CHAPTER 96
Acquired Disorders of Dermal Connective Tissue
Christopher R. Lovell
Department of Dermatology, Royal United Hospital and Royal National Hospital for Rheumatic Diseases, UK
CHANGES IN DERMAL CONNECTIVE TISSUE DUE TO AGEING AND PHOTODAMAGE
Introduction and general description
Both intrinsic ageing and UV exposure result in alterations in dermal connective tissue which affect the appearance of the skin in old age [1, 2]. The relative contributions of each vary in the individual from body site to body site and in the population at large according to environmental factors, particularly cumulative photodamage and cigarette smoking. The skin becomes increasingly thin and atrophic in elderly people. Changes in both the epidermis and the dermis result in age-related skin fragility characterized by translucent, lax and wrinkled skin with a tendency to easy bruising and stellate scars. The term ‘dermatoporosis’ has been coined to describe these changes [3]. The relative contribution of intrinsic ageing and environmental factors to the changes in dermal connective tissue which manifest as skin ageing determine the clinical appearance. Much of what is perceived as aged skin is due to photodamage with the development of actinic elastosis, lax skin and wrinkles. Recent research has helped to elucidate the pathomechanisms underlying these changes. There is reduced collagen biosynthesis and increased production of matrix metalloproteinases, which both inhibit collagen fibril synthesis and promote fragmentation of collagen fibrils, leading to a reduction in healthy collagen but accumulation of damaged collagen. The topic is discussed in detail in Chapter 155. Some specific clinical manifestations associated with aged and photoaged skin are described later.
Wrinkles
Definition and nomenclature
Wrinkles are a characteristic of ageing skin. They may be defined as creases or furrows in the skin surface.
Introduction and general description
Wrinkles are particularly prominent in hypertrophic skin photo-damage (see Chapter 155). Cigarette smoking is also a potent independent cause of wrinkling. The so-called ‘cigarette face’ is characterized by pale grey wrinkled skin with rather gaunt features, so that heavy smokers can often be recognized from their facial appearance alone. Heavy smokers are five times more likely to be wrinkled than non-smokers of the same age, and cigarette smoking probably has at least as much effect on facial wrinkles as sun exposure [1].
Clinical features
They are particularly prominent in hypertrophic skin photodamage (see Chapter 155). Cigarette smoking is also a potent independent cause of wrinkling. The so-called ‘cigarette face’ is characterized by pale grey wrinkled skin with rather gaunt features, so that heavy smokers can often be recognized from their facial appearance alone. Heavy smokers are five times more likely to be wrinkled than non-smokers of the same age, and cigarette smoking probably has at least as much effect on facial wrinkles as sun exposure [1].
Wrinkles can be classified into three morphological types [2] as follows.
- Crinkles. This is a very fine wrinkling which occurs in aged skin, even in areas protected from sunlight. These fine wrinkles disappear when the skin is slightly stretched. They are caused by deterioration of elastin, especially the vertical subepidermal fine elastic fibres which keep the epidermis in tight apposition to the dermis [3, 4]. Ultrastructural studies have shown that even in normal people the elastic fibres begin to deteriorate from the age of 30 years onwards, regardless of the amount of sun exposure, although sunlight undoubtedly increases the damage [5]. Crinkles are seen in a marked form in mid-dermal elastolysis.
- Glyphic wrinkles. These creases are an accentuation of the normal skin markings. They occur on skin which has been prematurely aged by elastotic degeneration caused by sunlight, for example on the sides and back of the neck (see Actinic elastosis).
- Linear furrows. These are long, straight or slightly curved grooves that are usually seen on the faces of elderly people. They include the horizontal frown lines along the forehead, the ‘crows’ feet’ radiating from the lateral canthus of the eye and the creases from the nose to the corners of the mouth.
Actinic elastosis
Definition and nomenclature
Actinic elastosis is another component of hypertrophic skin photodamage. It is characterized clinically by yellowish discoloration and thickening of the skin (Figure 96.1), and histologically by a reduction in collagen and an accumulation of amorphous masses of degenerate elastic fibres in the papillary and upper reticular dermis (Figure 96.2) [1].
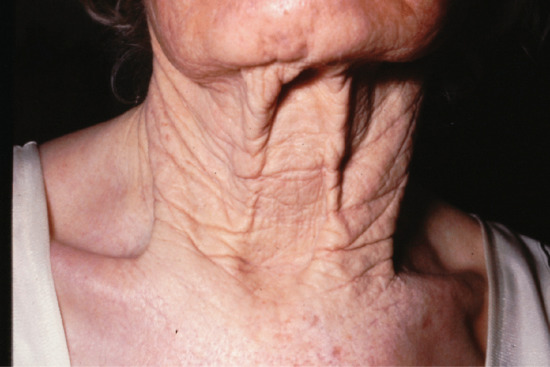
Figure 96.1 Actinic elastosis on the neck of an elderly female patient.
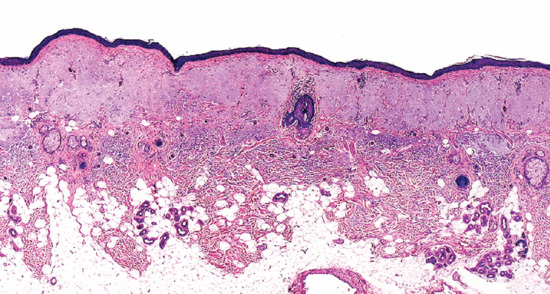
Figure 96.2 Actinic elastosis showing confluent masses of amorphous basophilic material in the papillary and upper reticular dermis with atrophy of the overlying epidermis.
Introduction and general description
Actinic elastosis usually results from prolonged exposure to sunlight [1], but it can also follow infrared (IR) radiation [2].
Epidemiology
Incidence and prevalence
It is related to the cumulative lifetime exposure to UV radiation rather than to episodes of intense UV exposure: it is more common in outdoor workers and in those living in sunny climates. There is, however, considerable variation in susceptibility between individuals.
Age
It does not usually present until the fourth decade or later but cumulative sun exposure is more important than chronological age alone.
Ethnicity
Fair-skinned people are the worst affected, although the condition can occur in black people [3].
Associated diseases
Severe elastosis may occur in photosensitized skin, for example in porphyria cutanea tarda.
Pathophysiology
Pathology
See Chapter 155.
Environmental factors
Cumulative UV exposure is the main exacerbating factor, although other factors, such as IR irradiation, may play a part [2].
Clinical features
History
The characteristic changes develop gradually over the course of years.
Presentation
The light-exposed areas are affected, particularly the forehead, bald scalp and the back of the neck. Mild degrees of elastosis may not be apparent until the skin is pinched up, when it may assume a wrinkled appearance. Elastosis is usually more advanced in the tissue than the clinical appearance would suggest.
The affected skin is diffusely thickened and yellowish (see Figures 96.1 and 155.2b), and on the neck it may be divided by well-defined furrows into an irregular rhomboidal pattern (cutis rhomboidalis nuchae) (see Figure 96.3). There may also be more sharply marginated, thickened plaques on the face or neck. These are usually, but not always, symmetrical. Recent studies suggest that the elastotic skin itself is protected from epithelial neoplasia [4].
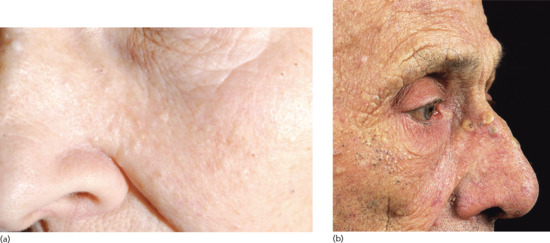
Figure 96.3 (a,b) Nodular actinic elastosis with comedones and cysts (Favre–Racouchot syndrome): early stages in a 78-year-old woman (a) and advanced stage in an elderly man (b). (Part b courtesy of Professor R. Marks, St Vincent's Hospital, Melbourne, Australia.)
Actinic elastosis may also be complicated by actinic granuloma (see later).
Clinical variants
Actinic comedonal plaque (synonyms Favre–Racouchot syndrome, nodular actinic elastosis with cysts and comedones). Actinic elastosis may form into confluent plaques studded with comedones. This is most commonly seen in periorbital skin (see Figure 96.296.3). It is usually symmetrical, but unilateral and circumscribed forms have been reported [5]. Rarely, a variant has been described with vesicular changes within zones of severe actinic elastosis [6]. Occasionally, similar plaques may form elsewhere than on facial skin, such as the forearm [7].
Elastotic nodules of the ear. In this variant of actinic elastosis, single or multiple firm papules occur on the anterior crus of the antihelix, usually in middle-aged or elderly males. Their significance is that they sometimes have a pearly edge, clinically suggesting basal cell carcinoma (BCC), but histology reveals large aggregates of amorphous elastotic material, sometimes with degradation of underlying cartilage [8–10].
Differential diagnosis
Plane xanthoma, pseudoxanthoma elasticum (PXE) and colloid milium may sometimes cause confusion, but the combination of the clinical and histological features is distinctive.
Classification of severity
Of cosmetic significance.
Disease course and prognosis
The process may be halted but not reversed by stringent photoprotection. Stopping smoking may be presumed to slow down progression [11].
Investigations
Skin biopsy if there is doubt about diagnosis. Histological changes may be more florid than the clinical appearance.
Management (see also Chapter 155)
Sunscreens protect against the development of photodamage both in humans and animals [12]. In hairless mice exposed to UVB radiation, synthesis of subepidermal collagen has been demonstrated in animals protected with a sunscreen [13]. Topical application of αhydroxy acids (‘fruit acids’), i.e. lactic, glycolic and citric acids, has been shown to lead to a modest improvement in photodamaged skin [14]. More impressive results have been obtained with topically applied tretinoin cream [15]. A double-blind study demonstrated a decrease in papillary dermal collagen type I in photodamaged skin, and subsequent treatment with 0.1% tretinoin cream for 10–12 months resulted in an 80% increase in dermal collagen [16]. Several studies have shown clinical and histological improvement after prolonged use [17]. Tretinoin may also repair skin changes due to intrinsic ageing [18]. Retinoids reduce matrix metalloproteinase 1 (MMP-1) expression in vitro, partially restoring levels of fibrillin 1 and collagens I and VII in the papillary dermis [19]. Similar results have been obtained in double-blind trials of topical isotretinoin [20] and tazarotene cream [21]. Antioxidants play a part in the prevention of photoageing [22] and may have a therapeutic role in established photodamage [23]. Non-ablative lasers, including the 1320 nm Nd : YAG and 1540 nm erbium glass lasers, are claimed to wound the upper dermis without epidermal damage [24]. Restoration of fibrillin I in the microfibrillar network of the papillary dermis may prove a useful ‘biomarker’ for the efficacy of topical products used in actinic elastosis [25, 26].
Collagenous and elastotic marginal plaques of the hands
Collagenous and elastotic marginal plaques of the hands is an acquired dermatosis affecting dermal connective tissue in which papules and plaques form on the dominant hand along the radial aspect of the index finger, the first web space and the ulnar aspect of the thumb (Figure 96.4a) [1, 2]. Histologically, there is hyperkeratosis, with sawtoothing of the rete ridges. The dermal collagen fibres are thickened and arranged haphazardly; there are basophilic elastotic masses, often containing calcium, in the upper reticular dermis (Figure 96.4b) [3]. Cases are sporadic, unlike the clinically similar disorders acrokeratoelastoidosis and focal acral hyperkeratosis (see p. 96.2872) [4].
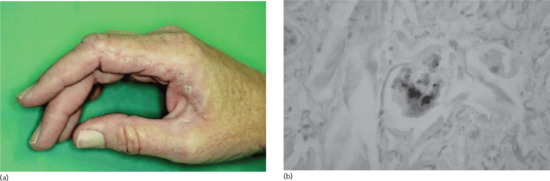
Figure 96.4 (a,b) Collagenous and elastotic marginal plaques of the hands: linear plaque involving radial aspect of the right index finger of a 49-year-old woman from Queensland, Australia (a); calcium deposits within collagen bundle (b). (From Mortimore and Conrad 2001 [7], with permission from John Wiley.)
Chronic friction and photodamage have been proposed as aetiological factors; the condition has been reported in manual workers and entirely from geographical areas with high solar irradiation. It is regarded as a variant of actinic elastosis [5] although actinic damage is not always observed clinically [6]; furthermore the papillary dermis is relatively spared by the elastotic process and the basophilic areas containing calcium differ from the changes normally seen in actinic elastosis.
Adult colloid milium and colloid degeneration of the skin
Definition and nomenclature
Colloid degeneration of the skin is defined histologically by the presence of colloid in dermal papillae and presents as yellowish, translucent papules, nodules or plaques on light-exposed skin. There are several clinical variants of which the commonest is adult colloid milium, which manifests as multiple milia-like papules on light-exposed skin, particularly on the face.
Introduction and general description
Colloid degeneration of the skin is a rare but probably underdiagnosed dermatosis which requires biopsy for definitive diagnosis [1]. It is defined histologically by the presence of colloid in dermal papillae and presents as yellowish translucent papules, nodules or plaques on light-exposed skin. There are several clinical variants of which the commonest is adult colloid milium, which manifests as multiple milia-like papules on light-exposed skin, particularly on the face. The differential diagnosis is presented in Table 96.1.
Table 96.1 Differential diagnosis of colloid milium.
| Deposition disorder | Clinical findings | Pathological characteristics | Staining pattern |
| Adult colloid milium | Multiple symmetrical yellow to flesh-coloured facial papules; associated with sun exposure | Homogeneous eosinophilic colloid masses in papillary dermis from degenerating elastic fibres, often with subepidermal Grenz zone | PAS + Congo red ±Cotton dye –Cytokeratin – |
| Juvenile colloid milium | Multiple translucent yellowish papules on cheeks, nose, perioral skin; onset before puberty; familial; associated with ligneous conjunctivitis | Secondary to UV-induced degeneration of keratinocytes | PAS + Congo red ± Cotton dye – Cytokeratin + |
| Nodular colloid degeneration | Flesh-coloured nodule on face, scalp or chest; usually solitary | May be associated with myeloma; lacks plasma cells | PAS + Congo red ± Cotton dye – |
| Acrokeratoelastoidosis of Costa | Multiple tiny skin-coloured umbilicated papules at sides of hands and feet; familial; commonest in black skin | Fragmentation and degeneration of elastic fibres | Congo red – Cotton dye – |
| Collagenous and elastotic marginal plaques of the hands | Skin-coloured papules and plaques along radial border of index finger and ulnar border of thumb | Fragmentation and degeneration of elastic fibres | Congo red – Cotton dye – |
| Nodular amyloid | Single flesh-coloured nodule | Deposition of monoclonal immunoglobulin light-chain fragments from localized plasma cell infiltrate | Congo red + Cotton dye + |
Adapted from Mehregan and Hooten 2011 [1].
PAS, periodic acid–Schiff.
Epidemiology
Incidence and prevalence
Rare but usually affects fair-skinned, outdoor workers living in sunny climates [1, 2].
Pathophysiology
The exact cause of adult colloid milium is uncertain but sunlight exposure is strongly implicated and actinic elastosis is usually evident as well [3]. Occupational exposure to mineral oils has also been implicated [4, 5]: an outbreak among refinery workers in the tropics was attributed to trauma and prolonged contact with photodynamic phenols in oxide fuel (gas oil) [4]. Cases have also been reported in association with ochronosis after the long-term application of strong hydroquinone bleaching creams [6].
Histopathology
The earliest histological change is the appearance of colloid globules at the tips of the dermal papillae. Homogeneous fissured masses of amorphous colloid occupy the upper dermis, each surrounded by bands of collagen. There is characteristically a subepidermal uninvolved Grenz zone. The colloid is usually eosinophilic but may be basophilic. Within it, small blood vessels and the nuclei of fibroblasts are well preserved. In the larger, plaque-like lesions, the colloid change occurs diffusely throughout the dermis. The source of the colloid material is uncertain. It could be a protein synthesized by fibroblasts or it could be derived from degraded elastic fibres [2, 7].
Environmental factors
UV exposure.
Clinical features
Presentation
Small dermal papules 1–5 mm in diameter, yellowish brown and sometimes translucent, develop slowly and more or less symmetrically in irregular groups in areas exposed to sunlight (Figures 96.5 and 96.6) [1]. They feel soft and may release their gelatinous contents when punctured. The most frequently involved sites are the face, especially around the orbits, the dorsa of the hands, the back and sides of the neck and the ears. There are usually other signs of actinic damage. The changes induced by prolonged light exposure are associated to varying degrees. Although colloid milium may become more severe and more extensive over the years, most cases reach their maximum development within 3 years and then remain unchanged.
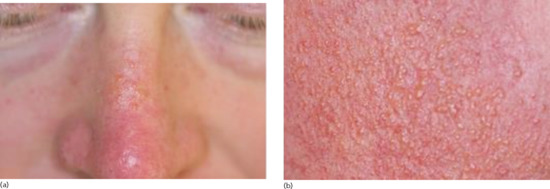
Figure 96.5 (a,b) Adult colloid milium with multiple tiny yellowish translucent papules on the dorsum of the nose (a) with close-up view of papules on the cheek (b). (From Mehregan and Hooten 2011 [1], with permission from John Wiley.)
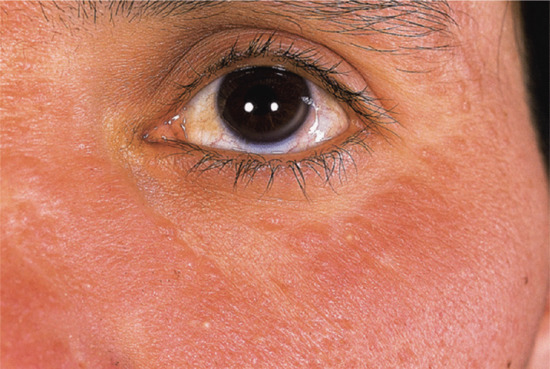
Figure 96.6 More advanced adult colloid milium manifesting as confluent plaques of the infraorbital region but with individual papules discernible at the margins.
Differential diagnosis (see Table 96.1)
The rare juvenile form manifests before puberty and is often familial [8, 9]. It is thought to derive from degeneration of keratinocytes rather than elastic fibres [10].
Clinical variants
Nodular colloid degeneration presents usually as a single nodule up to 5 cm in diameter though multiple nodules may occur. It may be associated with myeloma [11] (see Table 96.1).
Management
No completely satisfactory intervention has been found for this condition. Good results have been claimed for dermabrasion [12] and for the long-pulsed Er : YAG laser [13].
OTHER CAUSES OF CUTANEOUS ATROPHY
Introduction and general description
Atrophy of the skin is a term which is applied to the clinical changes produced by a decrease in the dermal connective tissue. It is characterized by thinning and loss of elasticity. The skin usually appears smooth and finely wrinkled, and it feels soft and dry. Veins or other subcutaneous structures may be unduly conspicuous. There is often associated loss of hair follicles, and telangiectasia may also be present, due to the loss of connective tissue support of the capillaries. There may or may not be an associated atrophy of the epidermis.
Atrophy of the skin occurs in varying degree in a large number of skin conditions, including naevi, and the underlying histological changes are also variable, because the several components of the connective tissue may be involved to a different degree. Atrophy that includes subcutaneous tissue or even deeper structures is referred to as panatrophy. Box 96.1 lists the main acquired disorders in which cutaneous atrophy is prominent.
Atrophy due to corticosteroids
Introduction and general description
Both systemic and topical glucocorticoid therapy can produce cutaneous atrophy by a dose-related pharmacological effect [1]. The effect is more severe with the more potent steroids (as assessed by the vasoconstrictor assay test) but both fluorinated and non-fluorinated topical steroids can cause atrophy. The effect is most marked when potent steroids are applied topically under an occlusive dressing. The skin becomes thin, fragile and transparent, and striae may develop (see later) (Figure 96.7).
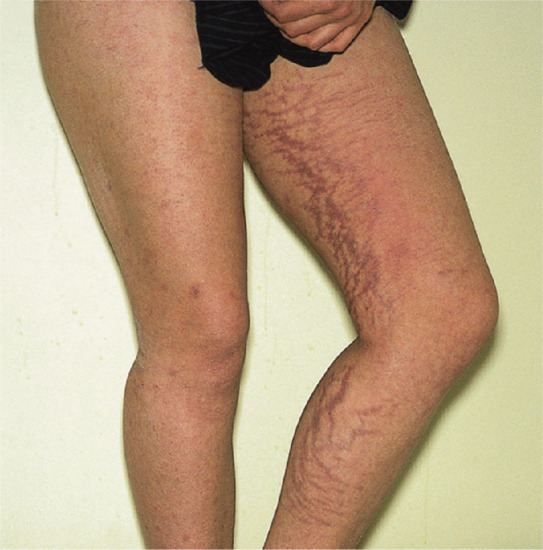
Figure 96.7 Striae of the legs due to long-term application of a potent topical steroid in a young woman with psoriasis.
Severe dermal atrophy can follow injection of intralesional steroids, such as triamcinolone acetonide (particularly if the higher concentration of 40 mg/mL is used, instead of the more usual 10 mg/mL, which is less likely to cause atrophy) (Figure 96.8). Inhaled corticosteroids also induce dermal thinning in adults and in children [2]. See Chapters 18 and 19, respectively, for more general discussions of topical and systemic glucocorticosteroids.
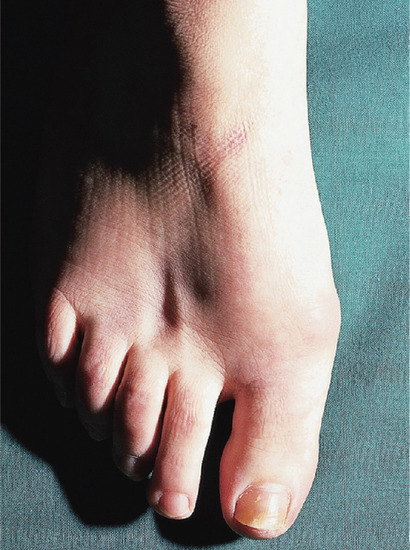
Figure 96.8 Localized atrophy due to injection of a steroid (triamcinolone 40 mg/mL) into the skin between the second and third metatarsals.
Pathophysiology
Predisposing factors
Steroids are known to inhibit the formation of glycosaminoglycans. Hyaluronate and the major cell surface hyaluronate receptor CD44 are depleted in atrophic skin [3]. Topical corticosteroids rapidly suppress hyaluran synthase 2 in the dermis; this precedes alteration of dermal collagen [4]. The fibroblasts become shrunken, although their numbers do not decrease, but the number of mast cells is markedly reduced. Topical steroids also inhibit the activity of enzymes involved in collagen biosynthesis [5], and they have been shown to depress synthesis of types I and III collagen in vivo [6, 7, 8]. Type III collagen synthesis is preferentially reduced in fibroblast cultures [7]. They can also depress collagenase production and collagen breakdown [9], and the rate of collagen turnover is probably decreased. Even a weak steroid, such as hydrocortisone, can suppress the stimulatory effect of cyclic nucleotides on collagenase production. Studies of the effect of topical steroids on collagen and elastic fibres in vivo have given conflicting results [10–12]. Capillaroscopic studies have shown that steroid-induced vasoconstriction involves the superficial capillary network, and prolonged superficial ischaemia could also play a role in producing atrophy [5].
Pathology
The earliest histological change is marked thinning of the epidermis, with flattening of the rete ridges and decreased corneocyte size [13]. This is followed a few weeks later by thinning of the dermis, which can be measured by skinfold calipers, ultrasonography or a radiographic technique [14–16].
The epidermal thinning probably results from a reduction of mitotic activity in the germinal layer [17], but the mechanism by which dermal thinning is produced is uncertain.
Loss of dermal ground substance leads to a reorganization of the dermal architecture. The spaces between the collagen and elastic fibres become smaller, so that the dermis becomes more compact but thinner [10].
Collagen microfibrils may form globular microfibrillar bodies, although the changes are not specific for steroid atrophy [18]. These ultrastructural changes can develop in the early stages before there is clinical or histological evidence of atrophy. Digestion of collagen fibrils in the endocytic vesicles of fibroblasts may be involved in the production of steroid-induced atrophy [9].
Environmental factors
Systemic, topical, intralesional or inhaled corticosteroids are implicated.
Clinical features
History
A careful history should be taken, including enquiry about the use of corticosteroid inhalers (Figure 96.9).
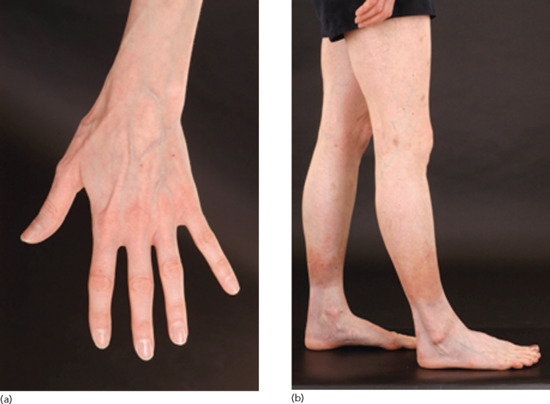
Figure 96.9 (a,b) Severe generalized cutaneous atrophy in a 29-year-old female as the result of using inhaled corticosteroids for asthma since the age of 7; (b) note haemosiderosis on the lower legs as a result of ready bruising of her atrophic skin.
Presentation
The skin becomes thin and fragile with easy bruising. Changes are generalized in patients on systemic corticosteroids, although the changes are more marked at sites of photodamage and trauma. Thinning due to topical corticosteroids may be localized to the site(s) of application. Severe dermal atrophy can follow injection of intralesional steroids.
Differential diagnosis
Other causes of cutaneous atrophy.
Complications and co-morbidities
Corticosteroid-induced skin thinning leads to delayed wound healing and easy bruising, often after trivial trauma. Measurement of bone density is advisable in at-risk patients, although extensive skin thinning is not necessarily associated with steroid-induced osteopenia [7].
Investigations
Consider measuring blood glucose and bone density, if systemic steroid toxicity is suspected.
Management
It has been suggested that local and oral vitamin C therapy might help restore the normal skin thickness [19]. Concurrent application of retinoic acid may partially prevent the epidermal atrophy due to steroids [20]. Intralesional saline injections can restore surface contour [21]. Hyaluronate fragments are reported to induce skin thickening in corticosteroid-induced atrophy [22].
Prevention is clearly the best approach, including the use of steroid-sparing systemic drugs and topical agents such as calcineurin inhibitors to treat skin disease. In the future, more selective corticosteroid receptor agonists, with potentially less atrophogenic effect may be developed [23].
Striae
Definition and nomenclature
Striae are visible linear scars which form in areas of dermal damage produced by stretching of the skin. They are characterized histologically by thinning of the overlying epidermis, with fine dermal collagen bundles arranged in straight lines parallel to the surface.
Introduction and general description
Aetiology
The factors which govern the development of striae are poorly understood. Many authors have suggested that striae develop as a result of stress rupture of the connective tissue framework [1], but others disagree. It has been suggested that they develop more easily in skin which has a critical proportion of rigid cross-linked collagen, as occurs in early adult life [2]. They are common during adolescence [3], and they seem to be associated with rapid increase in size of a particular region. They are very common over the abdomen and breasts in pregnancy, and they may develop on the shoulders in young male weight lifters when their muscle mass rapidly increases [4]. They are a feature of Cushing disease, and they may be induced by local or systemic corticosteroid therapy [2, 5]. The effects of glucocorticoids on the dermal connective tissue are outlined above. Together with other steroid effects, striae have been reported in HIV-positive patients receiving the protease inhibitor, indinavir [6].
Epidemiology
Incidence and prevalence
Striae are very common, and occur in most adult women, as they readily develop at puberty or during pregnancy.
Age
Adolescent striae may first develop soon after the appearance of pubic hair.
Sex
Abdominal striae gravidarum are extremely common in pregnancy.
Striae are often associated with growth spurts in adolescent males (Figure 96.10).
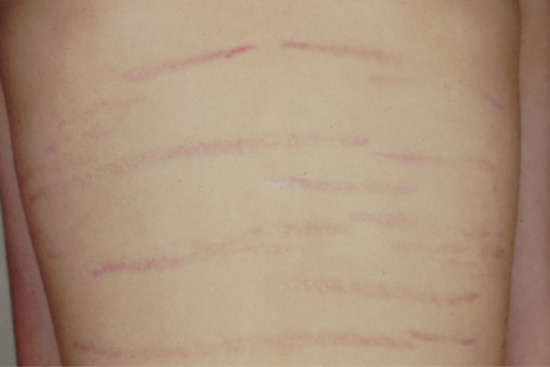
Figure 96.10 Pubertal growth striae across the back of an adolescent boy: note that these are normally all horizontally arranged right across the back (compare with Figure 96.11).
Associated diseases
Most striae occur in otherwise healthy individuals, although they are a feature of Cushing syndrome and Marfan syndrome.
Pathophysiology
Predisposing factors
Striae are associated with growth spurts, e.g. body building or pregnancy, more rarely they may reflect structural abnormalities of connective tissue such as Marfan syndrome or the effect of glucocorticoids.
Pathology
In the early stages, inflammatory changes may be conspicuous; the dermis is oedematous and perivascular lymphocytic cuffing is present. In the later stages, the epidermis is thin with flattening of the dermal papillae [7, 8]. The dermal collagen is layered in thin eosinophilic bundles, orientated in straight lines parallel to the surface in the direction of the presumed stress. Scanning electron microscopy shows amorphous sheet-like structures [9]. With Luna stain, the elastic fibres are numerous, close together, fine and straight, and in the same direction as the collagen bundles [10]. On scanning electron microscopy in collagen-free preparations there is an abundance of thin, curled and branched elastic fibres.
Genetics
The importance of genetic factors in determining susceptibility of connective tissue is emphasized by their presence as one of the (minor) diagnostic criteria for Marfan syndrome [11], and congenital arachnodactyly, associated with mutations of the fibrillin-1 and fibrillin-2 genes, respectively. Striae may occur in the absence of other phenotypic features of Marfan syndrome [12], and their presence may be predictors for aortic dissection [13]. They are commonly absent in pregnancy in Ehlers–Danlos syndrome.
Recent genome-wide association analysis of apparently otherwise normal individuals with striae has revealed associations with genes affecting expression of matrix proteins such as collagen, elastin and fibronectin [14].
Clinical features
The commonest sites for obesity-related striae are the outer aspect of the thighs and the lumbosacral region in boys (Figure 96.11), and the thighs, buttocks and breasts in girls, but there is considerable variation, and other sites, including the outer aspect of the upper arm, are sometimes affected. Pubertal growth striae are concentrated symmetrically over and on either side of the spine (see Figure 96.10).
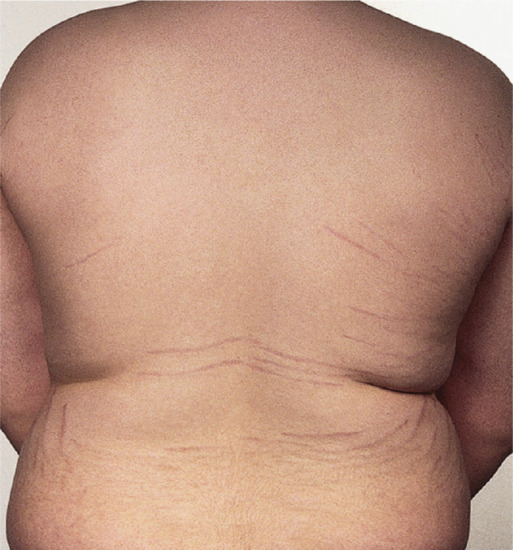
Figure 96.11 Striae due to obesity in a young man.
Early lesions may be raised and irritable, but they soon become flat, smooth and livid red or bluish in colour. Their surface may be finely wrinkled. They are commonly irregularly linear, several centimetres long and 1–10 mm wide. After some years, they fade and become inconspicuous. They are then generally paler than the surrounding skin.
The striae in Cushing syndrome or those induced by steroid therapy may be larger and more widely distributed, and involve other regions, including sometimes the face. In pregnancy, the striae appear first and are most conspicuous on the abdominal wall, and later on the breasts, but may involve most or all of the pubertal sites [15]. The striae induced by topical corticosteroid therapy occur particularly in the flexures, but may appear in other sites if occlusive plastic films increase absorption (see Figure 96.6) [16, 17].
Differential diagnosis
The diagnosis of striae is usually simple. The possibility of Cushing syndrome must be considered, although this is rarely the cause. Lay people may mistake adolescent growth striae for signs of physical abuse. In linear focal elastosis the lesions are yellow and palpable.
Complications and co-morbidities
Usually striae are no more than a cosmetic problem, but occasionally, if extensive, they may ulcerate or tear easily if traumatized.
Disease course and prognosis
Striae gravidarum generally improve after delivery and adolescent striae have an excellent prognosis. Even corticosteroid-induced striae may disappear or become less conspicuous when treatment is stopped.
Investigations
Exclude Cushing syndrome if suspected.
Management
In the case of common adolescent striae, the patient may be reassured that in time they will become less conspicuous. Numerous unproven remedies are available from cosmetic companies and there is no well substantiated evidence that topical therapies prevent or accelerate healing of striae [18, 19].
Some cases appear to respond to treatment with topical tretinoin cream (0.05% daily), although weekly superficial dermabrasion is claimed to be better tolerated [20]. The erythema of ‘younger’ striae is claimed to respond to the 585 nm pulsed dye and Nd : YAG lasers [21, 22]. Fractional photothermolysis has been used in chronic striae [23]. The application of silicone gel may be beneficial [24].
There is no proven treatment.
Acquired poikiloderma
Poikiloderma is a descriptive term, comprising atrophy, macular or reticulate pigmentation and telangiectasia. There may be associated areas of scaling, hypopigmentation and petechiae and signs of inflammation such as lichenoid papules. Congenital poikiloderma is a feature of several inherited disorders, including Kindler syndrome (see Chapter 71), dyskeratosis congenita, Rothmund–Thomson and Weary syndromes (see Chapter 77) and erythrokeratoderma variabilis (see Chapter 65).
Poikiloderma may occur as a pattern of cutaneous response to injury by cold, heat or ionizing radiation [1]. So-called poikiloderma of Civatte (see Chapter 88) is a similar reaction mediated by photosensitizing chemicals in cosmetics. Some inflammatory dermatoses, such as lichen planus, may also give rise to poikilodermatous changes.
Poikiloderma is a feature of some systemic autoimmune diseases, and is a marker of disease severity in dermatomyositis [2]. It is also seen in lupus erythematosus and rarely in systemic sclerosis. Poikiloderma atrophicans vasculare is an early presenting feature of cutaneous T-cell lymphoma (mycosis fungoides), typically stage IA–IIA; it predominantly affects males. It usually responds well to phototherapy and has a good prognosis [3] (Figure 96.12).
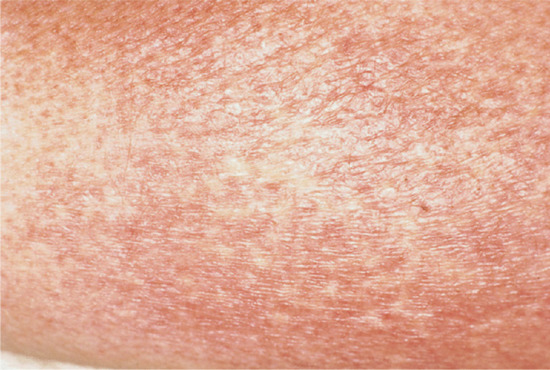
Figure 96.12 Poikilodermatous mycosis fungoides.
Atrophic scars
Definition
Scars resulting from the destruction of connective tissue by trauma or by inflammatory changes.
Introduction and general description
The distribution and character of the atrophic lesions may be so distinctive as to betray their origin, and is sometimes of considerable importance in diagnosis. Viral infections, such as varicella, can leave widespread small circular atrophic scars [1]. The scars left by tertiary syphilis, certain tuberculides and some deep mycoses, especially sporotrichosis, are usually completely atrophic. Onchocerciasis may result in extensive areas of dermal atrophy [2] (Figure 96.13). Areas of cutaneous lupus erythematosus may also leave atrophy without clinical evidence of sclerosis. Lupus vulgaris, the chronic follicular pyodermas and some cases of lupus erythematosus leave a combination of atrophy and sclerosis, in which the latter predominates. Lesions that have been treated by intralesional steroid injections may also leave atrophic scars.
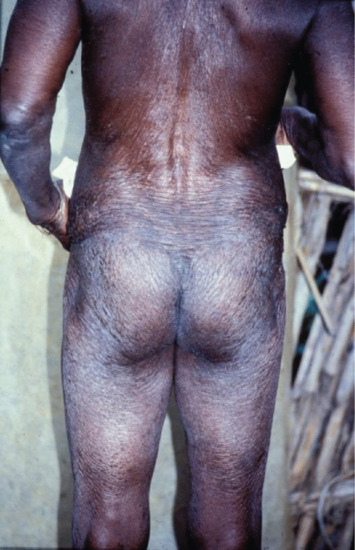
Figure 96.13 Atrophy due to onchocerciasis. (From Murdoch et al. 1993 [2], courtesy of Dr M. Murdoch, West Hertfordshire Hospitals NHS Trust, Hertfordshire, UK.)
Exposure to ionizing radiation gives rise to a very striking combination of atrophy, pigmentation and telangiectasia (poikiloderma).
The wide atrophic scars which follow injuries in Ehlers–Danlos syndrome (see Chapter 73) emphasize the importance of constitutional factors in determining the pattern of dermal response to a known external injury.
Stellate pseudoscars are white, irregular or ‘star-shaped’ atrophic scars (Figure 96.14). They are common on light-exposed skin, particularly on the extensor aspects of the forearms, often in association with purpura. These are seen in 20% of patients aged 70–90 years, and a much less common presenile form occasionally occurs before the age of 50 years. These pseudoscars are secondary to mild trauma, and are probably always preceded by haemorrhage into the dermis [3, 4].
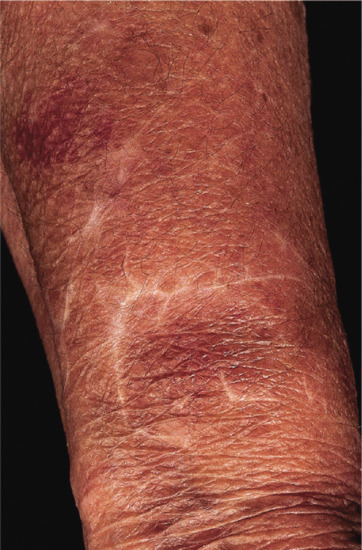
Figure 96.14 Stellate pseudoscars on the forearm of an elderly woman. There was no history of trauma.
Stellate scars following trivial trauma can also occur in other conditions which cause fragile skin, for example porphyria cutanea tarda and prolonged use of potent topical steroids.
Brown pseudoscars may also develop over the shins of diabetic patients with no history of trauma (diabetic dermopathy) (Figure 96.15) (see also Chapter 64). Histology reveals that the pigmentation is due to dermal deposition of haemosiderin and melanin [5].
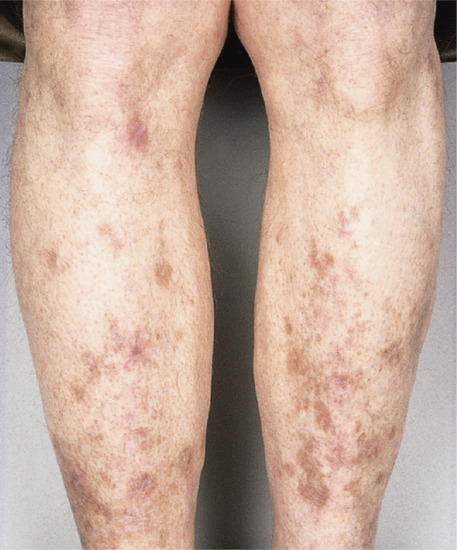
Figure 96.15 Brown pseudoscars of the legs due to diabetic dermopathy. There was no history of trauma.
