Dermatol 1997;108:129–32.
- Haider N, Neuman R, Foster CS, Ahmed AR. Report on the sequence of DQB1*0301 gene in ocular cicatricial pemphigoid patients. Curr Eye Res 1992;11:1233–8.
- Ahmed AR, Foster S, Zaltas M, et al. Association of DQw7 (DQB1*0301) with ocular cicatricial pemphigoid. Proc Natl Acad Sci U S A 1991;88:11579–82.
- Mondino BJ, Brown SI, Rabin BS. HLA antigens in ocular cicatricial pemphigoid. Br J Ophthalmol 1978;62:265–7.
- Marren P, Wojnarowska F, Venning V, Wilson C, Nayar M. Vulvar involvement in autoimmune bullous diseases. J Reprod Med 1993;38:101–7.
- Saw VP, Dart JK. Ocular mucous membrane pemphigoid: diagnosis and management strategies. Ocul Surf 2008;6:128–42.
- Kirtschig G, Wojnarowska F, Marsden RA, Edwards S, Bhogal B, Black MM. Acquired bullous diseases of childhood: re-evaluation of diagnosis by indirect immunofluorescence examination on 1 M NaCl split skin and immunoblotting. Br J Dermatol 1994;130:610–16.
- Farrell AM, Kirtschig G, Dalziel KL, et al. Childhood vulval pemphigoid: a clinical and immunopathological study of five patients. Br J Dermatol 1999;140:308–12.
- Foster CS, Wilson LA, Ekins MB. Immunosuppressive therapy for progressive ocular cicatricial pemphigoid. Ophthalmology 1982;89:340–53.
- Mondino BJ, Brown SI. Ocular cicatricial pemphigoid. Ophthalmology 1981;88:95–100.
- Mondino BJ, Ross AN, Rabin BS, Brown SI. Autoimmune phenomena in ocular cicatricial pemphigoid. Am J Ophthalmol 1977;83:443–50.
- Mondino BJ, Brown SI. Immunosuppressive therapy in ocular cicatricial pemphigoid. Am J Ophthalmol 1983;96:453–9.
- Tauber J, Jabbur N, Foster CS. Improved detection of disease progression in ocular cicatricial pemphigoid. Cornea 1992;11:446–51.
- Murrell DF, Marinovic B, Caux F, et al. Definitions and outcome measures for mucous membrane pemphigoid: recommendations of an international panel of experts. J Am Acad Dermatol 2015;72:168–74.
- Shklar G, McCarthy PL. Oral lesions of mucous membrane pemphigoid. A study of 85 cases. Arch Otolaryngol 1971;93:354–64.
- Anstey A, Wojnarowska F, Whitehead P, Leigh IM, Rutty GN. Oesophageal webs preceding carcinoma and rupture of the oesophagus in cicatricial pemphigoid. Clin Exp Dermatol 1991;16:395–8.
- Hardy KM, Perry HO, Pingree GC, Kirby TJ Jr. Benign mucous membrane pemphigoid. Arch Dermatol 1971;104:467–75.
- Person JR, Rogers RS 3rd. Bullous and cicatricial pemphigoid. Clinical, histopathologic, and immunopathologic correlations. Mayo Clin Proc 1977;52:54–66.
- Christophoridis S, Budinger L, Borradori L, Hunziker T, Merk HF, Hertl M. IgG, IgA and IgE autoantibodies against the ectodomain of BP180 in patients with bullous and cicatricial pemphigoid and linear IgA bullous dermatosis. Br J Dermatol 2000;143:349–55.
- Lazarova Z, Sitaru C, Zillikens D, Yancey KB. Comparative analysis of methods for detection of anti-laminin 5 autoantibodies in patients with anti-epiligrin cicatricial pemphigoid. J Am Acad Dermatol 2004;51:886–92.
- Hisamatsu Y, Nishiyama T, Amano S, Matsui C, Ghohestani R, Hashimoto T. Usefulness of immunoblotting using purified laminin 5 in the diagnosis of anti-laminin 5 cicatricial pemphigoid. J Dermatol Sci 2003;33:113–19.
- Kirtschig G, Murrell D, Wojnarowska F, Khumalo N. Interventions for mucous membrane pemphigoid/cicatricial pemphigoid and epidermolysis bullosa acquisita: a systematic literature review. Arch Dermatol 2002;138:380–4.
- El Darouti MA, Fakhry Khattab MA, Hegazy RA, Hafez DA, Gawdat HI. Pentoxifylline (anti-tumor necrosis factor drug): effective adjuvant therapy in the control of ocular cicatricial pemphigoid. Eur J Ophthalmol 2011;21:529–37.
- Munyangango EM, Le Roux-Villet C, Doan S, et al. Oral cyclophosphamide without corticosteroids to treat mucous membrane pemphigoid. Br J Dermatol 2013;168:381–90.
- Secchi AG, Tognon MS. Intraoperative mitomycin C in the treatment of cicatricial obliterations of conjunctival fornices. Am J Ophthalmol 1996;122:728–30.
- Donnenfeld ED, Perry HD, Wallerstein A, et al. Subconjunctival mitomycin C for the treatment of ocular cicatricial pemphigoid. Ophthalmology 1999;106:72–8, discussion 79.
- McCluskey P, Chang JH, Singh R, Wakefield D. Methotrexate therapy for ocular cicatricial pemphigoid. Ophthalmology 2004;111:796–801.
- Nottage JM, Hammersmith KM, Murchison AP, Felipe AF, Penne R, Raber I. Treatment of mucous membrane pemphigoid with mycophenolate mofetil. Cornea 2013;32:810–15.
- Foster CS, Ahmed AR. Intravenous immunoglobulin therapy for ocular cicatricial pemphigoid: a preliminary study. Ophthalmology 1999;106:2136–43.
- Letko E, Miserocchi E, Daoud YJ, Christen W, Foster CS, Ahmed AR. A nonrandomized comparison of the clinical outcome of ocular involvement in patients with mucous membrane (cicatricial) pemphigoid between conventional immunosuppressive and intravenous immunoglobulin therapies. Clin Immunol 2004;111:303–10.
- Ahmed AR, Colon JE. Comparison between intravenous immunoglobulin and conventional immunosuppressive therapy regimens in patients with severe oral pemphigoid: effects on disease progression in patients nonresponsive to dapsone therapy. Arch Dermatol 2001;137:1181–9.
- Kirtschig G, Murrell DF, Wojnarowska F, Khumalo NP. Interventions for mucous membrane pemphigoid and epidermolysis bullosa acquisita. Cochrane Database Syst Rev 2009:CD004056.
- Kennedy JS, Devillez RL, Henning JS. Recalcitrant cicatricial pemphigoid treated with the anti-TNF-alpha agent etanercept. J Drugs Dermatol 2010;9:68–70.
- Canizares MJ, Smith DI, Conners MS, Maverick KJ, Heffernan MP. Successful treatment of mucous membrane pemphigoid with etanercept in 3 patients. Arch Dermatol 2006;142:1457–61.
- Sacher C, Rubbert A, Konig C, Scharffetter-Kochanek K, Krieg T, Hunzelmann N. Treatment of recalcitrant cicatricial pemphigoid with the tumor necrosis factor alpha antagonist etanercept. J Am Acad Dermatol 2002;46:113–15.
- Heffernan MP, Bentley DD. Successful treatment of mucous membrane pemphigoid with infliximab. Arch Dermatol 2006;142:1268–70.
- Saw VP, Dart JK, Rauz S, et al. Immunosuppressive therapy for ocular mucous membrane pemphigoid strategies and outcomes. Ophthalmology 2008;115:253–61, e251.
- Chaidemenos G, Sidiropoulos T, Katsioula P, Koussidou-Eremondi T. Colchicine in the management of mucous membrane pemphigoid. Dermatol Ther 2011;24:443–5.
- Schmidt E, Seitz CS, Benoit S, Brocker EB, Goebeler M. Rituximab in autoimmune bullous diseases: mixed responses and adverse effects. Br J Dermatol 2007;156:352–6.
- Shetty S, Ahmed AR. Critical analysis of the use of rituximab in mucous membrane pemphigoid: a review of the literature. J Am Acad Dermatol 2013;68:499–506.
- Kasperkiewicz M, Shimanovich I, Ludwig RJ, Rose C, Zillikens D, Schmidt E. Rituximab for treatment-refractory pemphigus and pemphigoid: a case series of 17 patients. J Am Acad Dermatol 2011;65:552–8.
- Le Roux-Villet C, Prost-Squarcioni C, Alexandre M, et al. Rituximab for patients with refractory mucous membrane pemphigoid. Arch Dermatol 2011;147:843–9.
- Schmidt E, Goebeler M. CD20-directed therapy in autoimmune diseases involving the skin: role of rituximab. Exp Rev Dermatol 2008;3:259–78.
- Heelan K, Walsh S, Shear NH. Treatment of mucous membrane pemphigoid with rituximab. J Am Acad Dermatol 2013;69:310–11.
- Schmidt E, Goebeler M, Zillikens D. Rituximab in severe pemphigus. Ann N Y Acad Sci 2009;1173:683–91.
- Bedane C, Prost C, Ingen-Housz-Oro S, Joly P, Bernard P. [Mucous membrane pemphigoid. Guidelines for the diagnosis and treatment. Centres de reference des maladies bulleuses auto-immunes. Societe Francaise de Dermatologie.] Ann Dermatol Venereol 2011;138:259–63.
- Kourosh AS, Yancey KB. Therapeutic approaches to patients with mucous membrane pemphigoid. Dermatol Clin 2011;29:637–41.
- Arduino PG, Lopetuso E, Carcieri P, et al. Professional oral hygiene treatment and detailed oral hygiene instructions in patients affected by mucous membrane pemphigoid with specific gingival localization: a pilot study in 12 patients. Int J Dent Hyg 2012;10:138–41.
- Hertl M, Zillikens D, Borradori L, et al. Recommendations for the use of rituximab (anti-CD20 antibody) in the treatment of autoimmune bullous skin diseases. J Dtsch Dermatol Ges 2008;6:366–73.
- Zillikens D, Derfler K, Eming R, et al. Recommendations for the use of immunoapheresis in the treatment of autoimmune bullous diseases. J Dtsch Dermatol Ges 2007;5:881–7.
- Recke A, Shimanovich I, Steven P, Westermann L, Zillikens D, Schmidt E. [Treatment-refractory anti-laminin 332 mucous membrane pemphigoid. Remission following adjuvant immunoadsorption and rituximab.] Hautarzt 2011;62:852–8.
- Enk A, Fierlbeck G, French L, et al. Use of high-dose immunoglobulins in dermatology. J Dtsch Dermatol Ges 2009;7:806–12.
- Enk A. Guidelines on the use of high-dose intravenous immunoglobulin in dermatology. Eur J Dermatol 2009;19:90–8.
Linear IgA bullous dermatosis
- Schmidt E, Zillikens D. Pemphigoid diseases. Lancet 2013;381:320–32.
- Wojnarowska F, Marsden RA, Bhogal B, Black MM. Chronic bullous disease of childhood, childhood cicatricial pemphigoid, and linear IgA disease of adults. A comparative study demonstrating clinical and immunopathologic overlap. J Am Acad Dermatol 1988;19:792–805.
