CHAPTER 100
Other Acquired Disorders of Subcutaneous Fat
Amy Y.-Y.1 and ChenAmit Garg2
1Department of Dermatology, University of Connecticut School of Medicine, Canton, CT, USA
2Department of Dermatology, Hofstra NSLIJ School of Medicine, Manhasset, NY, USA
Introduction
This chapter addresses principally non-inflammatory acquired disorders of subcutaneous fat with an emphasis on acquired lipodystrophy, fat hypertrophy, subcutaneous lipomatosis and lipoedema. While some of the entities discussed are very common, such as cellulite and obesity, most are much more rare. Panniculitis and genetic disorders of subcutaneous fat are addressed in Chapters 99 and 74, respectively.
ACQUIRED LIPODYSTROPHY
Acquired lipodystrophy refers to a heterogeneous group of disorders in which there is localized, partial or generalized loss of subcutaneous fat (lipoatrophy), in certain cases accompanied by fat accumulation in other body sites.
Acquired generalized lipodystrophy
Definition and nomenclature
Acquired generalized lipodystrophy (AGL) is a rare disease characterized by a selective loss of adipose tissue from large regions of the body, occurring after birth [1].
Introduction and general description
After its initial report by Ziegler in 1928 [2], AGL was described in more detail through autopsy findings by Lawrence in 1946 [3]. Although loss of adipose tissue in AGL most often occurs during childhood and adolescence, a few cases report AGL in individuals over the age of 65 years [4, 5]. The pattern and extent of fat loss in AGL is variable. Most patients have generalized loss of fat, though fat may be spared in some areas, such as retro-orbital fat. In addition to the loss of adipose tissue, patients often develop severe hepatic steatosis and fibrosis, severe insulin resistance and hyperinsulinaemia, hypertriglyceridaemia and low serum high-density lipoprotein (HDL) levels [6–9].
Epidemiology
Incidence and prevalence
Acquired generalized lipodystrophy is a rare disease and thus its incidence and prevalence are difficult to estimate. Less than 100 cases were identified in a literature review published in 2003 [10]. More recently, in 2012, it was estimated that AGL affects approximately one in 100 000 people in the European Union.
Age
Most patients with AGL present during childhood or adolescence.
Sex
Women are affected three times more often than men [10].
Ethnicity
The majority of patients have been white, although AGL has also been reported in Hispanic and East Asian patients [10, 11].
Associated diseases
Most AGL patients have some degree of metabolic derangement, including fasting and/or postprandial hyperinsulinaemia, hypertriglyceridaemia, low serum levels of HDL, and low leptin and adiponectin [6–9, 10]. Diabetes most often occurs subsequent to the onset of AGL, though in some it may present before or at the time of onset [10]. AGL patients usually do not develop diabetic ketoacidosis [12]. Patients may also have increased basal metabolic rate and complain of fatigue and voracious appetite [10].
Hepatomegaly occurs in 70–100% of AGL patients. Hepatic steatosis or non-alcoholic steatohepatitis results in mild to moderate elevation of serum transaminases. Splenomegaly may result from portal hypertension and cirrhosis [10].
Cardiomyopathy may be associated with AGL. In a 2011 study of left ventricular mass in 13 patients with AGL, three had mild and three had moderate ventricular hypertrophy. Abnormalities were seen in five of 11 AGL patients whose electrocardiogram was available for analysis [13].
Muscle and neurological involvement have rarely been reported [5, 11].
Reproductive capacity is normal in male patients with AGL. Female AGL patients may have normal reproduction although irregular menses are common [10]. Primary or secondary amenorrhoea rarely occurs [10, 14, 15]. Single [16] or multiple [17, 18] bone lucencies and cysts have been reported in AGL patients, though the clinical significance of these lesions is not clear [18]. Lymphadenopathy has also been reported in some [7, 18–20].
Pathophysiology
The mechanism of fat loss in AGL is unknown. Despite reports of a variety of preceding infections, it is not clear that these infections directly cause AGL [21, 22]. The classic complement pathway is postulated to be involved in the pathogenesis among AGL patients with autoimmune hepatitis and low serum complement [23, 24]. Antibody-mediated destruction or cell-mediated lysis of adipocytes has also been considered [25].
The consequences are that there is an insufficient mass of adipose tissue to store excess energy, which is stored instead as triglyceride in the liver and skeletal muscle, and that there is a perpetual elevation of plasma free fatty acid (FFA), resulting in an impaired β-cell response to glucose and insulin resistance [26–29]. Low serum leptin and adiponectin levels, reflecting the low amount of body fat in these patients, may further contribute to severe insulin resistance and the metabolic complications observed in AGL [30–33].
