CHAPTER 57
Morphoea and Allied Scarring and Sclerosing Inflammatory Dermatoses
Catherine H. Orteu
Department of Dermatology, Royal Free London NHS Foundation Trust, London, UK
Definition and nomenclature
The term scleroderma derives from the Greek skleros, meaning hard, and derma, meaning skin. It has been used to encompass a spectrum of disorders that include systemic sclerosis (SSc), covered in Chapter 56, and localized forms of the disease. Morphoea, a term used in preference to ‘localized scleroderma’, encompasses a group of related conditions characterized by varying degrees of sclerosis, fibrosis and atrophy in the skin and subcutaneous tissues, sometimes extending deeply into muscle, bone and brain. Extracutaneous manifestations occur in up to 25% of cases but in contrast to SSc, no internal organ fibrosis or vascular changes occur. Antinuclear antibody (ANA) positivity is common but the specific autoantibodies seen in SSc are rarely present. There is no increased mortality, but substantial morbidity may occur as a result of joint contractures, facial and limb asymmetry, extracutaneous manifestations and the psychological impact of the condition [1–4].
Terminology
The nomenclature is confusing for patients and doctors. On hearing the term scleroderma, even when it is prefixed by localized, many patients assume they have SSc. This is compounded by the results of online searches that frequently find the terms used synonymously, and rarely give much information on localized forms of the disease. The terminology can be confusing for doctors too, for instance when terms such as ‘generalized localized scleroderma’ are used. Localized scleroderma is an umbrella term employed in the literature to encompass the various forms of morphoea – namely limited, generalized, linear and mixed forms of morphoea – which do not usually involve internal organs such as the lungs, heart and kidneys. The term morphoea is preferred by dermatologists for the various subtypes. In contrast, linear forms, which are commoner in childhood, are generally referred to in the paediatric and rheumatology literature as linear scleroderma. The term morphoea will be employed throughout this chapter.
Classification
There has been controversy as to which conditions should be included within the morphoea spectrum. This applies particularly to eosinophilic fasciitis and lichen sclerosis, and to atrophic variants, such as atrophoderma of Pasini–Pierini, linear atrophoderma of Moulin and progressive hemifacial atrophy. There has also been debate over whether deep and bullous morphoea should be considered as separate subtypes, and over what, exactly, constitutes generalized morphoea.
Several different classification systems have been proposed (Table 57.1) [5–7]. The most widely used in the literature has been that of Peterson et al. [5] based on the different clinical phenotypes. This classification remains controversial, firstly because it includes atrophoderma of Pasini–Pierini, lichen sclerosus and eosinophilic fasciitis which are not universally agreed to be within the morphoea spectrum, and secondly because it does not include a category for the 15–23% of patients who present a ‘mixed’ subtype (e.g. linear and plaque morphoea) [8, 9]. To address this, the European Society for Paediatric Rheumatology proposed a second classification excluding atrophoderma, lichen sclerosus and eosinophilic fasciitis, but including separate pansclerotic and mixed subtypes [6]. In 2009 the German Dermatological Society proposed a third classification subdividing morphoea into limited, generalized, linear and deep types, including atrophoderma and eosinophilic fasciitis within the classification, but not lichen sclerosus or a mixed subtype. Although this classification was helpfully based on the amount, extent and depth of fibrosis, the terminology employed was somewhat cumbersome (e.g. generalized localized scleroderma; linear localized scleroderma ‘en coup de sabre’ type) [7]. The classification employed here is an ‘inclusive’ one that incorporates elements from each of the previous classifications (Tables 57.1 and 57.2). It distinguishes limited, generalized, linear and mixed subtypes as well as a separate morphoea–lichen sclerosus overlap group. The level of depth of involvement can vary and it is understood that deep involvement can occur in any of the subtypes. The various atrophic variants are included since they frequently overlap with other forms of morphoea, although it is accepted that this remains controversial. Generalized morphoea is used as an umbrella term for those patients with widespread disease. It encompasses patients with multiple, typical morphoea plaques termed disseminated plaque morphoea, as well as a group with pansclerotic, near whole-body circumferential involvement as described by Kim et al. [10]. Eosinophilic fasciitis is another controversial inclusion, which interestingly can occur in a widespread fashion resembling pansclerotic disease, or more rarely in a limited fashion in association with plaques of morphoea elsewhere. This raises the question as to whether it could represent an intermediate condition between disseminated plaque and pansclerotic disease and it is included within the generalized group [11–14].
Table 57.1 Classification systems for morphoea
| Peterson et al. 1995 [5] | Laxer and Zulian 2006 [6] | Kreuter et al. 2009 [7] | Classification and terminology used in this chapter |
Plaque morphoea
|
Circumscribed morphoea
|
Limited type
|
Limited type
|
| Generalized morphoea(lesions at three or more anatomical sites) | Generalized morphoea | Generalized type
|
Generalized type
|
Linear morphoea
|
Linear morphoea
|
Linear type
|
Linear type
|
| Bullous morphoea | Pansclerotic morphoea | Deep type | Mixed type |
Deep morphoea
|
Mixed morphoea | Lichen sclerosus with morphoea |
Table 57.2 Proposed modified classification of morphoea and subtype characteristics
| Main division | Subtype | Description |
| Limited type | Limited plaque morphoea | Single or multiple round to oval lesions >1 cm in diameter in up to two anatomical regions. May be oedematous, erythematous to bruise-like, yellowish white, indurated ± a lilac ring, or atrophic and pigmented. Involves epidermis and dermis |
| Guttate morphoea | Multiple small <1 cm erythematous to yellowish white, round to oval lesions, usually on the trunk. Involves the papillary and superficial dermis | |
| Atrophoderma of Pasini–Pierini | Multiple, round to oval, non-indurated, sharply demarcated, depressed patches, ‘cliff-drop’ edge, and usually hyperpigmented. Involves the superficial reticular dermis | |
| Keloidal/nodular morphoea | Keloid-like or nodular lesions arising from normal or sclerodermatous skin, usually on the trunk | |
| Limited deep morphoea | Solitary or multiple lesions in up to two anatomical sites. Poorly defined, thickened and bound down, sometimes with cobblestone or peau d'orange appearance. Involves deep dermis and subcutis ± fascia and muscle. Overlying skin may appear normal or puckered | |
| Generalized type | Disseminated plaque morphoea | Occurrence of multiple plaques of morphoea at three or more anatomical sitesa Isomorphic pattern: plaques coalesce in the inframammary area and bra-line, waistband and around the hips and inguinal regions at sites of repeated minor trauma from clothing Non-isomorphic pattern: multiple individual plaques occur in a usually symmetrical distribution on the trunk and limbs. Deep involvement may occur |
| Pansclerotic morphoea | Circumferential involvement of the majority of body surface areas with sparing of fingers, toes and nipples. Affects the dermis and frequently the subcutis, muscle and/or bone. No internal organ fibrosis | |
| Eosinophilic fasciitis | Symmetrically involves the extremities, but spares the fingers and face, and infrequently affects the trunk. Painful, burning erythema followed by peau d'orange appearance and guttering around vessels and tendons. Skin is bound down to underlying structures. Involves deep fascia and muscle. Dermis may be sclerotic or normal. May coexist with plaque morphoea | |
| Linear type | ||
| Trunk/limb variant | Linear morphoea | Blaschkoid linear induration of the limbs or trunk involving the dermis, subcutaneous tissue ± underlying muscle and bone |
| Linear atrophoderma of Moulin | Blaschkoid hyperpigmented linear atrophic limb/trunk lesions. Involve superficial dermis | |
| Linear deep atrophic morphoea | Linear atrophic lesions involving the deep dermis and subcutis | |
| Head/neck variant | Morphoea en coup de sabre | Blaschkoid linear induration affecting the face and scalp, may involve underlying muscle, bone, eye and brain |
| Progressive hemifacial atrophy | Non-indurated skin, occasional bruise-like pigmentation with associated underlying atrophy on one side of the face. Probable Blaschkoid distribution. May involve the dermis, subcutaneous tissue, muscle, bone, eye and brain | |
| Mixed type | A combination of two or more of the above subtypes, most often linear and plaque | |
| Morphoea–lichen sclerosus overlap | Morphoea and extragenital lichen sclerosus lesions may occur at the same site or at different sites. Small patches of lichen sclerosus may arise within a larger plaque of morphoea. Usually truncal but may be widespread. Evidence of genital lichen sclerosus should be sought |
aThe seven anatomical sites include the head–neck, right and left upper limbs, right and left lower limbs, anterior trunk and posterior trunk.
The variations between classification systems has caused confusion and difficulty in interpretation of the published literature on morphoea. For example, eosinophilic fasciitis and disabling pansclerotic morphoea, which are usually extensive and involve structures deep to the skin, have been variably considered to be within the deep morphoea or generalized morphoea subtypes. The term ‘deep morphoea’ implies involvement of the deep dermis, subcutis, fascia, muscle and sometimes bone. Lesions may involve any combination of these structures. They may be confined to the deep dermis and subcutis, or to the fascia, or the underlying muscle, but can in other cases involve all of these structures. Lesions are poorly defined and ‘bound down’ and may have a peau d'orange or cobblestone appearance with ‘guttering’ along vessels and tendons. Since deep involvement can occur at varying levels in the context of any of the subtypes of morphoea, in this chapter it is not considered as a separate subtype. Similarly, bullous lesions can occur in any subtype of morphoea, usually in the context of the active inflammatory phase of the disease. They are most frequent on the lower legs. Their development has been linked to the presence of oedema in the papillary dermis, to lymphatic dilatation and to an increased release of major basic protein from eosinophils [15–18]. There are in addition some very rare linear, deep atrophic forms, and superficial forms of linear atrophoderma, which are thought to be within the morphoea spectrum, and are included within the linear subtype here. There is a lack of agreement as to what constitutes ‘generalized morphoea’. At least three different definitions have been proposed (see the section on generalized types in Clinical variants). From a clinical perspective, in addition to eosinophilic fasciitis, at least two other distinct phenotypes can be identified within this subtype. In the first, patients present with multiple, circumscribed plaques of morphoea at more than three body sites, most often over the trunk, with varying degrees of confluence and sometimes in an isomorphic distribution (see the trauma section in Predisposing factors) – herein referred to as disseminated plaque morphoea. In the second, rarer group, patients have extensive confluent morphoea, frequently with deep involvement, over a large part of the trunk and limbs, but sparing the areolae, fingers and toes. Whether they are adults or children, these patients are best referred to as having a pansclerotic subtype of morphoea. Laxer and Zulian [6] and Kim et al. [10] consider this to be a separate subtype of morphoea, however it is likely that amongst adults such cases have been included within the generalized morphoea group in a majority of the published literature. Whilst this group clearly has a distinctive and often severe form of the disease [10], it is retained within the generalized subtype in this text.
Introduction and general description
Morphoea comprises a group of related diseases that share a common underlying pathophysiology of increased collagen deposition in an autoimmune setting (see Table 57.2). They are largely confined to the skin and subcutaneous tissues, including the underlying fat, fascia, muscle, bone and joints and occasionally with involvement of the eye and brain [1–3]. Overall, one-fifth to one-quarter of patients experience extracutaneous manifestations, including musculoskeletal, neurological, ocular, vascular (including Raynaud phenomenon) and gastrointestinal complications [9, 19–22]. Autoantibodies such as ANA, antihistone and anti-ssDNA may be present, but the SSc-specific autoantibodies such as antitopoisomerase, anticentromere and anti-RNA polymerase are rarely found. It is distinguished from SSc by the absence of sclerodactyly and nail fold capillary changes. The internal organ involvement typical of SSc, namely pulmonary fibrosis and pulmonary hypertension, hypertensive renal crisis, and infiltration and fibrosis of the gastrointestinal tract, do not occur in morphoea. Although previously considered a self-limiting condition, there is now emerging evidence that a protracted, relapsing–remitting course may be common [22–24]. If left untreated, lesions may result in significant cosmetic and functional sequelae. In consequence, even though there is no increased mortality, significant morbidity can occur as a result of facial and limb asymmetry, flexion contractures, extracutaneous manifestations and psychological disability. As in SSc, there is an early, inflammatory, active phase, followed by a sclerotic and then atrophic damage phase. The key to successful treatment involves initiation during the active inflammatory stage, before significant damage has occurred. Factors that have hampered physicians’ evaluation and treatment of the condition include its frequently insidious nature, the propensity for spontaneous remission and a lack of validated methods to assess disease activity. There has been a recent international effort, not only to better understand the impact that morphoea can have on patients, but also to develop and validate clinical outcome measures and consensus treatment guidelines [25–27].
Epidemiology
Incidence and prevalence
Morphoea encompasses a rare group of conditions, with an overall incidence of 4–27 per million per year. In a landmark population-based study in the USA conducted between 1960 and 1993 by Peterson et al. [28] and employing his classification system (see Table 57.1), an annual age- and sex-adjusted incidence of 27 per million overall and of 5 per million for linear disease were documented. Prevalence was estimated at 0.05% at age 18 years and at 0.22% at age 80 years. In a more recent study of UK and Irish children, the reported incidence of morphoea was 3.4 per million children (<16 years) per year and 2.5 per million per year for linear disease [29].
Peterson found that overall, plaque morphea was the commonest subtype (56% of cases), followed in order of frequency by linear (20%), generalized (13%) and deep (11%) subtypes [28]. The frequencies of the various subtypes vary with age. In the UK/Irish childhood study 67% had linear disease (roughly equally distributed between head/neck and limb variants), 29% had non-linear forms and 4% a mixed pattern [29]. In the largest published childhood study to date, Peterson's classification was used in a series of 750 children from 70 centres worldwide. Linear morphoea was again the most frequent childhood subtype (65%), followed by plaque morphea (26%), generalized morphea (7%) and deep morphea (2%) [8]. These data are corroborated by two further large North American studies together covering 381 children. Linear morphoea occurred in 42–54%, followed by plaque in 15–28% and generalized forms in 7–11% [9, 30]. In these studies 15–23% of children had a mixed subtype, and linear–plaque was the most frequent combination (60–85%) [8, 9, 30]).
In adults, plaque morphoea is the commonest subtype (28–44% of cases), followed by generalized (24%) and linear (15%) forms [9, 22, 31]. There are fewer data regarding the frequency of mixed subtypes in adults, although one study identified them in 4% of 120 adult cases [9]. They may be seen more commonly in patients with childhood onset rather than adult onset of disease.
Age
Morphoea can occur at any age; however the peak age of onset differs for the different clinical subtypes of disease. Very rare cases of linear morphoea and atrophoderma of Pasini–Pierini presenting at birth have been described [32, 33]. In general, 75% of plaque disease occurs between the ages of 40 and 50 years, whereas 75% of linear disease occurs between the ages or 2 and 14 years. In Peterson's original study, the mean age at onset of disease was 12.2 years in linear, 31.5 years in plaque, 39.9 years in generalized and 45.1 years in deep forms of the disease [28]. The mean age at onset in adults is in the mid-forties [9, 22, 34]. In children the mean age at onset of disease is 7.3–8.3 years [8, 20, 30].
Sex
Most studies suggest that morphoea is commoner in women, with female to male ratios of between 7 : 1 and 2.6 : 1 [8, 9, 28, 31, 35]. This female preponderance may be less marked in children (2.4 : 1–3 : 1) [8, 9], particularly in those with disease onset under the age of 10 years (1.5 : 1). Adult pansclerotic morphoea appears to be more common in males [10].
Ethnicity
Although it affects all races, between 72.7% and 82% of published cases are in white people and a lower than expected prevalence has been identified in African Americans [8, 9, 34, 36]. In a study from Texas, USA, the racial distribution in 245 adult and paediatric morphoea subjects was 73% white, 14% Hispanic, 4% African American, 2% Asian and 6.5% other (Pacific Islander, Native American, etc.) [9].
Associated diseases
Autoimmune diseases
That morphoea lies within the autoimmune spectrum of disease is supported by the increased prevalence of autoimmune disease described in patients with morphoea and in their relatives [9, 19, 37]). Disorders reported to occur concomitantly with morphoea in case reports include psoriasis, vitiligo, alopecia areata, autoimmune hepatitis, primary biliary cirrhosis, inflammatory bowel disease, type 1 diabetes, autoimmune thyroid disease, polyglandular autoimmune disease type 2, Ménière disease, coeliac disease, multiple sclerosis, systemic lupus erythematosus, rheumatoid arthritis, Sjögren syndrome, antiphospholipid syndrome, Still disease and mixed connective tissue disease [38–40] (reviewed in [9, 19]). In a bid to understand which of these are true associations, some larger studies have been undertaken in recent years. Amongst 123 adults and 122 children from Texas, concomitant rheumatic or other autoimmune disorders were identified in 18% of patients, and were much commoner in adults (30%) than children (5%) [9]. This association was most marked amongst adults and children with generalized morphoea, occurring in 46% of this group versus 9% in the other subtypes combined. In this study the prevalence of psoriasis, systemic lupus erythematosus, multiple sclerosis and vitiligo appeared significantly higher in patients with morphoea than in the background population. In a multicentre worldwide study of 750 children, concomitant autoimmune disease was identified in 1.7% overall, and most frequently included vitiligo, insulin-dependant diabetes, autoimmune thyroid disease and ulcerative colitis [19]. In a retrospective German study of 472 patients (381 adults), the most frequently associated autoimmune diseases were Hashimoto thyroiditis, rheumatoid arthritis, alopecia areata and type 1 diabetes [41]. Interestingly, the offspring of parents with morphoea appear at greater risk of developing rheumatoid arthritis [42]. Furthermore, significant associations with the same group of autoimmune diseases – namely autoimmune thyroid disease, rheumatoid arthritis and systemic lupus erythematosus – have been found in SSc probands and their families, supporting a common pathogenesis (see Chapter 56) [43–45].
Lichen sclerosus–morphoea overlap
Cases of lichen sclerosus (also see Chapters 111 and 112) occurring in conjunction with morphoea are now well documented [46]. In earlier published cases most of the coexistent lichen sclerosus was extragenital, but more recently, larger studies have identified increasing numbers of patients with genital disease, which may present simultaneously or several years after the onset of morphoea [41, 47, 48]. In a retrospective study of 472 morphoea patients from Germany (381 adults, with a mean age of 46 years), lichen sclerosus was identified and histopathologically confirmed in 5.7% of morphoea cases, exclusively patients with plaque or generalized morphoea; 26/27 were adults and in eight females genital lesions were present. There was an increased prevalence of lichen sclerosus amongst patients with morphoea compared with the general population (1 : 300–1 : 1000), with an odds ratio of over 18 [41]. This may be an underestimate since the ano-genital area was only examined in this study in patients with suggestive clinical symptoms such as burning, itching, pain, dysuria or dyspareunia, and so asymptomatic patients, or those not admitting to symptoms, would have been missed. Accordingly, in a French study of 76 patients with morphoea (58 female, median age 59 years), who underwent a mandatory full clinical examination, 38% were found to have genital lichen sclerosus compared with 3% of the control group. Again, the condition was much commoner in patients with plaque morphoea (45%, 22/29 cases) [48]. Given the known risk of squamous cell carcinoma in genital lichen sclerosus, genital examination should be mandatory in all patients with morphoea, particularly those with plaque or generalized morphoea of the ‘disseminated plaque’ subtype.
That these two conditions coexist is clear, however their relationship is still in question. It has been argued by some that lichen sclerosus is a superficial, subepidermal form of morphoea [49], but others believe that the two conditions are completely distinct (see Chapters 111 and 112) [50]. A possible common genetic predisposition is suggested by their respective associations with other autoimmune diseases [9, 19, 51–53], the occurrence of familial cases [54–58), their coexistence in monozygotic twins [59, 60] and an association of lichen sclerosus with HLA-DQB1 haplotypes [61–63], which have also been associated with SSc [55, 64] (see Chapter 56). The coexistence of the two conditions at the same body site [39, 65, 66], sometimes in a segmental or Blashkoid distribution [67–69], and of histopathological specimens in which the features of both diseases have been found [66, 70–72] supports the concept of a common aetiology.
Clinically, the distinction between classic plaque morphoea (consisting of thicker, larger plaques with a peripheral, inflammatory, lilac ring or border) and lichen sclerosus (consisting of porcelain white papules and plaques with varying degrees of hyperkeratosis, follicular plugging, erythema, telangiectasia, purpura, sclerosis and atrophy) should be possible. In practice, however, it is often very difficult. Not all patients will have typical plaques of morphoea and typical lichen sclerosus lesions at extragenital or genital sites [73]. Some patients with typical plaque morphoea may develop porcelain white lichen sclerosus patches within their morphoea plaques, others may have widespread, thickened, waxy plaques with a crinkled, hyperkeratotic surface (Figure 57.1). Difficulties can also occur when trying to distinguish the two conditions histologically and a variety of means have been suggested including variations in the collagen and glycosaminoglycan production [74, 75]. Others have argued that the presence of normal upper dermal elastic fibres favours morphoea, whilst the loss of elastic fibres favours lichen sclerosus [76]. Patterson and Ackerman suggested that a lichenoid infiltrate in the papillary dermis and vacuolation of the basal layer was necessary to diagnose lichen sclerosus, and an infiltrate in the reticular dermis for morphoea (see Chapters 111 and 112) [50]. However, such features may not always be present in the later stages of either disease. Interestingly, patients with lichen sclerosus have antibodies to extracellular matrix protein 1 (EMP-1) [77, 78]. EMP-1 is involved in epidermal differentiation and angiogenesis, and through its binding to a variety of extracellular matrix components including perlecan (a heparin sulphate proteoglycan), type IV collagen, laminin 332, matrix metalloproteinase-9 (MMP-9) and fibulin (a calcium-binding proteoglycan) has a role in the structural organization of the dermis [79–81]. The gene encoding this protein is mutated in patients with lipoid proteinosis (see genetics section later in this chapter), an inherited disorder characterized clinically by skin and mucosal infiltration and scarring and histologically by reduplication of basement membranes and hyalinization of the underlying dermis [80, 82]. In both conditions the skin microvasculature is altered with reduplication of vascular basement membranes, loss of papillary dermal capillary loops and enlarged vessels in the deeper dermis [83]. The significance of these changes in the pathogenesis of lichen sclerosus is unclear, but the disruption of EMP-1-mediated control of MMP-9 activity may be important [84]. Antibodies to EMP-1 have not been documented in morphoea, although antibodies to another extracellular matrix microfibrillar protein, fibrillin 1, have been seen and implicated in pathogenesis in patients with linear and plaque morphoea as well as in SSc [85, 86].
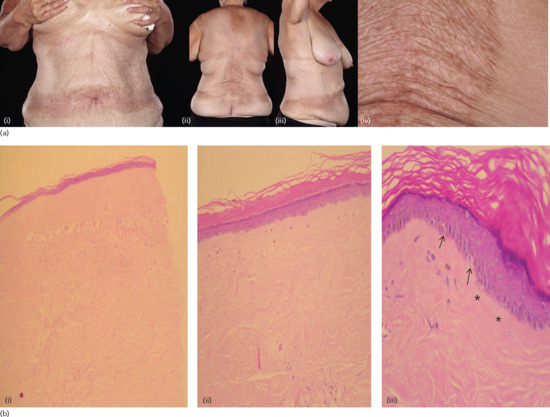
Figure 57.1 (a) Female patient with widespread, thickened, waxy plaques (i–iii) with a crinkled, hyperkeratotic surface (iv), occurring in an isomorphic pattern, i.e. at sites of pressure from clothing in the waistband, bra-line and inframammary areas (i–iii). (b) Histological appearances confirmed the presence of morphoea at low power (i) and features of lichen sclerosus (orthokeratosis, flattened epidermis, pigmentary incontinence (ii), colloid bodies (arrows) and hyalinization of papillary dermis (asterisks) (iii)) were seen at a higher power in the same biopsy specimen.
(Courtesy of Dr V. Swale, Department of Dermatology, Royal Free London NHS Foundation Trust, UK.)
Pathophysiology
Predisposing factors
The aetiology of morphoea is poorly understood. Trauma, radiation, medications and infection have all been proposed to act as triggering events in its development in susceptible individuals [1]. Morphoea is considered an autoimmune disease at least partially because of reported autoantibody and autoimmune disease associations. The coexistence of autoimmunity in patients and relatives, the very rare familial cases and the predominance of female gender suggest that underlying genetic factors are also involved. It seems likely that an environmental ‘signal’, on a background of genetic susceptibility, triggers a sequence of events including vascular activation, inflammation and subsequent fibrosis.
