CHAPTER 70
Genetic Disorders of Pigmentation
Alain Taïeb, Fanny Morice-Picard and Khaled Ezzedine
Service de Dermatologie et Dermatologie Pédiatrique, Hôpital St André, Bordeaux, France
Introduction and general description
Normal skin colour is determined by a number of variables, the most important of which is melanin. Differences in skin and hair colour are principally the result of differences in the melanin content of skin although other chromophores and skin thickness may also determine shade variation in the skin. Besides melanin, haemoglobin (in both the oxygenated and reduced state) and carotenoids contribute also significantly to skin colour (see Chapter 88).
Melanocytes are responsible for the synthesis of melanin, a complex quinone/indole-quinone-derived mixture of biopolymers. Melanocytes migrate from the neural crest into the epidermis during the first 2 months of gestation. They produce melanin within specialized vesicles known as melanosomes. Racial and ethnic differences in skin colour are related to variation in the number, size, composition and distribution of melanosomes to surrounding keratinocytes. Constitutive pigmentation refers to the amount of melanin pigmentation that is genetically determined in the absence of sun exposure and other influences. Facultative (inducible) pigmentation or ‘tan’, results from sun exposure (or other exogenous influences including drugs). The regulation of human pigmentation is complex and intertwined with other factors affecting epidermal or dermal–epidermal homeostasis. It involves both systemic and local factors secreted in the dermal or epidermal compartment. This is reflected in physiological pigmentary changes, such as normal palmar hypopigmentation more striking in black skin, pigmentary lines enhanced during pregnancy and systemic disorders with hyperproduction of pigmentation stimulators, such as α-melanocyte-stimulating hormone (MSH) in Addison disease.
The major precursor of melanins is tyrosine. Tyrosinase catalyses the hydroxylation of tyrosine to DOPA (3,4-dihydroxyphenylalanine). Once completely formed within melanocytes, melanosomes are transported along dendrites towards adjacent keratinocytes. The next step involves the extrusion of the melanosomes and their transfer into neighbouring keratinocytes, by a mechanism which is still debated. After being transferred to keratinocytes, melanosomes are translocated to the apical pole of the keratinocyte where they protect the nucleus from UV mutagenic damage. Keratinocyte terminal differentiation is accompanied by concomitant degradation of melanosomes so that no melanosomes are normally visible in the very upper part of the epidermis.
Hundreds of genes are known to modulate pigmentation type or pattern in the skin, hairs/coat and eyes in mammals during or after development by acting directly or indirectly on the pigment cell lineage. Among these, only a few have been also found to underlie inherited monogenic pigmentation disorders (Table 70.1). Of interest, some genes known to be important for normal variation in skin, eye or hair colour are in this list. These disorders can be classified into three major groups: disorders of hypopigmentation, disorders of hyperpigmentation and disorders featuring combined hypo- and hyperpigmentation, known as the dyschromatoses. This classical clinical classification is, however, in part misleading, since an overlap of phenotypes exist between the last two groups, as illustrated in this chapter.
Table 70.1 Major monogenic inherited pigmentation disorders
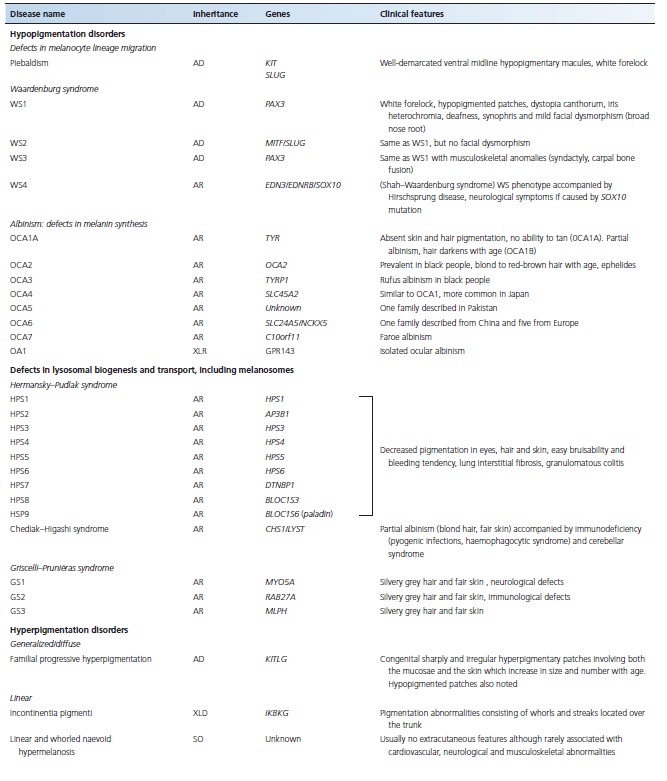
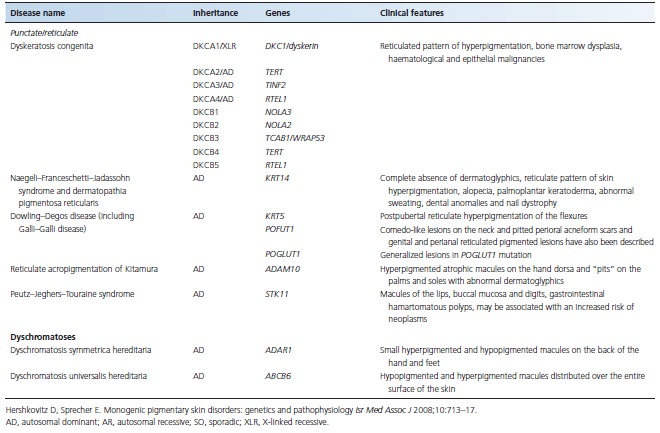
HYPOPIGMENTATION DISORDERS
Piebaldism
Introduction and general description
Piebaldism is a rare autosomal dominant trait characterized by well-demarcated irregular hypopigmented macules [1]. The incidence of piebaldism is estimated at less than 1 in 20 000. Both sexes are affected equally and no population is spared.
Pathophysiology
The disease results from heterozygous mutations in KIT, encoding c-KIT, a membranal tyrosine kinase receptor responsible for triggering cell proliferation and migration [2]. Dominant mutations in KIT result in impaired migration of melanocytes to the skin, as reflected by the absence of melanocytes and melanin in hypopigmented patches [2]. Piebaldism can also be caused by heterozygous mutation in the gene encoding the zinc finger transcription factor SNAI2 [3].
Morphological studies have shown either an absence of melanocytes and melanosomes in the hypomelanotic areas or sometimes reduced numbers of abnormal large melanocytes. In the hypermelanotic islands situated in areas of hypomelanosis, melanocytes produce normal melanosomes but also abnormal spherical and granular melanosomes.
Clinical features
The most typical and common clinical feature of the disease is a white forelock, often associated with a V-shaped area of leukoderma on the mid-forehead. The hypopigmented lesions of piebaldism have a predilection for the anterior part of the body and the mid-portion of the limbs (Figure 70.1). Often, white patches occur on the upper chest, abdomen and limbs, bilaterally but not necessarily symmetrically. Occasionally, they are found on the face, particularly the chin. The hands and feet, as well as the back, remain normally pigmented. In addition, small spots of hyperpigmentation arise subsequently within the hypopigmented lesions or even on the background of normal skin.
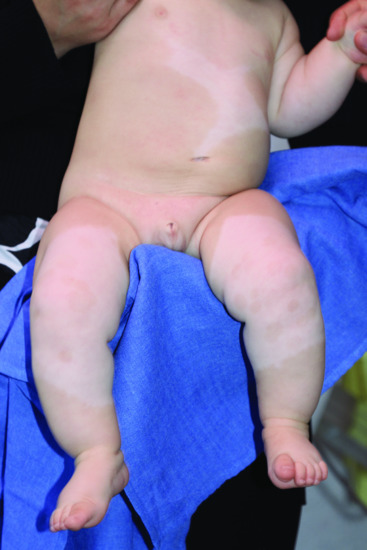
Figure 70.1 Typical body and limb pattern of hypopigmentation in piebaldism.
The extent of the areas of depigmentation is variable. Rarely, the white forelock may be the only lesion. Piebaldism is usually not associated with extracutaneous manifestations; however, mental retardation or deafness have been reported in the context of a large deletion [4].
Although the skin lesions in piebaldism are quite typical, the occurrence of isolated poliosis or skin hypopigmentation can suggest a number of inflammatory disorders affecting pigmentation (e.g. vitiligo, alopecia areata, Vogt–Koyanagi syndrome, Alezzandrini syndrome, Woolf syndrome), as well as monogenic (e.g. tuberous sclerosis, Waardenburg syndrome (WS)) or somatic (naevus depigmentosus) disorders.
Piebaldism can be distinguished from vitiligo because of the neonatal presence of white patches. In cases of fair skin and poorly detectable lesions at birth, the medial distribution of lesions is quite different from that of vitiligo; hyperpigmented patches which appear at the border or centre of hypopigmented patches are quite distinctive (Figures 70.2 and 70.3), and, overall, lesions remain stable in adults.
