CHAPTER 82
Inherited Immunodeficiency
Tim Niehues1 and Andrew R. Gennery2
1Centre for Child Health and Adolescence, HELIOS Klinikum, Krefeld; Academic Hospital, RWTH, Aachen; Immunodeficiency and Rheumatology Centre, Krefeld, Germany
2Institute of Cellular Medicine, Newcastle University, Newcastle upon Tyne, UK
Introduction
Definition
Primary immunodeficiencies (PIDs) have been traditionally defined as a group of inherited disorders resulting from defects in the immune system that lead to an increased susceptibility to infection, manifest by recurrent, persistent or opportunistic infectious episodes. With a greater understanding of the immune system and normal immunological responses, it has become clear that defects in immune responses can also present with the following:
- Susceptibility to single ubiquitous pathogens.
- Autoimmune disease.
- Autoinflammation.
- Failure of the inflammatory response.
- Failure of lymphocyte apoptosis following infection.
- Neoplasia.
Primary immunodeficiencies are differentiated from secondary immunodeficiencies, which result from HIV infection, use of immunosuppressive drugs (e.g. biologicals, chemotherapy agents, radiotherapy, ciclosporin, etc.), nutritional deficiencies, splenectomy, uraemia, protein-losing states such as enteropathy or nephrotic syndrome, hepatic failure or cirrhosis, metabolic disorders (e.g. diabetes), atopic eczema and autoimmune diseases (e.g. systemic lupus erythematosus (SLE)) and extremes of age. PIDs represent ‘experiments of nature’ and the clinical phenotype can help demonstrate the function of a particular molecule in the human immune system.
Given the complexity of disease, and wide variety of presentations, along with the specialized knowledge required to direct appropriate therapy, PID patients are best cared for by physicians with a high level of specialization and knowledge about the immune system, who are committed to an interdisciplinary approach.
Epidemiology
While each of the more than 200 genetic defects associated with PIDs is extremely rare, PIDs as a group are seen with increased frequency and have an estimated prevalence in Europe of 4.1/100 000 individuals [1].
Diagnosis
Clinical features suggesting primary immunodeficiency
Physiological susceptibility to infection is age dependent. While between 0 and 4 years of age, individuals experience almost five infections per year, this frequency decreases with increasing age from approximately three infections per year in the age group 5–19 years and two infections per year between 20 and 39 years of age and 1.5 infections above the age of 40 years. Some PIDs do not manifest before adulthood. The most common PID in adults is common variable immunodeficiency (CVID), with a twin peak incidence at 20–40 years of age and between 50 and 60 years of age.
The physician needs to differentiate between physiological or pathological susceptibility to infection. In adults, it has been defined that more than three infections per year which require treatment (including antibiotics), and each of which last more than 4 weeks, are defined as pathological susceptibility to infection. Other clues for pathological susceptibility to infection are unusual pathogens (e.g. pneumocystis), long duration of infection, unusual localizations (organ abscesses), unusually severe infection with common pathogens or recurring infections with the same infectious agent. Other manifestations of PID include signs of immune dysregulation often referred under the acronym of GARFIELD:
- Granuloma.
- Autoimmune disease.
- Recurring Fever and chronic Inflammation.
- Unusual Eczema.
- Lymphoproliferation.
- Chronic, inflammatory bowel Disease.
The 10 Warning Signs of the Jeffrey Modell Foundation were proposed by an expert panel but were never validated prospectively. In a retrospective analysis in Great Britain, 430 children with PID were tested for the predictive value of these warning signs. Only four signs had a positive predictive value (positive family history, more than 2 months of antibiotic treatment, failure to thrive, deeply seated abscesses). Thus, whilst they are used frequently in clinical practice, immunodeficiency should be considered even if the presentation does not fulfil these warning signs:
- Four or more new ear infections within 1 year (children), two or more new ear infections within 1 year (adults).
- Two or more serious sinus infections within 1 year (in the absence of allergy, adults).
- Two or more months of antibiotics with little effect (children).
- Two or more pneumonias within 1 year (children), one pneumonia for more than 1 year (adults).
- Failure of an infant to gain weight or grow normally.
- Recurring deep skin or organ abscesses.
- Persistent oro-pharyngeal Candida or fungal infection on the skin.
- Need for intravenous antibiotics to clear infections.
- Two or more deep-seated infections, including septicaemia.
- Family history of PID.
Other warning signs include recurrent bacterial or fungal infections, recurrent viral infections (e.g. herpes, warts, condylomata), in association with chronic diarrhoea with weight loss, one pneumonia per year for more than 1 year, lymphoreticular malignancy (e.g. lymphoma in diseases of increased chromosomal breakage, intestinal lymphoma in CVID), syndromal aspects (e.g. DiGeorge syndrome, autosomal dominant hyper-IgE syndrome), albinism (e.g. Chediak–Higashi syndrome) and a history suggestive of X-linked inheritance, or parental consanguinity. The type of microorganism involved, especially if atypical, should direct further investigation. In immunodeficient patients, cutaneous infections are either caused by microorganisms not usually pathogenic in normal individuals or follow a more severe course due to infection with a common microorganism as compared with those with normal immunity. Allergic/atopic manifestations are common in PIDs and may be unusually severe. Autoimmune and malignant diseases, though not common, have an increased incidence. Photosensitivity rashes may be a particular feature in some disorders, for example in immunodeficiencies associated with DNA repair disorders.
In some conditions, e.g. immunoglobulin A (IgA) deficiency, there may be a family history of collagen vascular or other immunopathological disease. Older relatives who are carriers of an inherited immunodeficiency or who are affected by milder variants of primary immune defects may have autoimmune manifestations (e.g. mouth ulcers and SLE variant in chronic granulomatous disease (CGD)) or have a history of malignant disease (lymphoma in X-linked lymphoproliferative disease (XLP) or Wiskott–Aldrich syndrome (WAS)).
Infectious disease related manifestations of primary immunodeficiencies
Furuncular lesions or abscesses can be an overlooked manifestation of a PID. They are most characteristically seen in neutrophil disorders such as CGD, Chediak–Higashi syndrome, leukocyte adhesion deficiency and neutrophil-specific granule deficiency but also in hyper-IgE syndrome and antibody deficiencies such as X-linked agammaglobulinaemia (XLA) or in complement disorders. In these disorders it is, however, usual for infections of the skin to be accompanied by infection at other sites.
Cutaneous and mucosal ulceration are features of several immunodeficiency states and are the hallmark of leucocyte adhesion deficiency. Although infection is believed to be the likely cause of skin ulceration, it can be difficult to identify the microorganisms responsible and the accumulation of neutrophils in small blood vessels to the point where blockage and tissue necrosis occurs probably plays an important part in their genesis. Such ulcers are a characteristic feature of disorders featuring neutropenia, including congenital neutropenia, cyclical neutropenia and the Chediak–Higashi syndrome. Gradually extending cutaneous ulcers due to herpes simplex virus (HSV) are suggestive of T-lymphocyte defects, but have also been reported in XLA.
Unusually severe or extensive infections with HSV or varicella-zoster virus, including the haemorrhagic vesicles seen in haemorrhagic chickenpox, are characteristic of T-lymphocyte defects. Bullous impetigo with clear blisters may be a presentation of neutropenia. A vesicular presentation of the hyper-IgE syndrome in infancy has been described.
Ordinary viral warts are virtually never indicative of immunodeficiency. However, exceptionally rapid growth of warts, exceptionally large size or unusually extensive infections are suggestive of underlying defects of immunity [2]. Severe extensive persistent molluscum contagiosum is seen in similar disorders such as WAS, CD40 ligand deficiency and DOCK8 deficiency.
Refractory mucosal and cutaneous Candida infections are a characteristic presenting sign of several immunodeficiency disorders, particularly severe combined immunodeficiency (SCID) and severe T-lymphocyte defects. Surprisingly, systemic Candida infections are rather rare in these conditions. Persistent mucosal and cutaneous Candida infection which responds poorly to systemic treatment suggests chronic mucocutaneous candidiasis. Invasive fungal infection is a hallmark of neutrophil deficiency [3].
Non-infectious non-specific manifestations of immunodeficiency
Eczema is a characteristic cutaneous feature of some primary immunodeficiencies disorders such as WAS, and has been recorded as occurring, more frequently than one would expect, in various other disorders, including selective IgA deficiency, selective IgM deficiency, ataxia telangiectasia and combined immunodeficiencies.
Morbilliform eruptions are sometimes caused by viral infections, as in other children, but in SCID, they are quite frequently manifestations of acute graft-versus-host reactions, due either to maternofetal engraftment, or to engraftment of viable lymphocytes from a third party after the transfusion of non-irradiated blood products after birth. A rash may be seen after vaccination with live vaccines such as measles, mumps and rubella or varicella, in patients with severe T-lymphocyte immunodeficiency, who develop disease.
Petechiae, due to thrombocytopenia, are a highly characteristic feature of WAS and may also occur in Fanconi anaemia, dyskeratosis congenita, Schwachman–Diamond syndrome and the Chediak–Higashi syndrome.
Vasculitic lesions may rarely be seen as an autoimmune manifestation and have been documented in XLP, autoimmune lymphoproliferative syndrome (ALPS) and immunodysregulation polyendocrinopathy enteropathy X-linked (IPEX) syndrome, as well as CVID.
Ichthyosis can be seen in nuclear factor κB essential modulator mutation (NEMO) deficiency and Comel–Netherton syndrome.
A combination of erythroderma of early onset with failure to thrive in early infancy is highly suggestive of immunodeficiency and often results from dysregulated activated T lymphocytes invading the skin. In some cases these are maternally derived and represent true graft-versus-host disease whilst in other cases aberrant clones of the infant's own T lymphocytes cause a similar reaction which results in the condition known as Omenn syndrome, a ‘leaky’ form of SCID.
Patients with PIDs can display indurated erythematous papules and plaques with central scaling, scarring, atrophy or ulceration, which demonstrated caseating granulomas histologically. The conditions in which these have been reported include common variable immunodeficiency, XLA, ataxia telangiectasia, CGD, recombinase activating gene (RAG) deficiency and RNA component of mitochondrial RNA processing endoribonuclease (RMRP) deficiency [4].
As well as classical systemic and discoid lupus erythematosus, a syndrome resembling SLE from the cutaneous point of view, but having only very mild non-cutaneous manifestations, and either absent or very low-titre plasma antinuclear antibodies, has been described in patients with a variety of complement deficiencies most commonly C2 deficiency but also C3, C4, C1q and C1 esterase inhibitor deficiency. In such patients, this syndrome can occasionally have its onset as early as the first year of life, and, because many of these patients are susceptible to certain infections, such as meningococcal meningitis, the association of a disorder resembling lupus erythematosus and recurrent infections of appropriate type is highly suggestive of a hereditary complement deficiency.
