CHAPTER 85
Neurological Conditions Affecting the Skin
David J. Eedy
Department of Dermatology, Craigavon Area Hospital, Craigavon, UK
Introduction
The relationship between the nervous system and the skin is complex. Neurocutaneous disorders may be divided broadly into those associated with sensory abnormalities, and those associated with autonomic abnormalities, although there is overlap between these two groups. Skin manifestations may occur where the pathology is predominantly located either in the central nervous system or in the peripheral nervous system. An understanding of the skin's sensory and autonomic innervation is essential to appreciate the clinical manifestations of neurodermatological disorders.
Skin innervation
The skin is innervated by a dense three-dimensional network of highly specialized afferent sensory and efferent autonomic nerve branches. The sensory system contains receptors for touch, temperature, pain, itch and various other physical and chemical stimuli [1, 2]. The autonomic system comprises post-ganglionic cholinergic parasympathetic nerves, and adrenergic and cholinergic sympathetic nerves. It plays a crucial role in maintaining cutaneous homeostasis by regulating vasomotor function, pilomotor activity and eccrine gland secretion. As many as 1000 such afferent neurons may innervate 1 cm2 of skin. The afferent sensory neurons are unipolar, and each branches off with a single axon travelling towards the skin. The autonomic nerves innervate the skin in a different pattern. Post-ganglionic fibres originate in the sympathetic chain and are co-distributed with the sensory neurons until they arborize into plexuses around sweat glands, blood vessels and arrector pili muscles [1, 2]. Skin nerves may contain myelinated and/or unmyelinated fibres: subgroups of sensory neurons are myelinated A fibres, whereas unmyelinated C fibres contain sensory and autonomic fibres. The sensory myelinated fibres can be further subdivided on the basis of diameter into rapidly conducting Aα nerves that transmit tactile sensitivity, and slowly conducting Aδ nerves that transmit temperature, noxious sensation and itch [2–4]. In the upper dermis, small myelinated nerves lose their nerve sheaths and, together with the unmyelinated nerves, end in either free nerve endings, or in association with receptors, such as Merkel cells or nerve-ending organs [2, 3].
Sensory innervation
Sensory innervation follows well-defined dermatomes, with some overlap between adjacent dermatomes. Sensory nerves not only function as an afferent system to conduct stimuli back from the skin to the central nervous system, but also act in an efferent neurosecretory fashion, releasing neuropeptides with important visceromotor, inflammatory and trophic effects on the skin. Unmyelinated type C fibres terminate as either free nerve receptor endings, or in association with receptors such as the Pacinian or Meissner's corpuscles. Pacinian corpuscles, each innervated by a single myelinated sensory axon, are most densely located on the palms and soles, where they act as mechanoreceptors. Meissner's corpuscles also occur in greatest density on the palms and soles; being innervated by one or more sensory nerve endings, they also act as mechanoreceptors, and in addition transmit touch sensation. Merkel cells, which occur at low density generally, with an increased density around hair follicles and at the palms, nail beds and lips, form synaptic-like contacts with sensory afferent terminals. The full function of Merkel cells is not yet fully understood, but recent molecular analysis has revealed that Merkel cells express dozens of presynaptic molecules that are essential for synaptic vesicle release in neurons [5]. Merkel cells produce and contain a wide range of neuropeptides, which may be important in the local regulation of inflammation [2].
Afferent sensory nerves, either unmyelinated C fibres or myelinated Aδ fibres, derive from the dorsal root ganglion and are capable of the release of a variety of neuropeptides in response to noxious stimuli [2–4]. Sensory impulses are conducted in the peripheral and central axon of the spinal ganglion cell in the dorsal root ganglion, and pass via the lateral spino-thalamic tract and the lemniscus spinalis to the thalamus [4]. From the thalamus, the information reaches consciousness via the thalamic radiation to the post-central gyrus of the parietal lobe.
C and Aδ fibres not only conduct nociceptive information to the dorsal root ganglion, but also have an important efferent function, in that they stimulate target tissues by releasing a range of neuropeptides in response to noxious stimuli, such as chemical, electrical, thermal and mechanical injury, or UV radiation. C fibres can also be stimulated as a result of psychological stress, which activates the hypothalamopituitary–adrenal axis, and sensory nerves in the brain and skin [6, 7]. Over 20 neuropeptides have been identified and characterized to a greater or lesser extent in the skin of various species [8]. Neuropeptides with an important neurotransmitter function contained in primary sensory neurons include three tachykinins: substance P (SP), neurokinin A (NKA) and calcitonin gene-related peptide (CGRP) [9]. These neuropeptides most frequently coexist in the same subpopulation of primary sensory neurons, the Aδ and C fibres, and are involved in the nerve transmission of impulses initiated by noxious stimuli. They usually occur in free nerve endings in the upper dermis (Figure 85.1) and epidermis throughout the body, but are at greatest density on the palms and soles, where some end in a plexus around Meissner's corpuscles (Figure 85.2) [9].
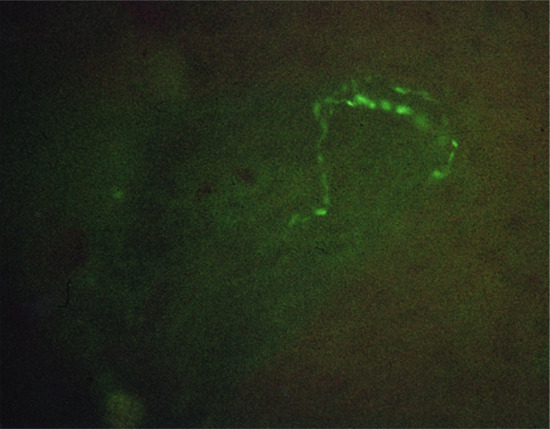
Figure 85.1 Substance P immunoreactive nerve endings in the epidermis of human skin.
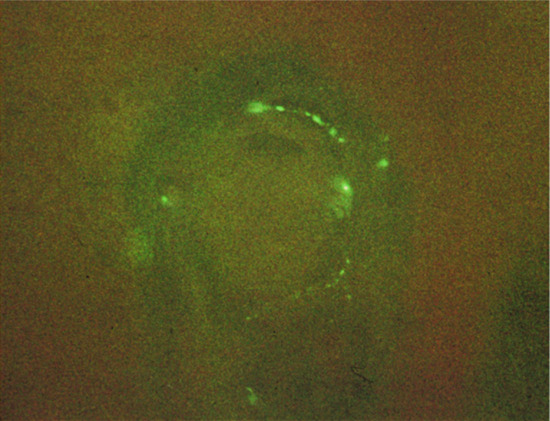
Figure 85.2 Substance P fibres ending in Meissner's corpuscles of skin.
Capsaicin, a vanilloid alkaloid found in red pepper fruit [10], functions in nociception by binding to a specific receptor called the vanilloid receptor, the name referring to a homovanillyl group found in the ligands of the receptor. Vanilloid receptors have been found on the unmyelinated C and thinly myelinated Aδ fibre types, which transmit painful stimuli. When capsaicin stimulates the transient receptor potential vanilloid receptor 1 (TRPV1) – one of the vanilloid receptors particularly implicated in neurogenic inflammation [10] – this causes Ca2+ ion influx which is rapidly followed by receptor desensitization. If calcium overload is sufficient, this can lead to cell death [10, 11]. TRPV1 links with TRPA1 (transient receptor potential ankyrin 1) channels on the surface of keratinocytes, and their interlinked function has recently been associated with pain and neurogenic inflammation [12]. It has been suggested that neurogenic inflammation is involved in the pathogenesis of the common chronic cutaneous vascular disorders such as rosacea. Due to their involvement in the pathophysiology of rosacea, TRPV1 and TRPA1 may act as therapeutic targets for the treatment of the inflammatory symptoms of rosacea [13]. TRPV1 and TRPA1 are also expressed on a multitude of non-neuronal sites, which has led to a plethora of research into possible functions of these receptors [12].
Release of neuropeptides often leads to neurogenic inflammation. The key components of neurogenic inflammation are precapillary vasodilation, plasma protein extravasation and leukocyte infiltration, which follow antidromic stimulation of peripheral nerves. Neuropeptides can regulate both acute and chronic inflammation by influencing vascular motility and cellular trafficking. After release, neuropeptides are metabolized by membrane-bound endopeptidases that occur on target structures such as blood vessels and eccrine sweat glands in skin [14]. Neutral endopeptidase (NEP) and angiotensin-converting enzyme (ACE) in particular seem to play an important role in terminating the action of neuropeptide agonists [8].
A key receptor on sensory nerves that is involved in neurogenic inflammation is the proteinase-activated receptor 2 (PAR-2). PAR-2, bound to G protein, occurs on keratinocytes (especially in the stratum granulosum), endothelial cells, hair follicles and in the myoepithelial cells of the sweat glands [10]. Tryptase released from degranulated mast cells cleaves PAR-2 in the sensory nerve endings. This leads to its activation and stimulates the release of CGRP and neurokinins SP and NKA from sensory nerve endings. CGRP interacts with CGRP-1 receptors to induce arteriolar dilation and hyperaemia; while SP interacts with NKA-1 receptors on the endothelial cells of postcapillary venules to induce plasma extravasation. The overall result is tissue oedema. SP may also stimulate degranulation of mast cells, leading to the release of tryptase, which produces positive feedback. Tryptase degrades CGRP and reduces its effects (CGRP inhibits SP degradation by NEP and enhances SP release). Other mediators from mast cells and other inflammatory cells stimulate the release of vasoactive peptides from sensory nerves [8, 10].
It can be seen that close interaction between the different neuromediators, target cells and neuropeptide-degrading enzymes is critical for the control of cutaneous neurogenic inflammation [8].
Autonomic nervous system
The autonomic nervous system innervates the skin through post-ganglionic fibres originating in sympathetic ganglia, and terminating in autonomic plexuses that supply sweat glands, blood vessels and arrector pili muscles [1]. Histochemically, there are two main groups of post-ganglionic nerve fibre in the skin. First, adrenergic fibres synthesize and store catecholamines and norepinephrine (noradrenaline). The second major group consists of the cholinergic fibres containing acetylcholine. Co-localizing with acetylcholine are ‘secretory’ neuropeptides, such as vasoactive intestinal peptide (VIP) and peptide histidine methionine (PHM). Nerves containing these should be regarded, at least physiologically, as parasympathetic. The secretory portions of the eccrine sweat glands, myoepithelial cells, and nearby blood vessels are innervated by a basket-weave pattern of nerves, containing predominantly acetylcholine but also significant numbers of fibres containing ‘secretory neuropeptides’ including VIP (Figure 85.3), PHM (Figure 85.4), neuropeptide Y (NPY), CGRP, galanin, atrial natriuretic peptide (ANP) and norepinephrine. Secretory neuropeptides, mainly VIP and ANP, together with norepinephrine, are not as effective as acetylcholine at sweat production, and probably synergistically amplify acetylcholine-induced adenosine 3',5'-cyclic monophosphate (cAMP) accumulation, which is the most important secondary messenger in sweat production. ANP may be responsible, at least in part, for the regulation of sodium and other electrolytes released in sweat [9]. NPY has been identified in the periarteriolar nerve fibres of the deep and superficial vascular plexus, and in eccrine sweat glands; and is likely to play a role in the regulation of skin blood flow and eccrine sweating [9]. Pituitary adenylate cyclase-activating polypeptide (PACAP) also plays a significant role in modulating cutaneous inflammation. It belongs to the VIP family and is capable of binding identical receptors to VIP in the same tissue but with different affinities. PACAP and VIP both participate in myelin maturation and synthesis [15]. PACAP leads to mast cell degranulation, and dilatation of small blood vessels [16]. PACAP may have a pro-inflammatory effect on endothelial cells during acute inflammation, yet it exerts anti-inflammatory effects under chronic inflammatory conditions [8].

Figure 85.3 Vasoactive intestinal peptide immunoreactive fibres surround secretory cells of eccrine sweat gland.
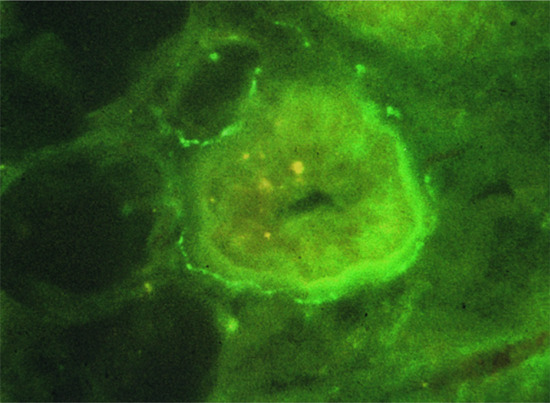
Figure 85.4 Peptide histidine methionine immunoreactive fibres surround eccrine sweat glands.
