CHAPTER 79
Syndromes with Premature Ageing
Alan D. Irvine1 and Jemima E. Mellerio2
1Our Lady’s Children’s Hospital, Crumlin; St James’s Hospital, Dublin; and Trinity College, Dublin, Ireland
2St John’s Institute of Dermatology, Guy’s and St Thomas’ NHS Foundation Trust, London; Great Ormond Street Hospital for Children NHS Foundation Trust, London, UK
Introduction
Progeroid syndromes comprise a heterogeneous group of conditions with variable cutaneous features that lead to a prematurely aged appearance, including poikiloderma, photosensitivity and pigmentation, sclerodermatous changes, alteration of the subcutaneous fat, or skin laxity and wrinkling. Broadly, these diseases can be grouped according to the underlying pathology. For example, conditions with impaired DNA repair mechanisms including the Bloom and Werner syndromes, and xeroderma pigmentosum, may be associated with photosensitivity, pigmentation and telangiectasia of the skin as well as an increased incidence of skin and other malignancies. Aberrant lamin A processing leading to an accumulation of prelamin A in the nucleus is another mechanism leading to a progeroid appearance, for example in Hutchinson–Gilford progeria syndrome and forms of mandibulo-acral dysplasia. In contrast, defects in genes involved in the formation of elastic fibres in the skin and other tissues are responsible for forms of cutis laxa. Importantly, many progeroid syndromes are associated with severe extracutaneous disease, notably a predisposition to early systemic ageing, malignancy, developmental and neurological impairment and premature death. Often, the cutaneous features of these rare disorders occur early and are key to helping identify the underlying disorder with important implications for genetic counselling, management and prognosis. Table 79.1 summarizes the major syndromes of premature ageing, some of which are discussed in other chapters.
Table 79.1 Progeroid disorders.
| Disease | MIM | Inheritance | Gene | Cutaneous features | Photo- sensitivity | Neurological | Immuno-deficiency | Growth restriction | Non-cutaneous malignancy | Death |
| Werner syndrome | 277700 | AR | RECQL2 | Scleroderma | − | − | − | From puberty | + (10%) | In fifties (malignancy and cardiovascular) |
| Bloom syndrome | 210900 | AR | RECQL3 | Poikiloderma, facial telangiectasia | + | − | + | Pre- and postnatal | + (15%), mean age 32 years | Early death from malignancy |
| XP-Aa | 278700 | AR | XPA | Freckles, telangiectasia, skin cancer | + | + | − | − | − | For all XP types, it depends on complementation and occurrence of skin malignancy and/or neurological involvement |
| XP-Bb | 610651 | AR | ERCC3 | Freckles, telangiectasia, skin cancer | + | + | − | + | − | |
| XP-Cc | 278720 | AR | XPC | Freckles, telangiectasia, skin cancer | − | − | − | − | − | |
| XP-Dd | 278730 | AR | ERCC2 | Poikiloderma, telangiectasia, skin cancer | + | + | − | − | − | |
| XP-Ee | 278740 | AR | DDB2 | Freckles, poikiloderma, skin cancer | − | − | − | − | − | |
| XP-Ff | 278760 | AR | ERCC4 | Freckles, skin cancer | + | +/− | − | +/− | +/− | |
| XP-Gg | 278780 | AR | ERCC5 | Mild freckling | + | +/− | − | − | − | |
| XP-Vh | 278750 | AR | POLH | Freckles, poikiloderma, skin cancer | − | − | − | − | − | |
| Cockayne syndrome type Ai | 216400 | AR | ECRR8 | Progeria, pigmentation, atrophy, brittle hair and nails, reduced subcutaneous fat | + | + | − | Pre- and postnatal | − | Usually death in childhood or adolescence |
| Cockayne syndrome type Bj | 133540 | AR | ECRR6 | Progeria, pigmentation, atrophy, brittle hair and nails, reduced subcutaneous fat | + | + | − | Pre- and postnatal | − | Death in childhood |
| Trichothiodystrophyk | 601675 | AD | ERCC3 GTF2H5 ERCC2 |
Thin dry hair, ichthyosiform erythroderma, brittle hair and nails, reduced subcutaneous fat | +/− | + | + | Postnatal | − | Variable |
| Classic HGPS | 176670 | AD | LMNA | Scleroderma, premature ageing, alopecia | − | − | − | Progressive from first year | - | Death in teens from cardiovascular disease, mean 13 years |
| Atypical HGPS and other progeroid laminopathies | 176670 | ADAR | LMNA LMNA |
Variable scleroderma, premature ageing, sparse hair | − | − | − | May be marked | - | Variable |
| MADA | 248370 | AR | LMNA | Scleroderma, pigmentation, alopecia | − | − | − | Postnatal | − | − |