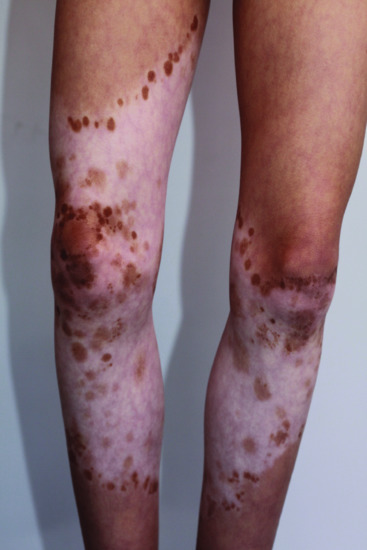
Figure 70.2 Partial repigmentation pattern on the legs in piebaldism.
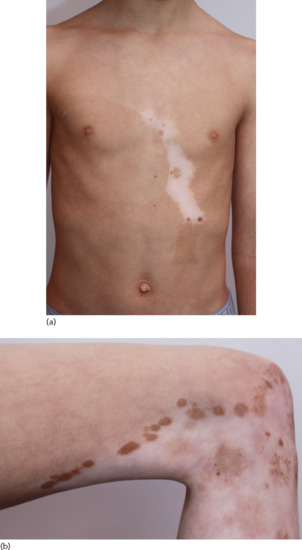
Figure 70.3 (a) Note the mosaic pattern on the trunk in piebaldism; (b) detail of repigmentation pattern (leg).
If the interpupillary distance is increased or the patient is deaf, the diagnosis of WS must be considered. The evolution of piebaldism is benign. Sun protection is recommended to protect the amelanotic areas from burning. Epidermal cell or skin grafting have been successfully tried in piebaldism [5]. In contrast, phototherapy is inefficacious, as are topical corticosteroids.
Waardenburg syndrome
Introduction and general description
Waardenburg syndrome is an autosomal dominant genetic disorder characterized by piebaldism and sensorineural deafness. A molecular classification is used (WS1–4) based on the mutant gene (see Table 70.1). It is an autosomal dominant condition except for the recessive WS4 with an estimated incidence of 1 in 20 000–40 000.
Pathophysiology
All abnormalities seen in WS involve the neural crest lineage. Type 1 and type 3 result from loss-of-function mutations in the PAX3 gene [1]. Mutations in MITF result accordingly in WS type 2 [2]. WS type 2 is characterized by genetic heterogeneity: it was shown to also result from mutations in SLUG encoding a zinc finger transcription factor [3].
Type 4 disease is caused by mutations in at least three genes including EDNRB, encoding the endothelin-B receptor, EDN3, encoding the ligand of EDNRB, and SOX10, which, like PAX3, regulates the expression of MITF. Bi-allelic mutations in EDNRB and EDN3 result in WS type 4 whereas heterozygous mutations in the same genes cause isolated Hirschsprung disease. Loss of SOX10 expression leads to abnormal expression of RET, which is known to cause Hirschsprung disease. In addition, lack of SOX10 expression is associated with abnormal myelination and neurological signs.
Clinical features
Waardenburg syndrome features congenital leukoderma reminiscent of piebald pattern in association with sensorineural deafness of varying severity. Areas of hypopigmentation may diminish in size or even disappear with time [4, 5].
Clinical variants
Waardenburg syndrome type 1 is inherited in an autosomal dominant fashion and is characterized by a white forelock, alopecia, hypopigmented patches, dystopia canthorum (increased distance between the inner canthi without any change in the distance between the pupils) and heterochromia irides associated with deafness in one third to one half of cases. Patients may also display dysmorphic features including a broad nasal root and synophrys (medial hyperplasia of the eyebrows) and mild skeletal anomalies (Figure 70.4). WS type 2 (Klein–Waardenburg syndrome) is inherited as an autosomal dominant trait. Clinical signs are similar to those seen in WS type 1 except for absence of facial dysmorphism and dystopia canthorum, and a higher frequency of deafness and heterochromia. WS type 3, an autosomal dominant disorder, is much rarer than the other types and presents the same clinical manifestations as type 1 in association with musculoskeletal anomalies. WS type 4 (Shah–Waardenburg syndrome) is inherited in an autosomal recessive fashion, features the white forelock and is accompanied by Hirschsprung disease.
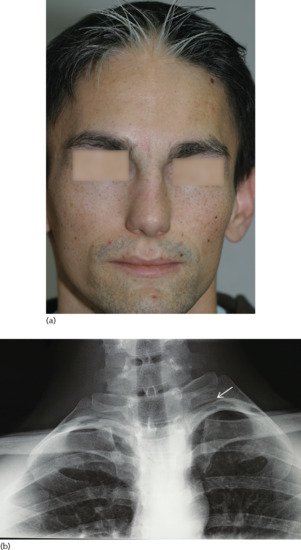
Figure 70.4 Typical facial dysmorphic features (a) and supernumerary ribs (b) in Waardenburg syndrome.
Management
Management of the hearing loss associated with WS1 depends on its severity. The hearing loss in WS1 is typically non-progressive. Hence, repeating the audiogram would typically be unnecessary.
Oculocutaneous albinism
Introduction and general description
Oculocutaneous albinism (OCA) is a rare genetic disorder characterized by generalized depigmentation of the skin, hair and eye, and by ophthalmological anomalies caused by a deficiency in melanin biosynthesis [1].
A molecular classification is used (OCA1–7) derived from mutant genes (see Table 70.1).
The appellation of the various OCA subtypes, initially centred on clinical findings, has moved towards a molecular classification based upon the identification of causative genes. OCA is a genetically heterogeneous disorder. Initially, four types of OCA were known and recently three additional forms of OCA have been reported. Altogether seven genes have been implicated in the pathogenesis of OCA: TYR in OCA1, OCA2 in OCA2, TYRP1 in OCA3, SLC45A2 in OCA4, SLC24A5 in OCA6 and C10orf11 in OCA7 [2–8]; OCA5 has been mapped to 4q24 but the gene still remains to be identified [2–8].
Epidemiology
Oculocutaneous albinism is the most frequent form of diffuse hypopigmentation worldwide with a prevalence estimated around 1/20 000.
The prevalence of the various OCA subtypes varies considerably from one continent to another, but OCA1 is the most frequent form worldwide. OCA2 is the most frequent form among African patients with a prevalence that reaches 1/1000 in some populations in Western Africa.
Pathophysiology and genetics
Oculocutaneous albinism is due to a deficiency of melanin biosynthesis but melanocytes are normally present and distributed. The reduction of the amount of melanin is responsible for an increased sensitivity to UV radiation and for a predisposition to skin cancers. The ophthalmological anomalies associated with albinism are not only a consequence of a lack of melanin but also of a lack of L-DOPA, an early intermediate of the synthesis of melanin, which has been shown to be required for normal retinal and visual development [9].
Oculocutaneous albinism type 1 is caused by mutations in TYR, encoding the tyrosinase gene [2, 10]. Total lack of tyrosinase activity results in OCA type 1A while partial activity causes OCA type 1B [11] (Figure 70.5a,b). Some patients with type 1B OCA show variations in hair and skin hypopigmentation with dark hairs being found in cooler areas of the body. This phenomenon has been related to the fact the underlying mutations in these cases are temperature sensitive. It has been shown that mutations in the mouse TYR gene cause the Tyr protein to be retained in the endoplasmic reticulum, with subsequent early degradation.

Figure 70.5 (a,b) Oculocutaneous albinism type 1B (OCA1B) and siblings/family. (a) The children in the centre and on the right are affected; the child on the left is unaffected. (b) Both children are affected.
Oculocutaneous albinism type 2 is caused by mutations in the OCA2 gene, encoding the P protein, a transmembrane protein of importance for melanin biosynthesis and for processing and transport of other melanosomal proteins such as tyrosinase. It seems that OCA2 exerts at least some of its effects by maintaining an acidic pH in melanosomes.
Mutations in TYRP1 are responsible for OCA type 3. The protein encoded by this gene catalyses the oxidation of dihydroxyindole-2-carboxylic acid (DHICA) monomers into eumelanin and serves also to stabilize tyrosinase. It is not required for phaeomelanin production, explaining the accumulation of the latter in the skin and hair in OCA3 [4].
Oculocutaneous albinism type 4 is caused by mutations in SLC45A2, encoding the membrane-associated transporter protein (MATP), a membrane transporter in melanosomes [5].
Oculocutaneous albinism type 6 is caused by mutations in SLC24A5, coding for a membrane-associated transporter protein (NCKX5). This protein is involved in proper maturation of melanosomes [6].
Clinical features
The pigmentation of the skin, hair and eyes is in general reduced but its degree varies with the type of albinism. It is important to check pigmentary characteristics in siblings and parents to consider the diagnosis of albinism in its subtle variants (see Figure 70.5a,b).
All types of OCA and ocular albinism (OA) have similar ocular findings, including various degrees of congenital nystagmus, hypopigmentation of the iris leading to iris translucency, reduced pigmentation of the retinal pigment epithelium, foveal hypoplasia, reduced visual acuity usually in the range 20/60 to 20/400 and refractive errors, and sometimes a degree of colour vision impairment [12, 13].
Photophobia may be prominent. Iris translucency is demonstrable by slit lamp examination. A characteristic finding is misrouting of the optic nerves, consisting in an excessive crossing of the fibres in the optic chiasma, which can result in strabismus and reduced stereoscopic vision [12]. The abnormal crossing of fibres can be demonstrated by monocular visual evoked potential [2]. Absence of misrouting excludes the diagnosis of albinism.
