CHAPTER 99
Panniculitis
Luis Requena
Department of Dermatology, Fundación Jiménez Díaz, Madrid, Spain
ANATOMY AND PHYSIOLOGY OF SUBCUTANEOUS FAT
Introduction
In order to appreciate how subcutaneous fat responds to inflammation, it is important to understand its structure and function. Subcutaneous tissue (subcutis) is composed predominantly of fat cells supported in a connective tissue framework (Figure 99.1). Subcutaneous fat is present almost universally over the body surface between the skin and the deep fascia and, in the normal state, constitutes about 10% of body weight (Figure 99.2). It forms a specialized closely regulated metabolic reserve capable of storing or rapidly releasing energy, typically providing sufficient for about 40 days’ requirements [1]. Subcutaneous fat also acts as an insulating layer against heat loss and a protective cushion against external injury. Subcutaneous fat also provides structural support to the overlying skin and has a cosmetic function, for example in the contours of the face.
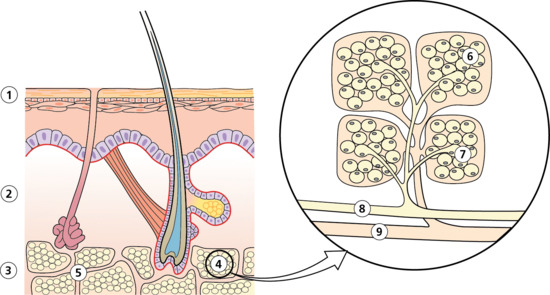
Figure 99.1 Schematic representation of the anatomy of subcutaneous fat with detailed view showing the vascular supply to the fat lobule and its constituent microlobules. (1) Epidermis. (2) Dermis. (3) Subcutis. (4) Fat lobule. (5) Connective tissue septum. (6) Adipocyte. (7) Arteriole. (8) Artery. (9) Vein.
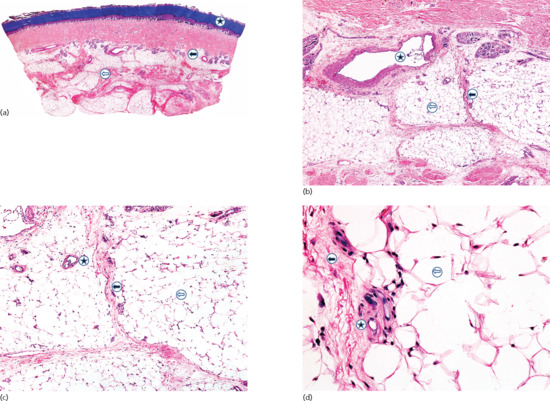
Figure 99.2 (a) Scanning power view of the normal skin of the sole. The epidermis is covered by a thick compact orthokeratotic horny layer (star). Numerous eccrine units are seen along the interface between the deep reticular dermis and subcutis (black arrow). The subcutis is organized into lobules of adipocytes and connective tissue septa (white arrow) surrounding and demarcating each fat lobule, and associated vessels and nerves. (b) At higher magnification, a large vein (star) may be identified in the deeper dermis because of the presence of valves within its lumen. The subcutis is composed of thin connective tissue septa (black arrow), which delimit lobules of adipocytes (white arrow). (c) At higher magnification, a small venule is seen within the fat lobule (star), as well as the thin septa of connective septa (black arrow) and mature adipocytes of the fat lobule (white arrow). (d) Still higher magnification shows that the connective tissue septa are mostly composed of thin collagen bundles (black arrow). A capillary is seen at the periphery of the fat lobule (star). With H&E stain, adipocytes appear as empty cells with signet-ring morphology. This is due to the fact that the lipid content dissolves in routinely processed specimens and the flat spindle nucleus is displaced to the periphery of the cell by a single large intracytoplasmic vacuole, which contains fat (white arrow).
Subcutaneous fat is absent from the eyelids and the male genitalia. There are obvious sexual differences in the distribution of fat around the body surface, with an increase in thickness resulting in the rounded contours of the female trunk, breasts, hips, pubis and thighs. Subcutaneous fat also varies in thickness with the race, age and endocrine and nutritional status of the individual. Fat also has great social importance as a major contributor to the sexual attractiveness of women on the one hand but, on the other hand, as a cause of misery when present in excess: fat children may be bullied or ostracised at school [2] and fat adults may find it more difficult to get certain jobs.
Brown fat in particular (see later) has a very important thermoregulatory role and acts by increasing the basal metabolic rate [3]. This is particularly important in infancy, and heat production in response to cold exposure is maximal in neonates, who have large quantities of brown fat.
In addition to the above functions, the obesity epidemic in westernized countries and the metabolic consequences of abnormal fat distribution have underlined the fact that the subcutaneous fat, comprising as it does innumerable adipocytes secreting a large variety of enzymes, hormones and cytokines, is also a major endocrine organ [4].
Cellular composition of subcutaneous tissue [5, 6]
The first fat-containing cell, the pre-adipocyte, appears in the mesenchyme around the 14th week of fetal life. The primitive mesenchymal cell that forms the determined pre-adipocyte is also capable of maturing to form a fibrocyte, myocyte, chondrocyte or osteoblast. Pre-adipocytes can terminally differentiate into either brown adipocytes or white adipocytes.
Brown fat is a special type of granular fat that differs from white fat in its distribution, histology and function. It is multilocular and is metabolically very active with many mitochondria, so that it is capable of transferring energy from food to produce heat. As it has a much greater capillary network surrounding it compared to white fat (which is partly responsible for the brown colour), heat can be rapidly transferred into the circulation. It is most prominent in the neck and upper thorax of the fetus, and it may be homologous to the hibernating gland fat found in some animals [7]. Brown fat is now known to persist into adult life [8], and it may have a role in preventing obesity [8]. Warm patches develop in the skin 1 h after taking ephedrine orally, and these warm patches may indicate the site of thermogenic brown fat.
Brown fat adipocyte mitochondria uniquely express uncoupling protein 1 (UCP-1), allowing confirmation that brown fat is present in adult white fat depots in variable amounts, and that transdifferentiation from white to brown adipocytes can occur. Development of brown fat begins at the 20th week of gestation, reaches its maximum at birth and then diminishes so that there are no large collections of brown fat in the adult, though fluorodeoxyglucose positron emission tomography (FDG PET) suggests that some adults have supraclavicular areas of brown fat [9]. Evidence for cold induction of brown fat as an adaptive response in humans is at present equivocal [9].
White fat adipocytes are the largest connective tissue cells in the body, with a diameter of up to 100 μm. Much of their differentiation occurs soon after birth. The mature adipocyte has a characteristic signet-ring appearance, because the flat oval nucleus is displaced to the side by a single, large, intracellular, fat-containing vacuole, which is surrounded by perilipin. Originally thought of as an inert store for emergency supplies of energy when necessary, it is now realized that the white adipocyte has a huge array of functions, secreting factors (adipokines) that affect lipid and glucose metabolism, endocrine functions, blood pressure control, coagulation, fibrinolysis, angiogenesis and inflammation. For a full review the reader is referred to Frühbeck [6].
Anatomy of subcutaneous tissue
Subcutaneous tissue is widely distributed throughout the body, forming a true organ as regards both structure and function [4]. Groups of adipocytes are arranged in lobules, each measuring approximately 1 cm in diameter; they are separated from each other by interlobular septa composed of collagen and reticulin fibres. Each lobule may be subdivided into 1 mm diameter microlobules, which represent the functional unit of the subcutaneous fat. Each microlobule is composed of a group of adipocytes arrayed around a central arteriole and surrounded by capillaries and postcapillary venules. Arteries and veins of the subcutis run along the septa. Each individual fat lobule is supplied by a small muscular artery (250–500 μm diameter) branching from the septa to form arterioles (up to 100 μm diameter) that supply every individual microlobule. Each arteriole branches to form a network of capillaries that surrounds each individual adipocyte. In addition to an abundant blood supply, subcutaneous fat also contains a rich lymphatic plexus, which receives vessels from the dermis. These lymph vessels traverse the subcutaneous layer parallel to the skin surface for some distance, before eventually penetrating the deep fascia and draining into the regional lymph nodes. The nature of the adipocyte and its relationship to blood vessels and lymphatics has been reviewed in detail by Ryan and Curri [10]. Both white fat and brown fat are innervated by noradrenergic fibres of the sympathetic nervous system and parasympathetic fibres.
The adipocytes may comprise only 25% of the total cell population of a lobule; the remainder, the stroma-vascular fraction, being macrophages, fibroblasts, mast cells, pericytes, endothelial cells and pre-adipocytes, enabling considerable cross-talk between cells by means of locally secreted cytokines including leptin and adiponectin (see later).
All fat tissue is composed of lobules of fat cells with their supporting connective and stroma-vascular tissue. In addition to the subcutaneous fat, approximately 20% of fat tissue occurs internally, in the mediastinal and retroperitoneal tissues, the mesentery and the bone marrow and in and around individual organs, including blood vessels. This tissue, although it is widely scattered throughout the body, forms a true organ as regards both structure and function [1] but in which depot-specific differences occur [11]. For example, increases in subcutaneous upper body and visceral fat are associated with an increased cardiovascular and metabolic risk but increases in gluteofemoral subcutaneous fat are not [12]. In addition, perivascular adipose tissue shows increased angiogenesis compared to subcutaneous fat [13]. The fact that some genetic lipodystrophy patients lose peripheral fat but fat padding for absorption of mechanical pressure is maintained, is further evidence for depot-specific differences.
The combination of the obesity epidemic and the advent of liposuction has rekindled interest in the structure of subcutaneous fat with MRI scanning as the investigative tool [14]. Subcutaneous fat is divided by the superficial fascia into two compartments, superficial and deep. The fat mass in the superficial (areolar) layer is compartmentalized into lobules by vertical and oblique fibrous septal planes and bands, whilst that of the deeper (lamellar) layer has its septae more horizontally positioned. The superficial layer is fairly constant, but the deeper is more variable, with an increase in fat mass accumulating between split horizontal septae. In females, subcutaneous fat is most abundant in the gluteofemoral region and breasts, resulting in the so-called gynaecoid distribution, whereas in males the android distribution of shoulders and upper arms, neck and lumbosacral area predominates.
Physiology of adipose tissue [5, 6, 15, 16, 17, 18]
Traditionally, adipose tissue was regarded as an inert energy store with insulating and padding properties. Whilst storage is still a major function, there is now an appreciation that adipocytes and their stroma-vascular tissue have many other highly complex and dynamic actions, including energy homeostasis, adipogenesis, insulin sensitivity and influences on immune and inflammatory responses (see also Chapter 149).
Energy homeostasis
A major function of white adipose tissue is to store energy at times of calorie excess and release it when needed, such as during exercise or starvation. The synthesis (anabolism) and catabolism of fat in the subcutaneous depot depends on many factors, including nourishment and endocrine and neural activity. The role of the autonomic nervous system in regulating fat metabolism is now well established [19], being particularly important for rapid energy need compared to the slower control exerted by neuroendocrine factors [20]. A decrease in parasympathetic activity results in increased lipolysis, as does an increase in sympathetic activity, with the opposites stimulating lipogenesis [21]. Hormones that may affect the energy metabolism of fat cells include insulin, cortisol, norepinephrine (noradrenaline) and several pituitary hormones, including somatotrophin, adrenocorticotrophic hormone (ACTH), thyrotrophin, lipotrophin and natrurietic peptide [22].
The fats contained within adipocytes are predominantly triglycerides (triacylglycerols), especially those of palmitic and stearic acids and the unsaturated oleic acid. All the fatty acids have an even number of carbon atoms, predominantly C16 and C18, with a few C14 and C12. Adipose tissue contains 10–30% of water with a small proportion of lipochromes, and less than 2% cholesterol. Fat-soluble substances are also present in varying amounts. These include fat-soluble vitamins and traces of chlorinated hydrocarbons (e.g. aldrin, dieldrin) ingested with the diet, as well as drugs such as acitretin. Adipose tissue in vitro has a metabolic rate similar to that of kidney tissue, and approximately half that of liver. Approximately half the triglyceride in the adipose tissue of rats and mice is catabolized and reconstituted in the course of a week or so.
The fat for storage enters the adipocyte as fatty acids, having been converted from lipoproteins by the extracellular enzyme lipoprotein lipase (Figure 99.3). The fatty acids combine with coenzyme A, using the energy of adenosine triphosphate (ATP), to form the corresponding acyl coenzyme A compounds. Some of these are then oxidized to provide energy for the regeneration of ATP, but most are converted to triglyceride by combination with glycerol-3-phosphate derived from glucose.
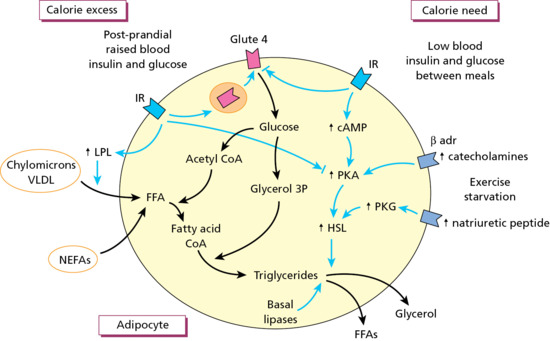
Figure 99.3 Simplified outline of lipogenesis in an adipocyte during energy excess and lipolysis during calorie need. Effects of hormones and enzymes are in blue. FFA, free fatty acid.
The adipocyte is one of the few cells to express the insulin-dependent glucose transporter receptor 4 (GLUT-4), which mediates the passage of glucose into the cell and thus facilitates triglyceride formation within the adipocyte via de novo lipogenesis, the latter providing only a small contribution to the pool. At the same time, insulin inhibits hydrolysis and breakdown of triglyceride, conserving the energy store.
When the body requires energy, lipolysis occurs. Triglyceride is hydrolysed in the adipocyte, converted to non-esterified fatty acids (NEFA) and glycerol, the rate-limiting enzyme being hormone-sensitive lipase (HSL). The NEFAs are conveyed in the blood to tissues such as liver and muscle, in which fatty acid oxidation readily takes place. In both tissues, the essential part of the process consists of the oxidation in the mitochondria of the long-chain fatty acids. The glycerol of the triglyceride molecule reacts with ATP to form glycerol phosphate, which is oxidized to glyceraldehyde-3-phosphate. This in turn may either be converted to glycogen by reversal of glycolysis, or it may be converted to pyruvate. Skeletal muscle readily oxidizes fatty acids but glucose, if available, is preferentially used. In cardiac muscle, fatty acids are a major source of energy. Lipolysis is regulated predominantly through insulin and catecholamines. The latter, elevated during a sudden energy demand, bind to β-adrenergic receptors on the adipocyte and activate HSL through the classic adenosine monophosphate-protein kinase A (AMP-PKA) pathway.
Role of leptin [23]
Leptin is an adipokine involved in energy homeostasis which may have evolved to help adaption from the starved to the adequately nourished state rather than to prevent obesity. Leptin, a product of the ob gene, is a 16-kDa polypeptide comprising 167 amino acids with a structural homology similar to other cytokine proteins such as tumour necrosis factor (TNF)-α and interleukin (IL)-6. It is secreted by adipocytes predominantly, but also by the stomach, aiding immediate appetite control. Leptin receptors are present in the hypothalamus, on adipocytes, skeletal muscle, liver, pancreatic β cells, ovary and endometrium. The main effect of leptin is via the satiety centres in the hypothalamus. If excess energy is being stored, rising leptin levels stimulate satiety centres to reduce appetite. Conversely, during starvation low leptin levels stimulate appetite. Circulating levels of leptin correlate with increasing body mass index (BMI), but have little effect on satiety centres, indicating an apparent leptin resistance. Leptin also influences several other functions, including neuroendocrine and reproductive functions, insulin secretion and blood pressure. Patients with congenital leptin deficiency (see Chapter 74) have gross obesity, hyperphagia, delayed pubertal development, abnormal T-cell number and function, and altered thyroid and growth hormone function [24]. In addition, leptin has a role in immune function and inflammation [25]. There is increased expression in chondrocytes and leptin may have a part to play in articular degenerative disease.
Adipogenesis
Adipogenesis refers to the recruitment from multipotent stem cells in the mesenchyme and stroma-vascular tissue, and proliferation of pre-adipocytes followed by their differentiation into mature fat cells. Culture of cell lines has led to the elucidation of many of the transcriptional factors involved in adipogenesis, the major ones being peroxisome proliferator-activated receptor-γ (PPARγ) and the CCAAT enhancer binding proteins (C/EBPs). The precise contribution of adipogenesis towards enlargement of the fat organ at different stages of human development and life changes is uncertain, but it seems maximal before and around birth before diminishing, then possibly continuing at a low rate throughout adult life. Glucocorticoids, growth hormone and insulin stimulate cells to terminal differentiation, but when mature fat cells reach a certain size, recruitment occurs so that the fat organ enlarges through hyperplasia (increased numbers of cells) rather than hypertrophy (increase in size of cells). Control of this hyperplastic response may come from the local adipocyte through paracrine effects involving local growth factors. During adipogenesis the local extracellular matrix also changes, the effects of which might play their own role in differentiation. This is supported by the fact that fat tissue repair is improved if elements of this matrix are included with the donor adipocytes.
Insulin sensitivity
Insulin secretion, stimulated by raised blood glucose levels after meals to reverse hyperglycaemia, has two major effects. It facilitates glucose uptake into most of the body's cells (liver, skeletal muscle and adipocytes) and it suppresses glucose output by the liver. Insulin resistance occurs when a target organ fails to respond normally to insulin, resulting in hyperinsulinaemia. The effect may be incomplete suppression of hepatic glucose output in the liver and/or impaired insulin-mediated glucose uptake in peripheral tissues, including adipocytes. If increased insulin secretion cannot prevent hyperglycaemia, type 2 diabetes results. Adipocytes secrete many factors, some of which have direct and indirect effects on insulin sensitivity.
Adiponectin [26, 27, 28]
Adiponectin is a 30-kDa protein composed of 244 amino acids with some structural similarity to both collagen and complement C1q and is currently thought to be secreted exclusively by adipocytes. It has autocrine/paracrine effects locally within adipose tissue as well as endocrine effects distantly. Locally, it can promote pre-adipocytes to become mature fat cells, which with increasing cell size down-regulate their adiponectin secretion to exert some feedback control. Adiponectin receptors are present in many tissues as well as adipocytes. It is likely that adiponectin receptor-activated AMPK (AMP-activated protein kinase) leads to enhanced insulin signalling and therefore insulin sensitivity. If BMI is elevated, the expression of adiponectin is reduced in visceral adipose tissue (VAT) adipocytes in comparison with that in subcutaneous adipose tissue (SAT). Serum adiponectin levels fall with weight gain and rise with weight loss.
Additionally, adiponectin exerts protective anti-inflammatory effects both locally and distantly. Local effects are mediated by inhibiting secretion of IL-6, IL-8, macrophage inflammatory protein 1 and monocyte chemotactic protein 1. It also has distant effects by its direct action on a range of cells including monocytes/macrophages, endothelial cells, hepatocytes and muscle cells, and indirectly by inhibition of TNF-α production.
There is an as yet unexplained paradox concerning adipo- nectin and its anti-inflammatory effects. Obesity is associated with macrophages in VAT which generate factors, particularly TNF-α, that suppress adiponectin secretion. However, low levels of adiponectin promote inflammation, generating a self-sustaining loop: thus in obesity adiponectin levels are inversely correlated with levels of inflammatory markers. In autoimmune states such as rheumatoid arthritis and systemic lupus erythematosus, adiponectin levels are raised, the level positively correlating with inflammatory markers. To explain this, it has been suggested that the adiponectin system has evolved as a mechanism for adaptation to starvation, a catabolic state [29]. It is therefore raised in other catabolic states such as autoimmune disease and inflammatory bowel disease, and did not evolve as a protective device against insulin resistance.
Other adipokines
Many other adipokines have been described [6, 18] and most are still being evaluated for their relevance to human biology. The stroma-vascular tissue itself is also responsible for a variety of cytokines. Macrophages secrete TNF-α, IL-1, IL-6, IL-8, IL-10, monocyte chemoattractant protein 1 (MCP-1), macrophage migration inhibitory factor (MIF), angiotensinogen, and endothelial and vascular growth factors. Therefore, as well as affecting energy homeostasis, insulin sensitivity and adipocyte differentiation, the fat organ has influences on inflammation, immune function, vascular inflammation and neoangiogenesis. All of this lends credence to the concept of the fat organ being an endocrine organ in its own right. Whilst these discoveries are of the utmost importance for worldwide obesity-associated morbidity and mortality, their relevance in disorders of subcutaneous fat other than lipodystrophies is unclear.
PANNICULITIS
Introduction and general description
Inflammatory diseases involving the subcutaneous fat comprise a heterogeneous group of disorders named generically panniculitis. These diseases have been classically considered diagnostically challenging both for clinicians and dermatopathologists; the reasons for this difficulty are varied. Firstly, dermatologists usually evaluate different morphological aspects of the skin anomalies to reach a specific diagnosis, but subcutaneous tissue is too deep to be visible to the examining eye. Moreover, cutaneous lesions of panniculitis usually show a disappointing monotony with completely different diseases involving the subcutaneous tissue showing the same clinical morphology, namely erythematous nodules located preferentially on the lower extremities. Secondly, because the lesions are situated deep in subcutaneous tissue, large incisional biopsies are necessary for diagnosis, which is usually based on the correct evaluation of the pattern of the inflammatory infiltrate and the involvement of blood vessels. This requires at the very least that the biopsy specimen should include a fat lobule and its surrounding connective tissue septa. Thirdly, many panniculitides are also histopathologically unsatisfactory, because subcutaneous fat has a limited range of responses and a variety of insults and panniculitic processes of entirely different aetiologies may produce very similar histopathological changes. Moreover, before an accurate histopathological diagnosis may be established, it must be remembered that panniculitides, like other inflammatory cutaneous disorders, are dynamic processes in which both the distribution and composition of the inflammatory cells of the infiltrate may change rapidly over the course of a few days: when biopsies are taken from late or resolving lesions, especially in predominantly lobular panniculitis, they may show completely non-specific findings. For the aforementioned reasons, some authorities have considered that ‘the histological septal-lobular dichotomy is sometimes diagnostically useful, but more often there is a mixed picture that adds to interpretative difficulties’ [1].
Despite these potential pitfalls, serial sections of an adequate biopsy enable the dermatopathologist in most cases to classify the panniculitic process as either a predominantly septal or a predominantly lobular panniculitis. This first classification step into one of the two general categories of panniculitis is very helpful for diagnostic purposes. However, classification of a panniculitis into a predominantly septal or predominantly lobular panniculitis is no more than an initial descriptive working classification and it should be followed by a search for additional histopathological clues to help reach a more specific clinically relevant final diagnosis. Thus, the next diagnostic step requires assessment of whether vasculitis is or is not present and, when it is present, of the size and nature of the involved blood vessels. The final diagnostic step requires the microscopic identification of the composition of the inflammatory infiltrate involving the septa and/or the fat lobule, the type of adipocyte necrosis and a search for additional histopathological features to enable a specific diagnosis to be reached. Table 99.1 provides a working classification of the panniculitides using this approach for diagnosis [2, 3].
Table 99.1 Classification of the panniculitides.
| Predominantly septal panniculitides | |
| With vasculitis | |
| Veins | Superficial migratory thrombophlebitis |
| Arteries | Cutaneous polyarteritis nodosa |
| No vasculitis | |
| Lymphocytes and plasma cells predominantly | |
| With granulomatous infiltrate in septa | Necrobiosis lipoidica |
| No granulomatous infiltrate in septa | Deep morphoea |
| Histiocytes predominantly (granulomatous) | |
| With mucin in centre of palisaded granulomas | Subcutaneous granuloma annulare |
| With fibrin in centre of palisaded granulomas | Rheumatoid nodule |
| With large areas of degenerate collagen, foamy histiocytes and cholesterol clefts | Necrobiotic xanthogranuloma |
| Without mucin, fibrin or degeneration of collagen, but with radial granulomas in septa | Erythema nodosum |
| Predominantly lobular panniculitides | |
| With vasculitis | |
| Small vessels | |
| Venules | Erythema nodosum leprosum |
| Erythema induratum of Bazin | |
| Large vessels | |
| Arteries | Erythema induratum of Bazin |
| No vasculitis | |
| Few or no inflammatory cells | |
| Necrosis at the centre of the lobule | Sclerosing panniculitis |
| With vascular calcification | Calcific uraemic arteriolopathy (calciphylaxis) |
| Lymphocytes predominant | |
| With superficial and deep perivascular dermal infiltrate | Cold panniculitis |
| With lymphoid follicles, plasma cells and nuclear dust of lymphocytes | Lupus panniculitisPanniculitis associated with dermatomyositis |
| Neutrophils predominant | |
| Extensive fat necrosis with saponification of adipocytes | Pancreatic panniculitis |
| With neutrophils between collagen bundles of deep reticular dermis | α1-antitrypsin deficiency panniculitis |
| With bacteria, fungi or protozoa | Infective panniculitis |
| With foreign bodies | Factitious panniculitis |
| Neutrophilic lobular panniculitis (subcutaneous Sweet syndrome) | |
| Histiocytes predominant (granulomatous) | |
| No crystals in adipocytes | Subcutaneous sarcoidosis |
| Traumatic panniculitis | |
| With crystals in histiocytes or adipocytes | Subcutaneous fat necrosis of the newborn |
| Poststeroid panniculitis | |
| Sclerema neonatorum | |
| Gouty panniculitis | |
| Fungal panniculitis due to zygomycosis, mucormycosis and aspergillosis | |
| With cytophagic histiocytes | Cytophagic histiocytic panniculitis and |
| subcutaneous panniculitis-like T-cell lymphomaa | |
| With sclerosis of the septa | Sclerosing post-irradiation panniculitis |
aAlthough these disorders are characterized by a neoplastic proliferation of cytotoxic T lymphocytes rather than an authentic panniculitic process, they are included in the classification of the panniculitides because they may mimic panniculitis both clinically and histopathologically.
There is probably no individual cell of the human body with a better vascular supply than the adipocyte. Postcapillary venules drain into veins which also run along the septa. In each microlobule, the arteriole occupies a central position, whereas the venule runs along the periphery [4]. As a consequence, interference with the arterial supply results in dramatic necrotic changes within the fat lobule (predominantly lobular panniculitis), while venous disorders manifest by alterations in the septal and paraseptal areas (predominantly septal panniculitis) [5]. This peculiar distribution of the vascularization in subcutaneous tissue explains why large vessel vasculitis involving the septal vessels is usually accompanied by little inflammation of the fat lobules, whereas vasculitis involving small blood vessels of the lobule usually causes extensive necrosis of the centrilobular adipocytes and a dense inflammatory response. In contrast with the dermal vascular network, the blood supply of each subcutaneous microlobule is terminal, implying there are no vascular connections between adjacent microlobules or between the dermis and subcutaneous fat. The septa of the subcutaneous fat also contain a prominent lymphatic plexus, which comes from the dermis and traverses the subcutis, first, parallel to the surface of the skin and then vertically penetrating the underlying fascia and draining into regional lymph nodes. The connective tissue septa, which are contiguous with the overlying reticular traverses and with the underlying fascia, provide stability to the subcutaneous tissue by compartmentalizing it. The normal septa are thin, from 200 to 300 μm, and are composed mostly of collagen bundles and thin elastic and reticulin fibres.
Mature normal individual adipocytes are relatively large cells with a diameter up to 100 mm and, in formalin-fixed and H&E-stained sections, appear as empty cells with signet-ring morphology. This is due to the fact that the lipid and triglyceride content dissolves in routinely processed specimens and the single large intracytoplasmic vacuole displaces the flat spindle nucleus without discernible nuclear features to the periphery of the cell. Frozen sections and special stains such as oil red O or Sudan B are required to demonstrate the lipid contents within the cytoplasm of mature adipocytes.
Perivascular adipocytes have been also demonstrated to be powerful endocrine cells capable of responding to metabolic changes and transducing signals to adjacent blood vessels. Cross-talk between perivascular adipose tissue and blood vessels is now being intensely investigated. There is evidence suggesting that perivascular adipose tissue regulates vascular function through a variety of mechanisms and plays an important role in inflammation and vasoreactivity in subcutaneous tissue [6]. Adipocytes also interact with the immune system. Normal subcutaneous fat contains T lymphocytes located between adipocytes of the fat lobule. They differ from those of other tissues and vary between different regions of the body [7]. It has recently been demonstrated that cytotoxic T lymphocytes precede the accumulation of macrophages during the process of inflammation of the fat lobule. In vitro co-cultures have shown a vicious cycle of interaction between cytotoxic T lymphocytes, macrophages and adipocytes, suggesting that adipocytes activate cytotoxic T lymphocytes with subsequent recruitment and activation of macrophages [8]. That there is an interaction between adipocytes and lymphocytes is also supported by the demonstration on human adipocytes of the inflammatory receptor CD40, which contributes to intercellular cross-talk between adipocyte and lymphocyte [9]. Co-cultures of adipocytes and lymphocytes have also shown up-regulation of pro-inflammatory cytokines, including IL-6, MCP-1 and plasminogen activator inhibitor 1 (PAI-1), but down-regulation of leptin and adiponectin [9].
Immunohistochemically, adipocytes express S-100 protein, with staining around the periphery of the cell, and vimentin [10]. In contrast with the multivacuolated cytoplasm of sebocytes and foamy histiocytes, which express adipophilin, the single large cytoplasmic vacuole of the adipocyte is adipophilin negative [11, 12].
Pattern-based histopathological classification of panniculitis with large vessel vasculitis also requires ascertainment of whether the involved vessel is an artery or a vein. A peculiarity of the veins in the subcutaneous fat of the lower limbs is that they have a thick muscular layer, conferring upon them an ‘arterial’ appearance [13]. However, it is usually possible to establish this distinction with confidence from H&E preparations, because the middle layer of subcutaneous veins is composed of several muscular fascicles separated by tiny unstained elastic fibres, whereas arteries show a more compact muscular layer. Nevertheless, many authors continue to promote the misleading notion that arteries of the subcutaneous fat of the lower legs have a thicker muscular layer than veins, when in fact veins often have a thicker muscular layer than arteries [13]. In difficult cases, elastic tissue staining allows definite discrimination between artery and vein, because arteries have a well-demarcated, thick and sharp internal elastic membrane, whereas veins have an ill-defined, thin and multilayered internal elastic lamina and tiny elastic fibres interspersed between muscular fascicles of the middle layer of the vessel wall. Some authors, however, believe that when inflammation is present within and around the wall of the vessel, all of the studied histological features become less reliable, and that the interobserver reliability of distinguishing arteritis from thrombophlebitis is low [14].
Types of necrosis of the adipocytes
The appearance of necrotic adipocytes is polymorphous and different from other necrotic cells [15, 16, 17]. In classical histopathology, nuclear abnormalities such as pyknosis, karyorrhesis and karyolysis are signs of cellular necrosis. In contrast, necrotic adipocytes, regardless of the aetiology of the cell death, show great variability and may appear as either anucleate cells or with complete disintegration of the cellular structure. Unfortunately, these distinctive forms of adipocyte necrosis have little value for diagnostic specificity.
Often, the only sign of necrosis of the adipocytes is the lack of nuclei in the involved cells, and dead fat cells appear as round empty bags with no inflammatory infiltrate among them. The most frequent type of adipocyte necrosis is lipophagic necrosis, which consists of the replacement of necrotic adipocytes by foamy macrophages formed by the engulfing of lipid products released from dead adipocytes by macrophages. These lipophages appear quite different from normal adipocytes, with large pale microvacuolated or granular-like cytoplasm and round central vesicular nuclei. Lipophagic granulomatous inflammation, however, is entirely non-specific and many lobular panniculitides show this pattern of fat necrosis at their late or resolving stages. It is usually seen in lipodermatosclerosis and traumatic panniculitis, but may also be present in erythema nodosum and erythema induratum of Bazin.
In contrast, liquefactive fat necrosis is a more specific pattern of adipocyte necrosis and it is more often seen in α1-antitrypsin deficiency panniculitis and in pancreatic panniculitis. Necrotic adipocytes injured by this mechanism appear as granular wisps of amphophilic detritus and their cellular structures are no longer evident. Enzymatic fat necrosis is a specific type of liquefactive fat necrosis characteristically observed in pancreatic panniculitis. It is due to saponification of the adipocyte lipid contents by pancreatic lipase, with secondary deposition of calcium salts, resulting in so-called ghost adipocytes, which consist of adipocytes with no nuclei and granular basophilic cytoplasm.
Hyalinizing fat necrosis is characteristically observed in lupus panniculitis and panniculitis associated with dermatomyositis. In this pattern, necrotic adipocytes appear as mummified anucleated cells, which are surrounded by glassy homogeneous proteinaceous material, effacing the architecture of the fat lobule.
Membranous fat necrosis is also a late-stage and non-specific type of necrosis of adipocytes, in which a leathery eosinophilic or amphophilic rim of collapsed cellular organelles with a crenulated or arabesque appearance is seen: periodic acid–Schiff (PAS) and Sudan III is positive. When membranous fat necrosis is extensive, formation of cystic structures devoid of cellular components and lined by hyaline-crenulated membrane can be observed. Membranous and membranocystic fat necrosis are almost always seen in lipodermatosclerosis, but like other types of fat necrosis, they may also be seen in late-stage lesions of several types of panniculitis.
Ischaemic fat necrosis is more frequently seen at the centre of fat lobules and is characterized by pallor of adipocytes, which are smaller than normal due to severe impairment of blood supply. Later stages of ischaemic necrosis also show lipophagic granulomata. Ischaemic fat necrosis is frequently seen in erythema induratum of Bazin, but may also be observed in other panniculitides, including calcific uraemic arteriolopathy (calciphylaxis), infectious panniculitis and cutaneous polyarteritis nodosa.
Finally, basophilic fat necrosis results from necrosis of adipocytes intermingled with nuclear dust of neutrophils and granular basophilic material, which represent aggregations of bacteria and is characteristically seen in cases of infectious panniculitis.
There are some disorders that should no longer be considered as specific variants of panniculitis. Weber–Christian disease is the term that has been classically used to refer to cases of predominantly lobular panniculitis without vasculitis in association with systemic manifestations including fever and involvement of visceral fat tissue. Additional terms such as idiopathic nodular panniculitis, nodular panniculitis and relapsing febrile non-suppurative nodular panniculitis have been used as synonyms for Weber–Christian disease. However, many cases originally considered as examples of Weber–Christian disease were later reclassified when other variants of lobular panniculitis, including erythema induratum of Bazin (nodular vasculitis), pancreatic panniculitis and α1-antitrypsin deficiency panniculitis were separated as specific diseases. White and Winkelmann [18] reviewed the clinical and histopathological features of 30 cases of panniculitis previously diagnosed as Weber–Christian disease at the Mayo Clinic and most of the cases could be reclassified: 12 cases as erythema nodosum, six cases as superficial thrombophlebitis (STP), five cases as factitious panniculitis, three cases as traumatic panniculitis, and individual cases as cytophagic histiocytic panniculitis, subcutaneous ‘panniculitic’ lymphoma and subcutaneous involvement by leukaemia. The authors concluded that the term Weber–Christian disease should be abandoned as a diagnosis for cases of lobular panniculitis because now a more specific diagnosis may be reached in the majority of cases. The same is true for Rothmann–Makai disease, a term that was used previously to describe cases of relapsing nodular panniculitis similar to that of Weber–Christian disease but with no fever or other systemic manifestations. These are now considered obsolete terms that should be no longer used.
Superficial migratory thrombophlebitis
Introduction and general description
STP is an inflammation and thrombosis of the superficial veins which presents as painful induration with erythema, often in a linear or branching configuration forming cords (Figure 99.4) [1, 2]. The clinical features are fully described in Chapter 103.
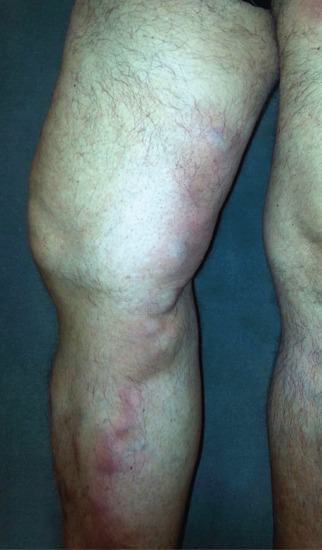
Figure 99.4 Superficial thrombophlebitis. Varicosities and erythematous nodules with linear arrangement involving the right lower extremity.
Pathophysiology
Predisposing factors
STP results from a hypercoagulable state, either primary [3] or secondary [4] (Box 99.1). The causes of secondary hypercoagulable states are varied, but in the majority of cases venous insufficiency of the lower extremities is the only precipitating factor.
Pathology
Histopathologically, cutaneous lesions of STP involve large veins of the septa in the upper subcutaneous tissue. The affected vein exhibits luminal thrombosis and an inflammatory infiltrate within its wall (Figure 99.5). In early lesions, the inflammatory cell infiltrate is composed mostly of neutrophils, whereas in later stages there are lymphocytes, histiocytes and occasional multinucleated giant cells. Granulomatous infiltration participates in the recanalization of the thrombus. A striking feature is that, in spite of the intense damage of the involved vein with dense inflammatory infiltrate in its wall and with marked septal thickening, there is little or no involvement of the adjacent fat lobule, and the process is more vasculitic than panniculitic. Intramural microabscesses in the wall of the involved vein have been described as characteristic of STP associated with Buerger disease [15].
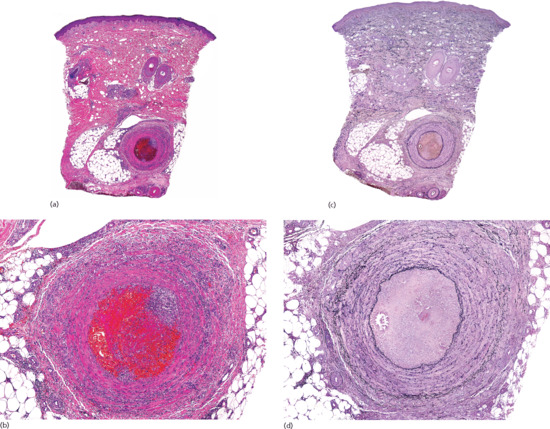
Figure 99.5 Histopathological features of superficial thrombophlebitis. (a) Scanning view showing involvement of a large vein in the septa of subcutaneous tissue. (b) The involved vein shows thrombosis of its lumen. (c) The same specimen stained for elastic tissue. (d) Several layers of internal elastic lamina are seen around the luminal thrombus.
Clinical features
Patients with STP should be appropriately investigated to rule out hypercoagulable states, paraneoplastic processes (Trousseau sign) and Behçet disease, although by far the most common cause of STP is chronic venous insufficiency of the lower limbs.
Differential diagnosis
The main histopathological differential diagnosis for STP is cutaneous polyarteritis nodosa. In contrast to STP, cutaneous polyarteritis nodosa is characterized by involvement of the small arteries and arterioles of the subcutaneous septa. The process is more inflammatory than thrombotic, with prominent fibrinoid necrosis of the tunica intima, resulting in the so-called target-like arteritis, in which an eosinophilic ring of fibrinoid necrosis replaces the intima of the affected arteriole. In doubtful cases, elastic tissue stain usually resolves any uncertainty, because in cutaneous polyarteritis nodosa the involved artery shows sharp and prominent internal elastic lamina, whereas in STP the damaged vessel is a vein with little or no discernible internal elastic membrane [16]. Some authors, however, believe that when inflammation is present within and around the wall of the vessel, the identification of the internal elastic lamina of the involved vessel is less reliable even with elastic tissue stains, and the smooth muscle pattern has the highest sensitivity and specificity for distinguishing arteries from veins [17]. The recently described type of tuberculid, nodular granulomatous phlebitis, may clinically resemble STP, but this tuberculid is histopathologically characterized by tuberculoid granulomas and multinucleate giant cells involving the walls of the veins of subcutaneous tissue [18, 19].
Cutaneous polyarteritis nodosa
Definition
A cutaneous vasculitis of poorly understood aetiology affecting subcutaneous arteries and arterioles (Figure 99.6). It is strongly associated with circulating antineutrophil cytoplasmic antibodies with peripheral staining (p-ANCA). In contrast to systemic polyarteritis nodosa, there is little or no evidence of systemic disease. It is fully described in Chapter 102.

Figure 99.6 Clinical appearance of cutaneous polyarteritis nodosa showing livedo reticularis of the lower extremities with ulcerated nodules on the right calf of a middle-aged woman.
Pathophysiology
The serum of patients with cutaneous polyarteritis nodosa is usually negative for p-ANCA by enzyme-linked immunosorbent assay (ELISA), but 84% of the patients reveal p-ANCA positivity by indirect immunofluorescence. Serum lysosomal-associated membrane protein 2 antibody (anti-LAMP-2) levels in cutaneous polyarteritis nodosa patients with p-ANCA are significantly elevated compared with those with negative p-ANCA, which suggests that anti-LAMP-2 antibodies might play an important role in the pathogenesis of the condition [1]. Immunoglobulin G (IgG) antiphosphatidylserine–prothrombin complex (anti-PS/PT) antibodies and/or IgG anticardiolipin antibodies have also been detected in the serum of some patients with cutaneous polyarteritis nodosa [2].
Pathology
Cutaneous lesions exhibit vasculitis involving medium-sized arteries and arterioles at the septa of the upper subcutis (Figure 99.7). Direct immunofluorescence studies of lesions of cutaneous polyarteritis nodosa have demonstrated IgM and complement deposition in the involved vessel walls and consistent absence of IgG [3]. The involved vessel appears with a thickened wall, within which an inflammatory infiltrate is seen. Its composition varies with the stage of evolution of the process. In early lesions, a neutrophilic infiltrate and leukocytoclasis are often seen and, in some cases, eosinophils may be prominent [4]. Characteristically, the intima of the involved artery exhibits an eosinophilic ring of fibrinoid necrosis, giving a target-like appearance to the damaged vessel. In older lesions, lymphocytes are predominant and in a still later stage there is fibrosis of the entire thickness of the vessel wall, leading to the obliteration of its lumen. A rare complication is the formation of periosteal new bone beneath the cutaneous lesions [5]. Although luminal thrombi may be present, they are less frequent than in lesions of superficial thrombophlebitis. Often, arterial involvement is segmental and serial sections throughout the entire specimen are required to demonstrate the pathology. As is the case in superficial thrombophlebitis, lesions of cutaneous polyarteritis nodosa show little or no involvement of the adjacent fat lobule, and the process is exclusively a septal arteritis.
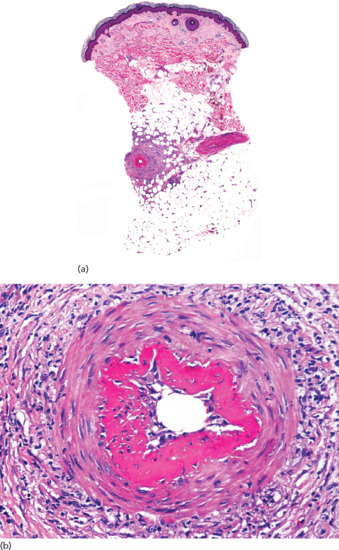
Figure 99.7 Histopathological features of cutaneous polyarteritis nodosa. (a) Scanning view of a punch biopsy showing involvement of a vessel of the septa of subcutaneous fat. (b) Higher magnification showing fibrinoid necrosis of the intima, giving a target-like appearance to the involved arteriole.
Necrobiosis lipoidica
Definition
Necrobiosis lipoidica is an uncommon skin condition in which degenerated dermal collagen is surrounded by a granulomatous inflammatory response to produce shiny, red-brown or yellowish plaques in the skin, particularly on the shins (Figure 99.8). In severe cases, the affected skin may ulcerate. It is associated in the majority of but not all cases with underlying diabetes, the onset of which it may precede. It is fully described in Chapter 97. It may involve the subcutis but does not cause a true panniculitis, because the palisading granulomatous process involving the subcutis is always a deep extension of the dermal process and, to our knowledge, there are no descriptions of necrobiosis lipoidica involving only subcutaneous fat.
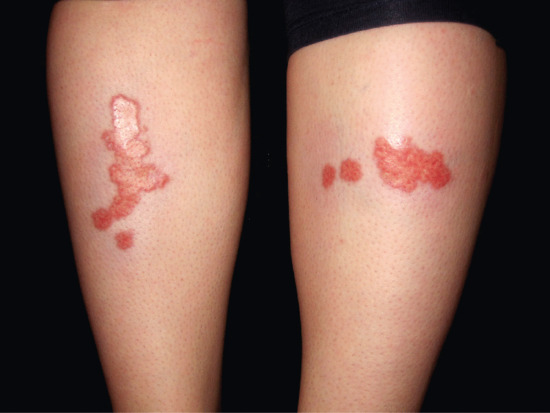
Figure 99.8 Necrobiosis lipoidica showing yellowish indurated plaques on the anterior aspect of the legs in a diabetic woman.
Pathology
Histopathologically, lesions of necrobiosis lipoidica involve the full thickness of the dermis, and often extend to the superficial subcutaneous tissue causing septal panniculitis [1] (Figure 99.9). There are palisading granulomas with histiocytes surrounding areas of degenerate collagen within widened septa. The most characteristic feature supporting a diagnosis of necrobiosis lipoidica as the cause of an inflammatory process involving the subcutis is the coexistence of similar lesions in the dermis, with alternating horizontal bands of inflammatory cells and fibrosis involving the entire dermis [2].
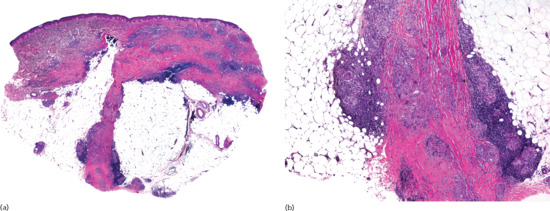
Figure 99.9 Histopathological features of necrobiosis lipoidica extending to subcutaneous tissue. (a) Scanning power showing involvement of the full thickness of the dermis and extension to the subcutaneous tissue throughout the septa. (b) Granulomas involving the thickened fibrous septa of the subcutaneous tissue.
Early lesions show an inflammatory infiltrate composed predominantly of neutrophils scattered within the septa, whereas in later lesions, histiocytes, lymphocytes and plasma cells, sometimes with lymphoid follicle formation [3], are predominant. Multinucleated giant cells involving the septa are sometimes prominent and in those cases histopathological findings resemble erythema nodosum. Differential diagnosis is, however, straightforward because in the latter condition there are no significant dermal changes other than a perivascular lymphocytic infiltrate.
In chronic longstanding lesions, the dermis and the superficial subcutaneous tissue are replaced by horizontal fibrosis with sclerotic collagen bundles arranged parallel to the epidermis and scattered by plasma cells, closely resembling the findings seen in morphoea. In these late-stage lesions, features of necrobiosis are no longer evident and elastic tissue stains demonstrate dramatic loss of elastic fibres. Some authors have postulated that the finding of vasculitis and leukocytoclasis in lesions of necrobiosis lipoidica is indicative of an underlying systemic disease [4]. Membranous fat necrosis has also been described in late-stage lesions of necrobiosis lipoidica extending to the subcutaneous tissue [5].
Direct immunofluorescence studies have demonstrated IgM and complement in the blood vessels of some lesions of necrobiosis lipoidica, suggesting that this process is an immune complex vasculitis [6], but extensive histopathological studies identified vascular involvement only in 30% of the cases [7]. The finding of GLUT-1 immunohistochemical expression in areas of sclerotic collagen of necrobiosis lipoidica raises the possibility that a disturbance in glucose transport by fibroblasts may contribute to the histogenesis of necrobiosis lipoidica [8].
Deep morphoea
Definition
A group of related diseases of poorly understood aetiology affecting principally skin and subcutaneous tissue and characterized by variable fibrosis, sclerosis and cutaneous atrophy (Figure 99.10). Within the deep morphoea group, three closely related processes are included, namely morphoea profunda, eosinophilic fasciitis and disabling pansclerotic morphoea of children [1]. Although classical morphoea often extends from the deep dermis to the subcutaneous tissue, morphoea is sometimes an entirely panniculitic process with no involvement of the epidermis, cutaneous adnexa or dermis. The process is known variously as morphoea profunda, nodular scleroderma or keloidal scleroderma. These conditions are fully described in Chapter 57.
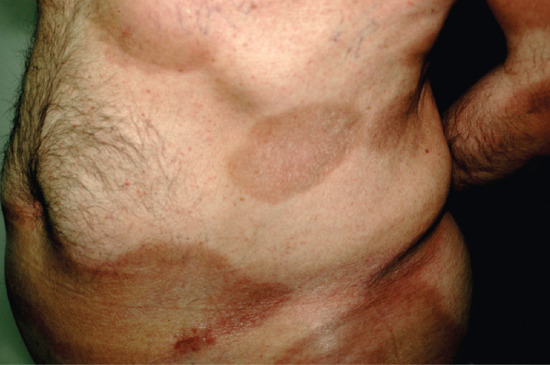
Figure 99.10 Morphoea profunda. The lesions consisted of indurated, hyperpigmented and slightly depressed plaques.
Investigations
Histopathologically, the lesions show a marked fibrous thickening of the septa of subcutaneous fat (Figure 99.11). As a consequence of thickening, collagen also replaces the fat normally present around and below the eccrine coils, giving the misleading impression that sweat glands have ascended into the dermis. When the sclerotic process involves both dermis and subcutis, the full thickness of the specimen appears homogeneously eosinophilic. Inflammatory infiltrate is present only in active lesions, consisting of aggregates of lymphocytes surrounded by plasma cells at the interface between the thickened septa and the fat lobules. Plasma cells may be also present arranged interstitially between the sclerotic collagen bundles [2, 3, 4]. Active lesions of deep morphoea usually show denser infiltrate than dermal morphoea [4, 5, 6].
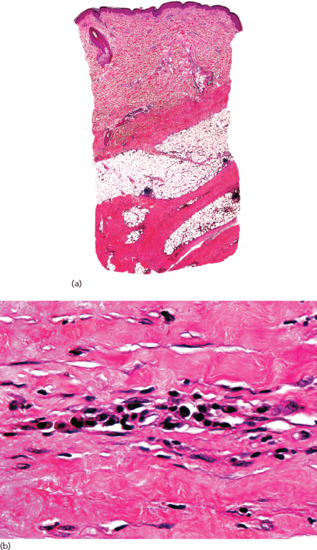
Figure 99.11 Histopathology of deep morphoea. (a) Scanning power showing sclerosis of the deeper reticular dermis and the septa of subcutaneous tissue. Note that the superficial and mid dermis were spared. (b) Thickened sclerotic collagen bundles with interstitial lymphocytes and plasma cells.
Eosinophilic fasciitis (Shulman syndrome) is regarded as a variant of deep morphoea in which the thick and sclerotic septa and the fascia show inflammatory infiltrate of lymphocytes, histiocytes, plasma cells and abundant numbers of eosinophils [7, 8, 9, 10, 11, 12, 13, 14]. Histopathological study of early stages of eosinophilic fasciitis shows oedema and infiltration by eosinophils, lymphocytes and plasma cells between the collagen bundles of the connective tissue septa of the subcutis and subcutaneous fascia. Lymphoid aggregates may be also present. In the later stages, there is fibrosis and hyalinization of the involved tissues [11].
Disabling pansclerotic morphoea in children is an aggressive clinical variant of morphoea which appears before 14 years of age [15], although adult onset has been also described [16]. The process involves not only the full thickness of the skin, but also the subcutaneous tissues, muscle and bone. Histopathological findings in cutaneous lesions of disabling pansclerotic morphoea show sclerotic replacement of the full thickness of the dermis and subcutaneous fat and the process extends to underlying fascia. In active lesions, a variable infiltrate of lymphocytes and plasma cells is seen between the sclerotic collagen bundles [15].
Subcutaneous granuloma annulare
Clinical features
Presentation
Subcutaneous granuloma annulare is a rare clinicopathological variant of granuloma annulare, characterized by subcutaneous nodules that may appear alone or in association with the classical dermal papular lesions [1, 2] (Figure 99.12). It typically presents in children or young adults [3, 4]. It is fully described in Chapter 97.
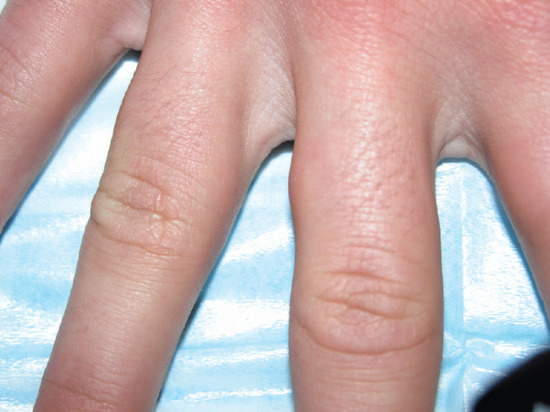
Figure 99.12 Subcutaneous granuloma annulare involving the lateral aspect of the first phalanx of the third right finger in a 14-year-old boy.
