CHAPTER 53
Dermatomyositis
Patrick Gordon1 and Daniel Creamer2
1Department of Rheumatology, King’s College Hospital, London, UK
2Department of Dermatology, King’s College Hospital, London, UK
Definition
Dermatomyositis (DM) is an autoimmune disorder affecting, predominantly, the skin and skeletal muscle. It is classified alongside polymyositis in the idiopathic inflammatory myopathies (IIMs).
The Bohan and Peter classification for dermatomyositis and polymyositis has been in use since 1975 (Table 53.1) [1]. Whilst these classification criteria remain the most cited they do not acknowledge amyopathic DM and fail to distinguish polymyositis from inclusion body myositis. Therefore the Griggs criteria for inclusion body myositis should be used in addition when this diagnosis is suspected. The Bohan and Peter criteria also perform poorly at discriminating polymyositis from some of its imitators.
Table 53.1 The Bohan and Peter classification for polymyositis and dermatomyositis
| Features | Polymyositis | Dermatomyositis |
|
|
|
Modified from Bohan and Peter 1975 [1].
Sontheimer defines amyopathic DM as ‘a subset of DM characterized by biopsy-confirmed hallmark cutaneous manifestations of classic DM occurring for 6 months or longer with no clinical evidence of proximal muscle weakness and no serum muscle enzyme abnormalities’ [2]. If more extensive muscle testing is carried out, the results should be within normal limits; however, if these investigations are positive or abnormal, the patient can be classified as having hypomyopathic DM. Exclusion criteria for amyopathic DM are: (i) treatment with systemic immunosuppressive therapy for two consecutive months or longer within the first 6 months after skin disease onset; and (ii) the use of drugs known to be capable of producing DM-like skin changes (e.g. hydroxyurea) [2]. Clinically amyopathic dermatomyositis (CADM) is a functional designation used to refer to either amyopathic DM or hypomyopathic DM [2].
In view of the limitations of the Bohan and Peter classification, a new set of criteria encompassing amyopathic DM and inclusion body myositis are in development (http://www.imm.ki.se/biostatistics/calculators/iim/; last accessed July 2015).
Introduction and general description
In DM, autoimmune inflammation damages the skin, muscle and, in certain cases, internal organs such as the lungs, joints, oro-pharynx and heart. At one end of the spectrum, myopathy predominates, at the other end, skin disease is prominent with minimal or absent myositis (amyopathic DM). Interstitial lung disease (ILD) may affect a patient with DM, irrespective of their muscle–skin phenotype. Myositis-specific antibodies can help predict phenotype and internal organ involvement although their availability remains limited.
Mortality is increased in DM; modern IIM series have a 10-year survival of up to 90%, with slightly higher mortality in DM than polymyositis [3, 4]. Although there is great variability in the precise figures between studies, the main causes of death in IIM are cardiac disease (15–64% deaths), respiratory disease (5–36% deaths) and malignancy (10–47% deaths) [5].
Epidemiology
Incidence/prevalence
The IIMs are rare with a reported annual incidence varying widely from 0.1 to 6.7 per 100 000 person-years [6–9], with the highest estimates coming from recent epidemiological studies from large administrative claims databases and lower estimates from studies based on hospital admission data confirmed by chart review.
The prevalence of IIM is in the region of 5.1–22 per 100 000 [6, 7, 10]. The relative proportion of DM within IIM appears to enlarge with increasing southerly latitude in Europe [11]. Similarly, there was an association between regional UV exposure and the proportion of female IIM patients with DM and Mi-2 antibodies in a study in the USA [12]. As such it would appear that UV radiation may have an influence on the presentation and phenotype of DM.
Age
The peak incidence of DM occurs around 50–60 years of age [13, 14]. DM occurs in children although it is much rarer in this age group with an incidence of around two cases per million children under 16 years per year, with a median age of onset of 6.8 years [15].
Sex
The incidence of adult DM is approximately twice as high in women [7, 8] with some studies suggesting an even greater female predominance in child-bearing years. Although the overall incidence of juvenile DM is much lower than for adults, the female preponderance in children is greater at 5 : 1 [15].
Ethnicity
As with systemic lupus erythematosus, the incidence of DM is greater in African Americans than in white Americans [6].
Associated diseases
There is a strong association between internal malignancy and adult DM. In a large Scandinavian population-based study, 32% of adult DM patients had an associated malignancy, with a standard incident ratio of 3 compared to the general population. However, the reported frequency in the literature varies between 7% and 60% [16–19]. Unlike adult DM, juvenile DM is not thought to be associated with an increased risk of malignancy, even though one study did suggest a possible association [20].
The risk of malignancy rises with increasing age, severe skin disease with necrosis, dysphagia, diaphragmatic weakness, lack of extramuscular systemic features (e.g. ILD) and antibodies to antitranscription intermediary factor 1γ (anti-TIF1γ) or nuclear matrix protein 2 (NXP-2) [21–24].
Neoplasms may be identified in patients before, at or after the onset of DM. In one study, 58% of the malignancies presented after the diagnosis of DM [16]. The risk of cancer is greatest in the first few years after the diagnosis of DM, but there remains an increased, albeit lower, risk more than 5 years after diagnosis [20].
Tumours of many types have been associated with DM although the majority are adenocarcinomas [16]. In a Scandinavian population-based study, carcinomas of the breast and ovary were the commonest malignancies in women, while lung cancer was the leading malignancy both overall and in men [16]. However, in two studies set in the Far East (one in Singapore and the other in Taiwan), naso-pharangeal carcinoma was the commonest myositis-associated malignancy, reflecting the high incidence of naso-pharangeal cancer in this region [17, 25].
Pathophysiology
An early histological feature of DM in muscle, which is not found in polymyositis, is the deposition of the membrane attack complex, the terminal complement component C5b-9 within capillaries [26, 27]. HLA class 1, which is usually absent on the cell membrane of muscle fibres, is diffusely up-regulated even in areas apparently unaffected, without inflammatory cell infiltration [28]. Capillary drop-out occurs with a compensatory increase in calibre of the remaining capillaries [27]. In the perifascicular areas muscle fibre atrophy occurs, which has been hypothesized to be a result of hypoxic stress, although models of ischaemic myopathy have not supported this theory [29, 30]. The inflammatory infiltrate, which comprises B cells, CD4+ T cells, macrophages and plasmocytoid dendritic cells, is found predominantly in the perivascular and perifascicular areas [31].
These features suggest a humorally mediated microvasculopathy in DM, in contrast to polymyositis where there is evidence of cell-mediated cytotoxicity with CD8+ T cells invading muscle fibres. Consistent with the notion of humoral activation in DM is the recognition that the majority of patients have disease-associated autoantibodies. These may be myositis-specific antibodies (MSAs), such as anti-Mi-2, TIF-1γ, MDA-5 and SUMO activating enzyme which are only found in myositis patients, or myositis-associated antibodies (MAAs), such as anti-52kD Ro, U1 RNP, PM-Scl and Ku which also occur in myositis-overlap syndromes and in other diseases [32].
For two of the MSAs, MDA-5 and anti-Jo-1, antibody titres show some correlation with disease activity suggesting a possible pathogenic role [33, 34]. Intriguingly the N-terminal proteolytic fragment of histidyl tRNA sythetase, the target antigen for anti-Jo-1 antibodies, is chemotactic to lymphocytes and activated monocytes [35].
The autoantigens in inflammatory myositis are not organ specific but are molecules ubiquitous to all human cells. Nonetheless, at least in some cases, they have been found in increased concentrations in target organs. Increased concentrations of the granzyme B-cleavable conformation of histidyl tRNA synthetase (Jo-1) are found in the lung (ILD), the target of anti-histidyl tRNA synthetase (Jo-1) disease [36]. Several myositis autoantigens including Mi-2, Ku and the aminoacyl tRNA synthetases are up-regulated in diseased muscle due to increased expression in regenerating fibres [37]. Thus target antigen expression is increased following muscle damage, potentially creating a positive feedback loop. Notably, Mi-2 expression is increased specifically in DM but not in polymyositis, showing disease specificity [37].
Type 1 interferon inducible gene expression is increased in DM in both muscle tissue and in the peripheral blood [31, 38]. Both anti-Jo-1 and anti-52kD Ro antibodies when combined with necrotic cell material can induce type 1 interferon production from peripheral blood mononuclear cells [39]. However, pretreatment of the necrotic cell debris with RNAse prevents type 1 interferon expression, demonstrating that immune complex formation containing RNA is required [39]. Immune complex internalization via Fc receptors with subsequent stimulation of Toll-like receptors by the ribonucleic acid component of the immune complexes has been proposed as a potential mechanism for this effect [39]. It is notable that other autoantigens in DM, aside from Jo-1 and Ro, bind directly or via macromolecules to nucleic acids and could potentially cause type 1 interferon induction by similar mechanisms [40].
