CHAPTER 62
Xanthomas and Abnormalities of Lipid Metabolism and Storage
Paul D. Flynn
Acute and Metabolic Medicine, Addenbrooke’s Hospital, Cambridge; and University of Cambridge, Cambridge, UK
Introduction
Disorders of lipid metabolism are heterogeneous. They include the very rare and the very common. They range from monogenic diseases with high penetrance through polygenic disorders to those that are paradigms of gene–environment interaction. Some are entirely or partially secondary to diabetes, hypothyroidism, renal failure or liver disease. Dyslipidaemias are associated with an increased risk of atherosclerosis and its complications. They are of particular relevance to dermatologists because they may present with subcutaneous lipid deposits (xanthomas). These may require treatment for the relief of the symptoms they cause, as well as enabling the identification and treatment of a dyslipidaemia before the premature onset of clinical cardiovascular disease.
This chapter includes an overview of the classification of the xanthomas and primary and secondary dyslipidaemias relevant to dermatologists, and the overall management of dyslipidaemia. More detailed information on the metabolic non-dermatological issues is available in the third edition of Hyperlipidaemia: Diagnosis and Management by P. N. Durrington [1] and the eighth edition of The Metabolic and Molecular Bases of Inherited Disease [2].
Classification of dyslipidaemias
There are a number of classifications of disordered lipid and lipoprotein metabolism, none entirely satisfactory. The most straightforward is the World Health Organization (WHO) classification, usually referred to as the Fredrickson classification [1], based on the class of lipoprotein present in excess (Table 62.1). However, this requires plasma ultracentrifugation or lipoprotein electrophoresis, does not include disorders characterized by low levels of high-density lipoprotein (HDL) cholesterol or secondary dyslipidaemias, and is not a diagnostic classification.
Table 62.1 World Health Organization classification of hyperlipoproteinaemias
| Type | Lipoprotein abnormality |
| I | Hyperchylomicronaemia |
| IIa | Elevated LDL |
| IIb | Elevated LDL and VLDL |
| III | Broad β-VLDL |
| IV | Elevated VLDL |
| V | Elevated chylomicrons and VLDL |
From Fredrickson et al. 1967 [1].
LDL, low-density lipoprotein; VLDL, very low-density lipoprotein.
For most patients, all that is available is a blood lipid profile comprising total cholesterol, triglycerides and high-density lipoprotein cholesterol measurements with a calculated low-density lipoprotein (LDL) cholesterol. Thus, in practice, patients are classified more broadly as having hypercholesterolaemia, hypertriglyceridaemia, combined (or mixed) dyslipidaemia, or other dyslipidaemia, with further characterization and diagnosis wherever possible based on further tests (Table 62.2).
Table 62.2 Working classification of dyslipidaemias
| Lipid abnormalities | WHO type | Primary dyslipidaemias | Secondary causes | Cutaneous features | |
| Hypercholesterolaemia | ↑ TC ↑ LDL-C Normal Tgs |
IIa | Familial hypercholesterolaemia Familial defective ApoB100 Polygenic hypercholesterolaemia |
Hypothyroidism Anorexia Cholestatic liver disease Nephrotic syndrome Acute intermittent porphyria Drugs: thiazide diuretics, corticosteroids |
Tendon xanthomas (FH) Xanthelasmas Interdigital planar xanthomas (homozygous FH) |
| Combined dyslipidaemia | ↑ TC ↑ LDL-C ↑ Tgs +/− ↓ HDL-C |
III IIb IV |
Familial dysbetalipoproteinaemia Familial combined hyperlipidaemia |
Diabetes Metabolic syndrome Lipodystrophies Hypothyroidism Hepatocellular liver disease Nephrotic syndromeChronic renal failure Paraproteinaemias Pregnancy Drugs: β-blockers, antiretrovirals, retinoic acid derivatives |
Tuberous xanthomas (in WHO type III) Palmar xanthomas (in WHO type III) |
| Hypertriglyceridaemia | ↑↑ Igs ↑ TC ↓ HDL-C |
I IV, V |
Lipoprotein lipase deficiency ApoCII deficiency |
Diabetes Alcohol excess Chronic renal failure Paraproteinaemias Pregnancy (esp. 3rd trimester) Drugs: retinoic acid derivatives, oral contraceptives |
Eruptive xanthomas |
| Other dyslipidaemias | ↓ HDL-C only ↑ HDL-C |
Tangier disease ApoAI Milano Hyperalphalipoproteinaemia |
Alcohol Anabolic steroids |
||
↓↓ LDL-C ↓ LDL-C |
Abetalipoproteinaemia Hypobetalipoproteinaemia |
FH, familial hypercholesterolaemia; HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol; TC, total cholesterol; Tgs, triglycerides.
XANTHOMAS
Introduction
The term xanthoma (Greek ‘xanthos’ meaning yellow) describes a variety of subcutaneous lipid deposits. All xanthomas contain macrophages loaded with cholesterol and cholesterol esters (‘foam cells’). There are a number of clinical types, some of which are associated with hyperlipidaemia.
Classification
Xanthomas are divided into those that are secondary to disorders of lipid metabolism (Box 62.1) and those that are classed under histiocytic disorders. Those due to histiocytic disorders are discussed elsewhere (see Chapter 136).
Tendon xanthoma
Definition and nomenclature
Tendon xanthomas occur as subcutaneous nodules or papules in relation to tendons.
Epidemiology
Associated diseases
Tendon xanthomas are most frequently seen in familial hypercholesterolaemia (FH) (see section later in this chapter), but are also a feature of the secondary hypercholesterolaemia seen in prolonged cholestasis. They also occur in the rare lipid disorders cerebrotendinous xanthomatosis and sitosterolaemia in which the tendon xanthomas are not associated with raised serum cholesterol levels.
Clinical features
These occur most commonly attached to the extensor tendons over the knuckles and in the Achilles tendon, although other tendons can sometimes be affected. In these sites they can usually be moved from side to side. Occasionally they can involve the periosteum at the site of insertion of the patellar tendon where they cannot be moved. As the accumulation of cholesterol is deep within the tendons, the overlying skin does not appear yellow. The xanthomas contain collagen in addition to foamy macrophages and so feel quite hard.