Pathology
Autoimmunity
Autoimmune mechanisms are thought to play an important role in the induction of morphoea and this is supported by the finding of increased serum levels of B-cell activating factor, a potent B-cell survival factor [87], and of a variety of autoantibodies in all subtypes of the disease. The published prevalence of ANA positivity in morphoea varies from 18% to 68% [9, 22, 41, 88–94]. In two large adult and childhood series, which have investigated a total of 110 adults and 748 children, ANA in significant titres was present in 34–42% of patients, and did not vary significantly between morphoea subtypes [8, 95]. The prevalence and distribution in a UK childhood incidence cohort was similar at 43% [20]. The significance of these autoantibodies is unclear. Previous studies had suggested that ANA positivity was commoner in patients with generalized, mixed or linear subtypes [9, 22, 89]. These findings may reflect smaller sample size in older studies, differences in study design and/or more frequent ANA testing in patients with these subtypes of the disease. Interestingly, children with one or more extracutaneous manifestation (see the section on complications and co-morbidities in this chapter) do appear more likely to have ANA positivity (51%) than those without any extracutaneous manifestations (39%) [19]. Speckled, homogeneous and nucleolar ANA staining patterns have been identified [90, 95, 96]; however the speckled pattern appears predominant, occurring in 81% in one study [95]. Many of these patients did not have antibodies to commonly identified extractable nuclear antigens, suggesting that as yet unrecognized antigens are involved in morphoea. ANA positivity may also be directed against a variety of recognized extractable nuclear antigens including histones, single-stranded or denatured DNA [97–99] and topoisomerase IIα [100]. An increased prevalence of antihistone antibodies (12% versus 2% in controls) [95] has been recorded in morphoea overall and in the linear subtype in particular. Although not the case in other subtypes, in linear disease the presence of ANA and antihistone antibodies has been correlated with more extensive disease, higher skin scores and greater functional impairment [93, 95, 101], suggesting that they may identify a group of patients who require more aggressive therapy. Although earlier studies suggested that titres of ANA and antihistone antibodies may parallel disease activity, this has not been borne out in larger studies [8, 95, 101].
Antibodies that may be more pathogenically specific to sclerotic skin diseases include antibodies to fibrillin 1, a major component of microfibrils in the extracellular matrix [85], and antibodies to MMP-I, which inhibit collagenase activity [102]. Both of these have been identified in patients with linear, plaque and generalized morphoea as well as in SSc.
Antibodies that are usually deemed to be specific markers of SSc, namely antitopoisomerase antibody (anti-Scl-70 or ATA) and anticentromere antibody (ACA) (see Chapter 56), have been identified at much lower frequency – for example in 3.2% and 1.7% of 750 children with morphoea, respectively [8]. This is comparable to published figures of 0.8–3% for ATAs [9, 22, 41]. The variable prevalence of 0–12% for ACAs in morphoea within the literature [41, 91, 92] may reflect the small numbers in some of the studies. Antibodies to dsDNA have also been reported in between 2.3% and 14% of cases [8, 9, 19, 41]. A variety of other autoantibodies, including anticardiolipin antibody [8], antimitochondrial antibodies [103] and antibodies to the ribonucleoproteins anti-U1-RNP [104], anti-U3-RNP [105] and anti-Th/To [106] may rarely be present. The significance of these autoantibodies is uncertain, since they do not usually appear to be associated with progression to systemic connective tissue diseases such as SSc or systemic lupus erythematosus. Nevertheless, such patients warrant increased surveillance.
Immunopathology
It has been proposed that Th1 and Th17 cytokines are activated during the early inflammatory stages of morphoea, whereas Th2 cytokines correlate with later stages of damage and fibrosis (Figure 57.2). Early presence of interleukin 2 (IL-2), tumour necrosis factor α (TNF-α) and the Th17 inducer IL-6 suggests that Th1 cytokines mediate the early inflammatory phase. This is then followed by a phase in which the Th17 effectors IL-17, IL-22 and transforming growth factor β (TGF-β) propagate inflammation and initiate fibrosis. During the later stages, fibrosis and tissue damage are favoured by Th2 effector cytokines IL-4 and IL-13 [107]. The importance of IL-13 is supported by initial gene expression profiling studies in three morphoea patients that suggest robust activation of IL-13 signalling [108].
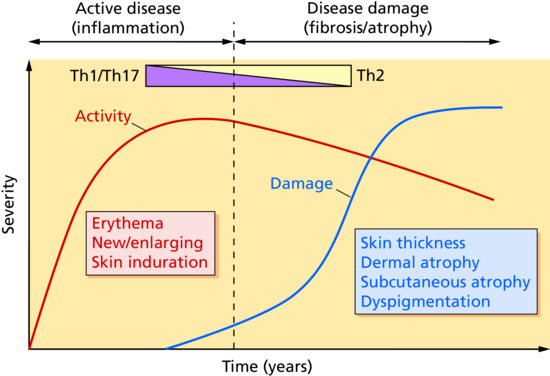
Figure 57.2 Proposed conceptual model of localized scleroderma: transition from Th1/Th17 to Th2 response. (From Kurzinski et al. 2011 [107].
Reproduced with permission of Elsevier.)
Although vascular damage is well recognized to be one of the early features involved in the pathogenesis of SSc, it is less well studied in morphoea. Nevertheless, endothelial cell swelling and apoptosis have been identified in early morphoea lesions [109, 110]. Direct immunofluorescence studies have shown immunoglobulin M (IgM) and C3 staining in the small blood vessels of the papillary dermis [111]. Antiendothelial cell antibody-mediated antibody-dependant cytotoxicity [109] and autologous complement activation [112] have been proposed as mechanisms for endothelial cell injury and activation [113]. Direct damage through trauma, infection and radiation may be contributory factors in some cases. There is evidence for vascular activation in morphoea with up-regulation of expression of the endothelial cell adhesion molecules vascular cell adhesion molecule 1 (VCAM-1), intercellular adhesion molecule 1 (ICAM-1) and E-selectin, and increased levels of IL-6 and chemokines including CCL2, -5, -7, -17, -22, -27, CCR7 and CXCL8 [114–118].
Another proinflammatory chemokine of interest in morphoea is interferon-γ-inducible protein 10 (IP10, CXCL10). IP10 is produced by keratinocytes, neutrophils, eosinophils, monocytes and macrophages and acts through CXCR3 receptors on lymphocytes and macrophages to attract them into the skin. IP10 levels were elevated significantly in the plasma of 69 children with morphoea compared with controls and correlated with objective measures of disease activity [119]. Increased levels of soluble IL-2 receptor (sIL-2r) reflecting immune activation have also been identified in patient sera [120, 121]. Increased CD4+ and CD8+ T cells and CD1a+ CD86+ dermal Langerhans cells are found in lesional skin in morphoea [122]. Increased serum levels of IL-2 have been identified in patients with generalized and linear disease and correlated with increased IL-4 and IL-6 concentrations in serum and degrees of skin sclerosis [123]. IL-4 stimulates B-cell and fibroblast proliferation, synthesis of extracellular matrix components and immunoglobulin and adhesion molecule production (reviewed in [107]). Increased serum IL-4 has been found in patients with generalized morphoea [123]. IL-6 is an inflammatory and profibrotic cytokine, involved in autoimmunity via its ability to regulate B cells, fibroblasts and Th17 cell differentiation. Increased serum levels of TNF-α (known to promote IL-6 production) have been identified in patients with the same morphoea subtypes and particularly in early disease [124]. TNF-α levels correlated with the presence of antihistone and anti-ssDNA antibodies and IL-6. In the same study, increased levels of IL-13, which has similar profibrotic properties to IL-4, were identified in the serum of patients with generalized and plaque morphoea.
The role of Th17 cells in skin fibrosis has increasingly come into focus. Increased serum levels of IL-17F and IL-22 have been identified in children with morphoea of between 24 and 48 months’ duration [107]. Levels of these cytokines correlated with ANA, antihistone and ssDNA antibody positivity, and it was suggested that the Th17 signature may reflect more severe or active disease. In a study of SSc fibroblasts, the production of IL-6, MMP-1 and monocyte chemoattractant protein-1 (MCP-1) was enhanced in a dose-dependent manner in the presence of IL-17E and IL-17F. In morphoea, a high dermal expression of IL-17E with concomitant low expression of IL-17C has been documented and may represent a ‘fibrosis-specific motif’ [125]. IL-8, a proinflammatory cytokine produced by endothelial cells, monocytes and fibroblasts, has shown variable results but in one study was increased in the sera of all morphoea subtypes [126]. Endogenous IL-1α from SSc fibroblasts stimulates collagen production by inducing IL-6 and platelet-derived growth factor (PDGF) [127]. Endothelial cell-derived IL-1α and IL-1β and fibroblast growth factor may be produced in response to cellular injury and are also capable of activating SSc fibroblasts [128]. There are currently no data regarding the effects of IL-1 in morphoea, but one may speculate that keratinocyte and/or endothelial cell IL-1 may play a role in fibroblast activation.
Fibrosis is thought to result from a combination of increased collagen deposition by fibroblasts and reduced extracellular matrix turnover in morphoea. Enhanced type I and type III collagen mRNA and protein expression has been identified in morphoea fibroblasts and lesional skin [129–131]. Glycosaminoglycan and fibronectin synthesis is also increased. Constitutive overexpression of connective tissue growth factor (CTGF) combined with increased TGF-β and PDGF production by dermal fibroblasts contribute to the sustained fibrotic response [132–138]. CTGF mRNA and protein have been identified in fibroblasts scattered throughout the dermis in morphoea skin sections [137, 138]. Skin biopsies and sera of morphoea patients have demonstrated increased TGF-β expression and co-localization with activated fibroblasts [134]. Up-regulation of TGF-β and TGF-β receptors I and II in perivascular lymphocyte-like cells and interstitial fibroblasts have been identified by immunohistochemistry and in situ hybridization in involved skin [134, 136]. Increased expression of the matrix-associated tissue inhibitor of metalloproteinase-3 (TIMP-3) in fibroblasts within fibrotic collagen fibres or in the vicinity of inflammatory cells suggests a role for reduced collagenolysis in addition to increased collagen synthesis in the development of fibrosis [139]. Fibrocytes are CD34+ cells expressing collagen that are recruited from the circulation into sites of injury. During tissue remodelling they lose CD34 expression and gain smooth muscle actin (SMA) expression to become myofibroblasts. Factor XIIIa is a protransglutaminase expressed on a population of dermal dendritic cells now thought to be macrophages [140], which is involved in cross-linking matrix proteins. CD34 positivity is lost, and factor XIIIa expression is increased in areas of fibrosis in morphoea [141–144]. Interestingly, enhanced dermal expression of α-SMA, as well as TGF-β1 and fibronectin, were identified in one small study in morphoea patients, suggesting that epithelial–mesenchymal transition may be involved in the fibrotic process [145]. Together, these findings suggest that a profibrotic wound-healing environment may develop in morphoea as a result of transformation of CD34+ fibrocytes to CD34– myofibroblasts. The increase in factor XIIIa-expressing cells may then function in cross-linking newly formed collagen fibrils and matrix proteins, thereby enhancing the fibrotic process [143].
By these mechanisms, vascular injury leading to recruitment and activation of lymphocytes and mononuclear cells, secretion of pro-inflammatory mediators and fibroblast activation are thought to lead to eventual fibrosis and damage.
Histopathology
All subtypes of morphoea share similar findings of an early active inflammatory phase, in which newer lesions demonstrate a lymphocytic infiltrate, with a variable number of plasma cells and eosinophils. As lesions evolve, the numbers of inflammatory cells are reduced as collagen bundles thicken and skin sclerosis increases in the later fibrotic phase. An intermediate picture is frequently found on skin biopsy (Figure 57.3).

Figure 57.3 Typical histopathological appearances in morphoea. (a) Low power view showing perivascular inflammation in the papillary and upper reticular dermis, dense horizontally orientated collagen bundles, loss of adnexal structures and a straight dermal–hypodermal junction. (b) Higher power view showing broad and thick collagen bundles, scattered aggregates of lymphocytes (bottom right) and atrophic sweat ducts and glands (top left). (Both H&E.)
(Courtesy of Dr F. Deroide, Department of Histopathology, Royal Free London NHS Foundation Trust, UK.)
Histopathological changes are similar in all subtypes of morphoea, but vary in relation to the depth of involvement. For example, in plaque morphoea they may be limited to the dermis, whereas in linear and deep types they may extend beyond the skin and into the underlying fascia, muscle and bone. In deep forms changes may be confined to the deep dermis and subcutis, or solely involve deeper structures such as the underlying fascia and muscle [146]. In some cases changes may be entirely superficial and confined to the reticular dermis [142].
The epidermis may be normal, flattened with loss of rete ridges or slightly acanthotic [147]. In the early inflammatory phase, oedema and a dense predominantly perivascular infiltrate of lymphocytes, plasma cells and macrophages, and occasional mast cells and eosinophils, is present in the reticular and occasionally the papillary dermis [148–150]. The infiltrate may extend into the lower dermis, around the eccrine glands, into the subcutaneous fat and beyond. The reticular dermis shows swollen collagen bundles running parallel to the skin surface. The subcutaneous fat may be replaced by thickened, wavy fibres of newly formed collagen, rich in type III collagen and fibrillin 1 [151, 152]. Vascular changes are mild in the dermis and subcutis and consist of endothelial swelling and oedema of the vessel walls [153].
In the sclerotic stage there are few recognizable fibroblasts and little inflammation. Collagen bundles are closely packed, highly eosinophilic and orientated horizontally. The dermal appendages and subcutaneous fat are progressively lost. Reduced numbers of eccrine glands are entrapped by collagen, and thus appear higher in the dermis. Fewer blood vessels are seen within the thickened hyalinized collagen; those that are present may show intimal thickening. The fascia and striated muscles underlying the lesions may likewise show fibrosis and sclerosis. Deeper structures including the eye and brain are involved in a significant number of patients with linear morphoea of the face or scalp. Brain biopsies performed in some patients with neurological involvement have shown dilated blood vessels, a perivascular lymphocytic infiltrate with features of vasculitis, gliosis and sclerosis of the leptomeninges and intravascular and intraparenchymal calcification [154].
The cutaneous histopathological findings in morphoea and SSc are similar [155]. Difficulties can sometimes arise in patients with the pansclerotic variant. Features that favour morphoea over SSc include more intense inflammation, the presence of perineural inflammation and more diffuse dermal sclerosis, simultaneously involving the papillary and deeper dermis, which is not usually seen in SSc [150, 156].
Causative organisms
A putative role for Borrelia species in triggering morphoea was initially proposed by Aberer et al. in 1985 [157]. It was suggested because of: (i) the clinical and histological similarities between morphoea and acrodermatitis chronica atrophicans, a cutaneous manifestation of late-stage Lyme disease (see Chapter 96) [158]; (ii) the finding that lichen sclerosus was observed to coexist with acrodermatitis chronica atrophicans in 12% of cases [159]; and (iii) the response of certain cases of morphoea to antibiotics. Since then, the proposed association has been studied extensively with different outcomes in Europe and North America. It has been suggested that the geographical differences reflect the fact that different subspecies of Borrelia predominate in different parts of the world. Borrelia burgdorferi sensu stricto is prevalent in the USA and B. afzelii and B. garinii predominate in Eurasia. High rates of Borrelia infection have been documented prior to the onset of morphoea in some European studies, but not in those from the USA [160–164]. In a retrospective review of 90 European morphoea patients, a statistically highly significant association between morphoea, serological evidence of Borrelia infection and high-titre ANA positivity was observed when disease onset was in childhood or adolescence, suggesting possible relevance in a subset of morphoea patients [165]. However, a significant number of studies from both sides of the Atlantic have found no association between the two entities [166–170]. The wide range of diagnostic tests used, which include immunoperoxidase, silver stain, focus-floating microscopy, tissue culture, serology and polymerase chain reaction, make it difficult to interpret the data. A literature review identified Borrelia DNA in only one of 49 morphoea cases investigated [170]. There is no conclusive evidence to date that morphoea is caused by Borrelia infection.
Genetics
Rare cases of familial linear morphoea have occurred in sisters and first-degree cousins [58, 171]. Recently, an association with HLA-DRB1*04:04 and HLA-B*37 has been demonstrated, particularly for the linear and generalized subtypes [172]. Interestingly, HLA-DRB1*04:04 has also been associated with SSc (see Chapter 56). A family history of rheumatic or autoimmune disease in first- or second-degree relatives seems commoner, and was reported in 12% of 750 children [8]. This family history was significantly more likely in patients with generalized morphoea (23.5%) than in those with linear (9%) or plaque (12.5%) disease. Rheumatoid arthritis, systemic lupus erythematosus, psoriasis, vitiligo, lichen sclerosus, autoimmune thyroiditis and insulin-dependent diabetes appear particularly associated [8, 9]. A family history of scleroderma was identified in 1.5% of children (although no mention is made whether this was systemic or localized disease) [8]. In a further study, including 123 adults and 122 children, 2% reported a family history of morphoea in a first- or second-degree relative [9]. At 18% overall, the prevalence of familial rheumatic and autoimmune disease was increased fourfold compared with that in the general population [28], and was higher in children (22%) than adults (11%). Children with generalized or mixed morphoea and adults with generalized disease had the highest frequencies. Taken together, the increased frequency of personal and familial autoimmunity in the generalized subtype may indicate a common susceptibility locus for this group of disorders.
There is still a great deal of work to be done to understand the genetic influences underlying morphoea, however genome-wide gene expression profiling of skin biopsies from three morphoea patients has demonstrated an inflammatory gene expression profile identical to that seen in limited and some diffuse cutaneous SSc patients [108] (see Chapter 56). This profile has been linked to T-cell infiltration, early growth response 1 (Egr-1) and IL-13 pathway activation [173, 174] and to CCL2 up-regulation [174].
Epigenetics has been a recent focus of attention in various fibrotic disorders. MicroRNAs (miRNAs) are small non-coding RNAs that bind to messenger RNAs inhibiting their translation into protein. Down-regulated miRNAs, in particular miR-7 [175], and miRNA-196a [176] in the serum and skin of morphoea patients may contribute to the pathogenesis of skin fibrosis by allowing increased type I collagen expression in morphoea fibroblasts. These recent findings suggest new therapeutic avenues to explore such as IL-13 or CCL2 blockade and miRNA up-regulation.
Environmental factors
Trauma and vaccination
A small number of case reports in children specifically document the onset of morphoea at the site of, and in a close temporal relationship to, vaccination for hepatitis B, MMR (measles, mumps and rubella), diphtheria, tetanus, pertussis, pneumococcus and BCG (bacille Calmette-Guérin) [177–182]. The onset of morphoea has also been reported after injection with vitamin B12 and K [183–185]. It has been suggested that morphoea may reflect an immunological response triggered by vascular injury and tissue hypoxia as a result of trauma at the injection site in susceptible individuals. Others have argued that since multiple vaccines have been implicated, it is the adjuvants in the vaccines that act as the trigger [186].
Anecdotal reports and early case series have suggested a potential role for trauma in the development of morphoea and particularly in linear disease [35, 187, 188]. This suggestion was supported more recently, when 13.3% of 750 children reported a specific potential triggering event, such as trauma, infection or exposure to a drug, occurring close to the time of disease onset [8, 36]. Mechanical trauma (including accidental trauma, insect bite reactions and vaccinations) accounted for two-thirds of these cases, infections for a quarter of cases, and drugs and psychological distress for 5% and 3%, respectively. Interestingly children with generalized morphoea had a lower reporting frequency for such events (6%). In contrast, there appeared to be a trend for mechanical factors to act as a trigger in linear and deep morphoea cases [8, 189].
The association of morphoea with skin trauma was systematically investigated in a cohort of 329 adult and childhood cases. Evidence of skin trauma or friction in the distribution of morphoea lesions at the onset of disease was identified in 52 patients (16%) [189]. The development of morphoea in the same area as previously healed skin disease or injury, also referred to as an isotopic response [190], occurred in 6%, and skin lesions occurring at sites of repeated current trauma, referred to as the isomorphic response of Koebner [191], were identified in 9% of patients [189]. Isotopic patients were defined as those who had trauma occur at the site of the initial lesion within 6 months of onset of morphoea. Isomorphic patients were those with lesions distributed exclusively in areas of friction in the bra-line, waistband area and inguinal creases. Both groups were female predominant and the mean age of onset was lower in the isotopic (44.4 years) versus the isomorphic (52.4 years) group. In contrast to the findings in children [8], in this predominantly adult group (48/52 cases), 87% of trauma-induced cases had generalized morphoea (defined in this study as the occurrence of indurated plaques that have become confluent on at least two anatomical sites), compared with 33% of the cohort overall. Isotopic patients also included some cases of linear morphoea and had more severe disease as measured by modified Rodnan skin score (MRSS; see Chapter 56) and dermatology life quality index scores [189]. The proposed triggering events in the isotopic group were surgery in 43%, penetrating trauma in 19%, injection in 14%, herpes zoster infection in 10% and radiotherapy, diagnostic X-ray and extreme exercise in 5% each. The majority of lesions occurred on the chest, breasts or abdomen. The underlying mechanism for such trauma-induced morphoea remains uncertain. However, the induction of an aberrant wound healing response with up-regulation of endogenous Toll-like receptor ligands, enhanced innate immune signalling and resultant fibroblast activation may be involved [192].
Radiation
A majority of published cases of post-irradiation morphoea have been linked to radiotherapy for breast cancer [193–195]. It is estimated to occur in 1/500 breast cancer patients [195]. More rarely, it has occurred after treatment for gynaecological and head and neck malignancies, subcutaneous lymphoma and metastatic adenocarcinoma [196]. Age, radiotherapy parameters and initial post-treatment reaction do not appear to influence the risk of developing post-irradiation morphoea, although a prior diagnosis of SSc may do. Most cases develop within a year of completing radiotherapy, but rarely delays of 10–32 years are reported [196, 197]. In most cases morphoea develops within the radiotherapy field, but in 20–25% of cases it can extend beyond this [193, 195]. The differential diagnosis can be challenging and includes chronic radiation dermatitis, cancer recurrence, post-irradiation recall dermatitis and cellulitis. Histological confirmation is usually necessary to exclude the possibility of cancer recurrence. In terms of pathogenesis, radiation-induced increases in IL-4, IL-5 and TGF-β have been implicated, with resultant fibroblast activation, collagen synthesis and fibrosis [198].
Drugs
A variety of drugs have been implicated in the development of morphoea-like lesions (see Table 57.3) [199–203]. The delay in onset ranges from 1 to 30 months and resolution upon withdrawal, although reported, is not invariable [201, 204]. Mechanisms suggested include the development of drug-specific lymphocyte responses and autoantibody production causing endothelial damage [203], direct vascular damage, generation of reactive oxygen species and up-regulation of IL-1, TNF-α and TGF-β [205]. The development of morphoea in nine patients on high-dose balicatib is of particular interest since this drug inhibits the collagenolytic activity of cathepsin K within lysosomes in skin fibroblasts [206, 207]. Cathepsin K is involved in intracellular collagen degradation. Its expression is up-regulated by IL-1α and inhibited by TGF-β1. This suggests a role for failed intracellular degradation of extracellular matrix proteins in the generation of fibrosis, and represents a different mechanism to the previously proposed impairment of metalloproteinase-mediated collagen degradation and/or increased collagen production in the extracellular space [207].
Table 57.3 Disorders associated with the development of skin sclerosis
| Disorder | Examples |
| Autoimmune disorders | Systemic sclerosis, sclerodermoid GVHD |
| Metabolic disorders | Porphyria cutanea tarda, phenylketonuria, muscle glycogenosis, hypothyroidism, carcinoid syndrome, diabetic cheiroarthropathy with skin thickening |
| Depositions disorders | Scleredema, scleromyxoedema, primary systemic amyloidosis |
| Genetic disorders | GEMSS (glaucoma, lens ectopia, microspherophakia, stiffness of joints, short stature), Werner syndrome, progeria, acrogeria and poikilodermatous epidermolysis bullosa, Moore Federman syndrome (short stature, stiff joints, characteristic facies), stiff skin syndrome, melorheosthosis, scleroatrophic Huriez syndrome (scleroatrophy hands and feet, nail hypoplasia, keratoderma hypohidrosis) |
| Occupational causes | Vinyl chloride disease, perchlorethylene, trichloroethylene, organic solvents, pesticides, epoxy resins, silicone |
| Chemically induced | Eosinophilia myalgia syndrome (l-tryptophan), toxic oil syndrome (ingestion of rapeseed oil contaminated with aniline), nephrogenic systemic fibrosis (gadolinium exposure on a background of renal failure, GFR <30 mL/min) |
| Drug induced | Bleomycin, pentazocine, progestin, vitamin B12, vitamin K, cocaine, d-penicillamine, peplomycin, interferon β-1a, uracil-tegafur, paclitaxel, methysergide, gemcitabine, bromocryptine, bisoprolol, l-5-hydroxytryptophan with carbidopa, balicatib, ibuprofen, mitomycin C |
| Associated with haematological disease | POEMS (polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, sclerodermoid skin changes), myeloma |
GFR, glomerular filtration rate; GVHD, graft-versus-host disease.