Histopathology
Skin
Despite striking clinical signs in the skin, the dermatopathology of DM is often subtle. A lichenoid tissue reaction with vacuolar changes in the basal layer and occasional Civatte bodies is typical (Figure 53.1) [41]. In some cases there is only a sparse superficial perivascular infiltrate of lymphocytes with upper dermal oedema and mucinous change. The basement membrane may be thickened. In acute DM the changes resemble those of subacute lupus erythematosus, although the dermal oedema may be more extensive and involve all layers of the dermis. In poikilodermatous DM there is epidermal atrophy, dilatation of superficial vessels and melanin incontinence. Hyperkeratosis, acanthosis and mild papillomatosis are seen in a biopsy from a Gottron's papule. Unusual skin biopsy findings in DM include subepidermal blistering, dystrophic calcification and lobular panniculitis.
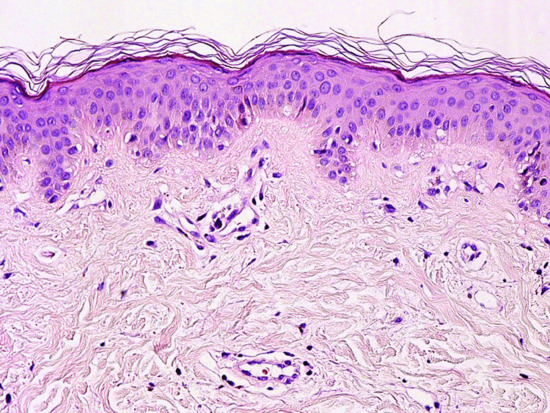
Figure 53.1 Skin histopathology (medium power). There is a mild lichenoid tissue reaction with subtle vacuolar change in the basal layer and occasional Civatte bodies. There is a sparse, superficial, perivascular infiltrate of lymphocytes with upper dermal oedema.
Muscle
Muscle biopsies in DM require specialist staining and should be performed in a specialist neuropathology laboratory experienced in the preparation and reporting of muscle biopsies. Cellular infiltrates containing B cells and CD4+ cells are seen predominantly in the perivascular and perifascicular areas. Muscle fibres show perifascicular atrophy (Figure 53.2a). An early change is C5b-9 deposition on the capillary walls (Figure 53.2b). Capillary drop-out is a common feature and rarely overt muscle infarcts may be seen. Tubuloreticular inclusions are found on electron microscopy within the capillary endothelial cells. HLA class I, which is not usually expressed on muscle fibres, is up-regulated, and whilst myositis is a patchy process, HLA class I up-regulation is also found in areas where no inflammatory infiltrates are seen (Figure 53.2c).
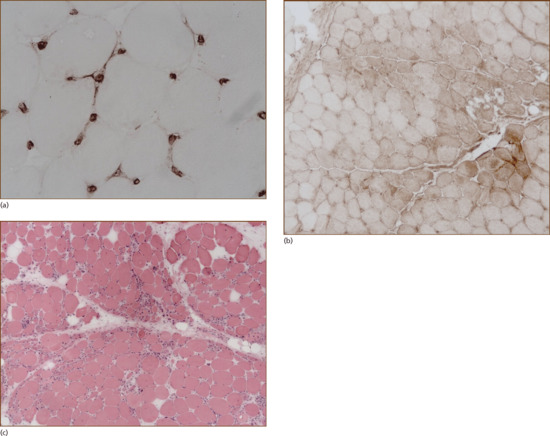
Figure 53.2 Muscle biopsy. (a) Capillary staining for C5b-9, the membrane attack complex, is an early pathological finding in dermatomyositis. (b) Diffuse up-regulation of HLA class I is another early pathological finding in the inflammatory myopathies, and can occur in the absence of a cell infiltrate. (c) In dermatomyositis muscle fibre atrophy occurs in a perifascicular distribution.
(Courtesy of Dr Safa Al-Sarraj, Brain Bank, King's College Hospital, London.)
Clinical features
History
A dermatosis is the presenting feature in over 50% of DM cases [42]. Most patients develop a facial rash initially. In some cases this is an acute inflammatory eruption accompanied by oedema; more typically the facial dermatosis begins insidiously and extends over a few weeks. The patient may also notice scalp involvement in the early stages, associated with hair loss. A rash on the hands, extensor surfaces of the limbs and upper torso tends to occur subsequently, although it may develop concurrently with the facial eruption. The eruption of DM can be both pruritic and sore [43]. Only a few patients give a clear history of photosensitivity [44].
Muscle disease in DM usually becomes clinically apparent in the weeks to months after the onset of skin signs. However, occasionally the dermatosis of DM occurs in isolation, when it is known as amyopathic DM (DM sine myositis). The initial symptoms of myositis are weakness and fatigue. Patients have difficulty in climbing stairs, raising their arms and standing from a sitting position. There may be myalgia but muscle tenderness is relatively rare. If untreated, the weakness will usually progress over a matter of weeks; severe loss of power may result in a patient becoming bed-bound. Respiratory muscle involvement can lead to the development of respiratory failure. Problems with swallowing and symptoms of aspiration reflect involvement of the pharyngeal and upper oesophageal musculature. Dysphagia occurring early in the disease indicates an aggressive course and is associated with a poor prognosis. Patients should be directly asked whether they have swallowing problems, which may present as a tendency to cough on their food and/or difficulty clearing food from their throats requiring water and several attempts to swallow a bolus. Other clues include recent weight loss, recurrent lower respiratory tract infections and a quiet, nasal voice.
Raynaud's phenomenon is relatively common and is particularly associated with the antisynthetase syndrome and mixed connective tissue disease, but is rare in children. Joint involvement occurs in approximately one-quarter of cases and presents as generalized arthralgia and morning stiffness.
Presentation
Skin signs
Dermatomyositis is distinguished by a broad spectrum of cutaneous manifestations. Diagnostically, the dermatologist needs to be aware of the atypical skin signs in DM, as well as the more common features. Variation in the severity and extent of skin involvement is usual.
The classic cutaneous signs in DM involve the skin of the face, hands and extensor surfaces of the limbs; however, all of the skin may be affected. Typically encountered are: facial rash, eyelid erythema and oedema, nail fold changes, Gottron's papules, involvement of the upper chest and back, an extensor surface dermatosis, scalp erythema and hair loss. Other less common signs are: oedema of the face and limbs, flagellate erythema, vasculopathic ulcers, ‘mechanic's hands’ (in the antisynthetase syndrome), poikiloderma and cutaneous calcinosis.
In the early phase of DM most patients develop a facial dermatosis. One pattern is characterized by confluent erythema of the whole face, extending onto the neck and upper chest. Alternatively, a lupus erythematosus-like malar rash can occur with fixed erythema over the nose and cheeks (Figure 53.3). Some patients have a seborrhoeic dermatitis-like eruption, with involvement of the hairline, facial margins and naso-labial folds [45]. Scalp involvement in DM is common and is characterized by erythema (sometimes poikiloderma) and a diffuse non-scarring hair loss (Figure 53.4) [46]. The facial eruption may be accompanied by slight scaling; scalp involvement may also be scaly.
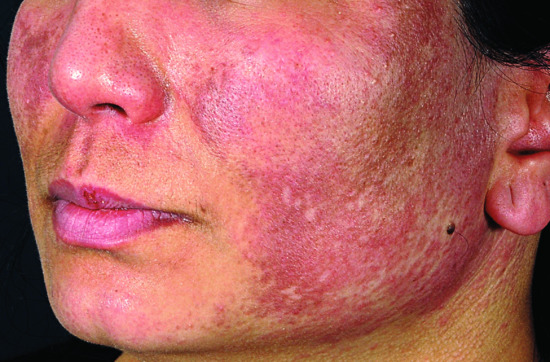
Figure 53.3 Facial erythema in dermatomyositis is often widespread and can mimic many dermatoses, including lupus erythematosus.
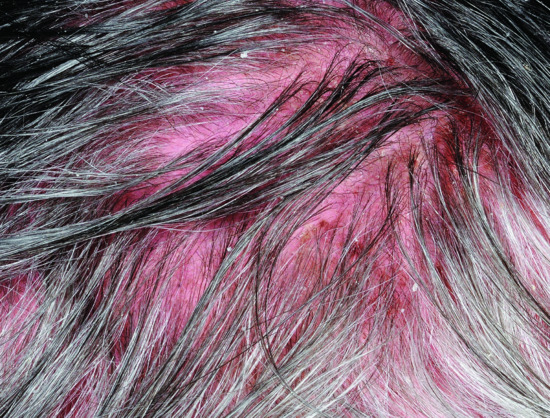
Figure 53.4 A diffuse, non-scarring alopecia is common in dermatomyositis. The scalp is inflamed with violaceous erythema and mild scaling.
Involvement of the eyelid skin is characteristic, even pathognomonic, of DM. The upper eyelids are preferentially affected with an erythema that is lilac (or heliotrope) in colour (Figure 53.5). This distinctive hue is only appreciable in fair-skinned patients. Oedema of the eyelids and periorbital skin also occurs. The presence of the eyelid dermatosis with oedema often reflects activity of the myositis. Fixed macular erythema commonly appears on the upper torso, both the ‘V’ of the chest (Figure 53.6), and across the shoulders, where it is termed the ‘shawl sign’ (Figure 53.7). If untreated the rash of DM often becomes poikilodermatous, with a prominent telangiectatic component (Figure 53.8).