Investigations
Investigations should include a full lipid profile (where an LDL cholesterol level of ≥4.9 mmol/L may indicate FH) and liver function tests.
Management
Tendon xanthomas sometimes improve with cholesterol reduction, but do not tend to resolve completely. Where the tendon xanthomas are painful, this symptom improves with LDL cholesterol reduction.
Tuberous xanthoma
Definition
Tuberous xanthomas are firm, yellow-red nodules that occur over sites of pressure, commonly the elbows and knees (Figure 62.1). Where a central tuberous xanthoma is surrounded by several smaller lesions it may be termed a ‘tubero-eruptive xanthoma’.
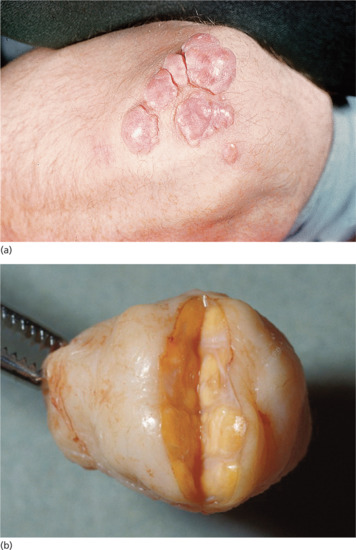
Figure 62.1 (a) Tuberous xanthomas.
(Courtesy of Addenbrooke's Hospital, Cambridge, UK.) (b) Lipid-laden lobules within a tuberoeruptive xanthoma excised from the elbow of a 40-year-old man with familial type III hyperlipidaemia (triglycerides up to 73 mmol/L; cholesterol >24 mmol/L).
Epidemiology
Associated diseases
Tuberous xanthomas usually indicate the presence of type III hyperlipoproteinaemia (see later in this chapter).
Clinical features
They start as small xanthomas, usually over the extensor aspects of the elbows and knees, but can develop into quite exuberant exophytic lesions several centimetres in diameter and height. They can develop over other pressure sites, particularly the heels and plantar surfaces of the feet. They can occasionally occur in the bone marrow. They tend to be painless although can be itchy and are susceptible to traumatic damage given their position.
Investigations
Initial investigations should include a lipid profile; further investigations could include apolipoprotein E (ApoE) genotyping and lipid electrophoresis or ultracentrifugation.
Management
They respond well to effective treatment of the combined dyslipidaemia with which they are associated.
Eruptive xanthoma
Definition
Eruptive xanthomas appear as multiple small papules over extensor surfaces (Figure 62.2).
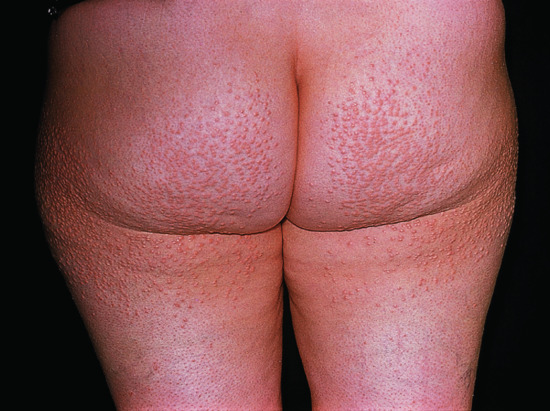
Figure 62.2 Eruptive xanthomas.
(Courtesy of Addenbrooke's Hospital, Cambridge, UK.)
Epidemiology
Associated diseases
Eruptive xanthomas can be associated with any cause of hypertriglyceridaemia.
Clinical features
These small xanthomas consist of yellow papules 2–5 mm in diameter arising on an erythematous base. They usually appear in large numbers over extensor surfaces, particularly the buttocks, back, legs and arms. In extreme cases they are pruritic and are more widely distributed. Their foamy macrophages contain triglycerides as well as cholesterol. Resulting from hypertriglyceridemia, they are almost always accompanied by lipaemia retinalis, a creamy yellow discoloration of the retinal blood vessels, and a lipaemic appearance of blood or serum samples.
Investigations
Investigations should include fasting lipids (especially triglycerides) and glucose.
Management
Eruptive xanthomas resolve within 2 weeks or so of the normalization of triglyceride levels.
Dyslipidaemic plane (planar) xanthoma
This group consists of xanthelasmas, plane xanthomas and palmar xanthomas.
Xanthelasma
Definition and nomenclature
Xanthelasmas are plane xanthomas that develop around the eyes (Figure 62.3).
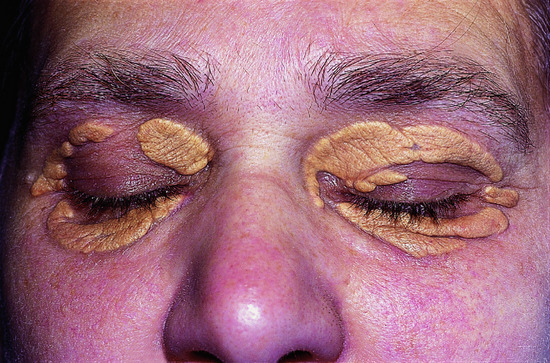
Figure 62.3 Extensive xanthelasmas palpebrarum.
(Courtesy of Addenbrooke's Hospital, Cambridge, UK.)
Epidemiology
Associated diseases
Xanthelasmas are seen in FH, type III hyperlipoproteinaemia and chronic cholestasis (especially primary biliary cirrhosis), but are also often seen in people with circulating lipid levels considered normal in the western population.
Clinical features
They most commonly affect the upper eyelids and the area around the medial canthus. They are relatively soft on palpation and range from pale yellow to yellow-orange in colour.
Complications and co-morbidities
Although previous small studies failed to identify xanthelasma as a risk factor for cardiovascular disease, recent analysis of the 12 745 participants in the Copenhagen City Heart Study, followed for 33 years, found xanthelasma to be an independent predictor of coronary heart disease, although not of ischaemic stroke [1].
