CHAPTER 69
Genetic Defects of Nails and Nail Growth
Adam Rubin1 and Amy S. Paller2
1Department of Dermatology, University of Pennsylvania, Philadelphia, PA, USA
2Departments of Dermatology and Pediatrics, Northwestern University, Chicago, IL, USA
Pachyonychia congenita
Definition and nomenclature
Nail changes are a component of a large number of genodermatoses, especially the ectodermal dysplasias. Included in this chapter are some of the more common hereditary nail disorders that are not described in detail elsewhere and have distinctive manifestations. A list of additional non-syndromic and syndromic genodermatoses with nail abnormalities and their features is given in Table 69.1.
Table 69.1 Key genetic syndromes with known mutations that have nail manifestations. (Modified from Harper J, Oranje A, Prose N, eds. Textbook of Pediatric Dermatology, 2nd edn. Blackwell Publishing, with permission.)
| Phenotypic characteristics | |||||
| Name (alternative names) | MIM number/primary ref. | Inh | Gene mutation | Nails | Other |
| Syndromic genodermatoses with nail anomalies | |||||
| Acro-dermato-ungual-lacrimal-tooth syndrome (adult syndrome) [1] | 103285 | AD | TP63 | Finger and toenail dysplasia | Skin: intensive freckling Other: lacrimal duct atresia; ectrodactyly, syndactyly; hypoplastic breasts and nipples |
| Adams–Oliver syndrome [2–5] | 614814 100300 614219 615297 |
AD | RBPJ EOGT DOCK6 ARHGAP31 |
Nail aplasia | Cutis aplasia, syndactyly, microcephaly, short palpebral fissures, short distal phalanges, absent toes, asymmetric shortening of the hands, asymmetric reductions of the feet, intellectual deficits |
| Anaemia, dyserythropoietic congenital, type i [6] | 224120 | AR | CDAN1 (codanin 1) | Nail hypoplasia | Growth retardation, acrodysostosis, scoliosis |
| Ankyloblepharon-ectodermal defects–cleft lip and palate (AEC) syndrome [7] | 106260 | AD | TP63 | Severe dystrophy | Skin: dry and smooth; palmoplantar hyperkeratosis with obliteration of dermatoglyphic patterns; occas reticulate hyperpigmentation; supernumerary nipples, severe recurrent scalp pustulation and erosion |
| Face: ankyloblepharon filiforme adnatum with partial fusion of eyelids at birth; broad nasal bridge; hypoplastic maxilla; auricular abnormalities; cleft lip | |||||
| Other: lacrimal duct atresia; photophobia | |||||
| Autoimmune polyendocrinopathy–candidiasis–ectodermal dystrophy (apeced) syndrome; autoimmune polyendocrinopathy syndrome, type I [8] | 240300 | AD | AIRE | Thickened and dystrophic | Oral candidiasis; autoimmune endocrinopathies (hypergonadotropic hypogonadism, insulin-dependent diabetes autoimmune thyroid diseases and pituitary defects); autoimmune or immunomediated gastrointestinal diseases (chronic atrophic gastritis, pernicious anaemia and malabsorption); chronic active hepatitis; autoimmune skin diseases (vitiligo and alopecia); keratoconjunctivitis; immunological defects (cellular and humoral); asplenia and cholelithiasis |
| Brachydactyly, type b1 [9] | 113000 | AD | ROR2 (receptor tyrosine kinase-like orphan receptor 2) | Nail aplasia, anonychia | Short middle phalanges, rudimentary or absent terminal phalanges, syndactyly |
| Coffin–Siris syndrome [10] | 135900 | AR?AD? | SMARCB1, SMARCA4, SMARCA2, ARID1A ARID1B | Abs to hypoplastic fifth fingernails and toenails; other nails occas hypoplastic or abs | Skin: dermatoglyphic changes; simian crease Other: coarse face with thick lips, wide mouth and nose, anteverted nostrils and low nasal bridge; retardation of psychomotor and growth development; hypotonia; lax joints; clinodactyly of the fifth fingers; general abs of terminal phalanges of fifth fingers and toes; general aplasia or variable hypoplasia of middle and prox phalanges of other fingers and toes; bilateral or unilateral dislocation of the radial heads; small or abs patella; frequent respiratory infections; umbilical and inguinal hernias; cleft palate; feeding problems in infancy; six lumbar vertebrae; short sternum; microcephaly |
| Corneal intraepithelial dyskeratosis and ectodermal dysplasia [11] | 615225 | AD | NLR family, pyrin domain containing 1 (NLRP1) | Dystrophic nails with prominent thickening of nail beds | Keratopathy with neovascularization and corneal opacification, palmoplantar hyperkeratosis, dyshidrosis, corneal dyskeratosis, pruritic hyperkeratotic scars, palmoplantar hyperkeratosis, chronic rhinitis, raspy voice. |
| Cranio-ectodermal syndrome (Levin syndrome I; Sensenbrenner syndrome) [12] | 218330 | AR | IFT122 | Broad and short | Skin: dimples over elbows and knees; bilateral hallucal creases; single flexion crease on each toe; bilateral single palmar creases |
| Skeletal: rhizomelic shortness (greatest in upper limbs); disproportionate shortness of the fibulae; pronounced shortness of middle and dist phalanges of toes and fingers; cutaneous syndactyly; clinodactyly; increased space between first and second toes; hallux valgus; dolichocephaly; gen osteoporosis; highly arched palate; sagittal suture synostosis; short and narrow thorax; pectus excavatum | |||||
| Other: hyperopia; myopia; nystagmus; frontal bossing; epicanthal folds and antimongoloid slant; full cheeks; posteriorly angulated pinnae with hypoplastic antihelix; hypotelorism; broad nasal bridge; anteverted nares; everted lower lip; capillary naevus on the forehead; multiple oral frenula; cong heart defects | |||||
| Deafness and onychodystrophy (door syndrome) [13] | 220500 | AR | TBC1D24 | Hypoplastic and dyst finger and toenails; anonychia | Skin: dermatoglyphic abnormalities |
| Other: sensorineural deafness; apparently low-set ears; seizures and mental retardation; triphalangy of both thumbs and halluces; hypoplasia or aplasia of terminal phalanges of fingers and toes; occas clinodactyly and camptodactyly | |||||
| Dyskeratosis congenita (X-linked) (Zinsser–Cole–Engman syndrome) [14] | 305000 | XR | DKC1 | Dystrophy with late-onset paronychia occas leading to anonychia; hypoplasia | Skin: hyper- and hypomelanosis, telangiectatic erythema; ulcers; dry desquamation; atrophy; hyperkeratotic plaques (palmoplantar and over joints); premalignant lesions; abs fingerprints |
| Eyes: blepharitis; ectropion of the lower lids; obliteration of the punctalacrimalia; bullous conjunctivitis; continuous lacrimation | |||||
| Other: sharp facial features; occas mental and growth retardation; Fanconi-like pancytopenia; premalignant leukoplakia on lips, mouth, anus, urethra and conjunctiva; frail skeletal structure; genital anomalies; oesophageal dysfunction and/or diverticulum; atrophic lingual papillae; gingivitis | |||||
| Dyskeratosis congenita (autosomal dominant) [15–17] | 127550 613990 613989 |
AD | TERC TINF2 TERT |
Dystrophic | Skin: hyper- and hypomelanosis, telangiectatic erythema; ulcers; dry desquamation; atrophy; hyperkeratotic plaques (palmoplantar and over joints); premalignant lesions; abs fingerprints |
| Eyes: blepharitis; ectropion of the lower lids; obliteration of the puncta lacrimalia; bullous conjunctivitis; continuous lacrimation | |||||
| Other: sharp facial features; occas mental and growth retardation; Fanconi-like pancytopenia; premalignant leukoplakia on lips, mouth, anus, urethra and conjunctiva; frail skeletal structure; genital anomalies; oesophageal dysfunction and/or diverticulum; atrophic lingual papillae; gingivitis | |||||
| Dyskeratosis congenita (autosomal recessive) [18–23] | 224230 613276 613987 613988 615190 613989 |
AR | NOLA3 C16orf57 NOLA2 WRAP53 RTEL1 TERT |
Dystrophic | Skin: hyper- and hypomelanosis, telangiectatic erythema; ulcers; dry desquamation |
| Other: pancytopenia; thrombocytopenia; small platelets; T-cell abnormalities; dystrophic fingers and toes | |||||
| Ectodermal dysplasia with ectrodactyly and macular dystrophy [24] | 225280 | AR | CDH3 | Dysplastic | Ectrodactyly; syndactyly; cleft hand; macular dystrophy |
| Ellis–van Creveld syndrome (chondroectodermal dysplasia, mesoectodermal dysplasia) [25] See Weyer acrofacial dysostosis, below | 225500 | AR | EVC (Ellis van Creveld syndrome), EVC2 (Ellis van Creveld syndrome 2) | Dysplastic (brittle, furrowed and underdeveloped) | Skin: eczema, petechiae are described in different patterns Short limbs, polydactyly, abnormal teeth |
| Other: occas strabismus, cataract, coloboma of the iris, microphthalmia; exophthalmia; short-limbed dwarfism; bilateral postaxial polydactyly (gen of the hands); brachymetacarpy; thick and short bones of limbs; fusion of the hamate and capitate; clubfoot; genua valga; syndactyly; occas mild mental retardation; cong heart disease; respiratory difficulties; gingivolabial fusion; cleft palate; epispadia; hypospadias; hypoplastic genitalia | |||||
| Face: broad nose; occas cleft lip, frontal bossing and hypertelorism | |||||
| Epidermolysis bullosa group | |||||
| Epidermolysis bullosa dystrophica, autosomal recessive [26] | 226600 | AR | COL7A1 (collagen, type VII, alpha 1) | Anonychia, nail dystrophy, nail atrophy | Severe scarring blisters, contractures of hands and feet, mitten deformities, abnormal teeth, milia, gastrointestinal strictures |
| Epidermolysis bullosa, junctional, non-herlitz type [27] | 226650 | AR | COL17A1 (collagen XVII, alpha-1 polypeptide), LAMA3 (alpha-3 laminin), LAMB3 (beta-3 laminin), LAMC2 (gamma-2 laminin gene), ITGB4 (integrin, beta 4) | Nail dystrophy, fragile nails | Non-scarring blisters, abnormal teeth, plantar hyperkeratosis |
| Epidermolysis bullosa, lethal acantholytic [28] | 609638 | NK | DSP (desmoplakin) | Nail loss | Skin: universal alopecia, skin fragility, large sheets of skin detached, histopathology similar to pemphigus vulgaris |
| Epidermolysis bullosa simplex with muscular dystrophy [29] | 226670 | AR | PLEC (plectin) | Nail dystrophy | Progressive muscular dystrophy, blisters, atrophic scarring, alopecia |
| Erythrokeratodermia variabilis et progressiva (EKVP) [30] | 133200 | AD | GJA1 (gap junction protein alpha 1), (connexin 43) | Prominent porcelain-white proximal nails without dystrophy | Skin: normal skin at birth. In childhood, develop hyperpigmentation and scaling at sites of friction. This progresses to near-confluent corrugated hyperkeratosis. There is also darkening of the periorificial areas, palmoplantar keratoderma and transient figurate erythema. Erythema was induced by stress and warm conditions |
| Laryngoonychocutaneous syndrome [30] | 245660 | AR | LAMA3 (laminin, alpha 3) | Dystrophic nails, loss of nails | Hoarseness, skin ulcers, abnormal teeth, conjunctival injection and erosions |