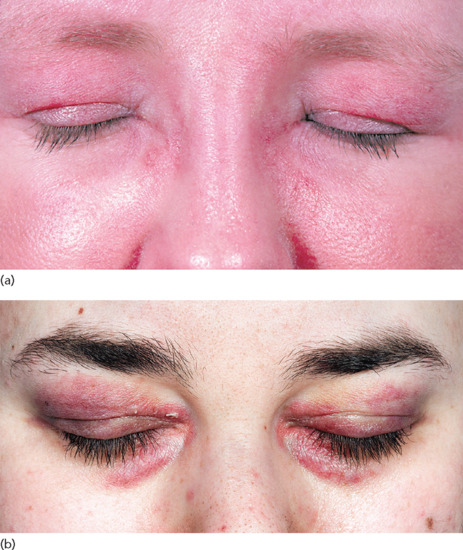
Figure 53.5 Eyelid involvement in DM. (a) There is lilac erythema of the upper eyelids, which are also oedematous. In this patient with DM the facial skin is generally red. (b) Dusky erythema affecting both the upper and lower eyelids. The rest of the facial skin is normal.
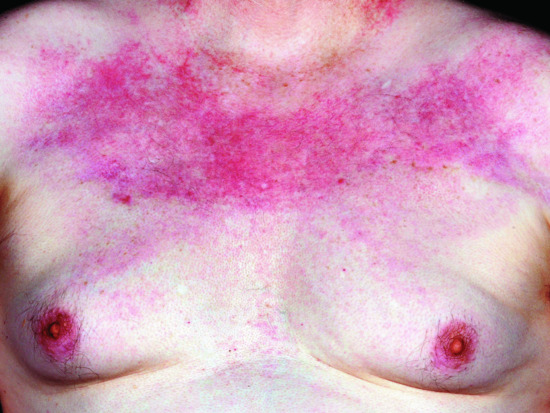
Figure 53.6 The upper chest is a common site of skin involvement in dermatomyositis.
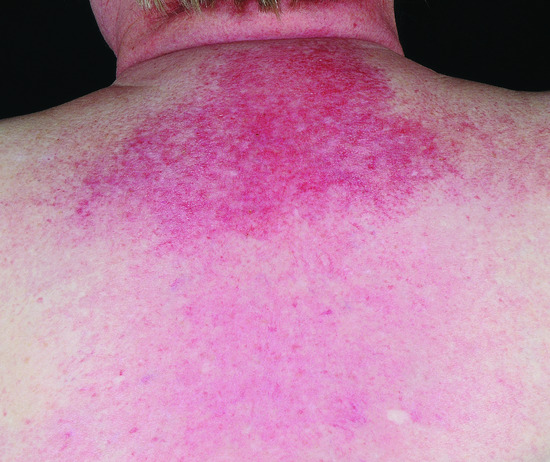
Figure 53.7 Erythema of the upper central back is known as the shawl sign.

Figure 53.8 Poikiloderma occurs in dermatomyositis; it is composed of telangiectatic erythema, epidermal atrophy and dyspigmentation.
Nail fold changes consist of periungual erythema with visible dilated capillary loops in the proximal nail fold (Figure 53.9a). The periungual vessels are best appreciated when viewed with the dermoscope [47]. Small haemorrhagic infarcts are also sometimes seen within the hyperaemic nail folds. Hypertrophic cuticles, which are dystrophic (‘ragged’), are typical (Figure 53.9b). The nail fold changes in DM are indistinguishable from those seen in other connective tissue diseases; cuticular overgrowth is also seen in scleroderma.
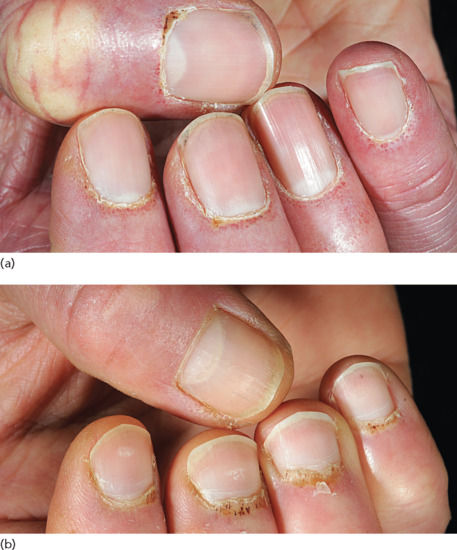
Figure 53.9 The nail folds and cuticles are usually affected in dermatomyositis. (a) Dilated nail fold capillary loops are visible. (b) The cuticles are hypertrophic and ragged. There are infarcts within the cuticles.
Gottron's papules are inflammatory, flat-topped, lichenoid lesions that occur on the skin overlying the distal and proximal interphalangeal joints of the fingers and the metacarpophalangeal joints (Figure 53.10). Gottron's papules are red or lilac in colour. Similar, larger lesions may develop over the elbows and knees. They may also occur, albeit rarely, on the palmar surfaces of the finger creases, where they are referred to as inverse Gottron's papules [48]. Examination of the backs of the hands may also reveal linear red streaks on the skin overlying the extensor tendons (Figure 53.11). Hand oedema may complicate acute DM. On the limbs, the rash of DM preferentially affects the extensor surfaces and is characterized by zones of livid erythema with mild scaling (Figure 53.12). The buttocks are often involved in DM; erythema over the hips and lateral thighs has been termed the ‘holster sign’ (Figure 53.13). In extremely active disease, lesional skin may become eroded. Rarely, the acute eruption of DM can be vesicular or bullous; this presentation is thought to herald an aggressive disease phenotype [49]. Distinctive vasculopathic ulcers, which tend to be punched-out and surrounded by a zone of dusky erythema, may occur on the fingers, dorsal aspect of the hands and extensor surfaces of the elbows and knees (Figure 53.14). Vasculopathic ulcers are particularly associated with the presence of anti-MDA-5 antibody [50].
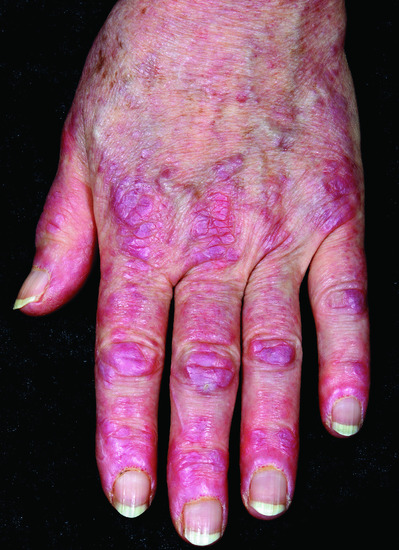
Figure 53.10 Gottren's papules: violaceous, flat-topped, shiny papules on the skin overlying the interphalangeal joints and metacarpophalangeal joints.
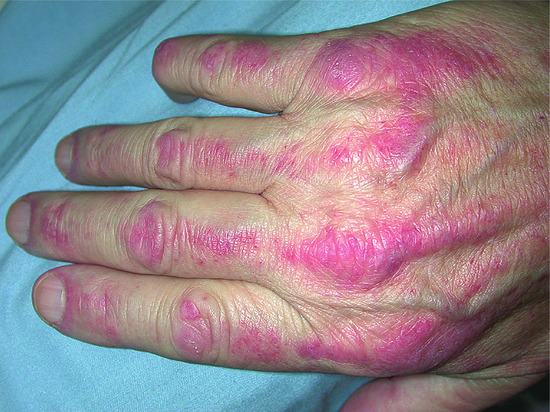
Figure 53.11 There are streaks of erythema on the dorsal aspects of the fingers, extending onto the backs of the hands.
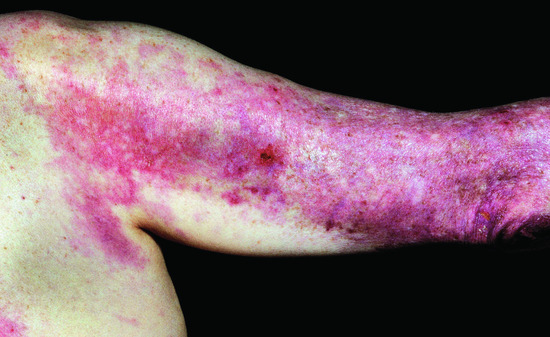
Figure 53.12 Involvement of the extensor surfaces of the arms is typical in dermatomyositis.
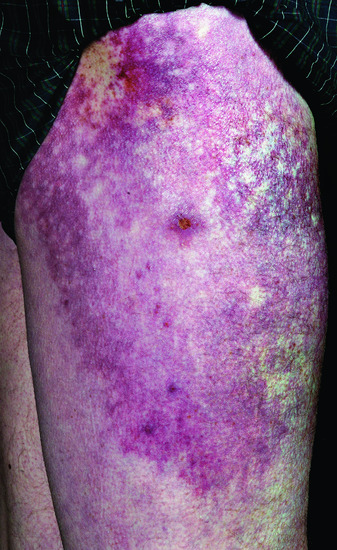
Figure 53.13 The dermatosis of dermatomyositis can affect the gluteal skin and proximal thighs. Involvement of the skin overlying the hips is known as the holster sign.

Figure 53.14 Vasculopathic ulcers, which have dusky margins and are ‘punched-out’, may occur in DM patients, particularly those who are positive for anti-MDA5 antibody. The fingers and knuckles are the usual sites of involvement.
