CHAPTER 68
Inherited Hair Disorders
Eli Sprecher
Department of Dermatology, Tel Aviv Sourasky Medical Center, Tel Aviv, Israel
Definition
Congenital and inherited disorders of hair growth and differentiation, also known as genotrichoses, can be subdivided into conditions associated with either excessive hair growth, known as hypertrichoses, or defective hair development termed alopecias [1]. Diseases in which hair is absent from the entire surface of the skin are known as atrichias whereas disorders featuring hair paucity are named hypotrichoses. Some of these disorders are associated with structural abnormalities of the hair shafts. Finally, inherited disorders of the hair follicle unit can present in isolation or as part of more complex clinical entities in which case they are known as syndromic genotrichoses. In the following sections, the clinical and pathogenetic features of representative genotrichoses are provided. A list of all major disorders associated with hypertrichoses and alopecias is provided in Tables 68.1, 68.2 and 68.3.
Table 68.1 Hypertrichoses
| Disease name | MIM | Inheritance | Gene or genetic defect | Cutaneous features | Extracutaneous features |
| A Hypertrichosis as main disease feature | |||||
| Hypertrichosis universalis congenital, Ambras type | 145701 | AD | Complex rearrangements on chromosome 8/possible involvement of TRPS1 |
Excessive vellus hair growth involving entire body surface | Facial dysmorphism Skeletal abnormalities |
| Hypertrichosis universalis | 145700 | AD | Unknown | Excessive lanugo-type hair growth Double eyebrows |
– |
| Generalized hypertrichosis terminalis | 135400 | AD | CNV variations on chromosome 17/possible involvement of SOX9 or ABCA5 (one case) | Excessive terminal hair growth involving face and body | Facial dysmorphism Gingival hyperplasia |
| Congenital generalized hypertrichosis | 307150 | RXL | Insertions on chromosome Xq27.1/possible involvement of FGF13 | Congenital generalized hypertrichosis in males;patchy hypertrichosis in females | Scoliosis Dental/palate anomalies Deafness |
| Cantu syndrome | 239850 | AD | ABCC9 | Hypertrichosis | Macrosomia Facial dysmorphism Gingival hyperplasia Bone abnormalities Cardiac defects Mild mental retardation |
| Histiocytosis and lymphadenopathy syndrome (H syndrome) | 602782 | AR | SLC29A3 | Hyperpigmentation, localized hypertrichosis | Hepatosplenomegaly, heart anomalies, hearing loss, hypogonadism, short stature, hyperglycaemia |
| Trichomegaly | 190330 | AR | FGF5 | Long eyelashes. Hypertrichosis of eyebrows, cheeks and forehead. |
– |
| B Hypertrichosis as minor disease feature | |||||
| Hurler syndrome | 607014 | AR | IDUA | Hypertrichosis since early infancy or childhood Prominent over the eyebrows Dermal melanocytosis |
Short stature, coarse face, gingival hyperplasia, abnormal dentition, macroglossia, eye problems, cardiac, gastroenterological, skeletal defects and neurological defects |
| Cornelia de Lange | 122470 300590 610759 614701 300882 |
AD RXL AD AD RXL |
NIPBL SMC1A SMC3 RAD21 HDAC8 |
Low posterior hairline Hypertrichosis Exuberant eyebrow growth Curly eyelashes Cutis marmorata Single transverse palmar crease |
Short stature, hearing loss, microcephaly, facial dysmorphism, cleft lip/palate, cardiac defects, kidney defects, mental retardation |
| Congenital generalized lipodystrophy (Berardinelli–Seip syndrome) | 608594 269700 612526 613327 |
AR | AGPAT2 BSCL2 CAV1 PTRF |
Hypertrichosis Acanthosis nigricans |
Decreased adipose tissue Diabetes Hypertriglyceridaemia Hepatic steatosis |
| Trisomy 18 | – | – | – | Generalized hypertrichosis | See Chapter 76 |
| Donohue syndrome (leprechaunism) | 246200 | AR | INSR | Hypertrichosis/hirsutism Acanthosis nigricans Dysplastic nails Keratoderma Loose and wrinkled skin due to loss of adipose tissue |
Severe failure to thrive Elfin facies Abdominal protuberance Oversized penis/clitoris Polycystic ovaries Gingival hyperplasia Large hands and feet Premature breast and nipple enlargement |
| Coffin–Siris syndrome | 135900 | AR | ARID1B | Generalized hypertrichosis with scalp hypotrichosis Nail dysplasia Cutis marmorata |
Short stature Psychomotor developmental delay Agenesis of the corpus callosum Coarse facial features Skeletal dysplasia Cardiac defects Kidney defects Gastrointestinal complications |
| Leigh syndrome | 256000 | AR | Mutations in genes encoding the various components of the mitochondrial respiratory chain complexes I–V | Generalized hypertrichosis | Progressive neurological deterioration, ophthalmoplegia, lactic acidosis |
| Zimmerman–Laband syndrome | 135500 | AD | ? | Generalized hypertrichosis Gingival fibromatosis Nail dysplasia |
Facial dysmorphism including bulbous soft nose, thick lips and thick floppy ears Skeletal dysplasia and joint hyperextensibility Hepatosplenomegaly Cardiovascular defects Mental retardation |
| Schinzel–Giedion midface retraction syndrome | 269150 | AD | SETBP1 | Hypertrichosis Facial haemangioma Hypoplastic dermal ridges Clubbing |
Failure to thrive Facial dysmorphism with proptosis due to shallow orbits, prominent forehead and macroglossia Genito-urinary abnormalities Skeletal dysplasia Neurological defects and mental retardation |
| Barber–Say syndrome | 209885 | AD | ? | Hypertrichosis Lax and atrophic skin |
Facial dysmorphism including prognathism, abnormal ears, bulbous nose, macrostomia and thin lips Hypoplastic nipples Abnormal external genitalia |
| Craniofacial dysmorphism, skeletal anomalies and mental retardation syndrome | 614132 | AR | TMCO1 | Hypertrichosis Low hairline Gingival hyperplasia |
Facial dysmorphism including wide nasal bridge with small nose, bushy eyebrows, flat face, low-set ears and macrocephaly Cleft lip/palate Skeletal dysplasia Neuropsychiatric manifestations |
| Lymphoedema–distichiasis syndrome | 153400 | AD | FOXC2 | Growth of extra row of eyelashes | Lymphoedema |
AD, autosomal dominant; AR, autosomal recessive; CNV, copy number variations; RXL, recessive X-linked.
Table 68.2 Atrichias
| Disease name | MIM | Inheritance | Gene or genetic defect | Cutaneous features | Extracutaneous features |
| A Non-syndromic atrichias | |||||
| Atrichia with papular lesions | 209500 | AR | HR | Atrichia Papular lesions |
None |
| Alopecia universalis congenital | 203655 | AR | HR | Atrichia | None |
| Ectodermal dysplasia 9, hair/nail type | 614931 | AR | HOXC13 | Atrichia (some cases hypotrichosis) Koilonychia and micronychia |
None |
| Lethal acantholytic epidermolysis bullosa | 609638 | AR | DSP | Generalized atrichia Generalized skin blistering Anonychia |
None |
| B Syndromic atrichias | |||||
| Vitamin D dependent rickets, type 2A | 277440 | AR | VDR | Atrichia or near-atrichia Papular lesions |
Growth retardation, rickets, dental abnormalities, delayed motor development, seizures |
| Ichthyosis follicularis, atrichia, photophobia syndrome (allelic to keratosis follicularis spinulosa decalvans) | 308205 | RXL | MBTPS2 | Atrichia or near-atrichia Dystrophic nails Erythroderma Ichthyosis Follicular hyperkeratosis Palmoplantar keratoderma |
Photophobia, vascularizing keratitis, BRESHECK syndrome |
| Keratitis–ichthyosis–deafness syndrome | 148210 | AD | GJB2 | Atrichia/hypotrichosis Ichthyosis Oral leukoplakia Palmoplnatar keratoderma Squamous cell carcinoma |
Keratitis Deafness |
| Growth retardation, alopecia, pseudoanodontia, optic atrophy (GAPO) syndrome | 230740 | AR | ANTXR1 | Scalp hair permanently lost in childhood | Facial dysmorphism with protruding ears and prominent lips Failure of tooth eruption Optic atrophy |
| T-cell immunodeficiency, alopecia and nail dystrophy syndrome | 601705 | AR | FOXN1 | Atrichia Dysplastic nails |
Severe T-cell immunodeficiency |
| Frontonasal dysplasia type 2 | 613451 | AR | ALX4 | Atrichia | Frontonasal dysplasia (large skull defect, coronal craniosynostosis, hypertelorism, severely depressed nasal bridge and ridge, bifid nasal tip) Cryptorchidism Agenesis of the corpus callosum and mental retardation |
AEC syndrome EEC syndrome Rapp-Hodgkin syndrome ADULT syndrome |
106260 604292 129400 103285 |
AD AD AD AD |
TP63 | Sparse, coarse, wiry hair Scalp erosions and red peeling skin Palmoplantar keratoderma Dystrophic nails |
Hearing loss Ankyloblepharon, lacrimal duct atresia, blepharoconjunctivitis Cleft lip/palate Abnormal dentition Syndactyly Cardiac defects Kidney defects Breast and mammary gland hypoplasia |
AD, autosomal dominant; ADULT, acrodermatoungual-lacrimal-tooth; AEC, ankyloblepharon-ectodermal defects–cleft lip/palate; AR, autosomal recessive; BRESHECK, brain anomalies, retardation, ectodermal dysplasia, skeletal malformations, Hirschsprung disease, ear/eye anomalies, cleft palate/cryptorchidism, and kidney dysplasia/hypoplasia; EEC, ectrodactyly, ectodermal dysplasia and cleft lip/palate; GAPO, growth retardation, alopecia, pseudoanodontia and optic atrophy; HR, hairless.