Clinical variants
In the severe OCA1a form, there is a complete absence of pigmentation with white hair and pink skin (Figure 70.6a,b). There is no tendency to tan. Naevi are achromic. The visual impairment is severe with nystagmus, photophobia and errors of refraction.
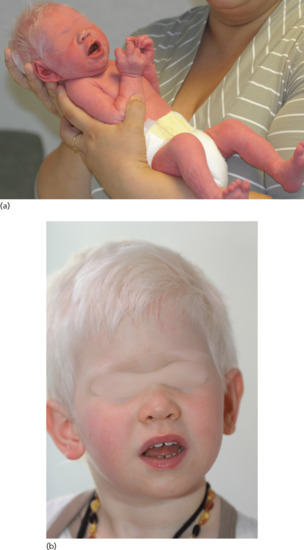
Figure 70.6 (a) Oculocutaneous albinism type 1A (OCA1A) at birth and (b) later in the same child.
In other types initially described as tyrosinase positive, pigmentation is highly variable and influenced by the phototype of the patient. Pigmentation can increase with age (Figure 70.7). Patients develop dark-brown freckles with age, particularly in sun-exposed areas. Visual acuity may improve as patients get older and they may have less severe nystagmus.

Figure 70.7 Oculocutaneous albinism type 2 (OCA2) in infant (shown with mother).
Rufous albinism (OCA3) was originally described in African patients with TYRP1 mutations [4, 14], but recent molecular investigations indicate that it is not restricted to African populations.
Differential diagnosis
The presence of OA excludes a large number of diseases associated with pigment dilution. Diagnoses to be considered in the case of ocular and cutaneous hypopigmentation include histidinaemia, homocystinuria, phenylketonuria, Hermansky–Pudlak and Chediak–Higashi syndromes, as well as Cross and Tietz syndromes. Pure OCA has to be distinguished from syndromic forms of OCA, especially Hermansky–Pudlak syndrome, and a platelet secretion test is recommended in the initial assessment of an OCA patient without family history of albinism.
Complications and co-morbidities
The incidence of skin cancer is increased in patients with OCA, especially spinous cell carcinoma which is a cause of mortality in Africans with OCA2. Melanoma is far less common, suggesting that melanin production is a key factor for melanoma risk in the context of UV exposure.
Disease course and prognosis
Lifespan in patients with OCA is normal, and medical problems are generally not increased compared to those in the general population. Skin cancers may occur and regular skin checks should be offered. Development and intelligence are normal. Persons with OCA have a normal fertility.
Investigations
The diagnosis of OCA is based on clinical findings of hypopigmentation of the skin and hair. Ophthalmological examination should include an examination with optical coherence tomography (OCT) of the retina showing characteristic foveal hypoplasia. Electrophysiological testing can demonstrate misrouting of the optic nerves, resulting in strabismus and impaired stereoscopic vision.
Due to the clinical overlap between the OCA subtypes, molecular diagnosis is necessary to establish a correct diagnosis, and subsequently provide patients and their families with prognostic information and genetic counselling.
Management
Sun protection is mandatory to avoid skin sunburns and skin cancers with a special emphasis in patients living in high UV risk environments. Early referral to an ophthalmologist is mandatory. Decreased visual acuity is usually managed with corrective lenses while strabismus requires eye patching or surgical correction. Dark glasses are important to protect the eyes and prevent photophobia.
Hermansky–Pudlak syndrome
Introduction and general description
Hermansky–Pudlak syndrome is a rare type of OCA associated with a haemorrhagic diathesis [1, 2]. Here too, a molecular classification has now been universally accepted recognizing nine clinicogenetic subtypes of the disease (see Table 70.1). The disorder is rare except in Puerto Rico.
Pathophysiology
The disease results from abnormal biogenesis of lysosome-related organelles with impaired melanosome maturation and absent dense bodies in thrombocytes [2]. Hermansky–Pudlak syndrome is associated with mutations in nine distinct genes: HPS1 (type 1) and HPS4 (type 4) encode components of the BLOC3 lysosomal complex, which is essential for the proper formation of lysosome-related organelles; AP3B1 (type 2) encodes a subunit of the AP3 complex, which is responsible for mediating protein sorting to lysosomes; HPS3 (type 3), HSP5 (type 5) and HSP6 (type 6) all encode components of BLOC2 and DTNBP1 (type 7), BLOC1S3 (type 8) and BLOC1S6 (type 9) encode components of BLOC1 which are all required for proper melanosome maturation [3–10].
Clinical features and complications
All subtypes of the syndrome share common clinical manifestations including decreased pigmentation in the eyes, hair and skin, easy bruisability and bleeding tendency, interstitial pulmonary fibrosis and granulomatous colitis [2].
The complications of Hermansky–Pudlak syndrome are secondary to bleeding problems, pulmonary fibrosis and colitis. Prognosis is guarded with a life expectancy of 30–50 years.
Investigations
Absence of dense bodies on whole mount electron microscopy of platelets constitutes a standard diagnostic test. Moreover upon stimulation of platelets, the dense bodies, which contain adenosine diphosphate (ADP), adenosine triphosphate (ATP), serotonin, calcium and phosphate, release their contents to attract other platelets, which can be tested to screen for Hermansky–Pudlak syndrome [7]. For detection and evaluation of lung fibrosis, pulmonary function tests should be performed on a regular basis in adulthood and computed tomography (CT) scans when necessary. Molecular analysis of the Hermansky–Pudlak syndrome genes helps to confirm the diagnosis and to give appropriate genetic counselling.
Chédiak–Higashi syndrome
Introduction and general description
Chédiak–Higashi syndrome is a rare autosomal recessive disorder characterized by hypopigmentation of the skin and eye, immunodeficiency and possibly neurological symptoms [1].
Pathophysiology and genetics
The disease results from loss-of-function mutations in CHS1 (LYST) encoding a protein known as lysosomal trafficking regulator. The melanocytes contain giant pigment granules which arise by autophagocytosis and defective fission of large melanosomes [2].
Clinical features
The skin is fair, the retinae are pale and the irides translucent. The diagnosis of Chédiak–Higashi syndrome is suspected in individuals with clinical criteria for OCA combined with a significant history of pyogenic infections (Figure 70.8). Neutropenia is noted. The most reliable diagnostic clinical criterion for Chédiak–Higashi syndrome is the finding of giant inclusions in polymorphonuclear neutrophils. Neurological manifestations (e.g. progressive intellectual decline, cranial nerve palsies, decreased deep tendon reflexes, tremor and abnormal gait, seizures) can appear any time from childhood to early adulthood.
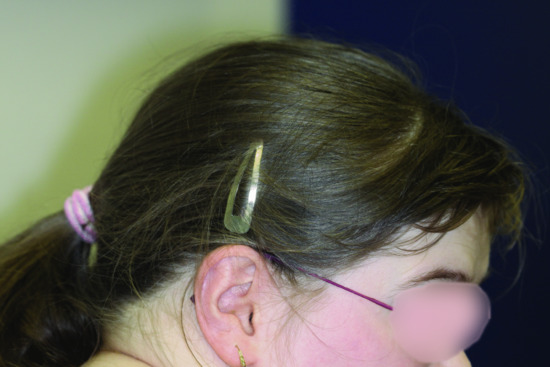
Figure 70.8 Chédiak–Higashi syndrome after successful bone marrow transplantation: mild mental retardation and hair pigment dilution.
Attenuated forms have been described with genotype–phenotype correlation. Loss-of-function mutations are associated with severe childhood-onset form [3, 4]. The accelerated phase corresponding to a haemophagocytic lymphohistiocytosis, occurs in 85% of individuals at any age and can be fatal [5, 6].
Investigations
Peroxidase-positive giant inclusions seen in leukocytes is a first line diagnostic test. Light microscopy of hairs shows pigment clumping.
Management
The only curative treatment available for Chédiak–Higashi syndrome is bone marrow transplantation. A case of complete remission after a combination therapy with rituximab and ciclosporin has been reported [7].
Griscelli–Pruniéras syndrome types I and II
Introduction and general description
Griscelli–Pruniéras syndrome is a rare autosomal recessive disorder that associates hypopigmentation, characterized by a silver-grey sheen of the hair and the presence of large clusters of pigment in the hair shaft, and the occurrence of either a primary neurological impairment or a severe immune disorder.
Pathophysiology and genetics
All genetic alterations associated with Griscelli–Pruniéras syndrome result in defective transport of melanosomes and consequently abnormal accumulation of melanosomes in melanocytes. Griscelli–Pruniéras syndrome type 1 results from mutations in MYO5A, encoding myosin 5a [1]. Type 2 disease results from mutations in the RAB27A gene whereas type 3 is caused by mutations in MLPH encoding melanophilin or by a specific genetic defect in MYO5A [2, 3].
Clinical features
In 1978, Griscelli et al. described two patients with partial albinism of the hair and skin, frequent pyogenic infections and acute episodes of fever, hepatosplenomegaly, neutropenia and thrombocytopenia [4].
Dermatological findings included pigmentary abnormalities of the hair variably described as silvery grey, silvery, greyish-golden or dusty [5]. Neurological involvement is a prominent feature. Patients with Griscelli–Pruniéras syndrome type 1 have primary central nervous system dysfunction, type 2 patients commonly develop haemophagocytic lymphohistiocytosis and type 3 patients have only partial albinism. The differential diagnosis of the disease in the patient presenting with silvery hair includes primarily the Griscelli–Pruniéras, Chédiak–Higashi, and Elejalde syndromes. Elejalde syndrome is inherited in an autosomal recessive fashion and is characterized by pigment dilution, silvery grey hair and neurological defects [6]. Some authors suggest that the disease may in fact be identical to Griscelli–Pruniéras syndrome type 1 [6].