Investigations
Investigations should include a full lipid profile and liver function tests.
Management
Given their prominent site xanthelasmas are often a cosmetic problem. Treatments include surgical excision, electrocautery, topical trichloracetic acid, silver nitrate or lasers. However, they often recur after treatment.
If LDL cholesterol levels are high, then treatment to lower the LDL cholesterol (e.g. with a statin) seems sensible even if not yet confirmed by clinical trial. Personal experience is that such treatment is often associated with regression of xanthelasmas in hypercholesterolaemic patients without the need for other intervention.
Plane xanthoma
Definition and nomenclature
Plane xanthomas are flat, smear-like lesions that can occur anywhere on the body (Figure 62.4).

Figure 62.4 Plane xanthomatosis.
(Courtesy of Addenbrooke's Hospital, Cambridge, UK.)
Epidemiology
Associated diseases
Plane xanthomas can be a feature of homozygous familial hypercholesterolaemia.
Clinical features
These xanthomas are wide based, flat and macular, although they may develop into raised plaques. They can occur anywhere on the body. Plane xanthomas affecting the interdigital web between the first and second fingers are only seen in homozygous familial hypercholesterolaemia. Diffuse dyslipidaemic plane xanthoma is less common and more widespread.
Differential diagnosis
Diffuse plane normolipidaemic xanthomatosis (see Chapter 136) is a rare form of histiocytosis, often associated with paraproteinemia or an underlying systemic disorder, usually of the haematological or lymphoproliferative type. Lipid levels are normal.
Necrobiotic xanthogranuloma (see Chapter 136) is a rare, chronic, progressive histiocytosis that is strongly associated with haematological malignant conditions.
Investigations
Familial hypercholesterolaemia and type III hyperlipoproteinaemia may be associated. Initial investigations should include a full lipid profile, serum electrophoresis, and autoimmune screen. Further tests could include a skeletal survey and bone marrow examination if necessary.
Management
Like tuberous xanthomas, they respond well to treatment of the dyslipidaemia.
Palmar xanthoma
Definition and nomenclature
Palmar xanthomas run in the palmar creases (Figure 62.5).
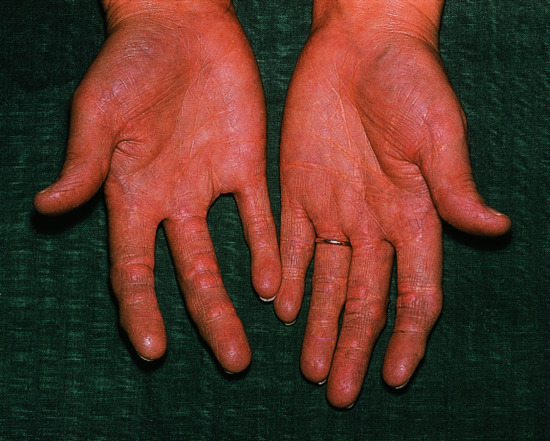
Figure 62.5 Xanthomatosis and yellow palmar creases.
(Courtesy of Addenbrooke's Hospital, Cambridge, UK.)
Epidemiology
Associated diseases
They are pathognomonic of type III hyperlipoproteinaemia.
Clinical features
Linear palmar xanthomas consist of an orange-yellow lipid deposition running along the palmar creases, and occasionally the flexor creases of the wrists.
Investigations
Initial investigations should include a full lipid profile, fasting glucose, urine and electrolytes, liver function tests and thyroid-stimulating hormone levels. Further specialist tests include ApoE genotyping and lipid electrophoresis or ultracentrifugation.
Management
They usually respond well to treatment of the causes of the type III hyperlipoproteinaemia.
PRIMARY DYSLIPIDAEMIAS: HYPERCHOLESTEROLAEMIA
Familial hypercholesterolaemia
Definition
Familial hypercholesterolaemia is an autosomal co-dominant condition characterized by the following:
- High total and LDL cholesterol, slightly low HDL cholesterol and normal triglyceride concentrations.
- Frequent tendon xanthomas (especially of the Achilles tendons).
- Premature onset of cardiovascular disease (CVD).
Epidemiology
Homozygotes number about one in 1 × 106 in white populations. Heterozygotes occur at a frequency of about one in 500 in most white populations, though with notably higher frequency in certain groups such as Afrikaners and the Lebanese, where there are marked founder gene effects.
Pathophysiology
Genetics
The familial clustering of the three features listed above (high total and LDL cholesterol, tendon xanthomas and premature CVD) has been recognized since at least the late 19th century and was the subject of detailed studies in the 1930s [1, 2]. In the 1950s the development of ultracentrifugation for quantifying lipoproteins revealed that the biochemical hallmark of FH is an accumulation of LDL [3], and in 1973 Goldstein and Brown discovered the LDL receptor and showed that its defective functioning was the underlying cause of the condition [4].
The majority of patients with FH have mutations in their LDL receptor associated with a variable degree of loss of function. As a result there is both defective clearance of very low-density lipoprotein (VLDL) from the circulation with consequent increased conversion to LDL, and impaired clearance of LDL resulting in the significant elevation of LDL cholesterol concentration that accounts for the raised plasma total cholesterol. Over 1000 disease-associated mutations have so far been described in the LDL receptor gene (which is one of the largest human genes). They have been classified on the basis of the effect they have on the synthesis, expression and processing of the LDL receptor:
- Type 1 mutations prevent receptor synthesis.
- Type 2 mutations impair transport of the receptor from the endoplasmic reticulum to the Golgi.
- Type 3 mutations affect receptor binding of circulating LDL.
- Type 4 mutations prevent clathrin-mediated receptor clustering and internalization.
- Type 5 mutations interfere with receptor recycling from the endosome.
Type 1 mutations tend to result in a complete absence of LDL receptor activity (‘receptor negative’), while the other classes of mutations usually cause markedly reduced but not completely absent activity (‘receptor defective’).
