CHAPTER 65
Inherited Disorders of Cornification
Vinzenz Oji, Dieter Metze and Heiko Traupe
Department of Dermatology, University Hospital Münster, Müster, Germany
Introduction
This chapter deals with ichthyoses, palmoplantar keratodermas (PPKs) and miscellaneous inherited or acquired cornification disorders. The majority of keratinization disorders are now referred to as Mendelian disorders of cornification (MeDOC) [1]. This is a very broad group that is clinically characterized by hyperkeratosis or visible scaling or both. The term ‘Mendelian’ implies that these diseases are genetically determined in nature. In many of them, distinct genetic defects can be elucidated revealing quite a few mechanisms and sometimes even pathways can be recognized, e.g. the hepoxilin pathway [2], that underlie their pathology. From a molecular perspective, it would be tempting to provide a classification based on molecular grounds, sorting out for instance keratinopathies relating to mutations in keratin genes from diseases relating to defects in lipid transport or cholesterol biosynthesis. However, mutations, e.g. in keratin genes, can cause very variable clinical phenotypes ranging from bullous diseases like epidermolysis bullosa simplex (KRT5 and KRT14) [3] to ichthyosis Curth–Macklin (ICM) (KRT1) [4] not to mention pigmentation disorders like Galli–Galli or Dowling–Degos disease relating to KRT5 mutations [5]. Since this textbook is aimed primarily at dermatologists and physician scientists who have to make a clinical diagnosis and provide adequate management for their patients, the authors have decided to adhere to a classification scheme that is based on clinicogenetic and morphological features. These clinical diagnoses are then discussed with their molecular pathology.
From a ‘classical dermatological’ perspective, we distinguish today among the many MeDOCs ichthyoses, PPKs and a remaining group of miscellanous keratinization disorders, such as the various forms of porokeratosis. Darier disease is dealt with elsewhere in this textbook (see Chapter 66). It should be noted that in the Ichthyosis Consensus Conference (in Sorèze 2009) it was decided not to make any longer a distinction between the group of erythrokeratodermas and ichthyoses, but rather to group them together [1]. In PPK, unlike ichthyosis, the cornification disorder is not widespread, but almost exclusively affects the hands and feet.
There are entities that could be regarded as either belonging to the ichthyosis or the PPK group. One such example is loricrin keratoderma (LK) [6, 7]. However, for the clinician, the distinction between ichthyosis and PPK is diagnostically valuable and helpful. It dates back more than 100 years and was first introduced by Peukert in 1899 [8]. Finally, this chapter will cover several cornification disorders that do not have a genetic basis, but are acquired or of unknown aetiology, e.g. acquired ichthyoses [9] or pityriasis rotunda [10].
ICHTHYOSES (see Tables 65.1, 65.2, 65.3, 65.4 and 65.6)
Table 65.1 Clinicogenetic classification of inherited ichthyoses: non-syndromic forms.
| Disease | Mode of inheritance | Genes |
| Common ichthyoses | ||
| Ichthyosis vulgaris (IV) | Semidominant | FLG |
| Non-syndromic recessive X-linked ichthyosis (RXLI) | XR | STS |
| Autosomal recessive congenital ichthyosis (ARCI) | ||
| Harlequin ichthyosis (HI) | AR | ABCA12 |
| Lamellar ichthyosis (LI) | AR | TGM1, NIPAL4, ALOX12B, ABCA12, PNPLA1, CERS3, LIPHa |
| Congenital ichthyosiform erythroderma (CIE) | AR | ALOXE3, ALOX12B, ABCA12, CYP4F22, NIPAL4, TGM1, PNPLA1, CERS3, LIPHb |
| Self-healing collodion baby (SHCB)c | AR | TGM1, ALOXE3, ALOX12B |
| Acral self-healing collodion baby | AR | TGM1 |
| Bathing suit ichthyosis (BSI) | AR | TGM1 |
| Keratinopathic ichthyosis (KPI) | ||
| Epidermolytic ichthyosis (EI) | AD | KRT1, KRT10 |
| Superficial epidermolytic ichthyosis (SEI) | AD | KRT2 |
| Congenital reticular ichthyosiform erythroderma (CRIE) | AD | KRT10 |
| Annular epidermolytic ichthyosis (AEI) | AD | KRT1, KRT10 |
| Ichthyosis Curth–Macklin (ICM) | AD | KRT1 |
| Autosomal recessive epidermolytic ichthyosis (AREI) | AR | KRT10 |
| Epidermolytic naevid | Somatic mutations | KRT1, KRT10 |
| Other non-syndromic forms | ||
| Loricrin keratoderma (LK) | AD | LOR |
| Erythrokeratodermia variabilis (EKV) | AD (AR) | GJB3, GJB4 |
| Inflammatory peeling skin disease (PSS type B) | AR | CDSN |
| Exfoliative ichthyosis | AR | CSTA |
| Keratosis linearis–ichthyosis congenita–keratoderma (KLICK) | AR | POMP |
Modified from Oji V, Tadini G, Akiyama M, et al. Revised nomenclature and classification of inherited ichthyoses: results of the First Ichthyosis Consensus Conference in Sorèze 2009. J Am Acad Dermatol 2010;63:607–41 (ref [4] under Ichthyoses).
1ARCI of late onset.
2Self-improving congenital ichthyosis (SICI).
3May indicate a gonadal mosaicism, which can cause generalized EI in the offspring generation.
AD, autosomal dominant; AR, autosomal recessive.
Table 65.2 Clinicogenetic classification of inherited ichthyoses: syndromic forms.
| Disease | Mode of inheritance | Genes |
| X-linked ichthyosis syndromes | ||
| Recessive X-linked ichthyosis (RXLI)a | XR | STS (and othersb) |
| Ichthyosis follicularis alopecia photophobia (IFAP) | XR | MBTPS2 |
| Conradi–Hünermann–Happle syndrome (CDPX2) | XD | EBP |
| Autosomal ichthyosis syndromes with prominent hair abnormalities | ||
| Netherton syndrome (NS) | AR | SPINK5 |
| Severe dermatitis–multiple allergies–metabolic wasting (SAM) | AR | DSG1 |
| Ichthyosis with hypotrichosisc | AR | ST14 |
| Neonatal ichthyosis–sclerosing cholangitis (NISCH)d | AR | CLDN1 |
| Autosomal ichthyosis syndromes with prominent neurological signs | ||
| Refsum syndrome (HMSN4) | AR | PHYH, PEX7 |
| Multiple sulphatase deficiency (MSD) | AR | SUMF1 |
| Gaucher syndrome type 2 | AR | GBA |
| Sjögren–Larsson syndrome (SLS) | AR | ALDH3A2 |
| Neutral lipid storage disease (NLSD) with ichthyosis | AR | ABHD5 |
| Trichothiodystrophy (TTD) | AR | C7ORF11 ERCC2, XPD ERCC3, XPB GTF2H5, TTDA |
| Cerebral dysgenesis–neuropathy–ichthyosis–palmoplantar keratoderma (CEDNIK) | AR | SNAP29 |
| Arthrogryposis–renal dysfunction–cholestasis (ARC) | AR | VPS33B |
| Autosomal ichthyosis syndromes with deafness | ||
| Keratitis–ichthyosis–deafness (KID) | AD | GJB2 (GJB6) |
| ELOVL4 deficiency | AR | ELOVL4 |
| Mental retardation–enteropathy–deafness–neuropathy–ichthyosis–keratodermia (MEDNIK) | AR | AP1S1 |
| Autosomal ichthyosis syndromes with transient neonatal respiratory distress | ||
| Ichthyosis–prematurity syndrome (IPS) | AR | SLC27A4 |
Modified from Oji V, Tadini G, Akiyama M, et al. Revised nomenclature and classification of inherited ichthyoses: results of the First Ichthyosis Consensus Conference in Sorèze 2009. J Am Acad Dermatol 2010;63:607–41 (ref [4] under Ichthyoses).
1In the context of a contiguous gene syndrome.
2Allelic variant: congenital ichthyosis–follicular atrophoderma–hypotrichosis–hypohidrosis (IFAH).
3Also known as ichthyosis–leukocyte vacuoles–alopecia–sclerosing cholangitis (ILVASC).
AD, autosomal dominant; AR, autosomal recessive; CDPX2, chondrodysplasia punctata type 2; HMSN4, hereditary motor and sensory neuropathy type 4.
Table 65.3 Keratinopathic ichthyoses.
| Epidermolytic ichthyosis (EI) | Superficial epidermolytic ichthyosis (SEI) | Ichthyosis Curth–Macklin (ICM) | Congenital reticular ichthyosiform erythroderma (CRIE)a | |
| MIM | 113800 | 146800 | 146600 | 609165 |
| Mode of inheritance | AD (rarely AR in KRT10) | AD | AD | AD |
| Gene | KRT1 or KRT10 | KRT2 | KRT1 | KRT10 |
| Onset | At birth | At birth | Early childhood | At birth |
| Initial clinical presentation | Large erosions, mild scaling, erythroderma at birth | Erythroderma, widespread blistering | Striate or diffuse PPK | Exfoliative CIE, larger areas forming a reticular pattern predominantly on the extremities |
| Disease course | Resolution of erosions replaced by hyperkeratosis in the first months Annular type: development of numerous annular polycyclic erythematous scaly plaques on the trunk and extremities that enlarge slowly, and then resolve (intermittent presentations of EI) |
Within weeks development of hyperkeratosis particularly over extensor sides of joints | Progressive worsening of PPK and development of hyperkeratotic plaques over joints and/or hyperkeratotic papules on the trunk and extremities | During childhood and puberty a characteristic patchy pattern starts to evolve |
| Cutaneous findings | ||||
| Distribution of scaling | Generalized, or predilection of friction areas, over joints | Friction areas | Palms and soles, large joints, rarely extremities and/or trunk | Generalized, later reticular ichthyosiform pattern |
| Scaling type | Adherent, moderate | Adherent, fine to moderate | Thick spiky hyperkeratosis | Fine |
| Scaling colour | White-brown | Brown (‘moulting’) | Yellow-brown hyperkeratoses | Yellow-brown |
| Erythema | Frequent | Initially, fades | Erythroderma possible | Pronounced |
| Palmoplantar involvement | KRT1: epidermolytic PPK KRT10: palms and soles are spared (exceptions possible) |
Usually no | Massive PPK leading to deep, bleeding and painful fissures, flexural contractures, constriction bands | Yes |
| Hypohidrosis | Possible | Possible | None | – |
| Scalp abnormalities | Scaling | – | None | Scaling |
| Other skin findings | Pruritus. Blisters after minor trauma, proneness to skin infections/impetigo | Pruritus, bullae may occur after minor mechanical trauma (often in summer) | – | – |
| Extracutaneous involvement | Growth failure with some severe phenotypes | Gangrene and loss of digits | Growth failure with some severe phenotypes | |
| Risk of death | Elevated during neonatal period | – | – | Elevated during neonatal period |
| Skin ultrastructure | EHK, aggregations and clumping of keratin filaments in suprabasal cells; partly cytolysis, lamellar body accumulation | Superficial EHK, cytolysis in granular cells of affected body areas; no keratin clumping | Binuclear cells, particular concentric perinuclear ‘shells’ of aberrant – putatively – keratin material | Vacuolization of superficial granular cells and filamentous material in vacuolated cells |
Modified from Oji V, Tadini G, Akiyama M, et al. Revised nomenclature and classification of inherited ichthyoses: results of the First Ichthyosis Consensus Conference in Sorèze 2009. J Am Acad Dermatol 2010;63:607–41 (ref [4] under Ichthyoses).
1Also known as ‘ichthyosis variegata’ and ‘ichthyosis en confetti’.
AD, autosomal dominant; AR, autosomal recessive; EHK, epidermolytic hyperkeratosis; PPK, palmoplantar keratoderma SC, stratum corneum; SG, stratum granulosum
Table 65.4 Neuro-ichthyotic syndromes.
| Gaucher syndrome type 2 | MEDNIK | CEDNIK | ARC | |
| MIM | 230900 | 609313 | 609528 | 208085 |
| Mode of inheritance | AR | AR | AR | AR |
| Gene | GBA | AP1S1 | SNAP29 | VPS33B |
| Onset | At birth, or later | At birth or within first weeks of life | After 5–11 months | At birth, can sometimes be late |
| Initial clinical presentation | CIE or less frequently mild collodion membrane | Erythematous rashes, similar to EKV | Until up to 1 year of age, normal skin; thereafter LI type | Xerosis and scaling within a few days of birth |
| Disease course | Ranging from mild to moderate | Progressive | Fatal | Fatal |
| Distribution of scaling | Generalized | Generalized | Generalized with sparing of skin folds | Generalized with sparing of skin folds |
| Scaling type | Fine or moderate; scaling may resolve after neonatal period | EKV-like | Coarse and large (plate-like) | Fine or plate-like (extensor sites) |
| Scaling colour | White or grey or brown | EKV-like | Whitish | White or brownish |
| Erythema | Unusual | EKV-like | Absent | Absent |
| Palmoplantar involvement | – | Not specifically | Yes | Spared |
| Scalp abnormalities | – | Not specifically | Fine sparse hair | Mild scarring alopecia |
| Other skin findings | – | Nail thickening, mucous membrane affected | None | Ectropion |
| Extracutaneous involvement | Hydrops fetalis; progressive neurological deterioration; hepatosplenomegaly, hypotonia, respiratory distress, arthrogryposis, facial anomalies | Congenital sensorineural deafness, peripheral neuropathy, psychomotor and growth retardation, chronic diarrhoea, mental retardation | Sensorineural deafness; cerebral dysgenesis; neuropathy; microcephaly; neurogenic muscle atrophy; optic nerve atrophy; cachexia | Arthrogryposis (wrist, knee or hip); intrahepatic bile duct hypoplasia with cholestasis; renal tubular degeneration; metabolic acidosis; abnormal platelet function; cerebral malformation |
| Risk of death | Death often by 2 years of age | Life-threatening congenital diarrhoea | Lethal within the first decade | Lethal within first year of life |
| Skin ultrastructure | Lamellar/non-lamellar phase separations in SC | Histology: hyperkeratosis with hypergranulosis | Impaired lipid loading onto LB and defective LB secretion | Defective LB secretion |
| Special analyses | Liver function tests; decreased β-glucocerebrosidase activity (leukocytes); Gaucher cells (bone marrow); increased acid phosphatase (serum) | Elevation of VLCFAs (blood), treatable by zinc acetate therapy | Absent SNAP29 protein on immunohistochemistry, magnetic resonance imaging | Liver and renal biopsy |
Modified from Oji V, Tadini G, Akiyama M, et al. Revised nomenclature and classification of inherited ichthyoses: results of the First Ichthyosis Consensus Conference in Sorèze 2009. J Am Acad Dermatol 2010;63:607–41 (ref [4] under Ichthyoses).
AR, autosomal recessive; ARC, arthrogryposis–renal dysfunction–cholestasis; CEDNIK, cerebral dysgenesis–neuropathy–ichthyosis–palmoplantar keratoderma; CIE, congenital ichthyosiform erythroderma; EKV, erythrokeratodermia variabilis; IV, ichthyosis vulgairs; LB, lamellar body; MEDNIK, mental retardation–enteropathy–deafness–neuropathy–ichthyosis–keratodermia (∼erythrokeratodermia variabilis 3, Kamouraska type); SC, stratum corneum; VLCFAs, very long chain fatty acids.
Table 65.5 Disorders associated with mutations in GJB2 (connexin 26).
| Syndrome | MIM | Inheritance | Mutation in GJB2 |
| Keratitis–ichthyosis–deafness (KID) | 148210 | AD | G12R, N14Y, S17F, G45E, D50N, D50Y |
| Hystrix-like ichthyosis–deafness (HID) | 602540 | AD | D50N |
| Bart–Pumphrey syndrome | 149200 | AD | N54H, N54K, G59S |
| Palmoplantar keratoderma–deafness | 148350 | AD | G59R, G59A, R75W, R75Q, E42, G130V |
| Vohwinkel syndrome | 124500 | AD (AR) | Y65H, D66H, G130V |
| Mucositis–deafness | AD | F142L | |
| Hypotrichosis–deafness | AD | N14K | |
| Vohwinkel-like papular palmoplantar keratoderma–deafness | AD | H73R | |
| Deafness | 220290 | AR (AD) | M34T, 35delG, 167delT, 235delC… |
| 220290 | AD | R75W, R75Q… |
Modified from de Zwart-Storm EA, Hamm H, Stoevesandt J, et al. A novel missense mutation in GJB2 disturbs gap junction protein transport and causes focal palmoplantar keratoderma with deafness. J Med Genet 2008;45:161–6 (ref [4] under Keratitis–ichthyosis–deafness).
AD, autosomal dominant; AR, autosomal recessive
Table 65.6 Selected syndromic ichthyoses with hair abnormalities and gastrointestinal or respiratory symptoms.
| Ichthyosis with hypotrichosisa | Neonatal ichthyosis–sclerosing cholangitis (NISCH)b | Ichthyosis–prematurity syndrome (IPS) | |
| MIM | 602400 | 607626 | 608649 |
| Mode of inheritance | AR | AR | AR |
| Gene | ST14 | CLDN1 | FATP4 |
| Onset | At birth | At birth (or shortly after) | At birth (polyhydramnion, prematurity, >6 weeks) |
| Initial clinical presentation | Lamellar ichthyosis, severe hypotrichosis, absent eyebrows and eyelashes | Mild scaling, neonatal jaundice with hepatomegaly; frontal alopecia in early childhood | Respiratory distress, generalized skin hyperkeratosis with focal accentuation on scalp, eyebrows |
| Disease course | Over time, scalp hair growth and appearance/colour may improve | Mild ichthyosis, liver involvement variable | Severe at birth, spontaneous improvement |
| Distribution of scaling | Generalized, including the scalp; face may be unaffected | Predominant on trunk | Focal accentuation (see above) |
| Scaling type | Coarse, plate-like, adherent | Fine to polygonal, thin | Caseous (vernix caseosa-like) |
| Scaling colour | Brown to dark | Normal | Whitish |
| Erythema | Unusual | Unusual | Mild to moderate |
| Palmoplantar involvement | No | No | Yes, initially |
| Hypohidrosis | Yes | No | No |
| Scalp abnormalities | Hypotrichosis in youth; sparse, unruly hair in adolescence; recessing frontal hair line in adults | Major criterion: coarse thick hair, frontotemporal scarring alopecia; hypotrichosis, curly/woolly hair | Extensive at birth |
| Other skin findings | Follicular atrophoderma | – | Follicular keratosis (‘toad skin’), atopic eczema, asthma, eosinophilia |
| Extracutaneous involvement | Sparse and curly eyebrows; occasionally photophobia and pingueculum | Major criterion: sclerosing cholangitis or congenital paucity of bile ducts | Pulmonary involvement and asphyxia at birth; later on atopic asthma, eosinophilia and occasionally hyper IgE |
| Risk of death | Normal | Not observed, but theoretically possible from liver involvement | Perinatally potentially fatal due to respiratory asphyxia; otherwise normal |
| Skin ultrastructure | High presence of intact corneodesmosomes in the upper SC, residues of membranous structures in the SC | Splitting of desmosomal anchoring plaques in the SG | Deposits of trilamellar membranous curved lamellae in swollen corneocytes and perinuclearly in oedematous granular cells |
| Special analyses | Hair microscopy may reveal dysplastic hair, pili torti or pili bifurcate | Liver function tests, cholangiography, liver biopsy | Blood cell count (eosinophilia) |
Modified from Oji V, Tadini G, Akiyama M, et al. Revised nomenclature and classification of inherited ichthyoses: results of the First Ichthyosis Consensus Conference in Sorèze 2009. J Am Acad Dermatol 2010;63:607–41 (ref [4] under Ichthyoses).
1Allelic variant: congenital ichthyosis–follicular atrophoderma–hypotrichosis–hypohidrosis (IFAH).
2Also known as ichthyosis–leukocyte vacuoles–alopecia–sclerosing cholangitis (ILVASC).
AR, autosomal recessive; SC, stratum corneum, SG, stratum granulosum.
Table 65.7 Keratolytic agents and moisturizers for topical treatment of ichthyoses.
| Agent | Concentration (%) | Comment |
| Urea | 5–10 | Classical humectant and keratolytic. Better avoided during first year of life because of possible systemic absorption |
| Lactic acid | 5–12 | Alternative to urea. Commercial preparations are often optimized by buffering |
| Sodium chloride | 3–10 | In ointments often adverse effects irritation/stinging, possible as bath additive |
| Dexpanthenol | 5–10 | Supporting normal epidermal differentiation |
| Macrogol 400 | 20–30 | Moisturizer and keratolytic |
| Propylene glycol | 15–20 | Moisturizer and keratolytic |
| Glycerol | 10–15 | Moisturizer |
| Vitamin E acetate | 5 | Moisturizer |
| Tretinoin | 0.025–0.05 | Frequent stinging/risk of absorption and teratogenicity in women of childbearing age |
| Calcipotriol | 0.05 × 10−3 | High risk of systemic absorption, treat less than 10% of body surface |
Warning: Salicylic acid might cause life-threatening poisoning in neonates and long-term toxicity in older patients.
Table 65.8 Resources and further information.
| Patient organizations for ichthyosis | |
Austria Belgium Denmark EU Finland France Germany Italy Japan Spain Sweden Switzerland UK US |
|
| Other databases and internet links | |
Web site hosted at NCBI Portal for rare diseases and orphan drugs Human intermediated filament database German guidelines for ichthyoses |
www.awmf.org/leitlinien/aktuelle-leitlinien/ll-liste/deutsche-dermatologische-gesellschaft-ddg.html |
All last accessed April 2015.
Definition
The term ‘ichthyosis’ was first introduced more than 200 years ago by Robert Willan in his textbook on cutaneous diseases [1]. The word ‘ichthyosis’ is derived from the Greek word ‘ichthys’, which means fish. It was coined at a time when the characteristics of human diseases were compared to those occurring in the animal kingdom. The literal translation ‘scaly fish disease’ is embarrassing and should be avoided. As a technical term, it is deeply entrenched in the medical literature and today refers to those MeDOCs that share a conspicuous scaling which is generalized and affects the whole integument [2, 3].
An important advance of the recent consensus conference on ichthyosis [4] was that we now differentiate between syndromic (Table 65.1) and non-syndromic (Table 65.2) forms of ichthyoses – a distinction already proposed by other authors [2, 5, 6]. In contrast, the onset of the disease, namely the distinction between ‘congenital onset’ and ‘non-congenital onset’, is no longer used as a major criterion, since some diseases such as recessive X-linked ichthyosis (RXLI) manifest at birth in some patients and after several months in others. Instead, it is suggested to distinguish between common and rare forms of the disease. Common forms include ichthyosis vulgaris (IV) and RXLI. However, one should keep in mind that RXLI has a prevalence of 1 : 2000–3000 in European populations and would be considered a rare disease according to the criteria of the European Union [7].
Specific pathophysiological aspects of all types of icthyoses are discussed with the respective entities. Generally, ichthyosis results in abnormal differentiation and/or abnormal desquamation showing for example impaired corneocyte shedding (retention hyperkeratosis) or accelerated keratinocyte production (epidermal hyperplasia/hyperproliferative hyperkeratosis). The development of hyperkeratosis in these diseases may be understood as a homeostatic repair response aimed at compensating for an abnormal epidermal barrier [3, 4].
COMMON ICHTHYOSES
Ichthyosis vulgaris
Definition and nomenclature
Ichthyosis vulgaris is a mild scaling disorder with a prevalence in Europe of 1 : 100 according to data from a population study in northern England [1].
Pathophysiology
Ichthyosis vulgaris is due to filaggrin mutations (FLG), which are inherited as an autosomal semidominant trait [2, 3]. Only a minority of those individuals who harbour only one FLG mutation actually develop clinically obvious ichthyosis, although they do exhibit accentuated palmar and plantar creases and may have somewhat dry skin. About two thirds of IV patients actually have two FLG mutations and present with a clear-cut clinical phenotype. In the third of the patients who have only one FLG mutation, disease expression is much milder [4]. This clinical difference relating to the mutation load is even reflected by functional studies concerning for example skin hydration or transepidermal water loss [5, 6]. Filaggrin mutations result in impaired epidermal barrier formation and a marked reduction of natural moisturizing factors (NMF) which play a critical role in hydration of the stratum corneum. Irrespective of the presence of IV, FLG mutations predispose to atopic eczema (AE), allergic rhinitis, asthma, food allergies, hand eczema, nickel sensitization and eczema herpeticatum in AE [1, 7, 8].
Clinical features
Ichthyosis is not present at birth. Usually it develops during the first months of life. Wells and Kerr [9] found demonstrable scaling in 40% of their patients at the age of 3 months. Even careful parents may have difficulties in reporting the exact time of disease onset and it should be noted that scaling may disappear or be reduced markedly in the summer time due to seasonal variation and increased humidity. IV patients present with light grey scales covering mainly the extensor surfaces of the extremities and the trunk (Figure 65.1a). The scales tend to be smaller than in RXLI, and the groin and larger flexures are always spared. Almost all IV patients exhibit accentuated palmar creases (Figure 65.1b), and this clinical feature is not influenced by factors such as season or humidity. A considerable number of patients indicate that they suffer from hypohidrosis and cannot perspire well [4]. Other frequent features of the disease are keratosis follicularis and mostly mild concomitant AE, and allergic rhinitis [1, 4, 7].
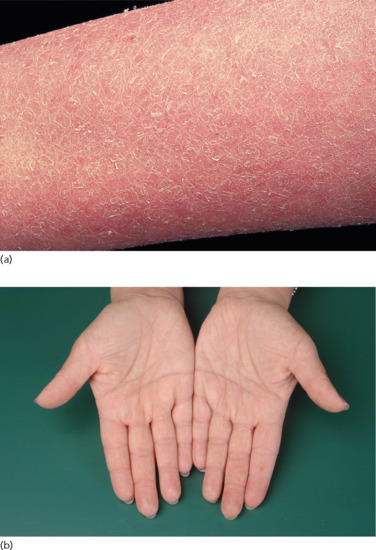
Figure 65.1 Ichthyosis vulgaris. (a) Fine scaling and (b) accentuated palmar creases. Courtesy of (a) Dr M. Judge, Salford Royal NHS Trust, UK; (b) the Department of Dermatology, University Hospital Münster, Münster, Germany.
Investigations
Histology reveals orthohyperkeratosis with a diminished or absent granular layer. Immunohistochemical studies show an absent or markedly reduced filaggrin signal. Ultrastructure reveals scarce and crumbly keratohyalin granules. The ultrastructural defects correlate very well with the number of FLG mutations in its severity [4, 6].
Management
Ichthyosis vulgaris patients benefit from ointments that hydrate the stratum corneum [10], and excellent experience, e.g. with creams containing glycerol [11], has been reported. In those patients without concomitant AE, urea containing creams (up to 10%) or creams containing lactic acid up to 12% also work well. In contrast to autosomal recessive congenital ichthyosis (ARCI), excessive bathing procedures are not necessary, but showering and subsequent application of ointments is advisable. In the future, topical protein substitution therapy may become a promising approach [12].
Recessive X-linked ichthyosis
Definition and nomenclature
Recessive X-linked ichthyosis is a rather mild scaling disorder. Based on systematic screening of pregnancies for steroid sulphatase deficiency, a prevalence in males of 1 : 1500 in Caucasian and Asian population has been determined [1].
Pathophysiology
Recessive X-linked ichthyosis is caused by mutations in the STS gene encoding steroid sulphatase [2]. Around 90% of cases are caused by deletions which in 75% span the whole gene sequence [3]. These deletions can extend to adjacent genes which may occasionally result in more complex phenotypes such as RXLI occurring together with Kallmann syndrome, with the recessive form of X-linked chondrodysplasia punctata or with brain abnormalities including mental retardation, unilateral polymicrogyria and retinitis pigmentosa [4]. Disease severity may be enhanced by a concomitant filaggrin mutation [5, 6] as well as by a further still unidentified modifier. As a result of enzyme deficiency, cholesterol sulphate accumulates in the epidermis. High concentrations of cholesterol sulphate inhibit proteases such as kallikrein 5 and kallikrein 7 that are pivotal for normal degradation of corneodesmosomes. Indeed, in RXLI skin, serine protease activity was found to be markedly reduced [7]. This in turn leads to decreased desquamation, and as a consequence hyperkeratosis. RXLI can thus be considered as a prototypic example of a retention hyperkeratosis. Measurements of transepidermal water loss have revealed a clear-cut increase of transepidermal water loss that is even more pronounced than for example in IV patients, while skin surface pH was not significantly altered [8].
Clinical features
The disease affects almost exclusively boys although female patients have been documented [9]. The mothers of affected children frequently report birth complications relating to the presence of the enzyme defect in the placenta. Insufficient cervical dilatation is often found in pregnant women with placental sulphatase deficiency and may cause prolonged delivery necessitating caesarean section or forceps delivery. Directly after birth, most patients present with a very fine scaling or peeling of the skin that often goes unnoticed and soon resolves. At the age of 2–6 months, usually large thick dark brown to yellow-brown hyperkeratoses develop covering the trunk, the extremities and the neck (Figure 65.2). The antecubital folds and the popliteal folds are usually spared as in IV. The palms of the hands and the soles remain unaffected. In around 30% of patients, the colour of the scale is light grey (Figure 65.2d). These patients are often misdiagnosed as having IV. Dark hyperkeratosis giving the lateral aspects of the trunk and the back of the neck a ‘dirty look’ is a further feature which is typical of RXLI, and is usually not present in IV.
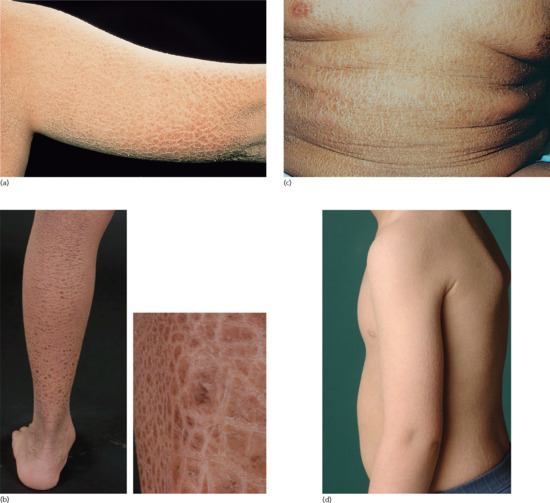
Figure 65.2 Recessive X - linked ichthyosis. (a) Scaling on the arm, (b) on the legs, (c) on the trunk and (d) patient with light grey scaling. Courtesy of (a)-(c) Dr M. Judge, Salford Royal NHS Trust, UK; (d) the Department of Dermatology, University Hospital Münster, Münster, Germany.
Deep stromal corneal opacity is a frequent finding if patients undergo an expert ophthalmological examination, but it usually does not affect visual acuity. RXLI may be associated with cryptorchidism in up to 20% [10], with attention-deficit hyperactivity syndrome (ADHD) in up to 40% or with autism in around 25% [11]. It is of note that steroid sulphatase deficient mice carrying a deletion of the STS gene exhibit behavioural abnormalities relevant to ADHD such as inattention and hyperactivity. Moreover, these mice display altered serotonergic function that may account for their abnormal behaviour [12].
Investigations
If multiple family members are affected, careful scrutiny of the pedigree tree is instrumental in establishing the mode of inheritance of the disease. The existence of an affected grandfather or of affected uncles on the maternal side is suggestive of the diagnosis of RXLI in a patient with ichthyosis.
Histology shows orthohyperkeratosis and a well-maintained, often thickened stratum granulosum. Marked follicular plugging can be noted in around 30% of patients although clinically there is no obvious keratosis follicularis. Ultrastructurally, a marked increase of persistent corneodesmosomes typical for retention hyperkeratosis can be seen.
Analysis making use of fluorescent in situ hybridization (FISH) or advanced technologies such as comparative genomic hybridization/comparative microarray analysis (CMA) allows rapid diagnosis in those cases who have large deletions [13], but will miss around 10% of cases. Standard sequencing techniques are used to identify point mutations. Likewise it is possible to measure steroid sulphatase activity biochemically, e.g. in fibroblasts or in plasma. Also lipoprotein electrophoresis is a simple, but useful tool revealing increased mobility of β-lipoproteins. Analysis of plasma levels for cholesterol sulphate by quantitative high-performance liquid chromatography (HPLC)/mass spectrometry is a very elegant method, but unfortunately currently available only for research purposes.
Management
Recessive X-linked ichthyosis patients benefit from the same therapeutic strategy that is applied for IV, namely the use of moisturizers. Again, excellent results can be achieved with a glycerol containing cream. In the summertime, often spontaneous marked improvement can be observed, while in winter the skin condition becomes worse. It is of note that treatment with moisturizers does not normalize the transepidermal water loss but rather tends to further increase it, whereas skin dryness does improve [8]. Systemic retinoids may be given at low dosage during periods of disease exacerbations, e.g. during the winter season [14].
NON-SYNDROMIC CONGENITAL ICHTHYOSES
(see Table 65.1)
Autosomal recessive congenital ichthyosis
Definition and nomenclature
Unlike for example IV or RXLI, the term ARCI does not denote a single MeDOC, but rather is an umbrella term that includes all non-syndromic autosomal recessive congenital forms of ichthyosis without a tendency toward blistering [1]. Thus the spectrum includes harlequin ichthyosis (HI), bathing suit ichthyosis (BSI), lamellar ichthyosis (LI), congenital ichthyosiform erythroderma (CIE), self-improving congenital ichthyosis (SICI), as well as transient manifestations, such as collodion baby [2, 3]. These diseases and their diverse genetic defects will be discussed separately.
Prevalence
Registry-based data from Spain [4] and Germany [5] show that the prevalence of ARCI in Europe is in the range of 1.6 : 100 000.
Pathophysiology
Autosomal recessive congenital ichthyosis is associated with mutations in a plethora of genes (see Table 65.1), which encode proteins involved in lipid transport, such as ABCA12 [6], in lipid biosynthesis such as CERS3 [7], in fatty acid metabolism or have a role in assembling suprastructures such as the cornified envelope. A ‘unified field theory’ explaining how these various proteins interact with each other and result in a barrier defect and in hyperkeratosis is lacking. Some of the molecules involved in ARCI have yet to be attributed a precise role. For example, it is still unclear whether ichthyin encoded by nipal4 acts as a magnesium transporter [8] or has a role in the hepoxilin pathway.
Genotype–phenotype correlations have been reported for some of these disorders. This is best illustrated by the ABCA12 gene. Missense mutations in this gene cause either LI [9] or CIE [10] whereas nonsense or frameshift mutations result in life-threatening HI [6]. The combination of the two types of mutations result in an intermediate phenotype [11]. Similarly, BSI has been associated with distinct temperature-sensitive mutations in TGM1. The numerous remaining ARCI types resolve into clinical phenotypes like LI, CIE or SICI, all of which can be caused by mutations in a number of different genes with no clear genotype/phenotype relationships. Around 10–20% of ARCI cases cannot be attributed to known genes [12].
Clinical features
See the sections ‘HI’, ‘collodion baby and SICI’, ‘BSI’ and ‘LI and CIE’.
Mangement
See the section ‘Management of congenital ichthyoses’.
Harlequin ichthyosis
Definition
Harlequin ichthyosis is the most devastating type of ARCI. It is still often lethal in around 44% of cases [1]. Based on preliminary data from the Network for Ichthyoses and Related Keratinization Disorders (NIRK) registry in Germany, the prevalence of HI is estimated to be in the region of 1 : 2 million. It appears to be roughly 10 times lower than transglutaminase-1 (TG1) deficient ARCI.
Pathophysiology
It has already been pointed out above that peculiar nonsense and/or frameshift mutations in the ABCA12 gene cause HI [2, 3]. This type of mutations usually results in mRNA decay and loss of expression of the protein [4, 5].
ABCA12 transfer lipids such as glucosylceramides, which are essential for epidermal barrier formation, into lamellar bodies. It plays an essential role in the formation of lamellar bodies that also transport proteases such as kallikrein 5, 7, and 14 and secrete these proteins into the intercellular space in the stratum corneum [6]. These proteases play an important role in desquamation by degrading corneodesmosomes [7], thus leading to retention hyperkeratosis [8].
Clinical features
Neonates are born with armour-like skin (truncal plates with fissuring) (Figure 65.3) which can considerably impair movement and the ability to drink and breath. Bilateral ectropion and eclabium are present and hyperkeratotic skin may result in ears lacking retroaural folds. Around 10% of children develop autoamputation of digits [1]. A major problem in early infancy is a proneness to infection of the skin, as well as other organs such as the lungs. Respiratory problems are the major cause of death in neonates. Data from an animal model seem to implicate that HI is not only a skin disease, but can also affect lung function causing alveolar collapse [9]. In those children who survive the critical initial phase of the disease, the thickening of the stratum corneum improves somewhat and large lamellar scales, accompanied by marked ichthyosiform erythroderma develop (Figure 65.3b,c). In later life persistent ectropion is a frequent major problem, and often these patients have problems achieving and maintaining normal body weight despite high-calorie supplementation. Vitamin D-deficiency causing rickets and osteomalacia can occur [1].

Figure 65.3 Harlequin ichthyosis. (a) Neonate, (b) aged 6 weeks on retinoid therapy and (c) 6 months. Courtesy of Dr M. Judge, Salford Royal NHS Trust, UK.
Investigations
Harlequin ichthyosis has a striking histology showing enormous thickness of the stratum corneum. There is parakeratosis and hypergranulosis and non-polar lipids are reduced, while expression of proteases like kallekrein 5 and cathepsin D are dramatically reduced [10]. Electron microscopy reveals numerous abnormal lamellar bodies in the stratum granulosum and accumulation of extruded irregular lamellar bodies as vesicular structures between the epidermal cornified cells. This defect of lamellar bodies is highly pathognomic for HI allowing its diagnosis (Figure 65.4a,b).
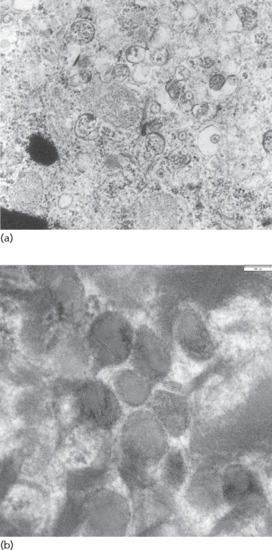
Figure 65.4 Ultrastructural diagnosis of harlequin ichthyosis. (a) Abnormal lamellar bodies in ABCA12 deficiency. (b) Morphology of normal lamellar bodies in granular layer cells with their diverse cargos: mostly lamellated, but also homogeneous areas (higher magnification).
(Courtesy of Dr I. Hausser, Heidelberg, Germany.)
Management
Management requires an interdisciplinary approach and will be discussed in the section on management of collodion baby in detail later; see also the review by Rajpopat and co-workers [1].
Collodion baby and self-improving congenital ichthyosis
Definition
The term collodion baby describes a transient condition in newborns. Except for HI, most ARCI patients present at birth as ‘collodion babies’ [1], but it should also be noted that several syndromic types of congenital ichthyosis such as trichothiodystrophy (TTD) or Gaucher syndrome type 2 typically present as collodion baby.
Pathophysiology
See sections on LI and CIE or BSI later.
Clinical features
The neonate is encased in a shiny parchment-like membrane, which cracks within a few days after birth (Figure 65.5) and usually peels off within the first 4 weeks of life. Initially, the clinical presentation can be quite severe and often includes ectropion and everted lips of different degrees. Afterwards for a brief time healthy skin becomes visible. Collodion babies look very much alike at birth, but later take different clinical courses. In around 80% of cases, collodion baby is then followed by the onset of an ARCI subtype. The clinical presentations may evolve into BSI or into the phenotypes of LI, e.g. in severe TG1 deficiency, or CIE, e.g. in lipoxygenase deficiency. However, around 10–20% of cases develop into SICI [2] or self-healing collodion baby (SHCB) [3].
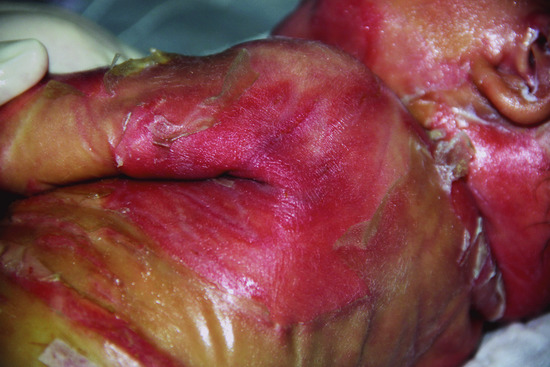
Figure 65.5 Autosomal recessive congenital ichthyosis. Sheddding of collodion membranes after 1 week.
Differential diagnosis
Ichthyosis prematurity syndrome (IPS) (see later) is an important differential diagnosis of SICI and SHCB.
Management
See the section ‘Management of congenital ichthyoses’.
Bathing suit ichthyosis
Definition
Bathing suit ichthyosis is a peculiar type of ARCI first recognized in South African Bantu of the Nguni ethnic group [1]. While children are born as collodion babies, they later develop a lamellar type of ichthyosis that spares the face and the extremities, and follows the distribution pattern of bathing suits.
Pathophysiology
Bathing suit ichthyosis was found to be due to peculiar missense mutations in TGM1 that render the enzyme TG1 temperature sensitive [2, 3]. Recombinant expression of the TGM1 mutations in BSI showed that they exhibit a marked shift in temperature optimum from 37°C to 31°C [4]. Deficient activity of BSI mutants could be rescued and even reconstituted by decreasing the temperature to below 33°C. All BSI mutations showed an activity above 10% at their temperature optimum at 31°C and a dramatic decrease at 37°C [4]. It is of note that the vast majority of BSI-causing mutations affect arginine residues, e.g. p.Arg315His, and often affects exons 5, 6 or 7 [5, 6]. A few of these patients heal eventually completely and could also be regarded as examples of SICI [5, 7].
Clinical features
The most striking aspect is the dynamic of the phenotype. Children are born as collodion babies involving the entire skin. Shedding of the collodion membrane is followed by the development of large dark grey/brownish scales affecting the trunk and the scalp, but sparing the face and extremities (Figure 65.6). Palms and soles are dry and diffusely mildly hyperkeratotic. Digital thermography validated a striking correlation between warmer body areas and the presence of scaling in patients [2]. The disease tends to become worse in the summer months and to improve in winter [8]. Hypohydrosis as is often seen in ARCI may play a crucial role in local heat accumulation that results in additional reduction of TG1 activity [9]. In our experience, hyperkeratoses can develop in the ear canal affecting the ability to hear.
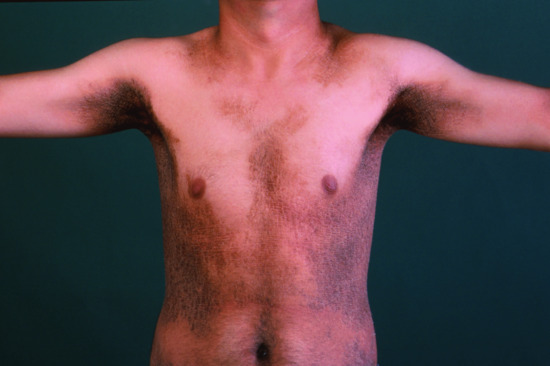
Figure 65.6 Bathing suit ichthyosis. Lamellar scaling on areas with high skin temperature.
(Courtesy of the Department of Dermatology, University Hospital Münster, Münster, Germany.)
Investigations
In situ assessment of TG1 activity reveals a deficiency only in affected skin, but sufficient activity in unaffected healthy appearing skin [2]. Likewise ultrastructural analysis reveals a massively thickened stratum corneum displaying multiple cholesterol clefts which are typical for TG1 deficiency, while stratum corneum is of normal thickness in healthy skin and shows no cholesterol clefts [2].
Management
Management corresponds to that of LI, but special attention should be given to the ears and to removing keratotic material from the ear canal.
Lamellar ichthyosis and congenital ichthyosiform erythroderma
Definition
When the term ‘lamellar ichthyosis’ was coined by the American dermatologist Frost [1] in the 1960s, it was used to denote a type of ARCI that is characterized by large plate-like dark-brown hyperkeratoses covering the entire body, but usually presenting rather mild palmoplantar involvement. At the other end of the clinical spectrum, ARCI patients may present with very marked erythroderma, and mostly fine often whitish or grey scales. These patients often have pronounced palmoplantar keratosis and have been referred to as having CIE (Figure 65.7).
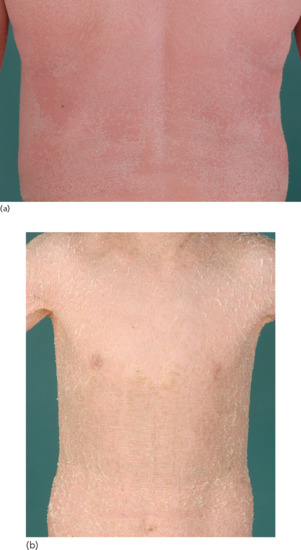
Figure 65.7 Autosomal recessive congenital ichthyosis. (a) Congenital ichthyosiform erythroderma. (b) Lamellar ichthyosis.
(Courtesy of the Department of Dermatology, University Hospital Münster, Münster, Germany.)
Prevalence
Deficiency of TG1 as the most frequent cause of ARCI is responsible for 32% of ARCI cases in Germany, while in the USA it has been found in up to 55% of the cases studied [2]. In Europe, mutations in ALOX12B account for 12% of ARCI, and ALOXE3 mutations are responsible for further 5% of cases. NIPAL4 mutations account for 16% of ARCI and are thus a frequent cause. Around 8% of ARCI cases are due to mutations in the CYP4F22 gene (previously known as FLJ39501) [3, 4].
Pathophysiology
Cell kinetic studies in the 1980s seemed to discriminate between the phenotypes of LI and CIE [5], which at that time were considered to represent distinct entities. However, genetic studies disclosed that the LI phenotype as well as the CIE phenotype is by no means specific for a certain gene, but mutations in the same gene may be associated with both phenotypes [6]. The specific pathogenesis may concern a malfunction of the following proteins.
- Transglutaminase-1 critically contributes to the the assembly of the cornified envelope by catalysing calcium-dependent cross-linking of proteins, such as involucrin, loricrin and proline-rich proteins and by binding Ω-hydroxy ceramides to proteins such as involucrin, thus connecting the lipid envelope with the CE [7, 8].
- The epidermal lipoxygenases E3 and 12B act on adjacent steps in the hepoxilin pathway and are believed to play a role in the secretion of lamellar bodies so that mutations in the genes encoding these enzymes result in impaired secretion of lipids and formation of the intercellular lipids in the stratum corneum [9–11].
- The NIPAL4 gene encodes for the protein ichthyin. Patients having a NIPAL4 mutation show a markedly increased expression of epidermal lipoxigenases and TG1 in their skin indicating a common metabolic pathway essential for skin barrier homeostasis [12, 13]. The precise function of this protein is not entirely understood. It appears to localize to desmosomes and keratins [14] and to interact with fatty acid transporter protein 4 [15], which is defective in IPS.
- This gene CYP4F2 encodes a cytochrome P450 polypeptide that is a homologue of a leukotriene B4 Ω-hydroxylase. The actual function of this gene for the epidermal barrier has not been established, but it has been hypothesized that it participates in the hepoxilin pathway by catalysing the conversion of trioxilin A3 to 20 hydroxy-(R) trioxilin [16].
- Mutations in the CERS3 gene encoding ceramide synthase 3 are a rare cause of ARCI [17, 18]. Inactivating mutations in this gene are associated with a loss of very long acyl chains from C26 up to C34 in terminally differentiating keratinocytes of affected patients and thus impair epidermal barrier formation.
- LIPN encodes an acid lipase that is involved in triglyceride metabolism in mammals and is expressed exclusively in the epidermis. A 2-bp deletion in LIPN was found to be associated with a mild form of CIE showing diffuse ichthyosis on the legs [19].
- PNPLA1 belongs to the patatin-like phospholipase family and is related to PNPLA2, which causes neutral lipid storage disease with myopathy but without ichthyosis. PNPLA1 mutations were first identified as a cause of ichthyosis in golden retriever dogs and afterwards in humans, who feature fine white scales and moderate erythroderma and PPK and a pseudosyndactyly of the second and third toes [20].
Clinical features
A definite genotype/phenotype relationship for LI and CIE has not yet been achieved, but in our experience there are clinical clues in ARCI that tend to be indicative for certain genes. The majority of patients having TGM1 mutations present with classical LI (see definition) often having complaints such as ectropion or alopecia ichthyotica. There is no obvious erythroderma, but beneath the thick scales some erythema can be present. Ears are often deformed and small. As indicated above, specific temperature-sensitive mutations in TGM1 are associated with BSI. Moreover, there is a group of patients who initially present as collodion babies, progress to mild CIE and later may present with a very mild or even absent scaling. This phenotype is referred to as SICI (Figure 65.8) [6, 21]. TGM1 patients who carry premature termination codon (PTC) mutations (e.g. nonsense or frameshift mutations) are more likely to report sweating abnormalities, such as hypohidrosis and overheating than those who have missense mutations [2].
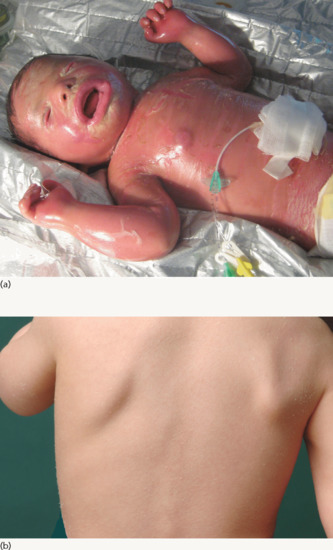
Figure 65.8 Self-improving congenital ichthyosis. (a) Collodion baby at birth. (b) Mild ichthyosis and minimal erythroderma at the age of 21 months.
(Courtesy of the Department of Dermatology, University Hospital Münster, Münster, Germany.)
Neonates with lipoxygenase mutations are often born with a mild type of collodion and in later life mostly present with the CIE phenotype, although some also present brownish scales. Typically, they show a striking palmoplantar hyperlinearity (Figure 65.9), which is reminiscent of the accentuated creases in IV [10]. However, mild keratotic lichenifications of the elbow fossa or of the dorsum of the hands help to rule out this differential diagnosis. Patients may progress to SICI [21]. Those with ALOX12B mutations more often exhibit pronounced palmoplantar keratosis than patients with ALOXE3 mutations [10]. Many of these patients report reduced or completely absent sweating ability [22], and many complain of pruritus.
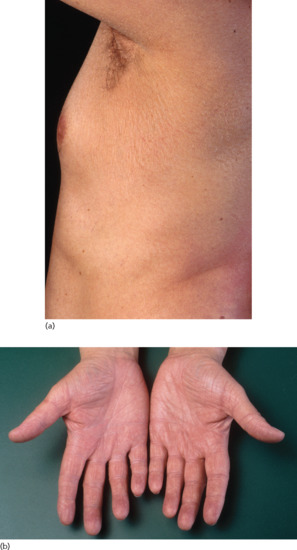
Figure 65.9 (a,b) Clinical phenotype of ALOXE3 mutations. Note palmoplantar hyperlinearity resembling accentuated creases in ichthyosis vulgaris (in b).
(Courtesy of the Department of Dermatology, University Hospital Münster, Münster, Germany.)
Patients with NIPAL4 mutation often present with a CIE/LI overlapping phenotype, ectropion, clubbing of the nails and a pronounced and diffuse yellowish keratoderma on the palms and soles [23]. This (Figure 65.10a) may be reminiscent of classical PPKs such as a focal non-epidermolytic type. Scaling may be reticulate on the trunk (Figure 65.10b).
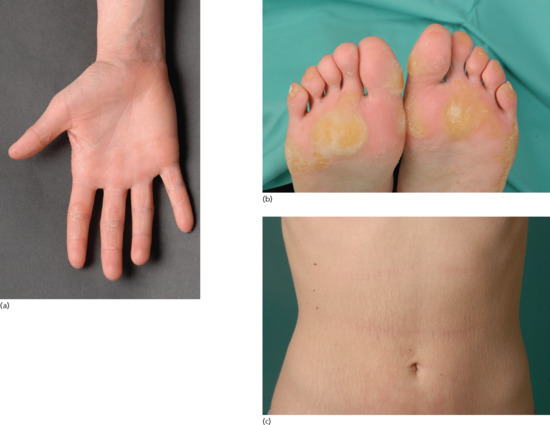
Figure 65.10 Clinical phenotype of NIPAL4 mutations. (a,b) Diffuse yellowish keratoderma on palms and soles. (c) Reticulate scaling on the trunk.
(Courtesy of the Department of Dermatology, University Hospital Münster, Münster, Germany.)
Most patients with CYP4F2 mutations present with a CIE or mild collodion baby phenotype at birth [24]. As the children grow older, they develop whitish-grey scales which are more pronounced in the periumbilical region [16]. Palms and soles show pronounced hyperlinearity or even PPK.
Patients with mutations in CERS3 are born as collodion babies and then progress to CIE often with improvement of the ichthyosis phenotype in the summer time. Also, patients have marked plantar hyperlinearity (Figure 65.11) and may experience pruritus and recurrent uncomplicated bacterial and Pityrosporum infections [17].
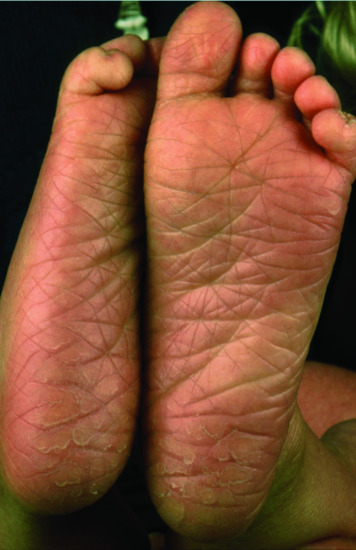
Figure 65.11 Clinical phenotype of CERS3 deficiency. Mild plantar keratoderma with hyperlinearity.
(Courtesy of the Department of Dermatology, University Hospital Münster, Münster, Germany.)
Rather remarkable LIPN mutations seem to cause a late-onset form of ichthyosis at the age of 5 years, so that it is not formally a congenital ichthyosis, although it belongs pathophysiologically to the ARCI spectrum [19].
Investigations
Diagnosis of TG1 deficiency can be made by sequencing [2, 3, 8] or by measuring in situ TG1 activity in cryostat sections [25]. Ultrastructural investigations reveal so-called cholesterol clefts in the stratum corneum (Figure 65.12) [26]. NIPAL4 deficiency may correlate with the ultrastructure of abnormal lamellar bodies and elongated membranes in the stratum granulosum classified as ARCI electron microscopy (EM) type III [27]. Direct sequencing is necessary for the diagnosis of lipoxygenase deficiency and other ARCI subtypes (CERS3, CYP4F2 or LIPN) that lack specific ultrastructural markers. Biochemical measurements of lipoxygenase activity is feasible but is currently available only in specialized research laboratories [28]. The same applies for ultrastructural methods with frozen sections or osmium tetroxide and ruthenium tetroxide postfixation that may enable an advanced electron microscopic diagnostics of all ARCI subtypes [29].
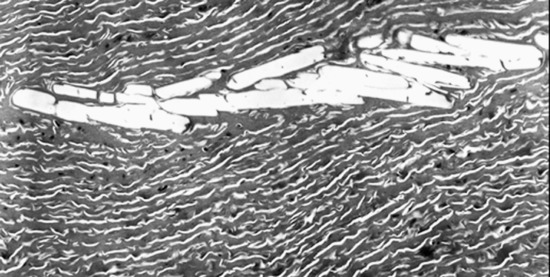
Figure 65.12 Ultrastructure of transglutaminase-1 deficient skin with typical cholesterol clefts in the stratum corneum.
(Courtesy of Dr I. Hausser, Heidelberg, Germany.)
Keratinopathic ichthyoses (see Tables 65.1 and 65.3)
Definition
Keratinopathic ichthyoses (KPI) are a group of very severe ultra rare cornification disorders having in Denmark a prevalence of 1 : 350 000 [1]. Patients often present at birth with erythroderma, scales and erosions. The term ‘keratinopathic’ was coined at the Sorèze Consensus Conference as an umbrella term for all types of ichthyoses which are caused by mutations in one of the keratin genes [2].
Pathophysiology
Epidermal keratins are intermediate filaments which contribute to the formation of the keratinocyte cell cytoskeleton. This cytoskeleton extends from the nucleus of the keratinocyte to the cell membrane where keratins attach either to desmosomes or hemidesmosomes [3]. Mutations in keratin genes like KRT1, KRT10, or KRT2 are usually associated with epidermolytic hyperkeratosis (EHK) on histology (Figure 65.13) and with the occurrence of cytoplasmic keratin aggregates (keratin clumps) or perinuclear shell formation, which can be seen not only with electron microscopy [4, 5].
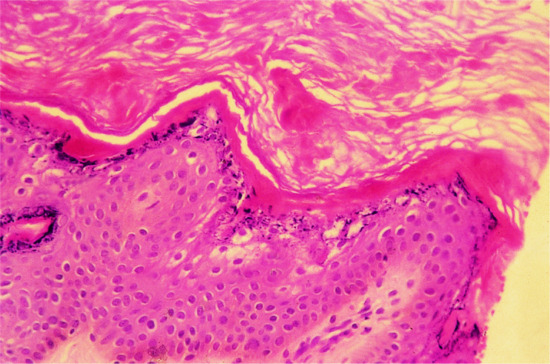
Figure 65.13 Histological diagnosis of epidermolytic hyperkeratosis.
These keratin aggregates can be induced by trauma or environmental conditions, e.g. high temperature, fever or skin infections, that are known modulators of disease severity. The keratin aggregates are reminiscent of those in classical protein folding disorders such as neurodegenerative diseases like Huntington disease [6]. Cells expressing mutant keratin aggregates have increased sensitivity to hyperosmotic stress which can be reduced for example by the chemical chaperone trimethylamine-N-oxide [7]. Likewise, it has been shown that retinoids reduce the formation of keratin aggregates in heat-stressed keratinocytes from an epidermolytic ichthyosis (EI) patient with a KRT10 mutation [8]. Keratin aggregates have been shown to interact with activated MAP kinases, molecular chaperones such as Hsp70 and components of the ubiquitin-proteasome system and may contribute to inflammatory changes seen in the disease [9]. Moreover, KRT1 knock-out mice release large amounts of the pro-inflammatory interleukin (IL) 18 and depletion of IL-18 partially rescued Krt1-/- mice [10], suggesting novel approaches to therapy.
Although KPI have been traditionally considered as autosomal dominant disorders [11], recessive and semidominant inheritance of KRT10 [12] and KRT1 [13] mutations have been reported, respectively. The vast majority of mutations in KPI consist of heterozygous single point mutations that are found in the highly conserved helix boundary motives of KRT1 and KRT10 that play a crucial role in filament formation [14–16]. Up to 75% of KPI-causing mutations are de novo mutations [17, 18].
Epidermolytic epidermal naevus results from somatic mutations in KRT1 or KRT10. Germ line mosaicism in these cases, which may be associated with a small epidermolytic naevus in a patient (Figure 65.14a), can result in full blown disease in his or her offspring [18, 19]. Occasionally, forms of EI can be seen in whom many Blaschko linear stripes of the skin are affected by widespread hyperkeratosis representing multiple epidermolytic naevi, resulting from extensive postzygotic mosaicism [20].
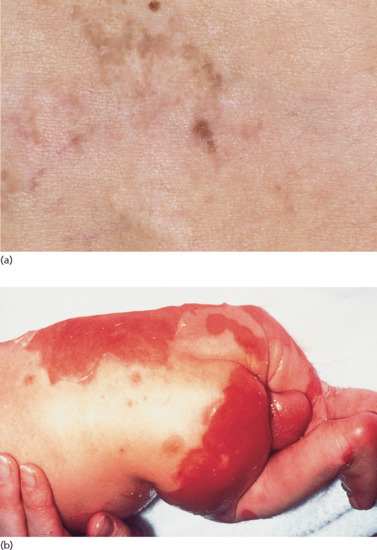
Figure 65.14 Special genetic aspect in keratinopathic ichthyosis. (a) Epidermolyticnaevus in a father of an affected boy with generalized epidermolytic ichthyosis due to keratin 10. (b) Neonatal presentation of epidermolytic ichthyosis (‘enfant brulé’). (b) Courtesy of Dr M. Judge, Salford Royal NHS Trust, UK.
Epidermolytic ichthyosis
Clinical features [1–6]
At birth, the presentation usually consists of CIE often associated with marked blistering. In the classic French literature, this presentation was referred to as ‘burned child/enfant brûlé’ (Figure 65.14b). In neonates, the differential diagnosis therefore often includes epidermolysis bullosa. In the first months of life, the blistering resolves and hyperkeratosis develops instead. However, fragility of the skin remains and when patients suffer from fever or skin infections or are exposed in the summer to high ambient temperature or mechanical friction, bouts of blistering can occur. The older child and adult patients usually present with marked keratotic lichenification meaning rippled keratotic ridges, in particular in the axilla, the elbows and the flexural aspects of the knees. It is striking that this severe involvement correlates with skin regions where the body temperature is somewhat elevated and thus this aggravation may be induced by differences in body temperature. On the knees and the lower legs, patients sometimes present with spiny hyperkeratosis (Figure 65.15a–c). In patients harbouring KRT10 mutations, the palms and soles are usually spared (Figure 65.15d) [1], and they tend to respond well to moderate dosages of systemic retinoids (see ‘Management of congenital ichthyoses’), while patients having KRT1 mutations usually have severe involvement of the palms and soles which can significantly impair walking so that some patients actually require a wheelchair. Moreover, the use of systemic retinoids may actually worsen their skin condition.
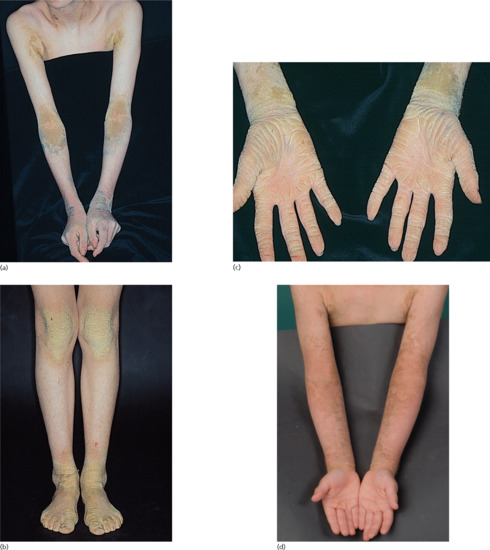
Figure 65.15 Epidermolytic ichthyosis without mosaicism.(a) Arms, (b) legs, (c) palms indicative of KRT1 and (d) palms indicative of KRT10 mutations. (a)-(c) Courtesy of Dr M. Judge, Salford Royal NHS Trust, UK.
Investigations [7–10, 11]
The above-mentioned types of KPI share a striking histology, namely EHK. In superficial EI, this finding is less marked and expressed mainly in the stratum granulosum and upper epidermis, and it may be important to take a biopsy from a site of maximal clinical involvement, e.g. from the knees. Similar considerations apply for annular EI. As mentioned above, the ultrastructure of these diseases is characterized by collapsed keratin aggregates (tonofilaments). These aggregates often form around the cell nucleus, have lost their connection to the desmosomes and therefore promote intraepidermal blistering. As already discussed, the presence of keratin aggregate links the pathology to that of protein folding diseases.
Superficial epidermolytic ichthyosis
Clinical features [1,2–7]
The clinical presentation of superficial epidermolytic ichthyosis (SEI) resembles that of EI. However, the course of the disease is milder and more localized, meaning that large parts of the body are clear. Typically, the keratosis is limited to the region around the navel and on the dorsal aspects of the hands and feet or the arm and the axillary region (Figure 65.16). A phenomenon that is quite typical is superficially denuded areas, e.g. on the back of the hand (Figure 65.17) [2]. For this phenomenon Siemens who first described the disease in 1937 coined the German term ‘Mauserung’ (moulting) [1].
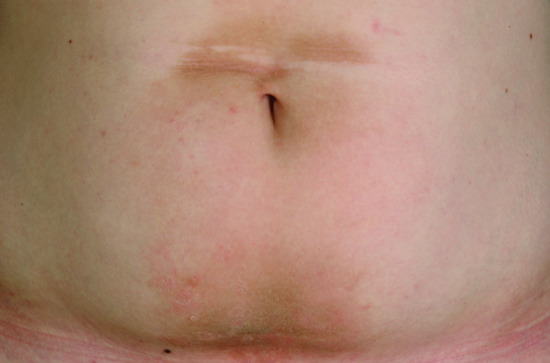
Figure 65.16 Superficial epidermolytic ichthyosis. Involvement around the navel.
(Courtesy of the Department of Dermatology, University Hospital Münster, Münster, Germany.)
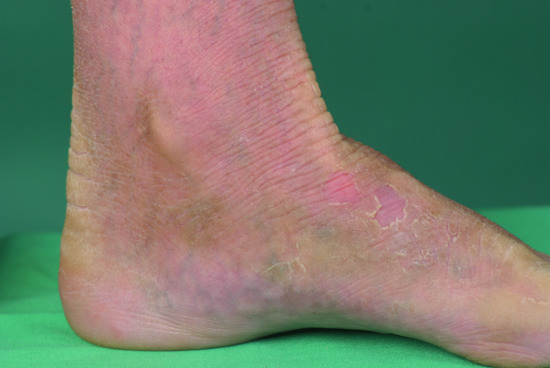
Figure 65.17 Moulting (‘Mauserung’) phenomenon in superficial epidermolytic ichthyosis.
(Courtesy of the Department of Dermatology, University Hospital Münster, Münster, Germany.)
Annular epidermolytic ichthyosis
Clinical features
Annular epidermolytic ichthyosis (AEI) is a rather mild variant of EI [1, 2], which shares a similar onset at birth, but later greatly improves and can feature bouts of disease activity associated with the development of numerous annular and polycyclic hyperkeratotic lesions especially on the trunk and extremities [1]. Outbreak of disease flares can be associated with high temperature in the summer, fever or pregnancy [3].
Congenital reticular ichthyosiform erythroderma
Pathophysiology
Congenital reticular ichthyosiform erythroderma (CRIE) is due to particular KRT10 mutations. Patients initially display generalized erythema and scaling with subsequent localized spontaneous healing which manifest with small pale white spots. The revertant phenotype is due to multiple recombination events in the KRT10 gene [1], which can be considered as a kind of natural gene therapy.
Clinical features
This disease was described independently in 1984 by a German group [2] and by a Swiss group [3], which coined the term ‘confetti ichthyosis’. It is characterized by very severe CIE and from the age of 3 years by the gradual onset of hundreds of pale normal-appearing confetti-like spots which can grow up to 2 cm in size (Figure 65.18). Many of these patients are severely ill and fail to thrive [4]. Often there is osteomalacia, for instance affecting the ankle joints.