Pathophysiology
An immunoglobulin-mediated vasculitis has been proposed as the underlying mechanism for necrobiotic areas in granuloma annulare [5], although direct immunofluorescence studies failed to demonstrate immune deposits within vessels walls [6]. Additional postulated pathogenic mechanisms include a cell-mediated immune response with increased helper/inducer T cells and CD1a-positive Langerhans cells [7], a Th1 inflammatory reaction with interferon (IFN)-γ-producing lymphocytes eliciting matrix degradation [8], increased collagen synthesis [9] and elastic tissue degeneration [10]. The inflammatory cells release cytokines, including macrophage inhibitor factor, which cause histiocytes to accumulate in the necrobiotic areas and release lysosomal enzymes resulting in degenerate connective tissue [11]. Usually, subcutaneous granuloma annulare is a true panniculitic process with no dermal involvement, although in 25% of patients subcutaneous nodular lesions coexist with the classical presentation of superficial papules [12, 13]. In rare instances, subcutaneous granuloma annulare may extend to involve deeper soft tissues and producing a destructive arthritis and limb deformity [14].
Differential diagnosis
Histopathological differential diagnosis of subcutaneous granuloma annulare includes rheumatoid nodule, necrobiosis lipoidica and epithelioid sarcoma.
In contrast with subcutaneous granuloma annulare, which usually exhibits a pale and mucinous centre with a tendency to be basophilic, the central necrobiotic areas of rheumatoid nodules appear homogeneous and eosinophilic with abundant fibrin deposits. Sometimes, however, the differential diagnosis between subcutaneous granuloma annulare and rheumatoid nodule may be impossible on histopathological grounds alone. Old rheumatoid nodules show extensive fibrosis in which necrobiotic areas persist.
Lesions of necrobiosis lipoidica involve the full thickness of the dermis and the subcutaneous involvement is just a deep extension from the dermis into the connective tissue septa of the subcutis. Plasma cells, aggregations of histiocytes and multinucleated giant cells are more common in necrobiosis lipoidica than in subcutaneous granuloma annulare. In the late stages of necrobiosis lipoidica, there is extensive fibrosis and degenerate collagen is no longer seen.
Epithelioid sarcoma (see Chapter 137) is a neoplastic process in which central areas of degenerate collagen are surrounded by epithelioid cells with hyperchromatic and pleomorphic nuclei, some of them showing atypical mitotic figures. Immunohistochemical studies demonstrate that, in contrast with the inflammatory cells in subcutaneous granuloma annulare, the neoplastic cells in the palisades of epithelioid sarcoma express immunoreactivity for low- and high-molecular-weight cytokeratins, epithelial membrane antigen and CD34; furthermore, their nuclei show no expression of integrase interactor 1 (INI-1) [15, 16, 17].
Investigations
Pathology
The histopathological changes seen in subcutaneous granuloma annulare consist of areas of basophilic degeneration of collagen bundles with peripheral palisading granulomas involving the connective tissue septa of the subcutis (Figure 99.13). Usually, the areas of collagen degeneration are larger than in the dermal counterpart of the process. The central necrobiotic areas contain increased amounts of connective tissue mucin and nuclear dust from neutrophils between the degenerated collagen bundles. Elastic tissue is usually absent within the foci of degenerate collagen. The peripheral ring is composed of epithelioid histiocytes arranged in a palisade fashion and multinucleated giant cells may also be seen [18, 19]. Eosinophils are more common in subcutaneous granuloma annulare than in the dermal superficial lesions [19]. The so-called incomplete or interstitial histopathological variant of granuloma annulare is characterized by histiocytes interstitially arranged between collagen bundles, with mucin deposition but no areas of degenerate collagen. This histopathological pattern, more frequent than the necrobiotic one in dermal lesions, has yet to be described in subcutaneous granuloma annulare and all reported patients with deep forms of the process showed the classical palisading necrobiotic pattern [20]. Immunohistochemical studies showed intense expression of CD68/PGM1 in the histiocytic population and a variable one of lysozyme. T-cell markers (CD3, CD4 and CD8) have been detected mainly in the perivascular lymphocytic infiltrate, with CD4+ T lymphocytes predominating over CD8+ [21].
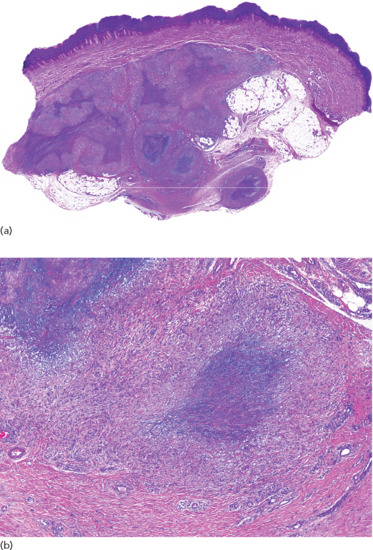
Figure 99.13 Histopathological findings in subcutaneous granuloma annulare. (a) Scanning power showing the involvement of deeper dermis and subcutaneous tissue. (b) There are several areas of degenerate collagen bundles surrounded by a palisade of histiocytes.
Rheumatoid nodule
Definition
Rheumatoid nodules are one of the extra-articular manifestations of rheumatoid arthritis. They are usually found in proximity to joints or extensor surfaces (Figure 99.14) and other areas subjected to mechanical pressure. They can also develop elsewhere, including in the pleura and meninges. Nodules vary in size and consistency and are rarely symptomatic. They are described more fully in Chapter 154.

Figure 99.14 Rheumatoid nodules involving the dorsum of the fingers in an adult woman with seropositive rheumatoid arthritis.
Pathophysiology
The pathogenesis of rheumatoid nodules remains unknown. Because the lesions develop at sites of trauma and pressure, mechanical factors have been postulated as pathogenic factors. Some genetic factor may also be involved, because patients with HLA-DRB1 present with severe rheumatoid arthritis and frequent rheumatoid nodules, whereas those with HLA-DRw2 have a mild articular disease and infrequent rheumatoid nodules [1–4]. Recently, microchimerism has been demonstrated almost in 50% of the cases of rheumatoid nodules of patients with rheumatoid arthritis. Since microchimerism is genetically disparate, it is possible that microchimerism in rheumatoid nodules serves as an allogeneic stimulus or allogeneic target [5]. Pro-inflammatory cytokines and cell adhesion molecules are very similar in rheumatoid nodules and the synovial lining in rheumatoid joints. The cytokine profile identified within the rheumatoid nodule showed the presence of IFN-γ, but not IL-2, and prominent expression of IL-1β and TNF-α together with IL-12, IL-18, IL-15 and IL-10. These findings support the hypothesis that the formation of rheumatoid nodules is driven by Th1 lymphocytes [6]. An immune complex-mediated mechanism has also been postulated: IgG and IgM have been detected by direct immunofluorescence in the vessel walls of rheumatoid nodules, suggesting that a vasculitic process may be involved [7, 8]. The mechanism for the central degeneration of the collagen bundles is also unknown. Although apoptosis has been demonstrated throughout the entire nodule [9], it seems that the proteases, collagenases and other chemotactic factors (e.g. granulocyte–macrophage colony-stimulating factor and fibronectin) secreted by lesional monocytes and macrophages are the main factors inducing degeneration of collagen, mucin deposition and palisading granuloma formation [7, 10].
Clinical variants
Accelerated rheumatoid nodulosis (ARN) is the term used to describe the development of new painful rheumatoid nodules in patients with chronic rheumatoid arthritis under treatment with methotrexate. These new nodules develop preferentially on the hands, feet and ears [11–15]. It seems that there is an individual susceptibility to ARN, because it develops more frequently in patients with HLA-DRB1 and seropositive rheumatoid arthritis [1, 15]. Genetically predisposed patients appear to be protected against the development of methotrexate-induced ARN by the concomitant administration of hydroxychloroquine [16], d-penicillamine [17], colchicine [18] or sulfasalazine [19]. The pathogenesis of ARN is unknown, although an adenosine A1 receptor promotion of multinucleated giant cell formation by human monocytes has been postulated [20]. ARN is not exclusively related to methotrexate therapy and identical lesions have also been reported in patients with rheumatoid arthritis receiving treatment with azathioprine [21], etanercept [22, 23], infliximab [24] and leflunomide [20, 25]. Neither is ARN found exclusively in rheumatoid arthritis: similar lesions have been described in patients with psoriatic arthritis [26] and systemic lupus erythematosus [27–30]. ARN has also been described in seropositive, polyarthritic-onset juvenile rheumatoid arthritis after methotrexate treatment [31, 32]. In all these patients, the condition causes minimal symptoms and regresses after methotrexate is withdrawn; it does, however, recur when methotrexate is reintroduced.
Differential diagnosis
Histopathological differential diagnosis of rheumatoid nodules includes other palisading granulomas, mainly necrobiosis lipoidica and subcutaneous granuloma annulare. Table 99.2 summarizes the main differential diagnostic features among these three necrobiotic disorders. Palisading necrobiotic granulomas have been classified into ‘blue’ and ‘red’ granulomas according to the colour of the central area of degenerate collagen stained with H&E [33, 34]. Blue granulomas, which show a basophilic centre due to mucin deposition and the presence of neutrophils and nuclear dust, are usually seen in subcutaneous granuloma annulare. Red granulomas exhibit an eosinophilic necrobiotic central area due to fibrin deposition and are seen predominantly in rheumatoid nodules (Figure 99.15). Necrobiosis lipoidica usually shows a more fibrotic pattern and the process always involves the dermis.
Table 99.2 Histopathological differential diagnosis of rheumatoid nodule, subcutaneous granuloma annulare and necrobiosis lipoidica.
| Rheumatoid nodule | Subcutaneous granuloma annulare | Necrobiosis lipoidica | |
| Location | Subcutaneous septa | Subcutaneous septa, often upper and mid reticular dermis involvement | Full thickness of the dermis with extension into subcutaneous septa |
| Pattern | Massive areas of degenerate collagen with fibrin deposition (eosinophilic necrobiotic granuloma) | Discrete foci of degenerate collagen with mucin deposition (basophilic necrobiotic granuloma) | Fibrosis and ill-defined areas of collagen degeneration (eosinophilic necrobiotic granuloma) |
| Collagen degeneration | Complete | Complete | Indistinct, elongated areas of degenerate collagen |
| Fibrosis | Common | Uncommon | Common |
| Histiocytes | Well-defined palisades of histiocytes | Well-defined palisades of histiocytes | Interstitial histiocytes, no palisading |
| Inflammatory components | Tuberculoid and sarcoid reaction common | Tuberculoid and sarcoid reaction uncommon | Tuberculoid and sarcoid reaction common |
| Vascular anomalies | Capillary hyperplasia at the periphery | Perivascular lymphocytes | Capillary wall thickening |
| Mucin Fibrin | Variable Common | Common No | Variable Variable |
Modified from Hewitt and Cole [38].
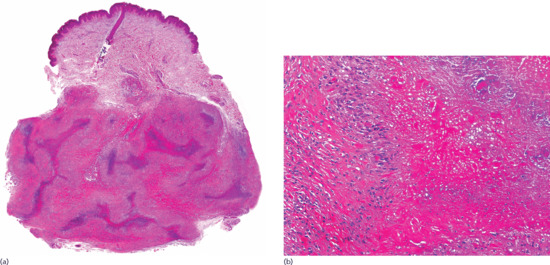
Figure 99.15 Histopathological features of rheumatoid nodule. (a) Scanning power showing a diffuse replacement of subcutaneous tissue by a fibrotic process with scattered areas of degenerate collagen. (b) The eosinophilic fibrinoid areas are surrounded by a palisade of histiocytes.
Investigations
Histopathological findings in rheumatoid nodules vary according to the age of the lesion. Early lesions show microscopic features of granulation tissue surrounded by mononuclear cells and fibroblasts [35]. In later stages, the lesions show a central area of degenerate collagen admixed with fibrinoid material and surrounded by a palisade of elongated mononuclear histiocytes. The inner central degenerated zone appears as intensely eosinophilic amorphous, granular or fibrillary material containing collagen fibrils, fibrin and cellular debris. Multinucleated giant cells, T lymphocytes, plasma cells, mast cells and eosinophils may also be seen at the periphery. Uncommonly, features of acute vasculitis have been described in the surrounding vessels and sometimes a necrotic blood vessel associated with nuclear debris and sparse neutrophils may be seen at the centre of necrobiotic areas, though these findings probably represent secondary vasculitis. In rare instances, superficial nodules may perforate the epidermis [36]. Longstanding rheumatoid nodules exhibit extensive fibrosis in which clefts and cystic degeneration appear due to liquefactive degeneration of the contents of the nodules [37].
Necrobiotic xanthogranuloma
Clinical features
Necrobiotic xanthogranuloma is a rare histiocytic disorder which causes progressive destruction of the involved cutaneous and extracutaneous tissues. It most commonly presents as multiple indurated yellow-red (Figure 99.16) or violaceous plaques or nodules, preferentially involving periorbital skin. It is described in detail in Chapter 136.
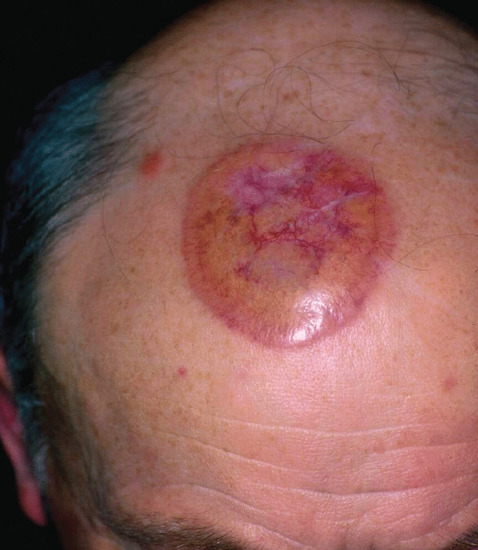
Figure 99.16 Necrobiotic xanthogranuloma. A plaque with yellowish hue involving the scalp.
Pathophysiology
The pathogenesis of necrobiotic xanthogranuloma is poorly understood. One proposed mechanism is that the monoclonal paraprotein behaves as a lipoprotein, binding to monocyte lipoprotein receptors to form xanthomata [1]. Intracellular accumulation of lipoprotein-derived lipids in skin macrophages may result from activation of monocytes [2], with both the paraprotein and immune complexes inducing granuloma formation [3]. It has been suggested that the central areas of necrobiosis in lesions of necrobiotic xanthogranuloma may be the consequence of ischaemia [4]. Another proposed pathogenetic mechanism is that an increase in circulating macrophage colony-stimulating factor (M-CSF) levels activates monocytes and favours the accumulation of large amounts of lipid and xanthoma formation [5, 6, 7]. The finding of Borrelia organisms in six of seven cases of necrobiotic xanthogranuloma using focus-floating microscopy has led some authors to propose an infectious aetiology for this process [8].
Differential diagnosis
The histopathological differential diagnosis of necrobiotic xanthogranuloma includes necrobiosis lipoidica, subcutaneous granuloma annulare, juvenile xanthogranuloma and deep xanthomas [5, 9]. Subcutaneous granuloma annulare occurs mainly in children and does not tend to ulcerate. Usually mucin deposits are evident at the centre of degenerate collagen; lymphoid follicles and cholesterol clefts are absent. Necrobiosis lipoidica may extend to subcutaneous tissue and then the differential diagnosis may be challenging. However, xanthomatization, lymphoid follicles and cholesterol clefts are less frequently seen than in necrobotic xanthogranuloma. Juvenile xanthogranuloma and deep xanthomas do not show large areas of degenerate collagen as seen in necrobiotic xanthogranuloma.
Investigations
From the histopathological point of view, necrobiotic xanthogranuloma is not a true panniculitis but a deeper extension of a predominantly dermal process (Figure 99.17). The most characteristic findings consist of a diffuse involvement of the dermis by foamy histiocytes and some Touton-like multinucleated giant cells. From the dermis, the infiltrate extends through the connective tissue septa of the subcutis and underlying soft tissues. Areas of degenerate collagen bundles and cholesterol clefts are often seen within the diffuse infiltrate [10, 11]. A palisading granuloma of epithelioid histiocytes is present, at least focally, around the areas of degenerate collagen [9]. Lymphoid aggregates, sometimes with germinal centre formation, and numerous plasma cells at the periphery are often seen around the deeper areas of collagen degeneration [12]. Although there is a diffuse infiltration of the dermis and subcutaneous tissue, some cases exhibit a multinodular pattern [13]. From the immunohistochemical point of view, histiocytes and foamy macrophages express immunoreactivity for lysozyme, CD68, Mac387 and CD11b [14]. In one case, intense histiocytic expression of CD10 was also observed [15].
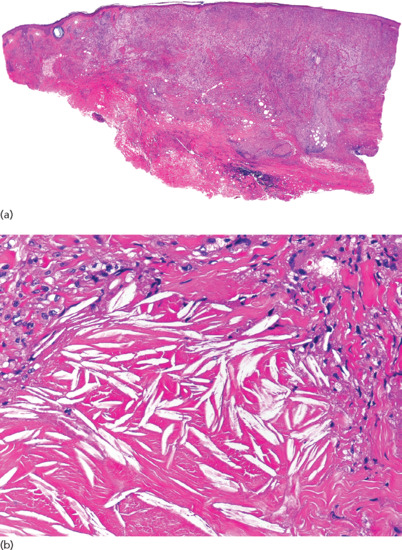
Figure 99.17 Histopathological features of necrobiotic xanthogranuloma. (a) Scanning power showing diffuse involvement of the entire thickness of the dermis and extension to subcutaneous tissue. (b) Areas of degenerate collagen with abundant cholesterol clefts.
Immunohistochemistry has demonstrated that, although necrobiotic xanthogranuloma is frequently associated with paraproteinaemia, the skin lesions represent reactive inflammation because the plasma cells present in the cutaneous lesions are polyclonal [16]. Vasculitis is not usually seen, although some lesions may show leukocytoclasis and thrombosis [13, 17]. Transepidermal and transfollicular elimination of degenerate collagen and cholesterol clefts have also been reported [18]. Reports of granuloma annulare with subsequent evolution into necrobiosis lipoidica or necrobiotic xanthogranuloma raise the possibility of a general granulomatous process accompanying paraproteinaemia [19, 20]. The coexistence of normolipaemic plane xanthoma and necrobiotic xanthogranuloma in the same patient also suggests that these two processes represent part of a spectrum of xanthomatous dermal reactions associated with paraproteinaemia and that they may be more closely related than previously recognized [21].
Erythema nodosum
Introduction and general description
Erythema nodosum is the most common panniculitis. The process usually shows an acute onset and self-limited course. It is clinically characterized by the sudden eruption of several erythematous, tender, non-ulcerating nodules and plaques, typically located on the shins. The condition normally resolves spontaneously without ulceration, scarring or atrophy, but recurrent episodes are common. Erythema nodosum is a cutaneous reactive process that may be triggered by a wide variety of infectious and inflammatory disorders and, less commonly, by malignant neoplasms and medications [1–7, 8, 9–127] (Table 99.3). The most common triggers are bacterial infections, sarcoidosis and inflammatory bowel disease.
Table 99.3 Aetiological factors in erythema nodosum [1–127].
| Infections | |
| Bacterial infections | Atypical mycobacterial infections [1] |
| Borrelia burgdorferi infections [2] | |
| Boutonneuse fever [3] | |
| Brucellosis [4] | |
| Campylobacter infections [5] | |
| Cat-scratch disease [6] | |
| Chancroid [1] | |
| Chlamydia psittaci infections [7] | |
| Corynebacterium diphteriae infections [1] | |
| Escherichia coli infections [8] | |
| Gonorrhoea [9] | |
| Klebsiella pneumoniae infections [10] | |
| Leprosy [11] | |
| Leptospirosis [12] | |
| Lymphogranuloma venereum [13] | |
| Meningococcaemia [14] | |
| Moraxella catarrhalis infections [15] | |
| Mycoplasma pneumoniae infections [16] | |
| Pasteurella pseudotuberculosis infections [17] | |
| Propionibacterium acnes [18] | |
| Pseudomona aeruginosa infections [19] | |
| Q fever [20] | |
| Rickettsiae [21] | |
| Salmonella infections [22] | |
| Shigella infections [23] | |
| Streptococcal infections [24] | |
| Syphilis [25] | |
| Tuberculosis [26] | |
| Tularaemia [27] | |
| Yersinia infections [28] | |
| Viral infections | Cytomegalovirus [29] |
| Epstein–Barr virus infection after living-donor liver transplantation [30] | |
| Hepatitis B [31] | |
| Hepatitis C [32] | |
| Herpes simplex [1] | |
| HIV infection [33] | |
| Infectious mononucleosis [34] | |
| Measles [35] | |
| Orf [36] | |
| Parvovirus B19 [37] | |
| Varicella [38] | |
| Fungal infections | Aspergillosis [39] |
| Blastomycosis [40] | |
| Coccidioidomycosis [41] | |
| Dermatophytes [42] | |
| Histoplasmosis [43] | |
| Sporotrichosis [44] | |
| Protozoal infections | Amoebiasis [45] |
| Ascariasis [46] | |
| Giardiasis [47] | |
| Hydatidosis [48] | |
| Hookworm infestation [1] | |
| Sparganum larva [49] | |
| Toxoplasmosis [50] | |
| Trichomoniasis [51] | |
| Visceral larva migrans [52] | |
| Drugs | Acetaminophen [53] |
| Actinomycin-D [53] | |
| All-trans-retinoic acid [54] | |
| Aminopyrine [1] | |
| Amiodarone [53] | |
| Amoxicillin [8] | |
| Ampicillin [8] | |
| Antimony [1] | |
| Arsphenamine [9] | |
| Azathioprine [53] | |
| Bromides [55] | |
| Busulfan [53] | |
| Cabergoline [56] | |
| Capecitabine [57] | |
| Carbamazepine [53] | |
| Carbenicillin [53] | |
| Carbimazole [58] | |
| Cefdinir [53] | |
| Certolizumab [59] | |
| Chlordiazepoxide [53] | |
| Chlorotrianisene [53] | |
| Chlorpropamide [53] | |
| Ciprofloxacin [53] | |
| Clomiphene [53] | |
| Codeine [53] | |
| Cotrimoxazole [53] | |
| D-penicillamine [60] | |
| Dapsone [53] | |
| Diclofenac [53] | |
| Dicloxacillin [53] | |
| Diethylstilboestrol [53] | |
| Disopyramide [53] | |
| Echinacea herbal therapy [61] | |
| Enoxacin [53] | |
| Erythromycin [8] | |
| Etanercept [62] | |
| Fluoxetine [53] | |
| Furosemide [53] | |
| Glatiramer acetate [63] | |
| Glucagon [53] | |
| Gold salts [64] | |
| Granulocyte colony-stimulating factor [65] | |
| Hepatitis B vaccine [66] | |
| HPV vaccine [67] | |
| Hydralazine [53] | |
| Ibuprofen [53] | |
| Imatinib mesylate [68] | |
| Indometacin [53] | |
| Infliximab [69] | |
| Interleukin 2 [70] | |
| Iodides [53] | |
| Isotretinoin [71] | |
| Leukotriene modifying agents (zileuton and rafirlukast) [72] | |
| Levofloxacin [53] | |
| Lidocaine [73] | |
| Meclofenamate [53] | |
| Medroxyprogesterone [53] | |
| Meprobamate [53] | |
| Mesalamine [53] | |
| Meticillin [53] | |
| Methimazole [53] | |
| Methyldopa [53] | |
| Mezlozillin [53] | |
| Minocycline [74] | |
| Naproxen [53] | |
| Nifedipine [53] | |
| Nitrofurantoin [1] | |
| Ofloxacin [53] | |
| Omeprazole [75] | |
| Oestrogens [53] | |
| Oral contraceptives [76] | |
| Oxacillin [53] | |
| Paroxetine [53] | |
| Penicillin [53] | |
| Phenylbutazone [53] | |
| Phenytoin [53] | |
| Piperacillin [53] | |
| Progestins [53] | |
| Propylthiouracil [77] | |
| Pyritinol [9] | |
| Rabies vaccine [78] | |
| Serotonin reuptake inhibitors [79] | |
| Sparfloxacin [53] | |
| Streptomycin [53] | |
| Sulfamethoxazole [53] | |
| Sulfisoxazole [53] | |
| Sulfonamides [80] | |
| Sulfasalazine [53] | |
| Thalidomide [81] | |
| Ticarcillin [53] | |
| Trimethoprim [82] | |
| Typhoid vaccination [83] | |
| Verapamil [53] | |
| Malignant diseases | Adenocarcinoma of the colon [84] |
| Carcinoid tumour [85] | |
| Carcinoma of the uterine cervix [86] | |
| Hepatocellular carcinoma [87] | |
| Hodgkin disease [88] | |
| Leukaemia [89] | |
| Lung cancer [90] | |
| Stomach cancer [8] | |
| Myelodysplastic syndrome [91] | |
| Non-Hodgkin lymphoma [92] | |
| Pancreatic carcinoma [93] | |
| Parathyroid carcinoma [94] | |
| Post-radiotherapy for pelvic carcinoma [95] | |
| Renal carcinoma [70] | |
| Sarcoma [9] | |
| Miscellaneous diseases | Acne fulminans [96] |
| Acupunture therapy and flu-like infection [97] | |
| Adult-onset Still disease [98] | |
| Ankylosing spondylitis [99] | |
| Antiphospolipid antibody syndrome [100] | |
| Behçet disease [101] | |
| Breast abscesses [102] | |
| Chronic active hepatitis [103] | |
| Coeliac disease [104] | |
| Colon diverticulosis [105] | |
| Crohn disease [116] | |
| Diverticulitis [105] | |
| Eosinophilic oesophagitis [107] | |
| Granulomatous mastitis [108] | |
| IgA nephropathy [109, 110] | |
| Intestinal bypass syndrome [111] | |
| Jellyfish sting [112] | |
| Lupus erythematosus [113] | |
| Pregnancy [114] | |
| Primary biliary cirrhosis [115] | |
| Radiotherapy [116] | |
| Relapsing polychondritis [117] | |
| Reactive arthritis [118] | |
| Rheumatoid arthritis [119] | |
| Sarcoidosis [120] | |
| Sjögren syndrome [121] | |
| Smoke inhalation in a house fire [122] | |
| Sweet syndrome [123] | |
| Systemic lupus erythematosus-like syndrome due to C4 deficiency [124] | |
| Takayasu arteritis [125] | |
| Ulcerative colitis [126] | |
| Vogt–Koyanagi disease [121] | |
| Granulomatosis with polyangiitis [127] |
The most well-documented associations are shown in bold.
Epidemiology
Incidence and prevalence
The population prevalence of erythema nodosum in a semirural area of England over a 2 year period was 2.4 cases per 1000 population per year [128]. Erythema nodosum accounted for about 0.5% of new cases seen in departments of dermatology in England [95] and about 0.38% of all patients seen in a Department of Internal Medicine in Spain [129]. The average annual incidence of biopsy proven erythema nodosum in persons aged 14 years or more at a hospital in north-western Spain was 52 cases per million population served [8], although this almost certainly underestimated the real incidence of the disease, because only biopsy confirmed cases were included. Most cases of erythema nodosum occur within the first half of the year [8], probably due to the increased frequency of streptococcal infections in this period; there is no difference in incidence between urban and rural areas [130]. Familial cases are usually attributable to infection; simultaneous occurrence in monozygotic twin sisters has been reported [131].
Age
Erythema nodosum may occur at any age, but most cases appear between the second and fourth decades of life, with the peak between 20 and 30 years of age, probably due to the high incidence of sarcoidosis at this age [132]. Racial and geographical variations in incidence may be explained by differences in the prevalence of aetiological factors.
Sex
Several studies have demonstrated that erythema nodosum occurs three to six times more frequently in women than in men [133], although the incidence before puberty is approximately equal in both genders [134].
Associated diseases
Concomitant presentation of Sweet syndrome and erythema nodosum has been repeatedly reported in the literature [10, 135, 136, 137, 138, 139, 140, 141, 142, 143, 144, 145, 146]. The simultaneous occurrence of these two reactive processes has been associated with sarcoidosis [136], throat infection [136, 137], acute myelogenous leukaemia [138, 139] and Crohn disease [139]. The association has been described in up to 15–30% of the patients in some series of biopsy-proven erythema nodosum [140–143], which suggests a common underlying pathogenetic mechanism for both processes [140, 146].
Pathophysiology
There are numerous recognized triggers of erythema nodosum [1–7, 8, 9–127] (see Table 99.3). Aetiological factors show considerable geographical variation related to specific endemic infections. In Europe, streptococcal infections, sarcoidosis and inflammatory bowel disease are important causes. In a significant percentage of cases (ranging between 37% and 60% in reported series), the aetiology of erythema nodosum cannot be determined despite extensive clinical and laboratory investigation [8, 38, 134, 147, 148, 149, 150].
A previous episode of upper respiratory infection by group A β-haemolytic Streptococcus is a frequent cause for erythema nodosum in children and young adults. Erythematous subcutaneous nodules usually develop 2–3 weeks after the throat infection and are accompanied by an elevation of the antistreptolysin O (ASO) titre; by the time cutaneous lesions appear, the cultures from throat swabs usually fail to detect microorganisms [8, 24]. Tuberculosis is a common cause of erythema nodosum in areas of high endemicity. This used to apply to many parts of Europe but no longer does so [8, 147, 151]. Erythema nodosum, when triggered by tuberculosis, presents mainly in children at the time of primary pulmonary infection and concomitant with the conversion of the tuberculin test [24].
Many medications have been implicated as the cause of erythema nodosum, although their real pathogenetic role is difficult to establish with confidence. Historically, sulfonamides, bromides and oral contraceptive pills have been the most commonly associated medications but a large number of drugs have been reported as triggers (see Table 99.3). Oral contraceptive pills have become a rare cause since their oestrogen content was greatly reduced. Where erythema nodosum has arisen in patients receiving antibiotics for infections it is difficult to discern whether the cutaneous reaction is due to the antiobiotic or the infectious process.
Sarcoidosis is one of the commonest disorders associated with erythema nodosum in adults [147]. Hilar adenopathy, arthritis around the ankles and erythema nodosum is characteristic of Löfgren syndrome [152]. However, erythema nodosum and bilateral hilar adenopathy do not occur exclusively in sarcoidosis but may also be seen in lymphomas, tuberculosis, streptococcal infections, coccidioidomycosis, histoplasmosis and acute infections by Chlamydophila pneumoniae [153, 154].
In adults, erythema nodosum is often associated with a flare of inflammatory bowel disease, although the cutaneous eruption may sometimes precede the bowel disease. Crohn disease [106] is more frequently associated than ulcerative colitis [126].
The large list of very disparate processes that may be associated with erythema nodosum indicates that this disorder is a cutaneous reactive process and that the skin has a limited response capacity to very different triggers. Most probably, erythema nodosum results from immune complex deposition in and around veins of the connective tissue septa of the subcutis. In support of this hypothesis is the demonstration of circulating immune complexes [155], complement activation [156, 157] and deposits of immunoglobulins in the blood vessels walls of the septa of subcutaneous fat in patients with erythema nodosum [158, 159]. Some investigators have, however, failed to demonstrate circulating immune complexes in erythema nodosum patients and a type IV delayed hypersensitivity reaction has been proposed as a possible pathogenetic mechanism [160].
Early lesions of erythema nodosum show a predominantly neutrophilic infiltrate. Recent investigations have demonstrated that patients with erythema nodosum have a fourfold higher percentage of reactive oxygen intermediates (ROI) produced by activated neutrophils in their peripheral blood compared with healthy volunteers. The percentage of ROI-producing cells correlates with the clinical severity of erythema nodosum and ROI may play their pathogenic role by oxidative tissue damage and by promoting tissue inflammation [161].
Patients with sarcoidosis-associated erythema nodosum secrete an uncommon tumour necrosis factor (TNF)-α II due to a nucleotide exchange (G-A) at position -308 in the human TNF-α gene promoter, whereas patients with erythema nodosum without underlying sarcoidosis do not have this genomic anomaly [162]. Conversely, polymorphism of the macrophage MIF gene at the position -173 has been associated with a significantly increased risk of developing sarcoidosis in patients with erythema nodosum [163]. These investigations support the hypothesis that sarcoidosis-associated erythema nodosum might be pathogenically linked to altered TNF-α or MIF production due to a genetic promoter polymorphism.
Other investigators have found that the pro-inflammatory cytokine pattern, both in infectious and non-infectious disease related erythema nodosum, is characterized by raised IL-6 serum concentrations [124]. High expression of Th1 cytokines (IL-2 and IFN-γ) has also been demonstrated in most skin lesions and in the peripheral blood of patients with erythema nodosum, whereas this cytokine gene expression pattern was absent or only minimally present in the skin and peripheral blood of control subjects. These results directly demonstrate that a polarized Th1 immune response occurs in the skin lesions of erythema nodosum patients regardless of the wide variety of provoking agents [164].
Recently, it has been demonstrated that adipocytes play an important role in activating inflammatory systems and adaptive immune systems, destroying pathogens throughout the secretion of multiple adipokines and adipocytokines [165]. This immunological role may explain why both coccidioidomycosis [166] and sarcoidosis appear to be less severe and of shorter duration in those patients who develop erythema nodosum, especially if they are carriers of the HLA-DRB1*03-positive leukocyte antigen [167].
Presentation
Clinically, the eruption is quite characteristic and consists of a sudden onset of symmetrical, bilateral, tender, erythematous, warm nodules and raised plaques usually involving the shins (Figure 99.18), ankles and knees. The nodules range from 1 to 5 cm or more in diameter and may become confluent resulting in erythematous plaques. Occasionally, lesions of erythema nodosum may appear in other areas, including the thighs, extensor aspects of the arms, neck and even the face. Early lesions show a bright red colour and are raised slightly above the skin. After a few days, they become flat, with a livid red or purplish colour. Finally, they show a yellow or greenish appearance, often taking on the look of a deep bruise, and for that reason the process was classically named ‘erythema contusiformis’. Nodules of erythema nodosum never ulcerate and the lesions regress without atrophy or scarring. Often, acute bouts of erythema nodosum are associated with a fever of 38–39°C, fatigue, malaise, arthralgia, headache, abdominal pain, vomiting, cough or diarrhoea. Episcleral lesions and phlyctenular conjunctivitis may also accompany the cutaneous lesions. Rare clinical manifestations associated with erythema nodosum include lymphadenopathy, hepatomegaly, splenomegaly and pleuritis [148]. The eruption persists for 3–6 weeks and regresses leaving no residual marks. Recurrences are common. Duration of erythema nodosum in children is shorter than in adults and arthralgias and fever are less frequent in paediatric cases [168, 169, 170].
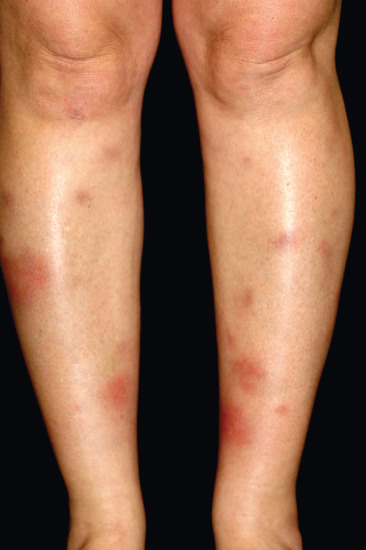
Figure 99.18 Characteristic eruption of erythema nodosum with lesions in different stages of evolution involving the anterior aspect of the legs of an adult woman. Some early lesions consist of bilateral erythematous nodules and plaques, whereas later stage lesions show a bruise-like appearance.
Clinical variants
The processes described under the names of erythema nodosum migrans [171–174], subacute nodular migratory panniculitis of Vilanova and Piñol [175, 176], and chronic erythema nodosum [151] are now all thought to be expressions of different stages of the evolution of erythema nodosum rather than separate entities [177]. In children, there is a rare variant of erythema nodosum characterized by unilateral erythematous nodules involving the palms or soles that appear after physical activity [178–181]. Histopathological study of these palmoplantar lesions shows features of classical erythema nodosum.
Differential diagnosis
Besides authentic erythema nodosum, patients with Behçet disease may present with eruptions that clinically resemble erythema nodosum [101]. However, histopathological study demonstrates that these are not erythema nodosum but show a predominantly lobular panniculitis with leukocytoclastic or lymphocytic vasculitis [182, 183]. Furthermore, the frequency of thrombophlebitis is higher in the erythema nodosum-like lesions of patients with Behçet disease compared with individuals with idiopathic erythema nodosum.
Investigations
To identify the responsible aetiological factor in a patient with erythema nodosum, a rational, cost-effective diagnostic approach should be initiated. A complete clinical history should include enquiry about previous diseases, medications, foreign travels, pets and hobbies, and possible familial cases. Initial laboratory investigations should include complete blood count, erythrocyte sedimentation rate, ASO titre, urinalysis, throat culture, intradermal tuberculin test and/or chest X-ray. When the aetiology is unclear, serological tests for those bacterial, viral, fungal or protozoal infections most prevalent in the area should be undertaken. The results of the tuberculin test should be evaluated in the context of the local prevalence of tuberculosis: where tuberculosis is endemic, a significant percentage of healthy adults show positive results for the tuberculin test. A stronger relationship with tuberculosis may be established using an IFN-γ release assay.
When the clinical and laboratory findings are characteristic, a tentative diagnosis of erythema nodosum may be established, but diagnostic confirmation requires biopsy.
Histopathologically, erythema nodosum is the prototype of a predominantly septal panniculitis without vasculitis. The connective tissue septa of the subcutis appear thickened and oedematous and are infiltrated by inflammatory cells that involve mainly the interface between the septa and the fat lobule. Usually, a superficial and deep perivascular inflammatory infiltrate composed predominantly of lymphocytes is also present in the overlying dermis.
As with other panniculitides, erythema nodosum is a dynamic process and the composition of the inflammatory cells involving the septa varies with the stage of the condition. Early lesions of less than 48 h duration show oedema and haemorrhage at the septa, and numerous neutrophils arranged interstitially between collagen bundles [149] (Figure 99.19). Sometimes, if these early lesions show extension of the infiltrate to the periphery of the fat lobule surrounding individual adipocytes in a lace-like fashion, the process may be misinterpreted as a predominantly lobular panniculitis. However, in contrast with a true lobular panniculitis, necrosis of the adipocytes at the centre of the fat lobule is never seen in erythema nodosum. In rare instances, eosinophils may be numerous in early lesions: this finding does not, however, correlate with any specific aetiological factor [184].
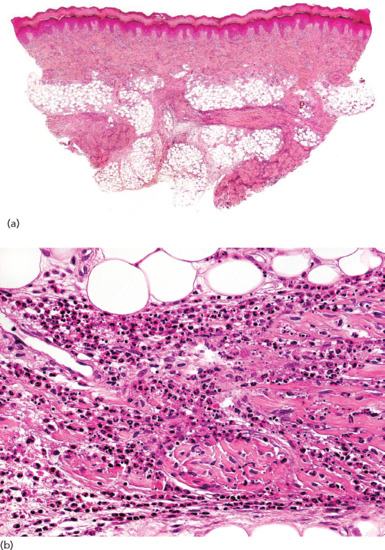
Figure 99.19 Histopathological features of an early lesion of erythema nodosum. (a) Scanning power showing a mostly septal panniculitis with thickened connective tissue septa of the subcutis. (b) The infiltrate at the septa is mostly composed of neutrophils interstitially arranged between collagen bundles.
A histopathological hallmark of erythema nodosum is the presence of so-called Miescher radial granulomas [185, 186, 187], that consist of small well-defined nodular aggregates of small histiocytic cells around a central stellate or banana-shaped cleft (Figure 99.20). The nature of the central cleft is unknown and, although some authors have considered them to be lymphatic spaces, immunohistochemical and ultrastructural studies have failed to demonstrate any endothelial cell lining. In early lesions, these granulomas are scattered in the septa and are surrounded by neutrophils. In older nodules, histiocytic cells group to form multinucleated giant cells, many of which still retain in their cytoplasm the stellate central cleft as found in Miescher radial granuloma.

Figure 99.20 Histopathological features of a fully developed lesion of erythema nodosum. (a) Scanning power showing thickened septa of the subcutaneous tissue. (b) Higher magnification shows the characteristic features of Miescher radial granuloma: Aggregations of small histiocytes around a central cleft.
Sometimes Miescher radial granulomas are conspicuous in the septa. Occasionally, however, serial sections are required to identify them: if carefully sought, they can be found in almost all stages of erythema nodosum lesions and such a search should always be undertaken to establish a specific diagnosis [187]. However, some authors consider that similar granulomas may be found in subcutaneous Sweet syndrome, erythema induratum of Bazin, Behçet disease and necrobiosis lipoidica [177]. Recent immunohistochemical studies have demonstrated that the cells around the central clefts of Miescher radial granulomas express myeloperoxidase [188], in a similar way to those of the small, elongated, twisted-appearing mononuclear cells of the infiltrate in so-called histiocytoid Sweet syndrome [189]. These findings demonstrate that the mononuclear cells that make up Miescher radial granuloma, and those seen in the so-called histiocytoid Sweet syndrome, are actually immature myeloid cells, providing a link between erythema nodosum and Sweet syndrome, two conditions in which neutrophils participate.
Late-stage lesions of erythema nodosum show septal fibrosis and periseptal granulation tissue, partially replacing the fat lobules, with an infiltrate composed of lymphocytes, histiocytes and multinucleated giant cells (Figures 99.21 and 99.22). Despite this fibrotic process involving the septa, it is striking that erythema nodosum resolves completely after some weeks or months.
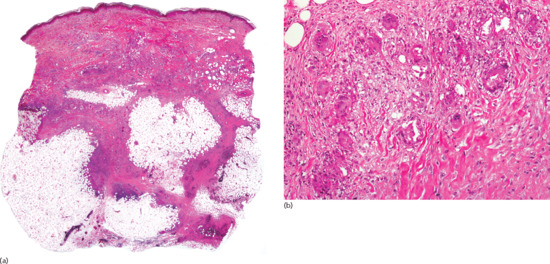
Figure 99.21 Histopathological features of a late stage lesion of erythema nodosum. (a) Scanning power showing a mostly septal panniculitis. (b) Numerous multinucleated giant cells are present in the infiltrate at the septa.
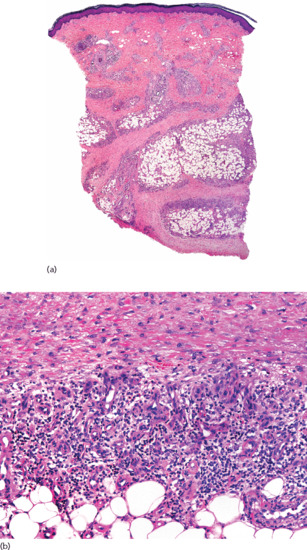
Figure 99.22 Histopathological findings of a very late stage lesion of erythema nodosum. (a) Scanning power showing very thick connective tissue septa at the subcutis. (b) The infiltrate at the interface shows features of granulation tissue.
Vasculitis is not normally seen at any stage in the course of erythema nodosum, although in rare cases a necrotizing small vessel vasculitis with fibrinoid necrosis of the small vessels in the septa has been described [190]. In a detailed histopathological study of a series of 79 cases of erythema nodosum, true leukocytoclastic vasculitis was not found [187]. The cases described as erythema nodosum with lobular neutrophilic panniculitis and vasculitis involving medium size arteries, are better interpreted as examples of STP, because the involved vessels were medium-sized veins rather than arteries [191]. Ultrastructural studies have also failed to demonstrate true vasculitis in lesions of erythema nodosum [192, 193]. Lipomembranous or membranocystic panniculitis [194] and encapsulated fat necrosis (‘mobile encapsulated lipoma’) [195] may be seen in late-stage lesions of erythema nodosum.
Management
Most cases of erythema nodosum regress spontaneously in 3–4 weeks, but relapses are common. Recurrent episodes of erythema nodosum are more frequent in patients with idiopathic erythema nodosum and erythema nodosum associated with streptococcal upper respiratory tract infections. Complications are uncommon, although cases of retrobulbar optic nerve neuritis during the acute episode of erythema nodosum [196] and erythema nodosum coexisting with erythema multiforme, lichen planus and concomitant reactivation of hepatitis C viral replication [197] have each been described.
Treatment primarily includes identification and management of the underlying cause, especially if infectious. Usually, nodules of erythema nodosum regress spontaneously within a few weeks: limiting physical exercise and bed rest should be recommended. Aspirin, as well as several other nonsteroidal anti-inflammatory drugs, such as indometacin 100–150 mg daily [198] or naproxen 500 mg daily [199] are helpful for pain and for hastening resolution. Non-steroidal anti-inflammatory drugs are contraindicated in patients with inflammatory bowel disease.
In more persistent cases, potassium iodide 400–900 mg daily or a saturated solution of potassium iodide, 2–10 drops in water or orange juice three times per day, may be administered [200, 201, 202]. The mechanism of action of potassium iodide in erythema nodosum is unknown, but it probably induces mast cells to release heparin, which suppresses delayed hypersensitivity reactions. The reported response in some patients with erythema nodosum to heparinoid ointment under occlusion also supports this mechanism of action [203]. In addition, potassium iodide inhibits neutrophil chemotaxis [204]. It should be remembered that potassium iodide is contraindicated during pregnancy, because it may induce goitre in the fetus. Severe hypothyroidism secondary to exogenous intake of iodide has also been described in patients with erythema nodosum treated with potassium iodide [205].
Systemic corticosteroids are not usually indicated in erythema nodosum and they should not be commenced unless an infectious aetiology has been excluded. When administered, prednisolone in a dosage of 40 mg/day is followed by resolution of the nodules in few days. Intralesional injection of triamcinolone acetonide at a concentration of 10 mg/mL into the centre of the nodules may also be helpful in solitary persistent lesions.
Some patients have responded to a course of colchicine, 0.6–1.2 mg twice a day [206, 207], and to hydroxychloroquine in a dosage of 200 mg twice a day [208].
Recently, cases of erythema nodosum have responded to treatment with anti-TNF biological agents including etanercept [209], adalimumab [210] and infliximab in patients treated with these drugs for inflammatory bowel disease [211]. Paradoxically, however, etanercept [62] and infliximab [69] have been reported to produce erythema nodosum as a cutaneous side effect.
Erythema nodosum leprosum
Definition
Erythema nodosum leprosum is a type II leprosy reaction which is characterized by a necrotizing vasculitis involving small to medium-sized vessels of the deep dermis and subcutis. It is provoked by an immune complex-mediated response to the release of mycobacterial antigens from effete bacilli in patients with multibacillary leprosy (lepromatous and borderline lepromatous), usually after initiation of treatment (Figure 99.23). Leprosy reactions are described in detail in Chapter 28.
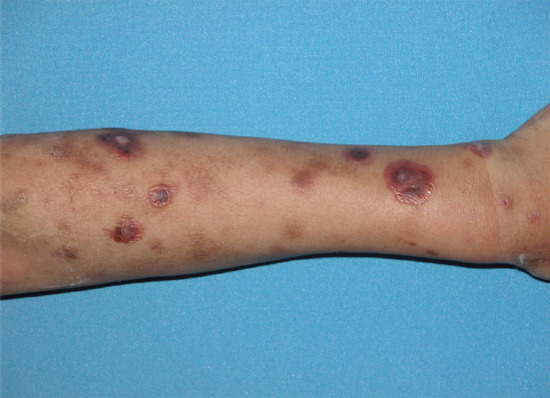
Figure 99.23 Clinical features of erythema nodosum leprosum. Acute onset of erythematous nodules involving the left upper extremity in a patient with lepromatous leprosy.
Introduction and general description
Type II reactions are due to the formation and deposit of immune complexes in association with excessive humoral reaction: the tissue expression of cytokines is mostly of IL-4 and IL-10. Erythema nodosum leprosum occurs only in patients with lepromatous or borderline lepromatous leprosy and usually occurs during treatment. It represents an immune complex-mediated vasculitis secondary to the deposition of large amounts of mycobacterial antigen, immunoglobulins and complement in the vessel walls [1]. During this type of reaction, patients show an increased CD4/CD8 ratio, due to an increase in T-helper and a decrease in T-suppressor lymphocytes, accompanied by a specific increase in Th2 lymphocytes, which induce plasma cell proliferation and release of immunoglobulins [2, 3]. In addition, these patients show a temporary recovery of Langerhans cell antigen-presenting function [4], which is directly proportional to the severity of the leprosy reaction [5, 6].
The term ‘erythema nodosum leprosum’ is inadequate because it may easily be confused with true erythema nodosum, the prototype of septal panniculitis without vasculitis. Erythema nodosum leprosum is a predominantly lobular panniculitis with vasculitis involving the large vessels of the subcutis [7, 8].
Histopathological findings in erythema nodosum leprosum vary according to the age of the lesions. Early nodules show oedema of the papillary dermis and a neutrophilic infiltrate with necrotizing vasculitis. Although the process is mostly centred in the dermis, it may occasionally extend to subcutaneous tissue, appearing as a predominantly lobular panniculitis with vasculitis involving vessels of the subcutis (Figure 99.24). Later stage lesions exhibit a lymphohistiocytic infiltrate, in which foamy macrophages with numerous mycobacteria are seen within the fat lobule [7, 8, 9].
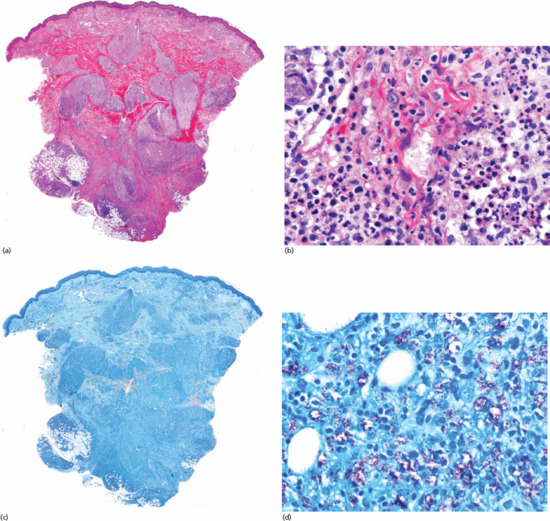
Figure 99.24 Histopathological features of erythema nodosum leprosum. (a) Scanning power showing dense nodular infiltrates in the dermis and a mostly lobular panniculitis. (b) Some of the small capillaries of the fat lobule show fibrin deposits and neutrophils involving their vessel walls. (c) A section of the same case stained with Ziehl–Neelsen stain. (d) Higher magnification of numerous microorganisms of Mycobacterium leprae within the cytoplasmic vacuoles of the histiocytes.
Lucio phenomenon is an uncommon variant of type II leprosy reaction with a unique clinical morphology [8, 10–12]. It occurs almost exclusively in Mexican and Central American patients with untreated, diffusely infiltrated, non-nodular lepromatous leprosy and is characterized by multiple haemorrhagic necrotic skin infarcts. It does not result in a panniculitis and subcutaneous tissue is not normally involved. Histologically, there is a necrotizing vasculitis involving the superficial and mid dermis [8, 12].
Erythema induratum of Bazin
Clinical features
History
Erythema induratum is a chronic recurrent reactive disorder characterized by subcutaneous nodules located preferentially on the posterior aspects of the lower legs of adult women. The nodules commonly develop after exposure to cold and may break down to form irregular ulcers (Figure 99.25). Its clinical features are discussed in greater detail in Chapter 27.
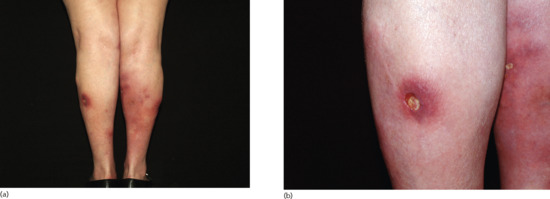
Figure 99.25 Clinical features of erythema induratum. (a) Erythematous nodules and plaques on the posterior aspect of the legs of an adult woman. (b) Some of the lesions are ulcerated.
Erythema induratum has historically been regarded as a tuberculid linked to the presence of a distant focus of tuberculosis and, when this is the case, the condition has been referred to as erythema induratum of Bazin. M. tuberculosis is not implicated in all cases, however, and the terms erythema induratum of Whitfield and nodular vasculitis have both been used to differentiate such cases from those linked to tuberculosis [1, 2, 3, 4, 5, 6, 7, 8, 9]. Whether it makes sense to maintain different terms for what is essentially an identical reactive process is open to question. Although there are still authors who regard erythema induratum of Bazin and nodular vasculitis as separate entities [10, 11], no significant clinicopathological differences have been consistently demonstrated between cases related to tuberculosis and those which are not. In recent years, most authors thus consider erythema induratum of Bazin and nodular vasculitis to be a single reactive process that may be provoked by a number of different mechanisms, one of which is active tuberculosis [12]. Box 99.2 shows other suspected aetiological associations which have been reported.
Presentation
From soon after its original description, the relationship between erythema induratum and M. tuberculosis has been a controversial issue, because mycobacteria cannot be cultured from the cutaneous lesions. Thus, evidence for the aetiological role of M. tuberculosis in erythema induratum has had to be established indirectly from a range of clinical and epidemiological data, including the strong hypersensitivity reaction to tuberculin [25], the presence of concomitant active distant (usually pulmonary) tuberculous infection [26, 27], the occasional coexistence in the same patient of erythema induratum and another tuberculid [28, 29], the frequent personal or family history of tuberculosis [30, 31] and the favourable response to antituberculous chemotherapy [32]. It has been suggested that adipose tissue might constitute a reservoir where the M. tuberculosis bacillus could persist for long periods of time and avoid both killing by antimicrobials and recognition by the host immune system [33]. It has, however, never been possible to isolate M. tuberculosis from lesions of erythema induratum [34].