Clinical features
History
The onset and progression of morphoea is usually insidious. Patients may describe changes in skin texture or colour, sometimes with associated itch, pain or numbness. When lesions extend across joints, reduced mobility or contractures and limb girth and length discrepancies may occur. In linear disease of the face and head, asymmetry of facial features, alopecia, indentation or grooves in the skull, ocular pain and dental abnormalities may develop. Headaches and seizures may occur. In generalized subtypes fatigue, myalgia and arthralgia are frequent. Occasionally, patients describe symptoms of gastro-oesophageal reflux. In cases where there is widespread chest wall sclerosis, respiratory symptoms may occur.
Presentation
Individual lesions of morphoea generally begin with an erythematous, oedematous, inflammatory phase, which may be subtle and ‘bruise-like’ in appearance. The onset is often slow and insidious. This is followed by the development of central sclerosis associated with a change in skin colour and texture to thickened, waxy, yellowish white. There may be loss of hair and absent sweating. This central sclerotic area may be surrounded by an erythematous to violaceous so-called ‘lilac ring’, widely thought to reflect ongoing active disease. Over months or years, lesions become atrophic and hyper- or hypopigmented. In some cases, no sclerotic phase is seen and lesions progress straight to the atrophic hyperpigmented stage. Depending on the depth and type of lesion, changes in the subcutis, muscle, fascia, bone and underlying brain may be present. The average delay between onset of symptoms and diagnosis ranges from 11 to 24 months in most childhood series [8, 20, 208]. In recent North American studies, 63% of 224 patients (129 adults) were given a diagnosis >6 months after onset of disease [34], and 37% of 259 children had a ≥5-year delay from symptom onset to their first paediatric rheumatology appointment [30]. Similar delays occur in the UK [208]. There is a suggestion that the delays in making a diagnosis may be greater in adults than in children and greater in patients with plaque or generalized morphoea rather than linear disease [34]. Such delays may have a significant impact on outcome, since they may result in physicians missing the early ‘active’ phase of disease that should be more amenable to treatment.
Limited type
Limited plaque morphoea
This commonest form of morphoea presents with round to oval lesions >1 cm in diameter, in up to two of seven anatomical regions (head–neck, each limb, anterior trunk, posterior trunk) (Figure 57.4). Histopathological changes are usually limited to the epidermis and dermis. Plaques are most frequently located on the trunk (41–74%). The breasts are often involved, but the nipples and areolae are uniformly spared. Plaques can occur anywhere, including the face and neck in 12–13% of patients [5, 35]. Although this type is referred to as circumscribed superficial morphoea in Laxer and Zulian's classification [6], in some cases plaques extend more deeply (Figure 57.5).
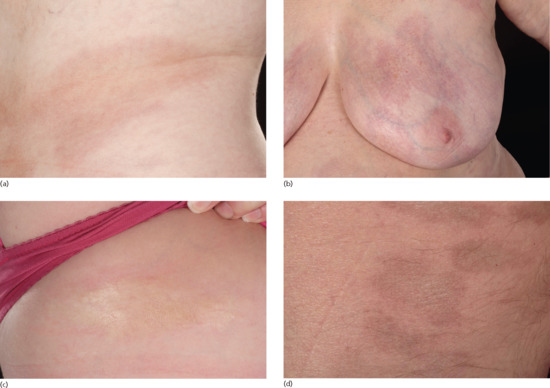
Figure 57.4 (a, b) Early, inflammatory, superficial plaque of morphoea with erythema and bruise-like appearance. (c) Sclerotic centre with inflammatory, peripheral lilac ring. (d) Hyperpigmented, atrophic late-stage disease.
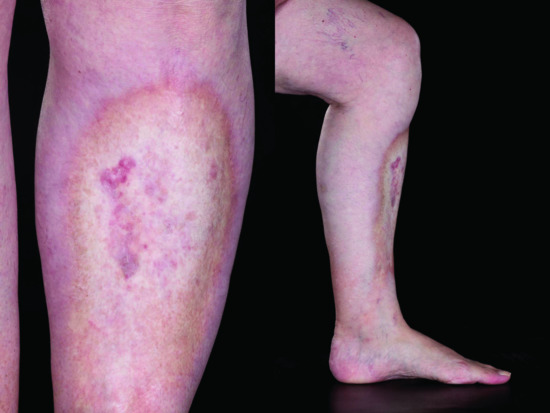
Figure 57.5 Plaque morphoea with deep involvement.
Guttate morphoea
This is a rare variant, similar to plaque morphoea, in which multiple, small (<1 cm), erythematous or yellowish white, mildly indurated lesions develop, most frequently on the trunk. Lesions are superficial and may have a shiny, crinkled surface, clinically resembling extragenital lichen sclerosus. It may be difficult to distinguish guttate morphoea from extragenital lichen sclerosis on clinical and histopathological grounds and some consider guttate morphoea to be a type of lichen sclerosis associated with morphoea [49, 209, 210]. In contrast to extragenital lichen sclerosus, however, lesions generally resolve leaving hyperpigmentation.
Atrophoderma of Pasini–Pierini
There are divergent opinions as to whether atrophoderma of Pasini–Pierini represents a separate entity or is a primarily superficial and atrophic variant of morphoea [142, 211–214] (Figure 57.6). It is a rare condition that represents 0.1% of childhood morphoea cases [8] and that usually occurs in adolescence and young adult life. Three congenital cases are published [33]. Symmetrically distributed truncal lesions are the most common but single lesions and zosteriform distributions are described [212, 215–217]. Typically, lesions are non-indurated, blue-grey to brown, hyperpigmented and sharply demarcated depressed patches, with a ‘cliff-drop’ border [212, 215]. A more recent publication of 16 cases found only 19% to be hyperpigmented, the remainder were either hypopigmented or skin coloured [213]. Histological appearances are variable: they may be normal or show mild lymphocytic infiltration, reduced dermal thickness and normal or sclerotic and hyalinized collagen [11, 211, 213]. Elastic stains may also show a spectrum of changes ranging from normal to severe diminution and fragmentation of the elastic fibre network [213]. The inclusion of atrophoderma of Pasini–Pierini within the spectrum of morphoea is supported by the coexistence of areas of induration more typical of morphoea in some patients [211, 212]. In a study of 139 patients followed for a mean of 10 years, areas of induration appeared within existing lesions in 17% and plaques of morphoea were found elsewhere on the body in 22% of cases [212].
Keloidal/nodular morphoea
The terms keloidal and nodular morphoea have been used interchangeably in the literature. This rare subtype is characterized by the presence of keloid-like nodules in patients with previous or coexistent morphoea or, in a majority of cases, SSc (Figure 57.7) [218–224]. Clinically, keloidal or nodular lesions arising from sclerodermatous skin may reveal a histological appearance typical of either keloid or morphoea [220]. More rarely histological features of hypertrophic scarring [222] or homogenization and thickening of collagen bundles with an increase in mucin are described. Lesions are most common on the upper body where they may coalesce or occur in a linear pattern [223, 225, 226]. In some cases nodules arise from normal skin in patients genetically predisposed to keloid formation [220]. Increased levels of epidermal growth factor and CTGF have been implicated in pathogenesis [138, 227]. In a histological and ultrastructural analysis in a patient with nodular morphoea on a background of diffuse cutaneous systemic sclerosis, Moinzadeh et al. found that increased density of immature collagen fibrils and absence of myofibroblasts characterized nodular lesions [228]. There was increased deposition of cartilage oligomeric matrix protein (COMP), collagen XII and fibrillin 1 within nodules in a distribution resembling that seen in keloids rather than normal skin. The authors suggested that COMP may promote fibroblast proliferation and increase production of extracellular matrix as a result of its ability to present TGF-β to fibroblasts and to bind to collagen I and XII.
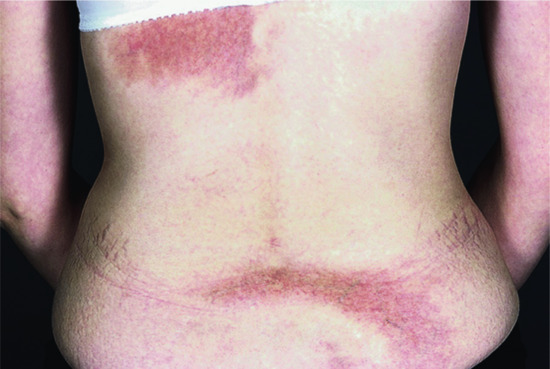
Figure 57.6 Atrophoderma of Pasini and Pierini.
(Courtesy of Dr D. A. Burns, Leicester Royal Infirmary, Leicester, UK.)

Figure 57.7 (a) Keloidal morphoea in a patient with limited cutaneous systemic sclerosis. (b) Low power view showing a thick dermis, dense collagen and reduced adnexal structures. Keloidal changes are just visible (circled). Magnification 40× (H&E). (c) Higher power view showing swollen collagen bundles of the keloidal collagen. Magnification 100× (H&E).
(Courtesy of Dr F. Deroide, Department of Histopathology, Royal Free London NHS Foundation Trust, UK.)
Limited deep morphoea
The term ‘deep morphoea’ describes a variant in which inflammation and sclerosis are found in the deep dermis, panniculus, fascia or muscle (synonym morphoea profunda). Deep involvement can occur in all subtypes of morphoea (Figure 57.8). In their description of morphoea profunda in 23 patients, Person and Su's diagnostic criteria included the presence of diffuse, taut, bound-down, deep cutaneous sclerosis, and of significant hyalinization and thickening of collagen bundles in the deep dermis, in the septa of subcutaneous fat and in the fascia [146, 229]. Although a majority of the cases they described had widespread, deep involvement, some had individual plaques in keeping with a diagnosis of limited deep morphoea (synonym solitary morphoea profunda). Solitary lesions of deep morphoea were first described by Whittaker et al. in five patients who had solitary, ill-defined, indurated and deeply tethered plaques with a peau d'orange appearance involving the upper trunk [230]. The main histopathological features are sclerosis and hyalinization of collagen, and a striking accumulation of inflammatory cells in the deep dermis as well as in the subcutaneous tissue, which is predominantly composed of plasma cells and T and B lymphocytes [230]. Involvement below the subcutaneous fat was not identified in Whittaker's cases, but only one had a deep biopsy. Increased numbers of eosinophils may be present in the skin and circulation [230, 231]. Rarely, a plasma cell panniculitis has been described (synonym morphoea panniculitis) [232]. The condition has been reported in children and adults [230, 233]. A number of cases have been documented post vaccination [178, 179, 234]. Occasionally, lesions present as non-inflammatory, cupuliform, depressed plaques with no associated induration, pigmentation or texture change, but with excessive dermal collagen deposition and thickened hyalinized collagen bundles in the deep dermis and subcutis on histology [234]. Deep morphoea can thus mimic lipoatrophy clinically and should be considered in patients presenting with asymptomatic atrophic lesions.
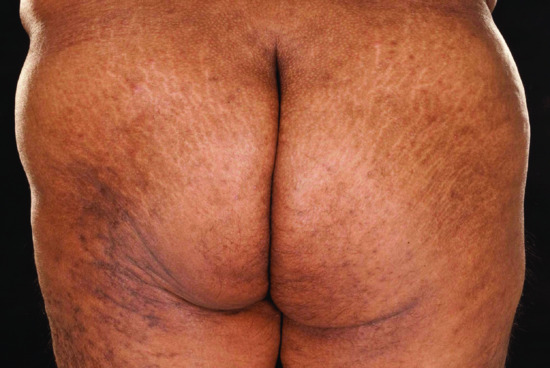
Figure 57.8 Deep involvement in morphoea. The skin is tethered to the underlying involved deep tissues giving a puckered appearance and bound-down feeling.
Generalized type
Generalized morphoea has been defined in a variety of ways in the literature. Falanga suggested five or more lesions, bilateral lesions and evidence of joining together of at least two individual patches [94]. Laxer and Zulian [6] defined generalized morphoea as induration of the skin starting as four or more individual plaques, larger than 3 cm, that become confluent and involve at least two out of seven anatomical sites (head–neck, right upper extremity, left upper extremity, right lower extremity, left lower extremity, anterior trunk, posterior trunk). Peterson et al. [5] and Kreuteret al. [7] define it more simply as plaques involving three or more of these same seven anatomical sites. Based on these definitions, the literature suggests that generalized morphoea accounts for 7–9% of childhood morphoea cases [8, 22, 30, 35] and between 13% and 52% of adult cases [9, 22, 28, 35]. Extracutaneous symptoms including myalgia, arthralgia and fatigue are common, dyspnoea and dysphagia may occur, and a higher prevalence of concurrent and familial autoimmune disease are documented (see the sections on genetics and complications and co-morbidities in this chapter).
These definitions do not take into account the fact that within this group there are distinct clinical presentations, which are described below.
Disseminated plaque morphoea
In the first type of presentation, patients gradually develop multiple plaques of morphoea at several anatomical sites, some of which may coalesce (Figure 57.9). This subtype is perhaps better described as disseminated plaque morphoea. Plaques may occur in different stages of evolution and are most frequently located on the trunk, thighs and lumbosacral area in adults. Multiple, isolated, often symmetrically distributed plaques may be present, or larger, coalescent plaques may develop in an isomorphic pattern due to minor trauma from clothing around the waistband, under the breasts and in the groins (see Predisposing factors in this chapter) [189, 235]. The isomorphic distribution pattern appears particularly common in middle-aged females [189]. In the largest multicentre study in children, co-involvement of the trunk and limbs occurred in 63% and of the trunk, limbs and head in 19% of cases [8, 19]. Both adults and children initially presenting with limited plaque morphoea may, as a result of ongoing or recurrent disease activity, progress to fulfil the criteria for the disseminated plaque subtype of generalized morphoea.
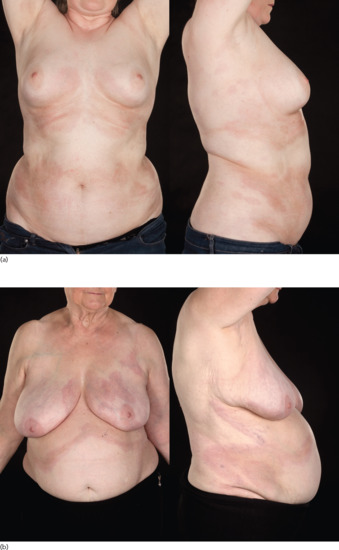
Figure 57.9 Disseminated plaque morphoea. (a) Active inflammatory stage with no deep involvement, and an isomorphic pattern of lesions around the bra, waistband area and groins. (b) Early central sclerosis and prominent peripheral inflammatory lilac ring with no deep involvement, and a symmetrical isomorphic pattern involving the inframammary and waistband areas.
Pansclerotic morphoea
This is a rarer presentation and is characterized by extensive, often circumferential involvement of the majority of body surface areas with sparing of the fingers and toes, and may best be referred to as pansclerotic morphoea (Figure 57.10) [10]. Pansclerotic morphoea has variably been included within either the generalized [7] or deep [5] subtypes or as a separate subtype of morphoea [6] in published classifications. This term has traditionally been used to describe a very rare, widespread and severe progressive disease occurring predominantly in children in which deep fibrosis progresses rapidly to involve muscle, fascia and underlying bone [236–238].
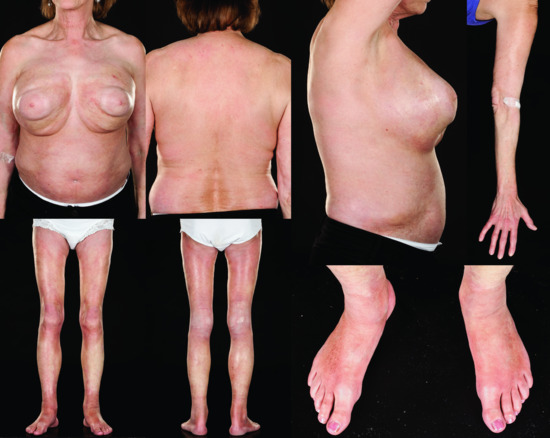
Figure 57.10 Pansclerotic morphoea in one patient showing circumferential involvement of the lower limbs and trunk with sparing of the areolae and hands.
It is mainly referred to as disabling pansclerotic morphoea of childhood in the literature and is frequently complicated by severe joint contractures, chronic ulceration and the development of squamous cell carcinoma [238–241]. Increased serum IgG, a positive ANA and peripheral eosinophilia are documented in some cases [236, 242, 243]. By virtue of the number of body sites involved, this subtype meets the classification criteria for generalized morphoea, but it is a clinically distinct condition. It is unclear whether the particularly severe phenotype described in children occurs as a consequence of growth impairment or because they have a more severe disease pattern than adults.
Whilst the previous descriptions of pansclerotic morphoea in childhood suggest deep tissue involvement, including subcutaneous tissue, muscle and bone [236, 238], the definition and frequency of involvement beyond the dermis has been inconsistent [237, 244–248]. In a recent cross-sectional study of the Morphoea in Adults and Children (MAC) cohort, an ongoing prospective registry of patients with morphoea in Texas, USA, the demographic and clinical features of patients with the pansclerotic subtype were described [10]. Because of the relative ambiguity as regards depth of involvement, in the literature, this criterion was removed, and pansclerotic morphoea was defined as the presence of near total body surface area involvement with sparing of the fingers and toes. The distinction from SSc was made based on the absence of sclerodactyly, nail fold capillary changes and internal organ involvment. Of the 360 patients (97 children, 263 adults) in the MAC cohort, 113 patients with generalized morphoea were identified. Thirteen of these patients, 3.6% of the cohort, met the above criteria for pansclerotic morphoea. A majority had onset of lesions on the trunk with rapid centrifugal spread and abrupt cut-off at the metacarpo- and metatarsophalangeal joints. None had demonstrable bony involvement. They had a more rapidly progressive and severe phenotype than the generalized morphoea group, but were distinct from SSc. Interestingly, there were no significant differences between the generalized morphoea and pansclerotic groups for age at onset of disease (mean 49 years, standard deviation 19), or prevalence of ANA (29–31%) and antihistone antibodies (7–10%). However, patients with the pansclerotic subtype were more likely to be male (46% versus 6% of generalized morphoea patients) and have a shorter time to diagnosis, higher rates of functional impairment (61% versus 16%) and higher skin sclerosis and damage scores. Even though it was not a defining factor, a high frequency of deep involvement on tissue biopsy (61% versus 17%) was observed. Restrictive defects on pulmonary function tests, dysphagia and/or hand oedema were identified in 4/13 patients and following investigation were attributed to severe, extensive skin sclerosis rather than internal organ involvement per se [10].
There is significant clinical overlap between pansclerotic morphoea as defined above and the various forms of deep morphoea described in the literature. The term subcutaneous morphoea was initially coined by Person and Su in 1979, who described 16 cases with biopsy-proven inflammatory sclerosis of the panniculus or facscia, 13 of whom had extensive, ill-defined, bound-down plaques with a rapid centrifugal progression [229]. Three years later they added seven cases and reviewed the published literature describing patients with deep involvement [146]. On the basis that involvement of the deep dermis, subcutaneous fat, fascia or muscle can be present alone or in any combination, they renamed the condition morphoea profunda, and included eosinophilic fasciitis within its spectrum [146]. The distinction between morphoea profunda, pansclerotic morphoea (as defined above) and eosinophilic fasciitis remains blurred. The terminology is confusing, as evidenced by the recent use of ‘disseminated morphoea profunda’ to describe a patient with an isomorphic pattern and deep involvement [249] and of ‘generalized deep morphoea’ to describe a case with a pansclerotic pattern of disease [250].
In order to avoid future confusion, it may be preferable to classify patients as having generalized morphoea of the disseminated plaque or pansclerotic type, accurately describing the extent and depth of involvement and accepting that the depth of involvement can vary and is best defined by a deep tissue biopsy and/or magnetic resonance imaging (MRI).
Eosinophilic fasciitis
Eosinophilic fasciitis (synonym Shulman syndrome) (Figure 57.11) was first described by Shulman in 1975 [251]. The inclusion of this subtype within the morphoea spectrum is debated but supported by the coexistence of other subtypes of morphoea in 29–41% of cases [252, 253, 254]. It is usually extensive and involves deep tissues. When included in previous classifications of morphoea it has variably been assigned to the generalized or deep groups. It symmetrically involves the extremities, particularly the lower limbs, but typically spares the fingers and face [255, 256]. Truncal involvement is described and there may be considerable clinical overlap with pansclerotic morphoea in such cases [11, 253]. Unaccustomed severe exercise may precede the onset of disease in up to 50% of cases [255]. In the early stages there is painful, burning erythema and pitting oedema of the limbs. This is replaced by induration and fibrosis resulting in a typical peau d'orange appearance, with tethering around vessels producing guttering referred to as the groove sign. The sclerotic process involves the fibrous septa of the subcutis and deep fascia and may extend into the underlying muscle [8, 253, 257, 258]. A significant eosinophilic infiltrate in the panniculus and deep fascia may be present in the early stage of disease [253, 257, 258] but is not invariable [146]. It can result in severe joint contractures and associated morbidity. A high erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP), hypergammaglobulinaemia and peripheral eosinophilia (present in 60–90% of cases) are typical [253, 254, 258, 259]. Haematological abnormalities including thrombocytopenia, aplastic anaemia and leukaemia have been associated [253, 254, 260]. A deep biopsy of the clinically affected tissue showing inflammation and thickening of deep fascia is essential to establish the diagnosis [256].
Linear type
Linear morphoea accounts for 15–20% of morphoea cases in adults [28, 35] and 42–67% of cases in children [8, 9, 31, 36]. It may be subdivided into limb/trunk and head variants. The limb/trunk variant occurs twice as often (54–65%) as the head variant (23–35%) [8, 23]. Linear head and trunk/limb variants have been found to coexist in 1% of children. It is quite common for the upper and lower limb on the same side to be affected. Although mostly unilateral, bilateral involvement occurs in 5–25% of cases [8, 36, 261, 262]. Linear morphoea appears to follow Blaschko lines (Figure 57.12) in the majority of cases suggesting that genetic mosaicism is important in pathogenesis [261, 263, 264]. Roughly a quarter of patients with linear disease have another form of morphoea elsewhere (mixed subtype) and most often this is plaque morphoea [8].
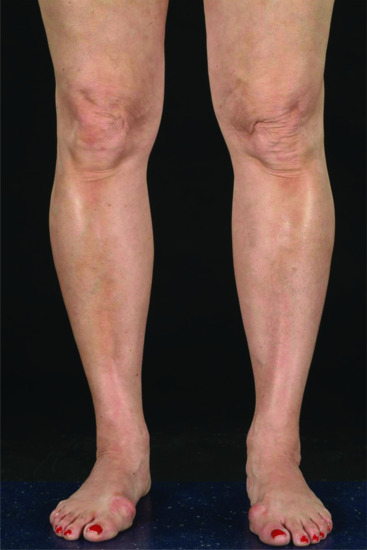
Figure 57.11 Eosinophilic fasciitis. Limited disease involving the lower legs and ankles. The skin appears shiny and taught
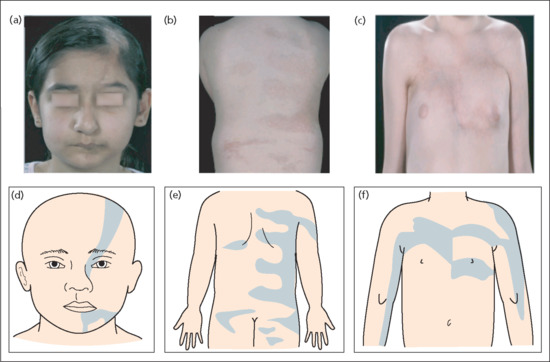
Figure 57.12 Blaschkoid nature of linear morphoea on the face (a, d), back (b, e) and chest (c, f). (From Weibel and Harper 2008 [261].
Reproduced with permission of Wiley.)
Head/neck variant
The head variant includes morphoea en coup de sabre (ECDS) and progressive hemifacial atrophy (PHA), also known as Parry–Romberg syndrome. In children, ECDS lesions are commoner than PHA, representing 87% of head variant lesions [8]. In a retrospective review of 54 patients diagnosed with morphoea ECDS or PHA at the Mayo Clinic from 1984 to 2004, 26 patients (48%) had morphoea ECDS, 13 (24%) had PHA and 15 (28%) had both [262]. Patients with sclerotic lesions, typical of morphoea ECDS, overlying atrophic areas of PHA are described [31, 265, 266]. Cases of typical morphoea ECDS have been reported to evolve into PHA, emphasizing the overlap and probable common pathogenesis which has been suggested previously [265–267]. Whilst morphoea ECDS occasionally has a sudden onset and rapidly progressive course, in a majority of cases the onset of both subtypes of disease is insidious and may progress slowly over many years.
