CHAPTER 54
Mixed connective tissue disease
Mark Goodfield
Department of Dermatology, Chapel Allerton Hospital, Leeds, UK
Definition and nomenclature
Mixed connective tissue disease (MCTD) [1], or overlap syndrome, describes the clinical situation in which symptoms of a number of connective tissue diseases (CTDs) occur in one patient. There may be specific antibody profiles associated with these constellations of symptoms, such as the presence of anti-U1-ribonucleoprotein (anti-U1-RNP) in Sharp's original description of MCTD in 1972 [2]. Progression to a specific CTD may occur, but many patients continue to have a mixed and static pattern of symptomatology.
Introduction and general description
Overlap syndromes, in which there are symptoms suggestive of more than one CTD, occur relatively commonly. This may be one stage in the development of a classic CTD, or a more persistent entity. More variants are being described, both with and without the U1-RNP antibody of Sharp's original description. Hence, some authors now regard MCTD as one among many ‘undifferentiated CTDs’ or ‘overlap syndromes’, and others doubt the validity of the concept, particularly in children [3]. Nevertheless, it is important to recognize this group of patients because of the generally good prognosis and the response to corticosteroid therapy. There have been a number of attempts at defining diagnostic criteria along the lines of those used for systemic lupus erythematosus (SLE) [4]. Some associations are more frequent than others: for example, systemic sclerosis combined with dermatomyositis is more frequent than systemic sclerosis combined with SLE [5].
Epidemiology
Incidence and prevalence
In a Norwegian study of 147 adults, the point prevalence of adult MCTD patients was 3.8 (95% confidence interval (CI) 3.2–4.4) per 100 000 adults. The incidence of adult-onset MCTD in Norway during the period from 1996 to 2005 was 2.1 (95% CI 1.7–2.5) per million per year [6].
Age
The mean age at diagnosis of adult-onset MCTD was 37.9 years (95% CI 35.3–40.4 years) in the Norwegian study quoted above [6]. The condition can occur in childhood and may be more severe in children, in whom cardiac renal disease and arthritis are common, and thrombocytopenia may be marked, although the overall prognosis may be quite good [7].
Sex
The condition is more common in females, with a female to male ratio of 3.3 : 1 in Norway in 2008.
Pathophysiology
In the classic form of MCTD, all patients have the speckled type of antinuclear antibody together with a high titre of antibody to extractable nuclear antigen (ENA). Different molecular forms of U1-RNP may be associated with different clinical variants [8]. Ro and La antibodies are found frequently, usually in association with sicca symptoms [9]. Precipitating antibodies designated PM-1 and Ku occur in polymyositis/systemic sclerosis overlap [10], and SL-Ki in patients with SLE, scleroderma and sicca syndrome [11]. Immune complexes occur in 90% of patients, and T cells are decreased. Complement levels are normal. Sometimes, anti-DNA antibody occurs in low titre, but this usually disappears with corticosteroid therapy. Anti-endothelial antibodies occur in approximately 50% of cases, and are associated with abnormal pulmonary, neurological and cardiac function, particularly pulmonary hypertension [12]. They are also related significantly to spontaneous abortion in female patients [13].
Predisposing factors
No predisposing factors have been identified.
Pathology
The microscopic appearances of the skin vary with the pattern of the disease. Immunohistology of uninvolved skin, where there is basement membrane staining with immunglobulin G (IgG) or IgM, may be helpful in distinguishing MCTD from uncomplicated systemic sclerosis where staining is absent [14]. Direct immunofluorescence study of apparently normal skin reveals particulate (‘speckled’) epidermal nuclear staining, and this correlates with high titres of anti-RNP. Epidermal nucleolar staining is associated with alopecia, hyper- and hypopigmentation, swollen hands with sclerodactyly, and lesions of discoid lupus erythematosus (DLE).
Genetics
Differences in HLA antigens between patients with MCTD and SLE indicate that they are genetically separate disorders [15]. HLA-DR4 is found more commonly in patients with arthritis than in normal controls, and is associated with a young age of onset of the condition [16]. Familial cases occur.
Environmental factors
Ultraviolet radiation may be relevant in precipitating and exacerbating lupus-like elements of MCTD [17]. Vitamin D deficiency has also been associated with the condition, although its relevance is unclear [18]. The role of silicone breast implants in the pathogenesis of the overlap syndromes is controversial; there is probably no relationship [19].
Clinical features
History
Patients, predominantly female, usually present with a history of joint symptoms, Raynaud phenomenon and skin changes – either erythematous skin rashes or skin swelling and thickening.
Presentation
Patients will show features of SLE, systemic sclerosis, dermatomyositis and polymyositis in varying combinations. Raynaud phenomenon, arthritis and arthralgia, sausage-shaped fingers and swelling of the dorsa of the hands are common presenting features. More than half have the abnormal nail fold capillaries seen in systemic sclerosis [20]. There may be prominent neurological or neuropsychiatric features [21], although these are less frequent and less severe than those seen in SLE. Other less frequent features include oro-genital ulceration, panniculitis and autonomic neuropathy [22].