| MADB | 608612 | AR | ZMPSTE24 | Atrophy, pigmentation, sparse hair | − | − | − | − | − | − |
| Neonatal progeria syndrome (Wiedemann−Rautenstrauch syndrome) | 264090 | AR | − | Sparse hair, aged appearance, reduced subcutaneous fat | − | + | − | IUGR | − | Death in childhood |
| Hallermann−Steiff syndrome | 234100 | Sporadic | − | Skin atrophy, telangiectasia, hypotrichosis | − | +/− | − | Short stature | − | − |
| Restrictive dermopathy | 275210 275210 |
ADAR | LMNA ZMPSTE24 |
Tight, sclerotic skin, erosions and fissures, thin and translucent | −− | −− | −− | IUGR IUGR |
−− | Often premature or stillborn. Death in first to seconnd week of life |
AR, autosomal recessive; AD, autosomal dominant; HGPS, Hutchinson–Gilford progeria syndrome; IUGR, intrauterine growth retardation; MADA, mandibulo-acral dysplasia with type A lipodystrophy; MADB, mandibulo-acral dysplasia with type B lipodystrophy; XP, xeroderma pigmentosum.
aSee also Chapter 78.
Werner syndrome
Definition
Clinically, the syndrome is characterized by premature ageing with sclerodermatous skin changes, subcutaneous calcification, short stature, prematurely aged facies, premature arteriosclerosis, diabetes and a predisposition to malignancy [1].
Pathophysiology
Werner syndrome is a rare, autosomal recessive disorder caused by mutations in gene RECQL2 (WRN) on 8p12-p11.2, encoding a DNA helicase [1–6]. Aberrant repair of double-stranded DNA damage in the absence of WRN helicase activity leads to an accumulation of DNA damage, telomere shortening, genetic instability and a reduction in cellular replicative lifespan [7, 8]. Clinically normal heterozygous carriers of WRN mutations have also been shown to have increased genetic instability compared with normal controls [9]. Tissues of mesenchymal origin are preferentially affected compared with tissues of neural origin, reflected phenotypically by absent neurological involvement such as Alzheimer or Parkinson diseases [10]. Werner syndrome occurs worldwide but there appears to be a higher frequency of reported cases from Japan where the incidence is estimated at approximately three per million [11].
Clinical features [1, 11–13]
Children with Werner syndrome generally have normal growth and development until puberty, when there is a lack of growth spurt and other clinical features usually start to manifest. The cutaneous features include thin, taut, sclerodermatous skin changes, particularly on the face and extremities, and subcutaneous calcification. There may be ulceration around the ankles and elbows. The face has a prematurely aged appearance with beaking of the nose, and hair may be sparse and prematurely grey. Stature is short with slender limbs. Patients tend to develop early arteriosclerosis, diabetes, osteoporosis, hypertension and hypogonadism, and are at risk of cataracts and retinal degeneration. Importantly, Werner syndrome patients are predisposed to developing malignancies, in particular myeloid disorders, meningiomas, melanomas, thyroid cancers and osteosarcomas. The median lifespan is in the mid-fifties with death usually the result of myocardial infarction or malignancy [13].
A subset of patients have ‘atypical’ Werner syndrome, characterized by more severe clinical features. Some individuals in this group have been shown to have heterozygous mutations of the LMNA gene encoding lamin A/C [14], and therefore this should probably be more correctly considered a milder, later onset variant of Hutchinson–Gilford progeria syndrome (HGPS) [13]. In other cases, however, mutations have not been identified in either RECQL2 or LMNA, indicating an alternative genetic aetiology [13].
Management
Complications of Werner syndrome such as cardiovascular disease, diabetes and cataract are treated conventionally as and when they arise. Recent in vitro research, however, suggests that the abnormal cellular phenotype in Werner syndrome might be amenable to treatment with mitogen-activated protein (MAP) kinase inhibitors [15] or rapamycin [16].
Bloom syndrome
Definition
This syndrome is characterized by photosensitivity, telangiectatic facial erythema, proportionate pre- and postnatal growth deficiency, distinctive facies, abnormal immune responses and a predisposition to malignancy [1–3].