Pathology
Acquired generalized lipodystrophy is a clinical diagnosis, though histopathology may help confirm the diagnosis. Tissue examination demonstrates a complete or near-complete absence of subcutaneous fat, with the dermis and fascia in direct apposition. If adipocytes are present, they are markedly reduced in number and size and they are arranged in small groups surrounded by abundant connective tissue [34].
Genetics
No known genetic mutation or familial cluster has been identified.
Clinical features
Presentation
In contrast to congenital lipodystrophy, patients with AGL have normal fat density and distribution at birth. The onset of fat loss is typically insidious over months to years (Figure 100.1), although rapid progression over weeks has been observed [10]. Rarely, the process of fat destruction may occur rapidly in one area and stay quiescent over months to years, only to become active again later and result in generalized fat loss [2, 10]. The extent and degree of fat loss is variable. Usual sites of involvement include the face (Figure 100.1a), trunk, abdomen and extremities. Underlying veins and musculature become prominent with severe fat loss (Figure 100.1b). In some, loss of fat may also involve the palms, soles and abdominal cavity. Generally, marrow and retro-orbital fat are preserved.

Figure 100.1 Physical examination revealed (a) lipoatrophy of the face and (b) generalized lipoatrophy of the torso with visible subcutaneous veins. (c) A photograph of the patient taken 5 years earlier, in 2008, is shown for comparison. (Reproduced with permission from Aslam A, Savage DB, Coulson IH. Acquired generalized lipodystrophy associated with peripheral T cell lymphoma with cutaneous infiltration. Int J Dermatol 2015;54:827–9.)
Acanthosis nigricans of the axillae, groin, neck, umbilicus and nipples is noted in 45–64% of AGL patients [10]. Other less common dermatological findings include localized [35, 36] or generalized hyperpigmentation [37, 38], telangiectasia [19] and hyperkeratosis of the palms and soles [16]. Women with AGL may have mild hirsutism [10, 37, 39, 40]. Rarely, virilization with temporal recession of hair and acne have been reported [14, 41]. Alopecia [6] and curly hair [7, 10, 16, 21, 37, 40, 42] have also been observed. Acromegaloid facial features with large hands and feet may rarely be seen [9, 10].
In 2003, Misra and Garg proposed new diagnostic criteria applicable to the broad spectrum of AGL patients. Essential criteria include selective loss of body fat affecting large regions of the body, beginning after birth but usually before adolescence. Supportive clinical criteria include loss of subcutaneous fat from the palms and soles, a preceding history of tender subcutaneous nodular swellings, histological confirmation from involved tissue, acanthosis nigricans, hepatosplenomegaly and the presence of other autoimmune diseases.
Supportive laboratory criteria include impaired glucose tolerance, severe fasting and/or postprandial hyperinsulinaemia, hypertriglyceridaemia, low serum HDL, low serum leptin and/or adiponectin, and evidence by magnetic resonance imaging (MRI) of fat loss from large regions of the body with preserved bone marrow fat [10]. The number of these secondary clinical or laboratory criteria needed to support a diagnosis of AGL has not been formally established.
Clinical variants
Acquired generalized lipodystrophy is subdivided into three types. Type I AGL, in which initially localized panniculitis precedes generalized fat loss, accounts for approximately 25% of cases [1]. Type II AGL, which accounts for another 25% of cases, is associated with autoimmune diseases including haemolytic anaemia, chronic autoimmune hepatitis, Hashimoto thyroiditis, juvenile rheumatoid arthritis, juvenile dermatomyositis and vitiligo [6, 23, 35, 36, 43, 44, 45]. Juvenile dermatomyositis may have the strongest association with AGL [10]. The remaining patients have the most common subtype, type III (idiopathic) AGL, in which no triggers or associated diseases are identified.
Differential diagnosis
Some AGL patients may initially present with localized or partial lipodystrophy and hence may be misclassified as having acquired partial lipodystrophy or localized lipodystrophy.
AGL patients are differentiated from congenital generalized lipodystrophy (CGL) patients who have near-complete generalized fat loss at birth (see Chapter 74). Furthermore, CGL patients have advanced bone age and mental retardation. As with AGL, CGL patients may have acromegaloid features, lytic bone lesions and cardiomyopathy [10]. Lytic bone lesions are, however, more common and the cardiomyopathy more severe in CGL [10, 13]. MRI studies of CGL patients show an absence of ‘metabolically active’ fat in intra-abdominal and intrathoracic regions, and in bone marrow, while marrow fat is preserved in AGL patients [10, 46]. Genetic analysis for known mutations may aid in the confirmation of CGL.
While patients with mandibuloacral dysplasia may have generalized fat loss, these patient also have skeletal anomalies (see Chapter 72). The identification of associated genetic mutations can help to confirm this diagnosis.
Family histories as well as genotyping will also help differentiate AGL from autosomal dominant familial partial lipodystrophy (see Chapter 74) [1].