Table 68.3 Hypotrichoses
| Disease name | MIM | Inheritance | Gene or genetic defect | Cutaneous features | Extracutaneous features |
| A Non-syndromic hypotrichoses | |||||
| Generalized hereditary hypotrichosis simplex (hypotrichosis type 1) | 605389 | AD | APCDD1 RPL21 |
Generalized hypotrichosis, progressing from childhood with sparing of eyebrows, eyelashes and facial hair | None |
| Hypotrichosis simplex of the scalp (hypotrichosis type 2) | 146520 | AD | CDSN | Scalp hypotrichosis, progressing from childhood | None |
| Hypotrichosis type 3 | 613981 | AD | KRT74 KRT71 |
Scalp hypotrichosis Woolly hair |
None |
| Marie Unna hereditary hypotrichosis (hypotrichosis type 4) | 146550 | AD | U2HR | Sparse to normal hair at birth;coarse, wiry, twisted hair in early childhood progressing to generalized hypotrichosis | None |
| Marie Unna hereditary hypotrichosis (hypotrichosis type 5) | 612841 | AD | EPS8L3 | None | |
| Localized, AR hypotrichosis, type 1 (hypotrichosis type 6) | 607903 | AR | DSG4 | Generalized hypotrichosis with sparing of secondary sexual hairs Monilethrix and other structural hair shaft abnormalities Follicular hyperkeratosis |
None |
| Localized, AR hypotrichosis, type 2 (hypotrichosis type 7) | 604379 | AR | LIPH | Sparse scalp, eyebrows, eyelashes, up to generalized hypotrichosis; woolly hair in some patients (as well as other hair shaft abnormalities) | None |
| Localized, AR hypotrichosis, type 3 (hypotrichosis type 8) | 278150 | AR | LPAR6 | Sparse and short scalp, eyebrows, eyelashes, up to generalized hypotrichosis; woolly hair in some patients (as well as other hair shaft abnormalities); fair hair in some patients; nail pitting or longitudinal ridging in some patients | None |
| Hypotrichosis type 9 | 614237 | AR | Mapped to 10q11.23-q22.3 | Hypotrichosis involving scalp, arms, and legs. Eyebrows and eyelashes are spared | None |
| Hypotrichosis type 10 | 614238 | AR | Mapped to 7p22.3-p21.3 | Generalized hypotrichosis Scalp papules |
None |
| Hypotrichosis type 11 | 615059 | AD | SNRPE | Generalized hypotrichosis with sparing of pubic hair | None |
| Keratoderma and woolly hair | – | AR | KANK2 | Generalized hypotrichosis Woolly hair Striate palmoplantar keratoderma Leukonychia Pseudoainhum |
None |
| Monilethrix | 158000 | AD | KRT81 KRT83 KRT86 |
Hypotrichosis (regional to generalized) due to hair fragility, starting during early childhood and typically improving over time;occipital area most severely involved Follicular hyperkeratosis Nail dystrophy |
None |
| Hypotrichosis and recurrent skin vesicles | 613102 | AR | DSC3 | Generalized hypotrichosis Skin vesicles (?) |
None |
| Skin fragility–woolly hair syndrome | 607655 | AR | DSP | Hypotrichosis Woolly hair Palmoplantar keratoderma Skin fragility and blistering Nail dystrophy |
None |
| Ectodermal dysplasia with skin fragility | 604536 | AR | PKP1 | Severe hypotrichosis Skin fragility Dystrophic nails Palmoplnatar erosive keratoderma |
None |
| Ectodermal dysplasia type 4, hair/nail type | 602032 | AR | KRT85 | Localized to generalized hypotrichosis (with pili torti) Dysplastic nails |
None |
| AD generalized follicular hamartoma syndrome | 605827 | AD | ? | Hypotrichosis Milia Palmar pits Hypohidrosis Basaloid follicular hamartomata |
None |
| Rombo syndrome | 180730 | AD | ? | Absent or dystrophic eyelashes and eyebrows Bluish discoloration of lips and hands Facial milia and telangiectasis Follicular atrophoderma Basal cell carcinomas |
None |
| B Syndromic hypotrichoses | |||||
| Naxos disease | 601214 | AR | JUP | Hypotrichosis Woolly hair Palmoplantar keratoderma |
Arrhythmogenic right ventricular dysplasia Dilated cardiomegaly |
| Dilated cardiomyopathy with woolly hair and keratoderma | 605676 | AR | DSP | Hypotrichosis Woolly hair Palmoplantar keratoderma |
Dilated left ventricular cardiomyopathy |
| AR ichthyosis with hypotrichosis | 610765 | AR | ST14 | Generalized hypotrichosis with curly hair and long eyelashes Multiple hair shaft abnormalities Ichthyosis |
Photophobia, corneal opacities, blepharitis Abnormal dentition |
| Netherton syndrome | 256500 | AR | SPINK5 | Hypotrichosis with abnormal hair shaft structure (trichorrhexis invaginata)Congenital erythroderma Ichthyosis linearis circumflexa |
Growth retardation Perinatal hypernatraemia Atopic diathesis Multiple food allergies Recurrent infections Gastroenteropathy High IgE levels |
| Neonatal ichthyosis-sclerosing cholangitis (NISCH) syndrome | 607626 | AR | CLDN1 | Hypotrichosis with loss of lateral eyebrows Sparse eyelashes Ichthyosis Jaundice |
Sclerosing cholangitis Hepatomegaly Hypodontia |
| Severe dermatitis, multiple allergies and metabolic wasting (SAM) syndrome | 615508 | AR | DSG1 | Hypotrichosis Congenital erythroderma with acantholysis Palmoplantar keratoderma |
Growth retardation Perinatal hypernatraemia Recurrent infections Oesophagitis Cardiac defects High IgE levels |
| Bazex syndrome | 301845 | XLD | Mapped to Xq25-27.1 | Hypotrichosis with pili torti and trichorrhexis nodosa Facial hyperpigmentation and milia (which disappear in adulthood) Follicular atrophoderma Comedones Keratosis pilaris Localized hypohidrosis Basal cell carcinomas in second decade of life |
Pinched long nose Joint hypermobility |
| Clouston syndrome | – | AD | GJB6 | Hypotrichosis with wiry, brittle and pale hair Palmoplantar keratoderma Dysplastic nails |
Short stature Ophthalmological manifestations including cataracts, conjunctivitis, strabismus, photophobia |
| Dermatopathia pigmentosa reticularis | 125595 | AD | KRT14 | Alopecia Reticulate hyperpigmentation Hypo-/hyperhidrosis Absent fingerprints Palmoplantar keratoderma Dystrophic nails |
Abnormal dentition |
| Hypotrichosis with juvenile macular dystrophy | 601553 | AR | CDH3 | Generalized hypotrichosis with pili torti Light-coloured hair |
Progressive retinal macular degeneration |
| Ectodermal dysplasia, ectrodactyly, and macular dystrophy | 225280 | AR | CDH3 | Generalized hypotrichosis Light-coloured hair |
Progressive retinal macular degeneration Abnormal dentition Ectrodactyly, syndactyly |
| Hypohidrotic ectodermal dysplasia types 1, 10A, 10B, 11A, 11B | 305100 129490 224900 614940 |
XLR AD AR |
EDA EDAR EDARADD |
Generalized hypotrichosis Hypohidrosis Dysplastic nails Dry skin |
Facial dysmorphism including pigmentation under the eyes, everted nose, prominent lips Conical teeth and hypodontia Heat stroke Hypoplastic mammary glands Hoarse voice |
| Ectodermal dysplasia-syndactyly syndrome 1 | 613573 | AR | PVRL4 | Generalized hypotrichosis with pili torti Dysplastic nails Palmoplantar keratoderma |
Syndactyly Abnormal dentition |
| Ectodermal dysplasia–syndactyly syndrome 2 | 613576 | AR | ? | Generalized hypotrichosis Dysplastic nails Palmoplantar keratoderma Hyperhidrosis |
Syndactyly Facial dysmorphism Abnormal dentition Cardiomegaly |
| Cleft lip/palate–ectodermal dysplasia syndrome | 225060 | AR | PVRL1 | Hypotrichosis Palmoplantar keratoderma Dystrophic nails |
Facial dysmorphism Cleft lip/palate Hypodontia Syndactyly |
| Oculodentodigital dysplasia | 164200 | AD | GJA1 | Hypotrichosis Dry hair Palmoplantar keratoderma |
Microphthalmia, cataracts, strabismus Abnormal dentition Cleft lip/palate Multiple skeletal abnormalities Hearing loss Lymphoedema Neurological defects |
| Hallerman–Streiff syndrome | 234100 | ? | (GJA1 ?) | Hypotrichosis Light hair Skin atrophy and telangiectases |
Short stature Multiple skeletal abnormalities Facial dysmorphism with small pointed nose, thin lips, microstomia, frontal bossing, micrognathia Microphthalmia, cataracts, strabismus, coloboma Abnormal dentition Tracheomalacia Pulmonary hypertension and infections Neurological defects |
| Odonto-onychodermal dysplasia | 257980 | AR | WNT10A | Hypotrichosis Dry and thin hair Dystrophic nails Palmoplantar keratoderma Hyperhidrosis Decreased number of tongue papillae |
Hypodontia |
| Schopf–Schulz–Passarge syndrome | 224750 | AR | WNT10A | Hypotrichosis Dry and thin hair Dystrophic nails Palmoplantar keratoderma Hyperhidrosis Decreased number of tongue papillae |
Hypodontia Bird-like facies Eyelid hydrocystomata Non-melanoma skin cancer |
| Hypotrichosis–lymphoedema–telangiectasia syndrome | 607823 | AD (?)AR | SOX18 | Progressive hair loss since childhood involving scalp, eyebrows and eyelashes | Telangiectases (acral) Leg lymphoedema, congenital eyelid oedema, hydrocele, scrotal oedema |
| Tricho-rhino-phalangeal syndrome types I, II, III | 190350 150230 190351 |
AD | TRPS1 | Hypotrichosis Dysplastic nails |
Growth retardation Skeletal dysplasia including various developmental abnormalities of hand and feet Late-onset osteopenia and osteoarthritis Facial dysmorphism including large ears and pear-shaped nose Abnormal dentition Hypotonia |
| Chondrodysplasia punctate 2 | 302960 | XLD | EBP | Patchy alopecia Coarse sparse hair Ichthyosis Follicular atrophoderma along Blaschko lines |
Short stature Frontal bossing Hearing loss Cataracts, microphtalmia Skeletal dysplasia with epiphyseal stippling Impaired mental function |
| Oro-facio-digital syndrome | 311200 | XLD | OFD1 | Alopecia Sparse, coarse hair Facial milia |
Short stature Microcephaly, frontal bossing, hypoplastic alar cartilage Hyperplastic oral frenulum, bifid/lobulated tongue, tongue hamartoma Cleft lip/palate Lip anomalies Abnormal dentition Liver, pancreas, ovarian, kidney cysts Clino-/syn-/brachy-/polydactyly Neuropsychiatric complications |
| Cranioectodermal dysplasia type 1 | 218330 | AR | IFT122 | Fine sparse hair Thin short nails |
Abnormal dentition Facial dysmorphism |
| Cranioectodermal dysplasia type 2 | 613610 | AR | WDR35 | Craniosynostosis Brachydactyly, short limbs |
|
| Cranioectodermal dysplasia type 3 | 614099 | AR | IFT43 | Bicuspid aortic valve Liver disease |
|
| Cranioectodermal dysplasia type 4 | 614378 | AR | WDR19 | Renal failure Osteoporosis |
|
| Cartilage–hair hypoplasia | 250250 | AR | RMRP | Generalized hypotrichosis with fair sparse and fine hair | Skeletal dysplasia and short stature Malabsorption and Hirschprung disease Haematological anomalies Cellular immunodeficiency Increased risk of lymphoma and skin cancer |
| Noonan syndrome-like disorder with loose anagen hair | 607721 | AD | SHOC2 | Sparse light-coloured hair Hyperpigmentation Wrinkled skin |
Developmental delay Macrocephaly Cardiac defects Hypotonia and mental retardation Brain abnormalities |
| Nicolaides–Baraitser syndrome | 601358 | ? | ? | Hypotrichosis Low anterior hairline Wrinkled or eczematous skin |
Short stature Skeletal dysplasia Facial dysmorphism Neuropsychiatric manifestations |
| Short stature, onychodysplasia, facial dysmorphism and hypotrichosis (SOFT) syndrome | 614813 | AR | POC1A | Generalized hypotrichosis Hypoplastic nails |
Short long bones and other skeletal abnormalities resulting in short stature Macrocephaly Facial dysmorphism Oligospermia High-pitched voice |
| Macrocephaly, alopecia, cutis laxa, scoliosis (MACS) syndrome | 613075 | AR | RIN2 | Alopecia Receding anterior hairline Cutis laxa |
Short stature Coarse facies Abnormal dentition Scoliosis Joint hypermobility |
| Costello syndrome | 218040 | AD | HRAS | Curly sparse hair Dystrophic nails Cutis laxa Periorifical papillomas Acanthosis nigricans |
Short stature Facial dysmorphism with full cheeks, macrocephaly, thick lips and macroglossia Cardiac defects Lung defects Pyloric stenosis Kidney defects Delayed psychomotor development and neurological defects Malignancies |
| Cardiofaciocutaneous syndrome | 115150 615278 615279 615280 |
AD | BRAF KRAS MAP2K1 MAP2K2 |
Sparse, curly hair Absence of eyebrows and eyelashes Ichthyosis Haemangiomata Keratosis pilaris Lentigines Abnormal palmoplantar creases |
Short stature Facial dysmorphism Eye defects Cardiac defects Osteopenia Neurological defects, including seizures and hypotonia Mental retardation |
| Yunis–Yaron syndrome | 216340 | AR | FIG4 | Hypotrichosis involving scalp, eyebrows and eyelashes Palmar crease Hypoplastic nails |
Growth retardation Facial dysmorphism including dysplastic ears and protruding eyes Abnormal dentition Heart defects Pyloric stenosis Micropenis and hypospadias Severe neurological defects |
| Alopecia–neurological defects–endocrinopathy (ANE) syndrome | 612079 | AR | RBM28 | Variable degress of alopecia Flexural hyperpigmentation |
Growth retardation Hypogonadism Addison disease Abnormal dentition |
| Woodhouse–Sakhati syndrome | 241080 | AR | DCAF17 | Scalp and eyebrows alopecia | Deafness Hypogonadism Diabetes Extrapyramidal abnormalities, mental retardation and other neurological defects |
| Bjornstad syndrome | 262000 | AR | BCS1L | Hypotrichosis Coarse dry and fragile hair with pili torti |
Nerve deafness Hypogonadism |
| Argininosuccinic aciduria | 207900 | AR | ASL | Dry, sparse brittle hair with trichorrhexis nodosa | Failure to thrive Hepatic fibrosis and hepatomegaly Encephalopathy Poor feeding, vomiting Neurological defects Mental retardation |
| Biotinidase deficiency | 253260 | AR | BTD | Alopecia Dermatitis Skin infections |
Hepatosplenomegaly Vomiting, diarrhoea Hearing loss Vision loss Breathing problems Neurological defects |
| Neonatal inflammatory skin and bowel disease | 614328 | AR | ADAM17 | Short and broken hair, disorganized eyebrows and eyelashes Perorificial erythema Pustular rash Dysplastic nails and paronychia |
Chronic bloody diarrhoea, malabsorption Left heart ventricular dilatation High IgE |
| Trichohepatoenteric syndrome | 222470614602 | AR | TTC37 SKIV2L |
Hypotrichosis with trichorrhexis nodosa and woolly hair | Failure to thrive Prominent cheeks and forehead Congenital heart defects Cholestatic jaundice, hepatomegaly, cirrhosis Severe secretory diarrhoea Mental retardation |
| Menkes disease | 309400 | XLR | ATP7A | Steely kinky sparse hair which can show pili torti, monilethrix and trichorrhexis nodosa Cutis laxa Hypopigmentation |
Short stature Microcephaly Joint laxity Osteoporosis Neurodegenerative manifestations Variable vascular pathologies Chronic diarrhoea Urogenital anomalies Skeletal dysplasia (including occipital horns) |
| Trichothiodystrophy, photosensitive type | 601675 | AR | ERCC2 ERCC3 GTF2H5 |
Hypotrichosis resulting from brittle sulphur-deficient hair Ichthyosis Photosensitivity Brittle nails Lack of adipose tissue |
Short stature Progeroid facies Microcephaly Various ocular manifestations including cataracts and microcornea Joint contractures Hypogammaglobulinaemia Recurrent infections Hypogonadism Asthma Mental retardation |
| Trichothiodystrophy, non-photosensitive type | 234050 | AR | MPLKIP | Hypotrichosis resulting from brittle sulphur-deficient hair | Short stature Mental retardation Decreased fertility |
| Rothmund–Thompson syndrome | 268400 | AR | RECQL4 | Hypotrichosis involving scalp, face, eyebrows and eyelashes. Poikiloderma mainly evident in sun-exposed areas Photosensitivity Hyperkeratotic lesions over soles and knees Nail dystrophy Non-melanoma skin cancers |
Short stature Skeletal dysplasia Hypogonadism Cataracts Abnormal dentition Risk of osteogenic sarcoma |
| Hutchinson–Gilford progeria syndrome | 176670 | AD | LMNA | Alopecia Absence of subcutaneous fat Wrinkled, atrophic and pigmented skin Scleroderma |
Growth retardation Osteoporosis Premature ageing Premature cardiovascular disease Insulin resistance |
| Werner syndrome | 277700 | – | RECQL2 | Premature balding Poikiloderma Scleroderma-like changes Loss of adipose tissue Hyperpigmentation Calcinosis cutis Ulcers |
Short stature Prematurely aged facies Cataracts Retinal degeneration Osteoporosis and avascular necrosis Premature cardiovascular disease Diabetes Hypogonadism Malignancies |
AD, autosomal dominant; AR, autosomal recessive; XLD, X-linked dominant.