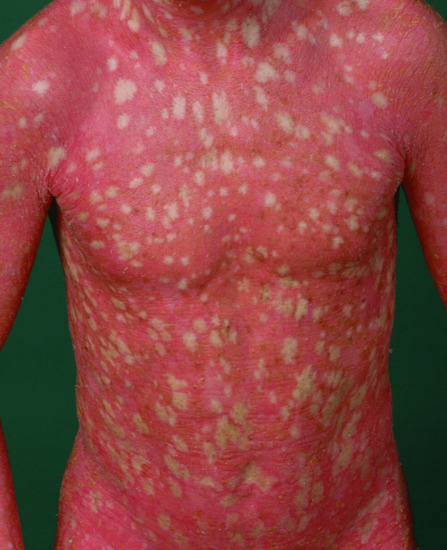
Figure 65.18 Congenital reticular ichthyosiform erythroderma. Note pale confetti-like spots representing localized spontaneous healing.
(Courtesy of the Department of Dermatology, University Hospital Münster, Münster, Germany.)
Investigations
Histology and ultrastructure differs from typical EI showing for example binuclear cells and perinuclear vacuolization, probably not the typical keratin aggregates [5].
Ichthyosis Curth-Macklin
Pathophysiology
Pathogenic mutations in KRT1 affect the variable tail domain (V2) of keratin 1 and result in a profoundly different abnormality of the cytoskeletal architecture than in EI [1, 2].
Clinical features
The skin of patients with ICM is characterized by extensive spiny hyperkeratosis (‘hystrix’-like) covering the entire body and involving the palms and soles (Figure 65.19) [3].
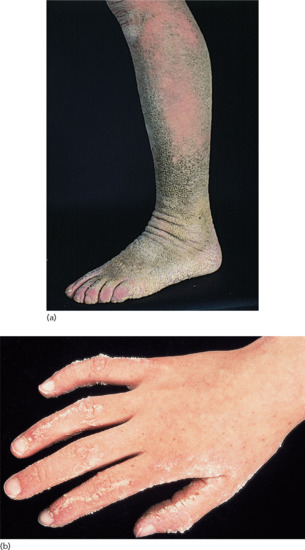
Figure 65.19 Ichthyosis. Hystrix with striate lesions on the hand. Courtesy of Dr M. Judge, Salford Royal NHS Trust, UK.
Investigations
Histology reveals perinuclear vacuolization and formation of bi-nucleated cells, without keratin aggregates, while ultrastructural studies usually reveal shell-like perinuclear arrangement of keratins [4, 5].
Erythrokeratoderma
In the past, it was common to distinguish between ‘true’ ichthyosis involving the entire body and more localized ichthyosiform conditions. When these localized conditions were characterized by erythema and hyperkeratosis they were called erythrokeratoderma or erythrokeratodermia [1–3]. In the Sorèze conference, it was decided that the various conditions that still carry the name erythrokeratoderma should also be considered as ‘ichthyosis’ [4]. Clinical expression of molecular defects can be quite variable as is seen for instance in superficial EI, which likewise would qualify as ‘erythrokeratoderma’. Moreover, some syndromic types of erythrokeratoderma such as the keratitis–ichthyosis–deafness (KID) syndrome have traditionally been regarded by most authors as an ichthyosis.
Erythrokeratoderma variabilis
Definition and nomenclature
Erythrokeratoderma variabilis (EKV) is a rare disease characterized by migrating polycyclic erythematous lesions accompanied by hyperkeratosis.
Pathophysiology
Inheritance of EKV is usually autosomal dominant. In many but certainly not all families, dominant negative mutations in GJB3 encoding connexin 31 or GJB4 encoding connexin 30.3 have been found [1–4]. Autosomal recessive mutations in GJB3 have likewise been reported [5]. Connexins form gap junctions, which are aqueous intercellular channels that are found in all tissues of the human body, including the skin, nervous tissue, heart and muscle [2, 6]. (For further disease mechanisms on connexinopathies see KID and Vohwinkel syndrome later.)
Clinical features [7, 8]
Onset is usually in infancy. The manifestations vary within a family and within the individual. There are two types of lesions: relatively fixed well-demarcated keratotic and erythematous plaques, often bizarrely shaped, which show a predilection for extensor surfaces, lateral trunk and buttocks and extend and regress in area thickness and degree of erythema; and transient erythematous, polycyclic or comma-shaped macular lesions occurring at any site (Figure 65.20).
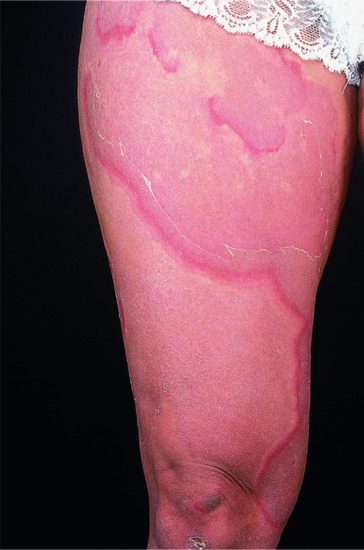
Figure 65.20 Erythrokeratoderma variabilis. Migrating polycyclic erythematous lesions.
Management
Acitretin treatment is the treatment of choice [9]. Likewise, the beneficial effect of low-dose isotretinoin has been reported [10].
Progressive symmetrical erythrokeratoderma
Definition and nomenclature
Progressive symmetrical erythrokeratoderma (PSEK) is a rare clinical variant of erythrokeratoderma with striking symmetrical appearance. It is unclear whether this type of erythrokeratoderma deserves the status of a distinct clinicogenetic entity or rather represents a manifestation of EKV.
Pathophysiology
From a genetic point of view, this clinical presentation is most likely also caused by the same connexin genes that underlie erythrokeratoderma variabilis. Actually, the same mutation G12D in the gene GJB4 has been identified in unrelated Dutch patients some of whom presented as EKV [2], while others were diagnosed as PSEK [3]. Moreover, occurrence of both types of erythrokeratoderma sharing the same ultrastructure has been reported in siblings [4]. It is conceivable that a different set of modifier genes (genetic background) decides whether a patient develops features of EKV or those of PSEK.
Clinical features
The skin is usually normal at birth. Large geographical but symmetrical fine scaly plaques with an orange-red erythema appear in infancy (Figure 65.21). There is little pruritus and the lesions are non-migratory in nature, as opposed to classical EKV. The shoulder girdle, cheeks and buttocks are most often affected. Keratoderma may be present.
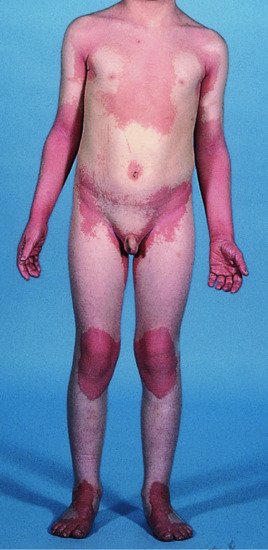
Figure 65.21 Symmetrical progressive erythrokeratoderma.
(Courtesy of Dr A. G. Smith, North Staffordshire Hospital, Stoke-on-Trent, UK.)
Management
See comment on EKV earlier.
Symmetrical acrokeratoderma
Clinical features
A Chinese group reported a study concerning 34 cases of symmetrical acrokeratoderma. Here, brown to black hyperkeratotic plaques were symmetrically distributed over the acral regions with marked worsening of the condition in the summer and improvement during winter. No genetic studies have been done so far and a relationship to IV has been suggested [1]. Clinically, there is whitish hyperkeratosis on the back of both hands and fingers and the wrists in particular after 5 minute water immersion reminiscent of aquagenic keratoderma. However, the authors emphasized that their patients did not suffer from palmoplantar involvement that could be typical for cystatin A deficiency [2].
Other non-syndromic forms of ichthyosis
There are some distinct generalized cornification disorders that are very much characterized by a palmoplantar phenotype. One example (LK) is discussed in the section on non-syndromic PPK later; others are described later.
Keratosis linearis-ichthyosis congenita-sclerosing keratoderma (KLICK)
Definition and nomenclature
This MeDOC form belongs to the group of ichthyoses but, like LK, it is dominated by keratoderma [1].
Pathophysiology
The ultrastructural phenotype of the skin indicated a disorder of keratohyaline granules [2]. Surprisingly, all affected individuals reported so far are carriers of a specific homozygous 1-bp deletion located upstream to the coding region of the POMP gene [3]. Immunohistochemical staining revealed an altered distribution of the proteasome subunits. Abnormal function of the POMP gene product may lead to increased stress of the endoplasmic reticulum (ER). The so-called ER stress interferes with epidermal differentiation as has been shown in connexin disorders [4].
Clinical features [1, 5, 6]
Clinically, the disorder manifests as a more sclerosing variant of loricrin PPK associated with mild congenital ichthyosis. In contrast with LK, it is inherited in an autosomal recessive fashion. Affected individuals demonstrate keratotic punctuate plugs or papules that are distributed in a linear pattern and are found on the flexural areas of the extremities – a distinct and probably pathognomonic phenotype (Figure 65.22). There are no associated features, but there is a report of secondary squamous cell carcinoma [7].
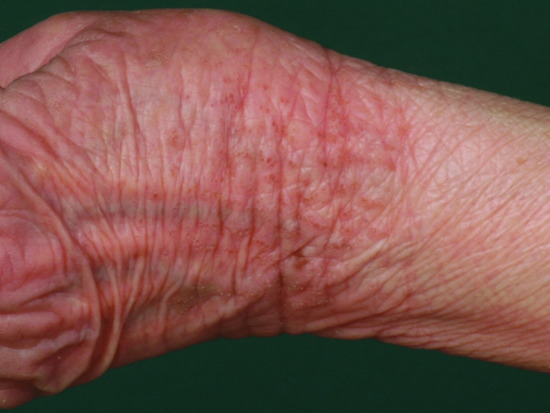
Figure 65.22 Keratosis linearis–ichthyosis congenita–sclerosing keratoderma (KLICK). Note the linear distribution of the keratotic papules.
Investigations
Ultrastructure confirms the histological finding of hypergranulosis and shows abnormally large keratohyaline granules.
Exfoliative ichthyosis
Pathophysiology
Loss-of-function mutations in the CSTA gene encoding the protease inhibitor cystatin A are the cause of this autosomal recessive disease [1]. Functional and ultrastructural data show that the defect manifests mainly within the basal and suprabasal layers of the epidermis characterized by expression of keratin 14.
Clinical features [2]
Exfoliative ichthyosis (EXI) is characterized by pronounced PPK (Figure 65.23), which tends to be sensitive to sweat and water exposure, similar to acral peeling skin syndrome (PSS) [3], the Bothnia type of non-epidermolytic PPK (NEPPK), or aquagenic PPK. Of note, EXI affects the entire integument showing mild dry and scaly skin, and as such fulfills the criteria of a non-syndromic form of congenital ichthyosis. Skin peeling may occur, easily elicited by moisture or minor trauma, and resemble the ‘moulting’ phenomenon in superficial EI [2].
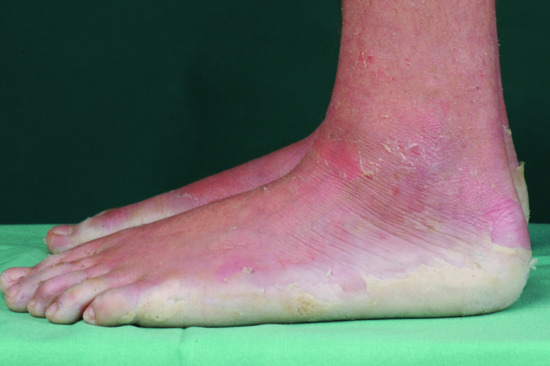
Figure 65.23 Exfoliative ichthyosis with pronounced plantar keratoderma.
(Courtesy of the Department of Dermatology, University Hospital Münster, Münster, Germany.)
Management
Efficient symptomatic treatment options seem completely lacking, because local therapy tends to increase humidity-associated sensibility.
SYNDROMIC ICHTHYOSES
(see Tables 65.2, 65.4 and 65.6)
X-linked syndromes concerning distal cholesterol biosynthesis
Conradi–Hünermann–Happle syndrome
Definition and nomenclature
Conradi–Hünermann–Happle syndrome (CHHS) is an extremely rare X-linked dominant skin disorder that usually affects only females and is lethal in males. Clinical hallmarks of CHHS are a mosaic presentation of linear ichthyosis, chondrodysplasia punctata, asymmetrically shortened limbs, unilaterial, sometimes sectorial cataracts and short stature.
Pathophysiology
Genetic mouse models for semidominant X-linked mutations were misleading for elucidating the genetics of this disease. Initially, the wrong mouse model, namely bare patches (Bpa) was considered to be a mouse model for the disease [1]. However, molecular studies, 16 years later, showed that a very similar mouse model, namely tattered (Td) is the true homologue of the human phenotype, mapping to the short arm of the X chromosome and not to the long arm as Bpa. The mouse model tattered is due to a mutation in the gene for emopamil binding protein (EBP) that functions as a δ8-δ7 sterol isomerase in the late steps of cholesterol biosynthesis. Mutations in the same gene were found to underlie CHHS mutations in humans [2–4]. The genetic defect is associated with metabolic alterations in the serum, namely markedly elevated levels of 8-dehydrocholesterol and of cholest-8(9)-en3-β-ol that can help to identify somatic mosaicism even in clinically unaffected males. However, the extent of the metabolic alterations detected in serum does not allow the prediction of the severity of the clinical phenotype [4]. The process of X-inactivation underlies the Blaschkoid pattern of distribution of skin lesions in CHHS. X-inactivation patterns of the patients showed no skewing, thus supporting the assumption that inactivation of EBP gene occurs at random [5]. Mosaicism in the parent generation has been reported several times [4–6]. The disease is characterized by anticipation, namely worsening of disease severity in subsequent generations [7]. It is of note that focal dermal hypoplasia can be associated with large submicroscopic deletions of the X-linked PORCN gene that also includes the adjacent EBP locus, although these patients with focal dermal hypoplasia did not exhibit features of CHHS [8]. It is believed that the accumulation of 8-dehydrocholesterol and other cholesterol precursors interferes with sonic hedgehog signalling and thus explains the developmental abnormalities in CHHS, such as facial dysmorphism, chondrodysplasia punctata or kyphoscoliosis [9]. The ichthyosis in CHSS is much more difficult to explain but it has been shown that lamellar bodies lack their normal lamellar structure [10].
Clinical features
Affected babies are typically female, premature and born with either partial collodion membrane or generalized ichthyosiform erythroderma. Within the first year, generalized linear and swirling patterns of erythroderma and scaling, following the lines of Blaschko, are established (Figure 65.24). Intervening areas of skin are unaffected. Palmoplantar hyperkeratosis and nail dystrophy may occur. Recurrent infections especially in the flexures, can be troublesome, and scalp and eyebrow hair is sparse and lustreless. The ichthyosis improves in early childhood and the residual signs are often so subtle in adult life that an affected mother may be missed. Signs to be sought in older children and adults include swirls of fine scales, linear pigmentary change, patchy atrophy, follicular atrophoderma mainly on the limbs and dorsal hands, and a striate cicatricial alopecia, all in a Blaschkoid pattern.
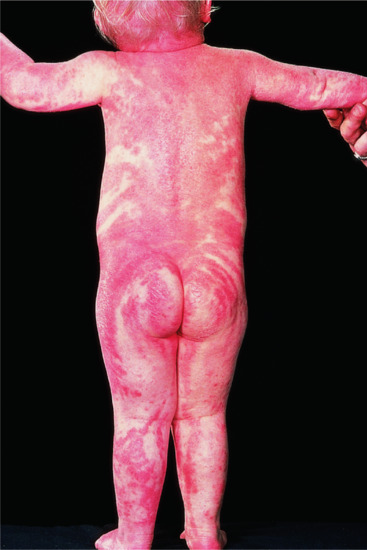
Figure 65.24 Conradi–Hünermann–Happle syndrome.
(Courtesy of Dr D. J. Atherton, London, UK.)
Other variable features include rounded or asymmetrical facies with frontal bossing and hypertelorism, a broad flat nasal bridge, congenital asymmetrical cataracts in 60% of patients, short stature, asymmetrical or, rarely, symmetrical shortening of limbs, kyphoscoliosis, supernumerary digits and other skeletal defects. Stippled calcification (asymmetrical) of long-bone epiphyses, vertebrae, pelvis, carpus and tarsus, and cartilage, including trachea, is a characteristic but not universal radiological finding in the neonatal period, and usually resolves by adulthood. Patients have normal or mildly impaired intellectual development and neural hearing loss have been reported.
Differential diagnosis
Milunsky and co-workers reported on a non-mosaic male with a mutation in EBP that presented a much milder phenotype characterized by failure to thrive, crossed renal ectopia and stenotic ear canals, but lacking chondrodysplasia punctata [11]. This unusual case was later discussed as a separate and novel clinicogenetic entity that is due to a hypomorphic mutation in EBP, a situation reminiscent of different phenotypes generated by hypomorphic NEMO mutations [12]. In the meantime, several such cases have been recorded and the term MEND syndrome (male EBP disorder with neurological defects) has been proposed for this distinct entity [13]. It is of note that MEND is inherited as an X-linked recessive trait with extreme behavioural symptoms, and female carriers of the hypomorphic EBP mutation seem to be unaffected [14]. This situation is reminiscent of the CK syndrome (MIM 300831) which is caused by hypomorphic temperature-sensitive mutations in NSDHL [15], while classic NSDHL mutations are associated with the X-linked dominant congenital hemidysplasia–ichthyosiform naevus–limb defect (CHILD) syndrome [16].
Management
Emollients are helpful in controlling ichthyosis and antimicrobial therapy may be needed for skin infections in infancy. The effect of retinoids is unknown and the need for treatment diminishes with age. Continued orthopaedic surveillance and appropriate procedures may be indicated for skeletal anormalies. Cataracts usually do not affect vision. The ichthyosis is probably caused by both cholesterol deficiency and accumulation of toxic sterol metabolites. Therefore, an approach similar to that used in treating hyperkeratosis in the metabolically related CHILD syndrome may be beneficial. In patients with CHILD syndrome, marked improvement of the cutaneous phonotype was observed following topical treatment twice daily with a 2% lovastatin/2% cholesterol lotion [17]. In CHHS no experience with this approach has been reported so far (April 2015).
Congenital hemidysplasia-ichthyosiform naevus-limb defect syndrome
Definition
This syndrome is a very rare X-linked dominant male lethal disorder of distal cholesterol biosynthesis featuring as a clinical hallmark the CHILD naevus.
Pathophysiology
The reader is also referred to the section on CHHS which also represents a disorder of distal cholesterol biosynthesis and is sometimes confused with CHILD syndrome. CHILD syndrome was fully delineated in 1980 as an X-linked dominant trait that is lethal in males by the German dermatologist Rudolf Happle [1]. In the initial report, the cutaneous phenotype was categorized as ichthyosis, but later the Happle group described it as an inflammatory naevus [2] showing a strikingly unilateral arrangement and differentiated the CHILD naevus from other epidermal naevi such as inflammatory linear verrucous epidermal naevus (ILVEN). In 2000, the same group established that the CHILD syndrome is due to mutations in the NSDHL gene encoding a 3 β-hydroxysteroid dehydrogenase [3]. Two mouse X-linked dominant male-lethal traits, bare patches (Bpa) and striated (Str) had previously been associated with mutations in Nsdhl and serve as animal models for this disease [4]. These mouse models revealed that Nsdhl deficiency has a deleterious effect on hedgehog signalling in early placental development, since male embryos for several mutant Nsdhl alleles die in mid-gestation with a thin and poorly vascularized placenta [5]. It is of interest that hypomorphic NSDHL mutations cause an X-linked recessive disease in males that has been termed CK syndrome (see also section on CHHS).
Clinical features
The hallmark of the CHILD syndrome is the CHILD naevus which is a peculiar inflammatory epidermal naevus having a unique lateralization pattern with a strict midline demarcation and ptychotropism (affinity to body folds) (Figure 65.25). This naevus shows hyperkeratosis which has a typical yellow wax-like scaling that is quite different from that seen in CHHS and allows the paediatric dermatologist to make the proper diagnosis easily. Associated ipsilateral extracutaneous defects in the form of hypoplasia or aplasia may involve the limbs and other skeletal structures as well as internal organs such as the lung, heart and kidney [1]. Although most reports deal with sporadic cases – around 60 cases had been reported by 2006 – this may be an underestimation of familial occurrence. Thus the molecular work-up of a large family revealed segregation of the causative NSDHL mutation through three generations affecting five patients of which four presented with very mild or minimal skin lesions such as periungual hyperkeratosis and onychodystrophy of the left index finger which may reflect extreme lyonization occurring at random [6]. It is of note that the majority of cases affect the right side of the body, but left-sided cases have also been reported [7].

Figure 65.25 Congenital hemidysplasia–ichthyosiform naevus–limb defect (CHILD) naevus with yellowish keratosis and ipsilateral limb defect.
(Courtesy of the Department of Dermatology, University Hospital Münster, Münster, Germany.)
Management
The cutaneous phenotype usually improves significantly in the first years of life; however, the large lesions that remain constitute a serious burden due to itching and oozing. Simple dermabrasion has been shown to fail and to be associated with recurrence of the naevus [8]. In contrast, dermabrasion followed by immediate covering with split-skin grafts from the unaffected contralateral side has been effective for long-term therapy and has been interpreted as ‘donor dominance’ that cures the CHILD naevus [9]. Recently, a pathogenesis-based topical therapy aiming at suppression of epidermal cholesterol biosynthesis and simultaneous application of topical cholesterol in a cream has been reported with excellent clinical response [10].
Ichthyosis follicularis-atrichia-photophobia syndrome
Definition
Ichthyosis follicularis–atrichia–photophobia syndrome (IFAP) syndrome is a rare X-linked recessive trait featuring ichthyosis follicularis, atrichia, photophobia and severe retardation of growth and psychomotor development. It has both clinical and molecular genetic overlap with two other disorders: brain anomalies, retardation, ectodermal dysplasia, skeletal malformations, Hirschsprung disease, ear/eye anomalies, cleft palate/cryptorchidism, and kidney dysplasia/hypoplasia (BRESEK/BRESHEK) syndrome and X-linked keratosis follicularis spinulosa (KFSDX; MIM 308800).
Pathophysiology
X-inked recessive inheritance of this condition was already suggested in 1991 [1] and was firmly established by the observation of functional cutaneous mosaicism showing Blaschko linear lesions reflecting lyonization in women heterozygous for IFAP syndrome [2]. Causative missense mutations were eventually identified in the X-linked gene MBTPS2 encoding membrane-bound transcription factor protease, site 2 [3]. Shortly afterwards, it was shown that keratosis follicularis spinulosa decalvans likewise is caused by different mutations affecting other sites in the MBTPS2 gene [4] and that the so-called BRESEK/BRESHEK syndrome that is characterized by brain anomalies, intellectual disability, ectodermal dysplasia, skeletal deformaties, ear or eye anomaly and renal anomalie with or without Hirschsprung disease is also due to specific mutations in MBTPS2 [5, 6]. Finally, even an X-linked variant of Olmsted syndrome (OLS) has been linked to a mutation in MBTPS2 [7].
Membrane-bound transcription factor protease site 2 is a zinc metalloprotease essential for cholesterol homeostasis as well as ER stress response [3, 6]. In cultured cells of IFAP patients, residual enzyme activity was only about 1/3 of wild-type activity and survival in cholesterol-depleted media was below 10%. It is of note that only missense mutations and intron mutations partially affecting transcription [8] are known so far. Most likely, total loss of MBTPS2 is not tolerated by male embryos and a residual enzyme activity is required for survival [6].
Clinical features
The full blown spectrum of IFAP syndrome is variable and seen only in males. All patients have the triad of follicular ichthyosis, congenital atrichia of the scalp (absence of hair) and photophobia. Children can be born as collodion babies. In particular as neonates, they present with generalized follicular keratosis over the entire body including the scalp. Follicular involvement can be very prominent, e.g. over the knees. It can improve markedly in early childhood. The most striking abnormality certainly is the congenital alopecia (atrichia) (Figure 65.26). A non-cicatricial complete body alopecia is almost a classical feature. Psoriasiform plaques, angular cheilitis, periungual inflammation, dystrophic nails, hypohidrosis and atopic eczema can be present [9]. In contrast, dental development is normal. Superficial corneal ulceration and vascularization leads to progressive corneal scarring and underlies photophobia, the third cardinal feature [9]. Neurological features include mental retardation and seizures as well as olivocerebellar atrophy, malformation of the temporal lobes, mild inner cerebral atrophy and hypoplasia of the corpus calllosum [10, 11]. The syndrome overlaps with BRESEK and BRESHEK syndromes [5]. Female carriers can present with much milder symptoms such as cutaneous hyperkeratotic lesions that follow the lines of Blaschko or asymmetrical distribution of body hair or Blaschko linear presentation of hypohidrosis that can be visualized by testing, but otherwise goes unnoticed [2, 11].
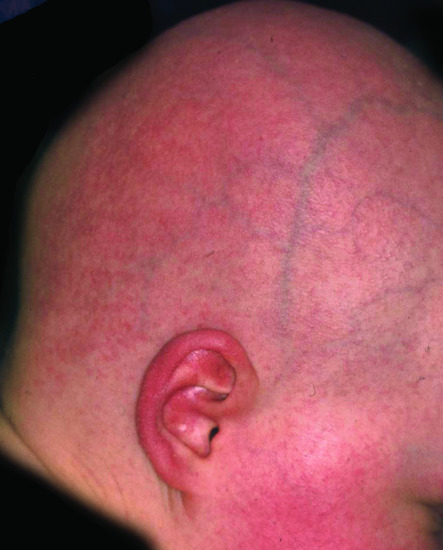
Figure 65.26 Ichthyosis follicularis–alopecia–photophobia (IFAP) syndrome.
(Courtesy of Dr A. S. Paller, Department of Dermatology and Pediatrics, Chicago, USA.)
Differential diagnosis
In the past, several cases of hereditary mucoepithelial dysplasia (HMD) were probably misdiagnosed as IFAP syndrome [12]. HMD can also feature photophobia and keratosis pilaris. However, no mutations in MBTPS2 have been reported in HMD. Moreover, in contrast to autosomal dominant HMD, X-linked recessive IFAP syndrome does not feature abnormal desmosomes (see earlier discussion).
Management
A moderate response to low-dose acitretin has been reported [13]. Emollients are helpful. Intensive lubrication of the ocular surface remains the mainstay of therapy for photophobia.
Exfoliative disorders of cornification
Comèl-Netherton syndrome
Definition and nomenclature
Comèl-Netherton syndrome (CNS) is a rare autosomal recessive disorder characterized by the triad of congenital ichthyosis, hair shaft anomalies and severe atopic diathesis. At birth, patients often display CIE, which in around 90% of cases, gradually evolves into a milder phenotype with polycyclic migrating plaques known as ichthyosis linearis circumflexa (ILC). The disease features associated symptoms such as life-threatening neonatal hypernatraemic dehydration, failure to thrive and recurrent infections [1–3].
Epidemiology
Worldwide prevalence is estimated at 1/50 000–200 000. CNS may account for up to 18% of all cases of infantile erythroderma; however, diagnosis is often delayed [4].
Pathophysiology
The disease is caused by recessive mutations in SPINK5 (serine protease inhibitor Kazal-type 5) [5], which encodes the multidomain serine protease inhibitor LEKTI (lymphoepithelial Kazal-type related inhibitor) expressed in the epidermis, thymus, oral and vaginal mucosa [6]. The protein is organized into 15 potential inhibitory domains with a four/six-cysteine residue pattern (Kazal-type like/Kazal-type). Subtilisin-like proprotein convertases like furin proteolytically cleave LEKTI full length protein. Subsequent processing creates several inhibitors with different target specificities [7, 8], e.g. domain 5 and 6 exhibit trypsin-inhibiting activity. The protein as such controls stratum corneum trypsin- and chymotrypsin-like enzymes (SCTE/SCCE) that are responsible for the processing of components such as corneodesmosin [9–12]. Reduction of serine protease inhibition not only leads to over-desquamation of corneocytes and degradation of desmosomal proteins (e.g. corneodesmosin and desmoglein 1), but also to induction of PAR-2 (protease-activated receptor 2) related pro-inflammatory responses [13]. TSLP as a biological marker correlates with disease activity [14, 15]. Another target of LEKTI is caspase 14 (involved in filaggrin processing), suggesting that it is a multifunctional protease inhibitor [16, 17].
Clinical features [1, 2, 17, 18–22]
Clinical diagnosis requires a combination of congenital erythroderma, neonatal failure to thrive and early development of atopy with high levels of IgE and hypereosinophilia. The hair shaft anomaly of trichorrhexis invaginata (‘bamboo hair’) confirms the diagnosis (Figure 65.27a) [3].
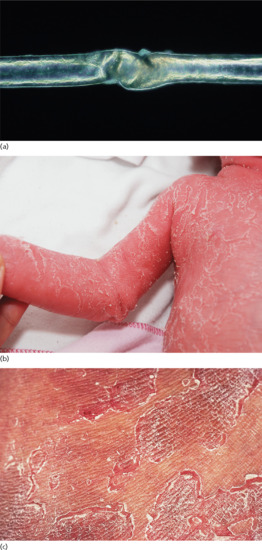
Figure 65.27 Comèl–Netherton syndrome. (a) Trichorrexisinvaginata. (b) Congenital ichthyosiform erythroderma with failure to thrive. (c) Ichthyosis lineariscircumflexa. (a), (c) Courtesy of Dr M. Judge, Salford Royal NHS Trust, UK.
Skin symptoms.
Collodion membrane is not a common feature, and erythroderma may also develop a few days after birth (Figure 65.27b). A typical ILC lesion is an erythematous, exfoliating or scaly, annular or polycyclic patch with an incomplete advancing double edge of peeling scale (Figure 65.27c). ILC is episodic, with lesions migrating often in a cephalocaudal direction, over several days. Lesions without the double-edged scaly margin are commonly seen.
Hair.
Although trichorrhexis invaginata is typical of CNS, other anomalies such as pili torti may also be seen. Hair, eyebrows and eyelashes can be present, sparse or absent at birth. Children may develop rare, thin, spiky and fragile hair of slow growth, but interindividual differences are striking.
Atopy.
Recurrent urticaria and facial angio-oedema, triggered by certain foods, are common complications, although the incidence varies [23]. The most common food allergens are nuts and fish. Food allergy can manifest in childhood or even in infancy. Pruritus is a constant feature.
Growth retardation.
Infants are usually born at term with average birth weight, but often develop failure to thrive, which might be explained by a frequent ‘dermopathic enteropathy’ and/or high energy loss through skin inflammation and hyperproliferation [24]. Nasogastric tube feeding might be required in severe cases in early life. However, most of these infants begin to gain weight in their second year, although they generally remain below the 25th centile for height and weight. One probably often unrecognized clinical feature is growth hormone (GH) deficiency [25] that shows good response to GH therapy [26].
Complications [27–32].
Perinatal complications include hypernatraemic dehydration, severe infections and malnutrition due to high calorie consumption and enteropathy. Later in life, bacterial (Staphylococcus aureus) and yeast colonization of the skin and human papilloma virus (HPV) associated viral warts (in older patients) are common. Severe complications include HPV-associated papillomatous skin lesions of the groin and perineal regions, spinous cell carcinoma, and giant condyloma of Buschke–Lowenstein [33].
Differential diagnosis
Erythrodermic forms of ARCI are important differential diagnoses, considering that atopy and hair shaft anomalies in CNS may not be present during the first months of life. Extreme eosinophilia could be diagnostic for Omenn syndrome (MIM 603554). Moreover, ectodermal dysplasia with eczema, inflammatory peeling skin disease and severe dermatitis–multiple allergies–metabolic wasting syndrome (SAM) syndrome need to be considered (see later).
Investigations
Loss of detection of LEKTI antigen in the epidermis is a useful diagnostic feature [20, 34, 35]. On histology, psoriasiform dermatitis with parakeratosis, acanthosis and a peculiar eosinophilic material just below the stratum corneum, are often observed. Often disregarded as artefactual, subcorneal or intracorneal separation can consistently be found and is a diagnostically useful hint [36]. Microscopic examination of the hairs usually leads to a rapid diagnosis: trichorrhexis invaginata refers to the protrusion of the distal part of the hair shaft into the cup shape of its proximal part [2]. Of note, this feature may not be apparent before 1 year of age. In addition, it is most likely to be found in eyebrow hair [37]. In addition, at the ultrastructural level NTS epidermis displays features such as altered lamellar body secretion and loss of corneocyte adhesion [38]. In around 90% of the cases, diagnosis can be confirmed by SPINK5 sequence analysis [17], which might be important for prenatal diagnosis of siblings [39, 40].
Management [3, 17, 41–53]
So far no effective therapy is available. The impaired epidermal barrier is a major clinical problem also including the risk of systemic toxicity from topically applied agents. Newborns are prone to hypernatraemic dehydration and/or systemic sepsis, and may need intensive medical care immediately after birth. Recurrent infections require antibiotic treatment. Consequent strengthening of the skin barrier relies on regular bathing, emollients and ointments (paraffin-based ointments, such as 50/50 white soft and liquid paraffin). Antiseptics might be added. Topical steroids should be avoided as far as possible or only used for a short period [43]; topical anti-inflammatory immunomodulators (pimecrolimus 1% or tacrolimus 0.05–0.1%) may be offered as an alternative, but systemic absorption is a concern [44–46]. Topical calcipotriol [44] and UV therapy can be tried [48, 49]. Type I hypersensitivity reactions, in particular food allergies to fish and nuts, should be prevented by dietary restrictions or may be treated specifically [50]. Hypotrichosis tends to improve after puberty; however, especially girls may profit from wearing a wig. Older patients with Netherton syndrome must be checked for HPV-induced skin cancers.
As a new and successful approach, intraveneous immunoglobulin (IVIG) therapy (0.4 g/kg/month) has demonstrated impressive results – especially in children with initial failure to thrive [51, 52]. Similarly, significant improvement has been reported by anti-tumour necrosis factor (TNF)-α monoclonal antibodies (infliximab) in a severe form of CNS [15]. So far, it is unclear whether ex vivo lentiviral gene therapy [53–55] or polypeptide replacement therapy might offer a specific therapy to patients in the future [17].
Severe dermatitis-multiple allergies-metabolic wasting syndrome
Definition
The acronym SAM recently introduced by Samuelov et al. (2013) refers to severe dermatitis, multiple allergies and metabolic wasting [1].
Pathophysiology [1, 2]
The disorder results from deficient membrane expression of desmoglein 1 which is also involved in the pathogenesis of pemphigus foliaceus, impetigo and is degraded in the absence of LEKTI in CNS [3, 4]. Loss-of-function mutations in DSG1 encoding desmoglein 1 (DSG1) causing SAM syndrome are inherited in a semidominant fashion as heterozygous carriers of DSG1 mutations showed focal palmoplantar keratoderma (see striate (and focal) palmoplantar keratoderma (SPPK) below).
Clinical features
This autosomal recessive disease is clinically reminiscent of ARCI, Netherton syndrome or PSS type B [1, 5].
Investigations
At the histological level SAM syndrome shows psoriasiform dermatitis and demonstrates subcorneal separation and acantholysis within the stratum spinosum and granulosum. At the ultrastructural level half-split desmosomes may be seen [1].
Peeling skin syndromes
Peeling skin syndromes (PSS) refer to a heterogenous group of generalized and/or palmoplantar disorders and overlap with other exfoliative forms of ichthyosis [1]. Clinical, ultrastructural, genetic and pathophysiological aspects demonstrate a fascinating relation between LEKTI deficiency (Netherton syndrome), desmoglein-1 deficiency (SAM syndrome) and corneodesmosin deficiency (inflammatory peeling skin disease) [2, 3].
Peeling skin syndrome type A
Definition
Fox reported the first case of generalized, non-inflammatory and asymptomatic skin shedding called ‘keratolysis exfoliativa congenita’ [1]. Other cases have been described as ‘familial continual skin shedding’ [2, 3] or ‘decidious skin’ [4].
Pathophysiology
A single recessive missense mutation in CHST8 has been described in a large consanguineous kindred with generalized PSS type A, but its pathogenic consequences are not well understood so far [5].
Clinical features [5, 6–8]
Non-inflammatory PSS type A usually starts between 3 and 6 years of age and is characterized by asymptomatic, generalized skin peeling with areas of hyperpigmentation, without skin blistering or erythema. There are no associated disorders.
Inflammatory peeling skin disease
Definition
The term ‘peeling skin syndrome’ was introduced by Levy and Goldsmith in 1982 [1]. Traupe differentiated PSS type A and B [2]. Inflammatory peeling skin disease initially described by Wile in 1921 [3] refers to PSS type B and is an ichthyosiform erythroderma characterized by lifelong patchy peeling of the skin with accompanying pruritus.
Pathophysiology
Peeling skin syndrome type B is due to autosomal recessive loss-of-function mutations in CDSN encoding corneodesmosin [4]. This finding has been independently confirmed by several groups [5–10]. CDSN is a secreted protein expressed in cornified epithelia and hair follicles [11]. It is specific to corneodesmosomes, cell–cell junction structures responsible for the stratum corneum (SC) cohesion [12]. Corneodesmosin adhesive properties are attributable to glycine-rich domains, which undergo sequential proteolysis leading to desquamation of corneocytes [13]. The essential role of CDSN for maintaining the integrity of the epidermis and hair follicle is demonstrated in mice [14]. Inactivation of the gene induces a lethal epidermal barrier disruption and hair follicle degeneration [15]. Interestingly, dominant nonsense mutations in CDSN, leading to the synthesis of an abnormal protein, cause hypotrichosis simplex due to the accumulation and toxic effect of abnormal corneodesmosin in the hair follicles [16, 17] (see Chapter 68).
Clinical features [1, 2, 3, 4, 18–23]
Inflammatory peeling skin disease presents at birth or a few days later. Infants develop ichthyosiform erythroderma with skin abnormalities consisting of spontaneous patchy peeling affecting the entire skin (Figure 65.28). Depending on mechnical stress, environmental factors, e.g. low humidity or temperature changes, patients experience recurrent episodes of peeling with severe pruritus. The presentation might be reminiscent of Netherton syndrome [24, 25], however, individuals do not show ‘bamboo hairs’ or hypotrichosis, and do not develop ichthyosis linearis circumflexa. The disease persists throughout life and is often accompanied by significant atopic manifestations.

Figure 65.28 Inflammatory peeling skin disease (peeling skin syndrome type B).
(Courtesy of the Department of Dermatology, University Hospital Münster, Münster, Germany.)
Investigations
Histopathology reveals subcorneal splitting and/or enhanced detachment of corneocytes. As such, Netherton syndrome and PSS type B appear similar at the histological and ultrastructural level [4, 22, 26]. Immunostaining for corneodesmosin and LEKTI may help to distinguish between these two disorders [4, 21].
Clinical features
Differential diagnosis
Hypotrichosis and failure to thrive seems unusual or less severe than in Netherton syndrome [4]. Other exfoliative inflammatory phenotypes should be distinguished including SAM syndrome and EXI.
Management
There is no effective treatment. Episodes of skin peeling are accompanied by severe and refractory pruritus. Allergies need to be prevented; tacalcitol cream [27], emollients with dexpanthenol and antiseptics or use of thermal water spray can be tried.
Acral peeling skin syndrome
Definition
Acral peeling skin syndrome (APSS) [1] is typically confused with epidermolysis bullosa simplex of localized type [2] and was recently reclassified as a type of epidermolysis bullosa [3].
Pathophysiology
This autosomal recessive condition [1] is caused by missense mutations in TGM5 encoding transglutaminase 5 [4].
Clinical features [1, 2, 5, 6]
The disease is characterized by superficial painless peeling of the skin predominantly on the dorsal aspects of the hands and feet. In infants, it frequently manifests with blistering on the palms and soles and is aggravated by mechanical factors and by humid warm environments. Distinction from epidermolysis bullosa simplex [2, 7] or EXI [8] may be difficult, but it is clearly different from generalized peeling skin diseases.
Differential diagnosis
Acral peeling skin syndrome should also be distinguished from keratolysis exfoliativa [9, 10]. This apparently common but underdiagnosed condition affects young adults, usually in the summer months and may be related to sweating. Lesions appear as tiny white rings or ‘air bubbles’, which soon rupture and peel off (‘ringed keratolysis’) (Figure 65.29). Attempts to identify a specific fungal or bacterial agent prove negative.
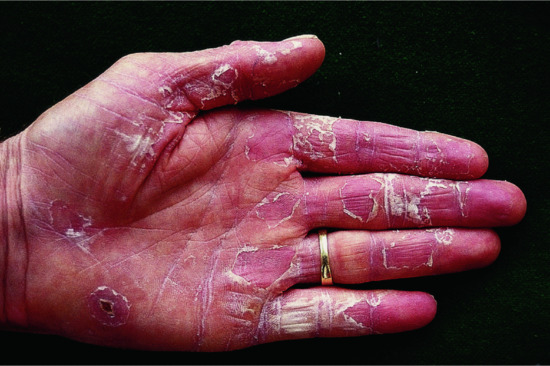
Figure 65.29 Keratolysis exfoliativa of the palms.
Neuro-ichthyotic syndromes
The combination of neurological manifestations and ichthyosis can be found in at least 16 distinct genetic disorders (reviewed in [1]). In mildly affected patients, cutaneous symptoms may not be apparent, or the ichthyosis may have a late onset, e.g. in Refsum syndrome. There is no effective therapy for the neurological symptoms of the diseases; however, special dietary restriction in Refsum syndrome (see later) or zinc acetate therapy in MEDNIK syndrome [2] are striking examples of successful pathogenesis-based treatment.
CEDNIK, MEDNIK, ARC, Gaucher disease type II, ELOVL4 deficiency and Stormorken syndrome
Gaucher disease type II represents a classic neuro-ichthyosis presenting at birth with collodion membranes [1, 2]. Diseases relatively new in the list of ichthyoses are the cerebral dysgenesis–neuropathy–ichthyosis–palmoplantar keratoderma (CEDNIK) syndrome [3–5], the arthrogryposis renal dysfunction cholestasis (ARC) syndrome [6–11] and the mental retardation–enteropathy–deafness–neuropathy–ichthyosis–keratodermia (MEDNIK) syndrome [12] (Table 65.4). ELOVL4 deficiency [13, 14] has been regarded as an erythrokeratoderma with sensorineural deafness [15] – clinically reminiscent of KID syndrome. In a five-generation Canadian pedigree, erythrokeratoderma appearing in infancy and clearing in later life was associated with late-onset ataxia and neuropathy [16]. Patients with Stormorken syndrome [17] (MIM 185070) may present with moderate thrombocytopenia, thrombocytopathia, muscle fatigue, asplenia, miosis, migraine, dyslexia and ichthyosis. The autosomal dominant disorder seems to belong to the group of channelopathies affecting calcium homeostasis in varies tissues [18, 19].
Refsum disease
Definition and nomenclature
Refsum disease (RD) is an ultra-rare autosomal recessive neurocutaneous lipid storage disorder featuring deteriorating vision and hearing, ataxia, neuropathy and usually mild ichthyosis.
Pathophysiology
In the early 1960s, accumulation of a storage product identified as a branched-chain 20 carbon fatty acid (phytanic acid) in plasma and tissues of patients with RD was reported [1]. Phytanic acid is derived from plant chlorophyll and cannot be synthesized by human tissues. It is under normal circumstances barely detectable in the serum, but in RD it accounts for 5–30% of serum lipids. It replaces other fatty acids in lipid-rich tissues thus interfering with membrane structure and function. RD was found to be caused by inactivating mutations in PHYH encoding a human phytanoyl-CoA hydroxylase which is responsible for α oxidation of phytanic acid [2]. Mutations in PEX7, encoding peroxin 7, were found to cause adult RD [3, 4]. This gene functions as a receptor for the PHYH protein and incorporates it into peroxisomes [5]. It is of note that accumulation of phytanic acid does not occur exclusively in RD, but can also be found in other diseases like Zellweger syndrome and in children with dysmorphic features, hepatomegaly, retinitis pigmentosa and hearing loss [6, 7]. This latter condition was called for some time ‘infantile RD’ because of the phytanic acid accumulation. However, in the meantime it was shown that ‘infantile RD’ actually is a hepatic peroxisome disorder and the designation ‘infantile RD’ has been abandoned [5].
Clinical features
Age of onset is usually in late childhood. Diagnosis is often delayed until early adult life. Progressive retinitis pigmentosa initially causes night blindness and later, failing vision and constricted visual fields. Neurological features develop in adolescence or in the early 20s. Anosmia and impaired taste is a frequent finding. Sensorineural deafness with tinnitus develops in more than 50%. A mixed sensorimotor polyneuropathy (type IV) with hypertrophic peripheral nerves and elevated cerebrospinal fluid (CSF) protein are characteristic findings. Cerebellar ataxia causes increasing disability. Ichthyosis occurs in around 25% of RD patients and coincides with or postdates the onset of neurological signs. It resembles IV. On histological examination many of the basal cells are often vacuolated and special lipid stains such as oil red O stain will reveal numerous fat globules within the basal cell layer and other keratinocytes [8].
Management
Early diagnosis is key to proper management of these patients [9]. Exclusion of sources of chlorophyll in the diet is mandatory in the treatment of RD. The major dietary exclusions are green vegetables (phytanic acids) and animal fat (phytol) and the aim of the dietary treatment is to reduce daily intake from the usual level of 50 mg/day to less than 5 mg/day. Rapid weight loss should be avoided as it mobilizes tissue phytanic acid, which can lead to acute clinical manifestations. Nowadays, lipid apheresis, that is the extracorporeal elimination of lipoprotein–phytanic acid complexes, is often used in initial treatment [10] and followed by a phytanic acid-poor diet [9].
Multiple sulphatase deficiency
Definition and nomenclature
Multiple sulphatase deficiency (MSD) is an exceedingly rare, autosomal recessive lysosomal storage disorder.
Pathophysiology
All known sulphatases are deficient and result in the accumulation of glycosaminoglycans (GAG) and sulphated lipids [1]. The responsible gene SUMF1 encodes a protein which is responsible for post-translational modification of sulphatases and catalyses the conversion of a conserved cysteine within the catalytic domain of various sulphatases into a C-α formyl glycine [2].
Clinical features
The enzyme deficiency can present as a very severe neonatal MSD, as severe late-infantile MSD with onset in the first year of life, or as mild late-infantile MSD with symptoms occurring between the age of 2 and 4 years and also as juvenile MSD presenting usually only a few of the symptoms such as mental retardation and ichthyosis [3, 4]. The disease is typically characterized by developmental delay and failure to thrive, and features of Hurler syndrome. A mild ichthyosis and progressive neurological degeneration evolve in the second or third year, but the phenotype varies according to the reduction in the enzyme activity [5]. A first neurological sign is often that children can no longer sit unsupported and lose their communication skills.
Management
No specific therapy is available. Children with predominant MPS II or MPS VI-like features may be candidates for enzyme replacement therapy.
Sjögren–Larsson syndrome
Definition
Sjögren–Larsson syndrome (SLS) is a rare autosomal recessive neurocutaneous condition featuring congenital ichthyotic hyperkeratosis, spastic diplegia and mild to moderate mental retardation. In the UK, prevalence is estimated to be 1 : 300 000, while in Sweden it is 1 : 100 000 and within the Province of Västerbotten even higher at 1 : 10 000.
Pathophysiology
Microsomal fatty aldehyde dehydrogenase (FALDH) deficiency underlies SLS [1]. FALDH catalyses oxidation of many different medium- and long-chain fatty aldehydes into fatty acids. Its deficiency results in the accumulation of fatty aldehydes and fatty alcohols in various tissues. FALDH has also been implicated in Ω-oxidation of the eicasanoid LTB4 [2]. SLS patients excrete large amounts of LTB4 in urine. It is believed that accumulation of LTB4 explains the marked pruritus in the disease. Further supporting this possibility, oral zileuton, which inhibits LTB4 synthesis, does improve the pruritus in SLS, but has no effect on ichthyosis or neurological disease [3]. The pathogenesis of SLS-associated ichthyosis has been linked to the hepoxilin pathway [4]. The neurological defect results from abnormal lipid composition of myelin. The gene that codes for FALDH is now called ALDH3A2. More than 72 ALDH3A2 mutations have been reported in SLS [5, 6].
Clinical features
Preterm birth is common and has been attributed to abnormal LTB4 inactivation [2]. Children are usually not born as collodion babies, but the skin is dry and mildly erythematous at birth and scaling develops within the first 3 month of life. Thereafter, a mild erythroderma persists and a variable degree of scaling develops (Figure 65.30) consisting of diffuse peeling on the trunk and more pigmented, lamellar-type ichthyosis on the lower limbs. Keratotic lichenification is often seen around the flexures, neck and mid-abdomen. The face is usually spared. Severe and persistent pruritus is a notable feature of SLS; scratch marks and dermographism are often seen on the trunk, but skin infections are rare. Neurological symptoms and signs appear during the first 2 years of life and consist of delay in reaching motor milestones due to spastic diplegia or much less commonly, of spastic tetraplegia. Seizures occur in up to 40% of patients. Delayed speech and dysarthria are common. A distinctive ophthalmological findings are so-called glistening white dots surrounding the fovea that are due to crystalline inclusions [7].
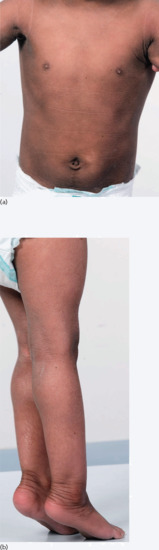
Figure 65.30 Sjögren–Larsson syndrome. A 3-year-old girl with spastic diplegia. Courtesy of Dr M. Judge, Salford Royal NHS Trust, UK.
With early physiotherapy, most patients learn to walk unaided or with crutches in childhood. Increased muscle tone leads to altered posture and movement which predispose to contractures (ankles, knees, hips), kyphoscoliosis and dislocated hips. Patchy leukodystrophy and myelination defects have been reported on computed tomography (CT) and magnetic resonance imaging (MRI) scanning studies in these patients.
Management
Dietary approaches with supplementation of medium-chain fatty acids have not been successful so far. Inhibition of LTB4 synthesis by zileuton improves pruritus in some patients, but not ichthyosis [3]. Retinoid therapy (etretinate, acitretin) has proven effective in relieving scaling and disabling keratotic lichenification, but less successful in controlling itching [8]. Intensive physiotherapy [9] and skills learning in early childhood clearly improve motor and social development in SLS. Bezafibrate, a lipid lowering agent, was shown to induce FALDH activity in fibroblasts in patients with residual enzyme activity, but clinical studies have not been carried out so far [10].
Keratitis–ichthyosis–deafness
Definition
Keratitis–ichthyosis–deafness syndrome and HID (hystrix-like ichthyosis and deafness) syndrome are clinically distinct types of congenital ichthyosis featuring severe sensorineural deafness. Despite their heterogeneous phenotypic manifestations, both conditions are related to mutations in GJB2 encoding connexin-26. Other diseases like Vohwinkel syndrome, as well as focal palmoplantar keratoderma with deafness and porokeratotic eccrine ostial duct naevus (PEN) – a unique type of epidermal naevus – likewise relate to GJB2 mutations (Table 65.5).
Pathophysiology
Connexins are universal membrane proteins that form inter- and intracellular channels for ion and molecule transfer, which is the basis of all cellular communication. Mutant connexin expression leads to abnormal cellular and calcium homeostasis and, in some cases, immune responses which cause cell dysfunction, lysis and death. Mutations in genes encoding gap junction proteins, namely connexins, have been reported in various epidermal diseases including hidrotic ectodermal dysplasia (connexin 30), erythrokeratoderma variabilis with and without deafness (connexin 31 and 30.3), Vohwinkel syndrome (connexin 26), KID syndrome (connexin 26), HID syndrome (connexin 26) and most recently in PEN [1–6].
Recurrent and novel GJB2 mutations have been recorded in a significant number of patients [7, 8]. The genotype/phenotype relationship between the various connexin 26 related conditions is not really understood. It is of note that the hearing deficiencies are often due to recessive mutations, whereas KID (and HID) syndrome are transmitted as autosomal dominant traits. In contrast, PEN result from mosaicism for GJB2 mutations, and therefore, a mother affected by PEN may give birth to a child affected by KID syndrome [5], a situation reminiscent of mosaicism in EI. Some mutations are closely associated with KID syndrome such as p.D50N mutation which was detected in 12 of 14 patients in one report [8]. Functional studies have highlighted that at least two GJB2 mutations, namely p.D50A and p.A88V, which are associated with the KID syndrome, do not simply inhibit channel formation, but rather result in high conductance hemichannels at the cell surface. Such gain of function due to GJB2 mutation thus creates channels that can result in toxicity by accelerating cell death in low extracellular calcium solutions [9]. It is tempting to speculate that modifier genes account for the considerable clinical differences between KID and HID syndromes, which can be caused by the same GJB2 mutation.
Clinical features
Many affected neonates have generalized erythema and some also have diffuse scaling and a leathery skin. The typical skin changes gradually develop during infancy with linear and spiny hyperkeratosis around the flexures, elbows and knees, and hystrix-like scaling on the limbs. Scattered follicular hyperkeratosis appears on the trunk. Typical features are the evolution of symmetrical, well-demarcated hyperkeratotic and warty plaques on the scalp, ears, face and occasionally the trunk and limbs. Some patients develop thick perioral rugae, and an aged or leonine facies. Keratotic, hyperplastic and inflammatory nodules may develop on the scalp, face, trunk and lower legs, and in situ and invasive squamous cell carcinoma arising within these dysplastic lesions have been reported in several KID patients in adult life (Figure 65.31). A 37-year-old man died of metastatic squamous carcinoma of the skin; his daughter also has KID syndrome [10]. Likewise squamous cell carcinoma of the tongue has occurred in three young patients [11–13] and a 28-year-old patient had a fatal malignant fibrous histiocytoma [14]. Multiple hair follicle tumours (including malignant progression to tricholemmal tumours) occurred in an adult with KID syndrome [15] and two further adult patients developed metastatic malignant pilar tumours [16].
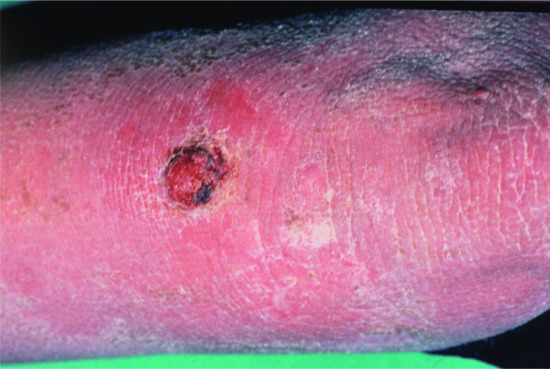
Figure 65.31 Keratitis–ichthyosis–deafness syndrome. Adult with squamous cell carcinoma on the arm.
(Courtesy of Dr D. J. Atherton, Great Ormond Street Hospital for Children, London, UK.)
Most patients have extensive scarring alopecia of the scalp and loss of the eyebrows, eyelashes and body hair resulting from follicular hyperkeratosis and atrophy. A reticulate PPK resembling grained leather is a characteristic feature, and progressive nail dystrophy and shedding may occur. Acneform eruptions and cysts on the upper trunk are common, and chronic deep abscesses and discharging sinuses (follicular occlusion triad) are a distressing late complication in some patients. Chronic cutaneous, granulomatous fungal and Candida infections may develop and contribute to the alopecia, nail dystrophy and body odour. Death in infancy from overwhelming infection (viral, bacterial and mycotic) has been reported in at least four patients with KID syndrome. Premature caries, oral leukoplakia, short stature, breast hypoplasia and cryptorchidism are occasional complications.
Congenital severe sensorineural deafness is evident during infancy in most patients. In typical KID syndrome, progressive corneal vascularization occurs in early childhood, often after a febrile illness, and leads to photophobia and blindness by adolescence in 75% of patients. A progressive peripheral neuropathy has occurred in several adults with KID.
The HID syndrome in contrast to the KID syndrome shows full blown CIE with hystrix-like keratosis in particular on the trunk and on the extremities and the entire body is affected, while in KID syndrome hyperkeratosis usually is confined to sites of predilection and the trunk is usually spared in later life.
Of note, the clinical appearance of PEN – caused by mosaic GJB2 mutations – can resemble that of KID syndrome showing a hyperkeratotic verrucous, hard epidermal naevus that features a peculiar histophathology including ortho- or parakeratotic plaques protruding form eccrine ducts [17].
Management
Early and frequent audiological and ophthamological assessment of patients with KID and HID syndrome is necessary to enable appropriate treatments such as hearing aids, cochlear implants and speech therapy to be instituted in a timely manner [18]. The ocular lesions are treated topically with lubricants, antibiotics, steroids or ciclosporin drops as indicated. Surgical procedures and corneal transplant to treat advanced keratitis of the eye have usually failed [19]. Antiseptic baths and cleansers, intermittent antibiotic and antiviral therapy and prolonged systemic antifungial agents play an important roll in controlling skin infections and odour [20]. Cleansing, debridement, dressing, excision of hyperplastic lesions and grafting may become necessary to reduce the potential for malignant transformation. Systemic retinoids like acitretin are helpful in controlling the disease, but isotretinoin may exacerbate corneal neovascularization [21]. In a recent case report, treatment with alitretinoin resulted in marked improvement of an extremely severe case of dissecting cellulitis of the scalp [22].
Neutral lipid storage disease with ichthyosis
Definition and nomenclature
Neutral lipid storage disease with ichthyosis (NLSDI) is a rare type of syndromic ARCI featuring CIE and lipid droplets in various tissues. Most patients originate from Mediterranean countries.
Pathophysiology
The disease is due to mutations in the ABHD5 (previously known as CGI-58) [1]. Another type of neutral lipid storage disease without ichthyosis, but with myopathy, has been designated NLSDM and is caused by mutations in the PNPLA2 gene [2]. ABHD5 is widely expressed in tissues such as the skin, muscle, liver and brain, but also lymphocytes [3] and localizes to the surface of cytoplasmic lipid droplets [4]. The genetic defect leads to acylceramide deficiency, likely contributing to the pathogenesis of ichthyosis [5]. For diagnostic purposes, it is useful to search for lipid droplets in leukocytes (Jordan anomaly) (Figure 65.32a) [6]. It is important to note that the characteristic lipid inclusions have to be specifically looked for in both affected patients and possible gene carriers. If an automated blood cell count is done, a ‘normal’ result will be handed out erroneously. Lipid deposition results in a combination of skin, hepatic, muscle and ocular abnormalities.

Figure 65.32 Neutral lipid storage disease with ichthyosis. (a) Lipid vacuoles (Jordan anomaly). (b) Ichthyosiform erythroderma. Courtesy of Dr M. Judge, Salford Royal NHS Trust, UK.
It is of note that the PNPLA2 gene – associated with NLSDM – encodes adipose triglyceride lipase which is necessary for lipolysis of cellular fat stores in non-cutaneous tissues [2]. Adipose triglyceride lipase requires the ABHD5 gene product as a co-factor, which explains the fact that NLSDI and NLSDM share common features in non-cutaneous tissues and also suggest the existence of an epidermal lipase, also utilizing ABHD5 as a co-factor [7].
Clinical features
Neutral lipid storage disease with ichthyosis is a multisystem disorder, but clinical features are variable [8, 9]. Affected newborns are either collodion babies or erythrodermic. The pattern of skin disease thereafter resembles mild to moderate CIE with fine white scales on an erythematous background and a lamellar scaling on the trunk and legs (Figure 65.32b). Scaling may diminish in warm weather and with advancing age. Pruritus is often troublesome and hypohidrosis may occur. Mild ectropion, flexural and neck lichenification and palmoplantar hyperkeratosis are common. Nail dystrophy and scalp alopecia have rarely been reported.
Muscle involvement ranges from an asymptomatic or subclinical myopathy with elevated muscle enzymes in most patients to marked proximal myopathy in few cases. Hepatomegaly, abnormal liver enzymes and fatty infiltration of the liver are common, even in childhood. Cirrhosis may evolve rapidly, even in childhood. Liver biopsy is more sensitive than biochemical markers in detecting the degree of involvement. Splenomegaly and malabsorption, resulting from intestinal mucosal lipid deposition, are occasional features. Cataracts of the nuclear type (subcapsular) may be detected from infancy in over 50% of cases, but rarely affect vision. Nystagmus may occur. Short stature, retinal disease, nerve deafness, ataxia, microcephaly, spasticity, neuropathy and developmental delay have been reported, but most patients are intellectually normal. Fetal renal complications occurred in an infant with NLSDI. Prognosis depends on the pattern and degree of organ involvement.
Management
Emollients are helpful and although liver function tests usually show abnormalities in these patients, administration of acitretin has been beneficial even in the presence of compromised liver function [10]. The effect of dietary approaches is doubtful [11].
Trichothiodystrophy/Tay syndrome
Definition and nomenclature
Trichothiodystrophy is a rare and heterogeneous group of neurocutaneous genodermatoses that have in common a hair defect termed TTD or sulphur-deficient brittle hair.
Pathophysiology
All types of TTD are autosomal recessive traits. From a genetic point of view, one can distinguish a photosensitive TTD group with (i) DNA repair anomalies involving various subunits of the transcription factor TFIIH; (ii) a non-photosensitive group without a DNA repair defect that features mutations in the C7ORF11 gene coding for TTDN1 protein; and (iii) a group without DNA repair defect and with still unclear underlying genetic defects [1, 2]. Since little is known on the function of the C7ORF11 gene that encodes the protein for non-photosensitive TTD (TTDN1), pathophysiological investigations focus on the photosensitive group, in which mutations in ERCC2 (XPD), ERCC3 (XPB) and GTF2H5 (TTDA) – all of which are subunits of the transcription/DNA repair factor IIH (TFIIH) – have been identified [2]. TFIIH is a complex of 10 proteins which are essential both for nucleotide excision repair and transcription [3]. Mutations in TTD-associated genes destabilize the superstructure of TFIIH and result in a low concentration of TFIIH, which in turn limits the level of transcription of a variety of target genes – in particular deregulation of thyroid hormone target genes in the brain occurs. In this way, the TTD phenotype is explained [4]. In contrast, TTDN1 is a nuclear protein that is not involved in DNA repair, but has several phosphorylation sites and is considered a regulator of mitosis [5].
Clinical features
Patients affected with the photosensitive forms of TTD are often born prematurely and typically present with a collodion membrane or with a CIE-like phenotype [3, 5]. Pregnancies of TTD neonates have to be considered as high-risk pregnancies and are frequently complicated by pre-eclampsia, decreased fetal movement, haemolysis, elevated liver enzymes and low platelets (HELLP syndrome) [6]. TTD neonates often have a low birth weight, are small for gestational age and require admission to a neonatal intensive care unit [6]. Congenital ichthyosis (collodion baby) – when present – often progresses to only mild ichthyosis, e.g. predominantly on the trunk (Figure 65.33), histologically showing a thin granular layer, thus resembling IV in later life [1]. It has been pointed out that the frequency of congenital ichthyosis with initial presentation as collodion baby is significantly higher in patients carrying mutations in genes encoding components of the TFIIH complex, while in contrast hypogonadism is significantly more frequent in the non-photosensitive group [1]. Additional features in some patients are eczema, palmoplantar hyperkeratosis, pulp atrophy, digital flexion contractures, fragile small nails and hypoplastic aural cartilage [7]. An elfin-like and aged (progeric) face resulting from fat atrophy might be seen. Scalp and eyebrow hair is fragile, sparse, short and unruly, but may improve with age. On polarizing microscopy, the typical ‘tiger-tail pattern’ can be noted that relates to a low content of the amino acid cysteine in the hair. Mild to moderate intellectual impairment is the rule and hypogonodism may lead to delayed puberty and infertility in adult patients [8]. Photosensitivity and photophobia occur in most patients having TFIIH-related mutations, but this feature can improve with age. Despite the DNA repair defect and in contrast to xeroderma pigmentosum, an increased risk of malignancy is not regarded as a feature of photosensitive TTD [9]. Important further characteristics of TTD are cataracts, otosclerosis, dental anomalies and neurological signs such as microcephaly, spasticity, cerebellar dysfunction, seizures and autism [10]. A temperature-dependent deterioration in hair, e.g. sudden loss of all hair, and worsening of skin and neurological features has been observed in several patients at the time of febrile illness [11] and has been attributed to increased instability in the TFIIH complex [12]. Early death from sepsis and recurrent infections with chronic neutropenia have been reported [2]. A friendly disposition and cuddlesome behaviour may be typical for TTD.
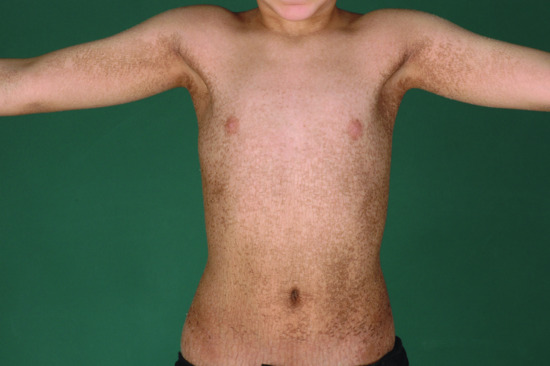
Figure 65.33 Trichothiodystrophy. Lamellar scaling similar to bathing suit ichthyosis.
Management
Emollients are helpful in skin comfort. The effect of retinoids on ichthyosis have been disappointing. Strict sun avoidance, protective measures and sunscreens are recommended for the photosensitive patients. Advice on avoidance of physical hair treatment is helpful in reducing further hair damage.
Neu–Laxova syndrome
Definition and nomenclature
This is an lethal autosomal recessive malformation syndrome, in which the cutaneous features (congenital ichthyosis) have never been studied adequately. The tight skin described in several reports is reminiscent of restrictive dermopathy [1–3].
Pathophysiology
Neu–Laxova syndrome (NLS) is heterogeneous and inherited as an autosomal recessive trait. It is caused by mutations in the PHGDH gene that lead to phosphoglycerate dehydrogenase deficiency [4]. This enzyme is involved in the first and limiting step of L-serine biosynthesis. Recently, it has been shown that mutations in PSAT1 and PSPH encoding two other enzymes of the L-serine biosynthesis pathway may lead into NLS [5]. The phenotype, albeit showing substantial variation in severity, may therefore represent the severe end of serine-deficiency disorders [5]. Histology of the skin reveals focal parakeratosis. No systematic analysis of the cutaneous phenotype with immunofluorescence studies or ultrastructure is available.