Complications and co-morbidities
Patients with AGL may suffer complications from metabolic disturbances and other associated co-morbid conditions in association with type II AGL. Retinopathy, nephropathy and neuropathy are common complications because of the patients’ longstanding diabetes. Hypertriglyceridaemia may lead to eruptive xanthomas, lipaemia retinalis and acute pancreatitis [10]. These metabolic abnormalities also predispose AGL patients to premature atherosclerosis and coronary heart disease [6, 10].
Disease course and prognosis
The loss of subcutaneous fat tissue in AGL patients is permanent. The prognosis of AGL patients largely depends on the course and management of co-morbidities.
Investigations
Laboratory and ancillary testing is pursued to establish the presence and monitor the course of co-morbid diseases. While serum leptin is not useful for establishing the diagnosis, it may predict response to replacement therapy with the synthetic recombinant analogue of human leptin, metreleptin [1].
Management
There is no established management algorithm for AGL. Subcutaneous fat loss is irreversible. To the extent feasible, cosmetic procedures such as filler injections, autologous adipose tissue transfer and muscle tissue transfers may help correct volume losses [1, 47, 48]. Optimal management of co-morbid conditions requires collaboration between primary care physicians and several specialists.
In one trial, which included three AGL patients among others with various lipodystrophies, 4 months of twice-daily subcutaneous metreleptin injections were shown to be safe and effective. There was a significant decrease in fasting blood glucose level and glycosylated haemoglobin in two patients. In three patients with hypertriglyceridaemia, fasting levels of plasma triglycerides decreased by 83%. In these patients, fasting plasma triglycerides increased soon after discontinuation of the injections and were corrected once again after reinitiation of the therapy [49]. Furthermore, liver volume and serum transaminases decreased significantly during metreleptin therapy, suggesting that it reduced hepatic steatosis [50]. Metreleptin has been approved under the orphan designation for the treatment of AGL in the European Union since July 2012; it remains investigational in the USA.
Acquired partial lipodystrophy
Definition and nomenclature
Acquired partial lipodystrophy (APL) is a rare disease characterized by symmetrical fat loss, usually occurring before the age of 15 years [1, 2].
Introduction and general description
Acquired partial lipodystrophy was first reported by Mitchell [3] and later by Barraquer [4] and by Simons [5]. Although rare, APL is the most common of the non-localized lipodystrophies, other than HIV-associated lipodystrophy. It is characterized by symmetrical and insidious although progressive fat loss starting from the face and scalp and gradually progressing downward to involve the neck, shoulders, upper extremities, thoracic region and upper abdomen. Involvement of the lower extremities is uncommon [1, 2].
Epidemiology
Incidence and prevalence
Because it is rare, the incidence and prevalence of APL is difficult to estimate. By 2000, there were approximately 250 cases reported in the English literature [1].
Age
Onset is typically before the age of 15 years, with a median of 8 years [1, 2].
Sex
Women are affected approximately three times more commonly than men [1].
Ethnicity
Most reported patients have been white.
Associated diseases
Approximately one-third of patients develop mesangiocapillary glomerulonephiritis (MCGN), usually more than 10 years after the onset of lipodystrophy [1]. Systemic lupus erythematosus has been associated with APL and has occurred 2–28 years after the onset of lipodystrophy [6–9]. Other autoimmune diseases, such as dermatomyositis [10], leukocytoclastic vasculitis [11], dermatitis herpetiformis and coeliac diseases [12], hypothyroidism, pernicious anemia [13], rheumatoid arthritis [8] and temporal arteritis, have also been reported. More recently, APL has been reported in the setting of chronic sclerodermatous graft-versus-host-disease, POEMS (polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy and skin changes) syndrome and extrinsic allergic alveolitis, as well as central nervous system (CNS) disorders including epilepsy, sensorineural deafness and mental retardation (see Chapter 148) [14–17].
Pathophysiology
There is evidence to support an autoimmune-mediated destruction of adipocytes in APL. Approximately 80–90% of APL patients have a serum immunoglobulin G named C3 nephritic factor [18, 19]. This blocks the degradation of the enzyme C3 convertase, which leads to excessive consumption of C3. As a result, serum C3 levels are low in more than 80% of APL patients [20]. Levels of C1q, C4, C5 and C6 and factors B and P are usually normal, suggesting selective activation of the alternative complement pathway [21]. Lysis of adipocytes may be related to the expression of several complement proteins such as factors D (adipsin), B, H and P [22, 23, 24]. For example, in vitro studies suggest that the C3 nephritic factor causes lysis in adipocytes expressing factor D [22]. Heterogeneity of factor D expression in adipose tissue in different anatomical locations has been postulated to explain the selective loss of upper body fat in APL [23].