Introduction and general description
The past 10 years have witnessed dramatic developments in the field of hair research and the number of inherited hair disorders, whose molecular basis has been discovered, has grown exponentially [2]. Two major reasons underlie this recent trend. First, the hair follicle is regarded today as a unique model for the study of complex developmental and regulatory interactions between epithelial and mesenchymal tissues [3–6]. Second, hair disorders are known to be the source of considerable morbidity. Absence (alopecia) or excess (hirsutism) of hair are obviously compatible with normal lifespan; yet individuals affected with these conditions often experience aberrant hair growth as a significantly detrimental event, affecting many aspects of their personal and social life [7–11].
Because most of these disorders are exceedingly rare, epidemiological data are mostly non-existent.
HYPERTRICHOSES
GENERALIZED HYPERTRICHOSES
This group of very rare disorders manifests generally at birth with generalized hypertrichosis (Figure 68.1). The various forms of generalized hypertrichosis are recognized on the basis of the presence of specific extracutaneous manifestations. Unfortunately, because the molecular basis of many of these diseases is still elusive, it is often not clear whether they are distinct entities or represent different clinical manifestations of common underlying genetic defects (see Table 68.1).
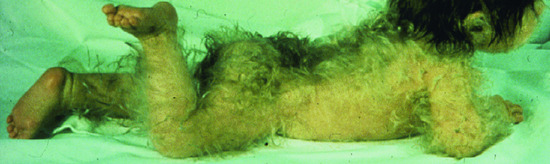
Figure 68.1 Generalized hypertrichosis.
(Courtesy of Professor Peter Itin.)
Hypertrichosis universalis congenita, Ambras type
Children affected by this disorder present with generalized hypertrichosis which consists of large amounts of velus hair over the entire surface of the skin, with some predilection for the face, ears and shoulders. Hypertrichosis of the external ears is typical. Facial dysmorphism and skeletal abnormalities have also been reported [12]. The disorder has been found to be due to chromosomal rearrangements over 8q23.1 leading to down-regulation of the TRPS1 gene [13], also known to be involved in the pathogenesis of tricho-rhino-phalangeal syndrome, another complex hair disorder (see Chapter 67). Of interest, a similar defect was also found in a murine model of the disease, the ‘Koala’ mouse [13].
Generalized hypertrichosis terminalis
This disorder is inherited in an autosomal dominant fashion and is characterized by excess of terminal hair and a variable age of onset. Many patients demonstrate facial dysmorphism and often severe gingival hyperplasia [14]. In several cases, genomic changes on chromosome 17q24.2-q24.3 were found to segregate with the disease phenotype [15]. It has been suggested that these changes may impact on the function of SOX9, which is known to play a role in the regulation of hair growth. More recently, a mutation in the ABCA5 gene has been identified in a single case [15a].
Cantu syndrome
Cantu syndrome is inherited in an autosomal dominant fashion and features congenital hypertrichosis with long curly eyelashes, macrosomia, facial dysmorphism, gingival hyperplasia, skeletal anomalies, cardiac defects and occasionally mild mental retardation [16]. The disease was found to be caused by activating mutations in ABCC9 encoding the sulfonylurea receptor 2 [17, 18]. This is of interest as this molecule is part of an adenosine triphosphate (ATP)-sensitive potassium channel complex, targeted by minoxidil [19] which is used to treat male and female pattern baldness.
LOCALIZED HYPERTRICHOSES
Congenital localized hypertrichosis has been reported as an isolated finding around the elbows (hypertrichosis cubiti) and the neck (posterior and anterior cervical hypertrichosis) [20, 21]. Congenital localized hypertrichosis has been described in association with spinal (faun-tail naevus) or cranial dysraphism (hair collar sign) [22–25]. Localized hypertrichosis often accompanies pigmentation abnormalities as in congenital melanocytic naevi and Becker's naevus. Localized hypertrichosis is also a classical feature of congenital porphyrias (see Chapter 60).
The histiocytosis and lymphadenopathy syndrome (also known as H syndrome) is a complex disorder featuring a wide and variable constellation of cutaneous and systemic manifestations including skin hyperpigmentation, localized hypertrichosis, hepatosplenomegaly, heart anomalies, hearing loss, hypogonadism, short stature and hyperglycaemia/diabetes [26, 27] (Figure 68.2). The disorder was found to be caused by mutations in SLC29A3 encoding the equilibrative nucleoside transporter (hENT3) [28, 29]. Although the exact mechanism underlying the disease pathogenesis remains elusive, it is of interest to note that the histiocytes present in dermal infiltrates in the skin of the patients resemble those seen in Rosai–Dorfman disease [30].
Figure 68.2 Hypertrichosis and hyperpigmented plaques localized to the lower limb in an adult patient with H syndrome.
(Courtesy of Professor Avraham Zlotogorski.)
DIFFERENTIAL DIAGNOSIS
The most important diagnosis to exclude in any form of hypertrichosis is hirsutism, which results from excessive growth of hair in a male pattern distribution and warrants thorough investigations in search for an endocrinological, drug or paraneoplastic cause (see Chapter 147). The late onset of hypertrichosis should always raise the possibility of an underlying malignancy [31].
TREATMENT
Hair removal using depilation or laser techniques has been successfully employed [32]. Surgical debulking of the gums and total tooth extraction have been reported to result in improved oral function in generalized hypertrichosis terminalis [33].
ATRICHIAS
Atrichias refer to a group of very rare disorders characterized by total or near total absence of visible scalp and body hair.
Atrichia with papular lesions
Clinical features
Children affected with atrichia with papular lesions (APL) are usually born with normal hair which is shed during the first months of life, never to regrow thereafter. During the first to second decade of life, patients develop a diffuse papular rash, which has been noted to be particularly prominent over the cheeks and scalp but can involve almost any part of the body [34–36] (Figure 68.3). Although APL has been described with mental retardation, gastrointestinal polyposis and delay in bone age, these associations are considered spurious. On histology, APL is characterized by the conspicuous presence of dermal cysts, which reflect abortive development of different regions of the hair follicles (Figure 68.3c) [37].
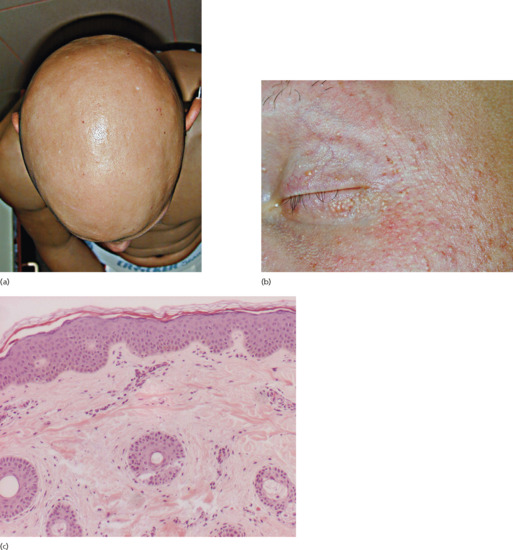
Figure 68.3 Total lack of hair (a) and follicular lesions (b) in atrichia with papular lesions. On histology, note the conspicuous presence of dermal cysts (c).
Pathogenesis
Atrichia with papular lesions is caused by mutations in the HR gene, encoding a transcription co-repressor factor called hairless [34, 36, 38]. Hairless has been shown to function as a histone H3K9 demethylase [39]. Hairless may regulate hair cycling through its effect on the WNT signalling pathway [40], through its effect on polyamine synthesis [41] or through additional targets [42]. Regardless of its exact mechanism of action, down-regulation of hairless is associated with abnormal catagen and interferes with normal hair cycle leading to the abnormal development of hair follicles into epidermal cysts that manifest at the clinical level as papules [43]. Of note, overexpression of hairless due to heterozygous mutations in a short regulatory open reading frame located upstream the HR gene, also leads to abnormal hair development in patients affected with Marie Unna hereditary hypotrichosis [44], indicating that absence or increased expression of hairless are equally detrimental to hair growth and development. Marie Unna hereditary hypotrichosis (also known as hypotrichoses 4 and 5) is in contrast with APL inherited in an autosomal dominant fashion. Affected individuals display sparse to normal hair at birth, develop coarse, wiry and twisted hair in early childhood, followed by the development of generalized alopecia in adulthood [45] (Figure 68.4).
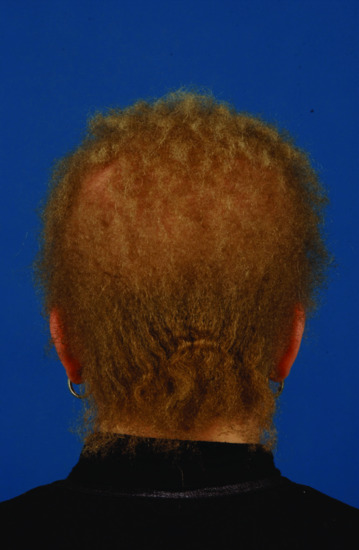
Figure 68.4 Wiry, coarse and sparse hair in Marie Unna hereditary hypotrichosis.
(Courtesy of Professor Maurice Van Steensel.)
Differential diagnosis
Another inherited form of congenital atrichia, termed alopecia universalis congenita (MIM203655), is also caused by mutations in HR and is clinically identical to APL except for the absence of skin papules [36, 38]. Vitamin D resistant rickets (VDRR; MIM277440) is inherited in an autosomal recessive fashion. It results from end organ unresponsiveness to 1,25-dihydroxycholecalciferol. Affected patients display normal serum 25-hydroxyvitamin D, high serum 1,25-(OH)2-cholecalciferol and profound hypocalcaemia, leading to rickets and loss of teeth [46]. VDRR with hair loss (type IIa) must be differentiated from VDRR without hair loss (type IIb). VDRR type IIa patients are born with normal hair, which is shed during the first year of life and never significantly regrows thereafter (Figure 68.5). In contrast, bony changes can improve with age. Milia-like lesions similar to those observed in APL have also been described in VDRR [46]. VDRR is caused by mutations in the gene encoding the vitamin D3 receptor (VDR). Most VDRR type IIa-causing mutations affect the N-terminal DNA-binding domain of the receptor, which harbours two zinc finger domains responsible for DNA binding and interactions with other proteins. In contrast, mutations in the vitamin D binding domain, situated at the C-terminus, do not cause alopecia [47]. Thus, vitamin D binding to the VDR is not necessary for normal hair development, which may explain why other forms of inherited rickets, with defective vitamin D binding, are not associated with alopecia [46, 47]. These studies emphasized the importance of VDR binding to DNA and its interactions with other transcription factors during hair cycling. The fact that hairless functions under physiological conditions in association with the VDR explains the clinical similarities of the two phenotypes [42].
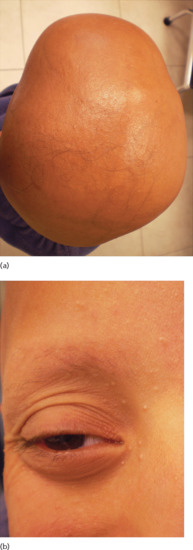
Figure 68.5 Near total absence of hair in a young patient with vitamin D resistant rickets type IIa (a); note the whitish papules over the central part of the face (b).
Bazex syndrome should also be included in the differential diagnosis of alopecia associated with a papular rash. This X-linked recessive disorder [48] manifests with hypotrichosis, milia, atrophoderma of the dorsa of the hands and feet, face and extensor surfaces of the elbows and knees, and hypohidrosis of the face [49, 50]. Hair microscopy can reveal trichorrhexis nodosa and pili torti. Basal cell neoplasms often develop after the second decade of life [50]. Generalized basaloid follicular hamartoma syndrome is clinically similar although basal cell carcinomas do not develop [51].
Another disorder featuring total absence of hair is the Growth retardation, Alopecia, Pseudoanodontia, Optic atrophy (GAPO) syndrome [52] (Figure 68.6). This autosomal recessive complex disorder is caused by mutations in the ANTXR1 coding for the anthrax toxin receptor, which seems to play an important role in actin assembly [53].