Clinical features [1–3]
Neu–Laxova syndrome is characterized by congenital ichthyosis, marked intrauterine growth retardation, microcephaly, short neck, central nervous system anomalies, limb deformities, hypoplastic lungs, oedema and abnormal facial features including severe proptosis with ectropion, hypertelorism, micrognathia, flattened nose and malformed ears. Prenatal ultrasound findings of marked ocular proptosis in a growth restricted, oedematous fetus are suggestive of the diagnosis [3, 6]. A polyhydramion is typical and most likely the result of the congenital ichthyosis as polyhydramion can also be seen in HI and IPS.
Diffential diagnosis
From a dermatological point of view, restrictive dermopathy is the most relevant differential diagnosis. Similar to NLS it is characterized by intrauterine growth retardation, thin tightly adherent translucent skin, typical facial dysmorphology with a mouth forming an O, generalized joint contractures, fetal akinesia and polyhydramion. Most cases are due to mutations in the ZMPSTE24 gene, while a few seem to be due to mutations in LMNA [7, 8].
Management
All reported cases have been lethal so far. Considering the metabolic basis of the disorder and similar to other serine deficiency therapies NLS might be a treatable condition when recognized and treated early enough [5, 9]. A supplement therapy may be provided for pregnant women who have previously had a child affected by NLS [4], which underscores the need for accurate diagnosis of neonates with congenital ichthyosis and lethal course.
Coloboma–heart defect–ichthyosiform dermatosis–mental retardation–ear anomalies syndrome
Definition and nomenclature
Coloboma–heart defect–ichthyosiform dermatosis–mental retardation–ear anomalies syndrome (CHIME) syndrome is an exceedingly rare autosomal recessive neurocutanous condition.
Pathophysiology
This syndrome is due to mutations in the gene PIGL which encodes the de-N-acetylase required for glycosyl phosphatidyl inisitol (GPI) anchor formation [1]. PIGL is an ER-localized enzyme that catalyses the second step of GPI biosynthesis. It is thus a congenital disorder of glycosylation. Glycosylation is the biosynthetic process of adding glycans to proteins and lipids and is an important modification of secretory and membrane-bound proteins [2]. Defects within the N-glycosylation or O-glycosylation biosynthesis pathway result in congenital disorders of glycosylation (CDG) [3].
Clinical features
In 1983, Zunich and Kaye [4] reported a child with migratory CIE, retinal colobomas, neurological disease, fine sparse hair and dental abnormalities. Further cases, featuring in addition cardiac abnormalities, have been described [4–7]. Generalized pruritus, erythema and scaling develop within the first month, and figurate, red scaly and itchy patches migrate on the head and body from early childhood. Palms and soles are thickened; the scalp hair is fine, sparse and hypopigmented. Cranial defects, alopecia, hypertelorism, a broad nasal bridge, wide mouth, full lips and wide-spaced teeth contribute to a characteristic facial appearance. Neurological features include hearing loss, seizures, developmental delay and outbursts of violent behaviour. A 4-year-old patient with CHIME syndrome developed leukaemia. The disease is considered to be a cancer-prone genodermatosis [8].
Management
For the ichthyosis, emollients (moisturizers) have been recommended [6].
Miscellaneous syndromic ichthyoses
Ichthyosis–prematurity syndrome (Table 65.6)
Clinical features
The disease is characterized prenatally by polyhydramnion and increased echogenic signals of amniotic fluid [1]. Affected neonates are often born between the 30th and 35th gestational week, and suffer from transient, potentially life-threating asphyxia, which is the result of reduced lung function and bronchial obstruction from keratin plugs [2–4]. The neonatal erythroderma (Figure 65.34a) evolves into a mild ichthyosis reminiscent of SHCB [5] (Figure 65.34b), but patients are prone to pruritus and atopic manifestations [4].
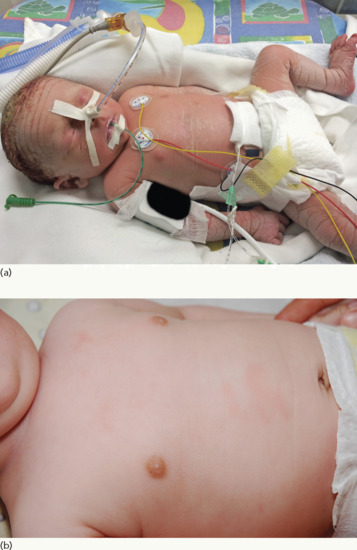
Figure 65.34 Ichthyosis prematurity syndrome. (a) Neonatal presentation. (b) Same patient after 3 months. Note mild velvet-like skin texture.
(Courtesy of the Department of Dermatology, University Hospital Münster, Münster, Germany.)
Pathophysiology
Ichthyosis prematurity syndrome is caused by recessive mutations in the FATP4 gene [6, 7], which encodes a fatty acid transporter and acyl coenzyme A synthetase expressed in the suprabasal layers of the epidermis [8]. Reduced function probably leads to disturbance of the intercellular lipid layer of the stratum corneum [9]. It can be concluded from the mouse model that FATP4 might be more important for the generation of the epidermal barrier than for its maintenance, which may explain the transient character of the disease [4, 10].
Investigations
Ultrastructure reveals the distinct phenotype of the so-called ‘ichthyosis congenita type IV’, which is characterized by irregular lentiform depositions of membrane-like material in granular cells and horny scales [11].
Management
Information about the risk of IPS, e.g. for subsequent pregnancies of parents who have previously had a child affected by IPS, may decide between a benign or unfavourable course of the condition. Optimal treatment, i.e. adequate bronchial suction, if initiated early enough, and optimal perinatal care will avoid life-threatening hypoxaemia and/or complications such as infantile cerebral paresis [4].
Ichthyosis with hypotrichosis (see Table 65.6)
Definition and nomenclature
The ichthyosis presents at birth, but without collodion membranes. Two allelic variants have been described as follows.
- Autosomal recessive ichthyosis with hypotrichosis (ARIH) features the ichthyosis and whole-body hypotrichosis, but no atrophoderma [1, 2].
- Congenital ichthyosis–follicular atrophoderma–hypotrichosis–hypohidrosis (IFAH) refers to a very similar phenotype associated in addition with follicular atrophoderma (e.g. follicular pitting on the dorsal aspects of the hands and fingers) [3, 4].
Pathophysiology
Both phenotypes are associated with autosomal recessive mutations in the ST14 gene [1, 2], which encodes matriptase, a novel key player of the epidermal protease network [5, 6]. The transmembrane serine protease is an efficient activator of epidermal pro-kallikreins. Matriptase deficiency leads to a decrease of filaggrin processing [7]. An elegant LEKTI-deficient mouse model showed that corneodesmosome integrity was restored when matriptase was ablated [8]. As such, ST14 deficiency might be seen as the functional counterpart to LEKTI deficiency, which leads to an increase of epidermal kallikrein activity (see Netherton syndrome earlier). Clinical heterogeneity may be caused by different types of ST14 mutations [1, 4, 9, 10]. The hair phenotype can be explained by the fact that matriptase is expressed in the cortex cells and shaft of the anagen hair [11].
Neonatal ichthyosis–sclerosing cholangitis (see Table 65.6)
Pathophysiology
Neonatal ichthyosis–sclerosing cholangitis (NISCH) belongs to the disorders of tight junctions being due to autosomal recessive mutations in CLDN1 encoding claudin-1 [1–3]. Claudin-1 is part of the epidermal tight junctions, but is also expressed in human cholangiocytes and hepatocytes [4]. The primary lack of claudin-1 leads to increased paracellular permeability between epithelial cells which may explain the phenotype of hypercholanaemia or epidermal barrier defect [1]. In mice, homozygeous deletion of CLDN1 leads to neonatal lethality because of transepidermal loss of water and desiccation. In humans, other tight junction components may partially compensate for claudin-1 deficiency [2].
Clinical features
Sclerosing cholangitis is a severe chronic condition characterized by inflammation and obliterative fibrosis of the intra- and extrahepatic bile ducts. NISCH syndrome is characterized by scalp hypotrichosis with scarring alopecia and ichthyosis, and shows a primary sclerosing cholangitis of variable severity [5–7]. Hence, infantile jaundice and ichthyosis may be an important clinical symptom of the disease, and early molecular analysis is recommendated as pathognomonic; histological and ultrastructural features are lacking [8].
MANAGEMENT OF CONGENITAL ICHTHYOSES
Inherited ichthyoses require lifelong management based on the establishment of the correct molecular diagnosis (Figure 65.35). This section focuses on ARCIs as well as KPIs; however, it may be applicable to other forms of hereditary ichthyosis.

Figure 65.35 Diagnostic management and clinical monitoring in ichthyosis. ENT, ear, nose and throat; GC-MS, gas chromatography–mass spectrometry; IgE, immunoglobulin E; LEKTI, lymphoepithelial Kazal-type related inhibitor; RBC, red blood cell; RXLI, recessive X-linked ichthyosis; WBC, white blood cell.
A recent review of clinical trials of treatment for congenital ichthyoses revealed only six trials that met the criteria of the methodology of the Cochrane collaboration [1] and therefore most of what is stated below cannot be considered to be evidence based. The difficulties that are encountered when carrying out randomized double-blind multinational placebo-controlled studies in these ultra-rare diseases are highlighted by a recent report on treatment with liarozol for moderate and severe LI. This retinoic acid metabolism-blocking agent resulted in good clinical improvement and was well tolerated, but the study failed to meet the endpoints possibly owing to the small sample size following premature termination [2]. National guidelines for the management of ichthyoses is available in Germany. The recommendations below are based on these guidelines and the long experience and exchange with patient organizations such as the German SI e.V. (Selbsthilfe Ichthyose e.V.) or the European Network for Ichthyosis (ENI).
Primarily non-specific measures are needed to alleviate symptoms [3], and one has to keep in mind that ichthyosis (hyperkeratosis) develops as a repair response to an inherent defect of the epidermal barrier. In a skin humanized mouse model of TG1 deficiency, topical enzyme replacement therapy restored TG1 activity and showed a healing of the ichthyosis [4]. However, as long as no targeted therapy is available for patients, most treatments have the goal of reducing scaling, but will enhance the barrier defect to some degree [5]. Thus transepidermal water loss increases after successful treatment of ARCI with either systemic retinoids [6] or topical keratolytics [7].
Management of collodion baby
Collodion baby as well as HI should be regarded as dermatological emergencies and require an interdisciplinary approach [1, 2]. Infants suffering from collodion baby as well as from HI [3, 4] have a profoundly disturbed epidermal barrier which is associated with increased transepidermal water loss [5] and may result in hypothermia and/or hypernatraemic dehydration. Further problems are proneness to infection, ectropion, poor sucking, restricted pulmonary ventilation and occasionally digital vascular constriction [6, 7]. These neonates are best taken care of in a neonatal intensive care unit [1, 6, 7]. A disease severity score may help to monitor response to treatment [8].
Collodion babies should be placed in a high humidity incubator with close monitoring of body temperature. Typically, it is recommended to start with humidity in the range of 60–80%, and to decrease every 3–4 days in order to reach normal humidity conditions, so that the children can be transferred to an open crib. In our experience, it is sufficient to use bland ointments, e.g. a dexpanthenol containing ointment two to four times a day. A report from the Netherlands cast doubt on the value of emollients arguing that in particular occlusive emollients, such as petrolatum or lanolin, predispose to skin infections. However, in most institutions, water in oil emollients are applied at least twice daily. Percutaneous absorption is very high and substances typically used in older children with ichthyosis, such as urea or lactic acid should be avoided in the first year of life. Salicylic acid is contraindicated as it may result in metabolic acidosis and death within 72 h even when used in low concentrations such as 3%.
The issue of bathing
A French study of 44 patients recently showed that a specific amount of hydrotherapy over 3 weeks, making use of thermal spring water and additional bathing with air bubbles at the Avène Hydrotherapy Centre, had a longlasting effect on the skin condition as well as on the quality of life [1]. The general beneficial effect of bathing for ARCI and KPI and in particular the effect of bath additives such as sodium bicarbonate (commonly known as baking powder) was established by the work of the late dermatologist Wolfgang Küster, who treated more than 300 in-patients with these diseases by this manner [2]. The mechanism behind sodium bicarbonate's beneficial effect is still unclear. It has been shown that two handfuls of baking soda to a bath tub will raise the pH from 5.5 to 7.9 [3]. Interestingly, most fresh water or lake water has a pH of around 5, while ocean water that many patients also report to be beneficial usually has a pH above 8.1 [3]. It is conceivable that mild alkalinization of the skin raises the pH to an optimum for serine protease activity such as KLK5 and KLK7 [4]. These serine proteases are required in normal desquamation for the dissolution of corneodesmosomes. Other popular bath additives are wheat corn or rice starch or bran, e.g. in Netherton syndrome. Antiseptics as bath additives can become necessary, e.g. in KPI, to overcome bad odour. Oils are usually messy and not recommended. Many patients have installed in their private homes a steam bath and benefit from daily steam bathing.
Practical treatment options for daily care
Patients with ARCI or KPI generally need to bath at least once a day. After soaking the skin for around 20–30 min, the patient or a parent can use a sponge, microfibre cloth or silk glove to rub the skin and thus provide mechanical scale removal as well as cleansing (i.e. removal of remaining ointment). This again may take another 20–30 min. Drying with a towel and the immediate application of ointments should be done while the skin is still wet. A variety of topical ointments can be used (Table 65.7), but local availability as well as composition will depend on the country and its traditions. Thus, in Germany and France ointments with higher water content are preferred for the treatment of ichthyoses, while in other countries mostly petrolatum-like ointments requiring bandages and not allowing the direct wearing of normal cloth are popular. The aim is to hydrate the stratum corneum by elevating the moisture level of the skin. Urea-based ointments (up to 10%) and lactic acid based ointments (up to 12%) as well as glycerol-based ointments are widely used. Substances such as macrogol 400 or propylene glycol can decrease the scaling and work as keratolytics. In Sweden, an ointment combining 5% lactic acid with 20% propylene glycol in the fatty cream locobase was shown to be effective in a controlled study [1]. Salicylic acid is contraindicated because of high penetration through the defective epidermal barrier and the danger of metabolic acidosis. Patients with a CIE phenotype often do not tolerate well ointments containing urea or lactic acid and do better when using substances that usually do not sting, such as glycerol, dexpanthenol or macrogol 400.
It is important to apply ointments twice a day so that the total time spent on body care for the average patient often is between 1 and 2 h/day. Special attention needs to be given to the face where less fatty creams may be used. Usually, it is possible to prevent or improve ectropion by topical treatment. The application of tazarotene gel (not available in all European countries) has been recommended for the treatment and prevention of ectropion [2]. A promising approach seems to be the topical use of n-acetylcysteine for ARCI [3–5]. N-acetylcysteine is a thiol derivative that has traditionally been used as a mucolytic agent and has been topically applied to prevent radiotherapy-induced skin irritation. It blocks cell-cycle progression in the G1 phase and has an antiproliferative effect. To our knowledge, there might be an unpleasant smell, and therefore it has not become popular among German patients when tried (personal communication, W. Küster).
Systemic treatment options
Retinoids such as acitretin or isotretinoin have been found to be helpful in ARCI and in some forms of KPI [1, 2], e.g. patients with TG1 deficiency – when presenting with LI – require rigorous treatment and usually benefit from systemic retinoids as well as from urea or lactic acid containing ointments. Patients with CIE may respond less well to systemic retinoids – some even can get worse – and also they tolerate moisturizers such as lactic acid or urea less well. Interestingly, EKV is known to respond rapidly to low-dose treatment with retinoids [3–5]. In patients with HI, the use of retinoids is warranted, even in newborns [6–8]. Patients suffering from EI due to KRT1 mutations have a strong risk of exacerbation of the disease when given retinoids, while those with KRT10 mutations may benefit when given a low dosage, e.g. up to 0.5 mg/kg body weight [1, 2, 9]. Likewise, superficial EI responds well to low retinoid doses [1].
In Europe, first etretinate and subsequently acitretin, the pharmacologically active metabolite of etretinate, have been used for severe congenital ichthyosis, while in the USA there is extensive experience also with the use of isotretinoin [2]. Doctors who wants to start a patient on a systemic retinoid should be aware of the relevant side effects, e.g. teratogenicity or hepatotoxicity, to name but a few. There is an excellent set of guidelines on the efficacy and use of acitretin in dermatology from the British Association of Dermatologists [10]. The attitude of physicians in the UK towards the use of acitretin is shared in most European countries [10, 11]. In Germany, there is a reluctance to use long-term retinoid therapy in children mostly because of fear of interference with bone development, such as premature closure of the epiphyseal growth lines. Patients with ichthyosis may be treated when they are over the age of 16 or who have stopped growing significantly. Only a detailed survey on a larger cohort of patients from several countries could provide definite data to answer satisfactorily whether there is a significant risk of bone growth toxicity for children.
In women of child-bearing age, a major advantage of isotretinoin over acitretin may be its short half-life. While isotretinoin is cleared within several months, acitretin can persist in the body for up to 2 years because of conversion to etretinate, in particular when taken with alcohol [10]. ‘Drug holidays’ are popular in particular when using isotretinoin. However, it can be very difficult convincing a patient that he/she should stop taking, e.g. acitretin, as it usually dramatically decreases the amount of time needed for personal care of the skin.
There is very limited experience in congenital ichthyosis with oral alitretion, a drug used for hand eczema. Alitretion is rapidly cleared and in contrast to acitretin and isotretinoin binds to both types of nuclear retinoid receptors (i.e. RARs and RXRs). In a recent clinical trial, two of four patients who had previously been on acitretin preferred to continue with alitretinoin [12]. A side effect of alitretinoin therapy may be hypothyroidism that can cause tiredness and it seems to be advisable to monitor thyroid-stimulating hormone (TSH) levels in these patients [12]. A dramatic improvement of dissecting cellulitis of the scalp has been reported in a patient with KID syndrome who had not responded to previous acitretin therapy [13].
Special aspects of treatment
Management of ARCI and KPI, even though they are non-syndromic diseases, requires a multidisciplinary approach and clearly extends beyond topical and systemic therapy [1]. The following issues should be addressed.
Eye.
Severe ectropion has to be addressed to avoid complications such as corneal perforation [2–4]. For severe cases, there are different methods for surgical corrective treatment of chronic ectropion [5–7]. Basal cell carcinoma may be masquerading as chronic ectropion in LI [8].
Ear.
Importantly, children need to be examined by an otorhinolaryngologist. Often during early infancy, especially in BSI, the external ear canal has to be professionally cleared of horny material (every 4 weeks) [9].
Hair.
Treatment of the scalp poses special problems and requires washable preparations.
Hypohydrosis.
A further issue is the inability to sweat (hypohidrosis) which often results in difficulties with thermoregulation, in particular in the summer time [1, 10]. This is a problem that is even experienced by ARCI patients with a mild phenotype, e.g. SICI. Interestingly, oral retinoid treatment can normalize sweat gland function and markedly improve the quality of life in ARCI patients [10].
Musculoskeletal system.
In many cases, in particular in infants and small children, physiotherapy is of enormous value, e.g. to treat flexural contractions.
Vitamin D.
There are several reports on Indian patients with ichthyosis, who developed severe signs of rickets [11–13]. Patients with ichthyosis and type IV–V skin [14] and/or malnutrition [15] may be more prone to vitamin D deficiency, but it seems that lamellar types of ichthyoses [16–18] as well as CIE [19] including Netherton syndrome [20] are important risk factors for 25-hydroxyvitamin D deficiency [21] with secondary hyperparathyroidism [22].
Superinfections.
Some ichthyoses such as forms of ARCI or KID syndrome may be associated with a higher incidence of dermatophytosis [23, 24] (Trichophyton rubrum infection may even mimic EI, if patients have not been seen before; authors’ own experience). KPI, KID syndrome, peeling skin disease or Netherton syndrome can be complicated by bacterial superinfections, often caused by Staphylococcus aureus [25, 26].
Patient organizations and other resources (Table 65.8)
Membership of a patient organization can be very helpful for patients and their families and should therefore be recommended. Many practical aspects, e.g. how to apply scalp treatment with occlusion overnight, are best explained by other patients who have become ‘experts’ in their disease. Moreover, membership of a patient organization can have a profound effect on psychosocial well-being. At the European level, national self-support groups have formed ENI (www.ichthyose.eu/ last accessed January 2015). At the national level, these groups are also active in patient associations caring for the special needs of rare diseases such as in Germany the Alliance for Chronic Diseases (ACHSE).
In the USA, the Foundation for Ichthyosis and Related Skin Types (FIRST; www.firstskinfoundation.org last accessed January 2015) has been active since 1981 and has the aim to educate, inspire and connect all those touched by ichthyosis and related disorders. General information on the diseases can be found on the homepage of the German network for ichthyoses (NIRK) (www.netzwerk-ichthyose.de last accessed January 2015) or of FIRST.
ACQUIRED ICHTHYOSES
Acquired or late-onset ichthyosis presents with features similar to IV and must be distinguished from xerosis, eczema, IV, RXLI and RD, in which scaling develops in early adult life [1].
Pathophysiology
Differential diagnoses of the underlying cause include malignancies, autoimmune, nutritional, metabolic, infectious and neurological diseases as well as medications [2].
Clinical features
Scaling is usually not associated with inflammation ranging from pityriasiform to lamellar, and it might be more obvious on the extensor aspects of the limbs.
Malignancies.
Increased expression of transforming growth factor α (TGF-α) or epidermal growth factor receptors (EGFR) mediated by a tumour may cause hyperproliferative paraneoplastic cutaneous conditions. The most common ichthyosis-associated malignancy is Hodgkin disease, and the skin changes may occur simultaneously, postdate or rarely precede the diagnosis [3–5]. The fine scaling affects the trunk and limbs, usually spares the flexures and histologically resembles IV with orthohyperkeratosis and reduced or absent granular layer. It clears with effective anticancer treatment and can be an early marker of subsequent recurrence. It may be associated with pruritus, an independent symptom of lymphoma. Reduced dermal lipogenesis, measured by radiolabelled carbon uptake, paralleled the severity of the ichthyosis and contrasted with results in IV in one study [6].
Acquired ichthyosis has also been reported in association with non-Hodgkin lymphoma [5, 7, 8], cutaneous T-cell lymphoma (Figure 65.36) [8, 9], lymphomatoid papulosis [10, 28], multiple myeloma [12], breast, lung, cervix, renal cell or liver carcinoma [13, 14], leiomyosarcoma [15], rhabdomyosarcoma [16] and Kaposi sarcoma [17]. It was the presenting sign of myelodysplasia [18], followed on from bone marrow transplant [19] or occurred with graft-versus-host disease [20].

Figure 65.36 Acquired ichthyosis in a man with cutaneous T-cell lymphoma.
Metabolic and distinct internal diseases.
Disturbances of lipid and vitamin absorption may cause those ichthyoses which develop with chronic metabolic derangement such as malnutrition or malabsorption (including coeliac disease and Crohn disease) [1, 2], essential fatty deficiency [21] and Shwachman syndrome (pancreatic insufficiency) [22]. Ichthyosis may be a presenting feature of inborn errors of metabolism, such as methylmalonic acidaemia [23], holocarboxylase and biotinidase deficiencies [24]. It may complicate renal failure with or without secondary hyperparathyroidism [25], primary hyperparathyroidism [26], autoimmune thyroiditis [27] and diabetes, where it affects the shins [28]. Ichthyosis may rarely occur with connective tissue diseases such as systemic lupus erythematosus [29], sarcoidosis (Figure 65.37) [34], dermatomyositis without associated malignancy [30], systemic sclerosis/lupus overlap [31], the so-called Haber syndrome [32] and eosinophilic fasciitis [33].
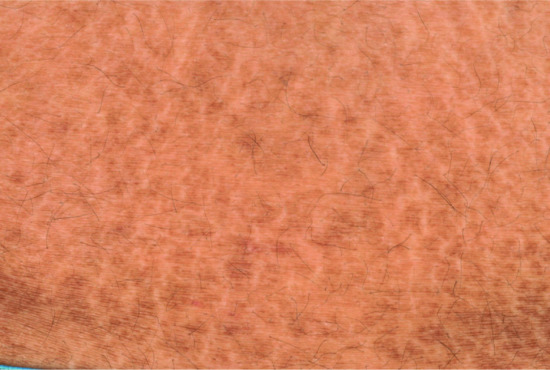
Figure 65.37 Acquired ichthyosis in an adult Asian man with multisystem sarcoidosis. Courtesy of Dr M. Judge, Salford Royal NHS Trust, UK.
Infections.
Ichthyosis has been noted in patients with leprosy [35], HIV infection with or without malignancies [2, 36, 37] and human T-cell lymphotropic virus type 1 (HTLV-1)-associated myelopathy [38].
Drugs.
The cholesterol-lowering drugs, triparanol and nicotinic acid, induce ichthyosis in a proportion of patients [39, 40]. Triparanol also causes poliosis and alopecia. Normal desquamation depends on the conversion of cholesterol sulphate (which maintains the structure of intercellular lipid lamellae in the subcorneal layers) to cholesterol, by cholesterol sulphatase, located on keratinocyte cell membranes. This enzyme is unaffected by hypocholesterolaemic agents. Keratinocytes lack low-density lipoprotein receptors, which may explain why so few treated patients show this complication. HMGCoA (hydroxymethylglutaryl coenzyme A) reductase inhibitors are also a rare cause of ichthyosis [41]. Ichthyosis and variable effects on hair have been attributed to certain butyrophenones, the phenothiazine dixyrazine, maprotiline [42], cimetidine (an antiandrogen) [43], allopurinol, hydroxyurea [44], clofazamine [45] and of course acitretin [46].
Pityriasis rotunda
Definition
Pityriasis rotunda describes a rare, persistent, sharply defined, circular patch of ichthyosiform scaling with no inflammatory changes. This name is preferred to pityriasis circinata, reported by Toyama in 1906, and to acquired pseudo-ichthyosis [1, 2].
Epidemiology
Pityriasis rotunda is relatively common in the Far East, especially Japan, where it accounts for some 0.2% of dermatological cases [2]. It has been reported also in South African Bantus [3], in an Egyptian [4], in Afro-Caribbeans living in London [5] and an African-Canadian woman [6]. It has been described in Caucasians [7] and in two cohorts from Sardinia [8, 9]. Its true incidence and geographical distribution are unknown. It is possible that genetic factors are involved and familial occurrence has been observed [8–12]. A familial association with IV has also been noted [2, 9, 12].
Pathophysiology
Systemic illnesses, particularly tuberculosis and malnutrition, coexisted in up to 50% of South African patients [2, 3, 13]. It may rarely be an acquired cutaneous marker of malignancy [14–16]. Pityriasis rotunda was observed in 16% of 63 South African black patients with hepatocellular carcinoma and nearly 5% of those with tuberculosis in one report [17]. However, cases reported from Sardinia had no associated systemic disorders and pityriasis rotunda tended to occur at a younger age [8, 9].
Clinical features
Typical lesions of pityriasis rotunda are circular sharply defined hyperpigmented patches of dry skin with ichthyosiform scaling, usually 2–3 cm in diameter but sometimes much larger, up to 14 cm (Figure 65.38). Lesion number ranges from four to 200. They rarely itch, slowly enlarge and coalesce. A hypopigmented halo has been described in some patients, and sometimes the entire lesion can be hypopigmented. Onset in childhood carries a better prognosis, with remission seen in later childhood in almost 50% [8, 9]. Lesions are commonly situated on the buttocks, thighs, abdomen, back or upper arms, and may be solitary or multiple. They most often develop between the ages of 20 and 45 years (2 and 76 years are the reported extremes) and may remain unchanged throughout life.
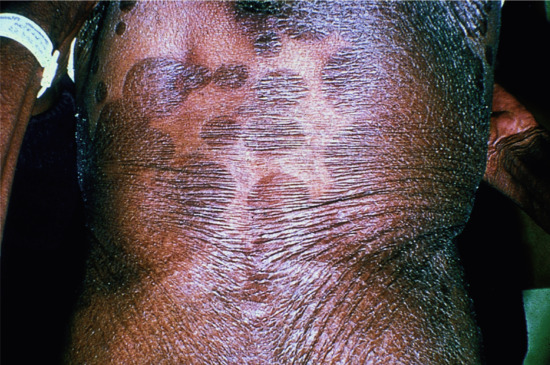
Figure 65.38 Pityriasis rotunda. Courtesy of Dr M. Judge, Salford Royal NHS Trust, UK.
Investigations
The age of onset, distribution, strikingly circular outline and absence of pruritus and inflammatory change should suggest the diagnosis. The histological changes resemble those of IV, with compact orthohyperkeratosis and reduced granular layer. There may be loss of the epidermal ridge pattern, pigmentary incontinence and mild perivascular lymphocytic infiltrate. Immunohistochemical studies have shown a marked reduction in loricrin and filaggrin expression in lesional skin [18], the latter caused by loss of the profilaggrin N-terminal domain [19]. Electron microscopy in one case revealed epidermal lipid vacuoles and other changes [8].
Management
Investigations for an underlying cause may be indicated. In prelymphomatous eruptions, the lesions show atrophy and telangiectasia. Dermatophytosis and pityriasis versicolor can be excluded based on negative mycological studies (microscopy and culture). Emollients, urea compounds and topical keratolytics may help. Topical tretinoin cream or systemic retinoids in more extensive disease can be useful to some extend but topical steroid and antifungal agents are not. Where malnutrition, infection or malignancy is the underlying cause, appropriate therapy should clear the lesions.
PALMOPLANTAR KERATODERMA
(see Tables 65.9, 65.11, 65.13, 65.14 and 65.15)
Definition and diagnostic approach
Palmoplantar keratodermas form a heterogeneous group of hereditary or acquired disorders defined by excessive epidermal thickening of the palms and soles [1, 2]. This clinical finding is observed as an isolated symptom and non-syndromic entity, but can also be part of a more complex (syndromic) phenotype, e.g. in PPK with cardiomyopathy. Of note, many generalized MeDOC forms manifest with PPK [3], for example, palmoplantar hyperkeratosis in autosomal recessive congenital ichthyosis (ARCI) is a ‘key feature’ of NIPAL4 deficiency [4, 5]. This section focuses on inherited ‘classic PPKs’ that are characterized by prominent or predominant palmoplantar involvement. A number of classifications of keratodermas have been published [1, 2, 6–11], none unite satisfactorily clinical presentation, pathology and molecular pathogenesis. However, syndromic and non-syndromic hereditary forms may be distinguished (Table 65.9) and differentiated from acquired PPKs. Finally, there are various other monogenetic diseases that may have a palmoplantar phenotype with features of hyperkeratosis (Table 65.10).
Table 65.9 Palmoplantar keratoderma.
| Disease | Mode of inheritance | Genes |
| Non-syndromic forms | ||
| Epidermolytic palmoplantar keratoderma (EPPK) | AD | KRT9 |
| Pachyonychia congenita (PC) | ||
|
AD AD AD AD AD |
KRT6A KRT16 KRT6B KRT17 KRT6c |
| Non-epidermolytic palmoplantar keratoderma (NEPPK) | ||
|
AR AR AR AD AD |
SLURP1 SLURP1 (variant) SERPINB7 AQP5 KRT1 (V1 domain) |
| Loricrin keratoderma (LK) | AD | LOR |
| Striate (and focal) palmoplantar keratoderma (SPPK) | ||
|
AD AD AD |
DSG1 DSP KRT1 (V2 domain) |
| Punctate palmoplantar keratoderma (PPPK) | AD | AAGAB COL14A1 (?) |
| Spiny keratoderma (SK) | AD? | Unknown |
| Marginal papular keratoderma (MPK) | ||
|
AD? AD? |
Unknown Unknown |
| Cole disease (CD) | AD | ENPP1 |
| Transient aquagenic keratoderma (TAK) | AD/AR? | Unknown |
| Syndromic forms | ||
| Palmoplantar keratoderma and cardiomyopathy | ||
|
AR (or AD) AR (or AD) |
JUP DSP |
| Palmoplantar keratoderma and hearing impairment | ||
|
AD AD AD AD/AR Maternal |
GJB2 GJB2 GJB2 GJB2 (see Table 65.5) MT-TS1 |
| Palmoplantar keratoderma and cancer | ||
|
AD AD AR |
Unknown RHBDF2 RSPO1 |
| Palmoplantar keratoderma in ectodermal dysplasia and related diseases | ||
|
AR AR AR AR AD XR |
GJB6 WNT10A CTSC CTSC TRPV3 MBTPS2 |
| Palmoplantar keratoderma and ophthalmic manifestations | ||
|
AR | TAT |
| Palmoplantar keratoderma and neurological manifestations | ||
|
||
AD, autosomal dominant; AR, autosomal recessive; XR, X-linked recessive
Table 65.10 Other inherited diseases with a palmoplantar phenotype.
| Disease | Genes | Cross reference |
Ichthyoses (see text for definitions) ARCI (NIPAL4, TGM1, CERS3, ALOXB12) EI SEI KRIE EKV LK KLICK EXI IFAP SAM CEDNIK SLS KID Neutral lipid storage disease with ichthyosis TTD |
See Table 65.1 See Table 65.1 See Table 65.1 See Table 65.1 See Table 65.1 See Table 65.1 See Table 65.1 See Table 65.1 See Table 65.1 See Table 65.2 See Table 65.2 See Table 65.2 See Table 65.2 See Table 65.2 See Table 65.2 See Table 65.2 |
|
| Naegeli–Franchescetti–Jadassohn syndrome | KRT14 | Chapter 70 |
| Epidermolysis bullosa simplex | KRT5, KRT14 | Chapter 71 |
| Kindler syndrome | KIND1 | Chapter 71 |
| Hypohidrotic ectodermal dysplasia | EDA | Chapter 67 |
| Autosomal recessive ectodermal dysplasia | GRHL2 | Chapter 67 |
| Ectodermal dysplasia–skin fragility syndrome | PKP1 | Chapter 67 |
| Dyskeratosis congenita | DKC1, TERC, TERT, TINF2, NOLA2, NOLA3, TCAB1, RTEL | Chapter 70 |
| Darier disease | ATP2A2 | Chapter 66 |
| Pityriasis rubra pilaris | CARD14 | Chapter 36 |
| Rapp–Hodgkin syndrome | TP63 | Chapter 67 |
| Hay–Wells syndrome | TP63 | Chapter 67 |
| ADULT syndromes syndrome | TP63 | Chapter 67 |
| Cowden syndrome | PTEN | Chapter 80 |
| Hypertrophic osteoarthropathy/pachydermoperiostosis | HPGD, SLCO2A1 | Chapter 72 |
The ‘clinical pattern’ – sometimes helpful.
Diffuse, focal/areate, striate or punctate patterns can be distinguished, but there are no absolute boundaries between these groupings. In diffuse keratodermas, the whole of the palmar or plantar epidermis, usually including the centripalmar skin and the instep, is uniformly thickened. In focal, areate or nummular keratoderma, the areas of palmoplantar skin under most pressure are disproportionately thickened. Striate keratoderma overlaps clinically with focal keratoderma, but the lesions are conspicuously longitudinal, particularly on the fingers, where keratoderma overlies flexor tendons. Punctate, papular or disseminated keratoderma consists of multiple scattered discrete round lesions. Transgredient keratoderma extends beyond palmoplantar skin, contiguously or as callosities on pressure points on the fingers or knuckles, or elsewhere. Confluent hyperkeratosis may extend round whole digits. Cicatrizing keratodermas (‘mutilating’) are those in which constricting bands appear around digits. Such ‘pseudo-ainhum’ is found in many severe transgredient keratodermas, and is not diagnostic of any one syndrome.
General diagnostic aspects and morphology.
Associated hyperhidrosis or fungal infections are common clinical symptoms, as many keratodermas, particularly on the feet, are spongy and malodorous. Water retention by abnormally keratotic and porous stratum corneum contributes to maceration and microbial overgrowth. Sometimes minor extrapalmoplantar signs such as insulate hyperkeratosis can be found. Therefore, the overall integument including mucous membranes, nails, hair, ability to sweat, etc., need to be examined. Diagnosis of suspected extracutaneous symptoms may require a multidisciplinary approach; a thorough family history is mandatory. Skin biopsy for histology and/or ultrastructure still plays a critical role in the diagnosis of keratodermas as it can provide essential clues for further genetic analyses [11], e.g. distinguishing between epidermolytic and non-EHK or demonstrating signs of disadhesion.
NON-SYNDROMIC PALMOPLANTAR KERATODERMA
Epidermolytic palmoplantar keratoderma
Definition and nomenclature
Palmoplantar keratoderma is associated with epidermolytic changes on histology and is due to mutations localized to a hotspot region of KRT9 or infrequently to specific domains of KRT1. Voerner described diffuse PPK with autosomal dominant inheritance, clinically indistinguishable from that described by Thost and Unna but with histological features of EHK in affected palms and soles [1]. Thost's original family in fact was apparently also affected with EPPK.
Epidemiology
Epidermolytic palmoplantar keratoderma (EPPK) is probably the most common form of diffuse keratoderma [2, 3]. A prevalence of 4.4/100 000 was found in Northern Ireland [4].
Pathophysiology
Epidermolytic palmoplantar keratoderma was initially found to map to the type I keratin gene cluster on chromosome 17 and subsequently shown to result from rare mutations in KRT1 and more common mutations in KRT9 [7–10], which is preferentially expressed in palmoplantar skin [5, 6]. Disruption of intermediate filament integrity due to these mutations is predicted to reduce the resilience of the cytoskeleton to minor external trauma, leading to blistering and hyperkeratosis as well as epidermolysis with tonofilament clumping. Most mutations identified to date affect the helix initiation peptide, but a 3-bp insertion in the helix termination motif has also been identified [11]. Infrequently, keratin 1 is involved: KRT1 gene mutations affecting the 2B domain, in the helix termination peptide and splice site mutations have been described [12–15]. EPPK with unusual ‘tonotubular’ filaments on electron microscopy [16] is due to mutations altering the 1B rod domain of keratin 1 [17, 18].
Clinical features [1, 19]
Diffuse keratoderma develops in infancy. In adults, there is confluent keratoderma (Figure 65.39), sparing dorsal surfaces, with a sharp demarcation and erythematous edge (Figure 65.40). Blistering is not a major feature, but a history of blisters or fissuring of the palms may hint at reduced structural strength. Hair, teeth and nails are normal, but knuckle pads and nail changes were found in Vörner's original families [1], many of whom have KRT9 mutations. Disabling pain especially on the palms might indicate the subtype of ‘tonotubular’ PPK [18]. Moreover, limited transgredient lesions, e.g. at the dorsum of the Achilles tendon often referred to as Greither keratoderma, may indicate an EPPK form associated with KRT1 mutations. Clinically, pure EPPK can be distinguished from epidermolytic ichthyoses which often involve the palms, soles and flexural areas [12].
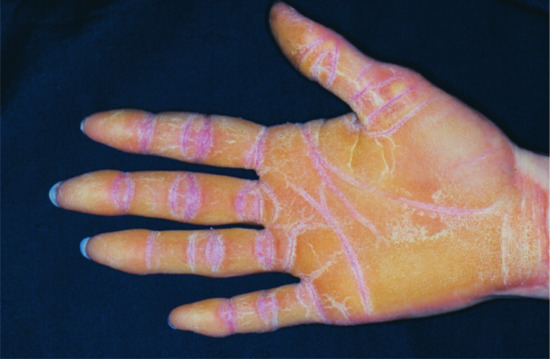
Figure 65.39 Epidermolytic palmoplantar keratoderma caused by a mutation in KRT9 encoding keratin 9.
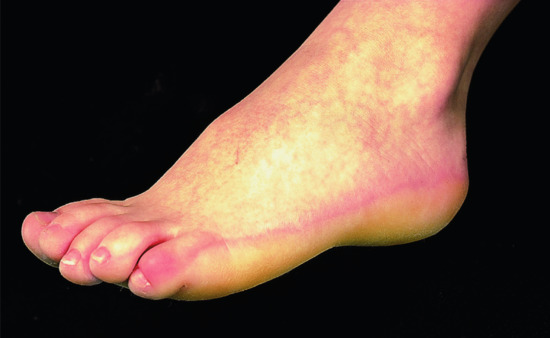
Figure 65.40 ‘Voerner–Unna–Thost’ keratoderma: even yellow hyperkeratosis of the sole with red border.
Investigations
Histologically, EPPK shows epidermolytic change in suprabasal keratinocytes. Round or ovoid eosinophilic inclusions may be detected [20], with large tonofilament aggregates visible on electron microscopy [21, 22]. Moreover, ultrastructure may reveal the peculiar finding of whorls of keratins containing tubular structures observed in transverse and longitudinal sections (‘tonotubules’) [16–18]. It may be advisable to take biopsies from different palmoplantar sites, because focal EHK may escape diagnosis in mild forms [15].
Management
The mainstay is mechanical debridement, followed by mild keratolytic re-lubrication to help avoid fissures. Oral retinoids can be tried at a low dose, but excessive peeling may be a problem [23]. Topical calcipotriol has reportedly been helpful [24].
Pachyonychia congenita
Definition and nomenclature
Pachyonychia congenita (PC) is a group of autosomal dominant keratinization disorders caused by a mutation in one of five keratin genes KRT6A, KRT16, KRT6B, KRT6C or KRT17. The variable clinical findings affect a number of ectodermal structures, including the nail bed, oral mucosae, palmoplantar skin, teeth and pilosebaceous unit [1–3].
Hypertrophic nail dystrophy gave rise to the name of PC [4]; however, the most problematic manifestation of the disease is focal plantar keratoderma with severe and often profoundly incapacitating pain [1]. Historically, PC has been classified under two major categories; PC type 1 (Jadassohn–Lewandowsky) and PC type 2 (Jackson–Lawler) [5–10], caused by mutations in genes encoding two pairs of dimerizing keratins, KRT6A/KRT16 and KRT6B/KRT17, respectively. PC type 1 was thought to be associated with PPK and oral leukokeratosis while PC type 2 was thought to be associated with pilosebaceous cysts and neonatal teeth [11]. A large-scale genotype/phenotype analysis of hundreds of patients did not confirm these associations [1, 12, 13], which led to a novel classification system that specifically relies on the causative keratin mutation, e.g. PC-6a, PC-6b, PC-6c, PC-16, PC-17 [11, 14–17]. Extensive clinicogenetic information on the diseases is provided by the International PC Consortium (IPCC) (www.pachyonychia.org last accessed January 2015).
Pathophysiology
Similar to several other keratin disorders, the majority of causative mutations in PC-related keratins are heterozygous missense mutations or small insertions/deletion mutations that disrupt cytoskeletal function via dominant-negative interference leading to cell fragility [14]. The variable distribution of lesions in PC corresponds to different expression patterns of the mutant keratins [11, 15–17]. For example, mutations in the prominent nail keratin K6a additionally affect oral mucosae [18]. In contrast, K17 is constitutively expressed in the pilosebaceous unit with lesser expression in palmoplantar skin and mucosae [18–20].
Clinical features [1, 3, 14]
A comprehensive analysis of clinical symptoms has been recently published [13]. Three clinical features are reported in more than 90% of patients across all mutation subtypes: toenail dystrophy, plantar keratoderma and plantar pain which in patients older than 3 years are highly diagnostic for PC (Figure 65.41).
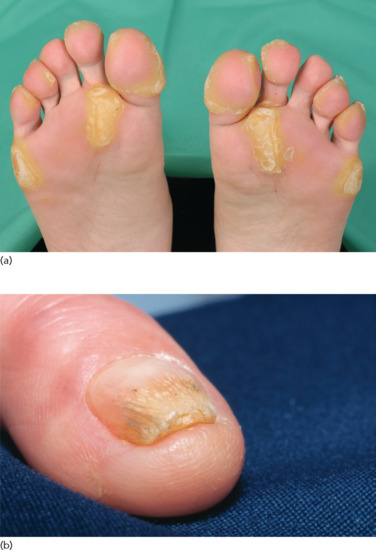
Figure 65.41 Pachyonychia congenita: (a) focal keratoderma on mechanically stressed areas; and (b) typical wedge-shaped nail.
Thickened toenails, i.e. hyperkeratosis of the nail bed, appear within the first year or up to 9 years of life. KRT6A mutation carriers show an early age of onset. Of note, in many cases not all 20 nails are involved.
Plantar keratoderma variably manifest as calluses, fissures and thickened skin. Thick yellow keratoses are found on sites of pressure. Frictional blisters may occur, especially in hot weather in childhood. Plantar pain is a very common symptom of PC and has an important impact on the quality of life. Around a quarter of patients with PC regularly require analgesic medication and many are confined to wheelchairs. The source of this exceptional pain is still a matter of debate but may be related to the conspicuous presence of blisters underneath plantar calluses [21].
Additional diagnostic findings include follicular hyperkeratoses on the knees and elbows, oral leukokeratosis, palmoplantar hyperhydrosis, cysts and natal teeth [22–26]. Patchy white thickened areas are seen on the tongue and oral mucosa. Severe oral lesions resemble candidiasis [27]. Laryngeal involvement may produce hoarseness and in infancy even fatal respiratory obstruction [28]. Some individuals – particularly if carrying KRT17 mutations – develop cysts in the form of steatocystomas and/or pilosebaceous cysts. The phenomenon of erupted teeth present at birth has primarily been reported in KRT17 mutation carriers [13]. Axillary cysts may mimic hidradenitis suppurativa [29].
Of note, mutations in KRT16 and in KRT17 have been reported in patients with isolated focal PPK [30] or steatocystoma multiplex [31], respectively, without any other PC manifestations. Mutations in K6c seem to be associated with a much milder focal keratoderma when compared with any other of the four PC types [32].
Investigations
Tonofilament aggregates seen on electron microscopy demonstrate the intermediate filament disorder [14, 22]. On histology, palmoplantar epidermis shows gross hyperkeratosis with alternating ortho- and parakeratosis. Acanthosis is present with patchy hypergranulosis, in which large and malformed keratohyalin granules are present, but there is no gross epidermolysis [33, 34]. Cysts may be keratinous epidermoid cysts, eruptive vellus hair cysts or true steatocysts.
Mutational analysis of the keratin genes may be provided by the IPCC (www.pachyonychia.org last accessed January 2015). Molecular differential diagnosis includes the frizzled 6 gene (FZD6), which is associated with autosomal recessive twenty nail dystrophy [35]. Heterozygous missense mutations in the connexin 30 gene, normally associated with Clouston syndrome, have been identified in a number of families with pachyonychia-like nail changes and no other phenotype [36].
Management
Mechanical reduction of hyperkeratosis and of nails, i.e. by filing, cutting, grinding, soaking supported by medical professional treatment, produces symptomatic benefit; attention to footwear and orthotics may reduce blistering and callosities. The effective treatment for patients having cysts is surgical removal/incision and drainage. Emollients and keratolytics may only help in milder cases of keratoderma [37, 38]. Systemic retinoid treatment is often unsatisfactory due to increased tenderness [37, 39]. Treatment of hyperhidrosis may reduce blistering [40]. The fact that PC-associated mutations exert a deleterious dominant negative effect has led to the development of different strategies aimed at eliminating mutant keratin from the skin using various strategies [41–43].
Resources
Patients or clinicians may contact the IPC consortium via email or register online (http://www.pachyonychia.org/ last accessed January 2015). Detailed information on disease expression (including photographs for each for of PC), diagnostics and updates on clinical studies are provided.
Painful hereditary callosities
Definition
The term painful hereditary callosities (PHC) most probably encompasses many different aetiological entities. In fact, plantar callosities of sufficient size are inevitably painful. Reported cases with blisters and tonofilament clumping are likely to correspond to an epidermolytic or keratinopathic disorder [1–4]. ‘Painful callosity’ is a clinical feature seen in PC; a novel minor variant has been described (‘PC-6c’) [5]. Painful keratoderma may also refer to desmosomal diseases [6] or to oculocutaneous tyrosinaemia (type II) [7].
Non-epidermolytic palmoplantar keratoderma (Table 65.11)
Table 65.11 Isolated nonepidermolytic palmoplantar keratoderma (NEPPK).
| Mal de Meledaa | NEPPK type Nagashima | NEPPK type Bothnia | NEPPK type Kimonis | ‘Greither keratoderma’b | |
| Synonyms | Keratosis palmoplantaris transgediens of Siemens | Keratosis palmoplantaris Nagashima | Non-epidermolytic palmoplantar tylosis | Non-epidermolytic keratinopathic PPK | Keratosis extremitatum hereditaria progrediens |
| MIM | 248300 | 603357 | 600231 | 600962 | 144200 |
| Inheritance | AR | AR | AD | AD | AD |
| Gene | SLURP1 | SERPINB7 | AQP5 | KRT1 (V1 domain) | e.g. KRT1 and others |
| Age of onset | Before 2 years, mainly under 20 years | At birth or in early infancy, mild and non-progressive | During childhood | At birth or in early infancy | After 2 years, may disappear in later life |
| Palmoplantar key features | Transgredient massive PPK, malodorous maceration, functional handicap with reduced mobility of hands and feet, nail involvement, asymmetrical presentations, hyperhidrosis, dermatophyte superinfection | Well-demarcated reddish and diffuse palmoplantar hyperkeratosis extending to the dorsal surfaces of the hands, feet, inner wrists, ankles, water sensitivity, hyperhidrosis | Diffuse hyperkeratosis with yellowish tint over the whole of the palms and soles, sometimes transgredient, white spongy appearance upon exposure to water, distinct margin between affected and normal skin, sometimes with papular border, hyperhidrosis, superinfection | Diffuse palmoplantar hyperkeratosis, discrete papules on the dorsa of fingers, hyperkeratotic pads over knuckles, erythematous halo separating hyperkeratotic from healthy skin, superinfection | Transgredient and progressive PPK, hyperkeratosis on ventral aspect of wrists, papules on the dorsa of fingers, knuckle pads, erythematous border separating hyperkeratotic from healthy skin, hyperhidrosis |
| Other clinical findings | Perioral erythema, insulate keratotic skin lesions particularly over the joints, amputation of digits possible | Achilles tendon, ankles, elbows and knees can be involved | – | Extending to the skin over the Achilles tendon, hyperkeratosis of the umbilicus and nipple areolae, very mild keratosis and dryness of the knees and elbows | Extending to the skin over the Achilles tendon, flexural areas, elbows and knees, amputation of digits or blistering describe |
| Histology and ultrastructure | Orthohyperkeratosis and papillomatosis, acanthosis without cytolysis, dermatophyte infectionc | Orthohyperkeratosis, acanthosis without cytolysis | Orthohyperkeratosis without cytolysis, dermatophyte infectiond | Orthohyperkeratosis without cytolysis | Unspecific and ill defined, keratin intermediate filament aberrations possible |
| Remarks | 1/100 000 in general population | High frequency in Japan and China (1.2–3.1/10 000) | Improvement after oral erythromycin | Similar to Unna–Thost keratoderma | If woolly hair is present exclude Carjaval syndrome |
1Keratoderma of Gamborg Nielsen 1985 (MIM 244850) represents a clinical variant of MDM.
2Greither keratoderma is not considered a clearly defined entity.
3Histology may show presence of spongiosis, vesiculation and neutrophils.
AD, autosomal dominant; AR, autosomal recessive; NEPPK, non-epidermolytic palmoplantar keratoderma.
Definition
Non-epidermolytic palmoplantar keratoderma refers to a heterogenous group of non-syndromic forms of diffuse keratoderma that histologically do not show cytolysis in the upper spinous or granular layers.
Historically, the eponym Thost–Unna keratoderma referred to NEPPK characterized by diffuse thick yellow hyperkeratosis [1, 2]. Meanwhile, the original Thost family was found to have EPPK [3]. Considering Mal de Meleda (MDM) as an important differential diagnosis, at least three different NEPPK entities can be distinguished: the Bothnia type [4–6], the Kimonis type [7] and the Nagashima type [8, 9]. Greither described an autosomal dominant PPK with gradual onset and a tendency to improve in the fifth decade [10]. This eponym now lacks a clear definition [11, 12] and it is likely to be genetically heterogeneous [13, 14], e.g. belonging to the EPPK group or overlapping with erythrokeratoderma [15].
Pathophysiology
Studies of a family from Bothnia in northern Sweden and three English pedigrees showed linkage centromeric to the type 2 keratin gene cluster on chromosome 12 [16–18]. Using linkage data and exome sequencing it has now been shown that this form of NEPPK is caused by dominant missense mutations in the AQP5 gene [19], encoding aquaporin 5 (AQP5). Aquaporins are a family of cell membrane proteins that allow the osmotic movement of water across the cell membrane. AQP5 is localized to the plasma membrane of the keratinocytes of the stratum granulosum. The causative mutations exert a gain-of-function effect leading to increased keratinocyte water uptake rather than transepidermal water loss [19]. Another autosomal dominant NEPPK entity has been described by Kimonis et al. [7], found in one family to be due to a mutation occurring in a highly conserved lysine residue in the V1 domain of keratin 1 [7]. In contrast, the Nagashima PPK (NPPK) is an autosomal recessive disorder almost exclusively observed in Asia and caused by nonsense mutations in the SERPINB7 gene. SERPINB7 codes for a serine protease inhibitor, whose target protease in the skin still needs to be identified [20].
Clinical features [5, 7, 8, 10, 21, 22] (see Table 65.11)
Non-epidermolytic palmoplantar keratoderma may present in the first few months of life and is usually obvious after 2 years of age. An even, variably thick, often yellow hyperkeratosis occurs over the whole surface of the foot, starting on the heel and anterior arch, spreading later to the palms (Figure 65.42). NEPPK can be transgredient and is characterized by a well-defined red border. Hyperhidrosis is usual, and dermatophyte infections and pitted keratolysis are frequent. The nails, hair and teeth are normal.
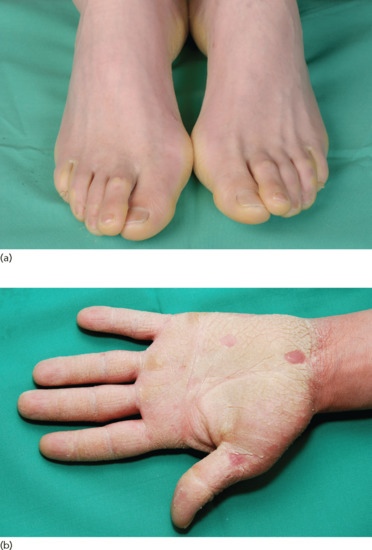
Figure 65.42 (a) Non-epidermolytic palmoplantar keratoderma suggestive of AQP5. (b) Palm of a 38-year-old man, who reported on extreme skin sensitivity to water exposure.
(Courtesy of the Department of Dermatology, University Hospital Münster, Münster, Germany.)
Differential diagnosis
Mal de Meleda as well as EXI and aquagenic PPK are important differential diagnoses.
Non-epidermolytic keratoderma reported by Sybert et al. [21] resembled MDM including autoamputation of toes, but dominant inheritance was suggested. The description of recessive non-epidermolytic keratoderma reported by Gamborg-Nielsen [6] also sounds similar to MDM.
Investigations
A biopsy serves mainly to exclude epidermolytic keratoderma. Because of the variable findings in EHK, detailed light and/or electron microscopic studies are useful in most cases of diffuse PPK [3]. Otherwise, histological changes are non-specific: orthokeratotic hyperkeratosis, hypergranulosis or normogranulosis and moderate acanthosis are often seen. Spongiosis due to dermatophyte infection may be a source of confusion.
Management
Keratolytic therapy, such as 5–10% salicylic acid in white soft paraffin, or a gel of 5–6% salicylic acid in 70% propylene glycol may be used. Occlusion with polythene for a few nights enhanced the efficacy of keratolytics. Low-dose systemic acitretin (0.2–0.5 mg/kg) may be tried – especially in patients with functional impairment. Importantly, fungal superinfection and bacterial overgrowth should be treated. Response to erythromycin has been described in NEPPK Bothnia type [22], and topical tacrolimus has been reported to be of benefit in NEPPK Nagashima type [23].
Mal de Meleda
Definition and nomenclature
Mal de Meleda is a rare autosomal recessive transgredient keratoderma named after the Croatian island of Meleda (Mljet) where it was first identified [1, 2]. The disease has been reported widely in the Mediterranean littoral.
Pathophysiology
Mal de Meleda is caused by bi-allelic mutations in SLURP1 (secreted Ly-6/PLAUR related protein 1) [3–6]. Almost all cases occur in consanguineous pedigrees and families from Croatia (including Meleda), Algeria, Israel and Tunisia share very few ancestral haplotypes, indicating founder mutations [3, 6–8]. Haplotype analysis in Western European patients with the disease showed a founder effect for the W15R mutation [9]. SLURP-1 represents an auto- and paracrine ligand of the α7 nicotinic acetylcholine receptor (nAChR) involved in the fine tuning of physiological cell activation and adhesion, e.g. by upregulated expression of transglutaminase 1, K10, p21 and caspase 3 [10]. The gene is expressed late in epidermal differentiation, in the granular layer, and particularly in association with the acrosyringium [3, 11].
Clinical features [2, 12–15]
Onset is in early childhood, and the development of hyperkeratosis is preceded by erythema. Patches of waxy ivory-yellow hyperkeratosis extend across the whole surface of the palms and soles (Figure 65.43), and on to the dorsal surfaces of the hands and feet (‘glove-and-socks’ distribution). Insular lesions of knees and elbows represent a key feature of this PPK. The erythematous component often persists in central palms and soles, typically with hyperhidrotic maceration and malodour. Fungal superinfection is common [16] (Figure 65.44). Circumferential hyperkeratosis of the fingers may lead to sclerodactyly and digital constrictions (pseudo-ainhum) (Table 65.12); nail changes include hypercurvature, thickening and koilonychia [14, 15]. Angular cheilitis and lip involvement is possible [17]. Hyperpigmented spots [18], melanoma arising within affected skin [19, 20] and Bowen disease of the sole have been reported [21]. Heterozygous female carriers may have a mild clinical phenotype [22], also pseudodominant inheritance has been reported [23].
Table 65.12 Inherited skin diseases and pseudo-ainhum.
| Keratoderma |
Vohwinkel syndrome (and Bart–Pumphrey syndrome) Clouston syndrome Mal de Meleda Papillon–Léfèvre syndrome Olmsted syndromeLoricrin keratoderma or KLICK (less common and severe than in Vohwinkel syndrome) |
| Generalized MeDOC |
Lamellar ichthyosis Erythrokeratodermia variabilis |
| Inherited epidermolysis bullosa |
Chronic epidermolysis bullosa Kindler syndrome |
| Others |
Tuberous sclerosis Erythropoietic protoporphyria |
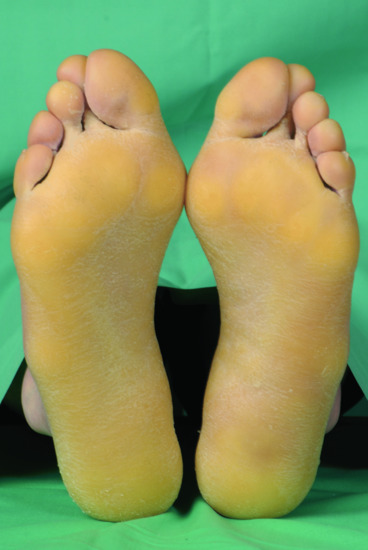
Figure 65.43 Mal de Meleda. Plantar phenotype without superinfection.
(Courtesy of the Department of Dermatology, University Hospital Münster, Münster, Germany.)
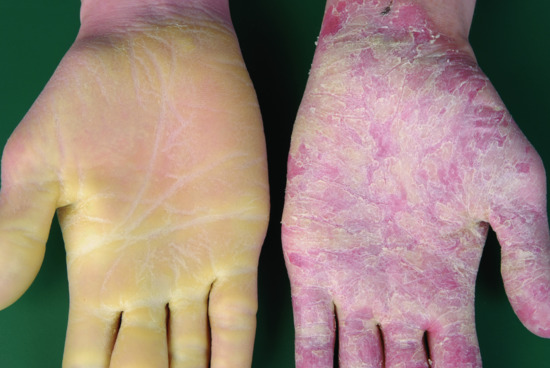
Figure 65.44 Mal de Meleda. Trichophyton rubrum superinfection of the left hand.
(Courtesy of the Department of Dermatology, University Hospital Münster, Münster, Germany.)
Differential diagnosis
See other NEPPKs (see Table 65.11). Keratoderma of Gamborg Nielsen 1985 (MIM 244850) represents a clinical variant of MDM. Keratoderma described by Sybert et al. [24] also resembles MDM.
Investigations
There is a greatly thickened corneal layer, increased stratum lucidum, with marked acanthosis, but without cytolysis [12]. The association of papillomatosis is a typical finding (authors’ experience). A prominent perivascular lymphohistiocytic infiltrate may be seen, sweat glands may be enlarged. Electron microscopy indicates a less abrupt transition from granular to cornified layers [18].
Management
Oral retinoids are effective but the hyperkeratosis may respond better than the erythema [25–29]. Excision of keratoses and split-thickness skin graft can be an option in order to relieve functional impairment. Long-term follow-up in one patient demonstrated no recurrence of keratosis on surgically treated areas [30]. Patients with a long history of MDM should be regularly seen for secondary malignancies [21].
Loricrin keratoderma
Definition and nomenclature
In two related pedigrees, Camisa and Rossana et al. delineated a diffuse transgredient honeycomb keratoderma with annular constrictions around the digits, accompanied by a mild ichthyosis [1–3]. In true Vohwinkel syndrome, there is impaired hearing but no generalized ichthyosis [4].
Pathophysiology
Loricrin keratoderma is caused by mutations in the LOR gene encoding loricrin, a glycine-rich cornified envelope protein [3, 4]. Several different single nucleotide insertions have been identified in this gene, which uniformly result in frame shift and lead to expression of an abnormal protein with an abnormal, arginine-rich C-terminal peptide containing nuclear recognition signals [3–11]. The mutant protein is transported to the nucleus, where it is thought to interfere with the regulation of cornification [12]. Direct or indirect consequences include moderate alterations on the cornified envelope, increased corneocyte fragility and abnormal epidermal barrier function with accelerated repair kinetics [13]. Transgenic mice in whom loricrin has been knocked out are largely asymptomatic [14], but mice expressing a pathogenic loricrin mutation showed generalized scaling, thickened footpads and a constricting band causing autoamputation of the tail [15, 16]. Progressive symmetrical erythrokeratoderma has been attributed in one case to a mutation in LOR [17], although these findings have not been reproduced [18] and are still debated.
Clinical features [4, 7–11]
Generalized desquamation may be noted at birth, and collodion babies are reported [6, 7, 10]. However, the ichthyosis is generally mild and may pass unnoticed. A rugose keratoderma develops during childhood, gradually extending to the confluent honeycomb pattern (Figure 65.45). The edges of the keratoderma are diffuse (in contrast to true Vohwinkel syndrome) and cicatricial bands (pseudo-ainhum) may develop around the digits. Knuckle pads and warty keratoses have been reported, but are not a prominent feature. Hence, the combination of honeycomb keratoderma with mild non-syndromic ichthyosis is typical [7], but in sporadic cases autosomal recessive KLICK syndrome (see earlier) should be considered an important differential diagnosis.
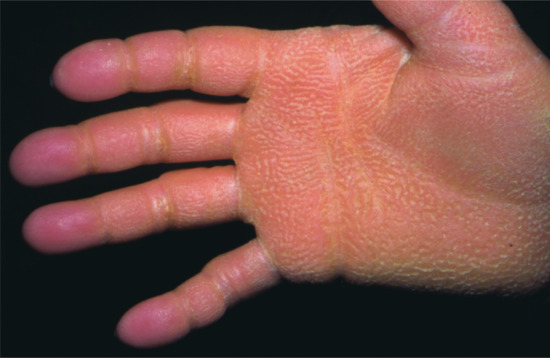
Figure 65.45 Loricrin keratoderma.
Investigations
In addition to hyperkeratosis, histological features include hypergranulosis and parakeratosis, i.e. nuclei may be retained in the stratum corneum [1, 7]. Electron microscopy shows dense intranuclear granules in granular cells, and a thin cornified cell envelope in the lower cornified layers with abnormal extracellular lamellae [13]. Immunoelectron microscopy shows the presence of loricrin in these nuclei [3, 4].
Management
The successful use of isotretinoin has been reported [1]. Results from a mutant loricrin HaCaT model demonstrate increased vascular endothelial growth factor (VEGF) release that may be the cause of pathogenic keratinocytic proliferation. Inhibitors of VEGF receptor 2 might be attractive to treat LK [19].
Striate (and focal) palmoplantar keratoderma (Table 65.13)
Definition and nomenclature
Isolated SPPK is usually transmitted as an autosomal dominant trait and is caused by defects in at least three different genes [2–4]. It is associated with a spectrum of Mendelian diseases of the desmosomes [5]. Most important, keratoderma with cardiomyopathy should be excluded.
Table 65.13 Isolated or syndromic palmoplantar keratoderma associated with keratinocytic disadhesion.
| Striate palmoplantar keratodermas (SPPK) | Naxos syndrome | Carvajal-Huerta syndrome | ||||
| Synonyms | SPPK1 | SPPK2 | SPPK3 | PPK with arrhythmogenic cardiomyopathy | PPK with left ventricular cardiomyopathy | |
| MIM | 148700 | 612908 | 607654 | 601214 | 605676 | |
| Inheritance | AD | AD | AD | AR (or AD) | AR (or AD) | |
| Gene | DSG1(Desmoglein 1) | DSP(Desmoplakin) | KRT1(V1 domain of K1) | JUP(Plakoglobin) | DSP(Desmoplakin) | |
| Age of onset | First or second decade of life | First or second decade of life | Early childhood | Early childhood,adolescence cardiomypathy | Early childhood, childhood cardiomypathy | |
| Palmoplantar key features | Striate hyperkeratosis on the flexure site of fingers and palms, more diffuse and focal changes on the soles; triggered by manual work/mechanical stress | Striate or focal keratoderma | Striate or focal keratoderma,can spread over the Achilles tendon | |||
| Other clinical findings | Hyperhidrosis and pain | Woolly hair without cardiomyopathy is possible | – | Woolly hair, dilated cardiomyopathy | Woolly hair, possibly short, keratoses on knees/elbows, right/biventricular cardiomyopathy | |
| Histology and ultrastructure | Disadhesion of keratinocytes and widening of the intercellular spaces, possibly less pronounced than in DSP mutations | Disadhesion of keratinocytes and widening of the intercellular spaces, cytoplasmic densities | Less electron dense IF within the SP, normal desmosomes, their insertion with IF network seems attenuated | Disadhesion of keratinocytes and widening of the intercellular spaces, clumping of desmosomes and IF | Wide intercellular spaces, clustering of desmosomes, and ultrastructural signs for IF disruption | |
| Remarks | Same gene as for severe dermatitis, atopy and metabolic wasting (SAM) syndrome | Same gene as for Carvajal syndrome, skin fragility woolly hair and lethal’ acantholytic EB | Similar mutation as in ichthyosis Curth–Macklin | Same gene as for acantholytic EB | Same gene as for SPPK2, skin fragility woolly hair, acantholytic EB | |
AD, autosomal dominant; AR, autosomal recessive, KIF, keratin intermediate filament; SG, stratum granulosum; SP, stratum spinosum.