Blood vessels in the skin are innervated by adrenergic fibres, which are vasoconstrictors, while acetylcholine and neuropeptides, such as VIP and PHM, act as vasodilators and increase vascular permeability [9, 17]. Thus, by increasing the release of acetylcholine and secretory neuropeptides, the body has a mechanism of increasing blood flow to the skin and increasing sweating, both of which act to reduce body temperature. If body temperature falls, this is detected in the preoptic region of the hypothalamus, which activates the sympathetic nervous system, which in turn reduces skin blood flow and sweating. Conversely, if warmer blood is detected in the hypothalamus, inhibition of sympathetic response allows skin sweating and blood flow to increase, thereby reducing core temperature [18, 19].
Adrenergic fibres mediate strong vasoconstriction and arrector pili muscle activity, thus diverting blood from the skin, and pulling hairs into the upright position, in the classical ‘fight or flight’ reaction [1].
The digital nerves in patients with Raynaud's phenomenon and with systemic sclerosis are deficient in CGRP fibres [20], and intravenous infusions of CGRP increase digital blood flow in such patients. A similar depletion of CGRP fibres has been described in digital nerves of patients with vibration white finger, and this may be responsible for both the vasoconstriction and the sensory abnormalities characteristic of this condition [21].
Neurophysiological testing for skin innervation
Sympathetic skin response
Sympathetic skin response (SSR) is a safe, simple and non-invasive electrophysiological test used to evaluate sudomotor function in a variety of clinical settings [22–24]. It can be used to assess autonomic function in patients with suspected sympathetic nerve dysfunction. The SSR is a polysynaptic reflex associated with the activation of sweat glands. It is usually performed by electrical stimulation of the median, ulnar, peroneal or sural nerves, and measuring the change in galvanic resistance on glabrous skin brought about by sweating using a conventional electromyograph apparatus. The afferent component may be activated by non-specific sensory stimuli or repeated electrical stimulation; the efferent component depends on the functioning of the sympathetic cholinergic fibres from the sympathetic chain to the sweat glands. In autonomic failure the SSR cannot be elicited.
Cold-induced vasodilation
Hand cooling is a cold pressor test in which an extremity is placed in cold water, leading to a rapid decrease in skin temperature, accompanied by initial strong vasoconstriction aimed at limiting heat transfer to the environment [25–29]. After a few minutes, skin temperature starts increasing as a consequence of cold-induced vasodilation. The precise mechanism of this reflex is unknown, but it is thought to be mediated through an increase in sympathetic stimulation, although factors independent of the sympathetic nervous system may also play a role. Exposure of the human body to cold stress elicits generalized cutaneous vasoconstriction. This is a response mediated by a sympathetic control process, triggered partly by stimulation of cutaneous cold receptors and partly by cold blood returning to the general circulation and stimulating the temperature-regulating centre in the hypothalamus. The tone of cutaneous vessels is controlled mainly by vasoconstrictor skin sympathetic nerve activity. A reduction in this response has been reported in diabetic and neuropathic patients, and in the immature and elderly, probably resulting from reduced sympathetic nerve vasoconstriction in the skin [28, 29].
Triple response of Lewis
Lewis described the capacity of the cutaneous microcirculation to vasodilate in response to direct stimulation with a firm mechanical stroke or with a dermographometer (the axon reflex) [29–31]. The amount of vasodilation can be measured by a Doppler flow meter. The axon reflex, known as ‘antidromic vasodilation’, does not occur in chronically denervated skin, or in skin in which neuropeptides have been depleted by capsaicin. The antidromic vasodilation and plasma extravasation, which occurs in the skin following stimulation of the dorsal nerve roots or peripheral sensory nerves, can be mimicked by intra-arterial infusion of SP.
Histamine is known to be a principal mediator of the triple response of Lewis, and to act via H1 and H2 receptors to produce vasodilation and increased vascular permeability. However, it is known that SP, NKA and CGRP from primary sensory fibres are also mediators in the skin flares of the triple response of Lewis [32, 33]. There is a bi-directional link between histamine and neuropeptides in neurogenic inflammation: neuropeptides induce the release of histamine from adjacent mast cells; and in turn, histamine evokes the release of SP and CGRP [34].
Neuropathic ulcer
Definition and nomenclature
A neuropathic ulcer is a form of chronic ulceration which develops in anaesthetic skin. Characteristically, neuropathic ulcers are painless, persistent and uninflamed, appearing on areas subject to trauma or pressure.
Introduction and general description
A neuropathic ulcer occurs as a result of skin ulceration in an area of skin anaesthesia. The ulcer is typically painless with well-defined margins surrounding the wound. Neuropathic ulcers usually occur on weight-bearing body surfaces such as the heels and metatarsal heads.
The underlying neuropathology is commonly a distal polyneuropathy encompassing motor, sensory and autonomic components: the vast majority of neuropathic ulcers occur in patients with type II diabetes [1, 2]. Other causes include peripheral nerve injury, peripheral neuropathy, renal failure, alcoholism, vitamin deficiencies, leprosy, pernicious anaemia, syringomyelia, tabes dorsalis, spinal dysraphism, spinal cord injury, and hereditary sensory and autonomic neuropathies (HSANs) [3, 4].
Epidemiology
Incidence and prevalence
Of people with diabetes, 25% develop foot ulcers at some point in their lives [5]. The annual incidence of diabetic foot ulcers is ∼3%, and the reported incidence in US and UK studies is as high as 10% [6].
Pathophysiology
The major underlying factors in the development of ulcers are peripheral neuropathy, and ischaemia secondary to peripheral vascular disease. In diabetic patients, hyperglycaemia leads to a complex of abnormal enzyme activity which results in a decrease in normal neuron conduction as well as nerve dysfunction and ischaemia, causing further injury to, and eventual death of, nerve cells [7].
As patients experience sensory loss, trauma to the affected site often goes unnoticed, and progressively worsens as the area is continuously subjected to repetitive pressure and shear forces.
Clinical features
Neuropathic ulcers typically occur on the foot, under the metatarsal heads or on the heel. They tend to be surrounded by thick hyperkeratosis, and have a pink punched-out base that bleeds easily and is painless (Figures 85.5 and 85.6). The changes of a neuropathic foot include pes cavus and clawed toes (Figure 85.7). The foot tends to be anaesthetic, warm with palpable pulses and dilated veins. The skin is dry and hyperkeratotic under the forefoot and heel. Primary neuropathic limbs tend to be warm, insensitive and prone to ulceration on the sole, while neuro-ischaemic limbs are usually cool, discoloured and prone to ulceration on the foot margins [2]. In diabetics, there may be a complex interplay of neuropathy and ischaemia, and when the latter is present, the foot may be cold with absent pulses, hyperkeratosis and a dark fibrotic base that does not bleed easily and is painful to touch. Continuing mechanical overload on established callus can result in fissures, increasing the risk of future ulcer formation [8].
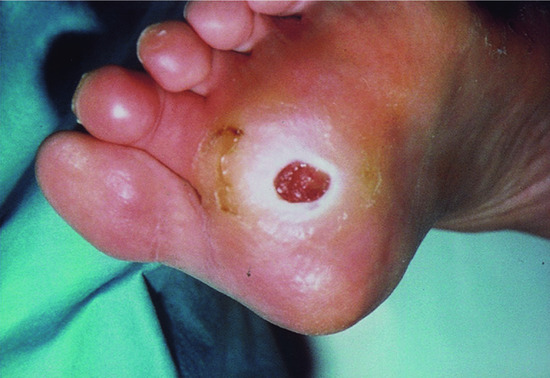
Figure 85.5 Typical neuropathic ulcers with surrounding hyperkeratosis under metatarsal heads.
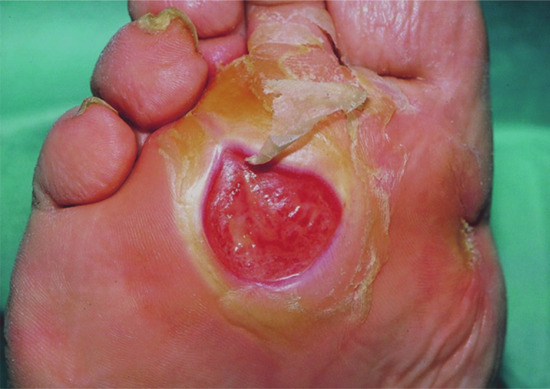
Figure 85.6 Neuropathic ulcer with hyperkeratosis removed.
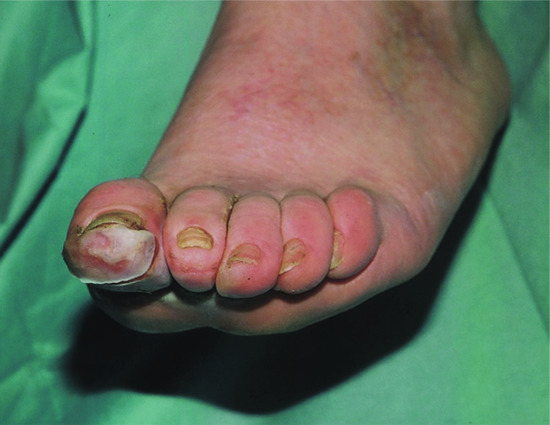
Figure 85.7 Clawing of feet occurring in neuropathic foot with painless damage to the skin on the great toe.
Classification of severity
A well-established system is the Wagner ulcer classification system, which ignores infection and ischaemia [7]. The wound classification developed by the International Consensus of the Diabetic Foot assesses patients for ischaemia, neuropathy, linear measurement of wound diameters, depth of wound and infection [9].
Complications and co-morbidities
A frequent complication is the presence of cellulitis or deep infection, with abscess formation or osteomyelitis. One study found that Gram-positive aerobic bacterial colonization predominates (84%), with Staphylococcus aureus being the commonest organism (79%). Meticillin-resistant Staphylococcus aureus (MRSA) was isolated in 30% of patients, a near twofold increase over the previous 3 years [10]. Bacterial colonization of the ulcer does not necessarily indicate infection. By contrast, infection may be present, especially in diabetics, without pyrexia. Leukocytosis, elevated erythrocyte sedimentation rate (ESR) and local signs may be less than expected. Cellulitis should alert one to the possibility of underlying osteomyelitis.
Investigations
In neuropathic ulcers, the reduced sensations of light touch and vibration, and sharp–blunt discrimination, can be demonstrated using a Bailey® nylon monofilament (which buckles in response to 10 g force) (Figure 85.8), a neurothesiometer and a Neurotip®, respectively [11].
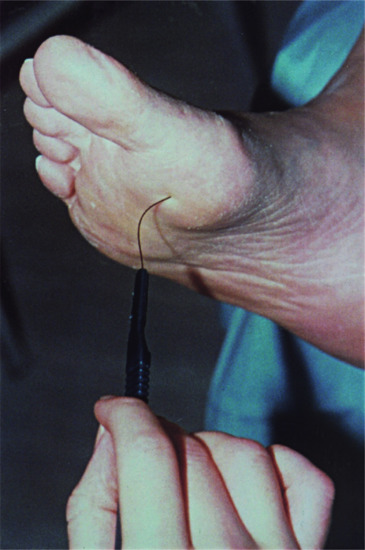
Figure 85.8 Bailey® nylon monofilament assessing sensory loss on neuropathic foot.
Palpable bone at the base of an ulcer on wound probing is an excellent predictor of osteomyelitis [12] (Figure 85.9). Plain X-rays may show periosteal reactions or osteolysis, and will also identify foreign bodies, tissue gas or bony abnormalities. A sinogram may be required to show communication of the sinus with a joint or a subfascial plantar abscess. A radio-isotope bone scan, or MRI scan, may help with the diagnosis [13].
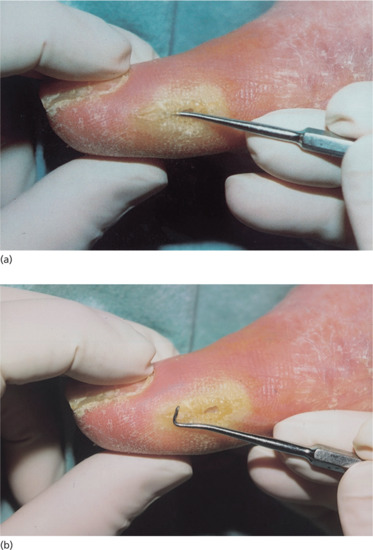
Figure 85.9 Probing a neuropathic wound: (a) probe in position, (b) with probe removed.