In those APL patients without C3 nephritic factor, other immune abnormalities are postulated to be relevant pathogenetic factors. In a paediatric APL patient without C3 nephritic factor, serum tumour necrosis factor α (TNF-α) and interleukin 6 (IL-6) were noted to be elevated [25]. TNF-α has been shown to cause apoptosis in adipocyte cultures, and IL-6 stimulates lipolysis in human adipocytes [26, 27]. TNF-α also influences the complement pathway by controlling factor D production in adipocytes [25].
Pathology
Acquired partial lipodystrophy is a clinical diagnosis based on the presentation and progression of disease. Histopathology, in common with AGL, shows complete or near-complete absence of subcutaneous fat, with the dermis and fascia in direct apposition. If adipocytes are present, they are markedly reduced in number and size, and they are arranged in small groups surrounded by abundant connective tissues [28].
Causative organisms
Although fat loss has been preceded by infection in some reported cases of APL, the relationship between APL and infection is unclear [23].
Genetics
LMNB2 mutations have been reported in five patients with APL, although some of these had atypical presentations [29].
Clinical features
Presentation
Fat loss in APL occurs symmetrically, starting on the face and scalp, then gradually spreading to involve the neck, shoulders, upper extremities, thoracic region and upper abdomen (Figure 100.2). Involvement of the inguinal region or thighs is uncommon. Fat in the hips and lower extremities is unaffected. In fact, affected women frequently accumulate excess fat in these regions after puberty. Fat loss typically progresses over a period of about 18 months, although it may continue for several years. Orbital, mediastinal, gluteal, intramuscular, intraperitoneal, perirenal and bone marrow fat is usually unaffected [1, 2].
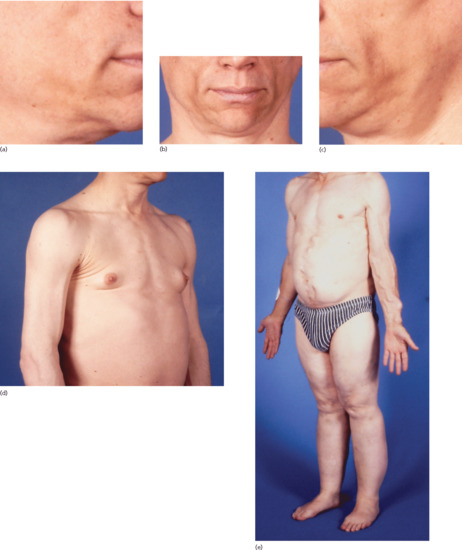
Figure 100.2 Acquired partial lipodystrophy (APL): (a–c) marked loss of facial fat resulting in a prematurely aged appearance and (d) prominence of arm veins, sternomastoid muscles and breast tissue resulting from subcutaneous fat loss in a 31-year-old man with APL and renal failure; (e) note preservation of subcutaneous fat in the lower half of the body contrasting with dramatic loss of subcutaneous fat in the upper half of the body in a 50-year-old man whose longstanding APL was not recognized until he presented with accelerated hypertension secondary to APL-related glomerulonephritis.
Clinical variants
Three phenotypic subtypes have been recognized: (i) upper body fat loss; (ii) upper body fat loss with hypertrophy of adipose tissue in the lower half of the body; and (iii) hemi-lipodystrophy in which only one side of the face or body is affected.
Differential diagnosis
Acquired partial lipodystrophy is differentiated from familial partial lipodystrophy in which there is a family history of similar lipodystrophy, variations in clinical presentation of fat loss and identified genetic mutations. A history of trauma and medication history can help differentiate other forms of acquired lipodystrophy from APL. APL is differentiated from AGL based on the extent of involvement as well as sparing of the intra-abdominal fat.
Complications and co-morbidities
Unlike in patients with AGL, insulin resistance, diabetes, dyslipidaemia, acanthosis nigricans, hirsutism or menstrual abnormalities are less common in APL patients [1].
Disease course and prognosis
Fat loss is irreversible in APL. Overall prognosis is driven by the presence of co-morbidities.
Investigations
The initial evaluation of APL patients should include serum C3 level and C3 nephritic factor, as well as baseline screening for metabolic derangements with a comprehensive metabolic panel, fasting glucose, lipid panels and insulin level. Evaluation should also include assessment of associated co-morbid conditions. Periodic and continued monitoring for renal disease and autoimmunity is warranted. MRI may demonstrate the extent of fat loss if needed.
Management
There is no established management algorithm for AGL. Subcutaneous fat loss is irreversible. To the extent feasible, cosmetic procedures such as filler injections, autologous adipose tissue transfer and muscle tissue transfers may help correct volume losses. However, the durable efficacy and safety of these approaches have not been extensively investigated. The identification of neutralizing antibodies against C3 nephritic factors in intravenous immunoglobulin (IVIg) has led to treatment of patients who have C3 nephritic factors and type II MCGN with IVIg with encouraging results [30, 31]. Optimal management of co-morbid conditions requires collaboration between primary care physicians and several specialists.