Introduction and general description
Historically, the occurrence of both insular and striate keratodermas within one family led Wachters [1] to suggest a single entity, keratoderma varians. Today, several distinct keratoderma with striate and/or focal pattern are recognized; moreover, some of them may be associated with a syndromic entity. Considering the influence of environmental factors, e.g. mechanical stress, recognition of predominantly striate and/or focal presentations may however still serve as a clinical clue for diagnosis: striate hyperkeratosis is particularly but not uniquely associated with ‘keratinocytic disadhesion’. This histological clue may support the diagnosis of a desmosomal defect.
Pathophysiology
Initially, autosomal dominant striate keratoderma was mapped to the desmosomal cadherin cluster on 18q12.1 [6]. SPPK has been shown to result from haploinsufficiency for desmoglein 1 due to heterozygous mutations in DSG1 [2, 3, 7–11]. Heterozygous mutations in DSG1 are not exclusively associated with a striate pattern of hyperkeratosis as they have also been found to cause focal and diffuse PPK [12, 13]. Moreover, since carriers of heterozygous mutations in DSG1 display PPK only while the offsprings of two such heterozygous carriers can be affected by a life-threatening condition known as SAM syndrome (see earlier), DSG1 mutations are actually inherited in a semidominant fashion [12, 14].
The second SPPK locus on 6p21 is linked to dominant nonsense mutations in the desmoplakin gene (DSP) also leading to haploinsufficiency [15, 16]. The majority of genetic reports on isolated striate keratoderma show dominant DSG1 mutations [2], whereas there are several reports on striate or focal keratoderma with cardiac involvement due to recessive (or dominant) DSP mutations (see Carjaval syndrome later) [17, 18]. Finally, there is one report on a frameshift mutation in the V2 tail domain of keratin 1 [19]. The mutation is similar to those being reported for ICM [20, 21].
Clinical features
As the name implies, there is a linear pattern of skin thickening on the palms and flexor aspects of the fingers [21–23]. However, lesions on the soles may be more areate or confluent (Figure 65.46), and striate lesions can be seen in affected members of pedigrees in which other patterns of keratoderma predominate. Mechanical stress is important; pain, hyperhidrosis and mild hyperkeratosis of the knees have been reported [2]. The presence of other features, especially woolly hair, should be specifically sought, and the possibility of cardiac disease considered. In summary, striate keratoderma is a striking key feature but not a specific one.
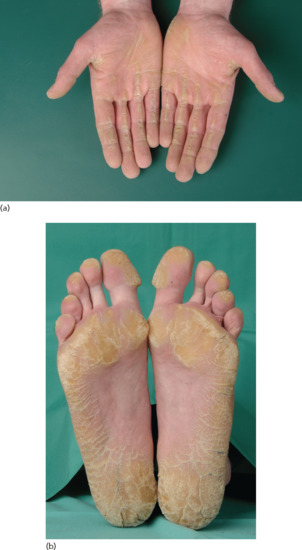
Figure 65.46 Striate and focal palmoplantar keratoderma. (a) Hands and (b) feet belong to the same patient.
(Courtesy of the Department of Dermatology, University Hospital Münster, Münster, Germany.)
Differential diagnosis
In addition to EPPKs, connexinopathies [24] and other syndromic diseases [25, 26] as well as acquired keratodermas [27] have to be considered. Importantly, Howel-Evans syndrome should be considered if focal and nummular keratoderma predominate and histology is unspecific (see Tylosis with oesophageal cancer later).
Investigations
Disadhesion of keratinocytes with widening of the intercellular spaces is an important histological clue for these cell–cell junction diseases [28]. Moreover, electron microscopy of keratoderma associated with DSP mutations show abnormal keratin filaments with loss of desmosome connections. Desmosome size seems reduced in DSG1, and perinuclear aggregation of keratin filaments seems more marked in DSP-associated disease [29].
Management
Several reports point to good response to systemic acitretin treatment and/or application of high percentage urea creams [30–32].
Punctate palmoplantar keratoderma
Definition and nomenclature
This autosomal dominant keratoderma is clinically characterized by small rounded papular lesions on the palms and soles that tend to coalesce over pressure points [1].
Prevalence
The incidence of palmoplantar keratoderma punctata (PPKP) is estimated as 1.17/100 000 individuals in Croatia [4].
Pathophysiology
The disorder has been mapped to two chromosomal regions 15q22 [5–7] and 8q24.13–8q24.21 [8]. The first locus harbours the AAGAB gene in which mutations have been found in patients with PPKP1 [9, 10]; AAGAB encodes the α- and γ-adaptin-binding protein p34 [9–16]. p34 deficiency results in increased epidermal growth factor signalling which in turn may drive keratinocyte proliferation [13]. The second locus on 8q was found to be associated with one missense mutation in the gene COL14A1 in a Chinese family [17], which needs to be confirmed for other families [15].
Clinical features [1, 2–4]
This autosomal dominant condition often has a later onset with lesions appearing in early adolescence but also up to the sixth decade of life [1]. Pinpoint keratotic papules, initially translucent and with a depression in the centre but later opaque and warty, appear on the palms and soles (Figure 65.47). Interfamilial variable severity is typical [13]. Enviromental factors and personal skincare regimes may affect the degree of hyperkeratosis [18]. In many families, small and large lesions coexist, including broader focal plantar callosities. Lesions are more florid in manual workers. There is no involvement of the dorsa of the hands or legs, nor of the knees or elbows. Hyperhidrosis and fungal infections are unusual [1]. It is not clear whether there is PPKP truly associated with an excess of malignancies [13].

Figure 65.47 Punctate palmoplantar keratoderma. Small even keratotic papules on the palms (a) and confluent hyperkeratosis on the feet of the same patient (b).
(Courtesy of the Department of Dermatology, University Hospital Münster, Münster, Germany.)
Differential diagnosis
Most important, focal keratoderma associated with malignancies such as breast and colonic adenocarcinoma [19, 20] should be differentiated (see Howel-Evans syndrome later). Autosomal dominant punctate porokeratosis (also referred to as PPKP2) may present with similar Clinical features [21]. In acrokeratoelastoidosis (referred to as PPKP3) disorganized elastic fibres are a histopathological hallmark [22].
Investigations
Punctate lesions are orthohyperkeratotic on histology with compact acanthosis and hypergranulosis with a depression in the centre of the lesion, but may also show hypogranulosis and (focal) parakeratosis [1].
Management
Careful choice of footwear and regular use of a pumice stone are useful in alleviating discomfort. Literature reports indicate no major beneficial effects of keratolytic ointments as well as topical retinoids or topical calcipotriol on the keratoses [1, 23, 24]. Systemic treatment with oral retinoids may yield a small effect, moreover depending on the dosage (0.5–1.0 mg/kg/day) [1, 3, 25]. Receptor tyrosine kinase inhibitors that are involved in p34 signalling are under development and of potential relevance for future treatment [9, 26].
Spiny keratoderma
Definition and nomenclature
Spiny keratoderma (SK) is a rare condition of unknown aetiology and exists as a hereditary/benign or idiopathic form [1–3]. Importantly, aquired digitate keratoses [4] need to be excluded. Brown [1] described the first case as ‘punctuate keratotic projections’ (‘punctate keratoderma’), and since then the disease has been described under a variety of terms, of which ‘porokeratosis’ is a misleading misnomer.
Clinical features
Fine 1–2 mm papules that project from the palmoplantar surface are described as filiform, spiked, prickly, minute digitate or music box-like spines (Figure 65.48). Unilateral presentations have been reported [9]. SK may be inherited as an autosomal dominant trait. The lesions may start in early childhood and gradually increase in number, i.e. they appear in the first up to the fifth decade [1–3]. Discomfort can be caused by a tendency to catch on clothing and other objects [9].
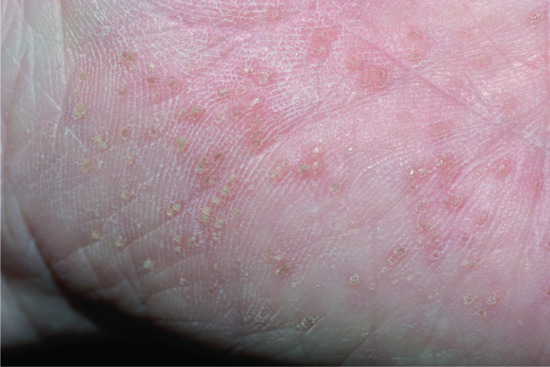
Figure 65.48 Spiny keratoderma. Present since the age of 20 on the palms and soles. Son and daughter also affected.
Differential diagnosis
Darier disease [10, 11], epidermodysplasia verruciformis [12], arsenic keratosis [4], multiple filiform verrucae, paraneoplastic follicular hyperkeratosis [13] and/or multiple minute digitate hyperkeratosis (reviewed in [4]) should be differentiated.
Investigations
Histology demonstrates dense columns of parakeratosis above a hypogranular epidermis [2, 3, 9, 14]. The absence of dyskeratosis and vacuolated keratinocytes below the parakeratotic columns differentiate the disease from porokeratosis [3, 15]. Autoimmune screen, blood investigations for full blood count, renal/liver function and radiological tests are necessary to exclude an associated condition, e.g. polycystic kidneys and liver [16], renal failure [17], tuberculosis [18], hyperlipidaemia [19], malignancy such as multiple myeloma [8, 20–23] or HIV-associated type 6 pityriasis rubra pilaris [24].
Treatment
Mechanical debridement, e.g. dermabrasion, may be more effective than topical keratolytics [9]. Etretinate or actitretin have a temporary effect [8, 12, 25]. Topical 5-fluorouracil ointment has been tried with differing results [14, 16].
Marginal papular keratoderma
Definition and nomenclature
Marginal papular keratoderma (MPK) is heterogeneous and refers to acrokeratoelastoidosis as well as focal acral hyperkeratosis, which are both characterized by papules, plaques and nodules located at the junction between the palmar and dorsal skin of the hands or feet along the thenar and hypothenar eminences (Figure 65.49) [1, 2].
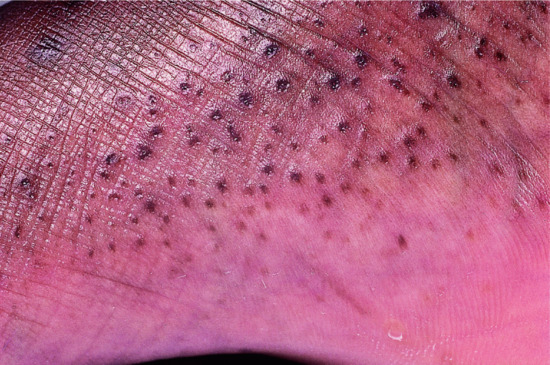
Figure 65.49 Marginal papular keratoderma (acrokeratoelastoidosis, punctate palmoplantar keratoderma type 3 (PPKP3)): crateriform punctate keratosis at the margin of the sole (Wallace line).
Pathophysiology
Autosomal dominant inheritance with linkage to chromosome 2p or sporadic occurrence has been described [4].
Clinical features and differential diagnoses
Costa [5, 6] reported 13 cases with cornified and umbilicated papules distributed along the borders of the hands and feet. He noted fragmentation and rarefaction of elastic fibres in the dermis, and introduced the term acrokeratoelastoidosis. Fiallo et al. [7] argued that the primary defect is in the elastic tissue. However, Dowd et al. [8] reported 15 cases, several familial, with similar oval or polygonal crateriform papules along the borders of the hands and feet in whom there was no solar damage or elastorrhexis. To distinguish this entity, it was termed focal acral hyperkeratosis. However, eight of Costa's 13 patients [5] appear identical. Rongioletti et al. [1] suggest unifying the marginal papular keratodermas with a pragmatic division into the hereditary type with elastorrhexis (acrokeratoelastoidosis), or without it (focal acral hyperkeratosis). The following entities may be differentiated from marginal papular keratodermas.
Punctate keratosis of the palmar creases.
This condition most commonly occurs in Afro-Caribbean or black individuals, may be transmitted in an autosomal dominant fashion and may be associated with AE and/or manual labour [9–14]. It clinically shows sharply defined 1–4 mm hyperkeratoses, i.e. hard warty lesions, confined to the palmar creases [15]. This location is possible but unusual for punctuate keratoderma [16]. Around one third of black individuals but rarely white may show hyperkeratotic pits involving the flexural creases of the palms [17, 18].
Keratoelastoidosis marginalis [19, 20]
is an acquired condition also known as digital papular calcific elastosis. It presents with degenerative collagenous plaques that are firm, sometimes concave, forming a linear band principally around the web of the thumb and index finger at the margin of the volar and dorsal surfaces. There is histological evidence of solar damage [21–23].
Mosaic acral keratosis [24]
reported in African patients with widespread polygonal papular lesions over the ankles and shins may present with marginal keratoderma [25].
Investigations
Histology shows focal orthohyperkeratosis. The key microscopic finding in acrokeratoelastoidosis is the presence of massive elastosis, whereas no elastic fibre changes are present in focal acral hyperkeratosis [1, 13].
Cole disease
Definition and nomenclature
This genodermatosis features punctuate keratoderma and pigmentary anomaly. Cole disease (CD) is characterized by congenital or early-onset punctate keratoderma with irregularly shaped hypopigmented macules of the extremities [1–4].
Pathophysiology
Ultrastructural analyses suggested a defective melanosome transfer from melanocyte to keratinocyte, as melanocytes show numerous melanosomes in the cytosol and dendritic extensions, whereas neighbouring keratinocytes are almost devoid of melanin [2]. Eytan et al. [4] recently demonstrated that specific missense mutations in the gene ENPP1 underlie the disorder. The gene product ENPP1 is a cell surface protein that catalyses the hydrolysis of adenosine triphosphate to adenosine monophosphate and generates extracellular inorganic pyrophosphate, which is a major inhibitor of mineralization. While recessive mutations in ENPP1 cause a range of inherited forms of ectopic calcifications, dominant mutations in this gene cause CD. CD-causing mutations affect a specific domain of ENPP1 which regulates insulin signalling. The fact that insulin signalling impact on epidermal growth factor signalling suggests a pathophysiological link between CD and punctate keratoderma due to AAGAB mutation [5].
Clinical features
The disease manifests during the first year of life. Affected individuals present with a relatively mild focal or punctuate keratoderma (Figure 65.50). In addition, they develop sharply demarcated irregular macules with varying degrees of hypopigmentation, mainly located over the extremities [1–3]. There might be an early-onset calcific tendinopathy or calcinosis cutis [4].
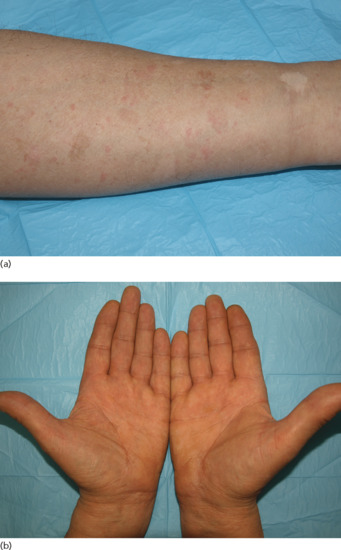
Figure 65.50 Cole disease. (a) Forearm with hypopigmentation and (b) mild keratoderma in the same patient.
(Courtesy of Dr Y. Gilaberte, Hospital San Jorge, Huesca, Spain.)
Differential diagnosis
Epidermolysis bullosa simplex with mottled pigmentation and Naegeli–Franceschetti–Jadassohn syndrome (see Chapter 70) may be confused with CD.
Investigations
Histology shows hyperorthokeratosis, hypergranulosis and acanthosis. Ultrastructure confirms a reduction of the melanin content of keratinocytes with disproportionately large melanosomes of melanocytes. The number of melanocytes is normal.
Transient aquagenic keratoderma
Definition and nomenclature
This peculiar, mainly palmar, disorder represents a mild keratoderma that is triggered or exacerbated by contact with water or sweat [1].
Pathophysiology
Most cases of transient aquagenic keratoderma (TAK) appear to be acquired, but autosomal recessive [1] or dominant cases have been described [2]. The disease may be associated with the use of cyclo-oxygenase 2 inhibitors [3]. The relationship with other diseases such as asthma is unknown [4]. It may be differentiated from hereditary papulotranslucent acrokeratoderma (MIM 101840), in which lesions are persistent once they appeared [5, 6].
Clinical features [1–4, 6, 7, 8–13]
Affected individuals aged 6–45 years, more often women, typically show a subtle keratoderma appearing after a few minutes of immersion of their hands in water or after sweating (Figure 65.51). Only in some patients are the soles affected. One characteristic sign might be that patients bring with them a vessel to immerse their hands in water (‘hands in the bucket’ sign) [13]. The painful, burning or itching, whitish papular lesions are associated with dilated acrosyringeal ostia, which can be seen by dermoscopy [14]. Lesions subside shortly after drying the hands, leaving minimal hyperkeratosis in the centre of the palms.
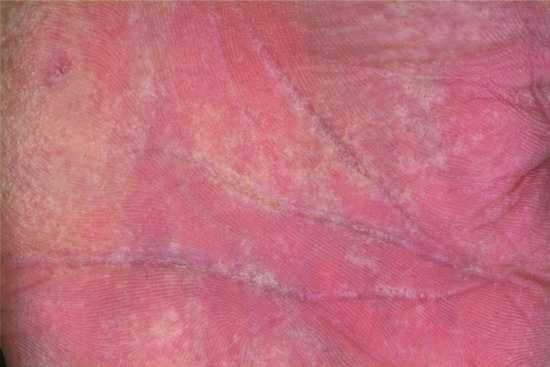
Figure 65.51 Transient aquagenic keratoderma showing whitish maceration of the palmar skin following immersion.
(Courtesy of Dr G. Kavanagh, Royal Infirmary of Edinburgh, UK.)
Differential diagnosis
Transient aquagenic keratoderma should be differentiated from other keratodermas that are sensitive to exposure to water, e.g. Bothnia type NEPPK [15, 16]. Importantly, aquagenic wrinkling of the palms (Figure 65.52) is associated with cystic fibrosis (in about 50% of affected patients) and can be observed in up to 10% of heterozygous CFTR gene mutation carriers [17–20].
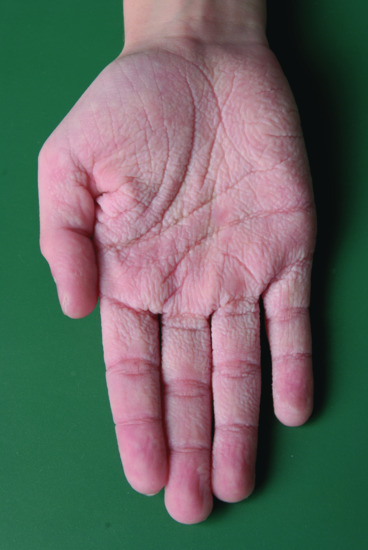
Figure 65.52 Aquagenic palmar wrinkling in cystic fibrosis.
Investigations
Histology might reveal hyperplasia of the eccrine sweat glands, with slight dilation of the lumen [4].
Treatment
Treatment with 20% aluminium chloride hexahydrate followed by urea cream [4, 11], or botulinum toxin [21] may lead to significant improvement.
SYNDROMIC KERATODERMA
Palmoplantar keratoderma and cardiomyopathy (see Table 65.13)
Whenever woolly hair is associated with any kind of PPK, especially in striate keratoderma, a search for possible cardiac abnormalities is recommended [1, 2]. Like isolated striate keratoderma (type I/II), the following syndromic disorders are histologically characterized by keratinocytic disadhesion [3].
Naxos syndrome
Definition
The disease is associated with arrhythmogenic right ventricular cardiomyopathy/cardiomyopathy [4] and is accompanied by PPK and woolly hair [5, 6].
Pathophysiology
The first reported seven pedigrees from the Greek islands of Naxos and Milos have been found to share a 2-bp deletion within the JUP gene encoding plakoglobin [7], a cell junction protein found in desmosomes in the epidermis and cardiac muscle. As can be seen in other desmosomal genodermatoses [8], different mutations of JUP show a large phenotypic spectrum, e.g. mutations allowing expression of altered plakoglobin may show mild skin fragility, keratoderma and woolly hair only [9]. Of note, autosomal dominant JUP mutations underlie isolated arrhythmogenic right ventricular cardiomyopathy [10] and a Naxos variant with leukonychia and oligodontia [11]. Finally, complete loss of plakoglobin due to homozygous nonsense mutations may lead to acantholytic epidermolysis bullosa [12].
Clinical features
Woolly hair develops from birth while diffuse or striate hyperkeratoses of the palms and soles appear during the first year of life, when the skin starts to become mechanically stressed. Cardiomypathy manifests by adolescence and shows 100% penetrance [13].
Carvajal-Huerta syndrome
Definition
The disease represents a cardiocutaneous syndrome with NEPPK, woolly hair and dilated cardiomyopathy [1].
Pathophysiology
Recessive desmoplakin (DSP) mutations producing a premature stop codon and leading to a truncated protein cause this disorder [14]. Other genetic defects in DSP have been found to generate a wide range of phenotypes. A 10 amino-acid insertion in DSP exerting a dominant negative effect on desmosomal assembly has been found to be associated with cardiomyopathy, and mild hyperkeratosis of the elbows and knees [15]. Heterozygous carriers of the missense mutation p.S299R showed isolated arrhythmogenic right ventricular cardiomyopathy without cutaneous phenotype [16]. Complete loss of the tail domain of desmoplakin presents as acantholytic epidermolysis bullosa [17]. Hence, dosage of desmoplakin is critical in maintaining epidermal integrity as illustrated by compound heterozygote patients carrying one null allele and one missense mutation, who developed pronounced skin fragility and alopecia without cardiac anomalies [18].
Clinical features
The three Equadorian pedigrees initially described by Carvajal-Huerta displayed recessive inheritance of a striate keratoderma with woolly hair and associated cardiomyopathy developing in the teenage years [1]. As such, the syndrome resembles Naxos disease, but it presents at a younger age with bilateral predominantly left ventricular involvement leading to early heart failure with cardiac enlargement and disrupted cardiac contraction [19, 20].
Investigations
Histological studies demonstrate large intercellular spaces of suprabasal keratinocytes (keratinocytic disadhesion). Ultrastructure may reveal clumping of desmosomes [3, 14] or signs for intermediate filament disruption such as perinuclear localization of keratin in suprabasal keratinocytes [1]. On resting electrocardiogram, affected patients exhibit repolarization and/or depolarization abnormalities; structural/functional abnormalities of the ventricle(s) will be found on echocardiography or cardiac MRI [15, 21].
Clinical features
Differential diagnosis
Of note, a novel entity combining woolly hair, hypotrichosis and PPK but no cardiac abnormalities was recently reported. The disease was found to be caused in one report by a mutation in KANK2 [22].
Management
Investigate cardiac function in patients with striate keratoderma, as early diagnosis and intervention may improve outcome. Depending on the myocardial symptoms, implantation of an automatic cardioverter defibrillator with/without antiarrhythmic drugs, or, at the end stages, heart transplantation is an option [23]. Screening of possibly affected family members should be initiated considering dominant as well as recessive inheritance [15].
Resources
Further information
http://anpat.unipd.it/ARVC/ (last accessed April 2015).
Palmoplantar keratoderma and hearing impairment
The association of hereditary keratoderma with hearing loss is most commonly caused by defective connexin function [1, 2], whereby PTCs in the gap junction beta 2 (GJB2) gene encoding connexin 26 are the most common cause of non-syndromic autosomal recessive deafness [3–5]. In several pedigrees with diffuse or transgredient autosomal dominant keratoderma and varying degrees of prelingual deafness, missense mutations in connexin 26 have been found (MIM 148350) [6–10].
Notwithstanding, in ‘palmoplantar keratoderma with hearing impairment’ establishment of a thorough family history is crucial considering that maternal inheritance possibly points to a rare mitochondrial type of keratoderma [11].
Vohwinkel syndrome and Bart–Pumphrey syndrome
Definition and nomenclature
Vohwinkel [12] and Wigley [13] independently reported honeycomb-like keratoderma associated with stellate keratoses on the knuckles and the formation of circumferential bands around digits (pseudo-ainhum). Vohwinkel's family had moderate sensorineural deafness [14], and subsequent cases have confirmed autosomal dominant inheritance [15–18].
Pathophysiology
In three unrelated English, Spanish and Italian pedigrees with ‘classic’ Vohwinkel syndrome, Maestrini et al. [19] found the same mutation, p.D66H, in GJB2 encoding connexin 26, a gene also implicated in Bart–Pumphrey syndrome [20] as well as KID syndrome [21]. The phenotypic variants related to mutations in GJB2 are reviewed in Table 65.5. A full list of dominant and recessive mutations in GJB2 can be found on the connexin–deafness homepage (http://davinci.crg.es/deafness/ last accessed April 2015) [22].
Connexin 26 is expressed in the cochlea where it may permit the recycling of potassium to endolymph [23, 24]. In skin, the protein is found in palmoplantar epidermis and sweat glands, and is up-regulated in conditions such as psoriasis [25, 26]. Patients homozygous for mutations preventing any expression of connexin 26 have no discernible skin phenotype, indicating that dominant negative or gain-of-function effects may be involved in the pathogenesis of cutaneous manifestations of mutations in GJB2.
There are various mechanisms which may underlie the phenotypic effects of connexin gene mutations [27, 28]: mutant proteins are sometimes unable to form functional gap junction channels; in other cases, aberrant trafficking of mutant proteins may prevent some of them reaching the cell surface membrane and lead to their accumulation in organelles triggering ER stress response [29]; finally, mutant proteins may exert deleterious effects on other connexins’ function (e.g. c.del42E has been shown to exert a dominant negative effect on the wild-type connexin 43) [30].
Clinical features [12, 13–18, 31]
Palmoplantar keratoderma begins in childhood as shiny or translucent papular hyperkeratosis, gradually becoming confluent on the hands and feet. Striate lesions may be seen. Warty papules on the knuckles and other extensor sites coalesce into the pathognomonic ‘starfish’ keratoses. The edge of the keratoderma at the wrists and Achilles tendon consists of spiky digitate hyperkeratotic projections onto normal skin. This contrasts with the diffuse edge seen in LK [32]. Multiple keratoses on the digits produce circumferential hyperkeratosis, which predisposes to the formation of cicatricial bands and autoamputation (Figure 65.53). The little finger and fifth toe are commonly affected. A high-tone sensorineural hearing loss [17] is probably present from birth, but may escape detection unless tested. It does not appear to be progressive. Focal epilepsy has been described in only one patient with Vohwinkel syndrome so far [33].
Figure 65.53 Vohwinkel syndrome: (a) ‘starfish’ lesions; and (b) pseudo-ainhum.
Bart–Pumphrey syndrome
Investigations
Histology may be non-specific, showing papillomatosis with prominent orthohyperkeratosis.
Management
Vohwinkel syndrome has been successfully treated by etretinate [18], acitretin [37] and the cicatricial bands released surgically [38–40]. Appropriate rehabilitation (hearing aids, speech therapy, language training, cochlear implantation, educational programmes) for the hearing loss are important to achieve auditory speech recognition effectively [41].
Mitochondrial palmoplantar keratoderma with hearing impairment (Table 65.14)
Table 65.14 Palmoplantar keratoderma in ectodermal dysplasias and mitochondrial diseases.
| Clouston syndrome | Odonto-onycho-dermal dysplasia | Papillon–Léfèvre syndrome | Mitochondrial PPK | |
| Synonyms | Hidrotic ectodermal dysplasia 2 (HED2) | PPK with cystic eylids, hypodontia and hypotrichosis, eccrine tumours with ectodermal dysplasia, Schöpf–Schulz–Passarge syndrome | Keratoderma with periodontitis | Mitochondrial sensorineural hearing loss with PPK |
| MIM | 129500 | 224750, 257980 | 245000 | 590080 |
| Inheritance | AD | AR (minor symptoms if heterozygous) | AR | Maternal |
| Gene | GJB6 Connexin 30 |
WNT10A Wingless-type MMTV integration site family, member 10A |
CTSC Cathepsin C |
MT-TS1 (A7445G, mtDNA) Mitochondrial tRNA serine 1 |
| Age of onset | Early life | Early life | Before second year of life | Early childhood |
| Palmoplantar key features | Combination of nail dystrophy, hypotrichosis and PPK (papillomatous, fissured and transgredient), pseudo-anihum possible | Macerated PPK with late onset, acral hyperhidrosis | Transgredient PPK, preceded by erythema, hyperhidrosis, pseudo-ainhum possible | PPK with early honeycomb appearance, mainly plantar, focal or diffuse, alongside the fingers, callus on the heels and toes, with or without erythematous border |
| Other clinical findings | Dystrophic nails, often small and thickened, with clubbing, pale and fine hair with (progressive) hypotrichosis, eyebrows sparse or absent, hyperpigmentation over the joints | Cysts of the eyelids (not necessarily in early age), reduced primary and/or lack of secondary dentition, nail dystrophy, hypotrichosis, smooth tongue, follicular and other adnexal tumours occur in older patients | Periodontosis, severe gingivitis, loss of deciduous teeth by the age of 4–5 years, psoriasiform plaques on the knees and elbows, tendency to pyogenic infection, hair may be sparse Haim–Munk syndrome (MIM 245010) +onychogryphosis, arachnodactyly, and acro-osteolysis |
Progressive sensorineural hearing loss with onset during childhood, variable severity, coincidence with warty linear epidermal naevus, ichthyosis vulgaris and/or cholesteatoma are described |
| Histology and ultrastructure | Targeted mutation analysis, exclude pachyonychia congenita | Histology of hidrocystomas | Hyperkeratosis with irregular parakeratosis and a moderate perivascular infiltrate, ultrastructural lipid-like vacuoles in granulocytes and corneocytes, tonofilament reduction, irregular keratohyalin granules | Increased granular layer, moderate acanthosis without epidermolytic changes |
| Remarks | Special hair care, wigs, artificial nails, keratolytics | Different manifestations may be regarded as allelic disorders | Good response to retinoids, combination with antibiotics and dental care lessening the gingival inflammation and saving the teeth | Targeted mutation analysis, hearing aids, cochlear implantation, speech therapy, educational programmes |
AD, autosomal dominant; AR, autosomal recessive; MMTV, mouse mammary tumour virus; PPK, palmoplantar keratoderma; SCC, squamous cell carcinoma.
In a Scottish family, Reid et al. [1] reported familial progressive post-lingual deafness with variable plantar keratoderma (Figure 65.54) caused by a specific mutation in mitochondrial DNA (mtDNA), encoding both a serine transfer RNA and the first subunit of cytochrome oxidase [2].
Figure 65.54 Mitochondrial keratoderma with deafness.
Pathophysiology
The same point mutation, namely m.7445A>G, was identified in a New Zealand family, in Japanese patients [3, 4] and other pedigrees [2, 5, 6], who demonstrated PPK, mainly plantar, and sensorineural deafness. Among those patients with clinical and audiological features of hearing loss due to mitochondrial mutations, around one quarter may have the A7445G substitution [7]. Individual differences in the mitochondrial contents of wild-type and mutant DNA (‘heteroplasmy’) affect the resulting phenotype including the severity of hearing loss, keratoderma and age of onset. Of note, mtDNA mutations may be responsible for ∼3.5% of patients with hereditary deafness; but other mutations, e.g. m.1555A>G, are not associated with keratoderma [8].
Palmoplantar keratoderma and cancer (Table 65.15)
Table 65.15 Palmoplantar keratoderma associated with cancer.
| Huriez syndrome | Tylosis oesophageal cancer (TOC) | Palmoplantar keratoderma, sex reversal and cancer | |
| Synonyms | Scleroatrophic and keratotic dermatosis of limbs | Howel-Evans syndrome | Palmoplantar hyperkeratosis with SSC and 46,XX sex reversal, true hermaphroditism with PPK |
| MIM | 181600 | 148500 | 610644 |
| Inheritance | AD | AD | AR |
| Gene | Unknown (Chromosome 4q23) |
RHBDF2 (iRhom2) |
RSPO1 (R-spondin protein 1) |
| Age of onset | Early infancy | Puberty | Infancy (?) |
| Palmoplantar key features | Accentuated scleroatrophy on the palms and fingers, hyperkeratosis and dry skin, soles less often affected, dermatoglyphics often absent | Focal non-epidermolytic PPK | Mild PPK with sclerodactyly, nail hypoplasia, hyperhidrosis |
| Other clinical findings | Malignant degeneration of the affected skin at a young age with a 100× higher risk of SCC | Oral leukokeratosis, follicular hyperkeratosis, high life time risk of squamous oesophageal cancer (95% by age of 65) | Male patient: hypospadia, hypogenitalism, gynaecomastia Female patient: true hermaphroditism (clitorial enlargement, ambiguous external genitalia, ovotesticular gonads) Premature loss of permanent teeth due to chronic periodontal disease, predisposition to SCC and laryngeal carcinoma |
| Histology and ultrastructure | Hypergranulosis and acanthosis, reduced numbers of Langerhans cells | Non-epidermolytic hyperkeratosis, abnormal cytoplasmic staining of iRhom2 in the epidermis | Non-epidermolytic hyperkeratosis, karyogram, shares features of Huriez syndrome |
| Remarks | Acitretin may reduce painful hyperkeratosis and incidence of skin cancer | Genetic counselling, screening of mutation carriers and preventive endoscopy | Karyogram in male or female patient: 46,XX (SRY–) |
AD, autosomal dominant; AR, autosomal recessive; PPK, palmoplantar keratoderma; SCC, squamous cell carcinoma.
Skin malignancies arising from PPK skin have been observed in mutilating keratoderma [1], porokeratosis [2] and Clouston syndrome [3] as well as non-syndromic focal or diffuse PPKs [4, 5]. Melanoma of the affected skin has been reported in various keratodermas [6–12]. It may be more common in Japanese patients, because of the higher incidence of acral melanoma in this population [13]. The reported association between EPPK and breast/ovarian cancer [14, 15] is not a general feature of the disease. Reports on punctate/focal keratoderma with malignancies [16–18] may include cases of Howel-Evans syndrome (see later) and should be differentiated from punctate PPK due to AAGAB mutation, which is very probably not associated with malignancies (see Buschke–Fischer–Brauer syndrome earlier).
Huriez syndrome
Description
This syndrome represents a prototypic form of a cancer-prone keratoderma [19–21], in which the risk of squamous cell carcinoma of the affected skin is increased by around 100-fold [22–24]. The disease is characterized by a triad of diffuse scleroatrophy of the hand, mild PPK and hypoplastic nail changes [25–28] (Figure 65.55). Kindler disease, cryptic forms of junctional epidermolysis bullosa as well as dyskeratosis congentia must be differentiated.
Figure 65.55 Tylosis with oesophageal cancer (Howel-Evans syndrome). (a) Keratoderma and (b) oral mucosal lesions.
(Courtesy of Professor W. R. Tyldesley, Liverpool University School of Dentistry, Liverpool, UK and Dr M. S. Lewis-Jones, Ninewells Hospital, Dundee, UK.)
Tylosis oesophageal cancer/Howel-Evans syndrome
Pathophysiology
The disorder maps to a 42.5-kb segment of chromosome 17q23 [29–31]. The region is commonly deleted in oesophageal carcinomas [32]. Missense mutations in RHBDF2 (rhomboid 5 homologue 2) encoding an intramembrane protease underlie the disease as first shown by Blaydon et al. [33] and confirmed recently [34, 35]. RHBDF2 regulates the maturation of TNF-α convertase (TACE) in the skin [36, 37]. Functional data suggest that mutant RHBDF2 increase signalling through EGFR resulting in hyperproliferation and dysregulation of wound repair, which might stimulate the subsequent development of precancerous lesions [33].
Clinical features
Howel-Evans described two families with autosomal dominant keratoderma associated with later development of oesophageal cancer [38]. Pressure points of the sole are predominantly affected, and less so the palms [39, 40] (Figure 65.56). There is variable oral leukokeratosis and follicular accentuation. Thirty-seven per cent of the original affected family members developed oesophageal cancer. A further, extensive German American family has been reported, with an increased (38-fold) risk of oesophageal cancer [29]. The disorder can be distinguished from PC by the absence of nail changes [17].

Figure 65.56 Huriez syndrome: (a) keratoderma, and (b) atrophic skin over the dorsa of the hand and sclerodactyly.
(Courtesy of Dr M. van Steensel, Department of Dermatology, University of Maastricht, the Netherlands.)
Palmoplantar keratoderma, sex reversal and cancer
Description
This form of PPK entity is inherited in an autosomal recessive fashion and is characterized by female to male sex reversal in females. Consequently, most affected individuals with this keratoderma present as male individuals with or without signs of hypogenitalism. (This is in contrast with true hermaphroditism being characterized by the coexistence of male and female gonadal structures.) Sex reversal is most often due to translocation of the SRY gene encoding the testis determinating factor (XX males SRY+) [1]. In PPK with sex reversal and cancer, male patients carry a normal female chromosome set without the SRY gene (XX male, SRY–) [2], but also XY males without sex reversal may display the keratoderma. Interestingly, the skin phenotype is reminiscent of Huriez syndrome; affected patients have a predisposition to squamous cell carcinoma [2–5].
Pathophysiology
Homozygous loss-of-function mutations in RSPO1, encoding respondin 1, were shown to cause scleratrophic PPK associated with either female-to-male sex reversal and squamous cell carcinoma [6] or true hermaphroditism, congenital bilateral corneal opacities, onychodystrophy and hearing impairment [7]. R-spondins are a small family of growth factors interacting with the FZD–LRP receptor complexes [8]. Mutation in RSPO1 were shown to be associated with larger intercellular spaces suggesting that the RSPO1 defect impairs desmosomal junctions [6]. Disruption of R-spondin 1 remarkably illustrates the fact that mutations in a single gene, namely RSPO1, can lead to complete female-to-male sex reversal even in the absence of the testis determining factor (SRY) [8]. Of note, the pedigree of the affected families may show a recessive mode of inheritance, in which only males seem to be affected due to the female-to-male sex reversal phenotype.
Palmoplantar keratoderma in ectodermal dysplasia and related diseases (see Table 65.14)
Clouston syndrome (hidrotic ectodermal dysplasia type 2)
Description
The combination of small dystrophic nails developing in early infancy with hypotrichosis in conjunction with papillomatous and fissured transgredient keratoderma [1–3] is suggestive [4], but the presentation may resemble PC [5].
Pathophysiology
This autosomal dominant ectodermal dysplasia, is caused by mutations in the gap junction β-6 (GJB6) gene encoding connexin 30 [6, 7], which is a potential target of p63 [8].
Odonto-onycho-dermal dysplasia
Description
This term encompasses a large and heterogeneous group of autosomal recessive disorders that are allelic sharing common various dental anomalies in association with PPK and nail dystrophy [9–11]. Several specific subtypes have been recognized based on specific clinical manifestations such as Schöpf–Schulz–Passarge syndrome. This presentation is characterized by hypotrichosis, nail fragility, early loss of deciduous teeth, hydrocystomas of the eyelids or other follicular and adnexal tumours occurring in older patients [12–15]. The diffuse PPK may develop due to multiple palmoplantar eccrine syringofibroadenoma [16].
Pathophysiology
The diseases are caused by mutations in the WNT10A gene encoding a signalling molecule expressed in skin critical for the development of ectodermal appendages [9, 10]. Phenotypic heterogeneity is the rule with mutations in WNT10A causing disorders ranging from monosymptomatic severe oligodontia to Schöpf–Schulz–Passarge syndrome. Of interest, heterozygotes (mostly males) can also display clinical features such as tooth and nail anomalies [10].
Resources
Further information
Ectodermal Dysplasia Society (www.ectodermaldysplasia.org).
National Foundation for Ectodermal Dysplasias (NFED) (www.nfed.org).
(All last accessed January 2015.)
Papillon-Léfèvre and Haim-Munk syndrome
Description
In Papillon–Léfèvre syndrome, redness and thickening of the palms and soles is associated with periodontitis and frequent bacterial skin infections (Figure 65.57). Hyperkeratotic lesions can also affect the elbows and knees, and pseudo-ainhum (see Table 65.12) has also been described in this syndrome. Dural and choroid plexus calcifications have also been reported. The prevalence has been estimated as 1–4 in 1 million [1–9]. Haim–Munk syndrome is allelic with Papillon–Léfèvre syndrome, combining its features with onychogryphosis, arachnodactyly and acro-osteolysis [10–12]; this latter condition has been reported mostly in a single family of Jews of South Indian origin (so-called ‘Cochin Jews’) [13]. Retinoids have been shown to be of benefit to patients, including in the treatment of oral disease and the prevention of autoamputation [14, 15].
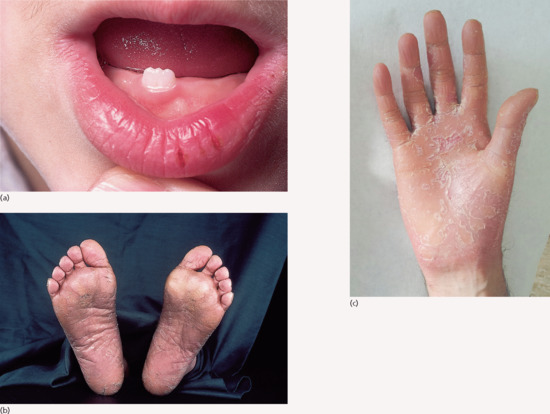
Figure 65.57 Papillon–Léfèvre syndrome: (a) Loss of dentition. (b) Diffuse plantar hyperkeratosis. (c) Psoriasiform palmar phenotype.
Pathophysiology
The conditions are both caused by homozygous mutations in the CTSC gene encoding the lysosomal protease cathepsin C [12, 16, 17]. Cathepsin C is expressed in various tissues, i.e. cells of the immune system such as nuclear leukocytes, in the lung, kidney and other epithelial tissues. Its main functions are protein degradation and proenzyme activation. Thus, the activity of several critical proteases is decreased in Papillon–Léfèvre syndrome [18]. Neutrophil phagocytosis and reactivity to T- and B-cell mitogens are impaired [19–21] explaining the predisposition to pyogenic infection, which may also involve internal organs [22–24]. Also high levels of oxidative stress markers have been confirmed [25] and natural killer (NK) cell cytotoxicity seems to be impaired [26]. Severe gingivitis and periodontitis affect both deciduous and permanent dentition leading to loss of teeth unless treated [5–7, 27, 28]. Virulent Gram-negative organisms invade the alveolar socket, usually including Actinobacillus actinomycetemcomitans [29–31]. Moreover, there are some reports on melanoma and/or squamous cell carcinoma associated with cathepsin C deficiency [32–34].
Olmsted syndrome
Definition and nomenclature
The disease often presents sporadically, and is clinically characterized by severe mutilating transgredient keratoderma with prominent periorificial hyperkeratosis. Recently, autosomal dominant, autosomal recessive and X-linked recessive forms have been confirmed and the responsible genes identified [1, 2, 7–10].
Epidemiology
A total of 65 cases have been reported worldwide, of which only 16 cases were familial with different modes of inheritance [8–12].
Pathophysiology
The disease may be added to the list of skin channelopathies: gain-of-function mutations in the transient receptor potential vanilloid 3 gene (TRPV3) were identified as a cause for autosomal dominant OLS [8]. Sporadic cases are caused by de novo dominant mutations [12]. Autosomal recessive inheritance of mutations in the same gene have also been reported [10]. TRPV3 encodes a critical element for a member of the TRP cation selective ion channels that are involved in the regulation of skin barrier formation, hair growth, epidermal differentiation (through TGF-α/EGFR signalling), skin inflammation, pain and pruritus [13, 14].
In contrast, patients suffering from an X-linked recessive OLS variant with alopecia universalis and severe nail dystrophy were shown to carry specific mutations in MBTPS2, the same gene that is associated with IFAP, KFSD and BRESEK/BRESHECK syndrome [9, 15–17] (see Ichthyosis follicularis–atrichia–photophobia syndrome). As such, it proved correct that there are a number of mutation-specific phenotypes due to this gene defect as predicted by Oeffner et al. [15]. MBTPS2 encodes a zinc metalloprotease essential for cholesterol homeostasis and ER stress response [15].
Clinical features
Onset is in the first year of life, with symmetrical sharply defined palmar and plantar keratoderma surrounded by erythema, and flexion deformities, constriction or spontaneous amputation of the digits [1] (Figure 65.58). The disease tends to have a slow, but progressive course. The keratotic lesions are pruritic and mildly painful with pressure [4, 7, 18]. Periorificial plaques present with erythema and warty hyperkeratosis involving the mouth and perianal regions. Massive hyperkeratosis and/or fissuring of the gluteal cleft may cause pain and considerable discomfort. Keratoses extending to the flexor sites of the forearms or knees may show a follicular and striate aspect [3, 4]. Alopecia, nail and tooth anomalies, joint laxity and corneal dystrophy have been observed [7, 19]. Recurrent skin infections [20] and the development of squamous cell carcinoma or malignant melanoma has been reported in a considerable number of cases of OLS [7, 8, 21, 22]. High IgE levels with eosinophilia, erythromelalgia and deafness have been rarely observed in association with TRPV3 mutations [23–26].
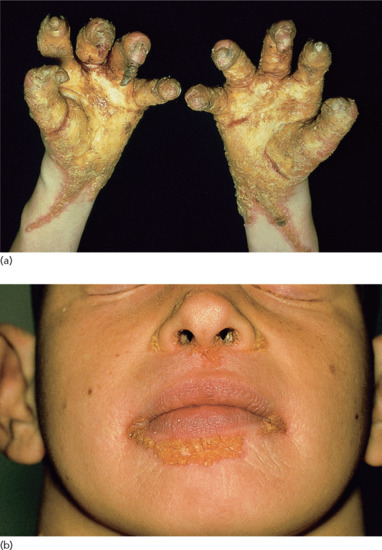
Figure 65.58 Olmsted syndrome: (a) gross keratoderma with striate features, and (b) periorificial hyperkeratosis.
(Courtesy of Professor R. K. Winkelmann, Mayo Clinic, Scottsdale, AZ, USA.)
Differential diagnosis
Palmoplantar keratodermas with periorificial lesions and digital constrictions may be reminiscent of MDM, acrodermatitis enteropathica, mucocutaneous candidiasis, psoriasis inversa, hidrotic ectodermal dysplasia (Clouston syndrome), PC, Papillon–Léfèvre syndrome and Vohwinkel keratoderma [6, 7].
Investigations
Histopathology reveals psoriasiform hyperplasia, orthohyperkeratosis and parakeratosis with perivascular inflammatory infiltration including increased numbers of mast cells [8, 27]. Cytochemical staining indicates hyperproliferation of the epidermis [26, 28].
Management
Treatment of the disorder is problematic. Variable improvement has been seen with etretinate [29, 30] or acitretin [12, 18]. Topical keratolytics, emollients and corticosteroid as well as calcineurin inhibitors may offer temporary relief of hyperkeratosis, pain or itching [7, 23, 31]. Excision and skin grafting of severe keratoderma showed favourable long-term clinical results in some patients [32]. Erlotinib (an EGFR inhibitior) led to thinning of keratoderma and resolution of perioral plaques within 3 weeks as shown in one female patient. The initial significant response was not fully maintained over the years [33].
Palmoplantar keratoderma and ophthalmic manifestations
Oculocutaneous tyrosinaemia (tyrosinaemia type II)
Definition and nomenclature
Tyrosine aminotransferase deficiency causes herpetiform corneal ulcers and painful palmoplantar keratoses with progressive mental impairment [1, 2].
Pathophysiology
This autosomal recessive condition is caused by bi-allelic mutations in TAT encoding tyrosine aminotransferase [3]. Tyrosine aminotransferase deficiency impacts on the degradation pathway of tyrosine and phenylalanine [4–6]. The keratinocytic ultrastructure reveals clumped tonofilaments with adherent globoid keratohyalin granules suggesting enhanced microfilament aggregation due to an excessive amount of intracellular tyrosine [7].
Clinical features
In the first year of life, photophobia and corneal ulcers occur. A year or two later, erythematous crusts appear on the pressure-bearing areas of the soles (Figure 65.59), followed by painful circumscribed hyperkeratosis [1, 2], typically making the child walk on the toes. The keratoses vary from gross keratoderma to patchy hyperkeratotic yellow-white papules. Bullous lesions and hyperhidrosis are sometimes seen. In incomplete forms, keratoderma may be the presenting feature [8], although conversely keratoderma may be delayed until the second decade [9]. Unless correctly treated, behavioural problems arise within a few years and progressively worsen, ending in inanition or death [10].

Figure 65.59 Oculocutaneous tyrosinaemia (Richner–Hanhart syndrome) with callosity-like hyperkeratoses.
Investigations
Elevated tyrosine levels in newborn screening by tandem mass spectrometry and analysis of the tyrosine aminotransferase gene can confirm the diagnosis. In urine, high levels of tyrosine and its metabolites are present [11]. Slit lamp examination may reveal tyrosine crystals in ocular lesions [12]. Histology shows acanthosis with hyperkeratosis with thickening of the granular layer [7].
Management
In general, reduction of plasma tyrosine can be achieved by restricting the intake of natural protein. To avoid deficiency of essential amino acids a phenylalanine- and tyrosine-free amino-acid formula is used (plasma tyrosine level below 600 μmol/L). Early initiation of diet causes prompt resolution of the ocular and cutaneous symptoms and prevents the development of mental manifestations [11, 13–15].
Palmoplantar keratoderma and neurological manifestations
This category largely overlaps with the neuro-ichthyotic syndromes, e.g. CEDNIK, SLS and Chanarin–Dorfmann syndromes as well as TTD (see Tables 65.2 and 65.4). It seems that there are only a few genetic diseases in which pure PPK manifests together with neurological symptoms. Some reports associate neuropathy or spastic paralysis with keratoderma. These include striate keratoderma with spastic paraplegia, pes cavus and mental retardation in four brothers [1]; autosomal dominant punctate keratoderma and spastic paralysis [2]; autosomal dominant focal keratoderma with nail dystrophy and motor and sensory neuropathy [3]; and Charcot–Marie–Tooth disease [4]. Atypical erythrokeratoderma with deafness has also been associated with peripheral neuropathy [5].
Clinical features
Differential diagnosis
As an example, acro-osteolysis with keratoderma (Bureau–Barrière syndrome) [6, 7] may show diffuse keratoderma with osteolysis in the forefoot area, polyneuropathy of the lower legs and painless ulcers of the feet. The disease is non-familial and often subsumed under the concept of neurotrophic ulcers [8].
ACQUIRED KERATODERMAS
This disease group refers to numerous underlying causes summarized and reviewed by Patel et al. [1].
Clinical features and pathology
Callosities or more extensive thickening of plantar epidermis commonly accompanies obesity and occurs with increasing age or orthopaedic problems [2].
Keratoderma climactericum (Haxthausen disease) [3, 4].
The specificity of this entitiy described in women over the age of 45 is uncertain. A strong association with obesity and hypertension has been noted, pressure areas of the heel and the forefoot are involved first (Figure 65.60). Erythema and heavy hyperkeratosis with fissuring make walking painful. The hyperkeratotic areas slowly extend to become confluent. Later, the central palms may be affected. Symptoms may be worse in winter. Deschamps et al. [4] excluded endocrine dysfunction, contact dermatitis and fungal infection, and found normal serum vitamin A levels. However, Wachtel [5] described three young women in whom an identical condition arising following bilateral oophorectomy was reversed by oestrogen replacement. Laurent et al. [6] implicated keratinization of the acrosyringium by the finding of composite keratohyaline granules in the granular cells of the interductal granular cells, believed to serve as a marker for acrosyringeal differentiation [7]. In one report, topical 0.05% oestradiol in a water-in-oil base was successful where keratolytics and emollients had failed [8]. Given the success of of etretinate, acitretine may be the treatment of choice [1].
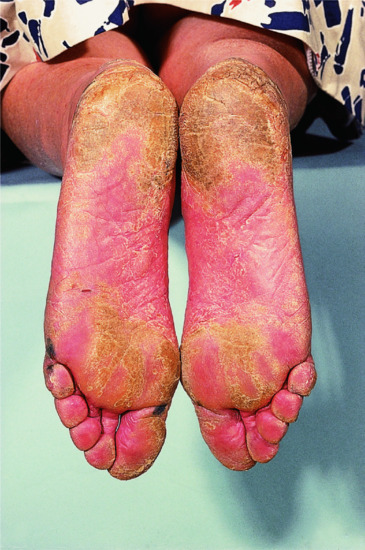
Figure 65.60 Keratoderma climactericum.
Inflammatory dermatoses.
In psoriasis, both diffuse gross and centripalmar hyperkeratosis are seen. A scalloped margin (‘festonné’), Caro–Senear lesions (depressed plaques) on the sides of the fingers and involvement of the knuckles may suggest the diagnosis. The lesions of reactive arthritis are compact, heaped up and resemble the heads of nails (keratoderma blenorrhagica). Extensive hyperkeratotic eczema may be difficult to distinguish on clinical and histological grounds but marked itching may indicate eczema. The even orange hyperkeratosis of pityriasis rubra pilaris is associated with an acute follicular eruption in adults and by lesions on the knees and elbows in children. Lupus erythematosus may show dry and atrophic [9], hypertrophic (Figure 65.61) [10] or ulcerative [11] palmar lesions. Keratoderma is also reported in association with acrocyanosis and livedo reticularis [12]. Antidesmocollin 3 antibodies were found in a patient with an immunobullous disorder and acquired diffuse PPK [13]. In lichen planus, warty lesions may be mistaken for viral warts; lichen planus and other lichenoid eruptions such as lichen nitidus may mimic punctate keratoderma [14].
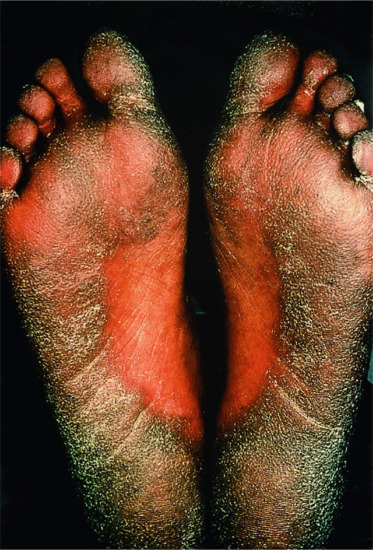
Figure 65.61 Keratoderma associated with lupus erythematosus.
(Courtesy of Dr I. Sarkany, Royal Free Hospital, London, UK.)
Infections.
Trichophytosis, especially resulting from Trichophyton rubrum, may be unilateral and lacking inflammatory signs. Keratoderma may be seen in crusted scabies (Figure 65.62). The tendency of secondary syphilis lesions to involve the palms is well known, and hyperkeratotic late syphilides may be warty or focal [15]. Tropical diseases such as late yaws may be complicated by keratoderma. In immunocompromised patients, viral warts may be confluent on the palms or soles.
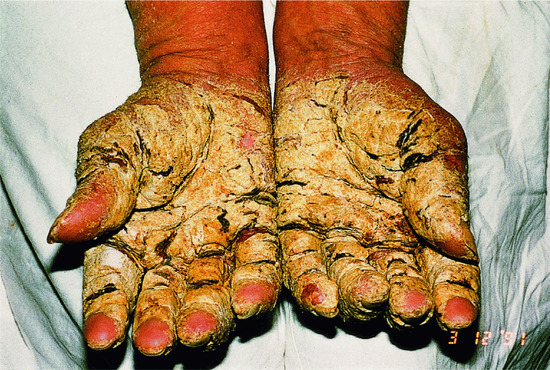
Figure 65.62 Norwegian scabies manifesting with keratoderma.
(Courtesy of Dr. N. Walker, Oxford Radcliffe Hospital, Oxford, UK.)
Myxoedema and lymphoedema.
Palmoplantar hyperkeratosis with myxoedema, improving with treatment, has been reported on several occasions [16, 17]. As such, hypothyroidism must be suspected in patients with acquired PPK [18]. In chronic lymphoedema, the skin overlying the lymphoedematous area first becomes diffusely thickened, and then develops into a velvety papillomatous surface, which is ultimately covered by large irregular warty projections (lymphostatic verrucosis; mossy foot) [19–21]. The condition may simulate chromoblastomycosis. Lymphoedematous keratoderma occurs most characteristically in filariasis, but may develop in the context of chronic lymphoedema of any origin. Histologically, there is hyperkeratosis, acanthosis and papillomatosis. The dermis is oedematous with dilated lymphatics, conspicuous new-vessel formation, some sclerosis and a variable infiltrate of inflammatory cells. Both the hyperkeratotic component and the lymphoedema improved in three cases given etretinate 0.6 mg/kg/day [19].
Malignancy.
In addition to ‘tripe palms’ (see Chapter 147), and Bazex acrokeratosis paraneoplastica (see Chapter 147), acquired diffuse PPK has been observed with cancer of the bronchus [22, 23], and filiform PPK has been reported with cancer of the breast, colon and kidney [24–26]. As for acquired ichthyoses, mycosis fungoides is an important differential diagnosis for acquired PPK [27]. Carcinogens, of which the best documented example is arsenic, may produce both keratoderma and internal malignancy [28, 29]. One survey showed that palmar keratoses occur four to five times more frequently in patients with cancer than in controls [30]. An increased incidence of keratoses in patients with lung or bladder cancer has been debated [31–33]. Smoking [31] and papillomavirus infection [32] are suggested culprits. Keratoses associated with cancer are histologically distinct from arsenical keratoses [34].
Drugs.
Keratoderma may be seen as a result of hypersensitivity to drugs such as iodine. Keratoderma may result from tegafur, glucan, lithium and halogenated weed-killers, and dioxin intoxication [35–39]. Arsenical-induced irregular warty keratoses, or more even glassy lesions, are still occasionally seen [40]. Agents used in cancer treatment commonly cause palmoplantar erythema (the hand–foot syndrome) and may cause keratoderma [41–43].
MISCELLANEOUS DISORDERS OF KERATINIZATION
Keratolytic winter erythema
Definition and nomenclature
This rare epidermal disorder, characterized by recurrent skin peeling, palmoplantar erythema and seasonal variation, was originally described as erythrokeratolysis hiemalis in 1977.
Epidemiology
It has been observed in at least 35 South African families of European descent originating from the Oudtshoorn district of Cape Provence [1, 2]. The incidence in this population is 1/7000. Cases have since been identified in several other countries, and a familial link to the Oudtshoorn cluster is evident in most.
Pathophysiology
It is an autosomal dominant disorder with variable penetrance and linkage to chromosome 8p22–p23 has been reported in five South African and one German kindred [3]. Until now, no pathogenic mutations have been found in candidate genes within the disease region, cathepsin B (CTSB) and farnesyl-diphosphate farnesyltransferase 1 (FDFT1) [4, 5]. The genetic alteration may have originated in a French immigrant in the late 1700s. Over 400 descendants are affected. A Norwegian family with four affected members did not show linkage to chromosome 8p22–p23, suggesting genetic heterogeneity [6]. A frequent precipitant is cold dry weather and, although in South Africa it is most active in winter months, it may be perennial in temperate climates. Other triggers include febrile illness, surgery, stress and menstruation, and it improves in pregnancy and with age. The father of an affected toddler was unaffected but paternal aunts and other family members had similar palmoplantar eruptions and her paternal great-great grandmother originated from Oudtshoorn [7], suggestive of partial penetrance.
Clinical features
Symmetrical keratolysis of the hands and feet may begin at any age from infancy to early adult life but it is not usually present at birth (Figure 65.63a). Cyclical centrifugal peeling (sometimes preceded by erythema multiforme-like papules) at several sites on the palms and soles is a constant feature, and may spread to the dorsal hands and feet, and the interdigital spaces. Episodes may be preceded by itch and hyperhidrosis and associated with pustulation. Palmoplantar erythema develops, and is followed by the evolution of painless superficial opaque dry blebs, which peel or can be pulled away, leaving a red base with intact markings. A second wave may begin at the centre of a lesion, resulting in gyrate and polycyclic annular erythema, which eventually resolves. Cycles repeat every few weeks and the palms and soles appear normal between attacks. Similar rosette lesions may arise on the lower legs, knees and rarely the thighs (Figure 65.63b), upper arms and shoulders. Truncal lesions were reported in one patient [2], and facial involvement in another [8].
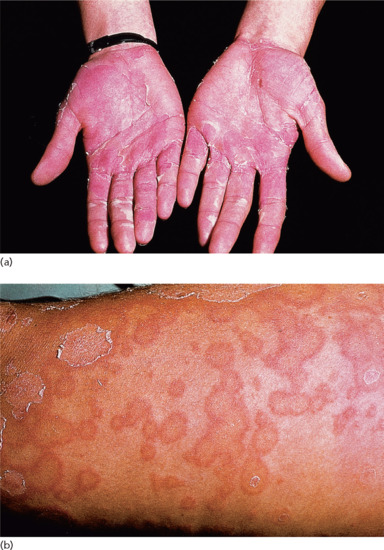
Figure 65.63 Oudtshoorn disease: (a) palmar lesions, and (b) truncal lesions.
Differential diagnosis
This includes familial PSSs, pustular bacterids, annular erythema, erythema multiforme, Hailey–Hailey disease, erythrokeratoderma and localized epidermolysis bullosa simplex. A similar phenotype affecting the palms, more active in summertime, was reported in two siblings (British) who had atypical autosomal recessive erythropoietic protoporphyria [9].
Investigations
Biopsy of the advancing edge of a lesion shows hyperplasia, spongiosis and, in the upper stratum spinosum, keratinocytes with pale cytoplasm, perinuclear vacuolization and pycnotic nuclei. In the abscence of a granular layer, the epidermis forms a parakeratotic wedge, which becomes sandwiched within the hyperkeratotic stratum corneum and is shed [2]. During regeneration, undifferentiated keratinocytes are not confined to the basal layer but appear in the lower half of the epidermis. Skin histology in the Norwegian family was non-specific [6].
Management
There is no effective treatment, and topical keratolytics, retinoids and steroids may aggravate the condition. Urea and tar compounds, antiperspirants, oral retinoids and photodynamic therapy have been tried [10].
Porokeratosis
Definition and nomenclature
Porokeratoses refer to a heterogeneous group of keratinization disorders, in which the presence of a so-called ‘cornoid lamella’ in a lesion can be seen. Clinical distinction between the various forms of disseminated porokeratosis may not be justified [1].
The disorders are characterized by marginate keratotic lesions, histologically showing a column of parakeratotic keratinocytes (the cornoid lamella). Various forms are recognized, but terminology and classification are still debated [2, 3]. Some forms appear to be premalignant [4].
Pathophysiology
Disseminated superficial (actinic) porokeratosis (DSAP) has been mapped in Chinese pedigrees to 12q [5], 15q [6],18p [7] and 16q [8]. Loss of heterozygosity at 12q and sequence variations in genes at this locus have been reported, but the significance of these findings is uncertain [9–13]. More recently, heterozygous mutations in the MVK gene have been reported to cause porokeratosis of Mibelli and in DSAP [14–17]. MVK encodes mevalonate kinase which is involved in the biosynthesis of cholesterol and isoprenoid. The enzyme seems to regulate keratinocyte differentiation. Of note, bi-allelic mutations in MVK are associated with a neurological disorder called mevalonic aciduria. It is still unclear why no cutaneous phenotype is seen in the parents of children with mevalonic aciduria, who are obligatory carriers of MVK mutations.
The centrifugal progress of individual lesions is thought to reflect the migration of a clone of abnormal cells [18]. There is keratinocyte dysplasia and Otsuka et al. [19, 20] have reported aneuploidy and chromosomal abnormalities in lesional keratinocytes. The tumour suppressor protein p53 is overexpressed in the cornoid lamella [21–24], but D'Errico et al. [24] found no evidence of p53 mutations or radiation hypersensitivity in DSAP-derived keratinocytes and fibroblasts. Cytogenetic anomalies in fibroblasts, particularly chromosome 3, are also recorded [25]. An association with immunosuppression [2] suggests impaired immunity is permissive, perhaps by reduced immune surveillance of dysplasia, but the possibility of an infective aetiology [26] remains. Esser et al. [27] found evidence of HPV types 66 and 14, respectively, in two patients with porokeratosis of Mibelli.
Clinical variants
Disseminated superficial actinic porokeratosis.
This form is the most common presentation, with multiple lesions of up to 10 mm predominantly found in sun-exposed sites in middle-aged individuals, in particular on the extremities of women, especially those with sun-sensitive skin (Figure 65.64). The papules are surrounded by a keratotic ridge albeit finer than in Mibelli porokeratosis. They are easily mistaken for actinic keratoses, with which they may coexist. Lesions are not induced by artificial light exposure [28], but have been provoked by photochemotherapy [29]. Radiation therepay has also been shown to exacerbate the disease. No evidence that skin cancer arises in the porokeratotic lesions was found in a study of 29 patients [30].
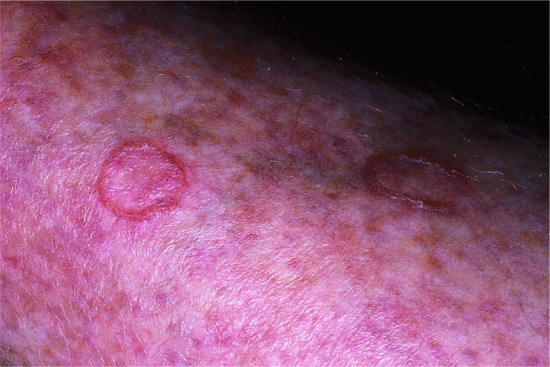
Figure 65.64 Disseminated superficial actinic porokeratoses: annular keratotic lesions with a raised margin.
Disseminated superficial porokeratoses of immunosuppression.
This variant has been reported after renal, hepatic, cardiac and bone marrow transplantation, and in AIDS [2]. The distribution of the lesions is similar to DSAP, but a history of sun exposure is less likely [31].
Disseminated superficial porokeratosis of childhood.
The condition may be inherited as an autosomal dominant disorder, but sporadic cases are seen. Widely disseminated flat lesions usually begin in childhood, the majority between the ages of 5 and 10 years, but they may be present at birth or may first appear at puberty or later. Palmoplantar lesions may be associated (porokeratosis palmaris et plantaris disseminata) [32]. Widespread lesions appeared first at the age of 1 month in a male infant with craniosynostosis and other congenital abnormalities [33].
Porokeratosis of Mibelli [34, 35].
The eponym Mibelli is sometimes used generically for porokeratoses, but usually refers only to the form with single or scanty and larger lesions. These develop as annular dry plaques (Figure 65.65) surrounded by a raised fine keratotic elevated border with a central groove. Lesions are most common on the limbs and by centrifugal spread may achieve several centimetres in diameter. The centre is usually atrophic but may be hyperkeratotic [36]. The face, scalp, nail, genitalia, oral mucosa and cornea may also be affected. The condition may be familial, inherited as an autosomal dominant trait with onset in childhood (MIM 175800), or sporadic and of later onset.