- Egan CA, Zone JJ. Linear IgA bullous dermatosis. Int J Dermatol 1999;38:818–27.
- Chorzelski TP, Jablonska S, Beutner EE, Maciejowska E, Jarzabek-Chorzelska M. Juvenile dermatitis herpetiformis versus “benign chronic bullous dermatosis of childhood.” Are these immunologic diseases? J Invest Dermatol 1975;65:447–50.
- Chorzelski TP, Jablonska S. Diagnostic significance of the immunofluorescent pattern in dermatitis herpetiformis. Int J Dermatol 1975;14:429–36.
- Jablonska S, Chorzelski TP, Beutner EH, Maciejowska E, Rzesa G. Dermatitis herpetiformis and bullous pemphigoid. Intermediate and mixed forms. Arch Dermatol 1976;112:45–8.
- Willsteed E, Bhogal BS, Black MM, McKee P, Wojnarowska F. Use of 1M NaCl split skin in the indirect immunofluorescence of the linear IgA bullous dermatoses. J Cutan Pathol 1990;17:144–8.
- Wojnarowska F, Collier PM, Allen J, Millard PR. The localization of the target antigens and antibodies in linear IgA disease is heterogeneous, and dependent on the methods used. Br J Dermatol 1995;132:750–7.
- Marinkovich MP, Taylor TB, Keene DR, Burgeson RE, Zone JJ. LAD-1, the linear IgA bullous dermatosis autoantigen, is a novel 120-kDa anchoring filament protein synthesized by epidermal cells. J Invest Dermatol 1996;106:734–8.
- Zone JJ, Taylor TB, Kadunce DP, Meyer LJ. Identification of the cutaneous basement membrane zone antigen and isolation of antibody in linear immunoglobulin A bullous dermatosis. J Clin Invest 1990;85:812–20.
- Wojnarowska F, Whitehead P, Leigh IM, Bhogal BS, Black MM. Identification of the target antigen in chronic bullous disease of childhood and linear IgA disease of adults. Br J Dermatol 1991;124:157–62.
- Hashimoto T, Ishiko A, Shimizu H, et al. A case of linear IgA bullous dermatosis with IgA anti-type VII collagen autoantibodies. Br J Dermatol 1996;134:336–9.
- Hayashi I, Shinkuma S, Shimizu S, et al. Mucous membrane pemphigoid with generalized blisters: IgA and IgG autoantibodies target both laminin-332 and type XVII collagen. Br J Dermatol 2012;166:1116–20.
- Natsuga K, Nishie W, Shinkuma S, et al. Circulating IgA and IgE autoantibodies in antilaminin-332 mucous membrane pemphigoid. Br J Dermatol 2010;162:513–17.
- Kanitakis J, Mauduit G, Cozzani E, Badinand P, Faure M, Claudy A. Linear IgA bullous dermatosis of childhood with autoantibodies to a 230 kDa epidermal antigen. Pediatr Dermatol 1994;11:139–44.
- Zambruno G, Manca V, Kanitakis J, Cozzani E, Nicolas JF, Giannetti A. Linear IgA bullous dermatosis with autoantibodies to a 290 kd antigen of anchoring fibrils. J Am Acad Dermatol 1994;31:884–8.
- Yamane Y, Sato H, Higashi K, Yaoita H. Linear immunoglobulin A (IgA) bullous dermatosis of childhood: identification of the target antigens and study of the cellular sources. Br J Dermatol 1996;135:785–90.
- Dmochowski M, Hashimoto T, Bhogal BS, Black MM, Zone JJ, Nishikawa T. Immunoblotting studies of linear IgA disease. J Dermatol Sci 1993;6:194–200.
- Bertram F, Brocker EB, Zillikens D, Schmidt E. Prospective analysis of the incidence of autoimmune bullous disorders in Lower Franconia, Germany. J Dtsch Dermatol Ges 2009;7:434–40.
- Nanda A, Dvorak R, Al-Saeed K, Al-Sabah H, Alsaleh QA. Spectrum of autoimmune bullous diseases in Kuwait. Int J Dermatol 2004;43:876–81.
- Wong SN, Chua SH. Spectrum of subepidermal immunobullous disorders seen at the National Skin Centre, Singapore: a 2-year review. Br J Dermatol 2002;147:476–80.
- Zillikens D, Wever S, Roth A, et al. Incidence of autoimmune subepidermal blistering dermatoses in a region of central Germany. Arch Dermatol 1995;131:957–8.
- Mulyowa GK, Jaeger G, Kabakyenga J, et al. Autoimmune subepidermal blistering diseases in Uganda: correlation of autoantibody class with age of patients. Int J Dermatol 2006;45:1047–52.
- Piamphongsant T, Sirimachan S, Himmunknan P. Juvenile blistering diseases: the problems of diagnosis and treatment. Asian Pac J Allergy Immunol 1986;4:133–7.
- Aboobaker J, Wojnarowska FT, Bhogal B, Black MM. Chronic bullous dermatosis of childhood–clinical and immunological features seen in African patients. Clin Exp Dermatol 1991;16:160–4.
- Adam BA. Bullous diseases in Malaysia: epidemiology and natural history. Int J Dermatol 1992;31:42–5.
- Ajithkumar K, Kurian S, Jacob M, Pulimood S. Linear IgA bullous dermatosis in south India. Int J Dermatol 1997;36:191–3.
- Denguezli M, Ben Nejma B, Nouira R, et al. [Iga linear bullous dermatosis in children. A series of 12 Tunisian patients.] Ann Dermatol Venereol 1994;121:888–92.
- Mahe A, Flageul B, Bobin P. [Bullous IgA linear dermatosis of children in Mali.] Ann Dermatol Venereol 1996;123:544–8.
- Godfrey K, Wojnarowska F, Leonard J. Linear IgA disease of adults: association with lymphoproliferative malignancy and possible role of other triggering factors. Br J Dermatol 1990;123:447–52.
- Paige DG, Leonard JN, Wojnarowska F, Fry L. Linear IgA disease and ulcerative colitis. Br J Dermatol 1997;136:779–82.
- McEvoy MT, Connolly SM. Linear IgA dermatosis: association with malignancy. J Am Acad Dermatol 1990;22:59–63.
- Pas HH, Kloosterhuis GJ, Heeres K, van der Meer JB, Jonkman MF. Bullous pemphigoid and linear IgA dermatosis sera recognize a similar 120-kDa keratinocyte collagenous glycoprotein with antigenic cross-reactivity to BP180. J Invest Dermatol 1997;108:423–9.
- Zone JJ, Taylor TB, Meyer LJ, Petersen MJ. The 97 kDa linear IgA bullous disease antigen is identical to a portion of the extracellular domain of the 180 kDa bullous pemphigoid antigen, BPAg2. J Invest Dermatol 1998;110:207–10.
- Hirako Y, Nishizawa Y, Sitaru C, et al. The 97-kDa (LABD97) and 120-kDa (LAD-1) fragments of bullous pemphigoid antigen 180/type XVII collagen have different N-termini. J Invest Dermatol 2003;121:1554–6.
- Franzke CW, Bruckner-Tuderman L, Blobel CP. Shedding of collagen XVII/BP180 in skin depends on both ADAM10 and ADAM9. J Biol Chem 2009;284:23386–96.
- Nishie W, Lamer S, Schlosser A, et al. Ectodomain shedding generates Neoepitopes on collagen XVII, the major autoantigen for bullous pemphigoid. J Immunol 2010;185:4938–47.
- Zillikens D, Herzele K, Georgi M, et al. Autoantibodies in a subgroup of patients with linear IgA disease react with the NC16A domain of BP1801. J Invest Dermatol 1999;113:947–53.
- Ishii N, Ohyama B, Yamaguchi Z, Hashimoto T. IgA autoantibodies against the NC16a domain of BP180 but not 120-kDa LAD-1 detected in a patient with linear IgA disease. Br J Dermatol 2008;158:1151–3.
- Georgi M, Scheckenbach C, Kromminga A, et al. Mapping of epitopes on the BP180 ectodomain targeted by IgA and IgG autoantibodies in patients with the lamina lucida-type of linear IgA disease. Arch Dermatol Res 2001;293:109–14.
- Lin MS, Fu CL, Olague-Marchan M, et al. Autoimmune responses in patients with linear IgA bullous dermatosis: both autoantibodies and T lymphocytes recognize the NC16A domain of the BP180 molecule. Clin Immunol 2002;102:310–19.
- Wojnarowska F, Bhogal BS, Black MM. Chronic bullous disease of childhood and linear IgA disease of adults are IgA1-mediated diseases. Br J Dermatol 1994;131:201–4.
- Zone JJ, Pazderka Smith E, et al. Antigenic specificity of antibodies from patients with linear basement membrane deposition of IgA. Dermatology 1994;189 Suppl. 1:64–6.
- Peters MS, Rogers RS. Clinical correlations of linear IgA deposition at the cutaneous basement membrane zone. J Am Acad Dermatol 1989;20:761–70.
- Kromminga A, Scheckenbach C, Georgi M, et al. Patients with bullous pemphigoid and linear IgA disease show a dual IgA and IgG autoimmune response to BP180. J Autoimmun 2000;15:293–300.
- Sheridan AT, Kirtschig G, Wojnarowska F. Mixed immunobullous disease: is this linear IgA disease? Australas J Dermatol 2000;41:219–21.
- Hendrix JD, Mangum KL, Zone JJ, Gammon WR. Cutaneous IgA deposits in bullous diseases function as ligands to mediate adherence of activated neutrophils. J Invest Dermatol 1990;94:667–72.
- Akahoshi Y, Kanda G, Anan S, Yoshida H. Dermo-epidermal blister formation by linear IgA dermatosis sera in normal human skin in organ culture. J Dermatol 1987;14:352–8.
- Zone JJ, Egan CA, Taylor TB, Meyer LJ. IgA autoimmune disorders: development of a passive transfer mouse model. J Investig Dermatol Symp Proc 2004;9:47–51.
- Ghohestani RF, Nicolas JF, Kanitakis J, Claudy A. Linear IgA bullous dermatosis with IgA antibodies exclusively directed against the 180- or 230-kDa epidermal antigens. J Invest Dermatol 1997;108:854–8.
- Paul C, Wolkenstein P, Prost C, et al. Drug-induced linear IgA disease: target antigens are heterogeneous. Br J Dermatol 1997;136:406–11.
- Buijsrogge JJ, Diercks GF, Pas HH, Jonkman MF. The many faces of epidermolysis bullosa acquisita after serration pattern analysis by direct immunofluorescence microscopy. Br J Dermatol 2011;165:92–8.
- Wozniak K, Hashimoto T, Fukuda S, et al. IgA anti-p200 pemphigoid. Arch Dermatol 2012;147:1306–10.
- Shimanovich I, Rose C, Sitaru C, Brocker EB, Zillikens D. Localized linear IgA disease induced by ampicillin/sulbactam. J Am Acad Dermatol 2004;51:95–8.
