CHAPTER 67
Ectodermal Dysplasias
Peter Itin
Department of Dermatology, University Hospital Basle, Basle, Switzerland
Ectodermal dysplasias
Definition and nomenclature
Ectodermal dysplasias, a large group of heterogeneous heritable conditions, are characterized by congenital defects in two or more ectodermal structures, one of which at least involves the hair (trichodysplasia), teeth (dental defects), nails (onychodysplasia) or sweat glands (dyshidrosis) [1]. Mesodermal and occasionally endodermal disturbances of development may coexist. The molecular basis of 80 out of approximately 220 known ectodermal dysplasias has been deciphered.
Traditional classification schemes based on purely clinical criteria lack practicality as, when applied strictly, they encompass many groups of diseases (e.g. keratodermas with additional skin or hair alterations or ichthyoses with associated features) [2], leading to endless lists of disorders of little use to the clinician. Recent evidence implicates genetic defects in different key pathways orchestrating ectodermal organogenesis. It is likely that with the increasing number of disorders of known aetiology, a better understanding of the pathogenesis of ectodermal dysplasia will lead to their reclassification based on a combination of clinical and molecular defining features.
Introduction and general description
Ectodermal dysplasias form a complex and highly diverse group of heritable disorders. Although several syndromes have very specific features, many of them have common clinical characteristics. By some authors’ estimations, the broader definition of ectodermal dysplasias encompasses some 200 to 220 individual conditions. Any approach to summarizing current knowledge about this group of conditions presents several challenges [3]. Firstly, and most importantly, how is an ectodermal dysplasia defined, and which distinct entities should be labelled under this broad term? The first well-documented patients with what we now call ectodermal dysplasia were described by Danz in 1793 [4]. He reported on two Jewish boys with congenital absence of hair and teeth. In 1875 Charles Darwin reported on a Hindu family with hypodontia and malformed teeth: ‘The men thus affected have very little hair on the body, and become bald early in life. They also suffer much during hot weather from excessive dryness of the skin’. This family would now be recognized as having X-linked hypohidrotic ectodermal dysplasia but the term ‘ectodermal dysplasia’ did not appear until 1929 [5]. Prior to this report, a small series of cases with hypotrichosis, hypodontia, onychodysplasia and anhidrosis had been described under various names such as ‘dystrophy of hair and nails’, ‘imperfect development of skin, hair and teeth’ and ‘congenital ectodermal defect’. The designation outlined by Weech specified three essential aspects of ectodermal dysplasias: (i) most of the disturbances must affect tissues of ectodermal origin; (ii) these disturbances must be developmental; and (iii) heredity plays a causal role. Weech had in mind the X-linked anhidrotic form of ectodermal dysplasia (Christ–Siemens–Touraine syndrome (CST) or hypohidrotic ectodermal dysplasia (HED); MIM: 305100) in males but noted that it had also been reported in females. He also noted that this pattern of involvement was occasionally inherited as a non-sex-linked trait [5]. For some authors and clinicians, the term ectodermal dysplasia is still used specifically with reference to the CST syndrome and the autosomal dominant and recessive forms of HED. As more clinical reports of patients with similar but subtly distinct patterns of anomalies were recorded, the term ‘ectodermal dysplasia’ became extended to include many different genetic entities. In an attempt to encapsulate this heterogeneity and diversity of symptoms seen, Touraine suggested the expression ‘ectodermal polydysplasia’. Attempts at more formal classification soon followed; initially conditions were classified as hidrotic or anhidrotic, but this simple classification failed to reflect the complexity of nail, hair and dental anomalies associated with the various forms of ectodermal dysplasias.
Currently, the most widely accepted and used definition of the ectodermal dysplasias is of a group of inherited disorders that share in common developmental abnormalities of two or more of the following: hair (Figure 67.1), teeth (Figure 67.2), nails (Figure 67.3), sweat glands and other ectodermal structures. Other organs derived from embryonic ectoderm include the mammary gland, central nervous system, external ear, melanocytes, cornea, conjunctiva, lacrimal gland and lacrimal duct. There are advantages and disadvantages of this approach to definition. One definite benefit is that the problems encountered by many patients and families are similar regardless of the specific subtype of ectodermal dysplasia; parents and children can benefit by being part of large support networks, exemplified by the Ectodermal Dysplasia Society (UK based; http://www.ectodermaldysplasia.org, last accessed August 2014) and the National Foundation for Ectodermal Dysplasias (US based; http://www.nfed.org, last accessed August 2014). Although the broader definition has the benefit of inclusivity, many conditions encompassed by this broad definition are not usually considered as primarily ectodermal dysplasias. For example, inherited conditions as diverse as incontinentia pigmenti, dyskeratosis congenita, trichothiodystrophies, cardiofaciocutaneous syndrome, pachyonychia congenita and Goltz syndrome by this definition are ectodermal dysplasias, but common practice has been to consider many of these as separate entities. More than 220 different conditions come under the umbrella term of ‘ectodermal dysplasia’ if the widest definition of ‘an inherited disorder involving two or more of the following ectodermal appendages: teeth, hair, eccrine sweat glands’ is used [6].
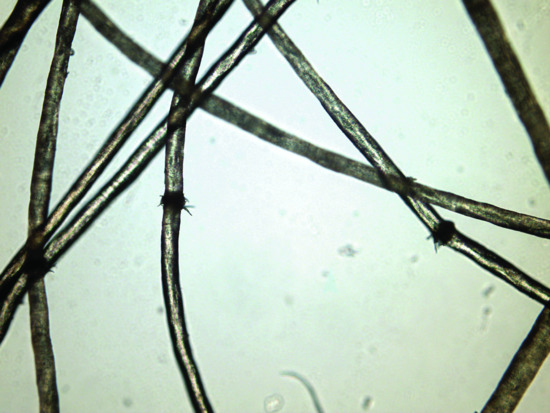
Figure 67.1 Hairshaft abnormalities such as trichorhexis nodosa in ectodermal dysplasia.
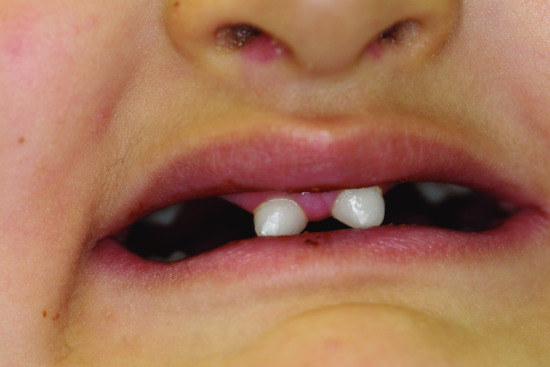
Figure 67.2 Hypodontia and dental malformations in ectodermal dysplasia.
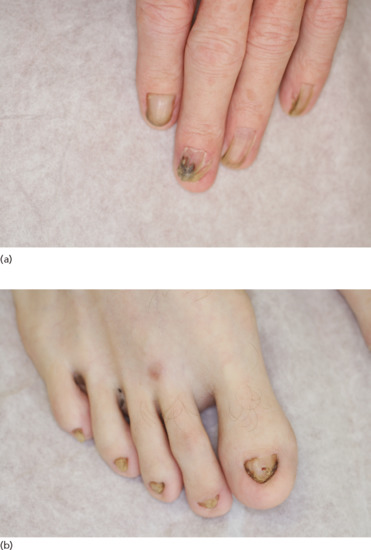
Figure 67.3 Nail dysplasia of the fingers (a) and toes (b) can be a marker of ectodermal dysplasia.
Classification: clinical approaches
Having accepted the broadest definition of an ectodermal dysplasia, the second challenge presented by this group of conditions is that of designing a meaningful and functional classification system. Until the end of the 20th century, classification systems for ectodermal dysplasias were based on clinical manifestations. Several authors addressed the issue of delineating nosological groups of conditions linked by shared phenotypical traits. The most comprehensive accounts of clinical phenotypes and inheritance patterns of ectodermal dysplasia were produced by Pinheiro and Freire-Maia in their classic monograph and in subsequent writings [7]. Their classification designated conditions by groups depending on the presence of hair, nail, tooth or sweat gland abnormalities, and assigned conditions to groups using a ‘1-2-3-4 system’ to collate conditions that had involvement of hair (1) (89%), teeth (2) (78%), nails (3) (73.5%) or sweat glands (4) (38%) (Figures 67.1, 67.2 and 67.3). This classification was a comprehensive attempt to bring order to an unwieldy group of conditions but was difficult to use and grouped together disparate clinical entities such as Goltz syndrome and pachyonychia congenita. In common with any other classification of ectodermal dysplasias based on clinical findings, this system is confounded by the subtleties of inheritance such as incomplete penetrance and variable expressivity of phenotype. This is especially true in the ectodermal dysplasias, in which sweating is often not formally measured and tooth or nail anomalies may be mild. A comprehensive contemporaneous consideration of the breadth of ectodermal dysplasia conditions in the tradition of Freire-Maia and Pinheiro is given by Visinoni et al. (Table 67.1) [2]. Clinical classifications now need to become more focused, user friendly and better integrated with emerging molecular data. Difficult decisions will need to be made to balance inclusiveness (of every possible ectodermal dysplasia) with accessibility and practicality of usage. In addition, the new classification should allow the inclusion of oligosymptomatic or even monosymptomatic variants of ectodermal dysplasia, which will be only possible by integrating clinical and molecular aspects.
Table 67.1 Classification of ectodermal dysplasias
| Ectodermal dysplasia (ED) | MIM | Inheritance |
| Subgroup hair–teeth–nails–sweat glands | ||
| 1 Acro-renal field defect–ED–lipoatrophic diabetes (AREDYLD) | 207780 | AR |
| 2 Alopecia–contractures–dwarfism–mental retardation syndrome | 20355 | AR |
| 3 Ankyloblepharon–ectodermal defects–cleft lip/palate (AEC) | 106260 | AD |
| 4 Anonychia with flexural pigmentation | 106750 | AD |
| 5 Arthrogryposis and ED | 601701 | AR |
| 6 Cleft lip/palate–ED (CLPED1) syndrome (Zlotogora–Ogur syndrome; Margarita Island syndrome) | 225060 | AR |
| 7 Curly hair–acral keratoderma–caries syndrome (Van Steensel et al. 2001 [61]) | 607656 | AD |
| 8 Dyskeratosis congenita, AD (dyskeratosis congenita, Scoggins type) | 127550 | AD |
| 9 Dyskeratosis congenita, AR | 224230 | AR |
| 10 Dyskeratosis congenita, X-linked (Zinsser–Cole–Engman syndrome) | 305000 | XR |
| 11 Ectrodactyly–ED–cleft lip/palate syndrome 1 (EEC1) | 129900 | AD |
| 12 Ectrodactyly–ED–cleft lip/palate syndrome 3 (EEC3) | 604292 | AD |
| 13 Hypohidrotic ED with acanthosis nigricans (Lelis syndrome) | 608290 | ? |
| 14 ED with natal teeth, Turnpenny type | 601345 | AD |
| 15 Hypohidrotic ED with hypothyroidism and agenesis corpus callosum | 225040 | AD? AR?; AD, X-linked, contiguous gene syndrome? |
| 16 Focal dermal hypoplasia (FDH) | 305600 | XD |
| 17 Hypohidrotic ED, AD (ADHED) | 129490 | AD |
| 18 Hypohidrotic ED, AR (ARHED) | 224900 | AR |
| 19 Hypohidrotic ED, X-linked (XLHED; Christ–Siemens–Touraine syndrome) | 305100 | XR |
| 20 Hypohidrotic ED with immune deficiency | 300291 | XD |
| 21 Hypohidrotic ED with immunodeficiency, osteopetrosis and lymphedema (OLEDAID) syndrome | 300301 | XD |
| 22 Hypomelanosis of Ito (HMI; incontinentia pigmenti type I (IP1)) | 300337 | XD |
| 23 Keratitis–ichthyosis–deafness (KID) syndrome, AD | 148210 | AD |
| 24 Keratitis-ichthyosis-deafness (KID) syndrome, AR | 242150 | AR |
| 25 Naegeli–Franceschetti–Jadassohn syndrome (NFJS) | 161000 | AD |
| 26 Odonto-onycho-dermal dysplasia (OODD) | 257980 | AR |
| 27 Odonto-trichomelic syndrome | 273400 | AR |
| 28 Pachyonychia congenita, type 1 (PC1) | 167200 | AD |
| 29 Pachyonychia congenita, type 2 (PC2) | 167210 | AD |
| 30 Papillon–Lefevre syndrome | 245000 | AR |
| 31 Rosselli–Gulienetti syndrome | 225000 | AR |
| 32 Scalp–ear–nipple syndrome (Finlay–Marks syndrome; ED with adrenal cyst) | 181270; 129550 | AD |
| Subgroup hair–teeth–nails | ||
| 33 Ackerman syndrome | 200970 | AR |
| 34 Acro-dermato-ungual–lacrimal–tooth (ADULT) syndrome | 103285 | AD |
| 35 Arthrogryposis–ED–cleft lip/palate–developmental delay | 301815 | XR |
| 36 Cardio-facio-cutaneous (CFC) syndrome | 115150 | AD |
| 37 Clouston syndrome | 129500 | AD |
| 38 Coffin–Siris syndrome | 135900 | AD? AR? XD? |
| 39 Costello syndrome | 218040 | AR |
| 40 Cranio-ectodermal dysplasia (Sensenbrenner syndrome) | 218330 | AR |
| 41 Dermo-odontodysplasia | 125640 | AD |
| 42 ED syndrome with distinctive facial appearance and preaxial polydactyly of feet (single case) | 129540 | AD? |
| 43 ED, tricho-odonto-onychial type | 129510 | AD |
| 44 Ellis–van Creveld (EVC) syndrome | 225500 | AR |
| 45 GOMBO syndrome | 233270 | AR |
| 46 Growth retardation–alopecia–pseudo-anodontia–optic atrophy (GAPO) syndrome | 230740 | AR |
| 47 Hidrotic ED, AR (Fried tooth and nail syndrome) | 602401 | AR |
| 48 Incontinentia pigmenti 2 (IP2) | 308300 | XD |
| 49 Oculo-trichodysplasia (OTD) | 257960 | AR |
| 50 Odonto-tricho-ungual–digital–palmar syndrome | 601957 | AD? XD? |
| 51 Pineal hyperplasia–insulin-resistant diabetes | 262190 | AR |
| 52 Rothmund–Thomson syndrome (RTS) | 268400 | AR |
| 53 Schinzel–Giedion midface–retraction syndrome | 269150 | AR? AD? |
| 54 Schoepf–Schulz–Passarge syndrome | 224750 | AR |
| 55 Sener syndrome | 606156 | ? |
| 56 Thumb deformity and alopecia | 188150 | AD |
| 57 Tricho-dento-osseus (TDO) syndrome | 190320 | AD |
| 58 Tricho-odonto-onychial dysplasia | 275450 | AR? |
| 59 Tricho-rhino-phalangeal syndrome, type 1 (TRPS1) | 190350 | AD |
| 60 Tricho-thio-dystrophy, photosensitive (TTDP) | 601675 | AR |
| 61 Witkop syndrome | 189500 | AD |
| Subgroup hair–teeth–sweat glands | ||
| 62 Böök syndrome | 112300 | AD |
| 63 Ichthyosis follicularis–atrichia–photophobia (IFAP) syndrome | 308205 | XR |
| 64 Johnson neuroectodermal syndrome | 147770 | AD |
| 65 Leukomelanoderma–infantilism–retardation–hypodontia–hypotrichosis | 246500 | AR |
| 66 Ulnar–mammary syndrome (UMS) | 181450 | AD |
| 67 Hypohidrotic ED with hypothyroidism and ciliary dyskinesia (HEDH syndrome) | 225050 | AR |
| 68 ED/skin fragility syndrome | 604536 | AR |
| Subgroup teeth–nails–sweat glands | ||
| 69 Amelo-onycho-hypohidrotic syndrome | 104570 | AD |
| 70 Limb–mammary syndrome (LMS) | 603543 | AD |
| Subgroup hair–teeth | ||
| 71 Barber–Say syndrome | 209885 | AR? AD? XD? |
| 72 Blepharo-cheilodontic syndrome | 119580 | AD |
| 73 Brachymetapody–anodontia–hypotrichosis–albinoidism | 211370 | AR |
| 74 Cataract–hypertrichosis–mental retardation (CAHMR) syndrome | 211770 | AR |
| 75 Cerebellar ataxia–ED | 212835 | AR |
| 76 Dubowitz syndrome | 223370 | AR |
| 77 ED–neurosensory deafness | 224800 | AR |
| 78 ED–ectrodactyly–macular dystrophy (EEM) syndrome | 225280 | AR |
| 79 Gingival fibromatosis with hypertrichosis | 135400 | AD |
| 80 Gorlin–Chaudhry–Moss syndrome | 233500 | AR |
| 81 Hallermann–Streiff syndrome (HSS) | 234100 | AR |
| 82 Hypertrichosis universalis | 145700 | AD |
| 83 Johanson–Blizzard syndrome (JBS) | 243800 | AR |
| 84 Oculo-dento-osseous dysplasia, AR | 257850 | AR |
| 85 Oculo-dento-digital dysplasia (ODDD) | 164200 | AD |
| 86 Oro-facio-digital syndrome 1 (OFD1) | 311200 | XD |
| 87 Pili torti | 261900 | AR |
| 88 Pilodental dysplasia with refractive errors | 262020 | AR |
| 89 Progeroid short stature with pigmented naevi (Mulvihill–Smith syndrome) | 176690 | AD |
| 90 Rodrigues blindness (microphthalmia, microcornea and sclerocornea with short stature and hair and dental abnormalities) | 268320 | AR |
| 91 Trichodental dysplasia | 601453 | AD |
| 92 Uncombable hair–retinal pigmentary dystrophy–dental anomalies–brachydactyly | 191482 | AD |
| 93 Zunich neuroectodermal syndrome | 280000 | AR |
| Subgroup hair–nails | ||
| 94 Alopecia congenita with keratosis palmoplantaris | 104100 | AD |