There is an increased incidence of SLE in patients with IgA deficiency, and skin lesions closely resembling discoid lupus erythematosus have occurred in female carriers of the gene for X-linked CGD.
Hypo- or hyperpigmented lesions are characteristic of a few immunodeficiency disorders, particularly those with an underlying DNA repair disorder, such as Bloom syndrome or Nijmegen breakage syndrome. Hypopigmentation of the skin or hair is also characteristic of Chediak-Higashi and Griscelli syndrome.
Diagnostic laboratory tests
Genetic diagnosis is best directed in the context of clinical and laboratory immunological findings. Although whole exome sequencing is becoming cheaper and more easily accessible, in the absence of clinical and laboratory immunological information, interpretation of the data can be difficult. Simple immunological investigations are useful before more specialized investigations are performed: IgG, IgA, IgM and IgE, blood count and a differential leukocyte count are of paramount importance in screening for immunodeficiency (e.g. neutropenia, lymphocytopenia, monocytopenia or eosinophilia may give important clues). In paediatrics, these values must be compared to age-related normal values. A titre of specific antibodies after vaccination indicates that there is sufficient B- and T-lymphocyte interaction as well as antibody production. Only rarely is there an indication to determine IgG subclasses; this is reserved for cases that show normal IgG but a classical picture of humoral immunodeficiency. Flow cytometric analysis of lymphocyte subpopulations is an important tool for the clinical immunologist, performed in a specialized laboratory that is experienced in the interpretation of results obtained from patients with PID.
Functional tests of the immune system include tests for complement function (e.g. CH50, AP50), lymphocyte proliferation cell cycle (e.g. ataxia telangiectasia), cytotoxicity (immunodeficiencies with immune dysregulation), apoptosis (e.g. ALPS) and simulation of peripheral blood mononuclear cells with cytokines as well as calcium mobilization assays. Genetic analyses and data can only be interpreted in the context of clinical and immunological findings, as finding a mutation does not necessarily imply that it is causing the disease.
Management
While a few PIDs do not require specific treatment (e.g. selective IgA deficiency), others necessitate highly intensive treatment (e.g. haematopoietic stem cell transplantation or gene therapy for SCID, CGD and other PIDs). In many PIDs, regular immunoglobulin substitution has changed the prognosis significantly, e.g. in CVID and agammaglobulinaemia. Once a sufficient IgG trough level is established, most infectious complications significantly diminish. Supportive treatment is key in some PIDs (e.g. CGD: antibacterial and antifungal prophylaxis). Autoimmune or autoinflammatory complications may require immunosuppressive or immunomodulatory treatment (e.g. steroids in granulomatous organ disease, anti-CD20 therapy in Epstein–Barr virus complications).
Primary immunodeficiencies with skin manifestations
Primary immunodeficiencies are classified on a regular basis by an expert committee of the International Union of Immunological Societies (IUIS) [1]. PID is classified as follows:
- I: Combined immunodeficiencies.
- II: Combined immunodeficiencies with associated or syndromic features.
- III: Predominantly antibody deficiencies.
- IV: Diseases with immune dysregulation.
- V: Congenital defects of phagocyte number and function.
- VI: Defects in innate immunity.
- VII: Autoinflammatory disorders.
- VIII: Complement deficiencies.
The skin is a critical mechanical barrier, and a vital component of the innate immune system and the site where many innate and adaptive immune responses to infection are seen. It is not surprising therefore that skin infections and other dermatological manifestations are common features of PID. It is estimated, that almost 50% of children who present with PID have skin manifestations. In this chapter, we will not cover all of the more than 200 genetically defined PIDs but will focus on PIDs with prominent skin manifestation as well as manifestations affecting the hair, nails and sweat glands (Table 82.1).
Table 82.1 Primary immune deficiencies with prominent skin manifestations as well as manifestations affecting the hair, nails and sweat glands (see text for definitions of abbreviations)
| Disease | MIM | Gene | Major immune defect | Major skin manifestations |
| Severe combined immune deficiencies | ||||
| X-linked | 300400 | IL2RG | SCID | Bacterial, viral, fungal skin infection, BCG nodules |
| JAK3-deficient | 600173 | JAK3 | SCID | Bacterial, viral, fungal skin infection, BCG nodules |
| IL7Ra-deficient | 146661 | IL7Ra | SCID | Bacterial, viral, fungal skin infection, BCG nodules |
| ADA-deficient | 608958 | ADA | SCID | Bacterial, viral, fungal skin infection, BCG nodules Dermatofibrosarcoma protuberans |
| Artemis-deficient | 602450 | DCLRE1C | SCID | Bacterial, viral, fungal skin infection, BCG nodules |
| RAG1-deficient | 601457 | RAG1 | SCID/CID | Bacterial, viral, fungal skin infection, BCG nodules, occasionally granuloma |
| RAG2-deficient | 179616 | RAG2 | SCID/CID | Bacterial, viral, fungal skin infection, BCG nodules Occasionally granuloma |
| ORAI-1 | 610277 | ORAI1 | SCID | Ectodermal dysplasia |
| STIM-1 | 605921 | STIM1 | SCID | Ectodermal dysplasia |
| FOXN1 | 601705 | FOXN1 | SCID | Congenital alopecia, nail dystrophy |
| DiGeorge syndrome | 188400 | TBX1 | SCID/CID/Omenn | Omenn features, autoimmune vitiligo |
| CHARGE syndrome | 214800 | CHD/ | SCID/CID/Omenn | Omenn features, autoimmune vitiligo |
| Omenn syndrome | 603554 | Commonly RAG1/2, but other SCID genes described | SCID/CID | Erythroderma, alopecia, lymphadenopathy |
| Combined immune deficiencies | ||||
| Wiskott–Aldrich syndrome | 301000 | WAS | CID | Eczema, petechiae, bruising, severe molluscum contagiosum and varicella-zoster infection |
| DOCK8 deficiency | 243700 | DOCK8 | CID | Atopic eczema, extensive, disfiguring, concurrently occurring cutaneous herpes simplex virus, human papillomavirus, molluscum contagiosum and varicella-zoster virus infections |
| X-linked hyper-IgM syndrome | 308230 | CD40LG | CID | Oral ulceration. Severe cutaneous Pseudomonas aeruginosa infections |
| CD40 deficiency | 606843 | CD40 | CID | Oral ulceration. Severe cutaneous Pseudomonas aeruginosa infections |
| MHC class I deficiency | 604571 | TAP1, TAP2, TAPBP | CID | Necrotizing granulomatous skin lesions, located on the extremities and midface |
| X-linked lymphoproliferative syndrome | 300490 | SH2D1A | CID | Vasculitis, polyarteritis nodosa |
| X-linked inhibitor of apoptosis | 300079 | BIRC4 | CID | Fistulating skin abscesses |
| Comel–Netherton syndrome | 256500 | SPINK5 | CID | Trichorrhexis invaginata, ichthyosiform erythroderma, and atopic eczema |
| Cartilage hair hypoplasia | 250250 | RMRP | CID/SCID | Sparse, thin hair, alopecia, short-limbed dwarfism, skin carcinoma, rarely cutaneous granulomas |
| DNA repair defects | ||||
| Ataxia telangiectasia | 607585 | ATM | CID | Telangiectasia, café-au-lait spots, granulomas (some patients) |
| Nijmegen breakage syndrome | 602667 | NBS1 | CID | Microcephaly, photosensitivity, psoriatic-like lesions and hypo- or hyperpigmented lesions |
| LIG4 syndrome | 606593 | LIG4 | CID/SCID | Photosensitivity, psoriatic-like lesions and hypo- or hyperpigmented lesions |
| SCID with microcephaly, growth retardation, and sensitivity to ionizing radiation | 611291 | NHEJ1 | CID/SCID | Photosensitivity, psoriatic-like lesions and hypo- or hyperpigmented lesions |
| Bloom syndrome | 604610 | BLM | CID | Photosensitivity, malar rash |
| Immunodeficiency, centromeric instability, facial anomalies (ICF) syndrome | 242860 | DNMT3B, ZBTB24 | CID | Extensive cutaneous warts and fungal infections |
| Dyskeratosis congenita | 305000, 127550, 613989, 604319, 606471, 613987, 604173, 613988, 612199, 615190 | DKC1, TERC, TERT, TINF2, NOP10, NHP2, C160rf57, TCAB1, CTC1, RETL1 | CID | Leucoplakia, oral ulceration and reticulated hyperpigmentation, palmar hyperkeratosis, perioral reticular hyperpigmentation, nail dystrophy |
| Fanconi anaemia | 227650, 300514, 227615, 605724, 227646, 600901, 603467, 614082, 609053, 609054, 614083, 614087, 610832, 613390, 613951, 615272 | FANCA, FANCB, FANCC, BRCA2, FANCD2, FANCE, FANCF, FANCG, FANCI, BRIP1, FANCL, FANCM, PALB2, RAD51C, SLX4, ERCC4 | CID | Cutaneous macular brownish hyperpigmentation, resembling freckles in sun-exposed areas or more diffusely. Guttate macular hypopigmentation. Hypopigmentation and café-au-lait spots |
| Antibody deficiencies | ||||
| X-linked agammaglobulinaemia | 300755 | BTK | Antibody deficiency | Skin sepsis, Stevens–Johnson syndrome, vitiligo, alopecia areata |
| Autosomal recessive agammaglobulinaemia | 601495, 613500, 613501, 613502, 613506, 612692, 615214 | IGHM, IGLL, CD79A, BLNK, LRCC8, CD79B, PIK3R1 | Antibody deficiency | Skin sepsis |
| PIK3CD | 602839 | PIK3CD | Antibody deficiency | Skin, salivary gland, lacrimal gland, dental abscess, orbital cellulitis |
| Common variable immunodeficiency | 607594, 240500, 613493, 613494, 613495, 613496, 614699, 614700, 615577, 615767 | ICOS, TNFRSF13B, CD19, TNFRSF13C, MS4A1, CD81, CR2, LRBA, NFKB2, IL21 | CID, autoimmunity | Vitiligo, eczema, vasculitis, petechiae, skin infections, sarcoid-like skin granulomas, dermatomyositis, SLE |