Pathophysiology
Bloom syndrome is a rare, autosomal recessive disorder. The gene for Bloom syndrome (RECQL3) lies on chromosome 15q26.1 and encodes a DNA helicase, RecQ protein-like-3 [4]. This helicase is involved in resolving abnormal structures that can arise during DNA replication, and may additionally be involved in transcriptional regulation [5]. Without normal DNA replication, there is chromosomal instability and this results in the observed predisposition to malignancy. Cultured lymphocytes and fibroblasts from patients with Bloom syndrome show a high incidence of chromosomal aberrations, and cells with abnormally high rates of sister chromatid exchange (SCE) are uniquely characteristic of this condition [6].
Bloom syndrome occurs with greater frequency in Ashkenazi Jews, accounting for approximately one-third of all cases. A common ancestral mutation has been identified in most of these patients, with an estimated carrier frequency of 1%. Other founder mutations have also been identified in patients from other geographical regions [7, 8].
Clinical features [1, 3, 9–11]
Cutaneous features
Telangiectatic erythema of the face develops during infancy or early childhood as red macules or plaques, which may simulate lupus erythematosus. They are most numerous on the ‘butterfly’ area of the nose and cheeks, but may involve the margins of the eyelids, the forehead, and the ears, and sometimes the dorsa of the hands and forearms. There may be slight scaling. Exacerbation after exposure to sunlight is usual but not invariable, and light may also provoke bullae, bleeding and crusting of the lips.
Extracutaneous features
Patients with Bloom syndrome have a characteristic appearance. There is moderate and proportionate growth deficiency both in utero and postnatally (Figure 79.1). Patients have a narrow, slender, delicate facies with a relatively prominent nose and small jaw. The limbs tend to be long with large hands and feet, and there may be reduced subcutaneous fat.
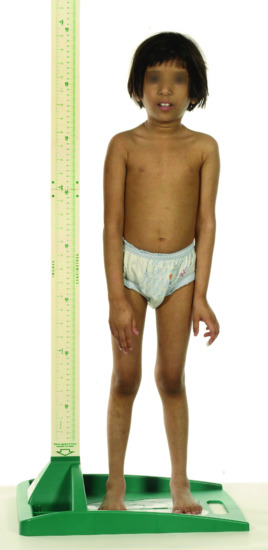
Figure 79.1 Child with Bloom syndrome showing short stature and characteristic facies.
(Courtesy of Dr H. Fassihi, St John's Institute of Dermatology, London, UK.)
Other associated abnormalities include café-au-lait patches, clinodactyly, syndactyly, congenital heart disease, annular pancreas and a high-pitched voice, possibly due to the cranio-facial anatomy and high-arched palate. Many associated developmental defects have been reported. T- and B-cell immunodeficiencies may occur, and IgA and IgM levels may be reduced predisposing to pneumonia, bronchiectasis and chronic lung disease. Gastro-oesophageal reflux is also common and may lead to aspiration, contributing to the risk of chronic lung disease.
Testicular atrophy is common and adult male patients are infertile. Although the tubular elements of the testes function poorly, the androgen-secreting portions are spared, thus permitting normal puberty [11]. Fertility in female patients appears reduced, although full-term pregnancies have been reported [12]. Bloom syndrome patients have an increased incidence of type II diabetes [13]. Neurological development is normal.
The mortality from neoplastic disease, particularly acute leukaemia, during the second or third decade is significantly increased [1, 3, 4, 6, 10]; it has been estimated that there is a 150–300 times increased risk of developing malignancy relative to normal. Cancers of the types and sites seen in the general population arise frequently and unusually early, as do rare cancers of early childhood [10]. They are predominantly internal such as those of the upper and lower gastrointestinal tract, genito-urinary cancers and haematological malignancies, but cutaneous malignancies are also more common with approximately 15% of patients in the Bloom Syndrome Registry having had skin cancer, with a mean age of 32 years [1, 9]. Although there is no increased frequency of malignancies in most heterozygous carriers of RECQL3 mutations, heterozygosity for a number of loss-of-function mutations have recently been shown to be associated with an increased breast cancer risk [14, 15].
Management
Early diagnosis of Bloom syndrome is important for awareness of the possible development of malignancies and for photoprotection. Chromosomal analysis may demonstrate increased numbers of SCEs and abnormal chromosomal configurations. Genetic testing of the RECQL3 gene will confirm the diagnosis. In the Ashkenazi Jewish population, where women have a one in 200 risk of having an affected child, targeted screening of the recurrent founder mutation may be indicated.