| 95 Anonychia-onychodystrophy with hypoplasia or absence of distal phalanges (Cooks syndrome) | 106995 | AD |
| 96 Cartilage–hair hypoplasia (CHH) | 250250 | AR |
| 97 Curly hair–ankyloblepharon–nail dysplasia syndrome (CHANDS) | 214350 | AR |
| 98 Hidrotic ED, Christianson–Fourie type | 601375 | AD |
| 99 ED, ‘pure’ hair–nail type | 602032 | AD? |
| 100 Hairy elbows (hypertrichosis cubiti) | 139600 | AD |
| 101 Ichthyosis and male hypogonadism | 308200 | XR? |
| 102 Ichthyosis with alopecia, eclabion, ectropion and mental retardation | 242510 | AR |
| 103 Lymphoedema–hypoparathyroidism syndrome | 247410 | AR? XR? |
| 104 Monilethrix | 158000 | AD |
| 105 Onycho-trichodysplasia and neutropenia | 258360 | AR |
| 106 Polyposis–skin pigmentation–alopecia–fingernail changes | 175500 | ? |
| 107 Popliteal pterygium syndrome, lethal type | 263650 | AR |
| 108 T-cell immunodeficiency–congenital alopecia–nail dystrophy | 601705 | AR? |
| 109 Trichomegaly with mental retardation, dwarfism and pigmentary degeneration of retina | 275400 | AR |
| 110 Tricho-thio-dystrophy, non-photosensitive 1 (TTDN1) | 234050 | AR |
| Subgroup hair–sweat glands | ||
| 111 Focal facial dermal dysplasia types I–IV | 136500; 227260 | AD, AR |
| 112 Tetra-amelia–ED–lacrimal duct abnormalities (single case) | 273390 | AR |
| Subgroup teeth–nails | ||
| 113 Corneo-dermato-osseous (CDO) syndrome | 122440 | AD |
| 114 Deafness, congenital, and onychodystrophy, AD | 124480 | AD |
| 115 Deafness–onychodystrophy–osteodystrophy–mental retardation (DOOR) syndrome | 220500 | AR? AD? |
| 116 Dermatoosteolysis, Kirghizian type | 221810 | AR |
| 117 Haim–Munk syndrome (HMS) | 245010 | AR |
| 118 Hearing loss, sensorineural, with enamel hypoplasia and nail defects (Heimler syndrome) | 234580 | AR |
| 119 Lacrimo-auriculo-dento-digital syndrome (LADD) | 149730 | AD |
| 120 Odonto-micronychial dysplasia | 601319 | AR |
| 121 Oto-palato-digital syndrome, type 1 (OPD1) | 311300 | XD |
| 122 Pycnodysostosis | 265800 | AR |
| 123 Weyers acrofacial dysostosis | 193530 | AD |
| 124 Williams–Beuren syndrome (WBS) | 194050 | AD |
| Subgroup teeth–sweat glands | ||
| 125 Kohlschutter–Tonz syndrome (epilepsy–dementia–amelogenesis imperfecta) | 226750 | AR? XR? |
| 126 Marshall syndrome I | 154780 | AD |
| Subgroup nail–sweat glands | ||
| 127 ED–absent dermatoglyphic pattern–changes in nails-simian crease | 129200 | AD |
| 128 Pachyonychia congenita, AR | 260130 | AR |
Adapted from Visinoni et al. 2009 [62].
AD, autosomal dominant; AR, autosomal recessive; XD, X-linked dominant; XR, X-linked recessive.
Classification: molecular approaches
The last decade has seen several important insights into the molecular basis of a number of the ectodermal dysplasias [1]. In some cases the molecular data have confirmed clinical impressions, for example the Hay–Wells syndrome and ectrodactyly–ectodermal dysplasia–cleft lip/palate (EEC) syndrome have ectodermal dysplasia and clefting of the palate and lip as common clinical findings and these conditions are now known to be allelic [8]. Prompted by the great advances in molecular knowledge, several authors have proposed new molecular-based approaches [1, 9, 10]. These proposed approaches classify conditions based on the class of molecule responsible for the disorder, for example categorizing together those with mutations in structural or developmental molecules (Box 67.1). This approach has many advantages, especially for the characterization of defects in preparation for molecular diagnostics and, hopefully, molecular therapy. These systems, however, need to be integrated with clinical findings and need to be accessible to all clinicians involved in the care of these patients. Importantly, it has to be realized that currently it is not possible to provide a molecular diagnosis for all patients, even in those with classic clinical features of conditions well characterized at a molecular level. In addition, access to and the affordability of molecular diagnostics often represent an obstacle to molecular characterization.
Epidemiology
Incidence and prevalence
A recent study documented an occurrence of 16 hypohidrotic ectodermal dysplasia patients in 100 000 people in Denmark. Therefore the prevalence rate of all ectodermal dysplasias must be higher. Ectodermal dysplasias are observed globally and this prevalence could apply to numerous places in the world [11].
Age
The diagnosis of ectodermal dysplasias are made within the first years of life, often after complications such as hyperthermia or pneumonia have occurred.
Sex
As all inheritance patterns occur, males and females are affected. In conditions inherited in an X-linked fashion, females often have less severe disease but are not free of manifestations.
Ethnicity
Ectodermal dysplasias have been observed in all parts of the world and in all races.
Associated diseases
Often atopic eczema, allergic asthma, rhinitis and food allergies are associated.
Pathophysiology
The skin and its appendages are mainly composed of ectodermal structures but the development of appendages is orchestrated by signals from the mesoderm with the help of placodes. A rather complicated network of signalling pathways coordinates the formation and function of ectodermal structures. In recent years much has been understood about the molecular mechanisms of ectodermal embryogenesis and this has allowed the establishment of a more rational basis for the classification of ectodermal dysplasia. Interestingly, not only full-blown ectodermal syndromes but also mono- or oligosymptomatic ectodermal malformations may result from a mutation in an ectodermal key gene. Embryogenesis occurs in distinct tissue organizational fields and specific interactions among the germ layers which may lead to a wide range of ectodermal dysplasias. Besides a typical clinical symptom complex or a single clinical key finding of ectodermal malformations, it is increasingly necessary to document a mutation in a key gene within a crucial signalling pathway. The skin is the product of ectodermal and mesodermal stem cell differentiation [12]. However, according to Spemann's organizer theory, even the endoderm has a role in all organ formations including the skin [13]. Orchestrated skin development is only possible by intensive information exchange especially between the mesoderm and ectoderm. Mesodermal signalling pathways such as wingless (Wnt) are crucial for the induction of appendages ofthe skin. Wnt signalling, for example, has influences on mesenchymal cells and on epithelial cells [14]. The same is true for the ectodysplasin cascade [14]. Whereas the mesoderm induces the placodes by Wnt, the ectoderm evolves into epidermis with the help of ectodysplasin and sonic hedgehog [15]. There is a cross-talk of several signalling cascades, such as the ectodysplasin and p53 signalling pathways. Genetic defects in signalling pathways, which disturb the interaction between the ectoderm and mesoderm, lead to ectodermal dysplasia. Dickkopf expression leads to the position and orientation of the placode and the sonic hedgehog cascade is responsible for the formation of hair germ. Ripply, which is a retinoic acid-inducible repressor, is required for setting the borders of the pre-placodal ectoderm [16]. The recent work by Mikkola and co-workers shows that numerous genes and gene pathways such as tumour necrosis factor (TNF), NF-κB (nuclear factor ‘kappa light chain enhancer’ of activated B-cells) and the Wnt pathway, transforming growth factor β (TGF-β) pathway, fibroblast growth factor pathway, chemokine pathway and other genes are involved in hair development [17]. Disease embryo development network analysis reveals a relationship between disease genes and embryo development–associated genes [18]. A similar transcription factor and microRNA profile is found during embryonic and disease development in numerous organs including the skin.
Ectodermal dysplasias manifest mainly in ectodermal structures and often involve a key gene affecting embryogenesis of the skin, such as ectodysplasin, wingless, p63 or sonic hedgehog (SHH) [1, 19]. The ectodysplasin A (EDA) pathway plays a conserved role in fine-tuning the size, spacing and position (and probably thereby shape) of ectodermal organs in vertebrates. EDA has a dual role in ectodermal organogenesis: inhibition of bone morphogenetic protein (BMP) activity and induction of SHH expression [20]. The Wnt pathway regulates both EDA and ectodysplasin A receptor (EDAR), and the activin and BMP pathways also regulate EDAR [19]. The role of Wnt in physiology and disease is well documented in numerous papers [21]. In the skin, Wnt signalling is important for the establishment of the dermatome architecture, dermis, skin regions with individual appendages, axial determination and intra-appendage regions [22]. Focal dermal hypoplasia, a multiorgan developmental disorder, underscores the importance of Wnt proteins in ectodermal–mesodermal communication as causative mutations were found in PORCN encoding a member of the Wnt cascade. WNT10A mutations are frequent causes of a broad spectrum of ectodermal dysplasias with teeth, nail, skin and hair abnormalities. Also the rare syndrome of odonto-onycho-dermal dysplasia is caused by mutations in the same gene. Schöpf–Schulz–Passarge syndrome is a consequence of a homozygous nonsense mutation in WNT10A [23]. The fact that isolated hypodontia can also be found in 50% of patients with WNT10A mutation is very important [24].
It has been shown, that only four genes (EDA1, EDAR, EDARAAD and WNT10A) account for 90% of hypohidrotic/anhidrotic ectodermal dysplasia cases [25]. p63 plays a crucial role in the maturation of the epidermis; it is important for barrier formation, terminal differentiation of keratinocytes, adhesion and proliferation as well as for basement membrane formation [26]. Because p63 has so many ligands and is linked with many other signalling pathways, the clinical features of p63 diseases are rather heterogeneous and broad [27]. p63 regulates human keratinocyte proliferation via the MYC–gene network [28]. Non-syndromic cleft lip has been observed with mutations in p63 [29], which underlines the fact that monosymptomatic ectodermal malformations may result from a mutation in an ectodermal key gene. In a recent study, fibroblasts from healthy donors and two EEC syndrome patients carrying two different point mutations in the DNA-binding domain of p63 were reprogrammed into induced pluripotent stem cell (iPSC) lines [30]. EEC syndrome iPSC from both patients showed early ectodermal commitment into K18(+) cells but failed to further differentiate into K14(+) cells (epidermis/limbus) or K3/K12(+) cells (corneal epithelium). APR-246 (PRIMA-1(MET)), a small compound that restores functionality of mutant p53 in human tumour cells, was able to revert corneal epithelial lineage commitment and reinstate a normal p63-related signalling pathway. This study shows a possible avenue for future therapy.
Signalling pathways in human skeletal dysplasias often also have an impact on the skin. They include TGF-β, Wnt, hedgehog, Notch, BMP, fibroblast growth factor (FGF) and others. Mutations in genes encoding FGF receptors cause Crouzon syndrome, Apert syndrome, Beare–Stevenson syndrome and numerous others that show cutaneous involvement. Sclerosteosis or Van Buchem disease also features nail alterations and the responsible gene is SOST (sclerostin) [31]. AXIN2 mutations can lead to tooth agenesis as well as to ectodermal dysplasia with malignant tumours [32]. Patched gene (PTCH) mutations or SHH mutations can lead to basal cell naevus syndrome and other malformations mainly in ectodermal structures. Numerous mutations in genes that encode for adhesion molecules can lead to ectodermal dysplasia, including mutations in connexin genes causing keratitis–ichthyosis–deafness syndrome, oculo-dento-digital dysplasias, Clouston syndrome, Vohwinkel syndrome, Bart–Pumphrey syndrome and others. However, mutations in the structural components of desmosomes can also lead to isolated heart diseases, which have no relation to ectodermal dysplasia. In addition mutations in desmosome-forming genes can also lead to combined skin and cardiac problems. There are numerous diseases with mutations in genes important for cell to cell adhesion that are placed within the ectodermal dysplasias but without clear systematics [33].
The following section reviews some of the commoner ectodermal dysplasias, classified according to the signalling pathway involved in their pathogenesis.
Ectodermal dysplasias due to mutations in TNF-like/NF-κB signalling pathways
The transcription factor NF-κB regulates the expression of multiple genes encoding important mediators of immune and stress responses, cell adhesion, protection against apoptosis and inflammatory reactions [34]. In addition, this transcription factor plays critical roles in the development and homeostasis of the epidermis and the proper function of lymphatic vessels [35]. NF-κB is composed of homo- or heterodimers of five proteins belonging to the Rel family. NF-κB is usually maintained in an inactive state within the cytoplasm when bound to inhibitory proteins of the IκB family: IκBα, IκBβ and IκBε. IκB molecules are phosphorylated on two critical serine residues in response to multiple stimuli such as cytokines, various stress signals and viral and bacterial infections. Phosphorylation at these sites enables recognition of IκB molecules by a ubiquitation complex. Following polyubiquitation, IκBs are degraded by the proteasome, thus releasing free NF-κB which then enters the nucleus and activates target genes [36]. The kinase that phosphorylates IκB has been designated IκK (for IκB kinase) and has been shown to consist of two catalytic subunits (IκKα/IκK1 and IκKβ/IκK2) and a third component IκKγ (more usually known as NEMO) that plays a structural and regulatory function within the complex. Cell lines lacking NEMO are unable to activate NF-κB in response to most stimuli. Extensive work with mouse models has confirmed the centrality of the NF-κB pathway in apoptosis, inflammatory and immune functions. A complete absence of NF-κB leads to prenatal death due to massive TNF-induced liver apoptosis, and more subtle knockouts that alter NF-κB activity all lead to immune defects. The NF-κB pathway has been found to be defective in a number of ectodermal dysplasias. In many cases these predominantly phenotype-driven, mouse–human comparison studies have yielded significant new insights into molecular pathways. One of the best characterized NF-κB-related system is the ectodysplasin pathway, which serves as an upstream activator of NF-κB. Since 1997, defects in this pathway have been demonstrated in the X-linked, autosomal dominant and recessive subtypes of HED. Subsequently, mutations in downstream components of the pathway have been shown to underlie familial incontinentia pigmenti and HED associated with immunodeficiency and/or osteopetrosis.