| Immune dysregulation | ||||
| Chediak–Higashi syndrome | 214500 | LYST | Immune deficiency with albinism | Oculocutaneous albinism, partial albinism |
| Griscelli syndrome type 2 | 607624 | RAB27A | Immune deficiency with albinism | Pigmentary dilution of skin, silvery-grey hair, cytophagic histiocytic panniculitis |
| Hermansky–Pudlak syndrome type 2 | 608233 | AP3B1 | Immune deficiency with albinism | Oculocutaneous albinism with neutropenia |
| Immunodysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome | 304790 | FOXP3 | Neonatal exanthema or eczema often with early-onset insulin-dependent diabetes, autoimmune enteropathy | |
| Autoimmune lymphoproliferative syndrome | 601859, 603909, 607271, | FASLG, TNFRSF6, CASP10, CASP8, | Rarely linear IgA disease, vasculitis and urticarial – more usually lymphadenopathy | |
| Phagocytic defects | ||||
| Chronic granulomatous disease | 306400, 233700, 233690, 233710, 613960, | CYBB, NCF1, CYBA, NCF2, NCF4 | Functional phagocytic defect | Neonatal pustulosis, non-specific, impetiginized periorificial rash, impetiginized or ecthymatous broken skin followed by nodular lesions forming necrotic ulcers. Translucent papular lesions around the nose, eyes, lips and cheeks. Subcutaneous nodules at immunization sites, which ulcerate. Poor healing of surgical wounds. Perianal abscesses, chronic suppurative paronychia, scalp folliculitis, ulcerative stomatitis. |
| Congenital neutropenia | 202700, 610738, 612541, 615285, 616022, 232220, 600871, 245000, 604173, 300299 | ELANE, HAX1, G6PC3, VPS45A, JAGN1, GFI1, GSD1b, CTSC, WAS, | Neutrophil differentiation defect | Omphalitis, skin abscesses, oral ulceration, gingivitis and early loss of permanent teeth |
| Neutrophil adhesion defects | 116920, 266265, 612840, | ITGB2, SLC35C1, KIND3 | Neutrophil migration defect | Delayed umbilical cord separation, omphalitis, rapidly progressive erosive perianal ulcers, gingivitis, ulcerative stomatitis, periodontitis, inflammatory lesions, affecting the skin and resembling pyoderma gangrenosum, |
| Defects in innate immunity | ||||
| NF-κB pathway-related immunodeficiencies | 300291, 164008, | IKBKG, IKBKA | Sparse scalp hair, conical teeth, absent sweat glands. | |
| Toll-like receptor pathway defects | 607676, 612260 | IRAK4, MYD88 | Skin sepsis | |
| Warts, hypogammaglobulinaemia, infections, myelokathexis (WHIM) syndrome | 193670 | CXCR4 | Severe, generalized verrucosis, involving all cutaneous and mucosal tissues, associated with hypogammaglobulinemia and neutropenia. | |
| Epidermodysplasia verruciformis | 226400 | EVER1 EVER2 | Similar to WHIM syndrome with increased susceptibility to human papillomavirus infections manifesting as widespread, flat warts and pityriasis versicolor-like lesions and verrucous skin carcinomas, neutropenia and hypogammaglobulinaemia are not features | |
| Autosomal dominant hyper-IgE syndrome | 147060 | STAT3 | Chronic dermatitis and repeated lung and skin infections, a non-specific, excoriated, papular and pustular eruption in infancy, over scalp, scalp margins, buttocks and proximal flexures, furunculosis and staphylococcal lung infections, oral candidiasis and Candida nail infections are common | |
| Chronic mucocutaneous candidiasis | 240300, 212050, 607644, 613108, 613953, 613956, 614162 | AIRE, CARD9, CANDN1, CLEC7A, IL17RA, IL17F, gain-of-function STAT1 | Ectodermal dystrophy with onychomycosis, superficial chronic mucocutaneous candidiasis, affecting particularly oro-pharyngeal mucosa, and the perineal region | |
| Complement diseases | ||||
| C1q | 613652 | C1QA, C1QB, C1QC | Systemic lupus erythematosus, recurrent skin lesions | |
| C1r | 216950 | C1R | Lupus-like symptoms | |
| C1s | 613783 | C1S | Systemic lupus erythematosus, | |
| C2 | 217000 | C2 | Henoch–Schoenlein purpura, systemic lupus erythematosus, polyarteritis, polymyositis, vasculitis | |
| C3 | 613779 | C3 | Recurrent bacterial infections, particularly with encapsulated bacteria, systemic lupus erythematosus | |
| C4 | 614380614379 | C4AC4B | Systemic lupus erythematosus, Henoch–Schoenlein purpura Bacterial meningitis |
|
| C5 | 609536 | C5 | Systemic lupus erythematosus, neisserial infection | |
| C6 | 612446 | C6 | Meningococcal infection | |
| C7 | 610102 | C7 | Recurrent meningococcal infection, pyoderma gangrenosum | |
| C8 | 613790 120690 | C8A C8B | Recurrent neisserial infection | |
| C9 | 613825 | C9 | Systemic meningococcal infection | |
| C1 esterase inhibitor | 106100 | C1NH | Episodic local subcutaneous oedema, rarely systemic lupus erythematosus | |
| Factor H | 609814 | HF1 | Recurrent bacterial infection, atypical haemolytic uraemic syndrome | |
| Factor I | 610984 | CFI | Recurrent bacterial infection, atypical haemolytic uraemic syndrome, rarely systemic lupus erythematosus | |
Combined immunodeficiencies
Severe combined immunodeficiency
Severe combined immunodeficiency comprises at least 12 genetic subtypes in all of which T-lymphocyte activity is compromised by either a lack of development or function of mature T lymphocytes. Moreover, B-lymphocyte activity and antibody production is severely impaired. Classical clinical manifestations are recurrent severe infections, chronic diarrhoea and associated failure to thrive. Unless haematopoietic stem cell transplantation (or, in selected diseases, gene therapy) is performed, children die within the first months of life. The skin manifestations of SCID may be one of the clues to clinical diagnosis.
Skin manifestations are multiple in SCID
Due to the decreased barrier function of the skin and the lack of adaptive immunity in SCID patients, skin infections can be very severe and ulcerating. A common finding in patients for whom the diagnosis is delayed is deep skin ulceration in the diaper area infected by Gram-negative bacteria or fungi (Figure 82.1). With the recent introduction in some countries of the live attenuated rotavirus infection, prolonged diarrhoea with subsequent localized skin inflammation is likely to become more common.
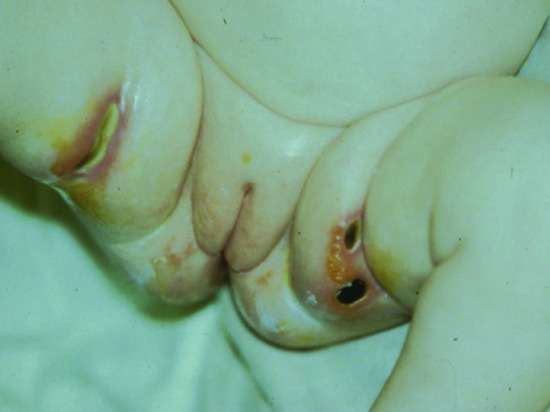
Figure 82.1 Ulcerated perineal region in an infant with severe combined immunodeficiency.
Chronic oro-perineal candidiasis
Due to the T-lymphocyte deficiency, sometimes associated with natural killer (NK) cell deficiency, persistent cutaneous Candida infection may be found.
Erythroderma
A morbilliform rash with fever, evolving into an erythematous exfoliating protein-losing rash which becomes a confluent erythema may be due to graft-versus-host disease. Whilst conventionally this is a complication of post-haematopoietic stem cell transplantation, in patients who have severe T-lymphocyte immunodeficiency, non-host T lymphocytes can cause severe, often fatal graft-versus-host disease. This is most commonly found following engraftment of transplacental maternal T lymphocytes (Figure 82.2), but transfusion of non-irradiated blood products may also transfer immune-competent HLA-reactive T lymphocytes. Histological examination will confirm the diagnosis, and genetic finger printing of circulating lymphocytes may help determine the origin of the cells.’
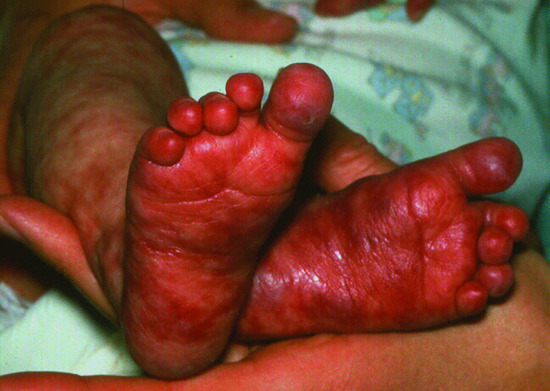
Figure 82.2 Maternofetal graft-versus-host disease in an infant with JAK3-deficient severe combined immunodeficiency.
Omenn syndrome is characterized by a generalized thickened erythematous rash, often with scaling and erythroderma [5]. The initial appearances may be papular, becoming confluent and the skin often becomes thickened with a ‘leathery’ consistency. Hair, including the eyebrows, is often lost as the rash evolves (Figure 82.3). The rash may be present at birth or evolve over the first few weeks of life. There is also lymphadenopathy particularly of the axillary and inguinal nodes as well as increased serum IgE levels with a marked eosinophilia and combined immunodeficiency. Children usually present in early infancy but may present later in the first year of life and suffer from diarrhoea, failure to thrive and persistent infection as seen in other forms of SCID. There are abnormally high numbers of activated oligoclonal poorly functional T lymphocytes, which have a restricted Vβ repertoire and there are high levels of circulating inflammatory cytokines. Peripheral B-lymphocyte numbers are low or absent, as are levels of immunoglobulin classes other than IgE. The clinical picture may resemble SCID with maternofetal engraftment. Histology of the skin shows a dense dermal perivascular lymphohistiocytic infiltrate, comprising activated T lymphocytes, with numerous eosinophils. S100-positive Langerhans cells are usually absent and there is no epidermotropism. Lymph node architecture is disordered, being replaced by a massive infiltrate of S100-positive interdigitating reticulum cells with absence of germinal centres, absent B cells and paucity of T lymphocytes. Activated oligoclonal lymphocytes in skin seemingly provoke Langerhans cells to migrate to lymph nodes, liver and spleen where lymphoid tissue architecture is severely disrupted. Molecular genetic studies to identify the origin of the T lymphocytes in the blood (maternal or autologous) will differentiate between Omenn syndrome and graft-versus-host reaction. Calcineurin inhibitors and interferon γ may ameliorate the clinical symptoms, but haematopoietic stem cell transplantation is the only curative treatment.
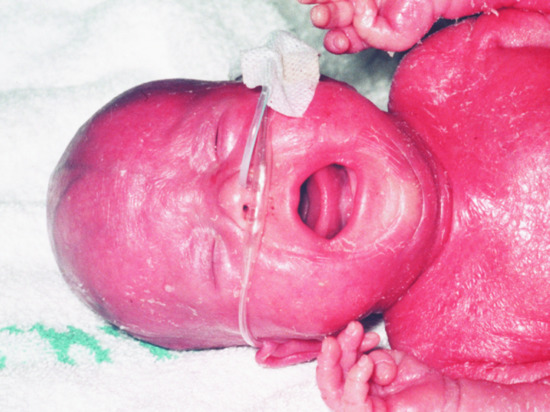
Figure 82.3 Omenn syndrome, with characteristic erythroderma and alopecia.
