CHAPTER 78
DNA Repair Disorders with Cutaneous Features
Hiva Fassihi
St John’s Institute of Dermatology, Guy’s and St Thomas’ NHS Foundation Trust, London, UK
Introduction
The human genome is made up of about 3 billion DNA base pairs containing an estimated 30 000 protein-encoding genes. This DNA is continually being damaged by a variety of endogenous sources (such as reactive oxygen species) and exogenous sources (such as ultraviolet and ionizing radiation).
Cells have evolved a number of complex and effective systems, including nucleotide excision repair, double strand break repair and mismatch repair, to recognize and repair this damage in actively transcribed genes. Successful repair of damaged DNA is also dependent on the unzipping of the DNA double helix by enzymes of the helicase group.
Defects in these DNA repair pathways result in a number of disorders, many with skin involvement, commonly photosensitivity, cancer and premature ageing (Table 78.1).
Table 78.1 DNA repair disorders with cutaneous features.
| Disorder | DNA repair defect |
| Xeroderma pigmentosum | Nucleotide excision repair |
| Cockayne syndrome | Nucleotide excision repair |
| Cerebro-oculo-facio-skeletal syndrome | Nucleotide excision repair |
| UV-sensitive syndrome | Nucleotide excision repair |
| Trichothiodystrophy | Nucleotide excision repair |
| Rothmund–Thomson syndrome | Recombination Q helicase |
| Bloom syndrome | Recombination Q helicase |
| Werner syndrome | Recombination Q helicase |
| Ataxia telangiectasia | Double strand break repair |
| Fanconi anaemia | Interstrand cross-link repair |
| Muir–Torre syndrome | Mismatch repair |
Xeroderma pigmentosum
Definition
Xeroderma pigmentosum (XP) is a rare autosomal recessive disorder of DNA repair. It is characterized by progressive pigmentary changes at exposed sites, an increased risk of ultraviolet radiation (UVR) induced skin and mucous membrane cancers, severe photosensitivity in about 50%, and neurodegeneration in approximately 30% of affected cases. It is divided into eight complementation groups: XP-A to XP-G and XP-V (XP variant), corresponding to the affected DNA repair gene. There is wide variability in clinical features both between and within XP complementation groups [1].
Introduction and general description
Xeroderma pigmentosum is a rare disorder of DNA repair, which presents clinically with progressive pigmentary abnormalities, and an increased incidence of UVR-induced skin and mucous membrane cancers at sun-exposed sites. About half of affected individuals also show an exaggerated and prolonged sunburn response on minimal exposure. In a minority of cases, there is progressive neurological degeneration [2].
The initial report of this disorder was made by Hebra and Kaposi in 1874 [3] and the term xeroderma pigmentosum, meaning pigmented dry skin, was introduced in 1882 [4]. The first neurological problems were reported in 1883 by Neisser [5], and in 1932 De Santis and Cacchione reported three brothers with XP, with severe progressive neurological degeneration and associated dwarfism and gonadal hypoplasia, and called this De Sanctis–Cacchione syndrome [6]. This term is no longer in general use as it is now appreciated that XP can be associated with neurological problems of widely varying severity, and that De Sanctis–Cacchione syndrome is at the extreme end of a continuous spectrum.
DNA repair abnormalities in XP were reported by Cleaver in 1968. He discovered deficient excision repair in cultured skin fibroblasts from these patients [7]. UVR-induced DNA photoproducts were identified by Setlow and Setlow [8] and XP cells were found to be defective in the excision repair of these photoproducts in vivo [9]. The excision repair-proficient form of XP was first described in 1971 [10], named ‘variant’ by Cleaver in 1972 [11]. These ‘variant’ cultured fibroblasts were able to repair UVR-induced damage but were found to be defective in another DNA repair pathway, post-replication repair [12]. This subtype is now known as XP variant (XP-V).
Cell fusion studies by De Weerd-Kastelein in 1972 demonstrated molecular heterogeneity in XP [13]. The fusion of fibroblasts from different XP patients to form heterokaryons was found to correct the defect in DNA repair. This suggested that patients had different defects in nucleotide excision repair, and one defect could be corrected by the fusion of cells from a patient with a different defect because of the availability of the protein that the other was lacking. This led to the characterization of XP into different complementation groups (XP-A through to XP-G).
Epidemiology
Incidence and prevalence
Estimates from the 1970s suggested an incidence of XP in the USA of one in 250 000 [14] and in Japan of one in 80 000 [15]. A more recent survey in western Europe suggests an incidence of approximately 2.3 per million live births [16]. The incidence of XP in Japan is significantly higher than in western countries, with the majority of XP patients in Japan belonging to the XP-A complementation group (90% of patients are homozygous for the XPA founder mutation, carried by 1% of the Japanese population [17]).
The prevalence is higher in North Africa and the Middle East, especially in communities in which consanguinity is common. Amid Indian and Middle Eastern areas, the incidence is quoted at one per 10 000–30 000 [18–21]. In 2010, Soufir et al. reported that 85% of XP families in the Maghreb region (Algeria, Tunisia and Morocco) carried a founder mutation in the XPC gene [22]. More recently, it has been reported that one in 5000 individuals of the black Mahori population in the Comoro Islands have XP-C. This is linked to another founder mutation [23].
Sex
Xeroderma pigmentosum affects males and females equally.
Ethnicity
It has been reported to occur in all ethnic groups worldwide.
Pathophysiology
Xeroderma pigmentosum is an autosomal recessive disorder and results from mutations in any one of eight genes. The products of seven of these genes (XPA through to XPG) are involved in the recognition and repair of UVR-induced photoproducts in DNA (cyclobutane pyrimidine dimers (CPDs) and pyrimidine (6-4) pyrimidone photoproducts (6-4PPs)) by the process of nucleotide excision repair (NER) [24].
NER is made up of two pathways (Figure 78.1): global genome nucleotide excision repair (GG-NER) in which damage to DNA not undergoing transcription is repaired, and transcription-coupled nucleotide excision repair (TC-NER) in which damage in transcribed regions of DNA is rapidly repaired [25,26]. In GG-NER, the photoproducts are recognized by the proteins XPC and XPE. In TC-NER, these bulky lesions block the progress of RNA polymerase II in a process involving two proteins not involved in XP, CSA and CSB (Cockayne syndrome proteins A and B). Following this initial damage recognition, the pathways converge. XPB and XPD are part of a protein complex, TFIIH, which opens up the structure of the DNA around the site of the photoproduct. XPA protein verifies that the proteins are in the correct position and then the endonucleases XPG and XPF cut the DNA on either side of the damage, so that the damaged section, in a fragment of about 30 nucleotides, can be removed. The gap is filled in by de novo DNA synthesis and the new stretch of DNA is finally joined up to the pre-existing strand. This is referred to as unscheduled DNA synthesis (UDS).

Figure 78.1 In global genome nucleotide excision repair (GG-NER), XPE (with its partner protein DDB1) binds to the photoproduct and recruits another protein, XPC, which recognizes and binds to the strand opposite the photoproduct. In transcription-coupled NER (TC-NER), RNA polymerase II stalls at the site of the photoproduct. This then leads to the recruitment of CSA and CSB protein (defective in Cockayne syndrome and not XP). The two pathways then converge. TFIIH (a complex containing 10 peptides including the helicases XPB and XPD) then opens up the DNA and subsequently XPA binds to verify the correct positioning of all the proteins. The heterodimeric nucleases ERCC1/XPF and XPG then cleave the damaged DNA strand at the 5' to 3' ends, on either side of the photoproduct. The gap is filled in by using the undamaged DNA strand as template. This process is referred to as unscheduled DNA synthesis. (From Sethi et al. 2013 [26].)
Mutation(s) in any of the seven genes encoding for these proteins (XPA to XPG) result in abnormal NER and the different XP complementation groups (XP-A to XP-G, respectively) (Table 78.2) [27]. More specifically, mutations in the XPA, ERCC3 (XPB), ERCC2 (XPD), ERCC4 (XPF) and ERCC5 (XPG) genes result in defects in both GG-NER and TC-NER, however mutations in the XPC and DDB2 (XPE) genes only affect GG-NER and therefore some preservation of NER via the TC-NER pathway.