Patients should minimize sun exposure and use a high-factor sunscreen. Regular screening for diabetes and malignancies (e.g. skin surveillance, colonoscopy and faecal occult blood testing in adults) may be indicated. Any symptoms suggestive of possible malignancy should prompt appropriate investigations. However, ionizing radiation from cancer surveillance or treatment and alkylating agents may predispose to further DNA damage and carcinogenesis in Bloom syndrome patients and should therefore be minimized as much as possible [16]. Aside from this, standard cancer treatments are recommended, as are treatments of associated diseases such as diabetes.
Prenatal testing for Bloom syndrome has been successfully undertaken by SCE analysis in chorionic villus cell cultures [17] as well as from RECQL3 screening from chorionic villi or amniotic cells. Preimplantation genetic diagnosis has also been carried out [1].
Progeroid laminopathies and related conditions
Lamins comprise a type of intermediate filament which collectively are important structural proteins of the nuclear lamina beneath the inner nuclear membrane; they have a role in maintaining the size and shape of the nuclear membrane. The LMNA gene encodes prelamin A, which undergoes post-translational modification to lamin A. Autosomal dominant or recessive LMNA mutations may result in a number of different syndromes, termed laminopathies, with variable phenotypic features depending on the site and nature of the mutations, but which essentially cause accumulation of prelamin A in the nucleus. Whilst some disorders are characterized by progeroid features, others involve cardiomyopathy, muscular dystrophy, peripheral neuropathy or lipodystrophy (see Table 79.1) [1, 2]. Mutations in a zinc metalloproteinase gene, ZMPSTE24, which is involved in post-translational lamin A processing, have also been implicated in clinically similar conditions, also due to the accumulation of unmodified prelamin A within nuclei.
Hutchinson–Gilford progeria syndrome
Definition
Growth retardation with premature and accelerated ageing from early childhood are the cardinal featires of HGPS [1]. Affected individuals usually die in childhood or teenage years due to cardiovascular causes.
Pathophysiology
Hutchinson–Gilford progeria syndrome is an autosomal dominant condition with a birth prevalence estimated at one in 4 million [2]. Almost all reported cases arise from de novo mutations in the gene encoding lamin A/C, LMNA, on chromosome 1q22 [3, 4]. In one report, the proband's mother was shown to have somatic and germline mosaicism for a causative LMNA mutation [5]. A single recurrent mutation, c.1824C>T, is responsible for the classic form of HGPS [3, 4, 6]; this mutation causes abnormal gene splicing and the deletion of 50 amino acids leading to an abnormal prelamin A protein termed progerin. This deletion interferes with the normal post-translational modification of prelamin A leading to persistent farnesylation of progerin, which accumulates at the inner nuclear membrane causing increased cellular damage with age. Other dominant LMNA mutations have been identified in cases of non-classic HGPS which may be more or less severe, dependent on the molecular consequences of the underlying mutation [6–9].
Fibroblasts from patients with classic HGPS show changes in nuclear morphology on light and electron microscopy, including nuclear envelope lobulation, nuclear lamina thickening, clustering of nuclear pores and loss of peripheral heterochromatin [10]. These changes are thought to be due to the accumulation of progerin increasing with cellular age and result in the progressive premature ageing seen in HGPS.
Histopathologically, during the early sclerodermatous phase in the skin, thickening of the dermis with hypertrophic collagen bundles but a normal epidermis have been described [11], although changes may be non-specific [12]. Later in life the epidermis may be thinned, the dermis replaced by fibrotic hyaline material, with a reduction of sweat glands and subcutaneous tissue [13].
Clinical features [1, 2, 14]
Intrauterine growth is usually normal but may be reduced; by the first year of life, however, failure to thrive with heights and weights below the third percentile are the norm and this growth retardation persists and worsens thereafter. Over the first few years of life, a number of characteristic changes occur. Individuals develop typical facies with a small receding mandible, and the nose develops a pointed tip and narrow bridge. The head appears large for the face and scalp veins are prominent, made more noticeable due to alopecia which may be partial or total, and which may also affect the eyebrows and eyelashes. The eyes tend to be prominent, the lips are thin and may have surrounding cyanosis, and earlobes may be absent. Also manifesting early are sclerodermatous changes of the skin, particularly on the trunk and limbs, with reduced subcutaneous fat and joint contractures. Nails are frequently dystrophic. The clavicles may be short, leading to sloping shoulders and a pear-shaped chest. Bony changes include osteolysis of the distal phalanges and delayed closure of the anterior fontanelle. Primary dentition is often late to erupt and may not be lost, leading to overcrowding. There may also be partial failure of eruption of secondary teeth.