Several recent polymerase chain reaction (PCR) studies have demonstrated the presence of M. tuberculosis DNA in cutaneous lesions of erythema induratum, with frequencies ranging from 25% to 77% of cases, supporting the pathogenic role of M. tuberculosis [35–39]. However, in a Spanish study of patients from a region with a high prevalence of tuberculosis, M. tuberculosis DNA was detected by PCR in only 14% of cases of erythema induratum [40]. Other investigators have failed to demonstrate the presence of DNA from M. tuberculosis or from any other Mycobacterium in lesions of erythema induratum [41], supporting the contention that there are likely to be other triggers than tuberculosis.
It has been suggested that erythema induratum results from an immune complex-mediated vasculitis [10], but most authors believe that the disorder results from a type IV, cell-mediated response to an antigenic stimulus [42]. Supporting a delayed hypersensitivity reaction is the presence of abundant number of S-100 positive dendritic cells within the granulomatous infiltrate, which are probably presenting antigens to T cells [43, 44]. The presence in lesional skin of M. tuberculosis DNA but not viable tuberculous bacilli suggests that erythema induratum is a hypersensitivity reaction to fragments of tuberculous bacilli.
Investigations
As in other panniculitides, the histopathological findings in lesional skin of erythema induratum vary with the age of the lesions. It is the prototype of a predominantly lobular panniculitis with vasculitis, but the histopathological picture and the composition of the infiltrate in the fat lobule greatly depend on the stage at which the biopsy has been taken [10, 26, 45, 46]. In early stages, the fat lobules are punctuated throughout by discrete collections of inflammatory cells, mostly neutrophils. There may be extensive necrosis of the adipocytes of the fat lobule (Figure 99.26). These necrotic adipocytes elicit a response from histiocytes, which phagocytose lipid and become lipophages. In fully developed lesions, epithelioid and foamy histiocytes, Langhans type or foreign-body multinucleated giant cells and lymphocytes contribute to the granulomatous appearance of the inflammatory infiltrate (Figure 99.27). When intense vascular damage occurs, large areas of caseous necrosis appear and the lesions show all the histopathological attributes of a tuberculoid granuloma. However, stains for acid-fast bacilli, such as Ziehl–Neelsen or Fite, and immunohistochemical stains for mycobacteria are negative. Caseous necrosis may extend to the overlying dermis and secondarily involve the epidermis with ulceration and discharge of liquefied necrotic fat. Previously, the presence of tuberculoid granulomas around the eccrine coils, was regarded as suggestive of a tuberculous aetiology [47], but these findings may also be seen in non-tuberculous cases.

Figure 99.26 Histopathological features of an early lesion of erythema induratum. (a) Scanning power showing a mostly lobular panniculitis. (b) Higher magnification showing necrotic adipocytes without nuclei and luminal thrombosis of a small blood vessel.
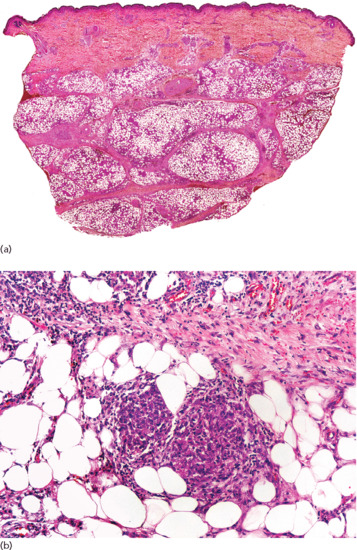
Figure 99.27 Histopathological features of a fully developed lesion of erythema induratum. (a) Scanning power showing a mostly lobular panniculitis. (b) Small granulomas involving the fat lobule.
In the literature, considerable controversy persists about whether or not vasculitis is a required histopathological criterion for establishing a diagnosis of erythema induratum. Furthermore, there is no agreement about the nature of the vessels involved in the vasculitis, because some authors have failed to report their nature or size [48], and even when the nature of the involved vessel was specifically stated, there has been disagreement as to whether it is arteries [49, 50], veins [51, 52] or both [52, 53, 54, 55, 56, 57] which are affected. In a recent large study of 101 skin biopsies from 86 patients with a clinicopathological diagnosis of erythema induratum, vasculitis was however present in 90% of cases [25] (Box 99.3).
Therefore, although vasculitis can be demonstrated in the majority of cases, there are some cases with all the other clinicopathological attributes of erythema induratum where vasculitis cannot be demonstrated despite a careful search: its presence should not be considered as an essential criterion for its histopathological diagnosis [25].
Sclerosing panniculitis
Definition and nomenclature
Sclerosing panniculitis is a relatively common form of long-term chronic panniculitis associated with chronic venous insufficiency and typically affecting the lower extremities of middle-aged or elderly women. This is manifested as a diffuse sclerosis and pigmentation of the skin and subcutaneous tissue (lipodermatosclerosis) (Figure 99.28). It is discussed in more detail in Chapter 103.
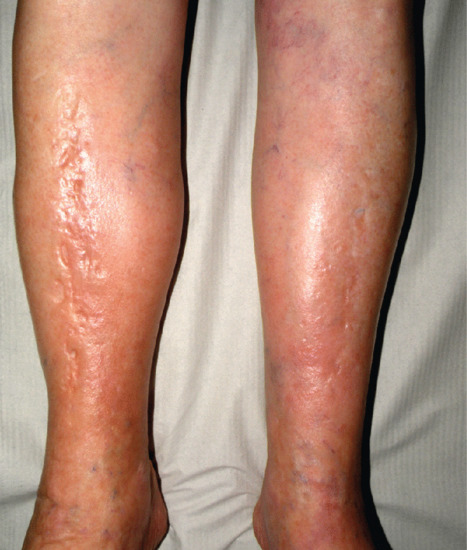
Figure 99.28 Late-stage sclerosing panniculitis showing depressed hard areas with woody induration involving the lower legs.
Clinical features
Presentation
The aetiological role of venous hypertension in the pathogenesis of sclerosing panniculitis is undisputed. Venous hypertension increases capillary permeability, which results in leakage of fibrinogen, its polymerization to form fibrin rings around vessels, with impedance of oxygen exchange and tissue anoxia [1]. Other factors such as trauma and recurrent episodes of cellulitis may also play a role [2, 3]. In a study of 128 patients with systemic sclerosis, patients with sclerosing panniculitis had pulmonary hypertension at a significantly higher incidence than those without. The authors suggested that thrombosis caused by venous hypertension of the leg may be the main cause of pulmonary hypertension in patients with systemic sclerosis and sclerosing panniculitis [4].
Reported findings which may be of pathogenetic relevance in sclerosing panniculitis include increased plasminogen activation in affected tissue [5, 6, 7], increased expression of vascular endothelial growth factor receptor 1 (VEGFR-1) and angiopoietin 2 (Ang-2) [8], deficiencies of protein C and S [9] and local increased synthesis of collagen [10, 11].
Differential diagnosis
Because initial stages of the disease involve only one leg, it may be confused clinically with bacterial cellulitis or erysipelas, although in the latter situation there is accompanying heat on palpation, fever or other systemic symptoms [12]. Early lesions may also mimic erythema nodosum [13]. In longstanding lesions, the differential diagnosis is with other sclerodermiform processes, but the absence of histopathological findings of dermal sclerosis, the peculiar distribution affecting only the distal part of one or both legs and the presence of features of chronic venous stasis exclude morphoea, scleroderma and acrodermatitis chronica atrophicans. A rare case of sarcoidosis clinically mimicked sclerosing panniculitis [14].
Investigations
Often, clinicians are reluctant to biopsy lesions of sclerosing panniculitis because it is not unusual to induce a chronic ulcer with poor healing at the site of the biopsy [3, 15]; most biopsies are thus obtained in the later stages of the disease. However, when a biopsy is performed at the early stages of the process, microscopic study demonstrates a sparse inflammatory infiltrate of lymphocytes in the septa and areas of ischaemic necrosis at the centre of the fat lobules. Necrosis of fat in these early stages is characterized by small pale anucleate adipocytes. The small blood vessels of the fat lobule appear congested, with extravasated erythrocytes, haemosiderin deposits and necrosis of endothelial cells [16]. In fully developed lesions, the septa show marked thickening and fibrosis, whereas lobules appear atrophic, often with lipophagic granulomata at their periphery. In this stage, the inflammatory infiltrate is composed of lymphocytes, histiocytes and foamy macrophages. Blood vessels appear prominent in the septa at all stages.
In late-stage lesions, the inflammatory infiltrate is sparse or absent, septal sclerosis is prominent and fat lobules appear atrophic, with microcysts and focal lipomembranous (membranocystic) changes (Figure 99.29). The latter consist of cystic structures lined by a lipomembrane, which appears as feathery amorphous eosinophilic material, sometimes with a crenellated or arabesque pattern. The thickened undulating membrane is PAS positive, stains for Sudan black and Luxol fast blue and expresses immunoreactivity for CD68 and lysozyme [17, 18, 19], suggesting the contribution of these macrophage-derived enzymes in its histogenesis. These changes are not, however, specific for sclerosing panniculitis, because they may be seen in longstanding lesions of various forms of septal and lobular panniculitis. Box 99.4 summarizes the conditions in which lipomembranous fat necrosis has been described in the literature [17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54]. Calcification and elastic tissue degeneration, sometimes with fragmented and calcified elastic fibres resembling those of pseudoxanthoma elasticum (PXE) may be seen in longstanding lesions [55]. These PXE-like fibres are positive for both von Kossa and Verhoeff van Gieson stains and may develop metaplastic ossification [56]. Elastic fibres with PXE-like changes are not specific for sclerosing panniculitis, because they have also been described in uraemic and non-uraemic calcific arteriolopathy (calciphylaxis) [57, 58], erythema nodosum, granuloma annulare, morphoea profunda [26, 55] and nephrogenic systemic fibrosis [59].

Figure 99.29 Histopathological findings in sclerosing panniculitis. (a) Scanning power showing thickened septa and cystic spaces replacing the fat lobules. (b) A cystic structure lined by a lipomembrane with a crenellated border. (c) Superficial dermis shows stasis changes. (d) Proliferation of thick-walled capillaries and venules in the superficial dermis. (e) A section of the same case stained with periodic acid–Schiff (PAS). (f) PAS positivity of the pseudopapillae with feathery projections into the cystic cavity.
The overlying superficial dermis shows venous stasis changes, with lobules of capillaries and venules of slightly thick-walled vessels in concert with extravasated erythrocytes and haemosiderin deposition. Increased melanin pigment along the basal layer of the epidermis as well as within melanophages in the superficial dermis also contribute to the characteristic hyperpigmentation [60].
Histopathological differential diagnosis of sclerosing panniculitis includes panniculitis of scleroderma and deep morphoea, but these conditions show predominantly septal panniculitis and, although lipodystrophy and lipophagic changes adjacent to the septa may be seen, they are not prominent.
Calcific uraemic arteriolopathy (calciphylaxis)
Definition
Calcific arteriolopathy (calciphylaxis) is strongly associated with end-stage chronic kidney disease and renal transplantation, particularly in diabetics, though a small proportion of cases arise in the absence of renal disease. It typically presents as irregular exquisitely tender patches of mottled dusky livedoid erythema with pale greyish areas of devitalization before progressing to full-thickness infarction of the skin with consequent necrotic ulceration. These changes commonly extend deeply into subcutaneous fat. Although the most common areas of involvement are the lower extremities and abdomen, the process may also involve other areas including the genitalia (Figure 99.30). The condition is discussed fully in Chapter 61.

Figure 99.30 Calciphylaxis involving the penis in a patient with end-stage renal disease.
Pathophysiology
Three-dimensional studies in calciphylaxis have shown vascular mural calcification as an early feature, which probably precedes endovascular fibrosis [1]. Recent studies in calciphylaxis have revealed the presence of the matrix Gla protein, osteopontin and bone morphogenic protein 2 (BMP-2) in pathologically calcified arteries [2, 3]. In involved skin, a significant up-regulation of BMP-2, its target gene Runx2 and its indirect antagonist sclerostin, as well as increased expression of inactive uncarboxylated matrix Gla protein (Glu-MGP) have been detected. The up-regulation of osteogenesis-associated markers is accompanied by an increased expression of osteopontin, fibronectin, laminin and collagen I indicating an extensive remodelling of the subcutaneous extracellular matrix. Electron dispersive X-ray analysis has revealed calcium/phosphate accumulations in the subcutis of calciphylaxis patients. Widespread medial calcification in cutaneous arterioles is associated with destruction of the endothelial layer and partial exfoliation of the endothelial cells. CD31 immunostaining has revealed aggregates of endothelial cells contributing to intraluminal obstruction and the consequent malperfusion which results in the clinical picture of ulcerative necrosis. These data indicate that vascular calcification in calciphylaxis is an active osteogenic process involving up-regulation of BMP-2 signalling, hydroxyapatite deposition and extensive matrix remodelling of the subcutis [4]. Matrix Gla protein is vitamin K dependent and inhibits vascular calcification. Therefore, oral anticoagulant therapy with warfarin, a vitamin K antagonist, favours vascular calcification in these patients [5]. Other triggering factors that have been recognized as inducers of vascular calcification in calciphylaxis include low levels of albumin, arterial hypertension, obesity, administration of systemic corticosteroids, vitamin D supplementation, blood transfusions, oral phosphate binders, metallic salts, calcitriol, local trauma, UV light treatment, malnutrition with weight loss and insulin injections [6–12].
Investigations
The most characteristic histopathological finding in calcific arteriolopathy consists in small and medium-sized vessel calcification (Figure 99.31). Small arteries, arterioles and venules may be involved. Vascular calcification is usually extensive within the vessel walls and often exhibits a concentric, circumferential, ring-like pattern. Detailed histopathological studies, however, have demonstrated that the earliest sites of calcification are the media and/or intima of cutaneous arterioles [13]. Vascular calcification in the arterioles of the deeper reticular dermis or subcutaneous tissue is usually evident with H&E staining but von Kossa stain may help to highlight these deposits. Sometimes the involved vessels also show luminal thrombosis. The affected vessels develop intimal hyperplasia with endovascular endothelial proliferation and intimal fibrosis, resulting in ischaemia of the areas they supply [13, 14]. Secondary ischaemic changes include epidermal ulceration and degeneration of dermal collagen. Focal lobular panniculitis is frequently seen [14], although some biopsies show few or no inflammatory infiltrate in the fat lobule adjacent to the calcified vessel. Some authors, however, have described a predominantly septal panniculitis in calciphylaxis [13]. Since vascular calcification may be seen in cutaneous biopsies from other disorders (Box 99.5), additional features may be needed to support a histological diagnosis of calciphylaxis: these include interstitial deposition of calcium in the dermis, fine calcium deposits in and around the adipocytes [15], epidermal and hair follicle calcification [16], perineural calcium deposits [17], and calcified elastic fibres with a PXE-like appearance [18, 19]. Perieccrine calcium deposition has been reported as a highly specific histopathological finding in calcific arteriolopathy [20].

Figure 99.31 Histopathological features of calciphylaxis. (a) Scanning power view showing involvement of the vessels of deeper reticular dermis. (b) Calcification of the vessel walls and occlusion of the vascular lumina. (c) A section of the same case stained with von Kossa stain. (d) Calcification of the vessel walls is positive with von Kossa stain.
Calcific arteriolopathy should be distinguished from metastatic cutaneous calcification, which is a rare phenomenon involving the dermis and subcutis and affecting predominantly uraemic patients with combined hyperphosphataemia and hypercalcaemia, often in the context of hyperparathyroidism (see Chapter 61). It typically presents as firm papules, nodules or plaques in the dermis or subcutis, particularly around large joints or flexural sites. Unlike calciphylaxis, metastatic cutaneous calcification does not lead to tissue necrosis and histopathologically deposits of calcium, appearing blue with H&E and black with von Kossa stains, are seen in the dermis and subcutis with variable surrounding inflammatory infiltrate, but vessel walls are spared [21].
Cold panniculitis
Definition
Cold panniculitis is a form of injury to subcutaneous fat induced by exposure to cold, either environmental [1] or as cold objects applied to the skin (e.g. ice packs) [2, 3, 4]. Infants are particularly susceptible. It is not uncommon in children in regions subject to low temperatures [5].
Pathophysiology
Predisposing factors
Cold panniculitis was originally described by Hochsinger in 1902 as submental nodules and plaques in children after cold exposure [6]. Lemez, in 1928, demonstrated infants to be more susceptible to fat necrosis by exposure to cold than adults [7]. Haxthausen in 1941 described similar cases on the cheeks of infants after cold temperatures and named the condition ‘adiponecrosis e frigore’ [8]. Adams et al. in 1954 clarified the pathogenesis of cold panniculitis demonstrating that pigs fed with a diet rich in saturated fatty acids showed higher cold susceptibility due to the higher ratio of saturated to unsaturated fatty acids, resulting in an elevated freezing point of fat [9]. In 1965 Hirsch confirmed than saturated fats solidified at a higher temperature than unsaturated fats [10]. Solomon and Beerman, in 1963, reported the occurrence of panniculitis in a 28-year-old Jamaican woman within hours of cold exposure with lesions which were readily inducible with local applications of ice [11]. Rotman reported similar cases involving the cheeks in two infants of five and eight months of age [12]. A similar process was described under the name ‘popsicle panniculitis’ on the cheeks of children a few hours after eating ice lollies [2, 3, 13]. The most extensive histopathological study of cold panniculitis was performed by Duncan et al. in 1966, who studied serial skin biopsies after exposing the skin of the child to ice for 2–4 min and performing sequential punch biopsies. Increased duration of ice exposure was required to produce cold panniculitis as the child aged and the reaction no longer occurred at the age of 22 months [4].
Presentation
Neonatal and infantile cold panniculitis
Neonates are particularly susceptible to cold panniculitis. This is thought to be because subcutaneous fat in newborns is rich in saturated fatty acids, particularly palmitic and stearic acids, which have a higher freezing point than unsaturated fatty acids, [10, 14] so that a small decrease in an infant's temperature may result in crystallization of subcutaneous fat [1]. The risk in neonates may be increased by cooling for management of birth asphyxia or for infants undergoing cardiac surgery [1, 15, 16]. Cold panniculitis has also been reported in neonates who are administered ice packs to control neonatal supraventricular tachycardia [17, 18]. Over the first 2 years of life the subcutaneous fat of children rapidly becomes less saturated, with an increase in the oleic acid content and a consequent lowering of the freezing point and lessening of the risk of cold injury.
In young children, the most commonly involved areas are the cheeks and chin, because they are rich in subcutaneous fat and are normally more exposed to the cold than other body areas. The lesions consist of indolent erythematous or violaceous indurated plaques or nodules with no systemic manifestations. The child is otherwise healthy and the lesions regress without treatment within weeks. Cold panniculitis of the cheeks may also be precipitated by sucking ice lollies (popsicle panniculitis) [2–4].
Cold panniculitis in adults
Adult patients with cold panniculitis are typically obese and predominantly female. The most commonly affected areas are the lateral upper thighs and gluteal region (Figure 99.32). The distribution of the lesions in adults has been postulated to be attributable to the effects of tight-fitting clothing compromising the blood flow in the upper lateral thighs, rendering the ischaemic fat more susceptible to cold injury [19]. These patients often have very cold skin in the affected areas even when they are seen in clinic and perhaps a more plausible explanation is that the skin and immediately subjacent fat is insulated from core body temperature by a thick layer of intervening fat. The tight-fitting clothing offers little protection from low external environmental temperature and the lesions are usually accompanied by some degree of vasodilatation as one would expect with perniosis. It has also been suggested that a diet rich in saturated fatty acids would result in a subcutaneous fat similar to that of a newborn [20]. Some such patients have been found to have cryofibrinogenaemia, which probably predisposed them to cold panniculitis [21].
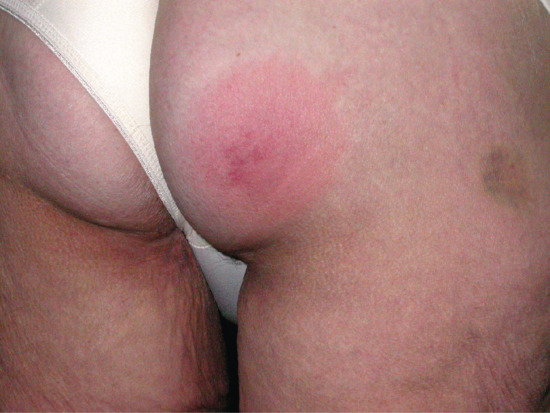
Figure 99.32 Erythematous nodule on gluteal region of a woman with equestrian cold panniculitis.
Cold panniculitis has also been described in healthy adult women with lesions involving the lateral upper thighs after prolonged horse-riding in cold weather. This variant may be regarded as a form of perniosis and has been named ‘equestrian cold panniculitis’ or horse rider's pernio [19]. Symptoms have been reported to be more pronounced in older women and appear to be aggravated by heavy smoking, by wearing tight riding clothes and by longer riding times [22]. Similar cases have been described in obese women engaged in sporting activities such as cycling and motorcycling in conditions of humidity and wind [23], or riding on open slow-moving vehicles in similar weather conditions [24].
Prolonged application of ice directly to the skin can produce local injury in a manner similar to popsicle panniculitis: ‘ice pack dermatosis’ has been described on the lower back of adults using ice packs for alleviation of chronic low back pain [25].
Investigations
Histopathologically, cold panniculitis consists of a predominantly lobular panniculitis [26], although a variable septal component is usually present [27]. Inflammation is denser in the lower reticular dermis and dermal–hypodermal interface (Figure 99.33). The infiltrate is composed mostly of lymphocytes and some histiocytes involving the fat lobules. The overlying dermis shows a superficial and deep perivascular lymphocytic infiltrate with no vasculitis [26]. Blood vessels of both septa and fat lobules may exhibit endothelial swelling and intramural oedema, but there are no fibrin deposits nor nuclear dust as would be seen in a true vasculitis. Interstitial mucin deposition may also be seen with a histopathological picture closely resembling lupus panniculitis [28].
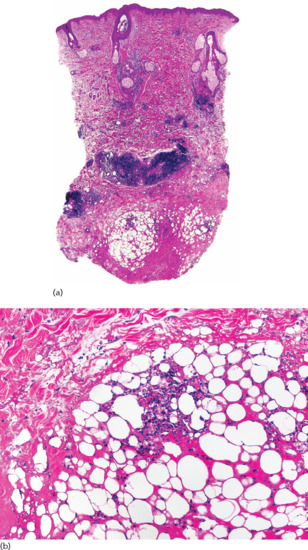
Figure 99.33 Histopathology of equestrian panniculitis. (a) Scanning power showing dense nodular infiltrates in deeper dermis. (b) Small aggregate of lymphocytes in the fat lobule.
Duncan et al. described the ‘lives of lesions’ of cold panniculitis in a 6-month-old black male infant by applying an ice cube to his buttocks and taking serial punch biopsies at 30 min, 6 h, 24 h, 48 h, 72 h, 6 days and 2 weeks [4]. Earliest changes were seen in the 24 h specimen, which showed a mild perivascular lymphohistiocytic infiltrate mostly located at the dermal–hypodermal interface, without associated fat necrosis. The changes were more intense in the specimens taken at 48 and 72 h, which showed a denser mixed infiltrate, composed of lymphocytes, scattered neutrophils, histiocytes and foamy macrophages mostly involving the fat lobule. At this time, features of adipocytic necrosis were evident in the form of lipophagic granulomas surrounding small cystic spaces. The inflammatory reaction progressively increased in biopsies taken in the next 2–3 days and then slowly decreased to regress completely by 2 weeks. The subcutaneous nodules regressed leaving no residual lesion.
Differential diagnosis
The clinicopathological differential diagnosis of cold panniculitis includes subcutaneous fat necrosis of the newborn, sclerema neonatorum, lupus panniculitis, poststeroid panniculitis, perniosis and frostbite.
Subcutaneous fat necrosis of the newborn usually appears in the first days of life and has a predilection for the thighs, buttocks, cheeks, back and arms. It may occasionally be associated with symptomatic hypercalcaemia. This panniculitis may be associated with hypothermia, obstetric trauma, maternal diabetes and maternal pre-eclampsia. Histopathology shows a lobular panniculitis with a mostly histiocytic infiltrate including multinucleate giant cells and adipocytes and histiocytes contain needle-shaped clefts that result from lipid crystallization [15, 16, 29, 30].
Sclerema neonatorum is an extremely rare disorder which was described in premature or debilitated children who developed a diffuse board-like stiffness due to generalized fat necrosis. The condition appeared in the first days after birth and was usually fatal. Histologically, the adipocytes contain needle-shaped clefts in radial arrays with no inflammatory response [26, 31]. There have been no recent reports of sclerema neonatorum and it is likely that the problem has disappeared in places with proper facilities for neonatal care of premature and debilitated newborns.
Lupus panniculitis is rare in children and the few described paediatric cases [32–36] do not differ significantly from lupus panniculitis in adults. Histopathology shows a mostly lobular panniculitis, with an infiltrate composed predominantly of lymphocytes and plasma cells, lymphoid aggregates with germinal centre formation and sclerotic collagen bundles at connective tissue septa. The process is chronic and longstanding lesions show hyaline necrosis of the fat lobule, which is not seen in the more acute and self-resolving process of cold panniculitis.
Poststeroid panniculitis occurs in children receiving high doses of systemic corticosteroids when the dose is rapidly decreased or suddenly withdrawn. The lesions appear as small painful nodules on the cheeks and posterior neck, the areas in which corticosteroid therapy has induced fat deposition. The histopathological picture is identical to that of subcutaneous fat necrosis of the newborn [27, 30].
Chilblains appear following cold exposure as bluish macules, papules and plaques involving mostly the acral areas of the skin, but they can also be found on the thighs and buttocks. Histopathologically, the picture may be very similar to cold panniculitis, although chilblains usually show oedema of the papillary dermis, the lymphocytic infiltrate is mostly arranged around eccrine coils and some cases show features of lymphocytic vasculitis in dermal blood vessels. Extension to subcutaneous fat is uncommon in chilblains [36, 37].
In early frostbite, there are erythematous and oedematous plaques that may be painful or anaesthetic. In fully developed severe frostbite, there is blistering and necrosis. Histopathology shows subepidermal oedema with blister formation, necrosis of epidermal keratinocytes and a superficial and deep perivascular lymphocytic infiltrate involving the full thickness of the dermis [36].
Management
Treatment of cold panniculitis is not usually required because the condition resolves spontaneously. Prevention of infantile cold panniculitis in children is achieved by avoiding cold exposure and direct contact with ice products [11, 16]. For equestrian cold panniculitis in adult women, the use of loose, warm clothing should be recommended when riding, with avoidance of tight-fitting clothes and, where possible, cold exposure [19, 38]. Nifedipine has been shown to be ineffective [38]. In one case, a dramatic response to tetracycline was observed, which was also effective prophylactically [39].
Lupus panniculitis
Definition and nomenclature
Lupus panniculitis is characterized by a destructive inflammation of subcutaneous fat. Clinically, lesions consist of indurated plaques which resolve with localized lipoatrophy. Depending on the intensity of inflammation a patient may first present with lipoatrophy rather than induration. The overlying skin may show changes of chronic cutaneous lupus erythematosus. The face, upper arms (Figure 99.34), upper trunk, breasts, buttocks and thighs are most commonly affected. The clinical features are described in detail in Chapter 51.
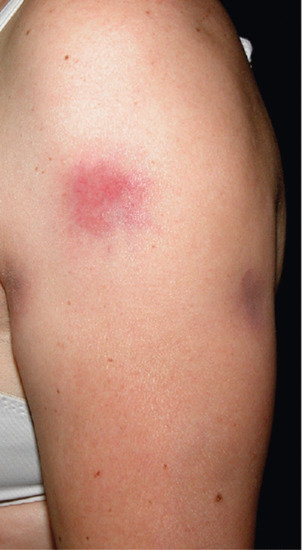
Figure 99.34 Clinical features of lupus panniculitis showing an active erythematous subcutaneous nodule and areas of hyperpigmented lipoatrophy secondary to regressed lesions.
Clinical features
Differential diagnosis
Panniculitis associated with dermatomyositis shares identical histopathological findings with lupus panniculitis [1–13]. Histopathological differential diagnosis of lupus panniculitis also includes cold panniculitis, deep morphoea, persistent nodules after injection of vaccines containing aluminium, panniculitis at the injection sites of glatiramer acetate and subcutaneous panniculitis-like T-cell lymphoma (SPTCL). Differential diagnosis with cold panniculitis is discussed earlier.
Deep morphoea may also display lymphoid nodules at the connective tissue septa of the subcutis, but in contrast with lupus panniculitis the process involves exclusively the septa and the fat lobule is spared.
Persistent nodules at injection sites of a vaccine containing aluminium show similar features to lupus panniculitis, including hyaline necrosis of the fat lobules and lymphoid aggregates, but the correct diagnosis may be suspected by the presence of histiocytes containing fine basophilic granules of aluminium and abundant number of eosinophils [14].
Several patients receiving daily glatiramer acetate injections for the treatment of multiple sclerosis developed localized panniculitis at the injection sites. The lesions consisted of a mostly lobular panniculitis, with lipophagic granulomata, namely histiocytes engulfing the lipids from necrotic adipocytes. In many areas, scattered neutrophils and eosinophils were seen both in the septa and in the fat lobules. Connective tissue septa showed widening and fibrosis in conjunction with many lymphoid follicles, presenting with germinal centre formation. Immunohistochemically, the inflammatory infiltrate of the fat lobule consisted of CD68-positive histiocytes and suppressor/cytotoxic T lymphocytes. In contrast, the lymphoid follicles in the septa and at the interface between septum and fat lobule were mainly composed of B lymphocytes. The clinical history with lesions localized only at the injection sites rules out lupus panniculitis [15, 16].
The most difficult histopathological differential diagnosis of lupus panniculitis is SPTCL and overlapping cases of lupus profundus and SPTCL have been described. SPTCL is a peculiar α/β T-cell lymphoma, usually CD8+, which involves subcutaneous fat, without epidermal and/or dermal involvement, and mimics a panniculitic process (see Chapter 140). Magro et al. [17] reviewed 32 cases of lymphocytic lobular panniculitis and classified them into three groups: lupus panniculitis, SPTCL and a third group with intermediate features that they named ‘indeterminate lymphocytic lobular panniculitis’. Lesions of this third group showed the involvement of subcutaneous fat by atypical lymphocytes with pleomorphic and hyperchromatic nuclei. The same group has recently proposed the term ‘atypical lymphocytic lobular panniculitis’ for these intermediate cases [18]. The infiltrate showed deletion of one or more pan-T-cell markers (CD3, CD5 and/or CD7) and monoclonal TCRγ gene rearrangement by PCR. The authors introduced the concept of subcutaneous lymphoid dyscrasia to encompass the cases with overlapping findings of lupus panniculitis and SPTCL. More recently, Pincus et al. [19] reported on five patients and Basisio et al. [20] described 11 patients with SPTCL who were unusual in that they also exhibited features of lupus erythematosus. In all cases, attributes indicating SPTCL included an infiltrate of lymphocytes with pleomorphic nuclei involving subcutaneous lobules exhibiting a cytotoxic T-cell (CD3/CD8/βF1) immunophenotype. Additionally, a high proliferation rate and a monoclonal TCRγ gene rearrangement were observed in most cases. The manifestations of lupus erythematosus in these patients included a spectrum of clinical and histopathological abnormalities. The clinical manifestations consisted in subcutaneous nodules that healed with lipoatrophy on the face and serological and/or extracutaneous end-organ abnormalities as seen in patients with systemic lupus erythematosus. Histopathological evidences of lupus erythematosus included vacuolar change at the dermal–epidermal interface, interstitial deposition of mucin in the reticular dermis, clusters of CD20+ B cells partially arranged within germinal centres, a few small clusters of CD123+ plasmacytoid dendritic cells within the adipose tissue and positive direct immunofluorescence test on clinically uninvolved and lesional skin. The authors concluded that some patients show overlap between SPTCL and lupus panniculitis and that patients with lupus panniculitis should be monitored for possible evolution into SPTCL. This recommendation is also supported by other reports of T-cell lymphomas with subcutaneous tissue involvement, probably cutaneous γ/δ T-cell lymphomas, with histopathological features similar to those of lupus panniculitis, including vacuolar interface dermatitis and dermal mucinosis [21, 22, 23]. In fact, one of these cases was erroneously diagnosed as lupus panniculitis [23]. The most useful criteria now for distinguishing lupus panniculitis from SPTCL are the presence in lupus panniculitis of epidermal involvement, superficial and deep perivascular lymphocytic infiltrate in the dermis, lymphoid follicles with reactive germinal centres, a mixed infiltrate with numerous plasma cells, clusters of B lymphocytes, clusters of CD123+ plasmacytoid dendritic cells [24], low proliferative index in lymphocytes and polyclonal TCRγ rearrangement [25].
Investigations
Histopathologically, lupus panniculitis is a predominantly lobular panniculitis in which the infiltrate in active lesions involves mainly the fat lobule [26]. Some authors find difficulty in classifying lupus panniculitis as predominantly lobular panniculitis because of the prominent septal component [25, 27, 28]. The septal component, however, consists of thickening and sclerosis of the collagen bundles in the septa, whereas most of the infiltrate is found in the fat lobule. Active lesions exhibit a picture of a predominantly lymphocytic panniculitis with numerous plasma cells. Longstanding lesions show hyaline necrosis of the fat lobule with little or no infiltrate and replacement by diffuse eosinophilic glassy remnants of adipocytes [25, 29–31]. Lymphoid aggregates, sometimes with germinal centre formation, are also frequently seen in the septa or at the periphery of the fat lobules (Figure 99.35). These lymphoid follicles, although characteristic, are not pathognomonic of lupus panniculitis because they may also be seen in deep morphoea, erythema nodosum, erythema induratum, necrobiosis lipoidica, panniculitis associated with dermatomyositis and necrobiotic xanthogranuloma [32].
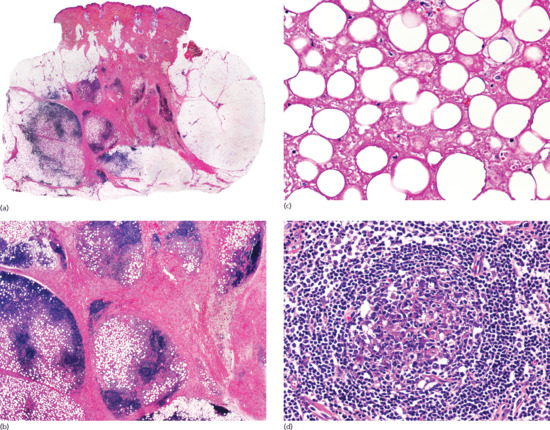
Figure 99.35 Histopathological features of lupus panniculitis. (a) Scanning magnification showing a predominantly lobular panniculitis. (b) Dense lymphoid aggregations at the interphase between the fat lobule and the thickened septa. (c) Hyaline necrosis involving the fat lobule. (d) Reactive germinal centre formation within the lymphoid aggregates.
Additional histopathological findings consist in calcification, interstitial mucin deposition and features of discoid lupus erythematosus in the overlying epidermis. An uncommon but, when seen, distinctive feature is the presence of nuclear dust within the infiltrate [28, 33, 34]. Eosinophils are not usually prominent in lesions of lupus panniculitis [25, 30] but, in contrast with other forms of cutaneous lupus erythematosus in which eosinophils are characteristically absent, the infiltrate of lupus panniculitis may contain some eosinophils [35].
As with other lobular panniculitides, longstanding and residual lesions of lupus panniculitis may show lipomembranous changes [36]. Lymphocytic vasculitis has been described in lupus panniculitis with variable frequency [37, 38, 39]. This vasculitis consists of the presence of lymphocytes in and around the vessel walls, mural fibrin deposition, luminal thrombosis and nuclear dust. Some authors consider that hyaline necrosis of the fat lobule results from the ischaemic process secondary to this lymphocytic vasculitis [40].
Histopathological features of discoid lupus erythematosus at the dermal–epidermal junction in lesions of lupus panniculitis have been described in varying proportions, ranging from 20% to 75% of cases [25, 26, 29, 31, 41, 42]. These changes include epidermal atrophy with hyperkeratosis, follicular plugging, vacuolar alteration of the basal layer of the epidermis and basement membrane thickening. Additional features of discoid lupus erythematosus are interstitial mucin deposition, telangiectasia, and superficial and deep perivascular dermal lymphocytic infiltrate. Calcification is also a frequent finding in chronic lesions of lupus panniculitis and consists of individual calcification of elastic fibres or large masses of calcium within the lobules and septa [31, 43].
Only a few direct immunofluorescence studies have been performed in lesions of lupus panniculitis. The lupus band test at the dermal–epidermal junction is often positive [1, 44]. Additional findings consist of IgG deposition at the periphery of adipocytes and around the vessels [42, 45].
Immunohistochemical studies have demonstrated that the infiltrate in lupus panniculitis is composed mostly of T lymphocytes, with a slight preponderance of CD4 over CD8 lymphocytes. All of them show α/β immunophenotype. Small aggregates of B lymphocytes are also present at the periphery of the lymphoid aggregates. PCR analyses for TCRγ gene rearrangement in the infiltrate has demonstrated its polyclonal nature in most cases [17, 28, 45–47]. Because of the presence of cytotoxic CXCR3+ lymphocytes in the fat lobules, it has been suggested that lupus panniculitis may be due to a type I interferon Th1 driven immune response [48].
Dermatomyositis-associated panniculitis
Clinical features
Presentation
Panniculitis is less frequent in dermatomyositis than in lupus erythematosus and systemic sclerosis [1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15]. In a series of 55 adult patients with dermatomyositis and cutaneous lesions studied histopathologically, panniculitis was only found in five cases [1]. Panniculitis has also been described in juvenile dermatomyositis [8, 11]. When the inflammation settles it leaves areas of lipoatrophy [4].
In some patients, panniculitis is associated with other characteristic cutaneous lesions of dermatomyositis [8], whereas in others panniculitis is the only cutaneous manifestation of the disease [3, 9]. Conversely, there is a report of a patient presenting with panniculitis and vesiculobullous skin lesions but no evidence of muscle involvement [15]. The clinical features of dermatomyositis are discussed in detail in Chapter 53.
Investigations
The histopathological features of dermatomyositis-associated panniculitis are similar to those of lupus panniculitis and consist of a predominantly lobular panniculitis with lymphocytes and plasma cells among the adipocytes. The septal collagen bundles show hyaline sclerosis, and there is progressive replacement of fat with fibrous tissue [2]. Additional histopathological findings include thickening of the blood vessels of the fat lobule, neutrophilic vasculitis with fibrinoid necrosis or lymphocytic vasculitis involving the arterioles of the septa, and calcification. Lymphoid follicles, with or without reactive germinal centre formation, have also been described [5], although this finding is less frequent than in lupus panniculitis or deep morphoea. As in lupus panniculitis, there may be vacuolar change at the dermal–epidermal junction and, in the late stages of the process, membranocystic changes [9, 12].
Direct immunofluorescence studies have been reported in only three cases: the results were negative in one case [5]; the second case had deposits of IgM, C3 and fibrinogen in the blood vessels walls of the dermis, but not at the dermal–epidermal junction [9]; and the third case showed deposits of C3 at the basement membrane zone of the dermal–epidermal junction and around the dermal blood vessels, but in the subcutaneous fat only deposits of fibrinogen were detected [5]. As with lupus panniculitis, patients with dermatomyositis-associated panniculitis seem to be a subgroup with a generally good prognosis and no obvious increase in the incidence of malignancy [10]: in fact, malignancy has been reported in only one patient with dermatomyositis-associated panniculitis [3].
More common than pure dermatomyositis-associated panniculitis is panniculitis occurring in association with calcification of muscle and deep tissue. In these cases, the fat lobule shows lipophagic granulomata, calcification and various degrees of acute and chronic inflammation [2].
Pancreatic panniculitis
Introduction and general description
Pancreatic panniculitis was originally described by Chiari in 1883 [1], but it was not until 1961 when Szymanski and Bluefarb reported the first case in the English literature [2]. There are only a few published series of patients with pancreatic panniculitis [3–7], and most reports include descriptions of no more than one or two cases.
Epidemiology
Incidence and prevalence
Pancreatic panniculitis is uncommon and appears in only about 2–3% of all patients with pancreatic disease [8], although its incidence is higher among males with alcoholism [9, 10].
Pathophysiology
The pathogenesis of pancreatic panniculitis remains unclear, but release of pancreatic enzymes, such as lipase, trypsin and amylase, seems to be the most important aetiological factor. It is not completely clear how pancreatic proenzymes become activated in the tissues to produce fat necrosis [11], but trypsin may increase the permeability of the microcirculation within lymphatic vessels [6], allowing lipase and amylase to enter the peripheral circulation. Within the fat lobules these enzymes hydrolyse neutral fat to form glycerol and free fatty acids, which results in adipocyte necrosis and an inflammatory response [12, 13]. This theory is supported by the finding of elevated enzyme levels in the blood, urine and skin lesions, even in the absence of detectable pancreatic disease [12], and by the positive intracellular immunostaining of adipocytes with a monoclonal antibody to pancreatic lipase in lesions of pancreatic fat necrosis [14]. However, other factors apart from pancreatic enzymes must also play some pathogenetic role, because there is clear discrepancy between the small number of cases of pancreatic panniculitis compared with the great number of patients with pancreatitis and pancreatic carcinoma who have increased serum levels of pancreatic enzymes but no panniculitis [13, 15]. Conversely, some patients with pancreatic panniculitis have had normal serum levels of all pancreatic enzymes [5]. Furthermore, in vitro investigations have failed to reproduce pancreatic panniculitis when normal human fat has been incubated with the serum of patients with high levels of pancreatic lipase, trypsin and amylase [5]. Other proposed mechanisms implicate vascular damage [12, 16], immune complexes [17], and adipocyte-generated cytokines and adipokines released in response to high levels of free fatty acids. Resistin and leptin have been shown to be potential markers of extrapancreatic fat necrosis [18].
Clinical features
Clinically, cutaneous lesions of pancreatic panniculitis consist of tender, erythematous or red-brown nodules that may spontaneously ulcerate, draining an oily brown, sterile and viscous material that results from liquefactive necrosis of adipocytes (Figure 99.36). These lesions show a predilection for the distal parts of the lower extremities, mostly around the ankles. Venous stasis may promote this process, although lesions in other areas including the knees, thighs, buttocks, arms, abdomen, chest and scalp may also be seen [19]. Ulceration and fistulization of necrotic fat to the skin surface are frequent clinical features in pancreatic panniculitis, but they may also occur in other panniculitides. Although there are no specific clinical findings, it seems that the panniculitis associated with pancreatic carcinoma tends to be more persistent, with more frequent recurrences and a greater tendency to ulceration, fistulization and involvement of cutaneous areas beyond the lower extremities than that related to inflammatory pancreatic disease [10]. Cases of pancreatic panniculitis with a single cutaneous nodule have, however, also been reported [20]. The association of panniculitis with fever, polyarthritis and abdominal pain should raise suspicion of pancreatic disease.

Figure 99.36 Pancreatic panniculitis in an alcoholic male. Nodular lesions, many of them ulcerated around the ankles.
Enzymatic fat necrosis induced by pancreatic enzymes is not confined to subcutaneous fat and often patients have other foci of fat necrosis: when this involves periarticular fat it may cause a mono- or oligoarticular arthritis [10, 21], which may be symmetrical and can be intermittent, migratory or persistent [16]. This enzymatic arthritis usually involves the small joints of the hands, wrists and feet, but also may affect larger joints such as the elbows, knees and ankles. Aspiration of the involved joint yields a creamy purulent sterile fluid with a few white cells [22]. Joint fluid may also contain lipid crystals with increased levels of amylase, lipase, free fatty acids, triglycerides and cholesterol [22]. In rare instances, this may progress to chondronecrosis and osteonecrosis of the involved joints [23]. Other less common extrapancreatic sites involved by enzymatic fat necrosis rarely cause symptomatology, but autopsies of patients with pancreatic carcinoma and panniculitis have demonstrated fat necrosis in abdominal fat [24] and bone marrow [25]. Radiological images are characteristic, showing osteolytic lesions, moth-eaten bone destruction and periostitis of the involved bone of the extremities due to extensive areas of bone marrow fat necrosis and trabecular bone destruction [26]. The association of panniculitis, polyarthritis and eosinophilia in a patient with pancreatic cancer is known as the Schmid triad and usually carries a poor prognosis [16].
Skin lesions may be the first sign of pancreatic disease and therefore represent an important clue for the diagnosis [1, 16, 27]. In the literature, there are several reports in which cutaneous nodules of pancreatic panniculitis preceded the detection of pancreatic carcinoma by several months, and the development of panniculitis may also predict progressive or metastatic malignant disease [28]. However, the most frequent underlying associated pancreatic disease is not pancreatic carcinoma, but acute [6] or chronic pancreatitis [29, 30] caused by alcohol abuse [6], trauma [31, 32], or cholelithiasis [33]. After acute and chronic pancreatitis, pancreatic carcinoma is the next most common cause of pancreatic panniculitis. Rarer disorders that may also cause this panniculitis are listed in Box 99.6.
Differential diagnosis
Clinically, the main differential diagnoses are erythema induratum, α1-antitrypsin deficiency panniculitis and infectious panniculitis [3, 12, 17]. Histopathology usually resolves any doubt by finding ghost adipocytes in the saponified fat lobule [12]. Lobular panniculitis at the site of subcutaneous IFN-β injections for the treatment of multiple sclerosis may histologically mimic pancreatic panniculitis [68].
Investigations
Histopathology of pancreatic panniculitis is almost pathognomonic, consisting in a predominantly lobular panniculitis without vasculitis [6, 17]. However, some authors have proposed that the earliest feature in pancreatic panniculitis is a predominantly septal panniculitis resulting from enzymatic damage to endothelial cells lining septal blood vessels. These damaged endothelial cells allow pancreatic enzymes to cross from the blood to fat lobules, resulting in necrosis of adipocytes [3, 6]. Regardless of this, early states of pancreatic panniculitis show a predominantly neutrophilic infiltrate, with occasional eosinophils and, as the most characteristic finding, ghost adipocytes resulting from coagulative adipocyte necrosis. These ghost adipocytes lose their nuclei and show a finely granular and basophilic material within their cytoplasm because of calcification. Often ghost adipocytes group in small clusters at the centre of the fat lobule, whereas the neutrophilic infiltrate is present at the periphery [69] (Figure 99.37). Dystrophic calcification in ghost adipocytes results from the hydrolytic action of pancreatic enzymes on fat cells with subsequent calcium deposition, a process known as saponification [5, 6]. Often, adjacent lobules show a different stage in the histopathological evolution of the process. In late stages, fat necrosis and ghost adipocytes are less evident and the inflammatory infiltrate is more granulomatous, containing foamy histiocytes, multinucleate giant cells and haemosiderin deposits [11]. Residual lesions show fibrosis and lipoatrophy. Although ghost adipocytes are very characteristic of pancreatic panniculitis, they are not pathognomonic and similar findings may be seen in mucocutaneous mucormycosis [70] and cutaneous aspergillosis [71]. It has been shown that fungi of the family Mucoraceae produce remarkable amounts of extracellular lipases [72, 73] and thus it is likely that the ghost adipocytes result from the local effect of these lipases.
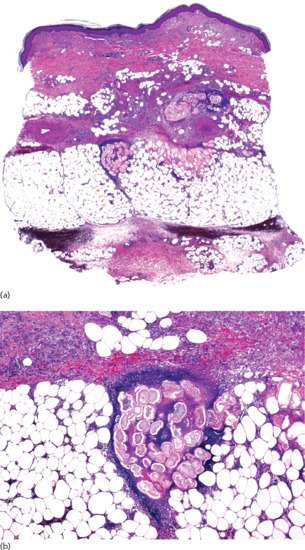
Figure 99.37 Histopathological features of pancreatic panniculitis. (a) Scanning power showing a mostly lobular panniculitis. (b) A group of ‘ghost’ adipocytes surrounded by neutrophils is seen at the periphery of the fat lobule.
Most patients, although not all, show elevated serum levels of amylase, lipase or trypsin though often one enzyme is within normal levels whilst others are elevated [10]. There is no correlation between the serum levels of pancreatic enzymes and the severity of the cutaneous lesions [74]. In rare instances, patients with pancreatic panniculitis may show high serum levels of pancreatic lipase with no evidence of underlying pancreatic disease [21, 75]. A leukaemoid reaction and eosinophilia in peripheral blood are also common haematological abnormalities, particularly in patients with pancreatic carcinoma [75, 76]. Other tumour markers such as carcinoembryonic antigen or Ca 19.9 are often elevated in these patients.
Management
Treatment of pancreatic panniculitis should be directed to the underlying pancreatic disease and usually the cutaneous lesions heal once the acute inflammatory pancreatic process has resolved or the pancreatic anomaly has been surgically corrected [3, 13, 53, 77]. Administration of the somatostatin analogue octreotide, a synthetic polypeptide which inhibits pancreatic enzyme production, has been reported to result in a significant improvement in pancreatic panniculitis in some [13, 38, 78] but not all [17, 30, 37, 48] patients with pancreatic carcinoma. Corticosteroids, non-steroidal anti-inflammatory drugs and immunosuppressive drugs are usually not effective treatments for pancreatic panniculitis [26].
Alpha-1 antitrypsin deficiency panniculitis
Definition
Alpha-1 antitrypsin deficiency is a genetic disorder that manifests as pulmonary emphysema, liver cirrhosis and, rarely, as cutaneous panniculitis. It is characterized by low serum levels of α1-antitrypsin, the main protease inhibitor in human serum.
Pathophysiology
Genetics
Alpha-1 antitrypsin is the most important serine protease inhibitor produced in the liver. Its principal function is to inhibit trypsin activity, but it also acts as a potent inhibitor of chymotrypsin, plasmin, thrombin, neutrophilic elastase, pancreatic elastase, serine proteases, collagenase, factor VIII, kallikrein, urokinase and cathepsin G. It may also inhibit complement activation, both through a direct effect on complement-related proteases and by inhibiting the neutrophil proteases that activate enzymes of the complement system. Additionally, it is thought to help regulate protease-stimulated activation of lymphocytes and phagocytosis by macrophages and neutrophils. It is an acute phase reactant which is released in stress situations.
Alpha-1 antitrypsin consists of 394 amino acids organized into three β-sheets and nine α-helices. The active site of the protein is a reactive central loop composed of 20 amino acids, which, when in contact with a serine protease, induces conformational changes resulting in inactivation of both α1-antitrypsin and protease [1]. The gene that encodes this protein, SERPINA 1 (formerly known as PI), is located at 14q32.1. More than 120 allelic variants have been described to date [1]. The most frequent allele, PiM, defined by its protein isoelectrophoretic mobility (M = medium mobility), is associated with homozygous PiMM phenotype and normal serum levels of α1-antitrypsin (120–200 mg/dL) [2]. Two alleles, PiS (S = slow mobility) and PiZ (Z = very slow mobility), each caused by a single nucleic acid substitution, are considered to be involved in pathological manifestations. In the Z variant, glutamic acid replaces lysine at position 342 [3]. Homozygosity for the Z allele (PiZZ) is associated with very low serum levels of α1-antitrypsin (20–45 mg/dL), whereas the heterozygous phenotypes PiMZ or PiMS result in a moderate reduction of α1-antitrypsin serum levels. A null allele variant, without any apparent gene alteration, but with no detectable mRNA produced, has also been described. In individuals who are homozygous for Pi null/null mutations, serum α1-antitrypsin is not detectable at all [4]. Heterozygosity for pathogenic α1-antitrypsin mutations (PiMS, MZ, SZ) is estimated to occur in about 10% of the general population [5], with 2% heterozygous for the Z allele [6]. The prevalence of the homozygous PiZZ phenotype is only about 1 in 3500 in northern Europe populations [3]. In heterozygous carriers of the S or Z allele, α1-antitrypsin production and function are apparently normal but only a small amount of α1-antitrypsin enters the circulation from its production site in the liver. Both mutations result in a high tendency for the protein to polymerize, especially in the Z variant. In both homozygous and heterozygous phenotypes, polymerized α1-antitrypsin cannot as a result be released from the liver. Z-type α1-antitrypsin polymers have been detected in lesional skin, which supports the inflammatory pathogenesis of panniculitis and the potential pro-inflammatory role of polymers [7].
In situations causing tissue injury, such as smoking for emphysema, trauma for panniculitis or hepatotoxins for cirrhosis, the absence or deficiency of α1-antitrypsin results in uncontrolled activation of lymphocytes and macrophages, lack of inhibition of the complement cascade including C3a–C5a neutrophilic chemotactic factors, and accumulation of neutrophils with release of proteolytic enzymes and secondary tissue damage [8]. The special susceptibility of subcutaneous fat to proteolytic degradation when not protected by α1-antitrypsin is due to its high fatty acid content. Fatty acids modify elastin conformation and render fat more susceptible to proteolytic degradation [9].