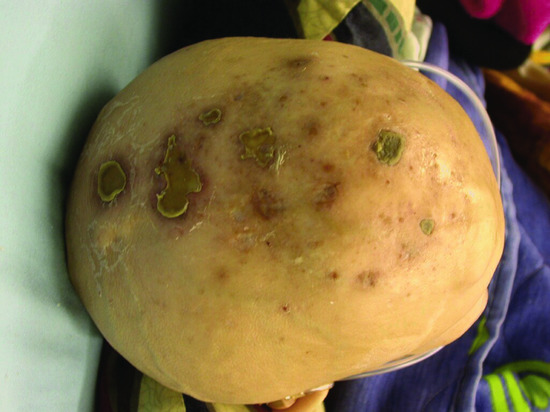
Figure 68.6 Alopecia totalis in a patient with growth retardation, alopecia, pseudoanodontia and optic atrophy (GAPO) syndrome.
(Courtesy of Professor David Enk.)
Treatment
An accurate diagnosis is critical to prevent unnecessary treatment of APL with systemic steroids when misdiagnosed as alopecia universalis. Oral or intravenous calcium and active vitamin D metabolites may attenuate the bone disease of patients with vitamin D resistant rickets but do not affect hair status [54].
Ichthyosis follicularis with atrichia and photophobia
Clinical features
This disorder manifests with congenital total or partial atrichia, severe and diffuse follicular hyperkeratosis, different degrees of scaling and vascularizing keratitis leading to photophobia and blindness [55]. Ichthyosis follicularis with atrichia and photophobia (IFAP) is transmitted as a recessive X-linked trait. Disease severity is highly variable. IFAP has been reported in association with a wide range of extracutaneous manifestations including the BRESHECK constellation of signs (Brain anomalies (including mental retardation, corpus callosum dysgenesis, olivopontocerebellar atrophy), growth Retardation, Ectodermal dysplasia, Skeletal deformities (scoliosis, rib and pelvic anomalies), Hirschsprung disease, Ear (hearing loss)/Eye anomalies, Cleft palate/cryptorchidism and Kidney dysplasia/hypoplasia) [56]. IFAP has been shown to overlap clinically with keratosis follicularis spinulosa decalvans (KFSD), which features scarring alopecia, typically prominent over the occiput and involving the eyelashes and eyebrows, facial erythema, follicular hyperkeratosis, keratoderma, blepharitis, conjunctivitis and keratitis [57].
Pathogenesis
IFAP, BRESHECK syndrome and KFSD have been found to result from mutations in the membrane-bound transcription factor protease, site 2 (MBTPS2) gene [56, 58, 59] which encodes a protein involved in endoplasmic reticulum stress response as well as in cholesterol homeostasis. MBTPS2 regulates the translocation of regulatory molecules and transcription activating factors to the nucleus [60]. As the MBTPS2 gene is located on the X chromosome, female carriers are either phenotypically normal or show patchy alopetic linear lesions of atrophoderma or follicular ichthyosis along the lines of Blaschko, reflecting Lyonization [61].
Treatment
Treatment is mainly aimed at maintaining visual function [62] as well as preventing or correcting systemic complications of the syndrome. A variable response to acitretin has been reported in several patients with improvement in ichthyosiform changes and corneal erosions but no change in alopecia or photophobia [55, 63, 64].
HYPOTRICHOSES
Hypotrichoses represent a very heterogeneous and vast group of disorders characterized by a reduced density of hair follicles. Phenotypic variability is the rule and can be striking even among affected members of the same family, which often complicates the diagnosis. These disorders are traditionally classified based on their mode of inheritance and based on the presence of extracutaneous features [2]. Table 68.3 provides a full list of this group of diseases. The major forms of hypotrichoses are described in details in the following subsections.
NON-SYNDROMIC AUTOSOMAL DOMINANT HYPOTRICHOSES
Autosomal dominant hypotrichosis comprises two major disorders known as generalized hereditary hypotrichosis simplex (also known as hypotrichosis type 1) and hypotrichosis simplex of the scalp (also known as hypotrichosis type 2).
Individuals with generalized hereditary hypotrichosis simplex typically show normal hair at birth, with progressive hair loss and thinning starting during early childhood and involving to a variable extent all parts of the body except for eyelashes, eyebrows and beard [65]. Hair pigmentation can be affected as well but hair shaft structure is normal. The disorder has been found to result from a recurrent mutation in the APCDD1 gene [65], encoding a regulator of the WNT signalling pathway, which is known to play a critical role during hair follicle development [40, 66, 67].
Similar clinical findings are observed in hypotrichosis simplex of the scalp except for the fact they are limited to the scalp (Figure 68.7). The disease has been found to result in several families from mutations in the CDSN gene encoding corneodesmosin, a component of the corneodesmosomes [68]. Of note, genetic alterations in the same gene have been linked to the inflammatory subtype of peeling skin syndrome [69] and to psoriasis [70, 71]. Each type of mutation seems to exert a different deleterious effect: dominant mutations associated with hypotrichosis result in the perifollicular accumulation of a toxic amyloidosis-like material [72]; recessive mutations causing peeling skin syndrome lead to absence of expression of corneodesmosin [69], while a polymorphism in the CDSN gene associated with an increased risk for psoriasis seems to result in increased CDSN mRNA stability [70].
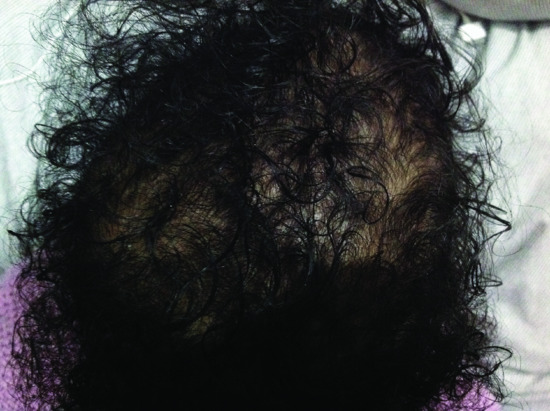
Figure 68.7 Autosomal dominant hypotrichosis of the scalp, caused by a mutation in CDSN.
NON-SYNDROMIC AUTOSOMAL RECESSIVE HYPOTRICHOSES
Autosomal recessive localized hypotrichosis (alias hypotrichosis types 7 and 8) features varying degrees of hair paucity over most parts of the body, with occasionally decreased pigmentation [73] (Figure 68.8). The disorder overlaps with woolly hair [74] which refers to the growth of fine and tightly curled hair (unlike normal curly hair, woolly hair grows slowly to a shorter distance than normal hair and can be associated with several structural anomalies such as trichorrhexis nodosa) [75]. The hypotrichosis is progressive and can be limited to the scalp or involve the whole body surface including the eyebrows, eyelashes and facial hair [75]. Autosomal recessive localized hypotrichosis has been found to be caused by mutations in two genes: LIPH [76] and LPAR6 [77, 78]. LIPH encodes lipase H, which promotes the synthesis of lysophosphatidic acid, the natural ligand of a G protein coupled receptor encoded by LPAR6 which is expressed in both the Henle and the Huxley layers of the inner root sheath of the hair follicle [79].

Figure 68.8 Sparse, short and light-coloured hair in a patient carrying a mutation in LIPH.
SYNDROMIC AUTOSOMAL DOMINANT HYPOTRICHOSES
Tricho-rhino-phalangeal syndrome
For a detailed description of the syndrome, see Chapter 67. Three subtypes of tricho-rhino-phalangeal syndrome (TRPS) have been described. TRPS I patients feature sparse scalp hair as well as thin and malformed nails associated with a typical bulbous nose, a long philtrum, abnormal dentition and large ears. Patients display short metacarpals and metatarsals, brachydactyly with cone-shaped epiphyses at the (middle) phalanges, scoliosis, lordosis, hip malformations, abnormal patellae, short stature and suffer from osteopenia and osteoarthritis in adulthood [80]. The disorder was found to be caused by mutations in the TRPS1 gene which codes for a GATA-type zinc finger transcription factor regulating cartilage, kidneys and hair follicle formation [81]. TRPS III is associated with more severe skeletal manifestations than TRPS I, but is also due to mutations in TRPS1 [82].
TRPS II (Langer–Giedion syndrome) is characterized by the same features as TRPS I apart from the conspicuous presence of multiple exostoses and borderline intelligence in some patients [83]. Eyebrows are thickened medially and absent laterally (signe du sourcil) in TRPS I and III but normal in TRPS II. TRPS II is a contiguous gene syndrome caused by deletion mutations affecting both TRPS1 as well as EXT1 encoding exostosin 1 [84].
Connexin disorders
A number of dominant syndromes caused by defective function of connexins are associated with various forms of alopecia [85]. Keratitis–ichthyosis–deafness syndrome can manifest with alopecia and dense follicular papules over the scalp (Figure 68.9) and result from mutations in the GJB2 gene encoding connexin-26 [86]. Clouston syndrome is characterized by focal to total alopecia (Figure 68.10) associated with palmoplantar keratoderma and nail dystrophy and results from mutations in the GJB6 gene encoding connexin-30 [87]. Mutations in the GJA1 gene coding for connexin-43, cause oculodentodigital dysplasia (ODDD) syndrome which presents with fine and slow-growing sparse hair, palmoplantar keratoderma, facial dysmorphism, a wide range of eye anomalies, abnormal teeth, cleft lip or palate, hyperostosis and other skeletal problems and numerous neurological defects [88, 89].
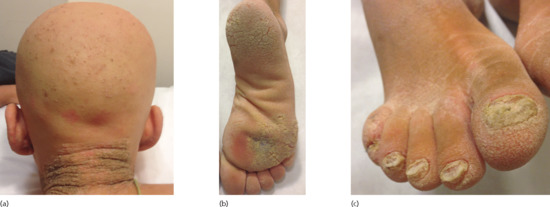
Figure 68.9 A patient with keratitis–ichthyosis–deafness syndrome demonstrates atrichia associated with follicular papules in the nape area (a), plantar keratoderma (b) and severe nail dystrophy (c).
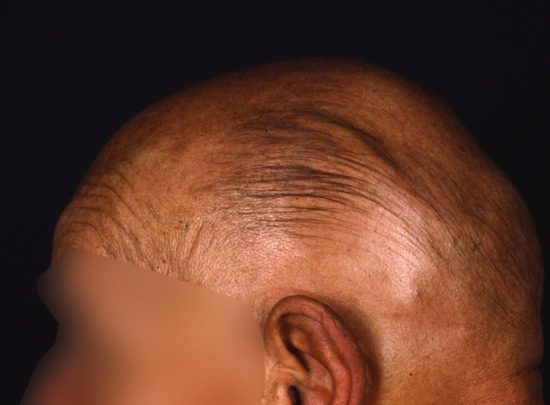
Figure 68.10 Diffuse hypotrichosis in an adult patient with Clouston syndrome.
(Courtesy of Professor Maurice Van Steensel.)
SYNDROMIC AUTOSOMAL RECESSIVE HYPOTRICHOSES
Hypotrichosis with juvenile macular dystrophy
In hypotrichosis with juvenile macular dystrophy (HJMD), short, light and sparse hair in early childhood is associated with the later development of progressive degeneration of the retinal macula leading to blindness during the second to third decade of life [90] (Figure 68.11). The disorder was found to result from mutations in the CDH3 gene [91], encoding P-cadherin, a component of the adherens junction which was shown to be expressed in the retinal pigment epithelium [92] and to regulate hair growth and pigmentation [93, 94]. HJMD has been shown to be allelic to Ectodermal dysplasia, Ectrodactyly, and Macular dystrophy (EEM) syndrome, which is characterized by the same clinical features as HJMD in association with abnormal development of the teeth and limbs [95].
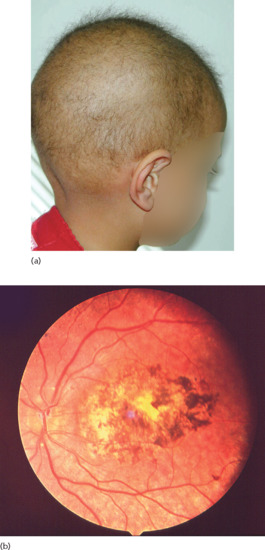
Figure 68.11 Sparse and short hair on the scalp of a young patient with hypotrichosis and juvenile macular dystrophy (a); fundus examination reveals severe degenerative pigmentary changes in the retinal macula (b).
Autosomal recessive ichthyosis with hypotrichosis
Affected individuals are born with sparse and short hair and display generalized scaling (Figure 68.12) with ophthalmological manifestations including photophobia, corneal opacities and pinguecula [96]. Abnormal dentition has also been noticed. Structural defects are observed on hair microscopy [96]. The disorder was found to result from mutations in the ST14 gene [96] which encodes matriptase, a serine protease that functions as a membrane-bound cell surface protein or as a soluble extracellular protease following release of its ectodomain [97]. Matriptase has been shown to have a role in filaggrin processing and regulates epidermal proliferation and differentiation [97, 98]. Mutations in the same gene have been found to cause a disorder called ichthyosis, follicular atrophoderma, hypotrichosis and hypohidrosis, which in many aspects resembles autosomal recessive ichthyosis with hypotrichosis [99].