Granulomatous skin lesions
Granulomas may occur in the skin and present in a livedo-like fashion [4]. These skin granulomas have been observed in different PIDs including atypical SCID, where residual function of autologous T and B lymphocytes is retained. Histopathologically, these granulomas are epitheloid and non-caseating (Figure 82.4).
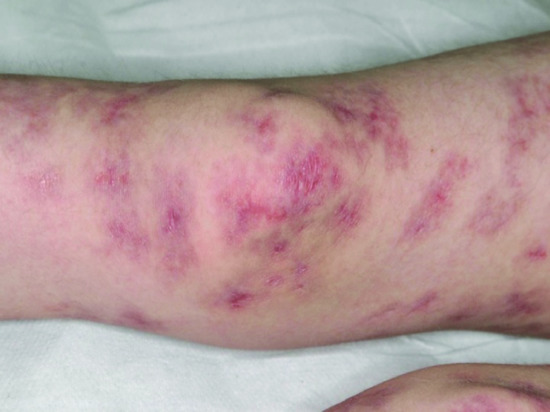
Figure 82.4 Inflammatory granulomatous skin lesions in a child with atypical severe combined immunodeficiency.
Bacille Calmette–Guérin infection
In certain geographical areas, infants with SCID may receive the live attenuated bacille Calmette–Guérin vaccine in the first week of life, before the immunological diagnosis is considered. Surprisingly, in many, the infection is latent, but a few present with disseminated BCG, which can affect many organs, and may manifest on the skin as discrete nodules, which upon biopsy will be found to contain acid-fast bacilli (Figure 82.5). Many of these lesions become inflamed following transplantation, as effective T-lymphocyte immunity is established, and may progress to form draining abscesses, particularly over lymph nodes.

Figure 82.5 Cutaneous lesion in a child with severe combined immunodeficiency who had been immunized with the live bacille Calmette–Guérin (BCG) vaccine. On biopsy, acid-fast bacilli were seen, and BCG strain mycobacteria were subsequently isolated.
Some particular SCID genotypes are associated with specific skin manifestations in addition to the ones described above.
- Adenosine deaminase (ADA) deficiency: these patients suffer from a rare malignant skin tumour, dermatofibrosarcoma protuberans. In addition, skeletal abnormalities (cupping deformities of the ends of the ribs, as well as abnormalities of the transverse vertebral processes and the scapulae) are reported in up to 50% of cases of ADA deficiency.
- Calcium-channel deficiencies (ORAI-1, STIM-1): some patients have ectodermal dysplasia and myopathy.
- Winged helix deficiency (nude phenotype – FOXN1): this form of SCID is associated with congenital alopecia and nail dystrophy.
- Complete DiGeorge syndrome and CHARGE syndrome: DiGeorge syndrome is a relatively common (1/4000) syndrome, classically with cardiac anomalies, hypoplastic thymus and hypocalcemia as well as typical facial dysmorphic features. CHARGE syndrome is a related disorder, with cardiac and oesophageal anomalies. In both cases, the complete form of the syndrome results in thymic aplasia and absence of T lymphocytes. Skin manifestations consist in an eczematous dermatitis with lymphadenopathy, similar to Omenn syndrome. In the partial forms of the disease, autoimmune phenomena may include vitiligo.
Other combined immunodeficiencies
Wiskott–Aldrich syndrome
Immunodeficiency, thrombocytopenia, eczema and an increased risk of autoimmune disorders and malignancy characterize this X-linked recessive condition. The gene responsible for coding for the Wiskott–Aldrich syndrome protein (WASP) is only found in bone marrow derived cells and is essential for actin cytoskeleton polymerization and consequently the correct assembly of cell surface receptors as well as cell movement. Patients who do express WASP, albeit in defective form, have a milder phenotype, known as X-linked thrombocytopenia, which is characterized by thrombocytopenia and bleeding tendency, but without eczema or immunodeficiency. Missense mutations in exons 1–3 lead to normal protein, whereas most other mutations result in the absence of WASP. Patients who do not express WASP at all usually exhibit the classical triad of thrombocytopenia, recurrent infections and eczema (Figure 82.6) but these vary in severity and in some patients the eczema is surprisingly mild. In general, it is indistinguishable from atopic eczema apart from the characteristic presence of purpura and bleeding from excoriation in many patients. The condition usually presents in early childhood with bruising, petechiae and bleeding: thrombocytopenia and bleeding episodes may require platelet transfusions. Herpesviruses, including herpes simplex and varicella-zoster virus, are poorly handled and may cause severe and recurrent disease. Impetigo, cellulitis and skin abscesses are surprisingly common, molluscum contagiosum and viral warts may be very extensive and together with excessive bruising help to clinically distinguish WAS from uncomplicated eczema. Indeed, very extensive molluscum contagiosum is quite characteristic of WAS. Infection exacerbates the bleeding tendency, and early death may result from bleeding. With increasing age, infectious complications replace bleeding as the major cause of death. Autoimmunity, particularly autoimmune haemolytic anaemia and vasculitis, and malignancy, particularly of the lymphoreticular system, become more common with increasing age and in many cases are related to abnormal persistence of Epstein–Barr viral infection. Thrombocytopenia with an abnormally small mean platelet volume (<5 femtolitres) is pathognomonic. The severity of immunodeficiency is variable, but progresses with age and affects cellular and humoral responses. Acute bleeding episodes may be controlled by platelet transfusions (irradiated to prevent graft-versus-host disease). Splenectomy and systemic steroids should be avoided if possible as they will increase the risk of infection and death. Topical steroids are required for the eczema. Intravenous immunoglobulin, with or without prophylactic antibiotics, reduces bacterial sinopulmonary infections and in high dose may help treat autoimmune phenomena. With only these supportive measures, the prognosis remains poor. Immunological and haematological reconstitution can be achieved by haematopoietic stem cell transplantation (HSCT) and despite a higher risk of Epstein–Barr virus driven lymphoproliferative disorders, results are good with a 5-year overall survival of 90% in transplants performed since 2000 [6].
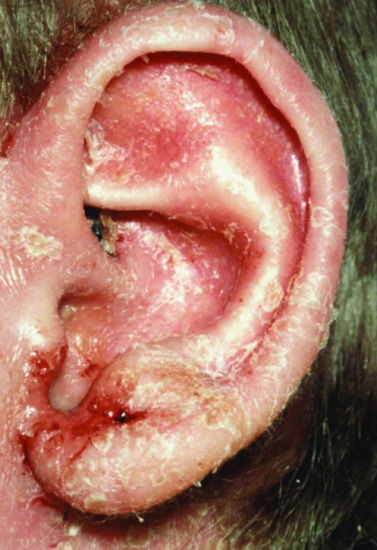
Figure 82.6 Severe eczema in a child with Wiskott–Aldrich syndrome.
DOCK8 deficiency
Mutations in dedicator of cytokinesis 8 (DOCK8) are the cause an autosomal recessive combined immunodeficiency with hyper-IgE. Cutaneous features include atopic eczema. The most significant feature of DOCK8 deficiency is susceptibility to extensive, disfiguring, concurrently occurring cutaneous viral infections, particularly HSV, human papillomavirus, molluscum contagiosum and varicella-zoster virus [7] (Figure 82.7). There are also Staphylococcus aureus skin abscesses and soft-tissue infections. Other invasive infections are described including recurrent sinopulmonary infection and meningitis from a wide spectrum of Gram-positive and Gram-negative bacteria, and intracellular fungi, such as Histoplasma capsulatum. Mucocutaneous candidiasis and recurrent gastrointestinal tract infections are common. Patients can also suffer from severe and extensive food allergies. Increased serum IgE levels, and eosinophilia are found. Patients with DOCK8 deficiency are at high risk of developing malignancies, particularly lymphomas and squamous carcinomas. HSCT is curative.
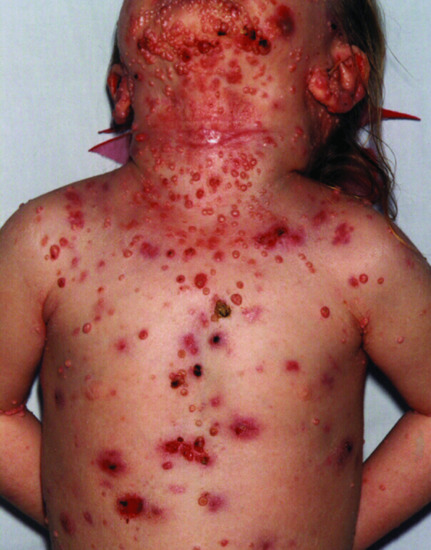
Figure 82.7 Severe molluscum contagiosum in a child with DOCK8 deficiency.
CD40 ligand and CD40 deficiencies
Skin manifestations are related to the coexisting neutropenia, and include oral ulceration. Severe cutaneous infections, particularly due to Pseudomonas aeruginosa may occur.
Major histocompatibility complex class I deficiency
Clinically, this disease has a milder phenotype than major histocompatibility complex (MHC) II deficiency with symptoms often not beginning until late childhood, and is caused by deficiencies in the transporter associated with antigen processing 1 or 2, or TAP binding proteins. Recurrent respiratory tract infections leading to bronchiectasis and sinus problems are common. Gastrointestinal disease is rare. The most striking clinical manifestation is necrotizing granulomatous skin lesions, which are located on the extremities (Figure 82.8) and also in the midface. This lesion begins with a small pustule or subcutaneous module, which slowly expands and ulcerates. The lesions are slow to heal, and usually leave hyperpigmented scars. Midface lesions can be particularly mutilating and resemble midline granuloma. Diagnosis is confirmed by showing absent HLA class I expression in peripheral blood. Treatment is directed towards prevention/limitation of lung disease with judicious use of antibiotics (directed by sputum cultures); physiotherapy and bronchodilators as required. Prophylactic continuous antibiotics are of unproven benefit, but may be helpful.
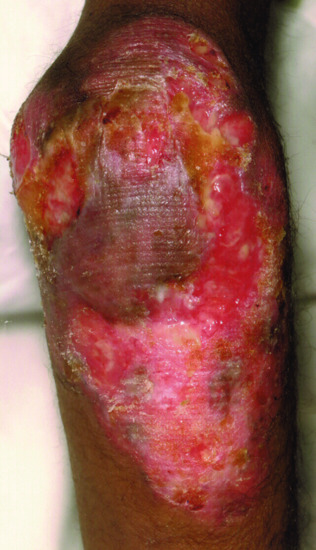
Figure 82.8 Sterile necrotizing granulomatous lesion on the knee of a patient with TAP1 deficiency.