Table 78.2 The genes, chromosomal locations, protein functions and the main clinical features in different XP complementation groups.
| XP complementation group | Gene | Chromosomal location | Protein function | Defective pathway | Clinical features |
| XP-A | XPA | 9q22.23 | Damage verification | NER | Severe subtype Common in Japan Exaggerated sunburn Mild to severe neurological abnormalities |
| XP-B | ERCC3 (XPB) | 2q14.3 | Helicase Part of TFIIH |
NER | Extremely rare subtype Exaggerated sunburn Mild neurological abnormalities (Mutations in XPB can also result in XP/CS and TTD) |
| XP-C | XPC | 3p25.1 | Damage recognition | NER (GG-NER) | Most common subtype No abnormal sunburn reaction Severely atypical and dense lentigines at exposed sites Severe ocular surface disease No neurological abnormalities |
| XP-D | ERCC2 (XPD) | 19q13.32 | Helicase Part of TFIIH |
NER | Severe exaggerated sunburn No neurological to severe neurological abnormalities (Mutations in XPD can also result in XP/CS and TTD) |
| XP-E | DDB2 (XPE) | 11p11.2 | Damage recognition | NER (GG-NER) | Extremely rare subtype No abnormal sunburn reaction Lentigines not prominent Large number of skin cancers from thirties onwards No neurological abnormalities |
| XP-F | ERCC4 (XPF) | 16p13.12 | Nuclease | NER | Mildly exaggerated sunburn Skin signs tend to be mild No neurological to severe neurological abnormalities (Mutations in XPF can also result in XP/CS) |
| XP-G | ERCC5 (XPG) | 13q33.1 | Nuclease | NER | Extremely rare subtype Exaggerated sunburn No neurological to severe neurological abnormalities (Mutations in XPG can also results in XP/CS) |
| XP-V | POLH (XPV) | 6p21.1 | Polymerase | TLS | No abnormal sunburn reaction Diagnosed in thirties to forties after multiple skin cancers No ocular disease No neurological abnormalities |
Adapted from Lehmann et al. 2011 [27].
GG-NER: global genome nucleotide excision repair subpathway; NER, nucleotide excision repair; TLS, translesion synthesis; TTD: trichothiodystrophy; XP/CS: xeroderma pigmentosum/Cockayne syndrome complex.
Defects in the eighth XP gene do not affect NER. About 20% of patients with XP, the so-called XP variant (XP-V), have problems replicating DNA containing UVR-induced damage [28]. They have defects in POLH (or XPV) gene, which encodes for DNA polymerase η, one of the specialized enzymes required for replication beyond the damaged sites (translesion synthesis) [12, 29].
Clinical features
History and presentation
Patients with XP are a clinically heterogeneous group with a wide variability in clinical features both between and within XP complementation groups (Table 78.2). The clinical manifestations, severity of disease and age of onset are in part dependent on the cumulative UVR exposure, the complementation group and the precise nature of the pathogenic mutation(s).
Exaggerated sunburn and pigmentary changes
The skin is normal at birth. The changes in XP are the result of exposure to UVR, therefore the severity of these changes is totally dependent on the amount of sun exposure and the degree of UVR protection. Delayed diagnosis and poor sun protection will exacerbate the cutaneous features, resulting in significant pigmentary changes, multiple skin cancers and a worse prognosis.
Acute and severe sunburn on minimal sun exposure (Figure 78.2a), taking weeks to resolve, was once considered a cardinal presenting feature of XP. However, it is now known that only 50% of XP patients suffer from severe and prolonged sunburn reactions [30], which have been blamed on neglect or labelled wrongly as impetigo in many cases. The remaining 50% have sunburn reactions that are normal for skin type and present with lentigines as well as hypopigmented macules (Figure 78.2b) at sun-exposed sites, initially on the face and dorsal aspect of the hands and later on other exposed parts. The lentigines are fixed and progress over time to become more dense and irregular.
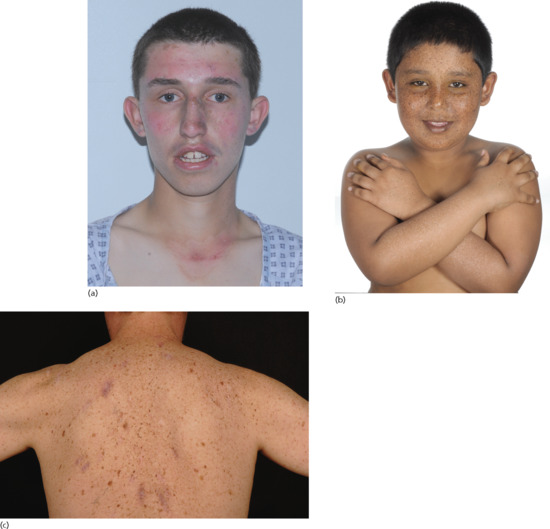
Figure 78.2 Xeroderma pigmentosum. (a) Severe and exaggerated sunburn on minimal sun exposure. (b) Lentigines and hypopigmented macules (seen on the forearms) at sun-exposed sites. (c) Pigmentary change and multiple surgical scars at sites of previous skin cancers.
Patients in complementation groups XP-A, XP-B, XP-D, XP-F and XP-G suffer from severe sunburn reactions, whereas those in groups XP-C, XP-E and XP-V have normal sunburn reactions for skin type [30]. XPA, XPB, XPD, XPF and XPG proteins are all required for the common stem of NER, whereas XPC and XPE proteins are only required for GG-NER. In XP-C and XP-E, the TC-NER pathway remains active, and so does the ability to selectively remove photoproducts from the transcribed strand of active genes with kinetics similar to those observed in normal cells [31]. XP-V patients with mutations affecting DNA polymerase η, also have normal functioning TC-NER (and GG-NER). It is therefore hypothesized that normal sunburn reactions in approximately half of the XP patients relate to the preservation of TC-NER [32] and this is supported by XPC knock-out mice model studies [33, 34].
Skin cancer
Absorption of UVR by DNA results in the formation of photoproducts (CPDs and 6-4PPs) that are recognized and repaired by NER. If left unrepaired, they can result in the classic ‘UVB signature’ mutations found in skin cancers [35, 36]. The molecular defects in XP cells result in abnormal recognition and repair of UVR-induced DNA damage, and a subsequent significantly increased induction of ‘UVB signature’ mutations in the sun-exposed skin of affected individuals. This increased mutation frequency is likely to account for the pigmentary changes and the skin cancers in XP patients [37]. Indeed, examination of mutations in the p53 gene in tumours from XP patients revealed mutations characteristic of UVR exposure in the majority of tumours [38].
XP patients have an over 10 000-fold increased risk of developing non-melanoma skin cancer (NMSC) and a 2000-fold increased risk of melanoma skin cancer under 20 years old [39]. The median age at diagnosis of first NMSC is 9 years, and the median age at diagnosis of first melanoma is 22 years [39]. UVR exposure to the oral cavity in XP patients can result in mucocutaneous malignancy, most commonly seen as squamous cell carcinoma of the tip of the tongue. In contrast to patients with XP complementation group XP-A to XP-G, who generally develop skin cancer before the age of 20, XP-V patients (Figure 78.2c) develop their first skin cancers in the second or third decade of life.
XP patient who have severe and exaggerated sunburn reactions generally have a lower frequency of skin cancer than those patients with sunburn reactions that are normal for skin type. This may be due to severe sunburn reactions prompting earlier diagnosis and an earlier age of initiating more rigorous photoprotection, therefore resulting in less cumulative UVR exposure.
Other malignancies
DNA damage caused by carcinogens in cigarette smoke (chemical compounds such as benzopyrene) is repaired by NER in normal cells [40] and therefore patients with XP are potentially at higher risk of smoking-induced lung cancers.
XP patients also have an approximately 50-fold increase in cancers of the brain, including medulloblastoma, glioblastoma, spinal cord astrocytomas [41] and schwannoma. These malignancies are not UVR related and the precise relationship to DNA damage is unknown. High-dose X-ray irradiation can be used for the treatment of some of these brain tumours as the pathways involved in the repair of DNA damage by X-rays are normal in patients with XP [41, 42].
Ocular manifestations
UVR exposure resulting in DNA damage of the eyelids and periocular skin can result in the development of cicatricial skin changes as well as skin cancers that require excision [43]. The ocular surface (conjunctiva and cornea) can develop UVR-related damage including dry eye, conjunctival injection and inflammation, as well as the development of premature pingueculae and pterygia. Prolonged corneal exposure can result in corneal scarring and visual impairment. Ocular surface cancers, mainly squamous cell carcinomas, have also been reported in patients with significant UVR exposure and poor ocular photoprotection [44]. Patients with XP-related neurodegeneration may also develop neuro-ophthalmological features, including sluggish pupils, nystagmus and strabismus. Photophobia is also common and often the earliest presenting ophthalmic symptom in XP.
Neurodegeneration
Approximately 30% of XP patients will develop neurodegeneration. The age at onset and rate of progression of the neurological abnormalities is variable between and within different complementation groups. Patients with XP-A, XP-B, XP-D, XP-F and XP-G have an increased susceptibility [1,30,32,39]. XP-C, XP-E and XP-V patients have not been reported to develop clinically detectable neurodegeneration.
The pathogenesis of the neurological abnormalities is poorly understood. It is not related to UVR exposure. Current theories suggest that oxidative DNA damage is generated during normal metabolism in the central nervous system, and that some of this damage, such as the generation of 8,5-cyclopurine-2-deoxynucleosides, is repaired by NER [45]. In the absence of functional repair, the lesions persist and result in neuronal cell death.