In a small proportion of patients with FH (around 2%), the mutation affects not the LDL receptor but the receptor-binding domain in the LDL ApoB100 (familial defective ApoB100). This is almost always due to a mutation in codon 3500 (R3500Q) resulting in a substitution of arginine for glutamine; rarely it is due to a different mutation in codon 3500 (R3500W – in which tryptophan replaces arginine) or in codon 3531. Such patients are phenotypically indistinguishable from those with LDL receptor mutations although there is some evidence that their cardiovascular risk is not as dramatically raised. In recent years a new genetic locus has been found to account for some of the previously mutation negative cases of FH. The gene codes for proprotein convertase subtilisin kexin 9 (PCSK9), a serine protease whose activity appears to decrease LDL receptor expression and LDL clearance and so increase LDL cholesterol concentrations [5, 6]. It has emerged that statins up-regulate PCSK9, so it represents an interesting target for agents to augment the LDL-lowering effect of these drugs. Finally, a small group of patients, all from Sardinia, have a phenotype similar to homozygous FH but inherited in a recessive pattern: autosomal recessive hypercholesterolaemia (ARH) [7, 8].
Clinical features
The presence of tendon xanthomas (see earlier in this chapter) is the principal cutaneous manifestation of FH, and may often be helpful in establishing the diagnosis. They are usually present in childhood in homozygotes and become more common with age in untreated heterozygotes. The prevalence rises from 12.5% in those aged 10–19, to 69.2% in those between 20 and 29, and up to 90% by the age of 40 years. This frequency has been much reduced by the earlier introduction of effective cholesterol-lowering medication. Planar and tuberous xanthomas may occasionally be seen in FH homozygotes, but not heterozygotes. Xanthelasmas are not commonly seen in FH.
Differential diagnosis
Very rarely, immunoglobulins directed against the LDL receptor can produce a hypercholesterolaemia with total and LDL cholesterol levels similar to those seen in FH homozygotes but without any tendon xanthomas. This has been referred to as ‘pseudo familial hypercholesterolaemia’.
Complications and co-morbidities
Homozygotes can have total cholesterol levels of 15 mmol/L or higher, be born with tendon xanthomas and suffer from the clinical effects of coronary heart disease as early as their teens. They can also develop a supravalvular aortic stenosis.
Heterozygotes have less marked elevations in their cholesterol than homozygotes (Table 62.3), but 50% of untreated males will have suffered a myocardial infarction and 24% will have died by the age of 50. Heterozygous females have a slightly delayed onset of CVD compared with males but 58% will have had a myocardial infarction and 15% have died by the age of 60 if not treated [9]. FH thus accounts for a significant proportion of premature CVD and of premature sudden cardiac death. There is a striking preponderance in FH of coronary arterial disease, as opposed to cerebrovascular or peripheral vascular disease.
Table 62.3 Lipid concentrations in familial hypercholesterolaemia
| Total cholesterol (mmol/L) | LDL cholesterol (mmol/L) | HDL cholesterol (mmol/L) | Triglycerides (mmol/L) | |
| Normal | 4.52 ± 0.72 | 2.84 ± 0.64 | 1.37 ± 0.33 | 0.68 ± 0.28 |
| Heterozygotes | 7.73 ± 1.63 | 6.23 ± 1.55 | 1.11 ± 0.31 | 0.93 ± 0.58 |
| Homozygotes | 17.5 ± 4.39 | 16.1 ± 4.13 | 0.88 ± 0.26 | 1.14 ± 0.58 |
From Kwiterovitch et al. 1974 [10].
HDL, high-density lipoprotein; LDL, low-density lipoprotein.
Investigations
Lipid concentrations in FH are listed in Table 62.3.
Given the genetic heterogeneity underlying FH, diagnosis has largely rested on clinical and biochemical criteria. In the UK the criteria used by the Simon Broome Register of patients with FH have been widely adopted (Box 62.2) [11]. Other sets of diagnostic criteria have also been developed, such as the Dutch Lipid Clinic Network Criteria and the Make Early Diagnosis to Prevent Early Disease on Medical Pedigree (MEDPED) system [12, 13]. Increasingly, advances in molecular biological techniques are proving helpful in diagnosing FH; this is particularly true where a disease-causing mutation has been identified in one family member. Genetic cascade screening for FH has been adopted in a number of countries, although not yet systematically in the UK.
Management
Statins have now become the mainstay of treatment and have played a large part in the reduction of cardiovascular mortality seen in recent years in FH. In comparison with the general population, statins usually have to be started at a much earlier age in patients with FH. To achieve the desired reduction in LDL cholesterol, the National Institute for Health and Care Excellence (NICE) in the UK has advised a greater than 50% reduction, so higher doses of the more potent statins may well have to be used. Referral to a specialist lipid clinic is advised to allow counselling of young patients starting life-long therapy, especially young women where the specific issue of the potential teratogenicity of the statins and family planning needs to be fully explored. Such clinics will have experience of other therapies that can prove helpful, including the bile acid sequestrants and LDL apheresis. Ezetimibe is a relatively new agent that works to reduce intestinal cholesterol absorption mediated by the recently described transporter NPC1L1 (Niemann–Pick C-1-like protein 1); it is well tolerated and can augment the LDL reduction achieved by statins by a further 20% but evidence that it is clinically effective in reducing CVD risk is sparse. Tendon xanthomas and xanthelasmas may respond to the reduction in LDL cholesterol; corneal arcus does not. FH homozygotes must be referred for specialist assessment and treatment. Their cholesterol does not respond well to statins or other drug therapies that work by up-regulating LDL receptor expression. These patients often require LDL apheresis and occasionally liver transplantation as well as coronary revascularization and aortic valve surgery. Promising new agents for treating homozygous FH include mipomersen, an antisense oligonucleotide directed against ApoB100 mRNA, and Lopitamide, an inhibitor of microsomal triacylglycerol transfer protein, both of which suppress the production of VLDL [14].