| Focal dermal hypoplasia syndrome (Goltz syndrome) [31] | 305600 | XL | PORCN | Thin, spooned, narrow, grooved hypopigmented or abs | Skin: abs of skin from various parts at birth; areas of underdevelopment or thinness; linear hypo- or hyperpigmentation; telangiectasia; herniation of subcutaneous fat; multiple papillomas of mucous membranes and periorificial skin; follicular hyperkeratotic papules; angiofibromatous nodules around lips and anus; palmoplantar hyperkeratosis; occas dermatoglyphic changes |
| Other: occas hearing loss; colobomas; microphthalmia; irregularity of pupils; clouding of cornea or vitreous; blue sclerae; lip papillomas; malformed auricles; asymmetry and notching of the alae nasi; pointed chin; triangular face; hypertelorism; mental retardation; short stature; syndactyly; polydactyly; hypoplasia of the external genitalia; umbilical and/or inguinal hernia; vertebral anomalies (scoliosis, spina bifida etc.) highly arched palate; gum papillomas; small breasts | |||||
| Growth retardation–alopecia–pseudoanodontia–optic atrophy (gapo) [32] | 230740 | AR | ANTXR1 | Hyperconvexity on fingers and toes is referred to in two patients | Skin: dry; redundant; fragile with inadequate wound healing (small, depressed scars); depigmented areas; unusual wrinkles; leather-like and thick on nape and upper back; abnormal dermatoglyphics |
| Face: ‘small’ and ‘characteristic’; asymmetrical; craniofacial dysostosis; micrognathia; protruding and thickened lips; protruding auricles; prom supraorbital ridges; depressed nasal bridge; minor auricular malformations | |||||
| Other: sensorineural hypoacusia; optic atrophy; glaucoma; keratoconus; nystagmus; photophobia; dwarfism; occas mental retardation; symmetrical prox shortening of humeri; hyperextensible fingers; second and third toes smaller than the fourth; wide gap between hallux and second toes; wide anterior fontanelle; prom scalp veins; hepatosplenomegaly; bilateral choanal atresia; hyperplasia of sublingual connective tissue; hypoplasia of mammary glands; pectus excavatum; umbilical hernia; delayed bone maturation through childhood and adolescence | |||||
| Haim-Munk syndrome [33] | 245010 | AR | CTSC (cathepsin C) | Onychogryphosis | Congenital palmoplantar keratosis, pes planus, periodontitis, arachnodactyly, acroosteolysis |
| Hyperphosphatasia with mental retardation syndrome 2 [34] | 614749 | AR | Phosphatidylinositol glycan anchor biosynthesis, class O (PIGO) | Nail hypoplasia, absent nails | Born with anal stenosis/anal atresia with perineal fistula; wide-set eyes, long palpebral fissures, short nose with broad nasal bridge and tip, tented mouth, Persistently elevated serum alkaline phosphatase (ALP), mental retardation |
| Hypohidrotic ectodermal dysplasia–X-linked (ED1; Christ–Siemens–Touraine (CST) syndrome) [35] | 305100 | XR | ED1 | Gen normal; sometimes dyst or abs at birth and/or fragile and brittle with incomplete development | Skin: thin, smooth and dry due to hypoplasia or abs of sebaceous glands; occas pigmentation and dermatoglyphic changes; abs or supernumerary nipples and areolae |
| Face: highly characteristic in persons who are severely affected (gen males) with thick and prom lips, depressed nasal bridge (saddle nose), frontal bossing, hypoplasia of the maxilla, wrinkles beneath the eyes, or around the eyes, nose and mouth and minor alterations of the auricles; the periorbital region is often more darkly pigmented than the rest of the body | |||||
| Other: occas conductive loss; photophobia; decreased function of the lacrimal glands; aplasia or hypoplasia of the lacrimal ducts; atrophic rhinitis; otitis media; decreased sense of taste and/or smell; atrophied mucous glands of the upper respiratory tract; respiratory difficulties; chronic pharyngitis and laryngitis; aplasia or hypoplasia of the mammary glands | |||||
| Hypohidrotic ectodermal dysplasia–autosomal dominant [36] | 129490 | AD | EDAR | Gen normal; sometimes hypoplastic | Skin: smooth, thin, dry and hypoplastic |
| Other: photophobia; hypoplasia of lacrimal ducts; decreased function of the lacrimal glands; saddle nose; thick and protruding lips; frontal bossing and prom auricles; chronic rhinitis, frequent respiratory infections | |||||
| Hypohidrotic ectodermal dysplasia (autosomal recessive) [37] | 224900 | AR | EDAR | Gen normal; sometimes hypoplastic | Skin: smooth, thin, dry and hypoplastic |
| Other: photophobia; hypoplasia of lacrimal ducts; decreased function of the lacrimal glands; saddle nose; thick and protruding lips; frontal bossing and prom auricles; chronic rhinitis, frequent respiratory infections | |||||
| HED with immune deficiency [38] | 300291 | XD | IKBKG | Gen normal; sometimes hypoplastic | Milder ectodermal dysplasia features than classic HED; failure to thrive, recurrent infections of digestive tract; recurrent respiratory infections; dysgammaglobulinaemia |
| HED with immune deficiency, osteopetrosis and lymphoedema [38] | 300301 | XD | IKBKG | Gen normal; sometimes hypoplastic | Milder ectodermal dysplasia features than classic HED; failure to thrive, recurrent infections of digestive tract; recurrent respiratory infections; dysgammaglobulinaemia; osteopetrosis. Generally more severe phenotype than 300291 |
| IFAP syndrome with or without BRESHECK syndrome [39] | 308205 | XL | MBTPS2 (membrane-bound transcription factor peptidase, site 2) | Dystrophic nails, paronychia, thick nails | IFAP: ichthyosis follicularis, atrichia and photophobia BRESHECK: brain anomalies, retardation, ectodermal dysplasia, skeletal deformities, Hirschsprung disease, ear/eye anomalies, cleft palate/cryptorchidism, and kidney dysplasia/hypoplasia |
| Incontinentia pigmenti (familial male-lethal type IP, Bloch–Sulzberger syndrome) [40] | 308300 | XL | IKBKG (inhibitor of kappa light polypeptide gene enhancer in B cells, kinase gamma) | Dystrophic in all or most of the fingers and toes in about one tenth of cases | Skin: vesicular-bullous eruption in the neonatal period followed or accompanied by verrucous lesions and bizarre pigmentation; pigmented macules may be present, alopecia |
| Other: occas cong hearing loss; ophthalmological alterations in about one fifth of the patients include blindness, strabismus, cataract, uveitis, retrolental fibroplasias, optic nerve atrophy, microphthalmia; occas clubfoot, cleft palate, microcephaly; about one-third of the cases present severe CNS anomalies: spastic tetraplegia, hemiplegia, diplegia; epilepsy; mental retardation; occas short stature | |||||
| Keratitis, ichthyosis and deafness (KID) syndrome [41] | 148210 | AD | GJB2 | Abs at birth; delayed development; leukonychia and thickening (most marked in the fingernails); destructive dystrophy | Skin: ichthyosiform erythroderma with sebaceous dysfunction; furrowing around mouth and chin; erythematous hyperkeratotic plaques on elbows, knees, and the dorsa of hands and feet; marked thickening (leather-like consistency) of palms and soles; increased susceptibility to squamous cell carcinoma |
| Other: cong sensorineural deafness; vascularisation of the cornea with pannus formation resulting in loss of vision; keratitis; occas decreased tear production; photophobia; bilateral flexion contractures at knees and elbows with tight heel cords | |||||
| Limb–mammary syndrome [42] | 603543 | AD | TP63 (tumour protein p63) | Nail dysplasia | Hypoplasia/aplasia of the mammary glands; cleft lip/palate +/– bifid uvula; lacrimal duct atresia |
| Margarita Island ED (allelic to cleft lip/palate–ectodermal dysplasia syndrome (CLEPD1)) [43] | 225060 | AR | PVRL1 | Dysplastic | Cleft lip/palate; syndactyly of fingers |
| Nail–patella syndrome [44] | 161200 | LMX1B (LIM homeobox transcription factor 1, beta) | Triangular lunulae, ulnar nail dystrophy, poorly formed lunulae, anonychia, dystrophic nails | Other: absent or hypoplastic patellae, elbow abnormalities, iliac horns, nephropathy, glaucoma, Lester sign (flower shape of pigmentation around the central portion of iris) | |
| Odonto-onychodermal dysplasia [45] | 257980 | AR | WNT10A (wingless-type MMTV integration site family, member 10A) | Dystrophic nails | Skin: thickening of the palms and soles; erythematous lesions of face; thickening of the palmar skin with painful chafing |
| Other: mild mental deficiency, peg-shaped incisor teeth, misshapen teeth, oligodontia, hypotrichosis, dry and sparse hair, thinning of the eyebrows. | |||||
| Pachyonychia congenita [46] | 167200 167210 |
AD | KRT6A KRT6B KRT6C KRT16 KRT17 |
Severe wedge-shaped thickening | Skin: focal palmoplantar keratoderma; verrucous lesions on the knees, elbows, buttocks, ankles, and popliteal regions; follicular keratoses/keratosis pilaris, multiple pilosebaceous cysts (steatocystomas) |
| Other: oral leukokeratosis; hoarseness | |||||
| Pallister–Hall syndrome [47] | 146510 | AD | GLI3 | Nail dysplasia, hypoplastic nails | Hypothalamic hamartoblastoma, polydactyly, pituitary dysfunction, internal organ abnormalities |
| Palmoplantar keratoderma, mutilating, with periorificial keratotic plaques (Olmsted syndrome) [48, 49] | 614594 | AD | Transient receptor potential cation channel, subfamily V, member 3 (TRPV3) | Various degrees of nail dystrophy | Painful and disabiling palmoplantar keratoderma, spontaneous autoamputations, or ainhum-constrictions of the fingers, erythematous keratotic scaly periorificial lesions |
| Papillon–Lefèvre syndrome [50] | 245000 | AR | CTSC | Occas dyst (spoon-shaped and striated; onychogryphosis) | Skin: hyperkeratosis of the palmar and plantar surfaces with a tendency towards fissuring and cracking; dry and dirty-appearing on the dorsal surface of the arms and the ventral surface of the legs; occas eczema and erythema of the face as well as of the sacral and gluteal regions |
| Other: severe gingivostomatitis; occas intracranial calcifications; abnormal liver function; renal abnormalities; gen osteoporosis | |||||
| Poikiloderma with neutropenia (Navajo poikiloderma) [51] | 604173 | AR | USB1 (U6 snRNA biogenesis 1), C16orf57 | Pachyonychia | Skin: eczematous rash at birth, becomes poikilodermatous over first 2 years of life |
| Other: non-cyclical neutropenia, recurrent upper respiratory tract infections and chest infections, neutrophil dysfunction | |||||
| Similarity to Rothmund–Thompson syndrome (see below) but no mutations in RECQL4 | |||||
| Popliteal pterygium syndrome [52] | 119500 | AD | IRF6 | Pyramidal skinfold extending from the base to the tip of the nails | Cleft palate/lip, popliteal webbing |
| Popliteal pterygium syndrome, lethal type [53] | 263650 | AR | RIPK4 (receptor-interacting serine-threonine kinase 4) | Anonychia, nail hypoplasia | Popliteal pterygia, ankyloblepharon, filiform bands between the jaws, cleft lip and palate syndactyly |