HIV-associated lipodystrophy
Definition and nomenclature
Lipodystrophy in patients affected with human immunodeficiency virus (HIV) is associated with highly active antiretroviral therapy (HAART) regimens containing protease inhibitor (PI) or nucleoside reverse transcriptase inhibitor (NRTI). It is now the most prevalent type of lipodystrophy, in which both lipoatrophy and lipohypertrophy may be observed (see Chapter 31).
Introduction and general description
The first report of fat redistribution in an HIV-infected individual undergoing antiretroviral therapy including a PI was in 1997 [1]. Lipodystrophy in HIV-infected patients usually appears after patients have been receiving PI- or NRTI-containing HAART regimens for at least 2 years [2]. Currently, there is no consensus on the definition or diagnostic criteria for lipodystrophy in the HIV-infected patient [3]. In 2003, Carr et al. established a diagnostic model that included the variables of age, sex, known duration of HIV infection, HIV disease stage, waist to hip ratio, anion gap, serum HDL cholesterol level and trunk to peripheral fat ratio [4]. Although follow-up prospective studies confirmed its high diagnostic sensitivity, the complexity of the model is thought to impede its use in daily clinical practice [5].
Epidemiology
Incidence and prevalence
Lipodystrophy in HIV-infected patients is the most prevalent among lipodystrophies. Due to limitations in the definition, selection of study population and duration of follow-up, there are considerable differences in its reported incidence and prevalence. Prevalence ranges from 8% to 84% with an average of 42% [6]. Average incidence ranges from 7.3 to 11.7 per 100 patient-years [7, 8]. Generally, higher prevalence is reported among patients receiving long-term therapy. In pooled analyses, the prevalence is 17% among adults treated with PI-containing therapy for less than 1 year and 43% in those treated for more than 1 year [6]. Each additional 6 months of treatment with HAART is associated with a 1.57 times increased risk of lipodystrophy [7]. It is expected that the prevalence will increase in the future with longer follow-up and continued use of HAART [6].
Age
The risk of developing HIV-associated lipodystrophy increases with age [6, 9]. Children receiving PI-containing HAART therapy exhibit a similar redistribution of body fat and metabolic derangements, although children may have a relatively smaller increase in visceral fat in the trunk [10–13].
Sex
Female sex is associated with an increased risk in some studies [3].
Ethnicity
White and East Asian races have been linked to having an increased risk of HIV-associated lipodystrophy [14].
Associated diseases
Patients with HIV-related lipodystrophy may develop hypertriglyceridaemia, although diabetes is less common [2]. There may also be a predisposition to coronary artery disease [15].
Pathophysiology
While the exact cause of HIV-associated lipodystrophy is unknown, both PIs and NRTIs are implicated in the pathogenesis. Because these drug classes are often given together as part of HAART, the individual effects of these drugs on the specific lipodystrophy phenotype remains unclear. It is speculated that while PIs induce peripheral lipodystrophy and metabolic abnormalities, NRTIs may be responsible for the fat accumulation in certain regions such as the buffalo hump [2].
First and second generation PIs have been shown to inhibit adipocyte differentiation and lipogenesis in vitro [16]. PIs may also induce insulin resistance by inhibiting glucose transporter-4 expressions [17]. NRTIs, especially zidovudine and stavudine, have been proposed to induce fat loss by inhibiting mitochondrial polymerase γ and causing mitochondrial toxicity [18, 19]. This results in production of reactive oxygen species (ROS), which have been linked to lipoatrophy [20]. Excess adipocyte apoptosis was also observed in ex vivo fat samples from lipoatrophic areas [21].
The mechanism for the increased amounts of visceral adipose tissue (VAT) observed in HIV-associated lipodystrophy is also unclear. VAT and subcutaneous adipose tissue (SAT) differ in their metabolism, gene expression and inflammatory status [22]. As a result, adipocytes or other cells in VAT and SAT may respond to stimuli (e.g. PIs) in different ways, resulting in hypertrophy in one area and atrophy in another [3].
Predisposing factors
Several factors have been postulated to influence the risk of developing HIV-associated lipodystrophy, including age, gender, ethnicity, duration and status of HIV infection, as well as exposure to both PI- and non-PI-containing HAART [3, 6]. Both high and low CD4 counts have been reported in affected patients [23–25].
There is some evidence that first generation unboosted PIs, such as indinavir, are associated with a higher risk of lipodystrophy than are booster PIs [3]. In one study, the incidence of lipoatrophy was lower in patients treated with ritonavir-boosted atazanavir than in those who received unboosted atazanavir [14]. In another study, patients treated with ritonavir-boosted lopinavir developed lipodystrophy by 96 weeks of treatment less frequently than those who received efavirenz, regardless of the type of NRTI used [26].