The X-linked HED gene, EDA, which maps to Xq12-13.1 and is also mutated in the mouse orthologue tabby, was first described in 1996 [37]. EDA encodes two isoforms of a transmembrane protein, ectodysplasin A, that has homology to the TNF family [38]. The extracellular domain of EDA has a collagen-like repeat and a furin cleavage site, unique among TNF proteins. Cleavage is necessary to enable solubility and functionality of EDA. The two longest isoforms, EDA-A1 and EDA-A2, bind to two different receptors: EDA-A1 binds to the EDAR protein and EDA-A2 binds to another X-linked receptor, XEDAR [19]. Mutations have been identified in all domains of EDA in patients with HED, and many of these mutations are thought to have an effect on solubility or cleavage of EDA, rendering it non-functional. The physiological role of EDA in hair follicle morphogenesis was demonstrated with the isolation of the gene for autosomal dominant/recessive HED. Patients with autosomal dominant or recessive HED are phenotypically identical to those with X-linked HED. The EDAR gene carrying mutations in this disorder is homologous to the downless (DL) mouse gene and encodes a member of the tumour necrosis factor receptor (TNFR) superfamily which functions as an ectodysplasin receptor (EDAR). Loss-of-function mutations in EDAR have been reported in autosomal recessive HED while dominant negative mutations affecting the death domain of EDAR cause autosomal dominant HED [39].
The EDA/EDAR pathway was further refined when the molecular basis of a third mouse homologue was identified. The crinkled mouse (cr) is a spontaneous mouse mutant with a phenotype identical to downless and tabby [19]. Using positional cloning techniques, the causative gene was found to encode an adapter protein termed EDARADD (EDAR-associated death domain) for the EDA/EDAR complex. Mutations in the human homologue of this gene, named EDARADD, were identified as a cause for autosomal recessive HED [40]. The EDARADD interacts with the intracellular death domain of EDAR, linking it to downstream signals, leading to NF-kB activation [40]. EDARADD associates with TRAF 1, 2 and 3. NF-κB activation by the EDAR pathway is NEMO dependent [41], and the relevance of this interaction to human ectodermal dysplasias became clearer when loss-of-function mutations were identified in the X-linked IKBKG gene (more usually known as NEMO) in incontinentia pigmenti [41]. Mutations in this gene are usually lethal in males. However, less severe (hypomorphic) mutations in IKBKG have been described in several male patients with an unusual phenotype of HED associated with immune deficiency (EDA-ID) [42]. Mutations in the coding region are associated with the EDA-ID phenotype, while specific mutations affecting the stop codon of IKBKG cause a more severe syndrome of osteopetrosis and/or lymphoedema associated with EDA-ID [43].
Two other EDAR-related members of the TNFR superfamily, XEDAR and TROY/TAJ have been reported [19]. Signals from each of these receptors were shown to activate NF-κB, providing further candidate genes and candidate signalling systems for human HED. The TNFR-associated factor 6 (TRAF-6) is a cytoplasmic adapter protein that links signals from members of the TNFR superfamily to activation of transcription factors such as NF-κB through IKK (inhibitor of κB kinase) complex activation. TRAF-6 –/– mice display HED, revealing yet more complexity to these signalling systems. Recently, the first patient with hypohidrotic ectodermal dysplasia and TRAF-6 mutation has been reported [44].
TP63-related phenotypes: overview of the molecular pathway
p63 plays a crucial role during the formation of the epidermis [45]. p63 is important for barrier formation, terminal differentiation of keratinocytes, adhesion and proliferation as well as for basement membrane assembly [26]; it is also involved in cleft palate. p63 has numerous target genes including EDAR, NOTCH, BMP, CTNNB1 and FAS as well as numerous others. Because p63 is linked with many other signalling pathways, the clinical features of p63-related diseases is rather heterogeneous and broad [27]. p63 regulates human keratinocyte proliferation via the MYC–gene network [28]. Numerous genes are differentially expressed in the absence of a fully functional p63. Accordingly, mutations in TP63 encoding p63 have been found in ankyloblepharon–ectodermal defects–cleft lip/palate (AEC) syndrome skin to disrupt several signalling pathways [46]. Epigenetic modifications have been shown to affect p63 regulation of terminal differentiation genes. p63 mutations can lead to altered stability and transcriptional activity in distinct ectodermal dysplasias [47]. Non-syndromic cleft lip has been observed with mutations in TP63 [29] which underlines the fact that monosymptomatic ectodermal malformations may result from a mutation in an ectodermal key gene. The E3 ligase itch mediates the degradation of p63, providing another level of regulation and thus suggesting novel therapeutic strategies for ectodermal dysplasia [48].
The p53 gene family comprises key regulators of the cell cycle, which are mutated in more than 50% of human cancers. p63 regulates human keratinocyte proliferation via a MYC-regulated gene network and differentiation commitment through a cell adhesion-related gene network [28]. p63 and p73 are related molecules that share high amino acid identity with p53. p63 and p53 are critical regulators of gene expression during embryogenesis. They regulate the development of facial prominence and limb buds, and are essential for cranial closure and development of the lens [27]. Limb defects are best explained by a failure of the apical ectodermal ridge to develop [49].The role of p63 and p73 in human cancers has been extensively studied. Whereas p53 has an important role in tumorigenesis, this is not as prominent in p63. The TP63 and TP73 genes also differ from TP53 in that they each encode several different isoforms by utilizing two different transcription initiation sites (for a review see [50]).
Mutations in p63 have now been identified in six distinct human phenotypes, all of which have ectodermal dysplasia as a key feature [26] (Figure 67.4). Some genotype–phenotype correlations have been identified (for a review see [50]. TP63 mutations account for most cases of EEC syndrome. In an authoritative paper, van Bokhoven et al. [51] were able to demonstrate mutations in 40 out of 43 families with EEC; all but one of these mutations were sited within the DNA-binding domain, and sequence alterations affecting five amino acid residues accounted for 75% of all mutations. In the AEC syndrome (also known as Hay–Wells syndrome), where the limb abnormalities are absent or minimal, mutations have been exclusively detected within the sterile alpha motif (SAM) domain-encoding region of the TP63 gene and are associated with complex gain-of-function as well as loss- and change-of-function effects [51]. In limb–mammary syndrome (LMS), TP63 frameshift mutations leading to truncations of p63 protein have been reported in exon 13 in two unrelated patients [51], and an N-terminal mutation in a further family [26]. Mutations in the acro-dermato-ungual–lacrimal–tooth (ADULT) syndrome have yielded interesting insights in that the first mutation was identified in exon 3’, which is only expressed in the transactivating (TA) isotypes of p63 and causes an amino acid substitution outside the DNA-binding domain [52]. A subsequent report has demonstrated a mutation that confers significant transactivation activity on ∆N-p63γ, an otherwise inert isoform of p63 [50]. Non-syndromic split-hand–split-foot malformation (SHFM) syndrome is a genetically heterogeneous group of conditions, but some cases (possibly around 10%) are attributable to TP63 mutations [51]. Some of these mutations seem specific for SHFM but others underlie both EEC and SHFM [50].
- Limb mammary syndrome (MIM: 603543). This autosomal dominant syndrome was first reported in a Dutch family with a constellation of features that had not previously been reported, including hand and foot anomalies and mammary gland aplasia/hypoplasia. The skin and hair were normal in all affected individuals, but some had lacrimal duct atresia, nail dysplasia, hypohidrosis, hypodontia or cleft palate [50] In addition, internal female genital dysplasias have been observed [54]. LMS is distinguished from EEC syndrome by the consistent finding of mammary anomalies in LMS (infrequent in EEC), and the much more frequent finding of skin, nail and tooth anomalies in EEC syndrome. The clefting in LMS is of the palate only, whereas in EEC syndrome the lip and palate are affected.
- Non-syndromic split-hand–split-foot malformation syndrome (MIM: 183600). This condition has no dermatological features and therefore is not discussed in detail here. However, the molecular basis of the isolated SHFM syndrome is also a mutation in p63 [29].

Figure 67.4 The central role of p63 in ectodermal dysplasia. (From Koster 2010 [26].
Reproduced with permission of Nature Publishing Group.)
Defects in transcription factors other than p63
In addition to the p63 pathway, several other ectodermal dysplasias have now been attributed to defects in transcription factors that control the expression of several target genes important in ectodermal morphogenesis [55]. In many cases, positional cloning studies have yielded the primary mutation in a specific gene but the detailed molecular signalling pathways have yet to be delineated.
Ellis–van Creveld (EvC) syndrome is a recessive ectodermal dysplasia that is characterized by a skeletal dysplasia with short limbs, short ribs, postaxial polydactyly and congenital heart defects. The gene has been identified [56]. There is evidence that the two ciliary proteins, Evc and Evc2, the products of human disease genes associated with the EvC syndrome, act downstream of smoothened to transduce sonic hedgehog signalling. Loss of Evc/Evc2 does not affect sonic hedgehog-induced smoothened phosphorylation and ciliary localization but impedes sonic hedgehog pathway activation mediated by constitutively active forms of smoothened [57].
Witkop syndrome (MIM: 189500) is an autosomal dominant ectodermal dysplasia with primary manifestation in the teeth (taurodontia and partial or complete anodontia) and nails (koilonychia, longitudinal ridging and nail pits) [58]. Mutations have been identified in the MSX1 gene, a member of the homeobox gene family and an important regulator of transcription.
Of the ectodermal dysplasias more likely to be seen by a dermatologist, tricho-dento-osseous (TDO) syndrome and tricho-rhino-phalangeal syndrome (TRPS) are both caused by mutations in genes coding for important transcription factors. The causative gene for TDO is DLX3, encoding a homeodomain transcription factor that is developmentally expressed in many structures derived from epithelial–mesenchymal interactions, such as the teeth, hair follicles and limb buds. A dominant mutation responsible for TDO syndrome was found to impair the ability of DLX3 to downregulate δNp63α [59]. The TRPS1 gene underlies TRPS types I and III and is important in the development and differentiation of bone, kidney and hair follicles [60]; a microdeletion syndrome (8q42.11 to 8q24.1), that includes TRPS1 and EXT1, underlies TRPS type II.
Genetics
As ectodermal dysplasias are such a heterogeneous group of diseases, all inheritance patterns have been described including autosomal recessive, autosomal dominant, X-linked and mitochondrial.
Environmental factors
Epigenetic factors seem to be important for the phenotypical expression of the disease.
Resources
Further information
Canadian Ectodermal Dysplasia Syndromes Association: http://ectodermaldysplasia.ca/.
Ectodermal Dysplasia Society: http://www.ectodermaldysplasia.org/.
Ectodermal Dysplasias – International Network: http://www.edsinfo.org/.
National Foundation for Ectodermal Dysplasias: http://nfed.org/.
(All last accessed August 2014.)
X-linked hypohidrotic ectodermal dysplasia with immunodeficiency
Definition and nomenclature
X-linked hypohidrotic ectodermal dysplasia with immunodeficiency (EDA-ID) is a rare disease with the clinical hallmarks of hypohidrosis, delayed teeth eruption, coarse hair and immunodeficiency complicated by frequent bacterial infections. Causative mutations have been identified in the IKBKG gene, which codes for the inhibitor of κ light polypeptide gene enhancer in B-cell kinase-γ, better known as NF-κB essential modulator (NEMO). NEMO phosphorylates and degrades IκB to activate NF-κB. NEMO dysfunction leads to an impaired antimicrobial response to polysaccharides, impaired NK-cell activity, hyper IgM syndrome and hypogammaglobulinaemia. Often patients have co-morbidity with inflammatory bowel disease and rheumatoid arthritis.
Introduction and general description
The observation of unusually severe recurrent infections in a small subset of patients with otherwise typical HED features led to the suggestion that there may be a specific syndrome of HED and immunodeficiency (EDA-ID) [1]. The EDA-ID syndrome was first reported in a boy with miliary tuberculosis, and the second reported case had recurrent life-threatening infections caused by Pseudomonas aeruginosa, Mycobacterium avium and cytomegalovirus. A third child had a milder phenotype with repeated infections due to Staphylococcus aureus and Streptococcus pneumoniae, and three further siblings from a different kindred had recurrent severe infections with S. pneumoniae, with impaired response to polysaccharide antigens. All of these cases were males, suggesting X-linked inheritance. Since these original reports, the disease phenotypic spectrum has been delineated and includes severe life-threatening or recurrent bacterial infections in the lower respiratory tract, skin, soft tissues, bones and gastrointestinal tract, as well as meningitis and septicaemia in early childhood. Overall, the causative pathogens have most often been Gram-positive bacteria (S. pneumoniae and S. aureus), followed by Gram-negative bacteria (Pseudomonas spp. and Haemophilus influenzae) and mycobacteria. Most patients have severe hypogammaglobulinaemia, with low serum IgG levels and varied levels of other immunoglobulin isotypes (IgA, IgM and IgE). Some patients have massively elevated IgM levels, and an impaired antibody response to polysaccharides is the most consistent feature of this condition.
Hypomorphic mutations in the X-linked IKBKG gene or somatic mutations in this gene result in various forms of anhidrotic ectodermal dysplasia with immunodeficiency [2]. Missense mutations in the gene encoding NEMO are associated with reduced signal-induced nuclear translocation of NF-kB proteins, resulting in defective expression of NF-kB target genes [3, 4]. It has been shown that the pathogenic mutation preferentially impairs the interaction with K63 and M1-linked di-Ub, which correlates with its ubiquitin-binding defect in vivo [5]. Impaired NK cell activity is reported in some, but not all, patients with EDA-ID; the degree and range of immunological abnormalities seen may relate to the type of NEMO mutation involved.
Epidemiology
Incidence and prevalence
Prevalence is not known in any detail. The incidence has been assumed to be approximately 1 in 250 000 live male births for the X-linked form.
Age
Typical signs and symptoms may develop early. Sparse hair with hypotrichosis or complete alopecia, abnormal teeth with conical shape or missing teeth and sweating impairment, combined with recurrent infections, are the leading clinical findings to make an early diagnosis. The disease can be associated with lymphoedema and osteopetrosis.
Sex
Transmission is X-linked recessive in patients with mutations in the coding regions of NEMO. This means that the disease usually occurs in males.
Ethnicity
There is no ethnic preponderance.
Associated diseases
Twenty-five per cent of patients have co-morbidity with inflammatory bowel disease and rheumatoid arthritis. There is a variant associated with osteopetrosis and lymphoedema. Recurrent bacterial infections are common [1].
Genetics
The disease is usually inherited in an X-linked recessive fashion. Somatic mosaicism is also frequently observed [2]. Fewer than 10 patients have been reported with an autosomal dominant form of the disease due to mutations in the NFKBIA gene encoding the NF-kB, subunit 1, which is related to the NF-kB signalling pathway as well.
Environmental factors
Absent or decreased sweating leads to heat intolerance.
Clinical features
History
The lack of normal hair growth is typical. Many patients fail to thrive. Fever of unknown origin as a child and later heat intolerance are typical constellations. Missing teeth or malformed teeth are also characteristic and a clue for diagnosis in children. Mutations in IKBKG should be considered in male infants with recalcitrant seborrhoeic or atopic eczema-like eruptions and intertrigo, especially when features of ectodermal dysplasia are present [6].
Differential diagnosis
Apart from recurrent bacterial infections, the clinical manifestations, differential diagnosis and natural course are very similar to those in patients with classic HED. The ectodermal dysplasia-related signs and symptoms, however, are less pronounced than in the classic form and osteopetrosis and lymphoedema are features uniquely found in X-linked EDA-ID. As mentioned, an autosomal dominant form of the disease can be distinguished on the basis of its mode of inheritance.