| Rapp–Hodgkin syndrome [54] | 129400 | AD? | TP63 | Small narrow and dysplastic | Skin: dry and coarse; thickened over the extensor surface of the elbows and knees; hypoplastic dermatoglyphics |
| Face: cleft lip; hypoplastic maxilla; mild frontal prominence; microstomia; mildly depressed nasal bridge; prom and malformed auricles | |||||
| Other: conductive loss (secondary to otitis media); chronic epiphora; corneal opacities; photophobia; atresia of puncta; ectropion; lacrimal papillae; short stature; occas syndactyly | |||||
| Absence of ankyloblepharon and severe erosive scalp dermatitis may distinguish Rapp–Hodgkin syndrome from AEC syndrome (see above) | |||||
| Robinow syndrome, autosomal recessive [55] | 268310 | AR | ROR2 (receptor tyrosine kinase-like orphan receptor 2) | Anonychia | Short stature, rib and vertebral abnormalities, hypoplastic genitalia, empty sella, gum hypertrophy, face with hypertelorism, wide palpebral fissures, broad based nose |
| Rothmund–Thomson syndrome [56] | 268400 | AR | RECQL4 | Frequently dyst | Skin: poikiloderma including atrophy, irregular pigmentation and telangiectasias beginning during the first 3–6 months; palmoplantar hyperkeratosis; sensitivity to sunlight; initial rash is red, elevated with oedematous patches appearing symmetrically on the cheeks, hands, forearms and buttocks and subsequently on the trunk and lower limbs; after a few years dry, scaling and atrophic skin develops with areas of hyperpigmentation, hypopigmentation and telangiectasia |
| Other: cataract, usually bilateral (onset 3–6 years); occas degenerative lesions of the cornea; small hands and feet; short terminal phalanges, syndactyly: abs of metacarpals, rudimentary ulna and radius; increased risk of osteosarcoma; short stature; occas mental retardation; hypogonadism; cryptorchidism; skull abnormalities; scoliosis | |||||
| Scalp–ear–nipple syndrome [57] (Finlay–Marks syndrome) | 181270 | AD | KCTD1 | Brittle fingernails | Skin: raised firm scalp nodules |
| Other: small tragi; cupped and protruding ears; absent/rudimentary nipples; breast aplasia; partial third/fourth finger syndactyly | |||||
| Schinzel–Giedion midface-retraction syndrome [58] | 269150 | AR? | SETBP1 | Narrow, deeply set, triangular and hyperconvex | Skin: abundant on the neck; hypoplastic nipples; hypoplastic dermal ridges; simian creases |
| Face: saddle nose with depressed root and short bridge; high/protruding forehead; orbital hypertelorism; small/malformed auricles; anteverted nostrils; midface hypoplasia; facial haemangiomata | |||||
| Limbs: mesomelic brachymelia; hypoplasia of dist phalanges in hands and feet; short metacarpals of thumbs; talipes | |||||
| Other: severe mental retardation; growth retardation; abnormal EEG and seizures; spasticity; recurrent apnoeic spells; multiple wormian bones; wide cranial sutures and fontanelles; broad ribs, broad cortex and increased density of long tubular bones and vertebrae, hypoplastic/aplastic pubic bones; choanal stenosis; short and broad neck; short penis with hypospadias | |||||
| Schopf–Schulz–Passarge syndrome [59] (cystic eyelids–palmoplantar keratosis–hypodontia–hypotrichosis) | 224750 | AR | WNT10A (wingless-type MMTV integration site family, member 10A) | Brittle with longitudinal and oblique furrows; onycholysis, hypoplastic nails, nail fragility, dystrophic nails | Skin: palmoplantar keratosis; telangiectatic facial skin; papules; multiple tumours with follicular differentiation Bird-like facies |
| Other: bilateral early senile cataract; arteriosclerotic fundi; myopia; cysts of eyelids developing late | |||||
| Sclerosteosis 1 [60] | 269500 | AR | SOST (sclerostin) | Nail dysplasia | Skeletal overgrowth, square appearance of jaw, syndactyly |
| Sclerosteosis 2 [61] | 614305 | NK | LRP4 (low density lipoprotein receptor related protein 4) | Hypoplastic nails, nails separated into 2 parts | Other: finger syndactyly, facial asymmetry, thickening of the skull and long bones |
| Short stature, onychodysplasia, facial dysmorphism, and hypotrichosis [62] | 614813 | AR | POC1 centriolar protein A (POC1A) | Hypoplastic fingernails | Severely short long bones, alopecia, growth retardation, macrocephaly, long, triangular face with prominent nose and small ears, unusual high-pitched voice. Clinodactyly, brachydactyly, hypoplastic distal phalanges, short and thick long bones |
| T-cell immunodeficiency, congenital alopecia, and nail dystrophy [63] | 601705 | AR | FOXN1 (forkhead box N1) | Ridging and pitting of nails | Other: congenital alopecia, severe T-cell immunodeficiency |
| Trichodento-osseous syndrome [64] | 190320 | AD | DLX3 | Flat, thickened, misshapen and striated; brittle | Face: occas frontal bossing |
| Others: occas clinodactyly; some of the calvarial sutures show evidence of premature fusion leading to mild to moderate dolichocephaly | |||||
| Trichorhinophalangeal syndrome types I and III [65, 66] | 190350 190351 |
AD | TRPS1 | Occas thin, short, with long longitudinal grooves; flattened, koilonychia-like and normal in colour, racket thumbnails | Other: pear-shaped nose; long and wide philtrum; large, prom ears; occas exotropia and photophobia; short stature; increased susceptibility to upper respiratory tract infections; narrow palate; scoliosis; lordosis or kyphosis; pectus carinatum |
| Limbs: brachymesophalangy; brachymetacarpy; brachymetatarsy; peripheral dysostosis with type 12 cone-shaped epiphyses at some of the middle phalanges of the hands (the joints are thickened); ulnar and radial deviation of the fingers; occas clinodactyly; winged scapulae; coxa valga; Perthes-like abnormalities | |||||
| (Type III: severe brachydactyly, short metacarpals, severe short stature) | |||||
| Trichorhinophalangeal syndrome type II (Langer–Giedion syndrome) [67] | 150230 | AD | Loss of TRPS1 and EXT1 genes | Occas thin, short, with long longitudinal grooves; flattened, koilonychia-like and normal in colour; racket thumbnails | As above plus multiple cartilaginous exostoses; mental retardation is common |
| Trichothiodystrophy, photosensitive [68] | 601675 | AR | ERCC2, ERCC3, or GTF2H5 | Brittle nails, dysplastic nails | Brittle hair, ichthyotic skin, short stature, mental retardation, photosensitivity |
| Ulnar-mammary syndrome [69] | 181450 | AD | TBX3 | Missing digits, partial duplication of the nail on the 5th digit | Limb defects, ectopic and hypoplastic teeth, mammary gland hypoplasia, genital abnormalities, delayed puberty in males |
| Weaver syndrome [70] | 277590 | AD | EZH2 (enhancer of zeste homolog 2 (Drosophila) | Flat, deep set nails, narrow nails | Broad forehead, face, ocular hypertelorism, prominent wide philtrum |
| Weyer acrofacial dysostosis [71] (? milder AD form of Ellis van Creveld, MIM 225500, see above) | 193530 | AD | EVC2 | Hypoplastic/ dysplastic | Other: short stature; postaxial polydactyly; short limbs; acrofacial dysostosis; abnormal mandible; hypotelorism; prominent ear antihelices |
| Witkop syndrome [72] (tooth and nail syndrome) | 189500 | AD | MSX1 | Koilonychia; longitudinal ridging; nail pits, toenails more affected than fingernails | Lip eversion, hypodontia |
| Yunis–Varon syndrome [73] | 216340 | AR | FIG4 | Absent thumbs, hypoplasia or loss of phalanx of other digits | Cleidocranial dysplasia, micrognathia, pelvic dysplasia, bilateral hip dislocation, retracted and poorly delineated lips, severe neurological involvement. Enlarged vacuoles are present in neurons, muscle, and cartilage |
| Non-syndromic genodermatoses with nail anomalies | |||||
| Candidiasis, familial, 7 (candidiasis, familial chronic mucocutaneous, autosomal dominant) [74] | 614162 | AD | STAT1 (signal transducer and activator of transcription 1) | Candida onychomycosis and paronychia | Other: Candida infections of skin and mucous membranes |
| Darier disease [75] | 124200 | AD | ATP2A2 | Nails with longitudinal erythronychia, longitudinal leukonychia and V-shaped nicks distally | Skin: verrucous papules and plaques in a seborrhoeic distribution, palmoplantar punctuate keratoses, thickening of the palms and soles, acrokeratosis verruciformis of Hopf Other: neuropsychiatric disorders including mild mental retardation, psychosis, and affective disorder |
| Ectodermal dysplasia 9, hair/nail type [76] | 614931 | AR | HOXC13 (homeobox C 13) | Micronychia and severe nail dystrophy | Other: congenital atrichia |
| Ectodermal dysplasia, pure hair and nail type [77] | 602032 | AD | KRT85 | Congenital onychodystrophy; micronychia; onycholysis; onychorrhexis | Skin: folliculitis decalvans of neck |
| Ectodermal dysplasia/skin fragility syndrome (McGrath syndrome) [78] | 604536 | NK | PKP1 (plakophilin 1) | Thick and dystrophic nails | Other: severe blistering, desquamation, short and sparse hair, fragile skin, cracking and hyperkeratosis of palms and soles |
| Epidermolysis bullosa simplex, autosomal recessive 2 [79] | 615425 | AR | DST (Dystonin) | Nail dystrophy | Skin: trauma-induced blistering mostly on the feet and ankles |
| Epidermolysis bullosa simplex, Dowling Meara type [80] | 131760 | AD | KRT5 KRT14 |
Nail dystrophy, nail shedding, pincer nail deformity | Skin: generalized blisters that develop in clusters, and have a herpetiform appearance, can be haemorrhagic; blisters are mostly acral and can be periungual; hyperkeratosis of palms and soles/palmoplantar keratoderma |
| Hailey–Hailey disease (benign chronic pemphigus) [81] | 169600 | AD | ATP2C1 | Longitudinal leukonychia of fingernails | Recurrent blisters in intertrigenous areas |
| Hidrotic ectodermal dysplasia (Clouston syndrome/ED2) [82] | 129500 | AD | GJB6 (connexin-30) | Hypoplastic nails, paronychia | Other: hair brittleness, alopecia, slow hair growth, palmoplantar hyperkeratosis |
| Kindler syndrome [83] | 173650 | AR | KIND1 | Nail dystrophy, long thick cuticles | Congenital blistering, poikiloderma, skin atrophy, photosensitivity |
| Monilethrix [84] | 158000 | AD | KRT81, KRT83, KRT86 | Occas dystrophic | Skin: keratosis pilaris, especially over nape of neck Hypotrichosis |
| Disorders with changes limited to the nails | |||||
| Anonychia congenita [85] | 206800 | AR | R-spondin 4 (RSPO4) | Congenital absence of the nails, involving fingernails and/or toenails | No other associated abnormalities |
| Leukonychia totalis and/or partialis [86] | 151600 | AD and AR | PLCD1 | White discoloration of a portion or all of multiple nails | No other confirmed associations |
| Nail disorder, non-syndromic congenital, 10 [87] | 614157 | AR | FZD6 (Frizzled family receptor 6) | Thick nails, hyponychia, onycholysis, claw-shaped nails | |
Abs, absent or absence; AD, autosomal dominant; AR, autosomal recessive; Dyst, dystrophic; Gen, generally or generalized; Occas, occasional; Prox, proximal; XR, X-linked recessive.