Morphoea en coup de sabre
Morphoea ECDS usually begins in childhood but occasional adult-onset cases are described [268, 269]. It most frequently involves the frontoparietal area of the face and scalp in a paramedian distribution and follows Blaschko's lines [261, 270] (Figure 57.12). Bilateral lesions [262, 271, 272] and concomitant linear limb/trunk and plaque lesions may occur [28, 187]. Sclerosis is thought to involve the skin and subcutis first and then later extend to the underlying fascia and bone. Varying degrees of sclerosis, hyperpigmentation and atrophy may be observed (Figure 57.13). Scarring alopecia of the eyelashes, eyebrows and scalp occur if they are involved in the band. Linear depressions in the skull bones are a common consequence. Neurological, ocular and auditory complications are well recognized [8, 262], and a variety of abnormalities are seen on computed tomography (CT) scan and MRI (see the section on complications and co-morbidities in this chapter) [154].
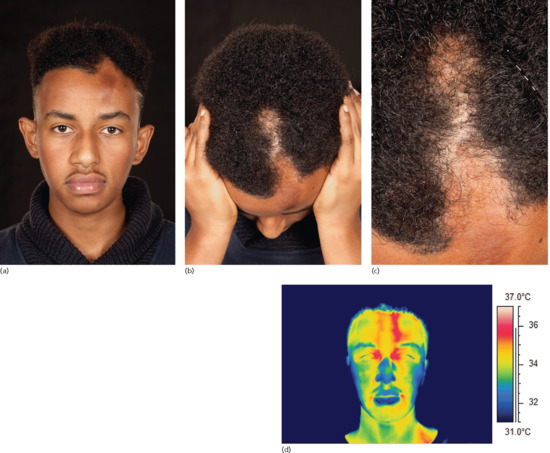
Figure 57.13 Linear morphoea en coup de sabre. (a) Hyperpigmention on the forehead with subcutaneous atrophy and a subtle indentation of bone. (b, c) Alopecia with more sclerotic changes on the scalp. Note the Blaschkoid distribution. (d) Thermographic image of the same patient showing increased temperature of the skin at a site corresponding to disease activity and extension on the left paramedian forehead.
Progressive hemifacial atrophy
Progressive hemifacial atrophy (synonym Parry–Romberg syndrome) has traditionally been described as affecting the area supplied by one or multiple branches of the trigeminal nerve [273]. More recently it has been suggested that lesions are Blaschkoid, as is the case for other types of linear morphoea [261]. A majority of cases begin in the first decade, but occasional adult-onset cases are described [274, 275]. It is a unilateral, progressive, primary atrophic disorder of the skin, subcutaneous tissue, muscle and underlying cartilage and bone (Figure 57.14) [262, 265, 266, 273, 276]. Altered pigmentation, usually a brownish or bruise-like change, but occasionally hypopigmentation, occurs at the affected sites. A progressive facial asymmetry develops as a result of a gradual loss of fat and muscle, and atrophy of the frontal, maxillary and/or mandibular bones. The mouth and nose become deviated towards the affected side. Enophthalmos is caused by a combination of progressive fat atrophy, shrinkage of the eyeball and thinning of the extraocular muscles [277]. Ocular complications including choroidal and retinal folding, hyperopia, uveitis, retinal vasculitis, glaucoma and third nerve palsies may occur as a result [276]. Neurological complications occur in 15% with headaches (9%) and epilepsy (13%) being the most common. Associated neuroimaging abnormalities occur in 19–44%, and may be ipsi-, contra- or bilateral [278]. Interestingly, their presence does not correlate with symptoms and they may develop years after the onset of disease [154]. Mandibular and intraoral involvement may lead to dental malocclusion and hemiatrophy of the tongue.
Figure 57.14 Progressive hemifacial atrophy involving the left side of the mandible and chin. Normal skin overlies atrophic deeper structures (arrows). Note the facial asymmetry. The patient had noticed progressive changes in facial contours for over 5 years before a diagnosis was reached.
In a study of 16 patients, vacuolar degeneration at the dermal–epidermal junction was a common histological feature in morphoea ECDS and a perivascular and/or periappendageal lymphocytic infiltrate and vacuolar degeneration of follicular epithelium were seen in early disease [279] suggesting that there may be an autoimmune attack of genetically different basal keratinocytes as a result of mosaicism.
Trunk/limb variant
As in morphoea ECDS, linear bands seem to follow Blaschko's lines and may exhibit varying degrees of erythema, sclerosis, atrophy and hyperpigmentation (Figure 57.15) [261, 263, 280]. Linear sclerotic bands may appear suddenly or insidiously and then progress. Coexistent or preceding plaque morphoea, most often on the trunk, is commonest in this form of linear disease. A majority of linear lesions are unilateral but bilateral lesions were found in 11% in the largest childhood study [8] and in 5.5–46% of other published cases [35, 36, 187]. The dermis, subcutis, underlying muscle and bone may be involved. In limb lesions, generalized arthralgias and oedema of the involved extremity can precede the onset of disease [8, 35, 187, 281]. Lesions extending across joints frequently result in flexion contractures [187]. Myopathic changes, atrophy and weakness of involved and adjacent muscles may occur [88]. In adults limb girth asymmetry results, and in children growth failure causes additional limb length discrepancies. Joint contractures, muscle atrophy and limb shortening cause pain and significant functional limitations, which in turn lead to a reduction in quality of life and to significant psychological morbidity [30, 281, 282].
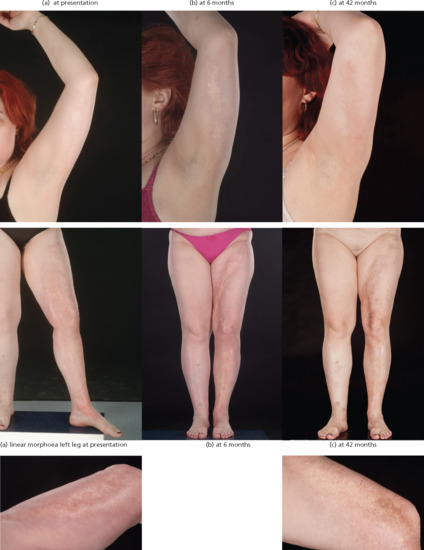
Figure 57.15 Linear morphoea of the left arm (top) and leg (below). (a, b) Active inflammatory phase showing (a) the shiny waxy appearance at presentation and (b) a marked increase in sclerosis with peripheral erythema at 6 months. (c) Softened, atrophic, hyperpigmented appearance of late-stage inactive disease at 42 months.
Linear atrophoderma of Moulin
The original 1992 publication described five patients with unilateral, depressed, hyperpigmented Blaschkoid plaques on the trunk and limbs, with onset between 6 and 20 years of age. Histology showed epidermal hyperpigmentation, but a normal dermis and normal collagen on histology [283]. The authors suggested that the impression of skin atrophy might therefore be due to atrophy of subcutaneous structures, and this has now been confirmed in one case by ultrasonography [284]. Subsequently, a variety of pathological findings have been described [285]. In one case, clinical overlap with atrophoderma of Pasini–Pierini and morphoea were seen and, histologically, perivascular inflammation and thickening of collagen fibres in the mid-reticular dermis constituted a significant overlap with atrophoderma of Pasini–Pierini and morphoea [286]. Although it is not universally agreed [285], linear atrophoderma of Moulin appears to lie within the morphoea spectrum and be akin to a linear Blaschkoid form of atrophoderma of Pasini–Pierini.
Linear deep atrophic morphoea.
Three cases of linear primary atrophic morphoea with no preceding clinical inflammation or sclerosis, involving the subcutis and deep dermis, and resembling PHA on a limb, are reported [287]. Although progressive in nature, they did not result in joint contractures. In one of these and two further cases, linear limb lesions occurred in association with PHA, confirming the relationship between these two presentations [287–289].
Mixed type
Mixed morphoea describes the 4% of adults and 15–23% of children with coexistence of more than one subtype of morphoea. The commonest combination is linear limb/trunk and plaque morphoea, but any combination can occur.
Differential diagnosis
Skin sclerosis may be seen in a number of conditions that are neither morphoea nor SSc. These include metabolic, deposition and genetic disorders, disease caused by exposure to chemicals, toxins and drugs, graft-versus-host disease and nephrogenic systemic fibrosis (Table 57.3) [290, 291]. The differential diagnosis of morphoea depends of the subtype and stage of disease and is summarized in Table 57.4 and Box 57.1.
Table 57.4 Differential diagnosis of the different phases of limited morphoea
| Early inflammatory phase | Sclerotic phase | Hyperpigmented phase | Atrophic phase |
Granuloma annulare Early extragenital lichen sclerosus Lyme disease–erythema migrans Mycosis fungoides Cutaneous mastocytosis Radiation dermatitis Fixed drug eruption |
Necrobiosis lipoidica Pretibial myxoedema |
Post-inflammatory hyperpigmentation Actinic lichen planus Café au lait macule |
Lipodystrophy Steroid-induced atrophy Lupus profundus Acrodermatitis chronica atrophicans Panniculitis (late stage) |
Classification of severity
Severity of disease in morphoea depends largely on the age at onset, extent and depth of disease, and whether there is bony, joint or central nervous system (CNS) involvement. Measures of severity should assess disease activity, the degree of irreversible damage caused by previously active disease, and the impact on quality of life. Until recently there has been no uniformly accepted scoring system for assessing disease severity in morphoea. A wide variety of imaging techniques and skin scoring systems have been employed, making it difficult to compare outcomes in different clinical studies and disease registries. The recent development and validation of the localized scleroderma cutaneous assessment tool or LoSCAT may help in this regard (see the section on outcome measures in Investigations) [27, 292, 293].
Severity levels for juvenile localized scleroderma were recently proposed by the Childhood Arthritis and Rheumatology Research Alliance (CARRA) [26]. High severity was defined as presentation with generalized or pansclerotic morphoea, craniofacial linear morphoea or another subtype with evidence of high morbidity such as CNS involvement, limb shortening or joint contracture. Moderate severity was defined as circumscribed (limited) deep morphoea or linear morphoea of the trunk or limb without evidence of high morbidity. Low severity disease was defined as superficial, circumscribed (limited) plaque morphoea. These measures of severity should be equally applicable to adult patients.
There is also some evidence that patients with extracutaneous manifestations, particularly children [19], appear to have more severe disease, as suggested by greater levels of systemic inflammation and a greater need for systemic immunosuppression. Interestingly, the presence of a positive rheumatoid factor may be associated with more severe articular involvement.
Functional limitation is defined as a clinically appreciable limited range of motion of a joint secondary to contracture or to skin and subcutaneous tissue involvement, but not due to abnormality of the joint itself and/or limb–length discrepancy [95]. This may also be considered a marker of disease severity since it is closely associated with quality of life scores and psychological morbidity.
Complications and co-morbidities
Extracutaneous manifestations
Although the classic description of morphoea suggests no internal organ or systemic manifestations, there is increasing evidence to challenge this assumption. Small series in adults and children have found extracutaneous features in 21–27% of patients [294, 295]. In these earlier studies, mild degrees of frequently asymptomatic oesophageal dysmotility were identified in 17–23% of cases, as well as mild gas transfer defects and restrictive changes in lung function in 14–18% [295, 296] and myositis in up to 34% [294, 295]. Electrocardiogram abnormalities, particularly incomplete right bundle branch block have been documented in adults and children with varying frequencies (8–88%) [19, 297–299]. In one study of 40 asymptomatic children with morphoea, 16 (40%) had echocardiographic abnormalities, particularly involving the mitral valve and left ventricle [297].
The frequent occurrence of extracutaneous manifestations has now been confirmed in larger studies. In a US review of 245 adults and children, the highest frequencies of arthralgia (24%), dysphagia (14%), dyspnoea (20%) and vascular complaints (mainly Raynaud phenomenon, 31%) were seen in patients with generalized morphoea. In contrast, the linear subtype was associated with neurological (31%) and ophthalmological (8%) complications related to the affected site on the face or scalp [9].
In the largest childhood study, 22.4% of 750 children had one or more extracutaneous manifestation including arthritis and neurological, ocular, vascular and gastrointestinal complications [19]. In this study, symptoms of arthritis were the most common, encountered in 11%, particularly in children with linear disease and rheumatoid factor positivity. Arthritis was unrelated to the site of the morphoea in a quarter of cases, suggesting that a systemic, rather than a local, inflammatory process is involved. Six per cent of children had an oligoarthritis (up to four joints) and 5% a polyarthritis more than four joints); 30% of the children with arthritis had a positive rheumatoid factor.
Neurological symptoms occurred in 4.4%, predominantly in children with linear disease involving the head. Most frequently these were seizures and headache, but peripheral neuropathy, vascular malformations and CNS vasculitis were also identified [19]. Asymptomatic CNS abnormalities were identified on neuroimaging or electroencephalography in approximately 1% of cases in this study [8]. In a third of cases neurological changes were unrelated to the site of skin involvement. These findings are similar to those of a systematic review of neurological involvement in 224 published cases of linear morphoea in which seizures (42%) and headache (19%) predominated, but cranial nerve palsies, hemiparesis and neuropsychiatric involvement were also documented [300, 301]. Both ipsilateral and contralateral bone thinning, cerebral atrophy, white matter lesions, focal subcortical calcifications and meningocortical changes have been described [301]. Scalp and calvarial abnormalities such as atrophy, T2-hyperintensities, calcifications and ipsilateral cerebral atrophy are the most commonly reported [154]. As in children, adults with head and neck disease are at risk [300]. Importantly, whilst some symptomatic patients have normal imaging – perhaps reflecting insensitive imaging techniques – substantial numbers of asymptomatic patients have significant CNS abnormalities on imaging, highlighting a need for screening in patients with disease involving the head and neck [154].
Oral and dental problems can also occur. In one study of 16 patients these included malocclusion (94%), an overgrowth tendency of the anterior lower third of the face (82%), abnormal mastication (69%), dental anomalies (63%), skeletal asymmetry (56%), bone involvement (50%) and temporomandibular joint involvement (19%) [302].
Ocular complications were seen in 2.1% of 750 children – most frequently episcleritis, anterior uveitis and keratitis, but also glaucoma, xerophthalmia, strabismus, mydriasis and papilloedema [19, 303]. In 25% of cases these were unrelated to the site of the skin lesions. Enophthalmos [277], choroidal and retinal folding, hyperopia, retinal vasculitis, glaucoma and third nerve palsies are also documented [276].
Of 750 children, 2.4% had vascular abnormalities. There was one case of cutaneous vasculitis and one of deep-vein thrombosis. The other 2.1% (16 patients) had Raynaud phenomenon which, in a minority, preceded the onset of morphoea. Ten of these patients had a positive ANA but none had any SSc-specific autoantibodies. Nine of the children with Raynaud phenomenon had more than one extracutaneous manifestation, including arthritis, gastro-oesophageal reflux and cardiac arrhythmia.
Symptomatic gastro-oesophageal reflux, confirmed on appropriate investigations, was identified in 1.6% of cases [304]. As with the Raynaud phenomenon, approximately 60% of these children had other concomitant extracutaneous features.
Respiratory (0.7%), cardiac (0.3%) and renal (0.3%) complications were rarer. Four per cent of children had more than one extracutaneous manifestation and again this occurred predominantly in patients with linear disease.
Together these findings suggest a more widespread inflammatory and/or autoimmune process in some forms of morphoea, as well as a need for more systematic multiorgan baseline investigations.
Psychological manifestations
There have been three studies that suggest that morphoea has a minimal impact on quality of life in children and that they have good emotional coping strategies [305–307]. Patients with childhood-onset disease, although they have a higher degree of disease-related damage, may experience less of an impact on their quality of life [308]. Two further studies, however, suggest that morphoea can have a significant impact on quality of life. In the first, 38% of 74 patients were at risk of anxiety or depression [309]. Psychological morbidity was highest in patients with generalized morphoea and eosinophilic fasciitis, and in those with more severe disease, greater levels of pain and fatigue and a greater impact of disease on daily life and social support [309]. The second study, in 277 adults and children, confirmed that reductions in quality of life correlated with functional impairment and symptoms of active disease such as pain and pruritus, independently of disease subtype, age and sex [282].
Disease course and prognosis
Early studies indicated that the duration of disease activity, although variable, was usually 3–5 years [35], with plaques generally resolving earlier than other subtypes. In Peterson et al.’s series [28], overall, 50% of the patients had at least 50% softening or resolution by 3.8 years after diagnosis. Fifty per cent resolution occurred on average at 2.7 years in the plaque group, at 5 years in the generalized and linear groups and at 5.5 years in the deep group. However, mean disease duration of childhood-onset morphoea may be twice as long as that for adult-onset disease (13.5 versus 5.8 years) [22].
There is now increasing evidence to suggest that in certain types of morphoea permanent remission is not the rule. In a retrospective study of 113 adults and 126 children referred over a 20-year period to 2001, children with mixed forms of disease were more likely to run a more protracted and complicated course, and relapse was more frequent in generalized, deep and mixed forms [22]. In a retrospective chart review of 52 paediatric patients with linear morphoea seen between 1990 and 2010, although disease stabilized after a mean duration of 5.4 years, 38% had functional limitations. Reactivation of disease was frequent, even after seemingly effective courses of methotrexate and corticosteroids, such that 31% of patients reported active disease after 10 years [23]. The most recently published retrospective chart review includes 344 patients of whom 119 had a childhood onset of disease. Disease recurrence occurred in 27% of the paediatric-onset group and 17% of the adult-onset group. The linear limb variant was associated with a higher risk of recurrence, irrespective of age at the onset of disease [310].
The long-term impact of paediatric-onset morphea was further highlighted in a group of 27 adult patients (mean age 30.6 years, range 18–78 years, median 26 years) with a mean age at onset of lichen sclerosus of 11.5 years (range 3–17 years, median 13 years) [24]. Seventy-four per cent had linear disease, and 18.5% had plaque morphoea at disease onset that progressed to meet the criteria for generalized disease over time. Overall, 81% had persistent symptoms of pain, itch or numbness; 89% of patients had persistent disease activity, continuous in 8/27, and with a remitting–relapsing course in 16/27. Seven of 27 (29%) patients described flares of activity triggered by trauma, pregnancy or reduction or discontinuation of systemic therapy. Fifteen of 20 patients with linear disease had permanent sequelae including reduced range of motion and deep atrophy. In 22% there was a ‘moderate to very large’ impact on quality of life. These studies highlight the need for careful counselling of patients and their families and continued vigilance.
There is still uncertainty regarding the relationship between morphoea and SSc. Concomitant morphoea, particularly plaque and nodular forms, have been reported in up to 6.7% of patients with SSc [219, 311]. Transition from morphoea to SSc has been reported in 0.13–1.3% of morphoea cases [35, 294, 312, 313]. SSc-specific antibodies have been identified in small numbers of morphoea patients (antitopoisomerase in 2–3% [8, 22], anticentromere in 1.7% [8]), however none of these cases have been reported to progress to SSc after 3 or more years of follow-up. Although such patients should be kept under close review, in the absence of nail fold capillary abnormalities and sclerodactyly a transition to SSc is extremely unlikely.
Investigations
The diagnosis of morphoea is largely clinical, but a number of investigations can be helpful in guiding management (Table 57.5). A biopsy can be undertaken if the diagnosis, the depth of involvement or the degree of activity are in doubt. An incisional ellipse through the violaceous/inflammatory edge of a lesion, into the sclerotic centre and extending down into the subcutis, should be taken and the site clearly indicated for the pathologist. If the depth of involvement is in question the biopsy should extend into the fascia and underlying muscle and referral to a plastics or general surgeon may be required. MRI can provide a useful tool for assessing the depth and extent of involvement, particularly tendon and muscle involvement in pansclerotic and linear limb lesions, and CNS involvement (frequently asymptomatic and unsuspected) in head variant lesions [314, 315]. An MRI and electroencephalogram should be undertaken routinely in all new cases of linear disease of the head. Serial MRI can be used to assess disease progression/stabilization. In some cases a CT scan can be more helpful than MRI if the extent of bony involvement is in question. There is no current recommendation as to whether or how often such investigations should be repeated, but since CNS involvement can occur years after the onset of disease, it may be appropriate to repeat the MRI every 2–3 years and/or if new symptoms develop. Serial ultrasonography can be used to evaluate skin thickness and loss of muscle and fat [316]. Disease activity can be correlated with echogenicity and tissue blood flow measured by Doppler [317] and laser Doppler [318, 319], ultrasound techniques [317, 320] and infrared thermography [321]. A durometer is a hand-held instrument that measures the depth of skin indentation after the application of a standardized amount of force. Durometry measures skin hardness, a recognized surrogate for skin thickness [322], and has been shown to objectively discriminate affected versus unaffected skin on the trunk and limbs in children with morphoea [323].
Table 57.5 Assessment of the patient with morphoea
| Clinical assessment | Investigations |
| Are lesions symptomatic (burning/pruritus)? | Measure LoSCAT score |
| Are lesions extending (new lesions/ extending lesions/lilac ring)? | Photography ± thermography ± ultrasonography ± scanning laser Doppler |
| Are there extracutaneous manifestations (headache, migraine, seizures, arthralgia, myalgia,dyspepsia, Raynaud phenomenon, etc.)? | Skin biopsy: deep incisional ellipse (to include fascia and muscle if deep involvement suspected) |
| Full skin examination to determine extent and activity of lesions | MRI (images brain and depth of extent on limbs), CT (images bony contours) for linear lesions |
| Examine for genital lichen sclerosus | Electroencephalogram (head variant) |
| Creatine kinase, aldolase (if muscle involvement suspected) | |
| Full blood count, eosinophils, immunoglobulins, TFT, erythrocyte sedimentation rate, C-reactive protein | |
| ANA, ENA, rheumatoid factor | |
| Borrelia serology | |
| Referral to allied specialties as appropriate: ENT, ophthalmology, oral medicine, orthopaedics, plastic surgery, physiotherapy |
ANA, antinuclear antibody; CT, computed tomography; ENA, extractable nuclear antigen; LoSCAT, localized scleroderma cutaneous assessment tool; MRI, magnetic resonance imaging.
Routine blood investigations are usually normal in limited subtypes. In patients with generalized, deep or linear disease, it may be helpful to measure eosinophils, serum immunoglobulins, creatine kinase, inflammatory markers (ESR/CRP), ANA, rheumatoid factor and, in newly presenting cases, Borrelia serology. Eosinophilia, hypergammaglobulinaemia and raised inflammatory markers may occur in active disease of any subtype, but are more frequent in generalized and linear forms, particularly if there is deep involvement. Increased levels of creatine kinase and aldolase have been associated with the development of new lesions, muscle atrophy and limb shortening, and may thus suggest muscle involvement and possibly disease activity [30]. Frequently associated autoimmune diseases such as autoimmune thyroid disease should be excluded. If there is concern about the possibility of SSc (e.g. in cases of pansclerotic morphoea or eosinophilic fasciitis) evidence of Raynaud phenomenon, puffy fingers, sclerodactyly, nail fold capillaroscopic abnormalities and SSc-specific antibodies (anticentromere, antitopoisomerase and anti-RNA polymerase) should be sought.
Referrals to allied specialties such as ophthalmology, ENT and maxillofacial should be considered for patients with head involvement and to orthopaedics and physiotherapy for linear limb disease, particularly in children.
Outcome measures
Defining outcome measures in morphoea has proved difficult because morbidity is caused by a combination of cutaneous and subcutaneous damage as well as disease activity. Trying to find accurate measures of disease activity has been a particular challenge. A variety of imaging techniques have been employed, including serial photography, infrared thermography, Doppler flowmetry, ultrasonography and MRI [324], and more recently a computerized skin scoring method [325]. Of these methods only the computerized skin score, ultrasonography and MRI have been validated. Variable frequency ultrasound (7–15 MHz linear probes) with colour Doppler is a non-invasive technique that reveals details of skin morphology and function. It has shown high specificity and sensitivity for assessing disease activity [317]. The most accurate sonographic signs of activity are increased subcutaneous echogenicity and increased cutaneous blood flow. Laser Doppler imaging measures blood flow within the dermis and is a more accurate reflection of skin inflammation than infrared thermography, which is difficult to interpret when there is soft tissue atrophy. Single-point laser Doppler flowmetry measurements correlate with clinical disease activity [318, 326]. Scanning laser Doppler imaging had a positive predictive value of 87% and negative predictive value of 94% in a small study evaluating its use in assessing activity and predicting progression [319].