Management
Management of neuropathic ulcers requires an integrated care approach from a multidisciplinary foot care team with the engagement of patients and carers, which can reduce amputations by up to 70% [14].
Prevention of ulceration is of the utmost importance. It is important to review the residential environment for safety, and to educate the patient and carers regarding the risks, and the use of protective footwear. Persons at risk should receive basic foot care instruction, preferably from a podiatrist, including regular self-inspection of the feet and correct nail-cutting techniques. Shoes should accommodate deformities and be tailored to the patient's needs [15, 16], with a follow-up programme to prevent further ulceration [17]. Urea-based products may help in managing callus [8]. Patients should be discouraged from removing callus themselves – it should be done by a podiatrist.
It is important to treat the underlying disease process: patients with critical limb ischaemia should be considered for arterial reconstruction [18]. Diabetic control should be meticulous, and the physical cause of the trauma must be addressed.
Ulcers are best treated with a combination of debridement, wound dressing and offloading. Regular local sharp debridement with scalpel, scissors and/or forceps is the gold standard. Infection prevention is paramount, and dressings should maintain a moist wound bed and absorb the exudate. Choice of dressing depends on a thorough assessment of the patient and wound [19, 20].
For chronic ulcers, some novel therapeutic approaches can be considered, such as microsurgical grafts for large defects, and tissue-engineered human skin, mainly as a transient method. Vacuum-assisted closure (VAC) devices improve the success of skin grafts [19]. Becaplermin, a growth factor with a proposed role in wound healing, has demonstrable efficacy [19, 21]. However, there is a possible increased cancer risk with higher doses of becaplermin [22].
Bone marrow derived cells may be effective in healing otherwise unresponsive wounds [23]. Hyaluronic acid dressings (Hyalofill®) are also routinely used in chronic ulcer treatment [24–26].
Studies suggest that what is removed from the foot (pressure, callus, infection and slough) is more important than what is applied to it (adjuvant treatments: hyperbaric oxygen, cytokines, growth factors) [27].
Offloading of areas of abnormal pressure on the foot is essential [28]. A contact casting (such as the Aircast Walkers® foam boot; Figure 85.10), is useful for healing ulceration over metatarsal heads, but requires expert application, and is contraindicated with infected ulcers, or where there is significant ischaemia [28]. A modified boot of layered adhesive foam may achieve complete removal of pressure points [29]. Plaster of Paris boots (with a rocker base) applied over weeks or months seem popular in some centres [30]. Non-removable offloading devices promote better ulcer healing than removable devices, presumably because of increased patient compliance [31].

Figure 85.10 Aircast Walker's® boot.
Resources
Patient resources
- www.woundsinternational.com/pdf/content_10803.pdf (last accessed June 2015).
Syringomyelia
Definition and nomenclature
Syringomyelia describes conditions involving abnormal fluid cavities within the spinal cord. A ‘syrinx’ is a fluid-filled cavity within the spinal cord parenchyma.
Introduction and general description
Syringomyelia is a rare disorder characterized by a longitudinal cyst in the cervical cord and/or medulla (syringobulbia) immediately anterior to the central canal which spreads, usually asymmetrically, to each side. The central nervous system disturbance causes dissociated sensory loss, with pain and temperature sensation being lost early in the fingers and upper limbs. The first clinical manifestation of syringomyelia is a tendency to painless burns and cuts on the hands and forearms.
Epidemiology
Incidence and prevalence
There are an estimated 8.4 cases per 100 000 population [1].
Age
Symptoms usually appear in young adults, and usually progress slowly over 20–30 years. Limited self-resolution of symptoms has occurred in adults [2] and children [3, 4].
Associated diseases
Chiari malformation type 1 (CM-1) is a developmental abnormality often associated with a syrinx, involving a congenital extension of the cerebellar tonsils below the foramen magnum [5].
Pathophysiology
The exact pathogenesis of syringomyelia is uncertain. The condition has several distinct cavitary patterns, which probably determine the pathogenesis and the clinical features of the condition [6]. The majority of lesions occur in association with type 1 Chiari syndrome. It is proposed that syrinx development, particularly in CM-1 patients, follows a differential between intracranial pressure and spinal pressure caused by a valve-like action at the foramen magnum [6]. Other causes include trauma and tumours [3].
Clinical features
History
Symptoms usually appear in young adults, and the disease generally slowly progresses over 20–30 years, although limited resolution has occurred in adults [3] and children [4, 5]. An association with other abnormalities, such as a short neck and a low hairline, suggests a developmental origin. Body asymmetry or hemi-hypertrophy is known to occur in syringomyelia.
Presentation
Early involvement of pain and temperature fibres leads to a characteristic dissociated sensory loss, where pain and temperature sensation is lost early in the upper limbs, while sensory modalities carried in the posterior columns (e.g. touch, vibration and position sense) remain relatively intact. The earliest manifestation of the disease is a tendency to painless burns and cuts on the hands and forearms. Later, upper motor neuron signs in the legs may accompany weakness, wasting and loss of reflexes in the arms. Morvan syndrome, involving progressive pain loss, resultant skin ulceration, soft-tissue loss, resorption of the phalanges and muscular atrophy, occasionally occurs.
Sixty per cent of patients report dyshidrosis (either hyperhidrosis or hypohidrosis) [7]. The dyshidrosis, usually over the face and upper arms, may be spontaneous or occur on consumption of hot or seasoned food [7]. Hyperhidrosis is probably caused by stimulation of the sympathetic preganglionic neurons and, as the disease progresses, hyperreactivity is replaced by hypoactivity. Focal hyperhidrosis may be a hallmark of a relatively intact, though slightly damaged, spinal cord. Asymmetry of scalp hair can also occur, with the denervated areas having less abundant slower-growing hair.
Complications and co-morbidities
Syringomyelia can coexist with other conditions. There is increased risk of scoliosis, and a reported coexistence with Guillain–Barré syndrome. Complications include infections and decubitus ulcers.
Disease course and prognosis
The disease slowly progresses over many years. Repeated burns, and other injuries, cause the skin of the fingers to become thickened, swollen, cyanotic and keratotic. Gangrene rarely occurs, but damage to, or loss of, terminal phalanges or nails can occur.
Extension of the syrinx into the medulla may disrupt the vestibular pathways, the descending trigeminal nerve, the sympathetic and taste pathways, and the hypoglossal nerve. Patients may then experience vertigo with nystagmus, dissociated facial sensory loss, loss of taste, a wasted tongue, Horner syndrome and occasionally vocal cord paralysis or facial oedema confined to areas of sensory loss [7].
Investigations
MRI scanning of the hindbrain and upper spinal cord is best for delineation of the syrinx [3–8].
Management
A neurosurgical and orthopaedic evaluation is warranted for all patients with a syrinx. Surgical indications have been stated as: progression of motor/sensory loss, scoliosis, associated pain and size of the syrinx [9].
Resources
Patient resources
- http://emedicine.medscape.com/article/1151685-overview (last accessed June 2015).
Spinal dysraphism
Definition and nomenclature
Neurological disorders involving the malformation of the spinal cord due to a failure of symmetrical fusion of the embryological spinal structures are referred to as spinal dysraphisms.
Introduction and general description
Spinal cord development occurs between weeks two and six of embryonic life. Dysraphism is the failure of fusion between symmetrical embryological structures (a raphe) [1], leading to malformations of the midline dorsal structures [2]. Early embryonic defects produce spinal dysraphisms, categorized clinically into two subsets: open – exposed to the environment (e.g. spina bifida, myelomeningocoele or MMC); and closed – covered by intact skin.
Congenital dermal sinuses (CDS) are epithelium-lined tracts resulting from incomplete separation of cutaneous ectoderm from the underlying neuroectoderm. CDS may be associated with dermoid cysts, and can cause complications by mass effect and by functioning as a pathway for infection [3].
Spina bifida and CDS are discussed in this section.
Epidemiology
Incidence and prevalence
For open dysraphism the incidence is around 1 : 1000 live births. The prevalence has declined worldwide over the past two decades, due to better maternal nutrition, timely folate replacement and better prenatal care with high-resolution ultrasound [4]. Spina bifida is one of the commonest congenital conditions [5].
Age
From birth.
Ethnicity
In Europe, the highest incidence rates are in Ireland and Wales (5 per 1000 live births) compared with south-eastern Europe (0.1–0.6 per 1000).
In Canada and the US, a higher incidence has been reported on the east coast, and in the US the Hispanic population has the highest risk.
Associated diseases
Open dysraphism is associated with certain genetic disorders, including trisomy 13 or 18, congenital hemidysplasia with ichthyosiform erythroderma and limb defects (CHILD syndrome), Fraser syndrome, Waardenburg syndrome and Meckel–Gruber syndrome [6].
Pathophysiology
Predisposing factors
The cause of neural tube defects is unknown. However, genetics, folate deficiency, antiepileptic drugs (sodium valproate, carbamazepine) and environmental conditions such as radiation can predispose [6]. Folate deficiency is a major cause of open dysraphism, largely preventable by folate supplementation before conception and during early pregnancy [7].
Pathology
Dysraphisms occur at three stages of embryogenesis: primary neurulation, secondary neurulation and gastrulation [4].
Clinical features
Presentation
A review of 200 published cases of spinal dysraphism [8] included 102 with cutaneous abnormalities, often in combination. A dermatologist may be the first physician to see such patients, and should be aware of possible associations with underlying neurological abnormalities. Coccygeal dimples may be unrelated to dysraphism, and not require aggressive investigation, whereas lumbo-sacral dimples present a higher risk for underlying problems [9, 10]. Correlation between sacral dimples and dysraphism has recently been questioned [11]. Patients with congenital giant melanocytic naevi overlying the scalp or dorsal spine can show brain abnormalities on MRI.
Many lumbo-sacral skin abnormalities have been reportedly associated with spinal dysraphism and cord tethering, including lipomas [12], port-wine stains, haemangiomas [13, 14], ‘faun tail’ (hypertrichosis), pigmented macules, and pits or dimples. Intraspinal dermoid cysts are usually associated with either lumbar spinal dysraphism or a dermal sinus tract [15].
For occult dysraphism, midline skin abnormalities have considerable diagnostic value, and can be divided into three groups of risk [6]:
- Group 1 (high risk): two or more lesions, subcutaneous lipoma, dermal sinus, ‘queue de faune’ (faun-tail).
- Group 2 (low risk): atypical dimple, aplasia cutis, gluteal fold deviation.
- Group 3 (very low risk): haemangioma, port-wine stain, hypertrichosis, fibroma pendulum, pigmentary naevus, coccygeal dimple.
Several varieties of spina bifida are described, differing in the nature and severity of the spinal defect. In the severe form, a sac protrudes through the vertebral opening and transmits an impulse on crying or coughing. In the mildest cases (spina bifida occulta) there is no such protrusion, but a defect in the vertebral lamina may be felt as a depression, and is sometimes covered by a tuft of hair or a dimple (Figure 85.11). The likelihood of a midline fusion defect is increased when the cutaneous findings are associated with a subcutaneous lesion such as a lipoma [12]. Spina bifida occulta may be a chance finding during routine examination.
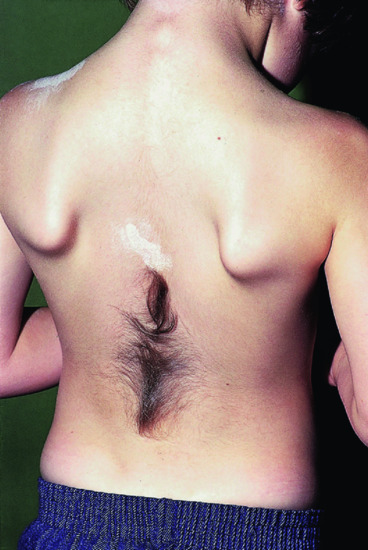
Figure 85.11 Tuft of hair in association with spina bifida.
Lesions preventing spinal cord ascent during normal growth can cause undue traction on the lower end of the cord and cauda equina. Neurological changes will then be those of a chronic cauda equina lesion. Such patients may be slow learning to walk. Sensation may be impaired over the areas innervated by the lowest sacral segments, causing a characteristic saddle-shaped analgesic area over the buttocks and dorsa of the thighs. Trophic changes are occasionally conspicuous. In milder cases, the feet are usually cold and cyanosed. Cutaneous injuries heal slowly and tend to ulcerate, particularly on the feet and in the analgesic skin of the buttocks and thighs. The most severe neurological abnormality is a flaccid paraparesis with sphincter paralysis.