- Nousari HC, Kimyai-Asadi A, Caeiro JP, Anhalt GJ. Clinical, demographic, and immunohistologic features of vancomycin-induced linear IgA bullous disease of the skin. Report of 2 cases and review of the literature. Medicine (Baltimore) 1999;78:1–8.
- Billet SE, Kortuem KR, Gibson LE, El-Azhary R. A morbilliform variant of vancomycin-induced linear IgA bullous dermatosis. Arch Dermatol 2008;144:774–8.
- Onodera H, Mihm MC Jr, Yoshida A, Akasaka T. Drug-induced linear IgA bullous dermatosis. J Dermatol 2005;32:759–64.
- Collier PM, Wojnarowska F. Drug-induced linear immunoglobulin A disease. Clin Dermatol 1993;11:529–33.
- Plunkett RW, Chiarello SE, Beutner EH. Linear IgA bullous dermatosis in one of two piroxicam-induced eruptions: a distinct direct immunofluorescence trend revealed by the literature. J Am Acad Dermatol 2001;45:691–6.
- Girao L, Fiadeiro T, Rodrigues JC. Burn-induced linear IgA dermatosis. J Eur Acad Dermatol Venereol 2000;14:507–10.
- Alberta-Wszolek L, Mousette AM, Mahalingam M, Levin NA. Linear IgA bullous dermatosis following influenza vaccination. Dermatol Online J 2009;15:3.
- Salmhofer W, Soyer HP, Wolf P, Fodinger D, Hodl S, Kerl H. UV light-induced linear IgA dermatosis. J Am Acad Dermatol 2004;50:109–15.
- Blenkinsopp WK, Haffenden GP, Fry L, Leonard JN. Histology of linear IgA disease, dermatitis herpetiformis, and bullous pemphigoid. Am J Dermatopathol 1983;5:547–54.
- Caproni M, Rolfo S, Bernacchi E, Bianchi B, Brazzini B, Fabbri P. The role of lymphocytes, granulocytes, mast cells and their related cytokines in lesional skin of linear IgA bullous dermatosis. Br J Dermatol 1999;140:1072–8.
- Terra JB, Pas HH, Hertl M, Dikkers FG, Kamminga N, Jonkman MF. Immunofluorescence serration pattern analysis as a diagnostic criterion in antilaminin-332 mucous membrane pemphigoid: immunopathological findings and clinical experience in 10 Dutch patients. Br J Dermatol 2011;165:815–22.
- Vodegel RM, Jonkman MF, Pas HH, de Jong MC. U-serrated immunodeposition pattern differentiates type VII collagen targeting bullous diseases from other subepidermal bullous autoimmune diseases. Br J Dermatol 2004;151:112–18.
- Collier PM, Wojnarowska F, Welsh K, McGuire W, Black MM. Adult linear IgA disease and chronic bullous disease of childhood: the association with human lymphocyte antigens Cw7, B8, DR3 and tumour necrosis factor influences disease expression. Br J Dermatol 1999;141:867–75.
- Powell J, Kirtschig G, Allen J, Dean D, Wojnarowska F. Mixed immunobullous disease of childhood: a good response to antimicrobials. Br J Dermatol 2001;144:769–74.
- Collier PM, Kelly SE, Wojnarowska F. Linear IgA disease and pregnancy. J Am Acad Dermatol 1994;30:407–11.
- Marsden RA, McKee PH, Bhogal B, Black MM, Kennedy LA. A study of benign chronic bullous dermatosis of childhood and comparison with dermatitis herpetiformis and bullous pemphigoid occurring in childhood. Clin Exp Dermatol 1980;5:159–76.
- Burge S, Wojnarowska F, Marsden A. Chronic bullous dermatosis of childhood persisting into adulthood. Pediatr Dermatol 1988;5:246–9.
- Kuechle MK, Stegemeir E, Maynard B, Gibson LE, Leiferman KM, Peters MS. Drug-induced linear IgA bullous dermatosis: report of six cases and review of the literature. J Am Acad Dermatol 1994;30:187–92.
- Zhou S, Wakelin SH, Allen J, Wojnarowska F. Blister fluid for the diagnosis of subepidermal immunobullous diseases: a comparative study of basement membrane zone autoantibodies detected in blister fluid and serum. Br J Dermatol 1998;139:27–32.
- Schmidt E, Skrobek C, Kromminga A, et al. Cicatricial pemphigoid: IgA and IgG autoantibodies target epitopes on both intra- and extracellular domains of bullous pemphigoid antigen 180. Br J Dermatol 2001;145:778–83.
- Schumann H, Baetge J, Tasanen K, et al. The shed ectodomain of collagen XVII/BP180 is targeted by autoantibodies in different blistering skin diseases. Am J Pathol 2000;156:685–95.
- Nie Z, Nagata Y, Joubeh S, et al. IgA antibodies of linear IgA bullous dermatosis recognize the 15th collagenous domain of BP180. J Invest Dermatol 2000;115:1164–6.
- Csorba K, Schmidt S, Florea F, et al. Development of an ELISA for sensitive and specific detection of IgA autoantibodies against BP180 in pemphigoid diseases. Orphanet J Rare Dis 2011;6:31.
- Ng SY, Venning VV. Management of linear IgA disease. Dermatol Clin 2011;29:629–30.
- Wozel VE. Innovative use of dapsone. Dermatol Clin 2010;28:599–610.
- Kasperkiewicz M, Meier M, Zillikens D, Schmidt E. Linear IgA disease: successful application of immunoadsorption and review of the literature. Dermatology 2010;220:259–63.
Anti-p200 pemphigoid
- Goletz S, Hashimoto T, Zillikens D, Schmidt E. Anti-p200 pemphigoid. J Am Acad Dermatol 2014;71:185–91.
- Zillikens D, Kawahara Y, Ishiko A, et al. A novel subepidermal blistering disease with autoantibodies to a 200-kDa antigen of the basement membrane zone. J Invest Dermatol 1996;106:1333–8.
- Chen KR, Shimizu S, Miyakawa S, Ishiko A, Shimizu H, Hashimoto T. Coexistence of psoriasis and an unusual IgG-mediated subepidermal bullous dermatosis: identification of a novel 200-kDa lower lamina lucida target antigen. Br J Dermatol 1996;134:340–6.
- Shimizu A, Funakoshi T, Ishibashi M, et al. Immunoglobulin G deposition to nonhemidesmosomal lamina lucida and early neutrophil involvement are characteristic features in a case of anti-p200 pemphigoid. Br J Dermatol 2013;168:647–55.
- Zillikens D, Ishiko A, Jonkman MF, et al VII collagen. Br J Dermatol 2000;143:1043–9.
- Egan CA, Yee C, Zillikens D, Yancey KB. Anti-p200 pemphigoid: diagnosis and treatment of a case presenting as an inflammatory subepidermal blistering disease. J Am Acad Dermatol 2002;46:786–9.
- Dilling A, Rose C, Hashimoto T, Zillikens D, Shimanovich I. Anti-p200 pemphigoid: a novel autoimmune subepidermal blistering disease. J Dermatol 2007;34:1–8.
- Dainichi T, Kurono S, Ohyama B, et al. Anti-laminin gamma-1 pemphigoid. Proc Natl Acad Sci USA 2009;106:2800–5.
- Hofmann SC, Voith U, Sasaki T, Trueb RM, Nischt R, Bruckner-Tuderman L. The autoantigen in anti-p200 pemphigoid is synthesized by keratinocytes and fibroblasts and is distinct from nidogen-2. J Invest Dermatol 2008;128:87–95.
- Ido H, Ito S, Taniguchi Y, et al. Laminin isoforms containing the gamma3 chain are unable to bind to integrins due to the absence of the glutamic acid residue conserved in the C-terminal regions of the gamma1 and gamma2 chains. J Biol Chem 2008;283:28149–57.
- Durbeej M. Laminins. Cell Tissue Res 2010;339:259–68.
- Groth S, Recke A, Vafia K, et al. Development of a simple enzyme-linked immunosorbent assay for the detection of autoantibodies in anti-p200 pemphigoid. Br J Dermatol 2011;164:76–82.
- Dainichi T, Koga H, Tsuji T, et al. From anti-p200 pemphigoid to anti-laminin gamma1 pemphigoid. J Dermatol 2010;37:231–8.
- Vafia K, Groth S, Beckmann T, et al. Pathogenicity of autoantibodies in anti-p200 pemphigoid. PLoS One 2012;7:e41769.
- Wozniak K, Hashimoto T, Fukuda S, Ohyama B, Ishii N, Koga H, Dainichi T, Kowalewski C. IgA anti-p200 pemphigoid. Arch Dermatol 2011;147:1306–10.
- Goto-Ohguchi Y, Nishie W, Akiyama M, et al. A severe and refractory case of anti-p200 pemphigoid resulting in multiple skin ulcers and scar formation. Dermatology 2009;218:265–71.
- Mitsuya J, Hara H, Ito K, Ishii N, Hashimoto T, Terui T. Metastatic ovarian carcinoma-associated subepidermal blistering disease with autoantibodies to both the p200 dermal antigen and the gamma 2 subunit of laminin 5 showing unusual clinical features. Br J Dermatol 2008;158:1354–7.
- Yamada T, Suzuki M, Koike Y, et al. A case of epidermolysis bullosa acquisita with autoantibody to anti-p200 pemphigoid antigen and exfoliative esophagitis. Dermatology 2006;212:381–4.
- Pastar Z, Rados J, Lipozencic J, Dobric I, Marinovic B, Ishii N, Hashimoto T. Case of concurrent epidermolysis bullosa acquisita and anti-p200 pemphigoid–how to treat it? Int J Dermatol 2007;46:295–8.
- Kasperkiewicz M, Hoppe U, Zillikens D, Schmidt E. Relapse-associated autoantibodies to BP180 in a patient with anti-p200 pemphigoid. Clin Exp Dermatol 2010;35:614–17.
- Kamata M, Fujita H, Hamanaka T, et al. Anti-laminin gamma1 pemphigoid accompanied by autoantibodies to laminin alpha3 and gamma2 subunits of laminin-332. JAMA Dermatol 2013;149:1437–9.
- Li X, Qian H, Ishii N, et al. A case of concurrent antilaminin gamma1 pemphigoid and antilaminin-332-type mucous membrane pemphigoid. Br J Dermatol 2014;171:1257–9.
- Iwata H, Hiramitsu Y, Aoyama Y, Kitajima Y. A case of anti-p200 pemphigoid: evidence for a different pathway in neutrophil recruitment compared with bullous pemphigoid. Br J Dermatol 2009;160:462–4.
- Koga H, Ishii N, Dainichi T, et al. An attempt to develop mouse model for anti-laminin gamma1 pemphigoid. J Dermatol Sci 2013;70:108–15.
- Florea F, Bernards C, Caproni M, et al. Ex vivo pathogenicity of anti-laminin gamma1 autoantibodies. Am J Pathol 2014;184:494–506.
- Colognato H, Yurchenco PD. Form and function: the laminin family of heterotrimers. Dev Dyn 2000;218:213–34.
- Rose C, Weyers W, Denisjuk N, Hillen U, Zillikens D, Shimanovich I. Histopathology of anti-p200 pemphigoid. Am J Dermatopathol 2007;29:119–24.