Patients with Griscelli–Pruniéras syndrome are predisposed to the occurrence of ‘accelerated phases’ similar to those encountered in Chédiak–Higashi syndrome. Griscelli–Pruniéras syndrome has been successfully treated with bone marrow transplantation.
Oculocerebral syndrome with hypopigmentation (Cross syndrome/Kramer syndrome)
The disorder was initially described in an inbred Amish family and is characterized by generalized hypopigmentation with white/silvery hair, severe mental retardation with spastic tetraplegia and athetosis [1]. Ocular anomalies include microphthalmos, a small opaque cornea and coarse nystagmus.
About 10 cases of Cross syndrome have been described in Amish and Gipsy families and in South Africa. It is an autosomal recessive disorder. The defective gene remains to be identified.
Albinism–deafness syndrome (Ziprkowski–Margolis syndrome/Woolf syndrome)
Albinism–deafness syndrome is an X-linked disorder consisting of congenital deafness and partial albinism (without OA) [1, 2]. The disease has been mapped to Xq26.3-q27.1 but the causative gene is unknown. [3]. Hypopigmentation has a piebald pattern, but more diffuse (Figure 70.9a,b). Deafness is of the subtotal sensorineural nerve type.
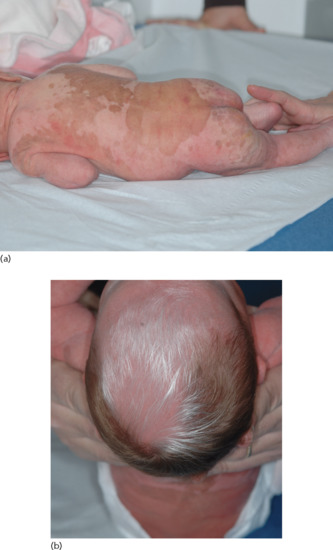
Figure 70.9 (a,b) Albinism–deafness syndrome.
(Courtesy Dr Odile Enjolras.)
Hypomelanosis of Ito
In 1952, Ito described the occurrence of a bilateral systematized depigmented naevus in a 22-year-old Japanese woman [1]. Hypomelanosis of Ito is a rare neuroectodermal disorder often associated with mental retardation and epilepsy [2, 3]. Prevalence is unknown but incidence has been estimated between 1/10 000 and 1/8500. The phenotypic presentation is highly variable. The skin abnormalities are characterized by unusual unilateral or bilateral cutaneous macular hypopigmented whorls, streaks and patches, corresponding to the lines of Blaschko (Figure 70.10) [4]. Extracutaneous findings include neurological, ophthalmological and skeletal defects. The skin lesions have to be distinguished from other mosaic depigmented lesions and from focal dermal hypoplasia where the skin shows atrophic changes. Nearly all cases are sporadic suggesting a postzygotic mutation, which is assumed to be lethal when transmitted to offspring. Various chromosomal anomalies have been identified in some patients and the current consensus is that the phenotype of hyper- or hypopigmentation following Blaschko lines (Figures 70.10 and 70.11) is the result of cutaneous mosaicism, either for a monogenic or a chromosomal disorder, rather than being a distinct disease [4–6].
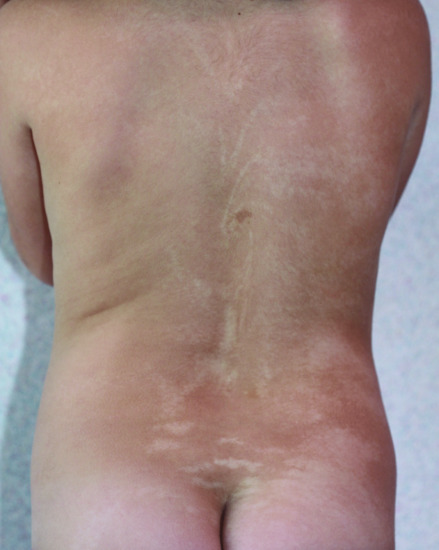
Figure 70.10 Hypomelanosis of Ito.
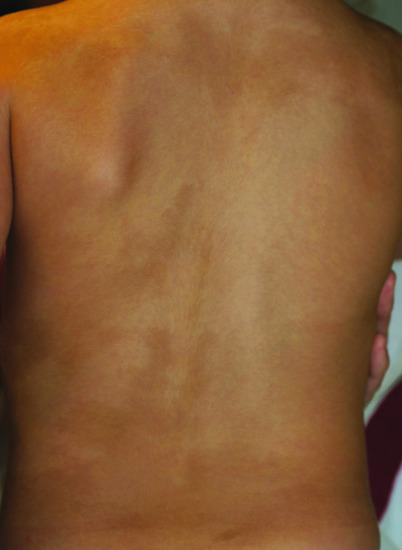
Figure 70.11 Linear and whorled naevoid hypermelanosis.
HYPERPIGMENTATION DISORDERS
Familial progressive hyperpigmentation/progressive hyperpigmentation and generalized lentiginosis without associated systemic symptoms/familial progressive hyper- and hypopigmentation
Familial progressive hyperpigmentation is a very rare autosomal dominant disorder. The disease is characterized by sharply and irregular hyperpigmented patches involving both the mucosae and the skin. These patches are present either at birth or in early infancy and increase in size and number with age.
Familial progressive hyperpigmentation-1 (FPH1) has been mapped to chromosome 19pter-p13.1 [1] whereas familial progressive hyperpigmentation-2 (FPH2; MIM 145250), is caused by mutation in the KITLG gene (MIM 184745) on chromosome 12q22 [2] which is also associated with the hyper–hypopigmented variant [3].
Incontinentia pigmenti
Definition
Incontinentia pigmenti (IP) is a rare X-linked dominant multisystemic ectodermal dysplasia that is usually lethal in males and that presents classically in females with skin lesions, teeth abnormalities, alopecia, nail dystrophy, and ocular and neurological findings [1, 2].
Epidemiology
Birth prevalence is 0.6–0.7/1 000 000. The female to male ratio is 20 : 1.
Pathophysiology and genetics
IP is caused by mutations of the IKBKG gene encoding the nuclear factor (NF)-κB essential modulator (NEMO), also known as the inhibitor of the NF-κB kinase subunit gamma (IKK-γ) [3]. NEMO (IKK-γ) is the regulatory subunit of the inhibitor of the I-κB kinase (IKK) complex, which activates NF-κB resulting in activation of genes involved in inflammation, immunity, cell survival and other pathways [4]. IP cells are highly sensitive to tumour necrosis factor-induced apoptosis that could explain the development of skin, retinal and neurological lesions in patients with IP.
IP is an X-linked dominant disorder. A recurrent exon 4–10 deletion of the gene underlies 85% of cases. The mutation can be inherited from an affected mother or occur de novo. When the mother of an affected female carries the mutant IKBKG, the risk to siblings of inheriting the mutant IKBKG allele at conception is 50%; most male conceptuses with loss-of-function mutation of IKBKG are miscarried. Fertility is normal except for the miscarriage of affected males. Genetic prenatal diagnosis is available [5].
Clinical features
IP cutaneous findings typically present perinatally with an erythematous vesicular rash (bullous stage I; Figure 70.12a) following Blaschko's lines. Stage I evolves within a few months to a verrucous stage II, occurring mainly on the limbs (Figure 70.12b). Stage III hyperpigmented streaks and whorls along Blaschko's lines begin within months and fade in adolescence (Figure 70.12c). Stage I rash can recur during febrile illness. Stage IV patients have pale, hairless, atrophic linear streaks or patches mostly on the lower extremities at adolescence (Figure 70.12d). Extracutaneous abnormalities observed in IP include delayed dentition, and missing or malformed cone-shaped teeth (Figure 70.13). Other manifestations include onychodystrophy (Figure 70.14), alopecia (Figure 70.15) and a wide range of ophthalmological abnormalities with retinal neovascularization. Central nervous system (CNS) abnormalities may comprise microcephaly, seizures and neurocognitive and motor impairments. The majority (>60%) of patients are neurologically normal [6].
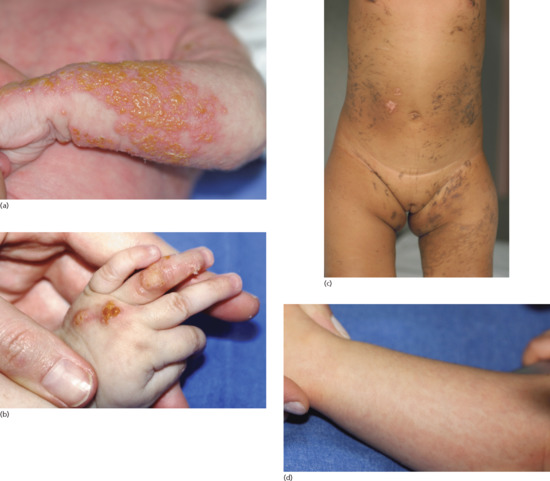
Figure 70.12 (a) Incontinentia pigmenti, vesiculobullous stage, frequently wrongly diagnosed as bullous impetigo. (b) Verrucous stage. (c) Pigmentary stage, with an obvious linear pattern following Blaschko's lines. (d) Hypopigmented stage with hair loss.