Complications and co-morbidities
Large skin defects produce complications including wound dehiscence, wound infection and cerebrospinal fluid leaks. The morbidities of wound healing and cerebrospinal fluid leaks during surgical management present significant challenges [4].
Children with spina bifida risk becoming overweight, which further reduces their mobility, increases difficulties with catheterization and toileting, adds pressure to already vulnerable skin and increases social isolation. Urinary tract infections are a common source of morbidity among children with spina bifida [16].
Spina bifida patients are prone to latex sensitivity [17] from exposure to latex products, both at the time of surgery and also with indwelling catheters. They should be assessed using latex-specific serum immunoglobulin E (IgE), the radioallergosorbent test (RAST), skin prick testing with latex suspension and a latex glove usage test [17, 18]. Latex-allergic patients may display urticaria, conjunctivitis, angio-oedema, rhinitis and bronchial asthma. They are at risk of anaphylaxis during healthcare procedures and – because of cross-sensitivity – can be allergic to fresh foods including kiwi, pear, orange, pineapple, tomato and banana.
Investigations
Estimation of α-fetoprotein in the amniotic fluid or maternal serum may successfully identify a fetus with severe central nervous system malformation, such as spina bifida cystica or anencephaly. Spinal ultrasound (SUS) is most helpful when supported by multiple clinical indications. It is significantly more likely to detect spinal dysraphism than imaging performed for isolated abnormalities or risk factors [11]. With antenatally diagnosed, open dysraphism caesarean section is often recommended to avoid additional trauma and infection during birth [19]. The decision to continue pregnancy or opt for termination raises many moral and ethical dilemmas for discussion with the parents.
Spinal sonography is a useful screening method in the first 4 months in newborns with a suggestive cutaneous lesion. Diagnosis is confirmed by radiography [20] which shows defective fusion of the laminae in the affected region, usually L5 and S1. Indications for ultrasound or MRI referral have recently been reviewed [9]. Because tethered spinal cord is treatable, but untreated can lead to progressive neurological degeneration, spinal MRI scanning of infants with giant melanocytic naevi involving the lumbo-sacral area is advocated [21–23].
Management
In open dysraphism, initial treatment is with saline gauze at 37oC, and non-permeable dressings [6]. Management aims to provide closure of the neural tube defect and the skin without any undesirable tension within 24–48 h. Early primary closure often has an excellent outcome, although the risk of neurological sequelae varies depending on the severity of the anomalies [4].
Postnatal surgery of open spina bifida aims at covering the exposed spinal cord, preventing infection and treating hydrocephalus with a ventricular shunt. Fetal surgery with the severest form of spina bifida, MMC, is also an option [24, 25].
Due to the risk of neurological deterioration, the recommended treatment of CDS with or without a concomitant intraspinal dermoid is prompt administration of antibiotics and definitive surgical intervention [3].
Resources
Patient resources
- http://www.spinabifidaassociation.org/site/c.evKRI7OXIoJ8H/b.8755685/k.F19B/Patient_Information.htm (last accessed June 2015).
Dermatoses associated with spinal cord injury
Definition
Spinal cord injury leading to a spinal cord defect results in multiple disabilities which can be complicated by skin problems.
Introduction and general description
The spinal cord may be injured directly by penetrating wounds or, more frequently, indirectly following dislocation or fracture dislocation of the vertebral column [1]. The causes of spinal cord injury are: trauma, including motor vehicle accidents (36–48%), violence (5–29%), falls (17–21%) and recreational activities (7–16%). The severity of the disability varies widely, mainly depending on the level of the injury: the worst scenario being at C1–C3, leading to ventilator dependence, limited talking ability and the need for a head- or chin-control wheelchair. As well as problems with pressure ulceration (see Chapter 124, the skin of patients with spinal cord injury is prone to a number of inflammatory dermatoses and disorders of sweating.
Clinical features
Seborrhoea and seborrhoeic dermatitis have been reported in quadriplegic patients [2, 3] with one study reporting around two-thirds developing the condition within a few weeks of injury. Nummular eczema may also occur below the level of the lesion [3], and acne on the back and buttocks may follow the onset of paralysis [3]. Changes in eccrine sweating after spinal cord injuries are complex [4]. Profuse sweating on the face, neck and upper trunk with lesions at or above T6 may occur in exaggerated response to stimuli such as bowel or bladder distension (autonomic dysreflexia). These episodes may involve facial flushing and headache. Other patients develop sweating of the face and arms after dizziness due to postural hypotension. Post-traumatic syringomyelia can lead to hyperhidrosis [5]. Dryness of the skin, particularly on the soles, is an effect of anhidrosis.
Management
Management of pressure ulcers is described in Chapter 124. Inflammatory dermatoses associated with spinal injury should be treated with appropriate topical and/or systemic therapy.
Resources
Further information
- http://guidance.nice.org.uk/index.jsp?action=byID&o=13897 (last accessed June 2015).
Patient resources
- http://www.spinalcord.org/resource-center/askus/index.php?pg=kb.page&id=392 (last accessed June 2015).
Hereditary sensory and autonomic neuropathies
Definition and nomenclature
The HSANs encompass a number of inherited disorders associated with sensory dysfunction (depressed reflexes, altered pain and temperature perception) and varying degrees of autonomic dysfunction (gastro-oesophageal reflux, postural hypotension, excessive sweating) [1].
The classification of HSANs was modified by Dyck into five main groups of syndromes, each with common clinical, pathophysiological and genetic phenotype [2].
Introduction and general description
For the dermatologist, an HSAN will usually be encountered in a patient with disordered sweating accompanied by severe sensory dysfunction. Advances have been made in identifying the specific loci and genes implicated in the different types of HSANs, and the resulting disease mechanisms.
Epidemiology
Incidence and prevalence
As a group, HSAN is a spectrum of rare genetic diseases affecting both sexes and tending to occur in families with parental consanguinity [1,2]. Several hundred cases of HSAN IV have been reported [1]. HSAN III is almost exclusive to individuals of Eastern European Jewish extraction, with 1 : 3600 live births. The worldwide prevalence of HSAN type II is very low. All types present at birth except HSAN I which presents in the second decade [1].
Hereditary sensory and autonomic neuropathy type I
HSAN I differs from all other HSANs in that its symptoms appear late, rather than from birth. It is the commonest HSAN, resulting in progressive degeneration of sensory dorsal root ganglia, with loss of both small myelinated and unmyelinated nerves leading to a distal sensory and motor loss, particularly in the lower limbs [3,5]. It presents in late childhood or adolescence, with progressive loss of all sensation in the lower extremities, particularly of pain and temperature. This often results in chronic trophic ulceration on weight-bearing areas of the feet, associated osteomyelitis, and mutilating deformity [6, 7]. Later manifestations include muscle wasting, weakness and lancinating pain in the lower limbs, and extension of sensory symptoms to the upper limbs. Neural deafness is frequent, and congenital cataracts and learning difficulties have been reported [8].
HSAN I is an autosomal dominant disorder, mapping to chromosome 9q22.1-q.22.3 [3]. The affected gene has been identified as serine palmitoyl transferase long-chain base subunit 1 (SPTLC1), leading to an impact on enzyme systems critical for sphingolipid synthesis [4].
Hereditary sensory and autonomic neuropathy type II
HSAN II is an autosomal recessive disorder with congenital or early onset [2]. There is loss of sensory, motor and autonomic motor neurons, and unmyelinated nerves [2,8]. Infantile hypotonia is common. Sensation loss occurs in the hands and feet, leading to ulceration at sites such as the forehead, trunk, tongue and lips, following repeated injury. Fingers can show ainhum-like constriction bands and spontaneous amputations. Loss of deep sensation results in deep ulceration, osteomyelitis, stress fractures and long-bone injury [8]. Tendon reflexes and anhidrosis occur in areas of decreased sensation, although anhidrosis is less prominent than in HSAN IV. Associated features include learning difficulties, neural deafness, impaired taste and smell, retinitis pigmentosa and cataracts.
High urinary excretion of sphingomyelin and lecithin suggest a disorder of phospholipid metabolism [7,9].
Hereditary sensory and autonomic neuropathy type III
HSAN III (previously familial dysautonomia) is the commonest type of HSAN [10, 11,12]. It is transmitted as an autosomal recessive trait, and is prevalent in Ashkenazi Jews. Scattered cases in non-Jewish individuals have been reported. Prenatal diagnosis is now possible [13]. Onset is congenital with hypotonia, sucking difficulties, a poor cry and vomiting: all present at birth. Autonomic features, such as impaired tear production, unstable temperature control, postural hypotension and excessive sweating (especially during infection) are prominent. Impaired oesophageal motility with resultant pneumonia, together with bouts of apnoea, can cause childhood death. Blotchy erythema occurs during overheating, and erythematous macules may appear on the trunk and limbs following emotional upset [13, 1415]. Diagnosis can be made in infancy by recognizing that the child cries without tears. Other diagnostic criteria include absence of fungiform papillae on the tongue, absent tendon and corneal reflexes, lack of axon flare after intradermal histamine, and miosis following instillation of methacholine. Later, the child may show delayed growth and invariably displays postural hypotension. Intelligence remains normal, but emotional lability is common. Progressive abnormality of cutaneous temperature discrimination and nociception is common [10]. Fifty per cent of patients now survive to adulthood [14]. Death may follow inhalation during vomiting attacks. Hypotension and cardiac arrest are general anaesthetic risks [15].
Hereditary sensory and autonomic neuropathy type IV
HSAN IV probably comprises a heterogeneous group, and is transmitted by autosomal recessive inheritance [16, 17]. Clinically, it is difficult differentiating between HSAN II and IV. HSAN IV is characterized by recurrent fevers, anhidrosis, absence of nociception, self-mutilation and learning difficulties [18, 19]. Infants are hypotonic and areflexic. High fevers, related to an inability to sweat, can be associated with seizures [18, 19]. Insensitivity to pain with self-inflicted injury results in multiple scars and bone fractures, auto-extraction of teeth, and ulceration of tongue, lips and buccal mucosa. The anomalous pain and temperature sensation and anhidrosis are due to, respectively, the absence of afferent neurons activated by tissue-damaging stimuli, and a loss of innervation of eccrine sweat glands [19].
Hereditary sensory and autonomic neuropathy type V
HSAN V is a rare autosomal recessive HSAN characterized by insensitivity to pain (it is also known as congenital insensitivity to pain), self-mutilating behaviour, anhidrosis with recurrent hyperpyrexia and learning difficulties [19]. HSAN V is caused by defective development of the sensory and autonomic peripheral nerves [20], with loss of unmyelinated and small myelinated fibres [20]. Signs present in early childhood when the infant starts to crawl. Affected children fail to learn the consequences of injury and do not cry with soft-tissue injury. Skin ulcerations on the fingers and toes are common from self-biting. There is heat intolerance due to anhidrosis, with hyperpyrexic episodes causing the death of up to 10% of patients in the first 3 years of life. Sunburn and frostbite may occur. The sympathetic skin response (SSR) is strikingly absent. Neurological examination often reveals complete analgesia, although tactile discrimination, sensation of cold, heat and vibration may be normal. Corneal reflexes are usually present, but deep tendon reflexes are absent. Neonatal hypotonia and psychomotor retardation are common, as are slow-healing fractures, joint deformities, osteomyelitis, avascular necrosis and acro-osteolysis of the fingers [21].
Differential diagnosis
Other disorders accompanied by self-mutilating behaviour can resemble some aspects of congenital HSANs. Examples are Lesch–Nyhan syndrome and untreated phenylketonuria. Diabetic neuropathy mimics some aspects of adult-onset HSAN.
Complications and co-morbidities
The commonest complications of HSAN IV are corneal scarring, multiple fractures, joint deformities, osteomyelitis and disabling self-mutilations. Malignant hyperthermia and sepsis are major causes of mortality. Mortality, behavioural disorders and learning difficulties significantly decrease after the age of 3 years [18].
Investigations
Diagnosis is based on clinical findings and molecular genetic testing of the mutated gene. Electrophysiology and neural histopathology are also useful [3].
Management
The ambient temperature must be controlled to help counter problems associated with sweating dysfunction. Protective aids can be used to reduce self-mutilation injuries. When necessary, wound care and antiseptic treatment should be initiated. A greasy emollient applied regularly to the skin of neuropathic limbs may moderate callus formation.
Resources
Patient resources
- http://www.ncbi.nlm.nih.gov/books/NBK49247/
- http://www.ncbi.nlm.nih.gov/books/NBK1769/
- For HSAN II, III and IV For HSAN II, III and IV http://www.ojrd.com/content/2/1/39 All last accessed June 2015.