Congenital erosive and vesicular dermatosis with reticulate scarring [6, 7, 8, 9]
This rare congenital condition, which was first described in 1985 [6], presents at birth with signs suggestive of congenital viral infection, including erythema, blistering, erosions and crusting often involving more than 75% of the skin surface. The skin heals over the course of a few months with soft reticulate scarring, which on the limbs tends to follow the long axis of the limbs (Figure 96.16). A recent review of 28 known cases [7] confirmed that it occurred predominantly in preterm infants (79%) and that there was often a history of maternal chorioamnionitis (43%). Neurodevelopmental problems were common. Histological examination in the early stages shows epidermal necrosis and subepidermal blistering but no evidence of viral infection or vasculitis. This is succeeded by scar formation with loss of appendageal structures, especially eccrine glands. The differential diagnosis includes Goltz syndrome, Rothmund–Thomson syndrome and aplasia cutis [7, 8]. An infant was treated successfully using a silicone sheet dressing [9].
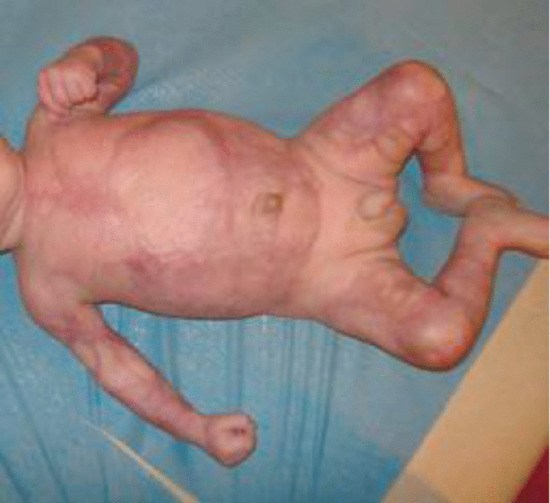
Figure 96.16 Congenital erosive and vesicular dermatosis with reticulate scarring. (From De Lange et al. 2009 [9], with permission from John Wiley.)
Spontaneous atrophic scarring of the cheeks
This is a very rarely reported condition in which spontaneous scars develop on the cheeks (Figure 96.17) in young adults [1, 2] or children [3]. It may, however, be much commoner than the lack of reports suggests. The shallow atrophic lesions have sharp margins and may be linear, rectangular or varioliform. They may be preceded by slight erythema and scaling. Histology shows mild loss of collagen or elastic fibres; there may be thickening of the stratum corneum [4]. Familial cases are recorded [1, 5]; inheritance is probably autosomal dominant [6]. The differential diagnosis includes atrophoderma vermiculatum (see Chapter 87), chickenpox scars and artefact.
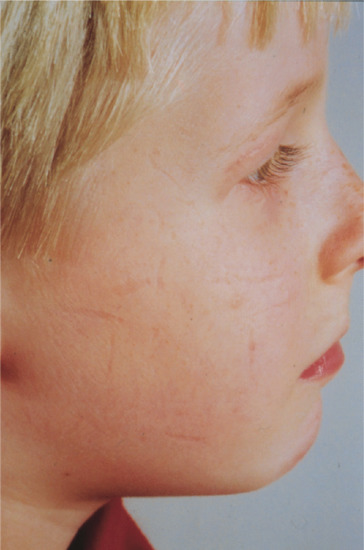
Figure 96.17 Spontaneous atrophic scarring of the cheeks (varioliform atrophy).
Acrodermatitis chronica atrophicans
Definition and nomenclature
This is a late skin manifestation of Lyme borreliosis (see Chapter 27). It is characterized by the insidious onset of painless, dull-red nodules or plaques on the extremities, which slowly extend centrifugally for several months or years, leaving central areas of atrophy.
Introduction and general description
The condition is due to infection with a spirochaete, Borrelia burgdorferi sensu lato, which is transmitted by ticks [1].
Epidemiology
Incidence and prevalence
The disease occurs mainly in northern or central Europe, Italy and the Iberian Peninsula. Occasional cases occur in other parts of Europe and Africa, but it is very rare in the UK, America, Australia and Asia [2]. These geographical variations are related to different strains of the organism [3, 5].
Age
Mostly between the ages of 30 and 60 years.
Pathophysiology
Pathology [6]
During the early stages, there is non-specific dermal oedema with perivascular inflammatory infiltration. Subsequently, the epidermis becomes atrophic and the epidermal appendages are destroyed. Beneath a subepidermal zone of degenerate connective tissue lies a dense, band-like infiltrate, predominantly consisting of lymphocytes, histiocytes and plasma cells. Ultimately, the infiltrate is reduced to narrow bands between collagen fibres. In some patients, scleroderma-like changes may develop [7, 8]. More typically, the dermis shows signs of atrophy; the swelling and homogenization of collagen and elastic fibres is followed by their disappearance [9]. Borrelia afzelii has been cultured from the atrophic skin [7] but culture is usually negative. Borrelia afzelii can be identified by polymerase chain reaction (PCR). The organism may be resistant to attack by the complement system and may lurk in immunologically protected areas such as fibroblasts and endothelial cells. Expression of pro-inflammatory cytokines, such as interferon-γ (IFN-γ), is increased [10].
Causative organisms
Borrelia afzelii is the predominant species associated with acrodermatitis chronica atrophicans [11]. This species is transmitted by ticks in Western Europe, but is rare in the USA, where Borrelia burgdorferi sensu stricto predominates [12].
Environmental factors
It is transmitted by bites from ticks, notably Ixodes spp., which favour scrubland [12].
Clinical features
History
Most cases occur in country-dwellers. There is usually a history of a tick bite. The onset is usually insidious, and constitutional symptoms are exceptional [13].
Presentation
Dull-red or bluish-red nodules or plaques, more or less infiltrated, develop on the feet or legs, and less often on the forearms and hands. The lesions themselves are typically painless, but there may be associated acral pain or paraesthesiae. Erythema chronicum migrans (see Chapter 27) may have been present at the same site some years earlier. Extension to the trunk and the greater part of the body, including the face, is sometimes seen. Single or multiple lesions may be present. They slowly extend centrifugally, the active inflammatory stage persisting for months, years or even decades. Marginal extension may continue once the central areas have already entered the atrophic phase, in which the skin is smooth, hairless and tissue-paper-like, dull red, pigmented or poikilodermatous (Figure 96.18).
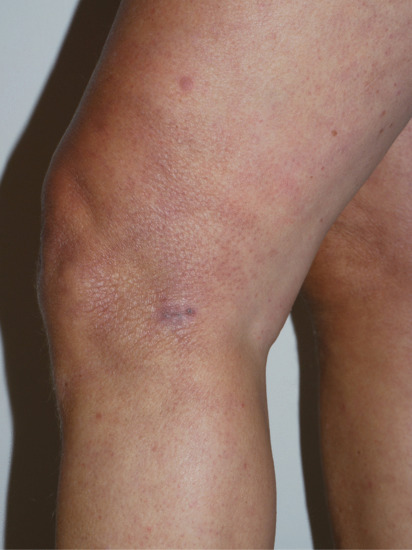
Figure 96.18 Acrodermatitis chronica atrophicans: image captured soon after commencement of antibiotic therapy; note atrophic wrinkled appearance of the skin at the side of the knee. (Courtesy of Dr Ian Coulson, Burnley Hospital, UK.)
Subcutaneous nodules may develop around the knees or elbows, and fibrous bands along the ulnar margin of the forearms. Gaiter-like sclerosis of the lower third of the legs, often accompanied by ulceration, is a further complication. Morphoea of the trunk and lichen sclerosus (both genital and extragenital) have also been reported in association [2, 14]. Conversely, Borrelia antibodies have been found in some patients with morphoea [14, 15], although this does not appear to be a common finding [16].
In some cases, involvement of the joint capsule or bone results in limitation of movement of the joints of the hands and feet, or of the shoulders.
Clinical variants
Occasional patients develop erythematous plaques, clinically and histologically suggestive of mycosis fungoides [17].
Differential diagnosis
The early cutaneous phase of Lyme borreliosis, erythema chronicum migrans, may be confused with other annular erythemas, although a history is often obtained of a recent tick bite at the site. When it occurs on the lower legs, it may mimic venous insufficiency [18], with thick cyanotic itchy skin.
Complications and co-morbidities
Very rarely, squamous carcinoma has developed in the atrophic skin, and lymphoma has also been reported in non-affected skin [19–21]. Other late manifestations of Lyme borreliosis (lymphocytoma, neurological, etc.) have been fully reviewed by Steere [1].
Disease course and prognosis
The bacteria can be eradicated with systemic antibiotics but some systemic features, such as neuroborreliosis, may persist.
Investigations
In the atrophic stage, diagnosis is usually readily made, and can be confirmed histologically.
Immunoblotting, using B. afzelii flagellar antigen (41 kDa) is confirmatory [5]. Serology is used to confirm the diagnosis of Lyme disease, but false-negative and false-positive results are common. In chronic atrophic acrodermatitis, however, the antibody titre is very high. Serology may be positive on enzyme-linked immunosorbent assay (ELISA) but negative on immunoblotting, particularly in patients with neurological disease [22]. A high titre of antibodies may reflect occult central nervous system involvement, when the antibodies can also be demonstrated in colony-stimulating factor [23].
Management
Oral antibiotics should be given for 1 month, for example doxycycline or amoxicillin in standard doses [1]. Improvement occurs gradually and may not become apparent until several weeks after the course of treatment. There may be no improvement if treatment is delayed until atrophy has already developed. If the antibody titre is high or there are clinical features of systemic disease (e.g. neuroborreliosis), intravenous benzylpenicillin, ceftriaxone or cefotaxime should be given for 3 weeks [23]. There may be a case to be made for introducing public health measures such as chemoprophylaxis programmes or eventually a vaccine in endemic areas [24].
Elevated immunoglobulin G (IgG) and IgM antibodies may persist after treatment; this does not reflect treatment failure [25].
Atrophodermas
Atrophoderma, follicular
Definition
This distinctive abnormality manifests as dimple-like depressions at the follicular orifices and is usually associated with one of a small number of genetic syndromes but may be sporadic. It may be manifest at birth but may not become apparent until late in childhood. It usually involves the backs of the hands (Figure 96.19) and the feet, and sometimes the elbow region. It may be associated with the following conditions [1]:
- Conradi–Hünermann–Happle syndrome (calcifying chondrodysplasia) (see Chapter 65) [2].
- Bazex–Dupré–Christol syndrome (see Chapter 68) [3].
- Hyperkeratosis palmoplantaris, follicular keratosis or palmoplantar hyperhidrosis.
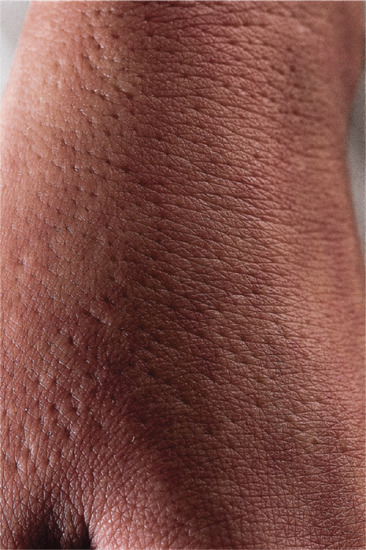
Figure 96.19 Follicular atrophoderma in Conradi syndrome.
It may also occur as an isolated defect of limited extent.
Pathophysiology
Pathology
Histology shows widened follicular ostia with thickening of the connective tissue sheath of the follicle.
Genetics
It appears to be associated with a variety of genetic defects.
Clinical features
Presentation
Follicular depressions on the backs of the hands (see Figure 96.19), feet and occasionally elbows.
Management
No proven treatment.
Atrophoderma, linear
Introduction and general description
It is probable that this and atrophoderma of Pasini and Pierini are atrophic variants of morphoea [2].
Epidemiology
Incidence and prevalence
Cases are sporadic and worldwide.
Age
Most cases are described in childhood and adolescence.
Associated diseases
Leuconychia has been associated [3].
Pathophysiology
Pathology
Histologically, the epidermis is normal apart from hyperpigmentation in the basal layer. There is a perivascular lymphocytic infiltrate in the dermis [4]. The collagen bundles are normal or thickened; there is diminished periadnexal and subcutaneous fat [5].
Genetics
The condition may reflect mosaicism following a postzygotic mutational event [4, 6].
Clinical features
History
Lesions are usually asymptomatic and insidious in onset.
Presentation
Linear atrophic hyperpigmented plaques in the distribution of Blaschko's lines, sometimes having a zosteriform appearance [5].
Differential diagnosis
Atrophic variants of morphoea strongly resemble this syndrome, and may be identical.
Investigations
Laboratory investigations are normal [5]. Skin biopsy is helpful if there is clinical doubt.
Management
One case of successful treatment with methotrexate is reported [7].
Atrophoderma of Pasini and Pierini
Definition
This condition is probably an atrophic variant of morphoea (see Chapter 57) in which one or more patches of skin become bluish and sharply depressed, with no surrounding erythema [11–13].
Epidemiology
Incidence and prevalence
Cases are mostly sporadic and rare.
Age
Most cases present in childhood or adolescence.
Associated diseases
There is a probable association with morphoea. Familial cases have been reported [14], together with an association with phenylketonuria [15].
Pathophysiology
Predisposing factors
The cause is unknown, although, as in morphoea, Borrelia burgdorferi has been implicated [16].
Pathology
The histological changes are often slight [13]. There may be increased pigmentation of the basal layer. During the earlier stages, the collagen in the lower dermis may be oedematous, and elastic tissue clumped and scanty. There may be a dermal perivascular infiltrate consisting of macrophages and T lymphocytes. Immunofluorescence studies may show IgM and C3 staining in the dermal blood vessels [17]. Later, the oedema subsides and there is some reduction in the total thickness of the dermis. Collagen bundles appear homogeneous and clumped in the reticular dermis. Eventually there may also be some epidermal atrophy.
Causative organisms
In common with morphoea, Borrelia burgdorferi has been implicated [16].
Genetics
No genetic factor has been reliably incriminated, although familial cases have been reported [14], and morphoea and atrophoderma of Pasini have occurred in siblings with phenylketonuria [15].
Clinical features
History
The lesions are generally asymptomatic.
Presentation [13, 18, 19]
The lesions, which may be single or multiple, range in size from 2 cm to many centimetres in diameter, and are round or oval in shape, but may become confluent to form irregular patches (Figure 96.20). They are smooth, slate-coloured or violet-brown, and are slightly depressed below the level of the entirely normal surrounding skin. The back is almost always involved, the chest and abdomen frequently, and the proximal parts of the limbs occasionally.
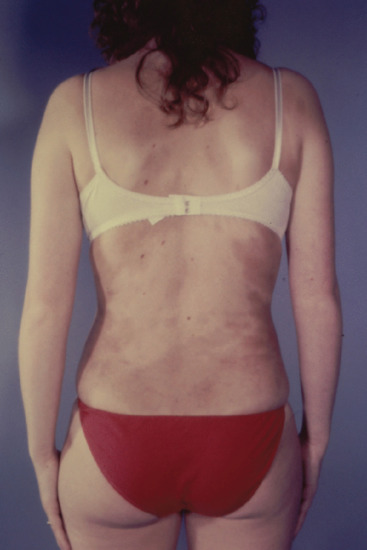
Figure 96.20 Atrophoderma of Pasini and Pierini.
Differential diagnosis
Atrophic morphoea and linear atrophoderma may represent the same condition. Clinical differentiation from morphoea, possibly an academic exercise, is based on the ivory-white indurated plaque with an oedematous lilac ring so characteristic of the latter. Histologically, sclerosis may be prominent in morphoea and is usually absent in atrophoderma.
Complications and co-morbidities
An overlap with juvenile idiopathic arthritis has been described [20].
Disease course and prognosis
The patches extend very slowly, increase in number for 10 years or more, and then usually persist unchanged. The eventual development of sclerodermatous changes within the patches has been observed, as has the presence in the same patient of lesions typical of atrophoderma and of morphoea.
Investigations
Serological tests for Borrelia burgdorferi are typically negative [13] although there are case reports of an association (e.g. see [16]).
Management
No treatment is of proven efficacy, but psoralen and UVA (PUVA) has helped some patients. Hydroxychloroquine has been used [21]. A case apparently associated with Borrelia burgdorferi responded to doxycycline [16].
Paroxysmal haematoma of the finger
Definition and nomenclature
This condition presents with the sudden spontaneous onset of one or more painful haematomas in the fingers (Figure 96.21).
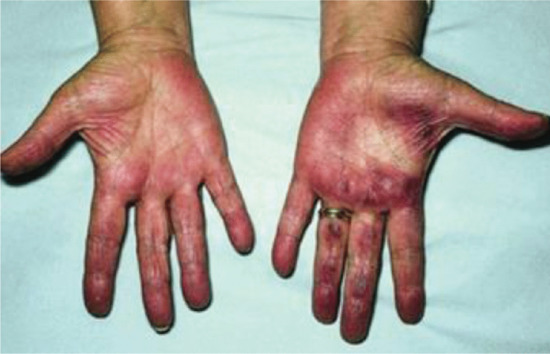
Figure 96.21 Paroxysmal haematoma of the finger. (Courtesy of Dr J. Verbov, Royal Liverpool University Hospitals, Liverpool, UK.)
Epidemiology
Age
Usually middle age.
Sex
Female predominance.
Associated diseases
None.
Pathophysiology
Predisposing factors
The cause is unknown but has been hypothesized to be due to a localized acquired fragility of vascular connective tissue.
Pathology
There is no evidence of vasculitis or amyloid on skin biopsy.
Clinical features
History
Sudden onset of often painful haematoma.
Presentation
Sudden bruising of the volar aspect of a finger may occur spontaneously or after minor trauma; the bruising resolves within days and the patient is asymptomatic between flares [1, 2, 3, 4]. The wrist may sometimes be involved [5]. There is no evidence of ischaemia [6].
Differential diagnosis
It may be mistaken for easy bruising due to steroid atrophy. The absence of ischaemic features and rapid improvement exclude occlusive vascular disease.
Complications and co-morbidities
There are no co-morbidities.
Disease course and prognosis
It may recur at intervals for several years. Although troublesome, it is a benign condition.
Investigations
Although subtle angiographic abnormalities have been described [7], investigation of the patient for significant vascular disease is unnecessary [8].
Panatrophy
Definition
Local panatrophy is a rare disorder involving partial or total loss of subcutaneous fat and atrophy of overlying skin, sometimes associated with atrophy or impaired growth of muscle or bone. A primary neurogenic disturbance has been postulated but not proved. The syndrome may represent the end result of more than one pathological process, but many cases may be due to a variant of morphoea. They are discussed further in Chapter 57.
The atrophic areas exhibit a reduced sympathetic response and aberrant production of non-esterified fatty acids after stimulation with norepinephrine (noradrenaline), and it has been suggested that there may be a primary abnormality of the sympathetic nervous system [1].
Two groups of cases can be differentiated.
- Panatrophy of Gower: no scleroderma or other sclerotic process accompanies or follows the loss of subcutaneous tissue. Most cases have occurred in women, usually in the second to fourth decades.
- Sclerotic panatrophy: either typical morphoea or similar sclerotic change in dermal collagen precedes the atrophy [2].
Clinical features
Clinical features of these two groups are as follows.
Panatrophy of Gower [3, 4]. Sharply defined areas of atrophy, irregular in size, shape and distribution, develop over a period of a few weeks, without preceding inflammatory stages. In each affected area, the subcutaneous tissue disappears and the overlying skin appears atrophic but is otherwise normal. There may be a single area of atrophy or two or more. In size they range from 2 to 20 cm across, and in shape they are very variable but are sometimes triangular or quadrangular. Most lesions have occurred on the back, buttocks, thighs or upper arms, but some have involved the forearms or lower legs. The atrophy reaches its maximum extent within a few months and then remains unchanged indefinitely.
Sclerotic panatrophy. Atrophy of the subcutis, and sometimes of underlying muscle and bone, may follow clinically and histologically typical morphoea, especially when the process begins in childhood and involves a limb (see Chapter 57).
Sclerotic panatrophy may also occur in the absence of morphoea. The sclerosis involves subcutaneous tissue and muscle, and dense sclerotic scar-like linear bands develop along a limb, or encircle the trunk in a metameric distribution, or encircle a limb. These lesions have also usually occurred in childhood. They cease to progress after a few months and, although new areas may be involved, most lesions have been solitary.
It is probable that Gower's panatrophy and linear morphoea are at the ends of a continuous disease spectrum. The histology of linear morphoea reveals thickened bundles of collagen, which appear to be intact on B-scan ultrasound imaging [5].
In the differential diagnosis of panatrophy, the various forms of panniculitis must be excluded. The preceding inflammatory changes are the single most distinctive feature, but they are not always easy to distinguish.
Facial defects can be corrected by autologous fat grafting [6].
Facial hemiatrophy (see also Chapter 57)
Introduction and general description
Facial hemiatrophy is an atrophic dysplasia of the superficial facial tissues, but the underlying muscles, cartilage and bone may also be affected [1].
Epidemiology
Age
This rare disease usually starts within the first two decades of life.
Sex
The sexes are equally affected.
Associated diseases
Some cases have been associated with syringomyelia, epilepsy or cerebrovascular disease, but in 90% of cases no such association is demonstrable.
Pathophysiology
Predisposing factors
The cause is unknown, but it may be a disorder of the sympathetic nervous system in some cases.
Genetics
There is no evidence that it is usually genetically determined, but it appears to be hereditary in a few pedigrees.
Clinical features
History
Occasionally, there may be premonitory muscle spasms or neuralgia [2] but often it is asymptomatic.
Presentation
The first manifestation is usually increased or decreased pigmentation in irregular patches on the cheeks, forehead or lower jaw. Progressive atrophy gradually develops in the affected sites, involving skin, subcutis, muscle and bone, and may extend in area — and sometimes in depth — for months or years with temporary remissions. The skin becomes dry, thin and atrophic, but may be scar-like and adherent in some areas. When the atrophy is fully developed, the contrast between the sunken, haggard, pigmented affected half of the face and the unaffected half is dramatic. The hair may be lost in the fronto-parietal region on the affected side but is often normal; occasionally, localized canities is an early change. A variety of neurological signs have been reported, of which Horner syndrome is the most frequent. Heterochromia of the iris has developed at the same time as the facial atrophy in about 5% of cases, and retinal changes may also be present [3], including central retinal artery occlusion [4]. There can be ipsilateral cerebral atrophy [5].
The degree of bone atrophy as established radiologically is usually much less than the clinical appearance suggests, and is severe only in some cases of early onset. In such cases, the cerebral cortex may also be affected, and contralateral epilepsy may result.
Differential diagnosis
When the cutaneous involvement is early and conspicuous, the diagnosis presents few difficulties. Hypoplasia following radiotherapy given in infancy, perhaps in treatment of a naevus in the region of the temporo-mandibular joint, could cause confusion. If the skin changes are slight, or of later onset, physiological asymmetry, unilateral mandibular agenesis, hemihypertrophy and atrophy secondary to facial paralysis must be excluded. Hemihypertrophy is always congenital. When the limbs are involved, infantile hemiplegia and lipodystrophy must also be considered.
Lupus panniculitis results in subcutaneous atrophy which can be hemifacial. Atrophic morphoea of the ‘coup de sabre’ paramedian form may be associated with some degree of facial hemiatrophy, especially if it begins early in life. However, it is generally a more superficial process than progressive facial hemiatrophy. The skin in scleroderma is bound down and adherent, and loss of hair and pigmentary changes are conspicuous. In progressive facial hemiatrophy, the skin may remain mobile and grossly normal. The two processes have been confused frequently in the literature, and may coexist [6].
Complications and co-morbidities
There may be associated segmental vitiligo [7]. Spontaneous fracture of the jaw has also been reported [8].
Disease course and prognosis
The atrophy may remain limited both in extent and depth. It may be confined to the distribution of one division of the trigeminal nerve or involve the whole of the side of the face, sharply demarcated at the midline. Rarely, it may be bilateral, and very rarely may involve half the body, usually on the same side as the face but exceptionally the opposite side — crossed hemiatrophy. The atrophy may, in such cases, begin on the trunk or a limb and only later involve the face.
Management
Plastic surgery using large buried pediculated flaps of dermis and fat, or silicone implants, offers some cosmetic benefit [9–11]. Autologous fat grafts have a variable ‘take’ although they remain the treatment of choice [12]; supplementation with stromal vascular fraction-supplemented cell therapy may improve the long-term result [13].
DISORDERS OF ELASTIC FIBRE DEGRADATION
Introduction and general description
The capacity of the skin to adapt to local or general changes in body size and contour, and to allow for movement of head and limbs and a wide range of facial expression, depends upon its tension, elasticity and tensile strength. These properties may be congenitally defective or modified by ageing or disease [1–3]. Acquired disorders of elastic tissue have been reviewed in detail by Lewis et al. [4, 5].
Elastic fibres are abundant in the skin, arteries, lungs and ligaments. They provide tissues with resilience and elasticity, enabling the skin to resume its original shape after deforming forces have ceased to act. There is wide individual variation, but a tendency for elastic fibres to become less plentiful with age. Cutaneous elasticity is also reduced in a variety of skin disorders including cutis laxa. Additionally, elastic fibres provide adhesion for cells and play a role in regulating growth factors (e.g. transforming growth factor β (TGF-β)) [2].
Tensile strength
The tensile strength of the skin is the degree to which it can be elongated before it tears. It is greatest in infancy and decreases with age, but is also abnormally low in diseases associated with qualitatively or quantitatively abnormal collagen such as Ehlers–Danlos syndrome and Cushing syndrome [3].
Lax skin
Increased laxity of the skin due to ageing (accelerated by dermal photodegradation) is extremely common, but cutaneous laxity can occasionally result from marked weight loss (especially after gross obesity) or can follow recovery from severe oedema. Less commonly, the skin may become lax due to localized or generalized defects in elastic tissue resulting from other causes, and these may be grouped as follows:
- Generalized elastolysis (cutis laxa):
- congenital (see Chapter 72): it may be a component of inherited disorders including PXE, SCARF syndrome (skeletal abnormalities, cutis laxa, craniostenosis, ambiguous genitalia, retardation and facial abnormalities), de Barsy syndrome, geroderma osteodysplastica;
- acquired: numerous associated disorders, e.g. inflammatory skin disease, multiple myeloma, systemic lupus erythematosus, hypersensitivity reactions, complement deficiency, penicillamine therapy.
- Localized elastolysis:
- anetoderma;
- blepharochalasis;
- chronic atrophic acrodermatitis (due to Borrelia) (see earlier);
- granulomatous slack skin (due to lymphoma);
- other localized lesions, including mid-dermal elastolysis, post-inflammatory elastolysis and cutis laxa (PECL), elastic tissue naevi, etc.
It is probable that many of the above conditions are variations of the same disease, and there is considerable overlap. They share a similar pathological process, namely elastophagocytosis (the phagocytosis of elastic fibres by histiocytes and/or multinucleate giant cells) [1].
Acquired cutis laxa
Definition and nomenclature
Cutis laxa presents clinically as lax skin which hangs in folds, together with loss of dermal elastic tissue histologically. Congenital forms are discussed in Chapter 72.
Introduction and general description
Cutis laxa may be acquired following inflammatory skin disease [1] or following exposure in utero to drugs such as penicillamine [2]. An immunological pathogenesis has been suggested in many cases.
Epidemiology
Associated diseases
Cutis laxa has been reported in association with urticarial eruptions, nephrotic syndrome [3], complement deficiency, sarcoidosis, syphilis, primary amyloidosis and multiple myeloma [4, 5], drug hypersensitivity and the Klippel–Trenaunay syndrome [6]. Focal elastolysis can also occur in association with lupus erythematosus [7], severe rheumatoid arthritis [8] and coeliac disease [9]. d-penicillamine disrupts elastic fibre formation and may cause cutis laxa, elastosis perforans serpiginosa and pseudoxanthoma-like changes [10, 11]. Congenital cutis laxa may also occur in offspring of mothers taking penicillamine [2].
Pathophysiology
Predisposing factors
Immunological or chemical disruption of dermal elastic fibres.
Pathology
In acquired cutis laxa, dermal elastic tissue is markedly reduced, although collagen is normal. Fibroblasts express increased elastolytic activity (cathepsin G). Levels of serum α1-antitrypsin and elastase inhibition are decreased [12].
Genetics
There may be an underlying genetic susceptibility, for example defects in the interaction of elastin and fibulin 5 results in elastic fibres that are more susceptible to degradation by matrix metalloproteinases [13].
Clinical features
History
Cutis laxa may rarely develop at any age following episodes of urticaria or angio-oedema, extensive inflammatory skin disease (such as systemic lupus erythematosus or erythema multiforme) or febrile illness (Figure 96.22). It may also follow hypersensitivity reactions such as penicillin allergy [14].
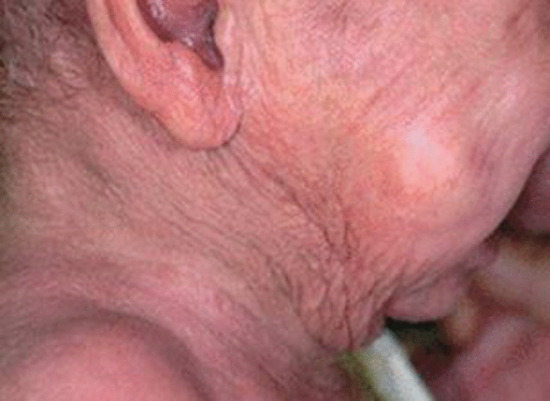
Figure 96.22 Acquired cutis laxa following a generalized inflammatory dermatitis in an 18-month-old child. (From Haider et al. [17], with permission from John Wiley.)
Presentation
There may be widespread massive folds of lax skin, or the changes may be mild and confined to a limited area, in which case it cannot be distinguished from anetoderma. Purpura may follow slight trauma and fibrotic nodules may form over bony prominences. Organs other than the skin may also be involved. Emphysema, gastric fibromas and tracheobronchomegaly have been reported [15].
Clinical variants
Post-inflammatory elastolysis and cutis laxa (Marshall syndrome) (Figure 96.23a–c) was originally described as a distinctive syndrome in African children but subsequently reported worldwide, Clinical features are intermediate between anetoderma and cutis laxa [16, 17, 18]. It is preceded by an inflammatory process, often with a neutrophilic component (e.g. Sweet syndrome [19, 20]) or an insect bite. The preceding inflammatory lesions may be urticaria-like or multiple red papules, which slowly enlarge to form rings 2–10 cm in diameter [17]. It has been associated with α1-antitryspin deficiency, which may enable matrix metalloproteinases to destroy dermal elastin, and screening for this enzyme deficiency is recommended [20].
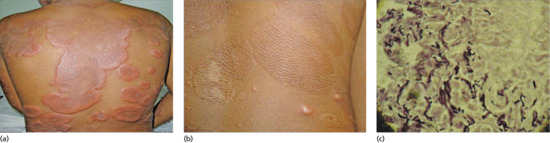
Figure 96.23 Post-inflammatory elastolysis and cutis laxa (Marshall syndrome) in a 6-year-old boy showing acute inflammatory phase (a) progressing to large plaques of lax wrinkled skin (b). Histology shows shortened and fragmented elastic fibres in the reticular dermis (c). (Reproduced from Fontenelle et al. 2013 [18] with permission from Sociedade Brasileira de Dermatologia.)
Differential diagnosis
The history should enable the condition to be distinguished from congenital cutis laxa. In Ehlers–Danlos syndrome, the skin is hyperextensible but not lax, and it recoils quickly. In PXE, the skin may be lax, but it is yellowish and the face is usually spared. It is distinguished histologically by the presence of calcification. There may be circumscribed folds of lax skin in neurofibromatosis, and loose folded skin may also occur in leprechaunism, Patterson syndrome and trisomy 18, but these conditions are distinguished by their associated features.
In severe actinic damage, there may be marked skin laxity due to damage to elastic fibres. There is doubtless considerable overlap with other elastolytic conditions described later.
Investigations
The diagnosis, which is suggested by finding loose skin that recoils only slowly after stretching, may be confirmed by histological confirmation of a reduction in elastic fibres. Investigations for emphysema may be indicated, with referral to a pulmonary physician if necessary. Underlying inflammatory disease may require investigation.
Management
Plastic surgery (‘face-lift’) may substantially reduce the cosmetic disability [18].
Anetoderma
Definition and nomenclature
The term anetoderma (anetos: slack) refers to a circumscribed area of slack skin associated with a loss of dermal substance on palpation and a loss of elastic tissue on histological examination. ‘Primary’ anetoderma implies that there is no associated localized underlying cutaneous disease, whereas ‘secondary’ anetoderma can be attributed to some associated condition.
Introduction and general description
Previously, cases of ‘primary’ anetoderma were divided into the Jadassohn–Pellizzari type, in which the lesions are preceded by erythema or urticaria, and the Schweninger–Buzzi type, in which there are no preceding inflammatory lesions. This is now of historical interest only, because in the same patient some lesions may be preceded by inflammation and others may not, and the prognosis and histology are identical in the two types [1, 2, 3].
Epidemiology
Incidence and prevalence
Rare.
Age
Mainly 20–40 years, but is occasionally reported in infants and older patients.
Sex
Mainly in women [2].
Associated diseases
Primary anetoderma is strongly associated with antiphospholipid syndrome [4, 5, 6] with or without features of systemic lupus. In older reports, this may have led to a misdiagnosis of syphilis in many cases, although there is a definite association with the disease and its treatment [7]. Secondary anetoderma has been reported in association with tuberculosis and leprosy [8], urticaria pigmentosa [9], pityriasis versicolor [10], granuloma annulare [11, 12], Stevens–Johnson syndrome [13], B- and T-cell lymphoma [14–16] and other conditions.
Some reported associations may be coincidental, but it is probable that many inflammatory diseases may occasionally be complicated by anetoderma.
Localized anetoderma may occur in premature infants, possibly due to the application of transcutaneous oxygen monitoring devices [17, 18]. Localized anetoderma-like changes on histology have been reported in association with pilomatricoma [19], dermatofibroma [20], juvenile xanthogranuloma [21] and hamartomatous congenital naevi [22]. Lesions resembling anetoderma occur in post-inflammatory elastolysis and cutis laxa (Marshall syndrome) (Figure 96.23b). Penicillamine-induced anetoderma has also been reported [23].
Pathophysiology
Predisposing factors
Primary anetoderma
Recently, it has become apparent that ‘primary’ anetoderma is strongly associated with antiphospholipid antibodies, with or without a prothrombotic state (Figure 96.24) [4, 5]. It is probable that these antibodies underly the association historically noted with syphilis, and more recently with borreliosis [27] and systemic lupus.
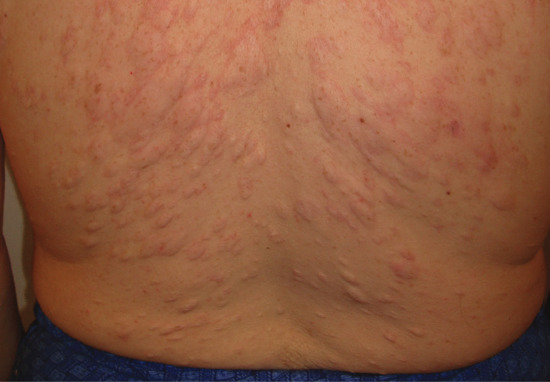
Figure 96.24 Primary anetoderma associated with antiphospholipid antibodies. (From Eungdamrong et al. [26], with permission from Dermatology Online Journal.)
In a few cases, there appears to be an underlying structural defect of connective tissue. Familial cases are reported [24–26] and there is an association with inherited bony or ocular abnormalities. The Blegvad–Haxthausen syndrome comprises anetoderma, blue sclerae and osteogenesis imperfecta (OI).
The histology of anetoderma suggests that the basic abnormality is focal elastolysis [1, 28, 29]. This may be secondary to the release of elastase from inflammatory cells which are probably always present in the early stages. Metalloproteinases are increased in lesional skin [30].
Complement activation may be involved, as C3 is deposited on the remaining elastic fibres [31]. It has been suggested that decay-accelerating factor (DAF) and vitronectin (an inhibitor of the membrane–attack complex) may protect elastic fibres against this type of damage [32]. Abnormalities in the protective system could play a role in primary anetoderma.
Secondary anetoderma
This is seen in association with another identifiable disease, and has occurred in association with systemic [33] or chronic cutaneous lupus erythematosus [34], not always in relation to the lesions. Anetoderma is also associated with lupus profundus [35, 36].
Some cases of primary anetoderma have direct immunofluorescence findings similar to those of either chronic cutaneous or systemic lupus erythematosus, even though there may be no other features of lupus erythematosus [38, 39]. Biopsy shows a focal loss of elastic tissue, and a perivascular infiltrate with prominent plasma cells [1, 2]. Generalized elastolysis (cutis laxa) has also occurred [37].
Antibodies have not been demonstrated against elastic fibres [39].
Pathology [2, 27]
During the early stages, the dermis is oedematous, and a lymphocytic infiltrate (predominantly helper T cells) surrounds the blood vessels and appendages [1, 29]. Plasma cells and histiocytes, with some granuloma formation, may also be seen. Later, the oedema and perivascular infiltrate subside and elastic fibres become scanty. The persistence of fine, irregular or twisted elastic fibres is common. The dermal collagen may also be diminished, but the fragmentation and disappearance of elastic tissue is the essential change, beginning superficially in the subpapillary zone and extending downwards. Electron microscopy shows phagocytosis of elastic fibres by macrophages [40–42].
Causative organisms
Serological evidence of Borrelia burgdorferi infection has been observed in some cases [27].
Genetics
Familial cases are reported [24–26].
Environmental factors
It is perhaps more frequent in central Europe than elsewhere, which suggests a possible relationship to chronic atrophic acrodermatitis (due to Borrelia spp.) in some cases [27].
Clinical features
History
There may be a history of a previous inflammatory, perhaps urticated, lesion at the site. Often lesions are asymptomatic.
Presentation
In primary anetoderma crops of round or oval pink macules 0.5–1.0 cm in diameter develop on the trunk, thighs and upper arms, less commonly on the neck and face and rarely elsewhere. The scalp, palms and soles are usually spared. Each macule extends for a week or two to reach a size of 2–3 cm. Sometimes, there are larger plaques of erythema, and nodules have also been reported as a primary lesion [43]. Slowly, each lesion fades and flattens from the centre outwards to leave a macule of wrinkled atrophic skin, which yields on pressure, admitting the finger through the surrounding ring of normal skin (Figure 96.25). The colour varies from skin colour to grey, white or blue. The number of lesions varies widely, from less than five to 100 or more.
Figure 96.25 Secondary anetoderma in chickenpox scar. (From Veraldi A, Schianchi R, Chickenpox, impetigo, and anetoderma Pediatric Dermatology 2006;23:305–6. With permission from John Wiley.)
In some cases, the lesions are initially urticarial weals which, after a succession of exacerbations and remissions, perhaps continuing for many weeks, are succeeded by atrophy. They may become confluent, to cover large areas, especially at the roots of the limbs and on the neck.
The atrophic areas in secondary anetoderma do not always develop at the sites of the known inflammatory lesions. They are soft, round or oval areas which occur mainly on the trunk.
Clinical variants
‘Confetti-like macular atrophy’ [44] may be a variant of anetoderma, although the lesions are not depressed or herniated. Hypopigmented shiny atrophic patches occur on the upper limbs and trunk. Histology shows an atrophic epidermis with disorganized, hyalinized coarse collagen bundles in mid-dermis, with elastic fibre loss and fragmentation in the upper dermis.
Differential diagnosis
Extragenital lichen sclerosus (see Chapter 57) presenting as white spots around the base of the neck and shoulders should not be confused with anetoderma. Histological examination establishes the diagnosis if there is doubt.