X-linked lymphoproliferative diseases
An X-linked immunodeficiency associated with fulminant fatal Epstein–Barr virus-driven infectious mononucleosis was first recognized in the Duncan kindred, a large midwestern American family in which six boys were affected, and after whom the disease was originally named (Duncan disease). There are three common clinical presentations, fulminant infectious mononucleosis (58%), dysgammaglobulinaemia, often evolving to CVID (31%) and Epstein–Barr virus-driven B lymphoma, usually extranodal, and affecting the gastrointestinal tract or central nervous system (20%). Less commonly, patients present with vasculitis, aplastic anaemia, haemophagocytic lymphohistiocytosis, pulmonary lymphomatoid granulomatosis or vasculitis. Few cases of XLP-associated vasculitis have been published (Figure 82.9). Polyarteritis nodosa (PAN) like vasculitis has been reported. Vasculitic changes in small and medium-sized muscular arteries have also been reported. A clinical picture of PAN in a boy with marked lymphadenopathy, erythrophagocytosis or recent infection should raise suspicion of this disease.
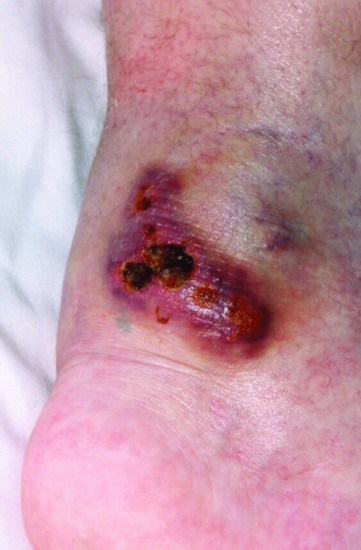
Figure 82.9 Vasculitis in a boy with X-linked lymphoproliferative disease.
The prognosis is poor with a high risk of death during initial Epstein–Barr virus infection and no recorded survivors after 40 years of age. Most patients are well until infected with Epstein–Barr virus, although other viruses may act as triggers. Confirmation of the diagnosis involves demonstrating Epstein–Barr virus genome in blood by polymerase chain reaction, together with immune defects outlined above and an abnormal response to the Epstein–Barr virus with absent antibody response to EB nuclear antigen (EBNA). Haemophagocytic lymphohistiocytic (HLH) episodes are treated as per the HLH 2004 protocol, a combination of dexamethasone, etoposide and intrathecal methotrexate if there is evidence of central nervous system disease, and immunosuppression with ciclosporin; intravenous immunoglobulin is beneficial particularly when hypogammaglobulinaemia is present.
More recently, a defect in BIRC4 has been described in patients with X-linked inhibitor of apoptosis protein (XIAP) defects. Patients also present with HLH, but other symptoms include Crohn-like bowel disease, isolated splenomegaly and fistulating skin abscesses. HSCT is the only curative treatment for either genetic defect.
Combined immunodeficiencies with associated or syndromic features
DNA repair defects (see also Chapter 78)
These include ataxia telangiectasia, Nijmegen breakage syndrome, ligase 4 (LIG4) syndrome and SCID with microcephaly, growth retardation and sensitivity to ionizing radiation (cernunnos–XLF deficiency), Bloom syndrome, and immunodeficiency, centromeric instability–facial anomalies (ICF) syndrome. Patients with ataxia telangiectasia, usually present with gait abnormalities before the development of characteristic telangiectasias, which usually appear first on the bulbar conjunctivae (Figure 82.10) but later elsewhere, particularly on the nose, ears and in the antecubital and popliteal fossae. The other DNA repair disorders are associated with facial dysmorphism, and often with small stature. Microcephaly is often a feature. Skin manifestations include photosensitivity, psoriatic-like lesions and hypo- or hyperpigmented lesions (Figure 82.11). Patients are at increased risk of developing lymphoreticular malignancy. Extensive warts or spreading cutaneous fungal infection are features of ICF syndrome (Figure 82.12).
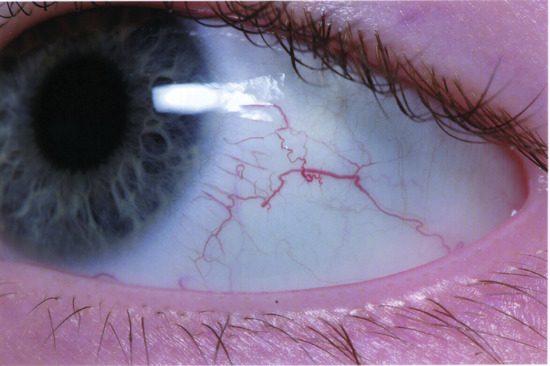
Figure 82.10 Bulbar telangiectasia in a patient with ataxia telangiectasia.
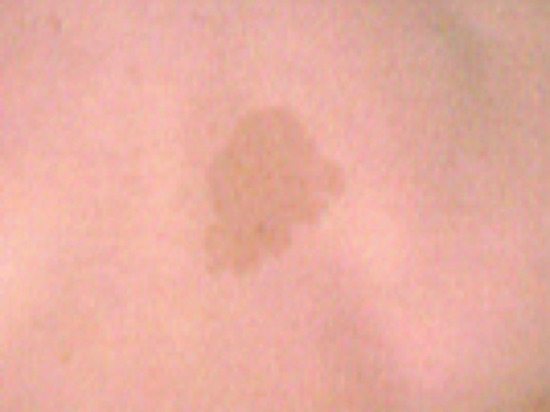
Figure 82.11 Hyperpigmented area on the back of a patient with Bloom syndrome.
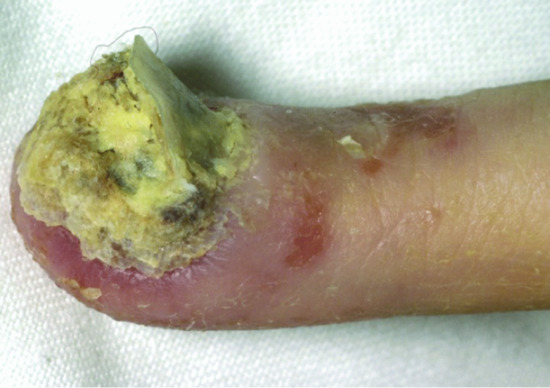
Figure 82.12 Extensive digital fungal infection in immunodeficiency, centromeric instability, facial dysmorphism syndrome.
Comel–Netherton syndrome (see also Chapter 65)
Patients with Comel–Netherton syndrome, due to mutations in SPINK5 encoding the serine protease inhibitor LEKTI, present with a triad of generalized infantile ichthyosiform erythroderma and atopic diathesis, diarrhoea and failure to thrive which may be associated with a variable immunodeficiency including mild lymphopenia and polysaccharide antibody deficiency. The clinical features may resemble those seen in Omenn syndrome or SCID with maternofetal engraftment, erythroderma and ichthyosis characterized by abnormal cornification, dryness and fish-like scaling of the skin. Hair shaft abnormalities (trichorrhexis invaginata or bamboo hair) are diagnostic, but may not be seen until several months of age. Distinguishing these entities is important as the other conditions are treated by haematopoietic stem cell transplantation, whereas Comel–Netherton syndrome is generally treated with conservative measures. Squamous cell carcinoma has been described in Comel–Netherton syndrome.
Cartilage hair hypoplasia
This is an autosomal recessive T-lymphocyte deficiency, usually associated with short-limbed dwarfism and alopecia. Characteristically, the hair of the scalp, eyebrows and eyelashes is sparse and upon hair microscopy there are characteristic findings of a lack of a central pigmented core. Cartilage hair hypoplasia, the best described of the immuno-osteochondrodysplasias, inherited in an autosomal recessive manner, is associated with mutations in RMRP, which encodes endoribonuclease Rnase MRP. Severe short-limbed short stature (–11.8 SD to 2.1 SD) with X-ray appearances of metaphyseal and spondyloepiphyseal dysplasia are characteristic, although not always present, and most patients have sparse light hair. Severe anaemia and Hirschsprung disease are less common but well-recognized associations, as are malignancies, notably lymphoma and skin carcinoma. Cutaneous granulomatous lesions are also described. The immunodeficiency is surprisingly variable; most patients have T lymphopenia, and impaired in vitro mitogen proliferative responses, but although half suffer from recurrent infections, a SCID-like presentation is well recognized. Patients are excessively vulnerable to viral infections, particularly varicella-zoster virus, Epstein–Barr virus and other human herpesvirus infections, and the risk of infective death is 300 times greater than normal. This condition should be considered in any child with severe chickenpox or herpes simplex infections who is short and has fine sparse hair. Severely affected patients should be assessed for HSCT, which has been successful in correcting the immunodeficiency.
Dyskeratosis congenita (see also Chapters 69 and 77)
Dyskeratosis congenita (DC) presents with recurrent infections, digestive tract involvement and pancytopenia. Cutaneous manifestations include leucoplakia, oral ulceration and reticulated hyperpigmentation, primarily in the regions of the neck, upper thorax and upper extremities. Some patients present with palmar hyperkeratosis and a characteristic perioral reticular hyperpigmentation (Figure 82.13). Nail dystrophy is typical and begins with longitudinal striations, increasing brittleness, deformation, onychoclasis and nail loss. Other cutaneous features include palmar and plantar hyperhidrosis, blistering, acrocyanosis and alopecia with sparse scalp hair and eyelashes. Mutations have been described in dyskerin (DKC1), NOLA2 (NHP2) or NOLA3 (NOP10), in TERC or in TERT as well as in TINF2, C160rf57, TCAB1, CTC1, RETL1 and WRAP53, which code for proteins essential for telomere maintenance. A particularly severe variant of DC, Hoyeraal–Hreidarsson syndrome, is characterized by microcephaly, cerebellar hypoplasia, aplastic anaemia and growth retardation as well as a progressive combined immunodeficiency, with hypogammaglobulinaemia and lymphopenia.
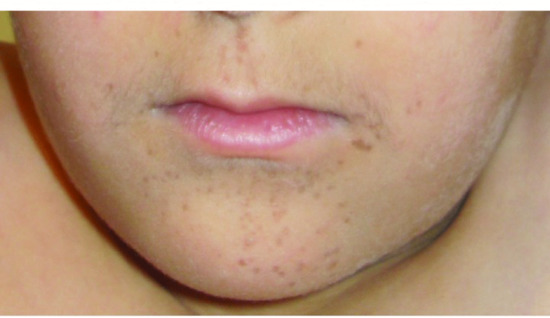
Figure 82.13 Characteristic perioral reticular hyperpigmentation in dyskeratosis congenita.