PRIMARY DYSLIPIDAEMIAS: COMBINED DYSLIPIDAEMIA
Type III hyperlipoproteinaemia
Definition and nomenclature
Type III hyperlipoproteinaemia is associated with combined dyslipidaemia, palmar and tuberous xanthomas and early-onset CVD and peripheral vascular disease.
Epidemiology
Incidence and prevalence
Estimates of the frequency of type III hyperlipoproteinaemia vary between about one and 10 in 10 000.
Pathophysiology
Patients with type III hyperlipoproteinaemia have a characteristic lipoprotein electrophoretic pattern that is the gold standard diagnostic test. There is an increase in small VLDL and intermediate-density lipoprotein (IDL) and a decrease in LDL. VLDL isolated from type III patients is uncharacteristically enriched with cholesterol esters and gives rise to a broad-β migrating band, rather than the usual pre-β band [1, 2]. This results from an accumulation of VLDL remnants and chylomicron remnants and produces a marked increase in plasma total cholesterol and triglycerides.
Genetics
The underlying defect is an abnormal ApoE [3], the apolipoprotein in remnant lipoproteins that mediates their binding to the LDL receptor and the LDL receptor-related protein. There are three common isoforms of ApoE: ApoE2, -E3 and -E4, each differing by one amino acid:
| E2 | E3 | E4 | |
| Residue 112 | Cysteine | Cysteine | Arginine |
| Residue 158 | Cysteine | Arginine | Arginine |
ApoE2 homozygosity represents the commonest abnormality associated with type III hyperlipoproteinaemia; its cysteine at residue 112 is within the receptor binding domain of the protein and its cysteine at residue 158 affects salt bridges within the protein and thereby the conformation of the receptor binding domain. Very occasionally other ApoE mutations are associated with type III hyperlipoproteinaemia, many of which severely reduce ApoE-mediated receptor binding and can cause the disorder in heterozygotes [4].
Environmental factors
Less than 10% of subjects homozygous for ApoE2 develop type III hyperlipoproteinaemia, and most of the rest have lower than average lipid levels. It is clear that a second factor is required for the development of type III hyperlipoproteinaemia, either environmental or genetic. Increased production of chylomicrons or VLDL can be one such factor and may result from excessive caloric intake, increased alcohol consumption, obesity, type 2 diabetes, the metabolic syndrome or from an interplay of multiple genetic influences on lipoprotein metabolism (as presumed to underlie familial combined hyperlipidaemia). Equally, increasing age, hypothyroidism or postmenopausal oestrogen deficiency can further impair ApoE-mediated receptor catabolism of lipoproteins and result in the development of the disorder.
Clinical features
The most characteristic skin manifestations are palmar xanthomas (see Figure 62.5). Tuberous xanthomata (see Figure 62.1) may also be present. Patients with particularly high triglyceride concentrations may also develop lipaemia retinalis and eruptive xanthomas over their buttocks (see Figure 62.2).
Complications and co-morbidities
There is evidence of an increased risk of both coronary artery and peripheral arterial disease, especially in those with all the clinical and biochemical features of the disorder – in whom up to 50% have premature CVD [5]. Gout also appears common in this group of patients [6]. Type III hyperlipoproteinaemia has been associated with lipoprotein glomerulopathy, manifesting as proteinuria or the nephrotic syndrome [7]. Renal biopsy shows glomerular foam cells; the condition usually resolves with successful treatment of the abnormal lipid profile.
Management
Identification and treatment of any contributing lifestyle factors or diseases is central to the management of these patients. Patients should be screened for obesity, diabetes and hypothyroidism, and treated accordingly. Dietary advice should be given and exercise encouraged as it increases the activity of lipoprotein lipase and thereby increases chylomicron and VLDL remnant clearance. Such measures are often successful in improving the lipid profile on their own; in their absence drug therapy is usually less effective. Normally both statins and fibrates work well, both in improving the lipid profile and in causing regression of the palmar and tuberous xanthomas [8, 9]. Occasionally, combined statin and fibrate therapy may be required.
PRIMARY DYSLIPIDAEMIAS: HYPERTRIGLYCERIDAEMIAS
Type I hyperlipoproteinaemia
Definition
Type I hyperlipoproteinaemia is an autosomal recessive condition in which there is an accumulation of chylomicrons alone due to a deficiency of lipoprotein lipase (LPL).
Epidemiology
Incidence and prevalence
The condition is extremely rare with a frequency of perhaps one per million in most populations, but with higher frequency in some populations such as French Canadians due to founder gene effects.
Pathophysiology
It results from a deficiency of LPL, and as such shows an autosomal recessive pattern of inheritance (although there is accumulating evidence that some heterozygotes for LPL deficiency may show a milder degree of hypertriglyceridaemia). An increasing number of mutations in the LPL gene have been described. Some cases result from ApoCII deficiency, an essential factor for LPL activation. This discovery followed from the observation that hyperchylomicronaemia was alleviated in some patients following plasma infusions.
It remains unclear why LPL deficiency can cause an isolated accumulation of chylomicrons, given the role of the enzyme in VLDL catabolism. However, with increasing age there is a progressive shift towards a type V hyperlipoproteinaemic pattern.
Clinical features
The characteristic skin finding in patients with type I hyperlipoproteinaemia is eruptive xanthomas (see Figure 62.2).
Complications and co-morbidities
The principal concern in type I hyperlipoproteinaemia is the increased risk of acute pancreatitis with its attendant morbidity and mortality. Whilst hypertriglyceridaemia may be a consequence of acute pancreatitis there is clear evidence it may also be a cause, perhaps accounting for up to 10% of cases. The risk increases with increasing triglyceride, with a threshold around fasting triglycerides of 10 mmol/L, rising markedly beyond 20 mmol/L. Other factors must play a part as some patients seem remarkably immune from attacks despite grossly elevated triglyceride concentrations. The mortality is further increased because the serum amylase, so often used as a sole diagnostic test for acute pancreatitis, may not be elevated.
Clearance of chylomicrons from the circulation by cells of the reticuloendothelial system can result in hepatomegaly and splenomegaly, with splenic infarcts in some cases.