Clinical scoring techniques have the advantage of not requiring expensive or cumbersome equipment. The MRSS, a measure of skin thickness which has been validated in SSc [327], has also been used in morphoea, but is not validated in this condition and does not assess damage. The DIET score (dyspigmentation, induration, erythema, telangiectasia; each scored 0–3, with a maximum score of 12) [328], has the advantage of simplicity, but may not provide an accurate assessment of damage. Four measures of disease activity and damage have recently been combined to develop a morphoea-specific skin scoring system –LoSCAT (Figure 57.16). Initially, two measures of disease activity, the physician global assessment of activity (PGA-A) and the modified localized scleroderma skin index (mLoSSI) were developed and validated in 2009 [27, 329]. The PGA-A is graded on a 100 mm analogue scale and includes the following cutaneous variables: the development of new lesions and/or enlargement of existing lesion within the previous month, erythema/violaceous colour and skin thickening/induration at the border of lesions. The mLoSSI includes the sum of three separate scores from the following domains: erythema (none, mild, moderate, severe), skin thickness (none, mild, moderate, severe) and new lesion/lesion extension (present, not present). A patient is classified as having active disease if they have a PGA-A of greater than 0 and a mLoSSI of greater than 0.

Figure 57.16 Localized scleroderma cutaneous assessment tool (LoSCAT). (From Arkachaisri et al. 2010 [292].
Reproduced with permission of Oxford University Press.)
Subsequently, two measures of disease damage, the localized scleroderma skin damage index (LoSDI) and the physician global assessment of disease damage (PGA-D), were developed. Disease damage was defined as irreversible or persistent changes of the lesion due to previous active disease or complications of therapy. The LoSDI was calculated by summing three scores for cutaneous features of damage: dermal atrophy, subcutaneous atrophy and dyspigmentation measured at 18 anatomical sites, recording the most severe score from each domain if multiple lesions were present within one anatomical site [292]. The PGA-D is graded on a 100 mm analogue scale and is anchored by ‘no damage’ at 0 and ‘markedly damaged’ at 100. Based upon consensus agreement, both cutaneous and extracutaneous manifestations are taken into account when scoring the PGA-D. The cutaneous manifestations include hyper/hypopigmentation and subcutaneous and dermal atrophy. The extracutaneous manifestations include: musculoskeletal involvement (skeletal muscle atrophy, bone atrophy, facial atrophy, limb length discrepancy, physical disability, joint contracture), neurological involvement (CNS symptoms, abnormal brain MRI, eye involvement) and psychosocial quality of life impairment [292].
The LoSCAT is now a validated tool to measure both disease activity and the more chronic and irreversible damage caused by morphoea [292, 293]. It is hoped that it will prove a useful tool for assessing clinical changes in routine patient care and outcomes in clinical trials.
Management
The management of morphoea has been challenging because of a lack of standardized evaluation methods and of evidence-based treatments. Linear and generalized forms of morphoea can lead to permanent cosmetic and functional impairment. Early intervention can limit disease progression and its consequences. However, treatment of patients with significant damage but inactive disease exposes them to the potential side effects of the medications, without providing significant benefit. Patient selection and appropriate timing of interventions is paramount.
Treatment choices should be based on the subtype of morphoea, the level of disease activity and, to some degree, on the age of the patient. In practice however, this has not always been the case and the treatment prescribed may depend more on the specialty of the treating physician than the patient or their type of disease. Thus, from a total of 531 prescriptions for 224 patients (95 children), dermatologists prescribed topical corticosteroids in 41%, other topical treatment in 25% and phototherapy in 16% of cases, and systemic corticosteroids in 5% and methotrexate in 4% of cases. Rheumatologists, in contrast, prescribed methotrexate in 34% and systemic corticosteroids in 31% of cases, and topical treatments in 10% and phototherapy in only 2% [34]. In this study, 68% of patients with linear morphoea received topical corticosteroids as the mainstay of treatment if they saw a dermatologist (4% received methotrexate), whereas 39% of linear morphoea patients seen by rheumatologists received methotrexate and 8% topical corticosteroids [34]. In a survey of UK dermatologists published in 2014 [330], clinicians were asked which treatment option produced the best outcome in active morphoea based on their experience. There was a wide range of responses: methotrexate with corticosteroids was the most frequent response (37.3%), followed by methotrexate alone (25.4%), phototherapy (18%) and topical steroids (13%). These surveys underscore the need for a joined up multidisciplinary approach to establish effective treatment guidelines and to ensure that patients are not undertreated.
The initial evaluation of a patient with morphoea should include an assessment of the type, extent and activity of disease (see Table 57.5). Treatment can then be based on the subtype, activity, extent and depth of disease, and patient symptoms [2, 25, 26].
Limited superficial forms of morphoea can generally be treated topically. Case reports and a prospective open-label study in 13 patients initially suggested a role for topical tacrolimus 0.1% [331–333]. Two small placebo-controlled trials support the use of twice-daily topical tacrolimus 0.1% ointment (10 patients) [334], and daily 5% imiquimod cream (22 patients) [335]. The latter was also found to be safe and beneficial when used 3–7 times weekly, over a 9-month period, in 21 adult and paediatric patients in two prospective open-label studies [328, 336]. Benefit in prospective but uncontrolled studies has been shown for twice-daily application of topical vitamin D analogues (calcipotriol/calcipotriene 0.005%), either alone under occlusion (12 patients) [337] or in combination with topical steroids (six patients) [338] or with low-dose UVA-1 phototherapy (19 children) [339]. Although there is no direct evidence to support their use, topical and intralesional corticosteroids are frequently used [330] with good effects, particularly in the early inflammatory stages of disease or if there are pronounced epidermal changes.
If there is no deep involvement, disseminated forms (e.g. disseminated plaque morphoea) can be treated with phototherapy, preferably UVA-1, but when this is not available broad-band UVA, narrow-band UVB or topical psoralen and UVA (PUVA) therapy are alternatives. The rationale for using phototherapy in morphoea is based on the ability of UVA and UVB to induce MMPs such as collagenase [340–342]. UVA wavelengths (320–400 nm) penetrate deeper into the dermis than UVB (280–320 nm). UVA-1 (340–400 nm) is less erythemogenic and penetrates deeper than UVA-2 (320–340 nm). In addition to collagenase, UVA-1 has been shown to up-regulate antifibrotic haem oxygenase-1 [343], to cause T-cell apoptosis, to deplete dermal Langerhans and mast cells, to induce interferon γ (INF-γ), IL-1 and IL-6 production, and to inhibit TGF-β production [344–346]. This latter effect may involve the SMAD family of proteins involved in TGF-β signal transduction [346] and a stimulatory effect on the antifibrotic protein decorin [347].
Although a variety of treatment doses and regimens have been employed, there is consensus that low-dose (10–20 J/cm2), medium-dose (>20–70 J/cm2) and high-dose (>70–130 J/cm2) UVA-1 have all shown efficacy, significantly reducing skin thickness and stiffness in adults and children with all forms of morphoea (reviewed in [25, 345, 348]). Early inflammatory and sclerotic lesions appear to respond most favourably, and cases with deep involvement least favourably. Outcome measures recorded in over 90% of cases include variable combinations of clinical examination, skin scores, ultrasound measurement of skin thickness, cutometer measurements and skin biopsies. UVA-1 appeared to be effective in all skin types in a retrospective review of treatment responses in 47 morphoea patients [349]. This is in contrast to Wang et al.’s findings that suggested that the antifibrotic effects of UVA-1 were determined by skin type, and that skin darkening as a result of UVA-1 treatment may attenuate its antifibrotic effects [350]. The duration of responses is variable and up to half of the patients may develop recurrence of active morphoea lesions at 2–3 years [351].
Significant benefit with low- and medium-dose UVA-1 has been confirmed in two randomized controlled studies [352, 353]. Medium doses appear more effective than low-dose UVA-1 based on ultrasound measurements of skin thickness [353] and may give better long-term results [354, 355]. It is preferred to high doses because of the lower cumulative UV exposure. The current recommendation, where available, is to use medium-dose UVA-1 (60 J/cm2) 3–5 times weekly for a total of 40 sessions [348]. Nevertheless, low-dose therapy may still have a role, particularly if combined with topical modalities such as vitamin D analogues [339].
UVA-1 is not widely available and this has led physicians to investigate the efficacy of other forms of phototherapy. Broad-band UVA has been used at varying doses in 114 patients in three controlled trials, with positive responses documented by clinical examination and skin biopsies [344, 356, 357]. Bath PUVA resulted in reductions in skin thickness and improvement in 15/19 patients and cream PUVA in 4/4 cases [358–361]. Nineteen patients treated with narrow-band UVB five times weekly showed improvement comparable with that in 27 patients receiving low-dose UVA-1 based on skin scores, visual analogue scales for itch and tightness, histology and 20 MHz ultrasound [352].
In patients with progressive disease despite topical agents and/or phototherapy, and in patients with linear, deep or disseminated forms of disease such as morphoea en coup de sabre, pansclerotic morphoea or eosinophilic fasciitis, systemic therapy is indicated. There is broad agreement that combinations of pulsed intravenous and/or oral steroids with methotrexate should be used first line.
An open study of 17 patients with severe morphoea (linear/generalized) found that oral corticosteroids (0.5–1 mg/kg/day for 6 weeks then reducing over a mean of 18 months) produced a marked improvement, with reduced inflammation, cessation of new lesion formation and skin softening. A third of patients relapsed on discontinuing therapy [362]. Systemic corticosteroids should be considered in patients with severe, active inflammatory disease and in patients with eosinophilic fasciitis who appear particularly steroid responsive [253].
Methotrexate is a cornerstone of morphoea management [363–370). Methotrexate is thought to exert its effects at multiple levels. It has been shown to enhance monocyte differentiation [371], reduce peripheral blood mononuclear cell production of IL-8 [372] and stimulate IL-1 receptor antagonist and soluble TNF receptor p75 in vitro in rheumatoid arthritis [373]. Reductions in circulating sIL-2R and IL-6 following successful therapy with methotrexate in children and adults with rheumatoid arthritis [374, 375] suggest a possible mechanism for its effects in morphoea, since reductions in serum IL-2, -4 and -6 have been found to parallel improvement in cutaneous sclerosis [123]. Furthermore, mast cell numbers and levels of tenascin – an extracellular matrix protein previously shown to be increased in the skin and circulation of morphoea patients [376, 377] – are both reduced in lesional skin after methotrexate therapy [378].
Two early uncontrolled case series (17 patients, 9 adults) suggested some improvement in skin lesions with methotrexate alone [262, 363]. Four retrospective reviews documented the response to methotrexate alone in 52 cases, and in combination with corticosteroids in 67 cases [365, 366, 379, 380]. Improvement was described in 79% of cases but was more variable in those treated with methotrexate alone. In three prospective studies, a total of 60 patients (15 adults) were treated with either monthly pulsed intravenous (1 g, 3 days per month, for 6 months in the adults and 30 mg/kg, 3 days per month, for 3 months in nine children) or daily oral corticosteroids (2 mg/kg/day (maximum dose 60 mg/day), tapered to 0.25 mg/kg/day for 12 months in 36 children) and methotrexate at doses of 0.3–1 mg/kg/week (maximum dose 25 mg) in children and 15 mg/week in adults [364, 368, 370]. In these studies, significant improvements were noted based on mLoSSI (mLoSSI score of 0 in 32/36 cases at 36 months) and PGA-A [368] or physician assessment [364] in children. A 50% reduction in skin scores, corroborated by biopsy and ultrasound measurements, was documented in 13/15 adults after a mean treatment duration of 9.8 months [370]. This benefit was confirmed in a randomized placebo-controlled trial in 70 children with active linear, generalized or mixed morphoea, comparing 12 months of oral methotrexate (15 mg/m2/week (maximum dose 20 mg/week), n = 46) with placebo (n = 24) [367]. All patients received a concomitant 3-month course of oral prednisolone (1 mg/kg/day (maximum dose 50 mg)). Response was assessed objectively with infrared thermography, a computerized skin scoring system, by physician global assessment of disease severity and the development of new lesions. Methotrexate was well tolerated, and resulted in a reduction in new lesion formation, which occurred in 6.7% of cases versus 17% of the corticosteroid-treated controls. As suggested by a previous study [362], corticosteroid monotherapy resulted in a sustained improvement in approximately 30% of patients at 12 months. However, the likelihood of experiencing a disease flare in the methotrexate-treated group was approximately one-third of that in the corticosteroid-treated placebo group. The percentage thermal change from baseline in a representative lesion was –44% in the treatment group compared with –12% in the placebo group at 12 months. Over two-thirds of patients were judged to have responded, and in over 50% the clinical improvement persisted long after the discontinuation of methotrexate. Fifteen per cent relapsed at 24 months [367, 369]. The open-label extension of this study suggested that 70% of responders maintained clinical remission off treatment for a mean of 25 months, but that treatment courses of at least 24 months were needed to ensure a sustained remission [369]. Others have suggested that 4 years of treatment may be required to reduce the risk of relapse [381].There is further evidence in patients with childhood-onset disease that methotrexate is effective in achieving disease inactivity and remission off treatment; however, up to 40% of cases may subsequently (21 months after stopping therapy) require a further course of treatment [382].
In response to the lack of consensus on treatment regimens amongst clinicians caring for patients with morphoea [34, 383], CARRA has recently developed consensus-derived standardized treatment plans for the initial 12 months of therapy for childhood morphoea of moderate or high severity [26]. These plans have established clinical assessment methods and treatment response criteria. They also recommend defined treatment protocols employing methotrexate alone or in combination with oral or intravenous corticosteroids, with mycophenolate mofetil used in addition to or as a replacement for methotrexate according to physician preference. Limiting the variability in medication used and methods of assessment may facilitate the evaluation of treatment strategies in future comparative effectiveness studies.
Based on the above, combinations of pulsed intravenous and/or oral corticosteroids with methotrexate should be used first line. There is increasing evidence for the antifibrotic effects of mycophenolate mofetil in SSc [384–386]. A small number of cases have been published that suggest benefit in morphoea, used alone (10 cases) or in combination with methotrexate in patients who fail on or are unsuitable for monotherapy [387]. In view of the possible role of Th17 cells in morphoea, newer T-cell-directed therapies are now being considered. The efficacy of ciclosporin was reported in a single case of childhood linear disease [388] and in two adults with pansclerotic disease [389]. Two cases of extensive deep morphoea treated with abatacept showed significant improvement [249]; abatacept is a cytotoxic T-lymphocyte antigen 4 (CTLA4) IgG1 recombinant fusion protein that selectively inhibits T-cell activation via competitive binding to CD80 or CD86. Imatinib has been used successfully in conjunction with methotrexate and prednisolone in one case [390]. Infliximab was reported to induce remission in a case of generalized morphoea with lichen sclerosus overlap unresponsive to conventional therapies [391]. Successful therapy with extracorporeal photopheresis has been reported in two cases [392, 393]. Based on early results in SSc, there may also be some rational for using high-dose intravenous immunoglobulin in resistant cases [394, 95]. Current evidence does not support the use of oral calcitriol [396], INF-γ [397], d-penicillamine or antimalarials [25].
The treatments discussed thus far are aimed at switching off active disease and preventing damage. Once damage such as dyspigmentation, atrophy or bony asymmetry has occurred, treatment should aim at improving the cosmetic appearances, provided that the disease is no longer active. To this end various techniques of autologous fat grafting alone or in combination with surgery have gained popularity in the treatment of tissue defects of the face [398].
Finally, ulceration occurring in the context of severe, deep or pansclerotic disease has shown improvement in a small cases series with sildenafil [238]. In one case improvement of limb ulcers, skin sclerosis and joint mobility was noted with the dual oral endothelin receptor antagonist bosentan [399].
It is hoped that the gradual introduction of consensus treatment plans [25, 26] and simple effective clinical outcome measures such as the LoSCAT [292], will help provide better evidence of therapeutic efficacy, and will facilitate management in the future. A therapeutic algorithm and suggested first, second and third line therapies are outlined with levels of evidence in Table 57.6 and Figure 57.17.
Table 57.6 Morphoea treatments and levels of evidence (in brackets)a
| Type of disease | First line therapies | Second line therapies | Third line therapies |
| Limited or superficial disease | Tacrolimus ointment 0.1% b.d. (± occlusion) (IIa) Imiquimod (IIa) Topical/ intralesional corticosteroids (III) |
Calcipotriol 0.005% b.d. under occlusion (IIb) Calcipotriol-betamethasone (IIb) Calcipotriol +low-dose UVA-1 (IIb) |
Phototherapy : UVA-1, broad-band UVA, narrow-band UVB (Ib) Psoralen and UVA (bath or cream) (IIb) |
| Disseminated plaque disease | UVA-1 phototherapy (medium dose recommended) (Ib) | Psoralen and UVA (bath or cream) (IIb) Calcipotriol + low-dose UVA-1 (IIb) Broad-band UVA (Ib) Narrow-band UVB five times per week (Ib) |
|
| Pansclerotic, linear or deep disease or disease unresponsive to other treatments and with significant impact on quality of life | Methotrexate (MTX) + IV corticosteroids (Ib) MTX + oral corticosteroids (Ib) |
Mycophenolate mofetil (MMF) (III) Abatacept (III) |
Combination oral medication, e.g. MTX + MMF (III) Ciclosporin (III) Combination oral medication with phototherapy (III) Extracorporeal photopheresis (III) |
aCategories of evidence:
Ia: evidence for meta-analysis of randomized controlled trials;
Ib: evidence from at least one randomized controlled trial;
IIa: evidence from at least one controlled study without randomization;
IIb: evidence from at least one other type of quasi-experimental study;
III: evidence from non-experimental descriptive studies, such as comparative studies, correlation studies and case–control studies;
IV: evidence from expert committee reports or opinions or clinical experience of respected authorities, or both.
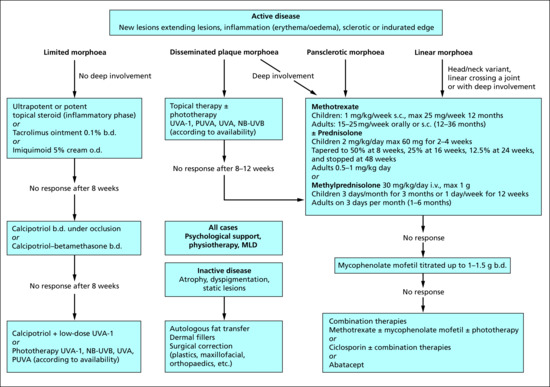
Figure 57.17 Therapeutic algorithm for morphoea based on existing evidence. b.d., twice a day; IV, intravenous; MLD, manual lymphatic drainage; NB, narrow band; o.d., once a day; PUVA, psoralen and UVA; s.c., subcutaneous; UV, ultraviolet.
Resources
Patient resources
- Fett NM. Morphea (localized scleroderma). JAMA Dermatol 2013;149:1124.
- JAMA Dermatology, Morphoea (localized scleroderma): http://archderm.jamanetwork.com/data/Journals/DERM/927631/dpg130102.pdf.
- Mayo Clinic, Morphea: http://www.mayoclinic.org/diseases-conditions/morphea/basics/definition/con-20028397.
- Patient, Morphoea: http://www.patient.co.uk/doctor/morphoea.
- Scleroderma Society: http://sclerodermauk.org
- UT Southwestern Medical Center, Morphea registry: http://www.utsouthwestern.edu/education/medical-school/departments/dermatology/research/morphea-registry/disease-faq.html.
(All last accessed July 2015.)
References
- Fett N, Werth VP. Update on morphea: part I. Epidemiology, clinical presentation, and pathogenesis. J Am Acad Dermatol 2011;64(2):217–28; quiz 29–30.
- Fett N, Werth VP. Update on morphea: part II. Outcome measures and treatment. J Am Acad Dermatol 2011;64(2):231–42; quiz 43–4.
- Fett N. Scleroderma: nomenclature, etiology, pathogenesis, prognosis, and treatments: facts and controversies. Clin Dermatol 2013;31(4):432–7.
- Kreuter A. Localized scleroderma. Dermatol Ther 2012;25(2):135–47.
- Peterson LS, Nelson AM, Su WP. Classification of morphea (localized scleroderma). Mayo Clinic Proc 1995;70(11):1068–76.
- Laxer RM, Zulian F. Localized scleroderma. Curr Opin Rheumatol 2006;18(6):606–13.
- Kreuter A, Krieg T, Worm M, et al. [AWMF Guideline no. 013/066. Diagnosis and therapy of circumscribed scleroderma.] J Deut Dermatol Gesell 2009;7(Suppl. 6):S1–14.
- Zulian F, Athreya BH, Laxer R, et al. Juvenile localized scleroderma: clinical and epidemiological features in 750 children. An international study. Rheumatology 2006;45:614–20.
- Leitenberger JJ, Cayce RL, Haley RW, Adams-Huet B, Bergstresser PR, Jacobe HT. Distinct autoimmune syndromes in morphea: a review of 245 adult and pediatric cases. Arch Dermatol 2009;145:545–50.
- Kim A, Marinkovich N, Vasquez R, Jacobe HT. Clinical features of patients with morphea and the pansclerotic subtype: a cross-sectional study from the Morphea in Adults and Children cohort. J Rheumatol 2014;41(1):106–12.
- Odhav A, Hoeltzel MF, Canty K. Pansclerotic morphea with features of eosinophilic fasciitis: distinct entities or part of a continuum? Pediatr Dermatol 2014;31(2):e42–7.
- Viraben R, Dupre A. Eosinophilic fasciitis (Shulman syndrome) in association with morphea, immunological disturbance and profuse achromia. Dermatologica 1987;174(2):93–5.
- Valentini G, Rossiello R, Gualdieri L, Tirri G, Gerster JC, Frenck E. Morphea developing in patients previously affected with eosinophilic fasciitis. Report of two cases. Rheum Int 1988;8(5):235–7.
- Hulshof MM, Boom BW, Dijkmans BA. Multiple plaques of morphea developing in a patient with eosinophilic fasciitis. Arch Dermatol 1992;128(8):1128–9.
- Su WP, Greene SL. Bullous morphea profunda. Am J Dermatopathol 1986;8(2):144–7.
- Daoud MS, Su WP, Leiferman KM, Perniciaro C. Bullous morphea: clinical, pathologic, and immunopathologic evaluation of thirteen cases. J Am Acad Dermatol 1994;30(6):937–43.
- Trattner A, David M, Sandbank M. Bullous morphea: a distinct entity? Am J Dermatopathol 1994;16(4):414–17.
- Kavala M, Zindanci I, Demirkesen C, Beyhan EK, Turkoglu Z. Intertriginous bullous morphea: a clue for the pathogenesis? Indian J Dermatol Venereol Leprol 2007;73(4):262–4.
- Zulian F, Vallongo C, Woo P, et al. Localized scleroderma in childhood is not just a skin disease. Arthritis Rheum 2005;52(9):2873–81.
- Herrick AL, Ennis H, Bhushan M, Silman AJ, Baildam EM. Clinical features of childhood localized scleroderma in an incidence cohort. Rheumatology 2011;50(10):1865–8.
- Pequet MS, Holland KE, Zhao S, et al. Risk factors for morphoea disease severity: a retrospective review of 114 paediatric patients. Br J Dermatol 2014;170(4):895–900.
- Marzano AV, Menni S, Parodi A, et al. Localized scleroderma in adults and children. Clinical and laboratory investigations on 239 cases. Eur J Dermatol 2003;13(2):171–6.
- Piram M, McCuaig CC, Saint-Cyr C, et al. Short- and long-term outcome of linear morphoea in children. Br J Dermatol 2013;169(6):1265–71.
- Saxton-Daniels S, Jacobe HT. An evaluation of long-term outcomes in adults with pediatric-onset morphea. Arch Dermatol 2010;146(9):1044–5.
- Zwischenberger BA, Jacobe HT. A systematic review of morphea treatments and therapeutic algorithm. J Am Acad Dermatol 2011;65(5):925–41.
- Li SC, Torok KS, Pope E, et al. Development of consensus treatment plans for juvenile localized scleroderma: a roadmap toward comparative effectiveness studies in juvenile localized scleroderma. Arthritis Care Res 2012;64(8):1175–85.
- Arkachaisri T, Vilaiyuk S, Li S, et al. The localized scleroderma skin severity index and physician global assessment of disease activity: a work in progress toward development of localized scleroderma outcome measures. J Rheumatol 2009;36(12):2819–29.
- Peterson LS, Nelson AM, Su WP, Mason T, O'Fallon WM, Gabriel SE. The epidemiology of morphea (localized scleroderma) in Olmsted County 1960–1993. J Rheumatol 1997;24(1):73–80.