Pachyonychia congenita (PC) is a group disorders caused by dominant mutations in one of five genes encoding nail keratins. The clinical features seen in virtually all affected patients are toenail thickening and plantar keratoderma often associated with plantar pain (Table 69.2). Interestingly, despite the origin of the name of the disorder (pachy = large, onyx = nail), about 2% of PC patients lack nail involvement. Approximately 5000–10000 affected individuals have been described globally. PC was originally classified into two subtypes based on clinical features, the Jadassohn–Lewandowsky type (PC type 1; MIM 167200) and the Jackson–Lawler type (PC type 2; MIM 167210). Based on genotype–phenotype analyses of more than 250 patients showing that correlations of clinical features with underlying genotype were not absolute, this old terminology has been abandoned.
Table 69.2 Clinical features of pachyonychia congenita
| Clinical feature | Overall % | PC-K6a % | PC-K6b % | PC-K6c % | PC-K16 % | PC-K17 % |
| Toenail dystrophy | 97 | 99 | 98 | 59 | 96 | 99 |
| Fingernail dystrophy | 79 | 99 | 51 | 0 | 62 | 87 |
| Plantar keratoderma | 91 | 89 | 96 | 94 | 99 | 80 |
| Plantar pain | 96 | 97 | 100 | 100 | 97 | 89 |
| Palmar keratoderma | 59 | 55 | 41 | 18 | 78 | 52 |
| Hoarseness | 28 | 45 | 20 | 0 | 35 | 23 |
| Oral leukokeratoses | 56 | 88 | 29 | 18 | 40 | 27 |
| Natal teeth | 14 | 2 | 0 | 0 | 0 | 76 |
| Cysts | 59 | 67 | 73 | 24 | 27 | 92 |
| Follicular hyperkeratoses | 45 | 60 | 47 | 0 | 14 | 68 |
Modified from http://www.pachyonychia.org/pc_data.php
Pathophysiology
The keratins affected in individuals with PC are expressed in the nail bed, palmoplantar epidermis and mucosae. The new classification is based on genotype with gene defects in one of five different genes encoding keratins (KRT6A, KRT6B, KRT6C, KRT16 and KRT17) underlying the disorder. The new designations are PC-K6a, PC-K6b, PC-K6c, PC-K16 and PC-K17 [1]. Because keratins provide structural integrity to epithelial structures, mutations in these genes lead to cell fragility and compensatory hyperkeratosis. The severity of nail changes may be mutation dependent. For example, individuals with PC-K16 and p.Leu132Pro mutations have dystrophy of all fingernails, whereas those with p.Asn125Ser or p.Arg127Cys mutations in the same gene generally have no fingernail changes [2].
Clinical features
Based on data from the Pachyonychia Congenita Project, information about the clinical features of PC has been elucidated and is constantly being updated (http://www.pachyonychia.org/pc_data.php last accessed July 2014). The earliest and most common clinical feature is toenail dystrophy, which occurs overall in 97% of patients and is present at birth in 56.7% of patients [1] (Figure 69.1a, b). The 5th and hallucal toenails are most often affected, and almost 70% have involvement of all 10 toenails, which usually become dystrophic concurrently. Fingernail dystrophy is noted in 79% of affected individuals [3] and is present at birth in almost 70% of patients [1] (Figure 69.1c). Most patients develop dystrophy simultaneously in all 10 fingernails and all patients with fingernail dystrophy also have toenail dystrophy. By 5 years of age, approximately 75% of patients have toenail and fingernail changes. The nails typically have marked subungual hyperkeratosis with a pinched V-shape, but can sometimes show premature nail termination. PC-K6c is the mildest form and may manifest only as plantar keratoderma without nail changes [4, 5]. PC-K6a and PC-K17 tend to be the most severe forms [6]. PC-K6a is the most common subtype, and affects 41.3% of PC patients [3]. Periungual infections (e.g. bacterial, candidal) have been reported in 93% of PC-K6a patients.
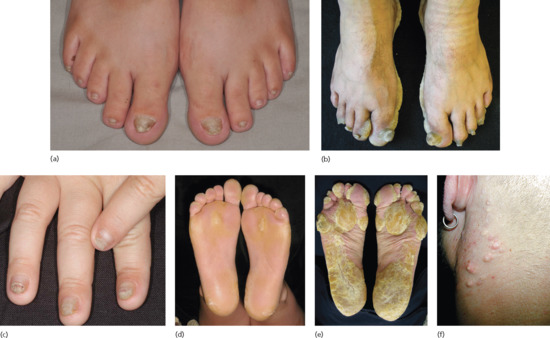
Figure 69.1 Nail dystrophy and plantar keratoderma are the principal features of pachyonychia congenita. (a) Toenails are dystrophic in this patient with PC-K17 (mutation K17 N92S). (b) Severe nail dystrophy and obvious plantar keratoderma in PC-K16 (mutation K16 L132P). (c) Fingernail dystrophy in PC-K6a (mutation K6a N172del). (d) Mild plantar keratoderma in PC-K6a (mutation K6a N171K). (e) Severe plantar keratoderma in PC-K16 (mutation K16 N125D). (Parts a–e courtesy of patients in the International Pachyonychia Congenita Research Registry (IPCRR) sponsored by the PC Project.) (f) Multiple sebaceous cysts on the neck of a patient with PC
(part f courtesy of Dr E. Sprecher).
Although the nail dystrophy can cause embarrassment, the often exquisitely painful plantar keratoderma exerts the greatest limitation on patients in terms of function and occurs overall in 96% of affected individuals [3] (Figure 69.1d, e). The plantar keratoderma is present in 10.4% of affected patients by 1 year of age, usually is present by 5 years of age in PC-K6a, -K16 and -K17 (comprising most patients), and occurs after 5 years of age in most individuals with milder forms, PC-K6b and -K6c [1]. Thickening tends to be focal and is most prominent at pressure points on the heel and ball of the foot; diffuse plantar involvement has occasionally been described [7]. Transgrediens spread (to the dorsal surface of the foot) has been described, especially with PC complicated by infection, trauma from standing or shoes, or exposure to foot moisture [8]. Plantar keratoderma is often complicated by painful erosions, fissures and/or bullae, which may be subcorneal and only detectable by imaging [9]. Palmar keratoderma occurs overall in fewer than 50% of patients, and only half of these show evidence of palmar keratoderma by 5 years of age [1].
Hoarseness and oral leukokeratoses typically present in the first year of life, particularly in babies with PC-K6a, leading to misdiagnosis of a variety of airway issues and thrush, respectively. Overall, 56% have oral leukokeratoses, with PC-K6a and PC-K17 having more severe or earlier onset of the oral leukokeratosis. Natal teeth in PC are virtually always a sign of PC-K17. Other features are pilosebaceous cysts (Figure 69.1f), found in most patients with PC-K6a, -K6b and -K17, and follicular hyperkeratoses. Cysts and hyperkeratoses most often develop in school-aged children, but can occur in younger children [1]. Hyperhidrosis affects more than 50% of PC patients, but usually is an issue in affected adults.
The nail changes lead to considerable embarrassment, particularly in adolescents, resulting in various strategies to conceal the nails [1]. The keratoderma impedes function, including walking, playing, schoolwork, a variety of other tasks and social development, especially in school-age patients and adults.
Nail trauma in children from shoe friction can lead to changes that resemble PC, but typically affects the 5th toenail without other nail involvement. The genetic disorder most often confused with PC is hidrotic ectodermal dysplasia (Clouston syndrome; MIM 604418; see Chapter 67), which results from mutations in GJB6, encoding connexin 30. Patients typically show toenail and fingernail alterations at birth and painful keratoderma that can be indistinguishable from PC. Hearing loss, if present, and thin sparse hair during childhood (not a feature of PC unless semidominantly inherited with two dominantly mutant keratin genes [10]) are distinguishing features of hidrotic ectodermal dysplasia. Mutations on both alleles of FZD6, which encodes frizzled 6, a Wnt-signalling pathway receptor in the nail matrix, similarly lead to hypertrophic nail dystrophy at birth [11, 12].
Investigations
Genotyping is currently performed at no cost on a research basis for patients registered with the International Pachyonychia Congenita Research Registry (IPCRR) (www.pachyonychia.org last accessed July 2014).
Management
Mechanical treatment of the nails, such as filing, grinding and cutting is most effective, particularly after soaking the nails [13]. While antibiotics and antifungal therapy are helpful in managing secondary paronychia, they do not improve the nail dystrophy. Mechanical intervention can also ameliorate the keratoderma, but topical agents such as topical retinoids, steroids, keratolytics and moisturizers have not been found too helpful. Given the often debilitating pain, pain control management is key. Injection of botulinum toxin led to approximately 3 months of relief in eight described cases with and other hyperhidrosis [14], but a study from the International Pachyonychia Congenita Consortium found botulinum injections relatively ineffective [13]. Oral retinoids can decrease plantar thickening in 50% of patients, but only decreased plantar pain in about one-third of cases, led to worsening or no improvement in nails in 87% of patients, and caused unacceptable side effects, especially at higher doses (e.g. more than 25 mg acitretin daily) [15]. Administration of systemic rapamycin, which suppresses activation of mTOR but also expression of keratin 6a, partially reversed the plantar thickening and pain in PC-K6a during a 12-week trial but had unacceptable side effects; oral rapamycin did not improve nails [16]. Statins have also been shown to decrease expression of KRT6A [17], suggesting that a trial of oral statins could suppress keratin 6a expression in PC-K6a and allow normal keratin 6b and 6c proteins to compensate. siRNA injections directed against a mutant KRT6A led to dramatic improvement in the plantar keratoderma at the localized site of injection in the proof-of-concept human trial [18], but could not be sustained; use of microneedles for delivery or a cream formulation that traverses the plantar epidermal barrier are under investigation [19].
Resources
Patient resources
Support group: International Pachyonychia Congenita Research Registry or IPCRR (www.pachyonychia.org last accessed July 2014).
Dyskeratosis congenita
Definition and nomenclature
Dyskeratosis congenita (DC) is a heterogeneous group of inherited disorders characterized by nail dysplasia, oral leukoplakia, reticulated hyperpigmentation and a tendency towards bone marrow failure and squamous cell carcinoma.