Hypohidrotic ectodermal dysplasia
Definition and nomenclature
X-linked hypohidrotic ectodermal dysplasia (Freire-Maia 1-2-3-4) is the most common of the ectodermal dysplasias. However, autosomal dominant and autosomal recessive forms have been described. The disorder is characterized by hypotrichosis with fine, slow-growing scalp and body hair, sparse eyebrows, hypohidrosis, nail anomalies and hypodontia. Peg-shaped primary and secondary teeth are typical. Decreased sweating leads to heat intolerance and enhances dryness of the skin. X-linked hypohidrotic ectodermal dysplasia 1 (ECTD1, HED/EDA) is caused by a mutation in the EDA gene, which encodes ectodysplasin. In addition, autosomal recessive forms (ECTD10B, ECTD11B) and autosomal dominant forms of HED (ECTD10A, ECTD11A) are caused by mutation in the EDAR and EDARADD genes, respectively. Treatment needs a multidisciplinary approach. Early diagnosis is imperative to avoid life-threatening complications induced by hyperthermia and infections.
Introduction and general description
X-linked HED was first described by Danz in 1792 and then by Charles Darwin in 1875. HED is a monogenic disorder inherited in an X-linked fashion, but autosomal dominant and recessive inheritance types have been described too. A fourth and rare type with immunodeficiency also exists. Hypohidrotic or anhidrotic ectodermal dysplasia is the most prevalent type within the spectrum of ectodermal dysplasias. Clinical hallmarks are hypotrichosis on the scalp but also of the eyebrows and eyelashes, hyperpigmentation around the eyes, dry skin and peg-shaped primary and secondary teeth with hypodontia (Table 67.2). Frontal bossing and a saddle-bridged nose may occur. The biological most important finding is anhidrosis or hypohidrosis. Affected individuals show heat intolerance with episodes of hyperpyrexia, which may result in seizures and neurological damage [1].
Table 67.2 Clinical features in patients with autosomal dominant hypohidrotic ectodermal dysplasia
| Complication | Occurrence (%) |
| Smooth, dry skin | 78 |
| Sparse hair | 89 |
| Sparse eyebrows | 100 |
| Sparse body hair | 62 |
| Decreased sweating | 85 |
| Heat intolerance | 50 |
| Onychodysplasia | 39 |
| Dental anomalies | 100 |
Adapted from Aswegan et al. 1997 [7].
Epidemiology
Incidence and prevalence
In a recent study by Nguyen-Nielsen et al., the population-based prevalence was 21.9 per 100 000, which is markedly higher than previously anticipated [2]. Teeth abnormalities occurred in 79% of all cases.
Age
The most frequent age at time of X-linked HED diagnosis occurred between the ages of 11 and 18 years.
Sex
Females with X-linked HED show less prominent findings, often displaying patchy lesions arranged along the lines of Blaschko, as opposed to the generalized presentation seen in males.
Ethnicity
There is no ethnic dominance.
Associated diseases
Immunodeficiency, sometimes associated with osteopetrosis and lymphoedema, may occur together in several subtypes of HED. Breast hypoplasia or aplasia might be an additional finding [3].
Pathophysiology
Hypohidrotic ectodermal dysplasia results from mutations in genes encoding members of the ectodysplasin/NF-kB pathway [4]. Mutations in EDA encoding the epithelial morphogen ectodysplasin A of the tumour necrosis factor family, cause X-linked HED (known as the Christ–Siemens–Touraine syndrome). Mutations in EDAR, which encodes the ectodysplasin A receptor, or EDARADD, encoding the EDARADD protein, cause both autosomal recessive and autosomal dominant HED. NEMO (also known as inhibitor of NF-κB kinase subunit γ (IKK-γ)) is a protein that in humans is encoded by the IKBKG gene located on Xq28. IKBKG mutations cause HED with immunodeficiency. Other forms of HED have been shown to result from mutations in: (i) WNT10A encoding wingless 10A; (ii) TRAF6 encoding TNFR-associated factor 6; (iii) NFKBIA encoding the nuclear factor of κ light polypeptide gene enhancer in B-cell inhibitor α; kand (iv) EDA2R (also known as XEDAR) encoding the ectodysplasin 2A receptor.
Predisposing factors
A positive family history is a predisposing factor.
Pathology
Histology shows missing or reduced sweat glands and also reduced sebaceous glands. The epidermis is thin with flattening of the rete ridges. Hair follicles and their sebaceous glands are variably reduced in number. Apocrine glands may be absent, sparse or even normal. Mucous glands of the upper respiratory tract may be sparse or absent. Hair shaft abnormalities are variable and include longitudinal clefts or grooves and transverse fissuring. Radiographs of the jaws may reveal dental hypoplasia or aplasia.
Genetics
X-linked, autosomal dominant and autosomal recessive traits exist.
Environmental factors
Patients should avoid hot places and extensive physical activity without cooling devices and special clothes. Patients should drink cool liquids in warm environments. Wearing a hat in sunny climates is obligatory.
History
A history of heat intolerance is characteristic but not obligatory. Sometimes other family members have been affected in the pedigree. Missing or malformed teeth can lead to the diagnosis.
- Hair. The hair is sparse, dry and lusterless with a light colour (Figure 67.5a). The bar code appearance that mirrors a microscopic artefact is often seen in patients with HED. There are parallel dark bands of different lengths running across the full width of the shaft. Scanning electron microscopy studies have shown follicular distortion, follicular ridging and distorted bulbs but these findings are non-specific. Eyebrows are scanty or absent (Figure 67.5b); occasionally just the outer two-thirds are missing. The eyelashes may be normal, sparse or completely absent. Secondary sexual hair in the beard, pubic and axillary regions is variably present and may be normal. Hair on the torso and extremities is usually absent. Approximately 70% of obligate female carriers of X-linked HED describe their hair as being sparse or fine. Light and electron microscopy shows variable hair shaft defects including twisting, pili canaliculi and trichorrhexis nodosa.
- Teeth. A wide range of dental abnormalities may be associated, ranging from a complete absence of teeth (anodontia) to sparse, abnormally shaped teeth (Figure 67.6). Studies reveal a mean of 24 missing teeth, out of a total of 28, in affected males. Dentition is delayed and the erupted teeth tend to be small, widely spaced and frequently conical or peg shaped. Both deciduous and permanent teeth are affected. The alveolar ridges are hypoplastic, which gives rise to full, everted lips. About 80% of obligate female carriers of X-linked HED have distinct dental abnormalities including absent permanent teeth and small or peg-shaped teeth.
- Nails. Often nails are normal although onychodysplasia has been reported in 39% of cases.
- Sweat glands. Sweating is often severely diminished or absent due to a paucity or absence of eccrine glands. An absence of sweating leads to an inability to thermoregulate by evaporative cooling, and hyperthermia can occur with physical exertion or in a warm environment. Due to a lower body surface area ratio, thermoregulation is most problematic in infants and young children who may experience recurrent bouts of fever as high as 42°C. Heatstroke is the most common cause of death in patients with anhidrotic ectodermal dysplasia within the first years of life. Heat intolerance does occur in older children and adults, but is less problematic as they are better able to control their body temperature by drinking cold liquids, wetting their skin or clothing and seeking out cool surroundings. Up to 25% of heterozygote females experience heat intolerance and almost half notice their ability to sweat is reduced. The hypohidrotic areas of skin in carrier females of X-linked HED occur in defined linear patterns corresponding to the lines of Blaschko.
- Skin. At birth, affected males may demonstrate marked scaling or peeling of their skin which may be mistaken for a collodion membrane. In children and adults, the skin is fine, smooth and dry. Periorbital hyperpigmentation and fine wrinkling around the eyes are characteristic features of the disorder. Eczema is common and is prominent in the flexural areas. Small milia-like papules may be found on the face. Dermatoglyphics are often reduced or even absent (Figure 67.7). Lower lips are often rather prominent and ears show a characteristic deformation (Spock ears) (Figure 67.8).
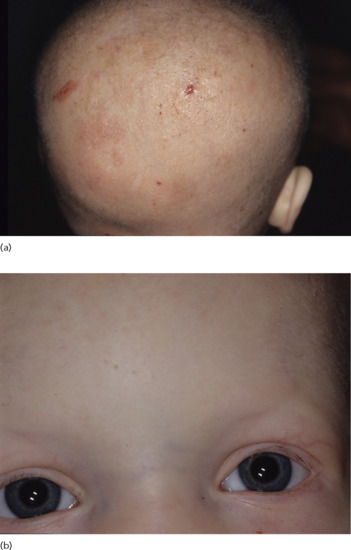
Figure 67.5 (a) Hypotrichosis and (b) reduced eyebrows and cilia in hypohidrotic ectodermal dysplasia.
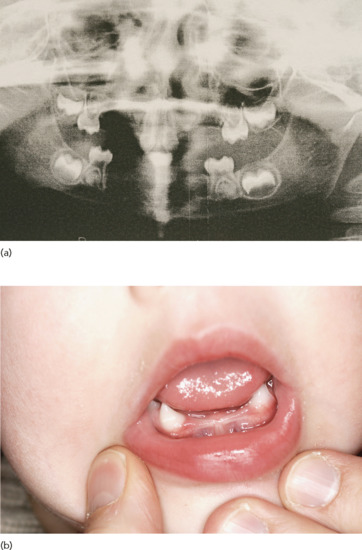
Figure 67.6 (a) Hypodontia and (b) conical teeth and hypodontia in hypohidrotic ectodermal dysplasia.
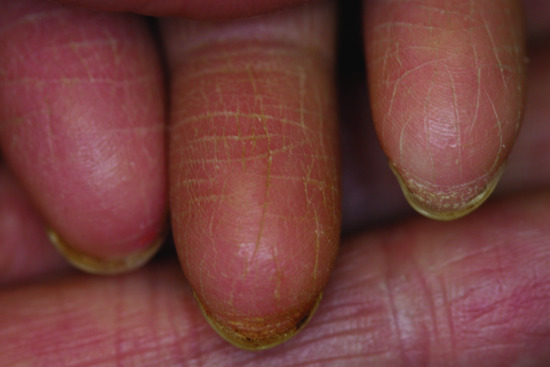
Figure 67.7 Rudimentary dermatoglyphics in hypohidrotic ectodermal dysplasia.
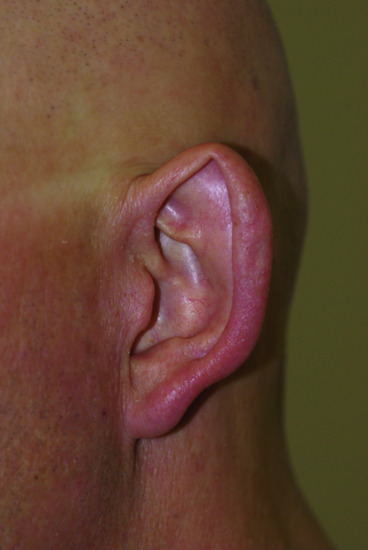
Figure 67.8 ‘Spock ears’ in hypohidrotic ectodermal dysplasia.
Clinical variants
Clinical manifestations are highly variable between individuals. Some have only slight, subjective heat intolerance although reduced to missing sweat glands are almost always identifiable. In some cases scalp hair can be black and rather light although most patients have reduced hair.
Differential diagnosis
Differential diagnosis of Christ–Siemens–Touraine syndrome includes other forms of HEDs including ectodermal dysplasia with immunodeficiency or deafness. In addition, Sjögren syndrome with symptomatic sicca syndrome should be ruled out in oligosymptomatic patients.
Classification of severity
Inter- and intrafamilial clinical heterogeneity have been observed in X-linked HED, suggesting that additional genetic factors may influence the severity of this condition. It has been shown that the EDAR 370A allele attenuates the severity of HED caused by EDA gene mutations.
Complications and co-morbidities
Anhidrotic ectodermal dysplasia is associated with mortality rates as high as 21%. Severe non-fatal illness was reported in 14 of 49 documented cases within the first year and total episodes of severe illness were observed in 43% [5]. Other complications in survivors were eczema in 71%, asthma and recurring wheezing in 65%, nasal crusting in 79%, recurring upper respiratory tract infections in 44% and feeding problems in infancy in 68% (Table 67.3).
Table 67.3 Complications of hypohidrotic ectodermal dysplasia at different ages
| Complication | Rate of occurrence |
| First year | |
| Mortality | 9/43 (21%) |
| Severe non-fatal illness | 14/49 (29%) |
| Total episodes of severe illness | 25/58 (43%) |
| Years 0–3 | |
| Mortality in years 0–3 | 12/43 (28%) |
| Severe non-fatal illness | 18/51 (49%) |
| Total with episodes of severe illness | 32/65 (49%) |
| Other complications in survivors | |
| Eczema | 39/55 (71%) |
| Asthma or recurring wheezing | 35/54 (65%) |
| Nasal crusting | 42/53 (79%) |
| Recurrent fevers in infancy | 27/50 (54%) |
| Recurrent upper respiratory tract infections in childhood | 21/47 (44%) |
| Feeding problems in infancy | 32/47 (68%) |
| Specific allergies | 14/53 (26%) |
Adapted from Clarke et al. 1987 [5]. Reproduced with permission of BMJ Publishing Group.
Disease course and prognosis
Early diagnosis is imperative to avoid life-threatening complications induced by hyperthermia and infections. Avoiding heat and physical overexertion is the most important preventative measure to recommend. Cooling the body with wet clothing and cool drinks is the only efficient way of treating hyperthermia. Orthodontic intervention is necessary, particularly for language development. Meticulous dental hygiene should be taught to the children as caries develop early.
Investigations
Gaide and Schneider [6] showed that treatment of pregnant Tabby mice, a mouse model for anhidrotic ectodermal dysplasia with a recombinant form of EDA1, engineered to cross the placental barrier, permanently rescued the Tabby phenotype in the offspring. Notably, sweat glands can also be induced by EDA1 after birth. The same group showed that in dogs postpartum treatment induced tooth growth; the first human study is currently ongoing.
Management
Exposure to heat must be monitored. Strict monitoring of body temperature is necessary for babies in an incubator. Older children should learn to handle heat and heat-generating activities with physical cooling measures, such as frequent drinking of cool liquids, wetting the clothes or wearing special cooling vests and caps. Early dental management may lead to restoring function and improving the appearance of the teeth. Orthodontic treatment often includes bone grafting or sinus-lift procedures followed by the placement of dental implants supporting dental prostheses. HED with immunodeficiency requires immune-based therapies plus aggressive management of infections or haematopoietic stem cell transplantation.
The high prevalence of asthma-like symptoms in X-linked HED patients as young as 6 years and a similar prevalence of dry eye problems indicate that screening evaluation, regular monitoring and consideration of therapeutic intervention should begin in early childhood.
Recently, 3% minoxidil 1 mL twice daily was applied with success in a patient with HED. After 1 month the patient showed growth of vellus hair and after 1 year dramatic hair growth was observed.
Resources
Further information
National Center for Technology Information, GeneReviews fact sheet: http://www.ncbi.nlm.nih.gov/books/NBK1112/.
National Foundation for Ectodermal Dysplasias, fact sheet: http://nfed.org/index.php/about_ed/types_of_ectodermal_dysplasias.
National Organization for Rare Diseases, fact sheet: http://www.rarediseases.org/rare-disease-information/rare-diseases/byID/804/viewAbstract.
Online Medelian Inheritance in Man, fact sheet: http://omim.org/entry/305100.
Orphanet: http://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=238468.
(All last accessed August 2014.)
Ankyloblepharon–ectodermal defect–cleft lip/palate syndrome
Definition and nomenclature
Ankyloblepharon–ectodermal defect–cleft lip/palate (AEC) syndrome is the result of missense mutations in TP63 affecting the p63 SAM of the gene, which is a protein–protein interaction domain. AEC syndrome is inherited in an autosomal dominant fashion. The syndrome is characterized by cleft lip/palate, severe scalp erosions and abnormalities of the epidermal appendages, including hypotrichosis, hypodontia, absent or dystrophic nails and mild hypohidrosis (Box 67.2). One distinctive feature is ankyloblepharon filiforme adnatum – partial thickness fusion of the eyelid margins.
Introduction and general description
In 1976, Hay and Wells described a syndrome inherited in an autosomal dominant fashion with ankyloblepharon, ectodermal defects and clefting of the lip and palate [1]. The disorder is today considered to be identical with Rapp–Hodgkin syndrome (MIM: 129400) [2].
Epidemiology
Incidence and prevalence
The AEC syndrome is rare and its precise incidence and prevalence are not known.
Age
The AEC syndrome manifests at birth with eroded scalp dermatitis and collodion membrane.