Fanconi anaemia (see also Chapter 78)
Fanconi anaemia is a chromosomal instability disorder characterized by developmental defects, progressive bone marrow failure and cancer susceptibility. Children with Fanconi anaemia are generally small and elfin-like with a history of low birth weight. The major clinical manifestation of Fanconi syndrome is progressive bone marrow failure during childhood and adolescence. Easy bruising is a common presenting symptom, most often apparent between the ages of 4 and 10 years. In addition to thrombocytopenia, anaemia and leucopenia are frequent, and the bone marrow is aplastic.
The principal skeletal abnormality is absence or hypoplasia of at least one of the thumbs; hypoplasia or absence of the radius is also common. Structural renal abnormalities, endocrinopathies, genital hypoplasia, microcephaly and microphthalmia are other common features.
Cutaneous hyperpigmentation is characteristic. Macular brownish pigmentation is typical, either resembling freckles and occurring mainly in sun-exposed areas or more diffusely, in which case the abdomen, genital area and flexures appear to be predominantly affected. Guttate macular hypopigmentation is often also present in affected areas. Hypopigmentation and café-au-lait spots are reported. Persistent and exceptionally severe viral wart infection can occur.
There is a high risk of leukaemia and other malignancies in these children, probably due to the structural instability of chromosomes observed in Fanconi anaemia, which leads to a high frequency of chromosomal breaks and rearrangements.
At least 16 complementation groups (A–Q) have been described and the gene defects have now been discovered. The Fanconi proteins are involved in the repair of DNA cross-linking damage and some interact with other DNA repair pathways. The outlook for untreated patients with Fanconi anaemia is poor, with death usually occurring within a few years of the first signs of marrow failure. Initially, bone marrow function can be stimulated with corticosteroids and with the androgenic steroid, oxymethalone. HSCT has been used successfully to treat patients, but care must be taken to avoid the use of alkylating agents in pretransplant conditioning. Patients are at increased risk of secondary malignancy post-transplantation.
Antibody deficiencies
X-linked agammaglobulinaemia (Bruton disease)
First described by Bruton in 1952, this X-linked intrinsic B-lymphocyte defect prevents B-lymphocyte development beyond the pre-B-lymphocyte stage. It is caused by mutations in a gene that encodes a cytoplasmic enzyme, Bruton tyrosine kinase (btk). Classically, affected boys demonstrate absence or severe depletion of all serum immunoglobulin classes, and antibody responses to vaccines are absent. There are normal numbers of T lymphocytes, but no B lymphocytes in peripheral blood, although pre-B lymphocytes (containing cytoplasmic μ chains) are found in bone marrow. Lymph nodes show absent follicles and germinal centres, and plasma cells cannot be demonstrated at any site. The diagnosis can be rapidly confirmed by demonstrating the absence of the btk protein in cell lysates. Since the molecular basis has been defined, milder phenotypes have been recognized where some antibody function is present. Typically, recurrent pyogenic infections commence in the latter half of the first year of life, once maternal IgG levels have declined. The diagnosis is often made surprisingly late; in one series, the average age at diagnosis was 3.5 years and 2 years even when there was a positive family history. Sinopulmonary infections are most common, but gastroenteritis, arthritis, meningitis and osteomyelitis may be presenting features. Boils or impetigo, usually associated with neutropenia, are the most common dermatological features, frequently due to Staphylococcus aureus or Pseudomonas (Figure 82.14), although other organisms are also described. Chronic ulcerative cutaneous HSV infection has been reported. Stevens–Johnson syndrome, vitiligo and total alopecia areata have also been described. Immunoglobulin replacement therapy is the mainstay of treatment. Chronic lung damage and sinus disease may progress on treatment and for this reason vigorous and early antibiotic therapy should be used for respiratory tract infections.
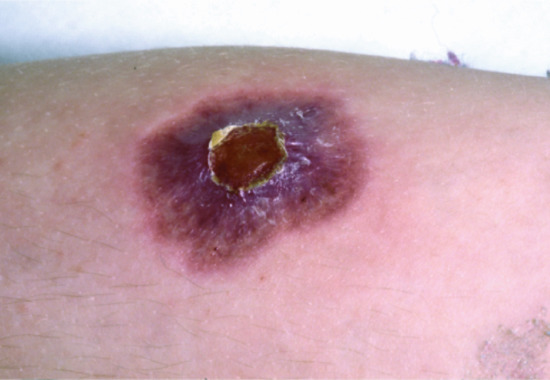
Figure 82.14 Cellulitis due to Pseudomonas infection in a patient with X-linked agammaglobulinaemia.
Autosomal recessive forms of agammaglobulinaemia
When hypogammaglobulinaemia is present in a female or a child with consanguineous parents, autosomal recessive genetic defects affecting B-lymphocyte differentiation should be considered. Mutations have been described so far in genes coding for: μ heavy chain, Ig α (part of the signal transduction complex of the B-lymphocyte antigen receptor), l5 light chain and BLNK (B-lymphocyte linker protein) molecules required for early B-lymphocyte development from pro-B lymphocyte to pre-B lymphocyte stage. Unlike XLA, pre-B lymphocytes are therefore not detectable in marrow samples. Other families have also been described in whom the molecular defect is yet to be identified. In all cases, the defect is B lymphocyte specific. Skin sepsis is described as a feature, but less commonly than in XLA.
PI3Kδ deficiency
PI3Kδ deficiency, a recently described, seemingly common, antibody deficiency due to dominant gain of function mutations in the gene encoding phosphoinositide 3 kinase δ predominantly presents with recurrent sinopulmonary infections. Cutaneous signs are seen in the skin, salivary glands and lacrimal glands, or in dental abscess formation, as well as orbital cellulitis.
Common variable immunodeficiency
Common variable immunodeficiency is defined by low total concentrations of IgG as well as low IgA and/or IgM. Patients demonstrate recurrent infections, chronic lung disease including bronchiectases, gastrointestinal inflammatory diseases and other manifestations. Cutaneous manifestations include vitiligo, eczema, vasculitis and petechiae (as a result of autoimmune dysregulation and cytopenia) and skin infections. Autoinflammation manifests as sarcoid-like granulomas of the skin. Dermatomyositis and SLE also have an increased incidence in these kindreds.
Diseases with immune dysregulation
Primary immunodeficiencies with hypopigmentation are due to the lack of formation or trafficking of cytotoxic granules. Chediak–Higashi, Griscelli type 2 and Hermansky–Pudlak syndromes are examples of these diseases. In Chediak–Higashi and Hermansky–Pudlak syndrome, there is partial albinism. Chediak–Higashi syndrome is characterized by oculocutaneous albinism, recurrent infections and neuropathy. Photophobia and nystagmus are regular features, due to ocular pigment dilution. Severe gingivitis and oral mucosal ulceration are well described. Deficient cutaneous pigmentation may be obvious, but, in some cases, is more subtle (Figure 82.15), and only apparent if nipples and genitalia are carefully examined. Under light microscopy, characteristic giant lysosomal granules are seen in the cytoplasm of all cells containing these organelles, and are easily detected on a peripheral blood film. Chediak–Higashi hair shafts show small aggregates of clumped pigmentation. The genetic defect is in LYST, which encodes a protein required for sorting endosomal resident proteins into late multivesicular endosomes by a mechanism involving microtubules. Patients usually, but not invariably, enter an accelerated lymphocyte and macrophage activation syndrome (similar to that seen in XLP), which if untreated is usually fatal. There is widespread tissue infiltration with activated lymphocytes and macrophages resulting in rapid enlargement of the liver, spleen and lymph nodes, together with jaundice, hepatic failure, respiratory distress, pancytopenia and bleeding. Death usually occurs in the first decade without HSCT, but survival into the second and third decades has been recorded. Progressive neurological deterioration is common in patients who survive early childhood, and is not prevented by HSCT.
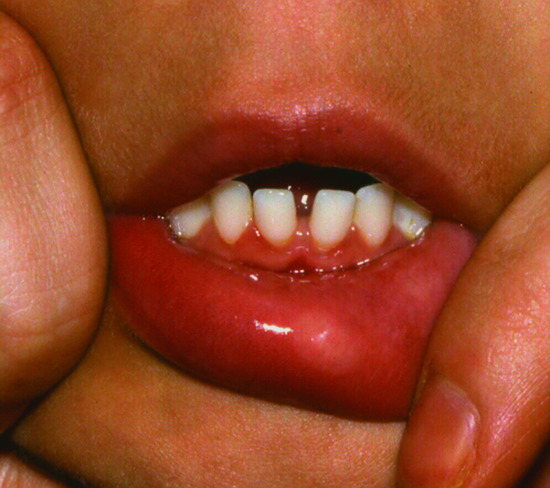
Figure 82.15 Partial albinism in Chediak–Higashi syndrome.
Hermansky–Pudlak syndrome is a disorder characterized by oculocutaneous albinism. Nine subtypes exist, but only type 2, caused by mutations in AP3B1, is also associated with congenital neutropenia. Other associations include platelet dysfunction, and, occasionally, interstitial lung disease, pulmonary fibrosis and inflammatory colitis.
In Griscelli syndrome, there is a peculiar pigmentary dilution of the skin and hair. Microscopy demonstrates an uneven distribution of large pigment granules in the hair shafts (Figure 82.16). Three genes have been identified as causing the disease. Defects in MYO5A which encodes myosin VA, and MLPH, encoding melanophilin, present with hypomelanosis, but no neurological or immunological defect. Patients with mutations in RAB27A present with similar features of hypopigmentation, characterized by typical silvery-grey hair, which persists even after haematopoietic stem cell transplantation, but also with recurrent pyogenic infections. In patients presenting with haemophagocytic lymphohistiocytosis, cytophagic histiocytic panniculitis has been rarely described.

Figure 82.16 Patient with Griscelli syndrome demonstrating a silver sheen to the hair which has persisted post haematopoietic stem cell transplantation.
Immunodysregulation polyendocrinopathy enteropathy X-linked syndrome
Immunodysregulation polyendocrinopathy enteropathy X-linked syndrome is a disease in which there is X-linked immune dysregulation due to mutations in FOXP3, encoding a transcription factor needed for the development of regulatory T lymphocytes. These children usually present in the neonatal or early infancy period with an impressive exanthema or eczema. There is often early-onset insulin-dependent diabetes, autoimmune enteropathy, thyroiditis and autoimmune cytopenias. Bruising may be apparent secondary to autoimmune thrombocytopenia. Defects with similar autoimmune manifestations, and particularly enteropathy include deficiencies of CD25, IL-10, IL-10 receptor subunits, in which folliculitis may be a feature, and the recently described gain-of-function mutations in STAT3.