Focal dermal hypoplasia and atrophic scars must also be considered.
Aquired cutis laxa (see earlier) and anetoderma are closely related, and may represent different forms of the same condition.
The diagnosis of ‘primary’ anetoderma can be established only by excluding the presence of any of the diseases known to be associated with ‘secondary’ atrophy, e.g. perifollicular elastolysis (see later).
Disease course and prognosis
The lesions remain unchanged throughout life, and new lesions often continue to develop for many years. If the lesions coalesce they form large atrophic areas, which are indistinguishable from acquired cutis laxa [2].
Investigations
In patients with primary anetoderma it is important to test for antiphospholipid syndrome and treat appropriately, e.g. with aspirin or warfarin.
Management
No specific treatment exists. In the case of secondary anetoderma, treatment should be directed against underlying disease or infections.
Penicillin and the antifibrinolytic drug ε-aminocaproic acid have been advocated [45], but Venencie et al. [2] studied 16 patients and found no treatment was beneficial once the atrophy had developed. Colchicine may prevent some atrophic changes [46]. Ablative (e.g. carbon dioxide) lasers may reduce scarring [13].
Mid-dermal elastolysis
Definition and nomenclature
Idiopathic loss of the elastic fibres in the mid-dermis leads to widespread wrinkling of the crinkle type in otherwise healthy young or middle-aged women (Figure 96.26) [1, 2]. The exact relationship between this condition and other elastolytic disorders such as acquired cutis laxa and anetoderma is uncertain. Localized areas may clinically resemble PXE, although they are histologically distinct [3, 4].
Figure 96.26 Idiopathic mid-dermal elastolysis. (Courtesy of Dr L. Ostlere, St George's Hospital, London, UK.)
Epidemiology
Incidence and prevalence
Sporadic cases.
Age
Young to middle age.
Sex
Mostly female.
Ethnicity
Fair-skinned.
Associated diseases
Cases have been associated with a prothrombotic state [5], suggesting a similarity to anetoderma.
Pathophysiology
Predisposing factors
It has been reported to follow granuloma annulare [6] and other inflammatory conditions.
Pathology
Ultrastructural studies of mid-dermal elastolysis demonstrate elastic fibres engulfed by macrophages [7]. In the perifollicular variant histology shows a non-inflammatory perifollicular loss of elastin fibres [8]. Immunological studies of affected skin show a non-specific profile of immune activation [9]. Cultured fibroblasts from lesional dermis exhibit increased elastolytic activity and reduced elastin mRNA compared with normal skin [10]. Maghraoui et al. [11] have distinguished post-inflammatory elastolysis, with or without features of cutis laxa, from non-inflammatory elastolysis.
The histology of idiopathic mid-dermal elastolysis is similar to that of PECL (see earlier). Those lesions are preceded by inflammatory lesions, but the lesions of idiopathic mid-dermal elastolysis may also occasionally be preceded by erythema, urticaria or a burning sensation, and the two conditions are similar, if not identical.
Causative organisms
Elastase-producing strains of Staphylococcus epidermidis have been implicated in the perifollicular variant [12].
Environmental factors
UV light may trigger elastophagocytosis [13]. The condition has also been reported in a patient receiving haemodialysis [14] and near the site of insertion of a pacemaker [15].
Inflammatory triggers may include UV radiation, insect bites, varicose veins, borreliosis and acute neutrophilic dermatosis [16, 17].
Clinical features
History
The condition is typically asymptomatic.
Presentation
Millimetre to centimetre large, well-circumscribed or net-like areas of crinkly skin (cigarette paper-like fine wrinkling).
Clinical variants
Three variants have been described [18] as follows:
- Type 1: cigarette paper-like fine wrinkling (crinkle) affecting the trunk and upper arms.
- Type 2: perifollicular papules [8, 12]. Lesions are small, grey–white, finely wrinkled, round or oval areas, each with a central hair follicle. Some exhibit a balloon-like bulge above the surface. They occur on the upper trunk, neck, earlobes and arms. Similar changes are more commonly seen in acne scars (see Chapter 91).
- Type 3: reticular variant; orange-red inflammatory papules precede net-like areas of atrophy, chiefly on the arms [15, 18]. Localized areas may clinically resemble PXE, although they are histologically distinct [3, 4]
In addition to these, a linear lumbar variant has also been described [19].
Upper dermal elastolysis
Definition and nomenclature
Selective loss of elastic tissue in the papillary dermis was originally described in an otherwise healthy 86-year-old woman, who presented with numerous yellowish papules on the neck and upper trunk, and associated coarse wrinkles [20]. Since, there have been several other reports, mostly in women aged 60–70 years [21, 22, 23]. This condition may be a unique variant or related to acquired PXE [23].
Differential diagnosis
The histology of idiopathic mid-dermal elastolysis is similar to that of PECL, which occurs in young African girls (see earlier). Those lesions are preceded by inflammatory lesions, but the lesions of idiopathic mid-dermal elastolysis may also occasionally be preceded by erythema, urticaria or a burning sensation, and the two conditions are similar, if not identical.
Management
No definite treatment exists but topical retinoic acid (0.01% gel) produced some cosmetic improvement in one patient [9]. Reduction of degradation by metalloproteinases would be desirable, as in other elastophagocytic disorders [24].
Blepharochalasis
Definition and nomenclature
Laxity of the eyelid skin due to a defect in the elastic tissue.
Epidemiology
Incidence and prevalence
Rare, mostly sporadic.
Age
Usually around the time of puberty.
Ethnicity
Most cases are reported in white people.
Associated diseases
Some cases may be a localized form of post-inflammatory elastolysis or follow angio-oedema [1].
Pathophysiology
Predisposing factors
Presumably inflammatory stimulus to elastophagocytosis.
Pathology
In the early stages, there may be a mild dermal lymphocytic infiltrate, and in the later stages the elastic fibres in the lids fragment and decrease [2]. Normal elastin gene expression suggests other factors may be involved in elastic fibre loss [3]. IgA deposition may be detected on fibres, implying an immunopathogenic mechanism may be relevant [4]. Disintegration of collagen fibres has also been observed in one case [5].
Causative organisms
None known.
Genetics
Some pedigrees show autosomal dominance, although most cases are sporadic.
Clinical features
History
There may be a history of previous transient episodes of painless eyelid swelling lasting for 2–3 days.
Presentation
Blepharochalasis is an uncommon condition that usually develops insidiously. Attacks of painless swelling of the eyelids are followed by laxity, atrophy, wrinkling and pigmentation, predominantly of the upper eyelids (Figure 96.27). There may be multiple telangiectases. These changes produce an appearance of tiredness, debauchery or premature ageing.
Figure 96.27 Blepharochalasis.
Reduplication of the mucous membrane of the upper eyelid is associated with blepharochalasis in about 10% of cases, and this may make the eyelids appear thick.
Clinical variants
Ascher syndrome
Ascher syndrome is the association of blepharochalasis with progressive enlargement of the upper lip due to hypertrophy and inflammation of the labial salivary glands [1–3, 6, 7, 9–11, 12, 13, 14]. The lip feels soft and lobulated and there may be excessive salivation. In some cases, the accessory lacrimal glands are also affected, with increased thickness of the eyelids. Goitre (enlargement of the thyroid) has also been reported as part of the syndrome [6].
Differential diagnosis
- The many other causes of eyelid swelling must be excluded (see Chapter 109). Ptosis is easily distinguished because the skin appears normal. Blepharochalasis is occasionally a manifestation of generalized cutis laxa, and it may form part of Ascher syndrome (see later).
- Laxity of the eyelid skin is most commonly an age-related phenomenon (dermatochalasis) due to degenerative changes in the connective tissue of the eyelid. Laxity also occurs in Ehlers–Danlos syndrome but other features of this syndrome will also be present. Occasionally laxity, particularly affecting the upper eyelid, occurs in otherwise healthy individuals [8].
Actinic granuloma and annular elastolytic giant cell granuloma
Definition and nomenclature
Actinic granuloma is an uncommon condition affecting actinically damaged skin that results from a low-grade reactive inflammatory process in which degenerate elastic fibres are phagocytosed by multinucleate giant cells and histiocytes. It is the commonest type of annular elastolytic giant cell granuloma, in which abnormal elastic fibres are progressively destroyed by an expanding ring of elastolysis and granulomatous inflammation.
Introduction and general description
Annular elastolytic giant cell granuloma (AEGCG) is an uncommon granulomatous cutaneous reaction pattern in which damaged dermal elastic fibres are slowly eliminated by a process of phagocytosis by multinucleate giant cells and histiocytes (Figure 96.28) [1]. The commonest form, actinic granuloma, occurs in sun-exposed skin and manifests as one or more slowly enlarging annular plaques with an elevated erythematous margin, leaving behind a central area of atrophy devoid of elastic fibres (Figure 96.29) [2]. Actinic granuloma is associated with diabetes in up to 40% of cases: it has been postulated that hyperglycaemia may alter the immunogenicity of elastic fibres [1, 3]. There are other less common variants of AEGCG including an annular form of sarcoidosis which typically presents around the temples and forehead and was originally described as atypical necrobiosis lipoidica [4, 5]; AEGCG in sun-protected skin [6, 7, 8]; and AEGCG occurring in burn scars [9, 10]. It has also been associated with prolonged doxycycline photosensitivity [11], prolonged sunbed exposure [12] and with the onset and recurrence of acute myeloid leukaemia [13]. The common theme would appear to be damage to elastic fibres provoking a granulomatous inflammatory response.
Figure 96.28 Annular elastolytic giant cell granuloma: high-power view showing fragments of degenerate elastic fibres engulfed by multinucleate giant cells. (Courtesy of Professor Luis Requena, Universidad Autónoma de Madrid, Spain.)
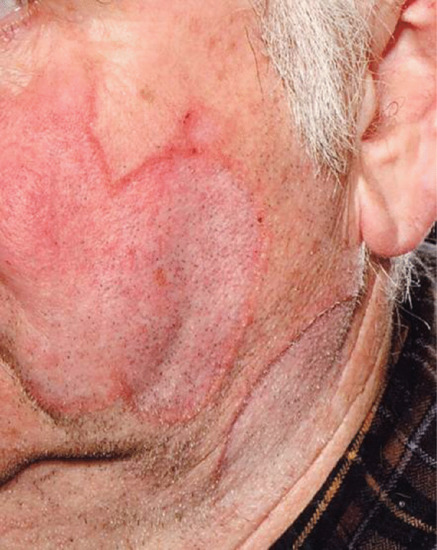
Figure 96.29 Typical actinic granulomas on the face and neck of an elderly man.
Epidemiology
Incidence and prevalence
The condition is more common in sunny countries.
Age
Usually over the age of 30.
Ethnicity
Fitzpatrick skin type I are particularly susceptible.
Associated diseases
Diabetes.
Pathophysiology
Pathology
The histological appearances are characteristic [1, 2, 14, 15, 16, 17, 18]. A biopsy taken radially across the thickened edge of the lesion and stained with elastic van Gieson stain shows three distinct zones in the dermis. In the external ‘normal’ skin, there is actinic elastosis. In the thickened annulus, there is a histiocytic and giant cell inflammatory reaction in relation to elastotic fibres (Figure 96.30a,b), and in the centre, within the annulus, little or no elastic tissue remains. The cellular infiltrate slowly expands outwards, leaving behind a central area from which elastic fibres have been removed by ‘elastoclasis’.
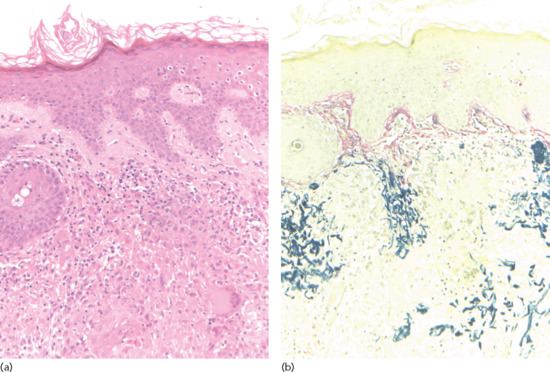
Figure 96.30 (a,b) Annular elastolytic giant cell granuloma: low-power view showing intense granulomatous inflammation (a) and elastorrhexis with loss of elastic fibres (b). (Courtesy of Dr Leigh Biddlestone, Royal United Hospitals, Bath, UK.)
The epidermis may be normal or it may show signs of actinic damage.
Environmental factors
Chronic photodamage.
Clinical features
History
Lesions are typically asymptomatic.
Presentation
Lesions may be single or multiple. They normally develop in sun-exposed skin such as the dorsa of the hands and forearms, the vee of the neck or the bald scalp (Figure 96.31). Fair-skinned or freckled subjects are particularly susceptible. The lesions start insidiously as small pink papules, which slowly extend centrifugally to form a ring of firm superficial dermal thickening which is smooth and slightly elevated (see Figure 96.29). The ring initially measures a few millimetres across but gradually expands, often attaining a diameter of several centimetres. The centre may become slightly atrophic and variable depigmentation may occur. The lesions are usually asymptomatic but a sunburn reaction may provoke severe erythema and irritation. Hair growth is not affected (see Figure 96.31).
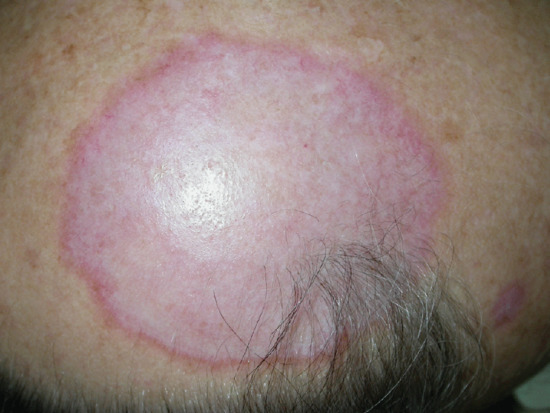
Figure 96.31 Actinic granuloma on a bald scalp. (Courtesy of Professor Luis Requena, Universidad Autónoma de Madrid, Spain.)
Clinical variants
See introduction: some cases of AEGCG may represent an annular form of cutaneous sarcoidosis [19].
Granuloma multiforme is a condition which many would regard as a variant of AECGC (see later).
Differential diagnosis
- Granuloma annulare
- Necrobiosis lipoidica
- Elastosis perforans serpiginosa
- Sarcoidosis
Complications and co-morbidities
Diabetes (see earlier).
Disease course and prognosis
The condition can improve with adequate sun protection.
Investigations
Skin biopsy.
Management
No treatment is of proven benefit. Topical steroids are generally unhelpful.
Anecdotal reports of successful treatment include intralesional triamcinolone, and oral hydroxychloroquine, isotretinoin (0.5 mg/kg/day) [20] acitretin 25 mg/day [21], dapsone, methotrexate and ciclosporin and topical tacrolimus [22].
Granuloma multiforme
Introduction and general description
Granuloma multiforme is a dermatosis reported in dark-skinned people mainly from Africa and India [1, 2, 3–8]. It shares many similarities with AEGCG in that it is characterized by annular plaques with giant cell granuloma formation at the periphery and loss of elastic tissue centrally [1]. As with AEGCG, it presents with papules which enlarge to form annular plaques with raised edges which may attain many centimetres in diameter. Histologically, the condition is difficult to distinguish from AEGCG except that focal necrobiosis and dermal mucin may be seen, which is not the case in AEGCG. Many authorities believe that granuloma multiforme should be regarded as a form of AEGCG [1].
Its importance lies in its superficial resemblance to tuberculoid leprosy, which is an important differential diagnosis. Leiker et al. [2, 3, 4] first described granuloma multiforme and distinguished it from tuberculoid leprosy. Leiker called it Mkar disease, after the town where it was first studied. The condition is endemic in certain villages in eastern Nigeria, where the local inhabitants refer to it in the Ibo tongue as ‘Ununo Enyi’ (elephant ringworm) [5, 8]. The disease appears to occur predominantly in females over the age of 40 years [5, 8, 9]. Intense sun exposure over many years in people able to withstand acute photodamage appears to be a common feature. The difference in skin type and other unknown factors may explain the rather minor histopathological differences from AEGCG as seen in fair-skinned individuals.
Clinical features
Presentation
The upper, uncovered parts of the body are predominantly affected. The initial lesions are small flesh-coloured papules which become aggregated into plaques or form the elevated rims of annular lesions. In larger annular lesions, the central area is often hypopigmented. Pruritus may be prominent. The condition lasts for many months or years, and may persist indefinitely.
Differential diagnosis
Leprosy is endemic in the same regions where granuloma multiforme is found, and can look very similar. However, there is no loss of sensation or sweating, or other evidence of neural involvement in granuloma multiforme.
Management
No treatment is known to be effective.
Other elastolytic conditions
Granulomatous slack skin is characterized by the slow development of pendulous folds of lax erythematous skin, which on histological examination contain a dense granulomatous dermal infiltrate, with destruction of dermal elastic tissue. It is now considered to be a type of cutaneous T-cell lymphoma (mycosis fungoides) (see Chapter 140) [1, 2].
Acquired pseudoxanthoma elasticum-like syndromes
Perforating pseudoxanthoma elasticum
Transepithelial elimination (TEE) of altered elastic fibres can occasionally occur in generalized hereditary forms of PXE (see Chapter 72), but it can also occur as a localized acquired defect in patients who do not have the other features of PXE [1]. These localized lesions usually occur in the periumbilical area in obese, multiparous black or Asian women, and it is possible that this represents a response to repeated cutaneous stretching (e.g. ascites or previous abdominal surgery) [2, 4]. Similar lesions on the breast have been reported in patients undergoing haemodialysis [2, 5].
Clinically, asymptomatic yellow macules and papules coalesce into well-demarcated hyperpigmented plaques which slowly enlarge. The surface may be atrophic, grooved, fissured or verrucous, and compression of the edge of the lesion may produce a liquid discharge.
It seems likely that most cases previously described as elastosis perforans serpiginosa in association with PXE were really examples of perforating PXE [6]. The histology of the two conditions is similar, but in perforating PXE there is transepidermal elimination of altered basophilic, calcified, elastic fibres [7], which are short, fragmented, curled and predominantly in the mid-dermis, whereas in elastosis perforans serpiginosa the fibres are abnormally large, non-calcified, eosinophilic and straight. The condition is similar to, or identical with, papillary dermal elastolysis (see earlier) [8].
Spontaneous resolution has been reported [9].
Acquired pseudoxanthoma elasticum
Iatrogenic
Skin changes which are virtually identical to those of PXE can rarely be produced by penicillamine (e.g. in the treatment of Wilson disease), although the systemic features do not occur [1, 2]. The skin changes can be explained by the known effect of penicillamine in inhibiting collagen and elastin cross-linking, with the production of vastly increased amounts of abnormal elastin in the dermis [3]. Transepidermal extrusion of elastin has been reported in this condition [4].
Toxic
Lesions clinically resembling PXE are reported in the eosinophilia–myalgia syndrome; dermal calcification is absent on histology [5]. Eosinophilia myalgia syndrome is defined by: (i) incapacitating myalgias; (ii) a blood eosinophil count greater than 1000 cells/μL; and (iii) no evidence of infection (e.g. trichinosis) or neoplastic conditions that could account for these findings, and related to toxic oil syndrome, caused by ingestion of contaminated l-tryptophan or other less well-characterized toxic substances (see Saltpetre disease later).
Depositions
Yellowish papules and plaques resembling PXE are seen in some patients with amyloidosis; amyloid deposits are seen and, again, dermal calcification is absent [6, 7].
Haematological disease
PXE–like lesions are described in several haemoglobinopathies, including congenital anaemia, sickle cell disease and thalassaemia [8]. These patients may develop systemic manifestations such as peripheral vascular occlusive disease and retinal neovascularization and haemorrhage [9].
Saltpetre disease [1, 2, 3]
A condition which resembles the skin changes of PXE clinically, histologically and ultrastructurally has been described in a group of elderly farmers, who decades earlier had spread a fertilizer containing calcium-ammonium nitrate (Norwegian saltpetre). The patients developed cutaneous ulcers at sites of exposure (including antecubital fossae). These quickly healed to leave yellowish-white papules and plaques. None of the patients had a positive family history or other signs of PXE.
Acrokeratoelastoidosis
Acrokeratoelastoidosis is an uncommon asymptomatic disorder which manifests as multiple tiny crateriform keratotic papules along the margins of the hands and feet, particularly in people of African descent (Figure 96.32a–c). The name derives from the histological appearances which include not only epidermal acanthosis and hyperkeratosis but also fragmentation of underlying dermal elastic fibres [1]. The mechanisms involved are not understood.
It is inherited in an autosomal dominant fashion but does not usually present until after puberty; sporadic cases also occur [2]. As elastorrhexis cannot always be demonstrated, alternative names for clinically indistinguishable cases have been proposed: these include focal acral hyperkeratosis and marginal papular acrokeratoderma [3, 4]. There is still controversy as to whether these should be regarded as separate entities.
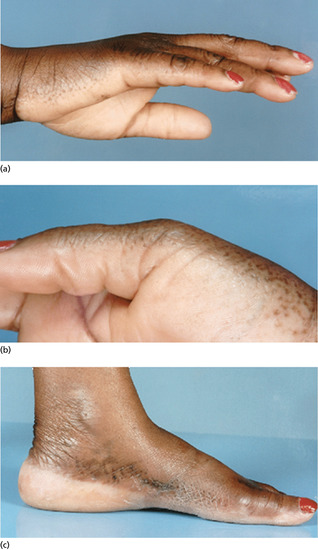
Figure 96.32 (a–c) Acrokeratoelastoidosis.
ACQUIRED DISORDERS OF ELASTIC TISSUE DEPOSITION
Linear focal elastosis
Definition and nomenclature
This condition is characterized by asymptomatic yellow linear bands arranged horizontally on the lower back [1, 2, 3, 4]. It less commonly occurs on the legs and shoulders.
Introduction and general description
This condition may represent a keloidal reaction to striae distensae.
Epidemiology
Incidence and prevalence
Case reports are mostly sporadic. It may be commoner than reports suggest.
Age
Initially reported in elderly males [1], although subsequently reported patients are chiefly adolescents [5, 6, 7].
Sex
Predominantly male.
Ethnicity
White people, Asiatic and Afro-Caribbean [5].
Pathophysiology
Predisposing factors
The lesions may be associated with a growth spurt, similar to adolescent growth striae [6].
Pathology
Ultrastructural studies reveal active elastogenesis. The middle and lower dermal collagen is separated by bluish grey fine fibrillar material, which is composed of thin wavy elastic fibres and fragmented elastic fibre bundles. Early lesions may, in contrast, show elastolysis, with decreased elastin and microfibrillar proteins [8]. The elastic fibres are near to or even in contact with fibroblasts [9]. Elastogenesis may occur in response to local trauma, UV light or perhaps following the development of striae distensae [10]. However, these mechanisms do not adequately explain the increasing number of cases reported, particularly in the young, and it may be that intrinsic defects of elastic fibre metabolism play a role [4].
Causative organisms
None known.
Genetics
There may be an underlying defect of elastic tissue, as yet unidentified [4]. Familial cases are reported [5, 11].
Clinical features
History
Insidious onset of asymptomatic lesions chiefly on the lower back.
Presentation
Superficially, the lesions resemble striae distensae, but they are palpable rather than depressed and yellow rather than purplish or white. Linear focal elastosis has been reported adjacent to striae distensae [10, 12] and in one case following potent topical corticosteroids [13].
Differential diagnosis
Striae (which often coexist).
Classification of severity
Of cosmetic importance only.
Complications and co-morbidities
Associated with striae in many cases.
Disease course and prognosis
Unknown. Lesions probably regress with time.
Investigations
None needed.
Late-onset focal dermal elastosis
Yellowish papules with a peau d'orange appearance appear on the flexures. Clinically and histologically, the lesions resemble elastomas, but this rare condition has only been reported in elderly Japanese men [1, 2].
Elastofibroma dorsi [1, 2–8]
Elastofibroma occurs predominantly in elderly women. Most cases are reported from Southern Japan. There may be a history of prolonged manual labour. The painless or slightly tender swelling beneath the lower angle of the scapula, from 2 to 10 cm in diameter, is often discovered fortuitously. It may enlarge slowly, displacing neighbouring structures, and it can be clinically confused with a sarcoma. This is a benign lesion, however, despite the fact that it is poorly circumscribed. The growth is composed of mature fibrous tissue, containing fibres which stain as elastic fibres. The lesions may be solitary or multiple.
Histologically, the lesion contains abundant large elastic fibres, some broken into irregular masses, and large amounts of relatively acellular collagen. The elastic f]ibres are composed of true elastin surrounded by a large amount of hydrophilic material forming an orderly array of tubules [4]. It is generally regarded either as a type of reactive hyperplasia, or as a hamartoma, arising either from dermis, subscapular connective tissue or periosteum [6]. It is cured by simple excision [7], although there is a high complication rate after surgery; postoperative suction is recommended [8] and there is a risk of recurrence [8, 9].
Elastoderma
Elastoderma is a very rare condition which is due to excessive elastogenesis, as distinct from acquired cutis laxa, where there is a loss of elastic tissue. A young woman developed a localized defect of the skin of one arm, which became pendulous and lax, but lost its elastic recoil. Histological and biochemical investigation showed this was due to accumulation of excessive elastin, with derangement of elastin fibrillogenesis [1].
In further cases, also affecting young women, clinically uninvolved skin showed thin elastic fibres on haematoxylin and eosin staining, without calcification [2]. Transmission electron microscopy showed irregular elastic tissue fibres with electron dense extensions; fibroblasts were abundant, possessing widened rough endoplasmic reticulum [3]. Despite the skin laxity, there is no joint hypermobility [3].
Papular elastorrhexis
This is a rare variant of connective tissue naevus. Adolescents or young adults present with multiple non-follicular oval white or yellowish papules on the trunk or limbs; dermal elastic fibres are decreased and fragmented on histology. Most case reports are sporadic, with no family history and no extracutaneous manifestations [1–3]. The differential diagnosis includes acne scars, in which the papules are follicular, with decreased elastin but no elastorrhexis; familial cutaneous collagenoma is histologically similar, but cases of papular elastorrhexis are sporadic [4]. Similar lesions are seen in some patients with Buschke–Ollendorff syndrome (see Chapter 15), in which osteopoikilosis is also a feature. To add to the confusion, abortive forms of Buschke–Ollendorff syndrome have been described, lacking osteopoikilosis [5]. A family has been described with this variant [6]. It is possible that papular elastorrhexis is not a separate entity [7]. Intralesional triamcinolone may be beneficial [8], if treatment is necessary.
FIBROMATOSES AND OTHER CAUSES OF DIFFUSE FIBROSIS
Introduction and general description
Fibrous overgrowth of dermal and subcutaneous connective tissue occurs most readily in certain sites and at certain ages, and some of the resulting syndromes are clinically and histologically distinctive and well defined. There are some cases, however, that defy precise classification, and others in which histological criteria may be a poor guide to prognosis. Invasiveness and a high local recurrence rate may or may not be associated with a tendency to metastasize. The borderline between simple overgrowth and a benign tumour may be equally difficult to define.
Abnormal fibrosis is a feature of many debilitating systemic disorders, such as cirrhosis and pulmonary fibrosis. A closer understanding of the myofibroblast and the regulatory pathways of cytokines and growth factors, such as TGF-β, should enable the development of effective and specific antifibrotic drugs [1].
Fibromatoses
Fibromatosis is a benign fibrous tissue proliferation, which is intermediate between benign fibroma and metastasizing fibrosarcoma. The lesions of fibromatosis tend to infiltrate and recur when removed, but they do not metastasize. The term should not be applied to reactive fibrous proliferation, or to keloid, which is usually secondary to injury. The lesions in fibromatosis may be single or multiple, and the likelihood of recurrence after surgical removal varies with the location of the lesion and the age of the patient. The fibromatoses occur in two major groups:
- Superficial fibromatoses (fascial fibromatoses):
- palmar (Dupuytren);
- plantar;
- penile (Peyronie);
- knuckle pads.
- Deep fibromatoses (non-metastasizing fibrosarcoma). These are rapidly growing tumours that usually involve the musculature or aponeuroses. Their tendon-like consistency accounts for their alternative name of desmoid tumours. They are discussed in more detail in Chapter 137.
Fibromatosis, palmar fascial
Definition and nomenclature
This is a fibromatous hyperplasia of the palmar aponeurosis, which is characterized by nodular thickening of the fascia with associated flexion contractures of one or more digits (Figure 96.33a,b).
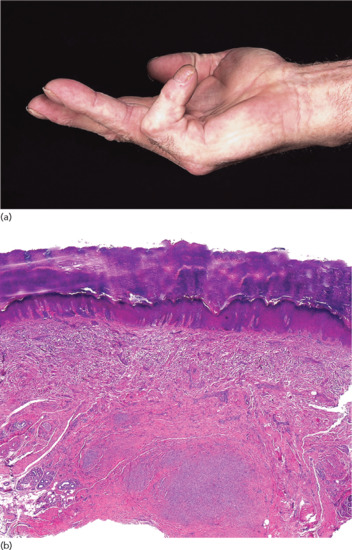
Figure 96.33 (a,b) Palmar fascial fibromatosis: clinical image illustrating (a) typical fixed contraction of the little finger, and (b) low-power photomicrograph showing grossly thickened deep fascia with nodules of proliferating fibroblasts surrounded by dense collagen. (Part b courtesy of Professor Luis Requena, Universidad Autónoma de Madrid, Spain.)
Introduction and general description
The condition seems to be due to a reactive proliferation of fibroblasts with no inflammatory component; the basic cause is obscure.
Epidemiology
Incidence and prevalence
The prevalence in the general adult population is around 2–6% [1], but it may approach 20% or more in elderly males [2, 3, 4], in diabetic patients and in patients with AIDS.
Age
Age of onset is generally 30–60 years.
Sex
It is generally commoner in men. Some families are described in which there is a predominantly female expression [5].
Ethnicity
Relatively uncommon in people of African or Asian descent.
Associated diseases
In about 5% of patients, the condition is associated with other fibromatoses such as knuckle pads or keloids. This has been termed the polyfibromatosis syndrome.
It occurs more commonly in patients with alcoholic cirrhosis, epilepsy [6] and diabetes [4, 7], but the prevalence is decreased in rheumatoid arthritis [8]. Other conditions which have been less convincingly claimed to be associated include periarthritis of the shoulder, chronic lung disease, gout, trauma and ulnar nerve damage [9].
Phenytoin appears to stimulate fibrosis in the polyfibromatosis syndrome [10] and it may also cause gingival hypertrophy by stimulating fibroblasts and increasing collagen production [6, 10, 11]. There is one case report of a girl aged 14 years who developed Dupuytren contracture while receiving growth hormone therapy for hypopituitarism [12].
High alcohol consumption, smoking and trauma, notably the use of vibrating hand tools, have also been implicated [13].
Pathophysiology
Free radical production secondary to ischaemia may be involved: the concentration of hypoxanthine substrate capable of releasing free radicals is greatly increased in the affected tissue [14]. Localized ischaemia has been thought to play a part, and in animal studies allopurinol (a competitive inhibitor of xanthine oxidase) has been shown to limit the damage associated with acute ischaemia [15]. High concentrations of free radicals are toxic, but in low concentration they stimulate fibroblast proliferation [14]. The contractures, which are a late complication, appear to follow the conversion of the fibroblasts to contractile myofibroblasts [16].
The presence of CD3 lymphocytes and the expression of major histocompatibility complex (MHC) class II proteins in the affected tissue imply that palmar fascial fibromatosis is a T-cell mediated autoimmune disorder [17].
Pathology
Fibroblasts in affected fascia appear to be identical to those in normal palmar fascia but their density is increased and they tend to be clustered around narrowed small vessels [18, 19]. In the early stages, there are nodules in the subcutaneous tissue or within the fascia: they are composed of proliferating fibroblasts with irregular hyperchromatic nuclei but there is no excess of collagen. Later stages are characterized by the presence of myofibroblasts, which have a fibrillary cytoplasmic ultrastructure and seem to have some other properties of smooth muscle. The nuclei are deeply indented, possibly due to the contractile properties of the cell. The cells also have altered surface membrane properties which enable attachment to neighbouring cells and stroma. Myofibroblasts have also been identified in the normal aorta and in granulation tissue, hypertrophic scars, keloids, liver fibrosis, dermatofibromata, etc. [16], in which their contractile properties may be important. The advanced stages of palmar fascial fibromatosis are characterized by dense fibrous connective tissue with a few elongated cells. An increased concentration of type III collagen is present in the nodules [20]. This may be due to decreased degradation resulting from increased levels of tissue metalloproteinase inhibitors in the lesions [21]. Structural abnormalities of glycosaminoglycans, notably dermatan sulphate, may predispose to abnormal fibrillogenesis [22].
Genetics
Palmar fascial fibromatosis is often familial, and may be inherited as an autosomal dominant trait [23], in which case the onset tends to occur at an earlier age [24]. Genome-wide studies show increased expression levels of metalloproteinases 1, 3 and 16, fibroblast growth factor and several collagen genes [25].
Environmental factors
Occupational exposure to hand-transmitted vibration may be an exacerbating factor [13].
Clinical features
History
Nodules may be painful initially, although the condition typically develops insidiously over several months or years.
Presentation
The earliest sign is the development of a palmar nodule, usually in the ulnar half of the hand. There are usually no symptoms, but there may be a dull ache or tingling. Insidious progression of the fibrosis over several years causes flexion contractures of the affected fingers. There is often puckering of the overlying skin.
Clinical variants
Plantar and penile fibromatosis are closely related conditions (see later).
Differential diagnosis
In most cases, the diagnosis is straightforward. There may be a histological resemblance to fibrosarcoma, but the latter is more pleomorphic, with larger nuclei and more mitoses. Juvenile aponeurotic fibroma may produce palmar or plantar nodules, but palmar fascial fibromatosis does not occur in young children.
Disease course and prognosis
The condition tends to progress more slowly in women [9]. Eventually, the function of the hand is impaired due to fixed flexion of one or more digits. If left untreated, there may be some improvement after many years.
Investigations
The possibility of one or more of the associated disorders, e.g. diabetes, should be considered and investigated if appropriate.
Management
The advice of an orthopaedic or hand surgeon should be sought. Traditionally complete removal of the palmar aponeurosis has been recommended [26], although minimally invasive subtotal fasciectomy and direct closure is more generally favoured [27, 28].
Initial encouraging placebo-contolled trials of collagenase injections [29] have been supported by subsequent experience, and the technique is now more widely practised [30].
Medical treatments are disappointing. Allopurinol may help by decreasing free radical production [31], and it has been suggested that vitamin C might prevent progression of the disease by acting as a free-radical scavenger [2].
Many other non-surgical approaches have been tried, including continuous slow skeletal traction, radiotherapy, dimethyl sulfoxide, vitamin E, steroid injections and interferon, although none has been proven to be clinically useful [32].
High-dose tamoxifen following minimally invasive surgery reduces the risk of recurrent fibrosis in the short term, but the effect is lost on discontinuing the drug [33]. Intriguing results have been reported from the use of relaxin gene therapy on Dupuytren myofibroblasts in vitro, with the potential for use in vivo [34].
Fibromatosis, plantar fascial [1, 2]
Definition and nomenclature
This is a rarer condition than palmar fascial fibromatosis, although often associated; a survey from Reykjavik found that 15% of men with the latter had plantar fibromatosis [3]. It most commonly affects the medial half of the mid-foot, presenting as one or more nodules which may become painful and may ulcerate (Figure 96.34). In 25% of cases it is bilateral. The fibromatosis rarely results in contractures but tends to be locally invasive and to recur.
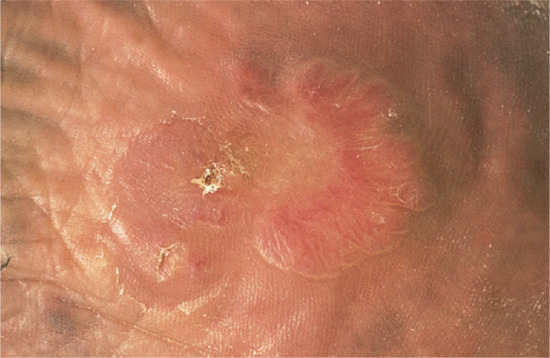
Figure 96.34 Plantar fibromatosis.
Clinical features
Differential diagnosis
The differential diagnosis includes keloid and fibrosarcoma. Magnetic resonance imaging may confusingly demonstrate the cerebriform pattern typically seen in fibromyxoid sarcoma [4].
In younger patients, aggressive infantile fibromatosis and aponeurotic fibroma must also be considered [5]. Complications are rare, although squamous carcinoma has been reported occurring within a lesion of plantar fibromatosis [6]. Similar nodules have been described symmetrically affecting the anteromedial aspects of the heel pad in children. They are asymptomatic and may resolve spontaneously [7, 8]: surgery is contraindicated.
Management
Conservative management may be best in the early stages [9]. High-energy shockwave therapy reduces pain [10]. Total excision of the lesion and the entire plantar fascia seems to give the best results, with the lowest incidence of recurrence.
Fibromatosis, penile
Definition and nomenclature
Penile fibromatosis is characterized by one or more irregular dense fibrous plaques in the penile shaft. These commonly result in painful erections and curvature of the erect penis.
Pathophysiology
Penile fibromatosis may occur as an isolated abnormality, or as one component of polyfibromatosis in association with palmoplantar fibromatosis, keloids and knuckle pads. Atheroma predisposes to the condition, and it is now thought that the reported association with the use of α-adrenoreceptor blocking drugs was probably attributable to concomitant atheroma [1, 2]. There may be a genetic factor, but reliable studies of the mode of inheritance are lacking. The condition is rare below the age of 20 years, and the highest incidence is between 40 and 60 years. It is much less common than palmar or plantar fibromatosis.
Histopathology [3]
The thickened plaque shows cellular fibroblastic proliferation surrounded by dense masses of collagen. Calcification and ossification may occur. The process appears to begin as a vasculitis in the areolar connective tissue beneath the tunica albuginea, whence it extends to adjacent structures.
Presentation
The disease presents with painful erections and curvature of the erect penis due to a thickened subcutaneous plaque, rubbery or hard, usually on the dorsal aspect of the penis in its distal third (Figure 96.35a,b). The erectile deformity may make vaginal penetration impossible, and pain or anxiety about performance may cause secondary impotence. Fibrosis of the underlying cavernous erectile tissue may lead to a constriction or ‘waisting’ of the penile shaft, leading to flaccidity of the distal portion.

Figure 96.35 (a) Normal erect penis. (b) Erect penis with deformation from Peyronie disease showing a fibrous plaque causing ‘waisting’.
The course is unpredictable [4]. The pain generally subsides within a few months, but the fibrous plaque may resolve, remain unchanged or progress [5].
Investigations
The severity of the disease and the response to treatment can now be evaluated by high-resolution ultrasonography [6], computed tomography [7] or magnetic resonance imaging of the erect penis [8]. If necessary, an erection can be induced by the intracavernosal injection of papaverine [9].
Management
Many treatments have been tried, but there is little evidence that vitamin E, potassium aminobenzoate, orgotein, radiotherapy, ultrasonic therapy or intralesional steroids affect the long-term outcome, although they may relieve the pain [4, 10]. There are case reports of success with more aggressive treatment using pulsed dexamethasone and low-dose cyclophosphamide [10]. Clostridial collagenase injections have given promising results, as in Dupuytren contracture [11–13]. A recent study suggests that iontophoresis with verapamil and dexamethasone (‘electromotive drug administration’) may reduce the curvature and is well tolerated [14]. Penile traction has its devotees [15].
Surgery is probably the treatment of choice: a bewildering variety of procedures have been employed, including Nesbit's procedure, in which ellipses of normal tunica albuginea are excised from the side of the shaft, opposite the point of maximum curvature. Alternatives include plaque incision and grafting [16] and venous grafting, using the deep dorsal vein [17] A semirigid penile prosthesis may also be inserted.