- Yamane N, Sawamura D, Nishie W, et al. Anti-p200 pemphigoid in a 17-year-old girl successfully treated with systemic corticosteroid and dapsone. Br J Dermatol 2007;156:1075–8.
- Myers DJ, Unwala RD, Xia L, Groth S, Zillikens D, Stratman EJ. Generalized annular bullous eruption–quiz case. Anti-laminin gamma1 pemphigoid with a linear IgA bullous dermatosis-like presentation. Arch Dermatol 2011;147:109–14.
- Majima Y, Yagi H, Tateishi C, et al. A successful treatment with ustekinumab in a case of antilaminin-gamma1 pemphigoid associated with psoriasis. Br J Dermatol 2013;168:1367–9.
- Kasperkiewicz M, Hoppe U, Zillikens D, Schmidt E. Relapse-associated autoantibodies to BP180 in a patient with anti-p200 pemphigoid. Clin Exp Dermatol 2010;35:614–17.
- Alloo A, Strazzula L, Rothschild B, et al. Refractory antilaminin gamma1 pemphigoid successfully treated with intravenous immunoglobulin and mycophenolate mofetil. J Eur Acad Dermatol Venereol 2014;28:1401–3.
- Ohata C, Fukuda S, Ishii N, et al. Refractory anti-laminin gamma1 pemphigoid with psoriasis vulgaris successfully treated by double-filtration plasmapheresis. Eur J Dermatol 2013;23:715–16.
Epidermolysis bullosa acquisita
- Woodley DT, Burgeson RE, Lunstrum G, Bruckner-Tuderman L, Reese MJ, Briggaman RA. Epidermolysis bullosa acquisita antigen is the globular carboxyl terminus of type VII procollagen. J Clin Invest 1988;81:683–7.
- Roenigk HH Jr, Ryan JG, Bergfeld WF. Epidermolysis bullosa acquisita. Report of three cases and review of all published cases. Arch Dermatol 1971;103:1–10.
- Gammon WR, Briggaman RA, Woodley DT, Heald PW, Wheeler CE Jr. Epidermolysis bullosa acquisita – a pemphigoid-like disease. J Am Acad Dermatol 1984;11:820–32.
- Dahl MG. Epidermolysis bullosa acquisita – a sign of cicatricial pemphigoid? Br J Dermatol 1979;101:475–84.
- Richter BJ, McNutt NS. The spectrum of epidermolysis bullosa acquisita. Arch Dermatol 1979;115:1325–8.
- Elliot GT. Two cases of epidermolysis bullosa. J Cutan Genitourin Dis 1895;13:10.
- Kushniruk W. The immunopathology of epidermolysis bullosa acquisita. Can Med Assoc J 1973;108:1143–6.
- Nieboer C, Boorsma DM, Woerdeman MJ, Kalsbeek GL. Epidermolysis bullosa acquisita. Immunofluorescence, electron microscopic and immunoelectron microscopic studies in four patients. Br J Dermatol 1980;102:383–92.
- Yaoita H, Briggaman RA, Lawley TJ, Provost TT, Katz SI. Epidermolysis bullosa acquisita: ultrastructural and immunological studies. J Invest Dermatol 1981;76:288–92.
- Woodley DT, Briggaman RA, O'Keefe EJ, Inman AO, Queen LL, Gammon WR. Identification of the skin basement-membrane autoantigen in epidermolysis bullosa acquisita. N Engl J Med 1984;310:1007–13.
- Komorowski L, Muller R, Vorobyev A, et al. Sensitive and specific assays for routine serological diagnosis of epidermolysis bullosa acquisita. J Am Acad Dermatol 2012;68:e89–95.
- Saleh MA, Ishii K, Kim YJ, et al. Development of NC1 and NC2 domains of type VII collagen ELISA for the diagnosis and analysis of the time course of epidermolysis bullosa acquisita patients. J Dermatol Sci 2011;62:169–75.
- Vodegel RM, Jonkman MF, Pas HH, de Jong MC. U-serrated immunodeposition pattern differentiates type VII collagen targeting bullous diseases from other subepidermal bullous autoimmune diseases. Br J Dermatol 2004;151:112–18.
- Ludwig RJ, Zillikens D. Pathogenesis of epidermolysis bullosa acquisita. Dermatol Clin 29:493–501, xi.
- Bernard P, Vaillant L, Labeille B, et al. Incidence and distribution of subepidermal autoimmune bullous skin diseases in three French regions. Bullous Diseases French Study Group. Arch Dermatol 1995;131:48–52.
- Bertram F, Brocker EB, Zillikens D, Schmidt E. Prospective analysis of the incidence of autoimmune bullous disorders in Lower Franconia, Germany. J Dtsch Dermatol Ges 2009;7:434–40.
- Zillikens D, Wever S, Roth A, Weidenthaler-Barth B, Hashimoto T, Brocker EB. Incidence of autoimmune subepidermal blistering dermatoses in a region of central Germany. Arch Dermatol 1995;131:957–8.
- Nanda A, Dvorak R, Al-Saeed K, Al-Sabah H, Alsaleh QA. Spectrum of autoimmune bullous diseases in Kuwait. Int J Dermatol 2004;43:876–81.
- Wong SN, Chua SH. Spectrum of subepidermal immunobullous disorders seen at the National Skin Centre, Singapore: a 2-year review. Br J Dermatol 2002;147:476–80.
- Buijsrogge JJ, Diercks GF, Pas HH, Jonkman MF. The many faces of epidermolysis bullosa acquisita after serration pattern analysis by direct immunofluorescence microscopy. Br J Dermatol 2011;165:92–8.
- Kim JH, Kim YH, Kim SC. Epidermolysis bullosa acquisita: a retrospective clinical analysis of 30 cases. Acta Derm Venereol 2011;91:307–12.
- Delgado JC, Turbay D, Yunis EJ, et al. A common major histocompatibility complex class II allele HLA-DQB1* 0301 is present in clinical variants of pemphigoid. Proc Natl Acad Sci U S A 1996;93:8569–71.
- Callot-Mellot C, Bodemer C, Caux F, et al. Epidermolysis bullosa acquisita in childhood. Arch Dermatol 1997;133:1122–6.
- Tanaka H, Ishida-Yamamoto A, Hashimoto T, et al. A novel variant of acquired epidermolysis bullosa with autoantibodies against the central triple-helical domain of type VII collagen. Lab Invest 1997;77:623–32.
- Bordier-Lamy F, Eschard C, Coste M, et al. [Epidermolysis bullosa acquisita of childhood.] Ann Dermatol Venereol 2009;136:513–17.
- Arpey CJ, Elewski BE, Moritz DK, Gammon WR. Childhood epidermolysis bullosa acquisita. Report of three cases and review of literature. J Am Acad Dermatol 1991;24:706–14.
- Edwards S, Wakelin SH, Wojnarowska F, et al. Bullous pemphigoid and epidermolysis bullosa acquisita: presentation, prognosis, and immunopathology in 11 children. Pediatr Dermatol 1998;15:184–90.
- Gammon WR, Heise ER, Burke WA, Fine JD, Woodley DT, Briggaman RA. Increased frequency of HLA-DR2 in patients with autoantibodies to epidermolysis bullosa acquisita antigen: evidence that the expression of autoimmunity to type VII collagen is HLA class II allele associated. J Invest Dermatol 1988;91:228–32.
- Zumelzu C, Le Roux-Villet C, Loiseau P, et al. Black patients of African descent and HLA-DRB1*15:03 frequency overrepresented in epidermolysis bullosa acquisita. J Invest Dermatol 2011;131:2386–93.
- Chen M, O'Toole EA, Sanghavi J, et al. The epidermolysis bullosa acquisita antigen (type VII collagen) is present in human colon and patients with Crohn's disease have autoantibodies to type VII collagen. J Invest Dermatol 2002;118:1059–64.
- Hundorfean G, Neurath MF, Sitaru C. Autoimmunity against type VII collagen in inflammatory bowel disease. J Cell Mol Med 14:2393–403.
- Oostingh GJ, Sitaru C, Zillikens D, Kromminga A, Luhrs H. Subclass distribution of type VII collagen-specific autoantibodies in patients with inflammatory bowel disease. J Dermatol Sci 2005;37:182–4.
- Ishii N, Recke A, Mihai S, et al. Autoantibody-induced intestinal inflammation and weight loss in experimental epidermolysis bullosa acquisita. J Pathol 2011;224:234–44.
- Ishii N, Hamada T, Dainichi T, et al. Epidermolysis bullosa acquisita: what's new? J Dermatol 2010;37:220–30.
- Chen M, Kim GH, Prakash L, Woodley DT. Epidermolysis bullosa acquisita: Autoimmunity to anchoring fibril collagen. Autoimmunity 2012;45:91–101.
- Ludwig RJ. Clinical presentation, pathogenesis, diagnosis, and treatment of epidermolysis bullosa acquisita. ISRN Dermatol 2013;2013:812029.
- Schulze F, Neumann K, Recke A, Zillikens D, Linder R, Schmidt E. Malignancies in pemphigus and pemphigoid diseases. J Invest Dermatol 2015;135:1445–7.
- Gammon WR, Murrell DF, Jenison MW, et al. Autoantibodies to type VII collagen recognize epitopes in a fibronectin-like region of the noncollagenous (NC1) domain. J Invest Dermatol 1993;100:618–22.
- Lapiere JC, Woodley DT, Parente MG, et al. Epitope mapping of type VII collagen. Identification of discrete peptide sequences recognized by sera from patients with acquired epidermolysis bullosa. J Clin Invest 1993;92:1831–9.
- Chen M, Doostan A, Bandyopadhyay P, et al. The cartilage matrix protein subdomain of type VII collagen is pathogenic for epidermolysis bullosa acquisita. Am J Pathol 2007;170:2009–18.
- Chen M, Chan LS, Cai X, O'Toole EA, Sample JC, Woodley DT. Development of an ELISA for rapid detection of anti-type VII collagen autoantibodies in epidermolysis bullosa acquisita. J Invest Dermatol 1997;108:68–72.
- Ishii N, Yoshida M, Hisamatsu Y, et al. Epidermolysis bullosa acquisita sera react with distinct epitopes on the NC1 and NC2 domains of type VII collagen: study using immunoblotting of domain-specific recombinant proteins and postembedding immunoelectron microscopy. Br J Dermatol 2004;150:843–51.
- Schmidt E, Hopfner B, Chen M, et al. Childhood epidermolysis bullosa acquisita: a novel variant with reactivity to all three structural domains of type VII collagen. Br J Dermatol 2002;147:592–7.
- Jonkman MF, Schuur J, Dijk F, et al. Inflammatory variant of epidermolysis bullosa acquisita with IgG autoantibodies against type VII collagen and laminin alpha3. Arch Dermatol 2000;136:227–31.
- Fairley JA, Woodley DT, Chen M, Giudice GJ, Lin MS. A patient with both bullous pemphigoid and epidermolysis bullosa acquisita: an example of intermolecular epitope spreading. J Am Acad Dermatol 2004;51:118–22.
- Furukawa H, Miura T, Takahashi M, et al. A case of anti-p200 pemphigoid with autoantibodies against both a novel 200-kD dermal antigen and the 290-kD epidermolysis bullosa acquisita antigen. Dermatology 2004;209:145–8.