Severe α1-antitrypsin deficiency is associated with a variety of clinical manifestations including disorders of blood coagulation and fibrinolysis, anomalies in the phagocytic mechanism of the immune response and anomalies in the activation of zymogens and release of hormonal peptides in addition to its effect on the lung, liver and subcutaneous fat.
Alpha-1 antitrypsin deficiency panniculitis is most severe in homozygous PiZZ individuals. It can, however, also develop in heterozygotes with PiMS [1, 9], PiMZ [10], PiSZ [11] genotypes and in homozygotes with PiSS [12] and PiM1M1 [13] genotypes. This suggests that other factors than α1-antitrypsin deficiency may be involved in the pathogenesis. Furthermore, some authors believe that there are patients with the phenotypic features of α1-antitrypsin deficiency but normal serum levels of α1-antitrypsin who develop panniculitis because of mutations which do not induce polymerization but which affect the reactive centre loop, resulting in the production of non-functional protein [1, 11].
Clinical features
History
Warter et al., in 1972, were the first authors to describe panniculitis in association with α1-antitrypsin deficiency, although these authors considered the process to be a manifestation of familial Weber–Christian disease [14]. Rubinstein et al., in 1977, described the first two cases of α1-antitrypsin deficiency-related panniculitis as a specific manifestation of this autosomal recessive inborn error of metabolism [15]. Panniculitis associated with α1-antitrypsin deficiency is rare, with equal incidence in both genders and the age of presentation ranging from 7 to 73 years, with a mean age of 39.7 years [16], although children may also be affected [17].
Presentation
Dermatologically, panniculitis is the most important clinical manifestation of α1-antitrypsin deficiency. Inflammation of subcutaneous fat may be the first sign of the disease, although subcutaneous nodules usually appear when other manifestations of the disorder have already developed. The panniculitis presents as erythematous nodules and plaques mainly located on the trunk and around the shoulders and hips [9]. The head and extremities may sometimes also be involved [18]. The earliest lesions resemble cellulitis [6] and show a tendency to ulcerate and exude oily material derived from necrotic adipocytes [18] (Figure 99.38). Healing of lesions leaves atrophic scars. A chronic relapsing course is characteristic. Antecedent trauma at the site of the lesion [9], surgical debridement [18], cryosurgery [19] or injections [20] can precipitate new lesions. Postpartum flares of the process have been attributed to an oestrogen-stimulated increase in proteinase inhibitor levels during pregnancy, followed by a precipitous decline to subnormal levels postpartum [21]. Uncommon cutaneous manifestations associated with α1-antitrypsin deficiency include vasculitis and acquired angio-oedema [2, 22, 23]. Visceral extension is uncommon, but involvement of perinephric fat as well as hepatic and splenic sterile abscesses have been reported [12].
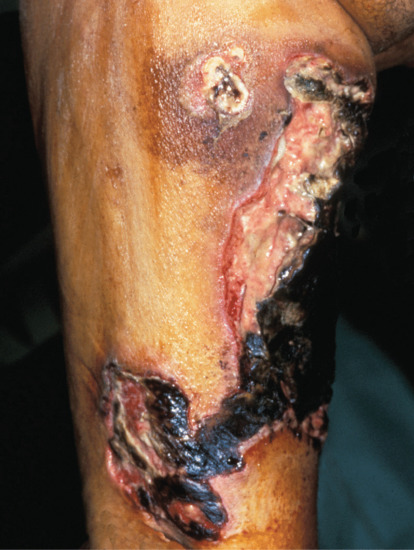
Figure 99.38 Panniculitis associated with α1-antitrypsin deficiency. Necrotic ulcers exudate oily material that result from necrotic adipocytes.
Differential diagnosis
Differential diagnosis of α1-antitrypsin deficiency panniculitis includes other neutrophilic panniculitides with a tendency to ulceration and fistula formation such as the early stages of erythema induratum, pancreatic panniculitis, factitial panniculitis and infective panniculitis. Most of these panniculitides show specific histopathological findings. When neutrophilic lobular panniculitis is found without indications of other specific diagnoses, measurement of serum levels of α1-antitrypsin and electrophoretic mobility studies should be undertaken.
Complications and co-morbidities
Systemic manifestations of α1-antitrypsin deficiency include panacinar emphysema, neonatal hepatitis, cirrhosis and liver disease resulting from retention of the abnormal polymerized protein within the liver, pancreatitis, membranoproliferative glomerulonephritis, c-ANCA-positive vasculitis and angio-oedema due to deficiency of protease inhibitor [9, 20]. Approximately 50% of ZZ patients will die from emphysema-related complications and 10% will develop liver disease; the MZ phenotype is associated with a slightly higher risk of both lung and liver disease; and no increased likelihood of either lung or liver disease is seen in MS patients [1]. The null/null phenotype is usually accompanied by emphysema, but no liver disease because there is no α1-antitrypsin synthesis and accumulation in the liver cannot therefore occur [4].
Investigations
Histopathological study of α1-antitrypsin deficiency panniculitis shows a predominantly lobular panniculitis with no vasculitis. In the early stages, the presence of neutrophils extending into the lower reticular dermis in an interstitial pattern between collagen bundles (‘splaying of neutrophils’) has been proposed by some authors as a specific clue for the diagnosis [24]. This is, however, a non-specific finding that may be found in any neutrophilic lobular panniculitis. A more specific finding consists in the focal nature of the damage with large clusters of normal adipocytes adjacent to areas of necrosis and dense neutrophilic and histiocytic infiltrates [18, 25] (Figure 99.39). Occasionally, the intense neutrophilic infiltrate may cause collagenolysis and elastic tissue destruction at the connective tissue septa and then necrotic fat lobules appear to be ‘floating’ and surrounded by neutrophils [26]. Transepidermal elimination of liquefied dermis may occur as a secondary phenomenon [27]. In late-stage lesions, neutrophils and necrotic adipocytes are less evident and the histopathological picture is dominated by non-specific lipophagic granulomata replacing fat lobules. Some macrophages may engulf nuclear dust of neutrophils and dystrophic calcification may develop [10]. Direct immunofluorescence studies have revealed deposits of complement C3 and IgM around the dermal blood vessels: these are of uncertain significance [9, 18].
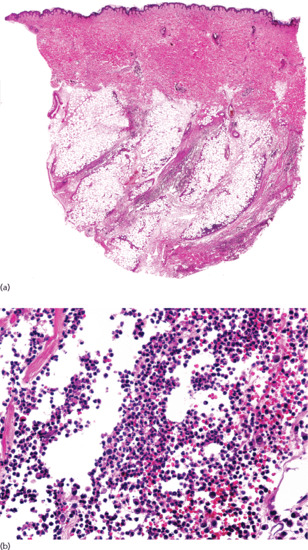
Figure 99.39 Histopathological features of α1-antitrypsin deficiency panniculitis. (a) Scanning power showing involvement at the septa and the periphery of the fat lobules. (b) Neutrophilic infiltrate and nuclear dust but no evidence of vasculitis.
Laboratory anomalies include absence or significantly reduced levels of α1 globulin on plasma protein electrophoresis and abnormally reduced levels of α1-antitrypsin. Sometimes an isoelectrophoretic mobility study for α1-antitrypsin may demonstrate an abnormal phenotype with normal serum levels. In chronic cases, normocytic normochromic anaemia and hypoalbuminaemia are frequently found [20].
Management
Trauma and surgical debridement should be avoided, as should smoking or exposure to hepatotoxins. Reduction of alcohol intake should be recommended. Several treatments including corticosteroids [9], immunosuppressive drugs, colchicine [19], danazol and antimalarials have shown poor or no response. Doxycycline or minocycline in a dose of 200 mg daily for at least 3 months, may be effective in mild cases, as tetracyclines have anticollagenase activity which may partly re-establish protease-antiprotease homeostasis [5]. Dapsone has also shown to be effective because it inhibits the migration of neutrophils [9, 12]. For severe cases with liver and lung involvement, the best option is replacement of α1-antitrypsin using human pooled plasma from normal donors (Prolastin®). Intravenous infusions in a dosage of 60–100 mg/kg per week, depending on the severity of the deficiency, over a period of 3–7 weeks [3, 27, 28, 29, 30, 31, 32, 33, 34]. Recurrence after discontinuation of therapy is common, but there is a good response to reinfusion [31]. Other interventions which have been used include plasma exchange [35] and liver transplantation [36]. In one patient α1-antitrypsin deficiency panniculitis appeared after liver transplant and was successfully treated with retransplant [37]. The role of genetic engineering in producing α1-antitrypsin is being investigated.
Infective panniculitis
Pathophysiology
Causative organisms
Several bacterial and fungal infections may cause panniculitis as their main clinical manifestation. Full details about cutaneous infections may be found in Chapters 25, 26, 27, 28, 29, 30, 31 and 32.
Bacteria implicated in subcutaneous panniculitis include Streptococcus pyogenes [1], Staphylococcus aureus [1], Pseudomonas spp. [1, 2, 3], Klebsiella [1], Nocardia spp. [1, 4], Brucella [5] and Borrelia burgdorferi [6, 7]. Most cases of mycobacterial panniculitis reported in the literature have been caused by non-tuberculous mycobacteria [8–19], especially rapidly growing mycobacteria such as M. chelonae [14–17] and M. fortuitum [18, 19], and less frequently by slow-growing mycobacteria such as M. avium intracellulare complex [9, 11] and M. marinum [8, 12]. There are exceptional cases due to Mycobacterium tuberculosis [12, 20] and panniculitis caused by Mycobacterium leprae is extremely rare [21]. M. ulcerans causes a well-defined clinicopathological entity known as Buruli ulcer, which involves predominantly the subcutaneous fat [22–27].
Fungal infections of the subcutaneous fat may be classified into two main categories: (i) panniculitis in the setting of a disseminated fungal infection; and (ii) classical subcutaneous mycosis. These two groups differ in their causative microorganisms, their pathogenesis, the setting in which they appear, their prognosis and their treatment. The most common disseminated fungal infections causing panniculitis are Candida spp. [1, 28, 29], Aspergillus spp. [30], Fusarium spp. [1] and Histoplasma capsulatum [31, 32], whereas the most common classical subcutaneous mycoses are sporotrichosis due to Sporothrix schenckii, eumycetoma caused by Madurella mycetomatis [6, 33] and chromoblastomycosis caused by pigmented fungi, the most common being Phialophora verrucosa, Fonsecaea pedrosoi, Fonsecaea compacta and Cladophialophora carrionii [34, 35]. Uncommon subcutaneous fungal infections include phaeohyphomycosis, lobomycosis, rhinosporidiosis and subcutaneous zygomycosis. A case of septal panniculitis caused by cytomegalovirus infection with many cytomegalovirus inclusions in endothelial cells has been described in an immunosuppressed patient [36] and another case of neutrophilic lobular panniculitis due to acanthamoebiasis has been reported in a patient with AIDS [37].
Presentation
With the exception of the classical subcutaneous mycoses, most of these infective panniculitides occur in immunosuppressed patients and are uncommon in immunocompetent hosts. The immunosuppressed population, which has been increasing in recent decades due to HIV infection, organ transplantation and the widespread use of immunosuppressive drugs, is at risk not only of common cutaneous infections but also of opportunistic infections with atypical clinical presentations [38, 39].
Bacterial panniculitis may appear in the setting of septicaemia, as the consequence of direct inoculation or by direct spread from an underlying infection. In patients with sepsis, solitary or multiple nodules and abscesses appear as a consequence of the haematogenous dissemination of bacteria. Constitutional symptoms are often absent, but the general condition of the patient is impaired by the underlying disease.
The clinical features of subcutaneous mycobacterial infections vary according to the immune state of the patient. In immunocompromised patients, lesions tend to be widespread due to haematogenous dissemination. Sporotrichoid spread is not uncommon in these patients [40, 41, 42] (Figure 99.40) and spread to internal organs may occur [9, 40]. In immunocompetent patients, the infection is usually localized and related to trauma, e.g. penetrating injury, surgical procedures, acupuncture, injections or postepilation folliculitis [10, 11, 15].
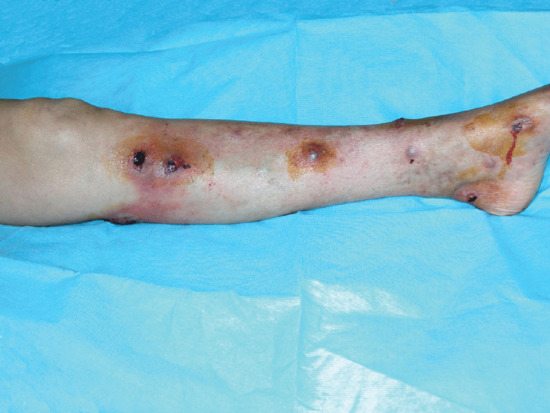
Figure 99.40 Sporotrichoid arrangement of subcutaneous nodules, several of them ulcerated and draining serous or oily discharge in an immunocompromised patient. Cultures isolated M. chelonae.
Panniculitis in immunosuppressed patients with disseminated fungal infection presents as multiple erythematous subcutaneous nodules, pustules or fluctuant abscesses [28, 30, 31]. In subcutaneous mycoses, the fungus enters the skin from the soil, plants or wood via a penetrating injury and the lesions are localized mostly to exposed areas of the skin, such as the face, hands, arms or feet [33]. These lesions consist of a solitary painless nodule that spreads slowly; with time, secondary nodules and papules may develop in adjacent skin and may be accompanied by sinuses exuding a serous or oily discharge.
Investigations
The histopathological features of infective panniculitis consist of a predominantly lobular neutrophilic panniculitis without vasculitis [1, 5, 41], although in some cases small vessel vasculitis has been described [1]. Apart from the neutrophilic infiltrate in the fat lobule, additional features suggestive of an infective aetiology of a lobular panniculitis are haemorrhage, proliferation of vessels, foci of basophilic necrosis and necrosis of sweat glands [1]. Special stains, including Gram, PAS, Ziehl–Neelsen and methenamine–silver, as well as tissue cultures should be performed.
Rarely, infective panniculitis may resemble SPTCL, as was reported in a case due to Borrelia burgdorferi where the lobular infiltrate was composed of atypical lymphocytes with cytotoxic immunophenotye. Borrelia burgdorferi aetiology was, however, proven by positive PCR findings, serology and a favourable response to antibiotics [7].
The histopathological findings in panniculitis caused by mycobacterial infections vary according to the organism involved and the immune state of the host. In most cases, the histopathological picture, as in other bacterial panniculitides, is a neutrophilic lobular panniculitis, but mycobacterial panniculitides often contain suppurative granulomata [14, 15, 18, 43] (Figure 99.41) or frank tuberculoid granulomata [12, 14, 44]. Caseous necrosis is suggestive of tuberculous panniculitis [12].
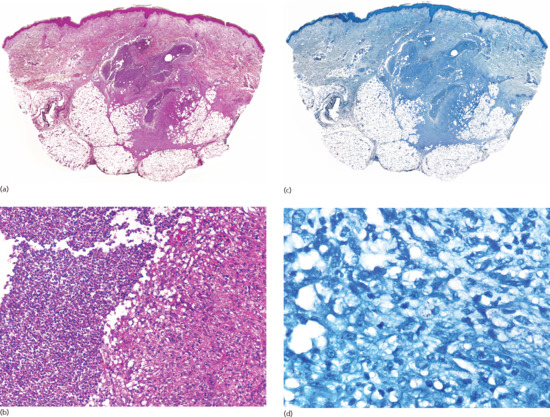
Figure 99.41 Histopathological features in a cutaneous infection by M. chelonae. (a) Scanning power showing involvement of the reticular dermis and subcutaneous fat. (b) Suppurative granuloma involving the fat lobule. (c) A section of the same case stained with Ziehl–Neelsen stain. (d) Several acid–alcohol resistant bacillus (BAAR) positive mycobacteria may be seen at the centre of the figure.
In Q fever due to Coxiella burnetii a ‘doughnut-like’ lobular granulomatous panniculitis, similar to the changes found in the liver and bone marrow, has been described [45].
Ghost adipocytes reminiscent of pancreatic panniculitis have been documented in cases of mucormycosis [46] and aspergillosis [47] involving subcutaneous fat.
Different histopathological patterns have been described according to the inoculation route of the microorganisms into the skin. Primary cutaneous infections arise either from direct physical inoculation or at the site of an occlusive dressing over an indwelling catheter, whereas secondary cutaneous infections develop either from direct extension to the chest wall in pulmonary infections, or from haematogenous dissemination. In primary cutaneous infections, the epicentre of the inflammation is the superficial dermis, and thrombosed vessels do not contain intravascular organisms. In contrast, in secondary cutaneous infections, the epicentre of inflammation is more deeply seated and involves only the deep reticular dermis and subcutaneous fat. The blood vessels are thrombosed and dilated with masses of organisms expanding their lumina [30]. In immunosuppressed patients microorganisms are numerous and they may be easily identified in tissue sections with routine H&E staining or with special stains, but in immunocompetent patients microorganisms are sparse and they may be difficult to detect. In these latter cases, the diagnosis may be established only from culture [1, 42]. In cases with few microorganisms, immunohistochemical staining with anti-BCG antibody was proposed as a helpful tool for screening, because this commercially available polyclonal antibody showed cross-reactivity with many bacteria, mycobacteria and fungi, and produced minimal background staining; it might therefore identify microorganisms that could not be seen using conventional stains [48]. However, due to over-purification by the manufacturer in recent years, anti-BCG antibody no longer has the original wide sensitivity for bacteria and fungi and currently it should be restricted to the search for mycobacteria in formalin-fixed and paraffin-embedded samples. PCR-based methods are also available to test formalin-fixed tissue for specific agents.
Factitious panniculitis
Definition
Factitious or artefactual panniculitides result from external injury to subcutaneous fat. Aetiological factors may be mechanical trauma, chemical substances and thermal injury; the reasons for the injury may be accidental, intentional or iatrogenic. Traumatic and cold panniculitis are covered in other sections of this chapter. Often factitious panniculitis is a manifestation of underlying psychiatric disorders [1]. In other instances, the process results from iatrogenic injections of drugs or immunization agents.
Pathophysiology
Causative organisms
Recently, the use of injectable filler agents has become widespread in aesthetic dermatology and plastic surgery for the treatment of wrinkles and soft-tissue augmentation. Biodegradable or resorbable agents may induce severe complications but these will usually disappear spontaneously in a few months. Slowly biodegradable or non-resorbable fillers may give rise to severe reactions that show little or no tendency to spontaneous improvement. They may appear several years after the injection, when the patient does not remember which product was injected. Previously, factitious panniculitis frequently resulted from subcutaneous injection of oily materials including mineral oil (paraffin) or vegetable oils (cottonseed and sesame oils) [2]. These products were used over many years to augment the size of breasts or genitalia but often induced subcutaneous foreign-body reactions known as paraffinoma or sclerosing lipogranuloma. Fortunately, most such fillers have now been abandoned by medical professionals, although complications may appear a long time after the injections, even 30 years later, and it still is possible to see cases of paraffinoma or sclerosing granuloma [3]. Although cosmetic fillers currently used for tissue augmentation, such as bovine collagen, silicone, PMMA microspheres (Artecoll®), polymethylsiloxane (Bioplastique®) and hydroxyethylmethacrylate particles in hyaluronic acid (Dermalive®) are better tolerated, they may also sometimes induce factitious panniculitis [4–5, 6, 7, 8]. In recent years, injections with Lipostabil®, a phosphatidylcholine-containing substance, have become a popular therapeutic technique for the treatment of localized fat accumulation and lipomas, causing factitious panniculitis of the injected fat tissue [9]. Mesotherapy injections in an attempt to produce reduction of the thickness of hypertrophic subcutaneous fat produce a granulomatous panniculitis with some cystic fat necrosis [10]. Panniculitis has also been reported at the sites of injection of several therapeutic drugs (Box 99.7)
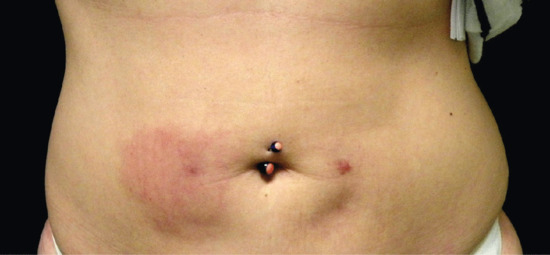
Figure 99.42 Panniculitis on the anterior abdominal wall secondary to subcutaneous glatiramer acetate injections for the treatment of multiple sclerosis.
The extravasation of cytostatic agents during antineoplastic chemotherapy also presents as a severe panniculitis [24, 25]. Cupping and acupuncture techniques for the relief of pain may induce factitious panniculitis on the limbs [26, 27]. Finally, patients with psychiatric disorders may present with self-inflicted panniculitis due to subcutaneous injections of a wide range of substances including acids, alkalis, farming products, mustard, milk, microbiologically contaminated material, urine and faeces [1, 28].
The exact mechanisms involved in factitious panniculitis are uncertain, but vasoconstriction with tissue ischaemia at injection sites, a local inflammatory response elicited by direct contact with drugs or noxious injected substances, immune mechanisms and trauma due to repeated injections may be implicated.
Clinical features
Presentation
The clinical features of factitious panniculitis are variable, depending upon the causative agent. Self-induced factitious panniculitis usually occurs in young adults or middle-aged women with a history of drug addiction or psychiatric disorders (see Chapter 86). Lesions tend to be localized to areas easily accessible to the hands, such as buttocks and thighs, and they are usually solitary or few; when multiple they tend to be grouped. The clinical appearance is bizarre and suspicions should be raised when they do not fit with any well-defined dermatosis. Lesions due to blunt trauma often appear bruised and frequently involve the arm or hand [29]. A particular presentation of self-induced traumatic panniculitis is the so-called Secretan syndrome (l'oedème bleu) [30, 31], which consists of a factitious oedema of the hand caused by the frequent application of a tourniquet or repeated trauma. The course is chronic and recurrent, leading to progressive fibrosis of the dorsum of the hand [32].
Self-inflicted injections with contaminated material produce an acute suppurative panniculitis, often with systemic symptoms [33]. Early lesions manifest as inflammatory nodules and plaques secondary to fat necrosis and suppuration. Some cases may show abscess formation and lymphangitic spread. A case of factitious panniculitis masquerading as florid pyoderma gangrenosum was reported in a depressed woman [34]. There is a risk of progression to granulomatous inflammation and fibrosis in longstanding lesions. Sclerosing lipogranuloma is the term used for factitious lesions of the male genitalia secondary to injections of liquid paraffin intended to augment the size of the penis [35]. Often, these patients deny previous injections, making diagnosis difficult. Lesions similar to sclerosing granuloma may appear in other locations such as the eyelids, lips or gluteal region after injection of liquid silicone [36].
Diagnosis of panniculitis secondary to injection of drugs is usually easy and the location of the lesions provides a clue to their cause. Pentazocine abuse has been described in patients with chronic pain or addiction: repeated injections may induce panniculitis and myositis. Pentazocine panniculitis presents as multiple nodulo-ulcerative lesions of long duration located bilaterally on the buttocks and shoulders [12]. These nodules slowly progress to fibrosis resulting in sclerodermoid plaques that extend to the underlying fascia and muscle [37]. Texier disease is another iatrogenic panniculitis due to vitamin K1 injections [16]: early lesions have an eczematous appearance, but longstanding reactions mimic morphoea [38]. Procaine povidone was used to treat chronic pain with local infiltrations. Povidone is a synthetic product now widely used in skin care products such as hair sprays and as a dispersing or suspending agent in drugs such as procaine and hormones. Povidone polymers cannot be excreted by the kidney and they are phagocytised and stored permanently in macrophages, resulting in the so-called ‘povidone storage disease’ [39]. In addition to panniculitis at the sites of injection, povidone may cause pulmonary lesions, lymphadenopathy and visceromegaly [14].
Subcutaneous extravasation of cytotoxic agents causes severe painful necrotic reactions of the subcutis and underlying muscles, which may disable a patient for months. Clinical lesions show red-brown painful oedema which may evolve into necrotic plaques that heal with sclerotic, indurated scars which may become bound to underlying muscle and bone [25].
Investigations
The histopathological findings in factitious panniculitis vary depending upon the causal agent. In most cases, early lesions show features of a predominantly neutrophilic lobular panniculitis, with severe fat necrosis and an intense inflammatory infiltrate. In some instances, eosinophils may be abundant [28], especially in panniculitis arising at the site of cancer vaccines [20], and in sclerosing lipogranulomata of the genitalia [40]. Superimposed infection often complicates self-induced panniculitis. Fully developed lesions show granulomatous infiltrates involving the fat lobule, whereas longstanding lesions are characterized by lipophagic granulomata and surrounding fibrosis. In some cases, polarized light will reveal the birefringent foreign bodies responsible for the panniculitis. In panniculitis due to injected substances, the dermis is also involved by the inflammatory process, which may be a clue to the correct diagnosis.
Sometimes, specific histopathological findings may be helpful in identifying the nature of the foreign material. Paraffinoma is characterized by a predominantly lobular panniculitis, in which the subcutaneous fat exhibits a ‘Swiss-cheese’ appearance, with cystic spaces of variable size and shape, surrounded by foamy histiocytes and multinucleated giant cells; intense fibrosis with sclerotic collagen bundles is seen surrounding the cystic spaces [41]. Similar findings have been described in the penis following a grease gun injury [42]. Exogenous oils may be highlighted by special stains such as oil red O and osmium tetroxide [2].
Histopathological findings in local reactions to implants of silicone are variable depending mainly on the form of the injected silicone. Solid elastomer silicone induces an exuberant foreign-body granulomatous reaction, whereas silicone oil and gel induce a sparser inflammatory response. Silicone particles appear as groups of round empty vacuoles of different sizes between collagen bundles or within macrophages. Silicone particles are not birefringent under polarized light, but sometimes translucent angulated foreign bodies that represent impurities in the silicone are also found [3]. Granulomas from polymethylsiloxane fillers consist of irregularly shaped cystic spaces containing translucent, jagged ‘popcorn’, non-birefringent particles of varying size dispersed in a sclerotic stroma, surrounded by abundant multinucleated foreign-body giant cells [43].
Granulomas from collagen-based cosmetic fillers containing polymethylmethacrylate microspheres show a nodular or diffuse granulomatous infiltrate surrounding rounded vacuoles of similar shape and size, which mimic normal adipocytes and correspond to the implanted microspheres [5].
Injections of lipomas with phosphatidylcholine-containing substances induce an early reaction characterized by neutrophilic infiltration with partially destroyed fat cells; late lesions show infiltration of T lymphocytes and macrophages with foamy histiocytes, accompanied by thickened septa and pseudocapsule formation surrounding the inflamed area [9].
Persistent reactions to aluminium at the site of injection of hyposensitization vaccines show abundant lymphoid follicles in the subcutaneous tissue with germinal centre formation. They may mimic lupus profundus, pseudolymphoma or deep morphoea, but the abundant eosinophils and the identification of characteristic histiocytes with basophilic granular cytoplasm are the key distinctive features allowing the correct diagnosis [19]. These macrophages contain lysosomes filled with aluminium salts that can be demonstrated with X-ray dispersion microanalysis.
Pentazozine panniculitis is manifested as sclerodermoid plaques that result from thrombosis of small vessels, endarteritis, granulomatous inflammation, lipophagic granulomata and pronounced fibrosis of the dermis and subcutaneous fat [11, 44].
Panniculitis secondary to vitamin K injections is also characterized by prominent sclerosis of the collagen bundles of the connective tissue septa of the subcutis and an inflammatory infiltrate of lymphocytes, mast cells and plasma cells, which raises the histopathological differential diagnosis with morphoea [16, 38]. In contrast with deep morphoea, vitamin K1 panniculitis usually also involves the fat lobule with lipophagic granulomata.
Povidone panniculitis shows granulomatous infiltration of the fat lobule with focal haemorrhage and necrosis. Many macrophages contain grey-blue foamy material in their cytoplasm, which is positive for Congo red and chlorazol-fast pink [39].
Extravasation of cytotoxic drugs shows lobular panniculitis, abundant adipocyte necrosis with little inflammatory infiltrate together with epidermal lesions attributable to direct cytotoxicity. More chronic cases show marked fibrosis and lipomembranous changes [24, 45].
Panniculitis secondary to subcutaneous glatiramer acetate injections for the treatment of multiple sclerosis consisted of a predominantly lobular panniculitis, with lipophagic granulomata and scattered neutrophils and eosinophils both in the septa and in the fat lobules; the connective tissue septa show widening and fibrosis in conjunction with many lymphoid follicles, some with reactive germinal centres (Figure 99.43). Immunohistochemistry demonstrates that the inflammatory infiltrate of the fat lobule consists of CD68-positive histiocytes and suppressor/cytotoxic T lymphocytes. In contrast, the lymphoid follicles in the septa and at the interface between septum and fat lobule are mainly composed of B lymphocytes [17].
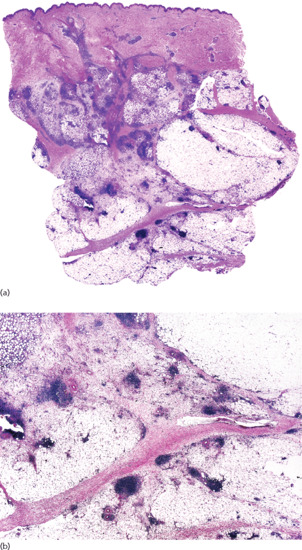
Figure 99.43 Histopathological features of panniculitis secondary to subcutaneous glatiramer acetate injections for the treatment of multiple sclerosis. (a) Scanning power showing a mostly lobular panniculitis. (b) Numerous lymphoid aggregates are seen at the periphery of the fat lobules.
Factitious panniculitis due to repeated trauma shows organizing haematomas, focal granulomas and haemosiderin deposition [29].
Management
Early lesions of factitious panniculitis should be treated with systemic antibiotics to cover a wide spectrum of microorganisms. If artefact is suspected, the affected area may be occluded for a week with a bandage: improvement would support a suspicion of self-induced factitious panniculitis, for which appropriate social and psychiatric care should be offered. Regrettably, these offers are usually rejected by patients.
Panniculitis secondary to cosmetic fillers usually require intralesional steroids and, if possible, removal of the implanted material. Panniculitis secondary to injection of drugs usually requires only supportive care and withdrawal of the responsible drug.
Neutrophilic lobular panniculitis
Definition
Neutrophilic lobular panniculitis incorporates a range of different panniculitides in which the fat lobule infiltrate is mostly composed of neutrophils (Box 99.8). According to Cohen, ‘neutrophilic lobular panniculitis is not a distinct entity but has a concise pathologic description of specific changes in the subcutaneous fat that have been observed in association with several conditions’ [1]. Some of these entities have been covered in other sections of this chapter. Included here is a small group of rare disorders characterized by neutrophilic lobular panniculitis.
Introduction and general description
Subcutaneous Sweet syndrome (see Chapter 49)
Acute febrile neutrophilic dermatosis or Sweet syndrome is a neutrophilic dermatosis characterized by an acute onset of oedematous, erythematous papules and plaques, often accompanied by fever and malaise, which was first described by Sweet in 1964 [2]. Histopathologically, cutaneous lesions show oedema of the papillary dermis and a dense band-like infiltrate of neutrophils involving mostly the superficial dermis, with no vasculitis [3]. Usually, Sweet syndrome is mainly a dermal process, but even in the original series Sweet reported that the neutrophilic infiltrate may extend into the underlying subcutaneous tissue with an associated neutrophilic panniculitis [2]. Subcutaneous involvement in classic Sweet syndrome is not rare and it has been described in some series with frequencies ranging from 25% to 50% of cases [4–8]. A diagnosis of subcutaneous Sweet syndrome should be made, however, only in those cases in which the neutrophilic infiltrate involves exclusively the subcutaneous tissue with few or no neutrophils in the dermis.
History
Only a few patients have been described with this peculiar variant of Sweet syndrome with exclusively subcutaneous involvement. Cullity et al. [9] were the first authors to use the term ‘Sweet's panniculitis’ or ‘acute febrile neutrophilic panniculitis’ to describe this entity, although similar cases had been previously reported [4, 10]. Most of the cases show a neutrophilic lobular panniculitis, although in rare instances a septal component may be predominant [11]. To date, only 22 well-documented cases of subcutaneous Sweet syndrome have been reported [9–20, 21, 22–26]. Several features support the relationship of neutrophilic lobular panniculitis to Sweet syndrome. In some cases, subcutaneous Sweet syndrome was followed by classical dermal Sweet syndrome [4], whereas another patient presented simultaneously with classical Sweet syndrome and Sweet panniculitis [17]. As in classical Sweet syndrome, many patients with subcutaneous Sweet syndrome had associated myelodysplastic syndromes and haematological neoplasms [9, 12, 13, 14, 18, 19, 23, 24, 26, 27, 28], and cutaneous lesions showed an excellent response to systemic corticosteroids.
Presentation
Subcutaneous Sweet syndrome, like classical Sweet syndrome, has a median age of onset during the sixth decade of life but appears not to show the female preponderance of the latter [3, 8]. Clinically, the lesions consist of erythematous nodules [10, 11, 12, 13, 14, 16, 17] or plaques [9, 11] (Figure 99.44). Often the nodules are tender or painful [14]. Frequently, the onset of subcutaneous nodules is preceded or accompanied by systemic symptoms such as fever and malaise [4, 9, 11, 13, 15, 16]; leukocytosis was found in several patients [11–13, 15, 16]. The most frequent locations are the lower extremities [10, 11, 12, 13, 14, 16, 17], followed by upper extremities [11, 12, 14], trunk [9, 11, 12, 14] and head [9]. In one patient, the lesions were associated with all-trans-retinoic acid chemotherapy for promyelocytic leukaemia [18]; another patient presented associated dacryoadenitis [19]; and another developed the condition when administered pegylated granulocyte colony-stimulating factor (GCSF) [25].
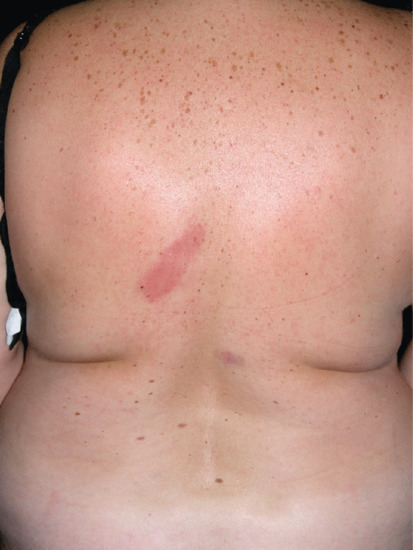
Figure 99.44 Neutrophilic lobular panniculitis showing erythematous plaques and nodules on the back of an adult woman.
Investigations
Histopathological study of the lesions demonstrates a dense infiltrate of mature neutrophils involving subcutaneous tissue. The neutrophilic infiltrate may involve septa, lobules or both [1], but in most cases the lobular component predominates [9, 11, 12, 14, 15, 16, 17] (Figure 99.45). Vasculitis is usually absent in all cases, but in two patients leukocytoclasia was noted [9, 12]. The dermis is, by definition, spared in all patients. Occasionally, some mononuclear cells may be found in the subcutaneous tissue [9, 13]. Rarely, infiltration of myeloperoxidase positive immature granulocytes has been described [20], representing the subcutaneous counterpart of the so-called histiocytoid Sweet syndrome [29]. In summary, Sweet syndrome may involve subcutaneous tissue with two different patterns: (i) with a mostly septal panniculitis and occasionally granulomatous infiltrate, as in classical erythema nodosum associated with Sweet syndrome [30]; and (ii) with a neutrophilic infiltrate mostly involving the fat lobules, as is the case in subcutaneous Sweet syndrome.
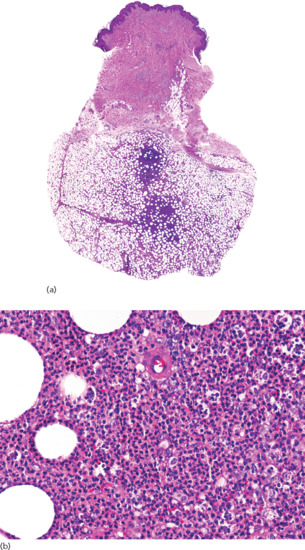
Figure 99.45 Histopathological features of neutrophilic lobular panniculitis. (a) Scanning power showing a predominantly lobular panniculitis. (b) The lobular infiltrate is composed mainly of neutrophils.
Management
Most patients with subcutaneous Sweet syndrome show dramatic response to systemic corticosteroids, such as 14 prednisolone [9, 10, 1315]. Dapsone has also been administered successfully in one patient [16].
Behçet disease (see Chapter 48)
In the differential diagnosis of subcutaneous Sweet syndrome are the ‘erythema nodosum-like’ lesions which occur in approximately 30% of patients with Behçet disease [31, 32]. They consist of nodules on the lower extremities [32, 33], and sometimes also on the arms [32, 33], identical to those of classical erythema nodosum [31]. However, histopathological study demonstrates involvement of both septa and fat lobules [32, 33], with necrotizing leukocytoclastic vasculitis involving arterioles and venules [31–34]. In some cases, neutrophilic abscesses are found within the fat lobules [31]. A variable degree of fat necrosis is nearly always present [31]. Therefore, the histopathological findings rule out a diagnosis of erythema nodosum and the vasculitis component allows subcutaneous Sweet syndrome to be ruled out [32].
Rheumatoid arthritis (see Chapter 154)
Several cases of neutrophilic (pustular) panniculitis have been described in patients with rheumatoid arthritis [35–41]. In these patients, subcutaneous nodules are mostly located on the lower extremities and they show a tendency to form draining fistulae with a yellowish discharge [36, 40–42]. Histopathologically, these lesions are characterized by a lobular infiltrate of neutrophils accompanied by lymphocytes, macrophages and multinucleate giant cells [36–39, 41, 42]. Fat necrosis has been found in nearly all cases [36, 37, 41, 42] and leukocytoclastic vasculitis was described in some [36, 42].
Bowel-associated dermatosis-arthritis syndrome (see Chapter 152)
The bowel-associated dermatosis–arthritis syndrome is characterized by recurrent fever, arthralgia and skin lesions after intestinal bypass or bariatric surgery [43, 44]. The most frequent skin lesions consist of erythematous papules or vesiculopustules. Lobular neutrophilic panniculitis with tender subcutaneous nodules on the lower extremities has, however, rarely been described in these patients [43–46].
Subcutaneous sarcoidosis
Definition and nomenclature
Minimal dermal involvement is acceptable for a histopathological diagnosis of subcutaneous sarcoidosis [1], but subcutaneous sarcoidosis is considered a specific clinicopathological variant of sarcoidosis involving exclusively the subcutaneous fat and should be differentiated from nodular dermal lesions of sarcoidosis with deep extension into the subcutaneous tissue [2]. Mostly, subcutaneous sarcoidosis is associated with systemic sarcoidosis, although with an indolent and non-aggressive form of the disease [3].
Introduction and general description
The most frequent subcutaneous disorder associated with sarcoidosis is not subcutaneous sarcoidosis, but classical erythema nodosum [3]. However, patients with systemic sarcoidosis may also develop sarcoidal granulomas involving the subcutaneous tissue as a specific cutaneous lesion and which is referred to as subcutaneous sarcoidosis. Subcutaneous sarcoidosis is a rare form of cutaneous sarcoidosis [3–6] (see Chapter 98).
Epidemiology
Incidence and prevalence
Subcutaneous sarcoidosis is the least common specific cutaneous manifestation of sarcoidosis. Its frequency varies, ranging in different series between 1.4% and 6% of patients with systemic sarcoidosis [7] and represents 11.76% of specific cutaneous lesions [8]. Its clinical features are described in detail in Chapter 98 (Figure 99.46).
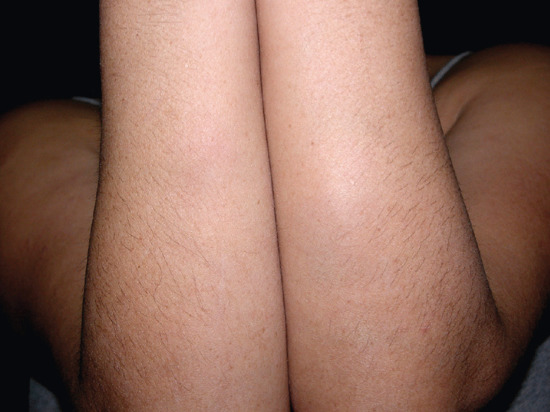
Figure 99.46 Subcutaneous sarcoidosis. Subcutaneous firm nodules on the forearms covered by normal appearing skin.
Investigations
Histopathologically, subcutaneous sarcoidosis is characterized by non-caseating granulomas involving fat lobules [3, 8] (Figure 99.47). At low-power magnification, the process appears as a predominantly lobular panniculitis, with minimal or no septal involvement. Sarcoidal granulomas in subcutaneous tissue are usually small, uniform in size, and mainly composed of epithelioid histiocytes, with limited numbers of multinucleate giant cells and a sparse lymphocytic component. Occasionally, small foci of eosinophilic necrosis may appear in the centre of regressing sarcoid granulomas [9], raising the differential diagnosis of tuberculosis [10]. In rare instances, caseating necrosis may be extensive [10]. The development of calcification in these sarcoidal granulomata has also been reported [11]. Foreign refractile particles under polarized light have been detected in some cases of subcutaneous sarcoidosis and this finding should not exclude the diagnosis [12, 13, 14].
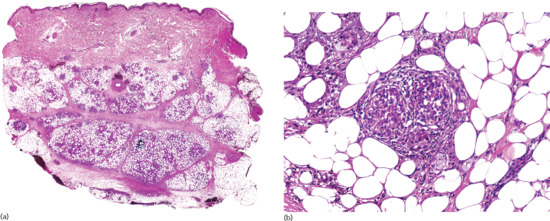
Figure 99.47 Histopathological features of subcutaneous sarcoidosis. (a) Scanning power showing a predominantly lobular panniculitis. (b) Small non-caseating granulomas involving the fat lobule.
Traumatic panniculitis
Definition
Traumatic panniculitis refers to damage of subcutaneous tissue induced by physical and chemical agents. The physical injuries may be from trauma, cold, electricity or chemicals. Cold panniculitis and chemical factitious panniculitis have been covered in other sections of this chapter. In general, there is no direct relationship between the severity of the injury and the severity of the resultant panniculitis: even minor trauma often causes nodules of panniculitis on the shins (Figure 99.48).
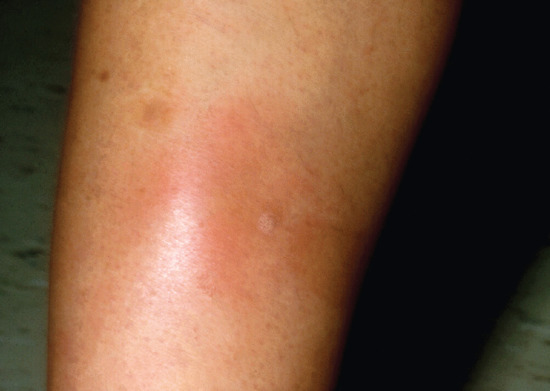
Figure 99.48 Traumatic panniculitis involving the shin.
Presentation
A specific form of traumatic panniculitis is common in women with large pendulous breasts. Large breast masses can form and may be misinterpreted as breast cancer [1].
Another peculiar variant of traumatic panniculitis is the so-called semicircular lipoatrophy that is probably induced by repeated microtrauma (see Chapter 100) [2, 3, 4, 5, 6, 7]. These patients, generally adult women, develop horizontal band-like or circular depressions around the anterolateral aspects of the thighs. Lipoatrophy in these areas develops over several weeks without symptoms. Trauma is the most probable cause because the affected areas of the thighs are vulnerable to repetitive trauma from sitting at desks and tables.
Post-traumatic panniculitis may present as a solitary or as multiple subcutaneous nodules caused by traumatic separation and consequent devascularization of pieces of subcutaneous fat from its blood supply. The resultant walling off of the necrotic fat by fibrous tissue results in so-called encapsulated fat necrosis, otherwise termed nodular cystic fat necrosis or mobile encapsulated lipoma [8–10, 11, 12–17].
Electrical injuries appear at the point of contact of different electrodes causing skin burns with superficial subcutaneous involvement.
Investigations
The histopathological findings in traumatic panniculitis are not specific. Early lesions show intense haemorrhage and an inflammatory infiltrate, mostly composed of lymphocytes and macrophages arranged around septal vessels. Fully developed lesions are associated with cystic areas of fat necrosis surrounded by histiocytes, lipophagic granulomata scattered with haemosiderophages, and some neutrophils and eosinophils. Late lesions show fibrotic replacement of the fat lobule with residual cystic fat necrosis surrounded by macrophages and foreign-body multinucleate giant cells. Lipomembranous changes are also common in late-stage lesions of traumatic panniculitis [18]. Dystrophic calcification may appear in longstanding nodules.
Biopsy from semicircular atrophy shows inflammatory perivascular changes in early lesions. Nodular-cystic fat necrosis shows extensive necrosis of the fat lobules, with cystic fat necrosis and lipomembranous changes, and, as the most characteristic finding, a thick eosinophilic peripheral fibrotic pseudocapsule with hyaline appearance encircling the nodule and separating it from the surrounding tissues [19] (Figure 99.49).
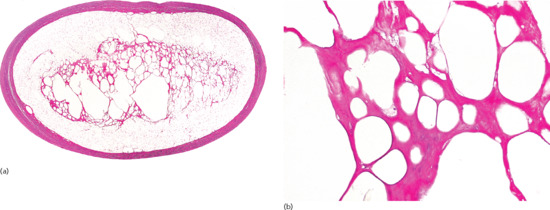
Figure 99.49 Histopathological features of encapsulated fat necrosis. (a) Scanning power showing an enucleated and very well-circumscribed lesion. (b) Necrotic adipocytes surrounded by hyaline material.
Management
Traumatic panniculitis is usually a self-limiting disorder and considerable improvement occurs with the time.
Lipoatrophic panniculitis of the ankles in childhood
Clinical features
History
Lipoatrophic panniculitis characteristically involving the ankles was initially reported by Shelley and Izumi in 1970 and they coined the expression ‘annular atrophy of the ankles’ to name this process [1]. Since then, only a handful of cases have been reported [2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19], some of them under the heading of connective tissue panniculitis [3].
Presentation
Many of the reported patients with lipoatrophic panniculitis of the ankles are children with antinuclear antibodies, autoimmune conditions or both, so that this disorder has been included under connective tissue panniculitis, together with morphoea, lupus panniculitis and dermatomyositis [4, 10].
Clinically, the characteristic annular or semicircular involvement of the ankles makes this a distinctive condition with few if any differential diagnoses. The lesions are tender and the onset of subcutaneous nodules is accompanied by fever, malaise and arthralgia of the ankles. Erythematous nodules and plaques resolve with annular scaling leaving lipoatrophy around the ankles (Figure 99.50).
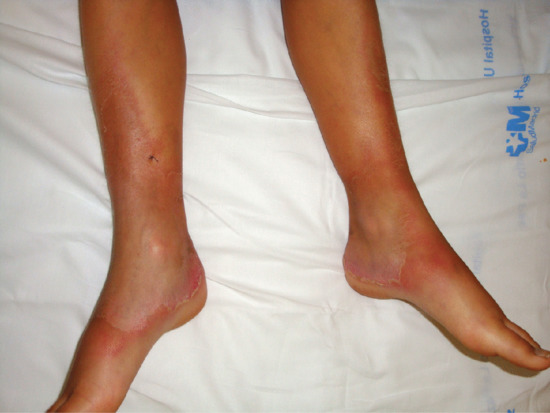
Figure 99.50 Lipoatrophic panniculitis of the ankles in a 12-year-old boy.
Investigations
In all except one case in which histopathology has been reported, a mainly lobular inflammation of the subcutaneous tissue has been described, with predominance of histiocytes, lipophagic granulomata and a smaller number of lymphocytes, neutrophils, plasma cells and eosinophils [1, 2, 3, 4, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18] (Figure 99.51). One reported case was remarkable for the relative abundance of lymphocytes, some of them mildly atypical [19].
Immunohistochemistry in this case demonstrated that most lymphocytes were positive for CD3 and CD7, with only occasional CD20+ B lymphocytes, and approximately equal numbers of CD4 and CD8 cells. PCR study for B- and T-cell clonality yielded polyclonal results [19]. The authors concluded that their findings of a mostly lymphocytic panniculitis in that case were due to a biopsy performed in an earlier stage of evolution of the lesions of the panniculitic process, compared to previously reported cases [19]. The partial ‘rimming’ of necrotic adipocytes by lymphocytes, as well as the relatively high proliferative activity disclosed by MIB-1 raised the differential diagnosis of SPTCL, but the other immunohistochemical results and PCR studies ruled out that diagnosis.
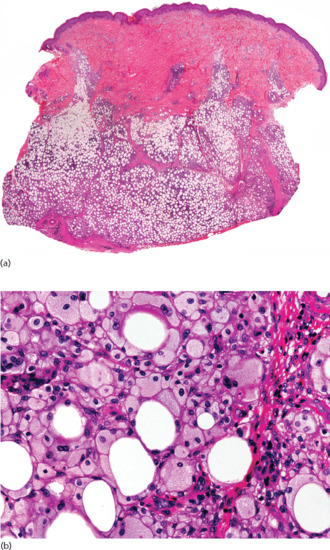
Figure 99.51 Histopathological findings in lipoatrophic panniculitis of the ankles in children. (a) Scanning power showing a mostly lobular panniculitis. (b) The fat lobule is replaced by a diffuse histiocytic infiltrate. Many histiocytes show a foamy cytoplasm as a consequence of phagocytosis of lipids, resulting in a lipophagic granuloma.
Management
Symptomatic treatment with non-steroidal anti-inflammatory drugs provides analgesia and helps resolution. Oral prednisolone usually leads to marked improvement, but some cases have recurred on tapering, and then other drugs such as hydroxychloroquine, methotrexate and azathioprine have been administered with good effect. Residual lipoatrophy of the ankles tends to improve slowly with time.
Subcutaneous fat necrosis of the newborn
Pathophysiology
Predisposing factors
The pathogenesis of this disorder is unknown. In many cases, perinatal complications are recorded, such as Rh factor incompatibility, meconium aspiration, umbilical cord prolapse, placenta praevia, birth asphyxia, seizures, congenital heart disease, intestinal perforation, hypothermia, sepsis, anaemia, obstetric trauma, gestational diabetes, pre-eclampsia or maternal abuse of drugs [1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12], but in many instances there is no history or any other associated anomaly. It has been suggested that localized and transient hypoxia may be an aetiological factor [13] and this might explain the predominant location of lesions on the shoulders and buttocks, cutaneous areas where mechanical pressure might compromise the circulation and contribute to hypoxia. Cold may also play a role, because there are several reports of subcutaneous fat necrosis of the newborn in children who have had cardiac surgery and the lesions appeared after cutaneous applications of ice to induce hypothermia [10, 14–17]. Cases of subcutaneous fat necrosis of the newborn have been described after whole-body cooling in an infant with polycythaemia and hypocalcaemia [18] and in other newborns after hypothermia treatment of hypoxic ischaemic encephalopathy [19]. Another important predisposing factor may be the particular subcutaneous fat composition in neonates, with a relatively high concentration of saturated fatty acids compared to unsaturated fatty acids, which results in a higher melting point for neonatal fat that confers on it a greater propensity to undergo crystallization under cold temperatures resulting in adipocyte necrosis [20]. It has also been suggested that newborns with subcutaneous fat necrosis might have a transitory protease inhibitor deficiency due to liver immaturity, similar to α1-antitrypsin deficiency [21]. The histopathological findings of subcutaneous fat necrosis of the newborn are, however, entirely different from those of α1-antitrypsin deficiency-associated panniculitis, which militates against this theory. Other authors have proposed that subcutaneous fat necrosis of the newborn is a disorder of brown fat, which is present in the most frequently involved areas [12]. Subcutaneous fat necrosis of the newborn has also been described after administration of prostaglandin E1 for the treatment of congenital heart disease, which supports some pathogenic role of prostaglandin E1 [22].
Approximately 25% of infants with subcutaneous fat necrosis of the newborn present with hypercalcaemia for unknown reasons: this anomaly is more frequent in infants with extensive lesions and when the trunk is involved [23]. The origin of this hypercalcaemia is not known, but increased calcium absorption due to extrarenal hyperproduction of 1,25-dihydroxyvitamin D3 (calcitriol) has been detected in several granulomatous processes, including sarcoidosis and subcutaneous fat necrosis of the newborn [23–27]. Elevated parathormone levels have been detected in some cases [28], but autopsy studies failed to reveal any parathyroid hyperplasia. Elevated urinary excretion of prostaglandin E1 suggests an increased calcium resorption from bone as the explanation for the hypercalcaemia [29, 30].
Clinical features
Presentation
Clinically, the lesions consist of multiple symmetrically distributed indurated, smooth, non-pitting mobile subcutaneous erythematous or violaceous nodules or plaques that appear in the first few weeks of life. The most common locations are the shoulders and buttocks (Figure 99.52), but lesions on the face, thighs, back and distal areas of the extremities have been also described [31, 32]. These lesions tend to spare the anterior trunk. In rare instances, the nodules may ulcerate, discharge oily contents and heal leaving atrophic scars. Confluence of the nodules results in extensive erythematous indurated plaques.