Knuckle pads
Definition and nomenclature
Knuckle pads are circumscribed thickenings overlying the finger joints. The term is a misnomer as most lesions occur over the proximal interphalangeal rather than the metacarpo-phalangeal joints (knuckles).
Introduction and general description
Knuckle pads were first described in the medical literature by Garrod [1] as an ‘unusual form of nodule upon the joints of the fingers’. However, they are probably not so unusual, and they feature in several of Michelangelo's works, including the statues of David and the Sleeping Slave [1]. They should be distinguished from the ‘pseudo-knuckle pads’ associated with trauma.
Epidemiology
Incidence and prevalence
The condition is not rare but the true prevalence is uncertain, as most patients ignore the lesions. Mikkelson reported a prevalence of 8.8% in the population of Haugesund, Norway, and of 44.3% in individuals with palmar fibromatosis [2].
Age
Onset is usually between 15 and 30 years of age; however, lesions typically develop slowly and asymmetrically and may not present significant cosmetic problems for several years.
Sex
Probably equal.
Ethnicity
Mostly reported in white people.
Associated diseases
There is a strong association with other fibromatoses such as palmar fibromatosis [2–4]. An association between Dupuytren contracture and other fibromatous lesions has been recorded in some families. In one large family, knuckle pads were associated with sensorineural deafness and with leukonychia (Bart–Pumphrey syndrome) [5]. Knuckle pads have also been associated with epidermolytic palmoplantar keratoderma in a Chinese family due to keratin 9 mutations [6].
Another family has been described with knuckle pads in association with oesophageal cancer, hyperkeratosis and oral leukoplakia [7].
Pathophysiology
Predisposing factors
Although trauma has been implicated, this is more closely linked to ‘pseudo knuckle pads’. Familial cases are reported.
Pathology
The epidermis is grossly hyperkeratotic and acanthotic, with elongated rete ridges. The dermal connective tissue is hyperplastic; a proliferative phase is followed by a fibrotic phase. individual collagen fibres may be obviously thickened and arranged in irregular bundles. Spindle-shaped myofibroblasts can be seen on electron microscopy [4, 8, 9]. Histologically, the changes resemble those of palmar fibromatosis.
Genetics
The condition is usually sporadic but several pedigrees have shown an autosomal dominant inheritance, e.g. the Bart–Pumphrey syndrome [5]. The age of onset and the distribution of the lesions tend to be more or less constant in each family, but show interfamily variation. Similar single nucleotide polymorphisms (SNPs) are found in familial Dupuytren contracture [10]. Knuckle pads are reported in families with palmoplantar keratodermas linked with keratin 9 mutations [6, 11]. A family has been reported with familial knuckle pads but no associated conditions [4].
Environmental factors
Trauma is more probably relevant in ‘pseudo knuckle pads’.
Clinical features
History
Usually asymptomatic, with insidious onset.
Presentation
Flat or convex, smooth, circumscribed nodules develop slowly and almost imperceptibly over the course of months or years, achieving 0.5–1.5 cm diameter. The lesions may be hypo- or hyperpigmented. In some patients, they become very much raised and obviously indurated, but in others the dermal component is not clinically apparent. They are most commonly seen over the dorsa of the proximal interphalangeal joints (Figure 96.36), but occasionally develop over the knuckles or the distal interphalangeal joints. Any single site or combination of sites may be involved [1, 4, 8].
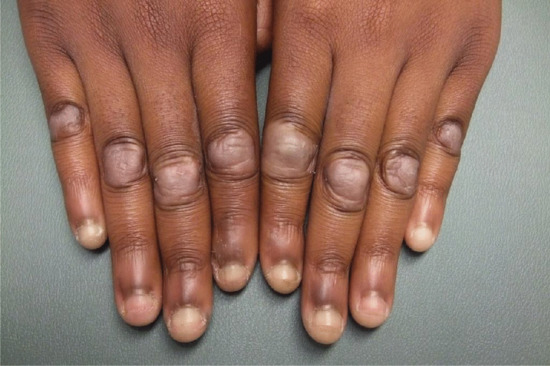
Figure 96.36 Knuckle pads. (From Hyman and Cohen [4], with permission from Dermatology Online Journal.)
Clinical variants
Sites other than the hands are not often affected, but similar lesions on the knees were also present in one family [3].
Differential diagnosis
Knuckle pads should be distinguished from the ‘pseudo knuckle pads’ associated with occupational trauma, such as in carpet layers, sheep shearers and tailors. Similar lesions are induced by children chewing or biting their fingers or ‘knuckle cracking’ [12] or playing video games [13]. Unlike true knuckle pads, these lesions tend to regress if the traumatic stimulus is removed, and may respond to topical keratolytics.
Heberden nodes of osteoarthritis, pachydermodactyly, granuloma annulare [14], erythema elevatum diutinum and rheumatoid nodules [15] should be excluded.
Classification of severity
A cosmetic embarrassment.
Complications and co-morbidities
Association with other fibromatoses (as noted earlier).
Disease course and prognosis
Lesions gradually enlarge to a maximal size and tend to persist.
Investigations
None usually necessary, unless another inflammatory condition needs to be excluded by skin biopsy.
Management
If the lesions do not bother the patient, conservative management is appropriate. Recurrence and keloidal scarring are common following excision. Intralesional triamcinolone or cryotherapy are ineffective and often painful. Intralesional 5-fluorouracil inhibits fibroblast proliferation and shows promise clinically [16].
Pachydermodactyly
Definition
This is a benign fibromatosis of the fingers (Figure 96.37).
Figure 96.37 Pachydermodactyly. (Courtesy of Dr A. Chamberlain, Churchill Hospital, Oxford, UK.)
Introduction and general description
This rare condition typically presents in adolescent males, who develop spade-like enlargement of the hands and occasionally the feet [1–9].
Epidemiology
Incidence and prevalence
Rare; incidence uncertain.
Age
Young adults.
Sex
Males are chiefly affected although it has been reported in women [4, 5] and two young girls, one of whom had tuberous sclerosis and the other Ehlers–Danlos syndrome [6].
Associated diseases
It may be associated with bilateral carpal tunnel syndrome [2] and varioliform atrophy (p. 96.12) [10].
Pathophysiology
Predisposing factors
It has been suggested that unconscious repeated rubbing and gripping of the fingers, repetitive movements or mechanical injury to the joints may contribute to the condition [3, 6, 11, 12], but pachydermodactyly must be distinguished from occupational callosities and obsessive ‘chewing pads’.
Pathology
Histology shows epidermal hyperplasia and marked dermal thickening, with extension of collagenous fibres into the subcutaneous tissue. Types III and V collagen are increased, and electron microscopy shows increased numbers of fine-diameter collagen fibres.
Genetics
Affected families have been reported [13].
Environmental factors
Possible local repetitive trauma.
Clinical features
History
Usually asymptomatic and insidious in onset. A few individuals describe pain of the long bones.
Presentation
It produces a symmetrical diffuse swelling of the skin around the dorsal and lateral aspects of the proximal interphalangeal joints of the index, ring and middle fingers.
Clinical variants
A distal variant has been described in an elderly woman, who also presented with nodules over the extensor aspects of the elbows [14].
Differential diagnosis
Patients may be referred to the rheumatologist with a diagnosis of juvenile idiopathic arthritis [12, 15]. It may be confused with knuckle pads [16, 17], which may coexist [18].
Classification of severity
The condition is benign.
Complications and co-morbidities
Knuckle pads and pachydermodactyly coexisted in one family [18].
Disease course and prognosis
The condition tends to persist.
Investigations
Patients with the condition can be spared the detailed investigations of a patient with suspected inflammatory arthritis [19].
Management
Most patients do not require treatment. Intralesional triamcinolone has been reported to be beneficial [20], although this is unlikely to be necessary.
White fibrous papulosis of the neck
Asymptomatic small white fibrous papules around the neck have been described in several Japanese [1, 2], Iranian and European patients [3, 4]. The number of papules ranges from 10 to 100; middle-aged to elderly men are predominantly affected. The papules are round to oval, clearly marginated and non-follicular (Figure 96.38). Histology is unremarkable, showing bundles of thickened collagen fibres in the mid-papillary dermis. Although lesions clinically resemble disorders of elastic tissue, such as anetoderma and Buschke–Ollendorff syndrome, elastic fibres are morphologically normal on histology. Acquired connective tissue naevi could exhibit similar features, although the late age of onset makes this diagnosis unlikely. The condition appears to have no prognostic significance, and may be underreported. It may reflect intrinsic ageing or photoageing [5].
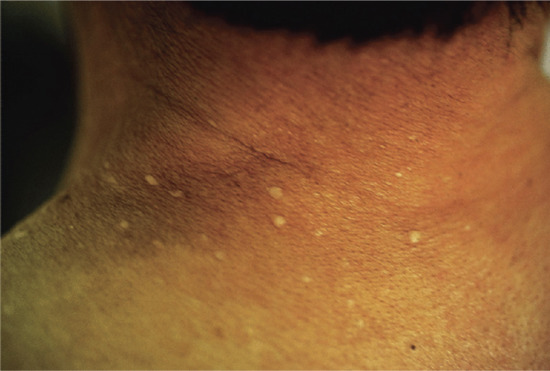
Figure 96.38 White fibrous papulosis of the neck. (Courtesy of Professor H. Shimizu, Sapporo Hospital, Tokyo, Japan.)
It has been suggested that there may be a relationship between fibrous papulosis of the neck and acquired elastolysis of the papillary dermis [6, 7], indeed lesions of PXE-like papillary dermal elastolysis coexisted in a patient with white fibrous papulosis, suggesting that they are part of the same disease spectrum [8]. These changes are attributed to ageing or photoageing [5, 9].
Camptodactyly
Definition
Camptodactyly is a non-traumatic flexion deformity affecting the proximal interphalangeal joint of one or more fingers (Figure 96.39) [1].
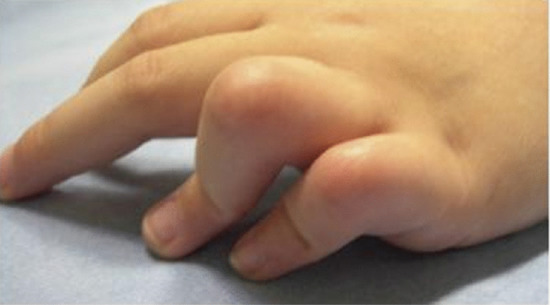
Figure 96.39 Camptodactyly in the ring and little fingers. (From Almeida SF, Monteiro AV, Lanes RCdS. Evaluation of treatment for camptodactyly: retrospective analysis on 40 fingers. Rev. Bras. Ortop. 2014;49:134–9, with permission.)
Introduction and general description
Camptodactyly should be distinguished from clinodactyly, which refers to bending or curvature of the finger in the plane of the hand. Camptodactyly is associated with numerous inherited disorders, the most important of which are described below. It is often depicted in Renaissance art [2].
Epidemiology
Incidence and prevalence
The prevalence of isolated camptodactyly is unclear. The most commonly associated syndrome is microdeletion of 1p36, which affects 1 : 5000 neonates [3].
Age
Most cases are congenital, although some familial cases present in adult life, associated with an erosive arthritis [4].
Sex
Probably equal sex incidence.
Ethnicity
All races may be affected.
Associated diseases
Camptodactyly may be a feature of a variety of syndromes of which several have had molecular defects identified. Clinical features of microdeletion of 1p36 include camptodactyly, facial dysmorphism and low-set ears, cardiac and central nervous system defects; it is responsible for around 1% of idiopathic mental retardation [3].
Congenital camptodactyly is most notably associated with non-inflammatory arthropathy [5]. Additionally, other serous membranes undergo fibrosis leading to constricting pericarditis, coxa vara and pleuritic (camptodactyly, arthropathy, coxa vara and pericarditis =CAP or CACP syndrome) [6–9]. Gordon syndrome encompasses camptodactyly, cleft palate and club foot [10]. The association of camptodactyly, tall stature and hearing loss has been termed the CATSHL syndrome [11]. Familial camptodactyly of later onset has been described in association with an inflammatory arthritis with erosive changes [4]. Blau syndrome encompasses familial camptodactyly, granulomatous arthritis, uveitis and an erythematous eruption with phenotypic overlap with early-onset sarcoidosis [12]. In one family, taurinuria was associated [13]. Bilateral camptodactyly is also part of an autosomal recessive disorder (Crisponi syndrome) characterized by muscular contractions of the face, trismus, facial anomalies and death due to fevers. The syndrome is caused by CRLF1 mutations and is allelic to cold-induced sweating syndrome type I [14].
Sporadic cases of camptodactyly have been linked with accelerated growth and osseous maturation, unusual facial appearance (including large ears, small mouth, broad forehead and hypertelorism), a hoarse, low-pitched cry and hypertonia (Weaver syndrome) [15]. Other associated features include pectus excavatum and scoliosis.
Pathophysiology
Predisposing factors
Several genetic disorders (see earlier).
Pathology
Histology of skin lesions may be unremarkable.
Genetics
Many cases are familial. Microdeletion of 1p36 is the most commonly associated abnormality. Mutations in NOD2/CARD15 have been shown to confer susceptibility to several chronic inflammatory disorders, including Crohn disease, Blau syndrome and early-onset sarcoidosis [16]. CAP (or CACP) syndrome is autosomal recessive, and related to the 1.9 cM interval in the human chromosome 1q25-31; this gene encodes for proteoglycan 4, a surface lubricant for tendons and joints [8]. CATSHL syndrome is associated with mutation in FGR3 [11], and Crisponi syndrome with a CRLF1 mutation [14].
Environmental factors
None apparent.
Clinical features
The deformity is asymptomatic.
Presentation
In most cases, the affected child will present with clinical features of a related syndrome.
Clinical variants
Streblodactyly [17, 18] (streblos = crooked) is inherited as a sex-linked autosomal dominant character. The affected females show from birth a flexion deformity at the metacarpo-phalangeal joints of the thumbs and the proximal interphalangeal joints of the little fingers. Some fingers show swan-neck deformities and hyperextensible metacarpo-phalangeal joints. In one family, there was an abnormal amino aciduria.
Differential diagnosis
Dupuytren disease (palmar fibromatosis) is associated with fibrous scarring affecting the fascia. Juvenile chronic arthritis is typically erosive, whereas the arthritis in CACP syndrome and related disorders is non-erosive [9].
Classification of severity
Morbidity relates to the associated syndrome, if any.
Complications and co-morbidities
Relates to any associated syndrome.
Disease course and prognosis
Lesions are persistent.
Management
Treatment, if required, is surgical [1, 19, 20]; however, results can be unsatisfactory [21]. Techniques include tendon transfer [21] and a flap with vascular reconstruction [22].
Juvenile fibromatoses
The term juvenile fibromatosis has been applied to a group of disorders occurring in infants and children, and characterized by proliferative activity of the fibroblasts [1–6]. There is a tendency to local recurrence but, unlike fibrosarcomas, they do not metastasize. The group includes a number of well-defined clinical entities that affect the skin as follows:
- Infantile myofibromatosis.
- Fibrous hamartoma of infancy.
- Juvenile hyaline fibromatosis.
- Infantile digital fibromatosis.
- Calcifying aponeurotic fibroma.
- Giant cell fibroblastoma.
Myofibromatosis, infantile
Definition
Solitary or multiple fibrous nodules developing in infancy in the skin, striated muscle, bone and occasionally viscera [1, 2].
Introduction and general description
Although rare, this is the commonest cause of fibrous nodules presenting in infancy. In around 50% of patients, lesions are solitary, predominantly affecting the head and neck. Multiple lesions can occur, with or without visceral involvement.
Epidemiology
Incidence and prevalence
Rare; incidence unknown.
Age
Around 60% of lesions are present at birth, and around 80% by the age of 2 years.
Sex
There is a slight male predominance, around 60% of patients are boys [2].
Ethnicity
May affect all races.
Pathophysiology
Predisposing factors
None known.
Pathology
Histology of a lesion shows characteristic zoning, with peripheral spindle-shaped cells in bundles surrounding a central zone of less poorly differentiated round and polygonal cells. Staining is positive for vimentin and α-smooth muscle elastin, negative for desmin and S-100 [1].
Genetics
Familial cases exhibit autosomal dominant inheritance. Mutations have been described in PDGFRB [3, 4] and NOTCH3 [5] genes.
Environmental factors
None known.
Clinical features
History
Lesions are typically asymptomatic.
Presentation
Solitary or multiple nodules, mostly on the head and neck, more rarely the arms. Lesions can occur anywhere on the body. Multiple lesions may be associated with visceral involvement. Lesions may ulcerate.
Clinical variants
Solitary lesions on the upper eyelid may cause amblyopia [6].
Differential diagnosis
The solitary lesions of fibrous hamartoma of infancy usually affect the hand or foot, histology is that of an organoid naevus containing mature adipose cells with a nodular aggregate of fibroblasts and interlacing collagen bands. Juvenile aponeurotic fibromatosis affects the fingers and palms of older children or adults; clinically, it may resemble Dupuytren disease (which is very rare in infants), but histology reveals large dark-staining nuclei in a background of bland fibrosis, with calcification. Bony lesions may be difficult to distinguish from fibrosarcoma.
Classification of severity
A benign process but the presence of systemic involvement considerably worsens the prognosis, with up to 30% mortality [2].
Complications and co-morbidities
Dependent on systemic involvement.
Disease course and prognosis
Many solitary and even multiple cutaneous lesions involute spontaneously [7, 8]. Systemic involvement carries a worse prognosis.
Investigations
Histology is essential to differentiate from other tumours and fibromatoses. Full clinical examination, chest and abdominal imaging are advisable in patients with multiple lesions.
Management
First line
Patients without systemic involvement can be managed conservatively. Debulking surgery, without attempting complete removal, may be necessary if the tumour compromises function.
Second line
Systemic disease warrants chemotherapy, e.g. with low-dose vinblastine and methotrexate [9].
Juvenile hyaline fibromatosis
Definition and nomenclature
This is a disorder of glycosaminoglycan synthesis, which is characterized clinically by skin papules or tumours, gingival enlargement, osteolytic lesions and joint contractures, and histologically by deposition of amorphous hyaline material [1–4].
Introduction and general description
This, together with systemic hyalinosis, is now regarded as part of the hyaline fibromatosis syndrome (see Chapter 72).
Epidemiology
Incidence and prevalence
The disease is very rare and occurs sporadically, but it has occurred in siblings [6].
Age
Onset in infancy.
Sex
Slight male predominance.
Ethnicity
Most case reports and series originate from the Indian subcontinent.
Pathophysiology
Predisposing factors
The cause is unknown, but increased chondroitin synthesis has been demonstrated in skin fibroblasts cultured from the tumour tissue [1].
Pathology [1–6]
The skin lesions contain ‘chondroid’ cells embedded in amorphous eosinophilic ground substance in the dermis. In the early lesions, this consists of glycosaminoglycans, but in the later lesions the matrix is mainly composed of chondroitin sulphate [7]. The dermal collagen is decreased and the collagen fibrils are fewer and thinner than in normal skin. The hyaline material may also be present in the muscles and bones. Absence of pro-α2 chains and type III collagen has been demonstrated in affected skin [8].
Genetics
Inheritance is autosomal recessive. The gene has been mapped to 4q21; there are also mutations in the capillary morphogenesis factor 2 gene [9]. Infantile systemic hyalinosis has been associated with mutations of the ANTXR2 gene [10, 11].
Clinical features
History
The condition presents at birth or early infancy.
Presentation
There may be small pearly papules or nodules, particularly on the face or neck. Large subcutaneous tumours may also occur, particularly on the scalp. These may be hard or soft, fixed or mobile, and they may ulcerate. Gingival hypertrophy is commonly present, and flexion contractures of the fingers, elbows, hips and knees may develop. Osteolytic lesions can occur in the skull, long bones or phalanges. The musculature is poorly developed [1, 9, 12–15].
Clinical variants
Infantile systemic hyalinosis is probably an extreme variant, often leading to death in infancy.
Differential diagnosis
Other infiltrative disorders, such as lipoid proteinosis may need to be excluded histologically.
Classification of severity
The condition is a severe disease, with considerable morbidity and reduced life expectancy.
Complications and co-morbidities
Joint contractures are disabling.
Disease course and prognosis
The condition persists into adult life. However, many patients die in infancy and rarely survive beyond the fourth decade [9].
Investigations
Histology is diagnostically helpful.
Management
No treatment is of proven benefit. Surgery may be the treatment of choice [5], although nodules may recur after excision [16]. The tumours do not respond to radiotherapy. Joint contractures may respond to intralesional steroid injections in the early stages and patients may benefit from systemic steroids and physiotherapy.
Other benign fibrous cutaneous nodules
Nodular fasciitis
In this condition, there is fibroblastic proliferation of one or more nodules, usually on the limbs or trunk.
Collagenoma
Collagenoma (collagen naevus) is a form of connective tissue hamartoma (see Chapter 75) which may manifest as a single or localized group of fibrous dermal papules or plaques: the shagreen patch of tuberous sclerosis is an example (see Chapter 80). Multiple fibrous dermal nodules with coarse collagen fibres may develop as sporadic cases (eruptive collagenoma) or as a genetic disorder with a dominant inheritance (familial cutaneous collagenoma).
Albopapuloid form of epidermolysis bullosa
This rare form of epidermolysis bullosa is characterized by the development of ivory-white papules on the trunk, which histologically show connective tissue hyperplasia. Epidermolysis bullosa is discussed in Chapter 71.
Buschke-Ollendorf syndrome
Extensive nodular fibrosis may occur in the Buschke–Ollendorff syndrome (see Chapter 75), in association with juvenile elastoma and osteopoikilosis.
Fibrous digital nodules
In addition to giant cell synovioma and infantile digital fibromatosis, fibrous nodules in the digits may be due to acquired digital fibrokeratoma, fibrous papule of the finger, dermatofibroma (see Chapter 137) or the Koenen tumour (see Chapter 95).
Nephrogenic systemic fibrosis
Definition and nomenclature
A rare fibrosing disorder which occurs in patients with renal impairment exposed to low-stability gadolinium-based contrast agents [1, 2].
Introduction and general description
The condition was first described in 1997 as nephrogenic fibrosing dermopathy [1]. Initially thought to be restricted to the skin, there are several reports of involvement of internal organs including lungs, myocardium and striated muscle, which contribute to a high mortality [3].
Epidemiology
Incidence and prevalence
Rare. With the development of guidelines on the use of gadolinium-based contrast agents [4], it is hoped that the condition will become a matter of historical importance only.
Age
Mostly in elderly adults but several cases have been reported in children [5].
Sex
Equal sex incidence.
Ethnicity
All races may be affected.
Associated diseases
Associated with renal impairment.
Pathophysiology
Predisposing factors
The condition is strongly associated with the prior administration of gadolinium-based magnetic resonance contrast agents, particularly in patients with severe renal disease, typically with a glomerular filtration rate below 30 mL/min/1.73 m2 or on dialysis [6]. Gadolinium chelates stimulate an NLRP3 inflammasome-dependent inflammatory response, leading to fibroblast growth, synthesis and differentiation into myofibroblasts [7, 8]. Non-ionic linear gadolinium-based contrast agents, particularly gadodiamide, are strongly implicated. Macrocyclic chelating agents, such as gadoterate, are more stable and considerably less likely to induce the syndrome [9]. Additional risk factors include an associated vascular repair (e.g. leaking aortic aneurysm), associated thrombosis or procoagulant state, and concurrent administration of intravenous iron [2]. High-dose erythropoietin is also implicated in some cases; it has a pro-inflammatory action, particularly in the presence of increased iron stores [10].
Pathology
Dermal mucin is detected with Alcian blue staining. Increased collagen and elastic fibres are laid down in haphazard bundles in the dermis and subcutis; there are increased numbers of CD68-positive fibroblasts in loose aggregates. Inflammatory changes may predominate, including a septal panniculitis [1, 11].
Causative organisms
None proven.
Genetics
None.
Environmental factors
Gadolinium-based contrast agents.
Clinical features
History
A history should be obtained of exposure to gadolinium chelates, although onset may be delayed by several years [7]. Patients may complain of myalgia.
Presentation
Irregular erythematous or brownish indurated plaques, with amoeba-like projections and islands of sparing, occur chiefly on the lower trunk and legs (Figure 96.40). Typically the face is spared. Sometimes the skin has a ‘peau d'orange’ texture, which can mimic carcinoma erysipeloides [12].
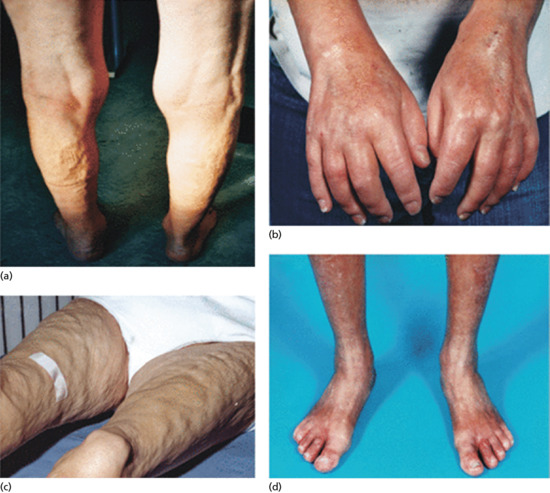
Figure 96.40 Nephrogenic systemic fibrosis: Deep involvement where fibrosis pulls down a linear band of skin on the thighs (a); tightness and hardness of the hands and feet and joint contractures b and d); and firm nodules producing a cobblestone appearance (c). (From Elmholdt TR, Pedersen M, Jørgensen B, et al. Nephrogenic systemic fibrosis is found only among gadolinium-exposed patients with renal insufficiency: a case–control study from Denmark. Br J Dermatol 2011;165:828–36, with permission from Wiley.)
Clinical variants
The extent of visceral involvement is variable; in some cases the process may involve the testes, myocardium and dura.
Differential diagnosis
Although initially described as ‘scleromyxoedema-like’, the lesions have a different distribution and morphology, and there is no associated paraproteinaemia [3].
Classification of severity
The condition can be severe, occasionally fatal.
Complications and co-morbidities
Fibrotic obstruction of structures, e.g. superior vena cava obstruction [13] may be a complicating factor. Associated metabolic abnormalities include hypophosphataemia [14].
Disease course and prognosis
Usually, the condition is progressive, although it may remit spontaneously, particularly with the correction of renal abnormalities.
Investigations
A careful history, together with skin or muscle biopsy, can usually confirm the diagnosis. Investigations such as magnetic resonance imaging may be needed to determine the extent of macroscopic visceral involvement.
Management
The most important aspect of management is to maximize renal function. No other treatment is of proven benefit, but thalidomide [15], hydroxychloroquine [16], corticosteroids, immune modulators (e.g. etanercept and rituximab), PUVA, intravenous sodium thiosulphate and extracorporeal photopheresis [17] have been used empirically. Alefacept appears to improve the skin disease [18]. Therapeutic plasma exchange offers pain relief [19]. Renal transplantation is sometimes associated with remission [2].
Prevention should be achieved by adherence to guidelines for the use of gadolinium chelates in radiology [4].
Diabetic thick skin
Some patients with diabetes have thick tight waxy skin and limited joint mobility which is thought to be related to altered collagen. This topic is discussed in Chapter 64.
Environmental and drug-induced scleroderma
A variety of environmental triggers, including drugs and occupational toxins, may stimulate a localized or diffuse scleroderma-like reaction in a genetically susceptible host. Important causes are listed in Box 96.2. Scleroderma-like lesions are seen in a photosensitive distribution in porphyria cutanea tarda. Lesions resembling generalized morphoea are seen in chronic graft-versus-host disease and paraneoplastic scleroderma is associated with neoplasms such as carcinoid. In most cases, the fibrotic process continues after withdrawal of the external stimulus. Sometimes, the ensuing clinical pattern resembles idiopathic forms of morphoea or systemic sclerosis (see Chapters 57 and 56 respectively).
Several occupational disorders resembling systemic sclerosis have been reported. In a Belgian study, men in construction-related occupations (notably electricians) were 10 times more likely to have systemic sclerosis than the general population [1]. Exposure to vinyl chloride monomer occurs in workers involved in polyvinyl chloride (PVC) production. One-third of male operatives in a British factory developed a clinical syndrome that included Raynaud phenomenon, dyspnoea, cutaneous sclerosis, pulp atrophy and radiological evidence of acro-osteolysis (Figure 96.41) [2]. Genetic marker studies have demonstrated an increased incidence of human leukocyte antigen (HLA)-DR5 in affected individuals; severe disease is linked with -B8 and -DR3 [3]. A similar syndrome has been reported in gold miners exposed to silica dust [4], which is the most commonly reported occupational association in the literature [5]. Organic solvents, such as trichlorethylene [6] and perchlorethylene [7], which are structurally similar to vinyl chloride, have also been implicated. Exposure to epoxy resin results in an acute syndrome of cutaneous sclerosis, muscle weakness, arthralgia, impotence, lung and oesophageal involvement [8]. The causative agent appears to be a cyclohexylamine. Acrylamide has also been implicated [9].
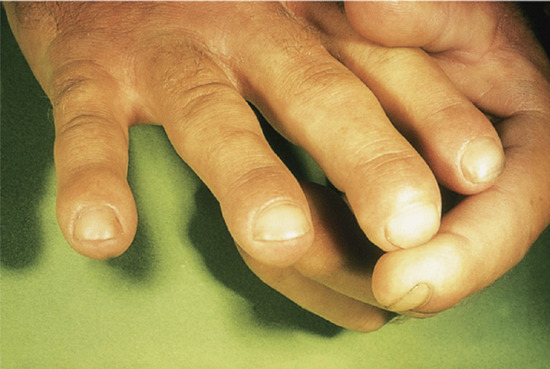
Figure 96.41 Vinyl chloride-induced osteolysis affecting fingertips.
Toxic oil syndrome is a multisystem illness, reported in Spain in 1981. Acute fever, severe but transient pulmonary oedema, myalgia and a pruritic exanthem and eosinophilia were followed after several months by widespread cutaneous sclerosis in 30% of cases [10, 11]. The syndrome was probably due to ingestion of imported rapeseed oil mixed with an aniline denaturant, designed to make the oil unfit for human consumption. Toxic oil syndrome bears a striking resemblance to the eosinophilia–myalgia syndrome [12–14], linked with consumption of L-tryptophan; this was used as a ‘food supplement’ to treat insomnia and depression. The offending batches of L-tryptophan contained impurities similar to the contaminants in toxic oil [15, 16].
In environmental fibrotic disorders, as in idiopathic scleroderma, subpopulations of fibroblasts appear to be activated to synthesize excess collagen; this property is perpetuated by fibroblasts in vitro, indicating that the elevated collagen gene expression is independent of extracellular stimuli [14]. Cytokines appear to stimulate the proliferation of these abnormal clones of fibroblasts; thus, TGF-β and platelet-derived growth factor (PDGF) are elevated in the eosinophilia–myalgia syndrome [17].
Numerous drugs have been reported to induce cutaneous sclerosis. Lesions resembling morphoea may follow injections of pentazocine [18], heparin [19] and vitamin K1 (phytomenadione) [20–23]; in the case of vitamin K1, the trigger may be a solvent rather than vitamin K1 itself [24]. Morphoea-like plaques have also been reported in patients taking penicillamine [25], valproate [26] and etanercept, even in areas remote from the injection site [27]. The case is not proven that silicone breast implants are associated with scleroderma-like disease [28].
Diffuse scleroderma-like changes have been reported following bleomycin therapy [29, 30]. A combination of L-5-hydroxytryptophan and carbidopa induced lesions resembling eosinophilia–myalgia syndrome [31]. Phenytoin and diltiazem both induce gingival hypertrophy [32, 33]. A patient on phenytoin developed florid hypertrophic retroauricular folds [34]. Thickened skin on the feet has been reported in a patient taking diltiazem [35].
Alcohol can provoke porphyria cutanea tarda, which can produce a sclerodermatous appearance in a photosensitive distribution (Figure 96.42).
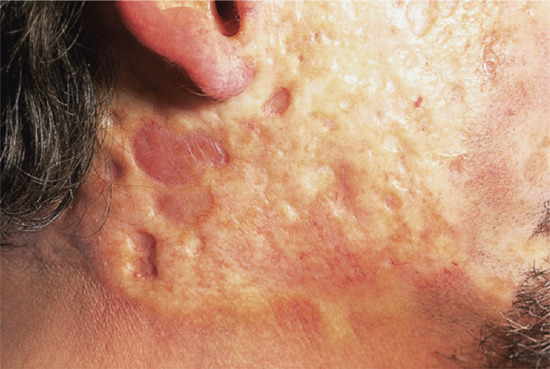
Figure 96.42 Scleroderma and scarring of the face due to porphyria cutanea tarda.
Constricting bands of the extremities
Definition and nomenclature
Constricting bands occur around a digit or limb. The bands may be shallow, involving only the skin, or deeper, involving fascia or bone, and in some cases amputation may result. The term ainhum (an African word meaning ‘to saw’ [1]) is applied to a specific type in which a painful constriction of the fifth toe occurs in adults, with eventual spontaneous amputation. Pseudo-ainhum is the term applied to other constricting bands which are congenital or secondary to another disease.
Introduction and general description
Constricting bands characteristically present in infants. Patterson [2] has provided a classification of congenital constrictions. Type I describes simple fibrotic rings; the limb distal to the ring is normal. In Type II, there is neurovascular or lymphatic disruption distal to the ring, causing atrophy, lymphoedema and maybe sensory deficit. Type III refers to acrosyndactyly (fenestrated syndactyly), where there is distal fusion of digits which are separated proximally, forming a ‘window’. In type IV, there is amputation of the digit or limb (ainhum).
Epidemiology
Incidence and prevalence
Sporadic cases of constricting bands occur rarely worldwide.
Age
Constricting bands typically present in infants and young children (Figure 96.43). Adults in resource-poor countries present with ainhum aged around 30–50 years, although the onset of the condition is probably in childhood [3–5].
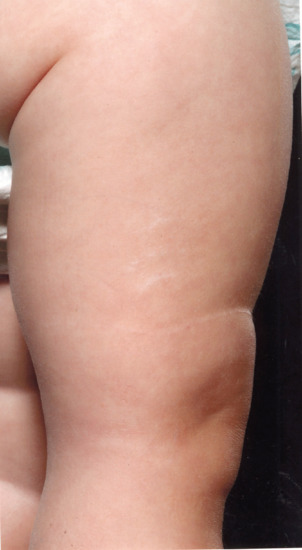
Figure 96.43 Constricting band across the thigh of a 6-month-old infant.
Sex
Both sexes are affected.
Ethnicity
Ainhum has been reported chiefly in black Africans and African Americans.
Associated diseases
Constricting bands are often associated with other congenital abnormalities [6].
Pathophysiology
Extrinsic and intrinsic factors are probably equally important. Disruption of the development of the germinal disc in the embryo may predispose to fibrotic bands and associated congenital abnormalities. Rupture of the amnion may result in loss of amniotic fluid and extrusion of all or part of the fetus into the chorionic cavity, with resultant trapping of limbs [6, 7]. In adults with ainhum, vascular damage appears to be important, resulting in hypoxia. In some patients, arteriography has shown that the posterior tibial artery is attenuated at the ankle, and the plantar arch and its branches are absent [3].
Predisposing factors
Vascular damage secondary to smoking or diabetes may exacerbate ainhum in adults [3].
Pathology
Fibrosis may be associated with distal degenerative change and osteoporosis, particularly in ainhum.
Causative organisms
Tropical infections have been implicated in ainhum, but are probably coincidental [3, 4].
Genetics
Most cases are sporadic, although familial cases of ainhum have been reported.
Environmental factors
Rupture of the amniotic membrane is likely to be an important factor in congenital constrictions. Mechanical factors, including trauma from walking barefoot, may precipitate the development of a groove in the ischaemic toe in ainhum.
Clinical features
Fibrous bands may be solitary or multiple, encasing the limb (usually the leg or foot).
Clinical variants
Ainhum represents the extreme form of the condition, resulting in spontaneous amputation of the digit.
Painful fissuring and hyperkeratosis on the medial aspect of the digit is followed by fibrosis, distal degeneration and osteoporosis. There may be secondary infection and osteomyelitis. The toe becomes dorsiflexed at the metatarso-phalangeal joint, and gradually becomes clawed. Rest pain, coolness and cyanosis of the digit distal to the groove suggest that ischaemia is present. Once the constricting band has encircled the toe, the condition tends to progress rapidly. The toe becomes globular, hangs by a thread of fibrous tissue and is eventually shed (Figure 96.44). Control of secondary infection and protection from trauma may prevent extension of the scarring process. If symptoms are severe, or the dangling digit is a disability, amputation is indicated.
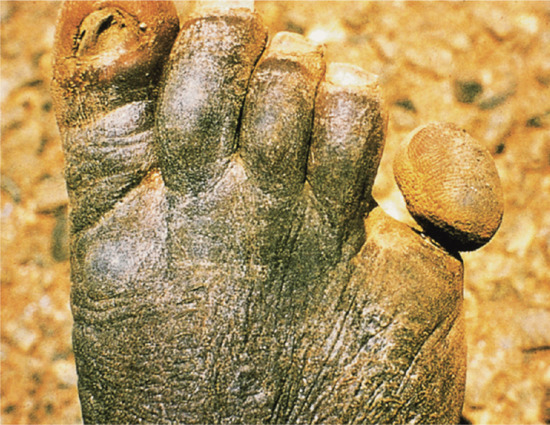
Figure 96.44 Ainhum, just before shedding of the fifth digit. (Courtesy of Dr D. Burley, Princess Margaret Hospital, Swindon, UK.)
Differential diagnosis
Pseudo-ainhum
Congenital. Congenital pseudo-ainhum may involve a digit, a limb or even the trunk, and it ranges in severity from a superficial groove to amputation in utero [6, 8–10]. The cause is unknown, but familial cases have been reported. Some cases of pseudo-ainhum may be due to amniotic bands [11] or adhesions in utero, which may arise as a result of tearing of the amnion some time after the 45th day of pregnancy [12]. Cases have occurred in Ehlers–Danlos syndrome and after amniocentesis [12, 13]. Several cases are reported where raised limb bands develop in the postnatal period, not always associated with amniotic tears; other possible causes include an early teratogenic insult [1, 14].
Histology of the affected digit or limb reveals broad, finger-like projections of collagen, and coarse elastic bundles that penetrate deep into the subcutaneous fat [10].
Congenital pseudo-ainhum must be distinguished from the following: aplasia of the limbs with rudimentary digits; acromelia (in which part of the limb does not develop); and hypoplasia (in which the parts, although formed, are poorly developed).
Acquired. Pseudo-ainhum may be acquired as a result of infection (particularly leprosy), trauma, cold injury, neuropathy (especially congenital sensory neuropathy), systemic sclerosis, etc. [15], chronic psoriasis [16] and it may occur in association with other hereditary diseases such as palmoplantar keratoderma, particularly Vohwinkel keratoderma (see Figure 96.57b) pachyonychia congenita, erythropoietic protoporphyria [17, 18] and Olmsted syndrome (see Chapter 65). Factitial pseudo-ainhum has also been reported due to the self-application of a rubber tourniquet.
Multiple skin creases resembling constrictions may be seen in the Michelin tyre baby syndrome and in ‘multiple benign annular creases of the extremity’.
Classification of severity
See Patterson severity grading earlier.
Complications and co-morbidities
Constricting bands may be associated with other congenital defects. In types II–IV, limb mutilation may be caused by fibrotic adhesions [6].
Ainhum results in spontaneous amputation of the digit.
Disease course and prognosis
Some children's constricting bands may involute spontaneously without functional deficit. Most will require surgery to prevent limb deformity or amputation.