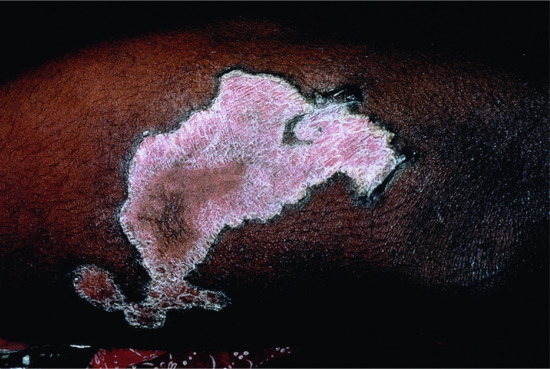
Figure 65.65 Porokeratosis of Mibelli.
Giant porokeratoses
(up to 20 cm in diameter with a surrounding elevated edge of 1 cm). These are very rare [37], and are most often found on the foot. Large lesions are said to have the highest potential for malignant transformation [4, 37, 38].
Palmoplantar porokeratosis (of Mantoux).
Parakeratotic hyperkeratosis histologically reminiscent of the cornoid lamella occurs in some punctate keratodermas, but the absence of marginate lesions distinguishes this entity from true porokeratosis [39]. Nonetheless, annular lesions of the palms and soles with a cornoid lamella are recognized [27, 40].
Linear porokeratosis [38, 41, 42].
Linear porokeratoses showing typical cornoid lamellae and following the lines of Blaschko usually appear in childhood. These lesions probably result from a predisposition to porokeratosis in an abnormal clone of epidermal precursors. Malignant degeneration and metastasis have been reported in this variant [38, 43]. Linear accentuation of disseminated actinic porokeratosis has been reported too [44–46].
Porokeratosis ptychotropica [47–49].
This rare type of porokeratosis is confined to body folds (‘ptyche’, Greek for a fold). Brownish to reddish macules or plaques usually develop symmetrically on the perianal region, and, as reported in one patient, on the scrotum. The typical presence of multiple cornoid lamellae as seen histologically (punctate type of porokeratosis) explains the keratotic or verrucous appearance and expansile papular growth. The highly pruritic disease is mostly confined to men ranging from 6 to 84 years of age. Linear porokeratoses or DSAP may coexist.
Investigations
The characteristic histopathology is seen on the edge of the lesion when cut at right angles. The stratum corneum is hyperkeratotic, and at the raised border a column of poorly staining parakeratotic stratum corneum cells, the cornoid lamella, is seen running obliquely through the surrounding normal-staining cells. The underlying keratinocytes are large, vacuolated, some of them dyskeratotic and pleomorphic. The granular layer is absent beneath the parakeratotic column. Beneath, a variably dense lichenoid lymphocytic infiltrate, less frequently colloid bodies and amyloid material may be present [50, 51]. These changes can also affect the hair follicles or acrosyringia. The involvement of the sweat pores explain the term ‘poro’-keratosis. The central area of a lesion is usually atrophic, but may occasionally show gross hyperkeratosis. The papillary dermis is fibrotic and contains melanophages [52]. Cornoid lamellae may also be found in other conditions, such as viral warts, some ichthyoses and naevoid hyperkeratoses but the characteristic changes of the underlying keratinocytes are absent.
Treatment
Treatment of disseminated superficial porokeratoses is usually unnecessary. Photoprotection should be recommended. Keratolytics offer little relief. Topical tacalcitol [56], topical retinoids, 5-fluorouracil ointment [57, 58], imiquimod cream [59] and oral etretinate [60] have been effective. Diclofenac gel was disappointing [61]. Cryotherapy, carbon dioxide, pulsed dye laser therapy and photodynamic treatment have all been used with variable results [53–55]. Deep dermabrasion by a dermatome is useful in porokeratosis ptychotropica [62].
Perforating keratotic disorders
The nature of perforating (epidermal elimination) disorders is uncertain [1]. They present as keratotic papules, but as epidermal involvement may be secondary to dermal disease they are probably not true disorders of keratinization. The unifying term ‘acquired perforating dermatosis’ and a subclassification has been proposed [2, 3].
Pathophysiology
The concept of a genuine perforation is increasingly disputed [4]. Trauma from scratching is thought to initiate the lesions. Familial occurrence, with ocular involvement, is reported (MIM 149500) [5, 6].
Clinical features
Follicular or non-follicular keratotic papules or nodules up to 1 cm in diameter are seen mainly on the limbs (Figure 65.66). Some of the lesions show a central depression containing an adherent necrotic plug. It is most often seen with diabetes or before, during or after dialysis for renal failure [7]. Eleven per cent of 72 British patients on renal dialysis developed a perforating dermatosis [8]. Many individual reports of association with malignancy, infection or inflammatory conditions have been published, but often also with renal failure or diabetes [9]. A patient who developed a perforating folliculitis with two anti-TNF agents, improved on withdrawal [10].
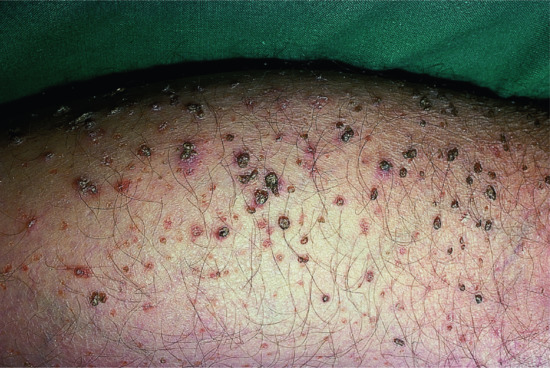
Figure 65.66 Kyrle disease/acquired perforating dermatosis: large keratinous plugs on the thigh.
Investigations
Histology of acquired perforating dermatoses may show follicular and non-follicular lesions with broad or narrow ulcer craters [9]. Degenerate collagen, elastic tissue and keratin are seen mixed with an unidentified clear material, which has been regarded by some as an accumulation of a metabolite [2]. Changes diagnosed as perforating folliculitis, reactive perforating collagenosis or Kyrle disease (hyperkeratosis follicularis et parafollicularis in cutem penetrans) may all be found [11–13].
Management
In most cases of acquired perforating dermatosis, lesions could be cleared by treatment with potent topical or intralesional steroids. Success with conventional or narrow-band UVB phototherapy [14–16] has also been recorded. Topical tretinoin may reduce the lesions. Other agents reported to be effective include allopurinol [17] and doxycycline [18].
Clinical variants
Necrotizing infundibular crystalline folliculitis.
Lucke et al. [19] reported two cases of a disorder characterized by transepidermal elimination of negatively birefringent needle-shaped crystals similar to monosodium urate. Kossard et al. [20] reported a similar case and suggested that the disorder was due to the initiation of crystal formation around microorganisms from follicular lipids at critical concentrations. Clinically, multiple waxy papules develop with a predilection for the forehead, neck and back. Histology reveals necrosis of the follicular epithelium and sometimes a perifollicular neutrophilic infiltrate. Crystalline deposits with yeasts and Gram-positive bacteria are found in the follicular ostia and are enclosed by parakeratotic columns. In addition, these histological changes can also be found as a coincidental finding in the vicinity of epithelial skin neoplasms [21]. Resolution of the lesions after topical or systemic antimycotic treatment suggests a microbial pathogenesis.
Elastosis perforans serpiginosa
(see Chapter 96). This presents as grouped arcuate or serpiginous keratotic papules and is associated with Down syndrome, disorders of connective tissue and penicillamine treatment. Histologically, amorphous masses that bind elastic tissue stains can be seen traversing the epidermis.
Reactive perforating collagenosis
(see Chapter 96). This mainly affects children, with the formation of 2–5-mm papules, usually on the limbs. Lesions in all stages of eruption and resolution are present at one time [22].
Multiple minute digitate hyperkeratoses
Definition
A number of entities have been described using names including minute and filiform keratoses, disseminated spiked hyperkeratosis, minute aggregate keratosis or digitate keratosis [1–4] (Figure 65.67). An inclusive approach to classification has been proposed under the name ‘multiple minute digitate hyperkeratoses’ [5]; and a useful algorithm is given in Cacetta et al. [6].
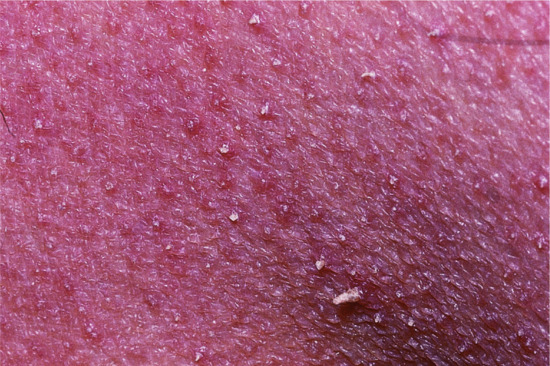
Figure 65.67 Digitate hyperkeratosis.
(Courtesy of Dr F. A. Ive, Dryburn Hospital, Durham, UK.)
Clinical features
Cases may be sporadic [3] or familial with probable autosomal dominant inheritance [2–8], early or late onset, transient or persistent. Non-follicular spiky keratoses develop on the trunk and limbs [6]. Reported associations include drugs, malignancy, especially haematological, and X-irradiation [4, 5, 8–13]. Hyperkeratotic spicules on the face, particularly on the nose, are follicular and often associated with paraproteinaemia, multiple myeloma and croyglobulinaemia, but may be also idiopathic [10, 11]. One case associated with carcinoma of the larynx cleared following surgery [14]. Filiform keratoses occur with a pityriasis rubra pilaris-like eruption and acne conglobata in association with HIV infection [15]. A familial form of filiform keratosis has been described to be associated with thickened nails, plantar hyperkeratosis, joint laxity and long fingers [16].
Differential diagnosis
Spiny palmoplantar keratosis [17, 18] is recognized as a particular type of PPK (see spiny keratoderma earlier).
Investigations
Histologically, there are focal areas of compact orthohyperkeratotic spicules mostly arising from a pointed epidermal elevation. The stratum granulosum usually is prominent [6, 19]. Parakeratosis may be present [16, 20] but the use of the term porokeratosis is misleading [5, 6]. Hyperkeratotic spicules associated with paraproteinaemia reveal eosinophilic inclusions in the hyperkeratotic columns that represent immunoglobulin deposits [10].
Management
Treatment including keratolytics and retinoids is often unsuccessful [6].
Flegel disease
Definition and nomenclature
Flegel disease is a rare and benign cornification disorder of older individuals characterized by multiple reddish-brown keratotic papules affecting the extremities [1].
Pathophysiology
It is inherited as an autosomal dominant condition [2]. Despite the strong genetic component in the disorder, no reports identifying a candidate gene have appeared to date. A low proliferation rate of keratinocytes together with downregulation of filaggrin, loricrin and high-molecular-weight keratins and loss of the keratin pattern in the horny layer suggests a retention hyperkeratosis and complex dysregulation of the epidermal differentiation [3].
Clinical features
Two to three millimetre keratotic red-brown papules with discrete irregular margins (‘cornflake sign’) appear over the dorsa of the feet and on the lower parts of the legs, not before the third or fourth decade. The lesions may spread to the upper part of the legs and thighs, also disseminating over the arms and trunk or concha of the ear. On the palms and soles, fine points may appear [1, 4] (Figure 65.68). Some patients complain of pruritus. The keratotic scale can be removed, leaving a non-exudative red bleeding base.
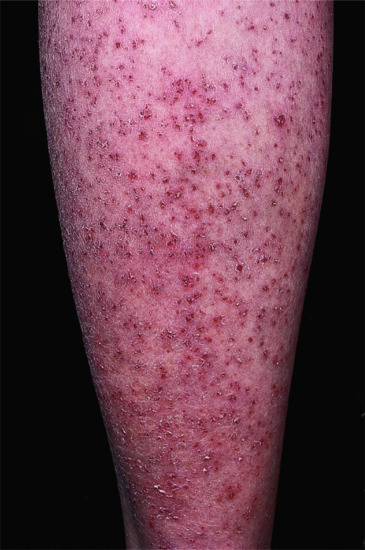
Figure 65.68 Flegel disease: polygonal keratotic lesions on the legs.
Investigations
Histologically, there is hyperkeratosis with a focal parakeratosis overlying a thinned flat epidermis with loss of keratohyalin granules. In the periphery of the lesions, the epidermis is acanthotic with collarette-like elongated rete ridges. A lymphocytic lichenoid infiltrate can be found in early lesions. Upon electron microscopy membrane coated granules (Odland bodies) appear reduced and malformed at least in evolving lesions [1, 2, 3, 5, 6].
Management
Treatment remains difficult and many modalities have been recommended, including cryotherapy, topical corticosteroids, topical and systemic retinoids, calcipotriol, PUVA and 5-fluorouracil albeit with variable efficacy [3, 7].
Circumscribed palmoplantar hypokeratosis
Pathophysiology
This condition is considered a localized keratinization disorder of an expanding clone of keratinocytes [1]. Acanthosis, dilated tortuous capillaries and coarse keratohyalin granules are also suggestive of a viral origin. Molecular studies failed to detect HPV apart from one report that found HPV type 4 specific DNA [2].
Clinical features
Circumscribed palmar or plantar hypokeratosis of the palms and soles is characterized by a solitary erythematous, sharply circumscribed depression on the thenar or hypothenar region of the palms or on the soles (Figure 65.69). Only rarely more than one lesion appears. Most patients are women between the ages of 42 and 84 years [3, 4]. Congenital cases are exceptional [5]. Sometimes there is a history of prior trauma or burn at the site.
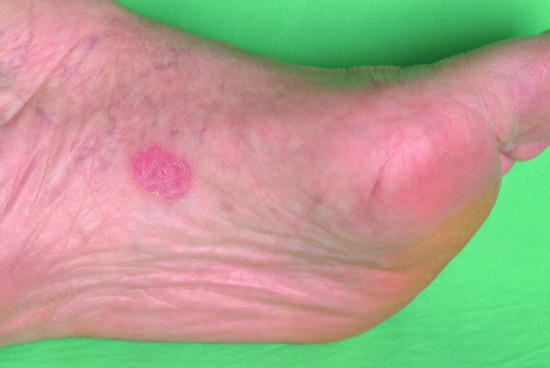
Figure 65.69 Circumscribed hypokeratosis on the inner part of the forefoot of a female patient.
(Courtesy of Dr F. Schedel, Department of Dermatology, University Hospital Münster, Münster, Germany.)
Investigations
Histologically, an abrupt thinning of the stratum corneum over a diminished granular layer forms a sharp stair between normal and involved skin [6]. Malignant transformation is unlikely although susceptibility to photocarcinogenesis has been assumed [7, 8].
Management
Topical calcipotriol [9], cryotherapy [10], photodynamic therapy [11] or fluorouracil cream [12] can be tried. However, total excision of the lesion is the definite treatment.
Waxy keratoses of childhood
Definition
This condition appear typically in children presenting with asymptomatic small hyperkeratotic papules, e.g. on the trunk or proximal limbs. The aetiology is unknown, and only a few cases have been reported so far [1].
Clinical features
Three children in two families showed generalized discrete domed keratotic papules, which were flesh coloured or yellowish [2]. Two young patients reported earlier with ‘disseminated hypopigmented keratoses’ appear to be identical [3]. Late manifestation in an adult has been observed [4]. The disorder has been reported in a linear form [5] and as a linear exacerbation in generalized disease [6], further supporting the notion that it could be a genodermatosis. The pattern resembles confluent and reticulated papillomatosis (Gougerot–Carteaud syndrome), but waxy keratosis shows more hyperkeratosis. Differential diagnosis includes the leukodermic macules in Darier disease in dark skin [7]. Larger papules with a mosaic pattern and acral distribution have been diagnosed as mosaic acral keratosis (see also Focal acral hyperkeratosis earlier) [8].
Investigations
Histological findings were marked orthokeratotic hyperkeratosis, tenting and papillomatosis of the epidermis, and mild acanthosis.
Hyperkeratosis of the nipple
Pathophysiology
The occurrence and aggravation around puberty, pregnancy or systemic hormone treatment suggests a hormonal influence [1].
Clinical features
The lesions are bilateral and involve predominantly the top of the nipple. Lesions may cause tenderness or discomfort, pruritus, sensitivity to touch or discomfort with breastfeeding [2]. Naevoid hyperkeratosis of the nipple and areola may either appear isolated or associated with an epidermal naevus and other dermatoses such as acanthosis nigricans, Darier disease, chronic eczema, chronic mucocutaneous candidiasis or cutaneous T-cell lymphoma [3, 4].
Differential diagnosis
The most important differential diagnosis is Paget disease.
Investigations
Histologically, papillomatosis, acanthosis and hyperkeratosis of the epidermis can be found. The rete ridges are filiform and anastomizing and the basal layer appears hyperpigmented. A sparse lymphocytic infiltrate and intraepidermal collections of lymphocytes must not be confused with T-cell lymphoma.
Management
Treatment includes topical agents (keratolytics, steroid, retinoic acid, calcipotriol) [5, 6] and ablative modalities (cryotherapy, carbon dioxide laser, radiofrequency and shave excision) [7–9].
References
Introduction
- Oji V, Tadini G, Akiyama M, et al. Revised nomenclature and classification of inherited ichthyoses: results of the First Ichthyosis Consensus Conference in Sorèze 2009. J Am Acad Dermatol 2010;63:607–41.
- Muñoz-Garcia A, Thomas CP, Keeney DS, et al. The importance of the lipoxygenase–hepoxilin pathway in the mammalian epidermal barrier. Biochim Biophys Acta 2014;401–8.
- Müller FB, Küster W, Wodecki K, et al. Novel and recurrent mutations in keratin KRT5 and KRT14 genes in epidermolysis bullosa simplex: implications for disease phenotype and keratin filament assembly. Hum Mutat 2006;27:719–20.
- Sprecher E, Ishida-Yamamoto A, Becker OM, et al. Evidence for novel functions of the keratin tail emerging from a mutation causing ichthyosis hystrix. J Invest Dermatol 2001;116:511–19.
- Hanneken S, Rütten A, Pasternack SM, et al. Systematic mutation screening of KRT5 supports the hypothesis that Galli–Galli disease is a variant of Dowling–Degos disease. Br J Dermatol 2010;163:197–200.
- Camisa C, Hessel A, Rossana C, Parks A. Autosomal dominant keratoderma, ichthyosiform dermatosis and elevated serum beta-glucuronidase. Dermatologica 1988;177:341–7.
- Gedicke MM, Traupe H, Fischer B, et al. Towards characterization of palmoplantar keratoderma caused by gain-of-function mutation in loricrin: analysis of a family and review of the literature. Br J Dermatol 2006;154:167–71.
- Peukert M. Über Ichthyosis. Eine Übersicht. Dermatol Z (Berlin) 1899:171–204.
- Patel N, Spencer LA, English JC 3rd, Zirwas MJ. Acquired ichthyosis. J Am Acad Dermatol 2006;55:647–56.
- Griffin LJ, Massa MC. Acquired ichthyosis and pityriasis rotunda. Clin Dermatol 1993;11:27–32.
Ichthyoses
- Willan R. On Cutaneous Diseases, Vol. 1, Chapter 4, Ichthyosis. London: Barnard, 1808: 197–212.
- Traupe H. The Ichthyoses. A guide to clinical diagnosis, genetic counseling, and therapy. Berlin: Springer-Verlag, 1989.
- Elias PM, Williams ML, Crumrine D, Schmuth M. Ichthyoses. Clinical biochemical, pathogenic and diagnostic assessment. Basel: Karger Verlag, 2010.
- Oji V, Tadini G, Akiyama M, et al. Revised nomenclature and classification of inherited ichthyoses: results of the First Ichthyosis Consensus Conference in Sorèze 2009. J Am Acad Dermatol 2010;63:607–41.
- Oji V1, Traupe H. Ichthyoses: differential diagnosis and molecular genetics. Eur J Dermatol 2006;16:349–59.
- Harper PS. Genetic heterogeneity in the ichthyoses. In: Marks R, Dylos PJ, ed. The Ichthyoses. Proceedings of the 2nd Annual Clinicaly Orientated Symposion of the ESDR. Lancaster: MTP Press, 1978:127–36.
- Traupe H, Fischer J, Oji V. Nonsyndromic types of ichthyoses – an update. J Dtsch Dermatol Ges 2013;12:109–21.
Ichthyosis vulgaris
- Brown SJ, Relton CL, Liao H, et al. Filaggrin haploinsufficiency is highly penetrant and is associated with increased severity of eczema: further delineation of the skin phenotype in a prospective epidemiological study of 792 school children. Br J Dermatol 2009;161:884–9.
- Smith FJ, Irvine AD, Terron-Kwiatkowski A, et al. Loss-of-function mutations in the gene encoding filaggrin cause ichthyosis vulgaris. Nat Genet 2006;38:337–42.
- Sandilands A, Terron-Kwiatkowski A, Hull PR, et al. Comprehensive analysis of the gene encoding filaggrin uncovers prevalent and rare mutations in ichthyosis vulgaris and atopic eczema. Nat Genet 2007;39:650–4.
- Oji V, Seller N, Sandilands A, et al. Ichthyosis vulgaris: novel FLG mutations in the German population and high presence of CD1a+ cells in the epidermis of the atopic subgroup. Br J Dermatol 2009;160:771–81.
- Perusquía-Ortiz AM, Oji V, Sauerland MC, et al. Complete filaggrin deficiency in ichthyosis vulgaris is associated with only moderate changes in epidermal permeability barrier function profile. J Eur Acad Dermatol Venereol 2013;27:1552–8.
- Gruber R, Elias PM, Crumrine D, et al. Filaggrin genotype in ichthyosis vulgaris predicts abnormalities in epidermal structure and function. Am J Pathol 2011;178:2252–63.
- Palmer CN, Irvine AD, Terron-Kwiatkowski A, et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet 2006;38:441–6.
- Thyssen JP, Godoy-Gijon E, Elias PM. Ichthyosis vulgaris: the filaggrin mutation disease. Br J Dermatol 2013;168:1155–66.
- Wells RS, Kerr CB. Clinical features of autosomal dominant and sex-linked ichthyosis in an English population. BMJ 1966;1:947–50.
- Oji V, Traupe H. Ichthyosis: clinical manifestations and practical treatment options. Am J Clin Dermatol 2009;10:351–64.
- Blanchet-Bardon C, Tadini G, Machado Matos M, Delarue A. Association of glycerol and paraffin in the treatment of ichthyosis in children: an international, multicentric, randomized, controlled, double-blind study. J Eur Acad Dermatol Venereol 2012;26:1014–19.
- Stout TE, McFarland T, Mitchell JC, et al. Recombinant filaggrin is internalized and processed to correct filaggrin deficiency. Invest Dermatol 2014;134:423–9.
Recessive X-linked ichthyosis
- Craig WY, Roberson M, Palomaki GE, et al. Prevalence of steroid sulfatase deficiency in California according to race and ethnicity. Prenat Diagn 2010;30:893–8.
- Koppe G, Marinković-Ilsen A, Rijken Y, et al. X-linked ichthyosis. A sulphatase deficiency. Arch Dis Child 1978;53:803–6.
- Cañueto J, Ciria S, Hernández-Martín A, et al. Analysis of the STS gene in 40 patients with recessive X-linked ichthyosis: a high frequency of partial deletions in a Spanish population. J Eur Acad Dermatol Venereol 2010;24:1226–9.
- Puri PK, Reddi DM, Spencer-Manzon M, et al. Banding pattern on polarized hair microscopic examination and unilateral polymicrogyria in a patient with steroid sulfatase deficiency. Arch Dermatol 2012;148:73–8.
- Liao H, Waters AJ, Goudie DR, et al. Filaggrin mutations are genetic modifying factors exacerbating X-linked ichthyosis. J Invest Dermatol 2007;127:2795–8.
- Ramesh R, Chen H, Kukula A, et al. Exacerbation of X-linked ichthyosis phenotype in a female by inheritance of filaggrin and steroid sulfatase mutations. J Dermatol Sci 2011;64:159–62.
- Elias PM, Crumrine D, Rassner U, et al. Basis for abnormal desquamation and permeability barrier dysfunction in RXLI. J Invest Dermatol 2004;122:314–19.
- Hoppe T, Winge MC, Bradley M, et al. X-linked recessive ichthyosis: an impaired barrier function evokes limited gene responses before and after moisturizing treatments. Br J Dermatol 2012;167:514–22.
- Hernández-Martín A, González-Sarmiento R, De Unamuno P. X-linked ichthyosis: an update. Br J Dermatol 1999;141:617–27.
- Traupe H, Happle R. Clinical spectrum of steroid sulfatase deficiency: X-linked recessive ichthyosis, birth complications and cryptorchidism. Eur J Pediatr 1983;140:19–21.
- Kent L, Emerton J, Bhadravathi V, et al. X-linked ichthyosis (steroid sulfatase deficiency) is associated with increased risk of attention deficit hyperactivity disorder, autism and social communication deficits. J Med Genet 2008;45:519–24.
- Trent S, Cassano T, Bedse G, et al. Altered serotonergic function may partially account for behavioral endophenotypes in steroid sulfatase-deficient mice. Neuropsychopharmacology 2012;37:1267–74.
- Ben Khelifa H, Soyah N, Ben-Abdallah-Bouhjar I, et al. Xp22.3 interstitial deletion: a recognizable chromosomal abnormality encompassing VCX3A and STS genes in a patient with X-linked ichthyosis and mental retardation. Gene 2013;527:578–83.
- Bruckner-Tuderman L, Sigg C, Geiger JM, Gilardi S. Acitretin in the symptomatic therapy for severe recessive x-linked ichthyosis. Arch Dermatol 1988;124:529–32.
Autosomal recessive congenital ichthyosis
- Oji V, Tadini G, Akiyama M, et al. Revised nomenclature and classification of inherited ichthyoses: results of the First Ichthyosis Consensus Conference in Sorèze 2009. J Am Acad Dermatol 2010;63:607–41.
- Vahlquist A, Bygum A, Gånemo A, et al. Genotypic and clinical spectrum of self-improving collodion ichthyosis: ALOX12B, ALOXE3, and TGM1 mutations in Scandinavian patients. J Invest Dermatol 2010;130:438–43.
- Traupe H, Fischer J, Oji V. Nonsyndromic types of ichthyoses – an update. J Dtsch Dermatol Ges 2014;12:109–21
- Hernández-Martín A, Garcia-Doval I, Aranegui B, et al. Prevalence of autosomal recessive congenital ichthyosis: a population-based study using the capture–recapture method in Spain. J Am Acad Dermatol 2012;67:240–4.
- Hartz T, Hennies H, Oji V, et al. The prevalence of autosomal recessive congenital ichthyosis and of transglutaminase-1 deficiency in Germany: Calculation of estimates using the three-source capture–recapture method. J Invest Dermatol 2013;133S:S91.
- Akiyama M, Sugiyama-Nakagiri Y, Sakai K, et al. Mutations in lipid transporter ABCA12 in harlequin ichthyosis and functional recovery by corrective gene transfer. J Clin Invest 2005;115:1777–84.
- Rabionet M, Gorgas K, Sandhoff R. Ceramide synthesis in the epidermis. Biochim Biophys Acta 2014;1841:422–34.
- Goytain A, Hines RM, Quamme GA. Functional characterization of NIPA2, a selective Mg2+ transporter. Am J Physiol Cell Physiol 2008;295:C944–53.
- Lefévre C, Audebert S, Jobard F, et al. Mutations in the transporter ABCA12 are associated with lamellar ichthyosis type 2. Hum Mol Genet 2003;12:2369–78.
- Sakai K, Akiyama M, Yanagi T, et al. ABCA12 is a major causative gene for non-bullous congenital ichthyosiform erythroderma. J Invest Dermatol 2009;129:2306–9.
- Akiyama M, Sakai K, Sugiyama-Nakagiri Y, et al. Compound heterozygous mutations including a de novo missense mutation in ABCA12 led to a case of harlequin ichthyosis with moderate clinical severity. J Invest Dermatol 2006;126:1518–23.
- Fischer J. Autosomal recessive congenital ichthyosis. J Invest Dermatol 2009;129:1319–21.
Harlequin ichthyosis
- Rajpopat S, Moss C, Mellerio J, et al. Harlequin ichthyosis: a review of clinical and molecular findings in 45 cases. Arch Dermatol 2011;147:681–6.
- Thomas AC, Cullup T, Norgett EE, et al. ABCA12 is the major harlequin ichthyosis gene. J Invest Dermatol 2006;126:2408–13.
- Akiyama M, Sugiyama-Nakagiri Y, Sakai K, et al. Mutations in lipid transporter ABCA12 in harlequin ichthyosis and functional recovery by corrective gene transfer. J Clin Invest 2005;115:1777–84.
- Akiyama M. ABCA12 mutations and autosomal recessive congenital ichthyosis: a review of genotype/phenotype correlations and of pathogenetic concepts. Hum Mutat 2010;31:1090–6.
- Yamanaka Y, Akiyama M, Sugiyama-Nakagiri Y, et al. Expression of the keratinocyte lipid transporter ABCA12 in developing and reconstituted human epidermis. Am J Pathol 2007;171:43–52.
- Scott CA, Rajpopat S, Di WL. Harlequin ichthyosis: ABCA12 mutations underlie defective lipid transport, reduced protease regulation and skin-barrier dysfunction. Cell Tissue Res 2013;351:281–8.
- Caubet C, Jonca N, Brattsand M, et al. Degradation of corneodesmosome proteins by two serine proteases of the kallikrein family, SCTE/KLK5/hK5 and SCCE/KLK7/hK7. J Invest Dermatol 2004;122:1235–44.
- Thomas AC, Tattersall D, Norgett EE, et al. Premature terminal differentiation and a reduction in specific proteases associated with loss of ABCA12 in Harlequin ichthyosis. Am J Pathol 2009;174:970–8.
- Yanagi T, Akiyama M, Nishihara H, et al. Harlequin ichthyosis model mouse reveals alveolar collapse and severe fetal skin barrier defects. Hum Mol Genet 2008;17:3075–83.
- Milner ME, O'Guin WM, Holbrook KA, Dale BA. Abnormal lamellar granules in harlequin ichthyosis. J Invest Dermatol 1992;99:824–9.
Collodion baby and self-improving congenital ichthyosis
- Van Gysel D, Lijnen RL, Moekti SS, et al. Collodion baby: a follow-up study of 17 cases. J Eur Acad Dermatol Venereol 2002;16:472–5.
- Vahlquist A, Bygum A, Gånemo A, et al. Genotypic and clinical spectrum of self-improving collodion ichthyosis: ALOX12B, ALOXE3, and TGM1 mutations in Scandinavian patients. J Invest Dermatol 2010;130:438–43.
- Raghunath M, Hennies HC, Ahvazi B, et al. Self-healing collodion baby: a dynamic phenotype explained by a particular transglutaminase-1 mutation. J Invest Dermatol 2003;120:224–8.
Bathing suit ichthyosis
- Jacyk WK. Bathing-suit ichthyosis. A peculiar phenotype of lamellar ichthyosis in South African blacks. Eur J Dermatol 2005;15:433–6.
- Oji V, Hautier JM, Ahvazi B, et al. Bathing suit ichthyosis is caused by transglutaminase-1 deficiency: evidence for a temperature-sensitive phenotype. Hum Mol Genet 2006;15:3083–97.
- Arita K, Jacyk WK, Wessagowit V, et al. The South African “bathing suit ichthyosis” is a form of lamellar ichthyosis caused by a homozygous missense mutation, p.R315L, in transglutaminase 1. J Invest Dermatol 2007;127:490–3.
- Aufenvenne K, Oji V, Walker T, et al. Transglutaminase-1 and bathing suit ichthyosis: molecular analysis of gene/environment interactions. J Invest Dermatol 2009;129:2068–71.
- Trindade F, Fiadeiro T, Torrelo A, et al. Bathing suit ichthyosis. Eur J Dermatol 2010;20:447–50.
- Bourrat E, Blanchet-Bardon C, Derbois C, et al. Specific TGM1 mutation profiles in bathing suit and self-improving collodion ichthyoses: phenotypic and genotypic data from 9 patients with dynamic phenotypes of autosomal recessive congenital ichthyosis. Arch Dermatol 2012;148:1191–5.
- Hackett BC, Fitzgerald D, Watson RM, et al. Genotype–phenotype correlations with TGM1: clustering of mutations in the bathing suit ichthyosis and self-healing collodion baby variants of lamellar ichthyosis. Br J Dermatol 2010;162:448–51.
- Yamamoto M, Sakaguchi Y, Itoh M, et al. Bathing suit ichthyosis with summer exacerbation: a temperature-sensitive case. Br J Dermatol 2012;166:672–4.
- Washio K, Fukunaga A, Terai M, et al. Hypohidrosis plays a crucial role in the vicious circle of bathing suit ichthyosis: a case with summer exacerbation. Acta Derm Venereol 2014;94:349–50.
Lamellar ichthyosis and congenital ichthyosiform erythroderma
- Frost P, Weinstein GD, Van Scott EJ. The ichthyosiform dermatoses. II. Autoradiographic studies of epidermal proliferation. J Invest Dermatol 1966;47:561–7.
- Farasat S, Wei MH, Herman M, Liewehr DJ, et al. Novel transglutaminase-1 mutations and genotype–phenotype investigations of 104 patients with autosomal recessive congenital ichthyosis in the USA. J Med Genet 2009;46:103–11.
- Fischer J. Autosomal recessive congenital ichthyosis. J Invest Dermatol 2009;129:1319–21.
- Eckl KM, de Juanes S, Kurtenbach J, et al. Molecular analysis of 250 patients with autosomal recessive congenital ichthyosis: evidence for mutation hotspots in ALOXE3 and allelic heterogeneity in ALOX12B. J Invest Dermatol 2009;129:1421–8.
- Hazell M, Marks R. Clinical, histologic, and cell kinetic discriminants between lamellar ichthyosis and nonbullous congenital ichthyosiform erythroderma. Arch Dermatol 1985;121:489–93.
- Vahlquist A. Pleomorphic ichthyosis: proposed name for a heterogeneous group of congenital ichthyoses with phenotypic shifting and mild residual scaling. Acta Derm Venereol. 2010 Sep;90(5):454–60.
- Candi E, Schmidt R, Melino G. The cornified envelope: a model of cell death in the skin. Nat Rev Mol Cell Biol 2005;6:328–40.
- Huber M, Rettler I, Bernasconi K, et al. Mutations of keratinocyte transglutaminase in lamellar ichthyosis. Science 1995;267:525–8.
- Jobard F, Lefèvre C, Karaduman A, et al. Lipoxygenase-3 (ALOXE3) and 12(R)-lipoxygenase (ALOX12B) are mutated in non-bullous congenital ichthyosiform erythroderma (NCIE) linked to chromosome 17p13.1. Hum Mol Genet 2002;11:107–13.
- Eckl KM, Krieg P, Küster W, et al. Mutation spectrum and functional analysis of epidermis-type lipoxygenases in patients with autosomal recessive congenital ichthyosis. Hum Mutat 2005;26:351–61.
- Akiyama M, Sakai K, Yanagi T, et al. Partially disturbed lamellar granule secretion in mild congenital ichthyosiform erythroderma with ALOX12B mutations. Br J Dermatol 2010;163:201–4.
- Lefèvre C, Bouadjar B, Karaduman A, et al. Mutations in ichthyin a new gene on chromosome 5q33 in a new form of autosomal recessive congenital ichthyosis. Hum Mol Genet 2004;13:2473–82.
- Li H, Loriè EP, Fischer J, et al. The expression of epidermal lipoxygenases and transglutaminase-1 is perturbed by NIPAL4 mutations: indications of a common metabolic pathway essential for skin barrier homeostasis. J Invest Dermatol 2012;132:2368–75.
- Dahlqvist J, Westermark GT, Vahlquist A, Dahl N. Ichthyin/NIPAL4 localizes to keratins and desmosomes in epidermis and Ichthyin mutations affect epidermal lipid metabolism. Arch Dermatol Res 2012;304:377–86.
- Li H, Vahlquist A, Törmä H. Interactions between FATP4 and ichthyin in epidermal lipid processing may provide clues to the pathogenesis of autosomal recessive congenital ichthyosis. J Dermatol Sci 2013;69:195–201.
- Lefèvre C, Bouadjar B, Ferrand V, et al. Mutations in a new cytochrome P450 gene in lamellar ichthyosis type 3. Hum Mol Genet 2006;15:767–76.
- Eckl KM, Tidhar R, Thiele H, et al. Impaired epidermal ceramide synthesis causes autosomal recessive congenital ichthyosis and reveals the importance of ceramide acyl chain length. J Invest Dermatol 2013;133:2202–11.
- Radner FP, Marrakchi S, Kirchmeier P, et al. Mutations in CERS3 cause autosomal recessive congenital ichthyosis in humans. PLOS Genet 2013;9:e1003536.
- Israeli S, Khamaysi Z, Fuchs-Telem D, et al. A mutation in LIPN, encoding epidermal lipase N, causes a late-onset form of autosomal-recessive congenital ichthyosis. Am J Hum Genet 2011;88:482–7.
- Grall A, Guaguère E, Planchais S, et al. PNPLA1 mutations cause autosomal recessive congenital ichthyosis in golden retriever dogs and humans. Nat Genet 2012;44:140–7.
- Vahlquist A, Bygum A, Gånemo A, et al. Genotypic and clinical spectrum of self-improving collodion ichthyosis: ALOX12B, ALOXE3, and TGM1 mutations in Scandinavian patients. J Invest Dermatol 2010;130:438–43.
- Haenssle HA, Finkenrath A, Hausser I, et al. Effective treatment of severe thermodysregulation by oral retinoids in a patient with recessive congenital lamellar ichthyosis. Clin Exp Dermatol 2008;33:578–81.
- Alavi A, Shahshahani MM, Klotzle B, et al. Manifestation of diffuse yellowish keratoderma on the palms and soles in autosomal recessive congenital ichthyosis patients may be indicative of mutations in NIPAL4. J Dermatol 2012;39:375–81.
- Sugiura K, Takeichi T, Tanahashi K, et al. Lamellar ichthyosis in a collodion baby caused by CYP4F22 mutations in a non-consanguineous family outside the Mediterranean. J Dermatol Sci 2013;72:193–5.
- Raghunath M, Hennies HC, Velten F, et al. A novel in situ method for the detection of deficient transglutaminase activity in the skin. Arch Dermatol Res 1998;290:621–7.
- Niemi KM, Kanerva L, Kuokkanen K. Recessive ichthyosis congenita type II. Arch Dermatol Res 1991;283:211–18.
- Dahlqvist J, Klar J, Hausser I, et al. Congenital ichthyosis: mutations in ichthyin are associated with specific structural abnormalities in the granular layer of epidermis. J Med Genet 2007;44:615–20.
- Eckl KM, Krieg P, Küster W, et al. Mutation spectrum and functional analysis of epidermis-type lipoxygenases in patients with autosomal recessive congenital ichthyosis. Hum Mutat 2005;26:351–61.
- Elias PM, Williams ML, Holleran WM, et al. Pathogenesis of permeability barrier abnormalities in the ichthyoses: inherited disorders of lipid metabolism. J Lipid Res 2008;49:697–714.
Keratinopathic ichthyoses
- Bygum A, Virtanen M, Brandrup F, et al. Generalized and naevoid epidermolytic ichthyosis in Denmark: clinical and mutational findings. Acta Derm Venereol 2013;93:309–13.
- Oji V, Tadini G, Akiyama M, et al. Revised nomenclature and classification of inherited ichthyoses: results of the First Ichthyosis Consensus Conference in Sorèze 2009. J Am Acad Dermatol 2010;63:607–41.
- Uitto J, Richard G, McGrath JA. Diseases of epidermal keratins and their linker proteins. Exp Cell Res 2007;313:1995–2009.
- Anton-Lamprecht I. Ultrastructural identification of basic abnormalities as clues to genetic disorders of the epidermis. J Invest Dermatol 1994;103(5 Suppl.):6S–12S.
- Ross R, DiGiovanna JJ, Capaldi L, et al. Histopathologic characterization of epidermolytic hyperkeratosis: a systematic review of histology from the National Registry for Ichthyosis and Related Skin Disorders. J Am Acad Dermatol 2008;59:86–90.
- Muchowski PJ, Wacker JL. Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci 2005;6:11–22.
- Lee D, Santos D, Al-Rawi H, et al. The chemical chaperone trimethylamine N-oxide ameliorates the effects of mutant keratins in cultured cells. Br J Dermatol 2008;159:252–5.
- Li H, Törmä H. Retinoids reduce formation of keratin aggregates in heat-stressed immortalized keratinocytes from an epidermolytic ichthyosis patient with a KRT10 mutation. Acta Derm Venereol 2013;93:44–9.
- Chamcheu JC, Wood GS, Siddiqui IA, et al. Progress towards genetic and pharmacological therapies for keratin genodermatoses: current perspective and future promise. Exp Dermatol 2012;21:481–9.
- Roth W, Kumar V, Beer HD, et al. Keratin 1 maintains skin integrity and participates in an inflammatory network in skin through interleukin-18. J Cell Sci 2012;125:5269–79.
- Jung EG, Schnyder UW. [“Erythroderma ichthyosiforme congenitum”: a heterogenic syndrome.] Dermatologica 1962;124:189–91.
- Müller FB, Huber M, Kinaciyan T, et al. A human keratin 10 knockout causes recessive epidermolytic hyperkeratosis. Hum Mol Genet 2006;15:1133–41.
- Nousbeck J, Padalon-Brauch G, et al. Semidominant inheritance in epidermolytic ichthyosis. J Invest Dermatol 2013;133:2626–8.
- Rothnagel JA, Dominey AM, Dempsey LD, et al. Mutations in the rod domains of keratins 1 and 10 in epidermolytic hyperkeratosis. Science 1992;257:1128–30.
- Cheng J, Syder AJ, Yu QC, et al. The genetic basis of epidermolytic hyperkeratosis: a disorder of differentiation-specific epidermal keratin genes. Cell 1992;70:811–19.
- Chipev CC, Korge BP, Markova N, et al. A leucine–proline mutation in the H1 subdomain of keratin 1 causes epidermolytic hyperkeratosis. Cell 1992;70:821–8.
- Lane EB, McLean WH. Keratins and skin disorders. J Pathol 2004;204:355–66.
- Arin MJ, Oji V, Emmert S, et al. Expanding the keratin mutation database: novel and recurrent mutations and genotype–phenotype correlations in 28 patients with epidermolytic ichthyosis. Br J Dermatol 2011;164:442–7.
- Paller AS, Syder AJ, Chan YM, et al. Genetic and clinical mosaicism in a type of epidermal nevus. N Engl J Med 1994;331:1408–15.
- Kiritsi D, Nanda A, Kohlhase J, et al. Extensive postzygotic mosaicism for a novel keratin 10 mutation in epidermolytic ichthyosis. Acta Derm Venereol 2014;94:346–8.
Epidermolytic ichthyosis
- DiGiovanna JJ, Bale SJ. Clinical heterogeneity in epidermolytic hyperkeratosis. Arch Dermatol 1994;130:1026–35.
- Jung EG, Schnyder UW. [“Erythroderma ichthyosiforme congenitum”: a heterogenic syndrome.] Dermatologica 1962;124:189–91.
- Lapière S. Epidermolyse ichthyosiforme congénitale (erythrodermie ichthyosiforme congénital forme bulleuse de Brocq). Ann Dermatol Syph 1932;3:401–15.
- DiGiovanna JJ, Bale SJ. Epidermolytic hyperkeratosis: applied molecular genetics. J Invest Dermatol 1994;102:390–4.
- Bale SJ, DiGiovanna JJ. Genetic approaches to understanding the keratinopathies. Adv Dermatol 1997;12:99–113.
- Traupe H, Fischer J, Oji V. Nonsyndromic types of ichthyoses – an update. J Dtsch Dermatol Ges 2013;12:109–21.
- Arin MJ. The molecular basis of human keratin disorders. Hum Genet 2009;125:355–73.
- Anton-Lamprecht I. Genetically induced abnormalities of epidermal differentiation and ultrastructure in ichthyoses and epidermolyses: pathogenesis, heterogeneity, fetal manifestation, and prenatal diagnosis. J Invest Dermatol 1983;81:149s–56s.
- Ishida-Yamamoto A, McGrath JA, Judge MR, et al. Selective involvement of keratins K1 and K10 in the cytoskeletal abnormality of epidermolytic hyperkeratosis (bullous congenital ichthyosiform erythroderma). J Invest Dermatol 1992;99:19–26.
- Ishida-Yamamoto A, Takahashi H, Iizuka H. Immunoelectron microscopy links molecules and morphology in the studies of keratinization. Eur J Dermatol 2000;10:429–35.
- Schmuth M, Yosipovitch G, Williams ML, et al. Pathogenesis of the permeability barrier abnormality in epidermolytic hyperkeratosis. J Invest Dermatol 2001;117:837–47.
Superficial epidermolytic ichthyosis
- Siemens WH. Dichtung und Wahrheit über die ‘Ichthyosis bullosa’, mit Bemerkungen zur Systemik der Epidermolysen. Arch Dermatol Syph 1937;175:590–608.
- Traupe H, Kolde G, Hamm H, Happle R. Ichthyosis bullosa of Siemens: a unique type of epidermolytic hyperkeratosis. J Am Acad Dermatol 1986;14:1000–5.
- Rothnagel JA, Traupe H, Wojcik S, et al. Mutations in the rod domain of keratin 2e in patients with ichthyosis bullosa of Siemens. Nat Genet 1994;7:485–90.
- Ang-Tiu CU, Nicolas ME. Ichthyosis bullosa of Siemens. J Dermatol Case Rep 2012;6:78–81
- Cervantes T, Pham C, Browning JC. Superficial epidermolytic ichthyosis: a report of two families. Pediatr Dermatol 2013;30:469–72.
- Nishizawa A, Toyomaki Y, Nakano A, et al. A novel H1 domain mutation in the keratin 2 gene in a Japanese family with ichthyosis bullosa of Siemens. Br J Dermatol 2007;156:1042–4.
- Akiyama M, Tsuji-Abe Y, Yanagihara M, et al. Ichthyosis bullosa of Siemens: its correct diagnosis facilitated by molecular genetic testing. Br J Dermatol 2005;152:1353–6.
Annular epidermolytic ichthyosis
- Joh GY, Traupe H, Metze D, et al. A novel dinucleotide mutation in keratin 10 in the annular epidermolytic ichthyosis variant of bullous congenital ichthyosiform erythroderma. J Invest Dermatol 1997;108:357–61.
- Sybert VP, Francis JS, Corden LD, et al. Cyclic ichthyosis with epidermolytic hyperkeratosis: A phenotype conferred by mutations in the 2B domain of keratin K1. Am J Hum Genet 1999;64:732–8.
- Sheth N, Greenblatt D, McGrath JA. New KRT10 gene mutation underlying the annular variant of bullous congenital ichthyosiform erythroderma with clinical worsening during pregnancy. Br J Dermatol 2007;157:602–4.
Congenital reticular ichthyosiform erythroderma
- Choate KA, Lu Y, Zhou J, et al. Mitotic recombination in patients with ichthyosis causes reversion of dominant mutations in KRT10. Science 2010;330:94–7.
- Marghescu S, Anton-Lamprecht I, Rudolph PO, Kaste R. [Congenital reticular ichthyosiform erythroderma.] [Article in German.] Hautarzt 1984;35:522–9.
- Camenzind M, Harms M, Chavaz P, Saurat JH. [Confetti ichthyosis.] [Article in French.] Ann Dermatol Venereol 1984;111:675–6.
- Burger B, Spoerri I, Schubert M, et al. Description of the natural course and clinical manifestations of ichthyosis with confetti caused by a novel KRT10 mutation. Br J Dermatol 2012;166:434–9.
- Brusasco A, Tadini G, Cambiaghi S, et al. A case of congenital reticular ichthyosiform erythroderma–ichthyosis ‘en confettis’. Dermatology 1994;188:40–5.
Ichthyosis Curth–Macklin
- Sprecher E, Ishida-Yamamoto A, Becker OM, et al. Evidence for novel functions of the keratin tail emerging from a mutation causing ichthyosis hystrix. J Invest Dermatol 2001;116:511–19.
- Fonseca DJ, Rojas RF, Vergara JI, et al. A severe familial phenotype of Ichthyosis Curth–Macklin caused by a novel mutation in the KRT1 gene. Br J Dermatol 2013;168:456–8.
- Curth H, Macklin MT. The genetic basis of various types of ichthyosis in a family group. Am J Hum Genet 1954;6:371–82.
- Ollendorff-Curth H, Allen FH, Jr, Schnyder UW, Anton-Lamprecht I. Follow-up of a family group suffering from ichthyosis hystrix type Curth–Macklin. Humangenetik 1972;17:37–48.
- Ishida-Yamamoto A, Richard G, Takahashi H, Iizuka H. In vivo studies of mutant keratin 1 in ichthyosis hystrix Curth–Macklin. J Invest Dermatol 2003;120:498–500.
Erythrokeratoderma
- Gottron H. Congenital angelegte symmetrische progressive erythrokeratodermie. Z Haut Ges 1922;4:493–4.
- Darier MJ. Erythro-kératodermie verruqueuse en nappes, symétrique et progressive. Bull Soc Fr Dermatol Syphiligr 1911;2:252–64.
- Richard G, Brown N, Rouan F, et al. Genetic heterogeneity in erythrokeratodermia variabilis: novel mutations in the connexin gene GJB4 (Cx30.3) and genotype–phenotype correlations. J Invest Dermatol 2003;120:601–9.
- Oji V, Tadini G, Akiyama M, et al. Revised nomenclature and classification of inherited ichthyoses: results of the First Ichthyosis Consensus Conference in Sorèze 2009. J Am Acad Dermatol 2010;63:607–41.
Erythrokeratoderma variabilis
- Richard G, Smith LE, Bailey RA, et al. Mutations in the human connexin gene GJB3 cause erythrokeratodermia variabilis. Nat Genet 1998;20:366–9.
- Avshalumova L, Fabrikant J, Koriakos A. Overview of skin diseases linked to connexin gene mutations. Int J Dermatol 2014;53(2):192–205.
- Nakamura M. Erythrokeratoderma variabilis without GJB3 or GJB4 mutation: a review of Japanese patients. Br J Dermatol 2007;157:410–11.
- Liu H, Liu H, Fu XA, et al. Mutation analysis of GJB3 and GJB4 in Chinese patients with erythrokeratodermia variabilis. J Dermatol 2012;39:400–1.
- Gottfried I, Landau M, Glaser F, et al. A mutation in GJB3 is associated with recessive erythrokeratodermia variabilis (EKV) and leads to defective trafficking of the connexin 31 protein. Hum Mol Genet 2002;11:1311–16.
- Xia K, Ma H, Xiong H, et al. Trafficking abnormality and ER stress underlie functional deficiency of hearing impairment-associated connexin-31 mutants. Protein Cell 2010;1:935–43.
- Rogers M. Erythrokeratodermas: a classification in a state of flux? Australas J Dermatol 2005;46:127–41.
- Itin P, Levy CA, Sommacal-Schopf D, Schnyder UW. [Family study of erythrokeratodermia figurata variabilis.] [Article in German.] Hautarzt 1992;43:500–4.
- van de Kerkhof PC, Steijlen PM, van Dooren-Greebe RJ, Happle R. Acitretin in the treatment of erythrokeratodermia variabilis. Dermatologica 1990;181:330–3
- Singh N, Thappa DM. Erythrokeratoderma variabilis responding to low-dose isotretinoin. Pediatr Dermatol 2010;27:111–13.
Progressive symmetrical erythrokeratoderma
- Gottron H. Congenital angelegte symmetrische progressive erythrokeratodermie. Z Haut Ges 1922;4:493–4.
- Richard G, Brown N, Rouan F, et al. Genetic heterogeneity in erythrokeratodermia variabilis: novel mutations in the connexin gene GJB4 (Cx30.3) and genotype–phenotype correlations. J Invest Dermatol 2003;120:601–9.
- van Steensel MA, Oranje AP, van der Schroeff JG, et al. The missense mutation G12D in connexin30.3 can cause both erythrokeratodermia variabilis of Mendes da Costa and progressive symmetric erythrokeratodermia of Gottron. Am J Med Genet A 2009;149A:657–61.
- Macfarlane AW, Chapman SJ, Verbov JL. Is erythrokeratoderma one disorder? A clinical and ultrastructural study of two siblings. Br J Dermatol 1991;124:487–91.
Symmetrical acrokeratoderma
- Liu Z, Zhou Y, Chen RY, et al. Symmetrical acrokeratoderma: A peculiar entity in China? Clinicopathologic and immunopathologic study of 34 new cases. J Am Acad Dermatol 2014;70:533–8.
- Blaydon DC, Nitoiu D, Eckl KM, et al. Mutations in CSTA, encoding Cystatin A, underlie exfoliative ichthyosis and reveal a role for this protease inhibitor in cell–cell adhesion. Am J Hum Genet 2011;89:564–71.
Other non-syndromic forms of ichthyosis
- Pujol RM, Moreno A, Alomar A, De Moragas JM. Congenital ichthyosiform dermatosis with linear keratotic flexural papules and sclerosing palmoplantar keratoderma. Arch Dermatol 1989;125:103–6.
- Vahlquist A, Pontén F, Pettersson A. Keratosis linearis with ichthyosis congenita and sclerosing keratoderma (KLICK-syndrome): a rare, autosomal recessive disorder of keratohyaline formation? Acta Derm Venereol 1997;77(3):225–7.
- Dahlqvist J, Klar J, Tiwari N, et al. A single-nucleotide deletion in the POMP 5' UTR causes a transcriptional switch and altered epidermal proteasome distribution in KLICK genodermatosis. Am J Hum Genet 2010;86:596–603.
- Tattersall D, Scott CA, Gray C, Zicha D, Kelsell DP. EKV mutant connexin 31 associated cell death is mediated by ER stress. Hum Mol Genet 2009 Dec 15;18(24):4734–45.
- Tattersall D, Scott CA, Gray C, Zicha D, Kelsell DP. EKV mutant connexin 31 associated cell death is mediated by ER stress. Hum Mol Genet 2009;18:4734–45.
- Chaves AJ, Merchán-García R, Fernández-Recio JM, Rodríguez-Nevado I, de Argila D. [Keratosis linearis with ichthyosis congenita and sclerosing keratoderma (KLICK syndrome).] Acta Dermosifiliogr 2006;97(5):342–4.
- Horev L, Murad S, Maly A, Zlotogorski A. Aggressive cutaneous squamous cell carcinoma in a patient with KLICK. J Am Acad Dermatol 2011 Jun;64(6):e128–30.
Exfoliative ichthyosis
- Blaydon DC, Nitoiu D, Eckl KM, et al. Mutations in CSTA, encoding Cystatin A, underlie exfoliative ichthyosis and reveal a role for this protease inhibitor in cell–cell adhesion. Am J Hum Genet 2011;89:564–71.
- Hatsell SJ, Stevens H, Jackson AP, et al. An autosomal recessive exfoliative ichthyosis with linkage to chromosome 12q13. Br J Dermatol 2003;149:174–80.
- Krunic AL, Stone KL, Simpson MA, McGrath JA. Acral peeling skin syndrome resulting from a homozygous nonsense mutation in the CSTA gene encoding cystatin A. Pediatr Dermatol 2013;30:e87–8.
X-linked syndromes concerning distal cholesterol biosynthesis
Conradi–Hünermann–Happle syndrome
- Happle R, Phillips RJ, Roessner A, Jünemann G. Homologous genes for X-linked chondrodysplasia punctata in man and mouse. Hum Genet 1983;63:24–7.
- Derry JM, Gormally E, Means GD, et al. Mutations in a delta 8-delta 7 sterol isomerase in the tattered mouse and X-linked dominant chondrodysplasia punctata. Nat Genet 1999;22:286–90.
- Braverman N, Lin P, Moebius FF, et al. Mutations in the gene encoding 3 beta-hydroxysteroid-delta 8, delta 7-isomerase cause X-linked dominant Conradi–Hünermann syndrome. Nat Genet 1999;22:291–4.
- Has C, Seedorf U, Kannenberg F, et al. Gas chromatography–mass spectrometry and molecular genetic studies in families with the Conradi–Hünermann–Happle syndrome. J Invest Dermatol 2002;118:851–8.
- Hellenbroich Y, Grzeschik KH, Krapp M, et al. Reduced penetrance in a family with X-linked dominant chondrodysplasia punctata. Eur J Med Genet 2007;50:392–8.
- Morice-Picard F, Kostrzewa E, Wolf C, et al. Evidence of postzygotic mosaicism in a transmitted form of Conradi–Hunermann–Happle syndrome associated with a novel EBP mutation. Arch Dermatol 2011;147:1073–6.
- Traupe H, Müller D, Atherton D, et al. Exclusion mapping of the X-linked dominant chondrodysplasia punctata/ichthyosis/cataract/short stature (Happle) syndrome: possible involvement of an unstable pre-mutation. Hum Genet 1992;89:659–65.
- Grzeschik KH, Bornholdt D, Oeffner F, et al. Deficiency of PORCN, a regulator of Wnt signaling, is associated with focal dermal hypoplasia. Nat Genet 2007;39:833–5.
- Porter FD, Herman GE. Malformation syndromes caused by disorders of cholesterol synthesis. J Lipid Res 2011;52:6–34
- Akiyama M, Sakai K, Hayasaka K, et al. Conradi–Hünermann–Happle syndrome with abnormal lamellar granule contents. Br J Dermatol 2009;160:1335–7.
- Milunsky JM, Maher TA, Metzenberg AB. Molecular, biochemical, and phenotypic analysis of a hemizygous male with a severe atypical phenotype for X-linked dominant Conradi–Hunermann–Happle syndrome and a mutation in EBP. Am J Med Genet A 2003;116A:249–54.
- Happle R. Hypomorphic alleles within the EBP gene cause a phenotype quite different from Conradi–Hünermann–Happle syndrome. Am J Med Genet A 2003;122A:279.
- Arnold AW, Bruckner-Tuderman L, Has C, Happle R. Conradi–Hünermann–Happle syndrome in males vs. MEND syndrome (male EBP disorder with neurological defects). Br J Dermatol 2012;166:1309–13.
- Hartill VL, Tysoe C, Manning N. An unusual phenotype of X-linked developmental delay and extreme behavioral difficulties associated with a mutation in the EBP gene. Am J Med Genet A 2014;164:907–14.
- McLarren KW, Severson TM, du Souich C, et al. Hypomorphic temperature-sensitive alleles of NSDHL cause CK syndrome. Am J Hum Genet 2010;87:905–14.
- Bornholdt D, König A, Happle R, et al. Mutational spectrum of NSDHL in CHILD syndrome. J Med Genet 2005;42:17.
- Paller AS1, van Steensel MA, Rodriguez-Martín M, et al. Pathogenesis-based therapy reverses cutaneous abnormalities in an inherited disorder of distal cholesterol metabolism. J Invest Dermatol 2011;131:2242–8.
Congenital hemidysplasia–ichthyosiform naevus–limb defect syndrome
- Happle R, Koch H, Lenz W. The CHILD syndrome. Congenital hemidysplasia with ichthyosiform erythroderma and limb defects. Eur J Pediatr 1980;134:27–33.
- Happle R, Mittag H, Küster W. The CHILD nevus: a distinct skin disorder. Dermatology 1995;191:210–16.
- König A, Happle R, Bornholdt D, et al. Mutations in the NSDHL gene, encoding a 3beta-hydroxysteroid dehydrogenase, cause CHILD syndrome. Am J Med Genet 2000;90:339–46.
- Liu XY, Dangel AW, Kelley RI, et al. The gene mutated in bare patches and striated mice encodes a novel 3beta-hydroxysteroid dehydrogenase. Nat Genet 1999;22:182–7.
- Jiang F, Herman GE. Analysis of Nsdhl-deficient embryos reveals a role for Hedgehog signaling in early placental development. Hum Mol Genet 2006;15:3293–305.
- Bittar M, Happle R, Grzeschik KH, et al. CHILD syndrome in 3 generations: the importance of mild or minimal skin lesions. Arch Dermatol 2006;142:348–51.
- Danarti R, Grzeschik KH, Radiono S, et al. Left-sided CHILD syndrome caused by a nonsense mutation in exon 7 of the NSDHL gene. Eur J Dermatol 2010;20:634–5.
- Happle R, Effendy I, Megahed M, et al. CHILD syndrome in a boy. Am J Med Genet 1996;62:192–4.
- König A, Skrzypek J, Löffler H, et al. Donor dominance cures CHILD nevus. Dermatology 2010;220:340–5.
- Paller AS, van Steensel MA, Rodriguez-Martín M, et al. Pathogenesis-based therapy reverses cutaneous abnormalities in an inherited disorder of distal cholesterol metabolism. J Invest Dermatol 2011;131:2242–8.
Ichthyosis follicularis–atrichia–photophobia syndrome
- Hamm H, Meinecke P, Traupe H. Further delineation of the ichthyosis follicularis, atrichia, and photophobia syndrome. Eur J Pediatr 1991;150:627–9.
- König A, Happle R. Linear lesions reflecting lyonization in women heterozygous for IFAP syndrome (ichthyosis follicularis with atrichia and photophobia). Am J Med Genet 1999;85:365–8.
- Oeffner F, Fischer G, Happle R, et al. IFAP syndrome is caused by deficiency in MBTPS2, an intramembrane zinc metalloprotease essential for cholesterol homeostasis and ER stress response. Am J Hum Genet 2009;84:459–67.
- Aten E, Brasz LC, Bornholdt D, et al. Keratosis Follicularis Spinulosa Decalvans is caused by mutations in MBTPS2. Hum Mutat 2010;31:1125–33.
- Naiki M, Mizuno S, Yamada K, et al. MBTPS2 mutation causes BRESEK/BRESHECK syndrome. Am J Med Genet A 2012;158A:97–102.
- Bornholdt D, Atkinson TP, Bouadjar B, et al. Genotype–phenotype correlations emerging from the identification of missense mutations in MBTPS2. Hum Mutat 2013;34:587–94.
- Haghighi A, Scott CA, Poon DS, et al. A missense mutation in the MBTPS2 gene underlies the X-linked form of Olmsted syndrome. J Invest Dermatol 2013;133:571–3.
- Oeffner F, Martinez F, Schaffer J, et al. Intronic mutations affecting splicing of MBTPS2 cause ichthyosis follicularis, alopecia and photophobia (IFAP) syndrome. Exp Dermatol 2011;20:447–9.
- Mégarbané H, Mégarbané A. Ichthyosis follicularis, alopecia, and photophobia (IFAP) syndrome. Orphanet J Rare Dis 2011;6:29.
- Bibas-Bonet H, Fauze R, Boente MC, et al. IFAP syndrome “plus” seizures, mental retardation, and callosal hypoplasia. Pediatr Neurol 2001;24:228–31.
- Keyvani K, Paulus W, Traupe H, et al. Ichthyosis follicularis, alopecia, and photophobia (IFAP) syndrome: clinical and neuropathological observations in a 33-year-old man. Am J Med Genet 1998;78:371–7.
- Rothe MJ, Lucky AW. Are ichthyosis follicularis and hereditary mucoepithelial dystrophy related diseases? Pediatr Dermatol 1995;12:195.
- Khandpur S, Bhat R, Ramam M. Ichthyosis follicularis, alopecia and photophobia (IFAP) syndrome treated with acitretin. J Eur Acad Dermatol Venereol 2005;19:759–62.
Exfoliative disorders of cornification
Comèl–Netherton syndrome
- Comèl M. Ichthyosis linearis circumflexa. Dermatologica 1949;98:133–6.
- Netherton EW. A unique case of trichorrhexis nodosa – ‘bamboo hairs’. Arch Dermatol 1958;78:483–87.
- Traupe H. The Comèl Netherton syndrome. In: Traupe H, ed. The Ichthyoses: a guide to clinical diagnosis, genetic counselling, and therapy. Berlin: Springer, 1989:168–78.
- Pruszkowski A, Bodemer C, Fraitag S, et al. Neonatal and infantile erythrodermas: a retrospective study of 51 patients. Arch Dermatol 2000;136:875–80.
- Chavanas S, Bodemer C, Rochat A, et al. Mutations in SPINK5, encoding a serine protease inhibitor, cause Netherton syndrome. Nat Genet 2000;25:141–2.
- Mägert HJ, Ständker L, Kreutzmann P, et al. LEKTI, a novel 15-domain type of human serine proteinase inhibitor. J Biol Chem 1999;247:21499–502.
- Komatsu N, Saijoh K, Otsuki N, et al. Proteolytic processing of human growth hormone by multiple tissue kallikreins and regulation by the serine protease inhibitor Kazal-Type5 (SPINK5) protein. Clin Chim Acta 2007;377:228–36.
- Bitoun E, Micheloni A, Lamant L, et al. LEKTI proteolytic processing in human primary keratinocytes, tissue distribution and defective expression in Netherton syndrome. Hum Mol Genet 2003;12:2417–30.
- Mitsudo, Jayakumar A, Henderson Y, et al. Inhibition of serine proteinases plasmin, trypsin, subtilisin A, cathepsin G, and elastase by LEKTI: a kinetic analysis. Biochemistry 2003;42:3874–81.
- Deraison C, Bonnart C, Lopez F, et al. LEKTI fragments specifically inhibit KLK5, KLK7, and KLK14 and control desquamation through a pH-dependent interaction. Mol Biol Cell 2007;18:3607–19.
- Descargues P, Deraison C, Prost C, et al. Corneodesmosomal cadherins are preferential targets of stratum corneum trypsin- and chymotrypsin-like hyperactivity in Netherton syndrome. J Invest Dermatol 2006;126:1622–32.
- Bonnart C, Deraison C, Lacroix M, et al. Elastase 2 is expressed in human and mouse epidermis and impairs skin barrier function in Netherton syndrome through filaggrin and lipid misprocessing. J Clin Invest 2010;120:871–82.
- Briot A, Deraison C, Lacroix M, et al. Kallikrein 5 induces atopic dermatitis-like lesions through PAR2-mediated thymic stromal lymphopoietin expression in Netherton syndrome. J Exp Med 2009;206:1135–47.
- Briot A, Lacroix M, Robin A, et al. Par2 inactivation inhibits early production of TSLP, but not cutaneous inflammation, in Netherton syndrome adult mouse model. J Invest Dermatol 2010;130:2736–42.
- Fontao L, Laffitte E, Briot A, et al. Infliximab infusions for Netherton syndrome: sustained clinical improvement correlates with a reduction of thymic stromal lymphopoietin levels in the skin. J Invest Dermatol 2011;131:1947–50.
- Denecker G, Ovaere P, Vandenabeele P, Declercq W. Caspase-14 reveals its secrets. J Cell Biol 2008;180:451–8.
- Hovnanian A. Netherton syndrome: skin inflammation and allergy by loss of protease inhibition. Cell Tissue Res 2013;351:289–300.
- Sprecher E, Chavanas S, DiGiovanna JJ, et al. The spectrum of pathogenic mutations in SPINK5 in 19 families with Netherton syndrome: implications for mutation detection and first case of prenatal diagnosis. J Invest Dermatol 2001;117:179–87.