Table 69.3 Underlying gene mutations in dyskeratosis congenita
| Inheritance | MIM number | Gene location | Gene | Protein name | Protein function | Percentage of casesa |
| XLR | 305000 | Xq28 | DKC1 | Dyskerin | Component of telomerase complex that converts uridine residues of ribosomal RNA to pseudouridine | 17–36 |
| AD | 613990 | 14q12 | TINF2 | TRF1-interacting nuclear factor 2 | Shelterin complex component that protects telomere ends during DNA replication | 11–24 |
| AD | 127550 | 3q26.2 | TERC | Telomerase RNA component | Acts as a template for adding telomere repeats | 6–10 |
| AD | 613989 | 5p15.33 | TERT | Telomerase | Catalytic component of telomerase, which synthesizes telomere repeats | 1–7 |
| AR | 613276 | 16q21 | C16orf57 | Mpn1 RNA exonuclease | Processes small nuclear RNAs posttranscriptionally | 2 |
| AR | 224230 | 15q14 | NOLA3 | NOP10: nuclear protein, Family A, member 3 | Small nucleolar RNA-binding protein that is associated with NHP2 and dyskerin in telomerase complex; involved in uridine to pseudouridine conversion | <1 |
| AR | 613987 | 5q35.3 | NOLA2 | NHP2: nuclear protein, Family A, member 2 | Small nucleolar RNA-binding protein that is associated with NOP10 and dyskerin in telomerase complex; involved in uridine to pseudouridine conversion | <1 |
| AR | 613988 | 17p13.1 | WRAP53 | WD repeat-containing protein encoding RNA Antisense to p53; also called TCAB1: telomerase Cajal body protein 1 | Important for telomerase trafficking | <1 |
| AR | 615190 | 20q13.33 | RTEL1 | Telomere replication | DNA helicase that is crucial for telomere maintenance, DNA repair and prevention of excess meiotic crossovers; interacts with TRF1 | <1 |
| AR | 613989 | 5p15.33 | TERT | Telomerase | Catalytic component of telomerase, which synthesizes telomere repeats | <1 |
*Not all cases have been associated with a particular genotype, so that the numbers do not add to 100%.
AD, autosomal dominant; AR, autosomal recessive; XLR, X-linked recessive.
Introduction and general description
The prevalence of DC is estimated to be 1 in 1000 000 individuals. Most patients are male, and X-linked recessive inheritance accounts for approximately half of the cases. Females with heterozygous DCK1mutations may show clinical findings if skewed X-inactivation leads to significant expression of the mutant allele [1]. Autosomal recessive and autosomal dominant inheritance patterns have also been reported (see Table 69.3). The median age of diagnosis is 15 years, as many patients first present as teens or young adults.
Pathophysiology
Dyskeratosis congenita is considered a ‘telomeropathy’, because most patients show severe shortening of telomeres. Mutations in one of 10 genes are found in approximately 60% of patients [2, 3], and the protein products of these genes affect telomerase maintenance (see Table 69.3). Telomeres provide a repetitive template (TTAGGG) for repair at the 3’ ends of chromosomes that prevents the loss of genetically encoded information after replication. When too short, telomeres signal the arrest of cell proliferation and lead to senescence and apoptosis, which particularly impacts rapidly dividing cells. Telomerase catalyses DNA synthesis to maintain telomere length; the telomerase catalytic unit contains telomerase reverse transcriptase (TERT) and its template, telomerase RNA component (TERC). Other components of the telomerase complex that are mutated in DC are DKC1, encoding dyskerin, a small nucleolar protein that binds to RNA and TERC, and the stabilizers NHP2 and NOP10 [4]. The most common cause of DC is mutations in DKC1, but features of DC have been described from mutations affecting other telomerase complex components. Mutations in TINF2, a component of the shelterin complex that is essential for telomere maintenance, cause at least 10% of cases of DC. More recently, mutations have been found in the TCAB1 gene [5], encoding a protein that is key for trafficking of telomerase to Cajal bodies, which is where modifications and assembly of nucleoprotein complexes occur; its deficiency misdirects telomerase RNA so it cannot elongate telomeres. Mutations have also been found in: (i) RTEL1, a helicase that interacts with shelterin [6, 7], in CTC1 (conserved telomere maintenance component 1) [8]; and (ii) the C16orf57 gene [9].
Clinical features
The phenotype of DC varies widely, including among DC patients with mutations in the same gene. The minimal clinical criteria for DC diagnosis include having at least two major features and two minor features [10] (Table 69.4). The thin dystrophic nails usually appear first, between the ages of 5 and 13 years (Figure 69.2a, b). Mildly affected nails show ridging and longitudinal grooving; severely affected nails are shortened and show pterygium formation. Cutaneous changes usually develop after the onset of nail changes, most commonly during late childhood to teenage years. A fine reticulated dusky brown hyperpigmentation, sometimes surrounding hypopigmented, atrophic, telangiectatic patches (‘poikilodermatous’), on the face, neck, shoulders, upper back and thighs is characteristic (Figure 69.2b, c). Other cutaneous changes may include the following.
- Telangiectasia of the trunk.
- Redness and atrophy of the face with irregular macular hyperpigmentation.
- Acrocyanosis.
- Palmoplantar hyperkeratosis.
- Hyperhidrosis and bullae of the palms and soles.
- Wrinkled atrophic skin over the elbows, knees and penis.
- A diffuse atrophic, transparent and shiny appearance on the dorsal aspects of the hands and feet.
Table 69.4 Clinical features of dyskeratosis congenita
Major features (>75% of patients) Nail dystrophy Leukoplakia Abnormal skin pigmentation Bone marrow failure |
Minor features 10–75% of patients Short stature Sparse hair or premature greying Hyperhidrosis Epiphora Developmental delay Pulmonary disease Oesophageal stricture Tooth decay and loss <10% of patients Squamous cell carcinoma Intrauterine growth retardation Gastrointestinal disease Liver disease, enteropathy Ataxia Genitourinary issues Hypogonadism, undescended testes, phimosis, urethral stricture Microcephaly Bone abnormalities Osteoporosis, scoliosis, aseptic necrosis Deafness |

Figure 69.2 Nail dystrophy, poikiloderma and oral leukoplakia are the major mucocutaneous findings of dyskeratosis congenita. (a) Pterygium formation. (b) Nail dystrophy with poikiloderma (telangiectasia, atrophy and dyspigmentation) of the underlying thigh. (c) Poikiloderma on the thighs of a young woman.
(Courtesy of Dr E. Sprecher.) (d) Leukoplakia on the tongue. (e) Squamous cell carcinoma on the hand; mucosa is a more typical location. (Courtesy of Dr E. Sprecher.)
The hair of the scalp, eyebrows and eyelashes is often sparse and lustreless.
Mucous membrane changes consist of leukoplakia (Figure 69.2d), and rarely blisters and erosions, of the oral and anal mucosae, oesophagus and urethra. Periodontitis may also develop. Similar changes of the tarsal conjunctivae may result in atresia of the lacrimal ducts, excessive lacrimation, chronic blepharitis, conjunctivitis and ectropion. The teeth tend to be defective and subject to early decay.
Overall, almost 90% of patients develop life-threatening bone marrow failure, characterized by severe aplastic anaemia with neutropenia, splenomegaly and a haemorrhagic diathesis. Acute myeloid leukaemia and myelodysplastic syndrome have also been described. Pulmonary fibrosis and hepatic cirrhosis are other life-threatening complications. Isolated idiopathic pulmonary fibrosis, liver disease, aplastic anaemia, myelodysplasia and leukaemia have each been described in patients found to have mutations in DC genes, especially single allele mutations in TERT and TERC. Epithelial tumours often first develop by the mid-teens, frequently in areas of mucosa with leukoplakia. Squamous cell carcinoma of the tongue is most common, but cutaneous squamous cell carcinoma can also occur (Figure 69.2e). Patients are at increased risk for the development of head and neck, as well as anogenital, squamous cell carcinomas. These carcinomas are not driven by human papillomavirus (HPV) infection and thus HPV vaccination is not predicted to decrease the risk. Rarely, common variable immunodeficiency is the initial presentation of DC [11].
A severe variant, Hoyeraal–Hreidarsson syndrome, shows the major features of DC, but in addition cerebellar hypoplasia, developmental delay, intrauterine growth retardation, and sometimes severe immunodeficiency, enteropathy and/or aplastic anaemia. Most patients have mutations in DKC1, but mutations in TERC, TERT, TINF2 or RTEL1 have been described [7]. Another severe variant of DC, Revesz syndrome, has bilateral exudative retinopathy, cerebellar hypoplasia and growth retardation, as well as nail dystrophy, fine hair and bone marrow hypoplasia [12].
The average life expectancy is 44 years. Patients usually die of bone marrow failure (∼60–70%), pulmonary disease (∼10–15%) or malignancy (∼10%).
The pterygium nail changes of DC can be seen in patients with lichen planus and graft-versus-host disease. The poikilodermatous change is sometimes confused with Rothmund-Thomson syndrome (RECQL4 mutations), epidermolysis bullosa simplex with mottled pigmentation (usually KRT5 mutation), Naegeli-Franceschetti-Jadassohn syndrome/dermatopathia pigmentosa reticularis (KRT14 mutation) or Clericuzio-type poikiloderma with neutropenia (another C16orf57 mutation) [13].
Investigations
Screening for DC is best performed by flow cytometry and fluorescence in situ hybridization (flow-FISH) of leukocyte subsets [10] to detect telomere shortening. Recommended disease surveillance includes biannual blood counts, bone marrow evaluations annually, hepatic ultrasounds, annual pulmonary functional tests and skin cancer screening.
Management
Treatment of bone marrow failure is recommended with a haemoglobin persistently below 8 g/dL, platelets <30 000/mm3 and neutrophils <1000/mm3. Allogeneic stem cell transplantation is usually performed for bone marrow failure, but the 10-year survival probability is 30% with mortality generally attributed to pulmonary or vascular complications related to disease progression [14] or toxicity related to myeloablative regimens. If a matched donor without genetic and clinical evidence of DC is available, stem cell transplant should be considered, but carries substantial risk, including an increased risk of pulmonary fibrosis with graft-versus-host disease. Long-term survival after transplantation has been poor. A trial of androgens, such as oxymetholone or danazol, may be considered before transplantation from an unrelated donor, and improves blood counts in 60% of treated patients [15]; however, androgens may cause hepatic tumours, and the combination of androgen and granulocyte colony-stimulating factor (G-CSF) has led to splenic rupture. Management of patients otherwise consists of bougienage for oesophageal stenosis; fulguration, curettage and surgical excision of leukokeratosis of the buccal and anal mucosae; and lifelong regular supervision for early detection of mucosal or cutaneous carcinomas.
Resources
Patient resources
Support group: Dyskeratosis Congenita Outreach (http://www.dcoutreach.org/ last accessed July 2014).
Nail–patella syndrome
Definition and nomenclature
Nail–patella syndrome is an autosomal dominant disorder classically characterized by nail changes and dystrophy of the patella and iliac horns.
Introduction and general description
Nail–patella syndrome, a disorder of nail and bone dysplasia, has an estimated prevalence of 1 in 50 000 individuals, although the prevalence could be higher, given the many affected individuals with a mild phenotype [1] (Table 69.5). De novo mutations occur in 12% of affected individuals (i.e. 88% have an affected parent). Interfamilial and intrafamiliar variability is seen. The diagnosis is often missed for several generations, although features tend to be present at birth. Males and females are affected equally.
Table 69.5 Clinical features of nail–patella syndrome
| Features | % Of patients |
| Nail changes, especially triangular lunula | 98 |
| Small or absent patella | 74 |
| Elbow deformity | 70 |
| Iliac horns | 70 |
| Renal dysfunction | 40 |
| End-stage renal disease | 5 |
| Ocular hypertension, glaucoma | 33 |
Pathophysiology
Nail–patella syndrome (NPS) results from mutations in LMX1B at chromosome 9q33.3, which encodes the LIM homeobox transcription factor 1-β. LIMX1B is induced by Wnt7a and is required for establishing dorsoventral polarity in the dorsal limb mesenchyme during development, as well as in the development of the kidneys, eyes and bones. For additional details on the anatomy and embryological development of the nails, the reader is referred to Chapter 95. Mutations in LMX1B can also lead to nail–patella-like renal disease, in which skeletal and nail findings are absent [2].