Flagellate erythema is a striking cutaneous sign that is strongly associated with a diagnosis of DM [51], although it is also seen as a side effect to bleomycin and as a toxicity manifestation of the ingestion of shitaki mushrooms [52]. It is characterized by multiple, red, macular streaks occurring on the trunk and proximal limbs. Each lesion is approximately 1 cm in diameter and may be several centimetres in length. The stripes are aligned diagonally, sometimes in a criss-cross pattern, giving the appearance of a scourging or whipping. The eruption may be either sore or pruritic. Erythema on the torso in DM can become confluent and extensive (Figure 53.15).
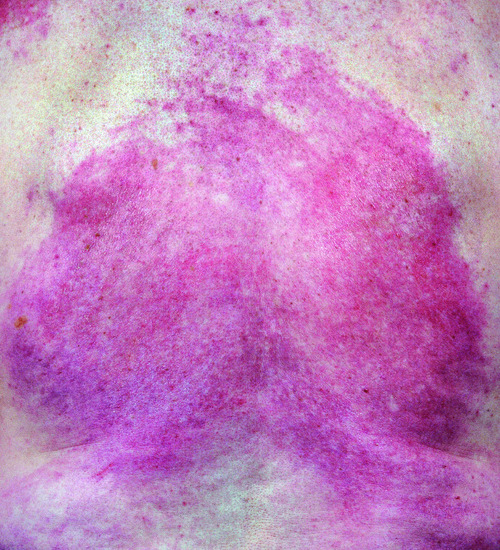
Figure 53.15 In severe dermatomyositis, skin involvement on the torso can become extensive and fixed.
In the antisynthetase syndrome, a skin change known as ‘mechanic's hands’ (or ‘machinist's hands’) can be seen. The term mechanic's hands denotes non-inflammatory hyperkeratosis occurring on the hands or feet, usually over the radial surfaces of the fingers and the outer border of the feet (Figure 53.16) [53]. The condition is reported to be similar to the callosities seen on the hands of manual workers.
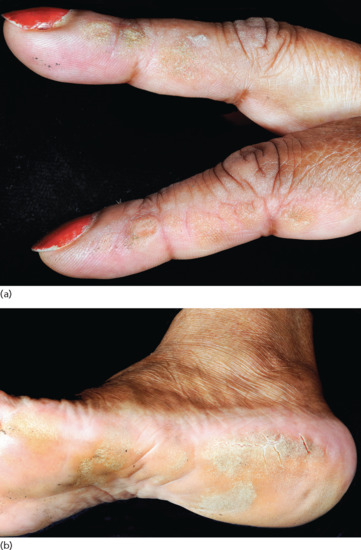
Figure 53.16 Non-inflammatory hyperkeratosis occurring on (a) the fingers and (b) the feet may be seen in Jo-1-positive DM. Involvement of the radial surfaces of the fingers resembles the callosities seen in manual workers, so-called mechanic's hands.
Calcinosis cutis (cutaneous calcinosis) is a complication of DM (and other connective tissue diseases, most notably systemic sclerosis) in which insoluble calcified material is deposited in the skin and subcutaneous tissue. Calcinosis occurs in around 10% of adult DM patients and is more common in patients with longer periods of sustained disease activity, digital ulceration and anti-NXP2 antibodies [54]. The areas of calcification are usually palpable to the patient and are felt as bony-hard nodules or sheets. These lesions are often painful and, depending on the site of involvement, may be functionally disabling. The skin may ulcerate over areas of calcinosis, although this is not typical.
Panniculitis is a rare cutaneous manifestation of DM [55]. Large zones of painful, deep-seated inflammation occur, typically on the proximal limbs. These lesions may ulcerate.
Muscle signs
Dermatomyositis is a proximal myopathy and as such patients frequently present with problems climbing stairs, getting out of chairs and performing tasks that requiring lifting the hands above the head such as brushing hair. However, since the onset of weakness can be quite insidious, patients may not appreciate that they are weak, particularly if there is associated arthritis or breathlessness. Even in patients who do not complain of weakness, muscle strength should still be assessed by manual muscle testing. A brief screen for muscle involvement can be made by assessing hip flexion, shoulder abduction and elbow extension. If the patient cannot be overcome on these assessments there is unlikely to be demonstrable muscle weakness on formal muscle testing. If there is evidence of weakness, formal manual muscle testing should be performed.
Muscle strength has traditionally been assessed using the Medical Research Council 0–5 grade scale for muscle strength, although the more extended 0–10 grade scale may be a more sensitive tool for assessing changes in patients with a moderate weakness [56]. The International Myositis Assessment and Clinical Studies (IMACS) group has introduced the MMT8 and MMT26 tools for IIM that grade the strength of specific muscle groups on a scale of 0–10. This produces an overall score that can be easily compared over different time points [57]. MMT8 and MMT26 are increasingly being used in research and clinical practice.
Patients with normal muscle strength on manual muscle testing may complain of ongoing weakness with lack of stamina and these more subtle changes may be picked up using the function index 2 scale that formally assesses muscle stamina in several muscle groups [58].
Respiratory and other systemic features
Respiratory compromise in DM may be caused by ILD, respiratory muscle weakness or aspiration in patients with dysphagia or immunosuppression. Although estimates vary, ILD is found in around 20–40% of IIM [59–64] –therefore all patients should be assessed for this at presentation to allow appropriate therapy and monitoring. Patients usually present with a dry cough and exertional dyspnoea and have fine, late inspiratory crepitations on chest auscultation. However, in mild or early disease these pointers may be absent and in the presence of significant muscle disease can be easily overlooked by both the patient and physician alike. Notably, patients with CADM commonly have coexistent ILD. As such it is sensible to perform lung function tests and in appropriate cases high-resolution computed tomography (CT) at presentation (see Investigations section).
The majority of ILD is non-specific interstitial pneumonitis [64]. Usual interstitial pneumonitis (UIP), cryptogenic-organizing pneumonia (COP) and diffuse alveolar damage (DAD) occur less frequently [64]. As its name suggests, COP may be difficult to distinguish from an infective pneumonia. Whilst these are histological subtypes of ILD they have characteristic features on CT, and in the context of dermatomyositis a lung biopsy is rarely required. COP and DAD tend to present the most acutely. Generally, COP has a very good prognosis, responding rapidly to steroids but in the context of DM a more sinister fibrosing form may occur. DAD is usually a severe disorder and carries a poor prognosis. A presenting carbon monoxide transfer factor (TLCO) of <45%, excess neutrophils on bronchoalveolar lavage fluid, and histology showing DAD or UIP are all associated with poor prognosis [65].
The oro-pharynx and the upper oesophageal sphincter are frequently affected in DM causing dysphagia. The severity of dysphagia does not correlate well with the severity of limb weakness and patients may not volunteer the symptoms of dysphagia. Proactive screening for the symptoms of dysphagia is necessary; early detection of this complication is important to prevent aspiration.
Whilst mild cardiac abnormalities such as minor rhythm disturbances can frequently be found in DM, clinically overt disease, which generally manifests as cardiac failure, is rare. Cardiac failure due to myocarditis responds to immunosuppression.
A symmetrical, non-deforming arthritis may develop, usually affecting the small joints of the hands, wrists and ankles. In the antisynthetase syndrome a more aggressive, erosive arthritis may occur and may be the presenting feature in these patients.
Clinical variants
Overlap syndromes
Mixed connective tissue disease is an overlap connective tissue disease characterized by high titre anti-RNP antibodies, Raynaud's phenomenon, sclerodactyly, inflammatory myositis, ILD and pulmonary hypertension.
Antisynthetase syndrome
The antisynthetase syndrome occurs in the presence of antisynthetase antibodies, most commonly anti-histidyl tRNA synthetase (anti-Jo-1) antibody, which is found in 10–20% of DM cases. There are seven other much rarer antisynthetase antibodies described: threonyl tRNA synthetase (anti-PL-7), alanyl tRNA synthetase (anti-PL-12), glycyl tRNA synthetase (anti-EJ), isoleucyl tRNA synthetase (anti-OJ), asparaginyl tRNA synthetase (anti-KS), tyrosyl tRNA synthetase and phenylalanyl tRNA synthetase (anti-Zo) [66].
The antisynthetase syndrome is characterized by myositis, arthritis, Raynaud's phenomenon, ILD, fever and acral skin changes, called ‘mechanic's hands’ (see Presentation section). ILD is particularly common, occurring in around 75% cases [67, 68]. The non-anti-Jo-1 patients have a slightly lower frequency of myositis and a slightly higher incidence of ILD than the Jo-1-positive patients [68].
Juvenile dermatomyositis
Juvenile dermatomyositis (JDM) may be superficially considered as DM presenting in childhood. However, there are a few subtle but important distinctions. Unlike adult DM there does not seem to be an increased risk of malignancy in JDM. However, calcinosis, which is relatively rare in adults, is found in 6–50% of children with JDM [69]. Calcinosis is associated with increased disease activity, being commoner in those with a delayed diagnosis and inadequately treated disease. In children four forms of calcinosis occur, a superficial cutaneous or subcutaneous calcinosis, intramuscular tumourous calcification, calcinosis of the myofacial planes and, in severe cases, an extensive encasing calcinosis or ‘exoskeleton’.