- Abrams ML, Smidt A, Benjamin L, Chen M, Woodley D, Mancini AJ. Congenital epidermolysis bullosa acquisita: vertical transfer of maternal autoantibody from mother to infant. Arch Dermatol 2010;147:337–41.
- Kim JH, Kim YH, Kim S, et al. Serum levels of anti-type VII collagen antibodies detected by enzyme-linked immunosorbent assay in patients with epidermolysis bullosa acquisita are correlated with the severity of skin lesions. J Eur Acad Dermatol Venereol 2012;27:e224–30.
- Borradori L, Caldwell JB, Briggaman RA, et al. Passive transfer of autoantibodies from a patient with mutilating epidermolysis bullosa acquisita induces specific alterations in the skin of neonatal mice. Arch Dermatol 1995;131:590–5.
- Sitaru C, Chiriac MT, Mihai S, Buning J, Gebert A, Ishiko A, Zillikens D. Induction of complement-fixing autoantibodies against type VII collagen results in subepidermal blistering in mice. J Immunol 2006;177:3461–8.
- Sitaru C, Kromminga A, Hashimoto T, Brocker EB, Zillikens D. Autoantibodies to type VII collagen mediate Fcgamma-dependent neutrophil activation and induce dermal-epidermal separation in cryosections of human skin. Am J Pathol 2002;161:301–11.
- Sitaru C, Mihai S, Otto C, et al. Induction of dermal–epidermal separation in mice by passive transfer of antibodies specific to type VII collagen. J Clin Invest 2005;115:870–8.
- Woodley DT, Chang C, Saadat P, Ram R, Liu Z, Chen M. Evidence that anti-type VII collagen antibodies are pathogenic and responsible for the clinical, histological, and immunological features of epidermolysis bullosa acquisita. J Invest Dermatol 2005;124:958–64.
- Woodley DT, Ram R, Doostan A, et al. Induction of epidermolysis bullosa acquisita in mice by passive transfer of autoantibodies from patients. J Invest Dermatol 2006;126:1323–30.
- Sitaru AG, Sesarman A, Mihai S, et al. T cells are required for the production of blister-inducing autoantibodies in experimental epidermolysis bullosa acquisita. J Immunol 2010;184:1596–603.
- Kasperkiewicz M, Muller R, Manz R, et al. Heat-shock protein 90 inhibition in autoimmunity to type VII collagen: evidence that nonmalignant plasma cells are not therapeutic targets. Blood 2011;117:6135–42.
- Muller R, Dahler C, Mobs C, et al. T and B cells target identical regions of the non-collagenous domain 1 of type VII collagen in epidermolysis bullosa acquisita. Clin Immunol 2010;135:99–107.
- Kasperkiewicz M, Nimmerjahn F, Wende S, et al. Genetic identification and functional validation of FcgammaRIV as key molecule in autoantibody-induced tissue injury. J Pathol 2012;228:8–19.
- Sadeghi H, Lockmann A, Hund AC, et al. Caspase-1-Independent IL-1 Release mediates blister formation in autoantibody-induced tissue injury through modulation of endothelial adhesion molecules. J Immunol 2015;194:3656–63.
- Iwata H, Pipi E, Mockel N, et al. Recombinant soluble CD32 suppresses disease progression in experimental epidermolysis bullosa acquisita. J Invest Dermatol 2015;135:916–19.
- Karsten CM, Pandey MK, Figge J, et al. Anti-inflammatory activity of IgG1 mediated by Fc galactosylation and association of FcgammaRIIB and dectin-1. Nat Med 2012;18:1401–6.
- Hirose M, Vafia K, Kalies K, et al. Enzymatic autoantibody glycan hydrolysis alleviates autoimmunity against type VII collagen. J Autoimmun 2012;39:304–14.
- Mihai S, Chiriac MT, Takahashi K, et al. The alternative pathway of complement activation is critical for blister induction in experimental epidermolysis bullosa acquisita. J Immunol 2007;178:6514–21.
- Samavedam UK, Kalies K, Scheller J, et al. Recombinant IL-6 treatment protects mice from organ specific autoimmune disease by IL-6 classical signalling-dependent IL-1ra induction. J Autoimmun 2012;40:74–85.
- Chiriac MT, Roesler J, Sindrilaru A, Scharffetter-Kochanek K, Zillikens D, Sitaru C. NADPH oxidase is required for neutrophil-dependent autoantibody-induced tissue damage. J Pathol 2007;212:56–65.
- Shimanovich I, Bohmke AK, Westermann L, Kramer J, Zillikens D, Schmidt E. Successful treatment of severe pemphigus with the combination of immunoadsorption and rituximab. J Invest Dermatol 2009;129:S97.
- Kasprick A, Yu X, Scholten J, et al. Conditional depletion of mast cells has no impact on the severity of experimental epidermolysis bullosa acquisita. Eur J Immunol 2015;45:1462–70.
- Ludwig RJ, Kalies K, Kohl J, Zillikens D, Schmidt E. Emerging treatments for pemphigoid diseases. Trends Mol Med 2013;19:501–12.
- Delbaldo C, Chen M, Friedli A, et al. Drug-induced epidermolysis bullosa acquisita with antibodies to type VII collagen. J Am Acad Dermatol 2002;46:S161–4.
- Jappe U, Zillikens D, Bonnekoh B, Gollnick H. Epidermolysis bullosa acquisita with ultraviolet radiationsensitivity. Br J Dermatol 2000;142:517–20.
- Baican A, Chiriac G, Baican C, et al. Metal sensitization precipitates skin blistering in epidermolysis bullosa acquisita. J Dermatol 2010;37:280–2.
- Wakelin SH, Allen J, Zhou S, Wojnarowska F. Drug-induced linear IgA disease with antibodies to collagen VII. Br J Dermatol 1998;138:310–14.
- Fine JD, Tyring S, Gammon WR. The presence of intra-lamina lucida blister formation in epidermolysis bullosa acquisita: possible role of leukocytes. J Invest Dermatol 1989;92:27–32.
- De Jong MC, Bruins S, Heeres K, et al. Bullous pemphigoid and epidermolysis bullosa acquisita. Differentiation by fluorescence overlay antigen mapping. Arch Dermatol 1996;132:151–7.
- Wozniak K, Kazama T, Kowalewski C. A practical technique for differentiation of subepidermal bullous diseases: localization of in vivo-bound IgG by laser scanning confocal microscopy. Arch Dermatol 2003;139:1007–11.
- Gammon WR, Kowalewski C, Chorzelski TP, Kumar V, Briggaman RA, Beutner EH. Direct immunofluorescence studies of sodium chloride-separated skin in the differential diagnosis of bullous pemphigoid and epidermolysis bullosa acquisita. J Am Acad Dermatol 1990;22:664–70.
- Allen J, Zhou S, Wakelin SH, Collier PM, Wojnarowska F. Linear IgA disease: a report of two dermal binding sera which recognize a pepsin-sensitive epitope (?NC-1 domain) of collagen type VII. Br J Dermatol 1997;137:526–33.
- Vodegel RM, de Jong MC, Pas HH, Jonkman MF. IgA-mediated epidermolysis bullosa acquisita: two cases and review of the literature. J Am Acad Dermatol 2002;47:919–25.
- Hashimoto T, Ishiko A, Shimizu H, et al. A case of linear IgA bullous dermatosis with IgA anti-type VII collagen autoantibodies. Br J Dermatol 1996;134:336–9.
- Zambruno G, Manca V, Kanitakis J, Cozzani E, Nicolas JF, Giannetti A. Linear IgA bullous dermatosis with autoantibodies to a 290 kd antigen of anchoring fibrils. J Am Acad Dermatol 1994;31:884–8.
- Lee CW, Kim SC, Han H. Distribution of HLA class II alleles in Korean patients with epidermolysis bullosa acquisita. Dermatology 1996;193:328–9.
- Ludwig RJ, Muller S, Marques A, et al. Identification of quantitative trait loci in experimental epidermolysis bullosa acquisita. J Invest Dermatol 2012;132:1409–15.
- Ludwig RJ, Recke A, Bieber K, et al. Generation of antibodies of distinct subclasses and specificity is linked to H2s in an active mouse model of epidermolysis bullosa acquisita. J Invest Dermatol 2011;131:167–76.
- Zhu XJ, Niimi Y, Bystryn JC. Epidermolysis bullosa acquisita. Incidence in patients with basement membrane zone antibodies. Arch Dermatol 1990;126:171–4.
- Briggaman RA, Gammon WR, Woodley DT. Epidermolysis bullosa acquisita of the immunopathological type (dermolytic pemphigoid). J Invest Dermatol 1985;85:79s–84s.
- Stewart MI, Woodley DT, Briggaman RA. Epidermolysis bullosa acquisita and associated symptomatic esophageal webs. Arch Dermatol 1991;127:373–7.
- Wieme N, Lambert J, Moerman M, Geerts ML, Temmerman L, Naeyaert JM. Epidermolysis bullosa acquisita with combined features of bullous pemphigoid and cicatricial pemphigoid. Dermatology 1999;198:310–13.
- Chen M, Kim GH, Prakash L, Woodley DT. Epidermolysis bullosa acquisita: autoimmunity to anchoring fibril collagen. Autoimmunity 45:91–101.
- Chan LS, Ahmed AR, Anhalt GJ, et al. The first international consensus on mucous membrane pemphigoid: definition, diagnostic criteria, pathogenic factors, medical treatment, and prognostic indicators. Arch Dermatol 2002;138:370–9.
- Brunsting LA, Perry HO. Benign pemphigold; a report of seven cases with chronic, scarring, herpetiform plaques about the head and neck. AMA Arch Derm 1957;75:489–501.
- Kurzhals G, Stolz W, Meurer M, Kunze J, Braun-Falco O, Krieg T. Acquired epidermolysis bullosa with the clinical feature of Brunsting-Perry cicatricial bullous pemphigoid. Arch Dermatol 1991;127:391–5.
- Tanaka N, Dainichi T, Ohyama B, et al. A case of epidermolysis bullosa acquisita with clinical features of Brunsting–Perry pemphigoid showing an excellent response to colchicine. J Am Acad Dermatol 2009;61:715–19.
- Minato H, Ishii N, Fukuda S, et al. Heterogeneity of Brunsting–Perry type pemphigoid: a case showing blister formation at the lamina lucida, immune deposition beneath the lamina densa and autoantibodies against the 290-kD polypeptide along the lamina densa. J Dermatol 2011;38:887–92.
- Schmidt E, Zillikens D. Pemphigoid diseases. Lancet 2013;381:320–32.
- Lee CW. Serum IgA autoantibodies in patients with epidermolysis bullosa acquisita: a high frequency of detection. Dermatology 2000;200:83–4.
- Lee CW. An extract of cultured A431 cells contains major tissue antigens of autoimmune bullous diseases. Br J Dermatol 2000;143:821–3.
- Oyama N, Bhogal BS, Carrington P, Gratian MJ, Black MM. Human placental amnion is a novel substrate for detecting autoantibodies in autoimmune bullous diseases by immunoblotting. Br J Dermatol 2003;148:939–44.