- Herrick AL, Ennis H, Bhushan M, Silman AJ, Baildam EM. Incidence of childhood linear scleroderma and systemic sclerosis in the UK and Ireland. Arthritis Care Res 2010;62(2):213–18.
- Wu EY, Li SC, Torok KS, et al. A28: description of the Juvenile Localized Scleroderma Subgroup of the CARRA Registry. Arthritis Rheumatol 2014;66(Suppl. 3):S43–4.
- Blaszczyk M, Janniger CK, Jablonska S. Childhood scleroderma and its peculiarities. Cutis 1996;58(2):141–4, 8–52.
- Zulian F, Vallongo C, de Oliveira SK, et al. Congenital localized scleroderma. J Pediatr 2006;149(2):248–51.
- Kang CY, Lam J. Congenital idiopathic atrophoderma of Pasini and Pierini. Int J Dermatol 2015;54(1):e44–6.
- Johnson W, Jacobe H. Morphea in Adults and Children cohort II: patients with morphea experience delay in diagnosis and large variation in treatment. J Am Acad Dermatol 2012;67(5):881–9.
- Christianson HB, Dorsey CS, Kierland RR, O'Leary PA. Localized scleroderma; a clinical study of two hundred thirty-five cases. AMA Arch Dermatol 1956;74(6):629–39.
- Christen-Zaech S, Hakim MD, Afsar FS, Paller AS. Pediatric morphea (localized scleroderma): review of 136 patients. J Am Acad Dermatol 2008;59(3):385–96.
- Harrington CI, Dunsmore IR. An investigation into the incidence of auto-immune disorders in patients with localized morphoea. Br J Dermatol 1989;120(5):645–8.
- Poojary SA. Vitiligo and associated autoimmune disorders: a retrospective hospital-based study in Mumbai, India. Allergol Immunopathol 2011;39(6):356–61.
- Natarajan S, Green ST. Generalized morphoea, lichen sclerosus et atrophicus and primary biliary cirrhosis. Clin Exp Dermatol 1986;11(3):304–8.
- Reed JR, De Luca N, McIntyre AS, Wilkinson JD. Localized morphoea, xanthomatosis and primary biliary cirrhosis. Br J Dermatol 2000;143(3):652–3.
- Kreuter A, Wischnewski J, Terras S, Altmeyer P, Stucker M, Gambichler T. Coexistence of lichen sclerosus and morphea: a retrospective analysis of 472 patients with localized scleroderma from a German tertiary referral center. J Am Acad Dermatol 2012;67(6):1157–62.
- Hemminki K, Li X, Sundquist J, Sundquist K. Familial associations of rheumatoid arthritis with autoimmune diseases and related conditions. Arthritis Rheum 2009;60(3):661–8.
- Caramaschi P, Biasi D, Volpe A, Carletto A, Cecchetto M, Bambara LM. Coexistence of systemic sclerosis with other autoimmune diseases. Rheum Int 2007;27(4):407–10.
- Avouac J, Airo P, Dieude P, et al. Associated autoimmune diseases in systemic sclerosis define a subset of patients with milder disease: results from 2 large cohorts of European Caucasian patients. J Rheumatol 2010;37(3):608–14.
- Arora-Singh RK, Assassi S, del Junco DJ, et al. Autoimmune diseases and autoantibodies in the first degree relatives of patients with systemic sclerosis. J Autoimmun 2010;35(1):52–7.
- Thomas RH, Ridley CM, McGibbon DH, Black MM. Anogenital lichen sclerosus in women. J R Soc Med 1996;89(12):694–8.
- Farrell AM, Marren PM, Wojnarowska F. Genital lichen sclerosus associated with morphoea or systemic sclerosis: clinical and HLA characteristics. Br J Dermatol 2000;143(3):598–603.
- Lutz V, Frances C, Bessis D, et al. High frequency of genital lichen sclerosus in a prospective series of 76 patients with morphea: toward a better understanding of the spectrum of morphea. Arch Dermatol 2012;148(1):24–8.
- Tuffanelli DL. Localized scleroderma. Semin Cutan Med Surg 1998;17(1):27–33.
- Patterson JA, Ackerman AB. Lichen sclerosus et atrophicus is not related to morphea. A clinical and histologic study of 24 patients in whom both conditions were reputed to be present simultaneously. Am J Dermatopathol 1984;6(4):323–35.
- Cooper SM, Ali I, Baldo M, Wojnarowska F. The association of lichen sclerosus and erosive lichen planus of the vulva with autoimmune disease: a case–control study. Arch Dermatol 2008;144(11):1432–5.
- Kreuter A, Kryvosheyeva Y, Terras S, et al. Association of autoimmune diseases with lichen sclerosus in 532 male and female patients. Acta Derm Venereol 2013;93(2):238–41.
- Meyrick Thomas RH, Ridley CM, McGibbon DH, Black MM. Lichen sclerosus et atrophicus and autoimmunity – a study of 350 women. Br J Dermatol 1988;118(1):41–6.
- Kuwana M, Kaburaki J, Okano Y, Inoko H, Tsuji K. The HLA-DR and DQ genes control the autoimmune response to DNA topoisomerase I in systemic sclerosis (scleroderma). J Clin Invest 1993;92(3):1296–301.
- Gladman DD, Kung TN, Siannis F, Pellett F, Farewell VT, Lee P. HLA markers for susceptibility and expression in scleroderma. J Rheumatol 2005;32(8):1481–7.
- Wadud MA, Bose BK, Al Nasir T. Familial localised scleroderma from Bangladesh: two case reports. Bangladesh Med Res Counc Bull 1989;15(1):15–19.
- Patrizi A, Marzaduri S, Marini R. A familial case of scleroderma en coup de sabre. Acta Derm Venereol 2000;80(3):237.
- Brownell I, Soter NA, Franks AG, Jr. Familial linear scleroderma (en coup de sabre) responsive to antimalarials and narrowband ultraviolet B therapy. Dermatol Online J 2007;13(1):11.
- Lis-Swiety A, Mierzwinska K, Wodok-Wieczorek K, Widuchowska M, Brzezinska-Wcislo L. Co-existence of lichen sclerosus and localized scleroderma in female monozygotic twins. J Pediatr Adolesc Gynecol 2014;27(6):133–6.
- Meyrick Thomas RH, Kennedy CT. The development of lichen sclerosus et atrophicus in monozygotic twin girls. Br J Dermatol 1986;114(3):377–9.
- Marren P, Yell J, Charnock FM, Bunce M, Welsh K, Wojnarowska F. The association between lichen sclerosus and antigens of the HLA system. Br J Dermatol 1995;132(2):197–203.
- Azurdia RM, Luzzi GA, Byren I, et al. Lichen sclerosus in adult men: a study of HLA associations and susceptibility to autoimmune disease. Br J Dermatol 1999;140(1):79–83.
- Powell J, Wojnarowska F, Winsey S, Marren P, Welsh K. Lichen sclerosus premenarche: autoimmunity and immunogenetics. Br J Dermatol 2000;142(3):481–4.
- Azzouz DF, Rak JM, Fajardy I, et al. Comparing HLA shared epitopes in French Caucasian patients with scleroderma. PLOS One 2012;7(5):e36870.
- Gordon W, Kahn LB, Dove J. Lichen sclerosus et atrophicus and scleroderma. S Afr Med J 1972;46(7):160–3.
- Uitto J, Santa Cruz DJ, Bauer EA, Eisen AZ. Morphea and lichen sclerosus et atrophicus. Clinical and histopathologic studies in patients with combined features. J Am Acad Dermatol 1980;3(3):271–9.
- Lampert A, Fortier-Beaulieu M, Thomine E, Young P, Lauret P. [Association of lichen sclerosus and monomelic scleroderma]. Ann Dermatol Venereol 1995;122(3):102–4.
- Kar BR, Dash K. Co-existence of lichen sclerosus et atrophicus and morphoea along lines of Blaschko. Indian J Dermatol 2014;59(1):77–9.
- Stewart L, Nuara A, Anand D, Kia K, Cockerell CJ. Perifollicular papules and hyperkeratotic plaques on the back in a blaschkoid distribution. Morphea with features of lichen sclerosus et atrophicus (LS). Arch Dermatol 2011;147(7):857–62.
- Kuchmeister B, Klemm-Mayer H. [Circumscribed scleroderma combined with lichen sclerosus et atrophicus]. Z Hautkr 1985;60(1–2):87–92.
- Nicolas JF, Perret-Liaudet P, Adam C, et al. [Bullous scleroderma with the histological appearance of lichen sclerosus et atrophicus]. Ann Dermatol Venereol 1988;115(4):461–6.
- Grekin J, Schwartz O, Mehregan A, Mendelson C, Burnham T. Lichen sclerosus et atrophicus and morphea. Arch Dermatol 1967;96(1):106.
- Kim DH, Lee KR, Kim TY, Yoon MS. Coexistence of lichen sclerosus with morphoea showing bilateral symmetry. Clin Exp Dermatol 2009;34(7):e416–18.
- Farrell AM, Dean D, Millard PR, Charnock FM, Wojnarowska F. Cytokine alterations in lichen sclerosus: an immunohistochemical study. Br J Dermatol 2006;155(5):931–40.
- Nishioka S. Histological comparison of morphea and lichen sclerosus et atrophicus. Kurume Med J 1997;44(2):83–90.
- Rahbari H. Histochemical differentiation of localized morphea-scleroderma and lichen sclerosus et atrophicus. J Cutan Pathol 1989;16(6):342–7.
- Chan I, Oyama N, Neill SM, Wojnarowska F, Black MM, McGrath JA. Characterization of IgG autoantibodies to extracellular matrix protein 1 in lichen sclerosus. Clin Exp Dermatol 2004;29(5):499–504.
- Oyama N, Chan I, Neill SM, et al. Autoantibodies to extracellular matrix protein 1 in lichen sclerosus. Lancet 2003;362(9378):118–23.
- Chan I. The role of extracellular matrix protein 1 in human skin. Clin Exp Dermatol 2004;29(1):52–6.
- Chan I, Liu L, Hamada T, Sethuraman G, McGrath JA. The molecular basis of lipoid proteinosis: mutations in extracellular matrix protein 1. Exp Dermatol 2007;16(11):881–90.
- Sercu S, Zhang M, Oyama N, et al. Interaction of extracellular matrix protein 1 with extracellular matrix components: ECM1 is a basement membrane protein of the skin. J Invest Dermatol 2008;128(6):1397–408.
- Hamada T, Wessagowit V, South AP, et al. Extracellular matrix protein 1 gene (ECM1) mutations in lipoid proteinosis and genotype–phenotype correlation. J Invest Dermatol 2003;120(3):345–50.
- Kowalewski C, Kozlowska A, Chan I, et al. Three-dimensional imaging reveals major changes in skin microvasculature in lipoid proteinosis and lichen sclerosus. J Dermatol Sci 2005;38(3):215–24.
- Fujimoto N, Terlizzi J, Aho S, et al. Extracellular matrix protein 1 inhibits the activity of matrix metalloproteinase 9 through high-affinity protein/protein interactions. Exp Dermatol 2006;15(4):300–7.
- Arnett FC, Tan FK, Uziel Y, et al. Autoantibodies to the extracellular matrix microfibrillar protein, fibrillin 1, in patients with localized scleroderma. Arthritis Rheum 1999;42(12):2656–9.
- Tan FK, Arnett FC, Reveille JD, et al. Autoantibodies to fibrillin 1 in systemic sclerosis: ethnic differences in antigen recognition and lack of correlation with specific clinical features or HLA alleles. Arthritis Rheum 2000;43(11):2464–71.
- Matsushita T, Hasegawa M, Matsushita Y, et al. Elevated serum BAFF levels in patients with localized scleroderma in contrast to other organ-specific autoimmune diseases. Exp Dermatol 2007;16(2):87–93.
- Uziel Y, Krafchik BR, Silverman ED, Thorner PS, Laxer RM. Localized scleroderma in childhood: a report of 30 cases. Semin Arthritis Rheum 1994;23(5):328–40.
- Vancheeswaran R, Black CM, David J, et al. Childhood-onset scleroderma: is it different from adult-onset disease. Arthritis Rheum 1996;39(6):1041–9.
- Rosenberg AM, Uziel Y, Krafchik BR, et al. Antinuclear antibodies in children with localized scleroderma. J Rheumatol 1995;22(12):2337–43.
- Takehara K, Moroi Y, Nakabayashi Y, Ishibashi Y. Antinuclear antibodies in localized scleroderma. Arthritis Rheum 1983;26(5):612–16.
- Ruffatti A, Peserico A, Glorioso S, et al. Anticentromere antibody in localized scleroderma. J Am Acad Dermatol 1986;15(4 Pt 1):637–42.
- Falanga V, Medsger TA, Reichlin M. High titers of antibodies to single-stranded DNA in linear scleroderma. Arch Dermatol 1985;121(3):345–7.
- Falanga V, Medsger TA, Jr, Reichlin M. Antinuclear and anti-single-stranded DNA antibodies in morphea and generalized morphea. Arch Dermatol 1987;123(3):350–3.
- Dharamsi JW, Victor S, Aguwa N, et al. Morphea in adults and children cohort III: nested case–control study – the clinical significance of autoantibodies in morphea. JAMA Dermatol 2013;149(10):1159–65.
- Guevara-Gutierrez E, Yinh-Lao J, Garcia-Gutierrez P, Tlacuilo-Parra A. Frequency of antinuclear antibodies in mestizo Mexican children with morphea. Clin Rheumatol 2010;29(9):1055–9.
- Sato S, Fujimoto M, Ihn H, Kikuchi K, Takehara K. Antigen specificity of antihistone antibodies in localized scleroderma. Arch Dermatol 1994;130(10):1273–7.
- Sato S, Ihn H, Soma Y, et al. Antihistone antibodies in patients with localized scleroderma. Arthritis Rheum 1993;36(8):1137–41.
- Parodi A, Drosera M, Barbieri L, Rebora A. Antihistone antibodies in scleroderma. Dermatology 1995;191(1):16–18.
- Hayakawa I, Hasegawa M, Takehara K, Sato S. Anti-DNA topoisomerase IIalpha autoantibodies in localized scleroderma. Arthritis Rheum 2004;50(1):227–32.
- Arkachaisri T, Fertig N, Pino S, Medsger TA, Jr. Serum autoantibodies and their clinical associations in patients with childhood- and adult-onset linear scleroderma. A single-center study. J Rheumatol 2008;35(12):2439–44.
- Tomimura S, Ogawa F, Iwata Y, et al. Autoantibodies against matrix metalloproteinase-1 in patients with localized scleroderma. J Dermatol Sci 2008;52(1):47–54.
- Fujimoto M, Sato S, Ihn H, et al. Autoantibodies to mitochondrial 2-oxo-acid dehydrogenase complexes in localized scleroderma. Clin Exp Immunol 1996;105(2):297–301.
- Yamane K, Ihn H, Kubo M, et al. Anti-U1RNP antibodies in patients with localized scieroderma. Arch Dermatol Res 2001;293(9):455–9.
- Yimane K, Ihn H, Kubo M, Asano Y, Yazawa N, Tamaki K. Anti-U3 snRNP antibodies in localised scleroderma. Ann Rheum Dis 2001;60(12):1157–8.
- Yamane K, Ihn H, Kubo M, et al. Antibodies to Th/To ribonucleoprotein in patients with localized scleroderma. Rheumatology 2001;40(6):683–6.
- Kurzinski K, Torok KS. Cytokine profiles in localized scleroderma and relationship to clinical features. Cytokine 2011;55(2):157–64.
- Milano A, Pendergrass SA, Sargent JL, et al. Molecular subsets in the gene expression signatures of scleroderma skin. PLOS One 2008;3(7):e2696.
- Sgonc R, Gruschwitz MS, Dietrich H, Recheis H, Gershwin ME, Wick G. Endothelial cell apoptosis is a primary pathogenetic event underlying skin lesions in avian and human scleroderma. J Clin Invest 1996;98(3):785–92.
- Kobayasi T, Serup J. Vascular changes in morphea. Acta Derm Venereol 1985;65(2):116–20.
- Berman A, Berman GD, Winkelmann RK. Atrophoderma (Pasini-Pierini). Findings on direct immunofluorescent, monoclonal antibody, and ultrastructural studies. Int J Dermatol 1988;27(7):487–90.
- Venneker GT, Das PK, Naafs B, Tigges AJ, Bos JD, Asghar SS. Morphoea lesions are associated with aberrant expression of membrane cofactor protein and decay accelerating factor in vascular endothelium. Br J Dermatol 1994;131(2):237–42.
- Stefanec T. Endothelial apoptosis in scleroderma: comment on the article by Black et al. Arthritis Rheum 1999;42(11):2491.
- Jones SM, Mathew CM, Dixey J, Lovell CR, McHugh NJ. VCAM-1 expression on endothelium in lesions from cutaneous lupus erythematosus is increased compared with systemic and localized scleroderma. Br J Dermatol 1996;135(5):678–86.
- Yamane K, Ihn H, Kubo M, et al. Increased serum levels of soluble vascular cell adhesion molecule 1 and E-selectin in patients with localized scleroderma. J Am Acad Dermatol 2000;42(1 Pt 1):64–9.
- Yamamoto T. Chemokines and chemokine receptors in scleroderma. Int Arch Allergy Immunol 2006;140(4):345–56.
- Gambichler T, Skrygan M, Labanski AA, Kolios AG, Altmeyer P, Kreuter A. Significantly increased CCL5/RANTES and CCR7 mRNA levels in localized scleroderma. Regul Pept 2011;170(1–3):4–6.
- Yamamoto T. Potential roles of CCL2/monocyte chemoattractant protein-1 in the pathogenesis of cutaneous sclerosis. Clin Exp Rheumatol 2003;21:369–75.
- Magee KE, Kelsey CE, Kurzinski KL, et al. Interferon-gamma inducible protein-10 as a potential biomarker in localized scleroderma. Arthritis Res Ther 2013;15:R188.
- Uziel Y, Krafchik BR, Feldman B, Silverman ED, Rubin LA, Laxer RM. Serum levels of soluble interleukin-2 receptor. A marker of disease activity in localized scleroderma. Arthritis Rheum 1994;37:898–901.
- Ihn H, Sato S, Fujimoto M, Kikuchi K, Takehara K. Clinical significance of serum levels of soluble interleukin-2 receptor in patients with localized scleroderma. Br J Dermatol 1996;134:843–7.
- Xie Y, Zhang X, Inoue Y, Wakasugi S, Makino T, Ihn H. Expression of CD1a and CD86 on scleroderma Langerhans cells. Eur J Dermatol 2008;18:50–4.
- Ihn H, Sato S, Fujimoto M, Kikuchi K, Takehara K. Demonstration of interleukin-2, interleukin-4 and interleukin-6 in sera from patients with localized scleroderma. Arch Dermatol Res 1995;287:193–7.
- Hasegawa M, Sato S, Nagaoka T, Fujimoto M, Takehara K. Serum levels of tumor necrosis factor and interleukin-13 are elevated in patients with localized scleroderma. Dermatology 2003;207:141–7.
- Lonati PA, Brembilla NC, Montanari E, et al. High IL-17E and low IL-17C dermal expression identifies a fibrosis-specific motif common to morphea and systemic sclerosis. PLOS One 2014;9(8):e105008.
- Ihn H, Sato S, Fujimoto M, Kikuchi K, Takehara K. Demonstration of interleukin 8 in serum samples of patients with localized scleroderma. Arch Dermatol 1994;130(10):1327–8.
- Kawaguchi Y, Hara M, Wright TM. Endogenous IL-1alpha from systemic sclerosis fibroblasts induces IL-6 and PDGF-A. J Clin Invest 1999;103(9):1253–60.
- Denton CP, Xu S, Black CM, Pearson JD. Scleroderma fibroblasts show increased responsiveness to endothelial cell-derived IL-1 and bFGF. J Invest Dermatol 1997;108(3):269–74.
- Scharffetter K, Lankat-Buttgereit B, Krieg T. Localization of collagen mRNA in normal and scleroderma skin by in-situ hybridization. Eur J Clin Invest 1988;18(1):9–17.
- Hatamochi A, Ono M, Arakawa M, Takeda K, Ueki H. Analysis of collagen gene expression by cultured fibroblasts in morphoea. Br J Dermatol 1992;126(3):216–21.
- Kahari VM, Sandberg M, Kalimo H, Vuorio T, Vuorio E. Identification of fibroblasts responsible for increased collagen production in localized scleroderma by in situ hybridization. J Invest Dermatol 1988;90(5):664–70.
- Leask A, Denton CP, Abraham DJ. Insights into the molecular mechanism of chronic fibrosis: the role of connective tissue growth factor in scleroderma. J Invest Dermatol 2004;122(1):1–6.
- Sonnylal S, Denton CP, Zheng B, et al. Postnatal induction of transforming growth factor beta signaling in fibroblasts of mice recapitulates clinical, histologic, and biochemical features of scleroderma. Arthritis Rheum 2007;56(1):334–44.
- Higley H, Persichitte K, Chu S, Waegell W, Vancheeswaran R, Black C. Immunocytochemical localization and serologic detection of transforming growth factor beta 1. Association with type I procollagen and inflammatory cell markers in diffuse and limited systemic sclerosis, morphea, and Raynaud's phenomenon. Arthritis Rheum 1994;37(2):278–88.
- Zheng XY, Zhang JZ, Tu P, Ma SQ. Expression of platelet-derived growth factor B-chain and platelet-derived growth factor beta-receptor in fibroblasts of scleroderma. J Dermatol Sci 1998;18(2):90–7.
- Kubo M, Ihn H, Yamane K, Tamaki K. Up-regulated expression of transforming growth factor beta receptors in dermal fibroblasts in skin sections from patients with localized scleroderma. Arthritis Rheum 2001;44(3):731–4.
- Igarashi A, Nashiro K, Kikuchi K, et al. Connective tissue growth factor gene expression in tissue sections from localized scleroderma, keloid, and other fibrotic skin disorders. J Invest Dermatol 1996;106(4):729–33.
- Yamamoto T, Sawada Y, Katayama I, Nishioka K. Nodular scleroderma: increased expression of connective tissue growth factor. Dermatology 2005;211(3):218–23.
- Mattila L, Airola K, Ahonen M, et al. Activation of tissue inhibitor of metalloproteinases-3 (TIMP-3) mRNA expression in scleroderma skin fibroblasts. J Invest Dermatol 1998;110(4):416–21.
- Haniffa M, Gunawan M, Jardine L. Human skin dendritic cells in health and disease. J Dermatol Sci 2015;77(2):85–92.
- Skobieranda K, Helm KF. Decreased expression of the human progenitor cell antigen (CD34) in morphea. Am J Dermatopathol 1995;17(5):471–5.
- McNiff JM, Glusac EJ, Lazova RZ, Carroll CB. Morphea limited to the superficial reticular dermis: an underrecognized histologic phenomenon. Am J Dermatopathol 1999;21(4):315–19.
- Sung JJ, Chen TS, Gilliam AC, McCalmont TH, Gilliam AE. Clinicohistopathological correlations in juvenile localized scleroderma: studies on a subset of children with hypopigmented juvenile localized scleroderma due to loss of epidermal melanocytes. J Am Acad Dermatol 2011;65(2):364–73.
- Aiba S, Tabata N, Ohtani H, Tagami H. CD34+ spindle-shaped cells selectively disappear from the skin lesion of scleroderma. Arch Dermatol 1994;130(5):593–7.
- Takahashi M, Akamatsu H, Yagami A, et al. Epithelial–mesenchymal transition of the eccrine glands is involved in skin fibrosis in morphea. J Dermatol 2013;40(9):720–5.
- Su WP, Person JR. Morphea profunda. A new concept and a histopathologic study of 23 cases. Am J Dermatopathol 1981;3(3):251–60.
- Morley SM, Gaylarde PM, Sarkany I. Epidermal thickness in systemic sclerosis and morphoea. Clin Exp Dermatol 1985;10(1):51–7.
- Fleischmajer R, Nedwich A. Generalized morphea. I. Histology of the dermis and subcutaneous tissue. Arch Dermatol 1972;106(4):509–14.
- Fleischmajer R, Perlish JS, Reeves JR. Cellular infiltrates in scleroderma skin. Arthritis Rheum 1977;20(4):975–84.
- Succaria F, Kurban M, Kibbi AG, Abbas O. Clinicopathological study of 81 cases of localized and systemic scleroderma. J Eur Acad Dermatol Venereol 2013;27(2):e191–6.
- Perlish JS, Lemlich G, Fleischmajer R. Identification of collagen fibrils in scleroderma skin. J Invest Dermatol 1988;90(1):48–54.
- Fleischmajer R, Jacobs L, Schwartz E, Sakai LY. Extracellular microfibrils are increased in localized and systemic scleroderma skin. Lab Invest 1991;64(6):791–8.