Acquired lipodystrophy, a rarely reported feature in adult DM, is a relatively common finding in patients with JDM occurring in the region of 10% children [70]. It is associated with insulin-resistant type 2 diabetes and acanthosis nigricans. It may be general, partial or focal. In focal disease there may be an associated panniculitis.
Antibody frequency and phenotype association differ somewhat between adult DM and JDM. For example, anti-Jo-1 occurs in around 20% of adult myositis cases but occurs in only a few percent of JDM patients [71]. Anti-TIF-1γ is associated with cancer in adult disease; whereas in juvenile disease there is an association with ulcerating skin lesions [72].
Clinically amyopathic dermatomyositis
Clinically amyopathic DM (CADM) is characterized by the presence of cutaneous manifestations of DM in the absence of clinical signs of muscle involvement (see classification earlier in this chapter). In a series of 103 DM cases from a single centre, eight were diagnosed with CADM; six of these had subclinical myositis and were subsequently classified as having hypomyopathic DM [73]. Despite the lack of myositis, patients with CADM appear to be at risk for developing cancer and ILD. In a number of studies from East Asia, anti-MDA-5 antibodies have been detected at a significantly higher frequency in CADM patients than in DM patients [74]. In Japanese populations a high incidence of rapidly progressive ILD with a mortality of around 50% has been reported in patients with clinically amyopathic disease and anti-MDA-5 antibodies [48]. Consequently it is important to make a thorough assessment for ILD in patients with amyopathic disease (see Investigations section).
Drug-induced dermatomyositis
Dermatomyositis has occasionally been attributed to an adverse drug reaction. A review of 70 reported cases of drug-induced DM demonstrated pathognomonic cutaneous findings in three-quarters of patients and compatible skin signs in one-quarter [75]. Hydroxycarbamide was identified as the culprit in 50% of cases. None of the hydroxycarbamide patients had evidence of myositis, whereas myositis was described in 80% of non-hydroxycarbamide cases [75]. Despite a large number of other drugs reported as inducing DM, no other specific agent has the same association as hydroxycarbamide. Clinically, the hydroxycarbamide cases are characterized by a violaceous, lichenoid and scaly eruption on the backs of the fingers and the dorsum of the hands, and the tops of the feet and elbows. The eruption can be atrophic and poikilodermatous. Histologically there is hydropic degeneration with scattered dyskeratotic cells and a lichenoid lymphocytic infiltrate; telangiectatic vessels occur within the dermis. Although similar to the dermatosis of DM, the features of this reaction are distinct, and it is best described as ‘hydroxycarbamide dermopathy’ [76]. Most reports occur in patients who have received hydroxycarbamide for many years, the commonest indication being myeloproliferative disorders such as polycythemia vera [76]. Hydroxycarbamide dermopathy tends to improve within 12 months of drug discontinuation [75].
Differential diagnosis
The skin signs in DM can mimic a number of other dermatoses. When myositis is prominent the diagnosis of DM is usually easily made. However, in amyopathic DM a number of differential diagnoses should be considered (Box 53.1).
Classification of severity
Various measures have been developed to assess myositis activity and damage in DM. The DM skin severity index (DSSI), developed by Carroll et al., is calculated from a visual inspection of lesional skin in four main body areas, quantifying redness, induration and scaliness in the same way used for the psoriasis activity and severity index (PASI) [77]. The DSSI has shown significant correlation to the physician's global assessment, assessments of poikiloderma and self-assessment of pruritus [77].
The IMACS group has also proposed composite measures for the measurement of disease activity in myositis – the myositis intention to treat index (MITAX) and the myositis disease activity assessment visual analogue scales (MYOACT), and the myositis damage index (MYODAM) for measuring damage for use in clinical trials [78].
Complications and co-morbidities
Patients with severe muscle weakness may develop hypoventilatory respiratory failure. In the acute setting, monitoring the forced vital capacity (FVC) serially at the bedside is the best way to monitor this, with blood gas measurements to assess for hypoxia and hypercapnia. Another measurement, the sniff nasal inspiratory pressure (SNIP), may be a more accurate predictor of ventilatory failure, with hypercapnic respiratory failure not usually occurring until the ventilatory muscle strength falls below 40% predicted. In contrast hypercapnia may occur with a FVC >55% predicted. Patients with less critical respiratory muscle weakness and normal daytime blood gases may develop hypercapnia overnight. Thus in patients with a low SNIP or FVC, overnight pulsoximetry should be performed to assess for nocturnal hypoventilation which may need non-invasive positive pressure ventilation.
Involvement of the oro-pharynx and the upper oesophageal sphincter in DM causes dysphagia. Aspiration pneumonia, exacerbated by respiratory muscle weakness, is the life-threatening complication of oro-pharyngeal involvement. Where dysphagia is prominent patients may present with dramatic weight loss in the absence of an underlying malignancy.
An inflammatory cardiomyopathy may occur in DM. Not surprisingly, cardiac involvement is a poor prognostic factor [59]. As with other autoimmune diseases there is evidence emerging that accelerated atherosclerosis is a cause of late mortality and morbidity [79, 80].
Several studies have suggested an increased incidence of cardiovascular disease in patients with DM. As such it is important to minimize modifiable risk factors in these patients such as obesity, hypertension, hypercholesterolaemia, steroid therapy and smoking.
Disease course and prognosis
The disease course is variable with the majority of patients requiring ongoing long-term immunosuppression. Around 20–35% of surviving myositis patients have a monophasic illness, with around 20–30% following a relapsing, remitting course and the remainder following a chronic continuous course [3, 81].
Prognosis relates closely to systemic features, with cancer and ILD being the two major causes of death. In modern series the 10-year survival is in the order of 90% [3, 59].
Investigations
Patients with suspected DM should have a full biochemical profile, full blood count, inflammatory markers and muscle enzymes performed. Inflammatory markers may be normal, as may creatine kinase levels, particularly in clinically amyopathic patients. As an inflammatory myositis may occur in lupus erythematosus and other connective tissue diseases, the following serology tests must be performed: antinuclear antibody (ANA), extractable nuclear antigen (ENA), double-stranded DNA (dsDNA), lupus anticoagulant, complement levels, anticardiolipin antibodies and β2-glycoprotein 1 antibodies. Approximately 80% of DM patients will have a positive ANA, often at a low titre, which would not generally be considered significant in other clinical scenarios [82]. However, many of the autoantibodies in DM are predominantly cytoplasmic and as such will not cause a positive ANA.
Myositis-associated antibodies (MAAs) are antibodies that occur in myositis but are not specific for it. As such they can support the diagnosis, not confirm it. Anti-52kD Ro antibodies are the commonest MAA, found in 20% of IIM cases, and may be found in conjunction with other myositis antibodies particularly anti-Jo-1 [83]. Anti-Ku, anti-U1-RNP, U3-RNP and PM-Scl antibodies may occur in myositis, usually in the context of overlap syndromes with sclerodermatous features.
Myositis-specific antibodies are specific for myositis and help define phenotype [84]. The antisynthetase antibodies are the commonest, occurring in approximately 20% of IIM patients, with anti-Jo-1 being by far the commonest and the other seven antisynthetase antibodies accounting for only a few percent of cases. They delineate a specific phenotype, as discussed earlier, with arthritis, fever, Raynaud's phenomenon, ILD, myositis and mechanic's hands. There is a spectrum of disease with some patients having little in the way of skin disease. With some of the rarer antisynthetase antibodies, lung disease may be the predominant clinical feature with clinically amyopathic disease, Raynaud's phenomenon and only subtle skin changes. Antisynthetase antibodies are extremely rare in JDM.
Antibodies to MDA-5 were first described in Japan, where they were found in 19–35% of DM patients and were associated with a high incidence of CADM (85–100%) and ILD (92%), with half developing rapidly progressive ILD [85, 86]. In western populations MDA-5 antibodies are less frequent, at around 13% of DM patients in an American cohort [50]. In the anti-MDA-5-positive patients there was a high incidence of clinically amyopathic disease (50%) and ILD (67%). Anti-MDA-5 antibodies are associated with a distinctive cutaneous phenotype characterized by vasculopathic ulcers on the fingers, knuckles, elbows and knees (see Figure 53.14) [87]. These ulcers are usually small in diameter but are deep, punched-out and surrounded by a dusky violaceous border. Painful red papules on the palmar surfaces of the metacarpophalangeal and interphalangeal joints are also observed with anti-MDA-5 antibodies [87].
Anti-TIF-1γ antibodies are specific to DM and are found in around 15–20% of cases [88]. They are associated with severe skin disease and a high incidence of cancer (50% in one series) [23, 88]. As such, these patients should have a thorough screen for malignancy.
Anti-NXP2 antibodies are frequent in JDM (11–23%) where they are associated with calcinosis [89]. In JDM, patients with anti-NXP2 appear to have a more severe disease course and worse functional status [89]. Anti-NXP2 antibodies are rare in adults.
Anti-Mi-2 antibodies are specific to DM and are found in around 10–20% of cases [90]. Patients with this antibody generally have classic cutaneous features of DM with Gottron's papules, heliotrope rash, upper chest erythema (‘V’ sign) and the shawl sign [90]. Muscle disease tends to be mild and ILD rare in patients with anti-Mi-2 antibodies.