- Vodegel RM, Kiss M, Cjm De Jong M, et al. The use of skin substrates deficient in basement membrane molecules for the diagnosis of subepidermal autoimmune bullous disease. Eur J Dermatol 1998;8:83–5.
- Prost C, Dubertret L, Fosse M, Wechsler J, Touraine R. A routine immuno-electron microscopic technique for localizing an auto-antibody on epidermal basement membrane. Br J Dermatol 1984;110:1–7.
- Kirtschig G, Murrell DF, Wojnarowska F, Khumalo NP. Interventions for mucous membrane pemphigoid and epidermolysis bullosa acquisita. Cochrane Database Syst Rev 2009:CD004056.
- Engineer L, Ahmed AR. Emerging treatment for epidermolysis bullosa acquisita. J Am Acad Dermatol 2001;44:818–28.
- Intong LR, Murrell DF. Management of epidermolysis bullosa acquisita. Dermatol Clin 2011;29:643–7.
- Prost-Squarcioni C, Ingen-Housz-Oro S, Joly P, Bernard P, Bedane C. [Epidermolysis bullosa acquisita. Guidelines for the diagnosis and treatment. Centres de reference des maladies bulleuses auto-immunes. Societe Francaise de Dermatologie.] Ann Dermatol Venereol 2011;138:274–9.
- Cunningham BB, Kirchmann TT, Woodley D. Colchicine for epidermolysis bullosa acquisita. J Am Acad Dermatol 1996;34:781–4.
- Megahed M, Scharffetter-Kochanek K. Epidermolysis bullosa acquisita – successful treatment with colchicine. Arch Dermatol Res 1994;286:35–46.
- Berbis P, Privat Y. [Value of colchicine in treating acquired epidermolysis bullosa.] Ann Dermatol Venereol 1989;116:301–7.
- Egan CA, Brown M, White JD, Yancey KB. Treatment of epidermolysis bullosa acquisita with the humanized anti-Tac mAb daclizumab. Clin Immunol 2001;101:146–51.
- Haufs MG, Haneke E. Epidermolysis bullosa acquisita treated with basiliximab, an interleukin-2 receptor antibody. Acta Derm Venereol 2001;81:72.
- Schmidt E, Zillikens D. Immunoadsorption in dermatology. Arch Dermatol Res 2010;302:241–53.
- Ishii N, Hashimoto K, Zillikens D, Ludwig RJ. High-dose intravenous immunoglobulin (IVIG) therapy in autoimmune skin blistering diseases. Ann N Y Acad Sci 2010;38:186–95.
- Schmidt E, Benoit S, Brocker EB, Zillikens D, Goebeler M. Successful adjuvant treatment of recalcitrant epidermolysis bullosa acquisita with anti-CD20 antibody rituximab. Arch Dermatol 2006;142:147–50.
- Schmidt E, Goebeler M. CD20-directed therapy in autoimmune diseases involving the skin: role of rituximab. Exp Rev Dermatol 2008;3:259–78.
- Gurcan HM, Ahmed AR. Current concepts in the treatment of epidermolysis bullosa acquisita. Exp Opin Pharmacother 2011;12:1259–68.
Bullous systemic lupus erythematosus
- Camisa C, Neff JC, Rossana C, Barrett JL. Bullous lichen planus: diagnosis by indirect immunofluorescence and treatment with dapsone. J Am Acad Dermatol 1986;14:464–9.
- Camisa C. Vesiculobullous systemic lupus erythematosus. A report of four cases. J Am Acad Dermatol 1988;18:93–100.
- Camisa C, Sharma HM. Vesiculobullous systemic lupus erythematosus. Report of two cases and a review of the literature. J Am Acad Dermatol 1983;9:924–33.
- Gammon WR, Woodley DT, Dole KC, Briggaman RA. Evidence that anti-basement membrane zone antibodies in bullous eruption of systemic lupus erythematosus recognize epidermolysis bullosa acquisita autoantigen. J Invest Dermatol 1985;84:472–6.
- Yell JA, Allen J, Wojnarowska F, Kirtschig G, Burge SM. Bullous systemic lupus erythematosus: revised criteria for diagnosis. Br J Dermatol 1995;132:921–8.
- Gammon WR, Briggaman RA. Bullous SLE: a phenotypically distinctive but immunologically heterogeneous bullous disorder. J Invest Dermatol 1993;100:28S–34S.
- Hall RP, Lawley TJ, Smith HR, Katz SI. Bullous eruption of systemic lupus erythematosus. Dramatic response to dapsone therapy. Ann Intern Med 1982;97:165–70.
- Woodley DT, Burgeson RE, Lunstrum G, Bruckner-Tuderman L, Reese MJ, Briggaman RA. Epidermolysis bullosa acquisita antigen is the globular carboxyl terminus of type VII procollagen. J Clin Invest 1988;81:683-7.
- Chan LS, Lapiere JC, Chen M, et al. Bullous systemic lupus erythematosus with autoantibodies recognizing multiple skin basement membrane components, bullous pemphigoid antigen 1, laminin-5, laminin-6, and type VII collagen. Arch Dermatol 1999;135:569–73.
- Doebelin B, Dalle S, Balme B, Kanitakis J, Thomas L. Bullous systemic lupus erythematosus with autoantibodies recognizing bullous pemphigoid antigen 1. Br J Dermatol 2005;153:232–3.
- Sebaratnam DF, Murrell DF. Bullous systemic lupus erythematosus. Dermatol Clin 2011;29:649–53.
- Loche F, Bernard P, Bazex J. Bullous pemphigoid associated with systemic lupus erythematosus. Br J Dermatol 1998;139:927–8.
- Tobon GJ, Toro CE, Bravo JC, Canas CA. Linear IgA bullous dermatosis associated with systemic lupus erythematosus: a case report. Clin Rheumatol 2008;27:391–3.
- Pedro SD, Dahl MV. Direct immunofluorescence of bullous systemic lupus erythematosus. Arch Dermatol 1973;107:118–20.
- Schmidt E, Zillikens D. Modern diagnosis of autoimmune blistering skin diseases. Autoimmun Rev 2010;10:84–9.
- Buijsrogge JJ, Diercks GF, Pas HH, Jonkman MF. The many faces of epidermolysis bullosa acquisita after serration pattern analysis by direct immunofluorescence microscopy. Br J Dermatol 2011;165:92–8.
- Vodegel RM, Jonkman MF, Pas HH, de Jong MC. U-serrated immunodeposition pattern differentiates type VII collagen targeting bullous diseases from other subepidermal bullous autoimmune diseases. Br J Dermatol 2004;151:112–18.
- Bernard P, Vaillant L, Labeille B, et al. Incidence and distribution of subepidermal autoimmune bullous skin diseases in three French regions. Bullous Diseases French Study Group. Arch Dermatol 1995;131:48–52.
- Bertram F, Brocker EB, Zillikens D, Schmidt E. Prospective analysis of the incidence of autoimmune bullous disorders in Lower Franconia, Germany. J Dtsch Dermatol Ges 2009;7:434–40.
- Zillikens D, Wever S, Roth A, Weidenthaler-Barth B, Hashimoto T, Brocker EB. Incidence of autoimmune subepidermal blistering dermatoses in a region of central Germany. Arch Dermatol 1995;131:957–8.
- Nanda A, Dvorak R, Al-Saeed K, Al-Sabah H, Alsaleh QA. Spectrum of autoimmune bullous diseases in Kuwait. Int J Dermatol 2004;43:876–81.
- Wong SN, Chua SH. Spectrum of subepidermal immunobullous disorders seen at the National Skin Centre, Singapore: a 2-year review. Br J Dermatol 2002;147:476–80.
- Yell JA, Mbuagbaw J, Burge SM. Cutaneous manifestations of systemic lupus erythematosus. Br J Dermatol 1996;135:355–62.
- Cardinali C, Caproni M, Bernacchi E, Amato L, Fabbri P. The spectrum of cutaneous manifestations in lupus erythematosus – the Italian experience. Lupus 2000;9:417–23.
- Gammon WR, Heise ER, Burke WA, Fine JD, Woodley DT, Briggaman RA. Increased frequency of HLA-DR2 in patients with autoantibodies to epidermolysis bullosa acquisita antigen: evidence that the expression of autoimmunity to type VII collagen is HLA class II allele associated. J Invest Dermatol 1988;91:228–32.
- Kole AK, Ghosh A. Cutaneous manifestations of systemic lupus erythematosus in a tertiary referral center. Indian J Dermatol 2009;54:132–6.
- Vassileva S. Bullous systemic lupus erythematosus. Clin Dermatol 2004;22:129–38.
- Gammon WR, Murrell DF, Jenison MW, et al. Autoantibodies to type VII collagen recognize epitopes in a fibronectin-like region of the noncollagenous (NC1) domain. J Invest Dermatol 1993;100:618–22.
- Borradori L, Caldwell JB, Briggaman RA, et al. Passive transfer of autoantibodies from a patient with mutilating epidermolysis bullosa acquisita induces specific alterations in the skin of neonatal mice. Arch Dermatol 1995;131:590–5.
- Sitaru C, Chiriac MT, Mihai S, et al. Induction of complement-fixing autoantibodies against type VII collagen results in subepidermal blistering in mice. J Immunol 2006;177:3461–8.
- Sitaru C, Kromminga A, Hashimoto T, Brocker EB, Zillikens D. Autoantibodies to type VII collagen mediate Fcgamma-dependent neutrophil activation and induce dermal-epidermal separation in cryosections of human skin. Am J Pathol 2002;161:301–11.
- Sitaru C, Mihai S, Otto C, et al. Induction of dermal–epidermal separation in mice by passive transfer of antibodies specific to type VII collagen. J Clin Invest 2005;115:870–8.
- Woodley DT, Chang C, Saadat P, Ram R, Liu Z, Chen M. Evidence that anti-type VII collagen antibodies are pathogenic and responsible for the clinical, histological, and immunological features of epidermolysis bullosa acquisita. J Invest Dermatol 2005;124:958–64.
- Woodley DT, Ram R, Doostan A, et al. Induction of epidermolysis bullosa acquisita in mice by passive transfer of autoantibodies from patients. J Invest Dermatol 2006;126:1323–30.
- Chen M, Doostan A, Bandyopadhyay P, et al. The cartilage matrix protein subdomain of type VII collagen is pathogenic for epidermolysis bullosa acquisita. Am J Pathol 2007;170:2009–18.
- Ishikawa O, Zaw KK, Miyachi Y, Hashimoto T, Tanaka T. The presence of anti-basement membrane zone antibodies in the sera of patients with non-bullous lupus erythematosus. Br J Dermatol 1997;136:222–6.
- Condon C, Phelan M, Lyons JF. Penicillamine-induced type II bullous systemic lupus erythematosus. Br J Dermatol 1997;136:474–5.
- Fleming MG, Bergfeld WF, Tomecki KJ, et al. Bullous systemic lupus erythematosus. Int J Dermatol 1989;28:321–6.
- Seo JY, Byun HJ, Cho KH, Lee EB. Methimazole-induced bullous systemic lupus erythematosus: a case report. J Korean Med Sci 2012;27:818–21.