Clinical variants
By its very nature, the condition is variable in its features, both at presentation and as the condition progresses. Features may lead to a diagnosis of a single CTD or may remain ‘mixed’ throughout the course of the illness.
Differential diagnosis
Any of the specific CTDs need to be considered in the differential diagnosis.
Classification of severity
There has been an attempt at staging the severity of MCTD, but this is not widely used [4].
Complications and co-morbidities
Thrombocytopenia is also occasionally found in adult cases, and may occur with thrombotic thrombocytopenic purpura [23]. Livedoid vasculitis [24], ankylosing spondylitis [25] and antineutrophil cytoplasmic antibody (ANCA) related glomerulonephritis [26] are reported to occur with MTCD. There may be an increased risk of malignancy, with cancer developing in 10% of cases [28]. Peripheral gangrene may occur due to obstruction of small blood vessels (Figure 54.1). Although the presence of anti-RNP is usually associated with a good prognosis, death occurs in approximately 4% from pulmonary hypertension, nephritis, myocarditis or widespread vasculitis [29].
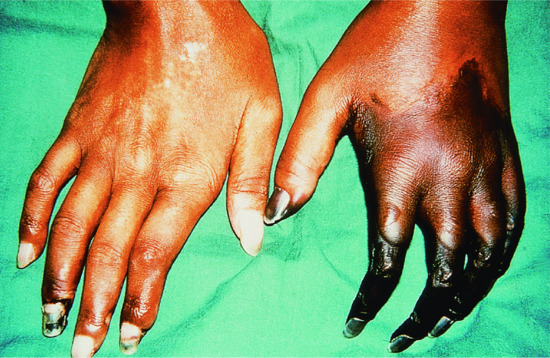
Figure 54.1 Gangrene of the hands in mixed connective tissue disease.
Approximately one-third of cases develop into a characteristic CTD, usually SLE or systemic sclerosis [30]. A few patients with HLA-DR4 develop rheumatoid arthritis. In patients whose disease differentiated into systemic sclerosis, there was an association with HLA-DR5 [31].
Investigations
The purpose of investigation is to establish the nature and extent of the disease. Autoantibody testing is fundamental, since the antibody profile helps to define the condition. Investigation of potential internal organ involvement should include routine assessment of renal and pulmonary function. Abnormal oesophageal motility, impaired pulmonary diffusing capacity and myositis are frequently found on investigation. The incidence of clinical renal disease is approximately 5%, but renal histology may be abnormal in 20% [32].
Management
First line
The response to treatment with corticosteroids is good and should be used early, but in controlled doses. Most reports use 0.5 mg/kg body weight or less, with rapid dose reduction [1]. As with the specific CTDs, symptomatic treatment for particular symptoms is needed: vascular symptoms should be treated with oral and intravenous vasodilators [33].
Second line
Other successful treatment options, including hydroxychloroquine and methotrexate, have been reviewed [1]. Haematological manifestations may need to be managed with anti-B-cell treatment [34]; renal and neurological disease may require treatment with plasmapheresis or more aggressive immunosuppression.
References
- Ortega-Hernandez OD, Shoenfeld Y. Mixed connective tissue disease: an overview of clinical manifestations, diagnosis and treatment. Best Pract Res Clin Rheumatol 2012;26:61–72.
- Sharp GC, Irvin WS, Tan EM, et al. Mixed connective tissue disease. Am J Med 1972;52:148–59.
- Mier R, Ansell B, Hall MA, et al. Long-term follow-up of children with mixed connective tissue disease. Lupus 1996;5:221–6.
- Alarcón-Segoviá D, Cardiel MH. Comparison between three diagnostic criteria for mixed connective tissue disease: study of 593 patients. J Rheumatol 1989;16:328–56.
- Minkin W, Rabhan N. Mixed connective tissue disease. Arch Dermatol 1976;112:1535–8.
- Gunnarsson R, Molberg O, Gilboe IM, et al. The prevalence and incidence of mixed connective tissue disease: a national multicentre survey of Norwegian patients. Ann Rheum Dis 2011;70:1047–51.
- Mier RJ, Shishov M, Higginns GC, et al. Paediatric onset mixed connective tissue disease. Rheum Dis Clin North Am 2005;31:483–96.
- Greidinger EL, Casciola-Rosen L, Morris SM, et al. Autoantibody recognition of distinctly modified forms of the U1-70-kD antigen is associated with different clinical disease manifestations. Arthritis Rheum 2000;43:881–8.
- Setty YN, Pittman CB, Mahale AS, et al. Sicca symptoms and anti-SSA/Ro antibodies are common in mixed connective tissue disease. J Rheumatol 2002;29:487–9.