Anti-SUMO-activating enzyme antibodies occur in approximately 8% of adult DM patients [91]. The majority of patients present with skin disease and in one series a high increased frequency of dysphagia was reported [91].
Electromyography can confirm a myopathic process with early recruitment of multiple, small-amplitude, short-duration motor unit action potentials [92]. However, the presence of spontaneous fibrillations, positive sharp waves and bizarre high-frequency discharges are more specific for the inflammatory myopathies. Estimates of the proportion of patients with spontaneous fibrillations varies from 45% to 100% in various studies, possibly relating to the number of muscles sampled [92]. Although relatively specific for myositis, spontaneous fibrillations may also be found in other primary myopathies with muscle fibre degeneration, such as muscular dystrophies and acid maltase deficiency [92].
Magnetic resonance imaging (MRI) of the proximal muscles may show oedema suggestive, but not diagnostic, of muscle inflammation. It can be helpful in assessing the extent of disease and guiding where the muscle biopsy should be taken when indicated; normal MRI does not exclude myositis.
In patients with the typical rash of DM a muscle biopsy is not required to confirm the diagnosis. When needed, muscle biopsy should, if possible, be performed prior to the start of systemic corticosteroid therapy to maximize diagnostic yield. Muscle biopsies should be assessed in an experienced specialist laboratory where immunohistochemistry can be performed on frozen muscle tissue.
Plain radiography is an effective method of detecting and monitoring calcinosis in DM.
ILD is a major cause of mortality in DM and should be actively screened for. Lung function tests should be performed at presentation to include FVC, FEV1 (forced expiratory voume in 1 s) and TLCO; in ILD these show a restrictive pattern with all reduced in equal proportion. If there is concern regarding respiratory muscle weakness, a SNIP or lying and sitting FVC should be performed. A high-resolution CT scan of the chest should be performed if lung function tests show a restrictive pattern. If lung function is normal in patients at high risk of developing ILD, such as those with anti-MDA-5, antisynthetase or anti-RNP antibodies, an initial high-resolution CT of the chest should be considered at presentation and annual lung function tests performed subsequently.
In view of the increased risk of an underlying tumour in DM, screening for malignancy is necessary. There is no agreed protocol on the set of investigations needed. CT scans of the neck, thorax, abdomen and pelvis should be performed on all adult patients. Further investigations should be organized as indicated following a full clinical assessment.
Higher rates of malignancy are found in the elderly and those with severe skin disease, dysphagia, diaphragmatic weakness and anti-TIF1γ and NXP-2 antibodies. If there is a high index of suspicion for an underlying malignancy a positron emission tomography scan should be performed.
Management
Multidisciplinary care is espoused in the management of patients with DM. A combined treatment programme delineated by specialists in dermatology and rheumatology is necessary in most cases. Clinical input from respiratory medicine and other specialities is often also necessary. Systemic corticosteroid is the primary treatment for DM when both skin and muscles are involved. Oral prednisolone, given at a dose of 0.5–1.0 mg/kg, is usually effective in controlling the symptoms; many patients report improvement in the rash and muscle weakness within 1–2 weeks of starting systemic corticosteroids. If oral prednisolone is lacking in efficacy, or the severity of the muscle disease is life-threatening, then pulsed intravenous methylprednisolone can be administered, 0.5–1 g/day for 3 consecutive days. The chronic nature of DM is such that systemic corticosteroids are usually required for a prolonged period. If the dose is not reduced then the patient will inevitably be at risk of the side effects of long-term glucocorticosteroid therapy. A number of immunosuppressant drugs are used to control the inflammatory process as the dose of prednisolone is slowly tapered [93]. There are very few controlled trials of immunosuppressant use in DM, however the following have been administered with varying benefit: azathioprine 100–200 mg daily, methotrexate 10–20 mg once per week, mycophenolate mofetil 500–1500 mg twice daily, ciclosporin 3.0–5.0 mg/kg/day and tacrolimus [93].
Intravenous immunoglobulin can be used as a disease-modifying therapy in DM and as a steroid-sparing agent. In 1993 Dalakaset al. reported a double-blind, placebo-controlled trial of intravenous immunoglobulin (IVIg) in 15 patients with DM [94]. The patients were maintained on prednisolone, at a mean dosage of 25 mg daily, and given either IVIg 2 g/kg body weight or placebo, each delivered monthly for 3 months. The patients who received IVIg demonstrated significant improvement in muscle strength and an observable improvement in skin disease [94]. However, the evidence for long-term IVIg therapy is based on case series only [95, 96].
A few published studies have investigated the role of rituximab in DM. An open-label pilot trial of seven patients studied outcome following 1 g of rituximab administered 2 weeks apart [97]. Three patients had a partial response with a reduction of muscle disease; the dermatosis did not improve. In a multicentre controlled trial, patients were randomized to receive rituximab at either weeks 0 and 1, with placebo at weeks 8 and 9, or placebo at weeks 0 and 1, with rituximab at weeks 8 and 9. Neither the primary nor the secondary end points, based on muscle parameters, were met. However, there was improvement in overall disease activity in both groups with 83% reaching the IMACS definition of improvement [98]. Additionally, in a subsequent post hoc analysis, anti-Jo-1 and Mi-2 antibodies strongly predicted clinical improvement [99].
Management of the skin involvement in DM is the prime concern for dermatologists. Systemic corticosteroid in combination with a steroid-sparing immunosuppressant is usually effective, however complete clearance of the dermatosis may take many months of therapy. In amyopathic DM, aggressive systemic immunosuppression may seem inappropriate, therefore topical therapy, or milder oral treatment, is usually tried. The efficacy of topical corticosteroid, including super-potent steroid, is often disappointing; however benefit from topical tacrolimus 0.1% ointment has been observed [100]. Uncontrolled studies of hydroxychloroquine, either alone or in combination with mepacrine, have been reported as effective in clearing the cutaneous signs of DM [101]. Methotrexate can also be beneficial in the management of skin disease in DM [102].
Calcinosis cutis is a troublesome and symptomatic complication of severe or poorly treated DM. Effective treatment of the underlying DM may lead to an improvement of calcinosis. Various therapeutic modalities have been tried, but studies are lacking to support the unequivocal benefit of any one particular treatment.
Resources
Further information
- International Myositis Assessment and Clinical Studies Group: www.niehs.nih.gov/research/resources/imacs.
- Myositis UK: www.myositis.org.uk.
(Both last accessed July 2015.)
References
- Bohan A, Peter JB. Polymyositis and dermatomyositis. N Engl J Med. 1975;292:344–7, 403–7.
- Sontheimer RD. Would a new name hasten the acceptance of amyopathic dermatomyositis (dermatomyositis siné myositis) as a distinctive subset within the idiopathic inflammatory dermatomyopathies spectrum of clinical illness? J Am Acad Dermatol 2002;46:626–36.
- Taborda AL, Azevedo P, Isenberg DA. Retrospective analysis of the outcome of patients with idiopathic inflammatory myopathy: a long-term follow-up study. Clin Exp Rheumatol 2014;32(2):188–93.
- Studynkova J, Mann H, Jarosova K, et al. A prospective, randomized, open-label, assessor-blind, multicenter study of efficacy and safety of combined treatment of methotrexate + glucocorticoids versus glucocorticoids alone in patients with polymyositis and dermatomyositis (prometheus trial). Ann Rheum Dis 2014;73(Suppl. 2):171.
- Lundberg IE, Forbess CJ. Mortality in idiopathic inflammatory myopathies. Clin Exp Rheumatol. 2008;26(5 Suppl. 51):S109–114.
- Smoyer-Tomic KE, Amato AA, Fernandes AW. Incidence and prevalence of idiopathic inflammatory myopathies among commercially insured, Medicare supplemental insured, and Medicaid enrolled populations: an administrative claims analysis. BMC Musculoskelet Disord 2012;13:103.
- Cooper GS, Stroehla BC. The epidemiology of autoimmune diseases. Autoimmun Rev. 2003;2:119–25.
- Oddis CV, Conte CG, Steen VD, Medsger TA. Incidence of polymyositis-dermatomyositis: a 20-year study of hospital diagnosed cases in Allegheny County, PA 1963–1982. J Rheumatol 1990;17(10):1329–34.
- Benbassat J, Geffel D, Zlotnick A. Epidemiology of polymyositis-dermatomyositis in Israel, 1960–76. Isr J Med Sci 1980;16(3):197–200.
- Bernatsky S, Joseph L, Pineau CA, et al. Estimating the prevalence of polymyositis and dermatomyositis from administrative data: age, sex and regional differences. Ann Rheum Dis 2009;68(7):1192–6.
- Hengstman GJ, van Venrooij WJ, Vencovsky J, Moutsopoulos HM, van Engelen BG. The relative prevalence of dermatomyositis and polymyositis in Europe exhibits a latitudinal gradient. Ann Rheum Dis. 2000;59(2):141–2.
- Love LA, Weinberg CR, McConnaughey DR, et al. Ultraviolet radiation intensity predicts the relative distribution of dermatomyositis and anti-Mi-2 autoantibodies in women. Arthritis Rheum 2009;60(8):2499–504.