Neurological manifestations of XP typically present after the skin signs in the natural history of the disease and do not arise before the age of 2 years [46]. XP patients are born normal. Parents notice mild cognitive impairment first, usually when the child is starting school. Cerebellar signs manifest usually between 4 and 16 years of age, commonly dysarthria and difficulties with balance. Ataxia and areflexia follow. Nerve conduction studies show evidence of axonal sensory and motor neuropathy although this is not usually seen before the second decade of life. Patients develop progressive microcephaly. Magnetic resonance imaging demonstrates atrophy of the cortex of the brain with concomitant dilatation of the ventricles, and a secondary thickening of the skull bones. Most XP patients with neurological abnormalities will also develop sensorineural deafness, and the degree of hearing loss has been shown to predict future neurological involvement [32]. Patients eventually become wheelchair- and then bed-bound, a few years before death [46]. The presence of neurological abnormalities is associated with a worse prognosis.
Psychological morbidity
The extreme sun protection and UVR-protective clothing restrictions (such as a facial visor) in many patients with XP can result in social isolation and inevitably predispose these patients to low mood, depression and anxiety. Compliance with extreme sun protection is also an issue.
Other psychological issues include the potential for developing anxiety and depression from the high risk of potentially fatal skin cancers, ongoing surgical procedures for skin cancers, many on the face, with associated disfigurement, and the possibility of neurodegeneration in some complementation groups.
Clinical variants
Xeroderma pigmentosum variant (XP-V).
Approximately 20% of XP patients have XP-V. This subtype is caused by mutations in the POLH (XPV) gene encoding DNA polymerase η, one of the specialized enzymes required for replication past UVR-damaged sites [12, 28]. The NER pathway is normal. XP-V is often diagnosed after the age of 30, once the patient has development multiple skin cancers. No neurological manifestations are observed.
Xeroderma pigmentosum/Cockayne syndrome complex (XP/CS).
This is a very rare autosomal recessive disorder characterized by cutaneous features of XP together with systemic and neurological features of CS such as short stature, microcephaly, cachexia, abnormal development, photosensitivity, premature ageing, retinal degeneration, sensorineural deafness and progressive neurological dysfunction (see the section on Cockayne syndrome). In contrast to the neurological abnormalities in XP, which are predominantly secondary to neuronal degeneration, in the XP/CS complex dysmyelination typical of CS is observed. Mutations in ERCC3 (XPB), ERCC2 (XPD), ERCC4 (XPF) and ERCC5 (XPG) genes have been reported in the XP/CS complex.
Xeroderma pigmentosum/trichothiodystrophy (XP/TTD) syndrome.
The XP/TTD syndrome, with mutations reported in the ERCC3 (XPB) and ERCC2 (XPD) genes, is characterized by phenotypic features of TTD such as sulphur-deficient hair with ‘‘tiger-tail’ banding of hair shafts, developmental delay/intellectual impairment, short stature and ichthyosis (see the section on trichothiodystrophy), with clinical and cellular findings of XP.
Differential diagnosis
Differential diagnoses include trichothiodystrophy, Cockayne syndrome, cerebro-oculo-facio-skeletal syndrome, UV-sensitive syndrome, erythropoietic protoporphyria and Rothmund–Thomson syndrome.
Complications and co-morbidities
The main complications of XP are related to the high risk of mucocutaneous cancers at sun-exposed sites and the possibility of neurodegeneration in some complementation groups.
Disease course and prognosis
There is no cure for XP. The overall median age of death is reported as 32 years [39], with skin cancer and neurodegeneration the main causes of death. Sun avoidance and regular follow-up to assess and treat any skin cancers increases life expectancy. For those without neurological disease and rigorous UVR protection, the prognosis is good. However, the neurological abnormalities are progressive and the median age at death in these patients (29 years) is significantly younger than in XP patients without neurological degeneration (37 years) [39].
Investigations
In most cases, a clinical diagnosis can be made on the presence of extreme and exaggerated sunburn reactions in those individuals who show this feature, or on the appearance and progressive development of lentigines on the face and other exposed site from an early age.
The diagnosis can be confirmed definitively by cellular tests for defective DNA repair. The most commonly used test is the measurement of unscheduled DNA synthesis in cultured skin fibroblasts. After UVR-induced DNA damage has been removed by NER, a patch of newly synthesized DNA replaces the damaged section. Synthesis of this new DNA is therefore referred to as unscheduled DNA synthesis (UDS) (see Figure 78.1). Skin fibroblast cultures are established from a 4 mm punch biopsy taken from an unexposed area of the skin. Fibroblasts are UV irradiated in a Petri dish, and UDS can be measured as incorporation of nucleotides into DNA of the irradiated cells either by autoradiography [47] or liquid scintillation counting [48], or more recently using a fluorescence assay [49]. A reduced level of UDS confirms the diagnosis of XP.
XP-V patients do not show this defect in UDS, as NER is unaffected. Furthermore XP-V cells are not hypersensitive to killing by UVR. However, it has been found empirically that caffeine specifically sensitizes XP-V cells to killing by UVR [50]. To diagnose XP-V cells, cultures are exposed to UVR, incubated in caffeine for a few days and their viability compared with that of normal cells. Specific sensitivity to UVR in the presence of caffeine together with normal UDS confirms the diagnosis of XP-V [51].
Subsequently, analysis of DNA, extracted from a blood sample, can identify the defective gene (confirming the complementation group) [52] and the causative mutation(s) in patients. This can give further insight into genotype–phenotype correlations and enable genetic counselling and prenatal testing if requested.
Management
The mainstay of management is the avoidance of UVR exposure from sunlight through the application of high-factor sunscreen, UVR-protective clothing, hats, gloves and sunglasses, and UVR-blocking window films (at home and in cars). Fluorescent light sources also emit UVR and should be avoided or covered in UVR filter film. Vitamin D deficiency is common and supplements should be prescribed. Smoking is prohibited. Retinoids may have a role in the prevention of skin cancer [53, 54].
Management requires a multidisciplinary approach. Regular skin and eye review and appropriate and early management of any cancers is essential. Topical 5-fluorouracil and imiquimod may be useful for early or premalignant lesions. Photodynamic therapy should not be used as the irradiation involved is likely to result in further skin damage and carcinogenesis [55]. The eyes may need to be treated with lubricating drops.
Patients should be given appropriate genetic counselling. Psychosocial issues including social isolation from peers at school and at home, limited career prospects and the impact of meticulous sun protection on the quality of life need to be addressed.
In vitro and ex vivo experiments have established that correction of the underlying genetic defect in different forms of XP is possible. Animal studies using viral vectors have also established that gene therapy approaches for patients with this disease may become possible [56, 57].
Resources
Further information
UK National Xeroderma Pigmentosum Service: www.guysandstthomas.nhs.uk/xp (last accessed November 2014).
Patient resources
Enfants de la lune: http://www.enfantsdelalune.org.
Xeroderma Pigmentosum Society: http://www.xps.org/.
XP Family Support Group: http://www.xpfamilysupport.org.
XP Freu(n)de: http://www.xerodermapigmentosum.de.
XP Support Group: http://xpsupportgroup.org.uk/.
(All last accessed November 2014.)
Cockayne syndrome
Definition
Cockayne syndrome (CS) is a rare autosomal recessive disorder of DNA repair. It is characterized by short stature, photosensitivity, a distinctive facial appearance, ocular defects, premature ageing and progressive neurological dysfunction associated with extensive demyelination.
Introduction and general description
CS is a rare disorder of DNA repair first described by Cockayne in 1936 [1]. The disease is defined by progressive postnatal growth failure, short stature, microcephaly, cachexia, abnormal development, photosensitivity, premature ageing, retinal degeneration and sensorineural deafness [2]. Patients have a characteristic facial appearance with enophthalmia. CS is divided into three clinical types (types I, II and III) based on severity of disease.
Epidemiology
The annual incidence of CS is about one in 200 000 in European countries.
Age
Photosensitivity is present from birth and the abnormal growth and development becomes evident within the first few years of life.
Sex
It affects males and females equally.
Ethnicity
CS has been reported to occur in many ethnic groups worldwide.
Pathophysiology
CS is an autosomal recessive disorder and results from mutations in one of two genes, ERCC8 (CSA) and ERCC6 (CSB). The products of these genes are involved in NER (see Figure 78.1) [3].
The skin fibroblasts of patients with CS are abnormally sensitive to UVR [4–6]. Unlike cells from patient with XP, the GG-NER process occurs normally in CS cells. However, CS cells are defective in the important transcription-coupled subpathway of NER (see Figure 78.1). Following DNA damage, it is of prime importance for the cell to remove damage from actively transcribed regions of DNA. This preferential repair is referred to as transcription-coupled NER (TC-NER), and it is this repair that is specifically defective in CS cells [7].