Figure 99.52 Subcutaneous fat necrosis of the newborn. Erythematous and violaceous plaques involving the shoulders, arms and buttocks.
Complications and co-morbidities
Most children remain otherwise healthy as the subcutaneous nodules develop, but approximately 25% develop complicating hypercalcaemia [23, 24, 25, 26, 27, 28, 29, 30, 33, 34]. The hypercalcaemia may persist for several weeks after the subcutaneous lesions have regressed, which requires calcium levels to be monitored and associated endocrinological disorders to be ruled out. Other reported laboratory anomalies include transient thrombocytopenia, probably due to platelet sequestration [5, 12, 13, 35], hypoglycaemia due to maternal diabetes and hypertriglyceridaemia [36]. In rare instances, the condition may be fatal, particularly when visceral fat is involved [4]. Nephrocalcinosis persisting several years after resolution of the skin nodules has been reported as a rare complication [37].
Investigations
Histopathologically, subcutaneous fat necrosis of the newborn shows a predominantly lobular panniculitis, with a dense inflammatory infiltrate composed of lymphocytes, histiocytes, lipophages, multinucleate giant cells and sometimes eosinophils interspersed among the adipocytes of the fat lobule. Many adipocytes are replaced by cells with finely eosinophilic granular cytoplasm that contain doubly refractile narrow needle-shaped clefts radially arranged, which represent triglyceride crystallization within adipocytes and stain with oil red O [38] (Figure 99.53). Some of these refractile clefts may also be found within the cytoplasm of multinucleate giant cells. The latter may also contain eosinophilic granules in their cytoplasm [39, 40, 41] and their origin from degranulating eosinophils has been postulated [39]. Fibrotic obliteration of small arterioles and calcium deposits in necrotic fat have also been described [42, 43, 44, 45]. In late-stage lesions, there is septal fibrosis and areas of calcification and lipoatrophy may appear within the fat lobule [11]. Adipocytes containing needle-shaped clefts radially arranged have been also found during autopsy studies in ventilator-associated tracheobronchitis (VAT) [4, 8]. In some cases, the diagnosis was established using touch preparation, which was prepared by pressing a fresh sample biopsy against a glass slide, and fine-needle aspiration techniques [46, 47].
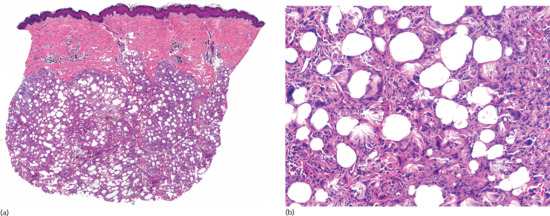
Figure 99.53 Histopathological features of subcutaneous fat necrosis of the newborn. (a) Scanning power showing a lobular panniculitis. (b) The fat lobule is replaced by a histiocytic infiltrate with some multinucleated giant cells. Many necrotic adipocytes and multinucleated giant cells contain needle-shaped clefts radially arranged.
Management
In most children with subcutaneous fat necrosis of the newborn, treatment is not required because the problem tends to resolve spontaneously. The main goals are the detection and treatment of hypercalcaemia [23, 24, 25, 26, 27, 28, 29, 30, 33, 34] with avoidance of calcium and vitamin D3. These measures should be instituted as early as possible with maintenance of adequate hydration with intravenous normal saline. Furosemide has been used to achieve increased calcium excretion by inhibiting calcium reabsorption, but a risk of dehydration and consequent worsening of hypercalcaemia exists. Prednisolone interferes with the metabolism of vitamin D to its active form and also inhibits the production of 1,25-dihydroxyvitamin D3 by the macrophages involved in the inflammatory process. Etidronate, a bisphosphonate that decreases bone resorption of calcium, has been helpful in the management of the associated hypercalcaemia [27, 28, 31–33]. It should be used as a second line drug, because its effects on bone production, growth plates and mineralization in infants are as yet unknown. Pamidronate is also helpful for treatment for hypercalcaemia and may reduce the risk of nephrocalcinosis [48, 49]. Calcitonin and citrate may be used as second line therapy for resistant cases.
Poststeroid panniculitis
Clinical features
History
In 1956, Smith and Good [1] reported 11 children with acute rheumatic fever receiving high doses of corticosteroids with rapid taper, of whom five developed erythematous subcutaneous nodules.
Presentation
Poststeroid panniculitis is a rare panniculitis of children and infants on prolonged systemic corticosteroid treatment, and is related to a rapid decrease or a sudden withdrawal of steroid therapy. Only about 20 cases have been described in the literature [2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15]. It develops in an older age group than sclerema neonatorum and subcutaneous fat necrosis of the newborn, with reported ages ranging from 20 months to 14 years [3]. Although the process is mostly seen in children, there are also a few reported cases in adults [12, 13, 15]. In one adult patient, the lesions involved the arms and legs and were painless [15].
The lesions vary in size from 0.5 to 4 cm and consist of asymptomatic firm subcutaneous nodules, often with overlying erythema, and tend to be localized in those areas where there is the greatest accumulation of fat from steroid therapy, such as the face, arms and posterior neck [5]. They usually appear 1–10 days after cessation of high doses of systemic corticosteroids.
The disorders for which treatment with high doses of corticosteroids have been administered are varied, including rheumatic fever, leukaemia, nephrotic syndrome, acute exacerbation of chronic obstructive pulmonary disease, erythema nodosum leprosum, autoimmune enteropathy, Sjögren syndrome and brain tumours. Conversely, different oral or intravenous corticosteroid drugs, including prednisolone and dexamethasone, have been associated with this panniculitis. Usually, reinstitution of corticosteroid administration induces improvement of panniculitis, although some authors believe that this is not necessary for its resolution [3].
Investigations
Histopathological findings in poststeroid panniculitis lesions are identical to those of subcutaneous fat necrosis of the newborn. They consist of a mostly lobular panniculitis with an inflammatory infiltrate of foamy histiocytes and lymphocytes involving the fat lobules [6]. Often, doubly refractile narrow needle-shaped clefts radially arranged are found within the cytoplasm of some histiocytes and necrotic adipocytes, although usually they are not as numerous as in subcutaneous fat necrosis of the newborn.
Management
Lesions of poststeroid panniculitis usually disappear gradually without residual scarring over the course of weeks or months, even without resuming steroid therapy and therefore no treatment is usually necessary. However, readministration of high doses of systemic corticosteroid and a slower and more gradual decrease of the dose is followed by a faster improvement and resolution of the lesions.
Sclerema neonatorum
Definition
Sclerema neonatorum is an uncommon condition which typically affects gravely ill, preterm neonates in the first week of life. It manifests as a diffuse hardening of skin and subcutaneous tissue such that the skin cannot be pitted or picked up and pinched into a fold. Histologically, there is minimal inflammation with extensive fat necrosis. It is associated with a high mortality (see also Chapter 116).
Clinical features
Presentation
Sclerema neonatorum is an extremely uncommon process and almost always appears during the first week of life, although there are also reports of infants born preterm with low birth weight who developed this condition later. In most cases, sclerema neonatorum is a grave illness and it has been associated with a mortality of up to 75% [1, 2, 3]. However, most case series date from 40 or more years ago, and now the process is very uncommon, probably due to better neonatal care. Prematurity and placental insufficiency have been proposed as pathogenetic factors for its development [3, 4]. Affected infants are almost always severely ill from conditions such as septicaemia or other disseminated infections, congenital heart disease, pneumonia, diarrhoea, dehydration, intestinal obstruction or other congenital developmental defects [3]. In a multivariate analysis of risk factors for sclerema neonatorum in preterm neonates in Bangladesh, lower maternal education, signs of jaundice and poor feeding on admission were the main risk factors, although the diagnosis in that study was not histopathologically confirmed [5]. Cold injury has also been proposed as an aetiological factor [6], but it does not appear to be important in most cases.
Lipolytic immaturity in infants born preterm [7] and the different composition of subcutaneous fat in newborns, with a higher proportion of saturated (palmitic and stearic) to unsaturated (oleic) fatty acids, may also be predisposing factors [8, 9], because they favour solidification of subcutaneous fat if there is a fall in the temperature of subcutaneous tissue as a result of peripheral circulatory collapse [10]. Sclerema neonatorum is characterized by increased blood lipid peroxidation and diminished superoxide dismutase activity, which raises the possibility that free radicals may also play some role in the pathogenesis of the process [11].
Clinically, infants with sclerema neonatorum typically appear severely ill from birth and during the first days of life they develop generalized woody induration of the skin [10, 12] (Figure 99.54). Usually, the process begins on the buttocks and thighs but rapidly extends to involve almost the entire skin surface with the exception of the palms, soles and genitalia. The involved skin has a hard consistency, is non-pitting and is cold to the touch; it is yellowish-white in colour, often with purplish mottling. There is immobility of the extremities and the face shows a mask-like expression. The prognosis is poor and most affected infants die within a few days. However, if the infant survives, the skin recovers its normal appearance and there are no long-term complications such as calcification.

Figure 99.54 Sclerema neonatorum in a newborn with generalized woody induration of the skin.
Differential diagnosis
The main differential diagnosis of sclerema neonatorum is subcutaneous fat necrosis of the newborn. This is important because they represent two distinctive clinicopathological processes with very different prognoses [12]. There is a single reported case of coexistence of both disorders in the same infant, but that is a doubtful case because histopathological study was lacking [13]. Usually this differential is straightforward, because subcutaneous fat necrosis of the newborn, which is a localized and self-healing process, does not develop in the first days of life and has characteristic histopathology consisting in a dense histiocytic infiltrate in the fat lobules with radially arranged needle-shaped refractile clefts within adipocytes and histiocytes.
Histopathological changes similar to those of sclerema neonatorum have been described in a patient with gemcitabine-associated livedoid thrombotic microangiopathy. The cutaneous biopsy showed small-vessel occlusion by intravascular fibrin and leukocytes, vessel wall thickening and endothelial cell swelling and some structures arranged radially with needle-shaped clefts resembling those of sclerema neonatorum [14].
Sclerema neonatorum should be not confused with scleredema neonatorum, an entirely different process seen in premature infants with congenital heart disease, which is characterized by distended skin with wax-like appearance that results from dermal oedema with increased amounts of mucin [12].
Investigations
Histopathologically, there is a striking contrast at low-power magnification between the severe damage in the subcutaneous fat and the sparse inflammatory response [9, 10, 15] (Figure 99.55). Most fat lobules appear to be replaced by amphophilic granular detritus and the cellular structures of the adipocytes are no longer evident. Surprisingly, there is sparse or no inflammatory infiltrate in spite of the intense fat necrosis. The most characteristic histopathological feature consists of the presence of radially arranged needle-shaped refractile clefts in adipocytes and, occasionally, in the few multinucleate giant cells, which result from crystallization of the lipid contents of the adipocytes. X-ray diffraction studies have demonstrated that the clefts in the fat cells are due to crystallization of tryglicerides [16]. In late-stage lesions, thickened connective tissue septa may be the only anomaly [15].
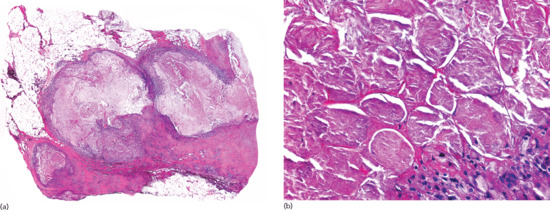
Figure 99.55 Histopathological features of sclerema neonatorum. (a) Scanning power showing necrosis of the entire fat lobules and thickened connective tissue septa. (b) An entire fat lobule is replaced by granular detritus and the cellular structures of the adipocytes are no longer evident. Many necrotic adipocytes contain needle-shaped clefts.
Management
Treatment of sclerema neonatorum is mainly directed to the underlying disease. Systemic corticosteroids have been demonstrated not to be effective [2]. Repeated exchange transfusions may substantially reduce mortality [17, 18], but the diagnosis in those cases was not histopathologically confirmed. Intravenous immunoglobulins were used in a newborn with sclerema neonatorum and sepsis, with transitory improvement of the skin induration, but the patient died of respiratory failure [19].
Gouty panniculitis
Definition
Panniculitis is a very uncommon complication of tophaceous gout (see Chapter 154).
Clinical features
Presentation
The usual clinical presentation consists of painful ulcerating nodules on the lower legs [1, 2, 3, 4, 5, 6, 7, 8, 9] (Figure 99.56). In most reported cases, patients had already sustained severe joint damage by the time panniculitis manifested as subcutaneous nodules, but in some cases panniculitis may be the first manifestation of hyperuricaemia [2]. One of the patients reported also had involvement of bone marrow fat [9].
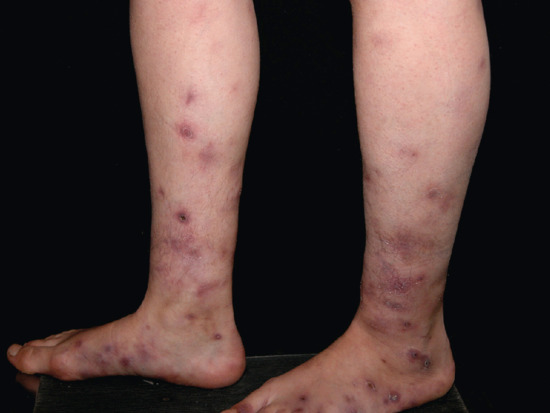
Figure 99.56 Gouty panniculitis. Ulcerated nodules on the lower legs of a patient with hyperuricaemia.
Investigations
Histopathology shows a lobular, sometimes neutrophilic panniculitis with deposition of needle-shaped refractile crystals within adipocytes (Figure 99.57) [2]. Frequently, histiocytes and multinucleate giant cells form a palisade around the urate crystals.
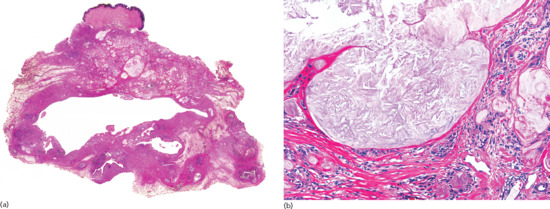
Figure 99.57 Histopathology of gouty panniculitis. (a) Scanning power showing involvement of the subcutaneous tissue, whereas the dermis is spared. (b) Basophilic amorphous deposits, which are fine needle-like shape urate crystals, appear surrounded by histiocytes.
Fungal panniculitis due to zygomycosis, mucormycosis and aspergillosis
A predominantly lobular panniculitis with fine needle-shaped refractile crystals within adipocytes has been described in subcutaneous fungal infections including zygomycosis [1], mucormycosis [2] and aspergillosis [3] (see Chapter 32). The presumed urate crystals are similar to those found in gouty panniculitis but the mechanism by which they are formed is unknown.
Cytophagic histiocytic panniculitis and subcutaneous panniculitis-like T-cell lymphoma
Introduction and general description
The term cytophagic histiocytic panniculitis was introduced to describe a rare entity characterized by the development of subcutaneous nodules containing lobular infiltrates of macrophages which had phagocytosed lymphocytes, erythrocytes and nuclear debris [1].
With the benefit of modern immunohistochemistry and genetic techniques, it has become evident that most patients who are found to have cytophagic histiocytic panniculitis have one of two types of primary lymphoma affecting subcutaneous tissue [2]: SPTCL or primary cutaneous γδ T-cell lymphoma. The former is a homogeneous entity with an α/β+ T-cell phenotype, indolent biological behaviour and good prognosis, whereas patients with the latter and a γ/δ+ T-cell phenotype are more heterogeneous and show a very aggressive clinical course with poor prognosis [3–7]. Both entities are discussed in detail in Chapter 140.
However, it seems that there is still a group of patients with cytophagic histiocytic panniculitis in whom a definitive diagnosis of lymphoma cannot be made and in whom molecular studies fail to demonstrate monoclonal rearrangements of infiltrating lymphocytes. Nevertheless the prognosis within this group is often poor, with pancytopenia and fatal outcome from haemophagocytic syndrome involving liver, spleen and bone marrow [8].
Presentation
SPTCL is rare, accounting for less than 1% of all primary cutaneous T-cell lymphomas, with equal incidence in both genders and preferentially involving adult patients, although there are also cases described in children. Clinically, patients present with erythematous subcutaneous nodules that may group into large plaques typically involving the lower extremities (Figure 99.58). A common feature consists of areas of lipoatrophy when the lesions resolve. Less commonly, SPTCL may involve the trunk, head or upper extremities. Approximately, 50% of patients have B symptoms, including fever, fatigue and weight loss; cytopenia and elevation of liver enzymes are frequently found, but a frank haemophagocytic syndrome is rare [9].

Figure 99.58 Subcutaneous pannicultis-like T-cell lymphoma. Erythematous plaques involving the anterior aspects of the lower extremities.
Investigations
Histopathologically, SPTCL and cytophagic histiocytic panniculitis may be indistinguishable, and only after immunohistochemical and molecular studies can they be differentiated. At low power both disorders mimic a predominantly lobular panniculitis. The fat lobule is involved by small and medium-size atypical lymphocytes with hyperchromatic nuclei. Numerous macrophages are also intermingled with the atypical lymphocytes. A characteristic finding in favour of the diagnosis of SPTCL consists in the sparing of the epidermis and dermis (Figure 99.59). In some areas, atypical lymphocytes are arranged in a circle around necrotic adipocytes, although this rimming is not entirely specific for SPTCL and it may also be seen in other lymphoid processes involving subcutaneous fat [10]. The presence of macrophages with cytophagic activity containing lymphocytes, neutrophils and nuclear debris within their cytoplasm is also a frequent finding in both processes, the so-called ‘bean bag’ cells. Foamy histiocytes are also frequently found in areas of fat necrosis.

Figure 99.59 Histopathological features of subcutaneous panniculitis-like T-cell lymphoma. (a) Scanning power showing features simulating a lobular panniculitis. (b) Numerous macrophages with cytophagic activity containing lymphocytes, neutrophils and nuclear debris within their cytoplasm, the so-called ‘bean bag’ cells.
A diagnosis of SPTCL can usually be established with confidence using immunohistochemistry and molecular techniques. Immunohistochemical studies have demonstrated that lymphocytes involving the fat lobules in SPTCL have a TRC-α/β+, CD3+, CD4–, CD8+, TIA-1+, perforin+, granzyme B+, CD30– T-cell immunophenotype. Evidence of Epstein–Barr virus (EBV) infection, either using PCR amplification to detect EBV-encoded RNA and EBV DNA or by immunohistochemical staining for latent membrane protein 1, are characteristically negative. As a consequence of its α/β+ T-cell phenotype, SPTCL expresses βF1 but is negative for CD56, in contrast with primary cutaneous γ/δ+ T-cell lymphoma and other lymphoproliferative processes involving subcutaneous fat. Monoclonal rearrangement of γ or β genes can usually be detected in lesions of SPTCL using PCR or Southern blot analysis, whereas these rearrangements are not found in patients with cytophagic histiocytic panniculitis.
Sclerosing postirradiation panniculitis
Definition and nomenclature
Sclerosing postirradiation panniculitis is a rare clinicopathological variant of panniculitis that appears months or years after radiotherapy in the irradiated skin. Clinically, lesions consist of subcutaneous indurated nodules.
Clinical features
History
In 1993 Winkelmann et al. [1] described the first four cases of pseudosclerodermatous panniculitis after irradiation and, since then, only six additional cases have been published [2–4, 5]. This process represents an unusual variant of panniculitis which may rarely occur as a cutaneous complication of radiotherapy.
Presentation
Clinically, sclerosing postirradiation panniculitis presents as an indurated asymptomatic subcutaneous nodule or plaque with little or no change in the epidermis and dermis at the site of previous radiotherapy (Figure 99.60). The most common location is the anterior chest wall following treatment for breast cancer. However, it may appear in any previously irradiated area of the skin [2]. The interval between the radiotherapy and the presentation of the condition varies from months to several years [1–4, 5].
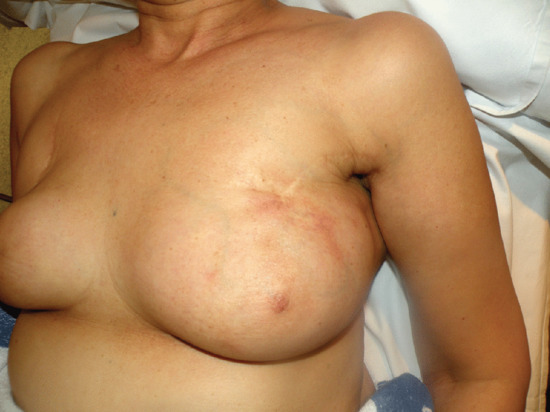
Figure 99.60 Sclerosing postirradiation panniculitis showing a depressed and indurate nodule on the previously irradiated area of the skin.
Differential diagnosis
The histopathological differential diagnosis includes other diseases showing a septal or lobular panniculitis with sclerosis of the connective tissue septa of the subcutis, such as deep morphoea [6, 7, 8] and lupus panniculitis [9]. Deep morphoea consists histopathologically of a septal panniculitis without significant lobular involvement in which there are sclerotic collagen bundles and aggregates of lymphocytes and especially plasma cells at the interface between the connective tissue septa and the fat lobules. In contrast with sclerosing postirradiation panniculitis, deep morphoea does not show lobular involvement and the inflammatory cells within the thickened septa are mainly plasma cells. Lupus panniculitis is a mostly lobular panniculitis with sclerotic septa, but, in contrast with sclerosing postirradiation panniculitis, the inflammatory infiltrate involving the lobules is mostly composed of lymphocytes and plasma cells.
Investigations
The histopathological findings in sclerosing postirradiation panniculitis include a predominantly lobular panniculitis with necrosis of the adipocytes at the centre of the fat lobule and dense inflammatory infiltrates composed mainly of foamy histiocytes. Lipophagic granulomas involving the periphery of the fat lobules and thickening and sclerosis of the connective tissue septa are the most characteristic features in the subcutaneous tissue (Figure 99.61). These histopathological findings may or may not be accompanied by dermal changes secondary to radiotherapy [10, 11, 12], namely sclerosis of the papillary dermis, atypical star-shaped fibroblasts scattered amongst dermal collagen bundles and dilated thrombosed blood vessels with endothelial cell swelling and hyaline sclerosis of their walls.

Figure 99.61 Histopathological features of sclerosing postirradiation panniculitis. (a) Scanning power showing a mostly lobular panniculitis. (b) Sclerotic collagen bundles at the septa of connective tissue of the subcutis.
Management
Treatment of sclerosing postirradiation panniculitis is not required and biopsy and subsequent histopathologic study are only performed when the possibility of a subcutaneous metastasis from the previously excised and/or irradiated breast cancer is raised.
References
Introduction
- Lundgren H, Bengtsson C, Blohme E, Lapidus L. Adiposity and adipose tissue distribution in relation to incidence of diabetes in women. Int J Obes 1989;13:413–18.
- Taitz LS. The Obese Child. Oxford: Blackwell Scientific Publications, 1983:21.
- Heaton JM. The distribution of brown adipose tissue in the human. Anatomy 1972;112:35–9.
- Bays HE, González-Campoy JM, Henry RR, et al. The Adiposopathy Working Group. Is adiposopathy (sick fat) an endocrine disease? Int J Clin Pract 2008;62:1474–83.
- Avram AS, Avram MM, James WD. Subcutaneous fat in normal and diseased states: 2. Anatomy and physiology of white and brown adipose tissue. J Am Acad Dermatol 2005;53:671–83.
- Frühbeck G. Overview of adipose tissue and its role in obesity and metabolic disorders. Methods Mol Biol 2008;456:1–22.
- Aherne W, Hull D. The site of heat production in the newborn infant. Proc R Soc Med 1964;57:1172–3.
- Jung RT, Shetty PS. Reduced thermogenesis in obesity. Nature 1979;279:322–3.
- Nedergaard J, Bengtsson T, Cannon B. Unexpected evidence for active brown adipose tissue in adult humans. Am J Physiol Endocrinol Metab 2007;293:E444–52.
- Ryan TJ, Curri SB, eds. Clinics in Dermatology, vol. 7. The Cutaneous Adipose Tissue. Philadelphia: JB Lippincott, 1989.
- Giorgino F, Laviola L, Eriksson JW. Regional differences of insulin action in adipose tissue: insights from in vivo and in vitro studies. Acta Physiol Scand 2005;183:13–30.
- Lafontan M, Berlan M. Do regional differences in adipocyte biology provide new pathophysiological insights? Trends Pharmacol Sci 2003;24:276–83.
- Thalmann S, Meier CA. Local adipose tissue depots as cardiovascular risk factors. Cardiovasc Res 2007;75:690–701.
- Johnson D, Cormack GC, Abrahams PH, et al. Computed tomographic observations on subcutaneous fat: implications for liposuction. Plast Reconstr Surg 1996;97:387–96.
- Bell GH. Textbook of Physiology and Biochemistry. Edinburgh: Churchill Livingstone, 1987.
- Frayn KN. Adipose tissue metabolism. In: Ryan TJ, Curri SB, eds. Clinics in Dermatology, vol. 7. The Cutaneous Adipose Tissue. Philadelphia: JB Lippincott, 1989:48–61.
- Avram MM, Avram AS, James WD. Subcutaneous fat in normal and diseased states 3. Adipogenesis: from stem cell to fat cell. J Am Acad Dermatol 2007;56:472–92.
- Wang P, Mariman E, Renes J, Keijer J. The secretory function of adipocytes in the physiology of white adipose tissue. J Cell Physiol 2008;216:3–13.
- Dalziel K. The nervous system and adipose tissue. In: Ryan TJ, Curri SB, eds. Clinics in Dermatology, Vol. 7. The Cutaneous Adipose Tissue. Philadelphia: JB Lippincott, 1989:62–77.
- Boden G, Hoeldtke RD. Nerves, fat, and insulin resistance. N Engl J Med 2003;349:1966–7.
- Romijn JA, Fliers E. Sympathetic and parasympathetic innervation of adipose tissue: metabolic implications. Curr Opin Clin Nutr Metab Care 2005;8:440–4.
- Langin D. Adipose tissue lipolysis as a metabolic pathway to define pharmacological strategies against obesity and the metabolic syndrome. Pharmacol Res 2006;53:482–91.
- Anubhuti, Arora S. Leptin and its metabolic interactions – an update. Diabetes Obes Metab 2008;10:973–93.
- Ranadive SA, Vaisse C. Lessons from extreme human obesity: monogenic disorders. Endocrinol Metab Clin North Am 2008;38:733–51.
- Lago R, Gómez R, Lago F, et al. Leptin beyond body weight regulation – current concepts concerning its role in immune function and inflammation. Cell Immunol 2008;252:139–45.
- Lara-Castro C, Fu Y, Chung BH, et al. Adiponectin and the metabolic syndrome: mechanisms mediating risk for metabolic and cardiovascular disease. Curr Opin Lipidol 2007;18:263–70.
- Fantuzzi G. Adiponectin and inflammation: consensus and controversy. J Allergy Clin Immunol 2008;121:326–30.
- Whitehead JP, Richards AA, Hickman IJ, et al. Adiponectin – a key adipokine in the metabolic syndrome. Diabetes Obes Metab 2006;8:264–80.
- Behre CJ. Adiponectin: saving the starved and the overfed. Med Hypotheses 2007;69:1290–2.
Panniculitis
- Patterson JW. Panniculitis. New findings in the “third compartment”. Arch Dermatol 1987;123:1615–18.
- Requena L, Sánchez Yus E. Panniculitis. Part I. Mostly septal panniculitis. J Am Acad Dermatol 2001;45:163–83.
- Requena L, Sánchez Yus E. Panniculitis. Part II. Mostly lobular panniculitis. J Am Acad Dermatol 2001;45:325–61.
- Lundgren H, Bengtsson C, Blone E, Lapidus L. Adiposity and adipose tissue distribution in relation to incidence of diabetic women. Int J Obes 1989;13:413–18.
- Reed RJ, Clark WH, Mihm MC. Disorders of the panniculus adiposus. Hum Pathol 1973;4:219–29.
- Hirsch J, Goldrick B. Metabolism of human adipose tissue in vitro. In: Handbook of Physiology. Washington DC: American Physiology Society, 1965:455–70.
- Caspar-Bauguil S, Cousin B, Galinier A, et al. Adipose tissues as an ancestral immune organ: site-specific change in obesity. FEBS Lett 2005;579:3487–92.
- Nishimura S, Manabe I, Nagasaki M, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med 2009;15:914–20.
- Poggi M, Jager J, Paulmyer-Lacroix O, et al. The inflammatory receptor CD40 is expressed on human adipocytes: contribution to crosstalk between lymphocytes and adipocytes. Diabetologia 2009;52:1152–63.
- Kanitakis J. Immunohistochemistry of normal human skin. Eur J Dermatol 1998;8:539–47.
- Muthusamy K, Halbert G, Roberts F. Immunohistochemical staining for adipophilin, perilipin and TIP47. J Clin Pathol 2006;59:1166–70.
- Ostler DA, Prieto VG, Reed JA, Deavers MT, Lazar AJ, Ivan D. Adipophilin expression in sebaceous tumors and other cutaneous lesions with clear cell histology: an immunohistochemical study of 117 cases. Mod Pathol 2010;23:567–73.
- Sánchez Yus E, Simón RS, Requena L. Vein, artery, or arteriole? A decisive question in hypodermal pathology. Am J Dermatopathol 2012;34:229–32.
- Hall LD, Dalton LS, Fillman LE, Dohse L, Elston DM. Re-examination of features to distinguish polyarteritis nodosa from superficial thrombophlebitis. Am J Dermatopathol 2013;35:463–71.
- White WL, Wieselthier JS, Hitchcock MG. Panniculitis: recent developments and observations. Semin Cut Med Surg 1996;15:278–99.
- Diaz Cascajo C, Borghi S, Weyers W. Panniculitis: definition of terms and diagnostic strategy. Am J Dermatopathol 2000;22:530–49.
- Segura S, Requena L. Anatomy and histology of normal subcutaneous fat, necrosis of adipocytes, and classification of the panniculitides. Dermatol Clin 2008;26:419–24.
- White JW, Winkelmann RK. Weber–Christian panniculitis: a review of 30 cases with this diagnosis. J Am Acad Dermatol 1998;39:56–62.
Superficial migratory thrombophlebitis
- Trousseau A. Phelegmasia alba dolens. Clin Med Hotel Dieu de Paris 1865;3:94.
- Salemis NS, Merkouris S, Kimpouri K. Mondor's disease of the breast. A retrospective review. Breast Dis 2011;33:103–7.
- Samlaska CP, James WD. Superficial thrombophlebitis: I. Primary hypercoagulable states. J Am Acad Dermatol 1990;22:975–89.
- Samlaska CP, James WD. Superficial thrombophlebitis. II. Secondary hypercoagulable states. J Am Acad Dermatol 1990;23:1–18.
- Sack GH, Levin J, Bell WR. Trousseau's syndrome and other manifestations of chronic disseminated coagulopathy in patients with neoplasms: clinical, pathophysiologic, and therapeutic features. Medicine 1977;56:1–37.
- Koc Y, Gullu I, Akpek G, et al. Vascular involvement in Behçet disease. J Rheumatol 1992;19:402–10.
- Giblin WJ, James WD, Benson PM. Buerger's disease. Int J Dermatol 1989;28:638–42.
- McColl MD, Ramsay JE, Tait RC, et al. Superficial vein thrombosis. Incidence in association with pregnancy and prevalence of thrombophilic defects. Thromb Haemost 1998;79:741–2.
- Alcaraz I, Revelles JM, Camacho D, et al. Superficial thrombophlebitis: A new clinical manifestation of the immune reconstitution inflammatory syndrome in a patient with HIV infection. Am J Dermatopathol 2010;32:846–9.
- Jordaan HF. Widespread superficial thrombophlebitis as a manifestation of secondary syphilis – a new sign. S Afr Med J 1986;70:493–4.
- Demitsu T. Yamada T, Usui K, et al. Superficial thrombophlebitis in a patient with secondary syphilis. Int J Dermatol 1996;35:821–4.
- Khan EA, Correa AG, Baker CJ. Suppurative thrombophlebitis in children: A ten years' experience. Pediatr Infect Dis 1997;16:63–7.
- Brook I, Frazier EH. Aerobic and anaerobic microbiology of superficial suppurative thrombophlebitis. Arch Surg 1996;131:95–7.
- Murray CK, Backius ML, McAllister K. Fusarium proliferatium superficial suppurative thrombophlebitis. Mil Med 2003;168:426–7.
- Tanaka K. Pathology and pathogenesis of Buerger's disease. Int J Cardiol 1998;66 Suppl. 1:S237–42.
- Sánchez Yus E, Simón RS, Requena L. Vein, artery, or arteriole? A decisive question in hypodermal pathology. Am J Dermatopathol 2012;34:229–32.
- Hall LD, Dalton LS, Fillman LE, Dohse L, Elston DM. Re-examination of features to distinguish polyarteritis nodosa from superficial thrombophlebitis. Am J Dermatopathol 2013;35:463–71.
- Hara K, Tsuzuki T, Takagi N, Shimokata K. Nodular granulomatous phlebitis of the skin: A fourth type of tuberculid. Histopathology 1997;30:129–134.
- Motswaledi HM, Schulz EJ. Superficial thrombophlebitic tuberculide. Int J Dermatol 2006;45:1337–40.
Cutaneous polyarteritis nodosa
- Kawakami T, Ishizu A, Arimura Y, Soma Y. Serum anti-lysosomal-associated membrane protein-2 antibody levels in cutaneous polyarteritis nodosa. Acta Derm Venereol 2013;93:70–3.
- Kawakami T, Soma Y. Correlation of livedo racemosa, cutaneous inflammatory plaques, and antiphospholipid antibodies in patients with cutaneous polyarteritis nodosa. Medicine (Baltimore) 2011;90:119–24.
- Díaz Perez JL, Schroeter AL, Winkelmann RK. Cutaneous periarteritis nodosa. Immunofluorescence studies. Arch Dermatol 1980;116:56–8.
- Díaz Pérez JL, Martínez de Lagrán Z, Díaz-Ramón JL, Winkelmann RK. Cutaneous polyarteritis nodosa. Semin Cutan Med Surg 2007;26:77–86.
- Kossard S, Lee JS, McGrath MA. Macular lymphocytic arteritis. J Cutan Pathol 2010;37:1114–15.
Necrobiosis lipoidica
- Peyri J, Moreno A, Marcoval J. Necrobiosis lipoidica. Semin Cutan Med Surg 2007;26:87–9.
- Ackerman AB. Histologic Diagnosis of Inflammatory Skin Diseases. Philadelphia: Lea & Febiger, 1978:424.
- Alegre VA, Winkelmann RK. A new histopathologic feature of necrobiosis lipoidica diabeticorum: lymphoid nodules. J Cutan Pathol 1988;15:75–7.
- Magro CM, Crowson AN, Regauer S. Granuloma annulare and necrobiosis lipoidica tissue reactions as a manifestation of systemic disease. Hum Pathol 1996;27:50–6.
- Díaz Cascajo C, Borghi S. Subcutaneous pseudomembranous fat necrosis. New observations. J Cutan Pathol 2002;29:5–10.
- Ullman S, Dahl MV. Necrobiosis lipoidica: An immunofluorescence study. Arch Dermatol 1997;113:1671–3.
- O'Toole EA, Kennedy U, Nolan JJ, et al. Necrobiosis lipoidica: Only a minority of patients have diabetes mellitus. Br J Dermatol 1999;140:283–6.
- Holland CH, Givens V, Smoller BR. Expression of the human erythrocyte glucose transporter glut-1 in areas of sclerotic collagen in necrobiosis lipoidica. J Cutan Pathol 2001;28:287–92.
Deep morphoea
- Peterson LS, Nelson Am, Su WPD. Classification of morphea (localized scleroderma). Mayo Clin Proc 1995;70:1068–76.
- Torres VM, George WM. Diffuse eosinophilic fasciitis. A new syndrome or a variant of scleroderma. Arch Dermatol 1977;113:1591–3.
- Jarrat M, Bybee JD, Ramsdell W. Eosinophilic fasciitis: an early variant of scleroderma. J Am Acad Dermatol 1979;1:221–6.
- Cramer SF, Kent L, Abramowsky C, Moskowitz RW. Eosinophilic fasciitis. Arch Pathol Lab Med 1982;106:85–91.
- Lee P. Eosinophilic fasciitis – new associations and current perspectives (editorial). J Rheumatol 1981;8:6–8.
- Michet CJ Jr, Doyle JA, Ginsburg WW. Eosinophilic fasciitis. Report of 15 cases. Mayo Clin Proc 1981;56:27–34.
- Barnes L, Rodnan GP, Medsger TA Jr, Short D. Eosinophilic fasciitis. A pathologic study of twenty cases. Am J Pathol 1979;96:493–518.
- Tamura T, Saito Y, Ishikawa H. Diffuse fasciitis with eosinophilia. Histological and electron microscopic study. Acta Derm Venereol 1979;59:325–31.
- Botet MW, Sanchez JL. The fascia in systemic scleroderma. J Am Acad Dermatol 1980;3:36–42.
- Lakhanpal S, Ginsburg WW, Michet CJ, et al. Eosinophilic fasciitis: Clinical spectrum and therapeutic response of 52 cases. Sem Arthritis Rheum 1988;17:221–31.
- Diaz Perez JL, Connolly SM, Winkelmann RK. Disabling pansclerotic morphea of children. Arch Dermatol 1980;116:169–73.
- Maragh SV, Davis MD, Bruce AJ, et al. Disabling pansclerotic morphea: Clinical presentation inn two adults. J Am Acad Dermatol 2005;53:115–19.
- Parodi PC, Riberti C, Draganic Stinco D, et al. Squamous cell carcinoma arising in a patient with long-standing pansclerotic morphea. Br J Dermatol 2001;144:417–19.
- Wollina U, Buslau M, Weyers W. Squamous cell carcinoma in pansclerotic morphea in childhood. Pediatr Dermatol 2002;19:151–4.
- Feldman SR, Silver RM, Maize JC. A histopathologic comparison of Shulman's syndrome (diffuse fasciitis with eosinophilia) and the fasciitis associated with the eosinophilia–myalgia syndrome. J Am Acad Dermatol 1992;26:95–100.
- Chosidow O, Bagot M, Vernant P, et al. Sclerodermatous chronic graft-versus-host disease. Analysis of seven cases. J Am Acad Dermatol 1992;26:49–55.
Subcutaneous granuloma annulare
- Requena L, Fernández-Figueras MT. Subcutaneous granuloma annulare. Semin Cutan Med Surg 2007;26:96–9.
- Grogg KL, Nascimento AG. Subcutaneous granuloma annulare in childhood: clinicopathologic features in 34 cases. Pediatrics 2001;107:E42.
- Salomon RJ, Gardepe SF, Woodley DT. Deep granuloma annulare in adults. Int J Dermatol 1986;25:109–12.
- Rubin M, Lynch FW. Subcutaneous granuloma annulare. Comment on familial granuloma annulare. Arch Dermatol 1966;93:416–20.
- Dahl MV, Ullman S, Goltz RW. Vasculitis in granuloma annulare: histopathology and direct immunofluorescence. Arch Dermatol 1977;113:463–7.
- Bergman R, Pam Z, Lichtig C, et al. Localized granuloma annulare. Histopathological and direct immunofluorescence study or early lesions, and the adjacent normal-looking skin of actively spreading lesions. Am J Dermatopathol 1993;15:544–8.
- Modlin RL, Vaccaro SA, Gottlieb B, et al. Granuloma annulare. Identification of cells in the cutaneous infiltrate by immunoperoxidase techniques. Arch Pathol Lab Med 1984;108:379–82.
- Fayyazi A, Schweyer S, Eichmeyer B, et al. Expression of IFN-gamma, coexpression of TNH-alpha and matrix metalloproteinases and apoptosis of T lymphocytes and macrophages in granuloma annulare. Arch Dermatol Res 2000;292:384–90.
- Kallioinen M, Sandberg M, Kinnunen T, Oikarinen A. Collagen synthesis in granuloma annulare. J Invest Dermatol 1992;98:463–8.
- Hanna WM, Moreno-Merlo F, Andrighetti L. Granuloma annulare: an elastic tissue disease? Case report and literature review. Ultrastructural Pathol 1999;23:33–8.
- Umbert P, Winkelmann RK. Histologic, ultrastructural, and histochemical studies of granuloma annulare. Arch Dermatol 1977;113:1681–6.
- Misago N, Narisawa Y. Subcutaneous granuloma annulare with overlying localized granuloma annulare. J Dermatol 2010;37:755–7.
- Draheim JH, Johnson LC, Helwig EB. A clinico-pathologic analysis of “rheumatoid” nodules occurring in 54 children. Am J Pathol 1959;35:678.
- Dabski K, Winkelmann RK. Destructive granuloma annulare of the skin and underlying soft tissues – report of two cases. Clin Exp Dermatol 1991;16:218–21.
- Patterson JW. Rheumatoid nodule and subcutaneous granuloma annulare: A comparative histologic study. Am J Dermatopathol 1988;10:1–8.
- Weedon D. Granuloma annulare. In: Weedon D, Skin Pathology. Edinburgh: Churchill-Livingstone, 1997:167–70.
- Stefanaki K, Tsivitanidou-Kakourou T, Stefanaki C, et al. Histological and immunohistochemical study of granuloma annulare and subcutaneous granuloma annulare in children. J Cutan Pathol 2007;34:392–6.
- Evans MJ, Blessing K, Gray ES. Pseudorheumatoid nodule (deep granuloma annulare) of childhood: clinicopathologic features of twenty patients. Pediatr Dermatol 1994;11:6–9.
- López Rios F, Rodríguez Peralto JL, Castaño E, Gil R. Epithelioid sarcoma masquerading as perforating granuloma annulare. Histopathology 1997;31:102–3.
- Miettinen M, Farnburg-Smith JV, Virolainen M, Shmookler BM, Fetsch JF. Epithelioid sarcoma: an immunohistochemical analysis of 112 classical and variant cases and a discussion of the differential diagnosis. Hum Pathol 1999;30:934–42.
- Hornick JL, Dal Cin P, Fletcher CD. Loss of INI1 expression is characteristic of both conventional and proximal-type epithelioid sarcoma. Am J Surg Pathol 2009;33:542–50.
Rheumatoid nodule
- Ahmed SS, Arnett FC, Smith CA, et al. The HLA-DRB1*0401 allele and the development of methotrexate-induced accelerated rheumatoid nodulosis: a follow-up study of 79 Caucasian patients with rheumatoid arthritis. Medicine 2001;80:271–8.
- MacGregor A, Ollier W, Thomson W, et al. HLA-DRB1*0401/0404 genotype and rheumatoid arthritis: increased association in men, young age of onset, and disease activity. J Rheumatol 1995;22:1032–6.
- Weyand CM, Xie C, Goronzy JJ. Homozygosity for the HLA-DRB1 allele selects for extraarticular manifestations in rheumatoid arthritis. J Clin Invest 1992;89:2033–9.
- Panayi GS, Wooley P, Batchelor JR. Genetic basis of rheumatoid disease: HLA antigens, disease manifestations, and toxic reaction to drugs. BMJ 1978;2:1326–8.
- Chan WF, Atkins CJ, Naysmith D, van der Westhuizen N, Woo J, Nelson JL. Microchimerism in the rheumatoid nodules of patients with rheumatoid arthritis. Arthritis Rheum 2012;64:380–8.
- Hessian PA, Highton J, Kean A, Sun CK, Chin M. Cytokine profile of the rheumatoid nodule suggests that it is a Th1 granuloma. Arthritis Rheum 2003;48:334–8.
- Kato H, Yamakawa M, Ogino T. Complement mediated vascular endothelial injury in rheumatoid nodules: a histopathological and immunohistochemical study. J Rheumatol 2000;27:1839–47.
- Rasker JJ, Kuipers FC. Are rheumatoid nodules caused by vasculitis? A study of 13 early cases. Ann Rheum Dis 1983;42:384–8.
- Highton J, Hessian PA, Kean A, Chin M. Cell death by apoptosis is a feature of the rheumatoid nodule. Ann Rheum Dis 2003;62:77–80.
- Miyasaka N, Sato K, Yamamoto K, et al. Immunological and immunohistochemical analysis of rheumatoid nodules. Ann Rheum Dis 1989;48:220–6.
- Segal R, Caspi D, Tishler M, et al. Accelerated nodulosis and vasculitis during methotrexate therapy for rheumatoid arthritis. Arthritis Rheum 1988;31:1182–5.
- Salaffi F, Carotti M, Sartini A, et al. A prospective study of the long-term efficacy and toxicity of low-dose methotrexate in rheumatoid arthritis. Clin Exp Rheumatol 1995;13:23–8.
- Filosa G, Salaffi F, Bugatti L. Accelerated nodulosis during methotrexate therapy for refractory rheumatoid arthritis. A case report. Adv Exp Med Biol 1999;455:521–4.
- Kerstens PJ, Boerbooms AM, Jeurissen ME, et al. Accelerated nodulosis during low dose methotrexate therapy for rheumatoid arthritis. An analysis of ten cases. J Rheumatol 1992;19:867–1.
- Williams FM, Cohen PR, Arnett FC. Accelerated cutaneous nodulosis during methotrexate therapy in a patient with rheumatoid arthritis. J Am Acad Dermatol 1998;39:359–62.
- Combe B, Guttierrez M, Anaya JM, et al. Possible efficacy of hydroxychloroquine on accelerated nodulosis during methotrexate therapy for rheumatoid arthritis. J Rheumatol 1999;20:755–6.
- Dash S, Seibold JR, Tiku ML. Successful treatment of methotrexate induced nodulosis with D-penicillamine. J Rheumatol 1999;26:1396–9.
- Abraham Z, Rozenbaum M, Rosner I. Colchicine therapy for low-dose-methotrexate-induced accelerated nodulosis in rheumatoid arthritis patient. J Dermatol 1999;26:691–4.
- Chatham WW. Methotrexate associated rheumatoid nodulosis: improvement with addition of sulfasalazine. Arthritis Rheum 1992;35(Suppl.):S148.
- Braun MG, van Rhee R, Becker-Capeller D. Neuauftreten und/oder Zunahme von Rheumaknoten unter Leflunomidtherapie bei RA-Patienten. Z Rheumatol 2004;63:84–7.
- Langevitz P, Maguire L, Urowitz M. Accelerated nodulosis during azathioprine therapy. Arthritis Rheum 1991;34:123–4.
- Cunnane G, Warnock M, Fye KH, et al. Accelerated nodulosis and vasculitis following etanercept therapy for rheumatoid arthritis. Arthritis Rheum 2002;47:445–9.
- Kekow J, Welte T, Kellner U, Pap T. Development of rheumatoid nodules during anti-tumor necrosis factor alpha therapy with etanercept. Arthritis Rheum 2002;46:843–4.
- Mackley CL, Ostrov BE, Ioffreda MD. Accelerated cutaneous nodulosis during infliximab therapy in a patient with rheumatoid arthritis. J Clin Rheumatol 2004;10:336–8.
- Rozin A, Yigla M, Guralnik L, et al. Rheumatoid lung nodulosis and osteopathy associated with leflunomide therapy. Clin Rheumatol 2006;25:384–8.
- Berris B, Houpt JB, Tenenbaum J. Accelerated nodulosis in a patient with psoriasis and arthritis during treatment with methotrexate. J Rheumatol 1995;22:2359–60.
- Rivero MG, Salvatore AJ, Gomez-Puerta JA, et al. Accelerated nodulosis during methotrexate therapy in a patient with systemic lupus erythematosus and Jaccoud's arthropathy. Rheumatology 2004;43:1587–8.
- Hahn BH, Yardley JH, Stevens MB. “Rheumatoid” nodules in systemic lupus erythematosus. Ann Intern Med 1970;72:49–58.
- DuBois EL, Friou GJ, Chandor S. Rheumatoid nodules and rheumatoid granulomas in systemic lupus erythematosus. JAMA 1972;220:515–18.
- Schofield JK, Cerio R, Grice K. Systemic lupus erythematosus presenting with “rheumatoid nodules”. Clin Exp Dermatol 1992;17:53–5.
- Muzaffer MA, Schneider R, Cameron BJ, et al. Accelerated nodulosis during methotrexate therapy for juvenile rheumatoid arthritis. J Pediatr 1996;128:698–700.
- Falcini F, Taccetti G, Ermini M, et al. Methotrexate-associated appearance and rapid progression of rheumatoid nodules in systemic-onset juvenile rheumatoid arthritis. Arthritis Rheum 1997;40:175–8.
- Lynch JM, Barrett TL. Collagenolytic (necrobiotic) granulomas: part 1 – the “blue” granulomas. J Cutan Pathol 2004;31:353–61.
- Lynch JM, Barrett TL. Collagenolytic (necrobiotic) granulomas: part II – the ‘red’ granulomas. J Cutan Pathol 2004;31:409–18.
- Sokoloff L, McCluskey RT, Bunim JJ. Vascularity of the early subcutaneous nodules of RA. Arch Pathol 1953;55:475–95.
- Patterson JW, Demos PT. Superficial ulcerating rheumatoid necrobiosis: a perforating rheumatoid nodule. Cutis 1985;36:323–8.
- Requena L, Sanchez-Yus E. Panniculitis. Part I. Mostly septal panniculitis. J Am Acad Dermatol 2001;45:163–83.
- Hewitt D, Cole J. Rheumatoid nodules without arthritis. Austr J Dermatol 2005;46:93–6.
Necrobiotic xanthogranuloma
- Bullock JD, Bartley GB, Campbell, et al. Necrobiotic xanthogranuloma with paraproteinaemia. Case report and a pathogenic theory. Ophthalmology 1986;93:1233–6.
- Matsuura F, Yamashita S, Hirano K, et al. Activation of monocytes in vivo causes intracellular accumulation of lipoprotein-derived lipids and marked hypocholesterolemia – a possible pathogenesis of necrobiotic xanthogranuloma. Atherosclerosis 1999;142:355–65.
- Langlois S, Brochot P, Reguiai Z, et al. Necrobiotic xanthogranuloma with multiple myeloma. Case report and pathogenic hypotheses. Joint Bone Spine 2006;73:120–2.
- Robertson DM, Winkelmann RK. Ophthalmologic features of necrobiotic xanthogranuloma with paraproteinaemia. Am J Ophthalmol 1984;97:173–83.
- Finan MC, Winkelmann RK. Necrobiotic xanthogranuloma with paraproteinaemia. A review of 22 cases. Medicine 1986;65:376–88.
- Ugurlu S, Bartley GB, Gibson LE. Necrobiotic xanthogranuloma: Long-term outcome of ocular and systemic involvement. Am J Ophthalmol 2000;129:651–7.
- Miguelez Hernández A, Vanaclocha F, Rodríguez Peralto JL, et al. Xantogranuloma necrobiótico: a propósito de un caso. Actas Dermosifiliogr 2000;91:511–15.
- Zelger B, Eisendle K, Mensing C, Zelger B. Detection of spirochetal micro-organisms by focus-floating microscopy in necrobiotic xanthogranuloma. J Am Acad Dermatol 2007;57:1026–30.
- Martínez Fernández M, Rodríguez Prieto MA, Ruiz González I, et al. Necrobiotic xanthogranuloma associated with myeloma. JEADV 2004;18:328–31.
- Kossard S, Winkelmann RK. Necrobiotic xanthogranuloma with paraproteinaemia. J Am Acad Dermatol 1980;3:257–70.
- Mehregan DA, Winkelmann RK. Necrobiotic xanthogranuloma. Arch Dermatol 1992;128:94–100.
- Valentine EA, Friedman HD, Zamkoff KW, et al. Necrobiotic xanthogranuloma with IgA multiple myeloma: a case report and literature review. Am J Hematol 1990;35:283–5.
- Johnston KA, Grimwood RE, Meffert JJ, et al. Necrobiotic xanthogranuloma with paraproteinemia: an evolving presentation. Cutis 1997;59:333–6.
- Goodman WT, Barrett TL. Histiocytosis. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. Barcelona: Elsevier, 2004:1429–79.
- Ferrara G, Palombi N, Lipizzi A, et al. Nonnecrobiotic necrobiotic xanthogranuloma. Am J Dermatopathol 2007;29:306–8.
- Wood AJ, Wagner VU, Abbott JJ, Gibson LE. Necrobiotic xanthogranuloma. A review of 17 cases with emphasis on clinical and pathologic correlation. Arch Dermatol 2009;145:279–84.
- Fortson JS, Schroeter AL. Necrobiotic xanthogranuloma with IgA paraproteinemia and extracutaneous involvement. Am J Dermatopathol 1990;12:579–84.
- Randell PL, Heenan PJ. Necrobiotic xanthogranuloma with paraproteinemia. Australas J Dermatol 1999;40:114–15.
- Dupré A, Viraben DA. Necrobiotic xanthogranuloma: a case without paraproteinemia but with transepithelial elimination. J Cutan Pathol 1988;15:116–19.