- Bohan A, Peter JB, Bowman RL, Pearson CM. Computer-assisted analysis of 153 patients with polymyositis and dermatomyositis. Medicine (Baltimore) 1977;56(4):255–86.
- Dobloug C, Garen T, Bitter H, et al. Prevalence and clinical characteristics of adult polymyositis and dermatomyositis; data from a large and unselected Norwegian cohort. Ann Rheum Dis 2014;1551–6.
- Symmons DP, Sills JA, Davis SM. The incidence of juvenile dermatomyositis: results from a nation-wide study. Br J Rheumatol 1995;34(8):732–6.
- Hill CL, Zhang Y, Sigurgeirsson B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet 2001;357:96–100.
- Leow YH, Goh CL. Malignancy in adult dermatomyositis. Int J Dermatol 1997;36(12):904–7.
- Goh CL, Rajan VS. Dermatomyositis in a skin clinic. Ann Acad Med Singapore 1983;12(1):6–12.
- Henriksson KG, Lindvall B. Polymyositis and dermatomyositis 1990 – diagnosis, treatment and prognosis. Prog Neurobiol 1990;35(3):181–93.
- Buchbinder R, Forbes A, Hall S, Dennett X, Giles G. Incidence of malignant disease in biopsy-proven inflammatory myopathy. A population-based cohort study. Ann Intern Med 2001;134(12):1087–95.
- Ponyi A, Borgulya G, Constantin T, Váncsa A, Gergely L, Dankó K. Functional outcome and quality of life in adult patients with idiopathic inflammatory myositis. Rheumatology (Oxford) 2005;44(1):83–8.
- András C, Ponyi A, Constantin T, et al. Dermatomyositis and polymyositis associated with malignancy: a 21-year retrospective study. J Rheumatol 2008;35(3):438–44.
- Chinoy H, Fertig N, Oddis CV, Ollier WE, Cooper RG. The diagnostic utility of myositis autoantibody testing for predicting the risk of cancer-associated myositis. Ann Rheum Dis 2007;66(10):1345–9.
- Fiorentino DF, Chung LS, Christopher-Stine L, et al. Most patients with cancer-associated dermatomyositis have antibodies to nuclear matrix protein NXP-2 or transcription intermediary factor 1γ. Arthritis Rheum 2013;65(11):2954–62.
- Chen YJ, Wu CY, Huang YL, Wang CB, Shen JL, Chang YT. Cancer risks of dermatomyositis and polymyositis: a nationwide cohort study in Taiwan. Arthritis Res Ther 2010;12(2):R70.
- Kissel JT, Mendell JR, Rammohan KW. Microvascular deposition of complement membrane attack complex in dermatomyositis. N Engl J Med 1986;314:329–34.
- Kissel JT, Halterman RK, Rammohan KW, Mendell JR. The relationship of complement-mediated microvasculopathy to the histologic features and clinical duration of disease in dermatomyositis. Arch Neurol 1991;48:26–30.
- McDouall RM, Dunn MJ, Dubowitz V. Expression of class I and class II MHC antigens in neuromuscular diseases. J Neurol Sci 1989;89(2–3):213–26.
- Hathaway PW, Engel WK, Zellweger H. Experimental myopathy after microarterial embolization; comparison with childhood x-linked pseudohypertrophic muscular dystrophy. Arch Neurol 1970;22(4):365–78.
- Karpati G, Carpenter S, Melmed C, Eisen AA. Experimental ischemic myopathy. J Neurol Sci 1974;23(1):129–61.
- Greenberg SA, Pinkus JL, Pinkus GS, et al. Interferon-alpha/beta-mediated innate immune mechanisms in dermatomyositis. Ann Neurol 2005;57(5):664–78.
- Ghirardello A, Bassi N, Palma L, et al. Autoantibodies in polymyositis and dermatomyositis. Curr Rheumatol Rep 2013;15(6):335.
- Stone KB, Oddis CV, Fertig N, et al. Anti-Jo-1 antibody levels correlate with disease activity in idiopathic inflammatory myopathy. Arthritis Rheum 2007;56(9):3125–31.
- Muro Y, Sugiura K, Hoshino K, Akiyama M. Disappearance of anti-MDA-5 autoantibodies in clinically amyopathic DM/interstitial lung disease during disease remission. Rheumatology (Oxford) 2012;51(5):800–4.
- Howard OM, Dong HF, Yang D, et al. Histidyl-tRNA synthetase and asparaginyl-tRNA synthetase, autoantigens in myositis, activate chemokine receptors on T lymphocytes and immature dendritic cells. J Exp Med 2002;196:781–91.
- Levine SM, Raben N, Xie D, et al. Novel conformation of histidyl-transfer RNA synthetase in the lung: the target tissue in Jo-1 autoantibody-associated myositis. Arthritis Rheum 2007;56(8):2729–39.
- Casciola-Rosen L, Nagaraju K, Plotz P, et al. Enhanced autoantigen expression in regenerating muscle cells in idiopathic inflammatory myopathy. J Exp Med 2005;201(4):591–601.
- Walsh RJ, Kong SW, Yao Y, et al. Type I interferon-inducible gene expression in blood is present and reflects disease activity in dermatomyositis and polymyositis. Arthritis Rheum 2007;56(11):3784–92.
- Eloranta ML, Barbasso Helmers S, Ulfgren AK, Rönnblom L, Alm GV, Lundberg IE. A possible mechanism for endogenous activation of the type I interferon system in myositis patients with anti-Jo-1 or anti-Ro 52/anti-Ro 60 autoantibodies. Arthritis Rheum 2007;56(9):3112–24.
- Suber TL, Casciola-Rosen L, Rosen A. Mechanisms of disease: autoantigens as clues to the pathogenesis of myositis. Nat Clin Pract Rheumatol 2008;4(4):201–9.
- Sontheimer RD. Lichenoid tissue reaction/interface dermatitis: clinical and histological perspectives. J Invest Dermatol 2009;129(5):1088–99.
- Callen JP. Dermatomyositis. Lancet 2000;355(9197):53–7.
- Hundley JL, Carroll CL, Lang W, et al. Cutaneous symptoms of dermatomyositis significantly impact patients' quality of life. J Am Acad Dermatol 2006;54:217–20.
- Cheong WK, Hughes GR, Norris PG, Hawk JL. Cutaneous photosensitivity in dermatomyositis. Br J Dermatol 1994;131(2):205–8.
- Okiyama N, Kohsaka H, Ueda N, et al. Seborrheic area erythema as a common skin manifestation in Japanese patients with dermatomyositis. Dermatology 2008;217(4):374–7.
- Kasteler JS, Callen JP. Scalp involvement in dermatomyositis. Often overlooked or misdiagnosed. JAMA 1994;272(24):1939–41.
- Hasegawa M. Dermoscopy findings of nail fold capillaries in connective tissue diseases. J Dermatol 2011;38(1):66–70.
- Quinter SD, Chiu YE, Lyon VB, Holland KE, Ruggeri SY, Drolet BA. Inverse Gottron's papules: an unusual cutaneous manifestation of juvenile dermatomyositis. Pediatr Dermatol 2012;29(5):641–4.
- Nishigori K, Yamamoto T, Yokozeki H. Vesiculo-bullous dermatomyositis: report of three cases. Dermatol Online J 2009;15(4):6.
- Fiorentino D, Chung L, Zwerner J, Rosen A, Casciola-Rosen L. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol 2011;65(1):25–34.
- Nousari HC, Ha VT, Laman SD, Provost TT, Tausk FA. “Centripetal flagellate erythema”: a cutaneous manifestation associated with dermatomyositis. J Rheumatol 1999;26(3):692–5.
- Yamamoto T, Nishioka K. Flagellate erythema. Int J Dermatol 2006;45(5):627–31.
- Sato Y, Teraki Y, Izaki S, Amano K. Clinical characterization of dermatomyositis associated with mechanic's hands. J Dermatol 2012;39(12):1093–5.
- Valenzuela A, Chung L, Casciola-Rosen L, Fiorentino D. Identification of clinical features and autoantibodies associated with calcinosis in dermatomyositis. JAMA Dermatol 2014;150(7):724–9.
- Solans R, Cortes J, Selva A, et al. Panniculitis: a cutaneous manifestation of dermatomyositis. J Am Acad Dermatol 2002;46:S148–50.
- Sultan SM. Clinical assessment in adult onset idiopathic inflammatory myopathy. Curr Opin Rheumatol 2004;16(6):668–72.
- Rider LG, Koziol D, Giannini EH, et al. Validation of manual muscle testing and a subset of eight muscles for adult and juvenile idiopathic inflammatory myopathies. Arthritis Care Res (Hoboken) 2010;62(4):465–72.
- Alexanderson H, Broman L, Tollbäck A, Josefson A, Lundberg IE, Stenström CH. Functional index-2: validity and reliability of a disease-specific measure of impairment in patients with polymyositis and dermatomyositis. Arthritis Rheum 2006;55(1):114–22.
- Dankó K, Ponyi A, Constantin T, Borgulya G, Szegedi G. Long-term survival of patients with idiopathic inflammatory myopathies according to clinical features: a longitudinal study of 162 cases. Medicine (Baltimore) 2004;83(1):35–42.