Management
Surgical treatments include staged Z-plasty [19]. Good results have been obtained from two-stage sine plasty with removal of the fascial groove and fasciotomy, treating half the limb initially and the other half a week later [20].
ABNORMAL FIBROTIC RESPONSES TO SKIN INJURY
Keloids and hypertrophic scars
Introduction and general description
Keloids and hypertrophic scars represent an excessive connective tissue response to injury, which may be trivial. A keloid is a benign well-demarcated overgrowth of fibrotic tissue which extends beyond the original boundaries of a defect (Figure 96.45a). A hypertrophic scar is similar, but remains confined to the original defect and tends to resolve after several months (Figure 96.45b). Both conditions may represent different stages of the same disorder [1].
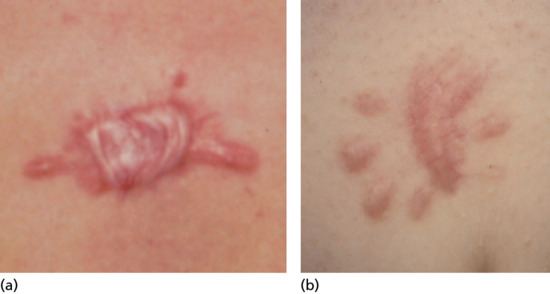
Figure 96.45 (a,b) Contrast between two scars from the presternal area: (a) spontaneous keloid, and (b) hypertrophic scar following excision of benign mole; the former shows partial involution after injection of triamcinolone.
Keloids and hypertrophic scars are cosmetically distressing (Figure 96.46), and often painful or pruritic. They appear to be unique to humans, and the lack of an animal model has hampered studies into their pathogenesis. A scar at any site has the potential to become keloidal or hypertrophic, although the earlobes (especially after ear piercing) (Figure 96.47), chin, neck, shoulders, upper trunk and lower legs are especially vulnerable. Burns, scars or tissue infection predispose to hypertrophic scarring. Lesions may follow trivial trauma or inflammatory conditions such as acne. Even chemical trauma, from irritant herbal remedies, can trigger keloid formation. Sometimes keloids appear to develop spontaneously, particularly on the upper chest.
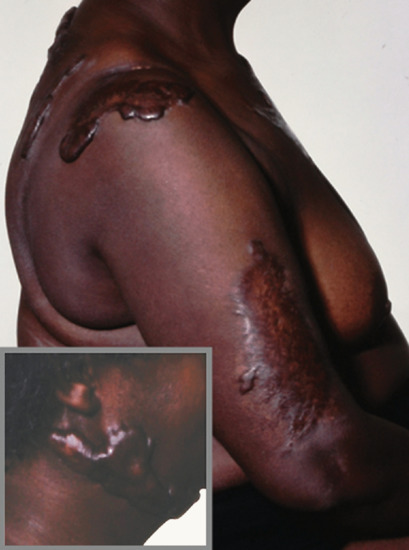
Figure 96.46 Extensive disfiguring keloids affecting an Afro-Caribbean woman.
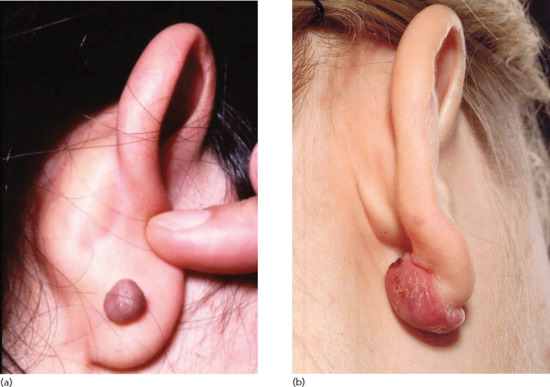
Figure 96.47 (a,b) Earlobe keloid.
Introduction of foreign material, either exogenous, such as suture material, or endogenous, such as embedded hairs, is another risk factor. Some African tribes introduce foreign bodies into tribal marks to induce scar hypertrophy. Scarring acne, particularly on the trunk, may become keloid-like (Figure 96.48).
Figure 96.48 Keloid nodules secondary to acne.
Isotretinoin has been reported to delay wound healing and induce keloids in patients who received argon laser or dermabrasion for acne or rosacea [2], although there is debate as to whether the association is real [3].
Epidemiology
Incidence and prevalence
Keloids or hypertrophic scars occur in 4.5–16.0% of people of African or Hispanic descent [4]. A survey of Taiwanese children reported a prevalence of keloids of 0.3–0.6% [5]. A positive family history is obtained in 5–10% of Europeans with keloids, particularly severe lesions. Family studies suggest an autosomal dominant inheritance with incomplete penetrance [6]. Keloids have been reported in identical twins [7].
Age
Keloids are rare in infancy and old age, occurring chiefly between puberty and age 30 years.
Sex
Women have a greater predisposition in some ethnic groups; keloids may appear or enlarge during pregnancy [8].
Ethnicity
Individuals of African, Hispanic or Asian descent are more prone to keloids (see Figure 96.46) [4].
Associated diseases
Keloids are associated with other fibromatoses such as palmar fibromatosis (Dupuytren contracture) [9], together with genetic disorders such as Ehlers–Danlos syndrome, pachydermoperiostosis [10], Rubinstein–Taybi syndrome [11] and Dubowitz syndrome [12].
Linear keloids occur in athletes abusing anabolic steroids [13]. They form readily in individuals with acromegaly and following thyroidectomy in young patients. Recently, it has been postulated that systemic hypertension may promote the development of keloids [14].
Pathophysiology
Biochemical studies confirm that synthesis of type I and type III collagen is increased in both keloids and hypertrophic scars [15]. Keloids differ from healthy skin in that there is greatly increased dermal cellularity, the ratio of type I to type III collagen is increased and there is greater dermal expression of several extracellular matrix proteins including fibronectin, versican, elastin and tenascin; conversely, there is decreased expression of fibrillin 1 and decorin [16, 17]. Hypertrophic scar collagen possesses the reducible keto cross-link, dehydrohydroxylysinonorleucine, normally associated with embryonic skin and granulation tissue [18]. Periostin, in particular, may play an important role in pathogenesis: it is expressed by keloid fibroblasts in hypoxic conditions and, among other actions, stimulates collagen synthesis [19]. Altered expression of proteoglycans may affect the three-dimensional organization of collagen fibres [20]. Keloid fibroblasts, unlike those from hypertrophic scar tissue, are hyperresponsive to TGF-β, which is abundant in healing wounds [21], and to PDGF [22]. They also express increased levels of heat shock protein (HSP) 47, another stimulus to collagen synthesis [23]. Neuropeptide-containing nerves are present [24] and the increased discomfort and itching which may be experienced in hypertrophic scars may be due to up-regulation of opioid receptors [25].
Pathology
Histology may resemble normal wound healing in the early stages, with increased cellularity. In a keloid of recent onset, endothelial proliferation is surrounded by increased numbers of fibroblasts, which form large, irregular nodules or whorls of hyalinized collagen. Later, the lesion matures into an acellular core, made up of thick, poorly vascularized bands of immature collagen [26] (Figure 96.49a,b). There may be focal deposition of mucinous material in keloids, but not in hypertrophic scars. Mast cell numbers are increased in hypertrophic scars [27]. The epidermis is normal or thinned by the underlying lesion in keloids, but may be thickened in hypertrophic scars [28]. The fibroblasts have a stellate morphology on transmission electron microscopy [28, 29]. Scanning electron microscopy reveals a more haphazard organization of collagen bundles than in normal skin or mature scars, with collagen fibrils about half the diameter of those of normal skin.
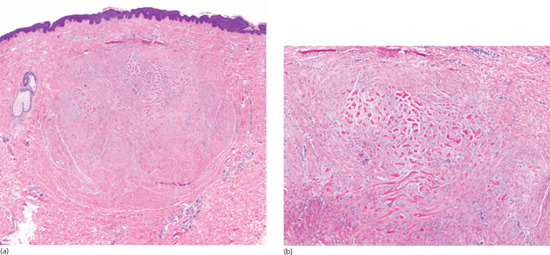
Figure 96.49 (a,b) Keloid nodule: large well-circumscribed dermal nodule sparing papillary dermis (a); higher-power view with haphazardly arranged thick sclerotic collagen surrounded by whorls of fibroblasts (b). (Courtesy of Professor Luis Requena, Universidad Autónoma de Madrid, Spain.)
Genetics
The genetic basis is unknown. Telomere shortening has been described in keloids, and attributed to oxidative stress [30].
Environmental factors
Local trauma, which may be trivial. Hypertrophic scars commonly follow deep burns. Tension on the scar, and the presence of foreign material are aggravating factors in keloids.
Clinical features
History
Hypertrophic scars and keloids typically become raised and thickened within 3–4 weeks of the provocative stimulus, although keloids may develop up to a year later. Lesions are often pruritic and hypersensitive, and sometimes exquisitely tender. They may continue to grow for months or years.
Presentation
Depending on skin colour, lesions may present as firm skin-coloured, pink or red plaques (Figure 96.50); hypertrophic scars remain within the boundaries of the initial wound (see Figure 96.45b), whereas keloids become smoother and rounder and extend outside the wound boundary (Figure 96.45a), often assuming a ‘dumb-bell’ configuration but sometimes becoming bizarre and irregular (see Figure 96.46).
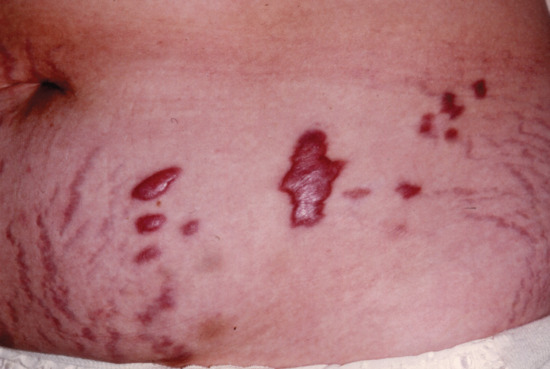
Figure 96.50 Fresh keloids arising in striae gravidarum 3 years after pregnancy.
Clinical variants
Keloids on the beard area sometimes undergo central suppurative necrosis.
Differential diagnosis
The diagnosis is usually straightforward if there is a history of trauma or an inflammatory skin lesion. Keloid scarring may follow surgical treatment of BCC, and a sclerotic BCC can mimic a keloid. Other differential diagnoses include fibrosarcoma, dermatofibrosarcoma protuberans, a malignancy developing in a scar or scar sarcoid. In endemic regions, blastomycosis and lobomycosis cause keloidal reactions.
Classification of severity
The Vancouver scar scale (VSS), recording pliability, height, vascularization and pigmentation, is used to quantify disease severity and response to treatment [31].
Complications and co-morbidities
Malignant degeneration is reported [32], although a fibrosarcoma can mimic a keloid clinically.
Disease course and prognosis
Hypertrophic scars typically regress spontaneously, although it may take a few years. Regression of keloids is a much slower process, and keloids can expand gradually over years.
Investigations
A diagnostic skin biopsy is mandatory if the diagnosis is in doubt.
Management
Non-essential surgery should be avoided in sites prone to keloids. Despite numerous small case series advocating a wide range of therapies, there is no level-one evidence for any single treatment [33]. Enthusiastic reports should be assessed critically as there may be racial variation in response to treatment and some studies include both keloids and hypertrophic scars. The follow-up period may be brief, and keloids have a high recurrence rate. Some modalities of treatment may exacerbate the condition. The optimal approach may involve a combination of different modalities of treatment.
First line
Intralesional corticosteroids (most commonly triamcinolone acetonide) inhibit fibroblast proliferation and collagen synthesis [34]. However, there is a risk of telangiectasia, atrophy and pigmentary change. Injections may need to be repeated monthly and recurrence rates can be up to 50% [35].
Second line
Self-adherent silicone gel sheeting may be effective for keloids and hypertrophic scars [36]; they may maintain skin hydration by occlusion but a meta-analysis of 13 trials showed only a weak preventative effect [37]. A cream made of 20% silicone oil applied under occlusion may be beneficial where it is impracticable to use silicone sheeting [38].
Third line
Other treatments include the following:
- Mechanical pressure with custom-made devices or garments can be beneficial, particularly on the earlobe [39] and bras or body corsets for truncal lesions.
- Surgical excision runs the risk of recurrence of an even bigger keloid. Intralesional (core) excision is preferable, and post-surgical intralesional steroids may prevent recurrence [40]. This topic is discussed in greater detail in Chapter 20
- 5-fluorouracil (5-FU), a pyrimidine analogue, inhibits keloid growth in vitro and in vivo. Two double-blind studies have demonstrated that intralesional 5-FU is more effective than silicone gel sheeting [41] and intralesional corticosteroids [42] in the treatment of keloids.
- A small case series has demonstrated benefit from bleomycin, using a multiple puncture injection method [43]. A recent Brazilian study reports favourable results from a combination of bleomycin (0.375 IU) and triamcinolone 4 mg injected 3-monthly [44]. Topical mitomycin C may reduce the risk of recurrence after shave biopsy[45]. Similarly, topical imiquimod may reduce the risk of post-surgical recurrence [46].
- Photodynamic therapy has a cytotoxic effect on keloid fibroblasts. A small case series demonstrates reduced blood flow, increased pliability and decreased collagen levels with no recurrence after 9 months [47].
- In a recent randomized study, intralesional verapamil (2.5 mg/mL) gave comparable results to triamcinolone, and was well tolerated [48].
- Laser and light-based treatments have been reviewed recently [49]. Ablative (e.g. carbon dioxide) laser monotherapy carries a high recurrence rate. Pulsed dye and Nd : YAG lasers appear to be more effective, particularly in combination with intralesional corticosteroids or 5-FU. Low-level red and IR light-emitting diodes (LEDs) suppress fibroblast synthesis and achieve cosmetic improvement. Other physical therapies include degenerate wave electrical stimulation [50].
- Inhibitors of pro-inflammatory cytokines such as TGF-β analogues [51] and IFN-α [52] show promise, perhaps in conjunction with intralesional triamcinolone. Recent reviews suggest that a combination of surgery with adjuvant radiotherapy [53] or intralesional 5-FU or corticosteroids [54] is preferable to monotherapy.
PERFORATING DERMATOSES
Definition
Skin disorders in which material is eliminated from the dermis by extrusion through the epidermis to the skin surface by a process of transepidermal (transepithelial) elimination. The primary perforating disorders are particularly associated with diabetes and chronic renal failure in the case of acquired perforating dermatosis and heritable disorders of connective tissue or Down syndrome in the case of elastosis perforans serpiginosum.
Introduction and general description
There has been considerable confusion over the terminology used to describe the perforating disorders, with an array of different terms used to denote what is now thought to represent essentially the same underlying process: biopsies taken at different sites or times from the same patient may show a variety of different patterns depending on whether the lesion involves a follicle and whether it has been modified by excoriation. Perforation is a histopathological construct signifying that material, usually degenerate collagen or elastin, has breached or perforated an epithelium, usually the epidermis, in which case the process is usually referred to as transepidermal elimination (TEE).
The term perforating folliculitis continues to be used in the published literature as a disease entity, though several authorities have recommended that it should be abandoned [1, 2]. The term acquired reactive perforating collagenosis fell out of favour some years ago to be replaced by acquired perforating dermatosis when it was demonstrated that both collagen and elastin were commonly involved [2]. Similarly, the distinction between acquired perforating dermatosis and Kyrle disease is not clear-cut [3] and separation of the two is no longer felt to be valid.
Many dermatoses occasionally exhibit the phenomenon of TEE, in which material from the dermis is extruded through the epidermis to the exterior with little or no disruption of the surrounding structures [4]. The extruded material may include inflammatory cells, red cells, microorganisms and extracellular substances, such as mucin or degenerate collagen and elastin [4–7]. In most of these conditions, the TEE is secondary to some underlying disease, such as granuloma annulare or PXE. There is also a rare hereditary disorder, familial reactive perforating collagenosis, which is characterized by TEE of collagen from an early age (see Chapter 72). The primary acquired perforating skin disorders which will be discussed here are therefore limited to acquired perforating dermatosis, which is strongly linked in the majority but not all cases to either diabetes, renal failure or both, and elastosis perforans serpiginosa, which is linked to heritable disorders of connective tissue and Down syndrome.
Acquired perforating dermatosis
Definition and nomenclature
An acquired disorder of transepidermal elimination of degenerate collagen, elastin and other connective tissue components. It is strongly associated with diabetes and chronic kidney disease.
Introduction and general description
Acquired perforating dermatosis is strongly linked with longstanding diabetes (Figure 96.51) and chronic kidney disease, often in association with haemodialysis [1, 2, 3, 4, 5, 6, 7, 8]. It is characterized by TEE of both collagen and elastin and presents as a chronic pruritic dermatosis with multiple keratotic crusted papules and nodules.
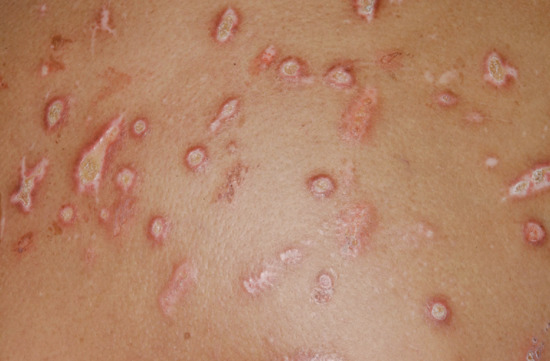
Figure 96.51 Acquired perforating dermatosis: close-up view of the back of a 65-year-old woman with longstanding diabetes.
Epidemiology
Incidence and prevalence
Relatively frequent in the commonly affected population with reported rates of 4–11% of patients on haemodialysis [7, 8].
Age
Generally occurs in the fifth to sixth decades of life.
Sex
Female to male ratio is 3 : 1 [9, 10].
Ethnicity
All races affected.
Associated diseases
Strong association with chronic kidney disease and diabetes. Case reports of association with dermatomyositis [11] and drugs such as natalizumab [12].
Pathophysiology
The bulk of the coarse granular basophilic material which is extruded by TEE appears to derive from the nuclei of polymorphonuclear leukocytes [13]. It has been suggested that lysosomal enzymes derived from leukocytes might be responsible for the altered staining of collagen fibres, the degradation of elastic fibres and the impairment of keratinocyte adhesion, which allows TEE of dermal components [3].
Most patients have chronic renal disease and/or longstanding diabetes.
Pathology
Histology reveals cup-shaped invagination of the epidermis, which is plugged with necrotic inflammatory debris. Collagen bundles are arranged vertically at the base of the lesion and there is transepidermal elimination of collagen fibres (Figure 96.52) [14].
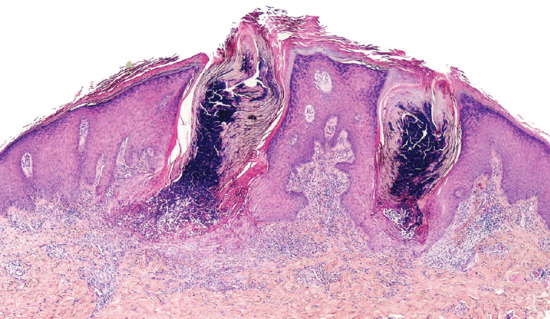
Figure 96.52 Acquired perforating dermatosis: invaginations of the epidermis enable columns of necrotic inflammatory debris to be extruded from the dermis. (Courtesy of Professor Luis Requena, Universidad Autónoma de Madrid, Spain.)
Causative organisms
Generally none, although there is one reported case associated with disseminated histoplasmosis [15].
Genetics
No known genetic factors.
Environmental factors
Clinical features
History
Pruritus, which may be intractable, is a common symptom.
Presentation
Keratotic dome-shaped papules with central crusts develop anywhere on the body but primarily on extensor aspects of the limbs and trunk (Figure 96.53). Dermoscopy with polarized light reveals bright white patches on a featureless grey background, surrounded by reticulate brown lines [1].
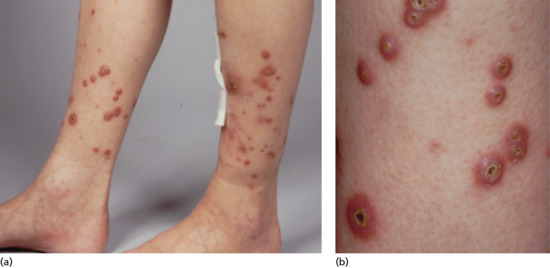
Figure 96.53 (a,b) Acquired perforating dermatosis: a 48-year-old woman with a 25-year history of type 1 diabetes with retinopathy and renal failure; and a 12-year history of skin ulceration with multiple tender crusted sores which were slow to heal.
Clinical variants
Familial reactive perforating collagenosis [2, 9, 13, 16, 17, 18] is a rare inherited form of TEE in which collagen is extruded through the epidermis. It is usually precipitated by environmental cold or trauma. The basic defect seems to be a type of focal damage to collagen, which is then extruded as a result of necrolysis of the overlying epidermis [19].
The lesion originates in the papillary dermis, where collagen is surrounded and engulfed by focal epidermal proliferation. The collagen appears normal on electron microscopy, but gives an abnormal staining pattern with trichrome and phosphotungstic acid haematoxylin. The central crater which develops contains inflammatory cells and keratinous debris. Elastic tissue is typically absent, and the abnormal collagen is eliminated by transepithelial migration [19, 20, 21].
It usually starts in early childhood as small papules on the extensor surface of the hands, the elbows and the knees following superficial trauma. Each skin-coloured papule increases to a size of about 6 mm over 3–5 weeks and then becomes umbilicated, with a keratinous plug [19]. The lesions regress spontaneously in 6–8 weeks to leave a hypopigmented area or slight scar, but new lesions may appear. Lesions can be produced experimentally, and the Koebner phenomenon may result in linear lesions [22]. The papules can also be provoked by inflamed acne lesions, but deep incisions do not produce the lesions. The condition persists into adult life. In some cases, the disease is associated with intolerance to cold and improves in warm weather.
The nosological relationship between familial reactive perforating collagenosis and the acquired reactive perforating dermatosis of chronic kidney disease remains uncertain [23].
Verrucous perforating collagenoma [24, 25, 26] (synonym collagénome perforant verruciforme). This rarely reported condition appears to be a reaction to the traumatic introduction of foreign materials including fibreglass, vegetable matter, calcium chloride and irritant drugs into the skin. Damaged collagen extruded to the surface by TEE, is manifest as verrucous papules.
Perforating disease due to exogenous agents. Occasionally, a chemical which has been applied to the skin topically or by intradermal injection can be eliminated by the transepidermal route. Eight cases have been reported following occupational exposure to a caustic drilling fluid used in the petrochemical industry [27]. Each patient noted skin irritation following exposure to the fluid and 1 or 2 days later developed tender papules with central umbilication followed by ulceration and crusting. Histological examination demonstrated TEE of altered collagen and debris which stained for calcium.
It is possible that the lesions were due to follicular penetration by the calcium present in the drilling mud. The drilling fluids contain many additives, but calcium carbonate or calcium chloride are often present in high concentrations in the mud. Similar cases have been reported following the use of calcium-containing electroencephalography paste [28].
TEE of altered collagen has also been reported following the use of intradermal steroid injections [29, 30].
Differential diagnosis
The condition may be mistaken clinically for molluscum contagiosum, papular urticaria or other perforating disorders, but the histology is characteristic [14, 18].
Management
Some patients improve spontaneously, particularly if renal function can be improved [7, 10]. No treatment is of proven benefit, although there are several reports of the use of allopurinol [31, 32].
Topical retinoids may reduce the number of lesions. Other treatments which may help include oral isotretinoin, methotrexate, rifampicin, emollient creams, intralesional steroids and topical steroids under occlusion [5, 9, 23]. Narrow-band UVB [33], PUVA [34] and photodynamic therapy [35] have all been used. Associated uraemic pruritus may improve with amitriptyline [36]. Complete remission has been reported after the use of topical tacalcitol [37], however spontaneous resolution can occur.
Elastosis perforans serpiginosa
Definition and nomenclature
A perforating dermatosis in which the material extruded through the epidermis is derived from elastic fibres in the upper dermis [1]. It is closely associated with heritable disorders of connective tissue and Down syndrome.
Epidemiology
Incidence and prevalence
Rare.
Age
Usually presents between the ages of 5 and 20 years.
Sex
Males are predominantly affected.
Ethnicity
All races affected.
Associated diseases
Some 40% of reported cases have been associated with heritable connective tissue disorders, such as PXE, Ehlers–Danlos syndrome, Marfan syndrome, OI and acrogeria [2, 3]. It has also been reported in otherwise healthy individuals and in association with Down syndrome [4–7].
It sometimes occurs in patients receiving penicillamine, which is known to cause the production of abnormal elastin [8–12], and there is an overlap with ‘pseudo-PXE’ (see earlier).
Pathophysiology
Predisposing factors
The altered elastin resembles that seen in experimental animals subjected to lathyrogens or copper deficiency.
It is probable that the primary abnormality is in the dermal elastin, which provokes a cellular response that ultimately leads to extrusion of the abnormal elastic tissue. It may be significant that the lesions are commonly seen in areas subjected to wear and tear.
Pathology
The earliest detectable change is the focal development of elastotic-staining tissue and basophilic debris in the dermis. This is followed by a reaction of the overlying epidermis, which grows down to engulf the elastotic material. The epidermis surrounding the fully developed lesion is acanthotic and hyperkeratotic (Figure 96.54). The papule consists of a circumscribed area of epidermal hyperplasia traversed by a channel communicating directly with the dermis and containing a mass of tissue, which projects above the surface. This plug consists of horny material in its upper third and of amorphous debris derived from elastin in its lower two-thirds [4, 13–16]. In the dermis beneath and around the lesion, there is a foreign-body giant cell reaction. The elastotic material is finally extruded, to leave irregular scarring and warty thickening. Electron microscopy shows an increase in elastic fibres, with fine filaments on the surface similar to those seen in normal embryos. In penicillamine-induced cases the elastic fibres have a characteristic ‘bramble bush’ or ‘lumpy-bumpy’ morphology [10, 17]. The hydroxylation of dermal collagen is similar to that of newborn skin [14].
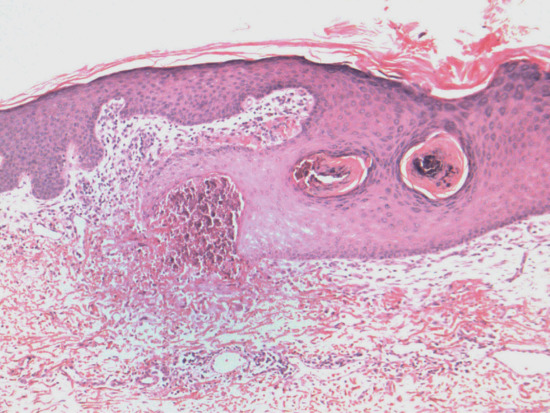
Figure 96.54 Elastosis perforans serpiginosa: note acanthotic epidermis growing downward in order to surround and engulf a focus of basophilic elastotic debris. (Courtesy of Dr Leigh Biddlestone, Royal United Hospitals, Bath UK.)
Causative organisms
No specific organism identified.
Genetics
The cause is unknown, but a genetically determined defect of elastic tissue may be involved [1]. Often associated with a known heritable disorder of connective tissue.
Environmental factors
The lesions may follow minor trauma such as an abrasion.
Clinical features
History
Lesions are generally asymptomatic.
Presentation
Small, horny or umbilicated papules are characteristically arranged in lines, circles or segments of circles in a serpiginous pattern (Figure 96.55). The individual papules may remain small or may enlarge slightly to assume a crateriform appearance with an elevated edge and a central plug, or further to leave an area of atrophic skin surrounded by smaller papules, each with a horny plug. The rings may reach a diameter of 15–20 cm but are usually smaller (Figure 96.56). The back and sides of the neck are most commonly affected, but the lesions may also occur on the cheeks or on the arms or thighs, and are sometimes bilaterally symmetrical [1, 16, 18, 19].
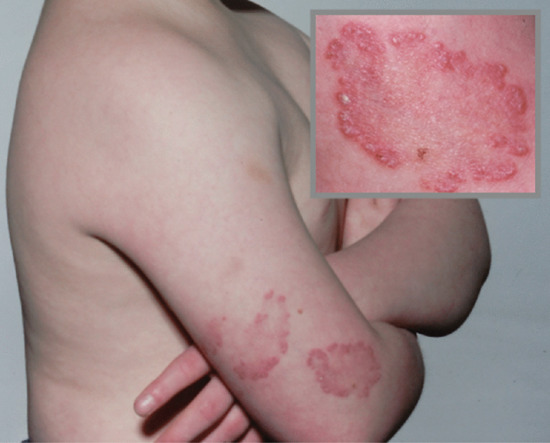
Figure 96.55 Elastosis perforans serpiginosa in a boy with Down syndrome.
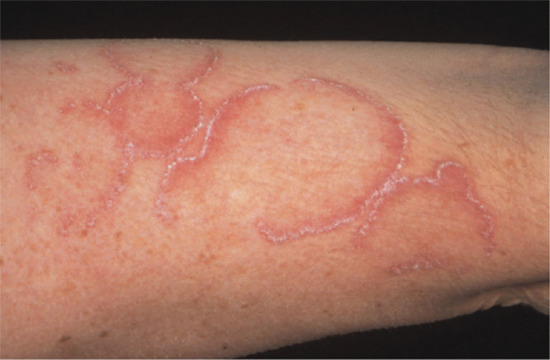
Figure 96.56 Elastosis perforans serpiginosa in a patient with vascular Ehlers–Danlos syndrome.
Differential diagnosis
The annular or linear arrangements of the papules and their distribution suggest the diagnosis, which is confirmed by the characteristic histology. Conditions which may cause confusion include porokeratosis of Mibelli, familial reactive perforating collagenosis and perforating granuloma annulare.
A similar histological appearance can occur in acquired perforating dermatosis (see earlier) [20].
Classification of severity
Not of prognostic significance in its own right, but may reflect an underlying heritable disorder of connective tissue.
Complications and co-morbidities
Associated connective tissue disease.
Disease course and prognosis
They may persist for several years, but eventually involute spontaneously to leave reticulate atrophic scars.
Investigations
Skin biopsy is diagnostic, but biopsy scars readily become keloidal.
Management
The condition tends to be self-limiting, and no treatment is of proven benefit [18]. However, careful removal of the nodules with a curette under local anaesthesia may give a reasonable cosmetic result. Freezing has been recommended [18, 21, 22]. Excision should be avoided, and dermabrasion may make the condition worse [4]. In a child with Down syndrome and associated vitamin A deficiency, clinical improvement was observed with oral retinoid therapy, even though the treatment produced side effects [6]. Isotretinoin has been used successfully in a patient with penicillamine-induced disease [23]. There are reports of improvement following Sellotape® stripping of the surface keratinous material [24], tazarotene [25], imiquimod [26] and calcipotriol [27]. Treatment with the pulsed dye [28], ultrapulsed carbon dioxide [29, 30] and Er : YAG [31] lasers have also been advocated [2].
References
Changes in dermal connective tissue due to ageing and photodamage
Introduction and general description
- Kligman AM, Lavker RM. Cutaneous aging. The difference between intrinsic aging and photoaging. J Cutaneous Aging Cosmetic Dermatol 1988;1:5–12.
- Yaar M, Gilchrest BA. Photoageing: mechanism, prevention and therapy. Br J Dermatol 2007;157:874–87.
- Kaya G, Saurat JH. Dermatoporosis: a chronic cutaneous insufficiency fragility syndrome. Clinicopathological features, mechanism, prevention and potential treatments. Dermatology 2007;215:284–294.
Wrinkles
- Smith JB, Fenske NA. Cutaneous manifestations and consequences of smoking. J Am Acad Dermatol 1996;34:717–32.
- Kligman AM, Zheng P, Lavker RM. The anatomy and pathogenesis of wrinkles. Br J Dermatol 1985;113:37–42.
- Tsuji T, Yorifuji T, Hamarta T, et al. Light and scanning electron microscopic studies on wrinkles in aged person's skin. Br J Dermatol 1986;114:329–35.
- Lavker RM. Structural alterations in exposed and unexposed skin. J Invest Dermatol 1979;73:59–69.
- Yin L, Morita A, Tsuji T. Skin aging induced by ultraviolet exposure and tobacco smoking: evidence from epidemiological and molecular studies. Photodermatol Photoimmunol Photomed 2001;17:178–83.
Actinic elastosis
- Kligman AM. Early destructive effects of sunlight on human skin. J Am Acad Dermatol 1969;210:2377–80.
- Finlayson GR, Sams WM Jr, Smith JG. Erythema ab igne: a histopathological study. J Invest Dermatol 1966;46:104–8.
- Braverman IM, Fonferko E. Studies in cutaneous aging. II. The microvasculature. J Invest Dermatol 1982;78:444–8.
- Bonkevitch F, Souza PR. Cutis rhomboidalis protects skin from malignant epithelial tumors. Med Hypotheses 2014;82:652–3.
- Wojno T, Tenzel RR. Actinic comedonal plaque of the eye. Am J Ophthalmol 1983;96:687–8.
- Lugo A, Montalvo l, González JR, et al. Bullous solar elastosis. Am J Dermatopathol 2005;27:34–5.
- John SM, Hamm H. Actinic comedonal plaque. A rare ectopic form of the Favre–Racouchot syndrome. Clin Exp Dermatol 1993;18:256–8.
- Carter VH, Constantine VS, Poole WL. Elastotic nodules of the antihelix. Arch Dermatol 1969;100:282–5.
- Requena L, Aguilar A, Sánchez YE. Elastotic nodules of the ears. Cutis 1989;44:452–4.
- Weedon D. Elastotic nodules of the ear. J Cutan Pathol 1981;8:429–33.
- Davis BE, Koh HK. Faces going up in smoke. A dermatologic opportunity for cancer prevention. Arch Dermatol 1992;128:255–62.
- Gilchrest BA. A review of skin ageing and its medical therapy. Br J Dermatol 1996;135:867–75.
- Kligman LH. Connective tissue photo-damage in hairless mice is potentially reversible. J Invest Dermatol 1987;88:S12–17.
- Moy LS, Murad H, Moy RL. Glycolic acid therapy, evaluation of efficacy and techniques in treatment of photodamaged lesions. Am J Cosmetic Surg 1993;10:9–13.
- Hubbard BA, Unger JG, Rohrich RJ. Reversal of skin aging with topical retinoids. Plast Reconstr Surg 2014;133;481e–490e.
- Singh M, Griffiths CEM. The use of retinoids in the treatment of photoaging. Dermatol Ther 2006;19:297–305.
- Kligman AM, Dogadkina D, Lavker RM. Effects of topical tretinoin on non-sun-exposed protected skin of the elderly. J Am Acad Dermatol 1993;29:25–33.
- Gilchrest BA. Turning back the clock: retinoic acid modifies intrinsic aging changes. J Clin Invest 1994;94:1711–12.
- Lateef H, Stevens MJ, Varani J. All-trans-retinoic acid suppresses matrix metalloproteinase activity and increases collagen synthesis in diabetic human skin in organ culture. Am J Pathol 2004;165:167–74.
- Maddin S, Laurharanta J, Agache P et al. Isotretinoin improves the appearance of photodamaged skin: results of a 36-week, multicenter, double-blind, placebo-controlled trial. J Am Acad Dermatol 2000;42:56–63.
- Phillips TJ, Gottlieb AB, Leyden JJ, et al. Efficacy of 0.1% tazarotene cream for the treatment of photodamage. Arch Dermatol 2002;138:1486–93.
- Pandel R, Poljšak B, Godic A, Dahmane R. Skin photoaging and the role of antioxidants in its prevention. ISRN Dermatol 2013; Article ID 930164.
- Farris P, Krutman J, Li YH, et al. Reservatrol: a unique antioxidant offering a multi-mechanistic approach for treating aging skin. J Drugs Dermatol 2013;12:1389–94.
- Ang P, Barlow RJ. Non-ablative laser resurfacing: a systematic review of the literature. Clin Exp Dermatol 2002;27:630–5.
- Watson REB, Craven NM, Kang S, et al. A short-term screening assay, using fibrillin-1 as a reporter molecule for photoageing repair agents. J Invest Dermatol 2001;116:672–8.
- Watson REB, Long SP, Bowden JJ, et al. Repair of photoaged dermal matrix by topical application of a cosmetic ‘antiageing’ product. Br J Dermatol 2008;158:472–7.
Collagenous and elastotic marginal plaques of the hands
- Burks JW, Wise LJ, Clark WH. Degenerative collagenous plaques of the hands. Arch Dermatol 1960;82:362–6.
- Jordaan HF, Rossouw DJ. Digital papular calcific elastosis: a histopathological, histochemical and ultrastructural study of 20 patients. J Cutan Pathol 1990;17:358–70.
- Menegesha YM, Kayal JD, Swerlick RA. Keratoelastoidosis marginalis. J Cutan Med Surg 2002;6:23–5.
- Rongioletti F, Betti R, Crosti C, et al. Marginal papular acrokeratodermas: a unified nosology for focal acral hyperkeratosis, acrokeratoelastoidosis and related disorders. Dermatology 1994;188:28–31.
- Calderone DC, Fenske NA. The clinical spectrum of actinic elastosis. J Am Acad Dermatol 1995;32:1016–24.
- Todd D, Al-Aboosi M, Hameed O, et al. The role of UV light in the pathogenesis of digital papular calcific elastosis. Arch Dermatol 2001;137:379–81.
- Mortimore RJ, Conrad RJ. Collagenous and elastotic marginal plaques of the hands. Australas J Dermatol 2001;42:211–13.
Adult colloid milium and colloid degeneration of the skin
- Mehregan D, Hooten J. Adult colloid milium: a case report and literature review. Int J Dermatol 2011;50:1531–4.
- Innocenzi D, Barduagni F, Cerio R, Wolter M. UV-induced colloid milium. Clin Exp Dermatol 1993;18:347–50.
- Dummer R, Laetsch B, Stutz S, et al. Elastosis colloidalis conglomerata (adult colloid milium, paracolloid of the skin) a maximal manifestation of actinic elastosis. Eur J Dermatol 2006;16:163–6.
- Holzberger PC. Concerning adult colloid milium. Arch Dermatol 1960;82:711–16.
- Muscardin LM, Bellocci M, Balus K. Papuloverrucous colloid milium: an occupational variant. Br J Dermatol 2000;143:884–7.
- Findlay GH, Morrison JGL, Simson IW, et al. Exogenous ochronosis and pigmented colloid milium from hydroquinone bleaching creams. Br J Dermatol 1975;93:613–22.
- Hashimoto K, Miller F, Bereston ES. Colloid milium: histochemical and electron microscopic studies. Arch Dermatol 1972;105:684–94.
- Handfield-Jones SE, Atherton D, Black M. Juvenile colloid milium. Br J Dermatol 1991;125:80–1.
- Chowdhury MMV, Blackford S, Williams S. Juvenile colloid milium. Br J Dermatol 1999;141(Suppl. 55):102–7.
- Hashimoto K, Nakayama H, Chimenti S, et al. Juvenile colloid milium. Immunohistochemical and ultrastructural studies. J Cutan Pathol 1989;16:164–74.
- Sanjuan EB, Planas G, Piquero J, et al. Colloid milium associated with multiple myeloma. Int J Dermatol 1994;33:793–5.
- Netscher DT, Sharma S, Kinner BM, Lyos A, Griego RD. Adult-type colloid milium of hands and face successfully treated with dermabrasion. South Med J 1996;89:1004–7.
- Ammirati CT, Giancola JM, Hruza GJ. Adult-onset facial colloid milium successfully treated with the long-pulsed Er:YAG laser. Dermatol Surg 2002;28:215–19.
Other causes of cutaneous atrophy
Atrophy due to corticosteroids
- Henqqe VR, Ruzicka T, Schwartz RA, et al. Adverse effects of topical corticosteroids. J Am Acad Dermatol 2006;54:1–15.