Investigations
Familial LPL deficiency causes extremely elevated levels of serum triglycerides, often greater than 20 mmol/L. Total cholesterol levels are often substantially elevated as well, reflecting the extreme concentrations of chylomicrons, which contain some cholesterol and cholesterol esters as well as their predominant triglycerides. Blood, serum or plasma from such patients will appear milky and a creamy layer will separate on top of the sample if centrifuged or allowed to stand for some hours. Hyperchylomicronaemia will be present from childhood onwards in homozygous LPL deficiency.
Management
The treatment of severe hypertriglyceridaemia involves strict restriction of all dietary fats to 20–25 g per day to reduce chylomicron production as much as possible. Attention must be paid to any other factors that may increase the production or decrease the clearance of the triglyceride-rich lipoproteins (TRLs). Good control of diabetes, weight reduction where appropriate, alcohol restriction and a review of all other medications are particularly important. Intake of refined carbohydrates should also be limited. Fibrates usually have little effect in genetic LPL deficiency. Nicotinic acid derivatives can be helpful. Bile acid sequestrants are contraindicated as they may increase the triglyceride level. Infusions of fresh frozen plasma are beneficial in those with ApoCII deficiency who present with acute pancreatitis. Antioxidant therapy has proved very effective in reducing the frequency of attacks of acute pancreatitis in a number of patients with severe hypertriglyceridaemia. All patients with severe hypertriglyceridaemia should have access to specialist lipid clinics.
Eruptive xanthomas respond to lowering of the triglycerides although they tend to last a few days longer than the lipid abnormality.
Type V hyperlipoproteinaemia
Definition
Type V hyperlipoproteinaemia is due to an accumulation of VLDL as well as chylomicrons.
Epidemiology
Incidence and prevalence
Type V hyperlipoproteinaemia affects one in every 5000–10 000.
Pathophysiology
Although it can result from LPL deficiency, this only accounts for a very small proportion of cases. Patients with type V hyperlipoproteinaemia may have relatives with hypertriglyceridaemia, though many of these will have a type IV lipoprotein pattern. This has led to the hypothesis that most type V patients have a reasonably marked defect in the catabolism of TRLs combined with some additional factor that either further decreases catabolism or additionally increases the production of TRLs. Such factors include obesity, poor diabetic control, alcohol consumption, pregnancy, drugs (oral contraceptives, highly active antiretroviral therapy, β-blockers and thiazide diuretics being the main offenders), hypothyroidism, lipodystrophy and renal failure.
Clinical features
Acute pancreatitis, eruptive xanthomas, hepatosplenomegaly, lipaemia retinalis and all the other clinical features of type I hyperlipoproteinaemia may be present. Additionally, hyperuricaemia, glucose intolerance and hepatic steatosis are often seen.
Investigations
Patient samples will demonstrate some milkiness with the separation of a creamy layer (made up of chylomicrons) after standing or centrifugation, but the serum or plasma will be opaque representing the presence of VLDLs. Lipid levels in type V hyperlipoproteinaemia may be as markedly elevated, as in type I.
Management
The treatment of type V hyperlipoproteinaemia is exactly as for type I, though fibrates are generally far more effective.
Type IV hyperlipoproteinaemia
Definition
In type IV hyperlipoproteinaemia, VLDL accumulate but without any increase in chylomicrons.
Pathophysiology
Patients with type IV hyperlipoproteinaemia are heterogeneous, the common abnormality being an increased production of VLDLs by the liver, often accompanied by a partial defect in their catabolism. As with other forms of hypertriglyceridaemia, LDLs tend to be smaller and denser, a pattern of abnormalities associated with increased cardiovascular risk.
Clinical features
Apart from those rare type IV patients with marked hypertriglyceridaemia, the clinical features of type I and type V hyperlipoproteinaemia are not present.
Investigations
The degree of hypertriglyceridaemia is usually much less marked than in type I or type V hyperlipoproteinaemia, with many patients having fasting triglycerides of less than 6 mmol/L and most less than 10 mmol/L.
Management
Lifestyle modification along the lines of that advocated for severe hypertriglyceridaemia (see earlier in this chapter) is appropriate for type IV hyperlipoproteinaemia. However, dietary fat restriction does not have to be so marked and can be mainly restricted to saturated fats. The need for drug therapy will largely be guided by estimations of the 10-year CVD risk. Many type IV patients will require such treatment and the first line will often be statins because of their proven efficacy in reducing that risk, even though they do not usually affect hypertriglyceridaemia much.
OTHER PRIMARY DYSLIPIDAEMIAS
Cerebrotendinous xanthomatosis
Definition
Cerebrotendinous xanthomatosis is an autosomal recessive condition and is a rare cause of tendon xanthomas and xanthelasmas.
Epidemiology
Presentation occurs during childhood or early adult life.
Pathophysiology
This condition results from a defect in sterol-27-hydroxylase with consequent increased production of cholestanol and 7-hydroxycholesterol, which accumulate in the plasma and throughout the body.
Clinical features
Cutaneous features include tendon xanthoma and xanthelasma. The accumulation of cholestanol and 7-hydroxycholesterol in the central nervous system causes myelin destruction leading to mental retardation, seizures, spasticity and ataxia; peripheral neuropathy results from similar pathology in the peripheral nervous system. Early-onset cataracts, diarrhoea and premature osteoporosis are additional features. Patients have an increased risk of CVD.
Investigations
Plasma cholesterol levels are normal.
Management
Treatment with chenodeoxycholate reduces plasma cholestanol concentrations and improves the neurological manifestations of the disease.
Sitosterolaemia
Definition
Sitosterolaemia is a rare autosomal recessive cause of tendon and tuberous xanthomas.
Pathophysiology
Genetics
This condition results from mutations in the genes ABCG5 or ABCG8, which encode the proteins sterolin-1 and sterolin-2 in enterocytes and hepatocytes. Sterolin-1 and sterolin-2 act together to form a lipid transporter that is thought to facilitate immediate excretion of any plant sterols absorbed across the small intestinal brush border. Defective function thereby allows a much greater absorption of plant sterols into the body, principally β-sitosterol but also sitostanol, campesterol and stigmasterol.