The associated failure of cells to restore normal levels of RNA synthesis after UVR exposure [8] has provided a means for carrying out complementation tests on CS cells. Two complementation groups have been identified, CS-A (accounting for approximately 20% of CS patients) and CS-B (Table 78.3).
Table 78.3 Genes, protein products and main clinical features in different Cockayne syndrome (CS) complementation groups and associated diseases.
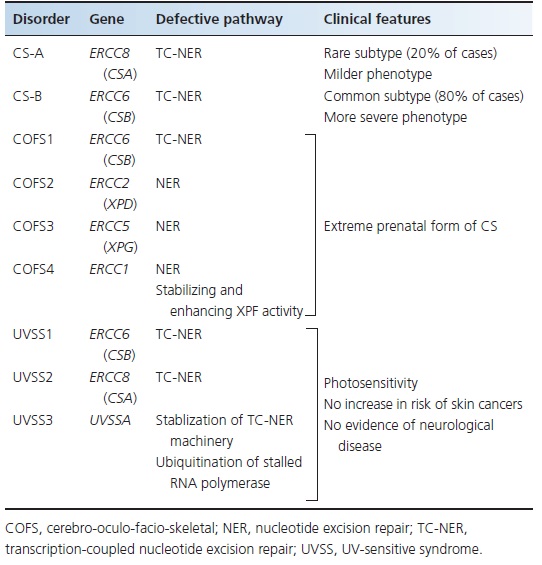
CS-A is caused by mutation in the ERCC8 gene and CS-B is caused by mutation in the ERCC6 gene [9, 10]. These genes encode proteins involved in TC-NER [10]. Among patients with CS, approximately 80% have mutations in the ERCC6 gene [11,12]. So far, no correlation has been found between the three clinical subtypes (I, II, III) of CS and the genes involved.
CS can rarely be found in combination with XP. Mutations in the XPB, XPD, XPF and XPG genes have been implicated in XP/CS complex.
Clinical features
History and presentation
CS is characterized by cutaneous photosensitivity (a cardinal symptom), progressive postnatal growth failure, short stature, microcephaly, characteristic bird-like facies (Figure 78.3) (prognathism, enophthalmia, a prominent thin nose, large ears and loss of subcutaneous fat), disproportionately large hands and feet, cachexia, premature ageing and dental caries. The skin is dry and thin, and the hair is often sparse and is sometimes prematurely grey [2,13, 14]. In contrast to XP, patients with CS have no increased incidence of skin cancer [2].
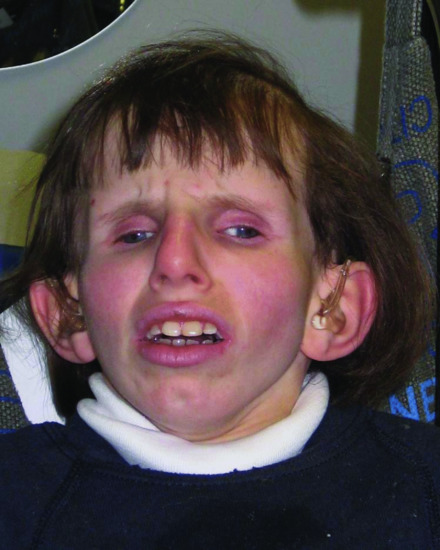
Figure 78.3 Cockayne syndrome demonstrating the characteristic bird-like facies with prominent enophthalmia.
Neurological features comprise of extensive demyelination of the peripheral and central nervous system, microcephaly, progressive cognitive decline, choreoathetosis, hydrocephalus and spasticity. Ophthalmological problems include retinal degeneration, cataracts and optic atrophy leading to loss of vision [15]. There is progressive sensorineural deafness and skeletal abnormalities with flexion deformity [16].
The age of onset and severity of disease are variable. In classic CS type I, the first symptoms usually appear during the first year of life. Type II refers to early-onset cases with more severe symptoms and type III are late-onset cases with milder symptoms. In general, CS-A is milder in clinical course than CS-B, but within each type, no clear genotype–phenotype correlation has been established [17,18, 19].
Clinical variants
Cerebro-oculo-facio-skeletal syndrome.
Cerebro-oculo-facio-skeletal (COFS) syndrome is a very rare autosomal recessive disorder of DNA repair and constitutes the prenatal extreme form of CS [20, 21]. Clinically it is characterized by congenital microcephaly, congenital cataract and/or microphthalmia, arthrogryposis, severe developmental delay, growth delay and facial dysmorphism (prominent metopic suture, micrognathism). The axial hypotonia contrasts with the peripheral hypertonia and is associated with feeding difficulties. Peripheral neuropathy, sensorineural hearing loss and pigmentary retinopathy can be observed. In COFS syndrome, photosensitivity is an inconstant feature [22]. Prenatal diagnosis can be suspected by the presence of cataracts, arthrogryposis and microcephaly, and is subsequently confirmed by examination of DNA repair in the chorionic villi or amniotic cells, or by checking for the familial mutation(s) if known.
In COFS, mutations have been identified in the ERCC6, ERCC2 (XPD), ERCC5 (XPG) and ERCC1 genes (see Table 78.3), designated as COFS1, COFS2, COFS3 and COFS4 syndromes respectively [23–26]. All these genes encode for proteins involved in NER (see Figure 78.1).
COFS syndrome is a severe disease with death in the first years of life, usually from respiratory tract infections [21].
UV-sensitive syndrome.
UV-sensitive syndrome (UVSS) is an autosomal recessive disorder of DNA repair characterized by sensitivity to UVR but without any increased risk of skin cancers or any evidence of neurological disease [27]. Despite this mild clinical phenotype, the cellular and biochemical responses of UVSS and CS cells to UVR are indistinguishable, and result from defective TC-NER of photoproducts in actively transcribed genes [28]. Mutations in the CS genes, ERCC6 and ERCC8, have been reported in UVSS cases, designated UVSS1 and UVSS2 respectively [29, 30]. More recently mutations in the UVSSA gene have been found in cases designated UVSS3 (see Table 78.3). The UVSSA gene encodes for a protein that interacts with the TC-NER machinery and stabilizes the CSB complex, and also facilitates ubiquitination of RNA polymerase stalled at DNA damage sites [31]. The mild phenotype and lack of neurological disease in these patients is not fully understood but may be associated with normal repair of oxidative damage in these patients [29].
Differential diagnosis
Differential diagnoses include progeria, XP, COFS syndrome, UVSS, Rothmund–Thomson syndrome, Werner syndrome, Bloom syndrome and Hartnup disease.
Classification of severity
CS is clinically classified into types I, II and III based on disease severity and the age of onset of symptoms and signs.
Disease course and prognosis
Patient with CS type I commonly die before the end of the second decade as a result of progressive neurological degeneration. Patients with type II have a worse prognosis, whereas patients with type III often survive into adulthood [32]. There is no cure for CS. Survival beyond the second decade is unusual and the mean age at death in reported cases is 12.5 years.
Investigations
A clinical diagnosis can be made based on the presence of short stature, photosensitivity, distinctive facial appearance, ocular defects and premature ageing. Early cerebral imagery is not very specific, but it can reveal cerebral and cerebellar atrophy. Diffuse myelinization anomalies and calcifications of the basal ganglia can appear secondarily.
The clinical diagnosis can be confirmed definitively by cellular tests for defective DNA repair, specifically TC-NER. A skin biopsy should be taken for fibroblast culture as in patients with XP. CS cells are specifically deficient in TC-NER, which results in a prolonged inhibition of RNA synthesis following UVR. The test for CS therefore involves ultraviolet irradiation of cells, followed by measurement of RNA synthesis 24 h later. In normal cells, RNA synthesis will have recovered to untreated levels, whereas in CS cells it remains depressed [8]. In CS cells there is no abnormality of UDS as seen in XP.
Subsequently, analysis of DNA extracted from the blood can identify the defective gene and the causative mutation(s) in patients [9, 10–12]. This can enable genetic counseling and prenatal testing if requested.
Management
Management is purely supportive and requires a multidisciplinary approach with input from clinical genetics, ophthalmology, neurology, dermatology, audiology (for hearing aids) and physiotherapy. Psychosocial issues for the patient and their family need to be addressed.
Patients should avoid UVR exposure because of their increased photosensitivity, therefore vitamin D deficiency is common and supplements should be given. There is no increased risk of skin cancers.
Patients should be given appropriate genetic counselling. Prenatal diagnosis is possible by amniocentesis or from chorionic villous samples [33, 34], using the RNA synthesis recovery test or by looking for causative familial mutation(s), if known.
Resources
Patient resources
Amy and Friends – Cockayne Syndrome Support: www.amyandfriends.org/.
Share & Care, Cockayne Syndrome Network: www.cockaynesyndrome.net/.
(Both last accessed November 2014.)