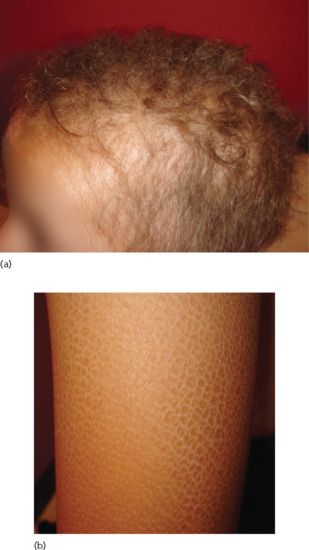
Figure 68.12 Hypotrichosis (a) and lamellar ichthyosis (b) in a patient with autosomal recessive ichthyosis with hypotrichosis.
HAIR SHAFT STRUCTURAL ABNORMALITIES
These disorders are easily identified when suspected since examination of a hair sample under a light microscope is very often sufficient to pose a diagnosis [100]. Rarely, scanning electron microscopy is needed to establish the diagnosis. More recently, dermoscopy has been recognized as a useful and cost-efficient adjunct technique for the rapid diagnosis of hair shaft abnormalities [101].
As a group, hair shaft disorders largely overlap at the clinical and molecular level with hypotrichoses as hair shaft anomalies often (but not always) result in hypotrichosis [75]. As with other genotrichoses, the first step in the diagnosis of hair shaft structural disorders is to determine whether the hair disease is isolated or is part of a more complex disorder. The following sections review those hair shaft disorders inherited as monogenic traits (see Table 68.3). This group of hair disorders is reviewed in details in Chapter 89.
Monilethrix
Clinical features
Beaded hair is the hallmark of monilethrix. Scalp hair is fragile at constricted sites leading to apparent hypotrichosis [74, 100] (Figure 68.13). Gradual improvement with age is the rule with hair looking sometimes normal by puberty or early adulthood. Improvement has also been noted during pregnancy and summertime. Body hair as well as eyelashes and eyebrows are less frequently involved. Intrafamilial phenotypic variability is common [102]. Associated features include follicular papules in the nape area as well as keratosis pilaris and nail dystrophy [74, 100].
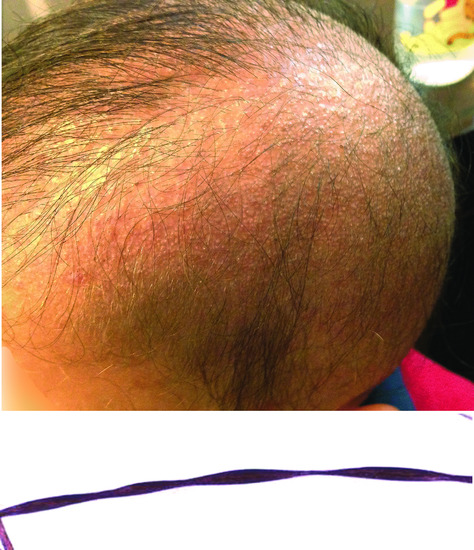
Figure 68.13 Short and sparse hair associated with follicular papules a patient with monilethrix; hair beading typical of monilethrix on microscopy.
Pathogenesis
Monilethrix is usually transmitted in an autosomal dominant fashion and is due to mutations in genes encoding several hair keratins (KRT81, KRT83, KRT86) [103–105]. Recessive inheritance of mutations in the DSG4 gene encoding desmoglein 4 have been reported in localized autosomal recessive hypotrichosis which is characterized by monilethrix-like hairs, fragile scalp and body hairs that break easily, and are associated with hyperkeratotic follicular papules [106]. Monilethrix has been observed in syndromic forms of hypotrichosis such as Menkes disease [107].
Differential diagnosis
Monilethrix must be distinguished from pili torti in which hair shaft twisting can generate the false impression of beading [100]. Pseudomonilethrix refers to a poorly defined form of hypotrichosis due to fragile and easily breakable hair shafts. On electron microscopy, irregular nodes represent the edges of depressions within the shafts; in other cases, the beaded appearance of the hair shafts reflect irregular twisting without flattening of the hair shafts as in pili torti;finally, pseudomonilethrix can result from breaks within the shafts. Although autosomal inheritance of pseudomonilethrix has been described, many authorities considered it as an artefactual finding due to trauma to the hair shafts [108].
Treatment
Monilethrix has been successfully treated in uncontrolled studies with minoxidil [109, 110] and with oral retinoids [111–113].
Woolly hair
Clinical features
Woolly hair refers to generalized or localized occurrence of curly hair, which usually demonstrates slow growth and/or easy breakage, sometimes associated with hypopigmentation (Figure 68.14). The disorder can appear as an isolated dominant trait, and in this case can be allelic to autosomal recessive localized hypotrichosis [75] but has also been reported in the context of various cardiocutaneous disorders [114].

Figure 68.14 Woolly hair in a white woman and her son.
(Courtesy of Professor Rudolf Happle.)
Patients with woolly hair may in some cases be affected with Noonan syndrome, especially in combination with ulerythema ophryogenes [115]. These patients tend to display a short stature, ptosis, borderline intelligence, a webbed neck and pulmonic stenosis.
Pathogenesis
Isolated autosomal recessive woolly hair has been shown to be allelic to autosomal recessive localized hypotrichosis (see Non-syndromic autosomal recessive hypotrichoses) caused by mutations in LPAR6 and LIPH [74, 78]. Autosomal dominant inheritance has also been described and linked to mutations in two hair keratin genes, KRT71 and KRT74 [116, 117].
Woolly hair has been described in the context of complex syndromes. Naxos disease and Carvajal syndrome manifest with woolly hair, hypotrichosis, diffuse palmoplantar keratoderma and cardiac disease, and are caused by recessive mutations in JUP and DSP, respectively, which encode two desmosomal proteins, plakoglobin and desmoplakin [118, 119].
Pili torti
Clinical features
Pili torti refer to hair showing 180 degree twists under the microscope [100] (Figure 68.15b). Patients with pili torti usually display breakable, short and sparse hair over the scalp (Figure 68.15a) and body. Isolated pili torti can be congenital but often becomes apparent in childhood only, with spontaneous improvement with time.
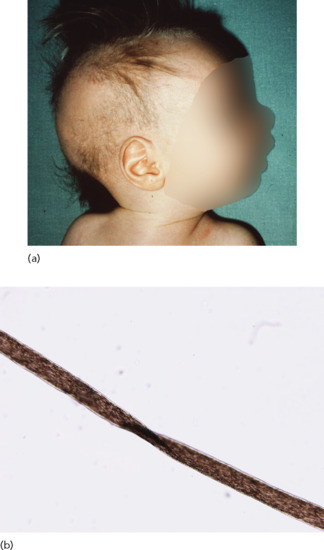
Figure 68.15 (a) Pale and sparse hair in a child with Menkes disease.
(Courtesy of Professor Rudolf Happle.) (b) Hair twisting along its axis, typical of pili torti. (Courtesy of Professor Reuven Bergman.)
Pathogenesis
Pili torti can be inherited in an autosomal dominant fashion. The molecular cause for isolated pili torti remains elusive.
Differential diagnosis
Pili torti has been reported in the context of complex syndromes. Pili torti is typically found in Menkes disease (see Chapter 63), an X-linked metabolic disorder usually fatal in early life [120]. Menkes disease is characterized by paucity of hair which is fine, wiry, fragile and silver or white. Phenotypic variability is the rule. Hair microscopy usually reveals pili torti as well as monilethrix and trichorrhexis nodosa. The skin can show a mottled discoloration and is often lax. The upper lip has an exaggerated ‘cupid bow’ configuration. Of interest, asymptomatic female carriers can display foci of pili torti and uneven skin pigmentation along the Blaschko lines. Systemic manifestations include early onset of neurological signs including hypotonia, seizures, psychomotor retardation as well as periodic hypothermia. Tortuous vessels in the central nervous system can be seen on magnetic resonance angiogram (MRA) and are characterized on histology by fragmentation of the internal elastic lamina. Osteoporosis, skeletal dysplasia and dental and ocular anomalies are also observed [121]. The disorder is caused by mutations in the ATP7A gene [122, 123] which encodes a trans–Golgi membrane bound copper transporter. Accordingly, serum copper and ceruloplasmin levels are low. As a result, numerous copper-dependent enzyme activities are lost. Among the enzymes most prominently affected are tyrosinase, lysyl oxidase, monoamine oxidase, cytochrome oxidase and ascorbate oxidase which explains the pigment defects, lax skin, hair abnormalities, hypothermic episodes and skeletal changes typically found in patients, respectively. Occipital horn disease is allelic to Menkes disease and manifests with more severe skin changes, less prominent neurological manifestations and the presence of exostoses on the occiput (hence the name of the syndrome) and other bones [124].
Björnstad syndrome features a combination of pili torti and progressive sensorineural hearing loss [125]. The syndrome is caused by mutations in the BCS1L gene, which plays an important role in mitochondrial function [126]. Pili torti has also been seen in Netherton syndrome [127] (see below), hypotrichosis with juvenile macular dystrophy (see above) [91], Bazex syndrome [128], citrullinaemia [129] and autosomal recessive ichthyosis with hypotrichosis [96] (see above).
In addition to the inherited conditions mentioned above, pili torti has been described in association with systemic acquired diseases such as lupus erythematosus and other forms of cicatricial alopecia [130].
Trichorrhexis nodosa
Clinical features
Trichorrhexis nodosa is diagnosed under the microscope as irregularly spaced swellings along the hair shaft which appear to be the consequence of cuticle loss and exposure of cortical fibres. These areas are prone to fracture. Trichorrhexis nodosa has seldom been described as an isolated finding. It rather accompanies conditions manifesting with hair fragility, inherited and acquired alike [131–134].
Pathogenesis
No single gene has been associated with isolated trichorrhexis nodosa. Trichorrhexis nodosa has been described in the context of Netherton syndrome [127] (see Chapter 65), Menkes syndrome [107] (see Chapter 81) and trichothiodystrophy [135]. It is also typically found in argininosuccinic aciduria [133] also featuring failure to thrive, liver disease and neurological manifestations and caused by mutations in ASL encoding argininosuccinate lyase [136]. Trichorrhexis nodosa has also been observed in association with hypotrichosis as part of the trichohepatoenteric (THE) syndrome, which features intractable diarrhoea and is caused by mutations in two regulatory genes, TTC37 and SKIV2L [137, 138] (Figure 68.16).
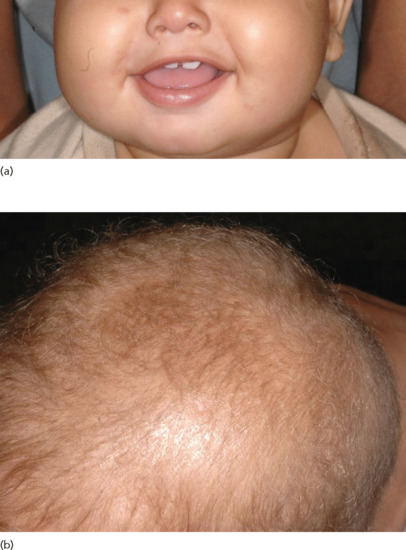
Figure 68.16 Typical cutaneous features of the trichohepatoenteric syndrome including puffy cheeks (a) and hypotrichosis (b).
Trichorrhexis invaginata
Clinical features
Hair is sparse, short, very thin and shows under the microscope the typical ball and socket (‘bamboo hair’) appearance in which the distal part of the hair shaft is compressed against the dilated and cupped proximal shaft. Trichorrhexis invaginata is considered as a hallmark of Netherton syndrome [139] (see Chapter 65) although it can be seen in other ichthyosiform disorders as well [140]. Trichorrhexis invaginata is often seen only after the first year of life. Examination of the eyebrows is recommended to demonstrate the hair shaft abnormality [141].
Pathogenesis
Netherton syndrome is caused by mutations in SPINK5, which encodes a serine protease inhibitor called LEKTI [142]. It is still unclear how LEKTI deficiency leads to hair shaft abnormality. LEKTI deficiency causes increased degradation of corneodesmosin and desmoglein 1 [143]; interestingly, corneodesmosin deficiency does not cause hair anomalies [69] and although defective desmoglein 1 has been linked to hypotrichosis in the context of severe dermatitis, multiple allergies and metabolic wasting (SAM syndrome) [144], hair microscopy did not reveal trichorrhexis invaginata, suggesting a specific role for LEKTI during hair shaft formation.
Trichothiodystrophy
Clinical features
Trichothiodystrophy refers to a strikingly heterogeneous group of disorders, all sharing in common brittle and fragile hair, which demonstrates on polarized hair microscopy a typical ‘tiger tail banding’ pattern [135, 145]. Various hair shaft defects such as pili torti and trichorrhexis nodosa have also been described. Hypotrichosis involves typically the scalp (Figure 68.17), eyebrows and eyelashes, but can also sometimes affect other areas. Nails demonstrate a wide range of dystrophic changes. Other clinical features include short stature, progeroid facies with loss of adipose tissue, microcephaly, various ocular manifestations including cataracts and microcornea, joint contractures and asthma [146]. The disorder has been reported in association with a number of clinical manifestations which demarcates distinct subsets including mental retardation and dental caries reported in Sabinas syndrome (MIM211390), infertility, developmental delay found in hair–brain syndrome (MIM234050), ichthyosis featured in Tay syndrome (MIM242170), photosensitivity seen in Photosensitivity Ichthyosis, Brittle hair, Intellectual impairment, Decreased fertility, and Short stature (PIBIDS syndrome) (MIM278720) and immune defects (MIM258360) (Table 68.4).