Clinical features
Patients with sitosterolaemia suffer from impaired growth, anaemia, thrombocytopenia and arthritis and are at risk of premature CVD. Cutaneous features include tendon and tuberous xanthomas.
Investigations
Diagnosis is made by measuring serum plant sterol concentrations.
Management
It has recently been shown that ezetimibe reduces plant sterol levels effectively in this condition, suggesting a role for the Niemann–Pick C1-like protein in plant sterol transport in the intestine.
SECONDARY DYSLIPIDAEMIAS
Dyslipidaemias are frequently secondary to, or exacerbated by, a range of other diseases or medications. The following account is far from exhaustive but will concentrate on those that may produce xanthomas or follow from the use of drugs used by dermatologists.
Secondary dyslipidaemia and diabetes
Perhaps the commonest cause of secondary dyslipidaemia is diabetes (see Chapter 64). People with type 1 diabetes frequently have quite high HDL cholesterol levels, but they also have a number of abnormalities of their LDLs and VLDLs and they are at high risk of CVD. People with type 2 diabetes have insulin resistance; their typical lipid profile includes relatively normal total and LDL cholesterol levels, but often somewhat increased triglyceride and reduced HDL cholesterol concentrations. These quantitative abnormalities are usually accompanied by qualitative changes in the LDLs with a preponderance of highly atherogenic, small, dense LDLs. As a result patients with type 2 diabetes are also at high risk of developing premature CVD. It is now recommended that statins should be considered for all people with diabetes over the age of 40, and in younger patients with other CVD risk factors including microalbuminuria. In some patients the development of type 2 diabetes, or worsening of its glycaemic control, can cause marked hypertriglyceridaemia (with type IV or V hyperlipoproteinaemia) with all its clinical features, including eruptive xanthomas, lipaemia retinalis and acute pancreatitis. This is most likely to occur on the background of some mild defect of triglyceride-rich lipoprotein clearance, and may be further exacerbated by the concomitant presence of obesity and excess alcohol intake. Diabetes can also precipitate the development of type III hyperlipoproteinaemia in those with the appropriate genetic background (usually homozygosity for ApoE2). Such patients may present to a dermatologist with the typical xanthomas of this condition. It follows that in any patient presenting with eruptive, tuberous or planar xanthomas a fasting glucose and/or and HbA1c should be requested along with the lipid profile and other appropriate tests.
Secondary dyslipidaemia and insulin resistance
Patients with insulin resistance, but without type 2 diabetes, have a similar combined dyslipidaemia. In some, a marked hypertriglyceridaemia can develop, sufficient to cause eruptive xanthomas. This is seen quite commonly in the lipodystrophies, a group of disorders with either partial or generalized loss of subcutaneous fat. They can be inherited (e.g. Dunnigan–Köbberling syndrome) or acquired. The latter is now most commonly seen in patients with HIV/AIDS treated with protease inhibitors (see Chapter 31), especially saquinavir and ritonavir. Management of lipodystrophy-associated dyslipidaemia is likely to require specialist input. The principles are lifestyle change, especially strict dietary fat restriction in hypertriglyceridaemia, optimization of the drug regimen in HIV, and judicious, monitored use of lipid-modulating drug therapy, given the potential for significant interactions between statins, fibrates and the antiretroviral agents.
Secondary dyslipidaemia due to chronic cholestasis
It has already been noted that xanthelasmas can be a feature of chronic cholestasis. This can occur, for example, in primary biliary cirrhosis, and the presence of xanthelasmas should prompt a request for blood tests including a bilirubin and an alkaline phosphatase at least. Longstanding cholestasis is associated with hypercholesterolaemia due to the accumulation of an abnormal lipoprotein, lipoprotein X. It remains unclear whether this dyslipidaemia increases CVD risk, but it can cause a wide range of xanthomas as well as xanthelasmas. Both the dyslipidaemia and the xanthomas improve with the relief of biliary obstruction; where this is not possible bile acid sequestrants can be helpful (and may also relieve the cholestasis-associated pruritus). Where necessary, statins must be used cautiously to avoid accumulation and toxicity. Hepatocellular liver disease leads to specific lipoprotein abnormalities, principally abnormal HDL secondary to progressive deficiency of lecithin-cholesterol acyl transferase with mild hypertriglyceridaemia, but these are not usually severe enough to cause xanthomas.
Secondary dyslipidaemia and the nephrotic syndrome
Nephrotic syndrome is complicated by hypercholesterolaemia or a combined dyslipidaemia that can prove difficult to treat. Hypertriglyceridaemia is not usually marked, although occasionally a type IV hyperlipoproteinaemia may occur. Xanthomas are not a frequent feature and the dyslipidaemia resolves if the nephrotic syndrome responds to therapy. Chronic renal failure leads to hypertriglyceridaemia, often exacerbated by haemodialysis or peritoneal dialysis, and this can sometimes lead to eruptive xanthomas, especially if other risk factors are present. Lipoprotein(a) levels are elevated and may contribute to the increased CVD risk in this population but do not produce any dermatological signs.
Secondary dyslipidaemia due to drugs
Many drugs can produce dyslipidaemia, alcohol perhaps being the commonest example and a frequent contributor to marked hypertriglyceridaemia. Of the prescription drugs used by dermatologists, it is worth considering corticosteroids, ciclosporin and retinoic acid derivatives.
Systemic corticosteroids increase total LDL and HDL cholesterol levels. Unless they precipitate diabetes in a susceptible individual they do not cause hypertriglyceridaemia.
Ciclosporin increases LDL cholesterol levels, sometimes quite significantly. It also inhibits cytochrome P450 3A4, the isoforms responsible for the metabolism of fluvastatin, simvastatin and atorvastatin, so patients taking these statins should be advised of the possible interaction and the risk of myalgia. In some patients (e.g. those with hepatic or renal impairment, or taking other drugs that affect CYP450 3A4) it may be appropriate to reduce the statin dose or change to an alternative (pravastatin or rosuvastatin are less dependent on CYP450 3A4).