- Montgomery H, O'Leary PA, Ragsdale WE, Jr. Dermatohistopathology of various types of scleroderma. AMA Arch Dermatol 1957;75(1):78–87.
- Chiu YE, Vora S, Kwon EK, Maheshwari M. A significant proportion of children with morphea en coup de sabre and Parry-Romberg syndrome have neuroimaging findings. Pediatr Dermatol 2012;29(6):738–48.
- Young EM, Jr, Barr RJ. Sclerosing dermatoses. J Cutan Pathol 1985;12(5):426–41.
- Torres JE, Sanchez JL. Histopathologic differentiation between localized and systemic scleroderma. Am J Dermatopathol 1998;20(3):242–5.
- Aberer E, Neumann R, Stanek G. Is localised scleroderma a Borrelia infection? Lancet 1985;2(8449):278.
- Aberer E, Klade H, Hobisch G. A clinical, histological, and immunohistochemical comparison of acrodermatitis chronica atrophicans and morphea. Am J Dermatopathol 1991;13(4):334–41.
- Asbrink E, Brehmer-Andersson E, Hovmark A. Acrodermatitis chronica atrophicans – a spirochetosis. Clinical and histopathological picture based on 32 patients; course and relationship to erythema chronicum migrans Afzelius. Am J Dermatopathol 1986;8(3):209–19.
- Dillon WI, Saed GM, Fivenson DP. Borrelia burgdorferi DNA is undetectable by polymerase chain reaction in skin lesions of morphea, scleroderma, or lichen sclerosus et atrophicus of patients from North America. J Am Acad Dermatol 1995;33(4):617–20.
- De Vito JR, Merogi AJ, Vo T, et al. Role of Borrelia burgdorferi in the pathogenesis of morphea/scleroderma and lichen sclerosus et atrophicus: a PCR study of thirty-five cases. J Cutan Pathol 1996;23(4):350–8.
- Goodlad JR, Davidson MM, Gordon P, Billington R, Ho-Yen DO. Morphoea and Borrelia burgdorferi: results from the Scottish Highlands in the context of the world literature. Mol Pathol 2002;55(6):374–8.
- Weide B, Walz T, Garbe C. Is morphoea caused by Borrelia burgdorferi? A review. Br J Dermatol 2000;142(4):636–44.
- Eisendle K, Grabner T, Zelger B. Morphoea: a manifestation of infection with Borrelia species? Br J Dermatol 2007;157(6):1189–98.
- Prinz JC, Kutasi Z, Weisenseel P, Poto L, Battyani Z, Ruzicka T. ‘Borrelia-associated early-onset morphea’: a particular type of scleroderma in childhood and adolescence with high titer antinuclear antibodies? Results of a cohort analysis and presentation of three cases. J Am Acad Dermatol 2009;60(2):248–55.
- Espinoza-Leon F, Arocha F, Hassanhi M, Arevalo J. [Using the polymerase chain reaction to Borrelia burgdorferi infection in localized scleroderma injure (morphea), in Venezuelan patients.] Invest Clin 2010;51(3):381–90.
- Weide B, Schittek B, Klyscz T, et al. Morphoea is neither associated with features of Borrelia burgdorferi infection, nor is this agent detectable in lesional skin by polymerase chain reaction. Br J Dermatol 2000;143(4):780–5.
- Colome-Grimmer MI, Payne DA, Tyring SK, Sanchez RL. Borrelia burgdorferi DNA and Borrelia hermsii DNA are not associated with morphea or lichen sclerosus et atrophicus in the southwestern United States. Arch Dermatol 1997;133(9):1174.
- Alonso-Llamazares J, Persing DH, Anda P, Gibson LE, Rutledge BJ, Iglesias L. No evidence for Borrelia burgdorferi infection in lesions of morphea and lichen sclerosus et atrophicus in Spain. A prospective study and literature review. Acta Derm Venereol 1997;77(4):299–304.
- Zollinger T, Mertz KD, Schmid M, Schmitt A, Pfaltz M, Kempf W. Borrelia in granuloma annulare, morphea and lichen sclerosus: a PCR-based study and review of the literature. J Cutan Pathol 2010;37(5):571–7.
- Anderson PJ, Molony D, Haan E, David DJ. Familial Parry–Romberg disease. Int J Pediatr Otorhinolaryngol 2005;69(5):705–8.
- Jacobe H, Ahn C, Arnett F, Reveille JD. Major histocompatibility complex class I and class II alleles may confer susceptibility to or protection against morphoea: findings from the Morphea in Adults and Children cohort. Arthritis Rheumatol 2014;66(11):3170–7.
- Bhattacharyya S, Sargent JL, Du P, et al. Egr-1 induces a profibrotic injury/repair gene program associated with systemic sclerosis. PLOS One 2011;6(9):e23082.
- Greenblatt MB, Sargent JL, Farina G, et al. Interspecies comparison of human and murine scleroderma reveals IL-13 and CCL2 as disease subset-specific targets. Am J Pathol 2012;180(3):1080–94.
- Etoh M, Jinnin M, Makino K, et al. MicroRNA-7 down-regulation mediates excessive collagen expression in localized scleroderma. Arch Dermatol Res 2013;305(1):9–15.
- Makino T, Jinnin M, Etoh M, et al. Down-regulation of microRNA-196a in the sera and involved skin of localized scleroderma patients. Eur J Dermatol 2014;24(4):470–6.
- Drago F, Rampini P, Lugani C, Rebora A. Generalized morphoea after antitetanus vaccination. Clin Exp Dermatol 1998;23(3):142.
- Torrelo A, Suarez J, Colmenero I, Azorin D, Perera A, Zambrano A. Deep morphea after vaccination in two young children. Pediatr Dermatol 2006;23(5):484–7.
- Benmously Mlika R, Kenani N, Badri T, et al. Morphea profunda in a young infant after hepatitis B vaccination. J Am Acad Dermatol 2010;63(6):1111–12.
- Khaled A, Kharfi M, Zaouek A, et al. Postvaccination morphea profunda in a child. Pediatr Dermatol 2012;29(4):525–7.
- Matsumoto M, Yamamoto T. Pediatric generalized morphea that developed at a BCG vaccination site. Actas Dermosifiliogr 2015;106(2):150–2.
- Viladomiu Edel A, Valls AT, Zabaleta BA, Moreno AJ, Perez NO. Deep morphea in a child after pneumococcal vaccination. Indian J Dermatol Venereol Leprol 2014;80(3):259–60.
- Morell A, Betlloch I, Sevila A, Banuls J, Botella R. Morphea-like reaction from vitamin K1. Int J Dermatol 1995;34(3):201–2.
- Alonso-Llamazares J, Ahmed I. Vitamin K1-induced localized scleroderma (morphea) with linear deposition of IgA in the basement membrane zone. J Am Acad Dermatol 1998;38(2 Pt 2):322–4.
- Ho J, Rothchild YH, Sengelmann R. Vitamin B12-associated localized scleroderma and its treatment. Dermatol Surg 2004;30(9):1252–5.
- Frances L, Leiva-Salinas M, Angelica MB, Marin I, Silvestre JF. Morphea as a sign of autoimmune syndrome induced by adjuvants (ASIA). Eur J Dermatol 2014;24(3):377–8.
- Falanga V, Medsger TA, Jr, Reichlin M, Rodnan GP. Linear scleroderma. Clinical spectrum, prognosis, and laboratory abnormalities. Ann Intern Med 1986;104(6):849–57.
- Yamanaka CT, Gibbs NF. Trauma-induced linear scleroderma. Cutis 1999;63(1):29–32.
- Grabell D, Hsieh C, Andrew R, et al. The role of skin trauma in the distribution of morphea lesions: a cross-sectional survey of the Morphea in Adults and Children cohort IV. J Am Acad Dermatol 2014;71(3):493–8.
- Wolf R, Ruocco V, Filioli FG. Isotopic response. Br J Dermatol 1997;136(3):466–7.
- Boyd AS, Neldner KH. The isomorphic response of Koebner. Int J Dermatol 1990;29(6):401–10.
- Ciechomska M, Cant R, Finnigan J, van Laar JM, O'Reilly S. Role of toll-like receptors in systemic sclerosis. Expert Rev Mol Med 2013;15:e9.
- Akay BN, Sanli H, Heper AO. Postirradiation linear morphoea. Clin Exp Dermatol 2010;35(4):e106–8.
- Colver GB, Rodger A, Mortimer PS, Savin JA, Neill SM, Hunter JA. Post-irradiation morphoea. Br J Dermatol 1989;120(6):831–5.
- Bleasel NR, Stapleton KM, Commens C, Ahern VA. Radiation-induced localized scleroderma in breast cancer patients. Australas J Dermatol 1999;40(2):99–102.
- Alhathlool A, Hein R, Andres C, Ring J, Eberlein B. Post-irradiation morphea: case report and review of the literature. J Dermatol Case Rep 2012;6(3):73–7.
- Spalek M, Jonska-Gmyrek J, Galecki J. Radiation-induced morphea – a literature review. J Eur Acad Dermatol Venereol 2015;29(2):197–202.
- Kumar S, Kolozsvary A, Kohl R, Lu M, Brown S, Kim JH. Radiation-induced skin injury in the animal model of scleroderma: implications for post-radiotherapy fibrosis. Radiation Oncology (London) 2008;3:40.
- Bouchard SM, Mohr MR, Pariser RJ. Taxane-induced morphea in a patient with CREST syndrome. Dermatol Rep 2010;2(1):e9.
- Kupfer I, Balguerie X, Courville P, Chinet P, Joly P. Scleroderma-like cutaneous lesions induced by paclitaxel: a case study. J Am Acad Dermatol 2003;48(2):279–81.
- Peroni A, Zini A, Braga V, Colato C, Adami S, Girolomoni G. Drug-induced morphea: report of a case induced by balicatib and review of the literature. J Am Acad Dermatol 2008;59(1):125–9.
- Runger TM, Adami S, Benhamou CL, et al. Morphea-like skin reactions in patients treated with the cathepsin K inhibitor balicatib. J Am Acad Dermatol 2012;66(3):e89–96.
- Kraigher O, Brenner S, Tur E. Anti-double-stranded DNA-positive unilateral generalized morphea in an adult, possibly exacerbated by ibuprofen. Arch Dermatol 2009;145(7):844–6.
- Verdelli A, Antiga E, Bonciolini V, Bonciani D, Volpi W, Caproni M. Drug induction in connective tissue diseases. G Ital Dermatol Venereol 2014;149(5):573–80.
- Calistru AM, Baudrier T, Mota A, et al. Pseudoscleroderma possibly induced by intravesical instillation of mitomycin C. J Am Acad Dermatol 2010;63(6):e116–18.
- Runger TM, Quintanilla-Dieck MJ, Bhawan J. Role of cathepsin K in the turnover of the dermal extracellular matrix during scar formation. J Invest Dermatol 2007;127(2):293–7.
- Quintanilla-Dieck MJ, Codriansky K, Keady M, Bhawan J, Runger TM. Expression and regulation of cathepsin K in skin fibroblasts. Exp Dermatol 2009;18(7):596–602.
- Weibel L, Laguda B, Atherton D, Harper JI. Misdiagnosis and delay in referral of children with localized scleroderma. Br J Dermatol 2011;165(6):1308–13.
- Connelly MG, Winkelmann RK. Coexistence of lichen sclerosus, morphea, and lichen planus. Report of four cases and review of the literature. J Am Acad Dermatol 1985;12(5 Pt 1):844–51.
- Blaya B, Gardeazabal J, de Lagran ZM, Diaz-Perez JL. [Patient with generalized guttate morphea and lichen sclerosus et atrophicu.] Actas Dermosifiliogr 2008;99(10):808–11.
- Buechner SA, Rufli T. Atrophoderma of Pasini and Pierini. Clinical and histopathologic findings and antibodies to Borrelia burgdorferi in thirty-four patients. J Am Acad Dermatol 1994;30(3):441–6.
- Kencka D, Blaszczyk M, Jablonska S. Atrophoderma Pasini-Pierini is a primary atrophic abortive morphea. Dermatology 1995;190(3):203–6.
- Saleh Z, Abbas O, Dahdah MJ, Kibbi AG, Zaynoun S, Ghosn S. Atrophoderma of Pasini and Pierini: a clinical and histopathological study. J Cutan Pathol 2008;35(12):1108–14.
- Jablonska S, Blaszczyk M. Is superficial morphea synonymous with atrophoderma Pasini–Pierini? J Am Acad Dermatol 2004;50(6):979–80; author reply 80.
- Canizares O, Sachs PM, Jaimovich L, Torres VM. Idiopathic atrophoderma of Pasini and Pierini. AMA Arch Dermatol 1958;77(1):42–58; discussion 60.
- Wakelin SH, James MP. Zosteriform atrophoderma of Pasini and Pierini. Clin Exp Dermatol 1995;20(3):244–6.
- Zhang RZ, Zhu WY. Two uncommon cases of idiopathic atrophoderma of pasini and pierini: multiple and giant. Indian J Dermatol Venereol Leprol 2011;77(3):402.
- Melani L, Caproni M, Cardinali C, et al. A case of nodular scleroderma. J Dermatol 2005;32(12):1028–31.
- Ohata C, Yasunaga M, Tsuruta D, et al. Nodular morphea (NM): report of a case of concurrent NM and morphea profunda associated with limited type systemic sclerosis, and overview and definition for NM. Eur J Dermatol 2013;23(1):87–93.
- Rencic A, Brinster N, Nousari CH. Keloid morphea and nodular scleroderma: two distinct clinical variants of scleroderma? J Cutan Med Surg 2003;7(1):20–4.
- Cannick L, 3rd, Douglas G, Crater S, Silver R. Nodular scleroderma: case report and literature review. J Rheumatol 2003;30(11):2500–2.
- Kauer F, Simon JC, Sticherling M. Nodular morphea. Dermatology 2009;218(1):63–6.
- Krell JM, Solomon AR, Glavey CM, Lawley TJ. Nodular scleroderma. J Am Acad Dermatol 1995;32(2 Pt 2):343–5.
- Micalizzi C, Parodi A, Rebora A. Morphoea with nodular lesions. Br J Dermatol 1994;131(2):298–300.
- Hsu S, Lee MW, Carlton S, Kramer EM. Nodular morphea in a linear pattern. Int J Dermatol 1999;38(7):529–30.
- Jain K, Dayal S, Jain VK, Aggarwal K, Bansal A. Blaschko linear nodular morphea with dermal mucinosis. Arch Dermatol 2007;143(7):953–5.
- Yamamoto T, Sakashita S, Sawada Y, Katayama I, Nishioka K. Possible role of epidermal growth factor in the lesional skin of nodular morphea. Acta Derm Venereol 1998;78(4):312–13.
- Moinzadeh P, Agarwal P, Bloch W, et al. Systemic sclerosis with multiple nodules: characterization of the extracellular matrix. Arch Dermatol Res 2013;305(7):645–52.
- Person JR, Su WP. Subcutaneous morphoea: a clinical study of sixteen cases. Br J Dermatol 1979;100(4):371–80.
- Whittaker SJ, Smith NP, Jones RR. Solitary morphoea profunda. Br J Dermatol 1989;120(3):431–40.
- Peters MS, Su WP. Eosinophils in lupus panniculitis and morphea profunda. J Cutan Pathol 1991;18(3):189–92.
- Tomb R, Soutou B, Chehadi S. [Plasma cell panniculitis: a histopathological variant of morphea profunda.] Ann Dermatol Venereol 2009;136(3):256–9.
- Kobayashi KA, Lui H, Prendiville JS. Solitary morphea profunda in a 5-year-old girl: case report and review of the literature. Pediatr Dermatol 1991;8(4):292–5.
- Khelifa E, Masouye I, Chavaz P, Hauser H, Grillet JP, Borradori L. Primary atrophic solitary morphea profunda. Dermatology 2008;217(3):207–10.
- Ahn JG, Kim YT, Lee CW. Trauma-induced isomorphic lesions in morphea – a brief case report. J Korean Med Sci 1995;10(2):152–4.
- Diaz-Perez JL, Connolly SM, Winkelmann RK. Disabling pansclerotic morphea of children. Arch Dermatol 1980;116(2):169–73.
- Wollina U, Looks A, Uhlemann C, Wollina K. Pansclerotic morphea of childhood – follow-up over 6 years. Pediatr Dermatol 1999;16(3):245–7.
- Wollina U, Buslau M, Heinig B, et al. Disabling pansclerotic morphea of childhood poses a high risk of chronic ulceration of the skin and squamous cell carcinoma. Int J Low Extrem Wounds 2007;6(4):291–8.
- Parodi PC, Riberti C, Draganic Stinco D, Patrone P, Stinco G. Squamous cell carcinoma arising in a patient with long-standing pansclerotic morphea. Br J Dermatol 2001;144(2):417–19.
- Wollina U, Buslau M, Weyers W. Squamous cell carcinoma in pansclerotic morphea of childhood. Pediatr Dermatol 2002;19(2):151–4.
- Petrov I, Gantcheva M, Miteva L, Vassileva S, Pramatarov K. Lower lip squamous cell carcinoma in disabling pansclerotic morphea of childhood. Pediatr Dermatol 2009;26(1):59–61.
- Scharffetter-Kochanek K, Goldermann R, Lehmann P, Holzle E, Goerz G. PUVA therapy in disabling pansclerotic morphoea of children. Br J Dermatol 1995;132(5):830–1.
- Todd DJ, Askari A, Ektaish E. PUVA therapy for disabling pansclerotic morphoea of children. Br J Dermatol 1998;138(1):201–2.
- Doede T, Wollina U, Hindermann W, Schier F, Bondartschuk M. Pansclerotic morphea in childhood: a case report. Pediatr Surg Int 2003;19(5):406–8.
- Tekin NS, Altinyazar HC, Tekin IO, Keskin SI, Kucukoglu R, Onsun N. Disabling pansclerotic morphoea: a case report. Int J Clin Pract 2010;64(1):99–101.
- Maragh SH, Davis MD, Bruce AJ, Nelson AM. Disabling pansclerotic morphea: clinical presentation in two adults. J Am Acad Dermatol 2005;53(2 Suppl. 1):S115–19.
- Sherber NS, Boin F, Hummers LK, Wigley FM. The “tank top sign”: a unique pattern of skin fibrosis seen in pansclerotic morphea. Ann Rheum Dis 2009;68(9):1511–12.
- Song P, Gocke C, Wigley FM, Boin F. Resolution of pansclerotic morphea after treatment with antithymocyte globulin. Nature Rev Rheumatol 2009;5(9):513–16.
- Stausbol-Gron B, Olesen AB, Deleuran B, Deleuran MS. Abatacept is a promising treatment for patients with disseminated morphea profunda: presentation of two cases. Acta Derm Venereol 2011;91(6):686–8.
- Neustadter JH, Samarin F, Carlson KR, Girardi M. Extracorporeal photochemotherapy for generalized deep morphea. Arch Dermatol 2009;145(2):127–30.
- Shulman LE. Diffuse fasciitis with eosinophilia: a new syndrome? Trans Assoc Am Phys 1975;88:70–86.
- Miller JJ, 3rd. The fasciitis–morphea complex in children. Am J Dis Child 1992;146(6):733–6.
- Lakhanpal S, Ginsburg WW, Michet CJ, Doyle JA, Moore SB. Eosinophilic fasciitis: clinical spectrum and therapeutic response in 52 cases. Semin Arthritis Rheum 1988;17(4):221–31.
- Lebeaux D, Frances C, Barete S, et al. Eosinophilic fasciitis (Shulman disease): new insights into the therapeutic management from a series of 34 patients. Rheumatology 2012;51(3):557–61.
- Anon. Eosinophilic fasciitis. BMJ 1980;280(6213):506.
- Pinal-Fernandez I, Selva-O'Callaghan A, Grau JM. Diagnosis and classification of eosinophilic fasciitis. Autoimmun Rev 2014;13(4–5):379–82.
- Barnes L, Rodnan GP, Medsger TA, Short D. Eosinophilic fasciitis. A pathologic study of twenty cases. Am J Pathol 1979;96(2):493–518.
- Michet CJ, Jr, Doyle JA, Ginsburg WW. Eosinophilic fasciitis: report of 15 cases. Mayo Clin Proc 1981;56(1):27–34.
- Falanga V, Medsger TA, Jr. Frequency, levels, and significance of blood eosinophilia in systemic sclerosis, localized scleroderma, and eosinophilic fasciitis. J Am Acad Dermatol 1987;17(4):648–56.
- Doyle JA. Eosinophilic fasciitis: extracutaneous manifestations and associations. Cutis 1984;34(3):259–61.
- Weibel L, Harper JI. Linear morphoea follows Blaschko's lines. Br J Dermatol 2008;159(1):175–81.
- Tollefson MM, Witman PM. En coup de sabre morphea and Parry-Romberg syndrome: a retrospective review of 54 patients. J Am Acad Dermatol 2007;56(2):257–63.
- Jue MS, Kim MH, Ko JY, Lee CW. Digital image processing for the acquisition of graphic similarity of the distributional patterns between cutaneous lesions of linear scleroderma and Blaschko's lines. J Dermatol 2011;38(8):778–83.
- Happle R. Mosaicism in human skin. Understanding the patterns and mechanisms. Arch Dermatol 1993;129(11):1460–70.
- Sommer A, Gambichler T, Bacharach-Buhles M, von Rothenburg T, Altmeyer P, Kreuter A. Clinical and serological characteristics of progressive facial hemiatrophy: a case series of 12 patients. J Am Acad Dermatol 2006;54(2):227–33.
- Blaszczyk M, Krolicki L, Krasu M, Glinska O, Jablonska S. Progressive facial hemiatrophy: central nervous system involvement and relationship with scleroderma en coup de sabre. J Rheumatol 2003;30(9):1997–2004.
- Jablonska S, Blaszczyk M. Long-lasting follow-up favours a close relationship between progressive facial hemiatrophy and scleroderma en coup de sabre. J Eur Acad Dermatol Venereol 2005;19(4):403–4.
- Miller K, Lehrhoff S, Fischer M, Meehan S, Latkowski JA. Linear morphea of the forehead (en coup de sabre). Dermatol Online J 2012;18(12):22.
- Mohan SV, Nittur V, Stevens KJ. Late-onset en coup de sabre of the skull. Skeletal Radiol 2013;42(10):1447–50.
- Soma Y, Fujimoto M. Frontoparietal scleroderma (en coup de sabre) following Blaschko's lines. J Am Acad Dermatol 1998;38(2 Pt 2):366–8.
- Rai R, Handa S, Gupta S, Kumar B. Bilateral en coup de sabre – a rare entity. Pediatr Dermatol 2000;17(3):222–4.
- Gambichler T, Kreuter A, Hoffmann K, Bechara FG, Altmeyer P, Jansen T. Bilateral linear scleroderma “en coup de sabre” associated with facial atrophy and neurological complications. BMC Dermatol 2001;1:9.
- Blaszczyk M, Jablonska S. Linear scleroderma en coup de sabre. Relationship with progressive facial hemiatrophy (PFH). Adv Exp Med Biol 1999;455:101–4.
- Stone J. Parry-Romberg syndrome: a global survey of 205 patients using the Internet. Neurology 2003;61(5):674–6.
- Mendonca J, Viana SL, Freitas F, Lima G. Late-onset progressive facial hemiatrophy (Parry-Romberg syndrome). J Postgrad Med 2005;51(2):135–6.
- El-Kehdy J, Abbas O, Rubeiz N. A review of Parry-Romberg syndrome. J Am Acad Dermatol 2012;67(4):769–84.
- Bandello F, Rosa N, Ghisolfi F, Sebastiani A. New findings in the Parry-Romberg syndrome: a case report. Eur J Ophthalmol 2002;12(6):556–8.
- Doolittle DA, Lehman VT, Schwartz KM, Wong-Kisiel LC, Lehman JS, Tollefson MM. CNS imaging findings associated with Parry-Romberg syndrome and en coup de sabre: correlation to dermatologic and neurologic abnormalities. Neuroradiology 2015;57(1):21–34.
- Taniguchi T, Asano Y, Tamaki Z, et al. Histological features of localized scleroderma ‘en coup de sabre’: a study of 16 cases. J Eur Acad Dermatol Venereol 2014;28(12):1805–10.
- Soma Y, Kawakami T, Yamasaki E, Sasaki R, Mizoguchi M. Linear scleroderma along Blaschko's lines in a patient with systematized morphea. Acta Derm Venereol 2003;83(5):362–4.
- Larregue M, Ziegler JE, Lauret P, et al. [Linear scleroderma in children (apropos of 27 cases).] Ann Dermatol Venereol 1986;113(3):207–24.