Sex
Both sexes are equally affected as the syndrome is inherited in an autosomal dominant pattern.
Ethnicity
No ethnic predominance is known.
Associated diseases
There are no associated disorders in AEC syndrome.
Pathophysiology
The AEC belongs to the TP63-related phenotypes (see overview of molecular pathology earlier in the chapter).
Predisposing factors
If one parent has the syndrome, the chance of disease transmission to the next generation is 50%.
Pathology
Histopathology of lesional skin shows mild atrophy, focal orthokeratosis and mild superficial perivascular lymphocytic infiltrates. Melanophages reflect postinflammatory changes. Examination of the hair shafts reveals thin hair and loss of melanin pigment in some patients. In addition, structural abnormalities include pili torti, pili trianguli et canaliculi and irregular indentation and shallow grooves [3]. Scanning electron microscopy of the affected hair shaft shows various defects, including fractures of the cuticle and pili torti, none of which are specific for the disorder. Nails may be hyperconvex and thickened, dystrophic or absent. Skin biopsy of involved scalp tissue shows a thin granular layer and stratum corneum. Hair follicles are reduced in size and arrector pili muscles appear hypertrophic. Sweat stimulation tests reveal a patchy loss of sweat glands over most of the body.
Genetics
The AEC syndrome follows an autosomal dominant mode of transmission. About 70% of cases are caused by a de novo mutation in TP63.
Clinical features (Box 67.2)
History and presentation
At birth about 90% of patients feature a collodion-like picture. In one series, 75% of patients had eroded skin at birth and 63% of patients continued to suffer from chronic scalp erosions and recurrent scalp infections [4]. Three of the seven initial patients had scalp infections or folliculitis of the scalp in association with hair loss. The scalp is commonly involved with severe and erosive dermatitis with secondary crusting and superinfection. Scalp hair is sparse in 89%, wiry in 100% and rather fair in 89% [5]. Focal or diffuse alopecia is common. Patients have rudimentary eyelashes and the eyebrows are also often affected. Absent fingernails occur in 39% and absent toenails in 11%. Dystrophic nails were found in 18 out of 18 patients. Ankyloblepharon refers to fusion of the eyelids, and in its mildest variant there is a partial thickness fusion of the central portion of the eyelid margins with sparing of the canthi (‘ankyloblepharon filiforme adnatum’). Conjunctivitis and blepharitis may result from eye abnormalities. The original family featured partial or complete hair loss, absent or dystrophic nails, widely spaced teeth and partial anhidrosis. Decreased sweat production was found in 16 out of 16 patients [5] Additional anomalies may include lacrimal duct atresia, supernumerary nipples, syndactyly and auricular deformities. Ichthyosiform scaling is found in 17% and hyperkeratosis in 39%. Pigment changes occurred in all patients with AEC.
Clinical variants
CHAND syndrome – which stands for curly hair, ankyloblepharon and nail disease – is most likely identical with AEC syndrome [6]. McGrath et al. [7] have shown that the syndrome is caused by heterozygous missense mutations in the SAM domain of p63. Mutations in TP63 have highly pleiotropic effects, as they can cause several allelic disorders including the EEC and ADULT syndromes, LMS and SHFM type 4. Rapp–Hodgkin syndrome is no longer considered as a separate entity, but is a clinical variant of AEC syndrome. Interestingly, in AEC syndrome patients the mutations give rise to amino acid substitutions in the SAM domain. However, in EEC syndrome mutations were found to result in amino acid substitutions in the DNA-binding domain. A newborn with overlapping features of AEC and EEC syndrome has been reported recently [8].
Differential diagnosis
Differential diagnosis includes epidermolysis bullosa simplex and hypohidrotic ectodermal dysplasia.
Complications and co-morbidities
Abnormalities of the external ear canals and palate frequently cause problems with chronic otitis media and secondary hearing loss. Atresia of the lacrimal duct can lead to epiphora, chronic conjunctivitis and photophobia. Scalp erosions and chronic scalp infections may be severe enough to warrant surgical intervention with skin grafting [9].
Disease course and prognosis
At birth, over three-quarters of affected newborns have red, eroded, peeling skin resembling a collodion membrane. This resolves over the first few weeks and the underlying skin is dry (Figure 67.9). Over two-thirds of individuals have chronic problems with severe, recurrent scalp erosions and scalp infections with eventual scarring which is a major feature of AEC syndrome. Palmoplantar keratoderma was reported in four of the original seven patients described by Hay and Wells and later palmoplantar changes were found in 18 out of 18 patients [5].
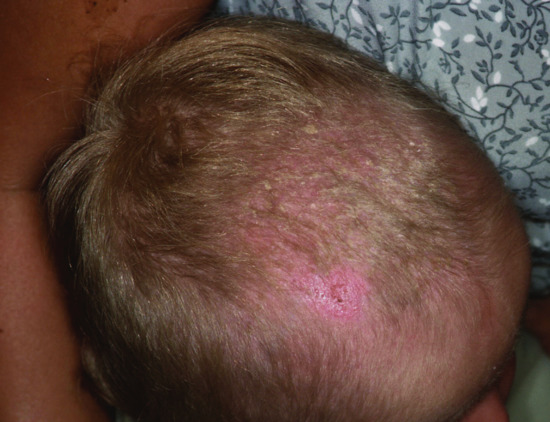
Figure 67.9 After erosive dermatitis on the vertex, marked hypotrichosis remains in the frontal part of the scalp. A mutation in p63 was documented in this patient.
Management
Emollients are appropriate for the collodion-like membrane in the newborn. Neonates with AEC often have extreme skin fragility and they should be handled with extreme care. Neonatal intensive care nursing protocols such as those existing for neonates with epidermolysis bullosa should be used. Ankyloblepharon filiforme adnatum may require surgical correction or may lyse spontaneously. Lacrimal duct atresia is surgically correctable. The scalp requires aggressive wound care and treatment with topical or systemic antibiotics as warranted. Other abnormalities such as cleft lip/palate, hypospadias and maxillary hypoplasia may be surgically corrected. Teeth preservation and restoration is imperative.
Resources
Further information
Ectodermal Dysplasia Society: http://www.ectodermaldysplasia.org/.
Genetic and Rare Diseases Information Center, fact sheet: http://rarediseases.info.nih.gov/gard/2076/eec-syndrome/resources/.
National Foundation for Ectodermal Dysplasias: http://www.nfed.org/.
(All last accessed August 2014.)
Ectrodactyly–ectodermal dysplasia–cleft lip/palate syndrome
Definition
The main features of the EEC syndrome are ectrodactyly (split-hand or -foot deformity), cleft lip/palate, tear duct anomalies and abnormalities of the epidermal appendages including hypotrichosis, hypodontia, dystrophic nails and occasional hypohidrosis.
Introduction and general description
Several genodermatoses have been mapped to chromosome 3q27 and are caused by mutations in the TP63 gene. The EEC syndrome was initially described by Eckholdt and Matens in 1804 and since then the clinical spectrum has been further delineated. Rosselli and Gulienetti described in 1961 a patient with typical features of the EEC syndrome. In 1970 Rüdiger et al. suggested the designation EEC for the syndrome. In more than 90% of cases, the syndrome is caused by mutations in the TP63 gene. The EEC syndrome is inherited in an autosomal dominant manner. It can be phenotypically highly variable with subtle limb and cranio-facial involvement even with apparently classic EEC mutations [1]. Management typically needs the cooperation of various specialists.
Epidemiology
Incidence and prevalence
The EEC syndrome is very rare and only about 300 cases have been reported. Out of 312 cases with oro-facial clefts included in the French registry between 1995 and 2006, only two patients with EEC syndrome were included [2].
Age
Normally the disease manifests at birth or at a very young age.
Sex
Both sexes are affected equally as the syndrome is inherited as an autosomal dominant trait.
Ethnicity
There is no specific ethnicity affected.
Associated diseases
Associated diseases include malformations of the genito-urinary system with renal agenesis, urethral atresia or hydronephrosis, conductive or sensorineural hearing loss, choanal atresia, mammary gland/nipple hypoplasia, several ophthalmological pathologies such as lacrimal duct alterations with atresia or hypoplasia of the lacrimal duct (which are seen in over 90% of affected individuals), photophobia, corneal ulcerations, keratitis, blepharitis, entropion and endocrine pathologies (hypoplastic thymus, hypopituitarism, growth hormone deficiency).
Pathophysiology
In about 90% of cases, EEC is the result of missense mutations in the TP63 gene (3q27) encoding the p63 transcription factor. This transcription factor is crucial for the development of the ectoderm and limbs. A complex series of genetic experiments revealed that the presence or absence of one variant type of the p63 protein, called TAp63, determines whether or not a child with a TP63 mutation will develop EEC. It has been reported recently that clefting and skin defects are caused by a loss of p63 function, and limb anomalies are due to gain- and/or dominant-negative mutations [3]. TAp63 is a strong modifier of EEC-associated phenotypes with regard to both penetrance and expressivity [3].
Predisposing factors
The presence of the disease in one of the parents confers a risk of 50% to a child to develop the disease.
Pathology
Radiographs of hand or foot deformities show missing or hypoplastic metacarpals and metatarsals. Scanning electron microscopic studies of the hair shafts of affected individuals show longitudinal grooves, distorted bulbs and cuticular defects. These findings can be seen in a number of other ectodermal dysplasias and are not specific to the EEC syndrome.
Genetics
The disease is inherited in an autosomal dominant fashion with incomplete penetrance and variable expression. Five hotspot mutations are responsible for almost 90% of all cases of EEC [4].
Environmental factors
As a reduction or absence of sweat glands may occur, hot and dry environments can be dangerous and lead to heat stroke, epilepsy or even death.
Clinical features
History
Often a positive family history is given.
Presentation
Patients present with the major signs of the syndrome: ectrodactylia (Figure 67.10), syndactylia of the hands and feet and clefting of the lips and palate (Box 67.3). In addition, numerous alterations in the ectoderm may be present such as hypopigmented dry skin, hypotrichosis with fine hairs of the scalp and eyebrows (Figure 67.11), small, malformed or missing teeth and nail dystrophies. Often sweat and sebaceous glands are missing or reduced. The clinical presentation can be highly variable and oligosymptomatic cases are common. Numerous associated findings are known as malformations of the uro-genital system, deafness, choanal atresia, hypoplasia of the breast or nipples, ophthalmological pathologies with abnormalities of the lacrimal ducts, photophobia, ulcerations of the cornea, keratitis, blepharitis and ectropion, and hormonal dysregulations. Prenatal diagnosis of the EEC syndrome based on the identification of the ‘lobster claw’ anomaly is possible by three dimensional ultrasound [5].
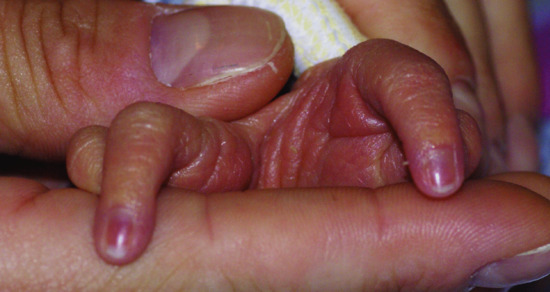
Figure 67.10 Ectrodactyly in the ectrodactyly–ectodermal dysplasia–cleft lip/palate syndrome.

Figure 67.11 Hair shaft alterations in the ectrodactyly–ectodermal dysplasia–cleft lip/palate syndrome.
Differential diagnosis
Differential diagnosis includes the whole spectrum of p63 syndromes including the AEC and ADULT syndromes, LMS and non-syndromic SHFM [4]. In addition, EvC syndrome has to be separated from the EEC syndrome [6]. Finally, van der Woude syndrome features cleft lip and cleft palate as with p63 syndrome [7]. Hypodontia and lower lip pits are typical. This syndrome was found to be caused by mutations in IRF6 [8], which is under the regulation of p63, explaining the phenotypic overlap. Of interest, lower lip pits were also recently reported in association with a TP63 mutation [9].
Classification of severity
There is a genotype–phenotype association of severity.
Complications and co-morbidities
Malformations of the kidneys with dilated urethers or urethral atresia, double urethers, hydronephrosis, multiple renal cysts and renal agenesia or dysplasia may coexist. Endocrine abnormalities include absent thymus, hypopituitarism, isolated growth hormone deficiency and pituitary diabetes insipidus.
Investigations
In patients with the typical signs and symptoms of EEC, the diagnosis is straightforward. However, molecular genetic confirmation by documentation of a TP63 mutation is recommended.
Management
Management is mainly symptomatic. However, molecular genetic research slowly improves gene therapy possibilities. Fibroblasts from healthy donors and EEC patients carrying two different point mutations in the DNA-binding domain of p63 were reprogrammed into iPSC lines in a recent study [10]. EEC iPSC from both patients showed early ectodermal commitment into K18(+) cells but failed to further differentiate into K14(+) cells (epidermis/limbus) or K3/K12(+) cells (corneal epithelium). APR-246 (PRIMA-1(MET)), a small compound that restores functionality of mutant p53 in human tumour cells, could revert corneal epithelial lineage commitment and reinstate a normal p63-related signalling pathway.
Resources
Further information
Ectodermal Dysplasia Society: http://www.ectodermaldysplasia.org/.
Genetic and Rare Diseases Information Center, fact sheet: http://rarediseases.info.nih.gov/gard/2076/eec-syndrome/resources/.
National Foundation for Ectodermal Dysplasias: http://www.nfed.org/.
(All last accessed August 2014.)
Tricho–dento–osseous syndrome
Definition and nomenclature
Tricho-dento-osseous syndrome is a rare autosomal dominant disease characterized by curly hair, enamel dysplasia with pitted teeth, enamel hypomineralization and sclerotic bones [1]. In addition, craniofacial morphology is highly variable [2]. Lichtenstein et al. [3] defined the features of this disorder in 107 individuals and proposed the name tricho-dento-osseous (TDO) syndrome.
Introduction and general description
Tricho-dento-osseous syndrome is a very rare ectodermal dysplasia with dense bones, especially in the skull.
Epidemiology
Incidence and prevalence
The disease is very rare and no epidemiological data exist.
Age
The disease manifests at birth with thick and kinky or curly hair on the scalp. The hair may straighten in later life.
Sex
It is an autosomal dominant disorder and therefore both sexes are equally affected.
Ethnicity
There is no ethnic predilection.
Associated diseases
Clinodactyly is rarely seen.
Pathophysiology
Pathology
On dental radiographs, unerupted teeth and taurodontia (increased size of the tooth pulp chamber) are found [1, 2]. Scanning electron microscopic analysis of affected teeth shows pits and depressions in the tooth enamel, uniformly thin tooth enamel and an abnormal collagenous membrane around the open apices. Radiographs of the skull reveal sclerosis and sometimes thickening of the calvarium. The long bones may also be sclerotic.
Genetics
Mutations in the distal-less homeobox 3 (DLX3) gene are causative for the syndrome [2].
Clinical features
History
Often there is a positive family history for the disease. Unruly hair at birth is typical.
Presentation
Patients have kinky hair at birth but, with age, patients show increasingly less weary and uncombable hair. There are several dental malformations, including enamel hypoplasia, which lead to pitted teeth, taurodontism and caries [1, 2]. Fingernails are thin, brittle and peel readily. Toenails may be thickened or normal. Sweat and sebaceous glands are normal and the skin is unremarkable. There is frontal bossing, the jaw is square and the head is elongated. Partial premature fusion of the cranial sutures occurs in three-quarters of affected individuals. The bones of the skull are radiographically dense and may be thick. This is not problematic for the patient and may be found incidentally when radiographs of the skull are obtained for unrelated reasons.
Clinical variants
Craniofacial variations in the tricho-dento-osseous syndrome are well documented [2].
Differential diagnosis
Curly hair and nail dysplasia are also seen in CHAND (curly hair, ankyloblepharon and nail dysplasia) syndrome, but ankyloblepharon makes this disorder distinct. Witkop syndrome (tooth and nail syndrome) lacks kinky or curly hair.
Disease course and prognosis
Hair alterations can ameliorate over time. Affected individuals are healthy, but lose most of their teeth by the age of 30 years.