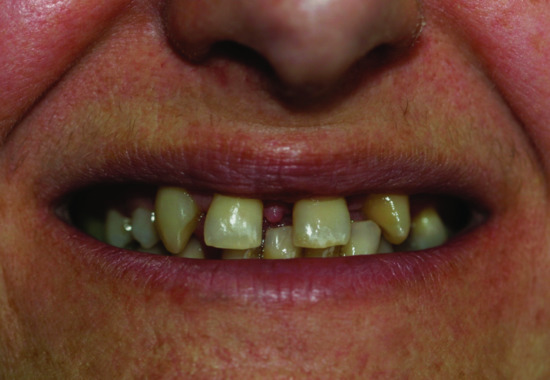
Figure 70.13 Dystrophic teeth in the carrier mother of a child with incontinentia pigmenti.
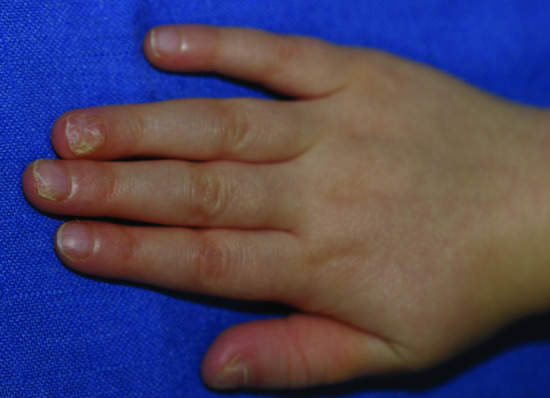
Figure 70.14 Nail dystrophy of incontinentia pigmenti.
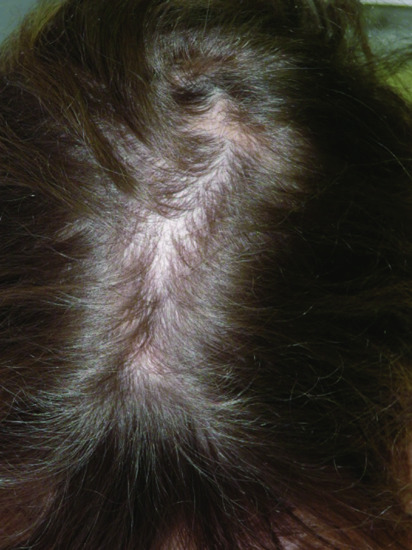
Figure 70.15 Cicatricial alopecia of incontinentia pigmenti.
Differential diagnosis
Stage I lesions have to be distinguished from other bullous dermatoses (bullous impetigo, epidermolysis bullosa, herpes or varicella). Differential diagnosis of stage II includes warts or epidermal naevus syndrome. Any condition with ‘linear and swirled’ pigmentation overlaps with stage III. Stage IV resembles scarring, Ito's hypomelanosis or other hypopigmentary disorders with localized alopecia.
Complications and co-morbidities
Life expectancy is normal. Patients without neonatal CNS abnormalities typically have normal physical and cognitive development.
Management
No specific treatment is available for IP. Symptomatic treatment includes standard management of blisters. Ophthalmological follow-up is required for retinal neovascularization monitoring and treatment (cryotherapy and laser photocoagulation) and treatment of retinal detachment if it occurs. Dental abnormalities should be managed by a paediatric orthodontist in combination with speech therapy and a paediatric nutrition programme. Patients should be referred to a paediatric neurologist for evaluation if microcephaly, seizures, spasticity or focal deficits are present. Brain magnetic resonance imaging is indicated in any child with functional neurological abnormalities or retinal neovascularization.
Linear and whorled naevoid hypermelanosis
Linear and whorled hypermelanosis (LWNH) is a benign skin condition characterized by the onset in infancy of hyperpigmented regions that follow the lines of Blaschko on the trunk and limbs. The soles, palms, face and mucous membranes are spared. In general, affected individuals have no accompanying extradermal features [1] although the condition has been rarely associated with cardiovascular, neurological and musculoskeletal abnormalities [2]. This phenotype is the reverse of hypomelanosis of Ito (see Figure 70.10). Mosaic chromosomal abnormalities have been detected [2]. As stated above for hypomelanosis of Ito, the phenotype of either hyper- or hypopigmentation following Blaschko lines (see Figures 70.10 and 70.11) is the result of cutaneous mosaicism, either for a monogenic or a chromosomal disorder, rather than being a distinct disease.
Dyskeratosis congenita
Dyskeratosis congenita (DKC) is a genetically heterogeneous disorder, showing autosomal recessive, autosomal dominant and X-linked inheritance (see Table 70.1). DKC is a disease with a highly variable phenotype that is classically defined by reticulated skin pigmentation, nail dystrophy and leukoplakia of the oral mucosa often undergoing malignant transformation [1]. The pigmentary change may be limited to the neck, upper chest and proximal parts of the limbs initially, but within affected areas the involvement is always diffuse. Overall, predisposition to malignancy is an important feature and bone marrow dysplasia, haematological and epithelial malignancies are frequent complications [2]. The disease is caused by mutations in multiple genes coding for proteins involved in telomere function and maintenance [3].
Differential diagnosis includes Fanconi anaemia, a clinically and genetically heterogeneous disorder that causes genomic instability. Fanconi anaemia is also sometimes associated with pigmentation anomalies including reticulated pigmentation and café-au-lait spots. The diagnosis is based mostly on haematological anomalies, especially pancytopenia and there is an increased risk of neoplasia [4].
Naegeli–Franceschetti–Jadassohn syndrome and dermatopathia pigmentosa reticularis
These two diseases are closely related autosomal dominant ectodermal dysplasia syndromes that are both caused by mutations specifically located at the start of the KRT14 gene sequence [1]. These mutations result in haploinsufficiency for keratin 14 and are associated with increased susceptibility of keratinocytes to pro-apoptotic stimuli [2].
The cardinal features of Naegeli–Franceschetti–Jadassohn syndrome are reticular cutaneous pigmentation (Figure 70.16) starting in early life without a preceding inflammatory stage, discomfort provoked by heat with diminished sweat gland function, poor teeth and moderate hyperkeratosis of the palms and soles [3]. Males and females are equally affected. In dermatopathia pigmentosa reticularis cutaneous findings include reticulate hyperpigmentation, non-cicatricial alopecia and onychodystrophy. These two diseases clinically share complete absence of dermatoglyphics, a reticulate pattern of skin hyperpigmentation mainly involving the trunk and face, palmoplantar keratoderma, abnormal sweating and other subtle developmental anomalies including plantar bullae in early childhood, dental anomalies and nail dystrophy.

Figure 70.16 Naegeli–Franceschetti–Jadassohn syndrome: case of the original family.
(Courtesy of P. Itin.)
Dowling–Degos disease
Dowling–Degos disease is an autosomal dominant form of reticulate pigmentary genodermatosis with variable penetrance that was first described by Dowling and Freudenthal [1]. The reticulate pigmentation usually has a flexural distribution. Comedo-like lesions on the neck and pitted perioral acneform scars and genital and perianal reticulated pigmented lesions have also been described (Figure 70.17) [2–4]. Onset is usually postpubertal and the reticulate hyperpigmentation is progressive and disfiguring. No abnormalities of the hair or nails have been reported. Generalized Dowling–Degos disease can also occur, with numerous hyperpigmented or erythematous macules and papules on the neck, chest and abdomen [5]. Histopathology from pigmented lesions discloses characteristic thin branch-like patterns of epidermal downgrowth.
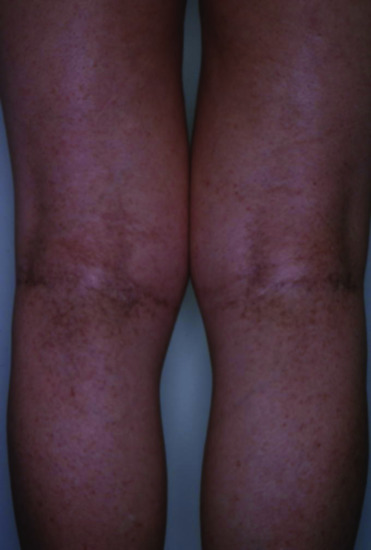
Figure 70.17 Dowling–Degos disease.
At least, three genes have been shown to be associated with Dowling–Degos disease. Flexural Dowling–Degos disease was initially found to be caused by loss of function mutations affecting the KRT5 gene region encoding the initial part of keratin 5 [4]. Galli–Galli disease which features acantholysis on histology in addition to clinical and histological signs of Dowling–Degos disease, was also shown to result from mutations in KRT5 [6]. More recently, generalized Dowling–Degos disease was found to be associated with mutations in POFUT1, which encodes protein O-fucosyltransferase 1 [7] and in POGLUT1 [8], which encodes protein O-glucosyltransferase 1. Knockdown of POFUT1 was shown to reduce the expression of KRT5 in keratinocytes. In addition, both POGLUT1 and POFUT1 are essential regulators of Notch activity.