Retinoic acid derivatives often induce hypertriglyceridaemia; this is usually mild and does not necessarily require anything other than dietary modification. Occasionally, however, marked hypertriglyceridaemia with a type IV or V hyperlipoproteinaemic pattern can be provoked, usually in a susceptible individual who may well have had evidence of dyslipidaemia prior to treatment. It is therefore sensible to include a full lipid profile on any pre-treatment blood tests and to repeat this several weeks after starting any retinoids.
References
Introduction
- Durrington PN. Hyperlipidaemia: Diagnosis and Management, 3rd edn. London: Hodder Arnold, 2007.
- Scriver CR, Beaudet AL, Sly WS, eds. The Metabolic and Molecular Bases of Inherited Disease, 8th edn. New York: McGraw Hill, 2001.
Classification of dyslipidaemias
- Fredrickson DS, Levy RI, Lees RS: Fat transport in lipoproteins – an integrated approach to mechanisms and disorders. N Engl J Med 1967;276:34–42, 94–103, 148–56, 215–52, 273–81.
Xanthomas
XanthomasDyslipidaemic plane (planar) xanthoma
Xanthelasma
- Christoffersen M, Frikke-Schmidt R, Schnohr P, et al. Xanthelasmata, arcus corneae, and ischaemic vascular disease and death in general population: prospective cohort study. BMJ 2011;343:d5497.
Primary dyslipidaemias: hypercholesterolaemia
Familial hypercholesterolaemia
- Thannhauser SJ, Magendanta H. The different clincal groups of xanthomatous diseases: a clinical physiological study of 22 cases. Ann Intern Med 1938;11:1662–746.
- Müller C. Angina pectoris in hereditary xanthomatosis. Arch Intern Med 1939;64:675–700.
- Gofman JW, De Lalla O, Glazier F, et al. The serum lipoprotein transport system in health, metabolic diseases, atherosclerosis and coronary artery disease. Plasma 1954;2:413–84.
- Goldstein JL, Brown MS. Familial hypercholesterolaemia: identification of a defect in the regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity associated with overproduction of cholesterol. Proc Natl Acad Sci USA 1973;70:2804–8.
- Timms KM, Wagner S, Samuels ME, et al. A mutation in PCSK9 causing autosomal dominant hypercholesterolaemia in a Utah pedigree. Hum Genet 2004;114:349–53.
- Abifadel M, Varret M, Rabes JP, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolaemia. Nat Genet 2003;34:154–6.
- Soutar AK, Nauomova RP, Traub LM. Genetics, clinical phenotype, and molecular cell biology of autosomal recessive hypercholesterolaemia. Arterioscler Thromb Vasc Biol 2003;23:1963–70.
- Fellin R, Zuliani G, Arca M, et al. Clinical and biochemical characterisation of patients with autosomal recessive hypercholesterolaemia (ARH). Nutr Metab Cardiovasc Dis 2003;13:278–86.
- Slack J. Risks of ischaemic heart disease in familial hyperlipoproteinaemia states. Lancet 1969;ii:1380–2.
- Kwiterovitch PO, Jr, Fredrickson DS, Levy RI. Familial hypercholesterolaemia (one form of familial type II hyperlipoproteinaemia). A study of its biochemical, genetic and clinical presentation in childhood. J Clin Invest 1974;53:1237.
- Scientific Steering Committee of the Simon Broome Register Group. Risk of fatal coronary heart disease in familial hypercholesterolaemia. BMJ 1991;303:893–6.
- World Health Organization. Familial Hypercholesterolaemia – Report of a Second WHO Consultation. WHO Publication No. WHO/HGN/FH/CONS/99.2. Geneva: WHO, 1999.
- Williams RR, Hunt SC, Schumacher MC, et al. Diagnosing heterozygous familial hypercholesterolaemia using new practical criteria validated by molecular genetics. Am J Cardiol 1993;72:171–6.
- Marais AD, Blom DJ. Recent advances in the treatment of homozygous familial hypercholesterolaemia. Curr Opin Lipidol 2013;24:288–94.
Primary dyslipidaemias: combined dyslipidaemia
Type III hyperlipoproteinaemia
- Gofman IW, Rubin L, McGinley JP, Jones HB. Hyperlipoproteinaemia. Am J Med 1954;17:514–20.
- Gofman IW, Rubin L, Lees RS. Fat transport in lipoproteins. An integrated approach to mechanisms and disorders. N Engl J Med 1967;276:32–44, 94–103, 148–56, 215–26, 273–81.
- Utermann G. Apolipoprotein E polymorphisms in health and disease. Am Heart J 1987;113;433–40.
- Mahley RW, Rall SC. Type III hyperlipoproteinaemia (dysbetalipoproteinaemia): the role of apolipoprotein E in normal and abnormal lipoprotein metabolism. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The Metabolic and Molecular Bases of Inherited Disease, 8th edn. New York: McGraw-Hill, 2001:2835–62.
- Morganroth J, Levy RI, Fredrickson DS. The biochemical, clinical and genetic features of type III hyperlipoproteinaemia. Ann Intern Med 1975;82:158–74.
- Brewer HB, Zech LA, Gregg RE, et al. Type III hyperlipoproteinaemia. Diagnosis, molecular defects, pathology and treatment. Ann Intern Med 1983;98:623–40.
- Yang AH, Ng YY, Tarng DC, et al. Association of apolipoprotein E polymorphism with lipoprotein glomerulopathy. Nephron 1998;78:266–70.
- Vega GL, East C, Grundy SM. Lovastatin therapy in familial dysbetalipoproteinaemia: effects on kinetics of apolipoprotein B. Atherosclerosis 1988;70:131–43.
- Kuo PT, Wilson AC, Kostis JB, et al. Treatment of type III hyperlipoproteinaemia with gemfibrozil to retard progression of coronary artery disease. Am Heart J 1988;116:85–90.