Sympathetic nerve injury
Definition
Interruption of the sympathetic innervation of the skin resulting in loss of both vasoconstrictor impulses (causing erythema) and sweating (causing anhidrosis).
Introduction and general description
Loss of sympathetic supply to the skin following nerve damage will result in vasodilation and anhidrosis.
Pathophysiology
Sympathetic nerve injury usually occurs when sympathetic axons are injured by trauma affecting major nerves. There can be dissociation of sudomotor and pilomotor activity after sympathetic ganglionectomy [1]. The areas of vasodilation generally match the areas of anhidrosis, suggesting close correspondence of sudomotor and vasoconstrictor fibres.
Clinical features
When the sympathetic supply to the skin is interrupted, loss of vasoconstrictor impulses leads to erythema with passive vasodilation. The denervated area is also anhidrotic. Affected skin may be scaly and fissured. In the denervated areas, there is no loss of cutaneous sensation, possibly due to regeneration of post-ganglionic cholinergic fibres. Some patients report hyperaesthesia.
When sympathetic denervation is combined with a loss of somatic sensation, as in peripheral nerve injury or severe peripheral neuropathy, neurotrophic ulcers may occur. These result from minor local trauma and are characteristically painless and slow healing. Sympathetic denervation may also slow or prevent the normal greying of the hair with ageing, and may cause hyperpigmentation of the skin in the affected area.
Investigations
The affected area of skin should be physically examined for sweating, temperature, allodynia and hyperalgesia. Pupillary examination is indicated. Measurements of sweating and vasomotor responses can help determine the extent of autonomic denervation [2].
Laboratory tests include the SSR, thermoregulatory sweat test (TST), quantitative sudomotor axon reflex test (Q-SART), skin wrinkling on water immersion, and microneurography.
Management
Appropriate neurological/neurosurgical management of damaged nerve(s).
Resources
Patient resources
- http://wiki.cns.org/wiki/index.php/Injury,_Sympathetic_Nerve (last accessed June 2015).
Complex regional pain syndrome
Definition and nomenclature
Complex regional pain syndrome (CRPS) is a debilitating, painful condition in a limb, commonly arising after injury, associated with sensory, motor, autonomic nerve problems, along with abnormalities in the bone and skin of the affected limb [1].
Introduction and general description
CRPS is a debilitating condition affecting a limb, which is generally caused by physical injury to that body part. There appears to be no relationship with the severity of trauma; indeed in a few cases there is no precipitating trauma. CRPS results in neurological, cutaneous and skeletal abnormalities in the affected limb. CRPS is a difficult condition to treat effectively; the primary aims are to reduce pain and to preserve or restore function.
Epidemiology
Incidence and prevalence
The European incidence rate of CRPS is 26/100 000 person-years [2].
Age
It commonly occurs in the 30–50-year age group.
Sex
Women predominate in a ratio of 4 : 1.
Pathophysiology
The pathogenesis is poorly understood, although abnormalities in the peripheral and central nervous systems have been described, with and without major nerve lesions. The earlier belief that the predominant problem is sympathetic dysfunction and that CRPS occurs in (stereotyped) stages is now obsolete. CRPS is not associated with a history of preceding psychological problems, or with somatization or malingering [3]. Hypotheses to explain the disorder include the release of neuropeptides from peripheral unmyelinated fibres which causes pain and vasodilation. Other proposed pathogenetic pathways are an enhanced α-adrenergic receptor activity, and up-regulation of afferent nocioreceptors in response to norepinephrine release from sympathetic efferents. Excessive inflammation at the injury site, and immune system or central nervous system involvement, may also contribute to the pathogenesis of CRPS [4].
Predisposing factors
Stroke, myocardial infarction, tuberculosis, herpes zoster and certain drugs may predispose to CRPS [4]. Dermatological conditions such as vasculitis and panniculitis may precede the condition. Excisional skin biopsies, including nail biopsies, have been reported as triggering CRPS [5].
Box 85.1 lists the dermatologically associated causes [4].
Clinical features
Presentation
CRPS is usually preceded by trauma, although there is no relationship with the severity of trauma [2]. CRPS can be divided into two types: in type I CRPS (the more common) a major nerve lesion is absent; in type II CRPS (the less common) a major nerve lesion is present [5]. CRPS usually affects one limb, but in 7% of cases can spread to others [6,7]. Onset of symptoms is usually within 1 month of the trauma or immobilization of the limb.
Symptoms fall into three stages. The first begins after several days or weeks and lasts about a month, involving spontaneous burning and stinging, or tearing or shooting, pain, precipitated by mechanical stimuli such as bathing, clothing on the skin or draughts [7].
Stage two occurs 1–7 months after injury and lasts 3–6 months. Symptoms relate to sympathetic hyperactivity and include cool, oedematous skin, hyperhidrosis and cyanosis, or livedo-like changes. Hair growth may increase or decrease. Nails may show decreased or increased growth or thickening, become brittle or develop striations. Pain is variable, and neuralgia may either spread or decrease.
Stage three, starting around 8 months after injury, involves progressive tissue damage, which can become permanent. The changes may be due to vasoconstriction (resulting in skin hypoxia) or decreased motion of the skin from inactivity of underlying joints, tendons or ligaments. Usually, the skin becomes shiny, atrophic and dry; fingertips may shrink. Some patients have no trophic skin changes [8]. Deeper structures, including fascia, can thicken, resulting in contractures. If the pain is worsened by physical stimuli, the patient may protect the limb leading to trophic changes in the bone, muscle and skin (Sudeck atrophy).
Box 85.2 lists dermatological manifestations of CRPS [4].
Differential diagnosis
The Royal College of Physicians, UK, provides an extensive list of conditions to consider in formulating a diagnosis, ranging from infections through arthritis and arthrosis, to bone, soft-tissue or neural injury [5].
Disease course and prognosis
Approximately 15% of sufferers will have unrelenting pain and physical impairment 2 years after CRPS onset, although more will suffer less ongoing pain and dysfunction [8].
Investigations
Electrodiagnostic studies (needle electromyography and nerve conduction) are normal in CRPS type I, but demonstrate a peripheral nerve lesion in type II [4]. Patchy osteoporosis may be seen on some plain radiographs in CRPS II. CT or MRI scans may show atrophy or soft-tissue swelling, and bone mineralization changes. Bone scintigraphy using three-phase bone scans may usefully identify early changes. Other tests include: pain relief on α1-adrenergic blockade with intravenous phentolamine; pain exacerbation on α2 stimulation by clonidine; and severe pain on cold stimulation. These tests help identify patients with sympathetically maintained pain (SMP). Despite having similar signs and symptoms, only some patients with CRPS type I show alleviation of the pain by selective sympathetic nervous system or α-adrenoreceptor blockade. SMP can also present in CRPS II patients, although the mechanism is probably different [5,8].
The Budapest criteria apply when diagnosing CRPS. These include the presence of allodynia and/or hyperalgesia; temperature asymmetry and/or skin colour changes/asymmetry; oedema and/or sweating changes/asymmetry; and decreased range of motion and/or motor dysfunction (weakness, tremor, dystonia) and/or trophic changes [5,8].
Management
Prompt diagnosis and early treatment are considered best practice to avoid secondary physical problems associated with disuse of the affected limb and the psychological consequences of living with undiagnosed chronic pain.
There is no proven cure for CRPS, and an integrated interdisciplinary treatment approach is recommended [5,8]. Primary aims are to reduce pain, to preserve or restore function, and to enable patients to manage their condition [8]. The four ‘pillars’ of care (education, pain relief, physical rehabilitation and psychological intervention) addressing these aims carry equal importance. However, full recovery can be elusive, even with early appropriate treatment [8]. Although there is no evidence that early physiotherapy can prevent CRPS, early diagnosis and treatment with physiotherapy and/or occupational therapy, delivered by specifically competent therapists, is recommended to improve function and to prevent complications such as contractures, and to speed recovery [8].
Medication is indicated to minimize pain and support physical rehabilitation. Patients waiting to see a pain specialist should be seen regularly and be advised about the use of simple analgesics.
If simple medication does not reduce the pain to a mild level after 3–4 weeks, medication should be considered according to the neuropathic pain guidelines [8]. Earlier use may be appropriate. The patient should also be referred to a pain clinic for multidisciplinary pain therapy treatment, which ideally should begin within 3 months of the condition's onset [8].
The best functional advice for patients with suspected CRPS, or CRPS for which concomitant pathology has not yet been ruled out, is currently unclear. Pragmatically, encouragement of gentle limb use and active lifestyle is recommended [8].
Psychological interventions, including biofeedback, cognitive behavioural and relaxation techniques are crucial, although often overlooked. If psychological factors (e.g. significant distress) are suspected, early referral to a psychologist specializing in pain may be advisable [5].
Surgery should only rarely be considered. Orthopaedic surgeons should be aware of specific treatments for chronic CRPS, such as specialist physiotherapy and occupational therapy, multidisciplinary pain management programmes, spinal cord stimulation and specialist rehabilitation programmes [5,8].
Resources
Patient resources
Horner syndrome
Definition and nomenclature
Horner syndrome follows partial or complete interruption of the sympathetic nerve pathways of the face. It is characterized by ptosis, miosis and anhidrosis.
Introduction and general description
Horner syndrome appears when the three-neuron sympathetic pathway is interrupted anywhere from the posteriolateral nuclei of the hypothalamus, through the spinal cord, to the eye [1]. It is characterized by ptosis, miosis and anhidrosis.
Pathophysiology
The sympathetic fibres supplying the facial skin travel from the hypothalamus via the spinal cord, to relay at the level of the first and second thoracic segments in the lateral column of the spinal grey matter. Preganglionic fibres emerge from the first and second thoracic spinal nerves, and pass up the cervical sympathetic chain to relay in the superior cervical ganglion. From here, post-ganglionic fibres pass to supply the eye and the skin of a small central area of the face via the internal carotid sympathetic plexus. Other fibres pass along the external carotid artery and its branches to innervate most of the facial skin with vasomotor and sudomotor fibres.
This pathway can be interrupted centrally in the spinal cord, for example by medullary infarction, syringomyelia, multiple sclerosis or intraspinal tumours. The peripheral fibres can be damaged by aortic aneurysm, cervical lymphadenopathy, surgery, regional anaesthetic procedures or tumours. Horner syndrome may follow sympathectomy for the treatment of palmar and axillary hyperhidrosis, and occurs in up to 40% of patients after open cervical sympathectomy, and 8% of those having transthoracic endoscopic sympathectomy [2]. In such cases, there may be resolution of longstanding pompholyx-type hand eczema ipsilaterally, suggesting a neurological pathogenesis for endogenous pompholyx in some patients [3, 4].
Clinical features
Horner syndrome is usually found in adults and only rarely in children, where it can be either congenital or acquired [1]. An irritative phase occurs rarely at the outset of Horner syndrome, characterized by transient unilateral hyperhidrosis and vasoconstriction. The paralytic phase involves drooping of the eyelid (ptosis) with narrowing of the palpebral fissure. The pupil is small, but shows normal reflex constriction to light and accommodation. Inflammation of the conjunctiva is often present. Sweating is absent on the ipsilateral side of the face. There may be slight retraction of the eyeball into the orbit (enophthalmos). Bilateral cases are rare. Sweat glands on the medial and lateral parts of the forehead are innervated separately, the former by fibres from the sympathetic plexus of the internal carotid, and the latter from the plexus surrounding the external carotid [5]. This explains the findings in Raeder syndrome, where damage involving the perivascular plexus of the internal carotid leads to anhidrosis only medially on the forehead [6].
Three cases of bilateral Horner syndrome, and three cases of contralateral Horner syndrome have been reported [7–10].
Investigations
Physical examination and pupil dilatation test using hydroxyamphetamine eyedrops can confirm the diagnosis [11]. To elicit the underlying cause, X-ray, MRI, and blood or urine tests might be needed.
Management
Treatment should be directed towards the underlying cause.
Resources
Further information
- http://www.mayoclinic.org/diseases-conditions/horner-syndrome/basics/definition/CON-20034650 (last accessed June 2015).
Gustatory hyperhidrosis
Definition and nomenclature
Gustatory hyperhidrosis describes excessive sweating occurring immediately after eating spicy or hot food. Gustatory sweating is localized to certain areas, including the scalp, upper lip, perioral region and sternum.
Introduction and general description
Gustatory hyperhidrosis is a disorder characterized by excessive sweating on the scalp, upper lip, perioral region or sternum, immediately after eating spicy or hot food. It can occur following damage to the sympathetic cervical trunk, the vagus nerve or the auriculotemporal nerve [1].