- Bitoun E, Chavanas S, Irvine AD, et al. Netherton syndrome: disease expression and spectrum of SPINK5 mutations in 21 families. J Invest Dermatol 2002;118:352–61.
- Raghunath M, Tontsidou L, Oji V, et al. SPINK5 and Netherton syndrome: novel mutations, demonstration of missing LEKTI, and differential expression of transglutaminases. J Invest Dermatol 2004;123:474–83.
- Israeli S, Sarig O, Garty BZ, et al. Molecular analysis of a series of israeli families with Comèl–Netherton syndrome. Dermatology 2014;228:183–8.
- Sprecher E, Tesfaye-Kedjela A, Ratajczak P, et al. Deleterious mutations in SPINK5 in a patient with congenital ichthyosiform erythroderma: molecular testing as a helpful diagnostic tool for Netherton syndrome. Clin Exp Dermatol 2004;29:513–17.
- Judge MR, Morgan G, Harper JI. A clinical and immunological study of Netherton's syndrome. Br J Dermatol 1994;131:615–21.
- Elias PM, Wakefield JS. Therapeutic implications of a barrier-based pathogenesis of atopic dermatitis. Clin Rev Allergy Immunol 2011;41:282–95.
- Komatsu N, Saijoh K, Otsuki N, et al. Proteolytic processing of human growth hormone by multiple tissue kallikreins and regulation by the serine protease inhibitor Kazal-Type5 (SPINK5) protein. Clin Chim Acta 2007;377:228–36.
- Aydın BK, Baş F, Tamay Z, et al. Netherton syndrome associated with growth hormone deficiency. Pediatr Dermatol 2014;31:90–4.
- Beljan G, Traupe H, Metze D, Sunderkötter C. [Comèl–Netherton syndrome with bacterial superinfection.] Hautarzt 2003;54:1198–202.
- Elbaum DJ, Kurz G, MacDuff M. Increased incidence of cutaneous carcinomas in patients with congenital ichthyosis. J Am Acad Dermatol 1995;33:884–6.
- Weber F, Fuchs PG, Pfister HJ, et al. Human papillomavirus infection in Netherton's syndrome. Br J Dermatol 2001;144:1044–9.
- Saghari S, Wollery-Lloyd H, Nouri K. Squamous cell carcinoma in a patient with Netherton's syndrome. Int J Dermatol 2002;41:415–16.
- Krasagakis K, Ioannidou DJ, Stephanidou M, et al. Early development of multiple epithelial neoplasms in Netherton syndrome. Dermatology 2003;207:182–4.
- Folster-Holst R, Swennson O, Stockfleth E, et al. Comel–Netherton syndrome complicated by papillomatous skin lesions containing human papillomaviruses 51 and 52 and plane warts containing human papillomavirus 16. Br J Dermatol 1999;140:1139–43.
- Li AL, Walsh S, McKay DR. Surgical management of a giant condyloma of Buschke–Löwenstein in a patient with Netherton syndrome using the pedicled anterolateral thigh flap – a case report. J Plast Reconstr Aesthet Surg 2011;64:1533–6.
- Ong C, O'Toole EA, Ghali L, et al. LEKTI demonstrable by immunohistochemistry of the skin: a potential diagnostic skin test for Netherton syndrome. Br J Dermatol 2004;151:1253–7.
- Leclerc-Mercier S, Hovnanian A, Fraitag S. Lekti immunochemistry for the diagnosis of netherton syndrome. Am J Dermatopathol 2012 Dec;34:853.
- Hausser I, Anton-Lamprecht I. Severe congenital generalized exfoliative erythroderma in newborns and infants: a possible sign of Netherton syndrome. Pediatr Dermatol 1996;13:183–99.
- Powell J, Dawber RP, Ferguson DJ, Griffiths WA. Netherton's syndrome: increased likelihood of diagnosis by examining eyebrow hairs. Br J Dermatol 1999;141:544–6.
- Ishida-Yamamoto A, Igawa S, Kishibe M. Order and disorder in corneocyte adhesion. J Dermatol 2011;38:645–54.
- Muller FB, Hauber I, Berg D, et al. Genetic analysis of a severe case of Netherton syndrome and application for prenatal testing. Br J Dermatol 2002;146:495–9.
- Bingol B, Tasdemir S, Gunenc Z, et al. Prenatal diagnosis of Comel–Netherton syndrome with PGD, case report and review article. J Assist Reprod Genet 2011;28:615–20.
- Oji V, Traupe H. Ichthyosis: clinical manifestations and practical treatment options. Am J Clin Dermatol 2009;10:351–64.
- Garty BZ, Nimri R. Hypothyroidism in Netherton syndrome. Pediatr Dermatol 2008;25:134–5.
- HalverstAm CP, Vachharajani A, Mallory SB. Cushing syndrome from percutaneous absorption of 1% hydrocortisone ointment in Netherton syndrome. Pediatr Dermatol 2007;24:42–5.
- Yan AC, Honig PJ, Ming ME, et al. The safety and efficacy of pimecrolimus, 1%, cream for the treatment of Netherton syndrome: results from an exploratory study. Arch Dermatol 2010;146:57–62.
- Oji V, Beljan G, Beier K, et al. Topical pimecrolimus: a novel therapeutic option for Netherton syndrome. Br J Dermatol 2005;153:1067–8.
- Allen A, Siegfried E, Silverman R, et al. Significant absorption of topical tacrolimus in 3 patients with Netherton syndrome. Arch Dermatol 2001;137:747–50.
- Godic A, Dragos V. Successful treatment of Netherton's syndrome with topical calcipotriol. Eur J Dermatol 2004;14:115–17.
- Capezzera R, Venturini M, Bianchi D, et al. UVA1 phototherapy of Netherton syndrome. Acta Derm Venereol 2004;84:69–70.
- Maatouk I, Moutran R, Tomb R. Narrowband ultraviolet B phototherapy associated with improvement in Netherton syndrome. Clin Exp Dermatol 2012;37:364–6.
- Pastore S, Gorlato G, Berti I, et al. Successful induction of oral tolerance in Netherton syndrome. Allergol Immunopathol (Madr) 2012;40:316–17.
- Renner ED, Hartl D, Rylaarsdam S, et al. Comèl–Netherton syndrome defined as primary immunodeficiency. J Allergy Clin Immunol 2009;124:536–43.
- Gallagher JL, Patel NC. Subcutaneous immunoglobulin replacement therapy with Hizentra® is safe and effective in two infants. J Clin Immunol 2012;32:474–6.
- Di WL, Larcher F, Semenova E, et al. Ex-vivo gene therapy restores LEKTI activity and corrects the architecture of Netherton syndrome-derived skin grafts. Mol Ther 2011;19:408–16.
- Roedl D, Oji V, Buters JT, et al. rAAV2-mediated restoration of LEKTI in LEKTI-deficient cells from Netherton patients. J Dermatol Sci 2011;61:194–8.
- Di WL, Mellerio JE, Bernadis C, et al. Phase I study protocol for ex vivo lentiviral gene therapy for the inherited skin disease, Netherton syndrome. Hum Gene Ther Clin Dev 2013;24:182–90.
Severe dermatitis–multiple allergies–metabolic wasting syndrome
- Samuelov L, Sarig O, Harmon RM, et al. Desmoglein 1 deficiency results in severe dermatitis, multiple allergies and metabolic wasting. Nat Genet 2013;45:1244–8.
- Ishida-Yamamoto A, Igawa S. Genetic skin diseases related to desmosomes and corneodesmosomes. J Dermatol Sci 2014;74:99–105.
- Caubet C, Jonca N, Brattsand M, et al. Degradation of corneodesmosome proteins by two serine proteases of the kallikrein family, SCTE/KLK5/hK5 and SCCE/KLK7/hK7. J Invest Dermatol 2004;122:1235–44.
- Descargues P, Deraison C, Bonnart C, et al. Spink5-deficient mice mimic Netherton syndrome through degradation of desmoglein 1 by epidermal protease hyperactivity. Nat Genet 2005;37:56–65.
- Has C, Jakob T, He Y, et al. Loss of desmoglein 1 associated with palmoplantar keratoderma, dermatitis and multiple allergies. Br J Dermatol 2014;172:157–61.
Peeling skin syndromes
- Bowden PE. Peeling skin syndrome: genetic defects in late terminal differentiation of the epidermis. J Invest Dermatol 2011;131:561–4.
- Ishida-Yamamoto A, Igawa S. Genetic skin diseases related to desmosomes and corneodesmosomes. J Dermatol Sci 2014;74:99–105.
- Ishida-Yamamoto A, Igawa S, Kishibe M. Order and disorder in corneocyte adhesion. J Dermatol 2011;38:645–54.
Peeling skin syndrome type A
- Fox H. Skin shedding (keratolysis exfoliativa congenita): report of a case. Arch Dermatol 1921;3:202.
- Abdel-Hafez K, Safer AM, Selim MM, Rehak A. Familial continual skin peeling. Dermatologica 1983;166:23–31.
- Kurban AK, Azar HA. Familial continual skin peeling. Br J Dermatol 1969;81:191–5.
- Beçhet PE. Deciduous skin. Ann Dermatol Syphilol 1938;37:267–71.
- Cabral RM, Kurban M, Wajid M, et al. Whole-exome sequencing in a single proband reveals a mutation in the CHST8 gene in autosomal recessive peeling skin syndrome. Genomics 2012;99:202–8.
- Traupe H. Peeling-skin syndrome: Clinical and morphological evidence for two types. In: Traupe H, ed. The Ichthyoses: a guide to clinical diagnosis, genetic counselling, and therapy. Berlin: Springer, 1989:207–10.
- Silverman AK, Ellis CN, Beals TF, et al. Continual skin peeling syndrome: an electron microscopic study. Arch Dermatol 1986;122:71–5.
- Köse O, Safali M, Koç E, et al. Peeling skin diseases: 21 cases from Turkey and a review of the literature. J Eur Acad Dermatol Venereol 2012;26:844–8.
Inflammatory peeling skin disease
- Levy SB, Goldsmith LA. The peeling skin syndrome. J Am Acad Dermatol 1982;7:606–13.
- Traupe H. Peeling-skin syndrome: Clinical and morphological evidence for two types. In: Traupe H, ed. The Ichthyoses: a guide to clinical diagnosis, genetic counselling, and therapy. Berlin: Springer, 1989:207–10.
- Wile, UJ. Familial study of three unusual cases of congenital ichthyosiform erythrodermia. Arch Dermatol Syph 1924;10:487–98.
- Oji V, Eckl KM, Aufenvenne K, et al. Loss of corneodesmosin leads to severe skin barrier defect, pruritus, and atopy: unraveling the peeling skin disease. Am J Hum Genet 2010;87:274–81.
- Israeli S, Zamir H, Sarig O, et al. Inflammatory peeling skin syndrome caused by a mutation in CDSN encoding corneodesmosin. J Invest Dermatol 2011;131:779–81.
- Ishida-Yamamoto A, Furio L, Igawa S, et al. Inflammatory peeling skin syndrome caused by homozygous genomic deletion in the PSORS1 region encompassing the CDSN gene. Exp Dermatol 2014;23:60–3.
- Mallet A, Kypriotou M, George K, et al. Identification of the first nonsense CDSN mutation with expression of a truncated protein causing peeling skin syndrome type B. Br J Dermatol 2013;169:1322–5.
- Mazereeuw-Hautier J, Leclerc EA, Simon M, et al. A novel mutationin CDSN causes peeling skin disease in a patient from Morocco. Br J Dermatol 2011;165:1152–5.
- Telem DF, Israeli S, Sarig O, Sprecher E. Inflammatory peeling skin syndrome caused a novel mutation in CDSN. Arch Dermatol Res 2012;304:251–5.
- Wada T, Matsuda Y, Muraoka M, et al. Alu-mediated large deletion of the CDSN gene as a cause of peeling skin disease. Clin Genet 2014;86:383–6.
- Lundstrom A, Serre G, Haftek M, Egelrud T. Evidence for a role of corneodesmosin, a protein which may serve to modify desmosomes during cornification, in stratum corneum cell cohesion and desquamation. Arch Dermatol Res 1994;286:369–75.
- Serre G, Mils V, Haftek M, et al. Identification of late differentiation antigens of human cornified epithelia, expressed in re-organized desmosomes and bound to cross-linked envelope. J Invest Dermatol 1991;97:1061–72.
- Simon M, Jonca N, Guerrin M, et al. Refined characterization of corneodesmosin proteolysis during terminal differentiation of human epidermis and its relationship to desquamation. J Biol Chem 2001;276:20292–9.
- Matsumoto M, Zhou Y, Matsuo S, et al. Targeted deletion of the murine corneodesmosin gene delineates its essential role in skin and hair physiology. Proc Natl Acad Sci U S A 2008;105:6720–4.
- Jonca N, Leclerc EA, Caubet C, et al. Corneodesmosomes and corneodesmosin: from the stratum corneum cohesion to the pathophysiology of genodermatoses. Eur J Dermatol 2011;21:35–42.
- Levy-Nissenbaum E, Betz RC, Frydman M, et al. Hypotrichosis simplex of the scalp is associated with nonsense mutations in CDSN encoding corneodesmosin. Nat Genet 2003;34:151–3.
- Caubet C, Bousset L, Clemmensen O, et al. A new amyloidosis caused by fibrillar aggregates of mutated corneodesmosin. FASEB J 2010;24:3416–26.
- Aras N, Sutman K, Tastan HB, et al. Peeling skin syndrome. J Am Acad Dermatol 1994;30:135–6.
- Dicken CH. Peeling skin syndrome. J Am Acad Dermatol 1985;13:158–60.
- Hacham-Zadeh S, Holubar K. Skin peeling syndrome in a Kurdish family. Arch Dermatol 1985;121:545–6.
- Komatsu N, Suga Y, Saijoh K, et al. Elevated human tissue kallikrein levels in the stratum corneum and serum of peeling skin syndrome-type B patients suggests an over-desquamation of corneocytes. J Invest Dermatol 2006;126:2338–42.
- Mevorah B, Frenk E, Saurat JH, Siegenthaler G. Peeling skin syndrome: a clinical, ultrastructural and biochemical study. Br J Dermatol 1987;116:117–25.
- Mevorah B, Salomon D, Siegenthaler G, et al. Ichthyosiform dermatosis with superficial blister formation and peeling: evidence for a desmosomal anomaly and altered epidermal vitamin A metabolism. J Am Acad Dermatol 1996;34:379–85.
- Farooq M, Kurban M, Abbas O, et al. Netherton syndrome showing a large clinical overlap with generalized inflammatory peeling skin syndrome. Eur J Dermatol 2012;22:412–13.
- Sárdy M, Fáy A, Kárpáti S, Horváth A. Comèl–Netherton syndrome and peeling skin syndrome type B: overlapping syndromes or one entity? Int J Dermatol 2002;41:264–8.
- Tsai K, Valente NY, Nico MM. Inflammatory peeling skin syndrome studied with electron microscopy. Pediatr Dermatol 2006;23:488–92.
- Mizuno Y, Suga Y, Hasegawa T, et al. A case of peeling skin syndrome successfully treated with topical calcipotriol. J Dermatol 2006;33:430–2.
Acral peeling skin syndrome
- Shwayder T, Conn S, Lowe L. Acral peeling skin syndrome. Arch Derm 1997;133:535–6.
- Kiritsi D, Cosgarea I, Franzke CW, et al. Acral peeling skin syndrome with TGM5 gene mutations may resemble epidermolysis bullosa simplex in young individuals. J Invest Dermatol 2010;130:1741–6.
- Fine JD, Bruckner-Tuderman L, Eady RA, et al. Inherited epidermolysis bullosa: Updated recommendations on diagnosis and classification. J Am Acad Dermatol 2014;70:1103–26.
- Cassidy AJ, van Steensel MA, Steijlen PM, et al. A homozygous missense mutation in TGM5 abolishes epidermal transglutaminase 5 activity and causes acral peeling skin syndrome. Am J Hum Genet 2005;77:909–17.
- Pavlovic S, Krunic AL, Bulj TK, et al. Acral peeling skin syndrome: a clinically and genetically heterogeneous disorder. Pediatr Dermatol 2012;29:258–63.
- Szczecinska W, Nesteruk D, Wertheim-Tysarowska K, et al. Underrecognition of acral peeling skin syndrome: 59 new cases with 15 novel mutations. Br J Dermatol 2014;171:1206–10.
- Blaydon DC, Nitoiu D, Eckl KM, et al. Mutations in CSTA, encoding Cystatin A, underlie exfoliative ichthyosis and reveal a role for this protease inhibitor in cell–cell adhesion. Am J Hum Genet 2011;89:564–71.
- Krunic AL, Stone KL, Simpson MA, McGrath JA. Acral peeling skin syndrome resulting from a homozygous nonsense mutation in the CSTA gene encoding cystatin A. Pediatr Dermatol 2013;30:87–8.
- Emmerson RW, Wilson-Jones E. Ringed keratolysis of the palms. Trans St John's Hosp Dermatol Soc 1967;53:165–7.
- Chang YY, van der Velden J, van der Wier G, et al. Keratolysis exfoliativa (dyshidrosis lamellosa sicca): a distinct peeling entity. Br J Dermatol 2012;167:1076–84.
Neuro-ichthyotic syndromes
- Rizzo WB, Jenkens SM, Boucher P. Recognition and diagnosis of neuro-ichthyotic syndromes. Semin Neurol 2012;32:75–84.
- Martinelli D, Travaglini L, Drouin CA, et al. MEDNIK syndrome: a novel defect of copper metabolism treatable by zinc acetate therapy. Brain 2013;136:872–81.
CEDNIK, MEDNIK, ARC, Gaucher disease type II, ELOVL4 deficiency and Stormorken syndrome
- Holleran WM, Ginns EI, Menon GK, et al. Consequences of beta-glucocerebrosidase deficiency in epidermis. Ultrastructure and permeability barrier alterations in Gaucher disease. J Clin Invest 1994;93:1756–64.
- Tsuji S, Choudary PV, Martin BM, et al. A mutation in the human glucocerebrosidase gene in neuronopathic Gaucher's disease. N Engl J Med 1987;316:570–5.
- Sprecher E, Ishida-Yamamoto A, Mizrahi-Koren M, et al. A mutation in SNAP29, coding for a SNARE protein involved in intracellular trafficking, causes a novel neurocutaneous syndrome characterized by cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma. Am J Hum Genet 2005;77:242–51.
- Fuchs-Telem D, Stewart H, Rapaport D, et al. CEDNIK syndrome results from loss-of-function mutations in SNAP29. Br J Dermatol 2011;164:610–16.
- Dereure O. [Differentiating between Mednik and Cednik syndromes.] Ann Dermatol Venereol 2009;136:850–1.
- Nezelof C, Martin G, Pruvost J. [A fatal syndrome associating a micromelic dwarfism, an ichtyosiform skin disorder and a severe combined immunologic deficiency. Report of a case and survey of the literature (author's transl).] Ann Pediatr (Paris) 1979;26:309–14.
- Gissen P, Johnson CA, Morgan NV, et al. Mutations in VPS33B, encoding a regulator of SNARE-dependent membrane fusion, cause arthrogryposis–renal dysfunction–cholestasis (ARC) syndrome. Nat Genet 2004;36:400–4.
- Hershkovitz D, Mandel H, Ishida-Yamamoto A, et al. Defective lamellar granule secretion in arthrogryposis, renal dysfunction, and cholestasis syndrome caused by a mutation in VPS33B. Arch Dermatol 2008;144:334–40.
- Smith H, Galmes R, Gogolina E, et al. Associations among genotype, clinical phenotype, and intracellular localization of trafficking proteins in ARC syndrome. Hum Mutat 2012;33:1656–64.
- Jang JY, Kim KM, Kim GH, et al. Clinical characteristics and VPS33B mutations in patients with ARC syndrome. J Pediatr Gastroenterol Nutr 2009;48:348–54.
- Dehghani SM, Bahador A, Nikeghbalian S, et al. Liver transplant in a case of arthrogryposis–renal tubular dysfunction–cholestasis syndrome with severe intractable pruritus. Exp Clin Transplant 2013;11:290–2.
- Montpetit A, Cote S, Burstein E, et al. Disruption of AP1S1, causing a novel neurocutaneous syndrome, perturbs development of the skin and spinal cord. Proc Natl Acad Sci USA 2009;4:e1000296.
- Aldahmesh MA, Mohamed JY, Alkuraya HS, et al. Recessive mutations in ELOVL4 cause ichthyosis, intellectual disability, and spastic quadriplegia. Am J Hum Genet 2011;89:745–50.
- Mir H, Raza SI, Touseef M, et al. A novel recessive mutation in the gene ELOVL4 causes a neuro-ichthyotic disorder with variable expressivity. BMC Med Genet 2014;26;15:25.
- Giroux JM, Barbeau A. Erythrokeratodermia with ataxia. Arch Dermatol 1972;106:183–8.
- Cadieux-Dion M, Turcotte-Gauthier M, Noreau A, et al. Expanding the clinical phenotype associated with ELOVL4 mutation: study of a large French-Canadian family with autosomal dominant spinocerebellar ataxia and erythrokeratodermia. JAMA Neurol 2014;71:470–5.
- Stormorken H, Sjaastad O, Langslet A, et al. A new syndrome: thrombocytopathia, muscle fatigue, asplenia, miosis, migraine, dyslexia and ichthyosis. Clin Genet 1985;28:367–74.
- Misceo D, Holmgren A, Louch WE, et al. A dominant STIM1 mutation causes Stormorken syndrome. Hum Mutat 2014;35:556–64.
- Morin G, Bruechle NO, Singh AR, et al. Gain-of-function mutation in STIM1 (p.R304W) is associated with Stormorken Syndrome. Hum Mutat 2014;35:1221–32.
Refsum disease
- Kahlke W, Richterich R. Refsum's disease (herecopathia a tactica polyneuritiformis): an inborn error of lipid metabolism with storage of 3,7,11,15-tetramethyl hexadecanoic acid. II. Isolation and identification of the storage product. Am J Med 1965;39:237–41.
- Jansen GA, Hogenhout EM, Ferdinandusse S, et al. Human phytanoyl-CoA hydroxylase: resolution of the gene structure and the molecular basis of Refsum's disease. Hum Mol Genet 2000;9:1195–200.
- van den Brink DM, Brites P, Haasjes J, et al. Identification of PEX7 as the second gene involved in Refsum disease. Am J Hum Genet 2003;72:471–7.
- Horn MA, van den Brink DM, Wanders RJ, et al. Phenotype of adult Refsum disease due to a defect in peroxin 7. Neurology 2007;68:698–700.
- Jansen GA, Waterham HR, Wanders RJ. Molecular basis of Refsum disease: sequence variations in phytanoyl–CoA hydroxylase (PHYH) and the PTS2 receptor (PEX7). Hum Mutat 2004;23:209–18.
- Scotto JM, Hadchouel M, Odievre M, et al. Infantile phytanic acid storage disease, a possible variant of Refsum's disease: three cases, including ultrastructural studies of the liver. J Inherit Metab Dis 1982;5(2):83–90.
- Poll-The BT, Saudubray JM, Ogier HA, et al. Infantile Refsum disease: an inherited peroxisomal disorder. Comparison with Zellweger syndrome and neonatal adrenoleukodystrophy. Eur J Pediatr 1987 Sep;146(5):477–83.
- Ramsay BC, Meeran K, Woodrow D, et al. Cutaneous aspects of Refsum's disease. J R Soc Med 1991 Sep;84(9):559–60.
- Kohlschütter A, Santer R, Lukacs Z, et al. A child with night blindness: preventing serious symptoms of Refsum disease. J Child Neurol 2012;27:654–6.
- Zolotov D, Wagner S, Kalb K, et al. Long-term strategies for the treatment of Refsum's disease using therapeutic apheresis. J Clin Apher 2012;27:99–105.
Multiple sulphatase deficiency
- Artigalás OA, da Silva LR, Burin M, et al. Multiple sulfatase deficiency: clinical report and description of two novel mutations in a Brazilian patient. Metab Brain Dis 2009;24:493–500.
- Dierks T, Dickmanns A, Preusser-Kunze A, et al. Molecular basis for multiple sulfatase deficiency and mechanism for formylglycine generation of the human formylglycine-generating enzyme. Cell 2005;121:541–52.
- Busche A, Hennermann JB, Bürger F, et al. Neonatal manifestation of multiple sulfatase deficiency. Eur J Pediatr 2009;168:969–73.
- Schlotawa L, Ennemann EC, Radhakrishnan K, et al. SUMF1 mutations affecting stability and activity of formylglycine generating enzyme predict clinical outcome in multiple sulfatase deficiency. Eur J Hum Genet 2011;19:253–61
- Loffeld A, Gray RG, Green SH, et al. Mild ichthyosis in a 4-year-old boy with multiple sulphatase deficiency. Br J Dermatol 2002;147:353–5.
Sjögren–Larsson syndrome
- Rizzo WB, Dammann AL, Craft DA. Sjögren–Larsson syndrome. Impaired fatty alcohol oxidation in cultured fibroblasts due to deficient fatty alcohol:nicotinamide adenine dinucleotide oxidoreductase activity. J Clin Invest 1988;81:738–44.
- Willemsen MA, Rotteveel JJ, de Jong JG, et al. Defective metabolism of leukotriene B4 in the Sjögren–Larsson syndrome. J Neurol Sci 2001;183:61–7.
- Willemsen MA, Lutt MA, Steijlen PM, et al. Clinical and biochemical effects of zileuton in patients with the Sjögren–Larsson syndrome. Eur J Pediatr 2001;160:711–17.
- Rizzo WB. Sjögren–Larsson syndrome: molecular genetics and biochemical pathogenesis of fatty aldehyde dehydrogenase deficiency. Mol Genet Metab 2007;90:1–9.
- De Laurenzi V, Rogers GR, Hamrock DJ, et al. Sjögren–Larsson syndrome is caused by mutations in the fatty aldehyde dehydrogenase gene. Nat Genet 1996;12:52–7.
- Rizzo WB, Carney G. Sjögren–Larsson syndrome: diversity of mutations and polymorphisms in the fatty aldehyde dehydrogenase gene (ALDH3A2). Hum Mutat 2005;26:1–10.
- Fuijkschot J, Cruysberg JR, Willemsen MA, et al. Subclinical changes in the juvenile crystalline macular dystrophy in Sjögren–Larsson syndrome detected by optical coherence tomography. Ophthalmology 2008;115:870–5.
- Gånemo A, Jagell S, Vahlquist A. Sjögren–Larsson syndrome: a study of clinical symptoms and dermatological treatment in 34 Swedish patients. Acta Derm Venereol 2009;89:68–73.
- Kathuria S, Arora S, Ramesh V. Sjögren–Larsson syndrome: importance of early diagnosis and aggressive physiotherapy. Dermatol Online J 2012 Sep 15;18(9):11.
- Gloerich J, Ijlst L, Wanders RJ, Ferdinandusse S. Bezafibrate induces FALDH in human fibroblasts; implications for Sjögren–Larsson syndrome. Mol Genet Metab 2006;89:111–15.
Keratitis–ichthyosis–deafness
- Richard G, Rouan F, Willoughby CE, et al. Missense mutations in GJB2 encoding connexin-26 cause the ectodermal dysplasia keratitis–ichthyosis–deafness syndrome. Am J Hum Genet 2002;70:1341–8.
- van Steensel MA, van Geel M, Nahuys M, et al. A novel connexin 26 mutation in a patient diagnosed with keratitis–ichthyosis–deafness syndrome. J Invest Dermatol 2002;118:724–7.
- van Geel M, van Steensel MA, Küster W, et al. HID and KID syndromes are associated with the same connexin 26 mutation. Br J Dermatol 2002;146:938–42.
- de Zwart-Storm EA, Hamm H, Stoevesandt J, et al. A novel missense mutation in GJB2 disturbs gap junction protein transport and causes focal palmoplantar keratoderma with deafness. J Med Genet 2008;45:161–6.
- Easton JA, Donnelly S, Kamps MA, et al. Porokeratotic eccrine nevus may be caused by somatic connexin26 mutations. J Invest Dermatol 2012;132:2184–91.
- Lazic T, Li Q, Frank M, et al. Extending the phenotypic spectrum of keratitis–ichthyosis–deafness syndrome: report of a patient with GJB2 (G12R) Connexin 26 mutation and unusual clinical findings. Pediatr Dermatol 2012;29:349–57.
- Yotsumoto S, Hashiguchi T, Chen X, et al. Novel mutations in GJB2 encoding connexin-26 in Japanese patients with keratitis–ichthyosis–deafness syndrome. Br J Dermatol 2003;148:649–53.
- Mazereeuw-Hautier J, Bitoun E, Chevrant-Breton J, et al. Keratitis–ichthyosis–deafness syndrome: disease expression and spectrum of connexin 26 (GJB2) mutations in 14 patients. Br J Dermatol 2007;156:1015–19.
- Mhaske PV, Levit NA, Li L, et al. The human Cx26-D50A and Cx26-A88V mutations causing keratitis–ichthyosis–deafness syndrome display increased hemichannel activity. Am J Physiol Cell Physiol 2013;304:1150–8.
- Grob JJ, Breton A, Bonafe JL, et al. Keratitis, ichthyosis and deafness (kid) syndrome: vertical transmission and death from multiple squamous cell carcinomas. Arch Dermatol 1987;123:777–82.
- Mazereeuw-Hautier J, Bitoun E, Chevrant-Breton J, et al. Keratitis–ichthyosis–deafness syndrome: disease expression and spectrum of connexion 26 (GJB2) mutations in 14 patients. Br J Dermatol 2007;156:1015–19.
- Lancaster L, Fournet LF. Carcinoma of the tongue in a child. J Oral Maxillofac Surg 1969;27:269–70.
- Baden EP, Alper JC. Ichthyosiform dermatosis, keratitis and deafness. Arch Dermatol 1977;113:1701–4.
- Grob JJ, Breton A, Bonafe JL, et al. Keratitis, ichthyosis and deafness (kid) syndrome: vertical transmission and death from multiple squamous cell carcinomas. Arch Dermatol 1987;123:777–82.
- Kim K-H, Kim J-S, Piao Y-J, et al. Keratitis ichthyosis and deafness syndrome with development of multiple hair follicle tumours. Br J Dermatol 2002;147:139–43.
- Nyquist GG, Mumm C, Grau R, et al. Malignant proliferating pilar tumours arising in KID syndrome: a report of two patients. Am J Med Genet 2007;143:734–41.
- Aloi FG, Pippione M. Porokeratotic eccrine ostial and dermal duct nevus. Arch Dermatol 1986;122:892–5.
- Smyth CM, Sinnathuray AR, Hughes AE, Toner JG. Cochlear implantation in keratitis–ichthyosis–deafness syndrome: 10-year follow-up of two patients. Cochlear Implants Int 2012;13:54–9.
- Messmer EM, Kenyon KR, Rittinger O, et al. Ocular manifestations of keratitis–ichthyosis–deafness (KID) syndrome. Ophthalmology 2005;112:1–6.
- Coggshall K, Farsani T, Ruben B, et al. Keratitis, ichthyosis, and deafness syndrome: a review of infectious and neoplastic complications. J Am Acad Dermatol 2013;69:127–34.
- Hazen PG, Carney JM, Langston RH, Meisler DM. Corneal effect of isotretinoin: possible exacerbation of corneal neovascularization in a patient with the keratitis, ichthyosis, deafness (“KID”) syndrome. J Am Acad Dermatol 1986;14:141–2.
- Prasad SC, Bygum A. Successful treatment with alitretinoin of dissecting cellulitis of the scalp in keratitis–ichthyosis–deafness syndrome. Acta Derm Venereol 2013;93:473–4.
Neutral lipid storage disease with ichthyosis
- Lefèvre C, Jobard F, Caux F, et al. Mutations in CGI-58, the gene encoding a new protein of the esterase/lipase/thioesterase subfamily, in Chanarin–Dorfman syndrome. Am J Hum Genet 2001;69:1002–12.
- Fischer J, Lefèvre C, Morava E, et al. The gene encoding adipose triglyceride lipase (PNPLA2) is mutated in neutral lipid storage disease with myopathy. Nat Genet 2007;39:28–30.
- Badeloe S, van Geel M, Nagtza I, et al. Chanarin–Dorfman syndrome caused by a novel splice site mutation in ABHD5. Br J Dermatol 2008;158:1378–80.
- Akiyama M, Sawamura D, Nomura Y, et al. Truncation of CGI-58 protein causes malformation of lamellar granules resulting in ichthyosis in Dorfman–Chanarin syndrome. J Invest Dermatol 2003;121:1029–34.
- Uchida Y, Cho Y, Moradian S, et al. Neutral lipid storage leads to acylceramide deficiency, likely contributing to the pathogenesis of Dorfman–Chanarin syndrome. J Invest Dermatol 2010;130:2497–9.
- Wollenberg A, Geiger E, Schaller M, Wolff H. Dorfman–Chanarin syndrome in a Turkish kindred: conductor diagnosis requires analysis of multiple eosinophils. Acta Derm Venereol 2000;80:39–43.
- Radner FP, Fischer J. The important role of epidermal triacylglycerol metabolism for maintenance of the skin permeability barrier function. Biochim Biophys Acta 2014;1841:409–15.
- Srebrnik A, Tur E, Perluk C, et al. Dorfman–Chanarin syndrome. A case report and a review. J Am Acad Dermatol 1987;17:801–8.
- Peña-Penabad C, Almagro M, Martínez W, et al. Dorfman–Chanarin syndrome (neutral lipid storage disease): new clinical features. Br J Dermatol 2001;144:430–2.
- Israeli S, Pessach Y, Sarig O, et al. Beneficial effect of acitretin in Chanarin–Dorfman syndrome. Clin Exp Dermatol 2012;37:31–3.
- Judge MR, Atherton DJ, Salvayre R, et al. Neutral lipid storage disease. Case report and lipid studies. Br J Dermatol 1994;130:507–10.
Trichothiodystrophy/Tay syndrome
- Morice-Picard F, Cario-André M, Rezvani H et al. New clinico-genetic classification of trichothiodystrophy. Am J Med Genet A 2009;149A:2020–30.
- Faghri S, Tamura D, Kraemer KH, Digiovanna JJ. Trichothiodystrophy: a systematic review of 112 published cases characterises a wide spectrum of clinical manifestations. J Med Genet 2008;45:609–21.
- Egly JM, Coin F. A history of TFIIH: two decades of molecular biology on a pivotal transcription/repair factor. DNA Repair (Amst) 2011;10:714–21
- Compe E, Malerba M, Soler L, et al. Neurological defects in trichothiodystrophy reveal a coactivator function of TFIIH. Nat Neurosci 2007;10:1414–22.
- Zhang Y, Tian Y, Chen Q, et al. TTDN1 is a Plk1-interacting protein involved in maintenance of cell cycle integrity. Cell Mol Life Sci 2007;64:632–40.
- Tamura D, Khan SG, Merideth M, et al. Effect of mutations in XPD(ERCC2) on pregnancy and prenatal development in mothers of patients with trichothiodystrophy or xeroderma pigmentosum. Eur J Hum Genet 2012;20:1308–10.
- Happle R, Traupe H, Gröbe H, Bonsmann G. The Tay syndrome (congenital ichthyosis with trichothiodystrophy). Eur J Pediatr 1984;141:147–52.
- Jorizzo JL, Atherton DJ, Crounse RG, Wells RS. Ichthyosis, brittle hair, impaired intelligence, decreased fertility and short stature (IBIDS syndrome). Br J Dermatol 1982;106:705–10.
- Stefanini M, Botta E, Lanzafame M, Orioli D. Trichothiodystrophy: from basic mechanisms to clinical implications. DNA Repair (Amst) 2010;9:2–10.
- Battistella PA, Peserico A. Central nervous system dysmyelination in PIBI(D)S syndrome: a further case. Childs Nerv Syst 1996;12:110–13.
- Kleijer WJ, Beemer FA, Boom BW. Intermittent hair loss in a child with PIBI(D)S syndrome and trichothiodystrophy with defective DNA repair–xeroderma pigmentosum group D. Am J Med Genet 1994;52:227–30.
- Vermeulen W, Rademakers S, Jaspers NG, et al. A temperature-sensitive disorder in basal transcription and DNA repair in humans. Nat Genet 2001;27:299–303.
Neu–Laxova syndrome
- Neu RL, Kajii T, Gardner LI, Nagyfy SF. A lethal syndrome of microcephaly with multiple congenital anomalies in three siblings. Pediatrics 1971;47:610–12.
- Laxova R, Ohara PT, Timothy JA. A further example of a lethal autosomal recessive condition in sibs. J Ment Defic Res 1972;16:139–43.
- Manning MA, Cunniff CM, Colby CE, et al. Neu–Laxova syndrome: detailed prenatal diagnostic and post-mortem findings and literature review. Am J Med Genet A 2004;125:240–9.
- Shaheen R, Rahbeeni Z, Alhashem A, et al. Neu–Laxova syndrome, an inborn error of serine metabolism, is caused by mutations in PHGDH. Am J Hum Genet 2014;94:898–904.
- Acuna-Hidalgo R, Schanze D, Kariminejad A, et al. Neu–Laxova syndrome is a heterogeneous metabolic disorder caused by defects in enzymes of the L-serine biosynthesis pathway. Am J Hum Genet 2014;95:285–93.
- Shapiro I, Borochowitz Z, Degani S, et al. Neu–Laxova syndrome: prenatal ultrasonographic diagnosis, clinical and pathological studies, and new manifestations. Am J Med Genet 1992;43:602–5.
- Smigiel R, Jakubiak A, Esteves-Vieira V, et al. Novel frameshifting mutations of the ZMPSTE24 gene in two siblings affected with restrictive dermopathy and review of the mutations described in the literature. Am J Med Genet A 2010;152A:447–52
- Loucks C, Parboosingh JS, Chong JX, et al. A shared founder mutation underlies restrictive dermopathy in Old Colony (Dutch-German) Mennonite and Hutterite patients in North America. Am J Med Genet A 2012;158A:1229–32.
- De Koning TJ. Treatment with amino acids in serine deficiency disorders. J Inherit Metab Dis 2006;29,347–51.
Coloboma heart defect–ichthyosiform dermatosis–mental retardation–ear anomalies syndrome
- Ng BG, Hackmann K, Jones MA, et al. Mutations in the glycosylphosphatidylinositol gene PIGL cause CHIME syndrome. Am J Hum Genet 2012;90:685–8.
- Jones MA, Ng BG, Bhide S, et al. DDOST mutations identified by whole-exome sequencing are implicated in congenital disorders of glycosylation. Am J Hum Genet 2012;90:363–8.
- Rymen D, Jaeken J. Skin manifestations in CDG. J Inherit Metab Dis 2014;37:699–708.
- Zunich J, Kaye CI. New syndrome of congenital ichthyosis with neurologic abnormalities. Am J Med Genet 1983;15:331–3, 335.
- Zunich J, Esterly NB, Holbrook KA, Kaye CI. Congenital migratory ichthyosiform dermatosis with neurologic and ophthalmologic abnormalities. Arch Dermatol 1985;121:1149–56.
- Shashi V, Zunich J, Kelly TE, Fryburg JS. Neuroectodermal (CHIME) syndrome: an additional case with long term follow up of all reported cases. J Med Genet 1995;32:465–9.
- Tinschert S, Anton-Lamprecht I, Albrecht-Nebe H, Audring H. Zunich neuroectodermal syndrome: migratory ichthyosiform dermatosis, colobomas, and other abnormalities. Pediatr Dermatol 1996;13:363–71.
- Schnur RE, Greenbaum BH, Heymann WR. Acute lymphoblastic leukemia in a child with the CHIME neuroectodermal dysplasia syndrome. Am J Med Genet 1997;72:24–9.
Miscellaneous syndromic ichthyoses
Ichthyosis–prematurity syndrome
- Blaas HG, Salvesen KÅ, Khnykin D, Jahnsen FL, Eik-Nes SH. Prenatal sonographic assessment and perinatal course of ichthyosis prematurity syndrome. Ultrasound Obstet Gynecol 2012;39:473–7.
- Bygum A, Westermark P, Brandrup F. Ichthyosis prematurity syndrome: a well-defined congenital ichthyosis subtype. J Am Acad Dermatol 2008;59(Suppl.):S71–4.
- Dereksson K, Kjartansson S, Hjartardóttir H, Arngrimsson R. Ichthyosis prematurity syndrome with separation of fetal membranes and neonatal asphyxia. BMJ Case Rep 2012;2012.
- Khnykin D, Rønnevig J, Johnsson M, et al. Ichthyosis prematurity syndrome: clinical evaluation of 17 families with a rare disorder of lipid metabolism. J Am Acad Dermatol 2012;66:606–16.
- Vahlquist A. Pleomorphic ichthyosis: proposed name for a heterogeneous group of congenital ichthyoses with phenotypic shifting and mild residual scaling. Acta Derm Venereol 2010;90:454–60.
- Klar J, Schweiger M, Zimmerman R, et al. Mutations in the fatty acid transport protein 4 gene cause the ichthyosis prematurity syndrome. Am J Hum Genet 2009;85:248–53.
- Inhoff O, Hausser I, Schneider SW, et al. Ichthyosis prematurity syndrome caused by a novel fatty acid transport protein 4 gene mutation in a German infant. Arch Dermatol 2011;147:750–2.
- Khnykin D, Miner JH, Jahnsen F. Role of fatty acid transporters in epidermis: Implications for health and disease. Dermatoendocrinology 2011;3:53–61.
- Lin MH, Khnykin D. Fatty acid transporters in skin development, function and disease. Biochim Biophys Acta 2014;1841:362–8.
- Lin MH, Hsu FF, Miner JH. Requirement of fatty acid transport protein 4 for development, maturation, and function of sebaceous glands in a mouse model of ichthyosis prematurity syndrome. J Biol Chem 2013;288:3964–76.
- Anton-Lamprecht I. Diagnostic ultrastructural of non-neoplastic diseases. In: Papadimitriou J, Henderson DW, Spagnolo DV, eds. The Skin. Edinburgh: Churchill Livingstone, 1992:459–550.
Ichthyosis with hypotrichosis
- Basel-Vanagaite L, Attia R, Ishida-Yamamoto A, et al. Autosomal recessive ichthyosis with hypotrichosis caused by a mutation in ST14, encoding type II transmembrane serine protease matriptase. Am J Hum Genet 2007;80:467–77.
- Avrahami L, Maas S, Pasmanik-Chor M, et al. Autosomal recessive ichthyosis with hypotrichosis syndrome: further delineation of the phenotype. Clin Genet 2008;74:47–53.
- Lestringant GG, Kuster W, Frossard PM, Happle R. Congenital ichthyosis, follicular atrophoderma, hyotrichosis, and hypohidrosis: a new genodermatosis? Am J Med Genet 1998;75:186–9.
- Alef T, Torres S, Hausser I, et al. Ichthyosis, follicular atrophoderma, and hypotrichosis caused by mutations in ST14 is associated with impaired profilaggrin processing. J Invest Dermatol 2009;129:862–9.
- Chen YW, Wang JK, Chou FP, et al. Matriptase regulates proliferation and early, but not terminal, differentiation of human keratinocytes. J Invest Dermatol 2014;134:405–14.
- Ishida-Yamamoto A, Igawa S. Genetic skin diseases related to desmosomes and corneodesmosomes. J Dermatol Sci 2014;74:99–105.
- List K, Szabo R, Wertz PW, et al. Loss of proteolytically processed filaggrin caused by epidermal deletion of Matriptase/MT-SP1. J Cell Biol 2003;163:901–10.
- Sales KU, Masedunskas A, Bey AL, et al. Matriptase initiates activation of epidermal pro-kallikrein and disease onset in a mouse model of Netherton syndrome. Nat Genet 2010;42:676–83.
- Désilets A, Béliveau F, Vandal G, et al. Mutation G827R in matriptase causing autosomal recessive ichthyosis with hypotrichosis yields an inactive protease. J Biol Chem 2008;283:10535–42.
- Dereure O. [Recessive autosomal ichthyosis with hypotrichosis with mutation in the ST14 gene.] Ann Dermatol Venereol 2007;134:798.
- List K, Currie B, Scharschmidt TC, et al. Autosomal ichthyosis with hypotrichosis syndrome displays low matriptase proteolytic activity and is phenocopied in ST14 hypomorphic mice. J Biol Chem 2007;282:36714–23.
Neonatal ichthyosis–sclerosing cholangitis
- Hadj-Rabia S, Baala L, Vabres P, et al. Claudin-1 gene mutations in neonatal sclerosing cholangitis associated with ichthyosis: a tight junction disease. Gastroenterology 2004;127:1386–90.
- Nagtza IF, van Geel M, Driessen A, Steijlen PM, van Steensel MA. Bile duct paucity is part of the neonatal ichthyosis–sclerosing cholangitis phenotype. Br J Dermatol 2010;163:205–7.
- Morita K, Miyachi Y, Furuse M. Tight junctions in epidermis: from barrier to keratinization. Eur J Dermatol 2011;21:12–17.
- Baala L, Hadj-Rabia S, Hamel-Teillac D, et al. Homozygosity mapping of a locus for a novel syndromic ichthyosis to chromosome 3q27-q28. J Invest Derm 2002;119:70–6.
- Feldmeyer L, Huber M, Fellmann F, et al. Hohl D. Confirmation of the origin of NISCH syndrome. Hum Mutat 2006;27:408–10.
- Shah I, Bhatnagar S. NISCH syndrome with hypothyroxinemia. Ann Hepatol 2010;9:299–301.
- Kirchmeier P, Sayar E, Hotz A, et al. Novel mutation in the CLDN1 gene in a Turkish family with neonatal ichthyosis sclerosing cholangitis (NISCH) syndrome. Br J Dermatol 2014;170:976–8.
Management of congenital ichthyoses
- Hernández-Martin A, Aranegui B, Martin-Santiago A, Garcia-Doval I. A systematic review of clinical trials of treatments for the congenital ichthyoses, excluding ichthyosis vulgaris. J Am Acad Dermatol 2013;69:544–9.
- Vahlquist A, Blockhuys S, Steijlen P, et al. Oral liarozole in the treatment of patients with moderate/severe lamellar ichthyosis: results of a randomized, double-blind, multinational, placebo-controlled phase II/III trial. Br J Dermatol 2014;170:173–81.
- Oji V, Traupe H. Ichthyosis: clinical manifestations and practical treatment options. Am J Clin Dermatol 2009;10:351–64.
- Aufenvenne K, Larcher F, Hausser I, et al. Topical enzyme-replacement therapy restores transglutaminase 1 activity and corrects architecture of transglutaminase-1-deficient skin grafts. Am J Hum Genet 2013;93:620–30.
- Vahlquist A, Gånemo A, Virtanen M. Congenital ichthyosis: an overview of current and emerging therapies. Acta Derm Venereol 2008;88:4–14.
- Kiistala R, Lauharanta J, Kanerva L. Transepidermal water loss and sweat gland response in lamellar ichthyosis before and during treatment with etretinate: report of three cases. Acta Derm Venereol 1982;62:268–70.
- Gånemo A, Virtanen M, Vahlquist A. Improved topical treatment of lamellar ichthyosis: a double-blind study of four different cream formulations. Br J Dermatol 1999;141:1027–32.
Management of collodion baby
- Prado R, Ellis LZ, Gamble R, et al. Collodion baby: an update with a focus on practical management. J Am Acad Dermatol 2012;67:1362–74.
- Van Gysel D, Lijnen RL, Moekti SS, et al. Collodion baby: a follow-up study of 17 cases. J Eur Acad Dermatol Venereol 2002;16:472–5.
- Rajpopat S, Moss C, Mellerio J, et al. Harlequin ichthyosis: a review of clinical and molecular findings in 45 cases. Arch Dermatol 2011;147:681–6.
- Buyse L, Graves C, Marks R, et al. Collodion baby dehydration: the danger of high transepidermal water loss. Br J Dermatol 1993;129:86–8.
- Eilers E, Stieler K, Thies C, et al. [Harlequin ichthyosis – medical and psychosocial challenges.] Klin Padiatr 2010;222:86–9.
- Harvey HB, Shaw MG, Morrell DS. Perinatal management of harlequin ichthyosis: a case report and literature review. J Perinatol 2010;30:66–72.
- Milstone LM, Choate KA. Improving outcomes for harlequin ichthyosis. J Am Acad Dermatol 2013;69:808–9.
- Rubio-Gomez GA, Weinstein M, Pope E. Development of a disease severity score for newborns with collodion membrane. J Am Acad Dermatol 2014;70:506–11.
The issue of bathing
- Bodemer C, Bourrat E, Mazereeuw-Hautier J, et al. Short- and medium-term efficacy of specific hydrotherapy in inherited ichthyosis. Br J Dermatol 2011;165:1087–94.
- Traupe H, Burgdorf WHC. Treatment of ichthyosis – There is always something you can do! In Memoriam: Wolfgang Küster. J Am Acad Dermatol 2007;57:542–7.
- Milstone LM. Scaly skin and bath pH: rediscovering baking soda. J Am Acad Dermatol 2010;62:885–6.
- Traupe H. Ichthyosis keeps surprising us. Acta Dermosifiliogr 2013;104:267–9.
Practical treatment options for daily care
- Gånemo A, Virtanen M, Vahlquist A. Improved topical treatment of lamellar ichthyosis: a double-blind study of four different cream formulations. Br J Dermatol 1999;141:1027–32.
- Craiglow BG, Choate KA, Milstone LM. Topical tazarotene for the treatment of ectropion in ichthyosis. JAMA Dermatol 2013;149:598–600.
- Redondo P, Bauzá A. Topical N-acetylcysteine for lamellar ichthyosis. Lancet 1999;354:1880.
- Sarici SU, Sahin M, Yurdakök M. Topical N-acetylcysteine treatment in neonatal ichthyosis. Turk J Pediatr 2003;45:245–7.
- Bassotti A, Moreno S, Criado E. Successful treatment with topical N-acetylcysteine in urea in five children with congenital lamellar ichthyosis. Pediatr Dermatol 2011;28:451–5.
Systemic treatment options
- Vahlquist A, Gånemo A, Virtanen M. Congenital ichthyosis: an overview of current and emerging therapies. Acta Derm Venereol 2008;88:4–14.
- Digiovanna JJ, Mauro T, Milstone LM, et al. Systemic retinoids in the management of ichthyoses and related skin types. Dermatol Ther 2013;26:26–38.
- Bilan P, Levy A, Sin C, et al. [Erythrokeratodermia variabilis.] Ann Dermatol Venereol 2013;140:129–33.
- Hunzeker CM, Soldano AC, Levis WR. Erythrokeratoderma variabilis. Dermatol Online J 2008;14:13.
- Ständer S, Stadelmann A, Traub O, et al. [Erythrokeratodermia variabilis (EKV) – a disorder due to altered epidermal expression of gap junction proteins.] J Dtsch Dermatol Ges 2005;3:354–8.
- Ahmed H, O'Toole EA. Recent advances in the genetics and management of harlequin ichthyosis. Pediatr Dermatol 2014;31:539–46.
- Koochek A, Choate KA, Milstone LM. Harlequin ichthyosis: neonatal management and identification of a new ABCA12 mutation. Pediatr Dermatol 2014;31:63–4.
- Milstone LM, Choate KA. Improving outcomes for harlequin ichthyosis. J Am Acad Dermatol 2013;69:808–9.
- Chamcheu JC, Wood GS, Siddiqui IA, et al. Progress towards genetic and pharmacological therapies for keratin genodermatoses: current perspective and future promise. Exp Dermatol 2012;21:481–9.
- Ormerod AD, Campalani E, Goodfield MJ, et al. British Association of Dermatologists guidelines on the efficacy and use of acitretin in dermatology. Br J Dermatol 2010;162:952–63.
- Lacour M, Mehta-Nikhar B, Atherton DJ, Harper JI. An appraisal of acitretin therapy in children with inherited disorders of keratinization. Br J Dermatol 1996;134:1023–9.
- Gånemo A, Sommerlund M, Vahlquist A. Oral alitretinoin in congenital ichthyosis: a pilot study shows variable effects and a risk of central hypothyroidism. Acta Derm Venereol 2012;92:256–7.
- Prasad SC, Bygum A. Successful treatment with alitretinoin of dissecting cellulitis of the scalp in keratitis–ichthyosis–deafness syndrome. Acta Derm Venereol 2013;93:473–4.
Special aspects of treatment
- Oji V, Traupe H. Ichthyosis: clinical manifestations and practical treatment options. Am J Clin Dermatol 2009;10:351–64.
- Chaudhary M, Shrestha GB, Keyal A. Lamellar ichthyosis presenting as bilateral spontaneous corneal perforation. Nepal J Ophthalmol 2013;5:117–19.
- Turgut B, Aydemir O, Kaya M, et al. Spontaneous corneal perforation in a patient with lamellar ichthyosis and dry eye. Clin Ophthalmol 2009;3:611–13.
- Chaurasia S, Das S, Ramamurthy B. Microbial keratitis in a case of lamellar ichthyosis. Int Ophthalmol 2008;28:367–8.
- Nayak S, Rath S, Kar BR. Mucous membrane graft for cicatricial ectropion in lamellar ichthyosis: an approach revisited. Ophthal Plast Reconstr Surg 2011;27:155–6.
- Das S, Honavar SG, Dhepe N, Naik MN. Maternal skin allograft for cicatricial ectropion in congenital icthyosis. Ophthal Plast Reconstr Surg 2010;26:42–3.
- Khan R, Arora S, El-Hindy N, Chang BY. Repair of cicatricial ectropion in a harlequin baby. J AAPOS 2009;13:415–16.
- Kampp JT, Kouba DJ, Fincher EF, Moy RL. Basal cell carcinoma masquerading as the chronic ectropion of lamellar ichthyosis. Dermatol Surg 2008;34:963–7.
- Huang JT, Mallon K, Hamill S, et al. Frequency of ear symptoms and hearing loss in ichthyosis: a pilot survey study. Pediatr Dermatol 2014;31:276–80.
- Haenssle HA, Finkenrath A, Hausser I, et al. Effective treatment of severe thermodysregulation by oral retinoids in a patient with recessive congenital lamellar ichthyosis. Clin Exp Dermatol 2008;33:578–81.
- Kothari D, Doshi B, Garg G, Khopkar US. Ichthyosis associated with rickets in two Indian children. Indian J Dermatol 2013;58:244.
- Kumar V, Kalra S, Mutreja D, Arya A. Rickets associated with ichthyosis. Paediatr Int Child Health 2012;32:119–20.
- Sethuraman G, Khaitan BK, Dash SS, et al. Ichthyosiform erythroderma with rickets: report of five cases. Br J Dermatol 2008;158:603–6.
- Chouhan K, Sethuraman G, Gupta N, et al. Vitamin D deficiency and rickets in children and adolescents with ichthyosiform erythroderma in type IV and V skin. Br J Dermatol. 2012;166:608–15.
- Thacher TD, Fischer PR, Pettifor JM, Darmstadt GL. Nutritional rickets in ichthyosis and response to calcipotriene. Pediatrics 2004;114:119–23.
- Sathish Kumar T, Scott XJ, Simon A, Raghupathy P. Vitamin D deficiency rickets with lamellar ichthyosis. J Postgrad Med 2007;53:215–17.
- Ali R, Aman S, Nadeem M. Lamellar ichthyosis with rickets. Pak J Med Sci 2013;292:660–2.
- Deka N, Sarma D, Saikia UK. Lamellar ichthyosis with genu valgum: unfolding the link. BMJ Case Rep 2012;22:2012.
- Bhagat SB, Bhagat SS, Sharma HK, et al. Severe bilateral rachitic genu valgum in patients with nonbullous congenital ichthyosiform erythroderma: a report of two cases and review of literature. J Pediatr Orthop B 2007;16:423–8.
- André E, Till M, Descargues P, et al. [Netherton syndrome: a type of infantile erythroderma with failure to thrive, immune deficiency, rickets. Report of 3 cases.] Arch Pediatr 2005;12:1364–7.
- Ingen-Housz-Oro S, Boudou P, Bergot C, et al. Evidence of a marked 25-hydroxyvitamin D deficiency in patients with congenital ichthyosis. J Eur Acad Dermatol Venereol 2006;20:947–52.
- Milstone LM, Ellison AF, Insogna KL. Serum parathyroid hormone level is elevated in some patients with disorders of keratinization. Arch Dermatol 1992;128:926–30.
- Freitas CF, Mulinari-Brenner F, Fontana HR, et al. Ichthyosis associated with widespread tinea corporis: report of three cases. An Bras Dermatol 2013;88:627–30.
- Machan M, Kestenbaum T, Fraga GR. Diffuse hyperkeratosis in a deaf and blind 48-year-old woman – quiz case. Diagnosis: keratitis–ichthyosis–deafness (KID) syndrome with secondary dermatophytosis. Arch Dermatol 2012;148:1199–200.
- Beljan G, Traupe H, Metze D, Sunderkötter C. [Comèl–Netherton syndrome with bacterial superinfection.] Hautarzt 2003;54:1198–202.
- Haruna K, Suga Y, Oizumi A, et al. Severe form of keratitis–ichthyosis–deafness (KID) syndrome associated with septic complications. J Dermatol 2010;37:680–2.
Acquired ichthyoses
- Patel N, Spencer LA, English JC 3rd, Zirwas MJ. Acquired ichthyosis. J Am Acad Dermatol 2006;55:647–56.
- Kutting B, Traupe H. Acquired ichthyosis-like skin disease: a challenge for diagnostic evaluation. Hautarzt 1995;46:836–40.
- Sneddon IB. Acquired ichthyosis in Hodgkin's disease. BMJ 1955;1:763–4.
- Akpinar TS, Ozkok A, Bakkaloglu OK, Saka B. Acquired ichthyosis as a presenting finding of Hodgkin's lymphoma. Int J Hematol 2012;96:401–2.
- Word AP, Cayce R, Pandya AG. Beware of underlying malignancy: acquired ichthyosis. Am J Med 2014;127:202–4.
- Dykes PJ, Marks R. Acquired ichthyosis: multiple causes for an acquired generalized disturbance in desquamation. Br J Dermatol 1977;97:327–34.
- Estines O, Grosieux-Dauger C, Derancourt C, et al. Paraneoplastic acquired ichthyosis revealing non-Hodgkin's lymphoma. Ann Dermatol Venereol 2001;128:31–4.
- Kutting B, Metze D, Luger TA, Bonsmann G. Mycosis fungoides presenting as an acquired ichthyosis. J Am Acad Dermatol 1996;34:887–9.
- Eisman S, O'Toole EA, Jones A, et al. Granulomatous mycosis fungoides presenting as an acquired ichthyosis. Clin Exp Dermatol 2003;28:174–6.
- Tokuriki A, Kiyohara T, Ido T, Kumakiri M. A case of lymphomatoid papulosis with extensive limb disease followed by extracutaneous involvement and acquired ichthyosis. Acta Derm Venereol 2012;92:278–9.
- Yokote R, Iwatsuki K, Hashizume H, et al. Lymphomatoid papulosis associated with acquired ichthyosis. J Am Acad Dermatol 1994;30:889–92.
- Bluefarb SM. Cutaneous manifestations of multiple myeloma. Arch Dermatol Syphilol 1955;72:506–22.
- Moore RL, Devere TS. Epidermal manifestations of internal malignancy. Dermatol Clin 2008;26:17–29.
- Levy O, Tishler M. Acquired ichthyosis as the primary manifestation of renal cell carcinoma. Isr Med Assoc J 2009;11:121–2.
- Majekodunmi AE, Fenii Pearse D. Ichthyosis: early manifestation of intestinal leiomyosarcoma. BMJ 1974;3:724.
- Grattan CE, Williams DM, Raafat F, et al. Acquired ichthyosis in a child with rhabdomyosarcoma. Pediatr Dermatol 1988;5:167–9.
- Krakowski A, Brenner S, Covo J, et al. Acquired ichthyosis in Kaposi's sarcoma. Dermatologica 1973;147:348–51.
- Tsochatzis E, Vassilopoulos D, Deutsch M, et al. Myelodysplastic syndrome presenting as acquired ichthyosis. Eur J Int Med 2006;5:368–9.
- Spelman LJ, Strutton GM, Robertson IM, et al. Acquired ichthyosis in bone marrow transplant recipients. J Am Acad Dermatol 1996;35:17–20.
- Huang J, Pol-Rodriguez M, Silvers D, et al. Acquired ichthyosis as a manifestation of acute graft-versus-host disease. Pediatr Dermatol 2007;24:49–52.
- Elias PM, Brown BE, Ziboh VA. The permeability barrier in essential fatty acid deficiency: evidence for a direct role for linoleic acid in barrier function. J Invest Dermatol 1980;74:230–3.
- Aggett PJ, Cavanagh HP, Matthew DJ, et al. Shwachman's syndrome: a review of 21 cases. Arch Dis Child 1980;55:331–47.
- Boggia P, Machado MC, De Oliveira Z, et al. Methylmalonic acidemia presenting with an ichthyosis vulgaris-like aspect. Pediatr Dermatol 2007;24:455–6.
- Arbuckle HA, Morelli J. Holocarboxylase synthetase deficiency presenting as ichthyosis. Pediatr Dermatol 2006;23:142–4.
- London RD, Lebwohl M. Acquired ichthyosis and hyperparathyroidism. J Am Acad Dermatol 1989;21:801–2.
- Dinnen JS, Greenwood RH, Jones JH, et al. Parathyroid carcinoma in familial hyperparathyroidism. J Clin Invest 1977;30:966–75.
- Brazzelli V, Larizza D, Muzio F, et al. Acquired ichthyosis in a child with autoimmune thyroiditis. Pediatr Dermatol 2010;27:413–14.
- Piérard GE, Seité S, Hermanns-Lê T, et al. The skin landscape in diabetes mellitus. Focus on dermocosmetic management. Clin Cosmet Investig Dermatol 2013;6:127–35.
- Font J, Bosch X, Ingelmo M, et al. Acquired ichthyosis with systemic lupus erythematosus. Arch Dermatol 1990;126:829.
- Inuzuka M, Tomita K, Tokura Y, et al. Acquired ichthyosis associated with dermatomyositis in a patient with hepatocellular carcinoma. Br J Dermatol 2001;144:416–17.
- Lee HW, Ahn SJ, Choi JC, et al. Acquired ichthyosis with an overlap syndrome of systemic sclerosus and systemis lupus erythematosus. J Dermatol 2006;33:52–4.
- Kikuchi J, Saita B, Inoue S. Haber's syndrome: report of a new family. Arch Dermatol 1981;117:321–4.
- de la Cruz-Alvarez J, Allegue F, Oliver J. Acquired ichthyosis associated with eosinophilic fasciitis. J Am Acad Dermatol 1996;34:1079–80.
- Kelley BP, George DE, LeLeux TM, Hsu S. Ichthyosiform sarcoidosis: A case report and review of the literature. Dermatol Online J 2010;16:5.
- Schulz EJ. Ichthyosiform conditions occurring in leprosy. Br J Dermatol 1965;77:151–7.
- Young L, Steinman HK. Acquired ichthyosis in a patient with acquired immunodeficiency syndrome and Kaposi's sarcoma. J Am Acad Dermatol 1987;16:395–6.
- Coldiron BM, Bergstresser PR. Prevalence and clinical spectrum of skin disease in patients infected with the human immunodeficiency virus. Arch Dermatol 1989;125:357–61.
- Okajima R, Oliveira AC, Smid J, et al. High prevalence of skin disorders among HTLV-1 infected individuals independent of clinical status. PLOS Negl Trop Dis 2013;7:2546.
- Winklemann RK, Perry HO, Achor RWP, Kirby TJ. Cutaneous syndromes produced as side-effects of triparanol therapy. Arch Dermatol 1963;87:372–7.
- Williams ML, Feingold KR, Grubauer G, et al. Ichthyosis induced by cholesterol-lowering drugs. Arch Dermatol 1987;123:1535–7.
- Sparsa A, Boulinguez S, Le-Brun V, et al. Acquired ichthyosis with pravastatin. J Eur Acad Dermatol Venereol 2007;21:549–50.
- Niederaurer HH, Bacharach-Buhles M, Altmeyer P. Ichthyosis and alopecia after maprotiline. Hautarzt 1991;42:455–8.
- Greist MC, Epinette WW. Cimetidine induced xerosis and asteatotic dermatitis. Arch Dermatol 1982;118:253–4.
- Kumar B, Saraswat A, Kaur I. Mucocutaneous adverse effects of hydroxyurea: a prospective study of 30 psoriasis patients. Clin Exp Dermatol 2002;27:8–13.
- Caver CV. Clofazamine induced ichthyosis and its treatment. Cutis 1982;29:341–3.
- Errichette E, Stinco G, Pegolo E, Patrone P. Acquired ichthyosis during acitretin therapy for psoriasis vulgaris. J Eur Acad Dermatol Venereol 2014 [epub].
Pityriasis rotunda
- Batra P, Cheung W, Meehan SA, Pomeranz M. Pityriasis rotunda. Dermatol Online J 2009;15:14.
- Ito M, Tanaka T. Pseudo ichthyose acquise en taches circulaires. Ann Dermatol Syphiligr 1960;87:26–37.
- Findlay GH. Pityriasis rotunda in the South African Bantu. Br J Dermatol 1965;77:63–4.
- El-Hefnawi H, Rasheed A. Pityriasis rotunda: report of study of first case in UAR. Arch Dermatol 1966;93:84–6.
- Sarkany I, Hare PJ. Pityriasis rotunda (pityriasis circinata). Br J Dermatol 1964;76:223–8.
- Zur RL, Shapero J, Shapero H. Pityriasis rotunda diagnosed in Canada: case presentation and review of the literature. J Cutan Med Surg 2013;17:426–8.
- Segal R, Hodak E, Sandbank M. Pityriasis rotunda in a Caucasian woman from the Mediterranean area. Clin Exp Dermatol 1989;14:325–7.
- Aste N, Pau M, Aste N, et al. Pityriasis rotunda: a survey of 42 cases observed in Sardinia, Italy. Dermatology 1997;194:32–5.
- Ena P, Cerimele D. Pityriasis rotaunda in childhood. Pediatr Dermatol 2002;19:200–3.
- Grimalt R, Gelmetti C, Brusasco A, et al. Pityriasis rotunda: report of a familial ccurrence and review of the literature. J Am Acad Dermatol 1994;31:866–71.
- Friedmann AC, Ameen M, Swale VJ. Familial pityriasis rotunda in black-skinned patients; a first report. Br J Dermatol 2007;156:1362–402.
- Guberman D, Lichtenstein DA, Gilead L, et al. Familial pityriasis rotunda. Acta Derm Venereol 1997;77:162.
- Swift PJ, Saxe N. Pityriasis rotunda in South Africa — a skin disease caused by undernutrition. Clin Exp Dermatol 1985;10:407–12.