- Das S, Bernstein I, Jacobe H. Correlates of self-reported quality of life in adults and children with morphea. J Am Acad Dermatol 2014;70(5):904–10.
- Moulin G, Hill MP, Guillaud V, Barrut D, Chevallier J, Thomas L. [Acquired atrophic pigmented band-like lesions following Blaschko's lines.] Ann Dermatol Venereol 1992;119(10):729–36.
- Norisugi O, Makino T, Hara H, Matsui K, Furuichi M, Shimizu T. Evaluation of skin atrophy associated with linear atrophoderma of Moulin by ultrasound imaging. J Am Acad Dermatol 2011;65(1):232–3.
- Ang GC, Lee JB. Linear atrophoderma of Moulin: is it a single disease? J Am Acad Dermatol 2005;52(5):923–4; author reply 4–5.
- De Golian E, Echols K, Pearl H, Davis L. Linear atrophoderma of Moulin: a distinct entity? Pediatr Dermatol 2014;31(3):373–7.
- Blaszczyk M, Krysicka-Janiger K, Jablonska S. Primary atrophic profound linear scleroderma. Report of three cases. Dermatology 2000;200(1):63–6.
- Chapman MS, Peraza JE, Spencer SK. Parry-Romberg syndrome with contralateral and ipsilateral extremity involvement. J Cutan Med Surg 1999;3(5):260–2.
- Duymaz A, Karabekmez FE, Keskin M, Tosun Z. Parry-Romberg syndrome: facial atrophy and its relationship with other regions of the body. Ann Plastic Surg 2009;63(4):457–61.
- Foti R, Leonardi R, Rondinone R, et al. Scleroderma-like disorders. Autoimmun Rev 2008;7(4):331–9.
- Boin F, Hummers LK. Scleroderma-like fibrosing disorders. Rheum Dis Clin North Am 2008;34(1):199–220; ix.
- Arkachaisri T, Vilaiyuk S, Torok KS, Medsger TA, Jr. Development and initial validation of the localized scleroderma skin damage index and physician global assessment of disease damage: a proof-of-concept study. Rheumatology 2010;49(2):373–81.
- Kelsey CE, Torok KS. The Localized Scleroderma Cutaneous Assessment Tool: responsiveness to change in a pediatric clinical population. J Am Acad Dermatol 2013;69(2):214–20.
- Dehen L, Roujeau JC, Cosnes A, Revuz J. Internal involvement in localized scleroderma. Medicine 1994;73(5):241–5.
- Luderschmidt C, Konig G, Leisner B, Scholz S, Albert EE. [Circumscribed scleroderma: internal manifestations and significant correlation to HLA-DR1 and DR5.] Hautarzt 1985;36(9):516–21.
- Konig G, Luderschmidt C, Hammer C, Adelmann-Grill BC, Braun-Falco O, Fruhmann G. Lung involvement in scleroderma. Chest 1984;85(3):318–24.
- Rokicki W, Dukalska M, Rubisz-Brzezinska J, Gasior Z. Circulatory system in children with localized scleroderma. Pediatr Cardiol 1997;18(3):213–17.
- Targa L, Cardin G, Cozzi F, Glorioso S, Rossi L, Todesco S. [Electrocardiographic disorders in diverse clinical variations of scleroderma.] G Clin Med 1990;71(1):17–24.
- Rokicki W, Rubisz-Brzezinska J, Dukalska M, et al. [The circulatory system in children with cutaneous forms of scleroderma. The results of routine as well as 24-hour ECG and physical performance test.] Pediatr Polska 1995;70(6):479–85.
- Amaral TN, Peres FA, Lapa AT, Marques-Neto JF, Appenzeller S. Neurologic involvement in scleroderma: a systematic review. Semin Arthritis Rheum 2013;43(3):335–47.
- Amaral TN, Marques Neto JF, Lapa AT, Peres FA, Guirau CR, Appenzeller S. Neurologic involvement in scleroderma en coup de sabre. Autoimmune Dis 2012;2012:719685.
- Trainito S, Favero L, Martini G, et al. Odontostomatologic involvement in juvenile localised scleroderma of the face. J Paediatr Child Health 2012;48(7):572–6.
- Zannin ME, Martini G, Athreya BH, et al. Ocular involvement in children with localised scleroderma: a multi-centre study. Br J Ophthalmol 2007;91(10):1311–14.
- Guariso G, Conte S, Galeazzi F, Vettorato MG, Martini G, Zulian F. Esophageal involvement in juvenile localized scleroderma: a pilot study. Clin Exp Rheumatol 2007;25(5):786–9.
- Orzechowski NM, Davis DM, Mason TG, 3rd, Crowson CS, Reed AM. Health-related quality of life in children and adolescents with juvenile localized scleroderma. Rheumatology 2009;48(6):670–2.
- Baildam EM, Ennis H, Foster HE, et al. Influence of childhood scleroderma on physical function and quality of life. J Rheumatol 2011;38(1):167–73.
- Szramka-Pawlak B, Danczak-Pazdrowska A, Rzepa T, Szewczyk A, Sadowska-Przytocka A, Zaba R. Health-related quality of life, optimism, and coping strategies in persons suffering from localized scleroderma. Psychol Health Med 2013;18(6):654–63.
- Condie D, Grabell D, Jacobe H. Comparison of outcomes in adults with pediatric-onset morphea and those with adult-onset morphea: a cross-sectional study from the morphea in adults and children cohort. Arthritis Rheumatol 2014;66(12):3496–504.
- Kroft EB, de Jong EM, Evers AW. Psychological distress in patients with morphea and eosinophilic fasciitis. Arch Dermatol 2009;145(9):1017–22.
- Mertens JS, Seyger MM, Kievit W, et al. Disease recurrence in localized scleroderma: a retrospective analysis of 344 patients with paediatric- or adult-onset disease. Br J Dermatol 2015;172(3):722–8.
- Soma Y, Tamaki T, Kikuchi K, et al. Coexistence of morphea and systemic sclerosis. Dermatology 1993;186(2):103–5.
- Curtis AC, Jansen TG. The prognosis of localized scleroderma. AMA Arch Dermatol 1958;78(6):749–55; discussion 55–7.
- Birdi N, Laxer RM, Thorner P, Fritzler MJ, Silverman ED. Localized scleroderma progressing to systemic disease. Case report and review of the literature. Arthritis Rheum 1993;36(3):410–15.
- Schanz S, Fierlbeck G, Ulmer A, et al. Localized scleroderma: MR findings and clinical features. Radiology 2011;260(3):817–24.
- Horger M, Fierlbeck G, Kuemmerle-Deschner J, et al. MRI findings in deep and generalized morphea (localized scleroderma). Am J Roentgenol 2008;190(1):32–9.
- Li SC, Liebling MS, Haines KA. Ultrasonography is a sensitive tool for monitoring localized scleroderma. Rheumatology 2007;46(8):1316–19.
- Wortsman X, Wortsman J, Sazunic I, Carreno L. Activity assessment in morphea using color Doppler ultrasound. J Am Acad Dermatol 2011;65(5):942–8.
- Weibel L, Howell KJ, Visentin MT, et al. Laser Doppler flowmetry for assessing localized scleroderma in children. Arthritis Rheum 2007;56(10):3489–95.
- Shaw LJ, Shipley J, Newell EL, Harris N, Clinch JG, Lovell CR. Scanning laser Doppler imaging may predict disease progression of localized scleroderma in children and young adults. Br J Dermatol 2013;169(1):152–5.
- Li SC, Liebling MS, Haines KA, Weiss JE, Prann A. Initial evaluation of an ultrasound measure for assessing the activity of skin lesions in juvenile localized scleroderma. Arthritis Care Res 2011;63(5):735–42.
- Martini G, Murray KJ, Howell KJ, et al. Juvenile-onset localized scleroderma activity detection by infrared thermography. Rheumatology 2002;41(10):1178–82.
- Seyger MM, van den Hoogen FH, de Boo T, de Jong EM. Reliability of two methods to assess morphea: skin scoring and the use of a durometer. J Am Acad Dermatol 1997;37(5 Pt 1):793–6.
- Poff S, Li SC, Kelsey C, Foeldvari I, Torok KS. A48: durometer measures discriminate affected versus normal skin in pediatric localized scleroderma. Arthritis Rheumatol 2014;66(Suppl. 3):S72–3.
- Zulian F, Cuffaro G, Sperotto F. Scleroderma in children: an update. Curr Opin Rheumatol 2013;25(5):643–50.
- Zulian F, Meneghesso D, Grisan E, et al. A new computerized method for the assessment of skin lesions in localized scleroderma. Rheumatology 2007;46(5):856–60.
- Serup J, Kristensen JK. Blood flow of morphoea plaques as measured by laser-Doppler flowmetry. Arch Dermatol Res 1984;276(5):322–5.
- Clements P, Lachenbruch P, Siebold J, et al. Inter and intraobserver variability of total skin thickness score (modified Rodnan TSS) in systemic sclerosis. J Rheumatol 1995;22(7):1281–5.
- Dytoc M, Ting PT, Man J, Sawyer D, Fiorillo L. First case series on the use of imiquimod for morphoea. Br J Dermatol 2005;153(4):815–20.
- Arkachaisri T, Pino S. Localized scleroderma severity index and global assessments: a pilot study of outcome instruments. J Rheumatol 2008;35(4):650–7.
- Warburton KL, McPhee MJ, Savage LJ, et al. Management of morphoea: results of a national survey of UK clinicians. Br J Dermatol 2014;171:1243–5.
- Mancuso G, Berdondini RM. Topical tacrolimus in the treatment of localized scleroderma. Eur J Dermatol 2003;13(6):590–2.
- Mancuso G, Berdondini RM. Localized scleroderma: response to occlusive treatment with tacrolimus ointment. Br J Dermatol 2005;152(1):180–2.
- Stefanaki C, Stefanaki K, Kontochristopoulos G, et al. Topical tacrolimus 0.1% ointment in the treatment of localized scleroderma. An open label clinical and histological study. J Dermatol 2008;35(11):712–18.
- Kroft EB, Groeneveld TJ, Seyger MM, de Jong EM. Efficacy of topical tacrolimus 0.1% in active plaque morphea: randomized, double-blind, emollient-controlled pilot study. Am J Clin Dermatol 2009;10(3):181–7.
- Dytoc M, Wat H, Cheung-Lee M, Sawyer D, Ackerman T, Fiorillo L. Evaluation of the efficacy and safety of topical imiquimod 5% for plaque-type morphea: a multicenter, prospective, vehicle-controlled trial. J Cutan Med Surg 2015;19(2):132–9.
- Pope E, Doria AS, Theriault M, Mohanta A, Laxer RM. Topical imiquimod 5% cream for pediatric plaque morphea: a prospective, multiple-baseline, open-label pilot study. Dermatology 2011;223(4):363–9.
- Cunningham BB, Landells ID, Langman C, Sailer DE, Paller AS. Topical calcipotriene for morphea/linear scleroderma. J Am Acad Dermatol 1998;39(2 Pt 1):211–15.
- Dytoc MT, Kossintseva I, Ting PT. First case series on the use of calcipotriol-betamethasone dipropionate for morphoea. Br J Dermatol 2007;157(3):615–18.
- Kreuter A, Gambichler T, Avermaete A, et al. Combined treatment with calcipotriol ointment and low-dose ultraviolet A1 phototherapy in childhood morphea. Pediatr Dermatol 2001;18(3):241–5.
- Wlaschek M, Heinen G, Poswig A, Schwarz A, Krieg T, Scharffetter-Kochanek K. UVA-induced autocrine stimulation of fibroblast-derived collagenase/MMP-1 by interrelated loops of interleukin-1 and interleukin-6. Photochem Photobiol 1994;59(5):550–6.
- Scharffetter K, Wlaschek M, Hogg A, et al. UVA irradiation induces collagenase in human dermal fibroblasts in vitro and in vivo. Arch Dermatol Res 1991;283(8):506–11.
- Hwang BM, Noh EM, Kim JS, et al. Curcumin inhibits UVB-induced matrix metalloproteinase-1/3 expression by suppressing the MAPK-p38/JNK pathways in human dermal fibroblasts. Exp Dermatol 2013;22(5):371–4.
- Nisar MF, Parsons KS, Bian CX, Zhong JL. UVA irradiation induced heme oxygenase-1 (HO-1): a novel phototherapy for morphea. Photochem Photobiol 2015;91(1):210–1.
- El-Mofty M, Mostafa W, Esmat S, et al. Suggested mechanisms of action of UVA phototherapy in morphea: a molecular study. Photodermatol Photoimmunol Photomed 2004;20(2):93–100.
- Breuckmann F, Gambichler T, Altmeyer P, Kreuter A. UVA/UVA1 phototherapy and PUVA photochemotherapy in connective tissue diseases and related disorders: a research based review. BMC Dermatol 2004;4(1):11.
- Kreuter A, Hyun J, Skrygan M, et al. Ultraviolet A1 phototherapy decreases inhibitory SMAD7 gene expression in localized scleroderma. Arch Dermatol Res 2006;298(6):265–72.
- Gambichler T, Skrygan M, Tomi NS, Altmeyer P, Kreuter A. Differential expression of decorin in localized scleroderma following ultraviolet-A1 irradiation. J Am Acad Dermatol 2007;56(6):956–9.
- Gambichler T, Terras S, Kreuter A. Treatment regimens, protocols, dosage, and indications for UVA1 phototherapy: facts and controversies. Clin Dermatol 2013;31(4):438–54.
- Jacobe HT, Cayce R, Nguyen J. UVA1 phototherapy is effective in darker skin: a review of 101 patients of Fitzpatrick skin types I–V. Br J Dermatol 2008;159(3):691–6.
- Wang F, Garza LA, Cho S, et al. Effect of increased pigmentation on the antifibrotic response of human skin to UV-A1 phototherapy. Arch Dermatol 2008;144(7):851–8.
- Vasquez R, Jabbar A, Khan F, Buethe D, Ahn C, Jacobe H. Recurrence of morphea after successful ultraviolet A1 phototherapy: a cohort study. J Am Acad Dermatol 2014;70(3):481–8.
- Kreuter A, Hyun J, Stucker M, Sommer A, Altmeyer P, Gambichler T. A randomized controlled study of low-dose UVA1, medium-dose UVA1, and narrowband UVB phototherapy in the treatment of localized scleroderma. J Am Acad Dermatol 2006;54(3):440–7.
- Sator PG, Radakovic S, Schulmeister K, Honigsmann H, Tanew A. Medium-dose is more effective than low-dose ultraviolet A1 phototherapy for localized scleroderma as shown by 20-MHz ultrasound assessment. J Am Acad Dermatol 2009;60(5):786–91.
- Su O, Onsun N, Onay HK, et al. Effectiveness of medium-dose ultraviolet A1 phototherapy in localized scleroderma. Int J Dermatol 2011;50(8):1006–13.
- Andres C, Kollmar A, Mempel M, Hein R, Ring J, Eberlein B. Successful ultraviolet A1 phototherapy in the treatment of localized scleroderma: a retrospective and prospective study. Br J Dermatol 2010;162(2):445–7.
- El-Mofty M, Zaher H, Bosseila M, Yousef R, Saad B. Low-dose broad-band UVA in morphea using a new method for evaluation. Photodermatol Photoimmunol Photomed 2000;16(2):43–9.
- El-Mofty M, Mostafa W, El-Darouty M, et al. Different low doses of broad-band UVA in the treatment of morphea and systemic sclerosis. Photodermatol Photoimmunol Photomed 2004;20(3):148–56.
- Kerscher M, Volkenandt M, Meurer M, Lehmann P, Plewig G, Rocken M. Treatment of localised scleroderma with PUVA bath photochemotherapy. Lancet 1994;343(8907):1233.
- Kerscher M, Meurer M, Sander C, et al. PUVA bath photochemotherapy for localized scleroderma. Evaluation of 17 consecutive patients. Arch Dermatol 1996;132(11):1280–2.
- Schiener R, Behrens-Williams SC, Gottlober P, Pillekamp H, Peter RU, Kerscher M. Eosinophilic fasciitis treated with psoralen-ultraviolet A bath photochemotherapy. Br J Dermatol 2000;142(4):804–7.
- Grundmann-Kollmann M, Ochsendorf F, Zollner TM, et al. PUVA-cream photochemotherapy for the treatment of localized scleroderma. J Am Acad Dermatol 2000;43(4):675–8.
- Joly P, Bamberger N, Crickx B, Belaich S. Treatment of severe forms of localized scleroderma with oral corticosteroids: follow-up study on 17 patients. Arch Dermatol 1994;130(5):663–4.
- Seyger MM, van den Hoogen FH, de Boo T, de Jong EM. Low-dose methotrexate in the treatment of widespread morphea. J Am Acad Dermatol 1998;39(2 Pt 1):220–5.
- Uziel Y, Feldman BM, Krafchik BR, Yeung RS, Laxer RM. Methotrexate and corticosteroid therapy for pediatric localized scleroderma. J Pediatr 2000;136(1):91–5.
- Fitch PG, Rettig P, Burnham JM, et al. Treatment of pediatric localized scleroderma with methotrexate. J Rheumatol 2006;33(3):609–14.
- Weibel L, Sampaio MC, Visentin MT, Howell KJ, Woo P, Harper JI. Evaluation of methotrexate and corticosteroids for the treatment of localized scleroderma (morphoea) in children. Br J Dermatol 2006;155(5):1013–20.
- Zulian F, Martini G, Vallongo C, et al. Methotrexate treatment in juvenile localized scleroderma: a randomized, double-blind, placebo-controlled trial. Arthritis Rheum 2011;63(7):1998–2006.
- Torok KS, Arkachaisri T. Methotrexate and corticosteroids in the treatment of localized scleroderma: a standardized prospective longitudinal single-center study. J Rheumatol 2012;39(2):286–94.
- Zulian F, Vallongo C, Patrizi A, et al. A long-term follow-up study of methotrexate in juvenile localized scleroderma (morphea). J Am Acad Dermatol 2012;67(6):1151–6.
- Kreuter A, Gambichler T, Breuckmann F, et al. Pulsed high-dose corticosteroids combined with low-dose methotrexate in severe localized scleroderma. Arch Dermatol 2005;141(7):847–52.
- Seitz M, Zwicker M, Loetscher P. Effects of methotrexate on differentiation of monocytes and production of cytokine inhibitors by monocytes. Arthritis Rheum 1998;41(11):2032–8.
- Seitz M, Dewald B, Ceska M, Gerber N, Baggiolini M. Interleukin-8 in inflammatory rheumatic diseases: synovial fluid levels, relation to rheumatoid factors, production by mononuclear cells, and effects of gold sodium thiomalate and methotrexate. Rheum Int 1992;12(4):159–64.
- Seitz M, Loetscher P, Dewald B, et al. Methotrexate action in rheumatoid arthritis: stimulation of cytokine inhibitor and inhibition of chemokine production by peripheral blood mononuclear cells. Br J Rheumatol 1995;34(7):602–9.
- Barrera P, Boerbooms AM, Janssen EM, et al. Circulating soluble tumor necrosis factor receptors, interleukin-2 receptors, tumor necrosis factor alpha, and interleukin-6 levels in rheumatoid arthritis. Longitudinal evaluation during methotrexate and azathioprine therapy. Arthritis Rheum 1993;36(8):1070–9.
- Rose CD, Fawcett PT, Gibney K, Doughty RA, Singsen BH. Serial measurements of soluble interleukin 2 receptor levels (sIL2-R) in children with juvenile rheumatoid arthritis treated with oral methotrexate. Ann Rheum Dis 1994;53(7):471–4.
- Lacour JP, Vitetta A, Chiquet-Ehrismann R, Pisani A, Ortonne JP. Increased expression of tenascin in the dermis in scleroderma. Br J Dermatol 1992;127(4):328–34.
- Inoue K, Jinnin M, Hara Y, et al. Serum levels of tenascin-C in collagen diseases. J Dermatol 2013;40(9):715–19.
- Seyger MM, van den Hoogen FH, van Vlijmen-Willems IM, van de Kerkhof PC, de Jong EM. Localized and systemic scleroderma show different histological responses to methotrexate therapy. J Pathol 2001;193(4):511–16.
- Cox D, O'Regan G, Collins S, Byrne A, Irvine A, Watson R. Juvenile localised scleroderma: a retrospective review of response to systemic treatment. Ir J Med Sci 2008;177(4):343–6.
- Kroft EB, Creemers MC, van den Hoogen FH, Boezeman JB, de Jong EM. Effectiveness, side-effects and period of remission after treatment with methotrexate in localized scleroderma and related sclerotic skin diseases: an inception cohort study. Br J Dermatol 2009;160(5):1075–82.
- Mirsky L, Chakkittakandiyil A, Laxer RM, O'Brien C, Pope E. Relapse after systemic treatment in paediatric morphoea. Br J Dermatol 2012;166(2):443–5.
- Koch SB, Cerci FB, Jorizzo JL, Krowchuk DP. Linear morphea: a case series with long-term follow-up of young, methotrexate-treated patients. J Dermatol Treat 2013;24(6):435–8.
- Strickland N, Patel G, Strickland A, Jacobe H. Attitudes and trends in the treatment of morphea: a national survey. J Am Acad Dermatol 2015;72(4):727–8.
- Mendoza FA, Nagle SJ, Lee JB, Jimenez SA. A prospective observational study of mycophenolate mofetil treatment in progressive diffuse cutaneous systemic sclerosis of recent onset. J Rheumatol 2012;39(6):1241–7.
- Tzouvelekis A, Galanopoulos N, Bouros E, et al. Effect and safety of mycophenolate mofetil or sodium in systemic sclerosis-associated interstitial lung disease: a meta-analysis. Pulm Med 2012;2012:143637.
- Hinchcliff M, Huang CC, Wood TA, et al. Molecular signatures in skin associated with clinical improvement during mycophenolate treatment in systemic sclerosis. J Invest Dermatol 2013;133(8):1979–89.
- Martini G, Ramanan AV, Falcini F, Girschick H, Goldsmith DP, Zulian F. Successful treatment of severe or methotrexate-resistant juvenile localized scleroderma with mycophenolate mofetil. Rheumatology 2009;48(11):1410–13.
- Strauss RM, Bhushan M, Goodfield MJ. Good response of linear scleroderma in a child to ciclosporin. Br J Dermatol 2004;150(4):790–2.
- Peter RU, Ruzicka T, Eckert F. Low-dose cyclosporine A in the treatment of disabling morphea. Arch Dermatol 1991;127(9):1420–1.
- Inamo Y, Ochiai T. Successful combination treatment of a patient with progressive juvenile localized scleroderma (morphea) using imatinib, corticosteroids, and methotrexate. Pediatr Dermatol 2013;30(6):e191–3.
- Diab M, Coloe JR, Magro C, Bechtel MA. Treatment of recalcitrant generalized morphea with infliximab. Arch Dermatol 2010;146(6):601–4.
- Schlaak M, Friedlein H, Kauer F, Renner R, Rogalski C, Simon JC. Successful therapy of a patient with therapy-recalcitrant generalized bullous scleroderma by extracorporeal photopheresis and mycophenolate mofetil. J Eur Acad Dermatol Venereol 2008;22(5):631–3.
- Pileri A, Raone B, Raboni R, Giudice V, Patrizi A. Generalized morphea successfully treated with extracorporeal photochemotherapy (ECP). Dermatol Online J 2014;20(1):21258.
- Takehara K, Ihn H, Sato S. A randomized, double-blind, placebo-controlled trial: intravenous immunoglobulin treatment in patients with diffuse cutaneous systemic sclerosis. Clin Exp Rheumatol 2013;31(2 Suppl. 76):151–6.
- Baleva M, Nikolov K. The role of intravenous immunoglobulin preparations in the treatment of systemic sclerosis. Int J Rheumatol 2011;2011:829751.
- Hulshof MM, Bouwes Bavinck JN, Bergman W, et al. Double-blind, placebo-controlled study of oral calcitriol for the treatment of localized and systemic scleroderma. J Am Acad Dermatol 2000;43(6):1017–23.
- Hunzelmann N, Anders S, Fierlbeck G, et al. Double-blind, placebo-controlled study of intralesional interferon gamma for the treatment of localized scleroderma. J Am Acad Dermatol 1997;36(3 Pt 1):433–5.
- Zanelato TP, Marquesini G, Colpas PT, Magalhaes RF, Moraes AM. Implantation of autologous fat globules in localized scleroderma and idiopathic lipoatrophy – report of five patients. An Bras Dermatol 2013;88(6 Suppl. 1):120–3.
- Roldan R, Morote G, Castro Mdel C, Miranda MD, Moreno JC, Collantes E. Efficacy of bosentan in treatment of unresponsive cutaneous ulceration in disabling pansclerotic morphea in children. J Rheumatol 2006;33(12):2538–40.