Pathophysiology
Gustatory hyperhidrosis may complicate surgery involving the parotid gland or the temporomandibular joint. Auriculotemporal nerve injury following closed treatment for maxillomandibular joint trauma can also lead to this form of focal hyperhidrosis.
The autonomic nervous system has a propensity for regrowth [2]. Damage to adjacent preganglionic parasympathetic fibres and post-ganglionic sympathetic fibres may result in parasympathetic fibres regrowing into the sympathetic nerves, thereby directly controlling sweat gland function. In the neck, for example, following damage to the sympathetic cervical trunk and the vagus (parasympathetic) during thyroidectomy or after trauma, such reinnervation may result in gustatory hyperhidrosis, even with bland foods [1]. A similar event may occur on the cheeks or chin following parotid or submandibular gland surgery – the auriculotemporal syndrome (Frey syndrome) [3]. Secretory sweating is now more frequently seen after endoscopic transthoracic sympathectomy [4, 5]. A recent review reported a rate of gustatory hyperhidrosis of 1.1% [6].
Clinical features
Immediately following the ingestion of spicy or hot food, sweating occurs which is localized to certain areas, typically the scalp, upper lip, perioral region or sternum. Frey syndrome describes gustatory sweating and facial flushing [7], and emerges between 3 and 24 months after surgery involving the parotid or temporomandibular joint [7,8].
Investigations
Subjective symptoms should be investigated. The Minor starch iodine test and infrared thermography can be used to investigate the symptoms and confirm the diagnosis [9]. With medical thermography, images are taken after the patient has chewed a sialagogue [9, 10].
Management
Topical preparations containing aluminium chloride hexahydrate can control the symptoms, but may produce an irritant dermatitis. Botulinum toxin has been shown to be effective and safe in gustatory sweating [11]. The use of a 0.5% aqueous solution of glycopyrronium bromide topically has been shown to be effective, safe, well tolerated and convenient in diabetes-associated gustatory sweating [12].
Resources
Further information
- https://www.neurology.org/content/65/11/E24.full (last accessed June 2015).
Restless legs syndrome/burning feet syndrome
Definition and nomenclature
Restless legs syndrome (RLS) is characterized by an uncomfortable twitching sensation in the leg muscles when sitting or lying down, which is only relieved by moving the legs.
Burning feet syndrome (BFS) is characterized by a burning and aching sensation of the feet (hyperaesthesia), accompanied by vasomotor changes causing excessive sweating.
Introduction and general description
RLS is a common sleep and movement disorder. It is characterized by leg paraesthesiae accompanied by an irresistible urge, occurring at rest, to move the legs. Movement of the legs gives relief. BFS is a poorly recognized and underdiagnosed condition consisting of a burning sensation of the feet which is accentuated by heat or cold [1]. Associated autonomic features include dry skin, eyes and mouth, and vasomotor symptoms with peripheral coldness, burning or flushing, hypertension and impotence [1].
Epidemiology
Incidence and prevalence
One population study estimated 7.2% to have had RLS symptoms at least weekly during the preceding year; 2.7% reported moderately or severely distressing RLS symptoms at least twice-weekly [2].
Age
The conditions appear on average at between 30 and 40 years.
Sex
Both conditions are more prevalent in females.
Predisposing factors
- Diabetes.
- Chronic kidney disease (uraemia).
- Vitamin deficiency (B12 and occasionally B6).
- Alcohol abuse.
- Hypothyroidism.
- Lyme disease.
- HIV/AIDS.
- Amyloid polyneuropathy.
- Drug side effects, including chemotherapy drugs, vitamin B6 overdose, HIV medicines, isoniazid, amiodarone, metformin and others.
- Heavy metal poisoning (lead, mercury, arsenic).
- Vasculitis.
- Sarcoidosis.
- Guillain–Barré syndrome.
- Arterial disease.
- Hypertension.
Pathophysiology
RLS is a clinically pleomorphic syndrome, probably reflecting multiple genetic and acquired factors. It may represent a subclinical sensory neuropathy [3]. Others have suggested a dopamine imbalance, due to the presence of dyskinetic movements and response to levodopa. Basal ganglia studies using positron emission tomography (PET) scanning have shown decreased binding of dopamine to its receptor, and involvement of the endogenous opioid system has recently been implicated [1]. Up to 30% of patients have iron deficiency [4]. RLS is common in haemodialysis patients [4] and associations have been noted with pregnancy, fibromyalgia, rheumatoid arthritis and multiple sclerosis [1].
Loss of small-fibre sensory nerves has been found both in type II diabetics [1] and idiopathic cases. Tests on the autonomic system have shown predominantly cholinergic defects, unlike with other autonomic neuropathies. There is a close correlation between quantitative abnormalities in the sudomotor axon reflex test and the loss of small-nerve fibres in the skin.
Genetics
There is a link between RLS and chromosome 12q [5].
A familial form of BFS has been described [6]. BFS families with autosomal dominant inheritance are recognized [7].
Clinical features
Presentation
RLS is characterized by leg paraesthesiae associated with an irresistible urge, occurring at rest, to move the legs. Movement of the legs provides relief [1]. Symptoms are worse at night – with both circadian rhythms and recumbency playing a role [1] – and often lead to disruption of normal life and chronic sleep deprivation. Children may struggle at school and display symptoms of attention deficit hyperactivity disorder (ADHD) [1].
Two subtypes of RLS are recognized. The first, with early onset, accounts for some 25% of cases, is often familial, and is associated with a childhood history of ‘growing pains’ [8]. The second (idiopathic RLS) lacks a family history, and has a late onset with milder paraesthesiae or dysaesthesia, and insomnia.
Sensory symptoms include painful legs and arms, and pain at ‘internal’ sites. Many patients with RLS have been diagnosed with a musculoskeletal disorder, such as joint and back pains. In RLS, there is a significant association with depression and other neuropsychiatric symptoms [1,2]. Morning and daytime headaches occur frequently, and hypertension and heart problems have been reported in men.
The International Restless Leg Syndrome Study Group rating scale (the international restless legs severity scale (IRLS)) is a validated measure for the presence and severity of RLS. All four essential criteria must be present for a positive diagnosis [9]:
- An urge to move the legs, usually with uncomfortable or unpleasant sensations.
- Unpleasant sensations or urges to move beginning or worsening during rest or inactivity.
- Unpleasant sensations or urges to move, partly or totally relieved by movement such as walking, bending, stretching, etc., at least during the activity.
- Unpleasant sensations or urges to move, worse (or exclusively) in the evening or night-time.
A recent fifth criterion stipulates that the four criteria must not be solely explained by another medical or behavioural condition [10].
The main clinical features of BFS are a burning sensation on the feet, accentuated by heat or cold. Associated autonomic features often accompany foot dysaesthesia, including dry skin, dry eyes and mouth, vasomotor symptoms with peripheral coldness, burning or flushing, hypertension and impotence [1].
Differential diagnosis
Restless legs syndrome
- Peripheral neuropathy
- Parkinsonism
- Nocturnal leg cramps
- Peripheral vascular disease
- ADHD in children
Burning feet syndrome
- Peripheral neuropathy
Investigations
To exclude peripheral neuropathy.
Management
Some believe the first choice of treatment for RLS should be dopaminergic agents such as levodopa, or the longer acting cabergoline [11]. Levodopa is efficacious in the short-term treatment of RLS [11]. Recent studies with gabapentin report significant improvement in both idiopathic RLS [12] and haemodialysis patients [4]. Other suggested second line options include opioids (oxycodone, codeine and methadone), and benzodiazepines (clonazepam or diazepam) [8]. Treatment with dopamine agonists can be complicated by augmentation, an iatrogenic effect whereby symptoms worsen with time [10].
Resources
Patient resources
- http://www.patient.co.uk/doctor/restless-legs-syndrome#ref-3 (last accessed June 2015)
References
Introduction
- Metze D, Luger T. Nervous system in the skin. In: Freinkel RK, Woodley DT, eds. The Biology of the Skin. New York: Parthenon, 2000:153–76.
- Lynn B. Cutaneous sensation. In: Goldsmith LA, ed. Physiology, Biochemistry and Molecular Biology of the Skin. New York: Oxford University Press, 1991:779–815.
- Sternberg EM. Neuroendocrine regulation of autoimmune/inflammatory disease. J Endocrinol 2001;169:429–35.
- Bridges B, Thompson SWN, Rice ASC. Mechanisms of neuropathic pain. Br J Anaesth 2001;87:12–26.
- Maksimovic S, Baba Y, Lumpkin EA. Neurotransmitters and synaptic components in the Merkel cell-neurite complex, a gentle-touch receptor. Ann NY Acad Sci 2013; 1279:13–21.
- Harvima IT, Nilsson G, Naukkarinen A. Role of mast cells and sensory nerves in skin inflammation. G Ital Dermatol Venereol 2010;145(2):195–204.
- Harvima IT, Nilsson G. Stress, the neuroendocrine system and mast cells: current understanding of their role in psoriasis. Expert Rev Clin Immunol 2012;8(3):235–41.
- Steinhoff M, Ständer S, Seeliger S, et al. Modern aspects of cutaneous neurogenic inflammation. Arch Dermatol 2003;139:1479–88.
- Eedy DJ. Neuropeptides in skin. Br J Dermatol 1993;128:597–605.
- Zegarska B, Leliñska A, Tyrakowski T. Clinical and experimental aspects of cutaneous neurogenic inflammation. Pharmacol Rep 2006;58:13–21.
- Garle MJ, Fry JR. Sensory nerves, neurogenic inflammation and pain: missing components of alternative irritation strategies? A review and a potential strategy. Altern Lab Anim 2003;31:295–316.
- Fernandes ES, Fernandes MA, Keeble JE. The functions of TRPA1 and TRPV1: moving away from sensory nerves. Br J Pharmacol 2012 May;166(2):510–21.
- Aubdool AA, Brain SD. Neurovascular aspects of skin neurogenic inflammation. J Investig Dermatol Symp Proc 2011;15(1):33–9.
- Pincelli C, Fantini F, Giannetti A. Neuropeptides and skin inflammation. Dermatology 1993;189:156–8.
- Castorina A, Scuderi S, D'Amico AG, et al. PACAP and VIP increase the expression of myelin-related proteins in rat schwannoma cells: Involvement of PAC1/VPAC2 receptor-mediated activation of PI3K/Akt signaling pathways. Exp Cell Res 2014; 322(1):108–21.
- Peters EMJ, Ericson ME, Hosoi J, et al. Neuropeptide control mechanisms in cutaneous biology: physiological and clinical significance. J Invest Dermatol 2006; 126:1937–47.
- Bonelli RM, Költringer P. Autonomic nervous function assessment using thermal reactivity of microcirculation. Clin Neurophysiol 2000;111:1880–8.
- Delis KT, Lennox AF, Nicolaides AN, Wolfe JH. Sympathetic autoregulation in peripheral vascular disease. Br J Surg 2001;88:523–8.
- Joyner MJ, Halliwill JR. Sympathetic vasodilation in humans. Topical review. J Physiol 2000;526:471–80.
- Bunker CB, Foreman J, O'Shaughnessy D, et al. Calcitonin gene-related peptide in the treatment of severe Raynaud's phenomenon. Br J Dermatol 1989;121:43–4.
- Goldsmith PC, Bunker CB, Leslie TA, et al. Cutaneous nerve fibre depletion in vibration finger. Br J Dermatol 1992;127:424 [Abstract].
- Delius W, Hagbarth K-E, Hongell A, Wallin BG. Manoeuvres affecting sympathetic outflow in human skin nerves. Acta Physiol Scand 1972;84:177–86.
- Shahani BT, Halperin JJ, Boulu P, Cohen J. Sympathetic skin response – a method of assessing unmyelinated axon dysfunction in peripheral neuropathies. J Neurol Neurosurg Psychiatry 1984;47:536–42.
- Vetrugno R, Liguori R, Cortelli P, Montagna P. Sympathetic skin response. Basic mechanisms and clinical applications. Clin Auton Res 2003;13:256–70.
- Sawasaki N, Iwase S, Tadaaki M. Effect of skin sympathetic response to local or systemic cold exposure on thermoregulatory functions in humans. Auton Neurosci 2001;87:274–81.
- Scott AR, MacDonald IA, Bennett T, Tattersall RB. Abnormal thermoregulation in diabetic autonomic neuropathy. Diabetes 1988;37:961–8.
- Inoue Y, Araki T, Tsujita J. Thermoregulatory responses of prepubertal boys and young men in changing temperature linearly from 28 to 15°C. Eur J Appl Physiol 1996;72:204–8.
- Khan F, Spence VA, Belch JJF. Cutaneous vascular responses and thermoregulation in relation to age. Clin Sci 1992;82:521–8.