Later in childhood, features of accelerated ageing become more wide ranging, including a pigmented, aged appearance to the skin, conductive hearing loss, osteoarthritis, atherosclerosis leading to ischaemic heart disease and cerebrovascular disease, Raynaud phenomenon and insulin resistance, although not usually diabetes. Growth delay means that individuals do not reach completion of secondary sexual characterics and do not, therefore, have children. Of note, motor and intellectual development are normal and there is no immunodeficiency or predisposition to malignancy.
In classic HGPS the usual survival is around 6–20 years with a mean of 13 years [2]; death usually results from myocardial infaction, heart failure or stroke.
Diagnosis
Diagnosis is usually suspected clinically from a combination of profound growth failure, typical physical appearances and consequences of premature ageing. If suspected, targeted sequencing of known LMNA mutations should be undertaken, followed, if negative, by full sequencing of the gene [1]. Other genes such as ZMPSTE24 may be implicated in other progeroid syndromes and may warrant testing if LMNA screening is negative.
Management
Management should be centred around monitoring disease progress, preventative care where possible, and treatment of complications as and when they arise [1, 2]. Patients should have 6–12-monthly growth, cardiovascular, neurological, musculoskeletal, dental, ear and eye assessments. Lipids, electrocardiogram, echocardiogram, carotid duplex scanning, hip X-rays and bone densiometry scans should also form part of annual care. Low-dose aspirin may be recommended in view of the increased risk of cardiovascular disease, and sun avoidance to limit cutaneous signs of increased ageing. Lipid levels are usually normal but may require statin treatment if elevated.
Recent research has highlighted some potential therapeutic agents to reverse the underlying molecular defect in HGPS, specifically by reducing the farnesylation of progerin which results in its nuclear accumulation [15]. Drugs of interest include pravastatin, lonafarnib and zolendronate, as well as rapamycin, which has been shown to reverse the cellular phenotypic deficit in vitro [16–18] and to improve survival in HGPS patients [19].
Mandibulo-acral dysplasia with type A and type B lipodystrophy
Definition
Mandibulo-acral dysplasia with type A (MADA) and type B (MADB) lipodystrophy are autosomal recessive conditions with growth delay, skeletal abnormalities, lipodystrophy and pigmentary skin changes [1–3].
Pathophysiology
MADA lipodystrophy is caused by homozygous or compound heterozygous LMNA mutations [1, 2, 4, 5], whereas MADB results from compound heterozygous mutations in the zinc metalloproteinase STE24 gene, ZMPSTE24, which is involved in post-translational modification of prelamin A [3, 6, 7].
Clinical features [1–7]
Affected individuals have postnatal growth retardation and develop progressive osteolysis of the clavicles and distal phalanges. Cranio-facial changes include mandibular hypoplasia, which leads to dental crowding, and delayed closure of the fonatanelles and cranial sutures, with a progeroid appearance in some. In general, MADB tends to present earlier in the first year or two of life with skeletal manifestations and skin atrophy compared with individuals with MADA. In MADA, lipodystrophy results in the loss of acral fatty tissue with an increase in subcutaneous fat in the trunk in some individuals, whereas in type B the lipodystrophy also tends to affect the trunk and neck. Impaired insulin resistance and diabetes may occur. Cutaneous changes include patchy pigmention, atrophy of the skin, sclerodermatous changes with stiff joints, sclerotic, clacified acral skin and alopecia.
Cutis laxa: autosomal dominant and autosomal recessive
Definition
Cutis laxa is a group of conditions in which the skin lacks elasticity leading to a loose, wrinkly and prematurely aged appearance. In addition to skin changes, these heterogeneous disorders are variably associated with connective tissue problems as a result of loss, disorganization or fragmentation of elastic tissues in other organs, notably the lungs, cardiovascular system, joints and gastrointestinal and genito-urinary systems. Some forms are also associated with developmental delay and neuromuscular compromise. Cutis laxa is very rare, with an estimated incidence of one in 4 million [1–3].