Trichothiodystrophy
Definition
Trichothiodystrophy (TTD) is a rare autosomal recessive disorder of DNA repair. It is a multisystem disorder characterized by short, brittle, sulphur-deficient hair with a pattern of alternating light and dark ‘tiger-tail’ bands under polarized light microscopy, along with photosensitivity, ichthyosis, developmental delay and short stature.
Introduction and general description
Trichothiodystrophy is a rare disorder of DNA repair that is characterized by sulphur-deficient hair with alternating dark and light banding on polarized light microscopy [1–4]. Other clinical features include photosensitivity in about half of the affected individuals, ichthyosis, developmental delay, short stature, haematological abnormalities, skeletal abnormalities and maternal pregnancy complications [5].
The term trichothiodystrophy was first introduced by Price et al. in 1979 and soon after they reported two patients with suphur-deficient, brittle hair and a variety of other features [6]. Since then many variants of TTD based on the different clinical associations have been proposed, including BIDS syndrome (TTD type D or Amish brittle hair syndrome), IBIDS syndrome (TTD type E or Tay syndrome or), PIBIDS syndrome (TTD type F), Sabinas syndrome (TTD type B), SIBIDS syndrome, ONMRS (Itin syndrome) and Pollitt syndrome (TTD type C) [7, 8, 9, 10]. However, a recent comprehensive review of 112 TTD patients reported in the literature demonstrated that many patients did not fit into these designated subtypes and that these acronyms were poor descriptors of TTD patients’ clinical manifestations [5]. These acronyms should therefore no longer be used, and TTD is now divided in photosensitive and non-photosensitive subtypes.
Epidemiology
Incidence and prevalence
The incidence of TTD is estimated as one per million live births.
Sex
It affects males and females equally.
Ethnicity
It has been reported to occur in many ethnic groups worldwide although it is more common in populations where consanguinity is frequent.
Pathophysiology
Trichothiodystrophy is an autosomal recessive disorder. Photosensitive TTD results from mutations in the DNA repair genes ERCC2 (XPD) [11], ERCC3 (XPB) [12] or GTF2H5 [13, 14]. Most patients carry mutations in the ERCC2 (XPD) gene. These three genes encode the XPD, XPB and p8/TTDA subunits, respectively, of the TFIIH complex in the NER pathway (see Figure 78.1) [15]. Mutations in these subunits can affect both DNA repair and transcription. Mutations in a gene of unknown function, MPLKIP (TTDN1), have been found in a few cases of non-photosensitive TTD [16, 17].
Although mutations in ERCC2 (XPD) and ERCC3 (XPB) genes are also found in XP, a disorder of DNA repair associated with a 10 000 fold increased risk of skin cancer, TTD patients have not been reported to have an increased risk of cancer. This is thought to be because the mutations in these genes in patients with XP predominantly affect DNA repair, while mutations in the same genes in patients with TTD predominantly affect transcription [5,18]. Therefore, the clinical features in XP are related to an abnormal repair of UVR-induced DNA damage in the skin, whereas TTD is primarily a disorder of development, related to transcriptional abnormalities. This would explain the multisystem involvement in TTD, particularly the fetal developmental abnormalities and the haematological features.
Clinical features
History and presentation
Trichothiodystrophy is characterized by short, brittle, sulphur-deficient hair with a pattern of alternating light and dark bands under polarized light microscopy (Figure 78.4a, b) [4]. About 50% have photosensitivity with severe and exaggerated sunburn reactions. However, unlike patients with XP, there is no increased risk of skin cancer. Other cutaneous features include ichthyosis (Figure 78.4c), dry skin and collodion membrane at birth. The nails may be brittle and demonstrate onychodystrophy. Pregnancy complications including intrauterine growth retardation, pre-eclampsia and eclampsia, low birth weight (<2500 g) and premature birth (before 37 weeks' gestation) occur in about 30% of cases [19, 20]. Other common features are developmental delay/intellectual impairment (85% of cases), short stature (73%) and facial dysmorphism with microcephaly, large ears and micrognathia. Neuroimaging shows dysmyelination and cerebral atrophy. Ocular abnormalities occur in about 50% of cases and include congenital cataracts, nystagmus and strabismus [21]. Patients gradually develop joint abnormalities with contractures and dislocations. Skeletal abnormalities include axial osteosclerosis and distal osteopenia. Many patients are anaemic and neutropenic and show haematological features of β-thalassaemia trait [22]. Severe and recurrent infections, especially respiratory, occur in the first year of life.
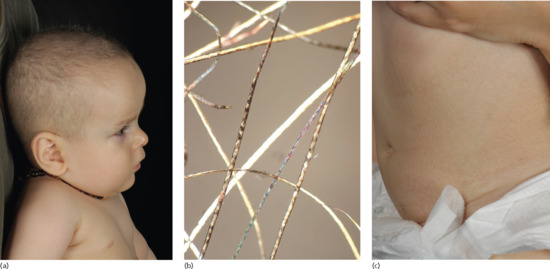
Figure 78.4 Trichothiodystrophy. (a) Short, brittle, sulphur-deficient hair. (b) The pattern of alternating light and dark ‘tiger-tail’ bands under polarized light microscopy. (c) Ichthyosis on the trunk.
The spectrum of clinical features varies from mild disease with only hair involvement to severe disease with profound developmental defects and recurrent infections [5,23]. The abnormalities at birth and during pregnancy suggest that the pathophysiology of TTD involves a developmental abnormality affecting pregnancy as well as transcriptional abnormalities after birth.
Clinical variants
Xeroderma pigmentosum/trichothiodystrophy (XP/TTD) syndrome.
XP/TTD syndrome with mutations reported in the ERCC2 (XPD) and ERCC3 (XPB) genes is characterized by phenotypic features of TTD with clinical and cellular findings of XP.
Differential diagnosis
Differential diagnoses include CS, XP and congenital alopecias, especially Menkes disease and Netherton syndrome.
Complications and co-morbidities
The main complications of TTD are related to the high risk of developing severe and potentially fatal infections.
Disease course and prognosis
There is no cure for TTD. There is significant morbidity and mortality in the neonatal and childhood years. The overall median age of death is reported as 3 years [5], with pneumonia and other infections (especially sepsis) being the main causes of death.
Investigations
A clinical diagnosis can be made on the presence of ‘tiger-tail’ bands of hair shafts under polarized light microscopy as well as the other clinical features.
The diagnosis can be confirmed definitively by cellular tests for defective DNA repair. Although the clinical features of TTD and XP are quite different, TTD cells, like XP cells, are defective in NER and this defect can be measured using UDS (see the section on XP investigation earlier in this chapter). A reduced level of UDS confirms the diagnosis of TTD.
Subsequent analysis of DNA extracted from the blood can identify the defective gene and the causative mutation(s) in patients. This can give further insight into genotype–phenotype correlations and enable genetic counseling and prenatal testing if requested.
Management
Management requires a multidisciplinary approach with input from obstetrics, paediatrics, genetics, ophthalmology, neurology, orthopaedics, infectious diseases and radiology. Patients and their families should be given appropriate genetic counselling and prenatal diagnosis offered if required.
There is no cure. Any infections should be aggressively treated as mortality from infections is significant (20-fold higher compared with the US population) [5]. Sun protection advice, in those patients who are photosensitive, should be given.
OTHER DISORDERS
Ataxia telangiectasia (Louis–Bar syndrome)
Ataxia telangiectasia (AT) is a rare autosomal recessive disorder of DNA repair. It was first reported by Denise Louis-Bar in 1941, but it was Boder and Sedgwick who introduced the term ataxia telangiectasia in 1958 [1]. It is a multisystem disorder characterized by ataxia and mucocutaneous telangiectasia. Telangiectasia initially appears in the conjunctiva and is most prominent in the facial areas. Additional cutaneous features include premature hair greying, café-au-lait spot and pigmentary changes, including poikiloderma. Progressive cerebellar degeneration is the first clinical manifestation in AT, starting at about 1 year of age. Immunodeficiency with increased susceptibility to infections, hypogonadism, sensitivity to ionizing radiation, insulin resistance and a predisposition to cancer are cardinal features in AT [2, 3].
AT is a chromosome instability disorder with inactivating mutations in the ATM gene. The ATM protein kinase plays a key role in the control of double strand break DNA repair [4, 5]. It is also involved in cell cycle regulation and telomere length maintenance. Management is symptomatic and involves a multidisciplinary approach with input from physiotherapy, speech therapy, neurology and early treatment of infections. As the cells of AT patients are sensitive to ionizing radiation, X-rays and radiotherapy should be used with caution. The prognosis is poor because of severe respiratory infections, progressive neurodegeneration and an increased risk of cancer.