- Turpeinen M, Raitio H, Pelkonen AS, et al. Skin thickness in children treated with daily or periodical inhaled budesonide for mild persistent asthma. The Helsinki early intervention childhood asthma study. Pediatr Res 2010;67:221–5.
- Sarrni H, Hopsu-Havu BK. The decrease of hyaluronate synthesis by anti-inflammatory steroids in vivo. Br J Dermatol 1978;98:445–9.
- Averbeck M, Gebhardt C Anderegg U, et al. Suppression of hyaluran synthase 2 expression reflects the atrophogenic potential of glucocorticoids. Exp Dermatol 2010;19:757–9.
- Risteli J. Effect of prednisolone on the activities of intracellular enzymes of collagen biosynthesis in rat skin. Biochem Pharmacol 1977;26:1295–8.
- Autio P, Oikarinen A, Melkko J, et al. Systemic glucocorticoids decrease the synthesis of type I and type III collagen in human skin in vivo, whereas isotretinoin has little effect. Br J Dermatol 1994;131:660–3.
- Oishi Y, Fu ZW, Ohnuki Y, et al. Molecular basis of the alteration in skin collagen metabolism in response to in vivo dexamethasone treatment: effects on the synthesis of collagen type I and III, collagenase, and tissue inhibitors of metalloproteinases. Br J Dermatol 2002;147:859–68.
- Werth VP, Kligman AM, Shi X, et al. Lack of correlation of skin thickness with bone density in patients receiving chronic glucocorticoid. Arch Dermatol Res 1998;290:388–93.
- Nuutinen P, Riekki R, Parikka M, et al. Modulation of collagen synthesis and mRNA by continuous and intermittent use of topical hydrocortisone in human skin. Br J Dermatol 2003;148:39–45.
- Koob TJ, Jeffrey JJ, Eisen AZ. Regulation of human skin collagenase activity by hydrocortisone and dexamethasone in organ culture. Biochem Biophys Res Commun 1974;61:1083–8.
- Lehmann P, Zheng P, Lavker RM, et al. Corticosteroid atrophy in human skin. A study of light scanning and transmission electron microscopy. J Invest Dermatol 1983;81:169–75.
- Jablonska S, Groniowska M, Dabrowski J. Comparative evaluation of skin atrophy in man produced by topical corticosteroids. Br J Dermatol 1979;100:193–206.
- Barnes L, Kaya G, Rollason V. Topical corticosteroid-induced skin atrophy: a comprehensive review. Drug Saf 2015;38:493–509.
- Burton JL, Winter GD. Experimentally induced steroid atrophy in the domestic pig and man. Br J Dermatol 1976;94(Suppl. 12):107–9.
- Dykes PF, Marks R. An appraisal of the methods used in the assessment of atrophy from topical corticosteroids. Br J Dermatol 1979;101:599–609.
- James MP, Black MH, Sparkes CG. Measurement of dermal atrophy induced by topical steroids using a radiographic technique. Br J Dermatol 1977;96:303–5.
- Marks R, Halprin K, Fukui K, et al. Topically applied triamcinolone and macromolecular synthesis by human epidermis. J Invest Dermatol 1971;56:470–3.
- Holze E, Plewig G. Effects of dermatitis, stripping and steroids on the morphology of corneocytes. J Invest Dermatol 1977;68:350–6.
- Pinnell S. Management of cutaneous atrophy after corticosteroid injection. J Am Acad Dermatol 1987;17:521.
- McMichael AJ, Griffiths CEM, Talwar HS, et al. Concurrent application of tretinoin (retinoic acid) partially protects against corticosteroid-induced epidermal atrophy. Br J Dermatol 1996;135:60–4.
- Shumaker PR, Rao J, Goldman MP. Treatment of local, persistent cutaneous atrophy following corticosteroid injection with normal saline infiltration. Dermatol Surg 2005;31:1340–3.
- Kaya G, Tran C, Sorg O, et al. Hyaluronate fragments reverse skin atrophy by a CD44-dependent mechanism. PLOS Med 2006;3:e493.
- Schäcke H, Zollner TM, Dӧcke WD, et al. Characterization of ZK245186, a novel selective glucocorticoid receptor agonist for the topical treatment of inflammatory skin diseases. Br J Pharmacol 2009;158:1088–103.
Striae
- Stevanovic DV. Corticosteroid-induced atrophy of the skin with telangiectasia. A clinical and experimental study. Br J Dermatol 1972;87:548–56.
- Shuster S. The cause of striae distensae. Acta Derm Venereol Suppl (Stockh) 1979;59(85):161–9.
- Herxheimer H. Cutaneous striae in normal boys. Lancet 1953;ii:204.
- Carr RD, Hamilton JF. Transverse striae of the back. Arch Dermatol 1969;99:26–30.
- Thiers H, Moulin G, Larive M. Les vergetures de la corticothérapie locale. Ann Dermatol Syphiligr (Paris) 1969;96:29–36.
- Darvay A, Acland K, Lynn W, Russell-Jones R. Striae formation in two HIV-positive persons receiving protease inhibitors. J Am Acad Dermatol 1999;41:467–9.
- Zheng PS, Lauker RM, Lehmann P, et al. Morphologic investigations on the rebound phenomenon after corticosteroid-induced atrophy in human skin. J Invest Dermatol 1984;82:345–52.
- Bertin C, Lopes-Da Cunha A, Nkengne A, et al. Striae distensae are characterized by distinct microstructural features as measured by non-invasive methods in vivo. Skin Res Technol 2014;20:81–6.
- Zheng P, Lavker RM, Kligman AM. Anatomy of striae. Br J Dermatol 1985;112:185–93.
- Arem AJ, Kischer CW. Analyses of striae. Plast Reconstr Surg 1980;65:22–9.
- De Paepe A, Devereux RB, Dietz HC, et al. Revised diagnostic criteria for the Marfan syndrome. Am J Med Genet 1996;62:417–26.
- Bergman R, Nevit MJ, Gescheidt-Shoshany H, Pimienta AL, Reinstein E. Atrophic skin patches with abnormal elastic fibres as a presenting sign of the MASS phenotype associated with mutation in the fibrillin-1 gene. JAMA Dermatol 2014;150(8):885–9.
- Agg B, Benke K, Szilveszter B, et al. Possible extracardiac predictors of aortic dissection in Marfan syndrome. BMC Cardiovasc Disord 2014;14:47.
- Tung JY, Kiefer AK, Mullins M, et al. Genome-wide association analysis implicates elastin microfibrils in the development of nonsyndromic striae distensae. J Invest Dermatol 2013;133:2628–31.
- Poidevin LOS. Striae gravidarum. Their relation to adrenal cortical hyperfunction. Lancet 1959;ii:436–8.
- Chernovsky ME, Knox JM. Atrophic striae after occlusive corticosteroid therapy. Arch Dermatol 1964;90:15–19.
- Kikuchi I, Horikawa S. Perilymphatic atrophy of the skin. A side effect of topical corticosteroid injection therapy. Arch Dermatol 1974;109:558–9.
- McAvoy BR. No evidence for topical preparations in preventing stretch marks in pregnancy. Br J Gen Pract 2013;63:212.
- Al-Himdani S, Ud-Din S, Gilmore S, et al. Striae distensae: a comprehensive revision and evidence-based evaluation of prophylaxis and treatment. Br J Dermatol 2014;170:527–47.
- Heixsal D, Soirefmann M, Porto MD, et al. Superficial dermabrasion versus topical tretinoin on early striae distensae: a randomized pilot study. Dermatol Surg 2014;40:537–44.
- Shokeir H, El Bedewi A, Sayed S, et al. Efficacy of pulsed dye laser versus intense pulsed light in the treatment of striae distensae. Dermatol Surg 2014;40:632–40.
- Goldman A, Rossato F, Prati C. Stretch marks: treatment using the 1,064nm Nd:YAG Laser. Dermatol Surg 2008;34:686–91.
- Kou BJ, Lee DH, Kim MN, et al. Fractional photothermolysis for the treatment of striae distensae in Asian skin. Am J Clin Dermatol 2008;9:33–7.
- Ud-Din S, McAnelly SL, Bowring A, et al. A double blind controlled clinical trial assessing the effect of topical gels on striae distensae (stretch marks): a morphological and immunohistochemical study. Arch Dermatol Res 2013;305:603–17.
Acquired poikiloderma
- Okazaki M, Kikuchi I. Radiodermatitis. An analysis of 43 cases. J Dermatol 1986;13:356–65.
- Carroll CL, Lang W, Snively B, et al. Development and validation of the Dermatomyositis Skin Severity Index. Br J Dermatol 2008;158:345–350.
- Bloom B, Marchbein S, Fischer M, et al. Poikilodermatous mycosis fungoides. Dermatol Online J 2012;18(12):4.
Atrophic scars
- Leung AKC, Pinkao C, Suave RS. Scarring resulting from chickenpox. Pediatr Dermatol 2001;18:378–80.
- Murdoch ME, Hay RJ, Mackenzie CD, et al. A clinical classification and grading system of the cutaneous changes in onchocerciasis. Br J Dermatol 1993;129:260–9.
- Colomb D. Stellate spontaneous pseudoscars. Senile and presenile forms: especially those forms caused by prolonged corticoid therapy. Arch Dermatol 1972;105:551–4.
- Zac FG, Pai SH, Kanshepolsky J. Stellate spontaneous pseudoscars (Colomb). Arch Dermatol 1968;98:499–501.
- McCash S, Emanuel PO. Defining diabetic dermopathy. J Dermatol 2011;38(10):988–92.
- Cohen BA, Esterley NB, Nelson PF. Congenital erosive and vesicular dermatitis healing with reticulated supple scarring. Arch Dermatol 1985;121:361–7.
- Tlougan BE, Paller AS, Schaffer JV, et al. Congenital erosive and vesicular dermatosis with reticulated supple scarring: unifying clinical features. J Am Acad Dermatol 2013;69(6):909–15.
- Gupta AK, Rasmussen JE, Headington JT. Extensive congenital erosions and vesicles healing with reticulate scarring. J Am Acad Dermatol 1987;17:369–76.
- De Lange A, Bayet B, Debauche C, et al. Congenital erosive and vesicular dermatosis with reticulated scarring in a newborn: an innovative treatment using a silicone dressing. Pediatr Dermatol 2009;26:735–8.
Spontaneous atrophic scarring of the cheeks
- Marks VJ, Miller OF. Atrophia maculosa varioliformis cutis. Br J Dermatol 1986;115:105–9.
- McCoriston LR, Roys HC. Atrophia maculosa varioliformis cutis. Arch Dermatol 1951;64:59–61.
- Paradisi M, Angelo C, Conti G, et al. Atrophia maculosa varioliformis cutis: a pediatric case. Pediatr Dermatol 2001;18:478–80.
- Kolenik SA, Perez MI, Davidson DM, et al. Atrophia maculosa varioliformis cutis. J Am Acad Dermatol 1994;30:837–40.
- Gordon PM, Doherty VR. Familial atrophia maculosa varioliformis cutis. Br J Dermatol 1996;134:982–3.
- Ou T, Wang B, Fang K. Familial atrophia maculosa varioliformis cutis: case report and pedigree analysis. Br J Dermatol 2005;153:821–4.
Acrodermatitis chronica atrophicans
- Steere AC. Lyme disease. N Engl J Med 1989;321:586–96.
- Coulson IH. Acrodermatitis chronica atrophicans with coexisting.morphoea. Br J Dermatol 1989;121:263–9.
- Aberer E, Kersten A, Klade H. Heterogeneity of Borrelia burgdorferi in the skin. Am J Dermatopathol 1996;18:571–9.
- Flisiak I, Schwartz RA, Chodynicka B. Clinical features and specific immunological response against Borrelia afzelii in patients with acrodermatitis chronica atrophicans. J Med 1999;30:267–78.
- Boehmer-Andersson E, Hovmark A, Asbrink E. Acrodermatitis chronica atrophicans: histopathologic findings and clinical correlation in 111 cases. Acta Derm Venereol 1998;78:207–13.
- Asbrink E, Hovmark A. Successful cultivation of spirochaetes from the skin lesions of patients with erythema chronicum migrans and acrodermatitis chronicum atrophicans. Acta Pathol Microbiol Immunol Scand B 1985;93:161–3.
- Asbrink E, Hovmark A. Early and late cutaneous manifestations of Ixodes borne borreliosis (Lyme borreliosis). Ann NY Acad Sci 1988;539:4–15.
- Müller KE. Damage of collagen and elastic fibres by Borrelia burgdorferi: known and new clinical and histological aspects. Open Neurol J 2012;6:179–86.
- Mullegger RR, McHugh G, Ruthazar R. Differential expression of cytokine mRNA in skin specimens from patients with erythema migrans or acrodermatitis chronica atrophicans. J Invest Dermatol 2000;115:1115–23.
- Grygorczuk S, Péter O, Kondrusik M, et al. Assessment of the different Borrelia burgdorferi sensu lato species in patients with Lyme borreliosis from north-east Poland by studying preferential serologic response and DNA isolates. Ann Agric Environ Med 2013;20:21–9.
- Stinco G, Ruscio M, Bergamo S, et al. Clinical features of 705 Borrelia burgdorferi seropositive patients in an endemic area of northern Italy. Sci World J 2014;2014(1)414505.
- Burgdorf WHC, Worret W, Schultka O. Acrodermatitis chronica atrophicans. Int J Dermatol 1979;18:595–601.
- Santos M, Ribiero-Rodrigues R, Talhari C, et al. Presence of Borrelia burgdorferi ‘sensu lato’ in patients with morphoea from the Amazonia region in Brazil. Int J Dermatol 2011;50:1373–8.
- Aberer E, Neumann R, Stanek G. Is localised scleroderma a Borrelia infection? Lancet 1985;ii:278.
- Halkier-Sorensen L. Antibodies to the Borrelia burgdorferi in patients with scleroderma. Acta Derm Venereol Suppl (Stockh) 1989;69:116–19.
- Tee SI, Martinez-Escanamé M, Zuriel D, et al. Acrodermatitis chronica atrophicans with pseudolymphomatous infiltrates. Am J Dermatopathol 2013;35:338–42.
- Fagrell B, Heiland RA, Howe TR. Acrodermatitis chronica atrophicans can mimic a peripheral vascular disorder. Acta Med Scand 1986;20:485–8.
- Goos W, Schwarz-Speck M. Acrodermatitis chronica atrophicans. Dermatologica 1972;145:287–90.
- Leverkas M, Finner AM, Pokrywka A, et al. Metastatic squamous carcinoma of the ankle in long-standing untreated acrodermatitis chronica atrophicans. Dermatology 2008;217:215–18.
- Garbe C, Stein H, Dienemann D, Orfanos CE. Borrelia burgdorferi-associated cutaneous B cell lymphoma. J Am Acad Dermatol 1991;24:584–90.
- Rees DHE, O'Connell S, Brown MM, et al. The value of serological testing for Lyme disease in the UK. Br J Rheumatol 1995;34:132–6.
- Aberer E, Breier F, Stanek G. Success and failure in the treatment of acrodermatitis chronica atrophicans. Infection 1996;24:85–7.
- Stanek G, Strle F. Lyme borreliosis. Lancet 2003;362:1639–47.
- Mullegger RR, Glatz M. Is serological follow-up useful for patients with cutaneous Lyme borreliosis? Curr Probl Dermatol 2009;37:178–82.
Atrophodermas
- Curth HO. The genetics of follicular atrophoderma. Arch Dermatol 1978;114:1479–83.
- Hartman RD, Molho-Pessach V, Schafer JV. Conradi–Hünermann–Happle syndrome. Dermatol Online J 2010;16(11):4.
- Viksnins P, Berlin A. Follicular atrophoderma and basal cell carcinoma. The Bazex syndrome. Arch Dermatol 1977;113:948–51.
- Moulin G, Hill MP, Guillaud V, et al. Bandes pigmentées atrophiques acquises suivant les lignes de Blaschko. Ann Dermatol Vénéréol 1992;119:729–36.
- de Golian E, Echols K, Pearl H, et al. Linear atrophoderma of Moulin: a distinct entity? Pediatr Dermatol 2014;31:373–7.
- Atasoy M, Aliagaoglu C, Sahin O, et al. Linear atrophoderma of Moulin together with leuconychia: a case report. J Eur Acad Dermatol Venereol 2006;20:337–40.
- Villani AP, Amini-Adlé M, Wagschal D, et al. Linear atrophoderma of Moulin: report of 4 cases and 20th anniversary case review. Dermatology 2013;227:5–9.
- Patsasti A, Kyriakou A, Chaidemenos G, et al. Linear atrophoderma of Moulin: a case report and review of the literature. Dermatol Pract Concept 2013;31:7–11.
- Danarti R, Bittar M, Happle R, et al. Linear atrophoderma of Moulin: postulation of mosaicism for a predisposing gene. J Am Acad Dermatol 2003;49:492–8.
- Zaouak A, Hammami Ghorbel H, Benmously-Mlika R, et al. A case of linear atrophoderma of Moulin successfully treated with methotrexate. Dermatol Ther 2014;27:153–5.
- Kee CE, Brothers WS, New W. Idiopathic atrophoderma of Pasini and Pierini with co-existent morphea. A case report. Arch Dermatol 1960;82:154–7.
- Miller RF. Idiopathic atrophoderma, report of a case and nosologic study. Arch Dermatol 1965;92:653–60.
- Beuchner SA, Rufli T. Atrophoderma of Pasini and Pierini. J Am Acad Dermatol 1994;30:441–6.
- Barsky S, Ke M. Congenital atrophoderma of the newborn. Arch Dermatol 1970;101:374–5.
- Lasser AE, Schultz BC, Beaff D, et al. Phenylketonuria and scleroderma. Arch Dermatol 1978;114:1215–17.
- Lee Y, Oh Y, Ahn SY, et al. A case of atrophoderma of Pasini and Pierini associated with Borrelia burgdorferi infection successfully treated with oral doxycycline. Ann Dermatol 2011;23:352–6.
- Berman A, Berman GD, Winkelmann RK. Atrophoderma (Pasini–Pierini) findings on direct immunofluorescent, monoclonal antibody and ultra-structural studies. Int J Dermatol 1988;27:487–90.
- Canizares O, Sachs PM, Jaimovich L, et al. Idiopathic atrophoderma of Pasini and Pierini. Arch Dermatol 1958;77:42–60.
- Jablonska S, ed. Scleroderma and Pseudoscleroderma, 2nd edn. Warsaw: Polish Medical Publishers, 1975.
- Kim YS, Lee CW. Overlap between atrophoderma Pasini–Pierini and juvenile idiopathic arthritis. Clin Exp Dermatol 2009;34:82–3.
- Carter JD, Valeriano J, Vasey FB. Hydroxychloroquine as a treatment for atrophoderma of Pasini and Pierini. Int J Dermatol 2006;45:1255–6.
Scleroatrophic syndrome of Huriez
- Huriez C, Agache P, Bombart M, et al. Épithéliomes spinocellulaires sur atrophie cutanée congénitale dans deux familles à morbidité cancéreuse élevée. Bull Soc Fr Dermatol Syphiligr 1963;70:24–8.
- Huriez C, Agache P, Souillart F, et al. Scléroatrophie familiale des extremités avec dégénérescences spinocellulaires multiples. Bull Soc Fr Dermatol Syphiligr 1963;70:743–4.
- Huriez C, Deminatti M, Agache P, et al. Une Génodysplasie non encore individualisée: la génodermatose scléroatrophiante et kératodermique des extrémités fréquemment dégénérative. Sem Hôp Paris 1968;44:481–8.
- Downs AMR, Kennedy CTC. Scleroatrophic syndrome of Huriez in an infant. Pediatr Dermatol 1998;15:207–9.
- De Berker D, Kavanagh G. Distinctive nail changes in scleroatrophy of Huriez [Abstract]. Br J Dermatol 1993;129(Suppl. 42):36.
- Hamm H, Traupe H, Bröcker E-B, et al. The scleroatrophic syndrome of Huriez: a cancer-prone genodermatosis. Br J Dermatol 1996;134:512–18.
- Lee YA, Stevens HP, Delaporte E, et al. A gene for an autosomal scleroatrophic syndrome predisposing to skin cancer (Huriez syndrome) maps to chromosome 4q23. Am J Hum Genet 2000;66:326–30.
- Delaporte E, N'Guyen-Mailfer C, Janin A, et al. Keratoderma with scleroatrophy of the extremities or sclerotylosis (Huriez syndrome): a reappraisal. Br J Dermatol 1995;133:409–16.
Localized abdominal wall atrophy
- Orvis BR, Bottles K, Kogan BA. Testicular histology in the ‘prune belly’ syndrome. J Urol 1988;139:335–7.
- Pagon RA, Smith DW, Shepherd TH. Urethral obstruction malformation complex. A cause of abdominal muscle deficiency and the ‘prune belly’. J Pediatr 1979;94:900–6.
- Fishman AI, Franco I. Laparoscopic-assisted surgical reconstruction of a rare congenital abdominal wall defect in two children misdiagnosed with prune belly syndrome. J Pediatr Urol 2013;9:448–52.
Paroxysmal haematoma of the finger
- Achenbach W. Das paroxysmale Handhämatom. Medizinische 1958;52:2138–40.
- Stieler W, Heinze-Werlitz C. Paroxysmales Fingerhämatom (Achenbach Syndrom). Hautarzt 1990;41:270–1.
- Layton AM, Cotterill JA. A case of Achenbach's syndrome. Clin Exp Dermatol 1993;18:60–1.
- Parslew R, Verbov JL. Achenbach syndrome. Br J Dermatol 1995;132:319.
- Huikeshoven M, de Priester JA, Engel AF. A case of spontaneous wrist haematoma in Achenbach syndrome. J Hand Surg Eur Vol 2009;34:551–2.
- Harper CM, Waters PM. Acute idiopathic blue finger: case report. J Hand Surg 2013;38A:1980–2.
- Robertson A, Liddington MI, Kay SP. Paroxysmal finger haematomas (Achenbach's syndrome) with angiographic abnormalities. J Hand Surg 2002;27B:391–3.
- Thies K, Beschorner U, Noory E, et al. Achenbach's syndrome revisited. Vasa 2012;41:366–70.
Panatrophy
- Nakano R, Wakamatsu N, Tsujui S. Juvenile Sandhoff disease with local panatrophy. A case report. Baillieres Clin Neurol 1989;29:1032–8.
- Jablonska S, ed. Scleroderma and Pseudoscleroderma, 2nd edn. Warsaw: Polish Medical Publishers, 1975.
- Barnes S. Gower's case of local panatrophy. Br J Dermatol 1939;51:377–80.
- Sakamoto T, Oku T, Takigawa M. Gowers' local panatrophy. Eur J Dermatol 1998;8:116–19.
- Levy JJ, Gassmuller J, Anding H, et al. Imaging subcutaneous atrophy in circumscribed scleroderma with 20 MHz B-scan ultrasound. Hautarzt 1993;44:446–51.
- Yoon J, Kim CW, Lee SH, et al. Correction of facial defects by autologous fat graft in local panatrophy. Eur J Dermatol 2012;22:710–11.
Facial hemiatrophy
- El-Kehdy J, Abbas O, Rubeiz N. A review of Parry–Romberg syndrome. J Am Acad Dermatol 2012;67:769–84.
- Dalla Costa G, Columbo B, Dalla Libera D, et al. Parry Romberg syndrome associated with chronic facial pain. J Clin Neurosci 2013;20:1320–2.
- Theodossiadis PG, Grigoropoulos VG, Emfietzoglou I, et al. Parry–Romberg syndrome studied by optical coherence tomography. Ophthalmic Surg Lasers Imaging 2008;39:78–80.
- Ehmann D, Riyaz R, Greve M. Central retinal artery occlusion in a child with Parry Romberg syndrome. Can J Ophthalmol 2014;49:e9–10.
- Chang S-E, Huh J, Choi J-H, et al. Parry–Romberg syndrome with ipsilateral cerebral atrophy of neonatal onset. Pediatr Dermatol 1999;16:487–8.
- Handfield-Jones SE, Peachey RDG, Moss ACH, et al. Ossification in linear morphoea with hemifacial atrophy: treatment by surgical excision. Clin Exp Dermatol 1988;13:385–8.
- Janowska M, Podolec K, Lipko-Godlewska S, et al. Coexistence of Parry–Romberg syndrome with homolateral segmental vitiligo. Postepy Dermatol Alergol 2013;30:409–11.
- Bramley P, Forbes A. A case of progressive hemiatrophy presenting with spontaneous fractures of the lower jaw. BMJ 1960;i:1476–8.
- Franz FP, Blocksma R, Brundage SR, et al. Massive injection of liquid silicone for hemifacial atrophy. Ann Plast Surg 1988;20:140–5.
- Sakamoto T, Oku T, Takigawa M. Gower's local panatrophy. Eur J Dermatol 1998;8:116–17.
- Guerrerosantos JG, Guerrerosantos F, Orozco J. Classification and treatment of facial tissue atrophy in Parry–Romberg disease. Aesthetic Plast Surg 2007;31:424–34.
- Rodby KA, Kaptein YE, Roring J, et al. Evaluating autologous lipofilling for Parry–Romberg syndrome-associated defects: a systematic literature review and case report. Cleft Palate Craniofac J 2015, August 21.
- Chang Q, Li J, Dong Z, et al. Quantitative volumetric analysis of progressive hemifacial atrophy corrected using stromal vascular fraction-supplemented autologous fat graft. Dermatol Surg 2013;39:1465–73.
Disorders of elastic fibre degradation
- Grahame R. A method for measuring human skin elasticity in vivo with observations on the effects of age, sex and pregnancy. Clin Sci 1970;39:223–9.
- Kielty CM. Elastic fibres in health and disease. Expert Rev Mol Med 2006;8:1–23.
- Uitto J. Biochemistry of collagen in diseases. Ann Intern Med 1986;105:740–56.
- Lewis KG, Bercovitch L, Dill SW, et al. Acquired disorders of elastic tissue: Part 1. Increased elastic tissue and solar elastotic syndromes. J Am Acad Dermatol 2004;51:1–21.
- Lewis KG, Bercovitch L, Dill SW, et al. Acquired disorders of elastic tissue: Part II. Decreased elastic tissue. J Am Acad Dermatol 2004;51:165–85.
Lax skin
- El Khoury J, Kurban M, Abbas O. Elastophagocytosis: underlying mechanisms and associated cutaneous entities. J Am Acad Dermatol 2014;70:934–44.
Acquired cutis laxa
- Nanko H, Jepson LV, Zachariae H, et al. Acquired cutis laxa (generalised elastolysis): light and electron microscopic studies. Acta Derm Venereol Suppl (Stockh) 1979;59:315–24.
- Harpey J-P. Cutis laxa and low serum zinc after antenatal exposure to penicillamine. Lancet 1983;ii:858–9.
- Tsuji T, Imajo Y, Sawabe M, et al. Acquired cutis laxa concomitant with nephrotic syndrome. Arch Dermatol 1987;123:1211–16.
- Ting HC, Foo MH, Wang F. Acquired cutis laxa and multiple myelomatosis. Br J Dermatol 1984;110:363–7.
- Lavorato, FG, de Alves M FGS, Maceira JMP, et al. Primary systemic amyloidosis, acquired cutis laxa and cutaneous mucinosis in a patient with multiple myeloma. An Bras Dermatol 2013;88(6 Suppl. 1):32–5.
- Marchase P, Holbrook K, Pinnell SR. A familial cutis laxa syndrome with ultrastructural abnormalities of collagen and elastin. J Invest Dermatol 1980;75:399–403.
- Randle EHW, Muller S. Generalised elastolysis associated with systemic lupus erythematosus. J Am Acad Dermatol 1983;8:869–73.
- Rongioletti F, Cutolo M, Bondavalli P, Rebora A. Acral localized acquired cutis laxa associated with rheumatoid arthritis. J Am Acad Dermatol 2002;46:128–30.
- García-Patos V, Pujol RM, Barnadas MA, et al. Generalized acquired cutis laxa associated with coeliac disease: evidence of immunoglobulin A deposits on the dermal elastic fibres. Br J Dermatol 1996;135:130–4.
- Hill VA, Seymour CA, Mortimer PS. Penicillamine-induced elastosis perforans serpiginosa and cutis laxa in Wilson's disease. Br J Dermatol 2000;142:560–1.
- Na SY, Choi M, Kim MJ, et al. Penicillamine-induced elastosis perforans serpiginosa and cutis laxa in a patient with Wilson's disease. Ann Dermatol 2010;22:468–71.
- Fornieri C, Quaglino D, Lungarella G, et al. Elastin production and degradation in cutis laxa acquisita. J Invest Dermatol 1994;103:583–8.
- Hu Q, Reymond J-L, Pinel N, et al. Inflammatory destruction of elastic fibers in acquired cutis laxa is associated with missense alleles in the elastin and fibulin-5 genes. J Invest Dermatol 2006;126:283–90.
- Kerl H, Berg G. Fatal penicillin-induced generalized post-inflammatory elastolysis (cutis laxa). Hautarzt 1975;26:191–8.
- Wanderer AA, Ellis EF, Goltz RW, et al. Tracheobronchomegaly and acquired cutis laxa in a child. Pediatrics 1969;44:709–15.
- Verhagen AR, Woerdemann MJ. Post-inflammatory elastolysis and cutis laxa. Br J Dermatol 1975;92:183–90.
- Haider M, Alfadley A, Kadry R, et al. Acquired cutis laxa type II (Marshall syndrome) in an 18-month-old child: a case report. Pediatr Dermatol 2010;27:89–91.
- Fontenelle E, de Almeida APM, de Almeida Souza GMA. Marshall's syndrome. An Bras Dermatol 2013;88:279–82.
- Timmer-de Mik L, Broekhuijsen van Henten DM, Oldhoff JM, De Geer DB, Sigurdsson V, Pasmans SG. Acquired cutis laxa in childhood Sweet's syndrome. Pediatr Dermatol 2009;26:358–60.
- Hwang ST, Williams ML, McCalmont TH, et al. Sweet's syndrome leading to acquired cutis laxa (Marshall's syndrome) in an infant with alpha-1 antitrypsin deficiency. Arch Dermatol 1995;131:1175–7.
Anetoderma
- Venencie PY, Winkelmann RK. Histopathologic findings in anetoderma. Arch Dermatol 1984;120:1040–4.
- Venencie PY, Winkelmann RK, Moore BA. Anetoderma: clinical findings, associations, and long term follow-up evaluations. Arch Dermatol 1984;120:1032–9.
- Karrer S, Szeimies RM, Stoltz W, Landthaler M. Primary anetoderma in children: report of two cases and literature review. Pediatr Dermatol 1996;13:382–5.
- Sparsa A, Piette JC, Wechsler B, et al. Anetoderma and its prothrombotic abnormalities. J Am Acad Dermatol 2003;49:1008–12.
- Hodak E, David M. Primary anetoderma and antiphospholipid antibodies: review of the literature. Clin Rev Allergy Immunol 2007;32:162–9.
- Eungdamrong J, Fischer M, Patel R, Meehan S, Sanchez M. Anetoderma secondary to antiphospholipid antibodies. Dermatol Online J 2012;18(12);26.
- Emer J, Roberts D, Sidhu H, et al. Generalized anetoderma after intravenous penicillin therapy for secondary syphilis in an HIV patient. J Clin Aesthet Dermatol 2013;6:23–8.
- Inemadar AC, Palit A, Athanikar SB, et al. Generalised anetoderma in a patient with HIV and dual mycobacterial infection. Lepr Rev 2003;74:275–8.
- Kalogeramitros D, Gregoriou S, Makris M, et al. Secondary anetoderma associated with mastocytosis. Int Arch Allergy Immunol 2007;142:86–8.
- Tatnall F, Rycroft R. Pityriasis versicolor with cutaneous atrophy. Clin Exp Dermatol 1985;10:258–61.
- Ozkan S, Fetil E, Izler F, et al. Anetoderma secondary to generalized granuloma annulare. J Am Acad Dermatol 2000;42:335–8.
- Kang HS, Paek JO, Lee MW, et al. Anetoderma developing in generalized granuloma annulare in an infant. Ann Dermatol 2014;26:283–5.
- Cho S, Jung JY, Lee JH. Treatment of anetoderma occurring after resolution of Steven–Johnson syndrome using an ablative 10,600-nm carbon dioxide fractional laser. Dermatol Surg 2012;38:677–9.
- Jubert C, Cosnes A, Clerici T, et al. Sjögren's syndrome and cutaneous B cell lymphoma revealed by anetoderma. Arthritis Rheum 1993;36:133–4.
- Sequrado M, Guerra-Tapia A, Zarco C, et al. Anetoderma secondary to cutaneous B-cell lymphoma. Clin Exp Dermatol 2006;31:130–1.
- Requena L, González-Guerra E, Angulo J, et al. Anetodermic mycosis fungoides: a new clinicopathological variant of mycosis fungoides. Br J Dermatol 2008;158:157–62.
- Prizant TL, Lucky AW, Frieden IJ, et al. Spontaneous atrophic patches in extremely premature infants. Anetoderma of prematurity. Arch Dermatol 1996;132:671–4.
- Cartlidge PH, Fox PE, Rutter N. The scars of newborn intensive care. Early Hum Dev 1990;21:1–10.
- Shames BS, Nassif A, Bailey CS, et al. Secondary anetoderma involving a pilomatrixoma. Am J Dermatopathol 1994;16:557–60.
- Page EH, Assaad M. Atrophic dermatofibroma and dermatofibrosarcoma protuberans. J Am Acad Dermatol 1987;17:947–50.
- Gamo R, Ortiz-Romero P, Sopeno J, et al. Anetoderma developing in juvenile xanthogranuloma. Int J Dermatol 2005;44:503–6.
- Cockayne SE, Gawkrodger DJ. Hamartomatous congenital naevi showing secondary anetoderma-like changes. J Am Acad Dermatol 1998;39:843–5.
- Davis W. Wilson's disease and penicillamine-induced anetoderma. Arch Dermatol 1977;113:976–7.
- Peterman A, Scheel M, Sams WM Jr, et al. Hereditary anetoderma. J Am Acad Dermatol 1996;35:999–1000.
- Zellman GL, Levy ML. Congenital anetoderma in twins. J Am Acad Dermatol 1997;36:483–5.
- Patrizi A, Neri I, Virdi A, et al. Familial anetoderma: a report of two families. Eur J Dermatol 2011;21:680–5.
- Hofer T, Golderberger D, Itin PH. Anetoderma and borreliosis: is there a pathogenic relationship? Eur J Dermatol 2003;13:399–401.
- Venencie PY, Winkelmann RK, Moore BA. Ultrastructural findings in the skin lesions of patients with anetoderma. Acta Derm Venereol Suppl (Stockh) 1984;64:112–20.
- Venencie PY, Winkelmann RK. Monoclonal antibody studies in the skin lesions of patients with anetoderma. Arch Dermatol 1985;121:747–9.
- Ghomrasseni S, Dridi M, Gogly B, et al. Anetoderma: an altered balance between metalloproteinases and tissue inhibitors of metalloproteinases. Am J Dermatopathol 2002;24:118–29.
- Kossard S, Kronman KR, Dicken CH, et al. Inflammatory macular atrophy. Immunofluorescence and ultrastructural findings. J Am Acad Dermatol 1979;1:325–34.
- Werth VP. Decay-accelerating factor in human skin is associated with elastic fibers. J Invest Dermatol 1988;91:511–16.
- Haider M, Alenazi M, Almutawa A, et al. Lupus erythematosus-associated primary and secondary anetoderma. J Cutan Med Surg 2012;16:64–7.
- De Bracco MM, Bianchi CA, Bianchi O, et al. Hereditary complement (C2) deficiency with discoid lupus erythematosus and idiopathic anetoderma. Int J Dermatol 1979;18:713–15.
- Ryll-Nardzewski C. Remarques sur le lupus érythémate profond et sur l'anetodermie érythématoide. Ann Dermatol Syphiligr (Paris) 1960;87:627–36.
- Schnitzler L, Sayag J. Pseudotumoral lupus anetoderma and infantile chorea. Ann Dermatol Vénéréol 1988;115:679–85.
- Randle HW, Muller S. Generalized elastolysis associated with systemic lupus erythematosus. J Am Acad Dermatol 1983;8:869–73.
- Bergman R, Friedman-Birnbaum R. An immunofluorescence study of primary anetoderma. Clin Exp Dermatol 1990;15:124–30.
- Hodak E, Shamai-Lubovitz O, David M, et al. Primary anetoderma associated with a wide-spectrum of autoimmune abnormalities. J Am Acad Dermatol 1991;25:415–18.
- Oikarinen AK, Palatsi R, Adomian GE, et al. Anetoderma: biochemical and ultrastructural demonstration of an elastin defect in the skin of three patients. J Am Acad Dermatol 1984;11:64–72.
- Zaki I, Scerri C, Nelson H. Primary anetoderma: phagocytosis of elastic fibres by macrophages. Clin Exp Dermatol 1994;19:388–90.
- Indianer L. Anetoderma of Jadassohn. Arch Dermatol 1970;102:697–8.
- Reiss F, Linn E. The therapeutic effect of epsilon-aminocaproic acid on anetoderma of Jadassohn. Dermatologica 1973;146:357–60.
- Braun RP, Borradori L, Chavaz P, et al. Treatment of primary anetoderma with colchicine. J Am Acad Dermatol 1998;38:1002–3.
Mid-dermal elastolysis, Upper dermal elastolysis
- Brenner W, Schmint FG, Konrad K, et al. Non-inflammatory dermal elastolysis. Br J Dermatol 1978;99:335–8.
- Rae V, Falanga V. Wrinkling due to mid-dermal elastolysis. Arch Dermatol 1989;125:950–1.
- El-Charif M, Mousani AM, Rubeiz NG, et al. Pseudoxanthoma elasticum-like papillary dermal elastolysis: a report of two cases. J Cutan Pathol 1994;21:252–5.
- Rongioletti F, Rebora A. Pseudoxanthoma elasticum-like papillary dermal elastolysis. J Am Acad Dermatol 1992;26:648–50.
- Cozzani E, Santoro F, Parodi A. Mid-dermal elastolysis with prothrombotic abnormalities: two cases. Br J Dermatol 2009;161:203–5.
- Lai JH, Murray SJ, Walsh NM Evolution of granuloma annulare to mid-dermal elastolysis: report of a case and review of the literature. J Cutan Pathol 2014;41:462–8.
- Harmon CB, Su WPD, Gagne EJ, et al. Ultrastructural evaluation of mid-dermal elastolysis. J Cutan Pathol 1994;21:233–8.
- Varadi DP, Saqueton AC. Perifollicular elastolysis. Br J Dermatol 1970;83:143–50.
- Sterling JC, Coleman N, Pye RJ. Mid-dermal elastolysis. Br J Dermatol 1994;130:502–6.
- Tajima S, Imazumi T, Kojiya H, et al. Elastin metabolism in skin fibroblasts explanted from a patient with mid-dermal elastolysis. Br J Dermatol 1999;140:752–4.
- Maghraoui G, Grossin M, Crickx B, et al. L'elastolyse acquise du derme moyen. Ann Dermatol Vénéréol 1994;121:259–65.
- Dick GF. Elastolytic activity of P. acnes and Staph. epidermidis in acne and normal skin. Acta Derm Venereol Suppl (Stockh) 1976;56:279–82.
- Wagner G, Sachse MM. Elastolysis mediodermalis: a case report and review of the literature. J Dtsch Dermatol Ges 2011;9:810–14.
- Eaton PA, Knable AL, Callen JP. Mid-dermal elastolysis in a patient undergoing hemodialysis. Cutis 2009;83:141–4.
- Cutillas E, Ferrando FJ, Marli ME, et al. Reticular variant of mid-dermal elastolysis after insertion of a pacemaker. Clin Exp Dermatol 2010;35:498–500.