Pathophysiology
The formation of elastic fibres is a stepwise process and involves interplay between a number of different proteins. Microfibrils consisting of fibulin, fibrillin and various glycoproteins act as a scaffold for soluble tropoelastin monomers, which are then cross-linked by lysyl oxidase into an insoluble elastin core [4]. The resulting elastic fibres are responsible for the resilience of organs such as the skin, blood vessels, intestine, bladder and joints. Defects in some of the proteins involved in elastic fibre formation, such as fibulins and elastin, result in different cutis laxa phenotypes. Histological and ultrastructural changes depend on the underlying defect.
Clinical features
Table 79.2 summarizes the different forms of cutis laxa, the main ones being autosomal dominant cutis laxa (ADCL) and autosomal recessive cutis laxa (ARCL).
Table 79.2 Cutis laxa subtypes, causative genes and predominant phenotypes
| Disease | MIM | Gene | Chromosome | Cutis laxa | Emphysema | Vascular involvement | Intellectual impairment | GI/GU diverticula | Skeletal manifestations |
| ADCL1 | 123700 | ELN | 7q11.23 | + | + | + | – | – | – |
| ADCL2 | 614434 | FBLN5 | 14q32.12 | + | + | + | – | – | – |
| ARCL1A | 219100 | FBLN5 | 14q32.12 | +++ | +++ | + | – | + | – |
| ARCL1B | 614437 | EFEMP2 (FBLN4) |
11q13.1 | ++ | ++ | +++ | – | – | – |
| ARCL1C | 613177 | LTBP4 | 19q13.2 | ++ | +++ | + | – | +++ | + |
| ARCL2A | 219200 | ATP6V0A2 | 12q24.31 | ++ | – | + | ++ | – | + |
| ARCL2B | 612940 | PYCR1 | 17q25.3 | ++ | – | – | ++ | – | ++ |
| ARCL2B | 219150 | ALDH18A1 | 10q24.1 | + | – | + | +++ | – | ++ |
| MACS | 613075 | RIN2 | 20p11.23 | ++ | – | – | – | – | ++ |
| Geroderma osteodysplasticum | 231070 | SCYL1BP1 | 1q24.2 | ++ | – | – | – | – | +++ |
| XLCL (OHS) | 304150 | ATP7A | Xq21.1 | + | – | + | + | + | +++ |
ADCL, autosomal dominant cutis laxa; ARCL, autosomal recessive cutis laxa; GI/GU, gastrointestinal/genito-urinary; MACS. macrocephlay, alopecia, cutis laxa, scoliosis syndrome; OHS, occipital horn syndrome; XLCL, X-linked cutis laxa.
Autosomal dominant cutis laxa
Autosomal dominant mutations of the elastin gene, ELN, cause lax skin and a prematurely aged appearance with onset between childhood and early adulthood, as well as gastrointestinal diverticula and inguinal hernias [5, 6]. Less common manifestations include emphysema, aortic aneurysm and aortic or mitral valve prolapse. Elastic fibres in the skin are sparse and fragmented with a paucity of amorphous elastin on electron microscopy [7]. ADCL has also been reported due to a heterozygous mutation of the fibulin 5 gene, FBLN5 [8].
Autosomal recessive cutis laxa
A number of different genes have been implicated in recessive forms of cutis laxa, including those encoding fibulins 5 and 4, FBLN5 and EFEMP2 (FBLN4), respectively [1, 2]. Other causative genes have less well characterized functions but encode proteins involved in amino acid metabolism, regulation of transforming growth factor β1 (TGF-β1) activity, or transport and/or modification of components of the extracellular matrix.
ARCL type 1 comprises severe forms with life-threatening complications and death occuring between infancy and young adulthood usually from cardiorespiratory compromise [1, 2]. Three main types are recognized depending on the underlying gene pathology (Table 79.2): in general terms, ARCL type 1A (FBLN5 mutations) is associated with more severe and early respiratory complications [9–11]; type 1B (EFEMP2 mutations) has greater vascular fragility and tortuosity [12, 13]; and type 1C (LTBP4 mutations) results in greater problems with diverticula of the gut or bladder [14]. In all types, the skin changes are generalized but may be particularly prominent over the axillae, neck and groins, and give the face a droopy, aged appearance. Atelectasis and emphysema usually present in early childhood and vascular anomalies, notably supravalvular aortic stenosis, pulmonary artery stenosis or aortic aneurysm, occur commonly. There may be multiple diverticula of the gastrointestinal and genito-urinary tracts and, less commonly, inguinal hernias, joint laxity, hip dislocation and delayed closure of the fontanelles. Facial dysmorphism and postnatal growth delay may be seen but intellectual development is normal. Histology shows normal or reduced elastic fibres with mild fragmentation in the dermis [14]; ultrastructurally there is a reduction in elastic fibres and accumulation of elastin globules [15].