- Nasongkhla P, Pratchyapruit W, Tagami H. Bullous systemic lupus erythematosus induced by UVB: report a case. J Med Assoc Thai 2012;95:969–73.
- Burrows NP, Bhogal BS, Black MM, et al. Bullous eruption of systemic lupus erythematosus: a clinicopathological study of four cases. Br J Dermatol 1993;128:332–8.
- Tsuchida T, Furue M, Kashiwado T, Ishibashi Y. Bullous systemic lupus erythematosus with cutaneous mucinosis and leukocytoclastic vasculitis. J Am Acad Dermatol 1994;31:387–90.
- Rappersberger K, Tschachler E, Tani M, Wolff K. Bullous disease in systemic lupus erythematosus. J Am Acad Dermatol 1989;21:745–52.
- Lalova A, Pramatarov K, Vassileva S. Facial bullous systemic lupus erythematosus. Int J Dermatol 1997;36:369–71.
- Piette EW, Werth VP. Dapsone in the management of autoimmune bullous diseases. Dermatol Clin 2011;29:561–4.
- Hamminga EA, Vermeer MH. Bullous systemic lupus erythematosus responding to mycophenolate mofetil. Eur J Dermatol 2010;20:844–5.
- Fernandez-Sanchez M, Charli-Joseph Y, Saeb-Lima M, Garcia-Hidalgo L, Orozco-Topete R. Are corticosteroids and immunosuppressors an efficacious treatment for bullous lupus erythematosus with systemic manifestations? Eur J Dermatol 2010;20:823–5.
- Alsanafi S, Kovarik C, Mermelstein AL, Werth VP. Rituximab in the treatment of bullous systemic lupus erythematosus. J Clin Rheumatol 2011;17:142–4.
- Malcangi G, Brandozzi G, Giangiacomi M, Zampetti M, Danieli MG. Bullous SLE: response to methotrexate and relationship with disease activity. Lupus 2003;12:63–6.
Very rare pemphigoid disorders
- Zaraa I, Mahfoudh A, Sellami MK, et al. Lichen planus pemphigoides: four new cases and a review of the literature. Int J Dermatol 2013;52:406–12.
- Davis AL, Bhogal BS, Whitehead P, et al. Lichen planus pemphigoides: its relationship to bullous pemphigoid. Br J Dermatol 1991;125:263–71.
- Willsteed E, Bhogal BS, Das AK, Wojnarowska F, Black MM, McKee PH. Lichen planus pemphigoides: a clinicopathological study of nine cases. Histopathology 1991;19:147–54.
- Zillikens D, Caux F, Mascaro JM, et al. Autoantibodies in lichen planus pemphigoides react with a novel epitope within the C-terminal NC16A domain of BP180. J Invest Dermatol 1999;113:117–21.
- Schmidt E, Zillikens D. Pemphigoid diseases. Lancet 2013;381:320–32.
- Chan LS, Ahmed AR, Anhalt GJ, et al. The first international consensus on mucous membrane pemphigoid: definition, diagnostic criteria, pathogenic factors, medical treatment, and prognostic indicators. Arch Dermatol 2002;138:370–9.
- Brunsting LA, Perry HO. Benign pemphigold; a report of seven cases with chronic, scarring, herpetiform plaques about the head and neck. AMA Arch Derm 1957;75:489–501.
- Joly P, Ruto F, Thomine E, et al. Brunsting–Perry cicatricial bullous pemphigoid: a clinical variant of localized acquired epidermolysis bullosa? J Am Acad Dermatol 1993;28:89–92.
- Kurzhals G, Stolz W, Meurer M, Kunze J, Braun-Falco O, Krieg T. Acquired epidermolysis bullosa with the clinical feature of Brunsting–Perry cicatricial bullous pemphigoid. Arch Dermatol 1991;127:391–5.
- Umemoto N, Demitsu T, Toda S, et al. A case of nonscarring subepidermal blistering disease associated with autoantibodies reactive with both type VII collagen and laminin 5. Dermatology 2003;207:61–4.
- Fukuda S, Tsuruta D, Uchiyama M, et al. Brunsting–Perry type pemphigoid with IgG autoantibodies to laminin-332, BP230 and desmoplakins I/II. Br J Dermatol 2011;165:433–5.
- Jedlickova H, Niedermeier A, Zgazarova S, Hertl M. Brunsting–Perry pemphigoid of the scalp with antibodies against laminin 332. Dermatology 2011;222:193–5.
- Minato H, Ishii N, Fukuda S, et al. Heterogeneity of Brunsting–Perry type pemphigoid: a case showing blister formation at the lamina lucida, immune deposition beneath the lamina densa and autoantibodies against the 290-kD polypeptide along the lamina densa. J Dermatol 2011;38:887–92.
- Tanaka N, Dainichi T, Ohyama B, et al. A case of epidermolysis bullosa acquisita with clinical features of Brunsting–Perry pemphigoid showing an excellent response to colchicine. J Am Acad Dermatol 2009;61:715–19.
- Daito J, Katoh N, Asai J, et al. Brunsting–Perry cicatricial pemphigoid associated with autoantibodies to the C-terminal domain of BP180. Br J Dermatol 2008;159:984–6.
- Demitsu T, Kakurai M, Yoneda K, et al. Localized pemphigoid (Brunsting–Perry type) with IgG antibody to BP180 NC16a domain resembling lupus erythematosus successfully treated with topical tacrolimus therapy. J Eur Acad Dermatol Venereol 2009;23:79–80.
- Buijsrogge JJ, Diercks GF, Pas HH, Jonkman MF. The many faces of epidermolysis bullosa acquisita after serration pattern analysis by direct immunofluorescence microscopy. Br J Dermatol 2011;165:92–8.
- Kim JH, Kim YH, Kim SC. Epidermolysis bullosa acquisita: a retrospective clinical analysis of 30 cases. Acta Derm Venereol 2011;91:307–12.
- Garcia-Martin P, Fraga J, Hashimoto T, Garcia-Diez A. Brunsting–Perry-type cicatricial pemphigoid with IgG autoantibodies to LAD-1. Br J Dermatol 2014;170:743–5.
- Leenutaphong V, von Kries R, Plewig G. Localized cicatricial pemphigoid (Brunsting–Perry): electron microscopic study. J Am Acad Dermatol 1989;21:1089–93.
- Baldwin H, Lynfield Y. Brunsting–Perry cicatricial pemphigoid precipitated by trauma. Arch Dermatol 1991;127:911–12.
- Takeichi S, Kubo Y, Arase S, Hashimoto T, Ansai S. Brunsting–Perry type localized bullous pemphigoid, possibly induced by furosemide administration and sun exposure. Eur J Dermatol 2009;19:500–3.
- Monihan JM, Nguyen TH, Guill MA. Brunsting–Perry pemphigoid simulating basal cell carcinoma. J Am Acad Dermatol 1989;21:331–4.
- Mittal RR, Kullar J, Sethi PS. Brunsting perry cicatricial pemphigoid. Indian J Dermatol Venereol Leprol 2000;66:304–5.
- Ghohestani RF, Hudson BG, Claudy A, Uitto J. The alpha 5 chain of type IV collagen is the target of IgG autoantibodies in a novel autoimmune disease with subepidermal blisters and renal insufficiency. J Biol Chem 2000;275:16002–6.
- Ghohestani RF, Rotunda SL, Hudson B, et al. Crescentic glomerulonephritis and subepidermal blisters with autoantibodies to alpha5 and alpha6 chains of type IV collagen. Lab Invest 2003;83:605–11.
- Chan LS, Fine JD, Briggaman RA, et al. Identification and partial characterization of a novel 105-kDalton lower lamina lucida autoantigen associated with a novel immune-mediated subepidermal blistering disease. J Invest Dermatol 1993;101:262–7.
- Cotell SL, Lapiere JC, Chen JD, et al. A novel 105-kDa lamina lucida autoantigen: association with bullous pemphigoid. J Invest Dermatol 1994;103:78–83.
- Chan LS, Cooper KD. A novel immune-mediated subepidermal bullous dermatosis characterized by IgG autoantibodies to a lower lamina lucida component. Arch Dermatol 1994;130:343–7.
- Chan LS, Wang XS, Lapiere JC, et al. A newly identified 105-kD lower lamina lucida autoantigen is an acidic protein distinct from the 105-kD gamma 2 chain of laminin-5. J Invest Dermatol 1995;105:75–9.
- Chan LS, Woodley DT. The 105-kDa basement membrane autoantigen p105 is N-terminally homologous to a tumor-associated antigen. J Invest Dermatol 1996;107:209–14.
- Mulyowa GK, Jaeger G, Sitaru C, Brocker EB, Zillikens D, Schmidt E. Scarring autoimmune bullous disease in a Ugandan patient with autoantibodies to BP180, BP230, and laminin 5. J Am Acad Dermatol 2006;54:S43–6.
- Murrell DF, Werth VP, Segall J, Zrnchik W, Stuart M, Sirois D. The International Pemphigus and Pemphigoid Foundation. Dermatol Clin 2011;29:655–7.
Other immunobullous diseases
Dermatitis herpetiformis
- Duhring LA. Landmark article, Aug 30, 1884: Dermatitis herpetiformis, by Louis A. Duhring. JAMA. 1983; 5 p.
- Bolotin D, Petronic-Rosic V. Dermatitis herpetiformis. Part I. Epidemiology, pathogenesis, and clinical presentation. J Am Acad Dermatol 2011;64:1017–24, quiz 1025–6.
- Bolotin D, Petronic-Rosic V. Dermatitis herpetiformis. Part II. Diagnosis, management, and prognosis. J Am Acad Dermatol 2011;64:1027–33, quiz 1033–4.
- Lundin KEA, Sollid LM. Advances in coeliac disease. Curr Opin Gastroenterol 2014;30:154–62.
- Guandalini S, Assiri A. Celiac disease: a review. JAMA Pediatr 2014;168:272–8.
- Moi H. Incidence and prevalence of dermatitis herpetiformis in a country in central Sweden, with comments on the course of the disease and IgA deposits as diagnostic criterion. Acta Derm Venereol 1984;64:144–50.
- Mobacken H, Kastrup W, Nilsson LA. Incidence and prevalence of dermatitis herpetiformis in western Sweden. Acta Derm Venereol 1984;64:400–4.
- West J, Fleming KM, Tata LJ, Card TR, Crooks CJ. Incidence and prevalence of celiac disease and dermatitis herpetiformis in the UK over two decades: population-based study. Am J Gastroenterol 2014;109:757–68.
- Hervonen K, Alakoski A, Salmi TT, et al. Reduced mortality in dermatitis herpetiformis: a population-based study of 476 patients. Br J Dermatol 2012;167:1331–7.
- Kárpáti S. Dermatitis herpetiformis. Clin Dermatol 2012;30:56–9.
- Smith JB, Tulloch JE, Meyer LJ, Zone JJ. The incidence and prevalence of dermatitis herpetiformis in Utah. Arch Dermatol 1992;128:1608–10.
- Hall RP, Clark RE, Ward FE. Dermatitis herpetiformis in two American blacks: HLA type and clinical characteristics. J Am Dermatol 1990;22:436–9.