Table 68.4 Trichothiodystrophy variants
| Type | Clinical features | Syndrome eponym |
| A | Hair ± nails | – |
| B | Hair ± nails + mental retardation | Sabinas |
| C | Hair ± nails + mental retardation, folliculitis, retarded bone age + caries | Pollitt |
| D | (Brittle) hair ± nails + infertility, developmental delay, short stature | BIDS |
| E | Ichthyosis + BIDS +mental retardation ± decreased gonadal function + cataracts + progeroid appearance + microcephaly ± ataxia ± basal ganglia calcifications | Tay/IBIDS |
| F | Photosensitivity and IBIDS | PIBIDS |
| G | Hair ± mental retardation + immune defects | Itin |
| H | Trichothiodystrophy with severe intrauterine growth retardation, developmental delay, recurrent infections, cataracts, hepatic angioendotheliomas | – |
PIBIDS, photosensitivity ichthyosis, brittle hair, intellectual impairment, decreased fertility and short stature.
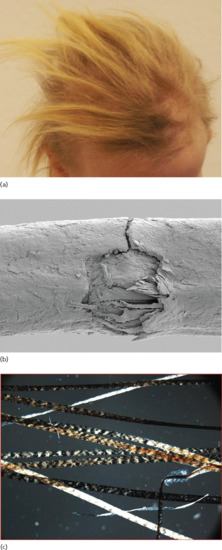
Figure 68.17 (a) Light-coloured and coarse hair in a patient with trichothiodystrophy.
(Courtesy of Professor Peter Itin.) (b) Defective cuticle visualized by scanning electron microscopy. (Courtesy of Professor Peter Itin.) (c) Tiger tail banding under polarizing light microscopy. (Courtesy of Professor Reuven Bergman.)
Pathogenesis
The hair defect results from a decrease in high sulphur protein contents in the hair shaft, which are a major component of the hair cuticle. The disorder has been found to be caused by mutations in a number of genes. Mutations in C7ORF11 (MPLKIP), encoding a protein whose function is poorly understood, cause non-photosensitive trichothiodystrophy [147]. Mutations in a number of genes encoding elements of the transcription factor complex, TTFIH [148], and involved in nucleotide excision repair cause photosensitive trichothiodystrophy including ERCC3, ERCC2 and GTF2H5 [149–151].
Pili triangulati et canaliculi
Clinical features
Isolated pili triangulati et canaliculi cause the uncombable hair syndrome [152]. This structural hair defect usually becomes apparent in early childhood as dry, coarse, frizzy and light hairs which stand straight up from the scalp and cannot be combed (Figure 68.18). Microscopic examination of pulled hair can be misleading as the triangular cross-sectional appearance (at the origin of the name of the hair shaft defect) and the typical longitudinal grooves, which confer to the hair its rigidity, are visible only on transverse section (or on scanning electron microscopy) [153].
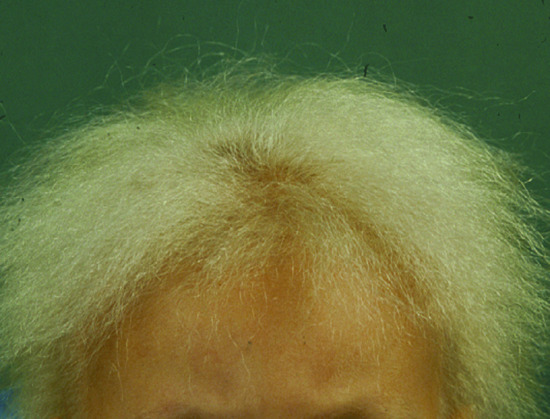
Figure 68.18 Uncombable hair syndrome.
(Courtesy of Professor Peter Itin.)
Pathogenesis
The uncombable hair syndrome can occur sporadically or be transmitted as a recessive or dominant trait [154]. Its molecular cause remains elusive. In addition, it has been described in association with skeletal dysplasias and other complex syndromes [153].
Other inherited hair shaft defects
Pili annulati and pseudoannulati
Pili annulati reflect the presence of air-filled cavities within the hair shaft, which results in alternating dark and light bands under the microscope [155, 156]. The disorder has been shown to be inherited as an autosomal dominant trait [157]. Pili pseudoannulati results from reflection of the light over flattened or twisted surfaces of the hair shaft, and is considered as a normal variant.
Loose anagen syndrome
This disorder starts in early childhood and features hair which is easily pulled away from the scalp. The disease is thought to result from poor adhesion between the cuticle and the inner root sheath. Autosomal dominant inheritance has been suggested. Patients often seek medical advice because of slow-growing hair and ensuing patchy alopecia. Hair can be short, sparse, unruly and is often light coloured. Increased shedding is noted [158] (Figure 68.19). Improvement over time is the rule. A diagnosis of loose anagen hair syndrome is based on the presence of 70% or more loose anagen hairs on a standard trichogram [159].

Figure 68.19 Loose anagen syndrome.
(Courtesy of Professor Peter Itin.)
Because symptoms are often mild, the disease may be underdiagnosed. However, because loose anagen hair can often be seen in normal children and adults, the disorder may also be overdiagnosed. A report suggesting that a keratin defect may underlie the disease [160] has not been subsequently replicated. Loose anagen syndrome has been reported in association with pili triangulati and canaliculi as well as with a Noonan-like phenotype [161].
Loose anagen syndrome should be distinguished from the short anagen syndrome [162] which is characterized by the inability to grow long hair because of an idiopathic short anagen phase. In contrast with loose anagen syndrome, it is not associated with hair unruliness, systemic diseases or skin disorders. Here too, some improvement is typical after puberty.
Kinky hair
Neonatal kinky hair is typical of the trichodento-osseous syndrome, caused by mutations in the DLX3 gene [163, 164]. The disorder is also associated with dysplastic nails, abnormal teeth and skeletal abnormalities including increased bone density and dolichocephaly due to premature fusion of the cranial sutures. Kinky hair is also found in Menkes disease, giant axonal neuropathy 1, Noonan syndrome and oculodentodigital dysplasia [165, 166].
Spiky hair
Spiky hair is a hallmark of the athyroidal hypothyroidism with spiky hair and cleft palate (Bamforth–Lazarus) syndrome, which also features choanal atresia and bifid epiglottis [167]. The disorder is transmitted in an autosomal recessive fashion and is caused by mutations in the FOXE1 gene [168], which is a downstream target of the Sonic hedgehog signalling pathway during hair follicle morphogenesis [169].
OVERALL APPROACH TO THE DIAGNOSIS OF GENOTRICHOSES
The most important step in the diagnosis of this group of disorders is to differentiate them from acquired diseases. Family history, neonatal or early onset, and associated clinical features can favour Mendelian inheritance although they cannot rule out an acquired form of hypertrichosis or alopecia. For example, the diagnosis of alopecia universalis congenita may require a skin biopsy as it is often difficult to distinguish this disorder from autoimmune alopecia areata.
Once it is clear that a patient is affected by an inherited form of hypertrichosis (see Table 68.1), atrichia (see Table 68.2) or hypotrichosis (see Table 68.3), it is important in order to make a diagnosis to establish: (i) the mode of inheritance;(ii) the absence (see Tables 68.1A, 68.2A and 68.3A) or presence (see Tables 68.1B, 68.2B and 68.3B) of extracutaneous manifestations; and (iii) the absence or presence of microscopic structural hair shafts abnormalities. Although evidence-based treatment options are mostly non-existent at this stage for most genotrichoses, a correct diagnosis may suggest the need for systemic work-up (see Tables 68.1B, 68.2B and 68.3B) and is essential to appropriately direct the subsequent molecular analysis, which in turn will set the stage for proper genetic counselling and prenatal diagnosis, when indicated.
References
- Duverger O, Morasso MI. To grow or not to grow: Hair morphogenesis and human genetic hair disorders. Semin Cell Dev Biol 2014;25–26:22–33.
- Betz RC, Cabral RM, Christiano AM, et al. Unveiling the roots of monogenic genodermatoses: genotrichoses as a paradigm. J Invest Dermatol 2012;132:906–14.
- Botchkarev VA, Kishimoto J. Molecular control of epithelial–mesenchymal interactions during hair follicle cycling. J Invest Derm Symp P 2003;8:46–55.
- Jamora C, DasGupta R, Kocieniewski P, et al. Links between signal transduction, transcription and adhesion in epithelial bud development. Nature 2003;422:317–22.
- Paus R, Cotsarelis G. The biology of hair follicles. N Engl J Med 1999;341:491–7.
- Stenn KS, Paus R. Controls of hair follicle cycling. Physiol Rev 2001;81:449–94.
- Firooz A, Firoozabadi MR, Ghazisaidi B, et al. Concepts of patients with alopecia areata about their disease. BMC Dermatol 2005;5:1.
- Hadshiew IM, Foitzik K, Arck PC, et al. Burden of hair loss: stress and the underestimated psychosocial impact of telogen effluvium and androgenetic alopecia. J Invest Dermatol 2004;123:455–7.
- Leavitt M. Understanding and management of female pattern alopecia. Facial Plast Surg 2008;24:414–27.
- Ruiz-Doblado S, Carrizosa A, Garcia-Hernandez MJ. Alopecia areata: psychiatric comorbidity and adjustment to illness. Int J Dermatol 2003;42:434–7.
- Williamson D, Gonzalez M, Finlay AY. The effect of hair loss on quality of life. J Eur Acad Dermatol Venereol 2001;15:137–9.
- Tadin M, Braverman E, Cianfarani S, et al. Complex cytogenetic rearrangement of chromosome 8q in a case of Ambras syndrome. Am J Med Genet 2001;102:100–4.
- Fantauzzo KA, Tadin-Strapps M, You Y, et al. A position effect on TRPS1 is associated with Ambras syndrome in humans and the Koala phenotype in mice. Hum Mol Genet 2008;17:3539–51.
- Canun S, Guevara-Sangines EG, Elvira-Morales A, et al. Hypertrichosis terminalis, gingival hyperplasia, and a characteristic face: a new distinct entity. Am J Med Genet A 2003;116A:278–83.
- Sun M, Li N, Dong W, et al. Copy-number mutations on chromosome 17q24.2-q24.3 in congenital generalized hypertrichosis terminalis with or without gingival hyperplasia. Am J Hum Genet 2009;84:807–13.
- DeStefano GM, Kurban M, Anyane-Yeboa K, et al. Mutations in the cholesterol transporter gene ABCA5 are associated with excessive hair overgrowth. PLoS Genet 2014;10:e1004333.
- Lazalde B, Sanchez-Urbina R, Nuno-Arana I, et al. Autosomal dominant inheritance in Cantu syndrome (congenital hypertrichosis, osteochondrodysplasia, and cardiomegaly). Am J Med Genet 2000;94:421–7.
- Harakalova M, van Harssel JJ, Terhal PA, et al. Dominant missense mutations in ABCC9 cause Cantu syndrome. Nat Genet 2012;44:793–6.
- van Bon BW, Gilissen C, Grange DK, et al. Cantu syndrome is caused by mutations in ABCC9. Am J Hum Genet 2012;90:1094–101.
- Shorter K, Farjo NP, Picksley SM, et al. Human hair follicles contain two forms of ATP-sensitive potassium channels, only one of which is sensitive to minoxidil. FASEB J 2008;22:1725–36.
- Martinez de Lagran Z, Gonzalez-Perez R, Asuncion Arregui-Murua M, et al. Hypertrichosis cubiti: another case of a well-recognized but under-reported entity. Pediatr Dermatol 2010;27:310–11.
- Reddy St, Antaya RJ. Two cases of isolated anterior cervical hypertrichosis. Pediatr Dermatol 2010;27:531–3.
- Harrington BC. The hair collar sign as a marker for neural tube defects. Pediatr Dermatol 2007;24:138–40.
- Stevens CA, Galen W. The hair collar sign. Am J Med Genet A 2008;146A:484–7.
- Chander R, Jain A, Jaykar K, et al. Faun tail nevus with aplasia cutis congenita. Pediatr Dermatol 2009;26:484–5.
- Kaptanoglu AF, Kaptanoglu E. Faun tail nevus and spinal dysraphism: cosmetic improvement with alexandrite laser epilation. Ann Dermatol 2011;23:S296–8.
- Molho-Pessach V, Ramot Y, Camille F, et al. H syndrome: the first 79 patients. J Am Acad Dermatol 2014;70:80–8.
- Senniappan S, Hughes M, Shah P, et al. Pigmentary hypertrichosis and non-autoimmune insulin-dependent diabetes mellitus (PHID) syndrome is associated with severe chronic inflammation and cardiomyopathy, and represents a new monogenic autoinflammatory syndrome. J Pediatr Endocrinol Metab 2013;26:877–82.
- Cliffe ST, Kramer JM, Hussain K, et al. SLC29A3 gene is mutated in pigmented hypertrichosis with insulin-dependent diabetes mellitus syndrome and interacts with the insulin signaling pathway. Hum Mol Genet 2009;18:2257–65.