- Lewis KG, Dill SW, Wilkel CS, et al. Mid-dermal elastolysis preceded by acute neutrophilic dermatosis. J Cutan Pathol 2004;31:72–6.
- Bayle-Lebey P, Periole B, Daste G, et al. Acquired localized elastolysis associated with varicose veins. Clin Exp Dermatol 1995;20:492–5.
- Posada C, No N, De la Torre C, Flórez A. Reticular variant of mid-dermal elastolysis. Australas J Dermatol 2013;54:69–71.
- Cohen PR, Tschen JA. Linear lumbar localised lysis of elastin fibres: a distinctive clinical presentation of mid-dermal elastolysis. J Clin Aesthet Dermatol 2013;6:32–9.
- Hashimoto K, Tye MJ. Upper dermal elastolysis; a comparative study with mid-dermal elastolysis. J Cutan Pathol 1994;21:533–40.
- Newlove T, Tzu J, Meehan S. Papillary dermal elastosis. Dermatol Online J 2011;17(10):12.
- Rongioletti F, Izakovic J, Romanelli P, et al. Pseudoxanthoma elasticum-like papillary dermal, elastolysis: a large case series with clinicopathological correlation. J Am Acad Dermatol 2012;67:128–35.
- Wang AR, Lewis K, Lewis M, et al. Papillary dermal elastosis: a unique elastic tissue disorder or an unusual manifestation of pseudoxanthoma elasticum-like papillary dermal elastolysis? J Cutan Pathol 2009;36:1010–13.
- Gambichler T. Mid-dermal elastolysis revisited. Arch Dermatol Res 2010;302:85–93.
Blepharochalasis, Ascher syndrome
- Jordan DR. Blepharochalasis syndrome: a proposed pathophysiologic mechanism. Can J Ophthalmol 1992;27:10–15.
- Tepaszto I, Liszkay L, Vass Z. Some data on the pathogenesis of blepharochalasis. Acta Ophthalmol (Copenh) 1963;41:167–75.
- Kaneoya K, Momota Y, Hatamochi A, et al. Elastin gene expression in blepharochalasis. J Dermatol 2005;32:26–9.
- Grasseger A, Romani N, Fritsch P, et al. Immunoglobulin A (IgA) deposits in lesional skin of a patient with blepharochalasis. Br J Dermatol 1996;135:791–5.
- Dózsa A, Károlyi ZS, Degrell P. Bilateral blepharochalasis. J Eur Acad Dermatol Venereol 2005;19:725–8.
- Brazin SA. Unilateral blepharochalasis. Arch Dermatol 1979;115:479–81.
- Harris WA, Dortzbach RK. Levator tuck. A simplified blepharoptosis procedure. Ann Ophthalmol 1975;7:873–8.
- Findlay GH. Idiopathic enlargements of the lips: cheilitis granulomatosa, Ascher's syndrome and double lip. Br J Dermatol 1954;66:129–38.
- Papanayotou PH, Hatzoitis JC. Ascher's syndrome: report of a case. Oral Surg Oral Med Oral Pathol 1973;35:467–71.
- Pitanguy I. Ascher's syndrome. Head Neck Surg 1988;10:309–10.
- Halling F, Sandrock D, Merten HA, et al. Das Ascher syndrome. Dtsch Z Mund Kiefer Gesichtschir 1991;15:440–4.
- Ali K. Ascher syndrome: a case report and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2007;103:e26–8.
- Ramesh BA. Ascher syndrome: review of literature and case report. Indian J Plast Surg 2011;44:147–9.
- Shah-Desai S, Sandy C, Collin R. Lax eyelid syndrome or ‘progeria’ of eyelid tissues. Orbit 2004;23:3–12.
Actinic granuloma and annular elastolytic giant cell granuloma
- Hanke CW, Bailin PL, Roenigk HH. Annular elastolytic giant cell granuloma: a clinicopathologic study of five cases and a review of similar entities. J Am Acad Dermatol 1979;1:413–21.
- Gutiérrez-González E, Pereiro M, Toribio J. Elastolytic actinic giant cell granuloma. Dermatol Clin 2015;33:331–41.
- Aso Y, Izaki S, Teraki Y. Annular elastolytic giant cell granuloma associated with diabetes mellitus: a case report and review of the Japanese literature. Clin Exp Dermatol 2011;36:917–19.
- Dowling GB, Wilson-Jones E. Atypical (annular) necrobiosis lipoidica of the face and scalp. Dermatologica 1967;135:11–26.
- Wilson-Jones E. Necrobiosis lipoidica presenting on the face and scalp. Trans St John's Hosp Dermatol Soc 1971;57:203–20.
- Campos-Munoz L, Diaz-Diaz RM, Quesada-Cortes A, et al. Annular elastolytic giant cell granuloma: a case report located in non-sun exposed areas. Actas Dermosifiliogr 2006;97:533–5.
- Ishibashi A, Yokoyama A, Hirano K. Annular elastolytic giant cell granuloma occurring in covered areas. Dermatologica 1987;174:293–7.
- Muramatsu T, Shirai T, Yamashina Y, et al. Annular elastolytic giant cell granuloma: an unusual case with lesions arising in non-sun-exposed areas. J Dermatol 1987;14:54–8.
- Pestoni C, Pereiro M, Toribio J. Annular elastolytic giant cell granuloma produced on an old burn scar and spreading after a mechanical trauma. Acta Derm Venereol 2003;83:312–13.
- Ozkaya-Bayazit E, Buyukbabani N, Baykal C, et al. Annular elastolytic giant cell granuloma: sparing of a burn scar and successful treatment with chloroquine. Br J Dermatol 1999;140:525–30.
- Davies MG, Newman P. Actinic granulomata in a young woman following prolonged sunbed usage. Br J Dermatol 1997;136:797–8.
- Lim DS, Triscott J. O'Brien's actinic granuloma in association with prolonged doxycycline phototoxicity. Australas J Dermatol 2003;44:67–70.
- Garg A, Kundu RV, Plotkin O, Aronson IK. Annular elastolytic giant cell granuloma heralding onset and recurrence of acute myelogenous leukemia. Arch Dermatol 2006;142:532–3.
- Al-Hoqail IA, Al-Ghamdi AM, Martinka M, et al. Actinic granuloma is a unique and distinct entity: a comparative study with granuloma annulare. Am J Dermatopathol 2002;24:209–12.
- McGrae JD. Actinic granuloma: a clinical, histopathologic and immunocytochemical study. Arch Dermatol 1986;122:43–8.
- O'Brien JP. Actinic granuloma: an annular connective tissue disorder affecting sun and heat-damaged (elastotic) skin. Arch Dermatol 1975;111:460–70.
- Prendiville J, Griffiths WAD, Russell-Jones R. O'Brien's actinic granuloma. Br J Dermatol 1985;113:353–8.
- Limas C. The spectrum of primary cutaneous elastolytic granulomas and their distinction from granuloma annulare: a clinicopathological analysis. Histopathology 2004;44:277–82.
- Berliner JG, Haemel A, Le Boit PE, et al. The sarcoidal variant of annular elastolytic granuloma. J Cutan Pathol 2013;49:917–20.
- Ratnavel RC, Grant JW, Handfield-Jones SE, Norris PG. O'Brien's actinic granuloma: response to isotretinoin. J R Soc Med 1995;88:528P–529P.
- Stefanaki C, Panagiotopoulos A, Kostakis P, et al. Actinic granuloma successfully treated with acitretin. Int J Dermatol 2005;44:163–6.
- Rongioletti F, Baldari M, Burlando M, et al. Papular elastolytic giant cell granuloma: report of a case associated with monoclonal gammopathy and responsive to topical tacrolimus. Clin Exp Dermatol 2010;35:145–8.
Granuloma multiforme
- Kumari R, Thappa DM, Chougule A, Adityan B. Granuloma multiforme: a report from India. Indian J Dermatol Venereol 2009;75:296–9.
- Leiker DL, Kok SH, Spaas JAJ. Granuloma multiforme: a new skin disease resembling leprosy. Int J Lepr 1964;32:368–76.
- Marshall J, Weber HW, Kok SH. Granuloma multiforme (Leiker). Dermatologica 1967;134:193–207.
- Leiker DL, Ziedses des Plantes M. Granuloma multiforme in Kenya. East Afr Med J 1967;44:429–36.
- Allenby CF, Wilson Jones E. Granuloma multiforme. Trans St John's Hosp Dermatol Soc 1969;55:88–98.
- Cherian S. Granuloma multiforme in India. Int J Lepr 1990;58:719–21.
- Verhagen ARHB, Koten JW, Chaddah VK, Patel RI. Skin diseases in Kenya: a clinical and histopathological study of 3168 patients. Arch Dermatol 1968;98:577–86.
- Garrett AS. Granuloma multiforme (called Nkanu disease in the 1940s and Mkar disease in 1964). Int J Lepr 1999;67:172–4.
- Cherian S. Is granuloma multiforme a photodermatosis? Int J Dermatol 1994;33:21–2.
Other elastolytic conditions
- Mattox AR, Marshall JA, Guo A. Granulomatous mycosis fungoides with clinical features of granulomatous slack skin. Cutis 2014;93:e4–5.
- Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T cell lymphoma (mycosis fungoides and Sézary syndrome): Part 1. Diagnosis: clinical and histopathologic features and new molecular and biologic markers. J Am Acad Dermatol 2014;70:205e1–16.
Acquired pseudoxanthoma elasticum-like syndromes
Perforating pseudoxanthoma elasticum
- Premathala S, Yesudian P, Thambiah AS. Periumbilical pseudoxanthoma elasticum with transepithelial elimination. Int J Dermatol 1982;10:604–5.
- Kazakis AM, Parish WR. Periumbilical perforating pseudoxanthoma elasticum. J Am Acad Dermatol 1988;19:384–8.
- Somarsundaram V, Premathala S, Rao NR, et al. Periumbilical perforating pseudoxanthoma elasticum. Int J Dermatol 1987;26:536–7.
- Budania A, De D, Saikia UN, Kanwar AJ, Mahajan R, Yadav S. Periumbilical perforating pseudoxanthoma elasticum: an acquired perforating disorder. Int J Dermatol 2012;51:439–41.
- Nickoloff BJ, Noodleman FR, Abel EA. Perforating pseudoxanthoma elasticum associated with chronic renal failure and haemodialysis. Arch Dermatol 1985;121:1321–2.
- Lund HZ, Gilbert CF. Perforating pseudoxanthoma elasticum. Its distinction from elastosis perforans serpiginosa. Arch Pathol Lab Med 1976;100:544–6.
- Pugashetti R, Shinkai K, Ruben BS et al. Calcium may preferentially deposit in areas of elastic tissue damage. J Am Acad Dermatol 2011;64:296–301.
- Rongioletti F, Izakovic J, Romanelli P, et al. Pseudoxanthoma elasticum-like papillary dermal, elastolysis: a large case series with clinicopathological correlation. J Am Acad Dermatol 2012;67:128–35.
- Sueki H, Amemiya M, Watanaba H, et al. Spontaneous resolution in a case of pseudo-pseudoxanthoma elasticum. Br J Dermatol 2001;144:213–15.
Acquired pseudoxanthoma elasticum
- Burge S, Ryan T. Penicillamine-induced pseudo-pseudoxanthoma elasticum in a patient with rheumatoid arthritis. Clin Exp Dermatol 1988;13:255–8.
- Na SY, Choi M, Kim MJ, Lee JH, Cho S. Penicillamine-induced elastosis perforans serpiginosa and cutis laxa in a patient with Wilson's disease. Ann Dermatol 2010;22:468–71.
- Light N, Meyrick-Thomas RH, Stephens A, et al. Collagen and elastin changes in d-penicillamine-induced pseudoxanthoma elasticum-like skin. Br J Dermatol 1986;114:381–8.
- Meyrick-Thomas RH, Kirby JDT. Elastosis perforans serpiginosa and pseudoxanthoma elasticum-like skin changes due to D-penicillamine. Clin Exp Dermatol 1985;10:386–91.
- Mainetti C, Masouyé I, Saurat JH. Pseudoxanthoma elasticum-like lesions in the L-tryptophan- induced eosinophilia-myalgia syndrome. J Am Acad Dermatol 1991;24:657–8.
- Winkelmann RK, Peters MS, Venencie PY. Amyloid elastosis. A new cutaneous and systemic pattern of amyloidosis. Arch Dermatol 1985;121:498–502.
- Vecchietti G, Masouyé I, Salomon D, et al. An unusual form of primary systemic amyloidosis: amyloid elastosis. Report of a case treated by haematopoietic cell transplantation. Br J Dermatol 2003;145:154–9.
- Fabbri E, Forni GL, Guerrini G, et al. Pseudoxanthoma elasticum-like syndrome and thalassaemias: an update. Dermatol Online J 2009;15(7):7.
- Barteselli G, Dell'arti L, Finger RP, et al. The spectrum of ocular alterations in patients with b-thalassaemia syndrome suggest a pathology similar to pseudoxanthoma elasticum. Ophthalmology 2014;121:709–18.
Saltpetre disease
- Christensen OB. An exogenous variety of pseudoxanthoma elasticum in old farmers. Acta Derm Venereol Suppl (Stockh) 1978;58:319–22.
- Neilson AO, Christensen OB, Hentzer B, et al. Saltpetre-induced dermal changes electron microscopically indistinguishable from pseudoxanthoma elasticum. Acta Derm Venereol Suppl (Stockh) 1978;58:323–7.
- Neri I, Marzaduri S, Berdazzi F, et al. Exogenous pseudoxanthoma elasticum: a new case in an old farmer. Acta Derm Venereol 1993;78:153–4.
Acrokeratoelastoidosis
- Costa OG. Ackrokeratoelastoidosis. Arch Dermatol Syphilol 1954;70:228–31.
- Rubegni P, De Aloe G, Romano C, Flori ML, Fimiani M. Acrokeratoelastoidosis: a report of two sporadic cases. Clin Exp Dermatol 1997;22:62–4.
- Rongioletti F, Betti R, Crosti C, Rebora A. Marginal papular acrokeratodermas: a unified nosography for focal acral hyperkeratosis, acrokeratoelastoidosis and related disorders. Dermatology 1994;188:28–31.
- Erkek E, Kocak M, Bozdoğan O, Atasoy P, Birol A. Focal acral hyperkeratosis: a rare cutaneous disorder within the spectrum of Costa acokeratoelastoidosis. Pediatr Dermatol 2004;21:128–30.
Acquired disorders of elastic tissue deposition
Linear focal elastosis
- Burket JM, Zelickson AS, Padilla RS. Linear focal elastosis (elastotic striae). J Am Acad Dermatol 1989;20:633–6.
- Vogel PS, Cardenas A, Ross EV, et al. Linear focal elastosis. Arch Dermatol 1995;131:855–6.
- Kanitakis J, Chouvet B, Dupin M, et al. Linear focal elastosis. Eur J Dermatol 1997;7:300–2.
- Péc J, Chromej I. Linear focal elastosis: what's new? J Eur Acad Dermatol Venereol 2004;18:247–9.
- Moiin A, Hashimoto K. Linear focal elastosis in a young black man: a new presentation. J Am Acad Dermatol 1994;30:874–7.
- Jang WS, Lee JW, Koo KH, et al. Could a growth spurt cause linear focal elastosis like striae distensae? Ann Dermatol 2012;24:81–3.
- Jeong JS, Lee JY, Kim MK, et al. Linear focal elastosis following striae distensae: further evidence of keloidal repair process in the pathogenesis of linear focal elastosis. Ann Dermatol 2011;23(Suppl. 2):S141–3.
- Choi SW, Lee JH, Woo HJ, et al. Two cases of linear focal elastosis: different histopathologic findings. Int J Dermatol 2000;39:207–9.
- Hagari Y, Mihara M, Morimura T, et al. Linear focal elastosis: an ultrastructural study. Arch Dermatol 1991;127:1365–8.
- Hashimoto K. Linear focal elastosis: keloidal repair of striae distensae. J Am Acad Dermatol 1998;39:309–13.
- Seo JK, Chen JS, Jung SY, et al. A case of linear focal elastosis with a family history. Ann Dermatol 2010;22:209–11.
- White G. Linear focal elastosis: a degenerative or regenerative process of striae distensae. J Am Acad Dermatol 1992;22:468.
- Neve S, Kirtschig G. Elastotic striae associated with striae distensae after application of very potent topical corticosteroids. Clin Exp Dermatol 2006;31:452–82.
Late-onset focal dermal elastosis
- Tajima S, Shimizu K, Izumi T, et al. Late-onset focal dermal elastosis: clinical and histological features. Br J Dermatol 1995;133:303–5.
- Limas C. Late onset focal dermal elastosis: a distinct clinicopathologic entity? Am J Dermatopathol 1999;21:381–3.
Elastofibroma dorsi
- Nagamine N, Nohara Y, Ito E. Elastofibroma in Okinawa: a clinicopathologic study of 170 cases. Cancer 1982;50:1794–805.
- Fukuda Y. Histogenesis of the unique elastinophilic fibres of elastofibroma. Hum Pathol 1987;18:424–9.
- Kapff PD. Elastofibroma of hand. J Bone Joint Surg 1987;69:468–9.
- Govoni E. Elastofibroma: an in vivo model of abnormal elastoneogenesis. Ultrastruct Pathol 1988;12:327–39.
- Gartmann H, Groth W, Kuhn A. Elastofibroma dorsi. Z Hautkr 1988;63:525–8.
- Yamazaki K. An ultrastructural and immunohistochemical study of elastofibroma: CD34, MEF-2, pronine 2 (CD33), and factor XIIIa-positive proliferating fibroblastic stromal cells connected by Cx43-type gap junctions. Ultrastruct Pathol 2007;31:209–19.
- Mortman KD, Hochheiser GM, Giblin EM, et al. Elastofibroma dorsi: clinicopathologic review of 6 cases. Ann Thorac Surg 2007;83:1894–7.
- Nagano S, Yokouchi M, Setoyama T, et al. Elastofibroma dorsi: surgical indications and complications of a rare soft tissue tumour. Mol Clin Oncol 2014;2:421–4.
- Karakurt O, Kaplan T, Gunal N, et al. Elastofibroma dorsi management and outcomes: review of 16 cases. Interact Cardiovasc Thorac Surg 2014;18:197–201.
Elastoderma
- Kornberg RL, Hendler SS, Oikarinen AI, et al. Elastoderma: disease of elastin accumulation within the skin. N Engl J Med 1985;312:771–2.
- Yen A, Wen J, Grau M, et al. Elastoderma. J Am Acad Dermatol 1995;33:389–92.
- de Waal AC, Blocx WA, Seyger MM, et al. Elastoderma: an uncommon cause of acquired hyperextensible skin. Acta Derm Venereol 2012;92:328–9.
Papular elastorrhexis
- Bordas X, Ferrandiz C, Ribera M, et al. Papular elastorrhexis: a variety of nevus anelasticus? Arch Dermatol 1987;123:433–4.
- Sears JK, Seabury Stone M, Argenyi Z. Papular elastorrhexis: a variant of connective tissue nevus. J Am Acad Dermatol 1988;19:409–14.
- Choonhakarn C, Jirarattanapochai K. Papular elastorrhexis. A distinct variant of connective tissue nevi or an incomplete form of Buschke–Ollendorff syndrome? Clin Exp Dermatol 2002;27:454–7.
- Choi Y, Jin SY, Lee JH, et al Papular elastorrhexis: a case and differential diagnosis. Ann Dermatol 2011;23(Suppl. 1):S53–6.
- Schorr WF, Opitz JM, Reyes CN. The connective tissue nevus–osteopoikilosis syndrome. Arch Dermatol 1992;106:208–14.
- Schirren H, Schirren CG, Stolz W, et al. Papular elastorrhexis: a variant of dermatofibrosis lenticularis disseminata (Buschke–Ollendorff syndrome). Dermatology 1994;189:368–72.
- Ryder HF, Antaya RJ. Nevus anelasticus, papular elastorrhexis and eruptive collagenoma: clinically similar entities with focal absence of elastic fibres in childhood. Pediatr Dermatol 2005;22:153–7.
- Lee SH, Park SH, Yoon TJ, et al. Papular elastorrhexis improved by intralesional injection of triamcinolone. J Dermatol 2001;28:569–71.
Fibromatoses and other causes of diffuse fibrosis
Fibromatoses
Palmar fascial fibromatosis
- Mikkelsen OA. The prevalence of Dupuytren's contracture in Norway. Acta Chir Scand 1972;138:695–700.
- Bower M, Nelson M, Gazzard BG. Dupuytren's contracture in patients infected with HIV. BMJ 1990;300:164–5.
- Evans RA. The aetiology of Dupuytren's disease. Br J Hosp Med 1986;35:198–9.
- Heathcote JG. Fibromatosis and diabetes mellitus. Lancet 1981;i:1420.
- Matthews P. Familial Dupuytren's contracture with predominant female expression. Br J Plast Surg 1979;32:120–3.
- Critchley EM, Vakil SD, Hayward HW, et al. Dupuytren's disease in epilepsy: result of prolonged administration of anticonvulsants. J Neurol Neurosurg Psychiatry 1976;39:498–50.
- Larkin JG, Frier BM. Limited joint mobility and Dupuytren's contracture in diabetic, hypertensive and normal populations. BMJ 1986;292:1494.
- Arafa M, Steingold RF, Noble J, et al. The incidence of Dupuytren's disease in patients with rheumatoid arthritis. J Hand Surg 1984;9B:165–6.
- Allen PW. The fibromatoses. A clinicopathologic classification based on 140 cases. Am J Surg Pathol 1977;1:255–70.
- Piérard GE, Lapière CM. Phenytoin dependent fibrosis in polyfibromatosis syndrome. Br J Dermatol 1979;100:335–41.
- Hassell TM, Page RC, Narayanan AS, et al. Diphenylhydantoin (Dilantin) gingival hyperplasia: drug-induced abnormality of connective tissue. Proc Natl Acad Sci USA 1976;73:2909–12.
- Kiess W, Butenandt O. Development of Dupuytren's contracture during growth hormone therapy. Lancet 1993;342:181–2.
- Palmer KT, D'Angelo S, Syddall H, et al. Dupuytren's contracture and occupational exposure to hand-transmitted vibration. Occup Environ Med 2014;71:241–5.
- Murrell GAC, Francis MJ, Bromley L, et al. Free radicals and Dupuytren's contracture. BMJ 1987;295:1373–5.
- Granger DN. Superoxide radicals in feline intestinal ischaemia. Gastroenterology 1981;81:22–9.
- James WD, Odom RB. The role of the myofibroblast in Dupuytren's contracture. Arch Dermatol 1980;116:807–11.
- Baird KS, Alwan WH, Crossan JF, Wojciak B. T-cell mediated response in Dupuytren's disease. Lancet 1993;341:1622–3.
- Gabbiani G, Manjo G. Dupuytren's contracture: fibroblast contraction. An ultrastructural study. Am J Pathol 1972;66:131–46.
- Murrell GA. The role of the fibroblast in Dupuytren's contracture. Hand Clin 1991;7:669–80.
- Bailey AJ, Sims TJ, Gabbiani G, et al. Collagen of Dupuytren's disease. Clin Sci Mol Med 1977;53:499–502.
- Ulrich D, Hrynyschyn K, Pallua N. Matrix metalloproteinases and tissue inhibitors of metalloproteinases in sera and tissue of patients with Dupuytren's disease. Plast Reconstr Surg 2003;112:1279–86.
- Koźna EM, Glowacki A, Olazyk K, et al. Dermatan sulphate remodeling associated with advanced Dupuytren's contracture. Acta Biochim Pol 2007;54:821–30.
- Ling RSM. The genetic factor of Dupuytren's disease. J Bone Joint Surg 1963;45B:709–18.
- Becker K, Tinschert S, Lienert A, et al. The importance of genetic susceptibility in Dupuytren's disease. Clin Genet 2015;87:483–7.
- Forrester HB, Temple-Smith B, Ham S, et al. Genome-wide analysis using exon arrays demonstrates an important role of expression of extracellular matrix, fibrotic control and tissue remodelling genes in Dupuytren's disease. PLOS One 2013;8(3):e59056.
- Rodrigo JJ, Niebauer JJ, Brown RL, et al. Treatment of Dupuytren's contracture. Long-term result after fasciotomy and fascial excision. J Bone Joint Surg Am 1976;58:380–7.
- Shaw DL, Wise D, Holms W. Dupuytren's disease treated by palmar fasciectomy and an open palm technique. J Hand Surg 1996;21B:484–5.
- Gelman S, Schlenker R, Bachoura A, et al. Minimally invasive partial fasciectomy for Dupuytren's contracture. Hand (NY) 2012;7:364–9.
- Badalamente MA, Hurst LC, Hentz VR. Collagen as a clinical target. Nonoperative treatment of Dupuytren's disease. J Hand Surg 2002;27A:788–98.
- Meals RA, Hentz VR. Technical tips for collagenase injection treatment for Dupuytren's contracture. J Hand Surg 2014;39A:1195–200.
- Murrell GAC. Hypothesis for the resolution of Dupuytren's contracture with allopurinol. Spec Sci Technol 1987;10:107–12.
- Hurst LC, Badalamente MA. Nonoperative treatment of Dupuytren's disease. Hand Clin 1999;15:97–107.
- Degreef I, Tejpar S, Sciot R, De Smet L. High-dosage tamoxifen as neoadjuvant treatment in minimally invasive surgery for Dupuytren's disease in patients with a strong predisposition toward fibrosis: a randomized controlled trial. J Bone Joint Surg Am 2014;96:655–62.
- Kang YM, Choi YR,Yun CO, et al. Down-regulation of collagen synthesis and matrix metalloproteinase expression in myofibroblasts from Dupuytren's nodule using adenovirus-mediated relaxin gene therapy. J Orthop Res 2014;32:515–23.
Plantar fascial fibromatosis
- Allen RA, Woolner LB, Ghormley RK, et al. Soft tissue tumours of the sole with special reference to plantar fibromatosis. J Bone Joint Surg Am 1995;37:14–26.
- Warthan TL, Rudolf RI, Gross PR, et al. Isolated plantar fibromatosis. Arch Dermatol 1973;108:823–5.
- Gudmundsson KG, Jónsson T, Arngrimsson R. Association of morbus ledderhose with Dupuytren's contracture. Foot Ankle Int 2013;34:841–5.
- Touraine S, Bousson V, Kaci R, et al. Plantar fibromatosis may adopt the brain gyriform pattern of a low-grade fibromyxoid sarcoma. Foot (Edinb) 2013;23;88–92.
- Fleischmajer R, Nedwich A, Reeves JR, et al. Juvenile fibromatoses. Arch Dermatol 1973;107:574–9.
- Motolese A, Mola F, Cherubino M, et al. Squamous cell carcinoma and Ledderhose disease: a case report. Int J Low Extrem Wounds 2013;12:297–300.
- Godette A, O'Sullivan M, Menelaus MB. Plantar fibromatosis of the heel in children: a report of 14 cases. J Pediatr Orthop 1997;17:16–17.
- Jacob CI, Kumm RC. Benign anteromedial plantar nodules of childhood: a distinct form of plantar fibromatosis. Pediatr Dermatol 2000;17:472–4.
- Veith NT, Tscherning T, Hirsting T, et al. Plantar fibromatosis: topical review. Foot Ankle Int 2013;34:1742–6.
- Knobloch K, Vogt PM. High-energy focused extracorporeal shockwave therapy reduces pain in plantar fibromatosis (Ledderhoses's disease). BMC Res Notes 2012;5:542.
Penile fibromatosis
- Chilton CP, Castle WM, Westwood CA, et al. Factors associated in the aetiology of Peyronie's disease. Br J Urol 1982;54:748–50.
- Pryor JP. Association between Peyronie's disease and chronic degenerative arterial disease rather than b-adrenoreceptor blocking agents. Prog Reprod Biol Med 1983;9:23–6.
- Smith BH. Peyronie's disease. Am J Clin Pathol 1966;45:670–8.
- Gingell JC, Desai KM. Peyronie's disease. Treatment should always restore sexual function. BMJ 1988;297:1489–90.
- Williams JL, Thomas GG. The natural history of Peyronie's disease. J Urol 1970;103:75–6.
- Balconi G, Angeli E, Nessi R, et al. Ultrasonic evaluation of Peyronie's disease. Urol Radiol 1988;10:85–8.
- Rollandi GA, Tentarelli T, Vespier M, et al. Computed tomographic findings in Peyronie's disease. Urol Radiol 1985;7:153–6.
- Bystrom J, Johansson B, Edgren J, et al. Induratio penis plastica (Peyronie's disease). Cavernosography in assessment of the disease process. Scand J Urol Nephrol 1974;8:155–61.
- Desai KM, Gingell JC. Outpatient assessment of penile curvature. Br J Urol 1987;60:470–1.
- Shaw EJ, Mitchell GL, Tan RB, et al. The non surgical treatment of Peyronie Disease: 2013 update. World J Mens Health 2013;31:183–92.
- Gupta R, Gupta S. Peyronie's disease treated with oral weekly dexamethasone and continuous low dose cyclophosphamide. Indian J Dermatol 2014;59:317.
- Gelbard MK, Lindner A, Kaufman JJ, et al. The use of collagenase in the treatment of Peyronie's disease. J Urol 1985;134:280–3.
- Honig SL. Intralesional collagenase in the treatment of Peyronie's disease. Ther Adv Urol 2014;6:47–53.
- Kokab A, Wylie K, Allen P, Shetty A, Davies-South D. Structured self-rated response to iontophoresis with verapamil and dexamethasone in Peyronie's disease. Adv Urol 2014; Article ID 957013 (epub).
- Chung E, Brock G. Penile traction therapy and Peyronie's disease: a state of the art review of the current literature. Ther Adv Urol 2013;5:59–65.
- Miranda AF, Sampaio FJ. A geometric model of plaque incision and graft for Peyronie's disease with geometric analyses of different techniques. J Sex Med 2014;11:1546–53.
- Hsu GC, Chen HS, Hsieh CH, et al. Long term results of autologous venous grafts for penile morphological reconstruction. J Androl 2007;28:186–93.
Knuckle pads
- Garrod AE. On an unusual form of nodule upon the joints of the fingers. BMJ 1893;29:157–61.
- Mikkelson OH. Knuckle pads in Dupuytren's disease. Hand 1977;9:301.
- Morginson WJ. Discrete keratodermas over the knuckle and finger articulations. Arch Dermatol 1955;71:349–53.
- Hyman CH, Cohen PR. Report of a family with idiopathic knuckle pads and review of idiopathic and disease-associated knuckle pads. Dermatol Online J 2013;19(5):18177.
- Bart RS, Pumphrey RE. Knuckle pads, leukonychia and deafness. N Engl J Med 1967;276:202–7.
- Lu Y, Guo C, Liu Q, et al. A novel mutation of keratin 9 in epidermolytic palmoplantar keratoderma combined with knuckle pads. Am J Med Genet 2003;120A:345–9.
- Ritter SB, Peterson G. Esophageal cancer, hyperkeratosis and oral leukoplakia. JAMA 1976;235:1723.
- Lagier R, Meineke R. Pathology of knuckle pads. Virchows Arch 1975;365:185–91.
- Kodama BF, Genty RH, Fitzpatrick JE. Paspules and plaques over the joint space. Arch Dermatol 1993;129:1044–5.
- Dolmans GH, de Bock GH, Werker PM. Dupuytren diathesis and genetic risk. J Hand Surg 2012;37A:2106–11.
- Codispoti A, Colombo E, Zocchi L, et al. Knuckle pads in an epidermal palmoplantar keratoderma patient with Keratin 9R163W transgrediens expression. Eur J Dermatol 2009;19:114–18.
- Peterson CM, Barnes CJ, Davis LS. Knuckle pads: does knuckle cracking play an etiologic role? Pediatr Dermatol 2000;17:450–2.
- Rushing ME, Sheehan DJ, Davis LS. Video game induced knuckle pads. Pediatr Dermatol 2006;23:455–7.
- Myeroff CM, Stern PJ. Granuloma annulare mimicking dorsal knuckle pads. J Hand Surg 2011;36A:1039–41.
- Bettoni L, Bani L, Airò P. Rheumatoid nodules: the importance of a correct differential diagnosis. Eur Ann Allergy Clin Immunol 2011;43:95–6.
- Weiss E, Amini S. A novel treatment for knuckle pads with intralesional fluorouracil. Arch Dermatol 2007;143:1458–60.
Pachydermodactyly
- Al Hammadi A, Hakim M. Pachydermodactyly: case report and review of the literature. J Cutan Med Surg 2007;11:185–7.
- Verbov J. Pachydermodactyly: a variant of the true knuckle pad. Arch Dermatol 1975;111:524.
- Meunier L, Pailler C, Barneon G, Meynadier J. Pachydermodactyly or acquired digital fibromatosis. Br J Dermatol 1994;131:744–6.
- Draluck JC, Kopf AU, Hodak E. Pachydermodactyly: first report in a woman. J Am Acad Dermatol 1992;27:303–5.
- Bardazzi F, Fanti PA, De Padova MP, et al. Localized pachydermodactyly in a woman. Acta Derm Venereol Suppl (Stockh) 1994;74:152–3.
- Bardazzi F, Neri I, Fanti PA, et al. Pachydermodactyly in two young girls. Pediatr Dermatol 1996;13:288–91.
- Curley RK, Hudson PM, Marsden RA. Pachydermodactyly: a rare form of digital fibromatosis. Report of four cases. Clin Exp Dermatol 1991;16:121–3.
- Hunt R, Mandal R, Walters R, Schaffer JV. Pachydermodactyly. Dermatol Online J 2010;16(11):5.
- Dias JM, Costa MM, Roman JC, et al. Pachydermodactyly in a 16 year old boy: a rare pathology affecting adolescents. J Clin Rheumatol 2012;18:246–8.
- Callot V, Wechsler J, Hovanian A, et al. Pachydermodactyly and atrophia maculosa varioliformis cutis. Dermatology 1995;190:56–8.
- Sagransky MJ, Pichardo-Geisinger RO, Muñoz-Ali D, et al. Pachydermodactyly from repetitive motion in poultry processing workers: a report of 2 cases. Arch Dermatol 2012;148:925–8.
- Dallos T, Oppl B, Kovács L, Zwerina J. Pachydermodactyly: a review. Curr Rheumatol Rep 2014;16:442.
- Russo F, Rodriguez-Picardo A, Camacho F. Familial pachydermodactyly. Acta Derm Venereol 1994;74:386–7.
- Tompkins SD, McNutt NS, Shea CR. Distal pachydermodactyly. J Am Acad Dermatol 1998;38:359–62.
- El-Hallak M, Lovell D. Pachydermodactyly mimicking juvenile idiopathic arthritis. Arthritis Rheum 2013;65:2736.
- Kopera D, Soyer HP, Kerl H. An update on pachydermodactyly and a report of three additional cases. Br J Dermatol 1995;133:433–7.
- Lautenschlager S, Itin PH, Rufli T. Pachydermodactyly: reflecting obsessive-compulsive behavior? Arch Dermatol 1994;130:387.
- Chamberlain AJ, Venning VA, Wojnarowska F. Pachydermodactyly: a forme fruste of knuckle pads? Australas J Dermatol 2003;44:140–3.
- Sinha NK, Ling SP, Nema SK, et al. Pachydermodactyly does not require rheumatologic work-up. J Postgrad Med 2013;59:335–6.
- Plana Pla A, Bassas Vila J, Toro Montecinos MA, et al. Pachydermodactyly successfully treated with triamcinolone injection. Actas Dermosifilogr 2014;105:319–21.
White fibrous papulosis of the neck
- Shimizu H, Nishikawa T, Kimura S. White fibrous papulosis of the neck: a review of 16 cases. Jpn J Dermatol B 1985;95:1077–84.
- Shimizu H, Kimura S, Harada T, et al. White fibrous papulosis of the neck: a new clinicopathologic entity? J Am Acad Dermatol 1989;20:1073–7.
- Cerio R, Gold S, Wilson-Jones E. White fibrous papulosis of the neck. Clin Exp Dermatol 1991;16:224–5.
- Redondo P, Vázquez-Doval J, de Alava E. White fibrous papulosis of the neck. Dermatology 1993;186:238–9.
- Balus L, Amanthea A, Donati P, et al. Fibroelastolytic papulosis of the neck: a report of 20 cases. Br J Dermatol 1997;137:461–6.
- Perrin CH, Castenet J, Lacour J-P, et al. Papulose blanche du cou. Aspects cliniques de pseudoxanthoma elastique. Ann Dermatol Vénéréol 1996;123:114–17.
- Kandhari R, Kandhari S, Jain S. White fibrous papulosis of the neck [Letter]. Indian J Derm Venerol Leprol 2015;81:224.
- Patterson AT, Beasley KJ, Kobayashi TT. Fibroelastolytic papulosis: histopathologic confirmation of disease spectrum variants in a single case. J Cutan Pathol 2015; doi: 10.1111/cup.12569 (epub).
- Siragusa M, Schepis C, Palazzo R, et al. Skin pathology findings in a cohort of 1500 adult and elderly subjects. Int J Dermatol 1999;38:361–6.
Camptodactyly
- Engbar WD, Flatt AF. Camptodactyly. An analysis of sixty-six patients and twenty-four operations. J Hand Surg 1977;2A:216–24.
- Johnson HA. The renaissance fifth finger. J Roy Soc Med 2005;98;87.
- Battaglia A, Hayme HE, Dallapiccola B, et al. Further delineation of deletion 1p36 syndrome in 60 patients: a recognisable phenotype and common cause of developmental delay and mental retardation. Pediatrics 2008;121:404–10.
- Gigante MC, Santori FS, Zoppini A, et al. Familial erosive arthritis associated with camptodactyly. Scand J Rheumatol 1970;19:239–44.
- Jacobs JC, Downey JA. Juvenile rheumatoid arthritis. In: Downey JH, Low NC, eds. The Child with Disabling Illness. Philadelphia: Saunders, 1974:5–24.
- Martinez-Lavin M, Buendia A, Delgardo E, et al. A familial syndrome of pericarditis, arthritis and camptodactyly. N Engl J Med 1983;309:224–5.
- Laxer RM, Cameron BJ, Chaisson D, et al. The camptodactyly–arthropathy–pericarditis syndrome: case report and literature review. Arthritis Rheum 1986;29:439–44.
- Choi B-R, Lim Y-H, Joo K-B. Camptodactyly, arthropathy, coxa vara, pericarditis (CACP) syndrome: a case report. J Korean Med Sci 2004;19:907–10.
- Kakkar RM, Soneji S, Badhe RR, Desai SB. Camptodactyly–coxa vara–pericarditis syndrome: important differential for juvenile idiopathic arthritis. J Clin Imaging Sci 2013;3:24.
- Halal F, Fraser FC. Camptodactyly, cleft palate and club foot (the Gordon syndrome). A report of a large pedigree. J Med Genet 1979;16:149–50.
- Toydemir RM, Brassington AF, Beyrak-Toydemir P. A novel mutation in FGR3 causes camptodactyly, tall stature, and hearing loss (CATSHL) syndrome. Am J Hum Genet 2006;79:935–41.
- Raphael SA, Blau EB, Zhang WH, et al. Analysis of a large kindred with Blau syndrome for HLA, autoimmunity and sarcoidosis. Am J Dis Child 1993;147:842–8.
- Nevin NC, Hurwitz LJ, Neill DW. Familial camptodactyly with taurinuria. J Med Genet 1966;3:265–8.
- Dagoneau N, Bellais S, Blanchet P, et al. Mutations in cytokine receptor-like factor 1 (CRLF1) account for both Crisponi and cold-induced sweating syndromes. Am J Hum Genet 2007;80:966–70.
- Weaver DD, Graham CB, Thomas IT, et al. A new overgrowth syndrome with accelerated skeletal maturation, unusual facies and camptodactyly. J Pediatr 1974;84:547–52.