- Marie I, Hachulla E, Chérin P, et al. Interstitial lung disease in polymyositis and dermatomyositis. Arthritis Rheum 2002;47(6):614–22.
- Chen YJ, Wu CY, Shen JL. Predicting factors of interstitial lung disease in dermatomyositis and polymyositis. Acta Derm Venereol 2007;87(1):33–8.
- Kang EH, Lee EB, Shin KC, et al. Interstitial lung disease in patients with polymyositis, dermatomyositis and amyopathic dermatomyositis. Rheumatology (Oxford) 2005;44:1282–6.
- Selva-O'Callaghan A, Labrador-Horrillo M, Muñoz-Gall X, et al. Polymyositis/dermatomyositis-associated lung disease: analysis of a series of 81 patients. Lupus 2005;14(7):534–42.
- Chua F, Higton AM, Colebatch AN, et al. Idiopathic inflammatory myositis-associated interstitial lung disease: ethnicity differences and lung function trends in a British cohort. Rheumatology (Oxford) 2012;51(10):1870–6.
- Kiely PD, Chua F. Interstitial lung disease in inflammatory myopathies: clinical phenotypes and prognosis. Curr Rheumatol Rep 2013;15(9):359.
- Gunawardena H, Betteridge ZE, McHugh NJ. Myositis-specific autoantibodies: their clinical and pathogenic significance in disease expression. Rheumatology (Oxford) 2009;48:607–12.
- Marie I, Hatron PY, Dominique S, et al. Short-term and long-term outcome of anti-Jo1-positive patients with anti-Ro52 antibody. Semin Arthritis Rheum 2012;41(6):890–9.
- Hervier B, Devilliers H, Stanciu R, et al. Hierarchical cluster and survival analyses of antisynthetase syndrome: phenotype and outcome are correlated with anti-tRNA synthetase antibody specificity. Autoimmun Rev 2012;12:210–17.
- Tugnet N, Rees DH. Calcinosis in juvenile dermatomyositis. Postgrad Med J 2010;86(1018):510.
- Huemer C, Kitson H, Malleson PN, et al. Lipodystrophy in patients with juvenile dermatomyositis – evaluation of clinical and metabolic abnormalities. J Rheumatol 2001;28(3):610–15.
- Shah M, Mamyrova G, Targoff IN, et al. The clinical phenotypes of the juvenile idiopathic inflammatory myopathies. Medicine (Baltimore) 2013;92(1):25–41.
- Gunawardena H, Wedderburn LR, North J, et al. Clinical associations of autoantibodies to a p155/140 kDa doublet protein in juvenile dermatomyositis. Rheumatology (Oxford) 2008;47(3):324–8.
- Neri R, Barsotti S, Simone B, et al. Cancer-associated myositis: a 35-year retrospective study of a monocentric cohort. Rheumatol Int 2014;34(4):565–9.
- Cao H, Pan M, Kang Y, et al. Clinical manifestations of dermatomyositis and clinically amyopathic dermatomyositis patients with positive expression of anti-melanoma differentiation-associated gene 5 antibody. Arthritis Care Res (Hoboken) 2012;64:1602–10.
- Seidler AM, Gottlieb AB. Dermatomyositis induced by drug therapy: a review of case reports. J Am Acad Dermatol 2008;59:872–80.
- Daoud MS, Gibson LE, Pittelkow MR. Hydroxyurea dermopathy: a unique lichenoid eruption complicating long-term therapy with hydroxyurea. J Am Acad Dermatol 1997;36(2 Pt 1):178–82.
- Carroll CL, Lang W, Snively B, Feldman SR, Callen J, Jorizzo JL. Development and validation of the Dermatomyositis Skin Severity Index. Br J Dermatol 2008;158(2):345–50.
- Isenberg DA, Allen E, Farewell V, et al. International consensus outcome measures for patients with idiopathic inflammatory myopathies. Development and initial validation of myositis activity and damage indices in patients with adult onset disease. Rheumatology (Oxford) 2004;43(1):49–54.
- Tisseverasinghe A, Bernatsky S, Pineau CA. Arterial events in persons with dermatomyositis and polymyositis. J Rheumatol 2009;36(9):1943–6.
- Zöller B, Li X, Sundquist J, Sundquist K. Risk of subsequent coronary heart disease in patients hospitalized for immune-mediated diseases: a nationwide follow-up study from Sweden. PLOS One 2012;7(3):e33442.
- Bronner IM, van der Meulen MF, de Visser M, et al. Long-term outcome in polymyositis and dermatomyositis. Ann Rheum Dis 2006;65:1456–61.
- Reichlin M, Arnett FC. Multiplicity of antibodies in myositis sera. Arthritis Rheum 1984;27(10):1150–6.
- Rutjes SA, Vree Egberts WT, Jongen P, Van Den Hoogen F, Pruijn GJ, Van Venrooij WJ. Anti-Ro52 antibodies frequently co-occur with anti-Jo-1 antibodies in sera from patients with idiopathic inflammatory myopathy. Clin Exp Immunol 1997;109(1):32–40.
- Tansley S, Gunawardena H. The evolving spectrum of polymyositis and dermatomyositis – moving towards clinicoserological syndromes: a critical review. Clin Rev Allergy Immunol 2014;47(3):264–73.
- Sato S, Hirakata M, Kuwana M, et al. Autoantibodies to a 140-kd polypeptide, CADM-140, in Japanese patients with clinically amyopathic dermatomyositis. Arthritis Rheum 2005;52(5):1571–6.
- Nakashima R, Imura Y, Kobayashi S, et al. The RIG-I-like receptor IFIH1/MDA5 is a dermatomyositis-specific autoantigen identified by the anti-CADM-140 antibody. Rheumatology (Oxford) 2010;49(3):433–40.
- Chaisson NF, Paik J, Orbai AM, et al. A novel dermato-pulmonary syndrome associated with MDA-5 antibodies: report of 2 cases and review of the literature. Medicine (Baltimore) 2012;91(4):220–8.
- Kaji K, Fujimoto M, Hasegawa M, et al. Identification of a novel autoantibody reactive with 155 and 140 kDa nuclear proteins in patients with dermatomyositis: an association with malignancy. Rheumatology (Oxford) 2007;46(1):25–8.
- Gunawardena H, Wedderburn LR, Chinoy H, et al. Autoantibodies to a 140-kd protein in juvenile dermatomyositis are associated with calcinosis. Arthritis Rheum 2009;60(6):1807–14.
- Targoff IN, Reichlin M. The association between Mi-2 antibodies and dermatomyositis. Arthritis Rheum 1985;28(7):796–803.
- Betteridge ZE, Gunawardena H, Chinoy H, et al. Clinical and human leucocyte antigen class II haplotype associations of autoantibodies to small ubiquitin-like modifier enzyme, a dermatomyositis-specific autoantigen target, in UK Caucasian adult-onset myositis. Ann Rheum Dis 2009;68(10):1621–5.
- Streib EW, Wilbourn AJ, Mitsumoto H. Spontaneous electrical muscle fiber activity in polymyositis and dermatomyositis. Muscle Nerve 1979;2(1):14–18.
- Gordon PA, Winer JB, Hoogendijk JE, Choy EH. Immunosuppressant and immunomodulatory treatment for dermatomyositis and polymyositis. Cochrane Database Syst Rev 2012;Issue 8:CD003643.
- Dalakas MC, Illa I, Dambrosia JM, et al. A controlled trial of high-dose intravenous immune globulin infusions as treatment for dermatomyositis. N Engl J Med 1993;329:1993–2000.
- Femia AN, Eastham AB, Lam C, Merola JF, Qureshi AA, Vleugels RA. Intravenous immunoglobulin for refractory cutaneous dermatomyositis: a retrospective analysis from an academic medical center. J Am Acad Dermatol 2013;69(4):654–7.
- Kampylafka EI, Kosmidis ML, Panagiotakos DB, Dalakas M, Moutsopoulos HM, Tzioufas AG. The effect of intravenous immunoglobulin (IVIG) treatment on patients with dermatomyositis: a 4-year follow-up study. Clin Exp Rheumatol 2012;30(3):397–401.
- Chung L, Genovese MC, Fiorentino DF. A pilot trial of rituximab in the treatment of patients with dermatomyositis. Arch Dermatol 2007;143(6):763–7.
- Oddis CV, Reed AM, Aggarwal R, et al. Rituximab in the treatment of refractory adult and juvenile dermatomyositis and adult polymyositis: a randomized, placebo-phase trial. Arthritis Rheum 2013;65(2):314–24.
- Aggarwal R, Bandos A, Reed AM, et al. Predictors of clinical improvement in rituximab-treated refractory adult and juvenile dermatomyositis and adult polymyositis. Arthritis Rheumatol 2014;66(3):740–9.
- Hollar CB, Jorizzo JL. Topical tacrolimus 0.1% ointment for refractory skin disease in dermatomyositis: a pilot study. J Dermatolog Treat 2004;15(1):35–9.
- Ang GC, Werth VP. Combination antimalarials in the treatment of cutaneous dermatomyositis: a retrospective study. Arch Dermatol 2005;141(7):855–9.
- Fendler C, Braun J. Use of methotrexate in inflammatory myopathies. Clin Exp Rheumatol 2010;28:S164–7.