- Leibowitz MR, Weiss R, Smith EH. Pityriasis rotunda: a cutaneous sign of malignant disease in two patients. Arch Dermatol 1983;119:607–9.
- Griffin LJ, Massa MC. Acquired ichthyosis and pityriasis rotunda. Clin Dermatol 1993;11:27–32.
- Ikada J, Oki M. Concurrent pityriasis rotunda and acquired ichthyosis with IgG myeloma. Br J Dermatol 1974;91:585–6.
- Berkowitz I, Hodkinson HJ, Kew MC, et al. Pityriasis rotunda as a cutaneous marker of hepatocellular carcinoma: a comparison with its prevalence in other disease. Br J Dermatol 1989;120:545–9.
- Hashimoto Y, Suga Y, Chikenji T, et al. Immunohistochemical characterization of a Japanese case of pityriasis rotunda. Br J Dermatol 2003;149:196–8.
- Yoneda K, Presland RB, Demitsu T, et al. The profilaggrin N-terminal domain is absent in pityriasis rotunda. Br J Dermatol 2012;166:227–9.
Palmoplantar keratoderma
- Greither A. Erbliche Palmoplantarkeratosen. Hautarzt 1977;28:395–403.
- Braun-Falco M. Hereditary palmoplantar keratodermas. J Dtsch Dermatol Ges 2009;7:971–84.
- Oji V, Tadini G, Akiyama M, et al. Revised nomenclature and classification of inherited ichthyoses: results of the First Ichthyosis Consensus Conference in Sorèze 2009. J Am Acad Dermatol 2010;63:607–41.
- Lefèvre C, Bouadjar B, Karaduman A, et al. Mutations in ichthyin a new gene on chromosome 5q33 in a new form of autosomal recessive congenital ichthyosis. Hum Mol Genet 2004;13:2473–82.
- Alavi A, Shahshahani MM, Klotzle B, Fan JB, Ronaghi M, Elahi E. Manifestation of diffuse yellowish keratoderma on the palms and soles in autosomal recessive congenital ichthyosis patients may be indicative of mutations in NIPAL4. J Dermatol 2012;39:375–81.
- Salamon T. An attempt at classification of inherited disorders of keratinization localized mainly, not exclusively on the palms and soles. Dermatol Monatsschr 1986;172:601–5.
- Zemstov A, Veitschegger M. Keratodermas. Int J Dermatol 1993;32:493–8.
- Lucker GPH, Van de Kerkhof PCM, Steiljen PM. The hereditary palmoplantar keratoses: an updated review and classification. Br J Dermatol 1994;131:1–14.
- Itin PH, Lautenschlager S. Palmoplantar keratoderma and associated syndromes. Semin Dermatol 1995;14:152–61.
- Ratnavel RC, Griffiths WAD. The inherited palmoplantar keratodermas. Br J Dermatol 1997;137:485–90.
- Schiller S, Seebode C, Hennies HC, et al. Palmoplantar keratoderma (PPK): acquired and genetic causes of a not so rare disease. J Dtsch Dermatol Ges 2014;12:781–8.
Epidermolytic palmoplantar keratoderma
- Vörner H. Zur Kenntnis des keratome hereditarium palmare et plantare. Arch Derm Syph (Berlin) 1901;56:3–31.
- Hamm H, Happle R, Butterfass T, et al. Epidermolytic palmoplantar keratoderma of Vörner: is it the most frequent type of hereditary palmoplantar keratoderma? Dermatologica 1988;177:138–45.
- Kuster W, Becker A. Indication for the identity of palmoplantar keratoderma type Unna–Thost with type Vörner. Acta Derm Venereol (Stockh) 1992;72:120–2.
- Covello SP, Irvine AD, McKenna KE, et al. Mutations in keratin K9 in kindreds with epidermolytic palmoplantar keratoderma and epidemiology in Northern Ireland. J Invest Dermatol 1998;111:1207–9.
- Knapp AC, Franke WW, Heid H, et al. Cytokeratin 9, an epidermal type 1 keratin, characteristic of a special programme of keratinocyte differentiation displaying body site specificity. J Cell Biol 1986;103:657–67.
- Langbein L, Heid H, Moll I, Franke WW. Molecular characterisation of the body site specific human epidermal cytokeratin 9, cDNA cloning, amino acid sequence, and tissue specificity of gene expression. Differentiation 1994;55:57–72.
- Reis A, Kuster W, Eckhardt R, Sperling K. Mapping of a gene for epidermolytic palmoplantar keratoderma to the region of the acidic keratin gene cluster at 17q12–q21. Hum Genet 1992;90:113–16.
- Reis A, Hennies H-C, Langbein L, et al. Keratin 9 gene mutations in epidermolytic palmoplantar keratoderma (EPPK). Nat Genet 1994;8:174–9.
- Irvine AD, McLean WH. Human keratin diseases: the increasing spectrum of disease and subtlety of the phenotype–genotype correlation. Br J Dermatol 1999;140:815–28.
- Kuster W, Reis A, Hennies HC. Epidermolytic palmoplantar keratoderma of Vorner: re-evaluation of Vorner's original family and identification of a novel keratin 9 mutation. Arch Derm Res 2002;294:268–72.
- Coleman CM, Munro CS, Smith FJ, et al. Epidermolytic palmoplantar keratoderma due to a novel type of keratin mutation, a 3-bp insertion in the keratin 9 helix termination motif. Br J Dermatol 1999;140:486–90.
- Terron-Kwiatkowski A, Paller AS, Compton J, et al. Two cases of primarily palmoplantar keratoderma associated with novel mutations in keratin 1. J Invest Dermatol 2002;119:966–71.
- Terron-Kwiatkowski A, Terrinoni A, Didona B, et al. Atypical epidermolytic palmoplantar keratoderma presentation associated with a mutation in the keratin 1 gene. Br J Dermatol 2004;150:1096–103.
- Tal O, Bergman R, Alcalay J, et al. Epidermolytic hyperkeratosis type PS-1 caused by aberrant splicing of KRT1. Clin Exp Dermatol 2005;30:64–7.
- Hatsell SJ, Eady RA, Wennerstrand L, et al. Novel splice site mutation in keratin 1 underlies mild epidermolytic palmoplantar keratoderma in three kindreds. J Invest Dermatol 2001;116:606–9
- Wevers A, Kuhn A, Mahrle G. Palmoplantar keratoderma with tonotubular keratin. J Am Acad Dermatol 1991;24:638–42.
- Terron-Kwiatkowski A, van Steensel MA, Van Geel M, et al. Mutation S233L in the 1B domain of keratin 1 causes epidermolytic palmoplantar keratoderma with ‘tonotubular’ keratin. J Invest Dermatol 2006;126:607–13.
- Grimberg G, Hausser I, Mueller FB, et al. Novel and recurrent mutations in the 1B domain of keratin 1 in palmoplantar keratoderma with tonotubules Br J Dermatol 2009;160:446–49.
- Kuster W, Zehender D, Mensing H, et al. Vörner keratosis palmoplantaris diffusa: clinical, formal genetic and molecular biology studies of 22 families. Hautarzt 1995;46:705–10.
- Bergman R, Khamaysi Z, Sprecher E. A unique pattern of dyskeratosis characterizes epidermolytic hyperkeratosis and epidermolytic palmoplantar keratoderma. Am J Dermatopathol 2008;30:101–5.
- Anton-Lamprecht I. Ultrastructural identification of basic abnormalities as clues to genetic disorders of the epidermis. J Invest Dermatol 1994;103:6S–12S.
- Hashimoto K, Mizuguchi R, Tanaka K, Dorman M. Palmoplantar keratoderma (Vörner) with composite keratohyalin granules: studies on keratinization parameters and ultrastructures. J Dermatol 2000;27:1–9.
- Braun-Falco M. Hereditary palmoplantar keratodermas. J Dtsch Dermatol Ges 2009;7:971–84.
- Lucker GPH, van de Kerkhof PCM, Steiljen PM. Topical calcipotriol in the treatment of epidermolytic palmoplantar keratoderma of Vörner. Br J Dermatol 1994;130:543–6.
Pachyonychia congenita
- McLean WH, Hansen CD, Eliason MJ, Smith FJ. The phenotypic and molecular genetic features of pachyonychia congenital. J Invest Dermatol 2011;131:1015–17.
- Smith FJ, Liao H, Cassidy AJ, et al. The genetic basis of pachyonychia congenita. J Investig Dermatol Symp Proc 2005;10:21–30.
- Munro CS. Pachyonychia congenita: mutations and clinical presentations. Br J Dermatol 2001;144:929–31.
- Jadassohn J, Lewandowski. Pachyonychia congenita: keratosis disseminata circumsripts (folliculitis). Tylomata. Leukokeratosis lingua. In: Neisser A, Jacobi E, eds. Ikonographia Dermatologica. Berlin: Urban and Schwarzenberg; 1906:29–31.
- Kumer L, Loos HO. [On pachyonychia congenital (type Riehl).] Wien Klin Wochenschr 1935;48:174–8.
- Buckley WR, Cassuto J. Pachyonychia congenita. Arch Dermatol 1962;85:397–402.
- Moldenhauer E, Ernst K. [The Jadassohn–Lewandowsky syndrome.] Hautarzt 1968;19:441–7.
- Kansky A, Basta-Juzbasic A, Videnic N, et al. Pachyonychia congenita (Jadassohn–Lewandowsky syndrome): evaluation of symptoms in 36 patients. Arch Dermatol Res 1993;285:36–7.
- Feinstein A, Friedman J, Schewach-Millet M. Pachyonychia congenita. J Am Acad Dermatol 1988;19:705–11.
- Paller AS, Moore JA, Scher R. Pachyonychia congenita tarda: a late-onset form of pachyonychia congenita. Arch Dermatol 1991;127:701–3.
- Smith FJ, Jonkman MF, van Goor H, et al. A mutation in human keratin K6b produces a phenocopy of the K17 disorder pachyonychia congenita type 2. Hum Mol Genet 1998:1143–8.
- Wilson NJ, Leachman SA, Hansen CD, et al. A large mutational study in pachyonychia congenita. J Invest Dermatol 2011;131:1018–24.
- Eliason MJ, Leachman SA, Feng BJ, et al. A review of the clinical phenotype of 254 patients with genetically confirmed pachyonychia congenita. J Am Acad Dermatol 2012;67:680–6.
- McLean WH, Smith FJ, Cassidy AJ. Insights into genotype–phenotype correlation in pachyonychia congenita from the human intermediate filament mutation database. J Investig Dermatol Symp Proc 2005;10:31–6.
- Munro CS, Carter S, Bryce S, et al. A gene for pachyonychia congenita is closely linked to the keratin gene cluster on 17q12-q21. J Med Genet 1994;31:675–8
- McLean WHI, Rugg EL, Lunny DP, et al. Keratin 16 and keratin 17 mutations cause pachyonychia congenita. Nat Genet 1995;9:273–8.
- Bowden PE, Haley JL, Kansky A, et al. Mutation of a type II keratin gene (KBa) in pachyonychia congenita. Nat Genet 1995;10:363–5
- De Berker D, Wojnarowska F, Sviland L, et al. Keratin expression in the normal nail unit: markers of regional differentiation. Br J Dermatol 2000;142:89–96.
- Smith FJD, Corden LD, Rugg EL, et al. Missense mutations in keratin 17 cause either pachyonychia congenita type 2 or a phenotype resembling steatocystoma multiplex. J Invest Dermatol 1997;108:220–3.
- McGowan KM, Coulombe PA. Keratin 17 expression in the hard epithelial context of the hair and nail, and its relevance for the pachyonychia congenita phenotype. J Invest Dermatol 2000;114:1101–7.
- Goldberg I, Sprecher E, Schwartz ME, Gaitini D. Comparative study of high-resolution multifrequency ultrasound of the plantar skin in patients with various types of hereditary palmoplantar keratoderma. Dermatology 2013;226:365–70.
- Leachman SA, Kaspar RL, Fleckman P, et al. Clinical and pathological features of pachyonychia congenita. J Investig Dermatol Symp Proc 2005;10:3–17.
- Murray FA. Congenital anomalies of the nails: four cases of hereditary hypertrophy of the nail-bed associated with a history of erupted teeth at birth. Br J Dermatol Syph 1991;23:409–11.
- Zamiri M, McLean WHI, Hodgins MC, Munro CS. Pachyonychia congenital: abnormal dentition extending into adulthood. Br J Dermatol 2008;159:500–1.
- Chang A, Lucker GPH, van de Kerkhof PCM, et al. Pachyonychia congenita in the absence of other syndrome abnormalities. J Am Acad Dermatol 1994;30:1017–18.
- Pryce DW, Verbov JL. A family with pachyonychia congenita affecting the nails only. Clin Exp Dermatol 1994;19:521–2.
- Mawhinney H, Creswell S, Beare JM. Pachyonychia congenita with candidiasis. Clin Exp Dermatol 1981;6:145–9.
- Stieglitz JB, Centerwall JW. Pachyonychia congenita (Jadassohn–Lewandowsky syndrome): a seventeen-member, four-generation pedigree with unusual respiratory and dental involvment. Am J Med Genet 1983;14:21–8.
- Todd P, Garioch J, Rademaker M, et al. Pachyonychia congenita complicated by hidradenitis suppurativa: a family study. Br J Dermatol 1990;123:663–6.
- Shamsher MK, Navsaria HA, Stevens HP, et al. Novel mutations in keratin 16 gene underly focal non-epidermolytic palmoplantar keratoderma (NEPPK) in two families. Hum Mol Genet 1995;4:1875–81.
- Covello SP, Smith FJ, Sillevis Smitt JH, et al. Keratin 17 mutations cause either steatocystoma multiplex or pachyonychia congenita type 2. Br J Dermatol 1998;139:475–80.
- Wilson NJ, Messenger AG, Leachman SA, et al. Keratin K6c mutations cause focal palmoplantar keratoderma. J Invest Dermatol 2010;130:425–9.
- Thomas DR, Jorizzo JL, Brysk MM, et al. Pachyonychia congenita: electron microscopic and epidermal glycoprotein assessment before and during isotretinoin treatment. Arch Dermatol 1984;120:1475–9.
- Bivolarevic I, Fartasch M, Diepgen TL. Pachyonychia congenita Syndrome. Z Hautkr 1991;66:588–96.
- Wilson NJ, Hansen CD, Azkur D, et al. Recessive mutations in the gene encoding frizzled 6 cause twenty nail dystrophy – expanding the differential diagnosis for pachyonychia congenita. J Dermatol Sci 2013;70:58–60.
- van Steensel MAM, Jonkman MF, van Geel M, et al. Clouston syndrome can mimic pachyonychia congenita. J Invest Dermatol 2003;121:1035–8.
- Goldberg I, Fruchter D, Meilick A, et al. Best treatment practices for pachyonychia congenita. J Eur Acad Dermatol Venereol 2013;28:279–85.
- Milstone LM, Fleckman P, Leachman SA, et al. Treatment of pachyonychia congenita. J Investig Dermatol Symp Proc 2005;10:18–20.
- Gruber R, Edlinger M, Kaspar RL, et al. An appraisal of oral retinoids in the treatment of pachyonychia congenita. J Am Acad Dermatol 2012;66:193–9.
- Tidman MJ, Wells RS. Control of plantar blisters in pachyonychia congenita with topical aluminium chloride. Br J Dermatol 1988;118:451–2.
- Smith FJ, Hickerson RP, Sayers JM, et al. Development of therapeutic siRNAs for pachyonychia congenita. J Invest Dermatol 2008;128:50–8.
- Leachman SA, Hickerson RP, Schwartz ME, et al. First-in-human mutation-targeted siRNA phase Ib trial of an inherited skin disorder. Mol Ther 2010;18:442–6.
- McLean WH, Moore CB. Keratin disorders: from gene to therapy. Hum Mol Genet 2011;20:189–97.
Painful hereditary callosities
- Roth W, Penneys NS, Fawcett N. Hereditary painful callosities. Arch Dermatol 1978;114:591–2.
- Wachters DHJ, Frensdorf EL, Hausman R, et al. Keratosis palmoplantaris nummularis (‘hereditary painful callosities’). J Am Acad Dermatol 1983;9:204–9.
- Baden HP, Bronstein BR, Rand RE. Hereditary callosities with blisters: report of a family and review. J Am Acad Dermatol 1984;11:409–15.
- Cambiaghi S, Morel P. Hereditary painful callosities with associated features. Dermatology 1996;193:47–9.
- Wilson NJ, Messenger AG, Leachman SA, et al. Keratin K6c mutations cause focal palmoplantar keratoderma. J Invest Dermatol 2010;130:425–9.
- Zamiri M, Smith FJ, Campbell LE, et al. Mutation in DSG1 causing autosomal dominant striate palmoplantar keratoderma. Br J Dermatol 2009;161:692–4.
- Bohnert A, Anton-Lamprecht I. Richner–Hanhart's syndrome: ultrastructural abnormalities of epidermal keratinization indicating a causal relationship to high intracellular tyrosine levels. J Invest Dermatol 1982;79:68–74.
Non-epidermolytic palmoplantar keratoderma
- Thost A. Über Erbliche Ichthyosis Palmaris et Plantaris Cornea. Heidelberg: Inaug diss, 1880.
- Unna PG. Über das Keratoma palmare et plantare hereditarum: eine Studie zur Kerato-Nosologie. Arch Derm Syph (Berlin) 1883;15:231–70.
- Kuster W, Becker A. Indication for the identity of palmoplantar keratoderma Unna Thost with type Vörner. Acta Derm Venereol (Stockh) 1992;72:120–2.
- Zhao L, Vahlquist A, Virtanen M, et al. Palmoplantar keratoderma of the Gamborg-Nielsen type is caused by mutations in the SLURP1 gene and represents a variant of Mal de Meleda. Acta Derm Venereol 2014;94:707–10.
- Hellström Pigg M, Gamborg Nielsen P, Hofer PA, Lagerholm B. The dominant form of hereditary palmoplantar keratoderma in the northernmost county of Sweden (Norrbotten). Dermatology 1994;188:188–93.
- Gamborg-Nielsen P. Two different clinical and genetic forms of hereditary palmoplantar keratoderma in the northernmost county of Sweden. Clin Genet 1985;28:361–6.
- Kimonis V, DiGiovanna JJ, Yang JM, et al. A mutation in the V1 end domain of keratin 1 in non-epidermolytic palmar-plantar keratoderma. J Invest Dermatol 1994;103:764–9.
- Mitsuhashi Y, Hashimato I. Keratosis palmoplantaris Nagashima. Dermatologica 1989;179:231.
- Kabashima K, Sakabe J, Yamada Y, Tokura Y. “Nagashima-type” keratosis as a novel entity in the palmoplantar keratoderma category. Arch Dermatol 2008;144:375–9.
- Greither A. Keratosis extremitatum hereditaria progrediens mit dominatem Erbgang. Hautarzt 1952;3:198–203.
- Kansky A, Arzensek J. Is palmoplantar keratoderma of Greither's type a separate nosologic entity? Dermatologica 1979;158:244–8.
- Fluckiger R, Itin PH. Keratosis extremitatum (Greither's disease): clinical features, histology, ultrastructure. Dermatology 1993;187:309–11.
- Richard G, Lin JP, Smith L, et al. Linkage studies in erythrokeratodermias: fine mapping, genetic heterogeneity and analysis of candidate genes. J Invest Dermatol 1997;109:666–71.
- Gach JE, Munro CS, Lane EB, et al. Two families with Greither's syndrome caused by a keratin 1 mutation. J Am Acad Dermatol 2005;53:S225–30.
- Wollina U, Knopf B, Schaaschmidt H, et al. Familiare Koexistenz von Erythrokeratodermia variabilis und Keratosis palmoplantaris transgrediens et progrediens. Hautarzt 1989;40:169–72.
- Lind L, Lundstrom A, Hofer PA, Holmgren G. The gene for diffuse palmoplantar keratoderma of the type found in northern Sweden is localized to chromosome 12q11–q13. Hum Mol Genet 1994;3:1789–93.
- Kelsell DP, Stevens HP, Ratnavel R, et al. Genetic linkage studies in non-epidermolytic palmoplantar keratoderma: evidence for heterogeneity. Hum Mol Genet 1995;4:1021–5.
- Kelsell DP, Stevens HP, Purkis PE, et al. Fine genetic mapping of diffuse non-epidermolytic palmoplantar keratoderma to chromosome 12q11–q13: exclusion of the mapped type II keratins. Exp Dermatol 1999;8:388–91.
- Blaydon DC, Lind LK, Plagnol V, et al. Mutations in AQP5, encoding a water-channel protein, cause autosomal-dominant diffuse nonepidermolytic palmoplantar keratoderma. Am J Hum Genet 2013;93(2):330–5.
- Kubo A, Shiohama A, Sasaki T, et al. Mutations in SERPINB7, encoding a member of the serine protease inhibitor superfamily, cause Nagashima-type palmoplantar keratosis. Am J Hum Genet 2013;93:945–56.
- Sybert VP, Dale BA, Holbrook KA. Palmar–plantar keratoderma: a clinical, ultrastructural, and biochemical study. J Am Acad Dermatol 1988;18:75–86.
- Wennerstrand LM, Lind LK, Hofer PA, Lundstrom A. Homozygous palmoplantar keratoderma type bothnia improved by erythromycin: a case report. Acta Derm Venereol 2004;84:405–6.
- Nonomura Y, Otsuka A, Miyachi Y, Kabashima K. Suspected Nagashima-type palmoplantar keratosis with atypical hyperkeratotic lesions on the ears. Eur J Dermatol 2012;22:392–3.
Mal de Meleda
- Stulli L. De una varieta cuanea. Antologia di Firenzi 1826: fasc. 71–2.
- Salamon T, Berberovic L, Topic B, et al. Mal de Meleda: data and remarks on a series. G Ital Dermatol Venereol 1988;123:649–55.
- Fischer J, Bouadjar B, Heilig R, et al. Mutations in the gene encoding SLURP-1 in mal de Meleda. Hum Mol Genet 2001;10:875–80.
- Eckl KM, Stevens HP, Lestringant GG, et al. Mal de Meleda (MDM) caused by mutations in the gene for SLURP-1 in patients from Germany, Turkey, Palestine, and the United Arab Emirates. Hum Genet 2003;112:50–6.
- Ward KM, Yerebakan Ö, Yilmaz E, Çelebi JT. Identification of recurrent mutations in the ARS (component B) gene encoding SLURP-1 in two families with mal de Meleda. J Invest Dermatol 2003;120:96–8.
- Marrakchi S, Audebert S, Bouadjar B, et al. Novel mutations in the gene encoding secreted lymphocyte antigen-6/urokinase-type plasminogen activator receptor-related protein-1 (SLURP-1) and description of five ancestral haplotypes in patients with mal de Meleda. J Invest Dermatol 2003;120:351–5.
- Bakija-Konsuo A, Basta-Juzbasic A, Rudan I, et al. Mal de Meleda: genetic haplotype analysis and clinicopathological findings in cases originating from the island of Mljet (Meleda), Croatia. Dermatology 2002;205:32–9.
- Bchetnia M, Laroussi N, Youssef M, et al. Particular Mal de Meleda phenotypes in Tunisia and mutations founder effect in the Mediterranean region. Biomed Res Int 2013:20 6803.
- Nellen RG, Steijlen PM, Hennies HC, et al. Haplotype analysis in western European patients with mal de Meleda: founder effect for the W15R mutation in the SLURP1 gene. Br J Dermatol 2013;168:1372–4.
- Arredondo J, Chernyavsky AI, Webber RJ, Grando SA. Biological effects of SLURP-1 on human keratinocytes. J Invest Dermatol 2005;125:1236–41.
- Favre B, Plantard L, Aeschbach L, et al. SLURP1 is a late marker of epidermal differentiation and is absent in Mal de Meleda. J Invest Dermatol 2007;127:301–8.
- Frenk E, Guggisberg D, Mevorah B, Hohl D. Meleda disease: report of two cases investigated by electron microscopy. Dermatology 1996;193:358–61.
- Niles HD, Klump M. Mal de Meleda: review of the literature and report of four cases. Arch Derm Syph 1939;39:409–21.
- Bouadjar B, Benmazouzia S, Prud'homme JF, et al. Clinical and genetic studies of three large, consanguineous, Algerian families with mal de Meleda. Arch Dermatol 2000;136:1247–52.
- Lestringant GG, Hadi SM, Qayed KI, et al. Mal de Meleda: recessive transgressive palmoplantar keratoderma with three unusual facultative features. Dermatology 1992;184:78–82.
- Ergin C, Ergin S, Arikan S. Prevalence of dermatomycoses in Mal de Meleda patients: a field study. Scand J Infect Dis 2002;34:753–5.
- Nath AK, Chaudhuri S, Thappa DM. Mal de meleda with lip involvement: a report of two cases. Indian J Dermatol 2012;57:390–3.
- Baroni A, Piccolo V, Di Maio R, et al. Mal deMeleda with hyperpigmented spots. Eur J Dermatol 2011;21:459–60.
- Mozzillo N, Nunziata CA, Caraco C, et al. Malignant melanoma developing in an area of hereditary palmoplantar keratoderma (Mal de Meleda). J Surg Oncol 2003;84:229–33.
- Sartore L, Bordignon M, Bassetto F, et al. Melanoma in skin affected with keratoderma palmoplantaris hereditaria (Mal de Meleda): Treatment with excision and grafting. J Am Acad Dermatol 2009;61:161–3.
- Tourlaki A, Bentivogli M, Boneschi V, Brambilla L. Genetically proven Mal de Meleda complicated by Bowen's disease of the sole. Eur J Dermatol 2011;21:292–4.
- Mokni M, Charfeddine C, Ben Mously R, et al. Heterozygous manifestations in female carriers of Mal de Meleda. Clin Genet 2004;65:244–6.
- Chao SC, Huang CY, Lai FJ, Yang MH. Pseudodominant inheritance with the G86R mutation in the ARS gene in Mal de Meleda. Int J Dermatol 2006;45:1456–8.
- Sybert VP, Dale BA, Holbrook KA. Palmar–plantar keratoderma: a clinical, ultrastructural, and biochemical study. J Am Acad Dermatol 1988;18:75–86.
- Brambilla L, Pigatto PD, Boneschi V. Unusual cases of Meleda keratoderma treated with aromatic retinoid, etretinate. Dermatologica 1984;168:283–6.
- Reed ML, Stanley J, Stengel F, et al. Mal de Meleda treated with isotretinoin. Arch Dermatol 1979;115:605–8.
- Jee S-H, Lee Y-Y, Wu Y-C, et al. Report of a family with mal de Meleda in Taiwan: a clinical, histopathological and immunological study. Dermatologica 1985;171:30–7.
- van de Kerkhof PC, van Dooren-Greebe RJ, Steijlen PM. Acitretin in the treatment of mal de Meleda. Br J Dermatol 1992;127:191–2.
- Bergman R, Bitterman-Deutsch O, Fartsach M, et al. Mal de Meleda keratoderma with pseudoainhum. Br J Dermatol 1993;128:207–12.
- Marchac A, Blanchet-Bardon C, Revol M, Servant JM. [Surgical treatment of keratosis palmaris in Mal de Meleda.] Ann Chir Plast Esthet 2009;54:152–5.
Loricrin keratoderma
- Camisa C, Rossana C. Variant of keratoderma hereditaria mutilans (Vohwinkel's syndrome): treatment with orally administered isotretinoin. Arch Dermatol 1984;120:1323–8.
- Camisa C, Hessel A, Rossana C, Parks A. Autosomal dominant keratoderma, ichthyosiform dermatosis and elevated serum β-glucuronidase. Dermatologica 1988;177:341–7.
- Maestrini E, Monaco AP, McGrath JA, et al. A molecular defect in loricrin, the major component of the cornified cell envelope underlies Vohwinkel's syndrome. Nat Genet 1996;13:70–7.
- Korge BP, Ishida-Yamamoto A, Punter C, et al. Loricrin mutation in Vohwinkel's keratoderma is unique to the variant with ichthyosis. J Invest Dermatol 1997;109:604–10.
- Armstrong DK, McKenna KE, Hughes AE. A novel insertional mutation in loricrin in Vohwinkel's keratoderma. J Invest Dermatol 1998;111:702–4.
- Matsumoto K, Muto M, Seki S, et al. Loricrin keratoderma: a cause of congenital ichthyosiform erythroderma and collodion baby. Br J Dermatol 2001;145:657–60.
- Gedicke MM, Traupe H, Fischer B, et al. Towards characterization of palmoplantar keratoderma caused by gain-of-function mutation in loricrin: analysis of a family and review of the literature. Br J Dermatol 2006;154:167–71.
- Song S, Shen C, Song G, et al. A novel c.545-546insG mutation in the loricrin gene correlates with a heterogeneous phenotype of loricrin keratoderma. Br J Dermatol 2008;159:714–19.
- Drera B, Tadini G, Balbo F, et al. De novo occurrence of the 730insG recurrent mutation in an Italian family with the ichthyotic variant of Vohwinkel syndrome, loricrin keratoderma. Clin Genet 2008;73:85–8.
- Yeh JM, Yang MH, Chao SC. Collodion baby and loricrin keratoderma: a case report and mutation analysis. Clin Exp Dermatol 2013;38(2):147–50.
- Corte LD, Silva MV, Oliveira CF, et al. Vohwinkel syndrome, ichthyosiform variant – by Camisa – Case report. An Bras Dermatol 2013;88:206–8.
- Ishida-Yamamoto A, Kato H, Kiyama H, et al. Mutant loricrin is not crosslinked into the cornified cell envelope but is translocated into the nucleus in loricrin keratoderma. J Invest Dermatol 2000;115:1088–94.
- Schmuth M, Fluhr JW, Crumrine DC, et al. Structural and functional consequences of loricrin mutations in human loricrin keratoderma (Vohwinkel syndrome with ichthyosis). J Invest Dermatol 2004;122:909–22.
- Jarnik M, De Viragh PA, Schärer E, et al. Quasi-normal cornified cell envelopes in loricrin knockout mice imply the existence of a loricrin backup system. J Invest Dermatol 2002;118:102–9.
- Koch PJ, De Viragh PA, Schärer E, et al. Lessons from loricrin-deficient mice: compensatory mechanisms maintaining skin barrier function in the absence of a major cornified envelope protein. J Cell Biol 2000;151:389–400.
- Suga Y, Jarnik M, Attar PS, et al. Transgenic mice expressing a mutant form of loricrin reveal the molecular basis of the skin diseases, Vohwinkel syndrome and progressive symmetric erythrokeratoderma. J Cell Biol 2000;151:401–12.
- Ishida-Yamamoto A, McGrath JA, Lam H, et al. The molecular pathology of progressive symmetric erythrokeratoderma: a frameshift mutation in the loricrin gene and perturbations in the cornified cell envelope. Am J Hum Genet 1997;61:581–9.
- Wei S, Zhou Y, Zhang TD, et al. Evidence for the absence of mutations at GJB3, GJB4 and LOR in progressive symmetrical erythrokeratodermia. Clin Exp Dermatol 2011;36:399–405.
- Yoneda K, Demitsu T, Nakai K, et al. Activation of vascular endothelial growth factor receptor 2 in a cellular model of loricrin keratoderma. J Biol Chem 2010;285:16184–94.
Striate (and focal) palmoplantar keratoderma
- Wachters DHJ. Over de Verschillende Morphologische Vormen van de Keratosis Palmoplantaris [Thesis]. Leyden: 1963.
- Zamiri M, Smith FJ, Campbell LE, et al. Mutation in DSG1 causing autosomal dominant striate palmoplantar keratoderma. Br J Dermatol 2009;161:692–4.
- Barber AG, Wajid M, Columbo M, et al. Striate palmoplantar keratoderma resulting from a frameshift mutation in the desmoglein 1 gene. J Dermatol Sci 2007;45:161–6.
- Itin PH, Fistarol SK. Palmoplantar keratodermas. Clin Dermatol 2005;23:15–22.
- Lai-Cheong JE, Arita K, McGrath JA. Genetic diseases of junctions. J Invest Dermatol 2007;127:2713–25.
- Hennies HC, Kuster W, Mischke D, Reis A. Localization of a locus for the striated form of palmoplantar keratoderma to chromosome 18q near the desmosomal cadherin gene cluster. Hum Mol Genet 1995;4:1015–20.
- Rickman L, Simrak D, Stevens HP, et al. N-terminal deletion in a desmosomal cadherin causes the autosomal dominant skin disease striate palmoplantar keratoderma. Hum Mol Genet 1999;8:971–6.
- Hunt DM, Rickman L, Whittock NV, et al. Spectrum of dominant mutations in the desmosomal cadherin desmoglein 1, causing the skin disease striate palmoplantar keratoderma. Eur J Hum Genet 2001;9:197–203.
- Hershkovitz D, Lugassy J, Indelman M, Bergman R, Sprecher E. Novel mutations in DSG1 causing striate palmoplantar keratoderma. Clin Exp Dermatol. 2009;34:224–8.
- Dua-Awereh MB, Shimomura Y, Kraemer L, et al. Mutations in the desmoglein 1 gene in five Pakistani families with striate palmoplantar keratoderma. J Dermatol Sci 2009;53:192–7.
- Hershkovitz D, Lugassy J, Indelman M, et al. Novel mutations in DSG1 causing striate palmoplantar keratoderma. Clin Exp Dermatol 2009;34:224–8.
- Milingou M, Wood P, Masouyé I, et al. Focal palmoplantar keratoderma caused by an autosomal dominant inherited mutation in the desmoglein 1 gene. Dermatology 2006;212:117–22.
- Keren H, Bergman R, Mizrachi M, et al. Diffuse nonepidermolytic palmoplantar keratoderma caused by a recurrent nonsense mutation in DSG1. Arch Dermatol 2005;141:62–8.
- Has C, Jakob T, He Y, et al. Loss of desmoglein 1 associated with palmoplantar keratoderma, dermatitis and multiple allergies. Br J Dermatol 2014;172:257–61.
- Armstrong DK, McKenna KE, Purkis PE, et al. Haploinsufficiency of desmoplakin causes a striate subtype of palmoplantar keratoderma. Hum Mol Genet 1999;8:143–8.
- Whittock NV, Ashton GH, Dopping-Hepenstal PJ, et al. Striate palmoplantar keratoderma resulting from desmoplakin haploinsufficiency. J Invest Dermatol 1999;113:940–6.
- Norgett EE, Hatsell SJ, Carvajal-Huerta L, et al. Recessive mutation in desmoplakin disrupts desmoplakin–intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum Mol Genet 2000;9:2761–6.
- Norgett EE, Lucke TW, Bowers B, et al. Early death from cardiomyopathy in a family with autosomal dominant striate palmoplantarkeratoderma and woolly hair associated with a novel insertion mutation in desmoplakin. J Invest Dermatol 2006;126:1651–4.
- Whittock NV, Smith FJ, Wan H, et al. Frameshift mutation in the V2 domain of human keratin 1 results in striate palmoplantar keratoderma. J Invest Dermatol 2002;118:838–44.
- Richardson ES, Lee JB, Hyde PH, Richard G. A novel mutation and large size polymorphism affecting the V2 domain of keratin 1 in an African-American family with severe, diffuse palmoplantar keratoderma of the ichthyosis hystrix Curth–Macklin type. J Invest Dermatol 2006;126:79–84.
- Brünauer SR. Zur Vererbung des Keratoma hereditarium palmare et plantare. Acta Dermato Venereol (Stockh) 1923;4:489–503.
- Fuhs H. Zur Kenntnis der herdweisen Keratosen an Händen und Füssen. Acta Dermato Venereol (Stockh) 1924;5:11–58.
- Siemens HW. Keratosis palmaris–plantaris striata. Arch Dermatol Syph 1929;157:392–408.
- Leonard NJ, Krol AL, Bleoo S, Somerville MJ. Sensorineural hearing loss, striate palmoplantar hyperkeratosis, and knuckle pads in a patient with a novel connexin 26 (GJB2) mutation. J Med Genet 2005;42:e2.
- Nakai K, Yoneda K, Moriue T, Kubota Y. Striate palmoplantar keratoderma in a patient with Rubinstein–Taybi syndrome. J Eur Acad Dermatol Venereol 2009;23:333–5.
- Van Steensel MA, Van Geel M, Steijlen PM. New syndrome of hypotrichosis, striate palmoplantar keratoderma, acro-osteolysis and periodontitis not due to mutations in cathepsin C. Br J Dermatol 2002;147:575–81.
- Verdolini R, Clayton N, Arkoumani E. Striate palmar keratoderma and antiretroviral treatment for human immunodeficiency virus infection: not just a coincidence. Clin Exp Dermatol 2013;38:556–8.
- Bergman R, Hershkovitz D, Fuchs D, et al. Disadhesion of epidermal keratinocytes: a histologic clue to palmoplantar keratodermas caused by DSG1 mutations. J Am Acad Dermatol 2010;62:107–13.
- Wan H, Dopping-Hepenstal PJ, Gratian MJ, et al. Striate palmoplantar keratoderma arising from desmoplakin and desmoglein 1 mutations is associated with contrasting perturbations of desmosomes and the keratin filament network. Br J Dermatol 2004;150:878–91.
- Casado M, Jimenez-Acosta F, Borbujo J, Martinez W, Almagro M, Soto-Melo J. Keratoderma palmoplantaris striata. Clin Exp Dermatol 1989;14:240–2.
- Fartasch M, Vigneswaran N, Diepgen TL, Hornstein OP. Abnormalities of keratinocyte maturation and differentiation in keratosis palmoplantaris striata. Immunohistochemical and ultrastructural study before and during etretinate therapy. Am J Dermatopathol 1990;12:275–82.
- Bragg J, Rizzo C, Mengden S. Striate palmoplantar keratoderma (Brunauer–Fuhs–Siemens syndrome). Dermatol Online J 2008;14:26.
Punctate palmoplantar keratoderma
- Emmert S, Kuster W, Hennies HC, et al. 47 patients in 14 families with the rare genodermatosis keratosis punctata palmoplantaris Buschke–Fischer–Brauer. Eur J Dermatol 2003;13:16–20.
- Heierli-Forrer E. Zur Klinik un Genetik der hereditaren papulosen Palmoplantarkeratosen. Dermatologica 1959;119:309–27.
- Hesse S, Berbis P, Privat Y. Keratodermia palmo-plantaris papulosa (Buschke–Fischer's disease): efficacy of acitretin. Br J Dermatol 1993;128:104–5.
- Stanimirovic A, Kansky A, Basta-Juzbasic A, et al. Hereditary palmoplantar keratoderma, type papulosa, in Croatia. J Am Acad Dermatol 1993;29:435–7.
- Martinez-Mir A, Zlotogorski A, Londono D, et al. Identification of a locus for type I punctate palmoplantar keratoderma on chromosome 15q22–q24. J Med Genet 2003;40:872–8.
- Gao M, Yang S, Li M, et al. Refined localization of a punctate palmoplantar keratoderma gene to a 5.06-cM region at 15q22.2–15q22.31. Br J Dermatol 2005;152:874–8.
- Mamaï O, Boussofara L, Adala L, et al. Reduction of palmoplantar keratoderma Buschke–Fischer–Brauer locus to only 0.967 Mb. J Dermatol Sci. 2012 Sep;67(3):210–12.
- Zhang XJ, Li M, Gao TW, et al. Identification of a locus for punctate palmoplantar keratodermas at chromosome 8q24.13–8q24.21. J Invest Dermatol 2004;122:1121–5.
- Pohler E, Mamai O, Hirst J, et al. Haploinsufficiency for AAGAB causes clinically heterogeneous forms of punctate palmoplantar keratoderma. Nat Genet 2012;44(11):1272–6.
- Giehl KA, Eckstein GN, Pasternack SM, et al. Nonsense mutations in AAGAB cause punctate palmoplantar keratoderma type Buschke–Fischer–Brauer. Am J Hum Genet 2012;91:754–9.
- Furniss M, Higgins CA, Martinez-Mir A, et al. Identification of distinct mutations in AAGAB in families with type 1 punctate palmoplantar keratoderma. J Invest Dermatol 2014;134:1749–52
- Eytan O, Sarig O, Israeli S, Mevorah B, Basel-Vanagaite L, Sprecher E. A novel splice-site mutation in the AAGAB gene segregates with hereditary punctate palmoplantar keratoderma and congenital dysplasia of the hip in a large family. Clin Exp Dermatol 2013;39:182–6.
- Pöhler E, Zamiri M, Harkins CP, et al. Heterozygous mutations in AAGAB cause type 1 punctate palmoplantar keratoderma with evidence for increased growth factor signaling. J Invest Dermatol 2013 Dec;133(12):2805–8.
- Li M, Yang L, Shi H, Guo B, Dai X, Yao Z, Zhang G. Loss-of-function mutation in AAGAB in Chinese families with punctate palmoplantar keratoderma. Br J Dermatol 2013 Jul;169(1):168–71.
- Cui H, Gao M, Wang W, et al. Six mutations in AAGAB confirm its pathogenic role in Chinese punctate palmoplantar keratoderma patients. J Invest Dermatol 2013 Nov;133(11):2631–4.
- Kiritsi D, Chmel N, Arnold AW, Jakob T, Bruckner-Tuderman L, Has C. Novel and recurrent AAGAB mutations: clinical variability and molecular consequences. J Invest Dermatol 2013;133:2483–6
- Guo BR, Zhang X, Chen G, et al. Exome sequencing identifies a COL14A1 mutation in a large Chinese pedigree with punctate palmoplantar keratoderma. J Med Genet 2012;49:563–8.
- Antonio JR, Oliveira GB, Rossi NC, Pires LG. Exuberant clinical picture of Buschke–Fischer–Brauer palmoplantar keratoderma in bedridden patient. An Bras Dermatol 2014;89:819–21.
- Bennion SD, Patterson JW. Keratosis punctata palmaris et plantaris and adenocarcinoma of the colon. J Am Acad Dermatol 1984;10:587–91.
- Stevens HP, Kelsall DP, Leigh IM, et al. Punctate palmoplantar keratoderma and malignancy in a four generation family. Br J Dermatol 1996;134:720–6.
- Patrizi A, Passarini B, Minghetti G, Masina M. Porokeratosis palmaris et plantaris disseminata: an unusual clinical presentation. J Am Acad Dermatol 1989;21:415–18.
- Stevens HP, Kelsell DP, Bryant SP, et al. Linkage of an American pedigree with palmoplantar keratoderma and malignancy (palmoplantar ectodermal dysplasia type III) to 17q24. Literature survey and proposed updated classification of the keratodermas. Arch Dermatol 1996;132:640–51.
- Horikoshi M, Kuroda K, Tajima S. Punctate palmoplantar keratoderma with pigmentary lesions on the dorsa of feet and ankles: successful treatment with a combination of low-dose oral etretinate and topical calcipotriol. J Dermatol 2004;31:469–72.
- Kong MS, Harford R, O'Neill JT. Keratosis punctata palmoplantaris controlled with topical retinoids: a case report and review of the literature. Cutis 2004;74:173–9.
- Christiansen JV. Keratodermia punctata hereditaria treated with etretinate (Tigason). Acta Derm Venereol (Stockh) 1983;63:181–2.
- Postel-Vinay S, Ashworth A. AXL and acquired resistance to EGFR inhibitors. Nat Genet 2012;44:835–6.
Spiny keratoderma
- Brown F. Punctate keratoderma. Arch Dermatol 1971;104:682–3.
- Friedman SJ, Herman PS, Pittelkow MR, Su WP. Punctate porokeratotic keratoderma. Arch Dermatol 1988;124:1678–82.
- Grillo E, Pérez-García B, González-García C, Vano-Galván S, Jaén-Olasolo P. Spiky keratotic projections on the palms and fingers. Spiny keratoderma. Dermatol Online J 2012;18:8.
- Caccetta TP, Dessauvagie B, McCallum D, Kumarasinghe SP. Multiple minute digitate hyperkeratosis: a proposed algorithm for the digitate keratoses. J Am Acad Dermatol 2012;67:e49–55.
- Lucker GP, Van de Kerkhof PC, Steijlen PM. The hereditary palmoplantar keratoses: an updated review and classification. Br J Dermatol 1994;131:1–14.
- Herman PS. Punctate porokeratotic keratoderma. Dermatologica 1973;147:206–13.
- Lestringant GG, Berge T. Porokeratosis punctata palmaris et plantaris: a new entity? Arch Dermatol 1989;125:816–19.
- Mehta RK, Mallett RB, Green C, Rytina E. Palmar filiform hyperkeratosis (FH) associated with underlying pathology. Clin Exp Dermatol 2002;27:216–19.
- Chambers CJ, Konia T, Burrall B, Fung MA, Sharon V. Unilateral prickly palmar papules. Punctate porokeratotic keratoderma (PPK). Arch Dermatol 2011;147:609–14.
- Zarour H, Grob JJ, Andrac L, et al. Palmoplantar orthokeratotic filiform hyperkeratosis in a patient with associated Darier's disease: classification of filiform hyperkeratosis. Dermatology 1992;185:205–9.
- Salmon-Ehr V, Grosieux C, Derancourt C, et al. Palmoplantar filiform hyperkeratosis with Darier's disease: association or coincidence? Eur J Dermatol 1998;8:519–20.
- Caputo R, Cavicchini S, Brezzi A, Grimalt R. Spiny hyperkeratosis of the fingers as an unusual sign of epidermodysplasia verruciformis. J Am Acad Dermatol 1995;32:523–4.
- Humphreys F, Spencer J, Benton C. Paraneoplastic follicular hyperkeratosis responsive to etretinate. Br J Dermatol 1992;127:62–3.
- Osman Y, Daly TJ, Don PC. Spiny keratoderma of the palms and soles. J Am Acad Dermatol 1992;26:879–81.
- McCallister RE, Estes SA, Yarbrough CL. Porokeratosis plantaris, palmaris, et disseminata. Report of a case and treatment with isotretinoin. J Am Acad Dermatol 1985;13:598–603.
- Anderson D, Cohen DE, Lee HS, et al. Spiny keratoderma in association with autosomal dominant polycystic kidney disease with liver cysts. J Am Acad Dermatol 1996;34:935–6.
- Feldmann R, Harms M. Multiple filiforme Hyperkeratosen. Hautarzt 1993;44:658–61.
- Gimenez-Arnau A, Camarasa JG. Palmar filiform or spiny hyperkeratosis associated with pulmonary tuberculosis. J Eur Acad Derm Venereol 1994;3:400–6.
- Urbani CE, Moneghini L. Palmar spiny keratoderma associated with type IV hyperlipoproteinemia. J Eur Acad Dermatol Venereol 1998;10:262–6.
- Handa Y, Sakakibara A, Araki M, Yamanaka N. Spiny keratoderma of the palms and soles: report of two cases. Eur J Dermatol 2000;10:542–5.
- Kaddu S, Soyer P, Kerl H. Palmar filiform hyperkeratosis: a new paraneoplastic syndrome? J Am Acad Dermatol 1995;33:337–40.
- Bernal AI, Gonzalez A, Aragoneses H, et al. A patient with spiny keratoderma of the palms and a lymphoproliferative syndrome: an unrelated paraneoplastic condition? Dermatology 2000;201:379–80.
- Bordel-Gómez M. Palmoplantar spiny keratoderma associated with chronic lymphoid leukaemia. J Eur Acad Dermatol Venereol 2008;22:1507–8.
- De D, Dogra S, Narang T, Radotra BD, Kanwar AJ. Pityriasis rubra pilaris in a HIV-positive patient (Type 6 PRP). Skinmed 2008;7:47–50.
- Tosti A, Morelli R, Fanti PA, et al. Nail changes of punctate keratoderma: a clinical and pathological study of two patients. Acta Derm Venereol (Stockh) 1993;73:66–8.
Marginal papular keratoderma
- Rongioletti F, Betti R, Crosti C, Rebora A. Marginal papular acrokeratodermas: a unified nosography for focal acral hyperkeratosis, acrokeratoelastoidosis and related disorders. Dermatology 1994;188:28–31.
- Hafner O, Gerstel C, Schroder B. Focal acral hyperkeratosis. Hautarzt 1999;50:586–9.
- Stevens HP, Kelsel, D, Bryant P, et al. Linkage of an American pedigree with palmoplantar keratoderma and malignancy (palmoplantar ectodermal dysplasia type III) to 17q24: literature survey and proposed updated classification of the keratodermas. Arch Derm 1996;132:640–51.
- Greiner J, Kruger J, Palden L, et al. A linkage study of acrokeratoelastoidosis: possible mapping to chromosome 2. Hum Genet 1983;63:222–7.
- Costa OG. Acroceratoses [Thesis]. University of Minas Gerais, Brazil, 1964.
- Costa OG. Acrokeratoelastoidosis. Arch Dermatol 1954;70:228–31.
- Fiallo P, Pesce C, Brusasco A, Nunzi E. Acrokeratoelastoidosis of Costa: a primary disease of the elastic tissue? J Cutan Pathol 1998;25:580–2.
- Dowd PM, Harman RRM, Black MM. Focal acral hyperkeratosis. Br J Dermatol 1983;109:97–103.
- Smith EB, Jetton RL. Punctate pits and keratoses of the palmar creases. South Med J 1970;63:1291–3.
- Weiss RM, Rasmussen JE. Dermatosis punctata of the palmar creases. Arch Dermatol 1980;116:669–71.
- Del Rio E, Vazquez-Vega H, Aguilar A, et al. Keratosis punctata of the palmar creases: a report on three generations, demonstrating an association with ichthyosis vulgaris and evidence of involvement of the acrosyringium. Clin Exp Dermatol 1994;19:165–7.
- Penas PF, Rios-Buceta L, Sanchez-Perez J, et al. Keratosis punctata of the palmar creases: case report and prevalence study in Caucasians. Dermatology 1994;188:200–2.
- Khera P, Shiferman G, English JC 3rd. Concurrent punctate keratosis of the palmar creases and focal acral hyperkeratosis. Cutis 2008;81:348–50.
- Rosen T, Martin S. Palmar oddities. In: Rosen T, Martin S, eds. Atlas of Black Dermatology. Boston: Little, Brown, 1981:12–13.
- Rustad OJ, Vance JC. Punctate keratoses of the palms and soles and keratotic pits of the palmar creases. J Am Acad Dermatol 1990;22:468–76.
- Lucker GP, Steijlen PM. Keratosis palmoplantaris varians et punctata: clinical variability of an single genetic defect? Hautarzt 1996;47:858–9.
- Kalter DC, Stone MS, Kettler A, et al. Keratosis punctata of the palmar creases: extremely uncommon? J Am Acad Dermatol 1986;14:510–11.
- Penas PF, Rios-Buceta L, Sanchez-Perez J, et al. Keratosis punctata of the palmar creases: case report and prevalence study in Caucasians. Dermatology 1994;188:200–2.
- Kocsard E. Keratoelastoidosis marginalis of the hands. Synonyms: marginal keratoderma of palms; degenerative collagenous plaques of the hands. Dermatologica. 1965;131(3):169–75.
- Mengesha YM, Kayal JD, Swerlick RA. Keratoelastoidosis marginalis. J Cutan Med Surg 2002;6:23–5.
- Burks JW, Wise LJ, Clark WH. Degenerative collagenous plaques of the hands. Arch Dermatol 1960;82:362–6.
- Ritchie EB, Williams HM. Degenerative collagenous plaques of the hands. Arch Dermatol 1966;93:202–3.
- Abulafia J, Vignale RA. Degenerative collagenous plaques of the hands and acrokeratoelastoidosis: pathogenesis and relationship with knuckle pads. Int J Dermatol 2000;39:424–32.
- Jacyk WK, Smith A. Mosaic acral keratosis. Clin Exp Dermatol 1990;15:361–2.
- Jacyk WK. Marginal papular acrokeratodermas: classification. Dermatology 1995;190:178.
Cole disease
- Cole LA. Hypopigmentation with punctate keratosis of the palms and soles. Arch Derm 1976;112:998–1000.
- Vignale R, Yusín A, Panuncio A, et al. Cole disease: hypopigmentation with punctate keratosis of the palms and soles. Pediatr Dermatol 2002;19:302–6.
- Moore MM, Orlow SJ, Kamino H, et al. Cole disease: guttate hypopigmentation and punctate palmoplantar keratoderma. Arch Derm 2009;145:495–7.
- Eytan O, Morice-Picard F, Sarig O, et al. Cole disease results from mutations in ENPP1. Am J Hum Genet 2013;93:752–7.
- Schmieder A, Hausser I, Schneider SW, et al. Palmoplantar hyperkeratoses and hypopigmentation. Cole disease. Acta Derm Venereol 2011;91:737–8.
Transient aquagenic keratoderma
- English JC, McCollough ML. Transient reactive papulotranslucent acrokeratoderma. J Am Acad Dermatol 1996;34:686–7.
- Luo DQ. Aquagenic acrokeratoderma: a case with family history and unusual involvements of the palms and soles, and the dorsum of fingers and toes. J Dermatol 2011;38:612–15.
- Carder KR, Weston WL. Rofecoxib-induced instant aquagenic wrinkling of the palms. Pediatr Dermatol 2002;19:353–5.
- Pastor MA, González L, Kilmurray L, et al. [Aquagenic keratoderma: 3 new cases and a review of the literature.] Acta Dermosifiliogr 2008;99:399–406.
- Onwukwe MF, Mihm MC, Toda K. Hereditary papulotranslucent acrokeratoderma: a new variant of familial punctate keratoderma? Arch Derm 1973;108:108–10.
- Sracic JK, Krishnan RS, Nunez-Gussman JK, et al. Hereditary papulotranslucent acrokeratoderma: a case report and literature review. Dermatol Online J 2005;11:17.
- Yan AC, Aasi SZ, Alms WJ, et al. Aquagenic palmoplantar keratoderma. J Am Acad Dermatol 2001;44:696–9.
- Ertürk-Özdemir E, Ozcan D, Seçkin D. Acquired aquagenic syringeal acrokeratoderma: A case series of 10 patients. Australas J Dermatol 2013 [epub].
- Yoon TY, Kim KR, Lee JY, Kim MK. Aquagenic syringeal acrokeratoderma: unusual prominence on the dorsal aspect of fingers? Br J Dermatol 2008;159:486–8.
- Falcón CS, Ortega SS. Aquagenic syringeal acrokeratoderma. J Am Acad Dermatol 2008;59:S112–13.
- Darlenski R, Tsankov N. Aquagenic syringeal acrokeratoderma. J Dtsch Dermatol Ges 2012;10:198.
- Luo DQ, Li Y, Huang YB, et al. Aquagenic syringeal acrokeratoderma in an adult man: case report and review of the literature. Clin Exp Dermatol 2009;34:e907–9.
- Itin PH, Lautenschlager S. Aquagenic syringeal acrokeratoderma (transient reactive papulotranslucent acrokeratoderma). Dermatology 2001;204:8–11.
- Sezer E, Erkek E, Duman D, Sahin S, Cetin E. Dermatoscopy as an adjunctive diagnostic tool in aquagenic syringeal acrokeratoderma. Dermatology 2012;225:97–9.
- Kabashima K, Shimauchi T, Kobayashi M, et al. Aberrant aquaporin 5 expression in the sweat gland in aquagenic wrinkling of the palms. J Am Acad Dermatol 2008;59:S28–32.
- Blaydon DC, Lind LK, Plagnol V, et al. Mutations in AQP5, encoding a water-channel protein, cause autosomal-dominant diffuse nonepidermolytic palmoplantar keratoderma. Am J Hum Genet 2013;93:330–5.
- Lowes MA, Khaira GS, Holt D. Transient reactive papulotranslucent acrokeratoderma associated with cystic fibrosis. Australas J Dermatol 2000;41:172–4.
- Grasemann H, Ratjen F, Solomon M. Aquagenic wrinkling of the palms in a patient with cystic fibrosis. N Engl J Med 2013;369:2362–3.
- Chinazzo C, De Alessandri A, Menoni S, et al. Aquagenic wrinkling of the palms and cystic fibrosis: an Italian study with controls and genotype–phenotype correlations. Dermatology 2014;228:60–5.
- Poletti ED, Muñoz-Sandoval R. Images in clinical medicine. Aquagenic keratoderma. N Engl J Med 2014;371:952.
- Diba VC, Cormack GC, Burrows NP. Botulinum toxin is helpful in aquagenic palmoplantar keratoderma. Br J Dermatol 2005;152:394–5.
Palmoplantar keratoderma and cardiomyopathy
- Hoeger PH, Yates RW, Harper JI. Palmoplantar keratoderma associated with congenital heart disease. Br J Dermatol 1998;138:506–9.
- Carvajal-Huerta L. Epidermolytic palmoplantar keratoderma with woolly hair and dilated cardiomyopathy. J Am Acad Dermatol 1998;39:418–21.
- Bergman R, Hershkovitz D, Fuchs D, et al. Disadhesion of epidermal keratinocytes: a histologic clue to palmoplantar keratodermas caused by DSG1 mutations. J Am Acad Dermatol 2010;62:107–13.
- Richardson P, McKenna W, Bristow M, et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of Cardiomyopathies. Circulation 1996;93:841–2.
- Barker JNWN, Protonatorios N, Tsatsopoulou A, et al. Palmoplantar keratoderma, curly hair and endomyocardial fibrodysplasia: a new syndrome. Br J Dermatol 1983;119 :13–14.
- Protonotarios N, Tsatsopoulou A, Fontaine G. Naxos disease: keratoderma, scalp modifications, and cardiomyopathy. J Am Acad Dermatol 2001;44:309–11.
- McKoy G, Protonotarios N, Crosby A, et al. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet 2000;355:2119–24.
- Petrof G, Mellerio JE, McGrath JA. Desmosomal genodermatoses. Br J Dermatol 2012;166:36–45.
- Cabral RM, Liu L, Hogan C, et al. Homozygous mutations in the 5' region of the JUP gene result in cutaneous disease but normal heart development in children. J Invest Dermatol 2010;130:1543–50.
- Asimaki A, Syrris P, Wichter T, et al. A novel dominant mutation in plakoglobin causes arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet 2007;81:964–73.
- Boulé S, Fressart V, Laux D, et al. Expanding the phenotype associated with a desmoplakin dominant mutation: Carvajal/Naxos syndrome associated with leukonychia and oligodontia. Int J Cardiol 2012;161:50–2.
- Pigors M, Kiritsi D, Krümpelmann S, et al. Lack of plakoglobin leads to lethal congenital epidermolysis bullosa: a novel clinico-genetic entity. Hum Mol Genet 2011;20:1811–19.
- Protonotarios N, Tsatsopoulou A. Naxos disease: cardiocutaneous syndrome due to cell adhesion defect. Orphanet J Rare Dis 2006;1:4.
- Norgett EE, Hatsell SJ, Carvajal-Huerta L, et al. Recessive mutation in desmoplakin disrupts desmoplakin–intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum Mol Genet 2000;9:2761–6.
- Norgett EE, Lucke TW, Bowers B, et al. Early death from cardiomyopathy in a family with autosomal dominant striate palmoplantar keratoderma and woolly hair associated with a novel insertion mutation in desmoplakin. J Invest Dermatol 2006;126:1651–4.
- Rampazzo A, Nava A, Malacrida S, et al. Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet 2002;71:1200–6.
- Jonkman MF, Pasmooij AM, Pasmans SG, et al. Loss of desmoplakin tail causes lethal acantholytic epidermolysis. Am J Hum Genet 2005;77:653–60.
- Whittock NV, Wan H, Morley SM, et al. Compound heterozygosity for nonsense and missense mutations in desmoplakin underlies skin fragility/woolly hair syndrome. J Invest Dermatol 2002;118:232–8.
- Williams T, Machann W, Kühler, et al. Novel desmoplakin mutation: juvenile biventricular cardiomyopathy with left ventricular non-compaction and acantholytic palmoplantar keratoderma. Clin Res Cardiol 2011;100:1087–93.
- Protonotarios N, Tsatsopoulou A. Naxos disease and Carvajal syndrome: cardiocutaneous disorders that highlight the pathogenesis and broaden the spectrum of arrhythmogenic right ventricular cardiomyopathy. Cardiovasc Pathol 2004;13:185–94.
- Mavrogeni S, Bratis K, Protonotarios N, et al. Cardiac magnetic resonance can early assess the presence and severity of heart involvement in Naxos disease. Int J Cardiol 2012;154:e19–20.
- Ramot Y, Molho-Pessach V, Meir T, et al. Mutation in KANK2, encoding a sequestering protein for steroid receptor coactivators, causes keratoderma and woolly hair. J Med Genet 2014;51:388–94.
- Gultekin N, Kucukates E. An unusual form of Naxos disease and its improvement by adjuvant low-dose colchicine therapy. Acta Cardiol 2013;68:433–7.
Palmoplantar keratoderma and hearing impairment
- Kelsell DP, Dunlop J, Hodgins MB. Human diseases: clues to cracking the connexin code. Trends Cell Biol 2001;11:2–6.
- Birkenhäger R, Lüblinghoff N, Prera E, et al. Autosomal dominant prelingual hearing loss with palmoplantar keratoderma syndrome: Variability in clinical expression from mutations of R75W and R75Q in the GJB2 gene. Am J Med Genet A 2010;152A:1798–802.
- Kelsell DP, Dunlop J, Stevens HP, et al. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature 1997;387:80–3.
- Kelley PM, Harris DJ, Comer BC, et al. Novel mutations in the connexin 26 gene (GJB2) that cause autosomal recessive (DFNB1) hearing loss. Am J Hum Genet 1998;62:792–9.
- White TW. Functional analysis of human Cx26 mutations associated with deafness. Brain Res Rev 2000;32:181–3.
- Richard G, White TW, Smith LE, et al. Functional defects of Cx26 resulting from a heterozygous missense mutation in a family with dominant deaf-mutism and palmoplantar keratoderma. Hum Genet 1998;103:393–9.
- Heathcote K, Syrris P, Carter ND, Patton MA. A connexin 26 mutation causes a syndrome of sensorineural hearing loss and palmoplantar hyperkeratosis (MIM 148350). J Med Genet 2000;37:50–1.
- Rouan F, White TW, Brown N, et al. Trans-dominant inhibition of connexin-43 by mutant connexin-26: implications for dominant connexin disorders affecting epidermal differentiation. J Cell Sci 2001;114:2105–13.
- Uyguner O, Tukel T, Baykal C, et al. The novel R75Q mutation in the GJB2 gene causes autosomal dominant hearing loss and palmoplantar keratoderma in a Turkish family. Clin Genet 2002;62:306–9.
- de Zwart-Storm EA, Hamm H, Stoevesandt J, et al. A novel missense mutation in GJB2 disturbs gap junction protein transport and causes focal palmoplantar keratoderma with deafness. J Med Genet 2008;45:161–6.
- Pandya A. Nonsyndromic hearing loss and deafness, mitochondrial. In: Pagon RA, Adam MP, Bird TD, Dolan CR, Fong CT, Stephens K, eds. GeneReviews. Seattle (WA): University of Washington, Seattle, 2013.
- Vohwinkel KH. Keratoma hereditarium mutilans. Arch Dermatol Syphilol 1929;158:354–64.
- Wigley JEM. A case of hyperkeratosis palmaris et plantaris associated with ainhum-like constriction of the fingers. Br J Dermatol 1929;41:188–91.
- Nockemann PE. Erbliche Hornhautverdickung mit Schnürfurchen an Fingern und Zehen mit Innenohrschwerhörigkeit. Med Welt 1961;37:1894–900.
- Gibbs RC, Frank SB. Keratoma hereditaria mutilans (Vohwinkel): differentiating features of conditions with constriction of digits. Arch Dermatol 1966;94:619–25.
- Ocaña-Sierra J, Blesa G, Montero E. Syndrome de Vohwinkel. Ann Derm Syph 1975;102:41–5.
- McGibbon DH, Watson RT. Vohwinkel's syndrome and deafness. J Laryngol Otol 1977;91:853–7.
- Wereide K. Mutilating palmoplantar keratoderma successfully treated with etretinate. Acta Derm Venereol Scand 1984;64:566–9.
- Maestrini E, Korge BP, Ocana-Sierra J, et al. A missense mutation in connexin 26, D66H, causes mutilating keratoderma with sensorineural deafness (Vohwinkel's syndrome) in three unrelated families. Hum Mol Genet 1999;8:1237–43.
- Richard G, Brown N, Ishida-Yamamoto A, Krol A. Expanding the phenotypic spectrum of Cx26 disorders: Bart–Pumphrey syndrome is caused by a novel missense mutation in GJB2. J Invest Dermatol 2004;123:856–63.
- Richard GA, Rouan F, Willoughby CE, et al. Mutations in GJB-2 encoding connexin-26 cause the ectodermal dysplasia keratitis–ichthyosis–deafness syndrome. Am J Hum Genet 2002;70:1341–8.
- Ballana E, Ventayol M, Rabionet R, et al. The Connexin-deafness homepage, 2011. http://davinci.crg.es/deafness/ last accessed January 2015.
- White TW. Functional analysis of human Cx26 mutations associated with deafness. Brain Res Rev 2000;32:181–3.
- Xu J, Nicholson BJ. The role of connexins in ear and skin physiology – functional insights from disease-associated mutations. Biochim Biophys Acta 2013;1828:167–78.
- Labarthe MP, Bosco D, Saurat JH, et al. Upregulation of connexin 26 between keratinocytes of psoriatic lesions. J Invest Dermatol 1998;111:72–6.
- Lucke T, Choudhry R, Thom R, et al. Upregulation of connexin 26 is a feature of keratinocyte differentiation in hyperproliferative epidermis, vaginal epithelium, and buccal epithelium. J Invest Dermatol 1999;112:354–61.
- Scott CA, Tattersall D, O'Toole EA, Kelsell DP. Connexins in epidermal homeostasis and skin disease. Biochim Biophys Acta 2012;1818:1952–61.
- de Zwart-Storm EA, Hamm H, Stoevesandt J, et al. A novel missense mutation in GJB2 disturbs gap junction protein transport and causes focal palmoplantar keratoderma with deafness. J Med Genet 2008;45:161–6.
- Tattersall D, Scott CA, Gray C, et al. EKV mutant connexin 31 associated cell death is mediated by ER stress. Hum Mol Genet 2009;18:4734–45.
- Rouan F, White TW, Brown N, et al. Trans-dominant inhibition of connexin-43 by mutant connexin-26: implications for dominant connexin disorders affecting epidermal differentiation. J Cell Sci 2001;114:2105–13.
- de Zwart-Storm EA, van Geel M, Veysey E, et al. A novel missense mutation in GJB2, p.Tyr65His, causes severe Vohwinkel syndrome. Br J Dermatol 2011;164:197–9.
- Korge BP, Ishida-Yamamoto A, Punter C, et al. Loricrin mutation in Vohwinkel's keratoderma is unique to the variant with ichthyosis. J Invest Dermatol 1997;109:604–10.
- Serrano Castro PJ, Naranjo Fernandez C, et al. Vohwinkel Syndrome secondary to missense mutation D66H in GJB2 gene (connexin 26) can include epileptic manifestations. Seizure 2010;19:129–31.