- Criado PR, Vasconcellos C, Pegas JR, et al. Necrobiotic xanthogranuloma with lambda paraproteinemia: case report of successful treatment with melphalan and prednisone. J Dermatol Treat 2002;13:87–9.
- Williford PM, White WL, Jorizzo JL, Greer K. The spectrum of normolipemic plane xanthoma. Am J Dermatopathol 1993;15:572–5.
Erythema nodosum
- White JM Jr. Erythema nodosum. Dermatol Clin 1985;3:119–27.
- Kramer N, Rickert RR, Brodkin RH, Rosenstein ED. Septal panniculitis as a manifestation of Lyme disease. Am J Med 1986;81:149–52.
- Jimenez Nacher JJ, Navarro Ibanez V, Nieto Garcia A, Salavert Lleti M, Ferrer Tuset C, Febrer I. Rickettsia conorii: una nueva causa de eritema nodoso. An Med Intern 1991;8:241–2.
- Perez Arellano JL, Martinez Martinez LM, Fernandez Lopez E, Luna Rodrigo G. Eritema nudoso y brucelosis. Med Clin (Barc) 1988;90:81.
- Ellis ME, Pope J, Mokashi A, Dunbar E. Campylobacter colitis associated with erythema nodosum. BMJ 1982;285:937.
- Sundaresh KV, Madjar DD, Camisa C, Carvallo E. Cat-scratch disease associated with erythema nodosum. Cutis 1986;38:317–19.
- Palmer JR. Psittacosis in man – recent developments in the UK: a review. Proc R Soc Med 1982;75:262–7.
- García-Porrúa C, González-Gay MA, Vázquez-Caruncho M, et al. Erythema nodosum. Etiologic and predictive factors in defined population. Arthritis Rheum 2000;43:584–92.
- Hannuksela M. Erythema nodosum. Clin Dermatol 1986;4:88–95.
- Vaccaro M, Guarneri F, Guarneri C, Borgia F, Cannavo SP. Sweet's syndrome and erythema nodosum after Klebsiella pneumoniae cystitis. Acta Derm Venereol 2003;83:290–1.
- Van Brakel WH, Khawas JB, Lucas SB. Reactions I leprosy: an epidemiologic study of 386 patients in west Nepal. Lepr Rev 1994;65:190–203.
- Derham RJL, Owens GG, Wooldridge MAW. Leptospirosis as a cause of erythema nodosum. BMJ 1976;2:403–4.
- Kousa M, Saikku P, Kanerva L. Erythema nodosum in chlamydial infections. Acta Derm Venereol 1980;60:319–22.
- Whitton T, Smith AG. Erythema nodosum secondary to meningococcal septicaemia. Clin Exp Dermatol 1999;24:97–8.
- Periyakoil V, Krasner C. Moraxella catarrhalis bacteremia as a cause of erythema nodosum. Clin Infect Dis 1996;23:650–1.
- Teyssandier R, Guidet B, Pinta B, Offenstadt G. Pneumopathie á mycoplasma pneumoniae avec anémie grave et érythème noueux. Presse Med 1985;14:1613.
- Wilkinson DS, Turner TW, Mair NS. Erythema nodosum due to Pasteurella pseudotuberculosis. BMJ 1969;2:226–7.
- Williamson DM, Cunliffe WJ, Gatecliff M, Scott DG. Acute ulcerative acne (acne fulminans) with erythema nodosum. Clin Exp Dermatol 1977;2:351–4.
- Watanakunakorn C. Multiple painful indurated erythematous nodular skin lesions associated with Pseudomonas aeruginosa septicemia. Clin Infect Dis 1998;27:662–3.
- Conget L, Mallolas J, Mensa J, Rovira M. Erythema nodosum and Q fever. Arch Dermatol 1987;123:867.
- Premaratna R, Chandrasena TG, Rajapakse RP, et al. Rickettsioses presenting as major joint arthritis and erythema nodosum: description of four patients. Clin Rheumatol 2009;28:867–8.
- Scott BB. Salmonella gastroenteritis – another cause of erythema nodosum. Br J Dermatol 1980;102:339–40.
- Tami LF. Erythema nodosum associated with Shigella colitis. Arch Dermatol 1985;121:590.
- Favour CB, Sosman MC. Erythema nodosum. Arch Intern Med 1947;80:435–53.
- Alinovi A, Lui P, Benoldi D. Syphilis – still a cause of erythema nodosum. Int J Dermatol 1983;22:310–11.
- Simila S, Pietilla J. The changing etiology of erythema nodosum in children. Acta Tuberc Scand 1965;46:159–68.
- Peter R, Banyai T. Erythema nodosum revealing oculoglandular tularemia. Dermatology 2001;202:79–80.
- Debois J, Vandepitte J, Degreef H.Yersinia enterocolitica as a cause of erythema nodosum. Dermatologica 1978;156:65–78.
- Spear JB, Kessler HA, Dworin A, Semel J. Erythema nodosum associated with acute cytomegalovirus mononucleosis in an adult. Arch Intern Med 1988;148:323–4.
- Yokoyama S, Kasahara M, Fukuda A, Sato S, Koda F, Nakagawa A. Epstein–Barr virus-associated erythema nodosum after living-donor liver transplantation: a case report. Liver Transpl 2009;15:446–8.
- Maggiore G, Grifeo S, Marzani MD. Erythema nodosum and hepatitis B virus (HBV) infection. J Am Acad Dermatol 1983;9:602–3.
- Domingo P, Ris J, Martinez E, Casas F. Erythema nodosum and hepatitis C. Lancet 1990;336:1377.
- Fegueux S, Maslo C, de Truchis P, Matheron S, Coulaud JP. Erythema nodosum in HIV-infected patients. J Am Acad Dermatol 1991;25:113.
- Bodansky HI. Erythema nodosum and infectious mononucleosis. BMJ 1979;2:1263.
- Anderson PC. Erythema nodosum. In: Demis JE, ed. Clinical Dermatology, vol. 2. Philadelphia: JB Lippincott, 1990:7–13.
- Kuokkanan K, Launis J, Mortinnen A. Erythema nodosum and erythema multiforme associated with milker's nodules. Acta Derm Venereol 1976;56:69–72.
- Imbert B, Brion JP, Janbon B, Gonzales M, Micoud M. Erytheme noueux associe a une infection par le parvovirus B19. Presse Med 1989;18:1753–4.
- Tay YK. Erythema nodosum in Singapore. Clin Exp Dermatol 2000;25:377–80.
- Miranda M, Fonseca E, Maza P. Eritema nodoso. Estudio de 133 casos. An Med Intern 1985;2:433–8.
- Miller DD, Davies SF, Sarosi GA. Erythema nodosum and blastomycosis. Arch Intern Med 1982;142:1839.
- Dickson EC. Erythema nodosum. JAMA 1937;109:36.
- Martínez Roig A, Llorens Teral J, Torres JM. Erythema nodosum and kerion on the scalp. Am J Dis Child 1982;13:440–2.
- Ozols II, Wheat LJ. Erythema nodosum in an epidemic of histoplasmosis in Indianapolis. Arch Dermatol 1981;117:709–12.
- Gutierrez Galhardo MC, de Oliveira Schubach A, deLima Barros MB, et al. Erythema nodosum associated with sporotrichosis. Int J Dermatol 2002;41:114–16.
- Harries AD, Taylor J. Erythema nodosum associated with invasive amoebiasis and giardiasis. Br J Dermatol 1986;114:394.
- De Paz Arranz S, Pérez Pimiento A, Santaolalla Montoya M, Trampal Gonzalez A, Rodriguez Mosquera M. Eritema nudoso asociado a infección por Ascaris lumbricoides. Actas Dermosifiliogr 1999;90:384–5.
- Lammintausta K, Kotilainen P, Hohenthal U, Talve L. A patient with mucocutaneous eruption and intestinal giardiasis. Acta Derm Venereol 2001;81:310–11.
- Cabeza F, Simal E, Mur M, Villalba MP. Eritema nudoso como primera manifestación de hidatidosis. Rev Clin Esp 1991;188:267–8.
- Sheskin J. Erupción tipo eritema nodoso por larva de Sparganum. Actas Dermosifiliogr 1977;68:269–72.
- Longmore HJA. Toxoplasmosis and erythema nodosum. BMJ 1977;1:490.
- Rockl H. Erythema nodosum bei Trichomoniasis. Hautarzt 1975;26:57.
- Hartleb M, Januszewski K. Severe hepatic involvement in visceral larva migrans. Eur J Gastroenterol Hepatol 2001;13:1245–9.
- Litt JZ. Drug Eruption Reference Manual 2000. New York: The Parthenon Publishing Group, 2000:628.
- Hakimian D, Tallman MS, Zugerman C, Caro WA. Erythema nodosum associated with all-trans-retinoic acid in the treatment of acute promyelocytic leukemia. Leukemia 1993;7:758–9.
- Eng AM, Aronson IK. Dermatopathology of panniculitis. Semin Dermatol 1984;3:1–13.
- Kaushik P, Soule MR, Ellison WA, Ahmed B, Kaushik R. Cabergoline-associated erythema nodosum. Ann Pharmacother 2008;42:284–7.
- Dalmau J, Roé E, Puig L, Alomar A. Granulomatous septal panniculitis associated with capecitabine (Xeloda). J Eur Acad Dermatol Venereol 2008;22:121–2.
- Marazuela M, Sanchez de Paco G, Jimenez I, et al. Acute pancreatitis, hepatic cholestasis, and erythema nodosum induced by carbimazole treatment for Graves' disease. Endocr J 2002;49:315–18.
- Biedermann L, Kerl K, Rogler G, Hofbauer GF. Drug-induced erythema nodosum after the administration of certolizumab in Crohn's disease. Inflamm Bowel Dis 2013;19:E4–6.
- Grauer JL, Fonteille J, Zaski JP, Gintz B, Phelip X, Cabanel G. Erythéme noueux et hépatite cholestatique au cors d'un traitment par D pénicillamine. Presse Med 1983;12:1997.
- Soon SL, Crawford RI. Recurrent erythema nodosum associated with Echinacea herbal therapy. J Am Acad Dermatol 2001;44:298–9.
- Rajakulendran S, Deighton C. Adverse dermatological reactions in rheumatoid arthritis patients treated with etanercept, an anti-TNF alpha drug. Curr Drug Saf 2006;1:259–64.
- Thouvenot E, Hillarie-Buys D, Bos-Thompson MA, et al. Erythema nodosum and glatiramer acetate treatment in relapsing-remitting multiple sclerosis. Mult Scler 2007;13:941–4.
- Stone RL, Claflin A, Penneys NS. Erythema nodosum following gold sodium thiomalate therapy. Arch Dermatol 1973;107:602–4.
- Nomiyama J, Shinohara K, Inoue H. Erythema nodosum caused by the administration of granulocyte colony-stimulating factor in a patient with refractory anemia. Am J Hematol 1994;47:333.
- Di Giusto CA, Bernhard JD. Erythema nodosum provoked by hepatitis B vaccine. Lancet 1986;2:1042.
- Longueville C, Doffoel–Hantz V, Hantz S, et al. Erytheme noueux induit par le Gardasil®. Rev Med Interne 2012;33:e17–18.
- Schienfeld N. Imatinib mesylate and dermatology. Part 2: a review of the cutaneous side effects of imatinib mesylate. J Drug Dermatol 2006;5:228–31.
- Rosen T, Martinelli P. Erythema nodosum associated with infliximab therapy. Dermatol Online J 2008;14:3.
- Weinstein A, Bujak D, Mittelman A, Davidian M. Erythema nodosum in a patient with renal cell carcinoma treated with interleukin 2 and lymphokine-activated killer cells. JAMA 1987;258:3120–1.
- Kellett JK, Beck MH, Chalmers RJE. Erythema nodosum and circulating immunocomplexes in acne fulminans after treatment with isotretinoin. BMJ 1985;290:820.
- Dellaripa PF, Wechsler ME, Roth ME, Drazen J. Recurrent panniculitis in a man with asthma receiving treatment with leukotriene-modifying agents. Mayo Clin Proc 2000;75:643–5.
- Rodríguez-Carreón AA, Vega-Memije E, Moreno-Coutiño G, López-García L, Domínguez-Cherit J, Arenas R. Simultaneous erythema nodosum and erythema multiforme after local lidocaine injection. Ann Pharmacother 2008;42:127–30.
- Bridges AJ, Graziano FM, Calhoun W, Reizner GT. Hyperpigmentation, neutrophilic alveolitis, and erythema nodosum resulting from minocycline. J Am Acad Dermatol 1990;22:959–62.
- Ricci RM, Deering KC. Erythema nodosum caused by omeprazole. Cutis 1996;57:434.
- Salvatore MA, Lynch PJ. Erythema nodosum, estrogens, and pregnancy. Arch Dermatol 1980;116:557–8.
- Keren G, Lehr V, Boichis H. Erythema nodosum related to propylthiouracil treatment for thyrotoxicosis. Isr J Med Sci 1985;21:62–3.
- Kaliyadan F, Dharmaratnam AM. Erythema nodosum – an association with rabies vaccination. Dermatol Online J 2008;14:22.
- Kraswoska D, Szymanek M, Schawartz RA, Myslinski W. Cutaneous effects of the most commonly used antidepressant medication, the selective serotonin reuptake inhibitors. J Am Acad Dermatol 2007;56:848–53.
- Beurey J, Jeandidier P, Bermont A. Les complications dermatologiques des traitments antidiabetiques. Ann Dermatol Syphiligr 1966;93:13–42.
- Viraben R, Dupre A. Erythema nodosum following thalidomide therapy for Behçet disease. Dermatologica 1988;176:107.
- Bartram R, Kastrup J, Andersen C. Erythema nodosum ved trimetoprimbehandling. Ugeskr Laeger 1983;145:1070.
- Thomson BJ, Nuki G. Erythema nodosum following typhoid vaccination. Scott Med J 1985;30:173.
- Lillo A, Gil MJ, Jimenez R, Monferrer R. Eritema nudoso y adenocarcinoma de colon. Med Clin (Barc) 1997;108:318.
- Lin JT, Chen PM, Huang DF, Kwang WK, Lo K, Wang WS. Erythema nodosum associated with carcinoid tumour. Clin Exp Dermatol 2004;29:426–7.
- Altomare GF, Capella GL. Paraneoplastic erythema nodosum in a patient with carcinoma of the uterine cervix. Br J Dermatol 1995;132:667–8.
- Glinkov S, Krasnaliev I, Atanassova M, Arnaudov P, Kirov K, Glinkova V. Hepatocellular carcinoma associated with paraneoplastic erythema nodosum and polyarthritis. J Hepatol 2003;39:656–7.
- Reynolds NJ, Kennedy CTC. Erythema nodosum and cutaneous vasculitis associated with recurrence of Hodgkin's disease. Br J Dermatol 1990;123(Suppl.):101–2.
- Sullivan R, Clowers-Webb H, Davis MD. Erythema nodosum: a presenting sign of acute myelogenous leukemia. Cutis 2005;76:114–16.
- Perez NB, Bernad B, Narvaez J, Valverde J. Erythema nodosum and lung cancer. Joint Bone Spine 2006;73:336–7.
- Then C, Langer A, Adam C, Oduncu FS. Erythema nodosum associated with myelodysplastic syndrome: a case report. Onkologie 2011;34:126–8.
- Parodi A, Costari R, Rebora A. Erythema nodosum as the presenting symptom of gastric centro-follicular lymphoma. Int J Dermatol 1989;28:336–7.
- Durden FM, Variyam E, Chren MM. Fat necrosis with features of erythema nodosum in a patient with metastatic pancreatic carcinoma. Int J Dermatol 1996;35:39–41.
- Hamzaoui A, Gassab E, Kochteli I, et al. Erythema nodosum revealing parathyroid carcinoma. Eur Ann Otorhinolaryngol Head Neck Dis 2011;128:272–4.
- Cox NH, Jorizzo JL, Bourke JF, Savage COS. Vasculitis, neutrophilic dermatosis and related disorder. In: Burns T, Breathnach S, Cox N, Griffiths C, eds. Rook's Textbook of Dermatology, 8th edn. Oxford: Wiley-Blackwell, 2010:50.1–50.95.
- Reizis Z, Trattner A, Hodak E, David M, Sandbank M. Acne fulminans with hepatosplenomegaly and erythema nodosum migrans. J Am Acad Dermatol 1991;24:886–8.
- Inoue T, Katoh N, Kishimoto S. Erythema nodosum induced by the synergism of acupuncture therapy and flu-like infection. J Dermatol 2005;32:493–6.
- Torinuki W, Funyu T. Adult Still's disease manifesting as erythema nodosum. J Dermatol 1996;23:216–17.
- Gillott TJ, Struthers GR. Cutaneous necrotizing vasculitis, erythema nodosum and ankylosing spondylitis. Rheumatology (Oxford) 1999;38:377–8.
- Nekhlyudov L, Gradzka M, Conti-Kelly AM, Greco TP. Erythema nodosum associated with antiphospholipid antibodies: a report of three cases. Lupus 2000;9:641–5.
- Behçet R. Immunological studies on aphthous ulcer and erythema nodosum-like eruptions in Behçet disease. Br J Dermatol 1985;113:303–12.
- Ujiie H, Sawamura D, Yokota K, Tateishi Y, Inokuma D, Shimizu H. Intractable erythema nodosum associated with severe breast abscesses: reports of two cases. Clin Exp Dermatol 2005;30:584–5.
- Cervia M, Parodi A, Rebora A. Chronic active hepatitis and erythema nodosum. Arch Dermatol 1982;118:878.
- Durand JM, Lefevre P, Weiller C. Erythema nodosum and coeliac disease. Br J Dermatol 1991;125:291–2.
- Ruiz-Rodriguez R, Winkelmann RK. Erythema nodosum and diverticulitis. Arch Dermatol 1990;126:1242–3.
- McCallum DI, Kinmont PDC. Dermatological manifestations of Crohn's disease. Br J Dermatol 1968;80:1–8.
- Ogden S, Denyer ME, Wilkinson SM. Erythema nodosum and eosinophilic oesophagitis: more than a chance association? Br J Dermatol 2007;156:1388–9.
- Adams DH, Hubscher SG, Scott DGI. Granulomatous mastitis – a rare cause of erythema nodosum. Postgrad Med J 1987;63:581–2.
- Glassey F, Saurat JH. Erythema nodosum and Berger's disease. Dermatologica 1988;177:327–8.
- Dux S, Grosskopf I, Rosenfeld JB. Recurrent erythema nodosum arthritis and IgA nephropathy. Dermatologica 1988;176:293–5.
- Katugampola RP, Patel GK, Farrell AM. Intestinal bypass syndrome presenting as erythema nodosum. Clin Exp Dermatol 2004;29:261–4.
- Auerbach PS, Hays JT. Erythema nodosum following a jellyfish sting. J Emerg Med 1987;5:487–91.
- Dabski K, Winkelmann RK. Histopathology of erythema nodosum in patients with coexisting lupus erythematosus. J Am Acad Dermatol 1988;19:131–2.
- Bombardieri S, Dimunno O, Dipunzio C, Pasero G. Erythema nodosum associated with pregnancy and oral contraceptives. BMJ 1977;1:1509–10.
- Gatselis NK, Zachou K, Dalekos GN. Early primary biliary cirrhosis: a new association with erythema nodosum of unknown origin. Gastroenterol Res Pract 2010;2010.
- Fearfield LA, Bunker CB. Radiotherapy and erythema nodosum. Br J Dermatol 2000;142:189.
- Ramos JM, Blazquez RM, Climent A, Pena MA. Meningitis aséptica, eritema nudoso y eritema anular centrífugo como primera manifestación de una policondritis recidivante. Med Clin (Barc) 2000;114:196–7.
- McMillan A. Reiter's disease in a female presenting as erythema nodosum. Br J Vener Dis 1975;51:345–7.
- Jorizzo JL, Daniels JC. Dermatologic conditions in patients with rheumatoid arthritis. J Am Acad Dermatol 1983;8:439–57.
- James DG, Neville E, Diltzbach LE. A worldwide review of sarcoidosis. Ann N Y Acad Sci 1976;278:321–34.
- Gouet D, Anquez M, Risse JF, Becq-Giraudon B. Association d'une maladie de Vogt–Koyanagi d'un syndrome de Goygerot–Sjögren et d'un érythéme noueux. Presse Med 1984;13:624.
- Srivastava S, Haddad R, Kleinman G, Manthous CA. Erythema nodosum after smoke inhalation-induced bronchiolitis obliterans organizing pneumonia. Crit Care Med 1999;27:1214–16.
- Blaustein A, Moreno A, Noguera J, de Moragas JM. Septal granulomatous panniculitis in Sweet's syndrome. Arch Dermatol 1985;121:785–8.
- Picco P, Gattorno M, Vignola S, et al. Clinical and biological characteristics of immunopathological disease-related erythema nodosum in children. Scand J Rheumatol 1999;28:27–32.
- Acha Arrieta V, Fuertes Perez J, Gonzalez de Zarate P, Aguirre Errasti C. Eritema nudoso y arteritis de células gigantes. Med Clin (Barc) 1987;88:171–2.
- Sams WM, Winkelmann RK. The association of erythema nodosum with ulcerative colitis. South Med J 1968;61:676–9.
- Barksdale SK, Hallahan CW, Kerr GS, Fauci AS, Stern JB, Travis WD. Cutaneous pathology in Wegener's granulomatosis. A clinicopathologic study of 75 biopsies in 46 patients. Am J Surg Pathol 1995;19:161–72.
- Vesey CMR, Wilkinson DS. Erythema nodosum. Br J Dermatol 1959;71:139–55.
- Hens M, Ruiz Moral R, Pérez Jiménez F. Eritema nudoso: ventajas de un protocolo para su estudio. Med Clin (Barc) 1987;89:638–40.
- McGibbon DH. Subcutaneous fat. In: Burns T, Breathnach S, Cox N, Griffiths C, eds. Rook's Textbook of Dermatology, 8th ed. Oxford: Wiley-Blackwell, 2010:46.1–46.49.
- Bobamahmoodi F, Babamahmoodi A, Barani H, Delavarian L. Simultaneous occurrence of erythema nodosum in monozygotic twin sisters. Case Rep Med 2012;2012:109247.
- James DG. Dermatological aspects of sarcoidosis. Q J Med 1959;28:109–24.
- Söderstrom RM, Krull EA. Erythema nodosum. A review. Cutis 1978;21:806–10.
- Gordon H. Erythema nodosum: a review of one hundred and fifteen cases. Br J Dermatol 1961;73:393–409.
- Cohen PR, Holder WR, Rapini R. Concurrent Sweet's syndrome and erythema nodosum: a report, world literature review, and mechanism of pathogenesis. J Rheumatol 1992;19:814–20.
- Wilkinson SM, Heagerty AHM, English JSC. Acute febrile neutrophilic dermatosis in association with erythema nodosum and sarcoidosis. Clin Exp Dermatol 1993;18:47–9.
- Ben-Noun L. Sweet's syndrome associated with erythema nodosum. Aust Fam Physician 1995;24:1867–9.
- Suzuki Y, Kuroda K, Kojima T, Fujita M, Iseki T, Shinkai H. Unusual cutaneous manifestations of myelodysplastic syndrome. Br J Dermatol 1995;133:483–6.
- Waltz KM, Long D, Marks JG, Billingsley EM. Sweet's syndrome and erythema nodosum. The simultaneous occurrence of 2 reactive dermatoses. Arch Dermatol 1999;135:62–6.
- Ginarte M, Toribio J. Association of Sweet syndrome and erythema nodosum. Arch Dermatol 2000;136:673–4.
- Mizoguchi M, Chikakare K, Goh K, Asahina Y, Masuda K. Acute febrile neutrophilic dermatosis (Sweet's syndrome) in Behçet disease. Br J Dermatol 1987;116:727–34.
- Zamora E, Martín L, de Castro A, Barat A. Síndrome de Sweet: estudio de diez casos y revisión de la literatura. Rev Clin Esp 1990;186:264–9.
- Sitjas D, Puig L, Cuatrecasas M, Moragas JM. Acute febril neutrophilic dermatosis (Sweet's syndrome). Int J Dermatol 1993;32:261–8.
- von den Driesch P. Sweet's syndrome (acute febrile neutrophilic dermatosis). J Am Acad Dermatol 1994;31:535–56.
- Ginarte M, García-Doval I, Toribio J. Síndrome de Sweet: estudio de 16 casos. Med Clin (Barc) 1997;109:588–91.
- Wasson S, Govindarajan G, Folzenlogen D. Concurrent occurrence of Sweet's syndrome and erythema nodosum: an overlap in the spectrum of reactive dermatoses. Clin Rheumatol 2006;25:268–72.
- Cribier B, Caille A, Heid E, Grosshans E. Erythema nodosum and associated diseases. A study of 129 cases. Int J Dermatol 1998;37:667–72.
- Psychos DN, Voulgari PV, Skopouli FN, Drosos AA, Moutsopoulos HM. Erythema nodosum: the underlying conditions. Clin Rheumatol 2000;19:212–16.
- Förstrom L, Winkelmann RK. Acute panniculitis: a clinical and histological study of 34 cases. Arch Dermatol 1977;183:909–17.
- More Monreal J, Rodríguez de la Serna A. Eritema nudoso: revisión de 68 casos. Rev Clin Esp 1983;171:405–8.
- Fine RM, Meltzer HD. Chronic erythema nodosum. Arch Dermatol 1969;100:33–8.
- Löfgren S. Primary pulmonary sarcoidosis. Acta Med Scand 1953;145:424–31.
- Löfgren S. Erythema nodosum studies on etiology and pathogenesis in 185 adult cases. Acta Med Scand 1946;174(Suppl.):1–197.
- Marie I, Lecomte F, Levesque H, et al. Lofgren's syndrome as the first manifestation of acute infection due to Chlamydia pneumoniae: a prospective study. Clin Infect Dis 1999;28:691–2.
- Hedfors E, Norberg R. Evidence for circulating immune complexes in sarcoidosis. Clin Exp Dermatol 1974;16:493–6.
- Baldock NE, Catterall MD. Erythema nodosum from Yersinia enterocolitica. Br J Dermatol 1975;93:719–20.
- Jones JV, Cumming RH, Asplin CM. Evidence for circulating immune complexes in erythema nodosum and early sarcoidosis. Ann NY Acad Sci 1976;278:212–19.
- Winkelmann RK, Fostrom L. New observations in the histopahology of erythema nodosum. J Invest Dermatol 1975;65:441–6.
- Niemi KM, Forstrom L, Hannuksela M. Mustakallio KK, Salo OP. Nodules on the legs. Acta Derm Venereol 1977;57:145–54.
- Nunnery E, Persellin RH, Pope RM. Lack of circulating immune complexes in uncomplicated erythema nodosum. J Rheumatol 1983;10:991–4.
- Kunz M, Beutel S, Brocker E. Leucocyte activation in erythema nodosum. Clin Exp Dermatol 1999;24:396–401.
- Labunski S, Posern G, Ludwig S, Kundt G, Brocker EB, Kunz M. Tumor necrosis factor alpha promoter polymorphism in erythema nodosum. Acta Derm Venereol 2001;81:18–21.
- Amoli MM, Donn RP, Thomson W, et al. Macrophage migration inhibitory factor gene polymorphism is associated with sarcoidosis in biopsy proven erythema nodosum. J Rheumatol 2002;29:1671–3.
- Llorente L, Richaud-Patin Y, Alvarado C, Reyes E, Alcocer-Varela J, Orozco-Topete R. Elevated Th1 cytokine mRNA in skin biopsies and peripheral circulation in patients with erythema nodosum. Eur Cytokine Netw 1997;8:67–71.
- Schäffler A, Müller-Ladner U, Schölmerich J, Büchler C. Role of adipose tissue as an inflammatory organ in human diseases. Endocr Rev 2006;27:449–67.
- Braverman IM. Protective effects of erythema nodosum in coccidioidomycosis. Lancet 1999;353:168.
- Grunewald J, Eklund A. Löfgren's syndrome: human leukocyte antigen strongly influences the disease course. Am J Respir Crit Care Med 2009;179:307–12.
- Labbe L, Perel Y, Maleville J, Taieb A. Erythema nodosum in children: a study of 27 patients. Pediatr Dermatol 1996;13:447–50.
- Hassink RI, Pasquinelli-Egli CE, Jacomella V, Laux-End R, Bianchetti MG. Conditions currently associated with erythema nodosum in Swiss children. Eur J Pediatr 1997;156:851–3.
- Kakourou T, Drosatou P, Psychou F, Aroni K, Nicolaidou P. Erythema nodosum in children. J Am Acad Dermatol 2001;44:17–21.
- Bafverstedt B. Erythema nodosum migrans. Acta Derm Venereol 1954;34:181–93.
- Hannuksela M. Erythema nodosum migrans. Acta Derm Venereol 1973;3(Suppl. 7):1–64.
- Rostas A, Lowe S, Smout MS. Erythema nodosum migrans in a young man. Arch Dermatol 1980;116:325–30.
- De Almeida Prestes C, Winkelmann RK, Su WPD. Septal granulomatous panniculitis: Comparison of the pathology of erythema nodosum migrans (migratory panniculitis) and chronic erythema nodosum. J Am Acad Dermatol 1990;22:477–83.
- Vilanova X, Piñol Aguade J. Hypodermyte nodulaire subaigue migratice. Ann Dermatol Syphiligr 1956;83:369–404.
- Perry HO, Winkelmann RK. Subacute nodular migratory panniculitis. Arch Dermatol 1964;89:170–9.
- White WL, Wieselthier JS, Hitchcock MG. Panniculitis: recent developments and observations. Semin Cutan Med Surg 1996;15:278–99.
- Hern AE, Shwayder TA. Unilateral plantar erythema nodosum. J Am Acad Dermatol 1992;26:259–60.
- Suarez SM, Paller AS. Plantar erythema nodosum: cases in two children. Arch Dermatol 1993;129:1064–5.
- Ohtake N, Kawamura T, Akiyama C, Furue M, Tamaki K. Unilateral plantar erythema nodosum. J Am Acad Dermatol 1994;30:654–5.
- Joshi A, Sah SP, Agrawal S, Agarwalla A, Jacob M. Palmar erythema nodosum. J Dermatol 2000;27:420–1.
- Chun SI, Su WPD, Lee S, III Rogers RS. Erythema nodosum-like lesions in Behçet's syndrome: a histopathologic study of 30 cases. J Cutan Pathol 1989;16:259–65.
- Kim B, LeBoit PE. Histopathologic features of erythema nodosum-like lesions in Behçet disease: a comparison with erythema nodosum focusing on the role of vasculitis. Am J Dermatopathol 2000;22;379–90.
- Winkelmann RK, Frigas E. Eosinophilic panniculitis: A clinicopathologic study. J Cutan Pathol 1986;13:1–12.
- Miescher G. Zur Histologie des Erythema nodosum. Acta Derm Venereol 1947;27:447–68.
- Miescher G. Zur Frage der Radiärknötchen beim Erythema nodosum. Arch Dermatol Syphl 1951;193:251–6.
- Sanchez Yus E, Sanz Vico MD, de Diego V. Miescher's radial granuloma. A characteristic marker of erythema nodosum. Am J Dermatopathol 1989;11:434–42.
- LeBoit PE. From Sweet to Miescher and back again. Am J Dermatopathol 2006;28:381–3.
- Requena L, Kutzner H, Palmedo G, et al. Histiocytoid Sweet syndrome: a dermal infiltration of immature neutrophilic granulocytes. Arch Dermatol 2005;141:834–42.
- White WL, Hitchcock MG. Diagnosis: Erythema nodosum or not? Semin Cutan Med Surg 1999;18:47–55.
- Thurber S, Kohler S. Histopathologic spectrum of erythema nodosum. J Cutan Pathol 2006;33:18–26.
- Haustein UF, Klug H. Ultrastrukturelle Untersuchungen der Blutgefässe beim Erythema nodosum. Dermatol Monatsschr 1977;163:13–22.
- Honma T, Bang D, Lee S, Saito T. Ultrastructure of endothelial cell necrosis in classical erythema nodosum. Hum Pathol 1993;24:384–90.
- Snow JL, Su WPD. Lipomembranous (membranocystic) fat necrosis: Clinicopathologic correlation of 38 cases. Am J Dermatopathol 1996;18:151–5.
- Ahn SK, Lee BJ, Lee SH, Lee WS. Nodular cystic fat necrosis in a patient with erythema nodosum. Clin Exp Dermatol 1995;20:263–5.
- Tanaka M, Inoue K, Yamasaki Y, Hatano H. Erythema nodosum complicated by retrobulbar optic nerve neuritis. Clin Exp Dermatol 2001;26:306–7.
- Calista D, Landi G. Lichen planus, erythema nodosum, and erythema multiforme in a patient with chronic hepatitis C. Cutis 2001;67:454–6.
- Ubogy Z, Persellin RM. Suppression of erythema nodosum by indomethacin. Acta Derm Venereol 1982;62:265–7.
- Lehman CW. Control of erythema nodosum with naproxen. Cutis 1980;26:66–7.
- Schulz EJ, Whiting DA. Treatment of erythema nodosum and nodular vasculitis with potassium iodide. Br J Dermatol 1976;94:75–8.
- Miyachi Y, Niwa Y. Effects of potassium iodide, colchicine and dapsone on the generation of the polymorphonuclear leukocyte-derived oxygen intermediates. Br J Dermatol 1982;107:209–14.
- Horio T, Imamura S, Danno K, Ofuji S. Potassium iodide in the treatment of erythema nodosum and nodular vasculitis. Arch Dermatol 1981;117:29–31.
- Bondi EE, Lazarus GS. Panniculitis. In: Fitzpatrick TB, Eisen AZ, Wolff K, Freedberg IM, Austen KF, eds. Dermatology in General Medicine, 3rd edn. New York: McGraw-Hill, 1987:1131–51.
- Honma K, Saga K, Onodera H, Takahashi M. Potassium iodide inhibits neutrophil chemotaxis. Acta Derm Venereol 1990;70:247–9.
- Johnson TM, Rapini RP. The Wolff–Chaikoff effect: Hypothyroidism due to potassium iodide. Arch Dermatol 1988;124:1184–5.
- Wallace SL. Erythema nodosum treatment with colchicine. JAMA 1967;202:1056.
- De Coninck P, Baclet JL, Di Bernardo C, Buschges B, Plouvier B. Traitment de l'erytheme noeux par la colchicine (Letter). Presse Med 1984;13:680.
- Jarret P, Goodfield MJD. Hydroxychloroquine and chronic erythema nodosum (Letter). Br J Dermatol 1996;134:373.
- Boyd AS. Etanercept treatment of erythema nodosum. Skinmed 2007;6:197–9.
- Ortego-Centeno N, Callejas-Rubio JL, Sanchez-Cano D, Caballero-Morales T. Refractory chronic erythema nodosum successfully treated with adalimumab. J Eur Acad Dermatol Venereol 2007;21:408–10.
- Hanauer SB. How do I treat erythema nodosum, apthous ulcerations and pyoderma gangrenosum? Inflamm Bowel Dis 1998;4:70.
Erythema nodosum leprosum
- Wemambu SNC, Turck JL, Waters MFR, et al. E.N.L. A clinical manifestation of the arthus phenomenon. Lancet 1969;2:933–5.
- Vieira LM, Sampaio EP, Nery JA, et al. Immunological status of ENL (erythema nodosum leprosum) patients: its relationship to bacterial load and levels of circulating IL-2R. Rev Inst Med Trop Sao Paulo 1996;38:103–11.
- Murphy GF, Sánchez NP, Flynn TC, et al. Erythema nodosum leprosum: Nature and extent of the cutaneous microvascular alterations. J Am Acad Dermatol 1986;14:59–69.
- Mathur NK, Mangal HN, Mathur D, et al. Langerhans cell and leprosy. Lepr India 1983;55:22–8.
- Cuevas-Santos J, Contreras F, McNutt NS. Multibacillary leprosy: lesions with macrophages positive for S100 protein and dendritic cells positive for Factor 13a. J Cutan Pathol 1998;25:530–7.
- Bjune G, Barnetson RS. Plasma factors in delayed type hypersensitivity augmentation of lymphocyte responses in borderline leprosy reactions. Clin Exp Immunol 1976;26:397–402.
- Vazquez-Botet M, Sánchez JL. Erythema nodosum leprosum. Int J Dermatol 1987;26:436–7.
- Rea TH, Ridley DS. Lucio's phenomenon: A comparative histological study. Int J Lepr 1979;47:161–6.
- Ridley MJ, Ridley DS. The immunopathology of erythema nodosum leprosum: the role of extravascular complexes. Lepr Rev 1983;54:95–107.
- Latapí F, Zamora AC. The “spotted” leprosy of Lucio. Int J Lepr 1948;16:421–9.
- Quismorio FP, Rea T, Chandors, Levan N, Friu G. Lucio's phenomenon: an immune complex deposition syndrome in lepromatous leprosy. Clin Immunol Immunopathol 1978;9:184–93.
- Magaña M. Eritema necrosante o fenómeno de Lucio. ¿Vasculitis? ¿De qué tipo, de qué vasos, qué mecanismo? Actas Dermatol Dermatopatol 2006;6:5–7.
Erythema induratum of Bazin
- Bazin E. Leçons Théoriques et Cliniques sur la Scrofule, 2nd edn. Paris: Delahaye, 1861.
- Audry CH. Étude de la lésion de l'érythème induré (de Bazin). Sur la notion du lymphatisme. Ann Dermatol Syph 1898;9:209–14.
- Whitfield A. On the nature of the disease known as erythema induratum scrofulosorum. Am J Med Sci 1901;122:828–34.
- Whitfield A. A further contribution to our knowledge of erythema induratum. Br J Dermatol 1905;17:241–7.
- Galloway J. Case of erythema induratum giving no evidence of tuberculosis. Br J Dermatol 1913;25:217–25.
- Telford ED. Lesions of the skin and subcutaneous tissue in diseases of the peripheral circulation. Arch Dermatol Syph 1937;36:952–63.
- Montgomery H, O'Leary PA, Barker NW. Nodular vascular diseases of the legs. Erythema induratum and allied condition. JAMA 1945;128:335–41.
- Vilanova X, Piñol J, Rubio J, et al. Vascularite nodulaire et érythème induré de Bazin. Étude statistique et diagnostique. Bull Soc Fr Derm Syph 1958;65:418–33.
- Vilanova X, Gimenez-Camarasa JM. Tubercúlides papulo-nodulares. Actas Dermosifiliogr 1965;56:306–10.
- Cho KH, Lee DY, Kim CW. Erythema induratum of Bazin. Int J Dermatol 1996;35:802–8.
- Cho KH, Kim YG, Yang SG, et al. Inflammatory nodules of the lower legs: a clinical and histological analysis of 134 cases in Korea. J Dermatol 1997;24:522–9.
- Requena L, Sánchez Yus E. Panniculitis. Part II. Mostly lobular panniculitis. J Am Acad Dermatol 2001;45:325–61.
- Cardinali C, Gerlini G, Caproni M, et al. Hepatitis C virus: a common triggering factor for both nodular vasculitis and Sjogren's syndrome? Br J Dermatol 2000;142:187–99.
- Ural I, Erel A, Ozenirler S, et al. Nodular vasculitis associated with chronic hepatitis C. J Eur Acad Dermatol Venereol 2002;16:298–9.
- Fernandes SS, Carvalho J, Leite S, et al. Erythema induratum and chronic hepatitis C infection. J Clin Virol 2009;44:333–6.
- Pozdnyakova O, Garg A, Mahalingam M. Nodular vasculitis – a novel cutaneous manifestation of autoimmune colitis. J Cutan Pathol 2008;35:315–19.
- Tanyel E, Tasdelen Fisgin N, et al. Panniculitis as the initial manifestation of brucellosis: a case report. Am J Dermatopathol 2008;30:169–71.
- Khachemoune A, Longo MI, Phillips TJ. Nodular vasculitis as a paraneoplastic presentation? Int J Dermatol 2003;42:639–42.
- Patterson JW, Brown PC, Broecker AH. Infection induced panniculitis. J Cutan Pathol 1989;16:183–93.
- Wolf D, Ben-Yehuda A, Okon E, et al. Nodular vasculitis associated with propylthiouracil therapy. Cutis 1992;49:253–5.
- Westers-Attema A, van Tubergen A, Plasschaert H, van Marion AM, Frank J, Poblete-Gutiérrez P. Nodular vasculitis in systemic lupus erythematosus. Int J Dermatol 2008;47 Suppl. 1:3–6.
- Sakuma H, Niiyama S, Amoh Y, Katsuoka K. Chlamydophila pneumonia infection induced nodular vasculitis. Case Rep Dermatol 2011;3:263–7.
- Inoue T, Fukumoto T, Ansai S, Kimura T. Erythema induratum of Bazin in an infant after Bacille Calmette–Guerin vaccination. J Dermatol 2006;33:268–72.
- Sughimoto K, Nakano K, Gomi A, et al. Aortic valve stenosis associated with Bazin's disease. J Heart Valve Dis 2007;16:212–13.
- Segura S, Pujol RM, Trindade F, Requena L. Vasculitis in erythema induratum of Bazin: A histopathologic study of 101 biopsy specimens from 86 patients. J Am Acad Dermatol 2008;59:839–51.
- Rademaker M, Lowe DG, Munro DD. Erythema induratum (Bazin's disease). J Am Acad Dermatol 1989;21:740–5.
- Shimizu A, Takahashi A, Negishi I, et al. The close association of lymphadenitis tuberculosa and erythema induratum of Bazin in Japanese patients. Dermatology 2003;207:426–7.
- Roblin D, Kelly R, Wansbrough-Jones M, et al. Papulonecrotic tuberculide and erythema induratum as presenting manifestations of tuberculosis. J Infect 1994;28:193–7.
- Chuang YH, Kuo TT, Wang CM, et al. Simultaneous occurrence of papulonecrotic tuberculide and erythema induratum and the identification of Mycobacterium tuberculosis DNA by polymerase chain reaction. Br J Dermatol 1997;137:276–81.
- Knap J. A re-examination of 63 cases of erythema induratum Bazin. Acta Derm Venereol 1943;24:456–71.
- Feiwel M. Erythema induratum Bazin. Incidence and views on aetiology related to the presence of an immigrant population in London. Acta Derm Venereol 1968;48:242–5.
- Forstrom L, Hannuksela M. Antituberculous treatment of erythema induratum Bazin. Acta Derm Venereol 1971;50:143–7.
- Neyrolles O, Hernández-Pando R, Pietri-Rouxel F, et al. Is adipose tissue a place for Mycobacterium tuberculosis persistence? PLOS One 2006;1:e43.
- Telford ED. Lesions of the skin and subcutaneous tissue in diseases of the peripheral circulation. Arch Dermatol Syph 1937;36:952–63.
- Schneider JW, Jordaan HF, Geiger DH, et al. Erythema induratum of Bazin. A clinicopathological study of 20 cases and detection of MTB DNA in skin lesions by polymerase chain reaction. Am J Dermatopathol 1995;17:350–6.
- Seckin D, Hizel N, Demirhan B, et al. The diagnostic value of polymerase chain reaction in erythema induratum of Bazin. Br J Dermatol 1997;137:1011–12.
- Degitz K, Messer G, Schirren H, et al. Successful treatment of erythema induratum of Bazin following rapid detection of mycobacterial DNA by polymerase chain reaction. Arch Dermatol 1993;129:1619–20.
- Yen A, Fearneyhough P, Rady P, et al. Erythema induratum of Bazin as a tuberculid: confirmation of MTB DNA polymerase chain reaction analysis. J Am Acad Dermatol 1997;36:99–101.
- Baselga E, Margall N, Barnadas MA, et al. Detection of MTB DNA in lobular granulomatous panniculitis (erythema induratum–nodular vasculitis). Arch Dermatol 1997;133:457–62.
- Vieites B, Suárez-Peñaranda, JM, Pérez del Molino ML, et al. Recovery of M. tuberculosis DNA in biopsies of erythema induratum – results in a series of patients using an improved polymerase chain reaction technique. Br J Dermatol 2005;152:1394–6.
- Bayer-Garner IB, Cox MD, Scott MA, et al. Mycobacteria other than M. tuberculosis are not present in erythema induratum/nodular vasculitis: a case series and literature review of the clinical and histologic findings. J Cutan Pathol 2005;32:220–6.
- Ollert MW, Thomas P, Korting HC, et al. Erythema induratum of Bazin. Evidence of T-lymphocyte hyperresponsiveness to purified protein derivative of tuberculin: report of two cases and treatment. Arch Dermatol 1993;129:469–73.
- Smolle J, Kneifel H. S-100-protein-positive dendritic cells in nodular vasculitis. Dermatologica 1986;172:139–43.
- Cribier B. Etude immunohistochimique de la vasculite nodulaire. Role possible d'une hypersensibilite retardee cellulaire. Ann Dermatol Venereol 1992;119:958–63.
- Requena L, Sánchez Yus E. Panniculitis. Part II. Mostly lobular panniculitis. J Am Acad Dermatol 2001;45:325–61.
- Schneider JW, Jordaan HF. The histopathologic spectrum of erythema induratum of Bazin. Am J Dermatopathol 1997;19:323–33.
- Forstrom L, Winkelmann RK. Granulomatous panniculitis in erythema nodosum. Arch Dermatol 1975;111:335–40.
- Barham Kl, Jorizzo JL, Grattan B, Cox NH. Nodular vasculitis. In: Burns T, Breathnach S, Cox N, Griffiths C, eds. Rook's Textbook of Dermatology, 7th edn. Oxford: Blackwell Publishing, 2004:49.18–49.19.
- Ackerman AB. Histologic Diagnosis of Inflammatory Skin Diseases. Philadelphia: Lea & Febiger, 1978:779–825.
- Sánchez Yus E, Simón P. Sanz Vico D. ¿Vena o arteria? Una cuestión decisiva en patología hipodérmica. Piel 1987;2:213–17.
- Degos R. Dermatologie. Paris: Flammarion, 1953:578.
- Black M. Panniculitis. J Cutan Pathol 1985;12:366–80.
- McKee PH, Calonje E, Granter SR. Nodular vasculitis. In: McKee PH, Calonje E, Granter SR, Pathology of the Skin with Clinical Correlations, 3rd edn. London: Elsevier-Mosby, 2005:366–9.
- Patterson JW. Panniculitis. In: Dermatology. Bolognia JL, Jorizzo J, Rapini RP, Callen JP, et al., eds. Barcelona, Mosby Elsevier, 2008:1515–35.
- Mehregan A, Hashimoto K, Mehregan D, Mehregan D. Nodular vasculitis. In: Pinkus' Guide to Dermatohistopathology, 6th edn. Norwalk, CT: Appleton & Lange, 1995:246–7.
- McNutt NS, Moreno A, Contreras F. Erythema induratum (nodular vasculitis). In Elder DE, Elenitas R, Johnson BL, Murphy GE, eds. Lever's Histopathology of the Skin, 9th edn. Philadelphia: Lippincott-Williams & Wilkins, 2005:529–30.
- Weedon D. Erythema induratum–Nodular vasculitis. In: Weedon D, Skin Pathology, 2nd edn. London: Churchill Livingstone, 2002:524–5.
Sclerosing panniculitis
- Stacey MC, Burnand KG, Bhogal BS, Black MM. Pericapillary fibrin deposits and skin hypoxia precede the changes of lipodermatosclerosis in limbs at increased risk of developing a venous ulcer. Cardiovasc Surg 2000;8:372–80.
- Fisher DA. Desideratum dermatologicum: eliminating lipodermatosclerosis; the term and the entities. Int J Dermatol 2000;39:490–2.
- Iyengar V, Hsu S, Pielop J. Progressive, painful hardening of the legs. Lipodermatosclerosis. Postgrad Med 2002;111:25–8.
- Jinnin M, Ihn H, Asano Y, Yamane K, Yazawa N, Tamaki K. Sclerosing panniculitis is associated with pulmonary hypertension in systemic sclerosis. Br J Dermatol 2005;153:579–83.
- Herouy Y, Nockowski P, Schöpf E, Norgauer J. Lipodermatosclerosis and the significance of proteolytic remodelling in the pathogenesis of venous ulceration (review). Int J Mol Med 1999;3:511–15.
- Herouy Y, Aizpurua J, Stetter C, et al. The role of the urokinase-type plasminogen activator (uPA) and its receptor (CD87) in lipodermatosclerosis. J Cutan Pathol 2001;28:291–7.
- Greenberg AS, Hasan A, Montalvo BM, Falabella A, Falanga V. Acute lipodermatosclerosis is associated with venous insufficiency. J Am Acad Dermatol 1996;35:566–8.
- Herouy Y, Kreis S, Mueller T, et al. Inhibition of angiogenesis in lipodermatosclerosis: implication for venous ulcer formation. Int J Mol Med 2009;24:645–51.
- Falanga V, Bontempo FA, Eaglstein WH. Protein C and protein S plasma levels in patients with lipodermatosclerosis and venous ulceration. Arch Dermatol 1990;126:1195–7.
- Kirsner RS, Pardes JB, Eaglstein WH, Falanga V. The clinical spectrum of lipodermatosclerosis. J Am Acad Dermatol 1993;28:623–7.
- De Giorgio-Miller AM, Treharne LJ, McAnulty RJ, Coleridge Smith PD, Laurent GJ, Herrick SE. Procollagen type I gene expression and cell proliferation are increased in lipodermatosclerosis. Br J Dermatol 2005;152:242–9.
- Miteva M, Romanelli P, Kirsner RS. Lipodermatosclerosis. Dermatol Ther 2010;23:375–88.
- Huang TM, Lee JY. Lipodermatosclerosis: a clinicopathologic study of 17 cases and differential diagnosis from erythema nodosum. J Cutan Pathol 2009;36:453–60.
- Huang CL, Mutasim DF. Sarcoidosis mimicking lipodermatosclerosis. Cutis 2005;75:322–4.
- Naschitz JE, Bejar J, Mogilner J, et al. Acute lipase-induced panniculitis in rats with ligated veins of the hind limb: a contribution to the role of acute panniculitis as a precursor of lipodermatosclerosis of venous disease. J Dermatol Sci 1999;19:9–16.
- Jorizzo JL, White WL, Zanolli MD, Greer KE, Solomon AR, Jetton RL. Sclerosing panniculitis. A clinicopathologic assessment. Arch Dermatol 1991;127:554–8.
- Diaz-Cascajo C, Borghi S. Subcutaneous pseudomembranous fat necrosis: new observations. J Cutan Pathol 2002;29:5–10.
- Nakamura T, Fujiwara M. Modes of participation of macrophages in the formation of membranocystic lesions. APMIS 2002;110:709–16.
- Pazzaglia UE, Benazzo F, Byers PD, Riboni L, Ceciliani L. Pathogenesis of membranous lipodystrophy. Case report and review of the literature. Clin Orthop Relat Res 1987;225:279–87.
- Dias Gonzalez F, Pedreira Magalhães F, Pontes Vilas Boas Freitas A, Castro Lima Filho H, Santiago MB. Lipodermatosclerosis in patients with diffuse connective tissue diseases. Eur J Intern Med 2006;17:288–9.
- Alegre VA, Winkelmann RK, Aliaga A. Lipomembranous changes in chronic panniculitis. J Am Acad Dermatol 1988;19(1 Pt 1):39–46.
- Segura S, Pujol RM. Lipomembranous fat necrosis of subcutaneous tissue. Dermatol Clin 2008;26:509–17.
- Machinami R. Membranous lipodystrophy-like changes in ischemic necrosis of the legs. Virchows Arch A Pathol Anat Histopathol 1983;399:191–205.
- Machinami R. Incidence of membranous lipodystrophy-like change among patients with limb necrosis caused by chronic arterial obstruction. Arch Pathol Lab Med 1984;108: 823–6.
- Ahn S, Yoo M, Lee S, et al. A clinical and histopathological study of 22 patients with membranous lipodystrophy. Clin Exp Dermatol 1996;21: 268–72.
- Snow JL, Su WPD. Lipomembranous (membranocystic) fat necrosis. Am J Dermatopathol 1996;18:151–5.
- Sueki H, Shinmura Y, Fujisawa R, et al. Ultrastructural study of the histogenesis of membranocystic lesions (Nasu) in diabetics. J Cutan Pathol 1986;13:390–401.
- Chun SI, Cheng KY. Membranous lipodystrophy: Secondary type. J Am Acad Dermatol 1994;31:601–5.
- Machinami R. Degenerative change of adipose tissue; the so-called membranous lipodystrophy. Virchows Arch A Pathol Anat Histopathol 1990;416:373–4.
- Snow JL, Su WPD, Gibson LE. Lipomembranous (membranocystic) changes associated with morphea: A clinicopathologic review of three cases. J Am Acad Dermatol 1994;31:246–50.
- Ishikawa O, Tamura A, Ryuzaki K, et al. Membranocystic changes in the panniculitis of dermatomyositis. Br J Dermatol 1996;134:773–9.
- Felipo F, Vaquero M, del Agua C. Pseudotumoral encapsulated fat necrosis with diffuse pseudomembranous degeneration. J Cutan Pathol 2004;31:565–7.
- Ramdial PK, Madaree A, Singh B. Membranous fat necrosis in lipomas. Am J Surg Pathol 1997;21:841–6.
- Takeda U, Kuroda K, Shinkai H. Encapsulated necrosis associated with Behçet syndrome. J Dermatol 1999;26:522–6.