Genetics
Polymorphisms in genes involved in adipocyte apoptosis and metabolism have also been implicated [3], including ones involved in the pyrimidine pathway or encoding potentially relevant enzymes, cytokines or peptides such as polymerase γ, matrix metalloproteinase 1, TNF-α, IL-1β, IL-6, resistin and mitochondrial haplogroups.
Clinical features
Presentation
Most patients present with a gradual loss of subcutaneous fat from the face, arms and legs. Facial involvement is present in 38–52% of HIV patients with lipodystrophy [3]. Fat loss from the suprazygomatic and temporal regions of the face can be severe enough to impart a stigmatizing emaciated appearance. Patients may also accumulate excess fat over the chin, breasts and waist, as well as over the upper back, producing a so-called ‘buffalo hump’ (Figure 100.3) [6].

Figure 100.3 Buffalo hump appearance in an HIV patient with lipodystrophy. (Courtesy of Professor L. Requena, Universidad Autónoma de Madrid, Spain.)
Differential diagnosis
Generalized loss of body fat is commonly seen in HIV-infected patients with AIDS wasting syndrome. Patients with AIDS wasting syndrome have decreased body weight and lean body mass, without accumulation of excess fat over the chin, upper back, breasts or waist. Those with AIDS wasting syndrome also do not develop glucose intolerance or hyperinsulinaemia, although they may have hypertriglyceridaemia [6]. Other differential diagnoses to consider include Cushing syndrome and iatrogenic lipodystrophy related to testosterone therapy.
Classification of severity
Scoring systems have been developed to quantify facial lipoatrophy. In one, the score ranges from 0 (absent) to 4 (severe), depending on the degree of malar depression, presence or absence of buccal extension and defined melolabial ridge [27]. Other scoring methods involve the use of various imaging modalities which may not be practical in the clinical setting [3].
Complications and co-morbidities
Complications are related to co-morbidities, including dyslipidaemia, impaired glucose intolerance, insulin resistance and coronary artery disease.
Disease course and prognosis
Lipodystrophy is progressive with ongoing HAART therapy [2]. While the possibility of subcutaneous fat recovery exists, it is typically slow and incomplete [3].
Management
There is no established algorithm for the management of HIV-associated lipodystrophy. Management is challenging and requires multidisciplinary input from infectious disease specialists, endocrinologists, nutritionists and primary care physicians. The stigma associated with facial lipodystrophy in particular may have a profound psychosocial impact and psychological support may be required.
Modification of previously successful antiretroviral therapy may increase the risk of treatment failure and must be done in consultation with an infectious disease specialist. Modest improvement in lipoatrophy has been reported with the removal of NRTIs [28]. While metabolic derangements such as dyslipidaemia and insulin resistance seem to be partly reversible if PI therapy is discontinued, lipodystrophy is usually unchanged [3].
Low-calorie or low-fat, high-fibre diets combined with aerobic exercise may improve central fat accumulation [29], although there is little evidence that diet or exercise significantly improves lipoatrophy [29, 30, 31]. Thiazolidinediones, which have been shown to induce adipocyte differentiation and increase subcutaneous fat mass, have been tried as pharmacological agents to treat lipoatrophy in HIV-infected patients with inconsistent results [32]. Uridine and pravastatin have been reported to improve lipoatrophy in small randomized trials, but these results need further confirmation [33, 34]. Facial fillers such as injectable poly-l-lactic acid as well as autologous fat transfers have both been used to correct severe facial lipoatrophy [35, 36].
Switching or discontinuing antiretroviral therapy has not been shown to reduce VAT [37, 38]. Since growth hormone deficiency has been associated with visceral adiposity in the general population as well as in HIV patients with lipodystrophy, growth hormone supplementation with doses ranging from 0.33 to 6 mg daily has been tried [39, 40]: patients with abdominal lipohypertrophy achieved a 10–20% reduction in VAT and 6–7% reduction in SAT in two studies involving the administration of growth hormone [41, 42]. The improvement was lost once therapy was discontinued. Treatment with tesamorelin, a growth hormone-releasing hormone analogue that closely mimics the physiological dose and function of its physiological counterpart, showed a 15.2% reduction in VAT, compared with a 5% increase in VAT in the placebo group (P <0.001), at 6 months in a large, randomized, double-blind, placebo-controlled study [43].
Surgical interventions such as liposuction have been used to remove excess fat from the anterior neck, breasts and abdominal compartment [44, 45, 46]. However, liposuction cannot remove intra-abdominal visceral fat. Furthermore, approximately 25% of patients will experience recurrence of lipohypertrophy after liposuction.
Localized lipoatrophy and/or lipodystrophy
Localized lipoatrophy and/or lipodystrophy is a heterogeneous group of disorders presenting as one or multiple depressions of various sizes, ranging from a few centimetres to greater than 20 cm in diameter.