- Molho-Pessach V, Lerer I, Abeliovich D, et al. The H syndrome is caused by mutations in the nucleoside transporter hENT3. Am J Hum Genet 2008;83:529–34.
- Colmenero I, Molho-Pessach V, Torrelo A, et al. Emperipolesis: an additional common histopathologic finding in H syndrome and Rosai–Dorfman disease. Am J Dermatopathol 2012;34:315–20.
- Slee PH, van der Waal RI, Schagen van Leeuwen JH, et al. Paraneoplastic hypertrichosis lanuginosa acquisita: uncommon or overlooked? Br J Dermatol 2007;157:1087–92.
- Rajpar SF, Hague JS, Abdullah A, et al. Hair removal with the long-pulse alexandrite and long-pulse Nd:YAG lasers is safe and well tolerated in children. Clin Exp Dermatol 2009;34:684–7.
- Cuestas-Carnero R, Bornancini CA. Hereditary generalized gingival fibromatosis associated with hypertrichosis: report of five cases in one family. J Oral Maxillofac Surg 1988;46:415–20.
- Sprecher E, Bergman R, Szargel R, et al. Identification of a genetic defect in the hairless gene in atrichia with papular lesions: evidence for phenotypic heterogeneity among inherited atrichias. Am J Hum Genet 1999;64:1323–9.
- Ahmad W, Panteleyev AA, Christiano AM. The molecular basis of congenital atrichia in humans and mice: mutations in the hairless gene. J Invest Derm Symp P 1999;4:240–3.
- Cichon S, Anker M, Vogt IR, et al. Cloning, genomic organization, alternative transcripts and mutational analysis of the gene responsible for autosomal recessive universal congenital alopecia. Hum Mol Genet 1998;7:1671–9.
- Bergman R, Schein-Goldshmid R, Hochberg Z, et al. The alopecias associated with vitamin D-dependent rickets type IIA and with hairless gene mutations: a comparative clinical, histologic, and immunohistochemical study. Arch Dermatol 2005;141:343–51.
- Ahmad W, Faiyaz ul Haque M, Brancolini V, et al. Alopecia universalis associated with a mutation in the human hairless gene. Science 1998;279:720–4.
- Liu L, Kim H, Casta A, et al. Hairless is a histone H3K9 demethylase. FASEB J 2014;28:1534–42.
- Thompson CC, Sisk JM, Beaudoin GM, 3rd. Hairless and Wnt signaling: allies in epithelial stem cell differentiation. Cell Cycle 2006;5:1913–17.
- Luke CT, Casta A, Kim H, et al. Hairless and the polyamine putrescine form a negative regulatory loop in the epidermis. Exp Dermatol 2013;22:644–9.
- Hsieh JC, Sisk JM, Jurutka PW, et al. Physical and functional interaction between the vitamin D receptor and hairless corepressor, two proteins required for hair cycling. J Biol Chem 2003;278:38665–74.
- Panteleyev AA, Paus R, Christiano AM. Patterns of hairless (hr) gene expression in mouse hair follicle morphogenesis and cycling. Am J Pathol 2000;157:1071–9.
- Wen Y, Liu Y, Xu Y, et al. Loss-of-function mutations of an inhibitory upstream ORF in the human hairless transcript cause Marie Unna hereditary hypotrichosis. Nat Genet 2009;41:228–33.
- Argenziano G, Sammarco E, Rossi A, et al. Marie Unna hereditary hypotrichosis. Eur J Dermatol 1999;9:278–80.
- Malloy PJ, Zhu W, Zhao XY, et al. A novel inborn error in the ligand-binding domain of the vitamin D receptor causes hereditary vitamin D-resistant rickets. Mol Genet Metab 2001;73:138–48.
- Malloy PJ, Feldman D. The role of vitamin D receptor mutations in the development of alopecia. Mol Cell Endocrinol 2011;347:90–6.
- Parren LJ, Abuzahra F, Wagenvoort T, et al. Linkage refinement of Bazex–Dupre–Christol syndrome to an 11.4-Mb interval on chromosome Xq25-27.1. Br J Dermatol 2011;165:201–3.
- Goeteyn M, Geerts ML, Kint A, et al. The Bazex–Dupre–Christol syndrome. Arch Dermatol 1994;130:337–42.
- Parren LJ, Frank J. Hereditary tumour syndromes featuring basal cell carcinomas. Br J Dermatol 2011;165:30–4.
- Wheeler CE, Jr., Carroll MA, Groben PA, et al. Autosomal dominantly inherited generalized basaloid follicular hamartoma syndrome: report of a new disease in a North Carolina family. J Am Acad Dermatol 2000;43:189–206.
- Manouvrier-Hanu S, Largilliere C, Benalioua M, et al. The GAPO syndrome. Am J Med Genet 1987;26:683–8.
- Stranecky V, Hoischen A, Hartmannova H, et al. Mutations in ANTXR1 cause GAPO syndrome. Am J Hum Genet 2013;92:792–9.
- Nicolaidou P, Tsitsika A, Papadimitriou A, et al. Hereditary vitamin D-resistant rickets in Greek children: genotype, phenotype, and long-term response to treatment. J Pediatr Endocrinol Metab 2007;20:425–30.
- Megarbane H, Megarbane A. Ichthyosis follicularis, alopecia, and photophobia (IFAP) syndrome. Orphanet J Rare Dis 2011;6:29.
- Naiki M, Mizuno S, Yamada K, et al. MBTPS2 mutation causes BRESEK/BRESHECK syndrome. Am J Med Genet A 2012;158A:97–102.
- Herd RM, Benton EC. Keratosis follicularis spinulosa decalvans: report of a new pedigree. Br J Dermatol 1996;134:138–42.
- Aten E, Brasz LC, Bornholdt D, et al. Keratosis follicularis spinulosa decalvans is caused by mutations in MBTPS2. Hum Mutat 2010;31:1125–33.
- Oeffner F, Fischer G, Happle R, et al. IFAP syndrome is caused by deficiency in MBTPS2, an intramembrane zinc metalloprotease essential for cholesterol homeostasis and ER stress response. Am J Hum Genet 2009;84:459–67.
- Rawson RB. The site-2 protease. Biochim Biophys Acta 2013;1828:2801–7.
- Tang L, Liang J, Wang W, et al. A novel mutation in MBTPS2 causes a broad phenotypic spectrum of ichthyosis follicularis, atrichia, and photophobia syndrome in a large Chinese family. J Am Acad Dermatol 2011;64:716–22.
- Traboulsi E, Waked N, Megarbane H, et al. Ocular findings in ichthyosis follicularis–alopecia–photophobia (IFAP) syndrome. Ophthalmic Genet 2004;25:153–6.
- Khandpur S, Bhat R, Ramam M. Ichthyosis follicularis, alopecia and photophobia (IFAP) syndrome treated with acitretin. J Eur Acad Dermatol Venereol 2005;19:759–62.
- Ming A, Happle R, Grzeschik KH, et al. Ichthyosis follicularis, alopecia, and photophobia (IFAP) syndrome due to mutation of the gene MBTPS2 in a large Australian kindred. Pediatr Dermatol 2009;26:427–31.
- Shimomura Y, Agalliu D, Vonica A, et al. APCDD1 is a novel Wnt inhibitor mutated in hereditary hypotrichosis simplex. Nature 2010;464:1043–7.
- Fu J, Hsu W. Epidermal Wnt controls hair follicle induction by orchestrating dynamic signaling crosstalk between the epidermis and dermis. J Invest Dermatol 2013;133:890–8.
- Myung PS, Takeo M, Ito M, et al. Epithelial Wnt ligand secretion is required for adult hair follicle growth and regeneration. J Invest Dermatol 2013;133:31–41.
- Levy-Nissenbaum E, Betz RC, Frydman M, et al. Hypotrichosis simplex of the scalp is associated with nonsense mutations in CDSN encoding corneodesmosin. Nat Genet 2003;34:151–3.
- Oji V, Eckl KM, Aufenvenne K, et al. Loss of corneodesmosin leads to severe skin barrier defect, pruritus, and atopy: unraveling the peeling skin disease. Am J Hum Genet 2010;87:274–81.
- Capon F, Allen MH, Ameen M, et al. A synonymous SNP of the corneodesmosin gene leads to increased mRNA stability and demonstrates association with psoriasis across diverse ethnic groups. Hum Mol Genet 2004;13:2361–8.
- Capon F, Munro M, Barker J, et al. Searching for the major histocompatibility complex psoriasis susceptibility gene. J Invest Dermatol 2002;118:745–51.
- Caubet C, Bousset L, Clemmensen O, et al. A new amyloidosis caused by fibrillar aggregates of mutated corneodesmosin. FASEB J 2010;24:3416–26.
- Sprecher E. Genetic hair and nail disorders. Clin Dermatol 2005;23:47–55.
- Shimomura Y, Wajid M, Petukhova L, et al. Mutations in the lipase H gene underlie autosomal recessive woolly hair/hypotrichosis. J Invest Dermatol 2009;129:622–8.
- Shimomura Y. Congenital hair loss disorders: rare, but not too rare. J Dermatol 2012;39:3–10.
- Kazantseva A, Goltsov A, Zinchenko R, et al. Human hair growth deficiency is linked to a genetic defect in the phospholipase gene LIPH. Science 2006;314:982–5.
- Pasternack SM, von Kugelgen I, Al Aboud K, et al. G protein-coupled receptor P2Y5 and its ligand LPA are involved in maintenance of human hair growth. Nat Genet 2008;40:329–34.
- Shimomura Y, Wajid M, Ishii Y, et al. Disruption of P2RY5, an orphan G protein-coupled receptor, underlies autosomal recessive woolly hair. Nat Genet 2008;40:335–9.
- Pasternack SM, von Kugelgen I, Muller M, et al. In vitro analysis of LIPH mutations causing hypotrichosis simplex: evidence confirming the role of lipase H and lysophosphatidic acid in hair growth. J Invest Dermatol 2009;129:2772–6.
- Carrington PR, Chen H, Altick JA. Trichorhinophalangeal syndrome, type I. J Am Acad Dermatol 1994;31:331–6.
- Momeni P, Glockner G, Schmidt O, et al. Mutations in a new gene, encoding a zinc-finger protein, cause tricho-rhino-phalangeal syndrome type I. Nat Genet 2000;24:71–4.
- Ludecke HJ, Schaper J, Meinecke P, et al. Genotypic and phenotypic spectrum in tricho-rhino-phalangeal syndrome types I and III. Am J Hum Genet 2001;68:81–91.
- Langer LO Jr, Krassikoff N, Laxova R, et al. The tricho-rhino-phalangeal syndrome with exostoses (or Langer–Giedion syndrome): four additional patients without mental retardation and review of the literature. Am J Med Genet 1984;19:81–112.
- Ludecke HJ, Johnson C, Wagner MJ, et al. Molecular definition of the shortest region of deletion overlap in the Langer–Giedion syndrome. Am J Hum Genet 1991;49:1197–206.
- Lai-Cheong JE, Arita K, McGrath JA. Genetic diseases of junctions. J Invest Dermatol 2007;127:2713–25.
- Mazereeuw-Hautier J, Bitoun E, Chevrant-Breton J, et al. Keratitis–ichthyosis–deafness syndrome: disease expression and spectrum of connexin 26 (GJB2) mutations in 14 patients. Br J Dermatol 2007;156:1015–19.
- Lamartine J, Munhoz Essenfelder G, Kibar Z, et al. Mutations in GJB6 cause hidrotic ectodermal dysplasia. Nat Genet 2000;26:142–4.
- Paznekas WA, Boyadjiev SA, Shapiro RE, et al. Connexin 43 (GJA1) mutations cause the pleiotropic phenotype of oculodentodigital dysplasia. Am J Hum Genet 2003;72:408–18.
- Paznekas WA, Karczeski B, Vermeer S, et al. GJA1 mutations, variants, and connexin 43 dysfunction as it relates to the oculodentodigital dysplasia phenotype. Hum Mutat 2009;30:724–33.
- Souied E, Amalric P, Chauvet ML, et al. Unusual association of juvenile macular dystrophy with congenital hypotrichosis: occurrence in two siblings suggesting autosomal recessive inheritance. Ophthalmic Genet 1995;16:11–15.
- Sprecher E, Bergman R, Richard G, et al. Hypotrichosis with juvenile macular dystrophy is caused by a mutation in CDH3, encoding P-cadherin. Nat Genet 2001;29:134–6.
- Burke JM, Cao F, Irving PE, et al. Expression of E-cadherin by human retinal pigment epithelium: delayed expression in vitro. Invest Ophthalmol Vis Sci 1999;40:2963–70.
- Samuelov L, Sprecher E, Sugawara K, et al. Topobiology of human pigmentation: P-cadherin selectively stimulates hair follicle melanogenesis. J Invest Dermatol 2013;133:1591–600.
- Samuelov L, Sprecher E, Tsuruta D, et al. P-cadherin regulates human hair growth and cycling via canonical Wnt signaling and transforming growth factor-beta2. J Invest Dermatol 2012;132:2332–41.
- Kjaer KW, Hansen L, Schwabe GC, et al. Distinct CDH3 mutations cause ectodermal dysplasia, ectrodactyly, macular dystrophy (EEM syndrome). J Med Genet 2005;42:292–8.