Reticulate acropigmentation of Kitamura
Reticulate acropigmentation of Kitamura (RAK) is a rare pigmentary disorder that has an autosomal dominant pattern of inheritance. Typical features include reticulate hyperpigmented atrophic macules on the dorsa of the hands (Figure 70.18a–c) and ‘pits’ on the palms and soles in the first or second decade of life [1–3]. The macules gradually darken and tend to spread to the proximal regions of the extremities with progression of the eruptions that stops in middle age. Histopathologically, the pigmented lesions show pigmentation in the tip of rete ridges with thinning of the epidermis, elongation and thinning of the rete ridges, and slight hyperkeratosis without parakeratosis [4]. RAK was found to be caused by mutations in ADAM10.
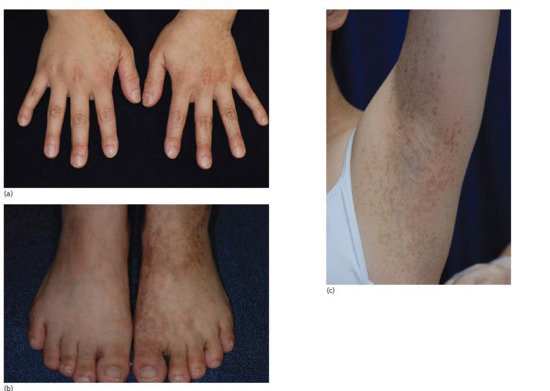
Figure 70.18 (a–c) Reticulate hyperpigmentation of Kitamura.
(Courtesy of Dr H. Ujiie and Professor H. Shimizu.) Molecular diagnosis was confirmatory.
Peutz–Jeghers–Touraine syndrome
Peutz–Jeghers–Touraine (PJT) syndrome is a rare but very distinctive lentiginosis syndrome with an autosomal dominant pattern of inheritance. Typical cutaneous features include lentigines of the lips of early onset (Figure 70.19). Associated melanocytic macules of buccal mucosa and digits are common.
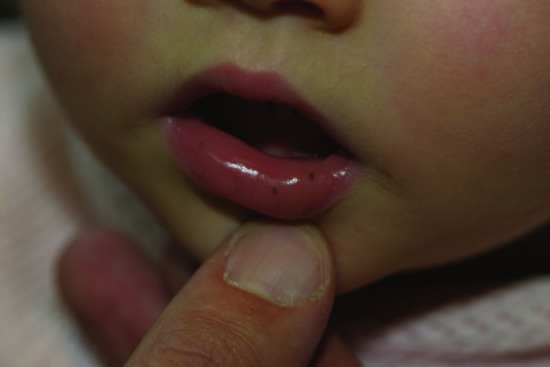
Figure 70.19 Peutz–Jeghers–Touraine syndrome. Lentigines on the lower lip of a 2-year-old girl.
Non-cutaneous features are dominated by hamartomatous polyps, which may occur in any part of the gastrointestinal tract but more consistently in the jejunum. They may lead to intussusception and bleeding. Malignant degeneration of the small intestinal polyps is rare [1]. Other polyps have been described in the kidney pelvis, ureter, bladder, bronchus and nose. Among non-gastrointestinal associated tumours in PJT syndrome, granulosa cell ovarian tumours are the most distinctive, but other malignant tumours (especially breast and pancreas) have been described [2].
Histologically, the oral mucosal lesions resemble lentigo simplex. In acral lesions there is an increased number of melanocytes with long dendrites filled with melanosomes, but few melanosomes in keratocytes, suggesting a pigment block [3]. Gastrointestinal hamartomas in PJT syndrome are distinct from those of other types and show an elongated, frond-like epithelium with cystic dilatation of glands overlying an arborizing network of smooth muscle bundles [1]. The disorder is due to mutations in the serine/threonine kinase (STK11) gene [4]. Mutations in the part of the gene involved in ATP binding and catalysis are rarely associated with cancer, whereas mutations in the part of the gene involved in substrate recognition are more frequently associated with malignancies. Patients with PJT syndrome with breast cancers have predominantly truncating mutations [5].
DYSCHROMATOSES
Dyschromatosis symmetrica hereditaria
Dyschromatosis symmetrica hereditaria (DSH) – also called symmetrical dyschromatosis of the extremities and symmetrical or reticulate acropigmentation of Dohi [1] – is characterized by freckle-like pigmented macules on the face associated with small hyper- and hypopigmented macules affecting the back of the hands and feet (Figure 70.20a,b). The condition usually begins in early childhood and has been predominantly reported in Japanese and Chinese individuals. The prevalence of DSH in the Japanese population is estimated to be 1.5 per 100 000 [2].
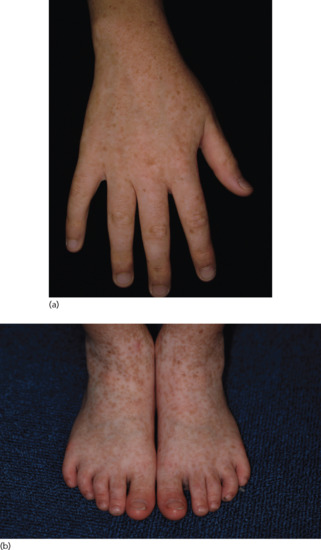
Figure 70.20 (a,b) Dyschromatosis symmetrica hereditaria.
(Courtesy of Dr H. Ujiie and Professor H. Shimizu.) Molecular diagnosis was confirmatory.
Dyschromatosis symmetrica hereditaria has a dominant pattern of inheritance and is caused by a heterozygous mutation in the ADAR1 gene (adenosine deaminase acting on RNA1) located on chromosome 1q21 [2]. ADAR1 mediates a post-transcriptional modification of the messenger RNA known as RNA editing. Miyamura et al. postulated that impaired RNA editing during melanoblast migration causes their differentiation into either hyper- or hypoactive melanocytes and that the most affected melanocytes are those that migrate farthest, to the extremities, i.e. the hands and feet [2].
Dyschromatosis universalis hereditaria
Dyschromatosis universalis hereditaria (DUH) is a rare autosomal genodermatosis characterized by hyper- and hypopigmented macules distributed over the entire surface of the skin (Figure 70.21a,b). The lesions usually appear in infancy or early childhood. Involvement of the palms or soles is unusual. Abnormalities of the hair and nails have also been reported. Also, DUH has been associated with neurological, ophthalmological and haematological complications [1].
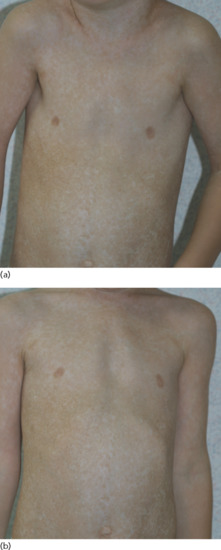
Figure 70.21 (a,b) Dyschromatosis universalis hereditaria (molecular diagnosis not done).
There is now evidence for genetic heterogeneity in DUH. Indeed, DUH1 maps to chromosome 6q24.2-q25.2 [2–4], DUH2 maps to chromosome 12q21-q23a [1, 5] and DUH3 is caused by mutation in the ABCB6 gene on chromosome 2q35 [6].
References
Hypopigmentation disorders
Piebaldism
- Thomas I, Kihiczak GG, Fox MD, Janniger CK, Schwartz RA. Piebaldism: an update. Int J Dermatol 2004;43:716–19.
- Spritz RA, Holmes SA, Ramesar R, Greenberg J, Curtis D, Beighton P. Mutations of the KIT (mast/stem cell growth factor receptor) proto-oncogene account for a continuous range of phenotypes in human piebaldism. Am J Hum Genet 1992;51:1058–65.**
- Sánchez-Martín M, Pérez-Losada J, Rodríguez-García A, et al. Deletion of the SLUG (SNAI2) gene results in human piebaldism. Am J Med Genet A 2003;122A(2):125–32.
- Sijmons RH, Kristoffersson U, Tuerlings JH, Ljung R, Dijkhuis-Stoffelsma R, Breed AS. Piebaldism in a mentally retarded girl with rare deletion of the long arm of chromosome 4. Pediatr Dermatol 1993;10:235–9.
- Van Geel N, Wallaeys E, Goh BK, De Mil M, Lambert J. Long term results of non cultured epidermal cellular grafting in vitiligo, halo nevi, piebaldism and nevus depigmentosus. Br J Dermatol 2010;163:1186–93.
Waardenburg syndrome
- Kubic JD, Young KP, Plummer RS, Ludvik AE, Lang D. Pigmentation PAX-ways: the role of Pax3 in melanogenesis, melanocyte stem cell maintenance, and disease. Pigment Cell Melanoma Res 2008;21:627–45.
- Tassabehji M, Newton VE, Read AP. Waardenburg syndrome type 2 caused by mutations in the human microphthalmia (MITF) gene. Nat Genet1994;8:251–5. **
- Sánchez-Martín M, Rodríguez-García A, Pérez-Losada J, Sagrera A, Read AP, Sánchez-García I. SLUG (SNAI2) deletions in patients with Waardenburg disease. Hum Mol Genet 2002;11:3231–6.
- Tomita Y, Suzuki T. Genetics of pigmentary disorders. Am J Med Genet C 2004;131C:75–81.
- Tamayo ML, Gelvez N, Rodriguez M, et al. Screening program for Waardenburg syndrome in Colombia: clinical definition and phenotypic variability. Am J Med Genet A 2008;146A:1026–31.