Semicircular lipoatrophy
Definition and nomenclature
Semicircular lipoatrophy (SL) is characterized by localized, often bilateral, symmetrical, transverse, semicircular depressions across the anterolateral aspects of the thighs due to atrophy of the underlying subcutaneous fat.
Introduction and general description
First described in the German literature in 1974 [1], SL is characterized by localized, symmetrical and often bilateral, transverse, semicircular depressions over the anterolateral thighs [1, 2, 3].
Epidemiology
Incidence and prevalence
Semicircular lipoatrophy is probably commoner than the small number of reported cases (about 100) would suggest. [2, 4, 5].
Age
Most SL cases present in the third or fourth decades of life [6], although it has also been reported among children and the elderly [4].
Sex
The majority of cases occur in women [3].
Pathophysiology
The aetiology of SL is unclear. An older theory related the presence of SL to impaired circulation in the upper leg as a consequence of a congenital abnormality in the lateral femoral circumflex artery [7]. However, patients with arteritis or those whose quadriceps artery has been ligated do not develop SL [8]. An anomaly of fat metabolism in these patients has also been proposed [8, 9].
A current and more plausible explanation associates SL with repeated mechanical pressure as a form of microtrauma to the thighs [10, 11]. Suggested mechanisms include repeatedly standing or sitting in an unvarying position in which the affected area is constantly compressed by or knocked against various objects [1, 12]. Wearing constricting jeans or use of an elastic girdle has also been implicated [3, 13–15].
In cases where clusters of co-workers are affected in the same company, repetitive pressure against desk furniture has been identified. In those cases, the height of the depression on the leg measured from the floor plus the height of the shoe heel were constant and the same as the height of the desk [12, 16]. In a recent company-wide case–control study, the only statistically significant variables for SL development are female sex and leaning of thighs against the edge of the table [6].
Pathology
The histopathology findings in SL are non-specific. There is partial or complete loss of fat in the affected area with replacement by newly formed collagen [5, 17].
Clinical features
Presentation
Semicircular lipoatrophy is characterized by localized, transverse, semicircular depressions, 2–4 cm in width, that are often symmetrical and bilateral. These depressions may also appear band-like, and when more than one is present, they may appear in a parallel arrangement [2, 3]. There is no preceding inflammation and the overlying skin is normal. The anterolateral thighs are commonly affected bilaterally. Unilateral cases have been described as well [2, 10, 11, 18]. Multilocular and progressive lesions affecting the trunk and limbs were seen in one patient [19]. Although SL is usually asymptomatic, some patients complained of heavy legs, a burning sensation, cramps or pain after exercising [2, 10, 18, 20].
Differential diagnosis
The differential diagnosis of SL includes other forms of localized lipoatrophy or lipodystrophy as described elsewhere in this chapter.
Disease course and prognosis
Semicircular lipoatrophy has an excellent prognosis as most cases resolve gradually upon withdrawal of repetitive trauma.
Investigations
Semicircular lipoatrophy is a clinical diagnosis and further investigation is rarely required [3, 6].
Management
There are no established management guidelines for SL. However, lesions usually regress spontaneously after the removal of microtrauma over months to as long as 8 years [12]. Recurrences are possible, however [7, 9].
Localized lipoatrophy due to injected drugs
Localized loss of subcutaneous fat can occur after intradermal, subcutaneous or intramuscular injection of certain drugs. The two most commonly implicated agents are insulin and corticosteroids and these are discussed in detail. Other injected medications or substances that have been implicated as causes of localized lipoatrophy include benzathine penicillin, vasopressin, human growth hormone, methotrexate, iron dextran and diphtheria–pertussis–tetanus (DPT) vaccine [1–4].
Insulin-induced localized lipoatrophy
Definition
Insulin-induced localized lipoatrophy is the loss of subcutaneous fat at the site of insulin injection [1].
Introduction and general description
Insulin-induced localized lipoatrophy was a common complication of insulin therapy prior to the development of purified insulin in the 1970s [2, 3]. With the advent of human insulin, the incidence lipoatrophy has decreased dramatically [4].
Epidemiology
Incidence and prevalence
Prior to the introduction of purified human insulin, lipoatrophy occurred in 25–55% of patients using insulin. Since the introduction of highly purified insulin, it is estimated that less than 10% of patients are affected [5, 6], although lipoatrophy is still reported with the use of recombinant human insulin, rapid-acting insulin analogues and continuous subcutaneous insulin infusion [4, 7, 8, 9, 10, 11, 12].
Age
Insulin-induced lipoatrophy occurs predominantly in children and young adults [13, 14, 15].
Sex
The risk is greater in females than males [16, 17].