Treatment
Treatment includes appropriate dental care and restoration.
Investigations
Confirmation of the clinical diagnosis can be made by mutation analysis.
Tricho-rhino-phalangeal syndrome
Definition and nomenclature
Tricho-rhino-phalangeal syndrome is an ectodermal dysplasia characterized by malformations of the hair and nail combined with facial abnormalities and cone-shaped epiphyses of the middle phalanges of some fingers and toes.
Introduction and general description
This syndrome was first described by Giedion in 1966 [1]. Three subtypes of TRPS with considerable clinical overlap can be distinguished.
Epidemiology
Incidence and prevalence
This syndrome is very rare and no prevalence and incidence numbers exist.
Age
Hypotrichosis, bulboid nose and short fingers may be obvious very early after birth depending on the severity of the phenotype.
Sex
Both sexes are equally affected in this autosomal dominant genodermatosis.
Genetics
Tricho-rhino-phalangeal syndrome is an autosomal dominant disorder caused by mutations in the TRPS1 gene which encodes a GATA-type transcription factor that has nine zinc-finger motifs [2]. Thus TRPS is today classified within the spectrum of ectodermal dysplasias as a transcription factor disease [3]. TRPS1 is a transcriptional repressor that mediates cell differentiation in several tissues [4, 5] including the chondrocyte and perichondrium [6].Patients with missense mutations in TRPS1 have a more severe phenotype (TRPS III) than patients carrying nonsense mutations in this gene (TRSP I).
Clinical features
Clinically, the syndrome is characterized by growth retardation, cranio-facial abnormalities, brachydactyly of the hands and feet, sparse and slow-growing hair, a pear-shaped nose, elongated philtrum and thin upper lip (Box 67.4). Typical radiological findings include cone-shaped epiphyses, shortening of the metacarpals and phalanges, hip malformations and short stature. Nails have been described as dystrophic, hypoplastic, brittle, slow growing, koilonychotic and leukonychotic. Shortening and broadening of the nails giving rise to racket nail appearance has been reported repeatedly [7]. In addition, longitudinal striation is often observed [8].
Three subtypes of TRPS with considerable clinical overlap can be distinguished (Table 67.4). Type I may feature, in addition to the above mentioned characteristics, moderate postnatal growth retardation, rarely mental retardation and moderate brachydactyly with shortened phalanges and metacarpals. Eyebrows are generally sparse or may be medially thick and laterally thin.
Table 67.4 Clinical findings in the major forms of tricho-rhino-phalangeal syndrome (TRPS)
| TRPS I | TRPS II | TRPS III | |
| Inheritance | Autosomal dominant | Mostly sporadic | Autosomal dominant |
| Exostoses | – | +++ | – |
| Eyebrows | Normal or sparse | Normal or bushy | Normal |
| Loose skin | – | + | – |
| Mental retardation | – (described in single cases) | Often present | – |
| Microcephaly | – | + | – |
| Growth retardation | (+) | – | +++ |
| Brachydactyly | + | + | +++ |
| Abnormalities of metacarpals and phalanges | + | + | +++ |
| Genetic defect | Nonsense mutation in TRPS1 gene | Deletion in 8q24.11-13 | Missense mutation in TRPS1 gene |
Adapted from Itin et al. 1996 [12].
Type II is usually called the Langer–Giedion syndrome. TRPS II is characterized by the presence of multiple cartilaginous exostoses which clearly distinguishes it from TRPS I and III [9]. Mental retardation and microcephaly are typical features of TRPS II. Eyebrows are generally thick, broad and bushy. Redundant and loose skin in infancy, disappearing during childhood, has been reported in TRPS II. Growth retardation is only moderate and mild brachydactyly may be present. Most cases are sporadic. It has been shown that Langer–Giedion syndrome is a true contiguous gene syndrome due to the loss of functional copies of at least two genes, TRPS1 and EXT1, encoding exostosin glycosyltransferase 1, which is also involved in the pathogenesis of hereditary multiple exostoses [10].
Type III, also known as Sugio–Kajii syndrome, is the rarest form [11, 12]. It is characterized by marked short stature, severe brachydacytyly due to shortening of the phalanges and metacarpals and pronounced cone-shaped epiphyses. The eyebrows are normal; exostoses and mental deficiency are absent. Inheritance is autosomal dominant. Sporadic cases have been reported. Lüdecke et al. [13] performed extensive mutation analysis and concluded that TRPS III is at the severe end of the TRPS spectrum and that it is caused by mutations in the TRPS1 gene.
Resources
Patient resources
TRPS Support Group UK: http://trpsuk.org/ (last accessed August 2014).
Hidrotic ectodermal dysplasia
Definition and nomenclature
Hidrotic ectodermal dysplasia is a rare genodermatosis within the spectrum of ectodermal dysplasias [1]. The disorder affects skin, nails and hair but sweating is normal in affected patients. Mutations in a connexin gene lead to the symptom complex characterized by hypotrichosis and brittle hair, dystrophic nails and clubbing with prominent dermal ridges on the finger pulps and variable keratoderma. Hyperpigmentation especially over the joints is common [2].
Introduction and general description
Hidrotic ectodermal dysplasia or Clouston disease is an autosomal dominant inherited disorder and is characterized by the clinical occurrence of nail dystrophy, alopecia and keratoderma.
Epidemiology
Incidence and prevalence
The prevalence of hidrotic ectodermal dysplasia is given at 1–9 per 100 000 although no exact epidemiological studies exist [3, 4].
Age
Sparse hair and dystrophic nails are seen in early life. Continuous hair loss may lead to partial or total alopecia until puberty. Hair is sparse and brittle from early childhood. Palmoplantar keratoderma begins in childhood but aggravates with age. There is marked variability in the phenotype expression even within the same family.
Sex
As the disease has an autosomal dominant inheritance pattern, both sexes are equally affected.
Ethnicity
Initially, most cases were described in French Canadian families. Later, a family of Chinese Malay and individuals of African, Spanish and Irelish descent have been described so the disease seems to occur globally.
Associated diseases
Secondary conjunctivitis and blepharitis are due to sparse or absent eyelashes. Strabismus and cataract have been reported sporadically but do not seem to be really associated. Photophobia, conjunctivitis, blepharitis and sparse eyelashes and eyebrows are often present. Thickened skull bones without functional consequences may be observed. In single cases Clouston syndrome has been reported with mild sensorineural deafness and photophobia, which illustrates that the keratitis–ichthyosis–deafness syndrome is caused by a closely related mutation [5].
Pathophysiology
A mutation in the β gap junction protein gene, which is located on chromosome 13q12 and codes for connexin 30 (Cx30), is responsible for the disorder.
Pathology
Conventional histology of palms and soles shows orthohyperkeratosis with a normal granular layer. Under electron microscopy an increased number of desmosomes can be seen in the stratum corneum.
Genetics
This is an autosomal dominant inherited disorder due to a mutation in Cx30 [6]. Several cases of eccrine syringofibroadenomas have been described in hidrotic ectodermal dysplasia. Immunostaining in normal human skin sections demonstrates a predominant expression of Cx30 in the hair follicles, nails and palmoplantar epidermis, which partially overlaps with p63 expression. In addition, the co-expression of Cx30 and p63 in developing mouse hair follicles and nail units were documented. In cultured cells, Cx30 protein expression was significantly up-regulated by the ΔNp63α isoform. Further in vitro analyses suggested that ΔNp63α was potentially involved in the Cx30 expression via binding to the sequences in intron 1 of the Cx30 gene [7].
Clinical features
History
A positive family history may be present in this autosomal dominant disorder. As the clinical manifestations are highly variable, the history is not straightforward. About one-third of patients have nail dystrophies only. Hypotrichosis is progredient but is also very heterogeneous and can be diffuse or patchy. Paronychial infections are rather common and persistent.
Presentation
Patients present with partial or total alopecia (Figure 67.12). There are sparse scalp hairs and in infancy hairs are twisted, brittle and pale. Hair loss may lead to total alopecia by puberty. Eyelashes are short and sparse and eyebrows as well as axillary and pubic hair are also sparse or absent. The nails may be whitish in early childhood and then have a tendency to thicken. The nails are slow growing and often have striations and thickening and distal onycholysis. Affected nails are a very constant finding. The nails may be malformed and also small. A variable expression of palmoplantar keratoderma is known, and pronounced dermatoglyphics exist on the finger tips (Figure 67.13). Hyperpigmentation especially over joints are common.

Figure 67.12 Hypotrichosis in hidrotic ectodermal dysplasia.
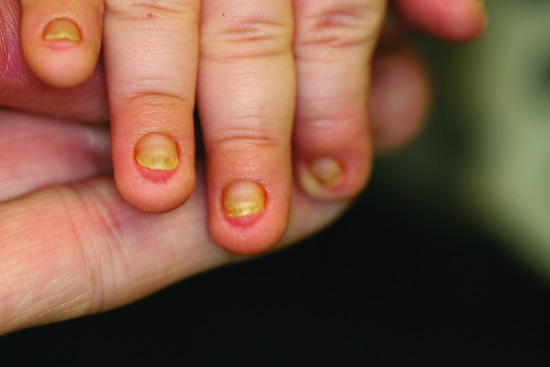
Figure 67.13 Hidrotic ectodermal dysplasia with micronychia, yellowish-coloured nail plates and pronounced dermatoglyphics.
Clinical variants
Phenotypic expression is highly variable.
Differential diagnosis
Differential diagnosis includes pachyonychia congenita and pure hair–nail ectodermal dysplasia. In 1996, an African family with six affected people over three generations was described with the association of short and sparse scalp hair and eyelashes. The nails were dystrophic and thickened but sweating, skin and teeth were normal.
Classification of severity
Mono- or oligosymptomatic expression of the disease is quite common. One-third of all patients with hidrotic ectodermal dysplasia have nail dystrophies as the only manifestation. The severity of disease expression is highly variable, even in the same family.
Disease course and prognosis
The clinical features increase over time but the expected life time is normal.
Investigations
To confirm the clinical diagnosis a gene mutation analysis for Cx30 is mandatory. However, insurance companies do not cover the costs for this analysis in all countries.
Management
There is no causal or specific treatment for this syndrome. Keratolytics and emollients should be used for palmoplantar keratoderma and in severe cases of alopecia wigs can be helpful. Professional pedicures and manicures can help to reduce the obvious aspects of nail dystrophy. It has been reported that tretinoin, which acts as mitogen by itself and also enhances the absorption of minoxidil – which acts by enlarging the miniaturized hair follicles – can be effective in hidrotic ectodermal dysplasia [8].
Resources
Further information
GeneReviews. Hidrotic ectodermal dysplasia 2. http://www.ncbi.nlm.nih.gov/books/NBK1200/ (last accessed August 2014).
Focal dermal hypoplasia
Definition and nomenclature
Focal dermal hypoplasia (FDH) belongs to the large group of ectodermal dysplasias. It is a rare genodermatosis that affects tissues of ectodermal and mesodermal origin. An X-linked dominant inheritance pattern is documented in numerous pedigrees with lethal effects to males which can only be survived by mosaicism. FDH has been associated with a mutation in the PORCN gene on the X chromosome that encodes a protein involved in the WNT signalling pathway. Clinically the syndrome is characterized by cutaneous, skeletal, dental, ocular and soft tissue defects.
Introduction and general description
In 1962 Goltz and co-workers defined a syndrome now known as FDH with numerous ectodermal and mesodermal malformations [1]. Only 200–300 cases have been described in the literature so far. Clinical key features are linear enamel hypoplasia according to the Blaschko linear pattern, periorificial papillomas, fat herniations and linear telangiectasias and pigmentation anomalies along Blaschko lines [2].
Epidemiology
Focal dermal hypoplasia is very rare and the prevalence is estimated at less than 1 in 1 000 000; 90% of affected patients are females. Mosaicism for mutations in PORCN or chromosomal anomalies underlie FDH in about 10% of affected males [3].
Age
Patients are born with multiple malformations and diagnosis is possible at birth.
Sex
The occurrence is 90% females and 10% males.
Ethnicity
The syndrome has been described all over the world and no ethnical or racial predisposition is known.
Associated diseases
It is an ectodermal–mesodermal malformation disease with numerous organs involved.
Pathophysiology
Focal dermal hypoplasia is inherited as an X-linked dominant trait, with 90% of affected individuals being female. Embryological development is programmed in development fields not only restricted to one tissue. During embryological differentiation, the growth process is controlled and coordinated in a spatially ordered and epimorphologically hierarchical manner. FDH is caused by mutations in the PORCN gene encoding an important element of the WNT signalling pathway. WNT proteins play important roles in embryo development, tissue homeostasis and stem cell maintenance. Consistent with the female-specific inheritance pattern of FDH, PORCN hemizygous Drosophila male embryos show growth arrest during early embryogenesis and fail to generate mesoderm, a phenotype previously associated with loss of Wnt activity and similar to the human homologue.
Pathology
Skin biopsies from the erythematous lesions show acanthosis and slight hyperkeratosis of the epidermis, sometimes with subepidermal cleft formation. In atrophic lesions the epidermis shows atrophy and hyperpigmentation of the basal cell layer. A wedge-shaped area of scar formation composed of hyaline collagen bundles in parallel arrangement and numerous fibroblasts is often found beneath the epidermis. The fatty tissue almost reaches the epidermis because collagen fibres of dermal tissue are scarce. This phenomenon has been described as heterotopic fat [4]. Blood vessels with prominent endothelial cells are found in large numbers within and at the periphery of lesional tissue. Some blood vessels may be densely filled with erythrocytes, and extravasations of red blood cells with small areas of haemorrhage can be seen. Small islands of adipose tissue are common in close vicinity to the fibrous tissue in the lower reticular dermis. A positive immunohistochemical reaction for vimentin, fibronectin and collagen type III is observed in the scar tissue. No collagen type IV is detected in the basement membrane zone of the epidermis covering the lesion [5].
Genetics
An X-chromosomal dominant inheritance pattern with lethality in males is typical in FDH [6]. Mutations in the PORCN gene lead to changes in the WNT cascade, which is very important during embryogenesis.
Clinical features
History
The clinical spectrum of FDH includes abnormalities of the skin and various defects of the eyes, bones, teeth, nails, oral structures and soft tissue [7]. The most prominent clinical features of the cutaneous disease are reddish or red-yellow cribriform atrophic skin lesions. Hyperpigmentation or hypopigmentation is common in atrophic sites. Telangiectases in atrophic skin is a common finding (Figure 67.14). A Blaschko linear distribution pattern is typical and the areas involved are usually the trunk and extremities but the lesions may be found on any part of the body. Linear enamel hypoplasia is an important key feature (Figure 67.15). Numerous oral alterations have been described including hypodontia, jaw cysts, clefting, hemihypoglossia of the tongue and papillomatosis. Other cutaneous abnormalities include lipomatous nodules and papillomatous lesions. Ocular abnormalities occur in about 40% and include coloboma, strabism, microphthalmia and nystagmus. Skeletal changes include syndactyly (Figure 67.16), polydactyly, lobster claw deformation and osteopathia striata.
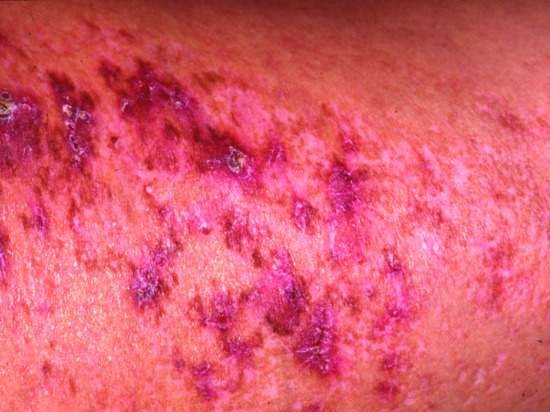
Figure 67.14 Striate atrophic and telangiectatic lesions in focal dermal hypoplasia.