Fanconi anaemia
Fanconi anaemia is an autosomal recessive disorder of DNA repair. It was first described in 1972 by the Swiss paediatrician, Guido Fanconi. It is clinically heterogeneous and characterized by congenital developmental defects, early-onset bone marrow failure and a high predisposition to cancer. There is cellular hypersensitivity to DNA interstrand cross-link agents such as mitomycin C [1]. Two-thirds of patients are born with congenital malformations of the kidneys, heart and skeleton (absent or abnormal thumbs and radii). Other features include a typical facial appearance with small head, eyes and mouth, hearing loss, hypogonadism and reduced fertility. Cutaneous abnormalities include reticulate or patchy hyper- or hypopigmentation and café-au-lait spots. Bone marrow failure usually presents in the first decade of life. Subsequently, patients develop tumours such as lymphomas, oesophageal carcinomas, squamous cell carcinomas of the head and neck, liver and brain tumours, and acute myeloid leukaemia [2].
So far mutations in at least 15 FANC genes, representing 15 Fanconi anaemia complementation groups, have been indentified. These gene products make up the ‘FA pathway’ involved in DNA repair, interstrand cross-link repair and maintenance of genome stability [3]. Diagnosis is based on the evaluation of chromosomal breakage induced by alkylating agents such as mitomycin C. Bone marrow failure and cancers results in a poor prognosis and the life expectancy of patients is reduced to an average of 20 years [2]. Management involves supportive care with transfusions as necessary. Haematopoietic stem cell transplant is curative for the haematological features but this increases the risk of solid tumours, which must be monitored.
Muir–Torre syndrome
Muir–Torre syndrome is a rare autosomal dominant disorder of DNA repair. It was independently reported by Muir in 1967 and Torre in 1968 [1, 2]. There is a lot of clinical variation but it is characterized by the occurrence of sebaceous gland neoplasms and/or keratoacanthomas associated with one or more visceral malignancies, in particular gastrointestinal or genito-urinary [3–5]. Although the malignancies are often multiple, they behave less aggressively and are often low grade. M–Torre syndrome occurs as a result of a mutation in one of the DNA mismatch repair genes (MSH-2, MLH-1 and more recently MSH-6). The management involves a multidisciplinary approach with genetic counselling, regular skin reviews and appropriate cancer screening.
References
Xeroderma pigmentosum
- Fassihi H. Spotlight on ‘xeroderma pigmentosum’. Photochem Photobiol Sci 2013;12:78–84.
- Kraemer KH, Lee MM, Scotto J. Xeroderma pigmentosum. Cutaneous, ocular, and neurologic abnormalities in 830 published cases. Arch Dermatol 1987;123:241–50.
- Hebra F, Kaposi M. On Diseases of the Skin Including the Exanthemata, Vol. 3 (translated by W. Tay). London: New Sydenham Society, 1874;61:252–8.
- Kaposi M. [Xeroderma pigmentosum.] Med Jahrb Wien 1882;619–33. (French translation, Ann Dermatol Syphiligr 1883;4:29–38.)
- Neisser A. Ueber das ‘Xeroderma pigmentosum’ (Kaposi): Lioderma essentialis cum melanosi et telangiectasia. Vierteljahrschr Dermatol Syphil 1883;47–62.
- De Sanctis C, Cacchione A. L'idiozia xerodermica. Riv Sper Freniatr 1932;56:269–92.
- Cleaver JE. Defective repair replication of DNA in xeroderma pigmentosum. Nature 1968;218:652–6.
- Setlow RB, Setlow JK. Evidence that ultraviolet-induced thymine dimers in DNA cause biological damage. Proc Natl Acad Sci USA 1962;48:1250–7.
- Epstein JH, Fukuyama K, Reed WB, Epstein WL. Defect in DNA synthesis in skin of patients with xeroderma pigmentosum demonstrated in vivo. Science 1970;168:1477–8.
- Burk PG, Lutzner MA, Clarke DD, Robbins JH. Ultraviolet-stimulated thymidine incorporation in xeroderma pigmentosum lymphocytes. J Lab Clin Med 1971;77:759–67.
- Cleaver JE. Xeroderma pigmentosum: variants with normal DNA repair and normal sensitivity to ultraviolet light. J Invest Dermatol 1972;58:124–8.
- Lehmann AR, Kirk-Bell S, Arlett CF, et al. Xeroderma pigmentosum cells with normal levels of excision repair have a defect in DNA synthesis after UV-irradiation. Proc Nat Acad Sci 1975;72:219-23.
- De Weerd-Kastelein EA, Keijzer W, Bootsma D. Genetic heterogeneity of xeroderma pigmentosum demonstrated by somatic cell hybridization. Nat New Biol 1972;238:80–3.
- Robbins JH, Kraemer KH, Lutzner MA, et al. Xeroderma pigmentosum: an inherited disease with sun- sensitivity, multiple cutaneous neoplasms, and abnormal DNA repair. Annals Internal Med 1974;80:221-248.
- Takebe H, Miki Y, Kozuka T, et al. DNA repair characteristics and skin cancers of xeroderma pigmentosum patients in Japan. Cancer Res 1977;37:490–5.
- Kleijer WJ, Laugel V, Berneburg M, et al. Incidence of DNA repair deficiency disorders in western Europe: xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. DNA Repair (Amst) 2008;7:744–50.
- Hirai Y, Kodama Y, Moriwaki S, et al. Heterozygous individuals bearing a founder mutation in the XPA DNA repair gene comprise nearly 1% of the Japanese population. Mutat Res 2006;601:171–8.
- Khatri ML, Bemghazil M, Shafi M, Machina A. Xeroderma pigmentosum in Libya. Int J Dermatol 1999;38:520–4.
- Fazaa B, Zghal M, Bailly C, et al. Melanoma in xeroderma pigmentosum: 12 cases. Ann Dermatol Vénéréol 2001;128:503–6.
- Zghal M, El-Fekih N, Fazaa B, et al. Xeroderma pigmentosum: manifestations cutanees, oculaires et neurologiques a partir de 49 patients tunisiens. Tunis Med 2005;83:760–3.
- Moussaid L, Benchikhi H, Boukind EH, et al. Tumeurs cutanees au cours du xeroderma pigmentosum au Maroc. Ann Dermatol Vénéréol 2004;131:29–33.
- Soufir N, Ged C, Bourillon A, et al. A prevalent mutation with founder effect in xeroderma pigmentosum group C from North Africa. J Invest Dermatol 2010;130:1537–42.
- Cartault F, Nava C, Malbrunot AC, et al. A new XPC gene splicing mutation has lead to the highest worldwide prevalence of xeroderma pigmentosum in black Mahori patients. DNA Repair (Amst) 2011;10:577–85.
- Cleaver JE, Trosko JE. Absence of excision of ultraviolet-induced cyclobutane dimers in xeroderma pigmentosum. Photochem Photobiol 1970;11:547–50.
- Cleaver JE, Lam ET, Revet I. Disorders of nucleotide excision repair: the genetic and molecular basis of heterogeneity. Nat Rev Genet 2009;10:756–68.
- Sethi M, Lehmann AR, Fassihi H. Xeroderma pigmentosum – a multidisciplinary approach. Eur Med J Dermatol 2013;1:54-63.
- Lehmann AR, McGibbon D, Stefanini M. Xeroderma pigmentosum. Orphanet J Rare Dis 2011;6:70.
- Friedberg EC. DNA damage and repair. Nature 2003;421:436–40.
- Masutani C, Kusumoto R, Yamada A, et al. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta. Nature 1999;399:700–4.
- Sethi M, Lehmann AR, Fawcett H, et al. Patients with xeroderma pigmentosum complementation groups C, E and V do not have abnormal sunburn reactions. Br J Dermatol 2013;169:1279–87.
- Venema J, van Hoffen A, Karcagi V, et al. Xeroderma pigmentosum complementation group C cells remove pyrimidine dimers selectively from the transcribed strand of active genes. Mol Cell Biol 1991;11:4128–34.
- Totonchy MB, Tamura D, Pantell MS, et al. Auditory analysis of xeroderma pigmentosum 1971–2012: hearing function, sun sensitivity and DNA repair predict neurological degeneration. Brain 2013;136:194–208.
- Berg RJ, Ruven HJ, Sands AT, et al. Defective global genome repair in XPC mice is associated with skin cancer susceptibility but not with sensitivity to UVB induced erythema and edema. J Invest Dermatol 1998;110:405–9.
- Garssen J, van Steeg H, de Gruijl F, et al. Transcription-coupled and global genome repair differentially influence UV-B-induced acute skin effects and systemic immunosuppression. J Immunol 2000;164:6199–205.
- Ziegler A, Jonason A, Simon J, et al. Tumor suppressor gene mutations and photocarcinogenesis. Photochem Photobiol 1996;63:432–5.
- Pleasance ED, Cheetham RK, Stephens PJ, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature 2010;463:191–7.
- Kraemer KH, Lee MM, Andrews AD, Lambert WC. The role of sunlight and DNA repair in melanoma and nonmelanoma skin cancer. The xeroderma pigmentosum paradigm. Arch Dermatol 1994;130:1018–21.