Oculocutaneous albinism
- Tomita Y, Suzuki T. Genetics of pigmentary disorders. Am J Med Genet C Semin Med Genet 2004;131C:75–81.
- Tomita Y, Takeda A, Okinaga S, Tagami H, Shibahara S. Human oculocutaneous albinism caused by single base insertion in the tyrosinase gene. Biochem Biophys Res Commun 1989;164:990–6. **
- Rinchik EM, Bultman SJ, Horsthemke B, et al. A gene for the mouse pink-eyed dilution locus and for human type II oculocutaneous albinism. Nature 1993;361:72–6. **
- Manga P, Kromberg JG, Box NF, Sturm RA, Jenkins T, Ramsay M. Rufous oculocutaneous albinism in southern African Blacks is caused by mutations in the TYRP1 gene. Am J Hum Genet 1997;61:1095–101. **
- Newton JM, Cohen-Barak O, Hagiwara N, et al. Mutations in the human orthologue of the mouse underwhite gene (uw) underlie a new form of oculocutaneous albinism, OCA4. Am J Hum Genet 2001;69:981–8. **
- Wei A-H, Zang D-J, Zhang Z, et al. Exome sequencing identifies SLC24A5 as a candidate gene for non-syndromic oculocutaneous albinism. J Invest Dermatol 2013;133:1834–40. **
- Grønskov K, Dooley CM, Østergaard E, et al. Mutations in c10orf11, a melanocyte-differentiation gene, cause autosomal-recessive albinism. Am J Hum Genet 2013;92:415–21. **
- Kausar T, Bhatti M, Ali M, Shaikh R, Ahmed Z. OCA5, a novel locus for non-syndromic oculocutaneous albinism, maps to chromosome 4q24. Clin Genet 2012;84:91–3. **
- Lavado A, Montoliu L. New animal models to study the role of tyrosinase in normal retinal development. Front Biosci J Virtual Libr 2006;11:743–52.
- Tomita Y. Tyrosinase gene mutations causing oculocutaneous albinisms. J Invest Dermatol 1993;100(2 Suppl.):186S–90S.
- Oetting WS, King RA. Molecular basis of type I (tyrosinase-related) oculocutaneous albinism: mutations and polymorphisms of the human tyrosinase gene. Hum Mutat 1993;2:1–6.
- Summers CG. Albinism: classification, clinical characteristics, and recent findings. Optom Vis Sci 2009;86:659–62.
- Grønskov K, Ek J, Brondum-Nielsen K. Oculocutaneous albinism. Orphanet J Rare Dis 2007;2:43.
- Kromberg JG, Castle DJ, Zwane EM, et al. Red or rufous albinism in southern Africa. Ophthalmic Paediatr Genet 1990;11:229–35.
Hermansky–Pudlak syndrome
- Hermansky F, Pudlak P. Albinism associated with hemorrhagic diathesis and unusual pigmented reticular cells in the bone marrow: report of two cases with histochemical studies. Blood 1959;14:162–9.
- Wei A-H, Li W. Hermansky–Pudlak syndrome: pigmentary and non-pigmentary defects and their pathogenesis. Pigment Cell Melanoma Res 2013;26:176–92.
- Li W, Zhang Q, Oiso N, et al. Hermansky–Pudlak syndrome type 7 (HPS-7) results from mutant dysbindin, a member of the biogenesis of lysosome-related organelles complex 1 (BLOC-1). Nat Genet 2003;35:84–9.
- Morgan NV, Pasha S, Johnson CA, et al. A germline mutation in BLOC1S3/reduced pigmentation causes a novel variant of Hermansky–Pudlak syndrome (HPS8). Am J Hum Genet 2006;78:160–6.
- Cullinane AR, Curry JA, Carmona-Rivera C, et al. A BLOC-1 mutation screen reveals that PLDN is mutated in Hermansky–Pudlak syndrome type 9. Am J Hum Genet 2011;88:778–87.
- Zhang Q, Zhao B, Li W, et al. Ru2 and Ru encode mouse orthologs of the genes mutated in human Hermansky–Pudlak syndrome types 5 and 6. Nat Genet 2003;33:145–53.
- Suzuki T, Li W, Zhang Q, et al. Hermansky–Pudlak syndrome is caused by mutations in HPS4, the human homolog of the mouse light-ear gene. Nat Genet 2002;30:321–4.
- Anikster Y, Huizing M, White J, et al. Mutation of a new gene causes a unique form of Hermansky–Pudlak syndrome in a genetic isolate of central Puerto Rico. Nat Genet 2001;28:376–80.
- Huizing M, Scher CD, Strovel E, et al. Nonsense mutations in ADTB3A cause complete deficiency of the beta3A subunit of adaptor complex-3 and severe Hermansky–Pudlak syndrome type 2. Pediatr Res 2002;51:150–8.
- Oh J, Bailin T, Fukai K, et al. Positional cloning of a gene for Hermansky–Pudlak syndrome, a disorder of cytoplasmic organelles. Nat Genet1996;14:300–6.
Chédiak–Higashi syndrome
- Shiflett SL, Kaplan J, Ward DM. Chediak–Higashi syndrome: a rare disorder of lysosomes and lysosome related organelles. Pigment Cell Res 2002;15:251–7.
- Durchfort N, Verhoef S, Vaughn MB, et al. The enlarged lysosomes in beige j cells result from decreased lysosome fission and not increased lysosome fusion. Traffic 2012;13:108–19.
- Karim MA, Suzuki K, Fukai K, et al. Apparent genotype–phenotype correlation in childhood, adolescent, and adult Chediak–Higashi syndrome. Am J Med Genet 2002;108:16–22. **
- Westbroek W, Adams D, Huizing M, et al. Cellular defects in Chediak–Higashi syndrome correlate with the molecular genotype and clinical phenotype. J Invest Dermatol 2007;127:2674–7.
- Introne WJ, Westbroek W, Golas GA, Adams D. Chediak–Higashi syndrome. In: Pagon RA, Adam MP, Bird TD, Dolan CR, Fong C-T, Stephens K, eds. GeneReviewsTM. Seattle (WA): University of Washington, Seattle, 1993. http://www.ncbi.nlm.nih.gov/books/NBK5188/ (last accessed August 2014).
- Eapen M, DeLaat CA, Baker KS, et al. Hematopoietic cell transplantation for Chediak–Higashi syndrome. Bone Marrow Transplant 2007;39:411–15.
- Ogimi C, Tanaka R, Arai T, Kikuchi A, Hanada R, Oh–Ishi T. Rituximab and cyclosporine therapy for accelerated phase Chediak–Higashi syndrome. Pediatr Blood Cancer 2011;57:677–80.
Griscelli–Pruniéras syndrome
- Pastural E, Barrat FJ, Dufourcq-Lagelouse R, et al. Griscelli disease maps to chromosome 15q21 and is associated with mutations in the myosin-Va gene. Nat Genet 1997;16:289–92. **
- Ménasché G, Pastural E, Feldmann J, et al. Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nat Genet 2000;25:173–6.
- Ménasché G, Ho CH, Sanal O, et al. Griscelli syndrome restricted to hypopigmentation results from a melanophilin defect (GS3) or a MYO5A F-exon deletion (GS1). J Clin Invest 2003;112:450–6.
- Griscelli C, Raux M, Attal C, Barthélémy C, Mozziconacci P. [Syndrome associating anemia, hepatomegaly, dwarfism, late puberty and geophagia. Geophagia syndrome]. Ann Pédiatrie 1970;17:214–19.
- Mancini AJ, Chan LS, Paller AS. Partial albinism with immunodeficiency: Griscelli syndrome: report of a case and review of the literature. J Am Acad Dermatol 1998;38(2 Pt 2):295–300.
- Lambert J, Vancoillie G, Naeyaert JM. Elejalde syndrome revisited. Arch Dermatol 2000;136:120–1.
Oculocerebral syndrome with hypopigmentation
- Cross HE, McKusick VA, Breen W. A new oculocerebral syndrome with hypopigmentation. J Pediatr 1967;70:398–406**.
Albinism–deafness syndrome
- Margolis E. A new hereditary syndrome – sex linked deafmutism associated with total albinism. Acta Genet Stat Med 1962;12:12–19. **
- Ziprkowski L, Krakowski A, Adam A, Costeff H, Sade J. Partial albinism and deaf–mutism due to a recessive sex-linked gene. Arch Dermatol 1962;86:530–9.
- Shiloh Y, Litvak G, Ziv Y, et al. Genetic mapping of X-linked albinism–deafness syndrome (ADFN) to Xq26.3-q27.I. Am J Hum Genet 1990;47:20–7.
Hypomelanosis of Ito
- Ito M. Studies on melanin XI: Incontinentia pigmenti achromians. A singular case of nevus depigmentosus systematicus bilateralis. Tohoku J Exp Med 1952;55:57–9. **
- Glover MT, Brett EM, Atherton DJ. Hypomelanosis of Ito: spectrum of the disease. J Pediatr 1989;115:75–80.
- Ruiz-Maldonado R, Toussaint S, Tamayo L, Laterza A, del Castillo V. Hypomelanosis of Ito: diagnostic criteria and report of 41 cases. Pediatr Dermatol 1992;9:1–10.
- Nehal KS, PeBenito R, Orlow SJ. Analysis of 54 cases of hypopigmentation and hyperpigmentation along the lines of Blaschko. Arch Dermatol 1996;132:1167–70.