- Wood C, Rupp M, Hafiz MA. Membranous lipodystrophy: a distinctive change in adipose tissue with many causes. J Cutan Pathol 1986;13:79.
- Ohtake N, Shimada S, Mizoguchi S, et al. Membranocystic lesions in a patient with cytophagic histiocytic panniculitis associated with subcutaneous T-cell lymphoma. Am J Dermatopathol 1998;20:276–80.
- Gouveia C, Soares Almeida LM. Paniculitis lipomembranosa: correlación clínico-patológica de 8 casos. Actas Dermosifiliogr 2006;97:379–84.
- Al-Brahim N, Ceballos K, Sur M, Daya D, Alowami S. Lipomembranous panniculitis: report of a case. Ann Diagn Pathol 2007;11:282–4.
- Biswas A, Byrne JP. Extensive endarteritis obliterans in a case of lupus panniculitis with membranocystic changes and dystrophic calcification: a pathogenic link? Eur J Dermatol 2007;17:550–1.
- Halvorson CR, Kwon SY, Kao GF, Germanas JP. Lipomembranous fat necrosis in a patient with mixed connective tissue disease. J Am Acad Dermatol 2011;64:1110–11.
- Khoury T, Arayssi T, Kibbi AG, Ghosn S. Extensive fat necrosis with lipomembranous changes and calcification in lupus erythematosus panniculitis is not necessarily associated with systemic lupus erythematosus. Am J Dermatopathol 2010;32:742–3.
- Yamamoto T, Furuhata Y, Tsuboi R. Lipomembranous changes and calcification associated with systemic lupus erythematosus. Clin Exp Dermatol 2007;32:278–80.
- Almeida MS, Lima SC, Carvalho LL, et al. Panniculitis – an unusual cutaneous manifestation of systemic sclerosis. J Cutan Pathol 2010;37:1170–3.
- Toritsugi M, Yamamoto T, Nishioka K. Nodular cystic fat necrosis with systemic sclerosis. Eur J Dermatol 2004;14:353–5.
- Pujol RM, Wang CY, Gibson LE, Su WP. Lipomembranous changes in nodular–cystic fat necrosis. J Cutan Pathol 1995;22:551–5.
- Weenig RH, Ng CS, Perniciaro C. Subcutaneous panniculitis-like T-cell lymphoma: an elusive case presenting as lipomembranous panniculitis and a review of 72 cases in the literature. Am J Dermatopathol 2001;23:206–15.
- Lee HJ, Ahn SK, Hong SP, Bak H. Nodular cystic fat necrosis with lipomembranous change observed in splinter granuloma. Int J Dermatol 2014;53:e135–7.
- Akay OM, Urer SM, Oner U, Gulbas Z. Lipomembranous panniculitis in a patient with acute leukemia induced by chemotherapy. Leuk Res 2008;32:669–71.
- Pincemaille B, Besançon C, Balme B, Devaux Y, Thomas L. Lipodystrophie membraneuse secondaire a une chimiotherapie. Ann Dermatol Venereol 1998;125:425–8.
- Nistal M, González-Peramato P, Paniagua R. Lipomembranous fat necrosis in three cases of testicular torsion. Histopathology 2001;38:443–7.
- Ramdial PK, Jogessar V, Dada MA. Membranous fat necrosis due to subcutaneous elemental mercury injections. Am J Forensic Med Pathol 1999;20:369–73.
- Ramdial PK, Bagratee JS. Membranous fat necrosis in mature cystic teratomas of the ovary. Int J Gynecol Pathol 1998;17:120–2.
- Ono T, Kageshita T, Hirai S, Kayashima K, Ishimaru Y. Coexistence of spherulocytic disease (myospherulosis) and membranocystic degeneration. Arch Dermatol 1991;127:88–90.
- Coyne JD, Parkinson D, Baildam AD. Membranous fat necrosis of the breast. Histopathology 1996;28:61–4.
- Bowen AR, Götting C, LeBoit PE, McCalmont TH. Pseudoxanthoma elasticum-like fibers in the inflamed skin of patients without pseudoxanthoma elasticum. J Cutan Pathol 2007;34:777–81.
- Walsh SN, Santa Cruz DJ. Lipodermatosclerosis: a clinicopathological study of 25 cases. J Am Acad Dermatol 2010;62:1005–12.
- Nikko AP, Dunningan M, Cockerell CJ. Calciphylaxis with histologic changes of pseudoxanthoma elasticum. Am J Dermatopathol 1996;18:396–9.
- Fernandez KH, Liu V, Swick BL. Nonuremic calciphylaxis associated with histologic changes of pseudoxanthoma elasticum. Am J Dermatopathol 2013;35:106–8.
- Lewis KG, Lester BW, Pan TD, Robinson-Bostom L. Nephrogenic fibrosing dermopathy and calciphylaxis with pseudoxanthoma elasticum-like changes. J Cutan Pathol 2006;33:695–700.
- Caggiati A, Rosi C, Franceschini M, Innocenzi D. The nature of skin pigmentations in chronic venous insufficiency: a preliminary report. Eur J Vasc Endovasc Surg 2008;35:111–18.
Calcific uraemic arteriolopathy (calciphylaxis)
- Sheila A, Crawford RI. Three-dimensional analysis of a calciphylaxis plaque: clues to pathogenesis. J Am Acad Dermatol 2002;47:53–7.
- Canfield AE, Farrington C, Dziobon MD, et al. The involvement of matrix glycoproteins in vascular calcification and fibrosis: an immunohistochemical study. J Pathol 2002;196:228–34.
- Griethe W, Schmitt R, Jurgensen JS, et al. Bone morphogenic pretein-4 expression in vascular lesions of calciphylaxis. J Nephrol 2003;16:728–32.
- Kramann R, Brandenburg VM, Schurgers LJ, et al. Novel insights into osteogenesis and matrix remodelling associated with calcific uraemic arteriolopathy. Nephrol Dial Transplant 2013;28:856–68.
- Hayashi M, Takamatsu I, Kanno Y, Yoshida T, Abe T, Sato Y; Japanese Calciphylaxis Study Group. A case–control study of calciphylaxis in Japanese end-stage renal disease patients. Nephrol Dial Transplant 2012;27:1580–4.
- Arseculeratne G, Evans AT, Morley SM. Calciphylaxis. A topical overview. J Eur Acad Dermatol Venereol 2006;20:493–502.
- Robinson-Bostom L, Di Giovanna JJ. Cutaneous manifestations of end-stage renal disease. J Am Acad Dermatol 2000;43:975–86.
- Fine A, Zacharias J. Calciphylaxis is usually non-ulcerating: risk factors, outcome and therapy. Kidney Int 2002;61:2210–17.
- Dahl PR, Winkelmann RK, Connoly SM. The vascular-calcification-cutaneous necrosis syndrome. J Am Acad Dermatol 1995;33:53–8.
- Ruggian JC, Maesaka JA, Fishbane S. Proximal calciphylaxis in four insulin-requiring diabetic haemodialysis patients. Am J Kidney Dis 1996;28:409–14.
- James LR, Lajoie G, Prajapati D, et al. Calciphylaxis precipitated by ultraviolet light in a patient with end-stage renal disease secondary to systemic lupus erythematosus. Am J Kidney Dis 1999;34:932–6.
- Elamin EM, McDonald AB. Calcifying panniculitis with renal failure: a new management approach. Dermatology 1996;192:156–9.
- Rudwaleit M, Schwarz A, Trautmann C, et al. Severe calciphylaxis in a renal patient on long-term anticoagulant therapy. Am J Nephrol 1996;16:344–8.
- Requena L, Sánchez-Yus E. Panniculitis. Part II. Mostly lobular panniculitis. J Am Acad Dermatol 2001;45:325–61.
- Dauden E, Ruiz-Genao D, Fraga J. Calcificación vascular cutánea. Correlación clínico-patológica y proposición de una nueva clasificación de las calcinosis cutáneas. Actas Dermosifiliogr 2002;93:22–34.
- Solomon AR, Comite SL, Headington JT. Epidermal and follicular calciphylaxis. J Cutan Pathol 1988;15:282–5.
- Ruiz-Genao D, García-F-Villalta M, Fraga J, et al. Perineurial and vascular calcification in a patient with chronic renal failure. Acta Derm Venereol 2005;85:72–3.
- Lewis KG, Lester BW, Pan TD, et al. Nephrogenic fibrosing dermopathy and calciphylaxis with pseudoxanthoma elasticum changes. J Cutan Pathol 2006;33:695–700.
- Nikko AP, Dunnigan M, Cockerell CJ. Calciphylaxis with changes of pseudoxanthoma elasticum. Am J Dermatopathol 1996;18:396–9.
- Mochel MC, Arakaki RY, Wang G, Kroshinsky D, Hoang MP. Cutaneous calciphylaxis: a retrospective histopathologic evaluation. Am J Dermatopathol 2013;35:582–6.
- Markova A, Lester J, Wang J, et al. Diagnosis of common dermopathies in dialysis patients: a review and update. Semin Dial 2012;25: 408–18.
- Dauden E, Oñate MJ. Calciphylaxis. Dermatol Clin 2008;26:557–68.
- Aherrahrou Z, Schunkert H. Genetics of atherosclerosis and vascular calcification go hand-in-hand. Atherosclerosis 2013;228:325–6.
- Lim SP, Batta K, Tan BB. Calciphylaxis in a patient with alcoholic liver disease in the absence of renal failure. Clin Exp Dermatol 2003;28:34–6.
- Goli AK, Goli SA, Shah LS, et al. Calciphylaxis: a rare association with alcoholic cirrhosis. Are deficiencies in protein C and S the cause? South Med J 2005;98:736–9.
- Ferreres JR, Marcoval J, Bordas X, et al. Calciphylaxis associated with alcoholic cirrhosis. J Eur Acad Dermatol Venereol 2006;20:599–601.
- Barri YM, Graves GS, Knochel JP. Calciphylaxis in a patient with Crohn's disease in the absence of end-stage renal disease. Am J Kidney Dis 1997;29:773–6.
- Mastruserio DN, Nguyen EQ, Neilsen T, et al. Calciphylaxis associated with metastatic breast carcinoma. J Am Acad Dermatol 1999;41:295–8.
- Reigert-Johnson DL, Kaur JS, Pfiefer EA. Calciphylaxis associated with cholangiocarcinoma treated with low-molecular heparin and vitamin K. Mayo Clin Proc 2001;76:749–52.
- Kutlu NO, Aydin NE, Aslan M, et al. Malignant melanoma of the soft parts showing calciphylaxis. Pediatr Hematol Oncol 2003;20:141–6.
- Raper RF, Ibels LS. Osteosclerotic myeloma complicated by diffuse arteritis, vascular calcification and extensive cutaneous necrosis. Nephron 1985;39:389–92.
- Goff HW, Grimwood RE. A case of calciphylaxis and chronic myelomonocytic leukaemia. Cutis 2005;75:325–8.
- Korkmaz C, Dunbar E, Zubaroglu I. Calciphylaxis in a patient with rheumatoid arthritis without renal failure and hyperparathyroidism: the possible role of long-term steroid use and protein S deficiency. Clin Rheumatol 2002;21:66–9.
- Ozbalkan Z, Calguneri M, Onat AM. Development of calciphylaxis after long-term steroid and methotrexate use in a patient with rheumatoid arthritis. Intern Med 2005;44:1178–81.
- Cockerell CJ, Dolan ET. Widespread cutaneous and systemic calcification (calciphylaxis) in patients with the acquired immunodeficiency syndrome and renal disease. J Am Acad Dermatol 1992;26:559–62.
- Wong JJ, Laumann A, Martinez M, et al. Calciphylaxis and antiphospholipid antibody syndrome J Am Acad Dermatol 2000;42:849.
- De Roma I, Filotico R, Cea M, et al. Calciphylaxis in a patient with POEMS syndrome without renal failure and/or hyperparathyroidism. A case report. Ann Ital Med Int 2004;19:283–7.
Cold panniculitis
- Collins HA, Stahlman M, Scott HW. The occurrence of subcutaneous fat necrosis in an infant following induced hypothermia used as an adjuvant in cardiac surgery. Ann Surg 1953;138:880–5.
- Collins HA, Stahlman M, Scott HW. The occurrence of subcutaneous fat necrosis in an infant following induced hypothermia used as an adjuvant in cardiac surgery. Ann Surg 1953;138:880–5.
- Rajkumar SV, Laude TA, Russo RM, et al. Popsicle panniculitis of the cheeks. Clin Pediatr 1976;15:619–21.
- Duncan WC, Freeman RG, Heaton CL. Cold panniculitis. Arch Dermatol 1966;94:722.
- Editorial. Cold panniculitis in children. JAMA 1968;204:924.
- Hochsinger C. Über eine acute kongelative Zellgewebsverhartnung in der Submentalregion bei Kinderm. Monatsschr Kinderth 1902;1:323–5.
- Lemez L. Beitrag zur pathogenese der subcutanen Fettgewebsnekrose Neugeborener (Sog. Sclerodermia neonatorum) and der Hand. Einer Kalterreaktion des subcutanen Fettgewebes bei Neugeborenen und jungen Sauglingen. Zeitung Kinderheilk 1928;46: 323–69.
- Haxthausen H. Adiponecrosis e frigore. Br J Dermatol 1941;53:83–9.
- Adams JE, Foster JH, Faulk WH. Experimental production of fat necrosis by general hypothermia: relation to the chemical composition of fat. Surgical Forum 1954;5:556–63.
- Hirsch J. Fatty acids pattern in human adipose tissue. In: Handbook of Physiology, Section 5: The Adipose Tissue. Baltimore: Williams and Wilkins, 1965:181–90.
- Solomon LM, Beerman HB. Cold panniculitis. Arch Dermatol 1963;88:265–8.
- Rotman H. Cold panniculitis in children: Adiponecrosis e frigore of Haxthausen. Arch Dermatol 1966;94:720–1.
- Sembrot JT. Practical point on popsicles. N Engl J Med 1970;282:1377.
- Smith CS. Chemical changes in the subcutaneous fat in sclerema neonatorum. J Cutan Dis 1918;36:436–40.
- Silverman A, Michels E, Rasmussen J. Subcutaneous fat necrosis in an infant, occurring after hypothermic cardiac surgery. J Am Acad Dermatol 1986;15:331–6.
- Wiadrowski T, Marshman G. Subcutaneous fat necrosis of the newborn following hypothermia and complicated by pain and hypercalcaemia. Australas J Dermatol 2001;42:207–10.
- Ter Poorten JC, Hebert AA, Ilkiw R. Cold panniculitis in a neonate. J Am Acad Dermatol 1995;33:383–5.
- Bolotin D, Duffy KL, Petronic-Rosic V, Rhee CJ, Myers PJ, Stein SL. Cold panniculitis following ice therapy for cardiac arrhythmia. Pediatr Dermatol 2011;28:192–4.
- Beacham B, Cooper P, Buchanan C, et al. Equestrian cold panniculitis in women. Arch Dermatol 1980;116:1025–7.
- Hultcrantz E. Haxthausen's disease. Cold panniculitis in children. J Laryngol Otol 1986;100:1329–32.
- Lowe LB. Cold panniculitis in children. Am J Dis Child 1968;115:709–13.
- Pekki A, Sauni R, Vaalasti P, Huotari-Orava R, Hasan T. Cold panniculitis in Finnish horse riders. Acta Derm Venereol 2011;91:463–4.
- Vickers R, Bleehen SS. Equestrian cold panniculitis in women. Arch Dermatol 1981;117:315–16.
- Grosshans E. Les panniculites de l'adulte. Ann Pathol 1992;12:250–4.
- West SE, McCalmont TH, North JP. Ice-pack dermatosis: a cold-induced dermatitis with similarities to cold panniculitis and perniosis that histopathologically resembles lupus. JAMA Dermatol 2013;149:1314–18.
- Requena L, Sánchez Yus E. Panniculitis. Part II. Mostly lobular panniculitis. J Am Acad Dermatol 2001;45:325–61.
- Patterson JW. Differential diagnosis of panniculitis. Adv Dermatol 1991;6:309–30.
- Yang AY, Schwartz L, Divers AK, Sternberg L, Lee JB. Equestrian chilblain: another outdoor recreational hazard. J Cutan Pathol 2013;40:485–90.
- Ryan T. Panniculitis. Clin Dermatol 1989;7:120–48.
- Labeille B. Panniculites de l'enfant. Ann Pediatr 1992;39:419–25.
- Ter Poorten MC, Thiers BH. Panniculitis. Dermatol Clin 2002;20:421–33.
- Bachmeyer C, Aractingi S, Blanc F, et al. Lupus érythémateux profound chez l'enfant. Ann Dermatol Venereol 1992;119:535–41.
- Muncaster A, Stewart G, Moss C, Southwood T. Facial lupus erythematosus profundus in a 9-year-old boy. J R Soc Med 1998;91:207–8.
- Nousari HC, Kimyai-Asadi A, Santana HM, Diglio GM, Tausk FA, Cohen BA. Generalized lupus panniculitis and antiphospholipid syndrome in a patient without complement deficiency. Pediatr Dermatol 1999;16:273–6.
- Nitta Y. Lupus erythematosus profundus associated with neonatal lupus erythematosus. Br J Dermatol 1997;136:112–14.
- Amblard P, Devani O, Berthod F. Les dermatoses dues au froid. Ann Dermatol Venéreol 1988;115:873–80.
- Wall LM, Smith NP. Perniosis: a histopathological review. Clin Exp Dermatol 1981;6:263–71.
- De Silva BD, McLaren K, Doherty VR. Equestrian perniosis associated with cold agglutinins: a novel finding. Clin Exp Dermatol 2000;25:285–8.
- Aroni K, Aivaliotis M Tsele E, et al. An unusual panniculitis appearing in winter with good response to tetracycline. J Dermatol 1998;25:677–81.
Lupus panniculitis
- Winkelmann RK. Panniculitis and systemic lupus erythematosus. JAMA 1970;211:472–5.
- Commens C, O'Neill P, Walker G. Dermatomyositis associated with multifocal lipoatrophy. J Am Acad Dermatol 1990;22(5 Pt 2):966–9.
- Winkelman WJ, Billick RC, Srolovitz H. Dermatomyositis presenting as panniculitis. J Am Acad Dermatol 1990;23:127–8.
- Fusade T, Belanyi P, Joly P, Thomine E, Mihout MF, Lauret P. Subcutaneous changes in dermatomyositis. Br J Dermatol 1993;128:451–3.
- Sabroe RA, Wallington TB, Kennedy CT. Dermatomyositis treated with high-dose intravenous immunoglobulins and associated with panniculitis. Clin Exp Dermatol 1995;20:164–7.
- Neidenbach PJ, Sahn EE, Helton J. Panniculitis in juvenile dermatomyositis. J Am Acad Dermatol 1995;33(2 Pt 1):305–7.
- Ishikawa O, Tamura A, Ryuzaki K, Kurosawa M, Miyachi Y. Membranocystic changes in the panniculitis of dermatomyositis. Br J Dermatol 1996;134:773–6.
- Molnár K, Kemény L, Korom I, Dobozy A. Panniculitis in dermatomyositis: report of two cases. Br J Dermatol 1998;139:161–3.
- Ghali FE, Reed AM, Groben PA, McCauliffe DP. Panniculitis in juvenile dermatomyositis. Pediatr Dermatol 1999;16:270–2.
- Lee MW, Lim YS, Choi JH, Sung KJ, Moon KC, Koh JK. Panniculitis showing membranocystic changes in the dermatomyositis. J Dermatol 1999;26:608–10.
- Chao YY, Yang LJ. Dermatomyositis presenting as panniculitis. Int J Dermatol 2000;39:141–4.
- Solans R, Cortés J, Selva A, et al. Panniculitis: a cutaneous manifestation of dermatomyositis. J Am Acad Dermatol 2002;46(5 Suppl.):S148–50.
- Nakamori A, Yamaguchi Y, Kurimoto I, et al. Vesiculobullous dermatomyositis with panniculitis without muscle disease. J Am Acad Dermatol 2003;49:1136–9.
- Chong H, Brady K, Metze D, et al. Persistent nodules at injection sites (aluminium granuloma) – clinicopathological study of 14 cases with a diverse range of histological reaction patterns. Histopathology 2006;48:182–8.
- Soares Almeida LM, Requena L, Kutzner H, Angulo J, de Sa J, Pignatelli J. Localized panniculitis secondary to subcutaneous glatiramer acetate injections for the treatment of multiple sclerosis: a clinicopathologic and immunohistochemical study. J Am Acad Dermatol 2006;55:968–74.
- Ball NJ, Cowan BJ, Moore GR, Hashimoto SA. Lobular panniculitis at the site of glatiramer acetate injections for the treatment of relapsing-remitting multiple sclerosis. A report of two cases. J Cutan Pathol 2008;35:407–10.
- Magro CM, Crowson AN, Kovatich AJ, et al. Lupus profundus, indeterminate lymphocytic lobular panniculitis and subcutaneous T-cell lymphoma: a spectrum of subcuticular T-cell lymphoid dyscrasia. J Cutan Pathol 2001;28:235–47.
- Magro CM, Crowson AN, Byrd JC, et al. Atypical lymphocytic lobular panniculitis. J Cutan Pathol 2004;31:300–6.
- Pincus LB, LeBoit PE, McCalmont TH, et al. Subcutaneous panniculitis-like T-cell lymphoma with overlapping clinicopathologic features of lupus erythematosus: coexistence of 2 entities? Am J Dermatopathol 2009;31:520–6.
- Bosisio F, Boi S, Caputo V, et al. Lobular panniculitic infiltrates with overlapping histopathologic features of lupus panniculitis (lupus profundus) and subcutaneous T-cell lymphoma: a conceptual and practical dilemma. Am J Surg Pathol 2015;39:206–11.
- Ma L, Bandarchi B, Glusac EJ. Fatal subcutaneous panniculitis-like T-cell lymphoma with interface change and dermal mucin, a dead ringer for lupus erythematosus. J Cutan Pathol 2005;32:360–5.
- Cassis TB, Fearneyhough PK, Callen JP. Subcutaneous panniculitis-like T-cell lymphoma with vacuolar interface dermatitis resembling lupus erythematosus panniculitis. J Am Acad Dermatol 2004;50:465–9.
- Garcia Gonzalez E, Selvi E, Lorenzini S, et al. Subcutaneous panniculitis-like T-cell lymphoma misdiagnosed as lupus erythematosus panniculitis. Clin Rheumatol 2006;11:1–3.
- Liau JY, Chuang SS, Chu CY, Ku WH, Tsai JH, Shih TF. The presence of clusters of plasmacytoid dendritic cells is a helpful feature for differentiating lupus panniculitis from subcutaneous panniculitis-like T-cell lymphoma. Histopathology 2013;62:1057–66.
- Massone C, Kodama K, Salmohofer W, et al. Lupus erythematosus panniculitis (lupus profundus): Clinical, histopathological, and molecular analysis of nine cases. J Cutan Pathol 2005;32:396–404.
- Requena L, Sanchez Yus E. Panniculitis. Part II. Mostly lobular panniculitis. J Am Acad Dermatol 2001;45:325–61.
- Black MM. Panniculitis. J Cutan Pathol 1985;12:366–80.
- Barnhill RL, Crowson AN. Textbook of Dermatopathology, 2nd edn. New York: McGraw-Hill, 2004:267–98.
- Nousari HC, Kimyai-Asadi A, Santana HM, Diglio GM, Tausk FA, Cohen BA. Generalized lupus panniculitis and antiphospholipid syndrome in a patient without complement deficiency. Pediatr Dermatol 1999;16:273–6.
- Peters MS, Su PD. Eosinophils in lupus panniculitis and morphea profunda. J Cutan Pathol 1991;18:189–92.
- Sanchez NP, Peters MS, Winkelmann RK. The histopathology of lupus erythematosus panniculitis. J Am Acad Dermatol 1981;5:673–80.
- Harris RB, Duncan SC, Ecker RI, et al. Lymphoid follicles in subcutaneous inflammatory disease. Arch Dermatol 1979;115:442–3.
- Ackerman AB, Jacobson M, Vitale P, eds. Clues to Diagnosis in Dermatopathology, vol. 1. Chicago: ASCP Press, 1991:397–400.
- Ackerman AB, Chongchinant N, Sanchez J, et al., eds. Histologic Diagnosis of Inflammatory Skin Diseases, 2nd edn. Baltimore: Williams & Wilkins, 1997:525–46.
- Sharon VR, Konia TH, Barr KL, Fung MA. Assessment of the ‘no eosinophils’ rule: are eosinophils truly absent in pityriasis lichenoides, connective tissue disease, and graft-vs.-host disease? J Cutan Pathol 2012;39:413–18.
- Alegre VA, Winkelmann RK, Aliaga A. Lipomembranous changes in chronic panniculitis. J Am Acad Dermatol 1988;19:39–46.
- Tuffanelli DL. Lupus panniculitis. Semin Dermatol 1985;4:79–81.
- De la Moneda Herrerin C, Conde Zurita JM, Guerra Tapia A, et al. El lupus paniculitis: Una paniculitis mixta. Actas Dermosifiliogr 1987;78:229–38.
- McCalmont TH, Kuo T, Scott GA, et al. Lymphocytic vasculitis in lupus panniculitis: an overlooked mechanism of pathogenic importance? J Cutan Pathol 1995;22:73.
- Kossard S. Defining lymphocytic vasculitis. Australas J Dermatol 2000;41:149–55.
- De Argila D, la Moneda C, Iglesias L. Epidermal changes are not unusual in lupus erythematosus profundus. Int J Dermatol 1996;35:680.
- McNutt NS, Fung MA. More about panniculitis and lymphoma. J Cutan Pathol 2004;31:297–9.
- Morgan KW, Callen JP. Calcifying lupus panniculitis in a patient with subacute cutaneous lupus erythematosus: Response to Diltiazem and Chloroquine. J Rheumatol 2001;28:2129–32.
- Tuffanelli DL. Lupus erythematosus panniculitis (profundus). Clinical and immunologic studies. Arch Derm 1971;103:231–42.
- Riccieri V, Sili Scavalli A, Spadaro E, et al. Lupus erythematosus panniculitis: An immunohistochemical study. Clin Rheumatol 1994;13:641–4.
- Patterson JW. Differential diagnosis of panniculitis. Adv Dermatol 1991;6:309–29.
- Park HS, Choi JW, Kim BK, Cho KH. Lupus erythematosus panniculitis: clinicopathological, immunophenotypic, and molecular studies. Am J Dermatopathol 2010;32:24–30.
- Wenzel J, Proelss J, Wiechert A, Zahn S, Bieber T, Tüting T. CXCR3-mediated recruitment of cytotoxic lymphocytes in lupus erythematosus profundus. J Am Acad Dermatol 2007;56:648–50.
Dermatomyositis-associated panniculitis
- Janis JF, Winkelmann RK. Histopathology of the skin in dermatomyositis. Arch Dermatol 1968;97:640–50.
- Winkelmann RK. Panniculitis in connective tissue disease. Arch Dermatol 1983;119:336–44.
- Raimer SS, Soloman AR, Daniels JC. Polymyositis presenting with panniculitis. J Am Acad Dermatol 1985;13:366–9.
- Commens C, O'Neill P, Walker G. Dermatomyositis associated with multifocal lipoatrophy. J Am Acad Dermatol 1990;22(5 Pt 2):966–9.
- Winkelman WJ, Billick RC, Srolovitz H. Dermatomyositis presenting as panniculitis. J Am Acad Dermatol 1990;23:127–8.
- Fusade T, Belanyi P, Joly P, Thomine E, Mihout MF, Lauret P. Subcutaneous changes in dermatomyositis. Br J Dermatol 1993;128:451–3.
- Sabroe RA, Wallington TB, Kennedy CT. Dermatomyositis treated with high-dose intravenous immunoglobulins and associated with panniculitis. Clin Exp Dermatol 1995;20:164–7.
- Neidenbach PJ, Sahn EE, Helton J. Panniculitis in juvenile dermatomyositis. J Am Acad Dermatol 1995;33(2 Pt 1):305–7.
- Ishikawa O, Tamura A, Ryuzaki K, Kurosawa M, Miyachi Y. Membranocystic changes in the panniculitis of dermatomyositis. Br J Dermatol 1996;134:773–6.
- Molnár K, Kemény L, Korom I, Dobozy A. Panniculitis in dermatomyositis: report of two cases. Br J Dermatol 1998;139:161–3.
- Ghali FE, Reed AM, Groben PA, McCauliffe DP. Panniculitis in juvenile dermatomyositis. Pediatr Dermatol 1999;16:270–2.
- Lee MW, Lim YS, Choi JH, Sung KJ, Moon KC, Koh JK. Panniculitis showing membranocystic changes in the dermatomyositis. J Dermatol 1999;26:608–10.
- Chao YY, Yang LJ. Dermatomyositis presenting as panniculitis. Int J Dermatol 2000;39:141–4.
- Solans R, Cortés J, Selva A, et al. Panniculitis: a cutaneous manifestation of dermatomyositis. J Am Acad Dermatol 2002;46(5 Suppl.):S148–50.
- Nakamori A, Yamaguchi Y, Kurimoto I, et al.Vesiculobullous dermatomyositis with panniculitis without muscle disease. J Am Acad Dermatol 2003;49:1136–9.
Pancreatic panniculitis
- Chiari H. Über die Sogenannte Fettnekrose. Prag Med Wochenschr 1883;8:255–6.
- Szymanski FJ, Bluefarb SM. Nodular fat necrosis and pancreatic diseases. Arch Dermatol 1961;83:224–9.
- Dahl PR, Su WP, Cullimore KC, Dicken CH. Pancreatic panniculitis. J Am Acad Dermatol 1995;33:413–17.
- Segurado Rodríguez A, Guerra Tapia A, Jaen Olasolo P, Cuevas Santos J. Paniculitis pancreática: estudio de 12 casos y valoración comparativa de sus caracteres epidemiológicos, clínicos, histopatológicos y terapéuticos. Actas Dermosifiliogr 1999;90:227–34.
- Ortiz Romero PL, Dorado Bris JM, Gil R, Lopez S, Iglesias Diez L. Estudio de 7 casos de necrosis grasa pancreática. Actas Dermosifiliogr 1989;80:557–64.
- Hudges SH, Apisarnthanarax P, Mullins F. Subcutaneous fat necrosis associated with pancreatic disease. Arch Dermatol 1975;111:506–10.
- Pott DE, Mass MF, Iseman MD. Syndrome of pancreatitis disease, subcutaneous fat necrosis and polyserositis. Am J Med 1975;58:417–23.
- Requena L, Sánchez Yus E. Panniculitis. Part II. Mostly lobular panniculitis. J Am Acad Dermatol 2001;45:325–61.
- Johnson MA, Kannan DG, Balachandar TG, Jeswanth S, Rajendran S, Surendran R. Acute septal panniculitis. A cutaneous marker of a very early stage of pancreatic panniculitis indicating acute pancreatitis. JOP 2005;6:334–8.
- Ball NJ, Adams SP, Marx LH, Enta T. Possible origin of pancreatic fat necrosis as a septal panniculitis. J Am Acad Dermatol 1996;34(2 Pt 2):362–4.
- Phelps RG, Shoji T. Update on panniculitis. Mt Sinai J Med 2001;68:262–7.
- Patterson JW. Disorders of subcutaneous fat: Panniculitis. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology, 1st edn. London: Mosby, 2003:1551–73.
- Heykarts B, Anseeuw M, Degreef H. Panniculitis caused by acinous pancreatic carcinoma. Dermatology 1999;198:182–3.
- Dhawan SS, Jimenez-Acosta F, Poppiti RJ Jr, Barkin JS. Subcutaneous fat necrosis associated with pancreatitis: histochemical and electron microscopic findings. Am J Gastroenterol 1990;85:1025–8.
- Berman B, Conteas C, Smith B, Leong S, Hornbeck L 3rd. Fatal pancreatitis presenting with subcutaneous fat necrosis. Evidence that lipase and amylase alone do not induce lipocyte necrosis. J Am Acad Dermatol 1987;17(2 Pt 2):359–64.
- Preiss JC, Faiss S, Loddenkemper C, Zeitz M, Duchmann R. Pancreatic panniculitis in an 88-year-old man with neuroendocrine carcinoma. Digestion 2002;66:193–6.
- Beltraminelly HS, Buechner SA, Hausermann P. Pancreatic panniculitis in a patient with an acinar cell cystadenocarcinoma of the pancreas. Dermatology 2004;208:265–7.
- Schäffler A, Landfried K, Völk M, et al. Potential of adipocytokines in predicting peripancreatic necrosis and severity in acute pancreatitis: pilot study. J Gastroenterol Hepatol 2007;22:326–34.
- Zellman GL. Pancreatic panniculitis. J Am Acad Dermatol 1996;35: 282–3.
- Sánchez MH, Fernández RS, Gomez-Calcerrada MR. Single-nodule pancreatic panniculitis. Dermatology 1996;193:269.
- Corazza M, Salmi R, Strumia R. Pancreatic panniculitis as a first sign of liver carcinoma. Acta Derm Venereol 2003;83:230–1.
- Chi ZC, Ma SZ. Rheumatologic manifestations of hepatic diseases. Hepatobiliary Pancreat Dis Int 2003;2:32–7.
- Kuwatani M, Kawakami H, Yamada Y. Osteonecrosis and panniculitis as life-threatening signs. Clin Gastroenterol Hepatol 2010;8:e52–3.
- Sorensen EV. Subcutaneous fat necrosis in pancreatic disease: a review and two new cases report. J Clin Gastroenterol 1988;10:71–5.
- Haller J, Greenway G, Rennick D, Kindynis P, Kang HS. Intraosseous fat necrosis associated with acute pancreatitis: MR imaging. Radiology 1989;179:193–5.
- Price-Forbes AN, Filer A, Udeshi UL, Rai A. Progression of imaging in pancreatitis panniculitis polyarthritis (PPP) syndrome. Scand J Rheumatol 2006;35:72–4.
- Hughes SH, Apisarnthanarax P, Mullins F. Subcutaneous fat necrosis associated with pancreatic disease. Arch Dermatol 1975;111:506–10.
- Marsh RW, Hagler KT, Carag HR, Flowers FP. Pancreatic panniculitis. Eur J Surg Oncol 2005;31:1213–15.
- Braun-Falco O, Hohenleutner U, von der Helm D, Landthaler M. Pankreatogenic pannikulitis. 2 case reports with a literature review. Hautarzt 1989;40:778–81.
- Mourad FH, Hannoush HM, Bahlawan M, Uthman I, Uthman S. Panniculitis and arthritis as the presenting manifestation of chronic pancreatitis. J Clin Gastroenterol 2001;32:259–61.
- Lee MS, Lowe PM, Nevell DF, Fryer J, L Guay J. Subcutaneous fat necrosis following traumatic pancreatitis. Autralas J Dermatol 1995;36:196–8.
- Levine N, Lazarus GS. Subcutaneous fat necrosis after paracentesis: Report of a case in a patient with acute pancreatitis. Arch Dermatol 1976;112:993–4.
- Cheng KS, Stansby G, Law N, Gardham R. Recurrent panniculitis as the first clinical manifestation of recurrent acute pancreatitis to cholelithiasis. J R Soc Med 1996;89:105–6.
- Lopez A, Garcia Estan J, Marras C, et al. Pancreatitis associated with pleural-mediastinal pseudocyst, panniculitis and polyarthritis. Clin Rheumatol 1998;91:1835–7.
- Lee SH, Bodensteiner D, Eisman S, Dixon AY, McGregor EH. Chronic relapsing pancreatitis with pseudocyst erosion into the portal vein and disseminated fat necrosis. Am J Gastroenterol 1985;80:452–8.
- Kirkland EB, Sachdev R, Kim J, Peng D. Early pancreatic panniculitis associated with HELLP syndrome and acute fatty liver of pregnancy. J Cutan Pathol 2011;38:814–17.
- Shehan JM, Kalaaji AN. Pancreatic panniculitis due to pancreatic carcinoma. Mayo Clin Proc 2005;80:822.
- Brown R, Buckley R, Kelley M. Pancreatic panniculitis. J Clin Oncol 1997;15:3418–19.
- Kuerer H, Shim H, Pertsemlidis D, Unger P. Functioning pancreatic acinar cell carcinoma: immunohistochemical and ultrastructural analyses. Am J Clin Oncol 1997;20:101–7.
- Durden FM, Variyam E, Chrem MM. Fat necrosis with features of erythema nodosum in a patient with metastatic pancreatic carcinoma. Int J Dermatol 1996;35:39–41.
- Hudson-Peacock MJ, Regnard CF, Farr PM. Liquefying panniculitis associated with acinous carcinoma of the pancreas responding to octreotide. J R Soc Med 1994;87:361–2.
- Good AE, Schnitzer B, Kawanishi H, Demetropoulos KC, Rapp R. Acinar pancreatic tumor with metastasis fat necrosis. Digest Dis 1976;21:978–87.
- Hasenöhrl K, Stein A, Wozel G, Meurer M. Pankreopathische pannikulitis bei neuroendokrinem karzino des pancreas. Hautarzt 2006;57:237–41.
- Berkovic D, Hallermann C. Carcinoma of the pancreas with neuroendocrine differentiation and nodular panniculitis. Onkologie 2003;26:473–6.
- Turner-Lewis C, Tschen A, Klima M. Subcutaneous fat necrosis associated with pancreatic islet cell carcinoma. Am J Dermatopathol 1991;13:52–6.
- Burdi C. Subcutaneous fat necrosis associated pancreatic islet cell tumor. Am J Dermatopathol 1992;14:181.
- Schwartz RA, Fleishman JS. Association of insulinoma with subcutaneous fat necrosis. J Surg Oncol 1981;16:305–11.
- Millns JL, Evans HL, Winkelmann RK. Association of islet cell carcinoma of the pancreas with subcutaneous fat necrosis. Am J Dermatopathol 1979;1:273–89.
- Freireich-Astman M, Segal R, Feinmesser M, David M. Pancreatic panniculitis as a sign of adenocarcinoma of unknown origin. Isr Med Assoc J 2005;7:474–5.
- Cox NH, Ramsay B, Dobson C, Comaish JS. Woody hands in a patient with pancreatic carcinoma: a variant of cancer-associated fasciitis–panniculitis syndrome. Br J Dermatol 1996;135:995–8.
- Wang MC, Sung JM, Chen FF, Lee WC, Huang JJ. Pancreatic panniculitis in a renal transplant recipient. Nephron 2000;86:550–1.
- Echeverría CM, Fortunato LP, Stengel FM, Laurini J, Díaz C. Pancreatic panniculitis in a kidney transplant recipient. Int J Dermatol 2001;40:751–3.
- Pike JL, Rice JC, Sanchez RL, Kelly EB, Kelly BC. Pancreatic panniculitis associated with allograft pancreatitis and rejection in a simultaneous pancreas-kidney transplant recipient. Am J Transplant 2006;6:2502–5.
- Prikis M, Norman D, Rayhill S, Olyaei A, Troxell M, Mittalhenkle A. Preserved endocrine function in a pancreas transplant recipient with pancreatic panniculitis and antibody-mediated rejection. Am J Transplant 2010;10:2717–22.
- Hu JC, Gutierrez MA. Pancreatic panniculitis after endoscopic retrograde cholangiopancreatography. J Am Acad Dermatol 2011;64:e72–4.
- Reynaud D, Alric L, Escourrou J, Bonnet E, Duffaut M. Traitment endoscopique d'une cystosteatonecrose secondaire a une fistule pancreaticovasculaire: a propos d'un case. Rev Med Interne 1998;19:123–7.
- Haber RM, Assaad DM. Panniculitis associated with a pancreas divisum. J Am Acad Dermatol 1986;14(2 Pt 2):331–4.
- Outtas O, Barthet M, De Troyer J, Franck F, Garcia S. Pancreatic panniculitis with intraductal carcinoid tumor of the pancreas divisum. Ann Dermatol Venereol 2004;131:466–9.
- Cabie A, Franck N, Gaudric M, Gorin I, Lessana-Leibowitch M, Escande JP. Recurrent nodular panniculitis associated with pancreas divisum. Ann Dermatol Venereol 1993;120:299–301.
- Haber RM, Assaad DM. Panniculitis associated with a pancreas divisum. J Am Acad Dermatol 1986;14:331–4.
- Feuer J, Spiera H, Phelps RG, Shim H. Panniculitis of pancreatic disease masquerading as systemic lupus erythematosus panniculitis. J Rheumatol 1995;22:2170–2.
- Cutlan RT, Wesche WA, Jenkins JJ 3rd, Chesney TM. A fatal case of pancreatic panniculitis presenting in a young patient with systemic lupus. J Cutan Pathol 2000;27:466–71.
- Martínez-Escribano JA, Pedro F, Sabater V, Quecedo E, Navarro V, Aliaga A. Acute exanthem and pancreatic panniculitis in a patient with primary HIV infection and haemophagocytic syndrome. Br J Dermatol 1996;134:804–7.
- Suwattee P, Cham PM, Pope E, Ho N. Pancreatic panniculitis in a 4-year-old child with nephrotic syndrome. Pediatr Dermatol 2007;24:659–60.
- Detlefs RL. Drug induced pancreatitis presenting as subcutaneous fat necrosis. J Am Acad Dermatol 1985;13:305–7.
- Ahmed B, Estey E, Manning J, et al. Anaplastic large cell lymphoma with involvement of the pancreas presenting as panniculitis in a patient with a history of acute myeloid leukemia – case report and review of the literature. Haematologica 2006;91(12 Suppl.):ECR55.
- Chiewchengchol D, Wananukul S, Noppakun N. Pancreatic panniculitis caused by L-asparaginase induced acute pancreatitis in a child with acute lymphoblastic leukemia. Pediatr Dermatol 2009;26:47–9.
- Ball NJ, Cowan BJ, Hashimoto SA. Lobular panniculitis at the site of subcutaneous interferon beta injections for the treatment of multiple sclerosis can histologically mimic pancreatic panniculitis. A study of 12 cases. J Cutan Pathol 2009;36:331–7.
- Cannon JR, Pitha JV, Everett MA. Subcutaneous fat necrosis in pancreatitis. J Cutan Pathol 1979;6:501–6.
- Requena L, Sitthinamsuwan P, Santonja C, et al. Cutaneous and mucosal mucormycosis mimicking pancreatic panniculitis and gouty panniculitis. J Am Acad Dermatol 2012;66:975–84.
- Colmenero I, Alonso-Sanz M, Casco F, Hernández-Martín A, Torrelo A. Cutaneous aspergillosis mimicking pancreatic and gouty panniculitis. J Am Acad Dermatol 2012;67:789–91
- Frikha F, Miled N, Bacha AB, Mejdoub H, Gargouri Y. Structural homologies, importance for catalysis and lipid binding of the N-terminal peptide of a fungal and a pancreatic lipase. Protein Pept Lett 2010;17:254–9.
- Nordén G, Björck S, Persson H, Svalander C, Li XG, Edebo L. Cure of zygomycosis caused by a lipase-producing Rhizopus rhizopodiformis strain in a renal transplant patient. Scand J Infect Dis 1991;23:377–82.
- Black MM, Cunliffe WJ. Subcutaneous fat: Inflammatory disorders of subcutaneous fat. In: Burns T, Breathnach S, Cox N, Griffiths C, eds. Rook's Textbook of Dermatology, 7th edn. Oxford: Blackwell, 2004:55.8–55.25.
- Förström TL, Winkelmann RK. Acute, generalized panniculitis with amylase and lipase in skin. Arch Dermatol 1975;111:497–502.
- Camilleri MJ, Daniel Su WP. Disorders of subcutaneous tissue: panniculitis. In: Freedberg IM, Eisen AZ, Wolff K, Austen KF, Goldsmith LA, Katz SI, eds. Fitzpatrick's Dermatology in General Medicine, 6th edn. New York: McGraw Hill, 2003:1047–67.
- Lambiase P, Seery JP, Taylor-Robinson SD, Thompsom JN, Hughes JM, Walters JR. Resolution of panniculitis after placement of pancreatic duct stent in chronic pancreatitis. Am J Gastroenterol 1996;91:1835–7.
- Francombe J, Kingsnorth AN, Tunn E. Panniculitis, arthritis and pancreatitis. Br J Rheumatol 1995;34:680–3.
Alpha-1 antitrypsin deficiency panniculitis
- Geraminejad P, DeBloom JR, Walling HW, et al. Alpha-1-antitrypsin associated panniculitis: The MS variant. J Am Acad Dermatol 2004;51:645–55.
- Patterson JW. Disorders of subcutaneous fat: Panniculitis. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology, 1st edn. London: Mosby, 2003:1551–73.
- Chowdhury MMU, Williams EJ, Morris JS, et al. Severe panniculitis caused by homozygous ZZα1-antitrysin deficiency treated successfully with human purified enzyme (Prolastin®). Br J Dermatol 2002;147:1258–61.
- Garver RI Jr, Mornex JF, Nukiwa T, et al. Alpha-1-antitrypsin deficiency and emphysema caused by homozygous inheritance of non-expressing alpha-1-antitrypsin genes. N Engl J Med 1986;314:762–6.
- Humbert P, Faivre B, Gibey R, et al. Use of Anti-collagenase properties of Doxycycline in treatment of α1 antitrypsin deficiency panniculitis. Acta Derm Veneorol 1991;71:189–94.
- McBean J, Sabie A, Maude J, et al. α1-Antitrysin deficiency panniculitis. Cutis 2003;71:205–9.
- Gross B, Grebe M, Wencker M, Stoller JK, Bjursten LM, Janciauskiene S. New findings in PiZZ alpha1-antitrypsin deficiency-related panniculitis. Demonstration of skin polymers and high dosing requirements of intravenous augmentation therapy. Dermatology 2009;218:370–5.
- Breit SN, Penny R. The role of alpha 1 protease inhibitor (α1-antitrypsin) in the regulation of immunologic and inflammatory reactions. Aust NZJ Med 1980;16:449–53.
- Smith KC, Pittelkow MR, Su WPD. Panniculitis associated with severe α1-antitrysin deficiency. Treatment and review of the literature. Arch Dermatol 1987;123:1655–61.
- Gaillard MC, Bothwell J, Dreyer L. A case of nodular panniculitis associated with M1 (Val213) phenotype of α1-protease inhibitor. Int J Dermatol 1997;36:276–301.
- Chng WJ, Henderson CA. Suppurative panniculitis associated with alpha 1-antitrypsin deficiency (PiSZ phenotype) treated with doxycycline. Br J Dermatol 2001;144:1282.
- Pinto AR, Maciel LS, Carneiro F, et al. Systemic nodular panniculitis in a patient with alpha-1 antitrypsin deficiency (PiSS phenotype). Clin Exp Dermatol 1993;18:154–5.
- Ortiz J, Borrego L, de Pablo P, et al. Paniculitis y defecto de alfa 1 antitripsina. Presentación de 3 casos. Actas Dermosifiliogr 1990;81:495–500.
- Warter J, Storck D, Grosshans E, et al. Weber–Christian syndrome associated with alpha-1-antitrypsin deficiency. Familial investigation. Ann Med Interne (Paris) 1972;123: 877–82.
- Rubinstein HM, Jaffer AM, Kudrna JC, et al. Alpha-1-antitrypsin deficiency with severe panniculitis: report of two cases. Ann Intern Med 1977;86:742–4.
- Requena L, Sánchez Yus E. Panniculitis. Part II: Mostly lobular panniculitis. J Am Acad Dermatol 2001;45:325–61.
- Edmonds BK, Hodge JA, Rietschel RL. Alpha 1-antitrypsin deficiency-associated panniculitis: case report and review of the literature. Pediatr Dermatol 1991;8:296–9.
- Smith KC, Su WPD, Pittelkow MR, et al. Clinical and pathologic correlations in 96 patients with panniculitis, including 15 patients with deficient levels of α1-antitrypsin. J Am Acad Dermatol 1989;21:1192–6.
- Linares-Barrios M, Conejo-Mir JS, Artola JL, et al. Panniculitis due to α1-antitrypsin deficiency induced by cryosurgery. Br J Dermatol 1998;138:552–3.
- Parr DG, Stewart DG, Hero I, et al. Panniculitis secondary to extravasation of clarithromycin in a patient with α1- antitrypsin deficiency (phenotype PiZ). Br J Dermatol 2003;149:410–13.
- Yesudian PD, Dobson CM, Wilson NJ. Alpha1-Antitrypsin deficiency panniculitis (phenotype PiZZ) precipitated postpartum and successfully treated with dapsone. Br J Dermatol 2004;150:1222–3.
- Black MM, Cunliffe WJ. Subcutaneous fat. In: Burns T, et al., eds. Rook's Textbook of Dermatology, 7th edn. Oxford: Blackwell Publishing, 2004: 55.14–55.15.
- Hwang ST, Williams ML, McCalmont TH, Frieden IJ. Sweet's syndrome leading to acquired cutis laxa (Marshall's syndrome) in an infant with alpha 1-antitrypsin deficiency. Arch Dermatol 1995;131:1175–7.
- Geller JD, Su WPD. A subtle clue to the histopathologic diagnosis of early α1-antitrypsin deficiency panniculitis. J Am Acad Dermatol 1994;31:241–5.
- Su WP, Smith KC, Pittelkow MR, Winkelmann RK. Alpha 1-antitrypsin deficiency panniculitis: a histopathologic and immunopathologic study of four cases. Am J Dermatopathol 1987;9:483–90.
- Hendrick SJ, Silverman AK, Solomon AR, Headington JT. Alpha 1-antitrypsin deficiency associated with panniculitis. J Am Acad Dermatol 1988;18:684–92.
- Pittelkow MR, Smith KC, Su WPD. Alpha-1-antitrypsin deficiency and panniculitis: perspectives on disease relationship and replacement therapy. Am J Med 1988;84:80–6.
- Kjus T, Lützow-Holm C, Christensen OB. Treatment of panniculitis associated with alpha-1-antitrypsin deficiency with alpha-1-protease inhibitor. Acta Derm Venereol 2003;83:462–3.
- Furey NL, Golden RS, Potts SR. Treatment of α1-antitrypsin deficiency, massive edema, and panniculitis with α1-protease inhibitor. Ann Intern Med 1996;125:699.
- Chowdhury MM, Williams EJ, Morris JS, et al. Severe panniculitis caused by homozygous ZZ alpha1-antitrypsin deficiency treated successfully with human purified enzyme (Prolastin). Br J Dermatol 2002;147:1258–61.
- Kjus T, Lützow-Holm C, Christensen OB. Treatment of panniculitis associated with alpha-1-antitrypsin deficiency with alpha-1-protease inhibitor. Acta Derm Venereol 2003;83:462–3.
- Blanco I, Lara B, de Serres F. Efficacy of alpha1-antitrypsin augmentation therapy in conditions other than pulmonary emphysema. Orphanet J Rare Dis 2011;6:14.
- Al-Niaimi F, Lyon C. Severe ulcerative panniculitis caused by alpha 1-antitrypsin deficiency: remission induced and maintained with intravenous alpha 1-antitrypsin. J Am Acad Dermatol 2011;65:227–9.
- Olson JM, Moore EC, Valasek MA, Williams LH, Vary JC. Panniculitis in alpha-1 antitrypsin deficiency treated with enzyme replacement. J Am Acad Dermatol 2012;66:e139–41.
- de Oliveira P, Paz-Melgar L, Takahashi MDF, et al. Alpha-1-antitrypsin deficiency associated with panniculitis treated with plasma exchange therapy. Int J Dermatol 2004;43:693–7.
- O'Riordan K, Blei A, Rao MS, et al. Alpha-1-antitrypsin deficiency-associated panniculitis: resolution with intravenous alpha-1-antitrypsin administration and liver transplantation. Transplantation 1997;63:480–2.
- Fernández-Torres R, García-Silva J, Robles O, Otero A, Vázquez MA, Fonseca E. Alfa-1-antitrypsin deficiency panniculitis acquired after liver transplant and successfully treated with retransplant. J Am Acad Dermatol 2009;60:715–16.
Infective panniculitis
- Patterson JW, Brown PC, Broecker AH. Infection-induced panniculitis. J Cutan Pathol 1989;16:183–93.
- Smith RA, Ross JS, Branfoot C. Panniculitis with Pseudomonas septicaemia in AIDS. J Eur Acad Dermatol Venereol 1995;4:166–9.
- Bagel J, Grossman ME. Subcutaneous nodules in Pseudomonas sepsis. Am J Med 1986;80:528–52.
- Douwes KE, Schmalzbauer E, Linde HJ, et al. Branched filaments no fungus, ovoid bodies no bacteria: two unusual cases of mycetoma. J Am Acad Dermatol 2003;49: S170–S3.
- Bartralot R, Garcia-Patos V, Repiso T, et al. Liquefactive panniculitis in the inguinal area as the first sign of chronic renal brucellosis. J Am Acad Dermatol 1996;35:339–41.
- Viljanen MK, Oksi J, Salomaa P, Skurnik M, Peltonen R, Kalimo H. Cultivation of Borrelia burgdorferi from the blood and a subcutaneous lesion of a patient with relapsing febrile nodular nonsuppurative panniculitis. J Infect Dis 1992;165:596–7.
- Kempf W, Kazakov DV, Kutzner H. Lobular panniculitis due to Borrelia burgdorferi infection mimicking subcutaneous panniculitis-like T-cell lymphoma. Am J Dermatopathol 2013;35:e30–3.