- Daya-Grosjean L, Sarasin A. The role of UV induced lesions in skin carcinogenesis: an overview of oncogene and tumor suppressor gene modifications in xeroderma pigmentosum skin tumors. Mutat Res 2005;571:43–56.
- Bradford PT, Goldstein AM, Tamura D, et al. Cancer and neurologic degeneration in xeroderma pigmentosum: long term follow-up characterises the role of DNA repair. J Med Genet 2010;48:168–76.
- Maher VM, Patton JD, Yang JL, et al. Mutations and homologous recombination induced in mammalian cells by metabolites of benzo[a]-pyrene and 1-nitropyrene. Environ Health Perspect 1987;76:33–9.
- DiGiovanna JJ, Patronas N, Katz D, et al. Xeroderma pigmentosum: spinal cord astrocytoma with 9-year survival after radiation and isotretinoin therapy. J Cutan Med Surg 1998;2:153–8.
- Giannelli F, Avery J, Polani PE, et al. Xeroderma pigmentosum and medulloblastoma: chromosomal damage to lymphocytes during radiotherapy. Radiat Res 1981;88:194–208.
- Brooks BP, Thompson AH, Bishop RJ, et al. Ocular manifestations of xeroderma pigmentosum: long-term follow-up highlights the role of DNA repair in protection from sun damage. Ophthalmology 2013;120:1324–36.
- Gaasterland DE, Rodrigues MM, Moshell AN. Ocular involvement in xeroderma pigmentosum. Ophthalmology 1982;89:980–6.
- Brooks PJ. The 8,5'-cyclopurine-2'-deoxynucleosides: candidate neurodegenerative DNA lesions in xeroderma pigmentosum, and unique probes of transcription and nucleotide excision repair. DNA Repair (Amst) 2008;7:1168–79.
- Anttinen A, Koulu L, Nikoskelainen E, et al. Neurological symptoms and natural course of xeroderma pigmentosum. Brain 2008;131:1979–89.
- Stefanini M, Keijzer W, Dalpra L, et al. Differences in the levels of UV repair and in clinical symptoms in two sibs affected by xeroderma pigmentosum. Hum Genet 1980;54:177–82.
- Lehmann AR, Stevens S. A rapid procedure for measurement of DNA repair in human fibroblasts and for complementation analysis of xeroderma pigmentosum cells. Mutat Res 1980;69:177–90.
- Limsirichaikul S, Niimi A, Fawcett H, et al. A rapid non-radioactive technique for measurement of repair synthesis in primary human fibroblasts by incorporation of ethynyl deoxyuridine (EdU). Nucleic Acids Res 2009;37:e31.
- Arlett CF, Harcourt SA, Broughton BC. The influence of caffeine on cell survival in excision-proficient and excision-deficient xeroderma pigmentosum and normal human cell strains following ultraviolet light irradiation. Mutat Res 1975;33:341–6.
- Broughton BC, Cordonnier A, Kleijer WJ, et al. Molecular analysis of mutations in DNA polymerase eta in xeroderma pigmentosum-variant patients. Proc Natl Acad Sci USA 2002;99:815–20.
- Chavanne F, Broughton BC, Pietra D, et al. Mutations in the XPC gene in families with xeroderma pigmentosum and consequences at the cell, protein and transcript levels. Cancer Res 2000;60:1974–82.
- Kraemer KH, DiGiovanna JJ, Moshell AN, et al. Prevention of skin cancer in xeroderma pigmentosum with the use of oral isotretinoin. N Engl J Med 1988;318:1633–7.
- Bettoli V, Zauli S, Virgili A. Retinoids in the chemoprevention of non-melanoma skin cancers: why, when and how. J Dermatolog Treat 2013;24:235–7.
- Procianoy F, Cruz AA, Baccega A, et al. Aggravation of eyelid and conjunctival malignancies following photodynamic therapy in DeSanctis–Cacchione syndrome. Ophthal Plast Reconstr Surg 2006;22:498–9.
- Marchetto MC, Muotri AR, Burns DK, et al. Gene transduction in skin cells in preventing cancer in xeroderma pigmentosum mice. Proc Natl Acad Sci USA 2004;101:17759–64.
- Marchetto MC, Correa RG, Menck CF, Muotri AR. Functional lentiviral vectors for xeroderma pigmentosum gene therapy. J Biotechnol 2006;126:424–30.
Cockayne syndrome
- Cockayne EA. Dwarfism with retinal atrophy and deafness. Arch Dis Child 1936;11:1–8.
- Nance MA, Berry SA. Cockayne syndrome: review of 140 cases. Am J Med Genet 1992;42:68–84.
- Cleaver JE, Lam ET, Revet I. Disorders of nucleotide excision repair: the genetic and molecular basis of heterogeneity. Nat Rev Genet 2009;10:756–68.
- Mayne LV, Lehmann AR, Waters R. Excision repair in Cockayne syndrome. Mutat Res 1982;106:179–89.
- Rainbow AJ, Howes M. A deficiency in the repair of UV and gamma-ray damaged DNA in fibroblasts from Cockayne's syndrome. Mutat Res 1982;93:235–47.
- Yatani R, Kusano I, Shiraishi T, et al. DNA synthesis and hypersensitivity to ultraviolet radiation in Cockayne's syndrome. Exp Mol Pathol 1982;36:361–72.
- Venema J, Mullenders LH, Natarajan AT, et al. The genetic defect in Cockayne syndrome is associated with a defect in repair of UV-induced DNA damage in transcriptionally active DNA. Proc Natl Acad Sci USA 1990;87:4707–11.
- Mayne LV, Lehmann AR. Failure of RNA synthesis to recover after UV-irradiation: an early defect in cells from individuals with Cockayne's syndrome and xeroderma pigmentosum. Cancer Res 1982;42:1473–8.
- Troelstra C, van Gool A, de Wit J, et al. ERCC6, a member of a subfamily of putative helicases, is involved in Cockayne's syndrome and preferential repair of active genes. Cell 1992;71:939–53.
- Henning KA, Li L, Iyer N, et al. The Cockayne syndrome group A gene encodes a WD repeat protein that interacts with CSB protein and a subunit of RNA polymerase II TFIIH. Cell 1995;82:555–64.
- Stefanini M, Fawcett H, Botta E, et al. Genetic analysis of twenty-two patients with Cockayne syndrome. Hum Genet 1996;97:418–23.
- Licht CL, Stevnsner T, Bohr VA. Cockayne syndrome group B cellular and biochemical functions. Am J Hum Genet 2003;73:1217–239.
- Macdonald WB, Fitch KD, Lewis IC. Cockayne's syndrome. A heredo-familial disorder of growth and development. Pediatrics 1960;25:997–1007.
- Frouin E, Laugel V, Durand M, et al. Dermatologic findings in 16 patients with Cockayne syndrome and cerebro-oculo-facial-skeletal syndrome. JAMA Dermatol 2013;149:1414–18.
- Lieberman WJ, Schimek RA, Snyder CH. Cockayne's disease. A report of a case. Am J Ophthalmol 1961;52:116–18.
- Riggs W, Seibert J. Cockayne's syndrome: roentgen findings. Am J Roentgenol 1972;116:623–33.
- Lehmann AR, Thompson AF, Harcourt SA, et al. Cockayne's syndrome: correlation of clinical features with cellular sensitivity of RNA synthesis to UV-irradiation. J Med Genet 1993;30:679–82.
- Bertola DR, Cao H, Albano LM, et al. Cockayne syndrome type A: novel mutations in eight typical patients. J Hum Genet 2006;51:701–5.
- Kraemer KH, Patronas NJ, Schiffmann R. Xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome: a complex genotype-phenotype relationship. Neuroscience 2007;145:1388–96.
- Pena SDJ, Shokeir MHK. Syndrome of camptodactyly, multiple ankyloses, facial anomalies, and pulmonary hypoplasia: a lethal condition. J Pediat 1974;85:373–5.
- Preus M, Fraser FC. The cerebro-oculo-facio-skeletal syndrome. Clin Genet 1974;5:294–7.
- Laugel V, Dalloz C, Tobias ES, et al. Cerebro-oculo-facio-skeletal syndrome: three additional cases with CSB mutations, new diagnostic criteria and an approach to investigation. J Med Genet 2008;45:564–71.
- Jaakkola E, Mustonen A, Olsen P, et al. ERCC6 founder mutation identified in Finnish patients with COFS syndrome. Clin Genet 2010;78:541–7.
- Graham JM, Jr, Anyane-Yeboa K, Raams A, et al. Cerebro-oculo-facio-skeletal syndrome with a nucleotide excision-repair defect and a mutated XPD gene, with prenatal diagnosis in a triplet pregnancy. Am J Hum Genet 2001;69:291–300.