- Sendowski I, Savourey G, Launay JC, et al. Sympathetic stimulation induced by hand cooling alters cold-induced vasodilatation in humans. Eur J Appl Physiol 2000;81:303–9.
- Lewis T. Blood Vessels of the Human Skin and their Responses. London: Shaw & Sons, 1927.
- Baraniuk JN. Neuropeptides and the skin. In: Bos JD, ed. Skin Immune System, 2nd edn. Boca Raton: CRC Press, 1997:311–23.
- Greaves MW, Sabroe RA. Histamine: the quintessential mediator. J Dermatol 1996;23:735–40.
- Anand P, Bannister R, McGregor GP, et al. Marked depletion of dorsal spinal cord substance P and calcitonin gene-related peptide with intact skin flare responses in multiple system atrophy. J Neurol Neurosurg Psychiatry 1988;51:192–6.
- Rosa AC, Fantozzi R. The role of histamine in neurogenic inflammation. Br J Pharmacol 2013;170(1):38–45.
Neuropathic ulcer
- Boulton A. Peripheral neuropathy and the diabetic foot. Foot 1992;2:67–72.
- Boulton A. The pathogenesis of diabetic foot problems: an overview. Diabet Med 1996;13:S12–16.
- Sumpio BE. Foot ulcers. N Engl J Med 2000;343:787–93.
- Phillips TJ. Leg ulcers. Postgrad Med 1999;105:165–73.
- Singh N, Armstrong DG, Lipsky BA. Preventing foot ulcers in patients with diabetes. JAMA 2005;293(2):217–28.
- Reiber GE, Vilerkyte L, Boyko EJ. Causal pathways for incident lower extremity ulcers in patients with diabetes from two settings. Diabetes Care 1999; 22:157–62.
- Clayton W, Elasy TA. A review of the pathophysiology, classification, and treatment of foot ulcers in diabetic patients. Clin Diabetes 2009;27(2):52–8.
- Pavicic T, Korting HC. Xerosis and callus formation as a key to the diabetic foot syndrome: dermatologic view of the problem and its management. J Deutsch Dermatol Gesell 2006;4: 935–41.
- Widatalla AH, Mahadi SE, Shawer MA, et al. Implementation of diabetic foot ulcer classification system for research purposes to predict lower extremity amputation. Int J Diabetes Dev Ctries 2009 Jan;29(1):1–5.
- Dang CN, Prasad YD, Boulton AJ, Jude EB. Methicillin-resistant Staphylococcus aureus in the diabetic foot clinic: a worsening problem. Diabet Med 2003;20:159–61.
- Klenerman L, McCabe C, Cogley D, et al. Screening for patients at risk of diabetic foot ulceration in a general diabetic outpatient clinic. Diabet Med 1996;13:561–3.
- Grayson ML, Gibbons GW, Balogh K, et al. Probing to bone in infected pedal ulcers: a clinical sign of underlying osteomyelitis in diabetic patients. JAMA 1995;273:721–3.
- Malone JM, Snyder M, Anderson G, et al. Prevention of amputation by diabetic education. Am J Surg 1989;158:520–3.
- Krishnan S, Nash F, Baker N, et al. Reduction in diabetic amputations over 11 years in a defined UK population: benefits of multidisciplinary team work and continuous prospective audit. Diabetes Care 2008;31(1):99–101.
- Catanzariti AR, Haverstock BD, Grossman JP, Mendicino RW. Off-loading techniques in the treatment of diabetic plantar neuropathic foot ulceration. Adv Wound Care 1999;12:452–8.
- Armstrong DG, Nguyen HC, Lavery LA, et al. Off-loading the diabetic foot wound: a randomized clinical trial. Diabetes Care 2001;24:1019–22.
- Rizzo L, Tedeschi A, Fallani E, et al. Custom-made orthesis and shoes in a structured follow-up program reduces the incidence of neuropathic ulcers in high-risk diabetic foot patients. Int J Low Extrem Wounds 2012 Mar;11(1):59–64.
- Boulton AJM. What you can't feel can hurt you. J Am Pod Med Assoc 2010;100(5): 349–52.
- Tsourdi E, Barthel A, Rietzsch H, et al. Current aspects in the pathophysiology and treatment of chronic wounds in diabetes mellitus. Biomed Res Int 2013 Mar:1–6.
- International Best Practice Guidelines: Wound management in diabetic foot ulcers. Wounds International, 2013 www.woundsinternational.com (last accessed June 2015).
- Bennett SP, Griffiths GD, Schor AM, et al. Growth factors in the treatment of diabetic foot ulcers. Br J Surg 2003;90:133–46.
- Papanas N, Maltezos E. Benefit-risk assessment of becaplermin in the treatment of diabetic foot ulcers. Drug Safety 2010;33(6):455–61.
- Badiavas EV, Falanga V. Treatment of chronic wounds with bone marrow-derived cells. Arch Dermatol 2003;139(4):510–16.
- Edmonds M, Bates M, Doxford M, et al. New treatments in ulcer healing and wound infection. Diabetes Metab Res Rev 2000;16 (Suppl. 1):S51–4.
- Vazquez JR, Short B, Findlow AH, et al. Outcomes of hyaluronan therapy in diabetic foot wounds. Diabetes Res Clin Pract 2003;59:123–7.
- Humbert P, Mikosinki J, Benchikhi H, et al. Efficacy and safety of a gauze pad containing hyaluronic acid in treatment of leg ulcers of venous or mixed origin: a double-blind, randomised, controlled trial. Int Wound J 2013 Apr;10(2):159–66.
- Ndip A, Ebah L, Mbako A. Neuropathic diabetic foot ulcers – evidence-to-practice. Int J Gen Med 2012;5:129–34.
- Larsson J, Apelqvist J, Agardh CD, Stenstrom A. Decreasing incidence of major amputation in diabetic patients: a consequence of a multidisciplinary foot care team approach? Diabet Med 1995;12:770–6.
- Myerly SM, Stavosky JW. An alternative method for reducing plantar pressures in neuropathic ulcers. Adv Wound Care 1997;10:26–9.
- Laing P. Diabetic foot ulcers. Am J Surg 1994;167 (Suppl. 1A):S31–6.
- Morona JK, Buckley ES, Jones S, et al. Comparison of the clinical effectiveness of different off-loading devices for the treatment of neuropathic foot ulcers in patients with diabetes: a systematic review and meta-analysis. Diabetes Metab Res Rev 2013;29(3):183–93.
Syringomyelia
- Sharma M, Coppa N, Sandhu FA. Syringomyelia: A Review. Semin Spine Surg 2006;18(3):180–4.
- Kastrup A, Nägele T, Topka H. Spontaneous resolution of idiopathic syringomyelia. Neurology 2001;57:1519–20.
- Jack CRJ, Kokmen E, Onofrio BM. Spontaneous decompression of syringomyelia: magnetic resonance imaging findings. Case report. J Neurosurg 1991;74:283–6.
- Yeager BA, Lusser MA. Spontaneous resolution of idiopathic syringomyelia: MR features. J Comput Assist Tomogr 1992;16:323–4.
- Krishna V, McLawhorn M, Kosnik-Infinger L, et al. High long-term symptomatic recurrence rates after Chiari-1 decompression without dural opening: A single center experience. Clin Neurol Neurosurg 2014;118:53–8.
- Williams B. Progress in syringomyelia. Neurol Res 1986;8(3):130–45.
- Sudo K, Fujiki N, Tsuji S, et al. Focal (segmental) dyshidrosis in syringomyelia. J Neurol Neurosurg Psychiatry 1999;67:106–8.
- Wolf M, Fürstenberg CH, Hähnel S, et al. Spinal cord injury and syringomyelia (Rückenmarktrauma und Syringomyelie). Radiologe 2013 Apr;53(4):353–66.
- Haroun RI, Guarnieri M, Meadow JJ, Kraut M, Carson BS. Current opinions for the treatment of syringomyelia and Chiari malformations: survey of the Pediatric Section of the American Association of Neurological Surgeons. Pediatr Neurosurg 2000 Dec;33(6):311–17.
Spinal dysraphism
- Storer JS, Hawk RJ. Cutaneous signs of spinal dysraphism. In: Schachner LA, Hansen RC, eds. Paediatric Dermatology, Vol. 1. New York: Churchill Livingstone, 1988: 275–7.
- Lichtenstein BW. Spinal dysraphism. Spina bifida and myelodysplasia. Arch Neurosurg Psychiatry 1940;44:792–809.
- Lee B, Jeelani Y, McComb JG. Congenital dermal sinus with an infected dermoid cyst in the cervico-thoracic spinal cord. Pediatr Neurosurg 2013;49(2):89–92.
- Lee B-J, Sohn M-J, Han S-R, et al. Analysis of risk factors and management of cerebrospinal fluid morbidity in the treatment of spinal dysraphism. J Korean Neurosurg Soc 2013 Sep;54(3):225–31.
- Mitchell LE, Adzick NS, Melchionne PS, et al. Spina bifida. Lancet 2004;364:1885–95.
- Zerah M, Kulkarni, AV. Spinal cord malformations. In: Dulac O, Lassonde M, Sarnat HB, eds. Handbook of Clinical Neurology, Vol. 112. New York: Barnes & Noble, 2013: 975–91.
- Czeizel AE, Dudas I. Prevention of the first occurrence of neural-tube defects by pericontraceptional vitamin supplementation. N Engl J Med 1992;327:1832–5.
- Tavafoghi V, Ghandchi A, Hambrick GW, et al. Cutaneous signs of spinal dysraphism. Arch Dermatol 1978;114:573–7.
- Schenk JP, Herweh C, Günther P, et al. Imaging of congenital anomalies and variations of the caudal spine and back in neonates and small infants. Eur J Radiol 2006;58:3–14.
- Guggisberg D, Hadj-Rabia S, Viney C, et al. Skin markers of occult spinal dysraphism in children. Arch Dermatol 2004;140(9):1109–15.
- McGovern M, Mulligan S, Carney O, et al. Ultrasound investigation of sacral dimples and other stigmata of spinal dysraphism. Arch Dis Child 2013;98(10):784–6.
- Pierre-Kahn A, Zerah M, Renier D, et al. Congenital lumbosacral lipomas. Child Nerv Syst 1997;13:298–334.
- Boyvat A, Yazar T, Ekmekci P, Gurgey E. Lumbosacral vascular malformation: a hallmark for occult spinal dysraphism. Dermatology 2000;201:374–6.
- Tubbs RS, Wellons JC, Iskandar BJ, Oakes WJ. Isolated flat capillary midline lumbosacral hemangiomas as indicators of occult spinal dysraphism. J Neurosurg 2004;100(Suppl. Pediatrics 2):86–9.
- Najjar MW, Kusske JA, Hasso AN. Dorsal intramedullary dermoids. Neurosurg Rev 2005;28:320–5.
- Madden-Fuentes RJ, McNamara ER, Lloyd JC, et al. Variation in definitions of urinary tract infections in spina bifida patients: a systematic review. Pediatrics 2013 Jul;132(1):132–9.
- Mertes PM, Mouton C, Fremont S, et al. Latex hypersensitivity in spinal cord injured adult patients. Anaesth Intensive Care 2001;29:393–9.
- Bernardini R, Novembre E, Lombardi E, et al. Risk factors for latex allergy in patients with spina bifida and latex sensitization. Clin Exp Allergy 1999;29:681–6.
- Hamrick SE. Cesarean delivery and its impact on the anomalous infant. Clin Perinatol 2008;35:395–406.
- Drugan A, Weissman A, Evans MI. Screening for neural tube defects. Clin Perinatol 2001;28:279–87.
- Foster RD, Williams ML, Barovich AJ, et al. Giant congenital melanocytic naevi: the significance of neurocutaneous melanosis in neurologically asymptomatic children. Plast Reconstr Surg 2001;107:933–41.
- Dawson HA, Atherton DJ, Mayou B. A prospective study of congenital melanocytic naevi: progress report and evaluation after 6 years. Br J Dermatol 1996;134:617–23.
- Tortori-Donati P, Rossi A, Brancheri R, Cama A. Magnetic resonance imaging of spinal dysraphism. Top Magn Reson Imaging 2001;12:375–409.
- Adzick NS. Fetal surgery for spina bifida: past, present, future. Semin Pediatr Surg 2013;22(1):10–17.
- Adzick NS. Prospects for fetal surgery. Early Hum Dev 2013;89(11):881–6.
Dermatoses associated with spinal cord injury
- McDonald JW, Sadowsky C. Spinal cord injury. Lancet 2002;359:417–25.