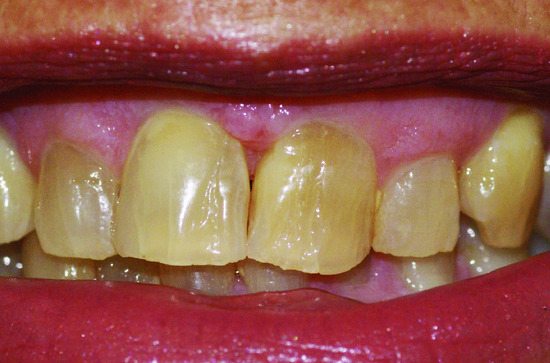
Figure 67.15 Blaschko linear enamel hypoplasia in focal dermal hypoplasia.
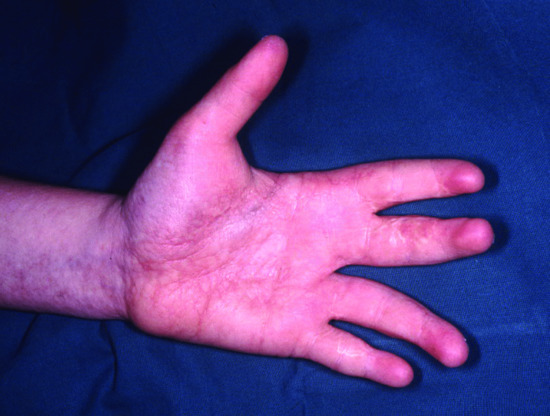
Figure 67.16 Operated syndactyly in focal dermal hypoplasia.
Differential diagnosis
Differential diagnoses include the group of focal facial dermal dysplasias types 1–4 and MIDAS syndrome (see following section).
Management
Clinical management of patients with FDH should be organized interdisciplinary. Children should be presented to the dentist and ophthalmologist as soon as possible. Orthopaedic specialists are also important in the treatment of affected patients. In addition psychological and behavioral management is necessary.
Resources
Patient resources
National Center for Advancing Translational Sciences, overview of organizations supporting patients with FDH: http://rarediseases.info.nih.gov/gard/6457/focal-dermal-hypoplasia/resources/5 (last accessed August 2014).
MIDAS syndrome
Definition and nomenclature
MIDAS syndrome (microphthalmia, dermal aplasia and sclerocornea), coined by Happle, is also named microphthalmia with linear skin defects syndrome. It is an X-linked dominant disorder with male lethality characterized by unilateral or bilateral microphthalmia with linear skin defects limited to the face and neck. It is a very rare disease with <1:1 000 000.
A related form of congenital linear skin defects with microcephaly, facial dysmorphism and additional malformations exists and is caused by a mutation in the COX7B gene on chromosome Xq21.
Introduction
Microphthalmia with linear skin defects syndrome is a rare X-linked male-lethal disorder with microphthalmia and other ocular malformations and linear aplastic skin lesions which are usually limited to the face and neck and develop hyperpigmented skin alterations in healed aplastic skin with age. In addition, microcephaly, intellectual disabilities, agenesis of corpus callosum and cardiac malformations may occur. Most patients have a cytogenetically visible deletion or unbalanced translocation leading to Xp22 monosomy. Heterozygous intragenic mutations in HCCS have been shown to be responsible for the disease [1]. HCCS encodes the holocytochrome c-type synthase, which catalyses the incorporation of heme moieties into cytochrome c and cytochrome c1. Cytochrome c plays a crucial role in oxidative phosphorylation, but also has been shown to regulate apoptosis. Cells lacking HCCS activity may therefore be prone to undergo apoptosis, which in turn may affect tissue formation. Further supporting a role for mitochondrial enzymes in the pathogenesis of MIDAS syndrome, mutations in another gene, COX7B on Xq21.1, encoding a structural subunit of cytochrome c oxidase, have been found in females with MIDAS phenotype [2].
Epidemiology
Incidence and prevalence
The disease is very rare with fewer than 1:1 000 000 cases.
Age
Clinical manifestations are partially apparent at birth and internal malformations or intellectual impairment unmask within the first years of life.
Sex
It is an X-linked dominant disorder with lethality in males.
Ethnicity
No racial predilections are known.
Associated diseases
The syndrome may be complicated by cardiac malformations (18%), hypoplastic corpus callosum and abnormal myelination, microcephaly, mild to severe mental retardation (25%), hearing loss, diaphragm herniation, anterior-placed anus or imperforate anus, and genitourinary malformations.
Clinical features
History
Family history may reveal a mother to daughter transmission.
Presentation
The most consistent clinical features were microphthalmia/anophthalmia (93%) and sclerocornea/corneal opacity and congenital linear skin defects in a Blaschko-line pattern on head and neck (95%). Additional manifestations are various ocular anomalies, cardiac defects, brain imaging abnormalities, microcephaly, postnatal growth retardation, and facial dysmorphism. Nail dystrophies may occur.
Clinical variants
A related form of congenital linear skin defects with microcephaly, facial dysmorphisms and additional malformations exists and is caused by a mutation in the COX7B gene on chromosome Xq21.
Differential diagnosis
Differential diagnoses include focal dermal hypoplasia, incontinentia pigmenti (Bloch–Sulzberger disease), oculocerebrocutaneous syndrome and Aicardi syndrome.
Investigations
Ophthalmological and dermatological examinations, brain MRI with and without contrast, hearing evaluation and cardiac evaluation are indicated. Clinically suggestive constellations for MIDAS syndrome should be confirmed by chromosome analyses and molecular genetic analyses which document a mutation in HCCS or COX7B.
Management
Eye and skin problems should be managed by specialized institutions. Patients with MIDAS syndrome should be checked for cardiac, urogenital and central nervous malformations. Learning disability should be evaluated in an early time point.
Focal facial dermal dysplasia
Definition and nomenclature
Focal facial dermal dysplasia (FFDD) represents a heterogeneous group of rare disorders with developmental defects on the bilateral facial areas. The lesional skin resembles aplasia cutis congenita. There are four types of FFDD [1]. Type 1 is also called Brauer syndrome and is inherited in an autosomal dominant pattern; the skin abnormalities in the bitemporal area resemble forceps marks. In addition sparse lateral eyebrows, a flattened nasal tip and distichiasis can be observed. Type 2 is also called Brauer–Setleis syndrome. The clinical features are very similar compared with type 1 but the periorbital skin is also affected with atrophic and wrinkling skin. Inheritance is also autosomal dominant. Type 3 is called Setleis syndrome and has the same clinical characteristics as type 2 but inheritance is autosomal recessive. The responsible gene defect has been located in the TWIST2 gene [2]. Type 4 shows isolated preauricular skin lesions and the inheritance pattern can be autosomal recessive or dominant [3]. It is caused by mutations in the CYP26C1 gene [4].
Epidemiology
Focal facial dermal dysplasia is a very rare disease group with less than 1 : 1 000 000 incidence.
Clinical features
The common denominator of all types in FFDD is an atrophy-like skin change in the bitemporal region.
References
Ectodermal dysplasias
- Itin PH. Ectodermal dysplasia: thoughts and practical concepts concerning disease classification – the role of functional pathways in the molecular genetic diagnosis. Dermatology 2013;226:111–14.
- Visinoni AF, Lisboa-Costa T, Pagnan NA, Chautard-Freire-Maia EA. Ectodermal dysplasias: clinical and molecular review. Am J Med Genet 2009;149A:1980–2002.
- Itin PH. Rationale and background as basis for a new classification of the ectodermal dysplasias. Am J Med Genet 2009;149A:1973–6.
- Danz DFG. Sechste Bemerkung. Von Menschen ohne Haare und Zähne. Arch Geb Frauen Neugeb Kinderkr 1793;4:684.
- Weech AA. Hereditary ectodermal dysplasia (congenital ectodermal defect). A report of two cases. Am J Dis Child 1929;37:766–90.
- Itin PH, Fistarol SK. Ectodermal dysplasias. Am J Med Genet 2004;131C:45–51.
- Pinheiro M, Freire-Maia N. Ectodermal dysplasias: a clinical classification and a causal review. Am J Med Genet 1994;53:153–62.
- Celik TH, Buyukcam A, Simsek-Kiper PO, et al. A newborn with overlapping features of AEC and EEC syndromes. Am J Med Genet 2011;155A:3100–3.
- DiGiovanna JJ, Priolo M, Itin P. Approach towards a new classification for ectodermal dysplasias: integration of the clinical and molecular knowledge. Am J Med Genet 2009;149A:2068–70.
- Nguyen-Nielsen M, Skovbo S, Svaneby D, Pedersen L, Fryzek J. The prevalence of X-linked hypohidrotic ectodermal dysplasia (XLHED) in Denmark, 1995–2010. Eur J Med Genet 2013;56:236–42.
- Strachan LR, Hadially R. Tiers of clonal organization in the epidermis: the epidermal proliferation unit revisited. Stem Cell Rev 2008;4:149–57.
- De Robertis EM, Larrain J, Oelgeschläger M, Wessely O. The establishment of Spemann's organizer and patterning of the vertebrate embryo. Nature Rev Genet 2000;1:171–81.
- Häärä O, Fujimori S, Schmidt-Ullrich R, Hartmann C, Thesleff I, Mikkola ML. Ectodysplasin and Wnt pathways are required for salivary gland branching morphogenesis. Development 2011;138:2681–91.
- Van Raamsdonk CD. Hereditary hair loss and the ancient signaling pathways that regulate ectodermal appendage formation. Clin Genet 2009;76:332–40.
- Janesick A, Shiotsugu J, Taketani M, Blumberg B. RIPPLY3 is a retinoic acid-inducible repressor required for setting the borders of the pre-placodal ectoderm. Development 2012;139:1213–24.
- Lefebvre S, Fliniaux I, Schneider P, Mikkola ML. Identification of ectodysplasin target genes reveals the involvement of chemokines in hair development. J Invest Dermatol 2012;132:1094–102.
- Gong B, Liu T, Zhang X, et al. Disease embryo development network reveals the relationship between disease genes and embryo development genes. J Theor Biol 2011;287:100–8.
- Sadier A, Viriot L, Pantalacci S, Laudet V. The ectodysplasin pathway: from diseases to adaptations. Trends Genet 2014;30(1):24–31.
- Pummila M, Fliniaux I, Jaatinen R, et al. Ectodysplasin has a dual role in ectodermal organogenesis: inhibition of Bmp activity and induction of Shh expression. Development 2007;134:117–25.
- Wend P, Wend K, Krum SA, Miranda-Carboni GA. The role of WNT10B in physiology and disease. Acta Physiol 2012;204:34–51.
- Widelitz RB. Wnt signaling in skin organogenesis. Organogenesis 2008;4:123–33.
- Bohring A, Stamm T, Spaich C, et al. WNT10A mutations are a frequent cause of a broad spectrum of ectodermal dysplasias with sex-biased manifestation pattern in heterozygotes. Am J Hum Genet 2009;85:97–105.
- Van den Boogaard MJ, Créton M, Bronkhorst Y, et al. Mutations in WNT10A are present in more than half of isolated hypodontia cases. J Med Genet 2012;49:327–31.
- Cluzeau C, Hadj-Rabia S, Jambou M, et al. Only four genes (EDA1, EDAR, EDARADD, and WNT10A) account for 90% of hypohidrotic/anhidrotic ectodermal dysplasia cases. Hum Mut 2011;32:70–2.
- Koster MI. p63 in skin development and ectodermal dysplasias. J Invest Dermatol 2010;130:2352–8.
- McDade SS, Henry AE, Pivato GP, et al. Genome-wide analysis of p63 binding sites identifies AP-2 factors as co-regulators of epidermal differentiation. Nucleic Acids Res 2012;40:7190–206.
- Wu N, Rollin J, Masse I, Lamartine J, Gidrol X. p63 regulates human keratinocyte proliferation via MYC-regulated gene network and differentiation commitment through cell adhesion-related gene network. J Biol Chem 2012;287:5627–38.
- Kantaputra PN, Malaivijtnond S, Vieira AR, et al. Mutation in SAM domain of TP63 is associated with nonsyndromic cleft lip and palate and cleft palate. Am J Med Genet 2012;155A:1432–6.
- Shalom-Feuerstein R, Serror L, Aberdam E, et al. Impaired epithelial differentiation of induced pluripotent stem cells from ectodermal dysplasia-related patients is rescued by the small compound APR-246/PRIMA-1MET. Proc Natl Acad Sci USA 2013;110:2152–6.
- Baldridge D, Shchelochkov O, Kelly B, Lee B. Signaling pathways in human skeletal dysplasias. Annu Rev Genomics Hum Genet 2012;11:189–217.
- Marvin ML, Mazzoni SM, Herron CM, Edwards S, Gruber SB, Petty EM. AXIN2-associated autosomal dominant ectodermal dysplasia and neoplastic syndrome. Am J Med Genet 2011;155A:898–902.
- Petrof G, Mellerio JE, McGrath JA. Desmosomal genodermatoses. Br J Dermatol 2012;166:36–45.
- Lindfors PH, Voutilainen M, Mikkola ML. Ectodysplasin/NF-kB signaling in embryonic mammary gland development. J Mammary Gland Biol Neoplasia 2013;18:165–9.
- Courtois G, Smahi A. NF-kappaB-related genetic diseases. Cell Growth Differ 2006;13:843–51.
- Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol 2000;18:621–63.
- Kere J, Srivastava AK, Montonen O, et al. X-linked anhidrotic (hypohidrotic) ectodermal dysplasia is caused by mutation in a novel transmembrane protein. Nature Genet 1996;13:409–16.
- Mathew SJ, Haubert D, Krönke M, Leptin M. Looking beyond death: a morphogenetic role for the TNF signalling pathway. J Cell Sci 2009;122:1939–46.
- Headon DJ, Overbeek PA. Involvement of a novel Tnf receptor homologue in hair follicle induction. Nature Genet 1999;22:370–4.
- Headon DJ, Emmal SA, Ferguson BM, et al. Gene defect in ectodermal dysplasia implicates a death domain adapter in development. Nature 2001;414:913–16.
- Smahi A, Courtois G, Rabia SH, et al. The NF-kappaB signalling pathway in human diseases: from incontinentia pigmenti to ectodermal dysplasias and immune-deficiency syndromes. Hum Mol Genet 2002;11:2371–5.
- Zonana J, Elder ME, Schneider LC, et al. A novel X-linked disorder of immune deficiency and hypohidrotic ectodermal dysplasia is alleleic to incontinentia pigmenti and due to mutations in IKK-gamma (NEMO). Am J Hum Genet 2000;67:1555–62.
- Doffinger R, Smahi A, Bessia C, et al. X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-kB signaling. Nature Genet 2001;27:277–85.
- Wisniewsky SA, Trzeciak WH. A rare heterozygous TRAF6 variant is associated with hypohidrotic ectodermal dysplasia. Br J Dermatol 2012;166:1353–6.
- Candi E, Cipollone R, Rivetti di Val Cervo P, Gonfloni S, Melino G, Knight R. p63 in epithelial development. Cell Mol Life Sci 2008;65:3126–33.
- Zarnegar BJ, Webster DE, Lopez-Pajares V, et al. Genomic profiling of a human organotypic model of AEC syndrome reveals ZNF750 as an essential downstream target of mutant TP63. Am J Hum Genet 2012;91:435–43.
- Browne G, Cipollone R, Lena AM, et al. Differential altered stability and transcriptional activity of Np63 mutants in distinct ectodermal dysplasias. J Cell Sci 2012;124:2200–7.
- Bellomaria A, Barbato G, Melino G, Paci M, Melino S. Recognition mechanism of p63 by the E3 ligase Itch: novel strategy in the study and inhibition of this interaction. Cell Cycle 2012;11:3638–48.
- Yang A, Schweitzer R, Sun D, et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 1999;398:714–18.
- Brunner HG, Hamel BCJ, van Bokhoven H. P63 gene mutations and human developmental syndromes. Am J Med Genet 2002;112:284–90.
- Van Bokhoven H, Hamel BCJ, Bamshad M et al. p63 gene mutations in EEC syndrome, limb-mammary syndrome, and isolated split hand-split foot malformation suggest a genotype-phenotype correlation. Am J Hum Genet 2001;69:481–92.
- Amiel J, Bougeard G, Francannet C, et al. TP63 gene mutation in ADULT syndrome. Eur J Hum Genet 2001;9:642–5.