- Hamel BCJ, Raams A, Schuitema-Dijkstra AR, et al. Xeroderma pigmentosum–Cockayne syndrome complex: a further case. J Med Genet 1996;33:607–10.
- Jaspers NGJ, Raams A, Silengo MC, et al. First reported patient with human ERCC1 deficiency has cerebro-oculo-facio-skeletal syndrome with a mild defect in nucleotide excision repair and severe developmental failure. Am J Hum Genet 2007;80:457–66.
- Spivak G. UV-sensitive syndrome. Mutat Res 2005;577:162–9.
- Itoh T, Ono T, Yamaizumi M. A new UV-sensitive syndrome not belonging to any complementation groups of xeroderma pigmentosum or Cockayne syndrome: siblings showing biochemical characteristics of Cockayne syndrome without typical clinical manifestations. Mutat Res 1994;314:233–48.
- Nardo T, Oneda R, Spivak G, et al. A UV-sensitive syndrome patient with a specific CSA mutation reveals separable roles for CSA in response to UV and oxidative DNA damage. Proc Natl Acad Sci USA 2009;106:6209–14.
- Horibata K, Iwamoto Y, Kuraoka I, et al. Complete absence of Cockayne syndrome group B gene product gives rise to UV-sensitive syndrome but not Cockayne syndrome. Proc Natl Acad Sci USA 2004;101:15410–15.
- Nakazawa Y, Sasaki K, Mitsutake N, et al. Mutations in UVSSA cause UV-sensitive syndrome and impair RNA polymerase IIo processing in transcription-coupled nucleotide-excision repair. Nat Genet 2012;44:586–92.
- Miyauchi H, Horio T, Akaeda T, et al. Cockayne syndrome in two adult siblings. J Am Acad Dermatol 1994;30:329–35.
- Lehmann AR, Francis AJ, Giannelli F. Prenatal diagnosis of Cockayne's syndrome. Lancet 1985;i:486–8.
- Cleaver JE, Volpe JP, Charles WC, et al. Prenatal diagnosis of xeroderma pigmentosum and Cockayne syndrome. Prenat Diagn 1994;14:921–8.
Trichothiodystrophy
- Baden HP, Jackson CE, Weiss L, et al. The physicochemical properties of hair in the BIDS syndrome. Am J Hum Genet 1976;28:514–21.
- Price VH, Odom RB, Ward WH, et al. Trichothiodystrophy: sulfur-deficient brittle hair as a marker for a neuroectodermal symptom complex. Arch Dermatol 1980;116:1375–84.
- Liang C, Kraemer KH, Morris A, et al. Characterization of tiger-tail banding and hair shaft abnormalities in trichothiodystrophy. J Am Acad Dermatol 2005;52:224–32.
- Liang C, Morris A, Schlucker S, et al. Structural and molecular hair abnormalities in trichothiodystrophy. J Invest Dermatol 2006;126:2210–16.
- Faghri S, Tamura D, Kraemer KH, et al. Trichothiodystrophy: a systematic review of 112 published cases characterises a wide spectrum of clinical manifestations. J Med Genet 2008;45:609–21.
- Price VH, Odom RB, Ward WH, Jones FT. Trichothiodystrophy: sulfur-deficient brittle hair as a marker for a neuroectodermal symptom complex. Arch Dermatol 1980;116:1375–84.
- Crovato F, Borrone C, Rebora A. Trichothiodystrophy – BIDS, IBIDS and PIBIDS? Br J Dermatol 1983;108:247.
- Jorizzo JL, Crounse RG, Wheeler CE, Jr. Lamellar ichthyosis, dwarfism, mental retardation, and hair shaft abnormalities. A link between the ichthyosis-associated and BIDS syndromes. J Am Acad Dermatol 1980;2:309–17.
- Jorizzo JL, Atherton DJ, Crounse RG, Wells RS. Ichthyosis, brittle hair, impaired intelligence, decreased fertility and short stature (IBIDS syndrome). Br J Dermatol 1982;106:705–10.
- Baden HP, Jackson CE, Weiss L, et al. The physicochemical properties of hair in the BIDS syndrome. Am J Hum Genet 1976;28:514–21.
- Stefanini M, Lagomarsini P, Arlett CF, et al. Xeroderma pigmentosum (complementation group D) mutation is present in patients affected by trichothiodystrophy with photosensitivity. Hum Genet 1986;74:107–112.
- Weeda G, Eveno E, Donker I, et al. A mutation in the XPB/ERCC3 DNA repair transcription gene, associated with trichothiodystrophy. Am J Hum Genet 1997;60:320–9.
- Stefanini M, Vermeulen W, Weeda G, et al. A new nucleotide-excision-repair gene associated with the disorder trichothiodystrophy. Am J Hum Genet 1993;53:817–21.
- Giglia-Mari G, Coin F, Ranish JA, et al. A new, tenth subunit of TFIIH is responsible for the DNA repair syndrome trichothiodystrophy group A. Nat Genet 2004;36:714–19.
- Hashimoto S, Egly JM. Trichothiodystrophy view from the molecular basis of DNA repair/transcription factor TFIIH. Hum Mol Genet 2009;18:R224–30.
- Nakabayashi K, Amann D, Ren Y, et al. Identification of C7orf11 (TTDN1) gene mutations and genetic heterogeneity in nonphotosensitive trichothiodystrophy. Am J Hum Genet 2005;76:510–16.
- Stefanini M, Botta E, Lanzafame M, et al. Trichothiodystrophy: from basic mechanisms to clinical implications. DNA Repair (Amst) 2010;9:2–10.
- Boyle J, Ueda T, Oh KS, et al. Persistence of repair proteins at unrepaired DNA damage distinguishes diseases with ERCC2 (XPD) mutations: cancer-prone xeroderma pigmentosum vs. non-cancer-prone trichothiodystrophy. Hum Mutat 2008;29:1194–208.
- Tamura D, Merideth M, DiGiovanna JJ, et al. High-risk pregnancy and neonatal complications in the DNA repair and transcription disorder trichothiodystrophy: report of 27 affected pregnancies. Prenat Diagn 2011;31:1046–53.
- Moslehi R, Signore C, Tamura D, et al. Adverse effects of trichothiodystrophy DNA repair and transcription gene disorder on human fetal development. Clin Genet 2010;77:365–73.
- Brooks BP, Thompson AH, Clayton JA, et al. Ocular manifestations of trichothiodystrophy. Ophthalmology 2011;118:2335–42.
- Viprakasit V, Gibbons RJ, Broughton BC, et al. Mutations in the general transcription factor TFIIH result in beta-thalassaemia in individuals with trichothiodystrophy. Hum Mol Genet 2001;10:2797–802.
- Kraemer KH, Patronas NJ, Schiffmann R, et al. Xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome: a complex genotype-phenotype relationship. Neuroscience 2007;145:1388–96.
Ataxia-telangiectasia (Louis-Bar syndrome)
- Boder E, Sedgwick RP. Ataxia-telangiectasia. (Clinical and immunological aspects). Psychiatr Neurol Med Psychol Beih 1970;13/14:8–16.
- Chun HH, Gatti RA. Ataxia-telangiectasia, an evolving phenotype. DNA Repair (Amst) 2004;3:1187–96.
- Knoch J, Kamenisch Y, Kubisch C, Berneburg M. Rare hereditary diseases with defects in DNA-repair. Eur J Dermatol 2012;22:443–55.
- Taylor AM. Unrepaired DNA strand breaks in irradiated ataxia telangiectasia lymphocytes suggested from cytogenetic observations. Mutat Res 1978;50:407–18.
- Goodarzi AA, Noon AT, Deckbar D, et al. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol Cell 2008;31:167–77.
Fanconi anaemia
- Deakyne JS, Mazin AV. Fanconi anemia: at the crossroads of DNA repair. Biochemistry 2011;76:36–48.
- Joenje H, Patel KJ. The emerging genetic and molecular basis of Fanconi anaemia. Nat Rev Genet 2001;2:446–57.
- Walden H, Deans AJ. The Fanconi anemia DNA repair pathway: structural and functional insights into a complex disorder. Annu Rev Biophys 2014;43:257–78.
Muir–Torre syndrome
- Muir EG, Bell AJY, Barlow KA. Multiple primary carcinoma of the colon, duodenum and larynx associated with keratoacanthoma of the face. Br J Surg 1967;54:191.
- Torre D. Multiple sebaceous gland tumors. Arch Dermatol 1968;98:549.
- Schwartz RA, Torre DP. The Muir–Torre syndrome: a 25 years retrospect. J Am Acad Dermatol 1995;33:90.
- Ponti G, Ponz de Leon M. Muir–Torre syndrome. Lancet Oncol 2005;6:980.
- Abbas O, Mahalingam M. Cutaneous sebaceous neoplasms as markers of Muir–Torre syndrome: a diagnostic algorithm. J Cutan Pathol 2009;36:613–19.