- Ritter CL, Steele MW, Wenger SL, Cohen BA. Chromosome mosaicism in hypomelanosis of Ito. Am J Med Genet 1990;35:14–17.
- Moss C, Larkins S, Stacey M, et al. Epidermal mosaicism and Blaschko's lines. J Med Genet 1993;30:752–5.
Hyperpigmentation disorders
Familial progressive hyperpigmentation
- Zhang C, Deng Y, Chen X, et al. Linkage of a locus determining familial progressive hyperpigmentation (FPH) to chromosome 19q13.1-pter in a Chinese family. Eur J Dermatol 2006;16:246–50.
- Wang ZQ, Si L, Tang Q, et al. Gain-of-function mutation of KIT ligand on melanin synthesis causes familial progressive hyperpigmentation. Am J Hum Genet 2009;84:672–7. **
- Amyere M, Vogt T, Hoo J, et al. KITLG mutations cause familial progressive hyper- and hypopigmentation. J Invest Dermatol 2011;131:1234–9.
Incontinentia pigmenti
- Kenwrick S, Woffendin H, Jakins T, et al. Survival of male patients with incontinentia pigmenti carrying a lethal mutation can be explained by somatic mosaicism or Klinefelter syndrome. Am J Hum Genet 2001;69:1210–17.
- Landy SJ, Donnai D. Incontinentia pigmenti (Bloch-Sulzberger syndrome). J Med Genet 1993;30:53–9.
- Smahi A, Courtois G, Vabres P, et al. Genomic rearrangement in NEMO impairs NF-kappaB activation and is a cause of incontinentia pigmenti. The International Incontinentia Pigmenti (IP) Consortium. Nature 2000;405:466–72.
- Smahi A, Courtois G, Rabia SH, et al. The NF-kappaB signalling pathway in human diseases: from incontinentia pigmenti to ectodermal dysplasias and immune-deficiency syndromes. Hum Mol Genet 2002;11:2371–5.
- Steffann J, Raclin V, Smahi A, et al. A novel PCR approach for prenatal detection of the common NEMO rearrangement in incontinentia pigmenti. Prenat Diagn 2004;24:384–8.
- O'Doherty M, McCreery K, Green AJ, et al. Incontinentia pigmenti: ophthalmological observation of a series of cases and review of the literature. Br J Ophthalmol 2011;95:11–16.
Linear and whorled naevoid hypermelanosis
- Kalter DC, Griffiths WA, Atherton DJ. Linear and whorled nevoid hypermelanosis. J Am Acad Dermatol 1988;19:1037–44. **
- Hong SP, Ahn SY, Lee WS. Linear and whorled nevoid hypermelanosis: unique clinical presentations and their possible association with chromosomal abnormality inv(9). Arch Dermatol 2008;144:415–16.
Dyskeratosis congenita
- Calado RT, Young NS. Telomere maintenance and human bone marrow failure. Blood 2008;111:4446–55.
- Ding YG, Zhu TS, Jiang W, et al. Identification of a novel mutation and a de novo mutation in DKC1 in two Chinese pedigrees with dyskeratosis congenita. J Invest Dermatol 2004;123:470–3. **
- Nelson ND, Bertuch AA. Dyskeratosis congenita as a disorder of telomere maintenance. Mutat Res 2012;730:43–51.
- Mathew CG. Fanconi anaemia genes and susceptibility to cancer. Oncogene 2006;25:5875–84.
Naegeli–Franceschetti–Jadassohn syndrome and dermatopathia pigmentosa reticularis
- Lugassy J, Itin P, Ishida-Yamamoto A, et al. Naegeli–Franceschetti–Jadassohn syndrome and dermatopathia pigmentosa reticularis: two allelic ectodermal dysplasias caused by dominant mutations in KRT14. Am J Hum Genet 2006;79:724–30. **
- Lugassy J, McGrath JA, Itin P, et al. KRT14 haploinsufficiency results in increased susceptibility of keratinocytes to TNF-alpha-induced apoptosis and causes Naegeli–Franceschetti–Jadassohn syndrome. J Invest Dermatol 2008;128:1517–24.
- Itin PH, Lautenschlager S, Meyer R, Mevorah B, Rufli T. Natural history of the Naegeli–Franceschetti–Jadassohn syndrome and further delineation of its clinical manifestations. J Am Acad Dermatol 1993;28:942–50.
Dowling–Degos disease
- Dowling GB, Freudenthal W. Acanthosis nigricans. Br J Dermatol 1938;50:467–71.
- Jafari R, Tronnier M, Vakilzadeh F. Morbus Dowling–Degos in genitoperianal localisation in a mother and daughter. Akt Derm 2003;29:240–2.
- Milde P, Goerz G, Plewig G. Morbus Dowling–Degos mit ausschliessich genitaler Manifestation. Hautarzt 1992;43:369–72.
- Betz RC, Planko L, Eigelshoven S, et al. Loss-of-function mutations in the keratin 5 gene lead to Dowling–Degos disease. Am J Hum Genet 2006;78:510–19. **
- Li M, Cheng R, Liang J, et al. Mutations in POFUT1, encoding protein O-fucosyltransferase 1, cause generalized Dowling–Degos disease. Am J Hum Genet 2013;92:895–903.
- Sprecher E, Indelman M, Khamaysi Z, Lugassy J, Petronius D, Bergman R. Galli–Galli disease is an acantholytic variant of Dowling–Degos disease. Br J Dermatol 2007;156:572–4.
- Li M, Cheng R, Liang J, et al. Mutations in POFUT1, encoding protein O-fucosyltransferase 1, cause generalized Dowling–Degos disease. Am J Hum Genet 2013;92:895–903.
- Basmanav FB, Oprisoreanu AM, Pasternack SM, et al. Mutations in POGLUT1, encoding protein O-glucosyltransferase 1, cause autosomal-dominant Dowling–Degos disease. Am J Hum Genet 2014;94:135–43.
Reticulate acropigmentation of Kitamura
- Kitamura K, Akamatsu S, Hirokawa K. Eine besondere form der akropigmentation: acropigmentatio reticularis. Hautarzt 1953;4:152–6.
- Kitamura K. Acropigmentatio reticularis, eine Allgemein in der Welt vorkommende Krankheit. Hautarzt 1976;27:352–4.
- Griffiths WAD. Reticulate acropigmentation of Kitamura. Br J Dermatol 1976;95:437–43.
- Kono M, Sugiura K, Suganuma M, et al. Whole-exome sequencing identifies ADAM10 mutations as a cause of reticulate acropigmentation of Kitamura, a clinical entity distinct from Dowling–Degos disease. Hum Mol Genet 2013;22:3524–33. **
Peutz–Jeghers–Touraine syndrome
- Gruber SB, Entius MM, Petersen GM, et al. Pathogenesis of adenocarcinoma in Peutz-Jeghers syndrome. Cancer Res 1998;58:5267–70.
- Giardiello FM, Welsh SB, Hamilton SR, et al. Increased risk of cancer in the Peutz-Jeghers syndrome. New Engl J Med 1987;316:1511–14.
- Yamada K, Matsukawa A, Hori Y, Kukita A. Ultrastructural studies on pigmented macules of the Peutz-Jeghers syndrome. J Dermatol 1981;8:367–7.
- Jenne DE, Reimann H, Nezu J, et al. Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nature Genet 1998;18:38–43.
- Schumacher V, Vogel T, Leube B, et al. STK11 genotyping and cancer risk in Peutz-Jeghers syndrome. J Med Genet 2005;42:428–35.
Dyschromatoses
Dyschromatosis symmetrica hereditaria
- Komaya G. Symmetrische Pigmentanomalie der Extremitaeten. Arch Dermatol Syph 1924;147:389–93.
- Miyamura Y, Suzuki T, Kono M, et al. Mutations of the RNA-specific adenosine deaminase gene (DSRAD) are involved in dyschromatosis symmetrica hereditaria. Am J Hum Genet 2003;73:693–9. **
Dyschromatosis universalis hereditaria
- Stuhrmann M, Hennies HC, Bukhari IA, et al. Dyschromatosis universalis hereditaria: evidence for autosomal recessive inheritance and identification of a new locus on chromosome 12q21-q23. Clin Genet 2008;73:566–72.
- Miyamura Y, Suzuki T, Kono M, et al. Mutations of the RNA-specific adenosine deaminase gene (DSRAD) are involved in dyschromatosis symmetrica hereditaria. Am J Hum Genet 2003;73:693–9.
- Suzuki N, Suzuki T, Inagaki K, et al. Mutation analysis of the ADAR1 gene in dyschromatosis symmetrica hereditaria and genetic differentiation from both dyschromatosis universalis hereditaria and acropigmentatio reticularis. J Invest Dermatol 2005;124:1186–92.
- Xing QH, Wang MT, Chen XD, et al. A gene locus responsible for dyschromatosis symmetrica hereditaria (DSH) maps to chromosome 6q24.2-q25.2. Am J Hum Genet 2003;73:377–82.
- Bukhari IA, El-Harith EA, Stuhrmann M. Dyschromatosis universalis hereditaria as an autosomal recessive disease in five members of one family. J Eur Acad Dermatol Venereol 2006;18:628–9.
- Zhang C, Li D, Zhang J, et al. Mutations in ABCB6 cause dyschromatosis universalis hereditaria. J Invest Dermatol 2013;133:2221–8.