- Bart RS, Pumphrey RE. Knuckle pads, leukonychia and deafness. A dominantly inherited syndrome. N Engl J Med 1967;276:202–7.
- Alexandrino F, Sartorato EL, Marques-de-Faria AP, Steiner CE. G59S mutation in the GJB2 (connexin 26) gene in a patient with Bart–Pumphrey syndrome. Am J Med Genet 2005;136:282–4.
- de Zwart-Storm EA, Hamm H, Stoevesandt J, et al. A novel missense mutation in GJB2 disturbs gap junction protein transport and causes focal palmoplantar keratoderma with deafness. J Med Genet 2008;45:161–6.
- Bondeson ML, Nyström AM, Gunnarsson U, Vahlquist A. Connexin 26 (GJB2) mutations in two Swedish patients with atypical Vohwinkel (mutilating keratoderma plus deafness) and KID syndrome both extensively treated with acitretin. Acta Derm Venereol 2006;86:503–8.
- Pisoh T, Bhatia A, Oberlin C. Surgical correction of pseudo-ainhum in Vohwinkel's syndrome. J Hand Surg 1995;20B:338–41.
- Sinha M, Watson SB. Keratoderma hereditarium mutilans (Vohwinkel syndrome). J Hand Surg Eur 2009;34:235–7.
- Bassetto F, Tiengo C, Sferrazza R, et al. Vohwinkel syndrome: treatment of pseudo-ainhum. Int J Dermatol 2010;49:79–82.
- Kong Y, Liu S, Wang SJ, et al. Cochlear implantation effect on deaf children with gap junction protein beta 2 gene mutation. Chin Med J (Engl) 2013;126:1298–301.
Mitochondrial palmoplantar keratoderma with hearing impairment
- Reid FM, Vernh GA, Jacobs HT. A novel mitochondrial point mutation in a maternal pedigree with sensorineural deafness. Hum Mutat 1994;3:243–7.
- Maasz A, Komlosi K, Hadzsiev K, et al. Phenotypic variants of the deafness-associated mitochondrial DNA A7445G mutation. Curr Med Chem 2008;15:1257–62.
- Hatamochi A, Nakagawa S, Ueki H, et al. Diffuse palmoplantar keratoderma with deafness. Arch Dermatol 1982;118:605–7.
- Sevior KB, Hatamochi A, Stewart IA, et al. Mitochondrial A7445G mutation in two pedigrees with palmoplantar keratoderma and deafness. Am J Med Genet 1998;75:179–85.
- Martin L, Toutain A, Guillen C, et al. Inherited palmoplantar keratoderma and sensorineural deafness associated with A7445G point mutation in the mitochondrial genome. Br J Dermatol 2000;143:876–83.
- Caria H, Matos T, Oliveira-Soares R, et al. A7445G mtDNA mutation present in a Portuguese family exhibiting hereditary deafness and palmoplantar keratoderma. J Eur Acad Dermatol Venereol 2005;19:455–8.
- Yelverton JC, Arnos K, Xia XJ, Nance WE, Pandya A, Dodson KM. The clinical and audiologic features of hearing loss due to mitochondrial mutations. Otolaryngol Head Neck Surg 2013;148(6):1017–22.
- Pandya A. Nonsyndromic hearing loss and deafness, mitochondrial. In: Pagon RA, Adam MP, Bird TD, Dolan CR, Fong CT, Stephens K, eds. GeneReviews™ [Internet]. Seattle (WA): University of Washington, Seattle, 2013.
Palmoplantar keratoderma and cancer
- Barnett JH, Estes SA. Multiple epitheliomata cuniculata occurring in a mutilating keratoderma. Cutis 1985;35:345–7.
- Abadir R, Zurowski S. Case report: squamous cell carcinoma of the skin in both palms, axillary node, donor skin graft site and both soles – associated hyperkeratosis and porokeratosis. Br J Radiol 1994;67:507–10.
- Ena P, Mazzarello V. Hydrotic ectodermal dysplasia (Clouston syndrome) with deafness, strabismus, nystagmus, cutaneous squamous cell carcinoma, oral leukoplakia and other anomalies: report of a previously undescribed case. G Ital Dermatol Venereol 1998;133:285–9.
- Rogozinski TT, Schwartz RA, Towpik E. Verrucous carcinoma in Unna–Thost hyperkeratosis of the palms and soles. J Am Acad Dermatol 1994;31:1061–2.
- Affleck AG, Leach IH, Littlewood SM. Carcinoma cuniculatum arising in focal plantar keratoderma. Dermatol Surg 2007;33:745–8.
- Hacham-Zadeh S, Goldberg L. Malignant melanoma and Papillon–Léfèvre syndrome. Arch Dermatol 1982;118:2.
- Seike T, Nakanishi H, Urano Y, Arase S. Malignant melanoma developing in an area of palmoplantar keratoderma (Greither's disease). J Dermatol 1995;22:55–61.
- Aygit AC, Baycin HN, Demiralay A. Malignant melanoma in association with palmoplantar keratoderma. Eur J Plast Surg 1999;22:49–50.
- Rubegni P, Poggiali S, Cuccia A, et al. Acral malignant melanoma and striated palmoplantar keratoderma (Brunauer–Fohs–Siemens syndrome): a fortuitous association? Dermatol Surg 2004;30:1539–42.
- Dessureault J, Poulin Y, Bourcier M, Gagne E. Olmsted syndrome–palmoplantar and periorificial keratodermas: association with malignant melanoma. J Cutan Med Surg 2003;7:236–42.
- Mozzillo N, Nunziata CA, Caraco C, et al. Malignant melanoma developing in an area of hereditary palmoplantar keratoderma (Mal de Meleda). J Surg Oncol 2003;84:229–33.
- Nakajima K, Nakano H, Takiyoshi N, et al. Papillon–Lefevre syndrome and malignant melanoma. A high incidence of melanoma development in Japanese palmoplantar keratoderma patients. Dermatology 2008;217:58–62.
- Chevrant-Breton J, Kerbrat P, Le-Marec B, et al. Keratodermie palmoplantaire epidermolytique, autosomique dominante et adenocarcinomes familiaux (etude de 4 generations). Ann Dermatol Vénéréol 1985;112:841–4.
- Blanchet-Bardon C, Nazzaro V, Chevrant-Breton J, et al. Hereditary epidermolytic palmoplantar keratoderma associated with breast and ovarian cancer in a large kindred. Br J Dermatol 1987;117:363–70.
- Torchard D, Blanchet-Bardon C, Serova O, et al. Epidermolytic palmo-plantar keratoderma cosegregates with a keratin 9 mutation in a pedigree with breast and ovarian cancer. Nat Genet 1994;6:106–10.
- Bennion SD, Patterson JW. Keratosis punctata palmaris et plantaris and adenocarcinoma of the colon: a possible familial association of punctate keratoderma and gastrointestinal malignancy. J Am Acad Dermatol 1984;10:587–91.
- Stevens HP, Kelsell DP, Leigh IM, et al. Punctate palmoplantar keratoderma and malignancy in a four-generation family. Br J Dermatol 1996;134:720–6.
- Kerdel FA, MacDonald DM. Palmo-plantar keratoderma associated with carcinoma of the bronchus. Acta Derm Venereol (Stockh) 1982;62:178–80.
- Huriez C, Agache P, Bombart M, Souilliart F. Spinocellular epitheliomas in congenital cutaneous atrophy in 2 families with high cancer morbidity. Bull Soc Fr Dermatol Syphiligr 1963;70:24–8.
- Delaporte E, N'guyen-Mailfer C, Janin A, et al. Keratoderma with scleroatrophy of the extremities or sclerotylosis (Huriez syndrome): a reappraisal. Br J Dermatol 1995;133:409–16.
- Lucker GPH, Zeedijk N, Steijlen PM. The Huriez syndrome. Scleroatrophic palmoplantar keratoderma Eur J Dermatol 1997;7:155–7.
- Lee YA, Stevens HP, Delaporte E, Wahn U. Reis A. A gene for an autosomal dominant scleroatrophic syndrome predisposing to skin cancer (Huriez syndrome) maps to chromosome 4q23. Am J Hum Genet 2000;66:326–330.
- Hamm H, Traupe H, Brocker EB, Schubert H, Kolde G. The scleroatrophic syndrome of Huriez: a cancer-prone genodermatosis Br J Dermatol 1996;134:512–18.
- Watanabe E, Takai T, Ichihashi M, Ueda M. A nonfamilial Japanese case of Huriez syndrome: p53 expression in squamous cell carcinoma Dermatology 2003;207:82–4.
- Guerriero C, Albanesi C, Girolomoni G, et al. Huriez syndrome: case report with a detailed analysis of skin dendritic cells Br J Dermatol 2000;143:1091–6.
- Al Aboud K, Khachemoune A. Claude Huriez and his syndrome. Skinmed 2011;9:313–14.
- Man XY, Li W, Chen JQ, et al. Huriez syndrome with squamous cell carcinoma. Eur J Dermatol 2011;21:294–5.
- Dumont LA, Gahagnon T, Martinot-Duquennoy V, et al. [Case report: squamous cell carcinoma, radial forearm flap and Huriez syndrome. Focus on a rare pathology.] Ann Chir Plast Esthet 2013;58:175–9.
- Stevens HP, Kelsell DP, Bryant SP, et al. Linkage of an American pedigree with palmoplantar keratoderma and malignancy (palmoplantar ectodermal dysplasia type III) to 17q24. Arch Dermatol 1996;132:1–12.
- Risk JM, Field EA, Field JK, et al. Tylosis oesophageal cancer mapped. Nat Genet 1994;8:319–21.
- Langan JE, Cole CG, Huckle EJ, et al. Novel microsatellite markers and single nucleotide polymorphisms refine the tylosis with oesophageal cancer (TOC) minimal region on 17q25 to 42.5 kb: sequencing does not identify the causative gene. Hum Genet 2004;114:534–40.
- Iwaya T, Maesawa C, Ogasawara S, Tamura G. Tylosis esophageal cancer locus on chromosome 17q25.1 is commonly deleted in sporadic human esophageal cancer. Gastroenterology 1998;114:1206–10.
- Blaydon DC, Etheridge SL, Risk JM, et al. RHBDF2 mutations are associated with tylosis, a familial esophageal cancer syndrome. Am J Hum Genet 2012;90:340–6.
- Dereure O. [RHBDF2 mutations in familial palmoplantar keratoderma associated with tylosis oesophageal cancer.] Ann Dermatol Venereol 2012;139:605–6.
- Saarinen S, Vahteristo P, Lehtonen R, et al. Analysis of a Finnish family confirms RHBDF2 mutations as the underlying factor in tylosis with esophageal cancer. Fam Cancer 2012;11:525–8.
- Issuree PD, Maretzky T, McIlwain DR, et al. iRHOM2 is a critical pathogenic mediator of inflammatory arthritis. J Clin Invest 2013;123:928–32.
- Adrain C, Zettl M, Christova Y, Taylor N, Freeman M. Tumor necrosis factor signaling requires iRhom2 to promote trafficking and activation of TACE. Science 2012;335:225–8.
- Howel-Evans W, McGonnell RB, Clarke GA, Sheppard PM. Carcinoma of the oesophagus with keratosis palmaris et plantaris (tylosis): a study of two families. QJM 1950;27:415–29.
- Ellis A, Field EA, Field JK, et al. Tylosis associated with carcinoma of the oesophagus and oral leukoplakia in a large Liverpool family: a review of six generations. Eur J Cancer Oral Oncol 1994;30:102–12.
- Howel-Evans W, McGonnell RB, Clarke GA, Sheppard PM. Carcinoma of the oesophagus with keratosis palmaris et plantaris (tylosis): a study of two families. QJM 1950;27:415–29.
Palmoplantar keratoderma, sex reversal and cancer
- McElreavey K, Vilain E, Abbas N, et al. A regulatory cascade hypothesis for mammalian sex determination: SRY represses a negative regulator of male development. Proc Nat Acad Sci 1993:3368–3372.
- Radi O, Parma P, Imbeaud S, et al. XX sex reversal, palmoplantar keratoderma, and predisposition to squamous cell carcinoma: genetic analysis in one family. Am J Med Genet 2005;138:241–6.
- Guerriero C, Albanesi C, Girolomoni G, et al. Huriez syndrome: case report with a detailed analysis of skin dendritic cells. Br J Derm 2000;143:1091–6.
- Vernole P, Terrinoni A, Didona B, et al. An SRY-negative XX male with Huriez syndrome. Clin Genet 2000;57:61–6.
- Micali G, Nasca MR, Innocenzi D, et al. Association of palmoplantar keratoderma, cutaneous squamous cell carcinoma, dental anomalies, and hypogenitalism in four siblings with 46,XX karyotype: a new syndrome. J Am Acad Derm 2005;53:234–9.
- Parma P, Radi O, Vidal V, et al. R-spondin1 is essential in sex determination, skin differentiation and malignancy. Nat Genet 2006;38:1304–9.
- Tomaselli S, Megiorni F, De Barnardo C, et al. Syndromic true hermaphroditism due to an R-spondin1 (RSPO1) homozygous mutation. Hum Mutat 2008;29:220–6.
- Capel B. R-spondin-1 tips the balance in sex determination. Nat Genet 2006;38:1233–4.
Palmoplantar keratoderma in ectodermal dysplasia and related diseases
- Clouston HR. A hereditary ectodermal dystrophy. Can Med Assoc J 1921;21:18–31.
- Patel RR, Bixler D, Norins AL. Clouston syndrome: a rare autosomal dominant trait with palmoplantar hyperkeratosis and alopecia. J Craniofac Genet Dev Biol 1991;11:176–9.
- Der Kaloustian VM. Hidrotic ectodermal dysplasia 2. 2005. In: Pagon RA, Adam MP, Bird TD, Dolan CR, Fong CT, Smith RJH, Stephens K, eds. GeneReviews™ [Internet]. Seattle (WA): University of Washington, Seattle, 1993–2013.
- Koch P, Foss P, Baum HP, Zaun H. [Verruciform palmoplantar keratoderma as a characteristic marker of hidrotic ectodermal dysplasia of the Clouston type.] Hautarzt 1995;46:272–5.
- van Steensel MAM, Jonkman MF, van Geel M, et al. Clouston syndrome can mimic pachyonychia congenita. J Invest Dermatol 2003;121:1035–8.
- Lamartine J, Essenfelder GM, Kibar Z, et al. Mutations in GJB6 cause hidrotic ectodermal dysplasia. Nat Genet 2000;26:142–4.
- Smith FJD, Morley SM, McLean WHI. A novel connexin 30 mutation in Clouston syndrome. J Invest Dermatol 2002;118:530–2.
- Fujimoto A, Kurban M, Nakamura M, et al. GJB6, of which mutations underlie Clouston syndrome, is a potential direct target gene of p63. J Dermatol Sci 2013;69:159–66.
- Kantaputra P, Kaewgahya M, Jotikasthira D, Kantaputra W. Tricho-odonto-onycho-dermal dysplasia and WNT10A mutations. Am J Med Genet A 2014;164:1041–8.
- Bohring A, Stamm T, Spaich C, et al. WNT10A mutations are a frequent cause of a broad spectrum of ectodermal dysplasias with sex-biased manifestation pattern in heterozygotes. Am J Hum Genet 2009;85:97–105.
- Mues G, Bonds J, Xiang L, et al. The WNT10A gene in ectodermal dysplasias and selective tooth agenesis. Am J Med Genet A 2014;164:2455–60.
- Schöpf E, Schulz HJ, Passarge E. Syndrome of cystic eyelids, palmo-plantar keratosis, hypodontia and hypotrichosis as a possible autosomal recessive trait. Birth Defects 1971;7:219–21.
- Monk BE, Pieris S, Soni V. Schöpf–Schulz–Passarge syndrome. Br J Dermatol 1992;127:33–5.
- Burket JM, Burket BJ, Burket D. Eyelid cysts, hypodontia, and hypotrichosis. J Am Acad Dermatol 1984;10:922–5.
- Verplancke P, Driessen L, Wynants P, Naeyaert JM. The Schöpf–Schulz–Passarge syndrome. Dermatology 1998;196:463–6.
- Hampton PJ, Angus B, Carmichael AJ. A case of Schopf–Schulz–Passarge syndrome. Clin Exp Dermatol 2005;30:528–30.
Papillon–Léfèvre and Haim–Munk syndrome
- Papillon MM, Lefèvre P. Deux cas de kératodermie palmaire et plantaire symétrique familiale (maladie de Meleda) chez le frère et la soeur. Coexistence dans les deux cas d'altérations dentaires graves. Bull Soc Fr Derm Venereol 1924;31:82–7.
- Haneke E. The Papillon–Léfèvre syndrome: keratosis palmoplantaris with periodontopathy. Hum Genet 1979;51:1–35.
- Vrahopoulos TP, Barber P, Kiakoni H, et al. Ultrastructure of the periodontal lesion in a case of Papillon–Léfèvre syndrome (PLS). J Clin Periodontol 1988;15:17–26.
- Nazzaro V, Blanchet-Bardon C, Mimoz C. Papillon–Léfèvre syndrome: ultrastructural study and successful treatment with acitretin. Arch Dermatol 1988;124:533–9.
- Hattab F, Rawashdeh MA, Yassin OM, et al. Papillon–Léfèvre syndrome: a review of the literature and report of four cases. J Periodontol 1995;66:413–20.
- Ullbro C, Crossner CG, Nederfors T, et al. Dermatologic and oral findings in a cohort of 47 patients with Papillon–Léfèvre syndrome. J Am Acad Dermatol 2003;48:345–51.
- Bergman R, Friedman-Birnbaum R. Papillon–Léfèvre syndrome: a study of the long-term clinical course of recurrent pyogenic infections and the effects of etretinate treatment. Br J Dermatol 1988;119:731–6.
- Sethuraman G, Malhotra AK, Khaitan BK, Sharma VK. Effectiveness of isotretinoin in Papillon–Lefevre syndrome. Pediatr Dermatol 2005;22:378–9.
- Ashwani P, Swapna K, Rani SM, Reddy BS. Papillon–Lefevre syndrome with pseudoainhum. Indian Dermatol Online J 2010;1:33–5.
- Haim S, Munk J. Keratosis palmo-plantaris congenita, with periodontosis, arachnodactyly and a peculiar deformity of the terminal phalanges. Br J Dermatol 1965;77:42–54.
- Puliyel JM, Iyer KSS. A syndrome of keratosis palmoplantaris congenita, pes planus, onychogryphosis, periodontosis, arachnodactyly and a peculiar acro-osteolysis. Br J Dermatol 1986;115:243–8.
- Hart TC, Hart PS, Michalec MD, et al. Haim–Munk syndrome and Papillon–Léfèvre syndrome are allelic mutations in cathepsin C. J Med Genet 2000;37:88–94.
- Selvaraju V, Markandaya M, Prasad PV, et al. Mutation analysis of the cathepsin C gene in Indian families with Papillon–Lefèvre syndrome. BMC Med Genet 2003;4:5.
- Ashwani P, Swapna K, Rani SM, Reddy BS. Papillon–Lefevre syndrome with pseudoainhum. Indian Dermatol Online J 2010;1:33–5.
- Lee MR, Wong LC, Fischer GO. Papillon–Lefèvre syndrome treated with acitretin. Australas J Dermatol 2005;46:199–201.
- Toomes C, James J, Wood AJ, et al. Loss-of-function mutations in the cathepsin C gene result in periodontal disease and palmoplantar keratosis. Nat Genet 1999;23:421–4.
- Moghaddasian M, Arab H, Dadkhah E, et al. Protein modeling of cathepsin C mutations found in Papillon-Lefèvre syndrome. Gene 2014;538:182–7.
- de Haar SF, Jansen DC, Schoenmaker T, et al. Loss-of-function mutations in cathepsin C in two families with Papillon–Lefèvre syndrome are associated with deficiency of serine proteinases in PMNs. Hum Mutat 2004;23:524.
- Djawari D. Deficient phagocytic function in Papillon–Léfèvre syndrome. Clin Exp Immunol 1980;40:407–10.
- Levo Y, Wollnier S, Hacham-Zadeh S. Immunological study of patients with the Papillon–Léfèvre syndrome. Clin Exp Immunol 1980;40:407–10.
- Van Dyke T, Taubman M, Ebersole J. The Papillon–Léfèvre syndrome: neutrophil dysfunction with severe periodontal disease. Clin Immunol Immunopathol 1984;31:419–29.
- Dhanawade SS, Shah SD, Kakade GM. Papillon–Lefevre syndrome with liver abscess. Indian Pediatr 2009;46:723–5.
- Mercy P, Singh A, Ghorpade AK, et al. Papillon–Lefevre syndrome: two siblings, one developing liver abscess. Indian J Dermatol 2013;58:410.
- Kanthimathinathan HK, Browne F, Ramirez R, et al. Multiple cerebral abscesses in Papillon–Lefèvre syndrome. Childs Nerv Syst 2013;29:1227–9.
- Bullón P, Morillo JM, Thakker N, et al. Confirmation of oxidative stress and fatty acid disturbances in two further Papillon–Lefèvre syndrome families with identification of a new mutation. J Eur Acad Dermatol Venereol 2014;28:1049–56.
- Lundgren T, Parhar RS, Renvert S, Tatakis DN. Impaired cytotoxicity in Papillon–Lefèvre syndrome. J Dent Res 2005;84:414–17.
- Nickles K, Schacher B, Ratka-Krüger P, et al. Long-term results after treatment of periodontitis in patients with Papillon–Lefèvre syndrome: success and failure. J Clin Periodontol 2013;40:789–98.
- Pimentel SP, Kolbe MF, Pereira RS, et al. Papillon–Lefèvre syndrome in 2 siblings: case report after 11-year follow-up. Pediatr Dent 2012;34:e231–6.
- Stabholz A, Taichman NS, Soskolne WA. Occurrence of Actinobacillus actinomycetemcomitans and anti-leukotoxin antibodies in some members of an extended family affected by Papillon–Léfèvre syndrome. J Periodontol 1995;66:653–7.
- Eickholz P, Kugel B, Pohl S, et al. Combined mechanical and antibiotic periodontal therapy in a case of Papillon–Léfèvre syndrome. J Periodontol 2001;72:542–9.
- Robertson KL, Drucker DB, James J, et al. A microbiological study of Papillon–Léfèvre syndrome in two patients. J Clin Path 2001;54:371–6.
- Nakajima K, Nakano H, Takiyoshi N, et al. Papillon–Lefèvre syndrome and malignant melanoma. A high incidence of melanoma development in Japanese palmoplantar keratoderma patients. Dermatology 2008;217:58–62.
- Al-Benna S, Hasler R, Stricker I, et al. Papillon–Lefèvre syndrome and squamous cell carcinoma: a case report. Cases J 2009;2:7067.
- Cook GP. Papillon–Lefèvre syndrome and malignant melanoma. Dermatology 2009;219:187–8.
Olmsted syndrome
- Olmsted HC. Keratoderma palmaris et plantaris congenitalis: report of a case showing associated lesions of unusual location. Am J Dis Child 1927;33:757–64.
- Perry HO, Su WP. Olmsted syndrome. Semin Dermatol 1995;14:145–51.
- Larregue M, Callot V, Kanitakis J, et al. Olmsted syndrome: report of two new cases and literature review. J Dermatol 2000;27:557–68.
- Cambiaghi S, Tadini G, Barbareschi M, et al. Olmsted syndrome in twins. Arch Dermatol 1995;131:738–9.
- Atherton DJ, Sutton C, Jones BM. Mutilating palmoplantar keratoderma with periorificial keratotic plaques (Olmsted's syndrome). Br J Dermatol 1990;120:245–52.
- Mevorah B, Goldberg I, Sprecher E, et al. Olmsted syndrome: mutilating palmoplantar keratoderma with periorificial keratotic plaques. J Am Acad Dermatol 2005;53:S266–72.
- Tao J, Huang CZ, Yu NW, et al. Olmsted syndrome: a case report and review of literature. Int J Dermatol 2008;47:432–7.
- Lin Z, Chen Q, Lee M, et al. Exome sequencing reveals mutations in TRPV3 as a cause of Olmsted syndrome. Am J Hum Genet 2012;90:558–64.
- Haghighi A, Scott CA, Poon DS, et al. A missense mutation in the MBTPS2 gene underlies the X-linked form of Olmsted syndrome. J Invest Dermatol 2013;133:571–3.
- Eytan O, Fuchs-Telem D, Mevorach B, et al. Olmsted Syndrome caused by a homozygous recessive mutation in TRPV3. J Invest Dermatol 2014;134:1752–4.
- Vosynioti V, Kosmadaki M, Tagka A, Katsarou A. A case of Olmsted syndrome. Eur J Dermatol 2010;20:837–8.
- Lai-Cheong JE, Sethuraman G, Ramam M, et al. Recurrent heterozygous missense mutation, p.Gly573Ser, in the TRPV3 gene in an Indian boy with sporadic Olmsted syndrome. Br J Dermatol 2012;167:440–2.
- Montell C. Preventing a perm with TRPV3. Cell 141:218–20.
- Nilius B, Biro T, Owsianik G. TRPV3: time to decipher a poorly understood family member! J Physiol 2014;592:295–304.
- Oeffner F, Fischer G, Happle R, et al. IFAP syndrome is caused by deficiency in MBTPS2, an intramembrane zinc metalloprotease essential for cholesterol homeostasis and ER stress response. Am J Hum Genet 2009;84:459–67.
- Bornholdt D, Atkinson TP, Bouadjar B, et al. Genotype–phenotype correlations emerging from the identification of missense mutations in MBTPS2. Hum Mutat 2013;34:587–94.
- Wang HJ, Tang ZL, Lin ZM, et al. Recurrent splice-site mutation in MBTPS2 underlying IFAP syndrome with Olmsted syndrome-like features in a Chinese patient. Clin Exp Dermatol 2014;39:158–61
- Tang L, Zhang L, Ding H, et al. Olmsted syndrome: a new case complicated with easily broken hair and treated with oral retinoid. J Dermatol 2012;39:816–17.
- Judge MR, Misch K, Wright P, et al. Palmoplantar and perioroficial keratoderma with corneal epithelial dysplasia: a new syndrome. Br J Dermatol 1991;125:186–8.
- Bergonse FN, Rabello SM, Barreto RL, et al. Olmsted syndrome: the clinical spectrum of mutilating palmoplantar keratoderma. Pediatr Dermatol 2003;20:323–6.
- Yoshizaki Y, Kanki H, Ueda T, et al. A further case of plantar squamous cell carcinoma arising in Olmsted syndrome. Br J Dermatol 2001;145:685–6.
- Dessureault J, Poulin Y, Bourcier M, Gagne E. Olmsted syndrome–palmoplantar and periorificial keratodermas: association with malignant melanoma. J Cutan Med Surg 2003;7:236–42.
- Danso-Abeam D, Zhang J, Dooley J, et al. Olmsted syndrome: exploration of the immunological phenotype. Orphanet J Rare Dis 2013;8:79.
- Duchatelet S, Pruvost S, de Veer S, et al. A new TRPV3 missense mutation in a patient with Olmsted syndrome and erythromelalgia. JAMA Dermatol 2014;150:303–6.
- Duchatelet S, Guibbal L, de Veer S, et al. Olmsted syndrome with erythromelalgia caused by recessive transient receptor potential vanilloid 3 mutations. Br J Dermatol. 2014;171:675–8.
- Requena L, Manzarbeitia F, Moreno C, et al. Olmsted syndrome: report of a case with study of the cellular proliferation in keratoderma. Am J Dermatopathol 2001;23:514–20.
- Elise Tonoli R, De Villa D, Hübner Frainer R, et al. Olmsted syndrome. Case Rep Dermatol Med 2012;2012:927305.
- Fonseca E, Pena C, Del Pozo J, et al. Olmsted syndrome. J Cutan Pathol 2001;28:271–5.
- Ueda M, Nakagawa K, Hayashi K, et al. Partial improvement of Olmsted syndrome with etretinate. Pediatr Dermatol 1993;10:376–81.
- Hausser I, Frantzmann Y, Anton-Lamprecht I, et al. Olmsted Syndrom: Erfolgreiche Therapie durch Behandlung mit Etretinat. Hautarzt 1993;44:394–400.
- Yaghoobi R, Omidian M, Sina N, et al. Olmsted syndromein an Iranian family: report of two new cases. Arch Iran Med 2007;10:246–9.
- Bédard MS, Powell J, Laberge L, et al. Palmoplantar keratoderma and skin grafting: postsurgical long-term follow-up of two cases with Olmsted syndrome. Pediatr Dermatol 2008;25:223–9.
- Kenner-Bell BM, Paller AS, Lacouture ME. Epidermal growth factor receptor inhibition with erlotinib for palmoplantar keratoderma. J Am Acad Dermatol 2010;63:e58–9.
Palmoplantar keratoderma and ophthalmic manifestations
Oculocutaneous tyrosinaemia (tyrosinaemia type II)
- Richner H. Hornhautaffektion bei Keratoma palmare et plantare hereditarium. Klin Monatsbl Augenheilkd 1938;100:580–8.
- von Hanhart E. Neue Sonderformen von Keratosis palmo-plantaris, u.a. einige regelmäßig-dominante mit systematisierten Lipomen, ferner 2 einfach-rezessive mit Schwachsinn und z. T. mit Hornhautveränderungen des Auges (Edtodermalsyndrom). Dermatologica 1947;94:286–308.
- Natt E, Westphal EM, Toth-Fejel SE, et al. Inherited and de novo deletion of the tyrosine aminotransferase gene locus at 16q22.1-q22.3 in a patient with tyrosinemia type II. Hum Genet 1987;77:352–8.
- Fellman JH, Vanbellinghen PJ, Jones RT, et al. Soluble and mitochondrial forms of tyrosine aminotransferase: relationship to human tyrosinaemia. Biochemistry 1969;8:615–22.
- Goldsmith LA, Thorpe JM, Roe CR. Hepatic enzymes of tyrosine metabolism in tyrosinemia. II. J Invest Dermatol 1979;73:530–2.
- Hunziker N. Richner–Hanhart syndrome and tyrosinemia type II. Dermatologica 1980;160:180–9.
- Bohnert A, Anton-Lamprecht I. Richner–Hanhart's syndrome: ultrastructural abnormalities of epidermal keratinization indicating a causal relationship to high intracellular tyrosine levels. J Invest Dermatol 1982;79:68–74.
- Viglizzo GM, Occella C, Bleidl D, Rongioletti F. Richner–Hanhart syndrome (tyrosinemia II): early diagnosis of an incomplete presentation with unusual findings. Pediatr Dermatol 2006;23:259–61.
- Podglajen-Wecxsteen O, Delaporte E, Piette F, et al. Tyrosinose oculo-cutanée de type II. Ann Dermatol Vénéréol 1993;120:139–42.
- Garibaldi LR, Siliato F, De Martini I, et al. Oculocutaneous tyrosinosis: report of two cases in the same family. Helv Paediat Acta 1977;32:173–80.
- Meissner T, Betz RC, Pasternack SM, et al. Richner–Hanhart syndrome detected by expanded newborn screening. Pediatr Dermatol 2008;25:378–80.
- Gipson IK, Burns RP, Wolfe-Lande JD. Crystals in corneal epithelial lesions of tyrosine-fed rats. Invest Ophthalmol 1975;14:937–41.
- Zaleski WA, Hill A, Krushniruk W. Skin lesions in tyrosinosis: response to dietary treatment. Br J Dermatol 1973;88:335–40.
- Ney D, Bay C, Schneider JA. Dietary management of oculocutaneous tyrosinaemia in an 11-year-old child. Am J Dis Child 1983;137:995–1000.
- Paige DG, Clayton P, Bowron A, et al. Richner–Hanhart syndrome (oculo-cutaneous tyrosinaemia, tyrosinaemia type II). J R Soc Med 1992;85:759–60.
Palmoplantar keratoderma and neurological manifestations
- Fitzsimmons JS, Fitzsimmons EM, McLachlan JI, et al. Four brothers with mental retardation, spastic paraplegia and palmoplantar hyperkeratosis: a new syndrome? Clin Genet 1983;23:329–35.
- Powell FC, Venencie PY, Gordon H, Winkelmann RK. Keratoderma and spastic paralysis. Br J Dermatol 1983;109:589–96.
- Tolmie JL, Wilcox DE, McWilli R, et al. Palmoplantar keratoderma, nail dystrophy, and hereditary motor and sensory neuropathy: an autosomal dominant trait. J Med Genet 1988;25:754–7.
- Rabbiosi G, Borroni G, Pinelli P, Cosi V. Palmoplantar keratoderma and Charcot–Marie–Tooth disease. Arch Dermatol 1980;116:789–90.
- Beare JM, Nevin NC, Froggatt P, et al. Atypical erythrokeratoderma with deafness, physical retardation and peripheral neuropathy. Br J Dermatol 1972;87:308–14.
- Rauch H-J, Neumayer K. Bureau–Barriere–Thomas-Syndrom eine seltene hereditare Palmoplantarkeratose mit assoziierten Symptomen. Z Hautkr 1980;56:102–8.
- Thoma E, Ruzicka T, Donhauser G, et al. Klinik und therapie des Bureau–Barriere–Syndroms: Beobachtungen an 17 Fallen mit Literaturubersicht. Hautarzt 1993;44:5–13.
- Dissemond J, Knab J, Goos M. [Bureau–Barrière syndrome of the hand. A case report on an unusual localization.] Hautarzt 2004;55:371–5.
Acquired keratodermas
- Patel S, Zirwas M, English JC 3rd. Acquired palmoplantar keratoderma. Am J Clin Dermatol 2007;8:1–11.
- Garcia-Hidalgo L, Orozco-Topete R, Gonzalez-Barranco J, et al. Dermatoses in 156 obese adults. Obesity Res 1999;7:299–302.
- Haxthausen H. Keratoderma climactericum. Br J Dermatol 1934;46:161–7.
- Deschamps P, Leroy D, Pedailles S, et al. Keratoderma climactericum (Haxthausen's disease): clinical signs, laboratory findings and etretinate treatment in 10 patients. Dermatologica 1986;172:258–62.
- Wachtel TJ. Plantar and palmar hyperkeratosis in young castrated women. Int J Dermatol 1981;20:270–1.
- Laurent R, Prost O, Nicollier M, et al. Composite keratohyaline granules in palmoplantar keratoderma: an ultrastructural study. Arch Dermatol Res 1985;277:384–94.
- Ishida-Yamamoto A, Iizuka A, Eady RAJ. Filaggrin immunoreactive composite keratohyalin granules specific to acrosyringia and related tumours. Acta Derm Venereol (Stockh) 1994;74:37–42.
- Zultak M, Bedeaux C, Blanc D. Keratoderma climactericum treatment with topical estrogen. Dermatologica 1988;176:151–2.
- Ashinoff R, Werth VP, Franks AG. Resistant discoid lupus erythematosus of palms and soles: successful treatment with azathioprine. J Am Acad Dermatol 1988;19:961–5.
- Buck DC, Dodd HJ, Sarkany I. Hypertrophic lupus erythematosus. Br J Dermatol 1988;119:72–4.
- Grossberg EB, Scherschun L, Fivenson DP. Ulcerating plantar keratoderma in association with systemic lupus erythematosus. Lupus 2001;10:650–2.
- Ohtake N, Sou K, Tsukamoto K, et al. Diffuse palmoplantar keratoderma associated with acrocyanosis and livedo reticularis: two sporadic cases. Acta Derm Venereol (Stockh) 1995;75.
- Bolling MC, Mekkes JR, Goldschmidt WF, et al. Acquired palmoplantar keratoderma and immunobullous disease associated with antibodies to desmocollin 3. Br J Dermatol 2007;157:168–73.
- Munro CS, Cox NH, Marks JM, et al. Lichen nitidus presenting as palmo-plantar hyperkeratosis and nail dystrophy. Clin Exp Dermatol 1993;18:381–3.
- Caumes E, Janier M, Janssen F, et al. Syphilis acquise au cours de l'infection par le virus de l'immunodeficience humaine. Six cas. Presse Med 1990;19:369–71.
- Tan OT, Sarkany I. Severe palmar keratoderma with myxoedema. Clin Exp Dermatol 1977;2:287–8.
- Hodak E, David M, Feuerman EJ. Palmoplantar keratoderma in association with myxoedema. Acta Derm Venereol (Stockh) 1986;66:243–5.
- Bouras M, Hali F, Khadir K, et al. [Palmoplantar keratoderma: a rare manifestation of myxoedema.] Ann Dermatol Venereol 2014;141:39–42.
- Zouboulis CC, Biczo S, Gollnick H, et al. Elephantiasis nostras verrucosa: beneficial effect of oral etretinate therapy. Br J Dermatol 1992;127:411–16.
- Richards RN. Verrucous and elephantoid lymphedema: morphologic spectrum and terminology. Int J Dermatol 1981;20:177–87.
- Mortimer PS. Lymphatics. In: Champion RH, Pye RJ, eds. Recent Advances in Dermatology. Edinburgh: Churchill Livingstone, 1990:175–92.
- Murata Y, Kumano K, Tani M, et al. Acquired diffuse keratoderma of the palms and soles with bronchial carcinoma: report of a case and review of the literature. Arch Dermatol 1988;124:497–8.
- Khanna SK, Agnone FA, Leibowitz AI, et al. Non-familial diffuse palmoplantar keratoderma associated with bronchial carcinoma. J Am Acad Dermatol 1993;28:295–7.
- Hillion B, Le-Bozec P, Moulonguet-Michaut I, et al. Hyperkeratose palmo-plantaire filiforme et cancer du sein. Ann Dermatol Vénéréol 1990;117:834–6.
- Fegueux S, Bilet S, Crickx B, et al. Hyperkeratose palmo-plantaire filiforme et cancer recto-sigmoidien. Ann Dermatol Vénéréol 1988;115:1145–6.
- Beylot C, Taieb A, Bioulac P, et al. Hyperkeratose palmo-plantaire filiforme et neoplasie viscerale. Ann Dermatol Vénéréol 1982;109:747–8.
- Kim J, Foster R, Lam M, Kumarasinghe SP. Mycosis fungoides: An important differential diagnosis for acquired palmoplantar keratoderma. Australas J Dermatol 2014;56:49–51.
- Junge J, Moll I. Multiple palmoplantarkeratosen, basaliome und porokarzinome nach Arsen-therapie. Hautarzt 1995;46:198–201.
- Jackson R, Grainge JW. Arsenic and cancer. Can Med Assoc J 1975;113:396–401.
- Dobson RL, Young MR, Pinto JS. Palmar keratoses and cancer. Arch Dermatol 1965;92:553–6.
- Cuzick J, Harris R, Mortimer PS. Palmar keratoses and cancers of the bladder and lung. Lancet 1984;i:530–3.
- Cartwright RA, Glashan RW. Palmar keratoses and bladder cancer. Lancet 1984;i:563.
- Rhodes EL. Palmar and plantar seed keratoses and internal malignancy. Br J Dermatol 1970;82:361–3.
- Woodside JR, Dobson RL. Histopathology of palmar keratoses associated with cancer. Arch Dermatol 1968;98:648–51.
- Jucglà A, Sais G, Navarro M, et al. Palmoplantar keratoderma secondary to chronic acral erythema due to tegafur. Arch Dermatol 1995;131:364–5.
- Duvic M, Reisman M, Finley V, et al. Glucan-induced keratoderma in acquired immunodeficiency syndrome. Arch Dermatol 1987;123:751–6.
- Labelle A, Lapierre YD. Keratodermia: side-effects of lithium. J Clin Psychopharmacol 1991;11:149–50.
- Poskitt LB, Duffill MB, Rademaker M. Chloracne, palmoplantar keratoderma and localized scleroderma in a weed sprayer. Clin Exp Dermatol 1994;19:264–7.
- Geusau A, Jurecka W, Nahavandi H, et al. Punctate keratoderma-like lesions on the palms and soles in a patient with chloracne: a new clinical manifestation of dioxin intoxication? Br J Dermatol 2000;143:1067–71.
- Sass V, Grosshans E, Simonart JM. Chronic arsenism: criminal poisoning or drug intoxication? Report of two cases. Dermatology 1993;186:303–5.
- Jucglà A, Sais G, Navarro M, et al. Palmoplantar keratoderma secondary to chronic acral erythema due to tegafur. Arch Dermatol 1995;131:364–5.
- Do JE, Kim YC. Capecitabine-induced diffuse palmoplantar keratoderma: is it a sequential event of hand–foot syndrome? Clin Exp Dermatol 2007;32:519–21.
- Lountzis NI, Maroon MS. Sorafenib-induced palmoplantar hyperkeratosis. J Drugs Dermatol 2008;7:588–9.
Keratolytic winter erythema
- Findlay GH, Nurse GT, Heyl T, et al. Keratolytic winter erythema or Oudtshoorn disease. S Afr Med J 1977;52:871–4.
- Findlay GH, Morrison JGL. Erythrokeratolysis hiemalis: keratolytic winter erythema or ‘Oudtshoorn skin’. Br J Dermatol 1978;98:491–5.
- Starfield M, Hennies HC, Jung M, et al. Localization of gene causing keratolytic winter erythema to chromosome 8p22-p23 and evidence for a founder effect in South African africaans speakers. Am J Hum Genet 1997;61:370–8.
- Hobbs A, Aron S, Hartshorne S, Hull PR, et al. Exclusion of CTSB and FDFT1 as positional and functional candidate genes for keratolytic winter erythema (KWE). J Dermatol Sci 2012;65:58–62.
- Hull PR, Hobbs A, Aron S, Ramsay M. The elusive gene for keratolytic winter erythema. S Afr Med J 2013;103:961–5.
- Huntington MK, Jassim AD. Genetic heterogeneity in keratolytic winter erythema (Oudtshoorn skin disease). Arch Dermatol 2006;142:1073–4.
- France DM, Xerri S. Erythrokeratolysis hiemalis: Oudtshoorn skin. Br J Dermatol 1984;113 (Suppl. 29):46.
- Degiovanni CV, Farrant PB, Howell S, et al. Keratolytic winter erythema with facial involvement: a novel presentation. Clin Exp Dermatol 2009;34:206–8.
- Holme SA, Whatley SD, Roberts AG, et al. Seasonal palmar keratoderma in erythropoietic protoporphyria indicates autosomal recessive inheritance. J Invest Dermatol 2009;129:599–605.
- Amin AN, DeGiovanni CV, Farrant PB, et al. Photodynamic therapy for the treatment of keratolytic winter erythema. Clin Exp Dermatol 2011;36:668–9.
Miscellaneous disorders of keratinization
Porokeratoses
- Sertznig P, von Felbert V, Megahed M. Porokeratosis: present concepts. J Eur Acad Dermatol Venereol 2012;26:404–12.
- Kanitakis J, Euvrard S, Faure M, Claudy A. Porokeratosis and immunosuppression. Eur J Dermatol 1998;8:459–65.
- Schamroth JM, Zlotogorski A, Gilead L. Porokeratosis of Mibelli. Acta Derm Venereol 1997;77:207–13.
- Sasson M, Krain AD. Porokeratosis and cutaneous malignancy: a review. Dermatol Surg 1996;22:339–42.
- Xia JH, Yang YF, Deng H, et al. Identification of a locus for disseminated superficial actinic porokeratosis at chromosome 12q23.2–24.1. J Invest Dermatol 2000;114:1071–4.
- Xia K, Deng H, Xia JH, et al. A novel locus (DSAP2) for disseminated superficial actinic porokeratosis maps to chromosome 15q25.1–26.1. Br J Dermatol 2000;147:650–4.
- Wei S, Yang S, Lin D, Li M, et al. A novel locus for disseminated superficial porokeratosis maps to chromosome 18p11.3. J Invest Dermatol 2004;123:872–5.
- Zhang ZH, Huang W, Niu ZM, et al. Two closely linked variations in actin cytoskeleton pathway in a Chinese pedigree with disseminated superficial actinic porokeratosis. J Am Acad Dermatol 2005;52:972–6.
- Luan J, Niu Z, Zhang J, Crosby ME, et al. A novel locus for disseminated superficial actinic porokeratosis maps to chromosome 16q24.1–24.3. Hum Genet 2011;129:329–34.
- Zhang Z, Niu Z, Wang Y, Huang W. Association of SSH1 mutations with disseminated superficial actinic porokeratosis in Chinese pedigrees (response to Frank, et al.). Hum Mutat 2007;28:1243–4.
- Zhang ZH, Niu ZM, Yuan WT, et al. A mutation in SART3 gene in a Chinese pedigree with disseminated superficial actinic porokeratosis. Br J Dermatol 2005;152:658–63.
- Zhang ZH, Niu ZM, Huang W, et al. Loss of heterozygosity analysis on chromosome 12q in disseminated superficial actinic porokeratosis. J Invest Dermatol 2007;127:482–5.
- Frank J, van Steensel MA, Van Geel M. Lack of SSH1 mutations in Dutch patients with disseminated superficial actinic porokeratosis: is there really an association? Hum Mutat 2007;28:1241–2.
- Zhang SQ, Jiang T, Li M, et al. Exome sequencing identifies MVK mutations in disseminated superficial actinic porokeratosis. Nat Genet 2012;44:1156–60.
- Zeng K, Zhang QG, Li L, Duan Y, Liang YH. Splicing mutation in MVK is a cause of porokeratosis of Mibelli. Arch Dermatol Res 2014;;306:749–55.
- Lu WS, Zheng XD, Yao XH, et al. Detection of a novel missense mutation in the mevalonate kinase gene in one Chinese family with DSAP. Int J Clin Exp Pathol 2014;15;7:728–32.
- Dai J, Chen M, Fu X, Yu et al. Mutation analysis of the MVK gene in Chinese patients with disseminated superficial actinic porokeratosis. J Dermatol Sci 2013;72:320–2.
- Reed RJ, Leone P. Porokeratosis: a mutant clonal keratosis of the epidermis. I. Histogenesis. Arch Dermatol 1970;101:340–7.
- Otsuka F, Shima A, Ishibashi Y. Porokeratosis has neoplastic clones in the epidermis. Microfluorometric analysis of DNA content of epidermal cell nuclei. J Invest Dermatol 1989;92:231S–3S.
- Otsuka F, Nashiro K, Kobayashi K, Ishibashi Y. Chromosome abnormalities of porokeratosis-cultured epidermal keratinocytes: comparison with those of cultured dermal fibroblasts. Cancer Genet Cytogen 1991;56:163–9.
- Magee JW, McCalmont TH, LeBoit PE. Overexpression of p53 tumor suppressor protein in porokeratosis. Arch Dermatol 1994;130:187–90.
- Urano Y, Sasaki S, Ninomiya Y, et al. Immunohistochemical detection of p53 tumor suppressor protein in porokeratosis. J Dermatol 1996;23:365–8.
- Nelson C, Cowper S, Morgan M. p53, mdm-2, and p21 waf-1 in the porokeratoses. Am J Dermatopathol 1999;21:420–5.
- D'Errico M, Teson M, Calcagnile A, et al. Characterization of the ultraviolet B and X-ray response of primary cultured epidermal cells from patients with disseminated superficial actinic porokeratosis. Br J Dermatol 2004;150:47–55.
- Scappaticci S, Lambiase S, Orecchia G, et al. Clonal chromosome abnormalities with preferential involvement of chromosome 3 in patients with porokeratosis of Mibelli. Cancer Genet Cytogenet 1989;43:89–94.
- Mizukawa Y, Shiohara T. Onset of porokeratosis of Mibelli in organ transplant recipients: lack of a search for transmissible agents in these patients. J Am Acad Dermatol 2001;44:143–4.
- Esser AC, Pittelkow MR, Randle HW. Human papillomavirus isolated from transplant-associated porokeratoses of mibelli responsive to topical 5% imiquimod cream. Dermatol Surg 2006;32:858–61.
- Ibbotson SH. Disseminated superficial porokeratosis: what is the association with ultraviolet radiation? Clin Exp Dermatol 1996;21:48–50.
- Allen AL, Glaser DA. Disseminated superficial actinic porokeratosis associated with topical PUVA. J Am Acad Dermatol 2000;43:720–2.
- Shumack SP, Commens CA. Disseminated superficial actinic porokeratosis: a clinical study. J Am Acad Dermatol 1989;20:1015–22.
- Bencini PL, Tarantino A, Grimalt R. Porokeratosis and immunosuppression. Br J Dermatol 1995;132:74–8.
- Patrizi A, Passarini B, Minghetti G, et al. Porokeratosis palmaris et plantaris disseminata: an unusual clinical presentation. J Am Acad Dermatol 1989;21:415–18.
- Judge MR, Michaels M, Sams VR, et al. Disseminated porokeratosis in an infant with craniosynostosis. Br J Dermatol 1996;123:249–54.
- Allegra F. The man behind the eponym: Vittorio Mibelli and the tale of porokeratosis. Am J Dermatopathol 1986;11:79–83.
- Virgili A, Strumia R. Annular hyperkeratosis: porokeratosis of Mibelli. Arch Dermatol 1986;122:586–7.
- Bacharach-Buhles M, Weindorf N, Altmeyer P. Porokeratosis Mibelli gigantea. Hautarzt 1990;41:633–5.
- Otsuka F, Someya T, Ishibashi Y. Porokeratosis and malignant skin tumors. J Cancer Res Clin Oncol 1991;117:55–60.
- Lucker GP, Steiljen PM. The coexistence of linear and giant porokeratosis associated with Bowen's disease. Dermatology 1994;189:78–80.
- Friedman SJ, Herman PS, Pittelkow MR. Punctate porokeratotic keratoderma. Arch Dermatol 1988;124:1678–82.
- Neumann RA, Knobler RM, Gebhart W. Unusual presentation of porokeratosis palmaris, plantaris et disseminata. J Am Acad Dermatol 1989;21:1131–3.
- Karadaglic DL, Berger S, Jankovic D, et al. Zosteriform porokeratosis of Mibelli. Int J Dermatol 1988;27:589–90.
- Veraldi S, Bocor M, Gasparini G. Zosteriform porokeratosis: a report of two cases. Cutis 1989;44:216–19.
- Lozinski AZ, Fisher BK, Walter JB, et al. Metastatic squamous cell carcinoma in linear porokeratosis of Mibelli. J Am Acad Dermatol 1987;16:448–51.
- Dover JS, Miller JA, Levene GM. Linear porokeratosis of Mibelli and DSAP. Clin Exp Dermatol 1986;11:79–83.
- Gautam RK, Bedi GK, Sehgal VN. Simultaneous occurrence of disseminated superficial actinic porokeratosis (DSAP), linear and punctate porokeratosis. Int J Dermatol 1995;34:71–2.
- Boente MC, Lopez-Baro AM, Frontini MV, Asial RA. Linear porokeratosis associated with disseminated superficial actinic porokeratosis: a new example of type II segmental involvement. Pediatr Dermatol 2003;20:514–18.
- Tallon B, Blumental G, Bhawan J. Porokeratosis ptychotropica: a lesser known variant. Clin Exp Dermatol 2009;34:895–7.
- Lucker GP, Happle R, Steijlen PM. An unusual case of porokeratosis involving the natal cleft: porokeratosis ptychotropical? Br J Dermatol 1995;132:150–1.
- Yeo J, Winhoven S, Tallon B. Porokeratosis ptychotropica: a rare and evolving variant of porokeratosis. J Cutan Pathol 2013;40:1042–7.
- Ginarte M, Leon A, Toribio J. Disseminated superficial porokeratosis with amyloid deposits. Eur J Dermatol 2005;15:298–300.
- Shumack S, Commens C, Kossard S. Disseminated superficial actinic porokeratosis: a histological review of 61 cases with particular reference to lymphocytic inflammation. Am J Dermatopathol 1991;13:26–31.
- Jacyk W, Esplin L. Hyperkeratotic form of porokeratosis of Mibelli. Int J Dermatol 1993;32:902–3.
- Barnett JH. Linear porokeratosis: treatment with the carbon dioxide laser. J Am Acad Dermatol 1986;14:902–4.
- Alster TS, Nanni CA. Successful treatment of porokeratosis with 585 nm pulsed dye laser irradiation. Cutis 1999;63:265–6.
- Cavicchini S, Tourlaki A. Successful treatment of disseminated superficial actinic porokeratosis with methyl aminolevulinate-photodynamic therapy. J Dermatolog Treat 2006;17:190–1.
- Bohm M, Luger TA, Bonsmann G. Disseminated superficial actinic porokeratosis: treatment with topical tacalcitol. J Am Acad Dermatol 1999;40:479–80.
- Hubler WR, Michaelson JD, Knox JM. Linear porokeratosis. Cutis 1974;14:61–4.
- McDonald SG, Peterka ES. Porokeratosis (Mibelli): treatment with topical 5-fluorouracil. J Am Acad Dermatol 1983;8:107–10.
- Agarwal S, Berth-Jones J. Porokeratosis of Mibelli: successful treatment with 5% imiquimod cream. Br J Dermatol 2002;146:338–9.
- Danno K, Yamamoto M, Yokoo T. Etretinate treatment in disseminated porokeratosis. J Dermatol 1988;15:440–4.
- Vlachou C, Kanelleas A, Martin-Clavijo A, Berth-Jones J. Treatment of disseminated superficial actinic porokeratosis with topical diclofenac gel: a case series. J Eur Acad Dermatol Venereol 2008;22:1343–5.
- Scheiba N, Enk A, Proske S, et al. Porokeratosis ptychotropica: successful treatment with the dermatome. Dermatol Surg 2010;36:257–60.
Perforating keratotic disorders
- Seghal VN, Jain S, Thappa DM, et al. Perforating dermatoses: a review and report of four cases. J Dermatol 1993;20:329–40.
- Patterson JW. The perforating disorders. J Am Acad Dermatol 1984;10:561–81.
- Rapini RP, Hebert AS, Drucker CR. Acquired perforating dermatosis: evidence for combined transepidermal elimination of both collagen and elastic fibers. Arch Dermatol 1989;125:1074–8.
- Schreml S, Hafner C, Eder F, et al. Kyrle disease and acquired perforating collagenosis secondary to chronic renal failure and diabetes mellitus. Case Rep Dermatol 2011;3:209–11.
- Tessler HH, Apple DJ, Goldberg MF. Ocular findings in a kindred with Kyrle disease. Hyperkeratosis follicularis et parafollicularis in cutem penetrans. Arch Ophthalmol 1973;146:101–6.
- Shivakumar V, Okade R, Rajkumar V, Prathima KM. Familial Kyrle's disease: a case report. Int J Dermatol 2007;46:770–1.
- De Mare S, Koopman RJJ, Steiljen PM. Acquired perforating dermatosis (Kyrle's disease). Br J Dermatol 1993;129:211.
- Morton CA, Henderson IS, Jones MC, et al. Acquired perforating dermatosis in a British dialysis population. Br J Dermatol 1996;135:671–7.
- Saray Y, Seckin D, Bilezikci B. Acquired perforating dermatosis: clinicopathological features in twenty-two cases. J Eur Acad Dermatol Venereol 2006;20:679–88.
- Gilaberte Y, Coscojuela C, Vazquez C, et al. Perforating folliculitis associated with tumour necrosis factor-alpha inhibitors administered for rheumatoid arthritis. Br J Dermatol 2007;156:368–71.
- Kyrle J. Uber einen ungewöhnlichen Fall von universeller follikularer und parafollikularer Hyperkeratose (Hyperkeratosis follicularis et parafollicularis in Cutem penetrans). Arch Dermatol Syphilol 1916;123:466–93.
- Carter VH, Constantine VS. Kyrle's disease. I. Clinical findings in five cases and review of literature. Arch Dermatol 1968;97:624–32.
- Constantine VS, Carter VH. Kyrle's disease. II. Histopathologic findings in five cases and review of the literature. Arch Dermatol 1968;97:633–9.
- Faver IR, Daoud MS, Su WP. Acquired reactive perforating collagenosis. Report of six cases and review of the literature. J Am Acad Dermatol 1994;30:575–80.
- Bayramgurler D, Apaydin R, Cetiner D, Zincirci C. Narrow-band ultraviolet B phototherapy for acquired perforating dermatosis. Australas J Dermatol 2003;44:76–8.
- Gambichler T, Altmeyer P, Kreuter A. Treatment of acquired perforating dermatosis with narrowband ultraviolet B. J Am Acad Dermatol 2005;52:363–4.
- Kruger K, Tebbe B, Krengel S, et al. Acquired reactive perforating dermatosis: successful treatment of two cases with allopurinol. Hautarzt 1999;50:115–20.
- Brinkmeier T, Schaller J, Herbst RA, Frosch PJ. Successful treatment of acquired reactive perforating collagenosis with doxycycline. Acta Derm Venereol 2002;82:393–5.
- Lucke TW, Fallowfield ME, Evans A, et al. Transepidermal elimination of urate-like crystals: a new perforating disorder? Br J Dermatol 1999;141:310–14.
- Kossard S, Scurry J, Killingsworth M. Necrotizing infundibular crystalline folliculitis. Br J Dermatol 2001;145:165–8.
- Denisjuk N, Hilty N, Pfaltz M, Kempf W. Necrotizing infundibular crystalline folliculitis: a clinicopathological study. J Am Acad Dermatol 2012;66:823–6.
- Mehregan AH, Schwartz OD, Livingood CS. Reactive perforating collagenosis. Arch Dermatol 1967;96:277–82.
Multiple minute digitate hyperkeratoses
- Folster-Holst R, Christophers E. Filiforme keratose. Hautarzt 1994;45:484–8.
- Frenk E, Mevorah B, Leu F. Disseminated spiked hyperkeratosis: an unusual discrete non-follicular keratinization disorder. Arch Dermatol 1981;117:412–14.
- Judd LE, Wood KP. Disseminated spiked hyperkeratosis. Int J Dermatol 1993;32:446–7.
- Carmichael AJ, Tan CY. Digitate keratoses: a complication of etretinate used in the treatment of disseminated superficial actinic porokeratosis. Clin Exp Dermatol 1990;15:370–1.
- Ramselaar C, Toonstra J. Multiple minute digitate hyperkeratoses report of two cases with an updated review and proposal for a new classification. Eur J Dermatol 1999;9:460–5.
- Caccetta TP, Dessauvagie B, McCallum D, et al. Multiple minute digitate hyperkeratosis: a proposed algorithm for the digitate keratoses. J Am Acad Dermatol 2012;67:49–55.
- Goldstein N. Multiple minute digitate hyperkeratoses. Arch Dermatol 1967;96:692–3.
- Balus L, Donati P, Amantea A, et al. Multiple minute digitate hyperkeratosis. J Am Acad Dermatol 1988;18:431–6.
- Izakovic J, Stanislaw A, Buchner M, et al. Haarartige Hyperkeratosen bei einem Nierentransplantierten: eine neue Cyclosporin-nebenwirkung. Hautarzt 1995;46:841–6.
- Paul C, Fermand J-P, Flageul B, et al. Hyperkeratotic spicules and monoclonal gammopathy. J Am Acad Dermatol 1995;33:346–51.
- Lukitsch O, Gebhardt K-P, Kovary PM. Follicular hyperkeratosis and cryocrystalglobulinemia syndrome: occurrence in a patient with multiple myeloma. Arch Dermatol 1985;121:795–8.
- Wilkinson SM, Wilkinson N, Chalmers RJ. Multiple minute digitate keratoses: a transient, sporadic variant. J Am Acad Dermatol 1994;31:802–3.
- Pimentel CL, Puig L, García-Muret MP, et al. Multiple minute digitate hyperkeratosis. J Eur Acad Dermatol Venereol 2002;16:422–4.
- Ferandiz C, Savall R, Baumann E. Hiperqueratosis multiple minuta y digitata (un sintoma paraneoplasico?). Med Cutan Ibero Lat Am 1978;6:279–83.
- Miralles ES, Nunez M, De La Heras ME, et al. Pityriasis rubra pilaris and human immunodeficiency virus infection. Br J Dermatol 1995;133:990–3.
- Schulz-Kiesow M, Metze D, Traupe H. Hystrix-like keratosis with nail and joint-involvement: a new genodermatosis? Dermatology 1996;192:321–4.
- McGovern TW, Gentry RH. Spiny keratoderma: case report, classification, and treatment of music box spine dermatoses. Cutis 1994;54:389–94.
- Torres G, Behshad R, Han A, et al. ‘I forgot to shave my hands’: a case of spiny keratoderma. J Am Acad Dermatol 2008;58:344–8.
- Feldmann R, Harms M. Multiple filiform hyperkeratosen. Hautarzt 1993;44:658–61.
- Pujol RM, Perez-Losada E, Matias-Guiu X, et al. Postirradiation multiple minute digitate porokeratosis: review. J Cutan Med Surg 2001;5:126–30.
Flegel disease
- Flegel H. Hyperkeratosis lenticularis perstans. Hautarzt 1958;9:362–4.
- Bean SF. Hyperkeratosis lenticularis perstans: a clinical, histopathological and genetic study. Arch Derm 1969;99:705–9.
- Metze D, Lübke D, Luger T. Hyperkeratosis lenticularis perstans (Flegel's disease) – a complex disorder of epidermal differentiation with good response to a synthetic vitamin D3 derivate. Hautarzt 2000;51:31–5.
- Zimmermann R. 40 years to Flegel's disease (hyperkeratosis lenticularis perstans). Hautarzt 2001;52:231–5.
- Price ML, Wilson Jones E, MacDonald DM. A clinicopathological study of Flegel's disease (hyperkeratosis lenticularis perstans). Br J Dermatol 1987;116:681–91.
- Ando K, Hattori H, Yamauchi Y. Histopathological differences between early and old lesions of hyperkeratosis Lenticularis Perstans (Flegel's disease). Am J Dermatopathol 2006;28:122–6.
- Sterneberg-Vos H, van Marion AM, Frank J, Poblete-Gutierrez P. Hyperkeratosis lenticularis perstans (Flegel's disease) – successful treatment with topical corticosteroids. Int J Dermatol 2008;47:Suppl. 1:38–41.
Circumscribed palmoplantar hypokeratosis
- Ishiko A, Dekio I, Fujimoto A, et al. Abnormal keratin expression in circumscribed palmar hypokeratosis. J Am Acad Dermatol 2007;57:285–91
- Böer A, Falk TM. Circumscribed palmar hypokeratosis induced by papilloma virus type 4. J Am Acad Dermatol 2006;54:908–9
- Pérez A, Rütten A, Gold R, et al. Circumscribed palmar or plantar hypokeratosis: a distinctive epidermal malformation of the palms or soles. J Am Acad Dermatol 2002;47:21–7.
- Urbina F, Pérez A, Requena L, et al. Circumscribed palmar or plantar hypokeratosis 10 years after the first description: what is known and the issues under discussion. Acta Dermosifiliogr 2013;731:262–7.
- Santamarina-Albertos A, Noguera-Morel L, Feito-Rodríguez M, et al. Congenital circumscribed acral hypokeratosis. Pediatr Dermatol 2013;30:e102–3.
- Lee SE, Kim YC, Kim SC. Circumscribed palmar or plantar hypokeratosis: report of a Korean case published work review. J Dermatol 2006;33:403–5.
- Kanitakis J, Lora V, Balme B, Roby J. Premalignant circumscribed palmar hypokeratosis: a new form of circumscribed palmar hypokeratosis? Case report and literature review. Dermatology 2010;220:143–6.
- Butler LK, Kiracofe EA, Marks VJ. Circumscribed acral hypokeratosis: successful treatment of a potentially premalignant condition. Arch Dermatol 2012;148:1427–8.
- Urbina F, Misad C, Gonzalez S. Circumscribed palmar hypokeratosis: clinical evolution and ultrastructural study after prolonged treatment with topical calcipotriol. J Eur Acad Dermatol Venereol 2005;19:491–4.
- Boffa MJ, Degaetano JS. Circumscribed palmar hypokeratosis: successful treatment with cryotherapy. J Eur Acad Dermatol Venereol 2007;21:420–1.
- Benoit S, Seitz CS, Hamm H, et al. Circumscribed palmar hypokeratosis: partial remission by photodynamic therapy. Br J Dermatol 2007;157:804–6.
- Wilk M, Zelger BG, Zelger B. Circumscribed palmar hypokeratosis: Successful treatment with fluorouracil cream. Dermatol Ther 2011;1:11–14.
Waxy keratoses of childhood
- Gönül M, Kiliç A, Gül U, et al. A case of waxy keratoses of childhood. Dermatology 2008;217(2):143.
- Coleman R, Malone M, Handfield-Jones S, et al. Waxy keratoses of childhood. Clin Exp Dermatol 1994;19:173–6.
- Morison WL, Kerker BJ, Tunnessen WW, et al. Disseminated hypopigmented keratoses. Arch Dermatol 1991;127:848–50.
- Tan C, Zhu WY. An adult case of waxy keratosis. Int J Dermatol 2013;52:1606–8.
- Mehrabi D, Thomas JE, Selim MA, Prose NS. Waxy keratoses of childhood in a segmental distribution. Pediatr Dermatol 2001;18:415–16.
- Happle R, Fleiner J, Loskamp U. Kerinokeratosis papulosa with a type 2 segmental manifestation. J Am Acad Dermatol 2004;50:84–5.
- Jacyk WK, Visser AJ. Leukodermic macules in keratosis follicularis (Darier's disease). Int J Dermatol 1992;31:715–17.
- Jacyk K, Smith A. Mosaic acral keratosis. Clin Exp Dermatol 1990;15:361–2.
Hyperkeratosis of the nipple
- Higgins HW, Jenkins J, Horn TD, et al. Pregnancy-associated hyperkeratosis of the nipple: a report of 25 cases. JAMA Dermatol 2013;149:722–6.
- Baykal C, Büyükbabani N, Kavak A, et al. Nevoid hyperkeratosis of the nipple and areola: A distinct entity. J Am Acad Dermatol 2002;46:414–18.
- Ahn SK, Chung J, Lee WS, et al. Hyperkeratosis of the nipple and areola simultaneously developing with cutaneous T-cell lymphoma. J Am Acad Dermatol 1995;32:124–5.
- Yang YH, Zhang RZ, Kang DH, et al. Three paraneoplastic signs in the same patient with gastric adenocarcinoma. Dermatol Online J 2013;19:18966.
- Okan G, Baykal C. Nevoid hyperkeratosis of the nipple and areola: Treatment with topical retinoic acid. J Eur Acad Dermatol Venereol 1999;13:218–20.
- Bayramgürler D, Bilen N, Apaydin R, et al. Nevoid hyperkeratosis of the nipple and areola: Treatment of two patients with topical calcipotriol. J Am Acad Dermatol 2002;46:131–3.
- Ghanadan A, Balighi K, Khezri S, et al. Nevoid hyperkeratosis of the nipple and/or areola: treatment with topical steroid. Indian J Dermatol 2013;58:408.
- Lee HW, Lee MW, Choi JH, et al. Unilateral nevoid hyperkeratosis of the nipple and areola: Excellent response to cryotherapy. Dermatol Surg 2005;31:611–12.
- Ozyazgan I, Kontaş O, Ferahbaş A. Treatment of nevoid hyperkeratosis of the nipple and areola using a radiofrequency surgical unit. Dermatol Surg 2005;31:703–5.