- Le Bourhis L, Benko S, Girardin SE. Nod1 and Nod2 in innate immunity and human inflammatory disorders. Biochem Soc Trans 2007;35:1479–84.
- Parish JG, Horn DB, Thompson M, et al. Familial streblodactyly with amino-aciduria. BMJ 1963;ii:1247–50.
- Donofrio P, Ayala F. Familial strebodactyly. Acta Derm Venerol 1983;63;361–3.
- Goffin D, Lenoble E, Marin-Braun F, et al. Camptodactyly: classification and therapeutic results. Ann Chir Main 1994;13:20–5.
- Foucher G, Loréa P, Khouri RK, et al. Camptodactyly as a spectrum of congenital deficiencies: a treatment algorithm based on clinical examination. Plast Reconstr Surg 2006;117:1897–905.
- Flatt AE. The troubles with pinkies. Proc Bayl Univ Med Cent 2005;18:341–4.
- Kakinoki R, Duncan SFM, Ohta S, et al. Simultaneous reconstruction of a palmar skin defect and the digital artery with an arterialized venous flap after correction of camptodactyly with severe flexion deformities. Hand (NY) 2011;6:645–9.
Juvenile fibromatoses
Infantile myofibromatosis
- Netscher DT, Baumholtz MA, Popek E. Non-malignant fibrosing tumors in the pediatric hand: a clinicopathologic case review. Hand (NY) 2009;4:2–11.
- Mashiah J, Hadj-Rabia S, Dompmartin A, et al. Infantile myofibromatosis: a series of 28 cases. J Am Acad Dermatol 2014;71:264–70.
- Martignetti JA, Tian L, Li D, et al. Mutations in PDGFRB cause autosomal-dominant infantile myofibromatosis. Am J Hum Genet 2013;92:1001–7.
- Cheung YH, Gayden T, Campeau PM, et al. A recurrent PDGFRB mutation causes familial infantile myofibromatosis. Am J Hum Genet 2013;92:996–1000.
- Lee JW Mutations in PDGFRB and NOTCH3 are the first genetic causes identified for autosomal dominant infantile myofibromatosis. Clin Genet 2013;84:3400–1.
- Gatibelza ME, Vazgucz BR, Bereni N, et al. Isolated infantile myofibromatoisis of the upper eyelid: uncommon localization and long-term results after surgical management. J Pediatr Surg 2012;47:1457–9.
- Kikuehi K, Abe R, Shinkuma S, et al. Spontaneous remission of solitary-type infantile myofibromatosis. Case Rep Dermatol 2011;3:181–5.
- Martin JM, Jordá E, Calduch L, et al. Self-healing generalized infantile myofibromatosis. J Eur Acad Dermatol Venerol 2008;22:236–8.
- Wu SY, McCavit TL, Cederberg K, Galindo RL, Leavey PJ. Chemotherapy for generalised infantile myofibromatosis with visceral involvement. J Pediatr Hematol Oncol 2015;37:402–5.
Juvenile hyaline fibromatosis
- Iwata S, Horiuchi R, Maeda H, et al. Systemic hyalinosis or juvenile fibromatosis. Ultrastructural and biochemical study of cultured skin fibroblasts. Arch Dermatol Res 1980;267:115–21.
- Chitale AR, Murthy AK, Maniar JK, et al. Juvenile hyaline fibromatosis. Ultrastruct Pathol 1987;11:771–5.
- Ishikawa H, Maeda H, Takamatsu H, et al. Systemic hyalinosis (juvenile hyaline fibromatosis). Ultrastructure of the hyaline with particular reference to the cross-banded structure. Arch Dermatol Res 1979;265:195–206.
- Finlay AY, Ferguson SD, Holt PJA, et al. Juvenile hyaline fibromatosis. Br J Dermatol 1983;108:609–16.
- Denadai R, Bertola DR, Raposo-Amaral CE. Hyaline fibromatosis syndrome. New unifying term and surgical approach. Indian J Pathol Microbiol 2012;55:262.
- Gupta CK, Singhi MK, Baisal M, et al. Juvenile hyaline fibromatosis in siblings. Indian J Dermatol Venereol Leprol 2005;71:115–18.
- Mayer DA, Silva A. Juvenile hyaline fibromatosis. A histologic and histochemical study. Arch Pathol Lab Med 1988;112:928–31.
- Winik B, Boente M, Asail R. Juvenile hyaline fibromatosis: ultrastructural study. Am J Dermatopathol 1998;20:372–8.
- Krishnamurthy J, Dalal BS, Sunita Gubanna MV. Juvenile hyaline fibromatosis. Indian J Dermatol 2011;56:731–3.
- Fong K, Rama Devi AR, Lai-Cheng JE, et al. Infantile systemic hyalinosis associated with a putative splice-site mutation in the ANTXR2 gene. Clin Exp Dermatol 2012;37:635–8.
- Rampoldi L. Different molecular consequences of frameshift mutations in the ANTXR2 gene. Hum Mutat 2013;34(7):v.
- Camarasa JG, Moreno A. Juvenile hyaline fibromatosis. J Am Acad Dermatol 1987;16:881–3.
- Fayad MN, Yacoub A, Salman S, et al. Juvenile hyaline fibromatosis. Two new cases and a review of the literature. Am J Med Genet 1987;26:123–31.
- Landing BH, Nadorra R. Infantile systemic hyalinosis: report of four cases of a disease, fatal in infancy, apparently different from juvenile systemic hyalinosis. Pediatr Pathol 1986;6:55–79.
- Remberger K, Krieg J. Fibromatosis hyalinica multiplex (juvenile hyaline fibromatosis). Cancer 1985;56:614–24.
- Quintal D, Jackson R. Juvenile hyaline fibromatosis. A 15-year follow up. Arch Dermatol 1985;121:1062–3.
Nephrogenic systemic fibrosis
- Cowper S, Robin H, Steinberg S, et al. Scleromyxedema-like cutaneous diseases in renal-dialysis patients. Lancet 2000;356:1000–1.
- Chopra T, Kandukurti K, Shah S, Ahmed R, Panesar M. Understanding nephrogenic systemic fibrosis. Int J Nephrol 2012; Article ID 912189.
- Swaminathan S, High WA, Ranville J, et al. Cardiac and vascular metal deposition with high mortality in nephrogenic systemic fibrosis. Kidney Int 2008;73:1413–18.
- Daftari Besheli L, Aran S, Shagdar K, et al. Current status of nephrogenic systemic fibrosis. Clin Radiol 2014;69:661–8.
- Weller A, Barber JL, Olsen OE. Gadolinium and nephrogenic systemic fibrosis: an update. Pediatr Nephrol 2013;29:1927–37.
- Grobner T. Gadolinium: a specific trigger for the development of nephrogenic fibrosing dermopathy and nephrogenic systemic fibrosis? Nephrol Dial Transplant 2006;21:1104–8.
- Edward M, Quinn J, Mukherjee S, et al. Gadodiamide contrast agent ‘activates’ fibroblasts: a possible cause for nephrogenic systemic fibrosis. J Pathol 2008;214:584–93.
- Schmidt-Lauber C, Bossaller L, Abujudeh HH, et al. Gadolinium-based compounds induce NLRP3-dependent IL-1b production and peritoneal inflammation. Ann Rheum Dis 2014; doi: 10.1136/annrheumdis-2013-204900 (epub).
- Nainani N, Panesar M. Nephrogenic systemic fibrosis. Am J Nephrol 2008;29:1–9.
- Swaminathan S, Ahmed I, McCarthy JT, et al. Nephrogenic fibrosing dermopathy and high-dose erythropoietin therapy. Ann Intern Med 2006;145:234–5.
- Naylor E, Hu S, Robinson-Bostom L. Nephrogenic systemic fibrosis with septal panniculitis mimicking erythema nodosum. J Am Acad Dermatol 2008;58:149–50.
- Solomon GJ, Wu E, Rosen PP. Nephrogenic systemic fibrosis mimicking inflammatory breast carcinoma. Arch Pathol Lab Med 2007;131:145–8.
- Holtzem KE, Nardone B, Lomasney JW, et al. Superior vena cava obstruction associated with nephrogenic systemic fibrosis. J Drugs Dermatol 2014;13:615–18.
- Bernstein EJ, Isakova T, Sullivan ME, Chibnik LB, Wolf M, Kay J. Nephrogenic systemic fibrosis is associated with hypophosphataemia: a case-control study. Rheumatology (Oxford) 2014;53:1613–17.
- Streams BN, Liu V, Liegois N, et al. Clinical and pathologic features of nephrogenic fibrosing dermopathy: a report of two cases. J Am Acad Dermatol 2003;48:42–7.
- Kalb RE, Helm TN, Sperry H, et al. Gadolinium-induced nephrogenic systemic fibrosis in a patient with an acute and transient kidney injury. Br J Dermatol 2007;158:607–10.
- Linfert DR, Schell JO, Fine DM . Treatment of nephrogenic systemic fibrosis: limited options but hope for the future. Semin Dial 2008;21:155–9.
- Robinson MR, Routhouska SB, Paspulati RM, et al. Alefacept therapy for nephrogenic systemic fibrosis: a case series. J Drugs Dermatol 2011;10:922–4.
- Poisson JL, Low A, Park YA. The treatment of nephrogenic systemic fibrosis with therapeutic plasma exchange. J Clin Apher 2013;28:317–20.
Environmental and drug-induced scleroderma
- Smith V, Vanthuyne M, Vander Cruysson B, et al. Over-representation of construction-related occupations in male patients with systemic sclerosis. Ann Rheum Dis 2008;67:1448–50.
- Ward AM, Udnoon S, Watkins J, et al. Immunological mechanism in the pathogenesis of vinyl chloride disease. BMJ 1976;i:936–8.
- Black CM, Pereira S, McWhirter A, et al. Genetic susceptibility to scleroderma-like syndrome in symptomatic and asymptomatic workers exposed to vinyl chloride. J Rheumatol 1986;13:1059–62.
- Sluis-Cremer GK, Hessel PA, Nizdo EH, et al. Silica, silicosis and progressive systemic sclerosis. Br J Ind Med 1985;42:838–43.
- Marie I, Gehanno JF, Bubenheim M, et al. Prospective study to evaluate the association between systemic sclerosis and occupational exposure and review of the literature. Autoimmun Rev 2014;13:151–6.
- Saihan EM, Burton JL, Heaton KW. A new syndrome with pigmentation, scleroderma, gynaecomastia, Raynaud's phenomenon and peripheral neuropathy. Br J Dermatol 1978;99:437–40.
- Sparrow GP. A connective tissue disease similar to vinyl chloride disease in a patient exposed to perchlorethylene. Clin Exp Dermatol 1977;2:17–22.
- Yamakage A, Ishikawa H, Saito Y, et al. Occupational scleroderma-like disorders occurring in men engaged in the polymerization of epoxy resins. Dermatologica 1980;161:33–44.
- Rothschild B Acrylamine-induced autoimmune phenomena. Clin Rheumatol 2010;29:999–1005.
- Noonan CW, Pfau JC, Carson TC, et al. Nested case-control study of autoimmune disease in an asbestos-exposed population. Environ Health Perspect 2006;114:1243–7.
- Iglesias JL, De Moragas JM. The cutaneous lesions of the Spanish toxic oil syndrome. J Am Acad Dermatol 1983;9:159–60.
- Phelps RG, Fleishmajer R. Clinical, pathologic, and immunological manifestations of the toxic oil syndrome. J Am Acad Dermatol 1988;18:313–24.
- Kaufman LD, Seidman RJ, Phillips ME, et al. Cutaneous manifestations of the L-tryptophan-associated eosinophilia–myalgia syndrome: a spectrum of sclerodermatous disease. J Am Acad Dermatol 1990;23:1063–9.
- Kilbourne EM, Posada de la Paz M, Borda IA, et al. Toxic oil syndrome: a current clinical and epidemiologic summary, including comparisons with the eosinophilia–myalgia syndrome. J Am Coll Cardiol 1991;18:711–17.
- Varga J, Jimenez SA. Chemical exposure-induced cutaneous fibrosis. Lessons from ‘experiments of nature’. Arch Dermatol 1994;130:97–100.
- Slutsker L, Hoesly FC, Miller L, et al. Eosinophilia–myalgia syndrome associated with exposure to tryptophan from a single manufacturer. JAMA 1990;264:213–17.
- Mayeno AN, Belongia EA, Lin F, et al. 3-(Phenylamino)alanine, a novel aniline-derived amino acid associated with the eosinophilia–myalgia syndrome: a link to the toxic oil syndrome. Mayo Clin Proc 1992;67:1134–9.
- Kaufman LD, Gruber BL, Gomez-Reion JJ. Fibrogenic growth factors in the eosinophilia–myalgia syndrome and the toxic oil syndrome. Arch Dermatol 1994;130:41–7.
- Palestine RF, Millas JL, Spigel GT, et al. Skin manifestations of pentazocine abuse. J Am Acad Dermatol 1980;2:47–55.
- Barthelemy H, Hermier C, Perrot H. Nécrose cutanée avec évolution sclérodermiforme aprés l'injection souscutanée d'heparinate de calcium. Ann Dermatol Vénéréol 1985;112:245–7.
- Brunskill NJ, Berth-Jones J, Graham-Brown RAC. Pseudosclerodermatous reaction to phytomenadione injection (Texier's syndrome). Clin Exp Dermatol 1988;13:276–8.
- Pujol RM, Puig L, Moreno A. Pseudoscleroderma secondary to phytomenadione (vitamin K1) injections. Cutis 1989;43:365–8.
- Morel A, Betlloch I. Morphea-like reaction from vitamin K1. Int J Dermatol 1995;34:201–2.
- Bourrat E, Moraillon I, Vignon-Pennamen MD. Placard sclérodermiforme de la cuisse de l'enfant aprés injection de vitamine K1 à la naissance. Ann Dermatol Vénéréol 1996;123:634–8.
- Bernstein RM, Hall MA, Gostelow BE. Morphoea-like reaction to D-penicillamine therapy. Ann Rheum Dis 1981;40:42–4.
- Goihman-Yahr M, Leal G, Essenfeld-Yahr E. Generalised morphea: a side effect of valproate sodium? Arch Dermatol 1980;116:621.
- Stewart FA, Gonino AC, Elewski BE. New side effect of TNF-alpha inhibitors: morphoea. Skinmed 2013;11:59–60.
- Dospinescu P, Jones GT, Basu N. Environmental risk factors in systemic sclerosis. Curr Opin Rheumatol 2013;25:179–83.
- Finch WR, Rodnan GP, Buckingham RB, et al. Bleomycin-induced scleroderma. J Rheumatol 1980;7:651–9.
- Liu S, Herault Y, Pavlovic G, et al. Skin progenitor cells contribute to bleomycin-induced skin fibrosis. Arthritis Rheum 2014;66:707–13.
- Sternberg EM, van Woert MH, Young SN, et al. Development of a scleroderma-like illness during therapy with L-5-hydroxytryptophan and carbidopa. N Engl J Med 1980;303:782–7.
- Hassell TM, Page RC, Narayanan AS, et al. Diphenylhydantoin (Dilantin) gingival hyperplasia: drug-induced abnormality of connective tissue. Proc Natl Acad Sci USA 1976;73:2909–12.
- Guistiniani S, Robustelli F, Marieni M. Hyperplastic gingivitis during diltiazem therapy. Int J Cardiol 1987;15:247–9.
- Trunnell TN, Waisman M. Hypertrophic retroauricular folds attributable to diphenylhydantoin. Cutis 1982;30:207–9.
- Ilia R, Goldfarb B, Gueron M. Skin thickening and sensory loss of the feet during diltiazem therapy. Int J Cardiol 1992;35:115.
Constricting bands of the extremities
- Meggitt ST, Harper J, Lacour M, et al. Raised limb bands developing in infancy. Br J Dermatol 2002;147:359–63.
- Patterson T. Congenital ring constrictions. Br J Plast Surg 1961;69:532–69.
- Dent DM, Fataar S, Rose AG. Ainhum and angiodysplasia. Lancet 1981;ii:396–7.
- Editorial. Ainhum. Lancet 1975;ii:19–20.
- Browne SG. Ainhum. Int J Dermatol 1976;15:348–50.
- Cignini P, Giorlandino C, Padula F, et al Epidemiology and risk factors of amniotic band syndrome or ADAM sequence. J Prenat Med 2012;6:59–63.
- Glessner JR. Spontaneous intra-uterine amputation. J Bone Joint Surg Am 1963;45:351–5.
- Petereka ES, Karon IM. Congenital pseudo-ainhum of the finger. Arch Dermatol 1964;90:12–14.
- Raque CJ, Stein KM, Lane JM. Pseudo-ainhum constricting bands of the extremities. Arch Dermatol 1972;105:434–8.
- Rushton DI. Amniotic band syndrome. BMJ 1983;286:919–20.
- Young ID, Lindenbaum RH, Thompson EM, et al. Amniotic bands in connective tissue disorders. Arch Dis Child 1985;60:1061–3.
- Lockwood C, Ghidini A, Romero R, et al. Amniotic band syndrome: reevaluation of its pathogenesis. Am J Obstet Gynecol 1989;160:1030–3.
- Moessinger AC. Amniotic band syndrome associated with amniocentesis. Am J Obstet Gynecol 1981;141:588–91.
- Latteo SA, Taylor AE, Meggitt SJ. Raised limb bands developing in infancy. Br J Dermatol 2006;154:791–2.
- Bockers M, Benes P, Bork K, et al. Persistent skin ulcers, mutilations and acro-osteolysis in hereditary sensory and autonomic neuropathy. J Am Acad Dermatol 1989;21:736–9.
- Almond SL, Curley RK, Feldberg L. Pseudoainhum in chronic psoriasis. Br J Dermatol 2003;149:1064–6.
- Christopher AP, Grattan CEH, Colvan MA. Pseudo-ainhum and erythropoietic protoporphyria. Br J Dermatol 1988;118:113–16.
- Schamroth JM. Mutilating keratoderma. Int J Dermatol 1986;25:249–51.
- Kamalan A. Ainhum trichosporosis Z-plasty. Dermatologica 1981;162:372.
- Hung NN. Congenital constriction ring in children: sine plasty combined with removal of fibrous groove and fasciotomy. J Child Orthop 2012;6:189–97.
Abnormal fibrotic responses to skin injury
Keloids and hypertrophic scars
- Köse O, Wasseem A. Keloids and hypertrophic scars: are they two different sides of the same coin? Dermatol Surg 2008;34:336–46.
- Zachariae H. Delayed wound healing and keloid formation following argon laser treatment or dermabrasion during isotretinoin treatment. Br J Dermatol 1988;118:703–6.
- Bagatin E, dos Santos Guadanhim LR, Yarak S, et al. Dermabrasion for acne scars during treatment with oral isotretinoin. Dermatol Surg 2010;36:483–9.
- Oluwasanmi JO. Keloids in the African. Clin Plast Surg 1974;1:179–95.
- Chen GY, Cheng YW, Wang CY, et al. Prevalence of skin diseases among schoolchildren in Magong, Penghu, Taiwan: a community-based clinical survey. J Formos Med Assoc 2008;107:21–9.
- Marneros AG, Norris JEC, Olsen BR, et al. Clinical genetics of familial keloids. Arch Dermatol 2001;137:1429–34.
- Brown JJ, Bayat A. Genetic susceptibility to raised dermal scarring. Br J Dermatol 2006;161:8–18.
- Moustafa MFH, Abdul Fattah AF. Presumptive evidence of the effect of pregnancy oestrogens on keloid growth. Plast Reconstr Surg 1975;56:450–3.
- González-Martínez R, Marín-Bertolín S, Amorrortu-Velayos J. Association between keloids and Dupuytren's disease: case report. Br J Plast Surg 1995;48:47–8.
- Ghatnatti V, Sarma D, Saikia U. Enlarged hands and feet: not always acromegaly. Indian J Endocrinol Metab 2012;16(Suppl. 2):S318–20.
- Kelly AP. Keloids: a review. Dermatol Clin 1988;7:130–9.
- Paradisi M, Angelo C, Conti G, et al. Dubowitz syndrome with keloidal lesions. Clin Exp Dermatol 1994;19:45–7.
- Scott MH Jr, Scott MJ, Scott AM. Linear keloids resulting from abuse of anabolic androgenic steroid drugs. Cutis 1994;53:41–3.
- Huang C, Ogawa R. The link between hypertension and pathological scarring: does hypertension cause or promote keloid and hypertrophic scar pathogenesis? Wound Repair 2014;22:462–6.
- Zhang K, Garner W, Cohen L, et al. Increased types I and III collagen and transforming growth factor-b1 mRNA and protein in hypertrophic burn scar. J Invest Dermatol 1995;104:750–4.
- Jumper N, Paus R, Bayat A. Functional histopathology of keloid disease. Histol Histopathol 2015;30:1033–57.
- Sidgwick GP, Bayat A. Extracellular matrix molecules implicated in hypertrophic and keloid scarring. J Eur Acad Dermatol Venereol 2012;26:141–52.
- Bailey AJ, Bazin S, Sims TJ, et al. Characterization of the collagen of hypertrophic and normal human scars. Biochim Biophys Acta 1975;405:412–21.
- Zhang Z, Nie F, Kang C, et al. Increased periostin expression affects the proliferation, collagen synthesis, migration and invasion of keloid fibroblasts under hypoxic conditions. Int J Mol Med 2014;34:253–61.
- Hunzelmann N, Anders S, Sollberg S, et al. Coordinate induction of collagen type I and biglycan expression in keloids. Br J Dermatol 1996;135:394–9.
- Younai S, Nichter LS, Wellisz T, et al. Modulation of collagen synthesis by transforming growth factor-b in keloid and hypertrophic scar fibroblasts. Ann Plast Surg 1994;33:148–51.
- Haisa M, Okochi H, Grotendorst GR. Elevated levels of PDGF and receptors in keloid fibroblasts contribute to an enhanced response to PGDF. J Invest Dermatol 1994;103:560–3.
- Chen JJ, Zhao S, Cen Y, et al. Effect of heat shock protein 47 on collagen accumulation in keloid fibroblast cells. Br J Dermatol 2007;156:1188–95.
- Crowe N, Parkhouse N, McGrouther D, et al. Neuropeptide-containing nerves in painful hypertrophic human scar tissue. Br J Dermatol 1994;130:444–52.
- Cheng B, Liu HW, Fu XB, et al. Coexistence and up-regulation of three types of opioid receptor, mu, delta and kappa, in human hypertrophic scars. Br J Dermatol 2008;158:713–20.
- Linares HA, Kischer CW, Dobrkovsky M, et al. The histiotypic organization of the hypertrophic scar in humans. J Invest Dermatol 1972;59:323–31.
- Kischer CW, Bunce H III, Shetlar MR. Mast cell analyses in hypertrophic scars, hypertrophic scars treated with pressure and mature scars. J Invest Dermatol 1978;70:355–7.
- Meenakshi J, Jayaraman V, Ramakrishnan KM, et al. Ultrastructural differentiation of abnormal scars. Ann Burns Fire Disasters 2005;18:83–8.
- Matsuoka LY, Uitto J, Wortsman J, et al. Ultrastructural characteristics of keloid fibroblasts. Am J Dermatopathol 1988;10:505–8.
- Granick M, Kimura M, Kim S, et al. Telomere dynamics in keloid. Eplasty 2011;11:e15.
- Bloemen MC, van der Veer VM, Ulrich MM, et al. Prevention and curative management of hypertrophic scar formation. Burns 2009;35:463–75.
- Kanaar P, Oort J. Fibrosarcomas developing in scar tissue. Dermatologica 1969;138:312–19.
- Ud-Din S, Bayat A. Strategic management of keloid disease in ethnic skin: a structured approach supported by the emerging literature. Br J Dermatol 2013;169(Suppl. 1):71–81.
- Jalali M, Bayat A. Current use of steroids in management of abnormal raised skin scars. Surgeon 2007;5:175–80.
- Gauglitz GG, Korting HC, Pavicic T, et al. Hypertrophic scarring and keloids: pathomechanisms and current and emerging treatment strategies. Mol Med 2011;17:113–25.
- Gold MH. A controlled clinical trial of topical silicone gel sheeting in the treatment of hypertrophic scars and keloids. J Am Acad Dermatol 1994;30:506–7.
- O'Brien L, Pandit A. Silicone gel sheeting for preventing and treating hypertrophic and keloid scars. Cochrane Database Syst Rev 2006;(1):CD003826.
- Wong T-W, Chiu H-C, Chen J-S, et al. Symptomatic keloids in two children: dramatic improvement with silicone cream occlusive dressing. Arch Dermatol 1995;131:775–7.
- Kischer CW, Shetlar MR, Shetlar CL. Alteration of hypertrophic scars induced by mechanical pressure. Arch Dermatol 1975;111:60–4.
- Tzitotzios C, Profyris C, Sterling J. Cutaneous scarring: pathophysiology, molecular mechanisms, and scar reduction therapeutics. Part II. Strategies to reduce scar formation after dermatologic procedures. J Am Acad Dermatol 2012;66:13–24.
- Hatamipour E, Mehrabi S, Hatamipour M, et al. Effects of combined intralesional 5-fluorouracil and topical silicone in prevention of keloids: a double-blind randomized clinical trial study. Acta Med Iran 2011;49:127–30.
- Sadeghinia A, Sadeghinia S. Comparison of the efficacy of intralesional triamcinolone acetonide and 5-fluorouracil tattooing for the treatment of keloids. Dermatol Surg 2012;38:104–9.
- España A, Solano T, Quintanilla E. Bleomycin in the treatment of keloids and hypertrophic scars by multiple needle punctures. Dermatol Surg 2001;27:23–7.
- Camacho-Martinez FM, Rey ER, Serrano FC, et al. Results of a combination of bleomycin and triamcinolone acetonide in the treatment of keloids and hypertrophic scars. An Bras Dermatol 2013;88:387–94.
- Bailey JN, Waite AE, Clayton WJ, et al. Application of topical mitomycin C to the base of shave-removed keloid scars to prevent their recurrence. Br J Dermatol 2007;156:682–6.
- Stashower ME. Successful treatment of earlobe keloids with imiquimod after tangential shave excision. Dermatol Surg 2006;32:380–6.
- Ud-Din S, Thomas G, Morris J, et al. Photodynamic therapy: an innovative approach to the treatment of keloid disease evaluated using subjective and objective non-invasive tools. Arch Dermatol Res 2013;305:205–14.
- Ahuja RB, Chatterjee P. Comparative efficacy of intralesional verapamil hydrochloride and triamcinolone acetonide in hypertrophic scars and keloids. Burns 2014;40:583–8.
- Mamalis AD, Lev-Tov H, Nguyen D-H, et al. Laser and light-based treatment of keloids: a review. J Eur Acad Dermatol Venereol 2014;28:689–99.
- Perry D, Colthurst J, Giddings P, et al. Treatment of symptomatic abnormal skin scars with electrical stimulation. J Wound Care 2010;19:447–53.
- Chu Y, Guo F, Li Y, Li X, Zhou T, Guo Y. A novel truncated TGF-beta receptor II down-regulates collagen synthesis and TGF-beta I secretion of keloid fibroblasts. Connect Tissue Res 2008;49:92–8.
- Lee JH, Kim SE, Lee AY. Effects of interferon-alpha-2b on keloid treatment with triamcinolone acetonide intralesional injection. Int J Dermatol 2008;47:183–6.
- van Leeuwen MC, Stokmans SC, Bulstra AE. Surgical excision with adjuvant irradiation for treatment of keloid scars: a systematic review. Plas Reconstr Surg Glob Opin 2015;3:e440.
- Jones CD, Guiot L, Samy M, Gorman M, Tehrani H. The use of chemotherapeutics for the treatment of keloid scars. Dermatol Reports 2015;7:5880.
Perforating dermatoses
- Burgdorf W, Plewig G, Wolff HH, Lamdthaler M, eds. Braun-Falco's Dermatology, 3rd edn. Berlin: Springer, 2008.
- Rapini RP, Herbert AA Drucker CR. Acquired perforating dermatosis. Evidence for combined transepidermal elimination of both collagen and elastic fibers. Arch Dermatol 1989;125:1074–8.
- Patterson JW. The perforating disorders. J Am Acad Dermatol 1984;10:561–81.
- Mehregan RH. Transepithelial elimination. Curr Probl Dermatol 1970;3:124–47.
- Patterson JW. Progress in the perforating dermatoses. Arch Dermatol 1989;125:1121–3.
- Akoglu G, Emre S, Sunqu N, et al. Clinicopathological features of 25 patients with acquired perforating dermatosis. Eur J Dermatol 2013;23:864–71.
- Zelger B, Hintner H, Aubock J, Fritsch PO. Acquired perforating dermatosis. Arch Dermatol 1991;127:695–700.
Acquired perforating dermatosis
- Ramirez-Fort MK, Khan F, Rosendahl CO, et al. Acquired perforating dermatosis: a clinical and dermatoscopic correlation. Derm Online J 2013;19(7):18958.
- Patterson JW. The perforating disorders. J Am Acad Dermatol 1984;10:561–81.
- Cochran RJ, Tucker SB, Wilkin JK, et al. Reactive perforating collagenosis of diabetes mellitus and renal failure. Cutis 1983;31:55–8.
- Poliak SC, Lebwohl MG, Parris A, et al. Reactive perforating collagenosis associated with diabetes mellitus. N Engl J Med 1982;306:81–4.
- Rapini RP, Herbert AA, Drucker CR, et al. Acquired perforating dermatosis: evidence of combined transepidermal elimination of both collagen and elastic fibres. Arch Dermatol 1989;125:1074–8.
- Stone RA. Kyrle-like lesions in two patients with renal failure undergoing dialysis. J Am Acad Dermatol 1981;5:707–9.
- Morton CA, Henderson IS, Jones MC, et al. Acquired perforating dermatosis in a British dialysis population. Br J Dermatol 1996;135:671–7.
- Gagnon IAL, Desai T. Dermatological diseases in patients with chronic kidney disease. J Nephropathol 2013;2:104–9.
- Patterson JW. Progress in the perforating dermatoses. Arch Dermatol 1989;125:1121–3.
- Akoglu G, Emre S, Sunqu N, et al. Clinicopathological features of 25 patients with acquired perforating dermatosis. Eur J Dermatol 2013;23:864–71.
- Kikuchi N, Ohtsuka M, Yamamoto T. Acquired perforating dermatosis: a rare association with dermatomyositis. Acta Derm Venereol 2013;93:735–6.
- Piqué-Dura E, Equia P, Garcia-Vázquez O. Acquired perforating dermatosis associated with natalizumab. J Am Acad Dermatol 2013;68:e185–7.
- Zelger B, Hintner H, Aubock J, Fritsch PO. Acquired perforating dermatosis. Arch Dermatol 1991;127:695–700.
- Kim SW, Kim MS, Lee JH, et al. A clinicopathological study of thirty cases of acquired perforating dermatosis in Korea. Ann Dermatol 2014;26:162–71.
- Choudhury N, Aggarwal I, Dutta D, Ghosh AGS, Chatterjee G, Chowdhury S. Acquired perforating dermatosis and Addison's disease due to disseminated histoplasmosis: presentation and clinical outcome. Dermatoendocrinology 2013;5:305–8.
- Mehregan AH, Schwartz OD. Reactive perforating collagenosis. Arch Dermatol 1967;96:277–82.
- Kanan MW. Familial reactive perforating collagenosis and intolerance to cold. Br J Dermatol 1974;91:405–14.
- Nair BKH, Sarojini PA, Basheer AM, et al. Reactive perforating collagenosis. Br J Dermatol 1974;91:399–403.
- Cerio R, Calnan CD, Wilson-Jones E. A clinico-pathological study of reactive perforating collagenosis: report of 10 cases [Abstract]. Br J Dermatol 1987;117(Suppl. 32):16–17.
- Fretzin DF, Beal DW, Jao W. Light and ultrastructural study of reactive perforating collagenosis. Arch Dermatol 1980;116:1054–8.
- Millard PR, Young E, Harrison DE, et al. Reactive perforating collagenosis: light, ultrastructural and immunohistological studies. Histopathology 1986;10:1047–56.
- Bovenmeyer DA. Reactive perforating collagenosis. Experimental production of the lesion. Arch Dermatol 1970;102:313–17.
- Chae KS, Park YM, Cho SH, et al. Reactive perforating collagenosis associated with periampullary carcinoma. Br J Dermatol 1998;139:548–50.
- Delacretaz J, Gattlen JM. Verrucous perforating collagenoma. Dermatologica 1976;152:65–6.
- Laugier P, Woringer F. Reflexions au sujet d'une collagenome perforant verruciforme. Ann Dermatol Syphiligr (Paris) 1963;90:29–32.
- Moulin G, Balme B, Musso M, Thomas L. Le collagénome perforant verruciforme, une dermatose par inclusion exogène? A propos d'un cas induit par le chlorure de calcium. Ann Dermatol Vénéréol 1995;122:591–4.
- Knox JM, Knox JM, Dinehart SM, et al. Acquired perforating disease in oil field workers. J Am Acad Dermatol 1986;14:605–11.
- Shoenfeld RJ, Grekin JN, Mehregan A. Calcium deposition in the skin. A report of four cases following electroencephalography. Neurology 1965;15:477–80.
- Goette DK. Transepidermal elimination of altered collagen after intralesional adrenal steroid injections. Arch Dermatol 1984;120:539–40.
- Katz R, Hood AF. Transepidermal elimination following the use of a topical adrenal steroid. Arch Dermatol 1985;121:412–13.
- Kruger K, Tebbe B, Krengel S, et al. Acquired reactive perforating dermatosis. Successful treatment with allopurinol in two cases. Hautzart 1999;50:115–20.
- Tilz H, Beck JC, Legat F, et al. Allopurinol in the treatment of acquired perforating dermatosis. An Bras Dermatol 2013;88:94–7.
- Mii S, Yatsa R, Hayashi R, et al. Acquired reactive collagenosis successfully treated with narrow-band ultraviolet B. Acta Derm Venereol 2009;89:530–1.
- Serrano G, Aliaga A, Lorente M, et al. Reactive perforating collagenosis responsive to PUVA. Int J Dermatol 1988;27:118–19.
- Sezar R, Erkek E. Acquired perforating dermatosis successfully treated with photodynamic therapy. Photodermatol Photoimmunol Photomed 2012;28:50–2.
- Yang A, Chong WS, Tay HL. Effective treatment of uraemic pruritus and acquired perforating dermatosis with amitriptyline. Australas J Dermatol 2014;55:e54–7.
- Escribano-Stablé JC, Doméneck C, Matarredona J, et al. Tacalcitol in the treatment of acquired perforating collagenosis. Case Rep Dermatol 2014;6:69–73.
Elastosis perforans serpiginosa
- Ayala F, Donofrio P. Elastosis perforans serpiginosa: report of a family. Dermatologica 1983;166:32–4.
- Reed WB, Pidgeon JW. Elastosis perforans serpiginosa with osteogenesis imperfecta. Arch Dermatol 1964;89:342–4.
- Mehta RK, Burrows NP, Payne CM, et al. Elastosis perforans serpiginosa and associated disorders. Clin Exp Dermatol 2001;26:521–4.
- Patterson JW. The perforating disorders. J Am Acad Dermatol 1984;10:561–81.
- O'Donnell B, Kelly P, Dervan P, et al. Generalized elastosis perforans serpiginosa in Down's syndrome. Clin Exp Dermatol 1992;17:31–3.
- Jan V, Saugier J, Arbeille B, et al. Elastome perforant serpigineux avec hypovitaminose A chez une enfant ayant une trisomie 21. Ann Dermatol Vénéréol 1996;123:188–90.
- Pereira AC, Baeta IG, Costa Júnior SR, et al. Elastosis perforans serpiginosa in a patient with Down's syndrome. An Bras Dermatol 2010;85:691–4.
- Pass F, Goldfischer S, Sternlieb I, et al. Elastosis perforans serpiginosa during penicillamine therapy for Wilson's disease. Arch Dermatol 1973;108:713–15.
- Kirsch N, Hukill PB. Elastosis perforans serpiginosa induced by penicillamine. Electron microscopic observations. Arch Dermatol 1977;113:630–5.
- Bardach H, Gebhart W. Elastic fiber changes induced by penicillamine. J Am Acad Dermatol 1982;6:398–9.
- Light N, Meyrick-Thomas RH, Stephens A, et al. Collagen and elastin changes in D-penicillamine-induced pseudoxanthoma elasticum-like skin. Br J Dermatol 1986;114:381–8.
- Na SY, Choi M, Kim MJ, et al. Penicillamine-induced elastosis perforans serpiginosa and cutis laxa in a patient with Wilson's disease. Ann Dermatol 2010;22:468–71.
- Hashimoto K, Hill WR. Elastosis perforans serpiginosa: histochemical and enzymic digestion studies. J Invest Dermatol 1960;35:7–14.
- Volpin D, Pasquali-Ronchetti I, Castellani I, et al. Ultrastructural and biochemical studies on a case of elastosis perforans serpiginosa. Dermatologica 1978;156:209–23.
- Bergman R, Friedman-Burnbaum R, Hazaz B. A direct immunofluorescence study in elastosis perforans serpiginosa. Br J Dermatol 1985;113:573–9.
- Lee SH, Choi Y, Kim SC. Elastosis perforans serpiginosa. Ann Dermatol 2014;26:103–6.
- Khatu SS, Dhurat RS, Nayak CS, et al. Penicillamine-induced elastosis perforans serpiginosa with abnormal ‘lumpy-bumpy’ elastic fibers in lesional and non-lesional skin. Indian J Dermatol Venereol Leprol 2011;77:55–8.
- Mehregan AH. Elastosis perforans serpiginosa. A review of the literature and report of 11 cases. Arch Dermatol 1968;97:381–93.
- Catterall MD, Padley NR. Elastosis perforans serpiginosa. Clin Exp Dermatol 1979;4:119–22.
- Schamroth JM. Elastosis perforans serpiginosa in a patient with renal disease. Arch Dermatol 1988;122:82–4.
- Whyte HJ, Winkelmann RK. Elastosis perforans: the association of congenital anomalies, and salient facts in histology. J Invest Dermatol 1960;35:113–22.
- Humphrey S, Hemmati I, Randhawa R, et al. Elastosis perforans serpiginosa treatment with liquid nitrogen cryotherapy and review of the literature. J Cutan Med Surg 2010;14:38–42.
- Ratnavel RC, Norris PG. Penicillamine-induced elastosis perforans serpiginosa treated successfully with isotretinoin. Dermatology 1994;189:81–3.
- Langeveld-Wildschut EG, Toonstra J, et al. Familial elastosis perforans serpiginosa. Arch Dermatol 1993;129:205–7.
- Outland JD, Brown TS, Callen JP. Tazarotene is an effective therapy for elastosis perforans serpiginosa. Arch Dermatol 2002;138:169–71.
- Kelly SC, Purcell SM. Imiquimod therapy for elastosis perforans serpiginosa. Arch Dermatol 2006;142:829–30.
- Mehta RK, Burrows NP, Payne CM, et al. Elastosis perforans serpiginosa and associated disorders. Clin Exp Dermatol 2001;26:521–4.
- Kaufman AJ. Treatment of elastosis perforans serpiginosa with the flashlamp pulsed dye laser. Dermatol Surg 2000;26:1060–2.
- Abdullah A, Colloby PS, Foulds IS, Whitcroft I. Localized idiopathic elastosis perforans serpiginosa effectively treated by the Coherent Ultrapulse 5000°C aesthetic laser. Int J Dermatol 2002;39:719–20.
- Yang JH, Han SS, Wou CH, et al. Treatment of elastosis perforans serpiginosa with the pinhole method using a carbon dioxide laser. Dermatol Surg 2011;37:524–6.
- Saxena M, Tope WD. Response of elastosis perforans serpiginosa to pulsed CO2, Er:YAG, and dye lasers. Dermatol Surg 2003;29:677–8.