Autoimmune lymphoproliferative syndrome
Apoptosis, or programmed cell death, is important for regulating immune responses once an infection has been countered. Defects in apoptosis lead to autoimmune and lymphoproliferative features which characterize ALPS. There are a number of pathways through which apoptosis can be induced; one of the most important is initiated through a cell surface molecule Fas (CD95). Ligation of CD95 initiates a cascade of intracellular reactions culminating in apoptosis induced by proteolytic enzymes including caspases. Mutations in molecules in this cascade result in genetically distinct but clinically similar forms of ALPS. Fas is expressed as a trimeric surface protein. Heterozyogotes with a Fas mutation in one allele often develop the full clinical syndrome as one abnormal protein chain is sufficient to significantly impair the trimer's function, a so-called dominant-negative effect. Most of the cases are due to heterozygous mutations though a few homozygous cases have also been reported.
Many patients present in early childhood, but adult presentation and asymptomatic cases may occur. Patients usually remain well, until the disease is triggered, often by human herpesvirus infection. Haematological autoimmunity is most common, but any system can be involved. Childhood linear IgA disease has been described (Figure 82.17). Other dermatological manifestations include vasculitis and urticaria. Lymphoproliferation leads to significant asymmetrical anterior cervical lymphadenopathy, with splenomegaly in nearly all cases and hepatomegaly in some. Malignant lymphoid disease (both Hodgkin and non-Hodgkin) is reported with increased frequency, although the histological picture of proliferation may be benign: clonality studies distinguish the two. Affected individuals usually have high lymphocyte counts and normal or high immunoglobulin levels. Autoantibodies are usually present. The occurrence of circulating CD3-positive T lymphocytes expressing the α/β receptor but not expressing CD4 or CD8 (so-called double-negative T lymphocytes) and usually constituting between 5 and 20% of the total CD3 cell count, is helpful in making a diagnosis of FAS deficiency, but raised vitamin B12 and soluble FAS ligand are more predictive. Autoimmunity usually responds to corticosteroids, high-dose intravenous immunoglobulin, and dapsone in the case of ALPS-associated juvenile bullous dermatosis. Splenectomy should be avoided if possible as severe infective complications may follow.
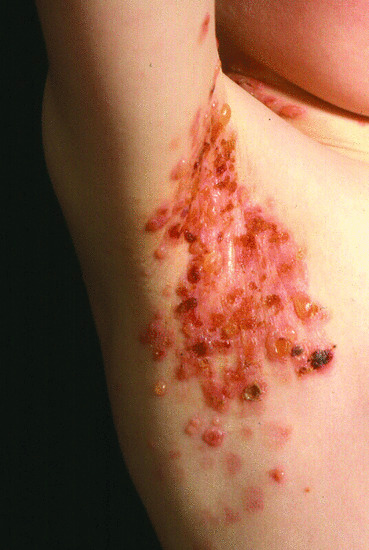
Figure 82.17 Childhood linear IgA disease in FAS-deficient autoimmune lymphoproliferative syndrome.
Congenital defects of phagocyte function, differentiation and adhesion
Functional neutrophil defects
The prototype of functional phagocyte deficiency is CGD, a result of an inherited defect in one of the six components of the phagocyte nicotinamide adenine dinucleotide phosphate (NADPH) oxidase enzyme complex which generates reactive oxygen species. The disease has numerous clinical manifestations, but the hallmark is acute, and potentially fatal, bacterial or fungal infection. Patients suffer from organ abscesses with staphylococci or fungi in the spleen, liver and lung, as well as lymph node abscesses. The earliest manifestations are often seen in the skin. Neonatal pustulosis is commonly the first sign of the disease. Subsequently, a rather non-specific, impetiginized periorificial rash is highly characteristic, most commonly seen around the nostrils, ears, mouth and eyes, and has sometimes been described as ‘eczematous’ or ‘seborrhoeic’.
Any area where the skin has been broken, by abrasion for example, tends to become impetiginized or ecthymatous. Nodular lesions may follow, and these frequently break down to form necrotic ulcers. Firm translucent papular lesions around the nose, eyes (Figure 82.18), lips and on the cheeks may mimic lupus vulgaris or sarcoidosis. Subcutaneous nodules may develop at immunization sites, and these also tend in time to ulcerate. Poor healing of surgical wounds, and of the discharging nodular skin lesions, is characteristic. Perianal abscesses are a regular feature. Other frequent findings include chronic suppurative paronychia, folliculitis of the scalp and ulcerative stomatitis. Acute febrile neutrophilic dermatosis (Sweet syndrome) has been rarely described, as has chronic bullous disease of childhood. The importance of non-infectious inflammatory complications is increasingly recognized. These include inflammatory bowel disease, which clinically and histologically can be indistinguishable from Crohn disease, restrictive lung defects, acute genitourinary obstruction and cutaneous granulomata particularly at vaccination sites. Female carriers not infrequently develop erythematous macular, papular and urticarial skin lesions following light exposure, and discoid lupus erythematosus or Jessner lymphocytic infiltrate (Figure 82.19).
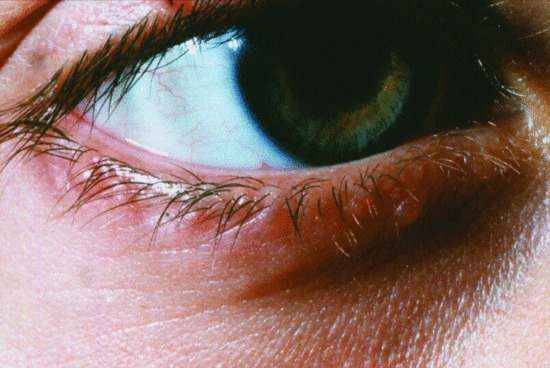
Figure 82.18 Translucent papular lesions around the eyelids of a patient with chronic granulomatous disease.
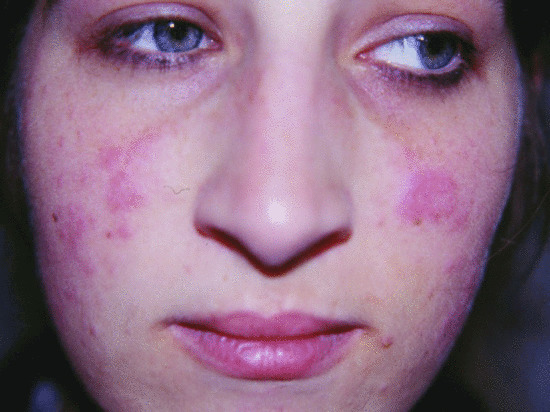
Figure 82.19 Malar erythematous photosensitive macular skin lesions in an X-linked carrier of chronic granulomatous disease.
Defects of neutrophil differentiation
There are at least nine subtypes of congenital neutropenia that have been described. Neutropenia may present as poorly healing, deep ulcerations of the skin and unusual abscesses. Patients with neutropenia present usually in infancy when omphalitis, skin and liver abscesses are most common. Moreover, there may be oral ulceration, gingivitis and early loss of permanent teeth. In Papillon–Lefèvre syndrome, there are mutations in CTSC encoding cathepsin C. Patients have aggressive periodontitis, leading to dental loss and palmoplantar hyperkeratosis.
Defects of neutrophil adhesion
In these disorders, adhesion molecules including CD18, FUCT1 (GDP fucose transporter) or other integrins are affected. Patients present with poor wound healing despite marked leucocytosis in their blood. The clinical picture is almost entirely explained by the way in which leukocytes are attracted to areas of infection and inflammation. Leukocytes normally attach to vessel walls at sites of inflammation but cannot pass out into the tissues. This leads to blockage of small vessels and rapidly expanding necrotic lesions without pus. Individuals with the most severe phenotype (<1% expression) present in the first weeks of life with delayed umbilical cord separation (the cord fails to shrink down and may not separate until 3–4 weeks of age), and omphalitis together with rapidly progressive erosive perianal ulcers (Figure 82.20). Gingivitis, ulcerative stomatitis and periodontitis are common and severe, leading to loss of teeth. Inflammatory lesions, particularly affecting the skin and resembling pyoderma gangrenosum, can occur in the partial forms of the deficiency and may respond to steroid treatment.
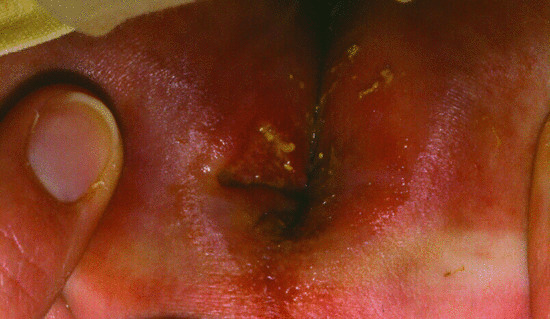
Figure 82.20 Erosive perianal ulcers in an infant with severe leukocyte adhesion deficiency type I.
Defects in innate immunity
NF-κB pathway-related primary immunodeficiencies
The NF-κB pathway is critical in the transduction of extracellular signalling from inflammatory cytokines and toll-like receptor ligands to the cell nucleus, in order to induce transcription of target genes involved in immunity, inflammation, cell survival and apoptosis regulation. The IκB kinase (IKK) complex, a key protein complex in this signalling pathway, is composed of two kinases (IKK-α and IKK-β) and the non-catalytic regulatory protein NEMO. Mutations in genes encoding these molecules can lead to immunodeficiency associated with ectodermal dysplasia.
Incontinentia pigmenti is a rare X-linked dominant condition characterized by developmental abnormalities in the skin, hair, teeth and central nervous system. Carrier mothers demonstrate well-recognized cutaneous features of Blashko linear skin lesions occurring in four successive sometimes overlapping stages: (i) erythema, vesicles, pustules; (ii) verrucous hyperkeratotic lesions; (iii) hyperpigmented whorls and streaks following lines of Blaschko; and (iv) pallor and scarring. In other respects, affected females are healthy. Previously, affected males were all reported to die before birth but it is now recognized that hypofunctional mutations in IKBKG encoding NEMO lead to X-linked anhidrotic ectodermal dysplasia (EDA) and immunodeficiency which is characterized by sparse scalp hair, conical teeth and absent sweat glands. Some patients experience recurrent sinopulmonary infection, often with encapsulated organisms, and have poor antibody responses’ to polysaccharide antigens, or frank hypogammaglobulinaemia. Affected infants share many of the cutaneous features of EDA, although they appear less severe than in children with ‘classical’ EDA without immunodeficiency, and the majority of patients have normal or sparse scalp hair. From early childhood, affected boys suffer from unusually severe life-threatening and recurrent bacterial infections of the lower respiratory tract, skin and soft tissues, bones, gastrointestinal tract, including meningitis, and septicaemia. Causative pathogens are most often Gram-positive bacteria (Streptococcus pneumoniae, Staphylococcus aureus), followed by Gram-negative bacteria (Pseudomonas species, Haemophilus influenzae) and Mycobacteria. Pneumocystis jiroveci infection has also been described. Other related features include osteopetrosis and lymphoedema (osteopetrosis lymphodema ectodermal dysplasia immunodeficiency).