- Wilson CL, Walshe M. Incidence of seborrhoeic dermatitis in spinal injury patients. Br J Dermatol 1988;119(S33):48.
- Thomas SE, Conway J, Ebling FJG, et al. Measurement of sebum excretion rate and skin temperature above and below the neurological lesion in paraplegic patients. Br J Dermatol 1985;112:569–73.
- Sato K, Kang WH, Saga K, Sato KT. Biology of sweat glands and their disorders II: Disorders of sweat gland function. J Am Acad Dermatol 1989;20(5):713–26.
- Stanworth PA. The significance of hyperhidrosis in patients with posttraumatic syringomyelia. Paraplegia 1982;20:282–7.
Hereditary sensory and autonomic neuropathies
- Axelrod F, Gold-von Simson G. Hereditary sensory and autonomic neuropathies: types II, III, and IV. Orphanet J Rare Dis 2007;2:39 www.ojrd.com/content/2/1/39 (last accessed June 2015).
- Dyck PJ. Neuronal atrophy and degeneration predominantly affecting peripheral sensory and autonomic neurones. In: Dyck PJ, Thomas PK, Griffin JW, et al., eds. Peripheral Neuropathy. Philadelphia: Saunders, 1993: 1065–93.
- Nicholson GA, Dawkins JL, Blair IP, et al. The gene for hereditary sensory neuropathy type I (HSN-1) maps to chromosome 9q22.1-q22.3. Nat Genet 1996;13:101–4.
- Bejaoui K, Uchida Y, Yasuda S, et al. Hereditary sensory neuropathy type 1 mutations confer dominant negative effects on serine palmitoyltransferase, critical for sphingolipid synthesis. J Clin Invest 2002;110:1301–8.
- Axelrod FB. Autonomic and sensory disorders. In:Emory AEH, Rimoin DL, eds. Principles of Medical Genetics. Edinburgh: Churchill Livingstone, 1996:397–411.
- Berginer V, Baruchin A, Ben-Yakar Y, Mahler D. Plantar ulcers in hereditary sensory neuropathy: a plea for conservative treatment. Int J Dermatol 1984;23: 664–8.
- Bockers M, Benes P, Bork K. Persistent skin ulcers, mutilations and acro-osteolysis in hereditary sensory and autonomic neuropathy with phospholipid excretion. Report of a family. J Am Acad Dermatol 1989;21:736–9.
- Heckmann JM, Carr JA, Bell N. Hereditary sensory and autonomic neuropathy with cataracts, mental retardation and skin lesions: five cases. Neurology 1995;45:1405–8.
- Ferrière G, Guzzetta F, Kulakowski S, et al. Nonprogressive type II hereditary sensory autonomic neuropathy: a homogeneous clinicopathologic entity. J Child Neurol 1992;7:364–70.
- Axelrod FB, Iver K, Fish I, et al. Progressive density loss in dysautonomia. Pediatrics 1981;67:517–22.
- Brunt PW, McKusick VA. Familial dysautonomia: a report of genetic and clinical studies with a review of the literature. Medicine (Baltimore) 1970;49:343–74.
- Mahloudji M, Brunt PW, McKusick VA. Clinical neurological aspects of familial dysautonomia. J Neurol Sci 1970;11:383–95.
- Eng CM, Slaugenhaupt SA, Blumenfeld A, et al. Prenatal diagnosis of familial dysautonomia by analysis of linked CA-repeat polymorphisms on chromosome 9q31-q33. J Med Genet 1995;59:349–55.
- Axelrod FB, Abularrage JJ. Familial dysautonomia: a prospective study of survival. J Pediatr 1982;101:234–6.
- Meridy HW, Creighton RE. General anaesthesia in eight patients with familial dysautonomia. Can Anaesth Soc J 1971;18:563–70.
- Rosemberg S, Marie SKN, Kliemann S. Congenital insensitivity to pain with anhidrosis (hereditary sensory neuropathy type IV). Pediatr Neurol 1994;11:50–6.
- Indo Y. Genetics of congenital insensitivity to pain with anhidrosis (CIPA) or hereditary sensory and autonomic neuropathy type IV. Clinical, biological and molecular aspects of mutations in TRKA (NTRK1) gene encoding the receptor tyrosine kinase for nerve growth factor. Clin Auton Res 2002;12 (Suppl.1):120–32.
- Verzé L, Viglietti-Panzica C, Plumari L, et al. Cutaneous innervation in hereditary sensory and autonomic neuropathy type IV. Neurology 2000;55:126–8.
- Shorer Z, Moses SW, Hershkovitz E, et al. Neurophysiological studies in congenital insensitivity to pain with anhidrosis. Pediatr Neurol 2001;25:397–400.
- Nolano M, Crisci C, Santoro L, et al. Absent innervation of skin and sweat glands in congenital insensitivity to pain with anhidrosis. Clin Neurophysiol 2000;111:1596–601.
- Schulman H, Tsodikow V, Einhorn M, et al. Congenital insensitivity to pain with anhidrosis (CIPA): the spectrum of radiological findings. Pediatr Radiol 2001;31:701–5.
Sympathetic nerve injury
- Brown GE, Adson AW. Physiologic effects of thoracic and of lumbar sympathetic ganglionectomy or section of the trunk. Arch Neurol Psychiatry 1929;22:322–57.
- Ravits JM. Autonomic nervous system testing. Muscle Nerve 1997;20:919–37.
Complex regional pain syndrome
- Veldman PH, Reynen HM, Arntz IE, Goris RJ. Signs and symptoms of reflex sympathetic dystrophy: prospective study of 829 patients. Lancet 1993;342(8878):1012–16.
- de Mos M, de Bruijn AG, Huygen FJ, et al. The incidence of complex regional pain syndrome: a population-based study. Pain 2007;129(1–2):12–20.
- Beerthuizen A, Stronks DL, Huygen FJ, et al. The association between psychological factors and the development of complex regional pain syndrome type 1 (CRPS1) – a prospective multicenter study. Eur J Pain 2011;15(9):971–5.
- Phelps RG, Wilentz S. Review. Reflex sympathetic dystrophy. Int J Dermatol 2000;39:481–6.
- Royal College of Physicians. Complex regional pain syndrome in adults: UK guidelines for diagnosis, referral and management in primary and secondary care. London: RCP, 2012 http://www.rcplondon.ac.uk/sites/default/files/documents/complex-regional-pain-full-guideline.pdf (last accessed June 2015).
- Harden RN. Complex regional pain syndrome. Br J Anaesth 2001;87:99–106.
- McBride A, Atkins B. Complex regional pain syndrome. Curr Orthop 2005;19:155–65.
- Turner-Stokes L, Goebel A. Complex regional pain syndrome in adults: concise guidance. Clin Med 2011;11:596–600.
Horner syndrome
- Lazar I, Cavari Y, Rosenberg E, Knyazer B. Horner's syndrome in patients admitted to the paediatric intensive care unit: epidemiology, diagnosis and clinical practice. Anaesth Intensive Care 2013 Jan;41(1):20–3.
- Chowdhury MM, Hedges R, Lanigan SW. Unilateral resolution of palmar eczema and hyperhidrosis complicated by Horner's syndrome following ipsilateral endoscopic cervical sympathectomy. Br J Dermatol 2000;143:653–4.
- Chowdhury MM, Hedges R, Lanigan SW. Intact nerve supply for eczema. Br J Dermatol 2001;144:1270–1.
- Möller H. Intact nerve supply for eczema. Br J Dermatol 2001;144:1270.
- Salvesen R. Innervation of sweat glands in the forehead. A study in patients with Horner's syndrome. Neurol Sci 2001;183:39–42.
- Sjaastad O, Elsäs T, Shen JM, et al. Raeder's syndrome: anhidrosis, headache and a proposal for a new classification. Funct Neurol 1994;9:215–34.
- Allen G, Samson B. Contralateral Horner's syndrome following stellate ganglion block. Can Anesth Soc J 1986;3:112–13.
- Manchikanti L. Bilateral Horner's syndrome following stellate ganglion block. Anesth Rev 1990;17:41–3.
- Wallace MS, Milholland AV. Contralateral spread of local anesthetics with stellate ganglion block. Reg Anesth 1993;18:55–9.
- Warrick JW. Stellate ganglion block in the treatment of Ménière's disease and symptomatic relief of tinnitus. Br J Anaesth 1969;41:699–702.
- Van der Wiel HL, Van Gijn J. Localization of Horner's syndrome. Use and limitations of the hydroxyamphetamine test. J Neurol Sci 1983 May;59(2):229–35.
Gustatory hyperhidrosis
- Cunliffe WJ, Johnson CE. Gustatory hyperhidrosis. A complication of thyroidectomy. Br J Dermatol 1967;79:519–26.
- Murray JG, Thompson JW. The occurrence and function of collateral sprouting in the sympathetic nervous system of the cat. J Physiol 1957;135:133–62.
- Bloor K. Post-parotidectomy gustatory sweating. BMJ 1958; ii:1295.
- Cartier B, Cartier P. Thoracoscopic cervicodorsal sympathectomy with diathermy. Ann Vasc Surg 1999;13:582–5.
- Collin J. Compensatory hyperhidrosis after thoracic sympathectomy. Lancet 1998;351:1136.
- Moya J, Ramos R, Morera R, et al. Thoracic sympathicolysis for primary hyperhidrosis. A review of 918 procedures. Surg Endosc 2006;20:598–602.
- Rustemeyer J, Eufinger H, Bremerich A. The incidence of Frey's syndrome. J Craniomaxillofac Surg 2008;36:34–7.
- Mellor TK, Shaw RJ. Frey's syndrome following fracture of the mandibular condyle: Case report and literature review. Injury 1996;27:359–60.
- Choi HG, Kwon SY, Won JY, et al. Comparisons of three indicators for Frey's syndrome: Subjective symptoms, minor's starch iodine test, and infrared thermography. Clin Exp Otorhinolaryngol 2013;6(4):249–53.
- Green RJ, Endersby S, Allen J, Adams J. Role of medical thermography in treatment of Frey's syndrome with botulinum toxin A. Br J Oral Maxillofac Surg 2014;52(1):90–2.
- Naumann M. Evidence-based medicine: botulinum toxin in focal hyperhidrosis. J Neurol 2001;248:31–3.
- Urman JD, Bobrove AM. Diabetic gustatory sweating successfully treated with topical glycopyrrolate: report of a case and review of the literature. Arch Intern Med 1999;159:877–8.
Restless legs syndrome/burning feet syndrome
- Walters AS. Restless legs syndrome and periodic limb movements in sleep. Continuum Lifelong Learning in Neurology 2007;13:115–38.
- Allen RP, Walters AS, Montplaisir J, et al. Restless legs syndrome prevalence and impact: REST general population study. Arch Intern Med 2005;165:1286–92.
- Polydefkis M, Allen RP, Hauer P, et al. Subclinical sensory neuropathy in late-onset restless legs syndrome. Neurology 2000;55:1115–21.
- Thorp ML, Morris CD, Bagby SP. A crossover study of gabapentin in treatment of restless legs syndrome among haemodialysis patients. Am J Kidney Dis 2001;38:104–8.
- Desautels A, Turecki G, Montplaisir J, et al. Identification of a major susceptibility locus for restless legs syndrome on chromosome 12q. Am J Hum Genet 2001;69:1266–70.
- Stögbauer F, Young P, Kuhlenbäumer G, et al. Autosomal dominant burning feet syndrome. J Neurol Neurosurg Psychiatry 1999;67(1):78–81.
- Kuhlenbaumer G, Young P, Kiefer R, et al. A second family with autosomal dominant burning feet syndrome. Ann NY Acad Sci 1999;883: 445–8.
- Allen RP, Earley CJ. Restless legs syndrome: a review of clinical and pathophysiological features. J Clin Neurophysiol 2001;18:128–47.
- Allen RP, Picchietti D, Hening WA, et al. Restless legs syndrome: diagnostic criteria, special considerations, and epidemiology. A report from the restless legs syndrome diagnosis and epidemiology workshop at the National Institutes of Health. Sleep Med 2003 Mar;4(2):101–19.
- Chokroverty S. Therapeutic dilemma for restless legs syndrome. N Engl J Med 2014;370:7.
- Scholz H, Trenkwalder C, Kohnen R, et al. Levodopa for the treatment of restless legs syndrome. Cochrane Database Syst Rev 2011 http://onlinelibrary.wiley.com/doi/10.1002/14651858.CD005504.pub2/abstract (last accessed June 2015).
- Garcia-Borreguero D, Larrosa O, de la Llave Y, et al. Treatment of restless legs syndrome with gabapentin: a double-blind cross-over study. Neurology 2002;59:1573–9.