Clinical features
The nails may be absent or underdeveloped and discoloured, split, ridged or pitted, but nail dysplasia with the characteristic triangular lunula instead of the crescent-shaped lunula occurs in most affected individuals and may be the only feature (Figure 69.3). Changes are usually bilateral, symmetrical and may be present at birth. The fingernails are more likely to be affected than the toenails, and the thumbnails are usually the most severely affected, with severity decreasing from the index toward the 5th fingers. The ulnar side of the fingernail is more severely affected than the radial side. Patients show flexion of the distal interphalangeal joints but hyperextension of the proximal interphalangeal joints, leading to ‘swan-necking’. Creases overlying the distal interphalangeal joints tend to be absent.
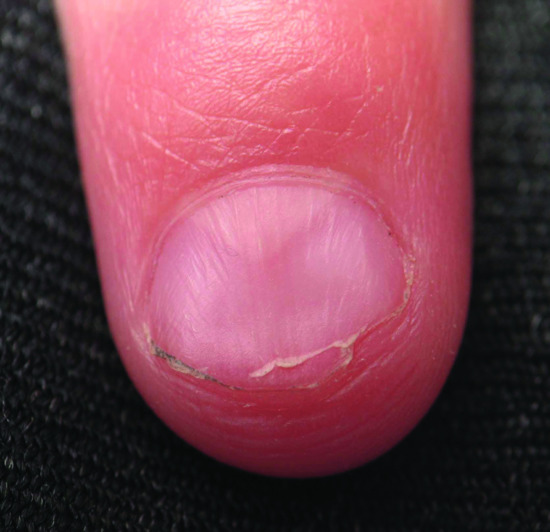
Figure 69.3 Nail dystrophy with triangular lunula of the nail–patella syndrome.
(Courtesy of Dr Mark Holzberg.)
Knees are involved in 74% of patients [3] and tend to have a flattened profile. Patellar involvement is often asymmetrical and commonly characterized by small irregularly shaped or absent patella with recurrent dislocation or subluxation. Flexion contractures and early degenerative arthritis are common; as a result, patients often develop knee pain, locking, instability and inability to straighten the knee. Elbow involvement also is often asymmetrical and occurs in 70% of individuals [3]. The characteristic iliac horns are bilateral conical bone projections that extend posteriorly and laterally from the centre of the pelvic iliac bones. They affect approximately 70% of affected individuals, are asymptomatic and are pathognomonic for the disorder. Renal changes occur in 30–50% of affected individuals, initially as proteinuria with or without haematuria and hypertension; only 5% of patients show end-stage renal disease, which may occur rapidly or develop gradually. Ocular hypertension and primary open-angle glaucoma occur overall in one-third of patients and more often and at a younger age. Crumbling teeth and thin dental enamel have been described [3]. Patients have an increased risk of attention deficit hyperactivity disorder and major depressive disorder, possibly because of mesencephalic dopaminergic neurology pathway abnormalities [4]. Sensorineural hearing loss has been described.
Investigations
The diagnosis of NPS is based on clinical findings, supplemented by laboratory testing to show the radiographic changes and evidence of renal disease, if present. Radiographic evaluation is necessary to detect the iliac horns, although large horns may be palpable. X-rays are also helpful to confirm the bony changes of the elbows and knees. Characteristic modifications of the glomerular basement membrane can be observed by electron microscopy.
Management
No treatment intervention is needed for the nail abnormalities. Orthopaedic complications are treated by physiotherapy, splinting or bracing, analgesics and occasionally surgery. However, chronic administration of non-steroidal anti-inflammatory drugs (NSAIDs) should be avoided because of the risk of renal disease. Annual monitoring for hypertension, renal disease (urinalysis) and glaucoma is recommended. Hypertension should be controlled medically but renal transplantation is occasionally necessary. Dual-energy X-ray absorptiometry (DEXA) bone density scans are recommended beginning in young adults.
Resources
Patient resources
Support groups: Nail Patella Syndrome Worldwide (www.npsw.org) and Nail Patella Syndrome UK (www.npsuk.org) (both last accessed July 2014).
Hereditary anonychia
Definition and nomenclature
Hereditary anonychia is an autosomal recessive disorder characterized by isolated congenital anonychia or hypoplasia of the fingernails and/or toenails.
General description of hereditary anonychia
In hereditary anonychia, anonychia or hypoplasia of the fingernails and/or toenails is congenital and usually involves all of the nails. The disorder results from mutations in the R-spondin 4 (RSPO4) gene on chromosome 20p13 [1, 2]. RSPO4 is only expressed in the mesenchymal tissue underlying the digit tip epithelium [3]. RSPO4 is a member of the R-spondin family of secreted proteins, which activates Wnt/β-catenin signalling and presumably Wnt7a.
References
- Avitan-Hersh E, Indelman M, Bergman R, Sprecher E. ADULT syndrome caused by a mutation previously associated with EEC syndrome. Pediatr Dermatol 2010 Nov–Dec;27(6):643–5.
- Hassed SJ, Wiley GB, Wang S, et al. RBPJ mutations identified in two families affected by Adams-Oliver syndrome. Am J Hum Genet 2012 Aug 10;91(2):391–5.
- Shaheen R, Aglan M, Keppler-Noreuil K, et al. Mutations in EOGT confirm the genetic heterogeneity of autosomal-recessive Adams-Oliver syndrome. Am J Hum Genet 2013 Apr 4;92(4):598–604.
- Shaheen R, Faqeih E, Sunker A, et al. Recessive mutations in DOCK6, encoding the guanidine nucleotide exchange factor DOCK6, lead to abnormal actin cytoskeleton organization and Adams-Oliver syndrome. Am J Hum Genet 2011 Aug 12;89(2):328–33.
- Southgate L, Machado RD, Snape KM, et al. Gain-of-function mutations of ARHGAP31, a Cdc42/Rac1 GTPase regulator, cause syndromic cutis aplasia and limb anomalies. Am J Hum Genet 2011 May 13;88(5):574–85.
- Ru Y, Liu G, Bai J, Dong S, Nie N, Zhang H, et al. Congenital dyserythropoietic anemia in China: a case report from two families and a review. Ann Hematol 2013 Nov 7.
- Sutton VR, Plunkett K, Dang DX, Lewis RA, Bree AF, Bacino CA. Craniofacial and anthropometric phenotype in ankyloblepharon–ectodermal defects–cleft lip/palate syndrome (Hay–Wells syndrome) in a cohort of 17 patients. Am J Med Genet A 2009 Sep;149A(9):1916–21.
- Faiyaz-Ul-Haque M, Bin-Abbas B, Al-Abdullatif A, et al. Novel and recurrent mutations in the AIRE gene of autoimmune polyendocrinopathy syndrome type 1 (APS1) patients. Clin Genet 2009 Nov;76(5):431–40.
- Lv D, Luo Y, Yang W, et al. A novel single-base deletion in ROR2 causes atypical brachydactyly type B1 with cutaneous syndactyly in a large Chinese family. J Hum Genet 2009 Jul;54(7):422–5.
- Tsurusaki Y, Okamoto N, Ohashi H, et al. Mutations affecting components of the SWI/SNF complex cause Coffin–Siris syndrome. Nat Genet 2012 Apr;44(4):376–8.
- Soler VJ, Tran-Viet KN, Galiacy SD, et al. Whole exome sequencing identifies a mutation for a novel form of corneal intraepithelial dyskeratosis. J Med Genet 2013 Apr;50(4):246–54.
- Walczak-Sztulpa J, Eggenschwiler J, Osborn D, et al. Cranioectodermal Dysplasia, Sensenbrenner syndrome, is a ciliopathy caused by mutations in the IFT122 gene. Am J Hum Genet 2010 Jun 11;86(6):949–56.
- Campeau PM, Kasperaviciute D, Lu JT, et al. The genetic basis of DOORS syndrome: an exome-sequencing study. Lancet Neurol 2014 Jan;13(1):44–58.
- Powell JB, Dokal I, Carr R, Taibjee S, Cave B, Moss C. X-linked dyskeratosis congenita presenting in adulthood with photodamaged skin and epiphora. Clin Exp Dermatol. 2014 Apr;39(3):310–14.
- Bessler M, Wilson DB, Mason PJ. Dyskeratosis congenita. FEBS Lett. 2010 Sep 10;584(17):3831–8.
- Sasa GS, Ribes-Zamora A, Nelson ND, Bertuch AA. Three novel truncating TINF2 mutations causing severe dyskeratosis congenita in early childhood. Clin Genet 2012 May;81(5):470–8.
- Basel-Vanagaite L, Dokal I, Tamary H, et al. Expanding the clinical phenotype of autosomal dominant dyskeratosis congenita caused by TERT mutations. Haematologica 2008 Jun;93(6):943–4.
- Walne AJ, Vulliamy T, Marrone A, et al. Genetic heterogeneity in autosomal recessive dyskeratosis congenita with one subtype due to mutations in the telomerase-associated protein NOP10. Hum Mol Genet 2007 Jul 1;16(13):1619–29.
- Walne AJ, Vulliamy T, Beswick R, Kirwan M, Dokal I. Mutations in C16orf57 and normal-length telomeres unify a subset of patients with dyskeratosis congenita, poikiloderma with neutropenia and Rothmund–Thomson syndrome. Hum Mol Genet 2010 Nov 15;19(22):4453–61.
- Vulliamy T, Beswick R, Kirwan M, et al. Mutations in the telomerase component NHP2 cause the premature ageing syndrome dyskeratosis congenita. Proc Natl Acad Sci U S A 2008 Jun 10;105(23):8073–8.
- Zhong F, Savage SA, Shkreli M, et al. Disruption of telomerase trafficking by TCAB1 mutation causes dyskeratosis congenita. Genes Dev 2011 Jan 1;25(1):11–16.
- Ballew BJ, Yeager M, Jacobs K, et al. Germline mutations of regulator of telomere elongation helicase 1, RTEL1, in dyskeratosis congenita. Hum Genet 2013 Apr;132(4):473–80.
- Marrone A, Walne A, Tamary H, et al. Telomerase reverse-transcriptase homozygous mutations in autosomal recessive dyskeratosis congenita and Hoyeraal-Hreidarsson syndrome. Blood 2007 Dec 15;110(13):4198–205.
- Kjaer KW, Hansen L, Schwabe GC, et al. Distinct CDH3 mutations cause ectodermal dysplasia, ectrodactyly, macular dystrophy (EEM syndrome). J Med Genet 2005 Apr;42(4):292–8.
- Kamal R, Dahiya P, Kaur S, Bhardwaj R, Chaudhary K. Ellis–van Creveld syndrome: a rare clinical entity. J Oral Maxillofac Pathol 2013 Jan;17(1):132–5.
- Lin Y, Chen XJ, Liu W, et al. Two novel mutations on exon 8 and intron 65 of COL7A1 gene in two Chinese brothers result in recessive dystrophic epidermolysis bullosa. PLOS One 2012;7(11):e50579.
- Yuen WY, Jonkman MF. Risk of squamous cell carcinoma in junctional epidermolysis bullosa, non-Herlitz type: report of 7 cases and a review of the literature. J Am Acad Dermatol 2011 Oct;65(4):780–9.