- Larson K, Glanz S, Bergfeld WF. Neutrophilic panniculitis caused by Mycobacterium marinum. J Cutan Pathol 1989;16:315.
- Sanderson TL, Moskowitz L, Henseley GT, Clearly TJ, Penneys NS. Disseminated Mycobacterium avium-intracellulare infection appearing as a panniculitis. Arch Pathol Lab Med 1982;106:112–14.
- Street ML, Umbert-Millet IJ, Roberts GD, Su WPD. Nontuberculous mycobacterial infections of the skin: Report of fourteen cases and review of the literature. J Am Acad Dermatol 1991;24:208–15.
- Escalonilla P, Esteban J, Soriano ML, et al. Cutaneous manifestations of infection by nontuberculous mycobacteria. Clin Exp Dermatol 1998;23:214–21.
- Santa Cruz, Strayer DS. The histologic spectrum of the cutaneous mycobacterioses. Hum Pathol 1982;13:485–95.
- Bartralot R, García-Patos V, Sitjas D. Clinical patterns of cutaneous nontuberculous mycobacterial infections. Br J Dermatol 2005;152:727–34.
- Mohty AM, Dereure O, Bessis D, et al. Panniculitis due to non-tuberculous mycobacteria in two immunocompromised patients. Acta Derm Venereol 2004;84:469–71.
- Ara M, Saenz de Santamaria C, Zaballos P. Mycobacterium chelonae infection with multiple cutaneous lesions after treatment with acupuncture. Int J Dermatol 2003;42:642–4.
- Lamb SR, Stables GI, Merchant W. Disseminated cutaneous infection with Mycobacterium chelonae in a patient with steroid-dependent rheumatoid arthritis. Clin Dermatol 2004;29:254–7.
- Chastain MA, Buckley J, Russo GG. Mycobacterium chelonae/abscessus complex infection in a liver transplant patient. Int J Dermatol 2001;40:769–74.
- Retief CR, Tharp MD. Mycobacterium fortuitum panniculitis in a steroid-dependent asthmatic patient. J Am Acad Dermatol 1998;39:650–3.
- Hendrick SJ, Jorizzo JL, Newton RC. Giant Mycobacterium fortuitum abscess associated with systemic lupus erythematosus. Arch Dermatol 1986;122:695–7.
- Langenberg A, Egbert B. Neutrophilic tuberculous panniculitis in a patient with polymyositis. J Cutan Pathol 1993;20:177–9.
- Alvarez-Ruiz SB, Delgado-Jimenez Y, Aragües M, Fraga J, Garcia-Diez A. Subcutaneous lepromas as leprosy-type presentation. JEADV 2006;20:344–5.
- Van der Werf TS, van der Graaf WT, Tappero JW et al. Mycobacterium ulcerans infection. Lancet 1999;354:1013–18.
- Sizaire V, Nackers F, Comte E, et al. Mycobacterium ulcerans infection: control, diagnosis, and treatment. Lancet Infect Dis 2006;6:288–96.
- George KM, Barrer LP, Welty DM, et al. Partial purification and characterization of biological effects of a lipid toxin produced by Mycobacterium ulcerans. Infect Immun 1998;66:587–93.
- Rondini S, Mensah-Quainoo E, Troll H, et al. Development and application of real-time PCR assay for quantification of Mycobacterium ulcerans DNA. J Clin Microbiol 2003;41:4231–7.
- Rondini S, Horsfield C, Mensah-Quainoo E, et al. Contiguous spread of Mycobacterium ulcerans in Buruli ulcer lesions analysed by histopathology and real-time PCR quantification of mycobacterial DNA. J Pathol 2006;208:119–28.
- Siegmund V, Adjei O, Racz P, et al. Dry- reagent-based PCR as a novel tool for laboratory confirmation of clinically diagnosed Mycobacterium ulcerans-associated disease in areas in the tropics where M. ulcerans is endemic. J Clin Microbiol 2005;43:271–6.
- Ginter G, Rieger E, Soyer P, Hoedl S. Granulomatous panniculitis caused by Candida albicans: a case presenting with multiple leg ulcers. J Am Acad Dermatol 1993;28:315–17.
- Galimberti RL, Flores V, Gonzalez Ramos MC, et al. Cutaneous ulcers due to Candida albicans in an immunocompromised patient – response to therapy with itraconazole. Clin Exp Dermatol 1989;14:295–7.
- Murakawa GJ, Harvell JD, Lubitz P, Schnoll S, Lee S, Berger T. Cutaneous aspergillosis and acquired immunodeficiency syndrome. Arch Dermatol 2000;136:365–9.
- Abildgaard WH Jr, Hargrove RH, Kalivas J. Histoplasma panniculitis. Arch Dermatol 1985;121:914–16.
- Silverman AK, Gilbert SC, Watkins D, Cooper B, Menter A. Panniculitis in an immunocompromised patient. J Am Acad Dermatol 1991;24:912–14.
- Lichon V, Khachenoun A. Mycetoma, a review. Am J Clin Dermatol 2006;7:315–21.
- Pang KR, Wu JJ, Huang DB, et al. Subcutaneous fungal infections. Dermatol Ther 2004;17:523–31.
- Hay RJ, Moore MK. Mycology. In: Burns T, Breathnach S, Cox N, Griffiths RCG, eds. Rook's Textbook of Dermatology, 7th edn. Oxford: Blackwell, 2004:31.75–31.81.
- Ballestero-Diez M, Alvarez-Ruiz SB, Aragües-Montañes M, Fraga J. Septal panniculitis associated with cytomegalovirus infection. Histopathology 2005;46:720–2.
- Rosenberg AS, Morgan MB. Disseminated acanthamoebiasis presenting as lobular panniculitis with necrotizing vasculitis in a patient with AIDS. J Cutan Pathol 2001;28:307–13.
- Berger TG, Greene I. Bacterial, viral, fungal and parasitic infections in HIV disease and AIDS. Dermatol Clin 1991;9:465–92.
- Köseoglu F, Emiroglu R, Karakayall H. Prevalence of mycobacterial infection in solid organ transplant recipients. Transplant Proc 2001;33:1782–4.
- Uslan DZ, Kowalski TJ, Wengenak NL, et al. Skin and soft tissue infections due to rapidly growing mycobacteria. Arch Dermatol 2006;142:1287–92.
- Requena L, Sanchez Yus E. Panniculitis. Part II. Mostly lobular panniculitis. J Am Acad Dermatol 2001;45:325–61.
- Aleman CT, Wallace ML, Blaylock WK, et al. Subcutaneous nodule caused by Pseudomonas aeruginosa without sepsis. Cutis 1999;63:161–3.
- Bartralot R, Pujol RM, Garcia-Patos V. Cutaneous infections due to nontuberculous mycobacteria: histopathological review of 28 cases. Comparative study between lesions observed in immunosuppressed patients and normal hosts. J Cutan Pathol 2000;27:124–9.
- Kelly SE. Multiple injection abscesses in a diabetic caused by Mycobacterium chelonei. Clin Exp Dermatol 1987;12:48–9.
- Galache C, Santos-Juanes J, Blanco S, Rodríguez E, Martínez A, Soto J. Q fever: a new cause of ‘doughnut’ granulomatous lobular panniculitis. Br J Dermatol 2004;151:685–7.
- Requena L, Sitthinamsuwan P, Santonja C, et al. Cutaneous and mucosal mucormycosis mimicking pancreatic panniculitis and gouty panniculitis. J Am Acad Dermatol 2012;66:975–84.
- Colmenero I, Alonso-Sanz M, Casco F, Hernández-Martín A, Torrelo A. Cutaneous aspergillosis mimicking pancreatic and gouty panniculitis. J Am Acad Dermatol 2012;67:789–91.
- Kutzner H, Argenyi ZB, Requena L, Rütten A, Hügel H. A new application of BCG antibody for rapid screening of various tissue microorganisms. J Am Acad Dermatol 1998;38:56–60.
Factitious panniculitis
- Requena L, Sanchez Yus E. Panniculitis. Part II. Mostly lobular panniculitis. J Am Acad Dermatol 2001;45:325–61.
- Darsow U, Bruckbauer H, Worret WI, et al. Subcutaneous oleomas induced by self-injection of sesame seed oil for muscle augmentation. J Am Acad Dermatol 2000;42: 292–4.
- Achauer BM. A serious complication following medical-grade silicone injection of the face. Plast Reconstr Surg 1983;71:251–4.
- Garcia-Domingo MI, Alijotas-Reig J, Cistero-Bahima A, et al. Disseminated and recurrent sarcoid-like granulomatous panniculitis due to bovine collagen injection. J Investig Allergol Clin Immunol 2000;10:107–9.
- Requena C, Izquierdo MJ, Navarro M, et al. Adverse reactions to injectable aesthetic microimplants. Am J Dermatopathol 2001;23:197–202.
- Sanmartín O, Requena C, Requena L. Factitial panniculitis. Dermatol Clin 2008;26:519–27.
- Requena L, Requena C, Christensen L, Zimmermann US, Kutzner H, Cerroni L. Adverse reactions to injectable soft tissue fillers. J Am Acad Dermatol 2011;64:1–34.
- Requena L, Cerroni L, Kutzner H. Histopathologic patterns associated with external agents. Dermatol Clin 2012;30:731–48.
- Bechara F, Sand M, Hoffmann K, et al. Fat tissue after lipolysis of lipomas: A histopathological and immunohistochemical study. J Cutan Pathol 2007;34:552–7.
- Davis MD, Wright TI, Shehan JM. A complication of mesotherapy: noninfectious granulomatous panniculitis. Arch Dermatol 2008;144:808–9.
- Vetter WL, Weiland AJ, Arnett FC. Factitious extension contracture of the elbow: case report. J Hand Surg [Am] 1983;8:277–9.
- Gandhi V, Agrawal SK, Chatterjee AK, et al. Pentazocine-induced cutaneous sclerosis and panniculitis in an Indian male. Int J Dermatol 2004;43:516–17.
- Farrant P, Creamer D, Fuller C. Extensive cutaneous fibrosis and ulceration caused by methadone injection. Clin Exp Dermatol 2005;30:87–8.
- Kossard S, Ecker RI, Dicken CH. Povidone panniculitis. Polyvinylpyrrolidone panniculitis. Arch Dermatol 1980;116:704–6.
- McCain J, West TW, Vasey FB, et al. Intramuscular aurothioglucose (Solganal) leading to panniculitis. J Rheumatol 1993;20:1632–3.
- Pang BK, Munro V, Kossard S. Pseudoscleroderma secondary to phytomenadione (vitamin K1) injections: Texier's disease. Australas J Dermatol 1996;37:44–7.
- Soares Almeida LM, Requena L, Kutzner H, Angulo J, de Sa J, Pignatelli J. Localized panniculitis secondary to subcutaneous glatiramer acetate injections for the treatment of multiple sclerosis: A clinicopathologic and immunohistochemical study.J Am Acad Dermatol 2006;55:969–75.
- Laws JW. Pyrexia of unusual origin. BMJ 1951;2:157–8.
- Chong H, Brady K, Metze D, et al. Persistent nodules at injection sites (aluminium granuloma) – clinicopathological study of 14 cases with a diverse range of histological reaction patterns. Histopathology 2006;48:182–8.
- Kaufman HL, Harandi A, Watson MC, et al. Panniculitis after vaccination against CEA and MUC1 in a patient with pancreatic cancer. Lancet Oncol 2005;6:62–3.
- O'Sullivan SS, Cronin EM, Sweeney BJ, et al. Panniculitis and lipoatrophy after subcutaneous injection of interferon beta-1b in a patient with multiple sclerosis. J Neurol Neurosurg Psychiatry 2006;77:1382–3.
- Prendiville J, Thiessen P, Mallory SB. Neutrophilic dermatoses in two children with idiopathic neutropenia: association with granulocyte colony-stimulating factor (G-CSF) therapy. Pediatr Dermatol 2001;18:417–21.
- Baars JW, Coenen JL, Wagstaff J, et al. Lobular panniculitis after subcutaneous administration of interleukin-2 (IL-2), and its exacerbation during intravenous therapy with IL-2. Br J Cancer 1992;66:698–9.
- Alfaro-Rubio A, Sanmartin O, Requena C, et al. Extravasación de agentes citostáticos: una complicación seria del tratamiento oncológico. Actas Dermosifiliogr 2006;97:169–76.
- Goolsby TV, Lombardo FA. Extravasation of chemotherapeutic agents: prevention and treatment. Semin Oncol 2006;33:139–43.
- Lee JS, Ahn SK, Lee SH. Factitial panniculitis induced by cupping and acupuncture. Cutis 1995;55:217–18.
- Jeong KH, Lee MH. Two cases of factitial panniculitis induced by electroacupuncture. Clin Exp Dermatol 2009;34:e170–3.
- Gomez Rodriguez N, Ortiz-Rey JA, de la Fuente Buceta A, et al. Paniculitis eosinofílica autoinducida. Un dilema diagnóstico. An Med Interna 2001;18:635–7.
- Winkelmann RK, Barker SM. Factitial traumatic panniculitis. J Am Acad Dermatol 1985;13:988–94.
- Cooper MA, Davies DM. Charcot's oedeme bleu des hysteriques. J Hand Surg [Br] 1985;10:399–400.
- Reading G. Secretan's syndrome: hard edema of the dorsum of the hand. Plast Reconstr Surg 1980;65:182–7.
- Ostlere LS, Harris D, Denton C, et al. Boxing-glove hand: an unusual presentation of dermatitis artefacta. J Am Acad Dermatol 1993;28:120–2.
- Dlesk A, Panush RS. Factitial Weber–Christian disease: a case report. J Rheumatol 1981;8:129–32.
- Oh CC, McKenna DB, McLaren KM, Tidman MJ. Factitious panniculitis masquerading as pyoderma gangrenosum. Clin Exp Dermatol 2005;30:253–5.
- Hohaus K, Bley B, Kostler E, et al. Mineral oil granuloma of the penis. J Eur Acad Dermatol Venereol 2003;17:585–7.
- Mason J, Apisarnthanarax P. Migratory silicone granuloma. Arch Dermatol 1981;117:366–7.
- Hertzman A, Toone E, Resnik CS. Pentazocine induced myocutaneous sclerosis. J Rheumatol 1986;13:210–14.
- Pujol RM, Puig L, Moreno A, et al. Pseudoscleroderma secondary to phytonadione (vitamin K1) injections. Cutis 1989;43:365–8.
- Chi CC, Wang SH, Kuo TT. Localized cutaneous polyvinylpyrrolidone storage disease mimicking cheilitis granulomatosa. J Cutan Pathol 2006;33:454–7.
- Watanabe K, Hoshi N, Baba K, et al. Immunohistochemical profile of primary sclerosing lipogranuloma of the scrotum: report of five cases. Pathol Int 1995;45:854–9.
- Oertel YC, Johnson FB. Sclerosing lipogranuloma of male genitalia. Review of 23 cases. Arch Pathol Lab Med 1977;101:321–6.
- Kalsi JS, Arya M, Peters J, Minhas S, Ralph DJ. Grease-gun injury to the penis. J R Soc Med 2002;95:254.
- Rudolph CM, Soyer HP, Schuller-Petrovic S, et al. Foreign body granulomas due to injectable aesthetic microimplants. Am J Surg Pathol 1999;23:113–17.
- Palestine RF, Millns JL, Spigel GT, et al. Skin manifestations of pentazocine abuse. J Am Acad Dermatol 1980;2:47–55.
- Bhawan J, Petry J, Rybak ME. Histologic changes induced in skin by extravasation of doxorubicin (adriamycin). J Cutan Pathol 1989;16:158–63.
Neutrophilic lobular panniculitis
- Cohen PR. Subcutaneous Sweet's syndrome. A variant of acute febrile neutrophilic dermatosis that is included in the histopathologic differential diagnosis of neutrophilic panniculitis. J Am Acad Dermatol 2005;52:927–8.
- Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol 1964;76: 349–56.
- von den Driesch P. Sweet's syndrome (acute febrile neutrophilic dermatosis). J Am Acad Dermatol 1994;31:535–56.
- Cooper PH, Innes DJ Jr, Greer KE. Acute febrile neutrophilic dermatosis (Sweet's syndrome) and myeloproliferative disorders. Cancer 1983;51:1518–26.
- Cho KH, Han KH, Youn SW, et al. Neutrophilic dermatoses associated with myeloid malignancy. Clin Exp Dermatol 1997;22:269–73.
- Sitjas D, Puig L, Cuatrecasas M, et al. Acute febrile neutrophilic dermatosis (Sweet's syndrome). Int J Dermatol 1993;32:261–8.
- Jordaan HF. Acute febrile neutrophilic dermatosis: a histopathological study of 37 patients and review of the literature. Am J Dermatopathol 1989;11:99–111.
- Cohen PR, Kurzrock R. Sweet's syndrome revisited: a review of disease concepts. Int J Dermatol 2003;42:761–78.
- Cullity J, Maguire B, Gebauer K. Sweet's panniculitis. Australas J Dermatol1991;32:61–4.
- Morioka N, Otsuka F, Nogita T, et al. Neutrophil dermatosis with myelodysplatic syndrome: Nuclear segmentation anomalies of neutrophils in the skin lesion and in peripheral blood. J Am Acad Dermatol 1990;23:247–9.
- Vignon Pennamen MD, Wallach D. Cutaneous manifestations of neutrophilic disease. A study of seven cases. Dermatologica 1991;183:255–64.
- Cooper PH, Frierson HF, Greer KE. Subcutaneous neutrophilic infiltrates in acute febrile neutrophilic dermatosis. Arch Dermatol 1983;119:610–11.
- Matsumura Y, Tanabe H, Wada Y, et al. Neutrophilic panniculitis associated with myelodysplastic syndromes. Br J Dermatol 1997;136:142–4.
- Soutra Loubet C, Carlotti A, Guillemette J, et al. Neutrophilic lobular dermatosis. J Am Acad Dermatol 2004;50:280–5.
- Chen HC, Kao WY, Chang DM, Gao HW, Lai WY, Lai JH. Neutrophilic panniculitis with myelodysplastic syndromes presenting as pustulosis: case report and review of the literature. Am J Hematol 2004;76:61–5.
- Carvalho P, Cordel N, Courville PH, et al. Cutaneous aseptic abscesses, manifestations of neutrophilic diseases. Ann Derm Venereol 2001;128:641–3.
- Florez A, Sanchez-Aguilar D, Roson E, et al. Sweet's syndrome associated with Salmonella enteritidis infection. Clin Exp Dermatol 1999;24:239–40.
- Jagdeo J, Campbell R, Long T, Muglia J, Telang G, Robinson-Bostom L. Sweet's syndrome-like neutrophilic lobular panniculitis associated with all-trans-retinoic acid chemotherapy in a patient with acute promyelocytic leukemia. J Am Acad Dermatol 2007;56:690–3.
- Maalouf T, Angioi K, Ssi-Yan-Kai I, Vernerey F, Witz B, George J. Dacryoadenitis associated with subcutaneous Sweet's syndrome in a patient with acute myeloid leukemia. Orbit 2005;24:55–7.
- Chow S, Pasternak S, Green P, et al. Histiocytoid neutrophilic dermatoses and panniculitides: variations on a theme. Am J Dermatopathol 2007;29:334–41.
- Guhl G, García-Díez A. Subcutaneous Sweet syndrome. Dermatol Clin 2008;26:541–51.
- Hood M, Yu K, Magro C, Reisacher W. Pathology quiz case 2. Subcutaneous Sweet syndrome of the neck. Arch Otolaryngol Head Neck Surg 2010;136:1038.
- Lin J, Zhang Q, Chen M. Subcutaneous histiocytoid Sweet's syndrome in a patient associated with myelodysplastic syndrome-refractory anemia. J Dermatol 2012;39:99–101.
- Shin MK, Oh YJ, Yoon HJ, Lee MH. Subcutaneous Sweet syndrome with nuclear segmentation anomalies: a diagnostic marker of myelodysplasia. Int J Dermatol 2012;51:976–8.
- Llamas-Velasco M, García-Martín P, Sánchez-Pérez J, Fraga J, García-Diez A. Sweet's syndrome with subcutaneous involvement associated with pegfilgrastim treatment: first reported case. J Cutan Pathol 2013;40:46–9.
- Chan MP, Duncan LM, Nazarian RM. Subcutaneous Sweet syndrome in the setting of myeloid disorders: a case series and review of the literature. J Am Acad Dermatol 2013;68:1006–15.
- Cohen PR, Kurzrock R. Sweet's syndrome and cancer. Clin Dermatol 1993;11:149–57.
- Wallach D. Neutrophilic dermatoses. Rev Med Interne 2005;26:41–53.
- Williams HJ, Samuelson CO, Zone JJ. Nodular nonsuppurative panniculitis associated with jejunoileal bypass surgery. Arch Dermatol 1979;115:1091–3.
- Stein HB, Schlappner OL, Boyko W, et al. The intestinal bypass: arthritis–dermatitis syndrome. Arthritis Rheum 1981;24:684–90.
- Vignon-Pennamen MD, Wallach D. Neutrophilic disease: a review of extracutaneous neutrophilic manifestations. Eur J Dermatol 1995;6:449–55.
- Rodot S, Lacour J, Van Elslande L, et al. Extracutaneous manifestations of neutrophilic dermatosis. Ann Derm Venereol 1996;123:129–34.
- Kim B, LeBoit PE. Histopathologic features of erythema nodosum-like lesions in Behçet disease. Am J Dermatopathol 2000;22:379–90.
- Chun SI, Su WPD, Lee S, et al. Erythema nodosum-like lesions in Behçet's syndrome: a histopathologic study of 30 cases. J Cutan Pathol 1989;16:259–65.
- Demirkesesn C, Tüzüner N, Mat C, et al. Clinicopathological evaluation of nodular cutaneous lesions of Behçet syndrome. Am J Clin Pathol 2001;116:341–6.
- Jorizzo JL, Abernethy JL, White WL, et al. Mucocutaneous manifestations for the diagnosis of Behçet disease: an analysis of clinicopathologic data from multiple international centers. J Am Acad Dermatol 1995;32:968–76.
- Tran TA, DuPree M, Carlson JA. Neutrophilic lobular (pustular) panniculitis associated with rheumatoid arthritis: a case report and review of the literature. Am J Dermatopathol 1999;21:247–52.
- Kuniyuki S, Shindow K, Tanaka T. Pustular panniculitis in a patient with rheumatoid arthritis. Int J Dermatol 1997;36:292–3.
- Anstey A, Wilkinson JD, Wojnarowska FT. Pustular panniculitis in rheumatoid arthritis. J R Soc Med 1991;84:307–8.
- Shindo H, Hide M. Neutrophilic lobular panniculitis with non-rheumatoid arthritis. Acta Derm Venereol 2005;85:262–3.
- Magro CM, Crowson AN. The spectrum of cutaneous lesions in rheumatoid arthritis: a clinical and pathological study of 43 patients. J Cutan Pathol 2003;30:1–10.
- Andre M, Piette JC, Frances C, et al. Neutrophilic dermatoses and aseptic abscesses: two sides of the same entity. Rev Med Interne 2005;26:5–7.
- Newton J, Wojnarowska FT. Pustular panniculitis in rheumatoid arthritis. Br J Dermatol 1988;119:97–8.
- Yaffee HS. A peculiar nodosity associated with arthritis. US Armed Med J 1955;6:1043–52.
- Caus F, Halimi C, Kevorkian JP, et al. Blind loop syndrome: an unusual cause of panniculitis. J Am Acad Dermatol 1997;37:824–7.
- Kennedy C. The spectrum of inflammatory skin disease following jejuno–ileal bypass for morbid obesity. Br J Dermatol 1981;105:425–36.
Subcutaneous sarcoidosis
- Ahmed I, Harstad S. Subcutaneous sarcoidosis: is it a specific subset of cutaneous sarcoidosis frequently associated with systemic disease? J Am Acad Dermatol 2006;54:55–60.
- Requena L, Sánchez Yus E. Panniculitis. Part II. Mostly lobular panniculitis. J Am Acad Dermatol 2001;45:325–61.
- Mañá J, Marcoval J, Graells J, et al. Cutaneous involvement in sarcoidosis. Relationship to systemic disease. Arch Dermatol 1997;133:882–8.
- James DG. Dermatological aspects of sarcoidosis. QJM 1959;28:109–24.
- Scadding JG, Mitchell DN. Sarcoidosis of the skin. In: Scadding JG, Mitchell DN, eds. Sarcoidosis. London: Chapman & Hall Medical, 1985:181–206.
- Elgart ML. Cutaneous sarcoidosis: definitions and types of lesions. Clin Dermatol 1986;4:35–45.
- Mayock RL, Bertrand P, Morrison CE, et al. Manifestations of sarcoidosis: analysis of 145 patients with a review of nine series selected from the literature. Am J Med 1963;35:67–89.
- Marcoval J, Maña J, Moreno A, et al. Subcutaneous sarcoidosis. Clinicopathological study of 10 cases. Br J Dermatol 2005;153:790–4.
- Barrier HJ, Bogoch A. The natural history of the sarcoid granuloma. Am J Pathol 1953;29:451–69.
- Kuramoto Y, Shindo Y, Tagami H. Subcutaneous sarcoidosis with extensive caseation necrosis. J Cutan Pathol 1988;15:188–90.
- Kroll JJ, Shapiro L, Koplon BS, et al. Subcutaneous sarcoidosis with calcification. Arch Dermatol 1972;106:894–5.
- Marcoval J, Mañá J, Moreno A, et al. Foreign bodies in granulomatous cutaneous lesions of patients with systemic sarcoidosis. Arch Dermatol 2001;137:485–6.
- Kim YC, Triffet MK, Gibson LE. Foreign bodies in sarcoidosis. Am J Dermatopathol 2000;22:408–12.
- Callen JP. The presence of foreign bodies does not exclude the diagnosis of sarcoidosis. Arch Dermatol 2001;137:485–6.
Traumatic panniculitis
- Hanif Z, Ahmad M. Subcutaneous fat necrosis presenting as a large mass. Eur J Emerg Med 2006;13:106–7.
- Gschwandter WR, Munzberger H. Lipoatrophia semicircularis. Wien Klin Wochenschr 1975;87:164–8.
- Bloch PH, Rune U. Lipotrophia semicircularis beim Mann: Zusammentreffen von Arterienvariet und Mikrotraumata als mögliche Krankheitsursache. Hautarzt 1978;29:270–2.
- Karkaritas C, Miller JA, Kirby JD. Semicircular lipoatrophy. Br J Dermatol 1981;105:591–3.
- Nagore E, Sánchez-Motilla JM, Rodríguez-Serna M, Vilata JJ, Aliaga A. Lipoatrophia semicircularis – a traumatic panniculitis: report of seven cases and review of the literature. J Am Acad Dermatol 1998;39(5 Pt 2):879–81.
- Gruber PC, Fuller LC. Lipoatrophy semicircularis induced by trauma. Clin Exp Dermatol 2001;26:269–71.
- Gomez-Espejo C, Perez-Bernal A, Camacho-Martinez F. A new case of semicircular lipoatrophy associated with repeated external microtraumas and review of the literature. J Eur Acad Dermatol Venereol 2005;19:459–61.
- Herbert DC, Degeus J. Post-traumatic lipomas on the abdominal wall. Br J Plast Surg 1975;28:303–6.
- Przyjemski CJ, Schuster SR. Nodular-cystic fat necrosis. J Pediatr 1977;91:605–7.
- Sahl WJ Jr. Mobile encapsulated lipomas: formerly called encapsulated angiolipomas. Arch Dermatol 1978;114:1684–6.
- Hurt MA, Santa Cruz DJ. Nodular-cystic fat necrosis. A reevaluation of the so-called mobile encapsulated lipoma. J Am Acad Dermatol 1989;21:493–8.
- Pujol RM, Wang CY, Gibson LE, et al. Lipomembranous changes in nodular-cystic fat necrosis. J Cutan Pathol 1995;22:551–5.
- Kiryu H, Rikihisa M, Furue M. Encapsulated fat necrosis: a clinicopathological study of eight cases and a literature review. J Cutan Pathol 2000;27:19–23.
- Felipo F, Vaquero M, del Augua C. Pseudotumoral encapsulated fat necrosis with diffuse pseudomembranous degeneration. J Cutan Pathol 2004;31:565–7.
- Toritsugi M, Yamamoto T, Nishioka K. Nodular cystic fat necrosis with systemic sclerosis. Eur J Dermatol 2004;14:353–5.
- Demitsu T, Yoneda K, Iida E, et al. A case of nodular cystic fat necrosis with systemic lupus erythematosus presenting the multiple subcutaneous nodules on the extremities. J Eur Acad Dermatol Venereol 2008;22:885–6.
- Sonmez E, Safak T, Kecik A. Giant nodular cystic fat necrosis: a report of a rare case. J Plast Reconstr Aesthet Surg 2009;62:152–4.
- Diaz-Cascajo C, Borghi S. Subcutaneous pseudomembranous fat necrosis: new observations. J Cutan Pathol 2002;29:5–10.
- Pujol RM, Wang C-Y, Gibson LE, Su WPD. Lipomembranous changes in nodular–cystic fat necrosis. J Cutan Pathol 1995;22:551–5.
Lipoatrophic panniculitis of the ankles in childhood
- Shelley WB, Izumi AK. Annular atrophy of the ankles. A case of partial lipodystrophy. Arch Dermatol 1970;102:326–9.
- Shen LY, Edmonson MB, Williams GP, et al. Lipoatrophic panniculitis: Case report and review of the literature. Arch Dermatol 2010;146:877–81.
- Roth DE, Schikler KN, Callen JP. Annular atrophic connective tissue panniculitis of the ankles. J Am Acad Dermatol 1989;21:1152–6.
- Corredera C, Iglesias M, Hernández-Martín A, et al. Annular lipoatrophic panniculitis of the ankles. Pediatr Dermatol 2011;28:146–8.
- Ferreira-Marques J. Lipoatrophia annularis; case of a so far not reported disease of panniculus adiposus. Arch Dermatol Syph 1953;195:479–91.
- Bruinsma W. Lipo-atrophia annularis, an abnormal vulnerability of the fatty tissue. Dermatology 1967;134:107–12.
- Jablonska S, Szczepanski A, Gorkiewicz A. Lipo-atrophy of the ankles and its relation to other lipo-atrophies. Acta Derm Venereol 1975;55:135–40.
- Billings JK, Milgraum SS, Gupta AK, et al. Lipoatrophic panniculitis: A possible autoimmune inflammatory disease of fat. Report of three cases. Arch Dermatol 1987;123:1662–6.
- Winkelmann RK, McEvoy MT, Peters MS. Lipophagic panniculitis of childhood. J Am Acad Dermatol 1989;21:971–8.
- Mirza B, Muir J, Peake J, Whitehead K. Connective tissue panniculitis in a child with vitiligo and Hashimoto's thyroiditis. Australas J Dermatol 2006;47:49–52.
- Nelson HM. Atrophic annular panniculitis of the ankles. Clin Exp Dermatol 1988;13:111–13.
- Melchiorre LP, Rose CD, Hyde PM, et al. Lipophagic granulomatous panniculitis with lipoatrophy mimicking arthritis with pitting edema. J Rheumatol 2000;27:504–6.
- Martinez A, Malone M, Hoeger P, et al. Lipoatrophic panniculitis and chromosome 10 abnormality. Br J Dermatol 2000;142:1034–9.
- Masala MV, Tedde G, Cottoni F. Annular atrophy of the ankles: An unusual case associated with bone anomaly. Dermatology 2001;203:81–2.
- Falcini F, Simonini G, Battini ML, et al. Lipophagic granulomatous panniculitis misdiagnosed as purpura in an 8-year old girl. Clin Exp Rheumatol 2002;20:432.
- Madasseri A, McDermott MB, Irvine AD. Lipoatrophic panniculitis of the ankles. Clin Exp Dermatol 2006;31:303–5.
- Dimson OG, Esterly NB. Annular lipoatrophy of the ankles. J Am Acad Dermatol 2006;54:S40–2.
- Kerns MJ, Stinehelfer S, Mutasim DF, et al. Annular lipoatrophy of the ankles: Case report and review of the literature. Pediatr Dermatol 2011;28:142–5.
- Santonja C, Gonzalo I, Feito M, Beato-Merino M, Requena L. Lipoatrophic panniculitis of the ankles in childhood: differential diagnosis with subcutaneous panniculitis-like T-cell lymphoma. Am J Dermatopathol 2012;34:295–300.
Subcutaneous fat necrosis of the newborn
- Holtzel A. Subcutaneous fat necrosis of the newborn. Arch Dis Child 1951;26:89–91.
- Mogilner BM, Alkaly A, Nissim F, et al. Subcutaneous fat necrosis of the newborn. Clin Pediatr (Phila) 1981;20:748–50.
- Tsuji T. Subcutaneous fat necrosis of the newborn: light and electron microscopic studies. Br J Dermatol 1976;95:407–16.
- Flory CM. Fat necrosis of the newborn. Report of a case with necrosis of the subcutaneous and visceral fat. Arch Pathol 1948;45:278–88.
- Ostwalt GC, Montes LF, Cassady G. Subcutaneous fat necrosis of the newborn. J Cutan Pathol 1978;5:193–9.
- Steiness I. Subcutaneous fat necrosis of the newborn (adiponecrosis subcutanea neonatorum) and maternal diabetes mellitus. Acta Med Scand 1961;170:411–16.
- McDonald R. Subcutaneous fat necrosis and sclerema neonatorum. S Afr Med J 1955;29:1007–12.
- Kohnstam GLS, Hernert FK. Sclerema neonatorum and its relation to fat necrosis. Arch Dis Child 1927;2:349–57.
- Noojin RO, Pace BF, Davis HG. Subcutaneous fat necrosis of the newborn: certain etiologic considerations. J Invest Dermatol1949;12:331–4.
- Blake HA, Goyette EM, Lyter CS. Subcutaneous fat necrosis complicating hypothermia. J Pediatr 1955;46:78–80.
- Duhn R, Schoen EJ, Siu M. Subcutaneous fat necrosis with extensive calcification after hypothermia in two newborn infants. Pediatrics 1968;41:661–4.
- Taieb A, Douard D, Sarlangue J, et al. Trois cas de cystostéatonécrose néonatale: discussion physiopathologique. Presse Méd 1986;15:2197–200.
- Chen TH, Shewmake SW, Hansen DD, et al. Subcutaneous fat necrosis of the newborn: a case report. Arch Dermatol 1981;117:36–7.
- Collins HA, Stahlman M, Scott HW. The occurrence of subcutaneous fat necrosis in an infant following induced hypothermia used as an adjuvant in cardiac surgery. Ann Surg 1953;133:880–5.
- Silverman AK, Michels EH, Rasmussen JE. Subcutaneous fat necrosis in an infant occurring after hypothermic cardiac surgery. J Am Acad Dermatol 1986;15:331–6.
- Glover MT, Catterall MD, Atherton DJ. Subcutaneous fat necrosis in two infants after hypothermic cardiac surgery. Pediatr Dermatol 1991;8:210–12.
- Chuang S-D, Chiu H-C, Chang C-C. Subcutaneous fat necrosis of the newborn complicating hypothermic cardiac surgery. Br J Dermatol 1995;132:805–10.
- Calisici E, Oncel MY, Degirmencioglu H, et al. A neonate with subcutaneous fat necrosis after passive cooling: does polycythemia have an effect? Case Rep Pediatr 2013;2013:254089.
- Akcay A, Akar M, Oncel MY, et al. Hypercalcemia due to subcutaneous fat necrosis in a newborn after total body cooling. Pediatr Dermatol 2013;30:120–3.
- Katz DA, Huerter C, Bogard P, Braddock SW. Subcutaneous fat necrosis of the newborn. Arch Dermatol 1984;120:1517–18.
- Silverman AK. Panniculitis in infants. Arch Dermatol1985;121:834.
- Sharata H, Postellon DC, Hashimoto K. Subcutaneous fat necrosis, hypercalcemia, and prostaglandin E. Pediatr Dermatol 1995;12:43–7.
- Burden AD, Krafchik BR. Subcutaneous fat necrosis of the newborn: a review of 11 cases. Pediatr Dermatol 1999;16:384–7.
- Cook JS, Stone MS, Hansen JR. Hypercalcaemia in association with subcutaneous fat necrosis of the newborn: studies of calcium regulating hormones. Pediatrics 1992;90:93–6.
- Finne PH, Sanderud J, Aksnes L, et al. Hypercalcemia with increased and unregulated 1,25-dihydroxyvitamin D production in a neonate with subcutaneous fat necrosis. J Pediatr 1988;112:792–4.
- Kruse K, Irle U, Uhlig R. Elevated 1,25-dihydroxy-vitamin D serum concentrations in infants with subcutaneous fat necrosis. J Pediatr 1993;122:460–3.
- Bachrach LK, Lum CK. Etidronate in subcutaneous fat necrosis of the newborn. J Pediatr 1999;135:530–1.
- Thomsen RJ. Subcutaneous fat necrosis of the newborn and idiopathic hypercalcaemia: report of a case. Arch Dermatol 1980;116:1155–8.
- Veldhuis JD, Kulin HE, Demers LM, et al. Infantile hypercalcaemia with subcutaneous fat necrosis: endocrine studies. J Pediatr 1979;95:460–2.
- Sharata H, Postellon DC, Hashimoto K. Subcutaneous fat necrosis of the newborn, hypercalcemia and prostaglandin E. Pediatr Dermatol 1995;12:43–7.
- Tran JT, Sheth AP. Complications of subcutaneous fat necrosis of the newborn: a case report and review of the literature. Pediatr Dermatol 2003;20:257–61.
- Borgia F, De Pasquale L, Cacace C, Meo P, Guarneri C, Cannavo SP. Subcutaneous fat necrosis of the newborn: be aware of hypercalcaemia. J Paediatr Child Health 2006;42:316–18.
- Wiadrowski TP, Marshman G. Subcutaneous fat necrosis of the newborn following hypothermia and complicated by pain and hypercalcaemia. Australas J Dermatol 2001;42:207–10.
- Norwood-Galloway A, Lebwohl M, Phelps RG, Raucher H. Subcutaneous fat necrosis of the newborn with hypercalcemia. J Am Acad Dermatol 1987;16:435–9.
- Wolach B, Raas-Rothchild A, Vogel R, et al. Subcutaneous fat necrosis with thrombocytopenia in a newborn infant. Dermatologica 1990;181:54–5.
- Vonk J, Janssens PMW, Demacker PNM, Folkers E. Subcutaneous fat necrosis in a neonate, in association with aberrant plasma lipid and lipoprotein values. J Pediatr 1993;123:462–4.
- Canpolat N, Ozdil M, Kuruğoğlu S, Calışkan S, Sever L. Nephrocalcinosis as a complication of subcutaneous fat necrosis of the newborn. Turk J Pediatr 2012;54:667–70.
- Fretzin DF, Arias AM. Sclerema neonatorum and subcutaneous fat necrosis of the newborn. Pediatr Dermatol 1987;4:112–22.
- Tajirian A, Ross R, Zeikus P, Robinson-Bostom L. Subcutaneous fat necrosis of the newborn with eosinophilic granules. J Cutan Pathol 2007;34:588–90.
- Montes LF. Subcutaneous fat necrosis of the newborn with eosinophilic granules. J Cutan Pathol 2008;35:699.
- Farinelli P, Gattoni M, Delrosso G, et al. Eosinophilic granules in subcutaneous fat necrosis of the newborn: what do they mean? J Cutan Pathol 2008;35:1073–4.
- Balazs M. Subcutaneous fat necrosis of the newborn with emphasis on ultrastructural studies. Int J Dermatol 1987;26:227–30.
- Pasyk K. Studies on subcutaneous fat necrosis of the newborn. Virchows Arch 1978;379:243–59.
- Friedman SJ, Winkelmann RK. Subcutaneous fat necrosis of the newborn: light, ultrastructural and histochemical microscopic studies. J Cutan Pathol 1989;16:99–105.
- Taieb A, Douard D, Maleville J. Subcutaneous fat necrosis and brown fat deficiency. J Am Acad Dermatol 1987;26:227–30.
- Lund JJ, Xia L, Kerr S, Stratman EJ, Patten SF. The utility of a touch preparation in the diagnosis of fluctuant subcutaneous fat necrosis of the newborn. Pediatr Dermatol 2009;26:241–3.
- Schubert PT, Razack R, Vermaak A, Jordaan HF. Fine-needle aspiration cytology of subcutaneous fat necrosis of the newborn: the cytology spectrum with review of the literature. Diagn Cytopathol 2012;40:245–7.
- Alos N, Eugène D, Fillion M, Powell J, Kokta V, Chabot G. Pamidronate: Treatment for severe hypercalcemia in neonatal subcutaneous fat necrosis. Horm Res 2006;65:289–94.
- Lombardi G, Cabano R, Bollani L, Del Forno C, Stronati M. Effectiveness of pamidronate in severe neonatal hypercalcemia caused by subcutaneous fat necrosis: a case report. Eur J Pediatr 2009;168:625–7.
Poststeroid panniculitis
- Smith RT, Good RA. Sequelae of prednisone treatment of acute rheumatic fever. Clin Res Proc 1956;4:156–7.
- Spagnuolo M, Taranta A. Post-steroid panniculitis. Ann Intern Med 1961;4:1181–90.
- Roenigk HH Jr, Haserick JR, Arundell FD. Poststeroid panniculitis. Arch Dermatol 1964;90:387–91.
- Browne SG. Nodular panniculitis and peripheral neuropathy following sudden withdrawal of corticosteroids. (Poststeroid nodular panniculitis.) Int J Lepr 1965;33:893–5.
- Browne SG. Poststeroid nodular panniculitis and the erythema nodosum of leprosy. Dermatol Int 1965;4:215–18.
- Jaffe N, Hann HWL, Vauter GF. Post-steroid panniculitis in acute leukaemia. N Engl J Med 1971;284:366–7.
- Hirokawa K, Nishido T, Okuda M. Post-steroid panniculitis with cerebral vascular and pulmonary complications – report of an autopsy case. Acta Pathol Jpn 1972;22:565–79.
- Saxena AK. Post steroid panniculitis. Australas J Dermatol 1986;27:143.
- Silverman RA, Newman AJ, LeVine MJ, Kaplan B. Poststeroid panniculitis: a case report. Pediatr Dermatol 1988;5:92–3.
- Reichel M, Diaz Cascajo C. Bilateral jawline nodules in a child with a brain-stem glioma. Poststeroid panniculitis. Arch Dermatol 1995;131:1448–9, 1451–2.
- Kwon EJ, Emanuel PO, Gribetz CH, Mudgil AV, Phelps RG. Poststeroid panniculitis. J Cutan Pathol 2007;34 Suppl. 1:64–7.
- Kim ST, Kim TK, Lee JW, et al. Post-steroid panniculitis in an adult. J Dermatol 2008;35:786–8.
- de-Andrés-del-Rosario A, Verea-Hernando MM, Yebra-Pimentel MT, Rosende-Maceiras L, Piñeyro-Molina F, Capdevila EF. Poststeroid panniculitis in an adult. Am J Dermatopathol 2011;33:e77–80.
- Peralta L, Morais P, Trindade E, Barreto F, Canelhas Á, Amil J. Poststeroid panniculitis: a rare complication of systemic corticosteroid therapy. Cutan Ocul Toxicol 2012;31:164–6.
- Marovt M, Miljković J. Post-steroid panniculitis in an adult. Acta Dermatovenerol Alp Panonica Adriat 2012;21:77–8.
Sclerema neonatorum
- Zeb A, Darmstadt GL. Sclerema neonatorum: a review of nomenclature, clinical presentation, histological features, differential diagnoses and management. J Perinatol 2008;28:453–60.
- Levin SE, Bakst CM, Isserow L. Sclerema neonatorum treated with corticosteroids. BMJ 1961;2:1533–6.
- Warwick W, Ruttenberg HD, Quie PG. Sclerema neonatorum: a sign not a disease. JAMA 1963;184:680–3.
- Molteni RA, Ames MR. Sclerema neonatorum and joint contractures at birth as a potential complication of chronic in utero hypoxia. Am J Obstet Gynecol 1986;155:380–1.
- Zeb A, Rosenberg RE, Ahmed NU, et al. Risk factors for sclerema neonatorum in preterm neonates in Bangladesh. Pediatr Infect Dis J 2009;28:435–8.
- Bower BD, Jones LF, Weeks MM. Cold injury of the newborn: a study of 70 cases. BMJ 1960;1:303–9.
- Rafstedt S. Studies in serum lipids and lipoproteins in infancy and childhood. Acta Paediatr Scand 1955;44 (Suppl. 102):1906–7.
- Channon HJ, Harrison GA. The chemical nature of the subcutaneous fat in the normal and sclerematous infant. Biochem J 1926;20:84–92.
- Kellum RE, Ray TL, Brown GR. Sclerema neonatorum: report of a case and analysis of subcutaneous and epidermal lipids by chromatographic methods. Arch Dermatol 1968;97:372–80.
- Hughes WE, Hammond ML. Sclerema neonatorum. J Pediatr 1948;32:676–92.
- Yao Y, Gong F, Xiong Y, Xiong F, Tang S. Observation on the changes of lipid peroxidation in neonates with sclerema. Hua Xi Yi Ke Da Xue Xue Bao 1997;28:440–1.
- Fretzin DF, Arias AM. Sclerema neonatorum and subcutaneous fat necrosis of the newborn. Pediatr Dermatol 1987;4:112–22.
- Jardine D, Atherton DJ, Trompeter RS. Sclerema neonatorum and subcutaneous fat necrosis of the newborn in the same infant. Eur J Pediatr 1990;50:125–6.
- Mir-Bonafé JM, Román-Curto C, Santos-Briz A, Cañueto J, Fernández-López E, Unamuno P. Gemcitabine-associated livedoid thrombotic microangiopathy with associated sclerema neonatorum-like microscopic changes. J Cutan Pathol 2012;39:707–11.
- Pasyk K. Sclerema neonatorum: light microscopic studies. Virchows Arch 1980;388:87–103.
- Horsfield GI, Yardley HJ. Sclerema neonatorum. J Invest Dermatol 1965;44:326–32.
- Pearse RG, Sauer PJ. Exchange transfusion in treatment of serious infection in the newborn and sclerema neonatorum. Arch Dis Child 1978;53:262.
- Pelet B. C3, factor B, alpha-1-antitrypsin in neonatal septicaemia with sclerema. Arch Dis Child 1980;55:782–8.
- Buster KJ, Burford HN, Stewart FA, Sellheyer K, Hughey LC. Sclerema neonatorum treated with intravenous immunoglobulin: a case report and review of treatments. Cutis 2013;92:83–7.
Gouty panniculitis
- Niemi KM. Panniculitis of the legs with urate crystal deposition. Arch Dermatol 1977;113:655–6.
- LeBoit PE, Schneider S. Gout presenting as lobular panniculitis. Am J Dermatopathol 1987;9:334–8.
- Conejo-Mir J, Pulpillo A, Corbi MR, et al. Panniculitis and ulcers in a young man. Arch Dermatol 1998;134:499–504.
- Snider AA, Barsky S. Gouty panniculitis: a case report and review of the literature. Cutis 2005;76:54–6.
- Dahiya A, Leach J, Levy H. Gouty panniculitis in a healthy male. J Am Acad Dermatol 2007;57(2 Suppl.):S52–4.
- Weberschock T, Gholam P, Hartschuh W, Hartmann M. Gouty panniculitis in a 68-year-old man: case report and review of the literature. Int J Dermatol 2010;49:410–3.
- Ochoa CD, Valderrama V, Mejia J, et al. Panniculitis: another clinical expression of gout. Rheumatol Int 2011;31:831–5.
- Peñaranda-Parada E, Bejarano NS, et al. Gouty arthritis and panniculitis: extensive involvement of the dermis and severe joint damage. J Clin Rheumatol 2012;18:142–3.
- Choi CM, Lew BL, Lee SH, Sim WY. Gouty panniculitis also involving the bone marrow. Acta Derm Venereol 2013;93:189–90.
Fungal panniculitis due to zygomycosis, mucormycosis and aspergillosis
- Vernon SE, Dave SP. Cutaneous zygomycosis associated with urate panniculitis. Am J Dermatopathol 2006;28:327–30.
- Requena L, Sitthinamsuwan P, Santonja C, et al. Cutaneous and mucosal mucormycosis mimicking pancreatic panniculitis and gouty panniculitis. J Am Acad Dermatol 2012;66:975–84.
- Colmenero I, Alonso-Sanz M, Casco F, Hernández-Martín A, Torrelo A. Cutaneous aspergillosis mimicking pancreatic and gouty panniculitis. J Am Acad Dermatol 2012;67:789–91.
Cytophagic histiocytic panniculitis and subcutaneous panniculitis-like T-cell lymphoma
- Winkelmann RK, Bowie EJ. Haemorrhagic diathesis associated with benign histiocytic, cytophagic panniculitis and systemic histiocytosis. Arch Intern Med 1980;140:1460–3.
- Wick MR, Patterson JW. Cytophagic histiocytic panniculitis – a critical reappraisal. Arch Dermatol 2000;136:922–4.
- Kumar S, Krenacs L, Medeiros J, et al. Subcutaneous panniculitic T-cell lymphoma is a tumor of cytotoxic T lymphocytes. Hum Pathol 1998;29:397–403.
- Salhany KE, Macon WR, Choi JK, et al. Subcutaneous panniculitis-like T-cell lymphoma: clinicopathologic, immunophenotypic, and genotypic analysis of alpha/beta and gamma/delta subtypes. Am J Surg Pathol 1998;22:881–93.
- Hoque SR, Child FJ, Whittaker SJ, et al. Subcutaneous panniculitis-like T-cell lymphoma: clinicopathological, immunophenotypic and molecular analysis of six patients. Br J Dermatol 2003;148:516–25.
- Santucci M, Pimpinelli MN, Massi D, et al. Cytotoxic/natural killer cell cutaneous lymphomas. Report of EORTC Cutaneous Lymphoma Task Force Workshop. Cancer 2003;97:610–27.
- Takeshita M, Imayama S, Oshiro Y, et al. Clinicopathologic analysis of 22 cases of subcutaneous panniculitis-like CD56- or CD56+ lymphoma and review of 44 other reported cases. Am J Clin Pathol 2004;121:408–16.
- Craig AJ, Cualing H, Thomas G, et al. Cytophagic histiocytic panniculitis – a syndrome associated with benign and malignant panniculitis: case comparison and review of the literature. J Am Acad Dermatol 1998;39:721–36.
- Willemze R, Jansen PM, Cerroni L, et al. Subcutaneous panniculitis-like T-cell lymphoma: definition, classification and prognostic factors: an EORTC Cutaneous Lymphoma Group Study of 83 cases. Blood 2008;11:838–45.
- Lozzi CP, Massone C, Citarella L, et al. Rimming of adipocytes by neoplastic lymphocytes: a histopathologic feature not restricted to subcutaneous T-cell lymphoma. Am J Dermatopathol 2006;28:9–12.
Sclerosing postirradiation panniculitis
- Winkelmann RK, Grado GL, Quimby SR, Connolly SM. Pseudosclerodermatous panniculitis after irradiation: an unusual complication of megavoltage treatment of breast carcinoma. Mayo Clin Proc 1993;68:122–7.
- Carrasco L, Moreno C, Pastor MA, et al. Postirradiation pseudosclerodermatous panniculitis. Am J Dermatopathol 2001;23:283–7.
- Dalle S, Skowron F, Ronger-Savlè S, Balme B, Thomas L. Pseudosclerodermatous panniculitis after irradiation and bronchiolitis obliterans organizing pneumonia: simultaneous onset suggesting a common origin. Dermatology 2004;209:138–4.
- Requena L, Ferrándiz C. Sclerosing postirradiation panniculitis. Dermatol Clin 2008;26:505–8.
- Pielasinski U, Machan S, Camacho D, et al. Postirradiation pseudosclerodermatous panniculitis: three new cases with additional histopathologic features supporting the radiotherapy etiology. Am J Dermatopathol 2013;35:129–34.
- Walsh N, Rheaume D, Barnes P, Tremaine R, Reardon M. Postirradiation morphea: an underrecognized complication of treatment for breast cancer. Hum Pathol 2008;39:1680–8.
- Schaffer JV, Carroll C, Dvoretsky I, Huether MJ, Girardi M. Postirradiation morphea of the breast presentation of two cases and review of the literature. Dermatology 2000;200:67–71.
- Davis DA, Cohen PR, McNeese MD, Duvic M. Localized scleroderma in breast cancer patients treated with supervoltage external beam radiation: radiation port scleroderma. J Am Acad Dermatol 1996;35:923–7.
- Fraga J, García-Díez A. Lupus erythematosus panniculitis. Dermatol Clin 2008;26:453–63.
- Liegner LM, Michaud NJ. Skin and subcutaneous reactions induced by supervoltage irradiation. Am J Roentgenol Radium Ther Nucl Med 1961;85:533–49.
- Fajardo LF, Berthrong M. Radiation injury in surgical pathology. Part I. Am J Surg Pathol 1978;2:159–99.
- Butler MJ, Lane RH, Webster JH. Irradiation injury to large arteries. Br J Surg 1980;67:341–3.