- Shibahara M, Nanko H, Shimizu M, et al. Dermatitis herpetiformis in Japan: an update. Dermatology 2002;204:37–42.
- Ohata C, Ishii N, Hamada T, et al. Distinct characteristics in Japanese dermatitis herpetiformis: a review of all 91 Japanese patients over the last 35 years. Clin Dev Immunol 2012;2012:562168–9.
- Collin P, Reunala T. Recognition and management of the cutaneous manifestations of celiac disease: a guide for dermatologists. Am J Clin Dermatol 2003;4:13–20.
- Kaplan RP, Callen JP. Dermatitis herpetiformis: autoimmune disease associations. Clin Dermatol 1991;9:347–60.
- Hervonen K, Vornanen M, Kautiainen H, Collin P, Reunala T. Lymphoma in patients with dermatitis herpetiformis and their first-degree relatives. Br J Dermatol 2005;152:82–6.
- Reunala T, Collin P. Diseases associated with dermatitis herpetiformis. Br J Dermatol 1997;136:315–18.
- Gaspari AA, Huang CM, Davey RJ, Bondy C, Lawley TJ, Katz SI. Prevalence of thyroid abnormalities in patients with dermatitis herpetiformis and in control subjects with HLA-B8/-DR3. Am J Med 1990;88:145–50.
- Hervonen K, Viljamaa M, Collin P, Knip M, Reunala T. The occurrence of type 1 diabetes in patients with dermatitis herpetiformis and their first-degree relatives. Br J Dermatol 2004;150:136–8.
- Reunala T, Salmi J, Karvonen J. Dermatitis herpetiformis and celiac disease associated with Addison's disease. Arch Dermatol 1987;123:930–2.
- Dieterich W, Ehnis T, Bauer M, et al. Identification of tissue transglutaminase as the autoantigen of celiac disease. Nat Med 1997;3:797–801.
- Sárdy M, Kárpáti S, Merkl B, Paulsson M, Smyth N. Epidermal transglutaminase (TGase 3) is the autoantigen of dermatitis herpetiformis. J Exp Med 2002 Mar 18;195:747–57.
- Hitomi K. Transglutaminases in skin epidermis. Eur J Dermatol 2005;15:313–19.
- Nakajima K. Recent advances in dermatitis herpetiformis. Clin Dev Immunol 2012;2012:914162–4.
- Kárpáti S. Dermatitis herpetiformis: close to unravelling a disease. J Dermatol Sci 2004;34:83–90.
- Balas A, Vicario JL, Zambrano A, Acuña D, García-Novo D. Absolute linkage of celiac disease and dermatitis herpetiformis to HLA-DQ. Tissue Antigens 1997;50:52–6.
- Spurkland A, Ingvarsson G, Falk ES, Knutsen I, Sollid LM, Thorsby E. Dermatitis herpetiformis and celiac disease are both primarily associated with the HLA-DQ (alpha 1*0501, beta 1*02) or the HLA-DQ (alpha 1*03, beta 1*0302) heterodimers. Tissue Antigens 1997;49:29–34.
- Marietta E, Black K, Camilleri M, et al. A new model for dermatitis herpetiformis that uses HLA-DQ8 transgenic NOD mice. J Clin Invest 2004;114:1090–7.
- Black KE, Murray JA, David CS. HLA-DQ determines the response to exogenous wheat proteins: a model of gluten sensitivity in transgenic knockout mice. J Immunol 2002 Nov 15;169:5595–600.
- Hervonen K, Hakanen M, Kaukinen K, Collin P, Reunala T. First-degree relatives are frequently affected in coeliac disease and dermatitis herpetiformis. Scand J Gastroenterol 2002;37:51–5.
- Reunala T. Incidence of familial dermatitis herpetiformis. Br J Dermatol 1996;134:394–8.
- Hervonen K, Karell K, Holopainen P, Collin P, Partanen J, Reunala T. Concordance of dermatitis herpetiformis and celiac disease in monozygous twins. J Invest Dermatol 2000;115:990–3.
- Molberg O, Mcadam SN, Körner R, et al. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nat Med 1998;4:713–17.
- Haffenden GP, Blenkinsopp WK, Ring NP, Wojnarowska F, Fry L. The potassium iodide patch test in the dermatitis herpetiformis in relation to treatment with a gluten-free diet and dapsone. Br J Dermatol 1980;103:313–17.
- Lear JT, English JS, Jones PW. Adult coeliac disease, dermatitis herpetiformis and smoking. Gut 1997;40:289.
- Smith JB, Smith SB, Zone JJ. Dermatitis herpetiformis and cigarette smoking. Gut 1998;43:732.
- Antiga E, Verdelli A, Calabrò A, Fabbri P, Caproni M. Clinical and immunopathological features of 159 patients with dermatitis herpetiformis: an Italian experience. G Ital Dermatol Venereol 2013;148:163–9.
- Marks R, Jones EW. Purpura in dermatitis herpetiformis. Br J Dermatol 1971;84:386–8.
- Tu H, Parmentier L, Stieger M, et al. Acral purpura as leading clinical manifestation of dermatitis herpetiformis: report of two adult cases with a review of the literature. Dermatology 2013;227:1–4.
- Lähteenoja H, Irjala K, Viander M, Vainio E, Toivanen A, Syrjänen S. Oral mucosa is frequently affected in patients with dermatitis herpetiformis. Arch Dermatol 1998;134:756–8.
- Aine L, Mäki M, Reunala T. Coeliac-type dental enamel defects in patients with dermatitis herpetiformis. Acta Derm Venereol 1992;72:25–7.
- Aine L, Reunala T, Mäki M. Dental enamel defects in children with dermatitis herpetiformis. J Pediatr 1991;118(4 Pt 1):572–4.
- Aine L. Coeliac-type permanent-tooth enamel defects. Ann Med 1996;28:9–12.
- Green PHR, Jabri B. Celiac disease and other precursors to small-bowel malignancy. Gastroenterol Clin North Am 2002;31:625–39.
- Bushara KO. Neurologic presentation of celiac disease. Gastroenterology 2005;128(4 Suppl. 1):S92–7.
- McKeon A, Lennon VA, Pittock SJ, Kryzer TJ, Murray J. The neurologic significance of celiac disease biomarkers. Neurology 2014 Nov 11;83:1789–96.
- Biagi F, Marchese A, Ferretti F, et al. A multicentre case control study on complicated coeliac disease: two different patterns of natural history, two different prognoses. BMC Gastroenterol 2014;14:139.
- Paek SY, Steinberg SM, Katz SI. Remission in dermatitis herpetiformis: a cohort study. Arch Dermatol 2011;147:301–5.
- Beutner EH, Chorzelski TP, Reunala TL, Kumar V. Immunopathology of dermatitis herpetiformis. Clin Dermatol 1991;9:295–311.
- Zone JJ, Meyer LJ, Petersen MJ. Deposition of granular IgA relative to clinical lesions in dermatitis herpetiformis. Arch Dermatol 1996;132:912–18.
- Cardones ARG, Hall RP. Management of dermatitis herpetiformis. Immunol Allergy Clin North Am 2012;32:275–81, vi–vii.
- Caproni M, Antiga E, Melani L, Fabbri P, Italian Group for Cutaneous Immunopathology. Guidelines for the diagnosis and treatment of dermatitis herpetiformis. J Eur Acad Dermatol Venereol 2009;23:633–8.
- Garioch JJ, Lewis HM, Sargent SA, Leonard JN, Fry L. 25 years' experience of a gluten-free diet in the treatment of dermatitis herpetiformis. Br J Dermatol 1994;131:541–5.
- Lewis HM, Renaula TL, Garioch JJ, et al. Protective effect of gluten-free diet against development of lymphoma in dermatitis herpetiformis. Br J Dermatol 1996;135:363–7.
- Booth SA, Moody CE, Dahl MV, Herron MJ, Nelson RD. Dapsone suppresses integrin-mediated neutrophil adherence function. J Invest Dermatol 1992;98:135–40.
- Zhu YI, Stiller MJ. Dapsone and sulfones in dermatology: overview and update. J Am Dermatol 2001;45:420–34.
- Jollow DJ, Bradshaw TP, McMillan DC. Dapsone-induced hemolytic anemia. Drug Metab Rev 1995;27(1–2):107–24.
- Barclay JA, Ziemba SE, Ibrahim RB. Dapsone-induced methemoglobinemia: a primer for clinicians. Ann Pharmacother 2011;45:1103–15.
- Coleman MD. Dapsone-mediated agranulocytosis: risks, possible mechanisms and prevention. Toxicology 2001 Apr 12;162:53–60.
- Lorenz M, Wozel G, Schmitt J. Hypersensitivity reactions to dapsone: a systematic review. Acta Derm Venereol 2012;92:194–9.
- Waldinger TP, Siegle RJ, Weber W, Voorhees JJ. Dapsone-induced peripheral neuropathy. Case report and review. Arch Dermatol 1984;120:356–9.
- Luzzatto L, Seneca E. G6PD deficiency: a classic example of pharmacogenetics with on-going clinical implications. Br J Haematol 2014;164:469–80.
- Coleman MD, Rhodes LE, Scott AK, et al. The use of cimetidine to reduce dapsone-dependent methaemoglobinaemia in dermatitis herpetiformis patients. Br J Clin Pharmacol 1992;34:244–9.
- Rhodes LE, Tingle MD, Park BK, Chu P, Verbov JL, Friedmann PS. Cimetidine improves the therapeutic/toxic ratio of dapsone in patients on chronic dapsone therapy. Br J Dermatol 1995;132:257–62.
- Goolamali SI, Macfarlane CS. The use of cimetidine to reduce dapsone-dependent haematological side-effects in a patient with mucous membrane pemphigoid. Clin Exp Dermatol 2009;34:e1025–6.
- McFadden JP, Leonard JN, Powles AV, Rutman AJ, Fry L. Sulphamethoxypyridazine for dermatitis herpetiformis, linear IgA disease and cicatricial pemphigoid. Br J Dermatol 1989;121:759–62.
- Cardones ARG, Hall RP. Management of dermatitis herpetiformis. Dermatol Clin 2011;29:631–5.
- Willsteed E, Lee M, Wong LC, Cooper A. Sulfasalazine and dermatitis herpetiformis. Australas J Dermatol 2005;46:101–3.
- Nieuwenhuis WP, Kneepkens CMF, Houwen RHJ, de Beer HJA, Mulder CJ, Mearin ML. [Guideline “Coeliac disease and dermatitis herpetiformis”.] Ned Tijdschr Geneeskd 2010;154:A1904.
- Ingen-Housz-Oro S, Joly P, Bernard P, Bedane C, Prost C, Centres de référence des maladies bulleuses auto-immunes. Société Française de Dermatologie. [Dermatitis herpetiformis. Guidelines for the diagnosis and treatment. Centres de référence des maladies bulleuses auto-immunes. Société Française de Dermatologie.] Ann Dermatol Vénéréol 2011; pp. 271–3.
- Herrero-González JE. [Clinical guidelines for the diagnosis and treatment of dermatitis herpetiformis.] Actas Dermosifiliogr 2010;101:820–6.