- Jonkman MF, Pasmooij AM, Pasmans SG, et al. Loss of desmoplakin tail causes lethal acantholytic epidermolysis bullosa. Am J Hum Genet 2005 Oct;77(4):653–60.
- Winter L, Wiche G. The many faces of plectin and plectinopathies: pathology and mechanisms. Acta Neuropathol 2013 Jan;125(1):77–93.
- McLean WH, Irvine AD, Hamill KJ, et al. An unusual N-terminal deletion of the laminin alpha3a isoform leads to the chronic granulation tissue disorder laryngo-onycho-cutaneous syndrome. Hum Mol Genet 2003 Sep 15;12(18):2395–409.
- Clements SE, Wessagowit V, Lai-Cheong JE, Arita K, McGrath JA. Focal dermal hypoplasia resulting from a new nonsense mutation, p.E300X, in the PORCN gene. J Dermatol Sci 2008 Jan;49(1):39–42.
- Stranecky V, Hoischen A, Hartmannova H, et al. Mutations in ANTXR1 cause GAPO syndrome. Am J Hum Genet 2013 May 2;92(5):792–9.
- Rai R, Thiagarajan S, Mohandas S, Natarajan K, Shanmuga Sekar C, Ramalingam S. Haim Munk syndrome and Papillon Lefevre syndrome – allelic mutations in cathepsin C with variation in phenotype. Int J Dermatol 2010 May;49(5):541–3.
- Krawitz PM, Murakami Y, Hecht J, et al. Mutations in PIGO, a member of the GPI-anchor-synthesis pathway, cause hyperphosphatasia with mental retardation. Am J Hum Genet 2012 Jul 13;91(1):146–51.
- Wawrzynek A, Slezak R, Wisniewski SA, Trzeciak WH. A novel mutation in the ED1 gene in a patient with X-linked hypohidrotic ectodermal dysplasia. Int J Dermatol 2013 Sep;52(9):1121–4.
- Callea M, Willoughby C, Nieminen P, et al. Identification of a novel frameshift mutation in the EDAR gene causing autosomal dominant hypohidrotic ectodermal dysplasia. J Eur Acad Dermatol Venereol 2014 Mar 18.
- Bashyam MD, Chaudhary AK, Reddy EC, et al. A founder ectodysplasin A receptor (EDAR) mutation results in a high frequency of the autosomal recessive form of hypohidrotic ectodermal dysplasia in India. Br J Dermatol 2012 Apr;166(4):819–29.
- Doffinger R, Smahi A, Bessia C, et al. X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-kappaB signaling. Nat Genet 2001 Mar;27(3):277–85.
- Naiki M, Mizuno S, Yamada K, et al. MBTPS2 mutation causes BRESEK/BRESHECK syndrome. Am J Med Genet Part A 2012 Jan;158A(1):97–102.
- Conte MI, Pescatore A, Paciolla M, et al. Insight into IKBKG/NEMO Locus: Report of new mutations and complex genomic rearrangements leading to incontinentia pigmenti disease. Hum Mutat 2013; Nov 12.
- Coggshall K, Farsani T, Ruben B, et al. Keratitis, ichthyosis, and deafness syndrome: a review of infectious and neoplastic complications. J Am Acad Dermatol 2013 Jul;69(1):127–34.
- van Bokhoven H, Hamel BC, Bamshad M, et al. p63 Gene mutations in eec syndrome, limb-mammary syndrome, and isolated split hand-split foot malformation suggest a genotype–phenotype correlation. Am J Hum Genet 2001 Sep;69(3):481–92.
- Sozen MA, Suzuki K, Tolarova MM, Bustos T, Fernandez Iglesias JE, Spritz RA. Mutation of PVRL1 is associated with sporadic, non-syndromic cleft lip/palate in northern Venezuela. Nat Genet 2001 Oct;29(2):141–2.
- Sweeney E, Fryer A, Mountford R, Green A, McIntosh I. Nail patella syndrome: a review of the phenotype aided by developmental biology. J Med Genet 2003 Mar;40(3):153–62.
- Adaimy L, Chouery E, Megarbane H, et al. Mutation in WNT10A is associated with an autosomal recessive ectodermal dysplasia: the odonto-onycho-dermal dysplasia. Am J Hum Genet 2007 Oct;81(4):821–8.
- Wilson NJ, O'Toole E, Milstone LM, et al. The molecular genetic analysis of the expanding pachyonychia congenita case collection. Br J Dermatol 2014; Mar 10.
- McPherson E, Cold C. Severe Pallister–Hall syndrome with persistent urogenital sinus, renal agenesis, imperforate anus, bilateral hypothalamic hamartomas, and severe skeletal anomalies. Am J Med Genet A 2013 Oct;161(10):2666–9.
- Mevorah B, Goldberg I, Sprecher E, et al. Olmsted syndrome: mutilating palmoplantar keratoderma with periorificial keratotic plaques. J Am Acad Dermatol 2005 Nov;53(5 Suppl. 1):S266–72.
- Larregue M, Callot V, Kanitakis J, Suau AM, Foret M. Olmsted syndrome: report of two new cases and literature review. J Dermatol 2000 Sep;27(9):557–68.
- Farkas K, Paschali E, Papp F, et al. A novel seven-base deletion of the CTSC gene identified in a Hungarian family with Papillon–Lefevre syndrome. Arch Dermatol Res 2013 Jul;305(5):453–5.
- Colombo EA, Bazan JF, Negri G, et al. Novel C16orf57 mutations in patients with poikiloderma with neutropenia: bioinformatic analysis of the protein and predicted effects of all reported mutations. Orphanet J Rare Dis 2012;7:7.
- Matsuzawa N, Kondo S, Shimozato K, et al. Two missense mutations of the IRF6 gene in two Japanese families with popliteal pterygium syndrome. Am J Med Genet A 2010 Sep;152A(9):2262–7.
- Gripp KW, Ennis S, Napoli J. Exome analysis in clinical practice: expanding the phenotype of Bartsocas–Papas syndrome. Am J Med Genet A 2013 May;161A(5):1058–63.
- Kantaputra PN, Hamada T, Kumchai T, McGrath JA. Heterozygous mutation in the SAM domain of p63 underlies Rapp–Hodgkin ectodermal dysplasia. J Dent Res 2003 Jun;82(6):433–7.
- Mazzeu JF, Pardono E, Vianna-Morgante AM, et al. Clinical characterization of autosomal dominant and recessive variants of Robinow syndrome. Am J Med Genet A 2007 Feb 15;143(4):320–5.
- Colombo EA, Fontana L, Roversi G, et al. Novel physiological RECQL4 alternative transcript disclosed by molecular characterisation of Rothmund–Thomson Syndrome sibs with mild phenotype. Eur J Hum Genet 2014; Feb 12.
- Marneros AG, Beck AE, Turner EH, et al. Mutations in KCTD1 cause scalp-ear-nipple syndrome. Am J Hum Genet 2013 Apr 4;92(4):621–6.
- Suphapeetiporn K, Srichomthong C, Shotelersuk V. SETBP1 mutations in two Thai patients with Schinzel–Giedion syndrome. Clin Genet 2011 Apr;79(4):391–3.
- Petrof G, Fong K, Lai-Cheong JE, Cockayne SE, McGrath JA. Schopf–Schulz–Passarge syndrome resulting from a homozygous nonsense mutation, p.Cys107X, in WNT10A. Australas J Dermatol 2011 Aug;52(3):224–6.
- Hamersma H, Gardner J, Beighton P. The natural history of sclerosteosis. Clin Genet 2003 Mar;63(3):192–7.
- Leupin O, Piters E, Halleux C, et al. Bone overgrowth-associated mutations in the LRP4 gene impair sclerostin facilitator function. J Biol Chem 2011 Jun 3;286(22):19489–500.
- Sarig O, Nahum S, Rapaport D, et al. Short stature, onychodysplasia, facial dysmorphism, and hypotrichosis syndrome is caused by a POC1A mutation. Am J Hum Genet 2012 Aug 10;91(2):337–42.
- Adriani M, Martinez-Mir A, Fusco F, et al. Ancestral founder mutation of the nude (FOXN1) gene in congenital severe combined immunodeficiency associated with alopecia in southern Italy population. Ann Hum Genet 2004 May;68(Pt 3):265–8.
- Wright JT, Hong SP, Simmons D, Daly B, Uebelhart D, Luder HU. DLX3 c.561_562delCT mutation causes attenuated phenotype of tricho-dento-osseous syndrome. Am J Med Genet A 2008 Feb 1;146(3):343–9.
- Jeon J, Kim JH, Oh CH. Trichorhinophalangeal syndrome type I – Clinical, microscopic, and molecular features. Indian J Dermatol Venereol Leprol 2014 Jan–Feb;80(1):54–7.
- Tariq M, Ahmad S, Ahmad W. A novel missense mutation in the TRPS1 gene underlies trichorhinophalangeal syndrome type III. Br J Dermatol 2008 Aug;159(2):476–8.
- Schinzel A, Riegel M, Baumer A, et al. Long-term follow-up of four patients with Langer–Giedion syndrome: clinical course and complications. Am J Med Genet A 2013 Sep;161(9):2216–25.
- Hashimoto S, Egly JM. Trichothiodystrophy view from the molecular basis of DNA repair/transcription factor TFIIH. Hum Mol Genet 2009 Oct 15;18(R2):R224–30.
- Linden H, Williams R, King J, Blair E, Kini U. Ulnar mammary syndrome and TBX3: expanding the phenotype. Am J Med Genet A 2009 Dec;149A(12):2809–12.
- Tatton-Brown K, Murray A, Hanks S, et al. Weaver syndrome and EZH2 mutations: clarifying the clinical phenotype. Am J Med Genet A 2013 Dec;161(12):2972–80.
- Shen W, Han D, Zhang J, Zhao H, Feng H. Two novel heterozygous mutations of EVC2 cause a mild phenotype of Ellis–van Creveld syndrome in a Chinese family. Am J Med Genet A 2011 Sep;155A(9):2131–6.
- Memarpour M, Shafiei F. Witkop tooth and nail syndrome: a report of three cases in a family. Pediatr Dermatol 2011 May–Jun;28(3):281–5.
- Campeau PM, Lenk GM, Lu JT, et al. Yunis–Varon syndrome is caused by mutations in FIG4, encoding a phosphoinositide phosphatase. Am J Hum Genet 2013 May 2;92(5):781–91.
- van de Veerdonk FL, Plantinga TS, Hoischen A, et al. STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. N Engl J Med 2011 Jul 7;365(1):54–61.
- Bae-Harboe YS, Mirzabeigi M, Gilchrest BA. JAAD Grand Rounds quiz: nail dystrophy and multiple hyperkeratotic papules on the face and neck. J Am Acad Dermatol 2013 Nov;69(5):847–9.
- Lin Z, Chen Q, Shi L, et al. Loss-of-function mutations in HOXC13 cause pure hair and nail ectodermal dysplasia. Am J Hum Genet 2012 Nov 2;91(5):906–11.
- Shimomura Y, Wajid M, Kurban M, Sato N, Christiano AM. Mutations in the keratin 85 (KRT85/hHb5) gene underlie pure hair and nail ectodermal dysplasia. J Invest Dermatol 2010 Mar;130(3):892–5.