Autosomal dominant gain-of-function mutations in IKBKA encoding IκBα cause a similar clinical picture of immunodeficiency associated with ectodermal dysplasia, clinically indistinguishable from patients with hypomorphic mutations in NEMO.
MyD88 and IRAK-4 molecules are activated through toll-like receptors, and signal through the nucleus via the NF-κB pathway. Patients with defects in these molecules experience severe pyogenic infection, most commonly meningitis or other deep-seated infections, but skin sepsis is reported.
WHIM syndrome
Gain-of-function mutations for the chemokine receptor CXCR4 cause warts, hypogammaglobulinemia, infections, myelocathexis (WHIM) syndrome, which results in a particular susceptibility to warts secondary to human papillomavirus infection, associated with neutropenia and hypogammaglobulinaemia. The generalized verrucosis may be severe and widespread, and involve all cutaneous and mucosal tissues. A similar clinical picture is apparent in epidermodysplasia verruciformis, with increased susceptibility to human papillomavirus infections manifesting as widespread flat warts and pityriasis versicolor-like lesions) and verrucous skin carcinomas. However, neutropenia and hypogammaglobulinaemia are not features of this disease. EVER1 and EVER2 genes are mutated in 75% of epidermodysplasia verruciformis cases.
Hyper-IgE syndrome
Autosomal dominant hyper-IgE syndrome (previously Job syndrome) is of special relevance to the dermatologist as the initial presentation may be cutaneous [9]. It is a complex disorder characterized by extreme elevation of the serum IgE level (usually in the range 2000–40 000 U/L), chronic dermatitis and repeated lung and skin infections. In the literature, these patients are frequently described as having eczema, although this is different from typical atopic eczema. Affected children develop a non-specific, excoriated, papular and pustular eruption in infancy, particularly over the scalp, scalp margins, buttocks and proximal flexures, such as the axillae, groins and neck. The rash may appear in the first few days of life, at which stage it may be vesicular, but crusting becomes a prominent feature. Typical eczematous features of lichenification or scales are absent or mild in hyper-IgE syndrome.
There is commonly a history of furunculosis and staphylococcal lung infections, abscesses and empyema. Many patients develop staphylococcal pneumatoceles, which strongly suggest the diagnosis. Although skin and lung infections predominate, infections of the ears, sinuses, joints and viscera are common. Lymphadenopathy may be complicated by the development of lymph node abscesses. Staphylococcus aureus is the predominant pathogen but infection is also seen with Haemophilus influenzae, pneumococci, group A streptococci and with Candida. Oral candidiasis and Candida nail infections are common. Pneumatoceles may provide the focus for the development of aspergillomas.
Non-immunological features which are variably present include abnormal, coarse often asymmetrical facies with a wide nasal bridge and large head; hypodense bones leading to frequent fractures; joint laxity; a high incidence of scoliosis; delayed resorption of primary dentition with consequent delayed eruption of secondary teeth; and cerebral aneuysms.
Peripheral blood eosinophilia may be marked, up to 50–60%. Serum IgE levels are consistently very high (more than 10 times the upper limit of normal), although they may be normal in infancy.
The disease is due to heterozygous loss of function mutations in the gene coding for STAT3.
The mainstay of treatment is long-term antistaphylococcal antibiotic prophylaxis usually with flucloxacillin. Attention to skin hygiene is important, with judicial use of topical antimicrobials. Candida infections should be treated topically, or when refractory, with oral ketoconazole or fluconazole. Persistent pneumatoceles should be excised.
Chronic mucocutaneous candidiasis
Chronic mucocutaneous candidiasis is a heterogenous group of syndromes with non-invasive Candida infections that affect the skin, nails and mucous membranes. In many forms of the disease, there is a lack of T-helper 17 (Th17) cells or cell function [8]. Defects include gain-of-function STAT1 mutations and mutations in CARD9, STAT3, IL17RA, IL17F, IL22, and IL12RB and CLEC7A encoding important mediators of Th17 cell development or function.
Autoimmune polyendocrine syndrome type 1, also known as autoimmune polyendocrinopathy candidiasis ectodermal dystrophy (APECED) syndrome is caused by mutations in AutoImmune REgulator (AIRE) encoding a transcription factor required to establish thymic self-tolerance. These patients suffer from many forms of autoimmunity, particularly involving parathyroid, adrenal and other endocrine organs. The cutaneous hallmark of this syndrome is ectodermal dystrophy with onychomycosis and superficial chronic mucocutaneous candidiasis, affecting particularly oro-pharyngeal mucosa, and the perineal region (Figure 82.21). The defect is due to autoantibodies, which impair the development or function of Th17 cells, which are important in mucosal immunity and defects of which are associated with susceptibility to candidiasis.
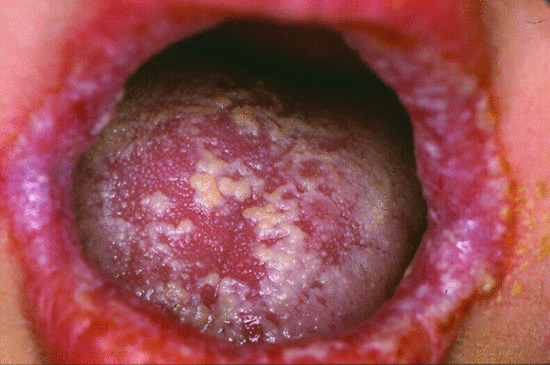
Figure 82.21 Oro-pharyngeal mucocutaneous Candida infection in a patient with a gain-of-function STAT1 mutation.
Complement diseases
Deficiencies of isolated complement components are rare, deficiency of C2 being the most frequent, and with the exception of properdin and C1 esterase inhibitor deficiency, which are X-linked, are generally transmitted in an autosomal recessive manner. However, heterozygosity results in approximately half normal levels of the protein which can sometimes be clinically important. A number of clinical patterns can occur depending upon which factor is deficient [10].
Complement defects predisposing to recurrent pyogenic infection
Recurrent pyogenic infections are a major feature of complement deficiencies. Encapsulated organisms such as streptococci and Haemophilus influenzae are the main problem as opsonization of antibody and complement to bacteria is critical for microbial elimination. C3 deficiency is the most severe, thus meningococcal meningitis and pneumococcal pneumonia have been major problems. The clinical picture is similar to that of hypogammaglobulinaemia. Deficiency of the classical pathway components C1q and C2 and of factor D in the alternative pathway also predisposes to infection. Deficiencies of the alternative pathway control proteins, factors H or I, lead to uncontrolled consumption of C3 resulting in increased susceptibility to pyogenic infections including meningococcal disease, as well as atypical haemolytic uraemic syndrome. Defects in properdin also lead to neisserial disease. Defects in the terminal complement cascade (C5–C9), lead to recurrent meningococcal meningitis and disseminated gonococcal infections, the principal clinical consequence of deficiencies of all these complement components. Cutaneous infections are generally not a problem in these patients.
Complement defects predisposing to autoimmunity
A variety of clinical manifestations have been described in patients with C1q deficiency, including cutaneous vasculitis, SLE, membranous glomerulonephritis and problems with infections, particularly meningitis and septicaemia, but also including stomatitis, pyoderma and persistent candidiasis of the mouth and nails.
Systemic lupus erythematosus or a disorder clinically suggestive of SLE, but lacking confirmatory serological findings, and/or membranous glomerulonephritis have similarly been reported in patients with deficiencies either of C1r or C1s. The majority of patients reported with C4 deficiency have been children or adolescents. Their principal clinical abnormalities have comprised SLE, or an SLE-like syndrome, Henoch–Schönlein purpura or Sjögren syndrome.
C2 deficiency is the most common complement deficiency, shown to be associated with a variety of diseases, but deficient individuals are often entirely healthy. Disorders occurring in C2-deficient patients have included SLE, discoid lupus erythematosus, membranous glomerulonephritis, Henoch–Schönlein purpura, rheumatoid arthritis, dermatomyositis, Crohn disease and idiopathic thrombocytopenic purpura.
Transient maculopapular rashes have been reported to occur in association with infections in patients with C3 deficiency; histologically, these have shown the features of leukocytoclastic vasculitis. Other manifestations such as SLE and membranous glomerulonephritis have also been reported. Whilst recurrent infection is the most common manifestation of defects in the terminal complement cascade (C5–C9), SLE, discoid lupus erythematosus, Sjögren syndrome, rheumatoid arthritis and ankylosing spondylitis have also been associated with these deficiencies.
Defects in the regulation of complement activation
Several proteins have regulatory effects on the complement system, and deficiencies in these components can lead to disease. The best known of these is C1 esterase inhibitor, deficiency of which results in hereditary angio-oedema (see Chapter 43). Clinically, this manifests as angio-oedema, typically affecting the bowel mucosa, face or extremities, and, most seriously, laryngeal oedema, which may be life threatening.
Deficiency of factor 1, previously termed C3b inactivator, leads to unchecked cleavage of C3, and therefore to clinical manifestations closely resembling those seen in C3-deficient individuals. In this condition, plasma infusions may provoke anaphylaxis, because the contained C3 is so rapidly cleaved to form the anaphylotoxin, C3a.
References
- Al-Herz W, Bousfiha A, Casanova JL, et al. Primary immunodeficiency diseases: an update on the classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency. Front Immunol 2014 Apr 22;5:162.
- Leiding JW, Holland SM. Warts and all: human papillomavirus in primary immunodeficiencies. J Allergy Clin Immunol 2012;130:1030–48.
- Lanternier F, Cypowyj S, Picard C, et al. Primary immunodeficiencies underlying fungal infections. Curr Opin Pediatr 2013;25:736–47.
- Rose CD, Neven B, Wouters C. Granulomatous inflammation: The overlap of immune deficiency and inflammation. Best Pract Res Clin Rheumatol 2014;28:191–212.
- Marrella V, Maina V, Villa A. Omenn syndrome does not live by V(D)J recombination alone. Curr Opin Allergy Clin Immunol 2011;11:525–31.
- Buchbinder D, Nugent DJ, Fillipovich AH. Wiskott-Aldrich syndrome: diagnosis, current management, and emerging treatments. Appl Clin Genet 2014;7:55–66.
- Chu EY, Freeman AF, Jing H, et al. Cutaneous manifestations of DOCK8 deficiency syndrome. Arch Dermatol 2012;148:79–84.
- Puel A, Cypowyj S, Maródi L, Abel L, Picard C, Casanova JL. Inborn errors of human IL-17 immunity underlie chronic mucocutaneous candidiasis. Curr Opin Allergy Clin Immunol 2012;12:616–22.
- Yong PF, Freeman AF, Engelhardt KR, Holland S, Puck JM, Grimbacher B. An update on the hyper-IgE syndromes. Arthritis Res Ther 2012;14:228.
- Lipsker D, Hauptmann G. Cutaneous manifestations of complement deficiencies. Lupus 2010;19:1096–106.