- Basel-Vanagaite L, Attia R, Ishida-Yamamoto A, et al. Autosomal recessive ichthyosis with hypotrichosis caused by a mutation in ST14, encoding type II transmembrane serine protease matriptase. Am J Hum Genet 2007;80:467–77.
- List K, Bugge TH, Szabo R. Matriptase: potent proteolysis on the cell surface. Mol Med 2006;12:1–7.
- Chen YW, Wang JK, Chou FP, et al. Matriptase regulates proliferation and early, but not terminal, differentiation of human keratinocytes. J Invest Dermatol 2014;134:405–14.
- Alef T, Torres S, Hausser I, et al. Ichthyosis, follicular atrophoderma, and hypotrichosis caused by mutations in ST14 is associated with impaired profilaggrin processing. J Invest Dermatol 2009;129:862–9.
- Cheng AS, Bayliss SJ. The genetics of hair shaft disorders. J Am Acad Dermatol 2008;59:1–22;quiz 3–6.
- Miteva M, Tosti A. Dermatoscopy of hair shaft disorders. J Am Acad Dermatol 2013;68:473–81.
- Zlotogorski A, Horev L, Glaser B. Monilethrix: a keratin hHb6 mutation is co-dominant with variable expression. Exp Dermatol 1998;7:268–72.
- Winter H, Rogers MA, Gebhardt M, et al. A new mutation in the type II hair cortex keratin hHb1 involved in the inherited hair disorder monilethrix. Hum Genet 1997;101:165–9.
- Winter H, Rogers MA, Langbein L, et al. Mutations in the hair cortex keratin hHb6 cause the inherited hair disease monilethrix. Nat Genet 1997;16:372–4.
- van Steensel MA, Steijlen PM, Bladergroen RS, et al. A missense mutation in the type II hair keratin hHb3 is associated with monilethrix. J Med Genet 2005;42:e19.
- Schweizer J. More than one gene involved in monilethrix: intracellular but also extracellular players. J Invest Dermatol 2006;126:1216–19.
- Taylor CJ, Green SH. Menkes' syndrome (trichopoliodystrophy): use of scanning electron-microscope in diagnosis and carrier identification. Dev Med Child Neurol 1981;23:361–8.
- Zitelli JA. Pseudomonilethrix. An artifact. Arch Dermatol 1986;122:688–90.
- Rossi A, Iorio A, Scali E, et al. Monilethrix treated with minoxidil. Int J Immunopathol Pharmacol 2011;24:239–42.
- Saxena U, Ramesh V, Misra RS. Topical minoxidil in monilethrix. Dermatologica 1991;182:252–3.
- de Berker D, Dawber RP. Monilethrix treated with oral retinoids. Clin Exp Dermatol 1991;16:226–8.
- Karincaoglu Y, Coskun BK, Seyhan ME, et al. Monilethrix: improvement with acitretin. Am J Clin Dermatol 2005;6:407–10.
- Sivasundram A. A case of monilethrix treated with etretinate. Dermatology 1995;190:89.
- Brooke MA, Nitoiu D, Kelsell DP. Cell–cell connectivity: desmosomes and disease. J Pathol 2012;226:158–71.
- Neild VS, Pegum JS, Wells RS. The association of keratosis pilaris atrophicans and woolly hair, with and without Noonan's syndrome. Br J Dermatol 1984;110:357–62.
- Fujimoto A, Farooq M, Fujikawa H, et al. A missense mutation within the helix initiation motif of the keratin K71 gene underlies autosomal dominant woolly hair/hypotrichosis. J Invest Dermatol 2012;132:2342–9.
- Shimomura Y, Wajid M, Petukhova L, et al. Autosomal-dominant woolly hair resulting from disruption of keratin 74 (KRT74), a potential determinant of human hair texture. Am J Hum Genet 2010;86:632–8.
- McKoy G, Protonotarios N, Crosby A, et al. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet 2000;355:2119–24.
- Norgett EE, Hatsell SJ, Carvajal-Huerta L, et al. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum Mol Genet 2000;9:2761–6.
- Collie WR, Moore CM, Goka TJ, et al. Pili torti as marker for carriers of Menkes disease. Lancet 1978;1:607–8.
- Tumer Z, Moller LB. Menkes disease. Eur J Hum Genet 2010;18:511–18.
- Chelly J, Tumer Z, Tonnesen T, et al. Isolation of a candidate gene for Menkes disease that encodes a potential heavy metal binding protein. Nat Genet 1993;3:14–19.
- Vulpe C, Levinson B, Whitney S, et al. Isolation of a candidate gene for Menkes disease and evidence that it encodes a copper-transporting ATPase. Nat Genet 1993;3:7–13.
- Tumer Z. An overview and update of ATP7A mutations leading to Menkes disease and occipital horn syndrome. Hum Mutat 2013;34:417–29.
- Scott MJ Jr, Bronson DM, Esterly NB. Bjornstad syndrome and pili torti. Pediatr Dermatol 1983;1:45–50.
- Hinson JT, Fantin VR, Schonberger J, et al. Missense mutations in the BCS1L gene as a cause of the Bjornstad syndrome. N Engl J Med 2007;356:809–19.
- Sprecher E, Chavanas S, DiGiovanna JJ, et al. The spectrum of pathogenic mutations in SPINK5 in 19 families with Netherton syndrome: implications for mutation detection and first case of prenatal diagnosis. J Invest Dermatol 2001;117:179–87.
- Glaessl A, Hohenlautner U, Landthaler M, et al. Sporadic Bazex–Dupre–Christol-like syndrome: early onset basal cell carcinoma, hypohidrosis, hypotrichosis, and prominent milia. Dermatol Surg 2000;26:152–4.
- Patel HP, Unis ME. Pili torti in association with citrullinemia. J Am Acad Dermatol 1985;12:203–6.
- Karadag Kose O, Gulec AT. Clinical evaluation of alopecias using a handheld dermatoscope. J Am Acad Dermatol 2012;67:206–14.
- Burkhart CG, Burkhart CN. Trichorrhexis nodosa revisited. Skinmed 2007;6:57–8.
- Fabre A, Andre N, Breton A, et al. Intractable diarrhea with “phenotypic anomalies” and tricho-hepato-enteric syndrome: two names for the same disorder. Am J Med Genet A 2007;143:584–8.
- Fichtel JC, Richards JA, Davis LS. Trichorrhexis nodosa secondary to argininosuccinicaciduria. Pediatr Dermatol 2007;24:25–7.
- Lurie R, Hodak E, Ginzburg A, et al. Trichorrhexis nodosa: a manifestation of hypothyroidism. Cutis 1996;57:358–9.
- Liang C, Morris A, Schlucker S, et al. Structural and molecular hair abnormalities in trichothiodystrophy. J Invest Dermatol 2006;126:2210–16.
- Stone RL, Aimi J, Barshop BA, et al. A mutation in adenylosuccinate lyase associated with mental retardation and autistic features. Nat Genet 1992;1:59–63.
- Fabre A, Charroux B, Martinez-Vinson C, et al. SKIV2L mutations cause syndromic diarrhea, or trichohepatoenteric syndrome. Am J Hum Genet 2012;90:689–92.
- Hartley JL, Zachos NC, Dawood B, et al. Mutations in TTC37 cause trichohepatoenteric syndrome (phenotypic diarrhea of infancy). Gastroenterology 2010;138:2388–98, 98 e1–2.
- Frenk E, Mevorah B. Ichthyosis linearis circumflexa comel with trichorrhexis invaginata (Netherton's syndrome): an ultrastructural study of the skin changes. Arch Dermatol Forsch 1972;245:42–9.
- Gyure KA, Kurczynski TW, Gunning W, et al. Autosomal recessive neurodegenerative disorder with trichorrhexis invaginata and ectodermal dysplasia. Pediatr Neurol 1992;8:469–72.
- Powell J, Dawber RP, Ferguson DJ, et al. Netherton's syndrome: increased likelihood of diagnosis by examining eyebrow hairs. Br J Dermatol 1999;141:544–6.
- Chavanas S, Bodemer C, Rochat A, et al. Mutations in SPINK5, encoding a serine protease inhibitor, cause Netherton syndrome. Nat Genet 2000;25:141–2.
- Hovnanian A. Netherton syndrome: skin inflammation and allergy by loss of protease inhibition. Cell Tissue Res 2013;351:289–300.
- Samuelov L, Sarig O, Harmon RM, et al. Desmoglein 1 deficiency results in severe dermatitis, multiple allergies and metabolic wasting. Nat Genet 2013;45:1244–8.
- Liang C, Kraemer KH, Morris A, et al. Characterization of tiger-tail banding and hair shaft abnormalities in trichothiodystrophy. J Am Acad Dermatol 2005;52:224–32.
- Itin PH, Sarasin A, Pittelkow MR. Trichothiodystrophy: update on the sulfur-deficient brittle hair syndromes. J Am Acad Dermatol 2001;44:891–920;quiz 1–4.
- Nakabayashi K, Amann D, Ren Y, et al. Identification of C7orf11 (TTDN1) gene mutations and genetic heterogeneity in nonphotosensitive trichothiodystrophy. Am J Hum Genet 2005;76:510–16.
- Seroz T, Hwang JR, Moncollin V, et al. TFIIH: a link between transcription, DNA repair and cell cycle regulation. Curr Opin Genet Dev 1995;5:217–21.
- Takayama K, Salazar EP, Broughton BC, et al. Defects in the DNA repair and transcription gene ERCC2(XPD) in trichothiodystrophy. Am J Hum Genet 1996;58:263–70.
- Weeda G, Eveno E, Donker I, et al. A mutation in the XPB/ERCC3 DNA repair transcription gene, associated with trichothiodystrophy. Am J Hum Genet 1997;60:320–9.
- Giglia-Mari G, Coin F, Ranish JA, et al. A new, tenth subunit of TFIIH is responsible for the DNA repair syndrome trichothiodystrophy group A. Nat Genet 2004;36:714–19.
- Calderon P, Otberg N, Shapiro J. Uncombable hair syndrome. J Am Acad Dermatol 2009;61:512–15.
- Hicks J, Metry DW, Barrish J, et al. Uncombable hair (cheveux incoiffables, pili trianguli et canaliculi) syndrome: brief review and role of scanning electron microscopy in diagnosis. Ultrastruct Pathol 2001;25:99–103.
- Hebert AA, Charrow J, Esterly NB, et al. Uncombable hair (pili trianguli et canaliculi): evidence for dominant inheritance with complete penetrance based on scanning electron microscopy. Am J Med Genet 1987;28:185–93.
- Amichai B, Grunwald MH, Halevy S. Hair abnormality present since childhood. Pili annulati. Arch Dermatol 1996;132:575, 8.
- Price VH, Thomas RS, Jones FT. Pili annulati. Optical and electron microscopic studies. Arch Dermatol 1968;98:640–7.
- Giehl KA, Rogers MA, Radivojkov M, et al. Pili annulati: refinement of the locus on chromosome 12q24.33 to a 2.9-Mb interval and candidate gene analysis. Br J Dermatol 2009;160:527–33.
- Olsen EA, Bettencourt MS, Cote NL. The presence of loose anagen hairs obtained by hair pull in the normal population. J Invest Derm Symp P 1999;4:258–60.
- Tosti A, Peluso AM, Misciali C, et al. Loose anagen hair. Arch Dermatol 1997;133:1089–93.
- Chapalain V, Winter H, Langbein L, et al. Is the loose anagen hair syndrome a keratin disorder? A clinical and molecular study. Arch Dermatol 2002;138:501–6.
- Cordeddu V, Di Schiavi E, Pennacchio LA, et al. Mutation of SHOC2 promotes aberrant protein N-myristoylation and causes Noonan-like syndrome with loose anagen hair. Nat Genet 2009;41:1022–6.
- Antaya RJ, Sideridou E, Olsen EA. Short anagen syndrome. J Am Acad Dermatol 2005;53:S130–4.
- Price JA, Bowden DW, Wright JT, et al. Identification of a mutation in DLX3 associated with tricho-dento-osseous (TDO) syndrome. Hum Mol Genet 1998;7:563–9.
- Price JA, Wright JT, Kula K, et al. A common DLX3 gene mutation is responsible for tricho-dento-osseous syndrome in Virginia and North Carolina families. J Med Genet 1998;35:825–8.
- Kjaer KW, Hansen L, Eiberg H, et al. Novel Connexin 43 (GJA1) mutation causes oculo-dento-digital dysplasia with curly hair. Am J Med Genet A 2004;127A:152–7.
- Tazir M, Nouioua S, Magy L, et al. Phenotypic variability in giant axonal neuropathy. Neuromuscul Disord 2009;19:270–4.
- Carre A, Hamza RT, Kariyawasam D, et al. A novel FOXE1 mutation (R73S) in Bamforth–Lazarus syndrome causing increased thyroidal gene expression. Thyroid 2014;24:649–54.
- Clifton-Bligh RJ, Wentworth JM, Heinz P, et al. Mutation of the gene encoding human TTF-2 associated with thyroid agenesis, cleft palate and choanal atresia. Nat Genet 1998;19:399–401.
- Brancaccio A, Minichiello A, Grachtchouk M, et al. Requirement of the forkhead gene Foxe1, a target of sonic hedgehog signaling, in hair follicle morphogenesis. Hum Mol Genet 2004;13:2595–606.