- McGrath JA, Hoeger PH, Christiano AM, et al. Skin fragility and hypohidrotic ectodermal dysplasia resulting from ablation of plakophilin 1. Br J Dermatol 1999 Feb;140(2):297–307.
- Groves RW, Liu L, Dopping-Hepenstal PJ, et al. A homozygous nonsense mutation within the dystonin gene coding for the coiled-coil domain of the epithelial isoform of BPAG1 underlies a new subtype of autosomal recessive epidermolysis bullosa simplex. J Invest Dermatol 2010 Jun;130(6):1551–7.
- Pfendner EG, Sadowski SG, Uitto J. Epidermolysis bullosa simplex: recurrent and de novo mutations in the KRT5 and KRT14 genes, phenotype/genotype correlations, and implications for genetic counseling and prenatal diagnosis. J Invest Dermatol 2005 Aug;125(2):239–43.
- Bel B, Jeudy G, Vabres P. Dermoscopy of longitudinal leukonychia in Hailey–Hailey disease. Arch Dermatol 2010 Oct;146(10):1204.
- Lamartine J, Munhoz Essenfelder G, Kibar Z, et al. Mutations in GJB6 cause hidrotic ectodermal dysplasia. Nature Genet 2000 Oct;26(2):142–4.
- Sadler E, Klausegger A, Muss W, et al. Novel KIND1 gene mutation in Kindler syndrome with severe gastrointestinal tract involvement. Arch Dermatol 2006 Dec;142(12):1619–24.
- Wu J, Lin Y, Xu W, Li Z, Fan W. A mutation in the type II hair keratin KRT86 gene in a Han family with monilethrix. J Biomed Res 2011 Jan;25(1):49–55.
- Bruchle NO, Frank J, Frank V, et al. RSPO4 is the major gene in autosomal-recessive anonychia and mutations cluster in the furin-like cysteine-rich domains of the Wnt signaling ligand R-spondin 4. J Invest Dermatol 2008 Apr;128(4):791–6.
- Kiuru M, Kurban M, Itoh M, et al. Hereditary leukonychia, or porcelain nails, resulting from mutations in PLCD1. Am J Hum Genet 2011 Jun 10;88(6):839–44.
- Frojmark AS, Schuster J, Sobol M, et al. Mutations in Frizzled 6 cause isolated autosomal-recessive nail dysplasia. Am J Hum Genet 2011 Jun 10;88(6):852–60.
Pachyonychia congenita
- Shah S, Boen M, Kenner-Bell B, Schwartz M, Rademaker A, Paller AS. Pachyonychia congenita in pediatric patients: natural history, features, and impact. JAMA Dermatol 2014;150(2):146–53.
- Wilson NJ, Leachman SA, Hansen CD, et al. A large mutational study in pachyonychia congenita. J Invest Dermatol 2011;131(5):1018–24.
- Eliason MJ, Leachman SA, Feng BJ, Schwartz ME, Hansen CD. A review of the clinical phenotype of 254 patients with genetically confirmed pachyonychia congenita. J Am Acad Dermatol 2012; Jan 18.
- Akasaka E, Nakano H, Nakano A, et al. Diffuse and focal palmoplantar keratoderma can be caused by a keratin 6c mutation. Br J Dermatol 2011;165(6):1290–2.
- Wilson NJ, Messenger AG, Leachman SA, et al. Keratin K6c mutations cause focal palmoplantar keratoderma. J Invest Dermatol [Research Support, Non-US Gov't]. 2010;130(2):425–9.
- Spaunhurst KM, Hogendorf AM, Smith FJ, et al. Pachyonychia congenita patients with mutations in KRT6A have more extensive disease compared with patients who have mutations in KRT16. Br J Dermatol [Research Support, Non-US Gov't]. 2012;166(4):875–8.
- Du ZF, Xu CM, Zhao Y, et al. Two novel de novo mutations of KRT6A and KRT16 genes in two Chinese pachyonychia congenita pedigrees with fissured tongue or diffuse plantar keratoderma. Eur J Dermatol 2012;22(4):476–80.
- Harris K, Hull PR, Hansen CD, et al. Transgrediens pachyonychia congenita (PC): case series of a nonclassical PC presentation. Br J Dermatol 2012;166(1):124–8.
- Goldberg I, Sprecher E, Schwartz ME, Gaitini D. Comparative study of high-resolution multifrequency ultrasound of the plantar skin in patients with various types of hereditary palmoplantar keratoderma. Dermatology 2013;226(4):365–70.
- Wilson NJ, Perez ML, Vahlquist A, et al. Homozygous dominant missense mutation in keratin 17 leads to alopecia in addition to severe pachyonychia congenita. J Invest Dermatol 2012;132(7):1921–4.
- Naz G, Pasternack SM, Perrin C, et al. FZD6 encoding the Wnt receptor frizzled 6 is mutated in autosomal-recessive nail dysplasia. Br J Dermatol 2012;166(5):1088–94.
- Frojmark AS, Schuster J, Sobol M, et al. Mutations in Frizzled 6 cause isolated autosomal-recessive nail dysplasia. Am J Hum Genet 2011;10;88(6):852–60.
- Goldberg I, Fruchter D, Meilick A, Schwartz ME, Sprecher E. Best treatment practices for pachyonychia congenita. J Eur Acad Dermatol Venereol. 2014;28(3)279–85.
- Swartling C, Karlqvist M, Hymnelius K, Weis J, Vahlquist A. Botulinum toxin in the treatment of sweat-worsened foot problems in patients with epidermolysis bullosa simplex and pachyonychia congenita. Br J Dermatol 2010;163(5):1072–6.
- Gruber R, Edlinger M, Kaspar RL, et al. An appraisal of oral retinoids in the treatment of pachyonychia congenita. J Am Acad Dermatol 2012;66(6):e193–9.
- Hickerson RP, Leake D, Pho LN, Leachman SA, Kaspar RL. Rapamycin selectively inhibits expression of an inducible keratin (K6a) in human keratinocytes and improves symptoms in pachyonychia congenita patients. J Dermatol Sci 2009;56(2):82–8.
- Zhao Y, Gartner U, Smith FJ, McLean WH. Statins downregulate K6a promoter activity: a possible therapeutic avenue for pachyonychia congenita. J Invest Dermatol 2011;131(5):1045–52.
- Leachman SA, Hickerson RP, Schwartz ME, et al. First-in-human mutation-targeted siRNA phase Ib trial of an inherited skin disorder. Mol Ther 2010;18(2):442–6.
- Hickerson RP, Flores MA, Leake D, et al. Use of self-delivery siRNAs to inhibit gene expression in an organotypic pachyonychia congenita model. J Invest Dermatol 2011;131(5):1037–44.
Dyskeratosis congenita
- Alder JK, Parry EM, Yegnasubramanian S, et al. Telomere phenotypes in females with heterozygous mutations in the dyskeratosis congenita 1 (DKC1) gene. Hum Mutat 2013;34(11):1481–5.
- Ballew BJ, Savage SA. Updates on the biology and management of dyskeratosis congenita and related telomere biology disorders.Expert Rev Hematol 2013;6(3):327–37.
- Dokal I. Dyskeratosis congenita. Hematol Am Soc Hematol Educ Program 2011;2011:480–6.
- Calado RT, Young NS. Telomere diseases. N Engl J Med 2009;10;361(24):2353–65.
- Zhong F, Savage SA, Shkreli M, et al. Disruption of telomerase trafficking by TCAB1 mutation causes dyskeratosis congenita. Genes Dev 2011;25(1):11–16.
- Ballew BJ, Joseph V, De S, et al. A recessive founder mutation in regulator of telomere elongation helicase 1, RTEL1, underlies severe immunodeficiency and features of Hoyeraal Hreidarsson syndrome. PLOS Genet 2013;9(8):e1003695.
- Deng Z, Glousker G, Molczan A, et al. Inherited mutations in the helicase RTEL1 cause telomere dysfunction and Hoyeraal–Hreidarsson syndrome. Proc Natl Acad Sci U S A 2013;110(36):E3408–16.
- Keller RB, Gagne KE, Usmani GN, et al. CTC1 Mutations in a patient with dyskeratosis congenita. Pediatr Blood Cancer 2012;59(2):311–14.
- Walne AJ, Vulliamy T, Beswick R, Kirwan M, Dokal I. Mutations in C16orf57 and normal-length telomeres unify a subset of patients with dyskeratosis congenita, poikiloderma with neutropenia and Rothmund–Thomson syndrome. Human Mol Genet 2010;19(22):4453–61.
- Savage SA, Alter BP. Dyskeratosis congenita. Hematol Oncol Clin North Am 2009;23(2):215–31.
- Allenspach EJ, Bellodi C, Jeong D, et al. Common variable immunodeficiency as the initial presentation of dyskeratosis congenita. J Allergy Clin Immunol 2013;132(1):223–6.
- Revesz T, Fletcher S, al-Gazali LI, DeBuse P. Bilateral retinopathy, aplastic anaemia, and central nervous system abnormalities: a new syndrome? J Med Genet 1992;29(9):673–5.
- Piard J, Holder-Espinasse M, Aral B, et al. Systematic search for neutropenia should be part of the first screening in patients with poikiloderma. Eur J Med Genet 2012;55(1):8–11.
- Gadalla SM, Sales-Bonfim C, Carreras J, et al. Outcomes of allogeneic hematopoietic cell transplantation in patients with dyskeratosis congenita. Biol Blood Marrow Transplant 2013;19(8):1238–43.
- Islam A, Rafiq S, Kirwan M, et al. Haematological recovery in dyskeratosis congenita patients treated with danazol. Br J Haematol 2013;162(6):854–6.
Nail–patella syndrome
- Sweeney E, Hoover-Fong JE, McIntosh I. Nail–patella syndrome. In: Pagon RA, Adam MP, Ardinger HH, Bird TD, et al., eds. GeneReviews®. Seattle (WA): University of Washington, Seattle, 1993–2014, updated 2009 Jul 28, PMID:20301311 (last accessed July 2014).
- Isojima T, Harita Y, Furuyama M, et al. LMX1B mutation with residual transcriptional activity as a cause of isolated glomerulopathy. Nephrol Dial Transplant 2013 Sep 15.
- Sweeney E, Fryer A, Mountford R, Green A, McIntosh I. Nail patella syndrome: a review of the phenotype aided by developmental biology. J Med Genet 2003;40(3):153–62.
- Lopez-Arvizu C, Sparrow EP, Strube MJ, et al. Increased symptoms of attention deficit hyperactivity disorder and major depressive disorder symptoms in nail–patella syndrome: potential association with LMX1B loss-of-function. Am J Med Genet B Neuropsychiatr Genet 2011;156B(1):59–66.
Hereditary anonychia
- Bergmann C, Senderek J, Anhuf D, et al. Mutations in the gene encoding the Wnt-signaling component R-spondin 4 (RSPO4) cause autosomal recessive anonychia. Am J Human Genet 2006;79(6):1105–9.
- Blaydon DC, Ishii Y, O'Toole EA, et al. The gene encoding R-spondin 4 (RSPO4), a secreted protein implicated in Wnt signaling, is mutated in inherited anonychia. Nature Genet 2006;38(11):1245–7.
- Ishii Y, Wajid M, Bazzi H, et al. Mutations in R-spondin 4 (RSPO4) underlie inherited anonychia. J Invest Dermatol 2008;128(4):867–70.