- Szodoray P, Hajas A, Kardos L, et al. Distinct phenotypes in mixed connective tissue disease: subgroups and survival. Lupus 2012;21:1412–22.
- Parodi A, Nigro A, Rebora A. Anti-SL-Ki antibody in a patient with fatal connective tissue overlap disease. Br J Dermatol 1989;121:243–6.
- Vegh J, Szodoray P, Kappelmayer J, et al. Clinical and immunoserological characteristics of mixed connective tissue disease associated with pulmonary hypertension. Scand J Immunol.2006;64:69–76.
- Bodolay E, Bojan F, Szegedi G, et al. Cytotoxic endothelial cell antibodies in mixed connective tissue disease. Immunol Lett 1989;20:163–8.
- Winkelmann RK, Carapeto FJ, Jordon RE. Direct immunofluorescence in the diagnosis of scleroderma syndromes. Br J Dermatol 1977;96:231–8.
- Black CM, Maddison PJ, Welsh KI, et al. HLA and immunoglobulin allotypes in mixed connective tissue disease. Arthritis Rheum 1988:31:131–4.
- Ruuska P, Hameenkorpi R, Forsberg S, et al. Differences in HLA antigens between patients with mixed connective disease and systemic lupus erythematosus. Ann Rheum Dis 1992;51:52–5.
- Zandman-Goddard G, Solomon M, Rosman Z, Peeva E, Shoenfeld Y. Environment and lupus-related diseases. Lupus 2012;21:241–50.
- Hajas A, Sandor J, Csathy L, et al. Vitamin D insufficiency in a large MCTD population. Autoimmun Rev 2011;10:317–24.
- McLaughlin JK, Lipworth L, Murphy DK, Walker PS. The safety of silicone gel-filled breast implants: a review of the epidemiologic evidence. Ann Plast Surg 2007;59:569–80.
- Kondo H. Vascular disease in mixed connective tissue disease (MCTD). Intern Med 2001;40:1176.
- Nowicka-Sauer K, Czuszynska Z, Majkowicz M, et al. Neuropsychological assessment in mixed connective tissue disease: comparison with systemic lupus erythematosus. Lupus 2012;21(9):927–33.
- Pope JE. Other manifestations of mixed connective tissue disease. Rheum Dis Clin North Am.2005;31:519–33.
- Poullin P, Lefevre P, Durand JM. Mixed connective tissue disease with haemolytic anemia and severe thrombocytopenia due to thrombotic thrombocytopenic purpura. Am J Hematol 1999; 61:275.
- Oh YB, Jun JB, Kim CK, et al. Mixed connective tissue disease associated with skin defects of livedoid vasculitis. Clin Rheumatol 2000;19:381–4.
- Lee JK, Jung SS, Kim TH, et al. Coexistence of ankylosing spondylitis and mixed connective tissue disease in a single patient. Clin Exp Rheumatoid 1999;17:63.
- Makita N, Katori H, Takemoto F, et al. A case of mixed connective tissue disease (MCTD) complicated with MPO-ANCA-related necrotising glomerulonephritis. Clin Nephrol 2000;54:164–8.
- Black KA, Zilko PJ, Dawkins RL, et al. Cancer in connective tissue disease. Arthritis Rheum 1982;25:1130–3.
- Lundberg IE. The prognosis of mixed connective tissue disease. Rheum Clin North Am.2005;31:535–47.
- Bodoly E, Csiki Z, Ben T, et al. Five year follow up of 665 Hungarian patients with undifferentiated connective tissue disease (UCTD). Clin Exp Rheumatol.2003;21:313–20.
- Gendi NS, Welsh KI, Van-Venrooij WJ, et al. HLA type as a predictor of mixed connective tissue disease differentiation: 10 year clinical and immunogenetic follow-up of 46 patients. Arthritis Rheum 1995;38:259–66.
- Konstantinov KN, Harris AA, Barry M, Murata GH, Tzamaloukas AH. Sustained remission of antineutrophil cytoplasmic antibody-mediated glomerulonephritis and nephrotic syndrome in mixed connective tissue disease. J Clin Med Res 2013;5(4):316–21.
- Jovancevic B, Lindholm C, Pullerits R. Anti B-cell therapy against refractory thrombocytopenia in SLE and MCTD patients: long-term follow-up and review of the literature. Lupus 2013;22(7):664–74.
- Seguchi M, Soejima Y, Tateishi A, Iida H, et al. Mixed connective tissue disease with multiple organ damage: successful treatment with plasmapheresis. Intern Med 2000;39:1119–22.
- Poullin P, Lefevre P, Durand JM. Mixed connective tissue disease with hemolytic anemia and severe thrombocytopenia due to thrombotic thrombocytopenic purpura. Am J Haematol 1999;61:275.