Type 2 ARCL comprises a number of different forms of cutis laxa with variable severity but overlap in some phenotypic features. It results from mutations in genes encoding different proteins involved in the function of mitochondria, endosomes and Golgi apparatus. Most cases result from mutations in the ATP6V0A2 ATPase gene, and less often from mutations in PYCR1 and ALDH38A1 [3, 16–20]. The cutaneous phenotype may vary between wrinkly skin and more pronounced cutis laxa with excess folds of skin over the face, large flexures and dorsa of the hands and feet, which may improve over time. Unlike type 1 ARCL, there is often pronounced developmental delay, seizures and neurological impairment with most individuals showing cerebral and cerebellar malformations. Other feaures include delayed growth, congenital hip dislocation, joint laxity, inguinal hernias, osteoporosis, high myopia and facial dysmorphism. De Barsy syndrome is a form of type 2 ARCL in which there is generalized cutis laxa giving a progeroid appearance, pre- and postnatal growth delay, clouding of the corneas or cataracts, and severe developmental delay in the majority [20–23]. There may also be agenesis of the corpus callosum, joint laxity, hypotonia, tortuosity of blood vessels and facial dysmorphism. Causative mutations have been identified in ALDH38A1 [2, 22] and PYCR1 [23]. Macrocephaly, alopecia, cutis laxa and scoliosis (MACS) syndrome is another variant of type 2 ARCL with coarse facial sagging and developmental delay. It results from mutations in RIN2 which encodes a Rab5 effector protein of endosomal vesicles. At the milder end of the phenotypic spectrum, geroderma osteodysplasticum is characterized by progeroid skin wrinkling mainly on the hands, feet and abdomen presenting in infancy or early childhood [27, 28]. There is osteoporosis and bony fractures, and there may be hernias or hip dislocation. Intellectual development is normal. The causative gene, SCYL1BP1, encodes a protein thought to be involved in Golgi trafficking and structure [28]. Skin histology in type 2 ARCL may be normal but ultrastructurally there is a reduced density of small and misshapen elastic fibres [3].
Occipital horn syndrome (OHS), so named because of characteristic calcifications over the occipital bone at the sites of muscle insertion, also features lax skin, inguinal hernias, joint laxity, tortuous blood vessels and, in some, intellectual impairment [24–26]. This X-linked disorder is caused by mutations in the copper-transporting ATPase gene, ATP7A. Mutations in the same gene can also cause Menke disease; in contrast to OHS, these patients have severe neurological degeneration, growth retardation and death in early childhood, with milder skin laxity and sparse, kinky hair [26].
Management [1–3]
Investigation of an individual with cutis laxa should be directed to the identification of possible systemic complications. Hip dislocation should be checked for clinically, and with ultrasonography if indicated. Clinical examination should also look for inguinal herniation and any abnormalities of the cardiovascular system. An echocardiogram, chest computed tomography scan, magnetic resonance (MR) angiogram and pulmonary function tests may be indicated. Similarly, a renal ultrasound, voiding cystourethrogram and barium enema may demonstrate evidence of diverticular disease. A full ophthalmological examination may be needed, particularly if ARCL type 3 is suspected, and neurological evaluation, electroencephalograph and brain MR scan if indicated.
Routine management is indicated for complications such as refractive errors, emphysema, hip dislocation, inguinal hernias and seizures.
A β-blocker or angiotensin-converting enzyme inhibitor may be indicated if dilatation of the aortic root is identified. Following encouraging results wth the angiotensin II type 1 receptor antagonist losartan in Marfan syndrome, results of studies in fibulin 4-deficient mice showing reductions in elastic fibre fragmentation and increased survival are likely to lead to future clinical trials in man [30]. Copper histidine therapy may diminish the phenotypic severity of Menke disease if given within the first 2 months of life [31]. Surgical management of aneurysms and other cadiovascular anomalies may also be warranted, as may botulinum toxin injections and plastic surgery for facial involvement by cutis laxa [32, 33].