- Cummings CJ, Zoghbi HY. Fourteen and counting: unraveling trinucleotide repeat diseases. Hum Mol Genet 2000;9:909–16.
- Guazzarotti L, Caprio C, Rinne TK, et al. Limb-mammary syndrome (LMS) associated with internal female genitalia dysgenesia: a new genotype/phenotype correlation? Am J Med Genet 2008;146:2001–4.
- Gerstein MB, Kundaje A, Hariharan M, et al. Architecture of the human regulatory network derived from ENCODE data. Nature 2012;489:91–100.
- Ruiz-Perez VL, Ide SE, Strom TM, et al. Mutations in a new gene in Ellis–van Creveld syndrome and Weyers acrodental dysostosis. Nat Genet 2000;24:283–6.
- Yang C, Chen W, Chen Y, Jiang J. Smoothened transduces Hedgehog signal by forming a complex with Evc/Evc2. Cell Res 2012;22:1593–604.
- Memarpour M, Shafiei F. Witkop tooth and nail syndrome: a report of three cases in a family. Pediatr Dermatol 2011;28:281–5.
- Di Costanzo A, Festa L, Roscigno G, et al. A dominant mutation etiologic for human tricho-dento-osseous syndrome impairs the ability of DLX3 to downregulate Np63alfa. J Cell Physiol 2011;226:2189–97.
- Gai Z, Gui T, Muragaki Y. The function of TRPS1 in the development and differentiation of bone, kidney, and hair follicles. Histol Histopathol 2011;26:915–21.
- Van Steensel MA, Koedam MI, Swinkels OQ, Rietveld F, Steijlen PM. Woolly hair, premature loss of teeth, nail dystrophy, acral hyperkeratosis and facial abnormalities: possible new syndrome in a Dutch kindred. Br J Dermatol 2001;145(1):157–61.
- Visinoni AF, Lisboa-Costa T, Pagnan NA, Chautard-Freire-Maia EA. Ectodermal dysplasias: clinical and molecular review. Am J Med Genet 2009;149A:1980–2002.
- Visinoni AF, Lisboa-Costa T, Pagnan NA, Chautard-Freire-Maia EA. Ectodermal dysplasias: clinical and molecular review. Am J Med Genet 2009;149A:1980–2002.
X-linked hypohidrotic ectodermal dysplasia with immunodeficiency
- Kawai T, Nishikomori R, Heike T. Diagnosis and treatment in anhidrotic ectodermal dysplasia with immunodeficiency. Allergol Int 2012;61:207–17.
- Kawai T, Nishikomori R, Izawa K, et al. Frequent somatic mosaicism of NEMO in T cells of patients with X-linked anhidrotic ectodermal dysplasia with immunodeficiency. Blood 2012;119:5458–66.
- Doffinger R, Smahi A, Bessia C, et al. X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-kB signaling. Nature Genet 2001;27:277–85.
- Temmerman ST, Ma CA, Zhao Y, et al. Defective nuclear IKK alfa function in patients with ectodermal dysplasia with immune deficiency. J Clin Invest 2012;122:315–26.
- Ngadjeua F, Chiaravalli J, Traincard FC, Raynal B, Fontan E, Agou F. Two-sided ubiquitin binding of NF-kB essential modulator (NEMO) zinc finger unveiled by a mutation associated with anhidrotic ectodermal dysplasia with immunodeficiency syndrome. J Biol Chem 2013;288(47):33722–37.
- Mancini AJ, Lawley LP, Uzel G. X-linked ectodermal dysplasia with immunodeficiency caused by NEMO mutation: early recognition and diagnosis. Arch Dermatol 2008;144:342–6.
Hypohidrotic ectodermal dysplasia
- Itin PH, Fistarol SK. Ectodermal dysplasias. Am J Med Genet 2004;131C:45–51.
- Nguyen-Nielsen M, Skovbo S, Svaneby D, Pedersen L, Fryzek J. The prevalence of X-linked hypohidrotic ectodermal dysplasia (XLHED) in Denmark, 1995–2010. Eur J Med Genet 2013;56:236–42.
- Haghighi A, Nikuei P, Haghighi-Kakhki H, et al. Whole-exome sequencing identifies a novel missense mutation in EDAR causing autosomal recessive hypohidrotic ectodermal dysplasia with bilateral amastia and palmoplantar hyperkeratosis. Br J Dermatol 2013;168:1353–6.
- Itin PH. Ectodermal dysplasia: thoughts and practical concepts concerning disease classification – the role of functional pathways in the molecular genetic diagnosis. Dermatology 2013;226:111–14.
- Clarke A, Phillips DIM, Brown R, Harper PS. Clinical aspects of X-linked hypohidrotic ectodermal dysplasia. Arch Dis Child 1987;62:989–96.
- Gaide O, Schneider P. Permanent correction of an inherited ectodermal dysplasia with recombinant EDA. Nat Med 2003;9(5):614–18.
- Aswegan AL, Josephson KD, Mowbray R, Pauli RM, Spritz RA, Williams MS. Autosomal dominant hypohidrotic ectodermal dysplasia in a large family. Am J Med Genet 1997;72:462–7.
Ankyloblepharon–ectodermal defect–cleft lip/palate syndrome
- Mancini AJ, Paller AS. What syndrome is this? Ankyloblepharon-ecteodermal defects-cleft lip and palate (Hay-Wells) syndrome. Pediatr Dermatol 1997;14:403–5.
- Cambiaghi S, Tadini G, Barbareschi M, Menni S, Caputo R. Rapp-Hodgkin syndrome and AEC syndrome: are they the same entity? Br J Dermatol 1994;130:97–101.
- Dishop MK, Bree AF, Hicks MJ. Pathologic changes of skin and hair in ankyloblepharon-ectodermal defects-cleft lip/palate (AEC) syndrome. Am J Med Genet 2009;149A:1935–41.
- Vanderhooft SL, Stephan MJ, Sybert VP. Severe skin erosions and scalp infections in AEC syndrome. Pediatr Dermatol 1993;10:334–40.
- Julapalli MR, Scher RK, Sybert VP, Siegfried EC, Bree AF. Dermatologic findings of ankyloblepharon-ectodermal defects-cleft lip/palate (AEC) syndrome. Am J Med Genet 2009;149A:1900–6.
- Bertola DR, Kim CA, Sugayama SMM, Albano LMJ, Utagawa CY, Gonzalez CH. AEC syndrome and CHAND syndrome: further evidence of clinical overlapping in the ectodermal dysplasias. Pediatr Dermatol 2000;17:218–21.
- McGrath JA, Duijf PH, Doetsch V, et al. Hay-Wells syndrome is caused by heterozygous missense mutations in the SAM domain of p63. Hum Mol Genet 2001;10:221–9.
- Celik TH, Buyukcam A, Simsek-Kiper PO, et al. A newborn with overlapping features of AEC and EEC syndromes. Am J Med Genet 2011;155A:3100–3.
- Cole P, Hatef DA, Kaufman Y, et al. Facial clefting and oroauditory pathway manifestations in ankyloblepharon-ectodermal defects-cleft lip/palate (AEC) syndrome. Am J Med Genet 2009;149A:1910–15.
- Camacho F, Ferrando J, Pichardo AR, Sotillo I, Jorquera E. Rapp-Hodgkin syndrome with pili canaliculi. Pediatr Dermatol 1993;10:54–7.
Ectrodactyly–ectodermal dysplasia–cleft lip/palate syndrome
- Maclean K, Holme SA, Gilmour E, et al. EEC syndrome, Arg227Gln TP63 mutation and micturition difficulties: is there a genotype–phenotype correlation? Am J Med Genet 2007;143A:1114–19.
- Doray B, Badila-Timbolschi D, Schaefer E, et al. Epidemiology of orofacial clefts (1995–2006) in France (Congenital Malformations of Alsace Registry). Arch Pediatr 2012;19:1021–9.
- Vernersson Lindahl E, Garcia EL, Mills AA. An allelic series of Trp63 mutations defines TAp63 as a modifier of EEC syndrome. Am J Med Genet 2013;161A:1961–71.
- Rinne T, Hamel B, van Bokhoven H, Brunner HG. Pattern of p63 mutations and their phenotypes – update. Am J Med Genet 2006;140A:1396–406.
- Rios LT, Araujo Junior E, Caetano AC, Nardozza LM, Moron AF, Martins MG. Prenatal diagnosis of EEC syndrome with “lobster claw” anomaly by 3D ultrasound. J Clin Imaging Sci 2012;2:40.
- Ruiz-Perez VL, Thompson SW, Blair HJ, et al. Mutations in two nonhomologous genes in a head-to-head configuration cause Ellis–van Creveld syndrome. Am J Hum Genet 2003;72:728–32.
- Birnbaum S, Reutter H, Lauster C, et al. Mutation screening in the IRF6-gene in patients with apparently nonsyndromic orofacial clefts and a positive family history suggestive of autosomal-dominant inheritance. Am J Med Genet 2008;146A:787–90.
- Kondo S, Schutte BC, Richardson RJ, et al. Mutations in IRF6 cause Van der Woude and popliteal pterygium syndromes. Nature Genet 2002;32:285–9.
- Goldsmith T, Eytan O, Magal L, et al. A mutation in TP63 causing a mild ectodermal dysplasia phenotype. J Invest Dermatol 2014;134:2277–80.
- Shalom-Feuerstein R, Serror L, Aberdam E, et al. Impaired epithelial differentiation of induced pluripotent stem cells from ectodermal dysplasia-related patients is rescued by the small compound APR-246/PRIMA-1MET. Proc Natl Acad Sci USA 2013;110:2152–6.
Tricho-rhino-phalangeal syndrome
- Giedion A. Das tricho-rhino-phalangeale syndrom. Helv Paediatr Acta 1966;5:475–82.
- Gai Z, Gui T, Muragaki Y. The function of TRPS1 in the development and differentiation of bone, kidney, and hair follicles. Histol Histopathol 2011;26:915–21.
- Lamartine J. Towards a new classification of ectodermal dysplasias. Clin Exp Dermatol 2003;28:351–5.
- Momeni P, Glöckner G, Schmidt O, et al. Mutations in a new gene, encoding a zinc-finger protein, cause tricho-rhino-phalangeal syndrome type I. Nature Genet 2000;24:71–4.
- Malik TH, Von Stechow D, Bronson RT, Shivdasani RA. Deletion of the GATA domain of TRPS1 causes an absence of facial hair and provides new insights into the bone disorder in inherited tricho-rhino-phalangeal syndromes. Mol Cell Biol 2002;24:8592–600.
- Napierla D, Sam K, Morello R, et al. Uncoupling of chondrocyte differentiation and perichondrial mineralization underlies the skeletal dysplasia in tricho-rhino-phalangeal syndrome. Hum Mol Genet 2008;17:2244–54.
- Seitz CS, Lüdecke HJ, Wagner N, Bröcker EB, Hamm H. Trichorhinophalangeal syndrome I. Clinical and molecular characterization of 3 members of a family and 1 sporadic case. Arch Dermatol 2001;137:1437–42.
- Lalevic BM, Nikolic MM, Polic DJ. Etude des cheveux du syndrome trichorhinophalangien type I. Ann Dermatol Venereol 1994;121:618–22.
- Hall BD, Langer LV, Giedion A, et al. Langer-Giedion syndrome. Birth Defects 1974;10:147–64.
- Hou J, Parrish J, Lüdecke HJ, et al. A 4-megabase YAC contig that spans the Langer-Giedion syndrome region on human chromosome 8q24.1: use in refining the location of the trichorhinophalangeal syndrome and multiple exostoses genes (TRPS1 and EXT1). Genomics 1995;29:87–97.
- Niikawa N, Kamei T. The Sugio-Kajii syndrome, proposed tricho-rhino-phalangeal syndrome type III. Am J Med Genet 1986;24:759–60.
- Itin PH, Bohn S, Mathys D, Guggenheim R, Richard G. Trichorhinophalangeal syndrome type III. Dermatology 1996;193:349–52.
- Lüdecke HJ, Schaper J, Meinecke P, et al. Genotypic and phenotypic spectrum in tricho-rhino-phalangeal syndrome types I and III. Am J Hum Genet 2001;68:81–91.
Hidrotic ectodermal dysplasia
- Fraser FC, Der Kaloustian VM. A man, a syndrome, a gene: Clouston's hidrotic ectodermal dysplasia (HED). Am J Med Genet 2001;100:164–8.
- Fulk CS. Hidrotic ectodermal dysplasia. Report of a case with reticulated acropigmentation. J Am Acad Dermatol 1982;6:476–80.
- Koch P, Foss P, Baum HP, Zaun H. Warzenförmige palmoplantare Keratodermie als charakteristisches Merkmal der hidrotischen ektodermalen Dysplasie vom Typ Clouston. Hautarzt 1995;46:272–5.
- Ando Y, Tanaka T, Horiguchi Y, Ikai K, Tomono H. Hidrotic ectodermal dysplasia: a clinical and ultrastructural observation. Dermatologica 1988;176:205–11.
- Suigura K, Teranishi M, Matsumoto Y, Akiyama M. Clouston syndrome with heterozygous GJB6 mutation p.Ala88Val and GJB2 variant p.Val27Ile revealing mild sensorineural hearing loss and photophobia. JAMA Dermatol 2013;epub.
- Lamartine J, Essenfelder GM, Kibar Z, et al. Mutations in GJB6 cause hidrotic ectodermal dysplasia. Nature Genet 2000;26:142–4.
- Fujimoto A, Kurban M, Nakamura M, et al. GJB6, of which mutations underlie Clouston syndrome, is a potential direct target gene of p63. J Dermatol Sci 2013;69:159–66.
- Melkote S, Duhrat RS, Palav A, Jerajani HR. Alopecia in congenital hidrotic ectodermal dysplasia responding to treatment with a combination of topical minoxidil and tretinoin. Int J Dermatol 2009;48:184–5.
Focal dermal hypoplasia
- Goltz RW, Peterson WC, Gorlin RJ, Ravits HG. Focal dermal hypoplasia. Arch Dermatol 1962;86:708–17.
- Itin P. Alterations in nails and teeth as a clue for genodermatoses. Hautarzt 2014;65:513–19.
- Lombardi MP, Bulk S, Celli J, et al. Mutation updat for the PORCN gene. Hum Mutat 2011;32:723–8.
- Howell JB, Freeman RG. Cutaneous defects of focal dermal hypoplasia: an ectomesodermal dysplasia syndrome. J Cutan Pathol 1989;16:237–58.
- Büchner SA, Itin P. Focal dermal hypoplasia syndrome in a male. Case report, histologic and immunohistochemical studies. Arch Dermatol 1992;128:1078–82.
- Hamm H. Cutaneous mosaicism of lethal mutations. Am J Med Genet 1999;85:342–5.
- Wang L, Jin X, Zhao X, et al. Focal dermal hypoplasia: updates. Oral Dis 2014;20:17–24.
MIDAS syndrome
- Wimplinger I, Morelo M, Rosenberger G, et al. Mutations of the mitochondrial holocytochrome c-type synthase in X-linked dominant microphthalmia with linear skin defects syndrome. Am J Hum Genet 2006;79:878–89.
- Indrieri A, van Rahden VA, Tiranti V, et al. Mutations in COX7B cause microphthalmia with linear skin lesions, an unconventional mitochondrial disease. Am J Hum Genet 2012;91:942–9.
Focal facial dermal dysplasia
- Kowalski DC, Fenske NA. The focal facial dermal dysplasias: report of a kindred and a proposed new classification. J Am Acad Dermatol 1992;27:575–82.
- Turkel T, Sosic D, Al-Gazali LI, et al. Homozygous nonsense mutations in TWIST2 cause Setleis syndrome. Am J Hum Genet 2014;87:289–96.
- Coughlin CC, Dunbar SW, Bayliss SJ, Berk DR. Focal preauricular dermal dysplasia in a newborn. Pediatr Dermatol 2013;30:e259–60.
- Slavotinek AM, Mehrotra P, Nazarenko I, et al. Focal facial dermal dysplasia, type IV, is caused by mutations in CYP26C1. Hum Mol Genet 2013;22:696–03.