Pathophysiology
The pathogenesis of insulin-induced lipoatrophy is not completely understood. Lipolytic components or impurities in certain insulin preparations may have resulted in local allergic or immunological reactions. For example, a local immune reaction to insulin crystals with resultant dedifferentiation of adipocytes has been suggested [14, 18]. An immune-mediated inflammatory process with release of lysosomal enzymes promoting lipoatrophy has also been proposed [19]. It has also been suggested that mast cells may play a pathogenetic role [12]: in one case series involving five patients with human insulin analogue-induced lipoatrophy, an elevated number of tryptase-positive, chymase-positive degranulated mast cells were seen in the subcutaneous tissue.
Predisposing factors
This form of lipoatrophy is more common among those with prior dermal reactions to insulin [1]. The use of older, less purified forms of insulin such as bovine or porcine insulin conveys a higher risk of lipoatrophy [20]. Repeated use of the same injection site also increases the risk.
Pathology
Histopathology shows lobules of small adipocytes and lipomembranous changes [21]. A discrete lymphoid infiltration abutting the blood vessels in the hypodermis may also be observed [22].
Clinical features
Presentation
Insulin-induced lipoatrophy presents as a depressed plaque due to loss of subcutaneous fat at the site of insulin injection. The overlying and surrounding skin appear normal. Lipoatrophy normally presents after 6–24 months of insulin treatment [1]. Interestingly, there is concomitant lipohypertrophy in approximately 25% of patients [23].
Differential diagnosis
Other forms of localized lipoatrophy should be considered.
Complications and co-morbidities
Because insulin absorption from the lipoatrophic area is erratic, continued injection of insulin into affected areas may result in poor glycaemic control [22].
Management
The risk of lipoatrophy may be reduced by regular rotation of insulin injection sites. The importance of this should be explained to all patients who require insulin. Once lipoatrophy develops, continued injection into the lipoatrophic site should be avoided due to erratic absorption of insulin. Spontaneous resolution of established insulin-induced localized lipoatrophy is rare.
There is no generally agreed management algorithm but, if feasible, a change to purified human insulin is recommended. Several authors describe success in restoring fat by co-administration of a corticosteroid such as dexamethasone with the insulin [4, 18, 24, 25]. Other strategies have included using an insulin jet-injection device [26] or a continuous insulin infusion pump [27], although lipoatrophy with the latter has also been reported [7, 8]. Twice-daily application of 4% sodium cromoglycate prepared in petrolatum has been claimed to reverse early lipoatrophy and prevent new lesions in one small case series [12].
Localized lipoatrophy due to injected corticosteroid
Definition
Localized lipoatrophy due to injected corticosteroid is the localized loss of subcutaneous fat that occurs after intramuscular or intralesional injection of corticosteroids [1].
Introduction and general description
Injected depot preparations of corticosteroids have been widely used for their sustained systemic antiallergic and anti-inflammatory action for more than half a century. It was recognized at an early stage that they were capable of producing profound lipoatrophy if they were injected into subcutaneous fat.
Epidemiology
Incidence and prevalence
An overall incidence of local reactions following intralesional corticosteroid injection is reported to be 0.5%. These reactions include pain, panniculitis, haemorrhage, secondary infection, pigment alteration, hypersensitivity and atrophy [2]. Of all the persistent local reactions due to corticosteroids, atrophy is the most common [3].
Sex
Women appear to be at significantly greater risk than men [1, 4]. In one early study, it was observed that lipoatrophy occurred in six of 14 women but in none of 13 men who received repeated intramuscular or deep subcutaneous injections of triamcinolone diacetate [4].
Pathophysiology
The exact pathogenesis is not clear but several factors are thought to be involved. It is believed that intramuscular injection of triamcinolone has a direct traumatic and a hormonally mediated destructive effect on fat cells [5]. In addition, decreased type I collagen and glycosaminoglycan synthesis has been noted following the injection of corticosteroid [6]. One common finding from histological analyses is the identification of a granular basophilic material in the dermis, thought to represent altered ground substance associated with deposits of corticosteroid crystals [5, 7, 8]. Cutaneous atrophy has been noted to resolve in parallel with the gradual disappearance of corticosteroid crystals from the tissue [7].
Predisposing factors
Compounds with low solubility, such as triamcinolone acetonide, injected at higher concentrations appear to be associated with greater risks of atrophy. One group of investigators noted that intralesional injections of triamcinolone acetonide at concentrations above 5 mg/cm3 were associated with increased risks of cutaneous atrophy [9]. However, no lipoatrophy was observed at 6 and 12 weeks after injection in a series of 14 patients with dermatological conditions who had received one or two 30 mg or 60 mg doses of intramuscular triamcinolone acetonide [10].
Pathology
In addition to the presence of granular basophilic material associated with the deposition of corticosteroid, other histological findings include epidermal atrophy, homogenization of collagen, degeneration of sebaceous glands, decreased elastin and involution of subcutaneous fat lobules with small lipocytes separated by hyaline material [1, 5, 7, 8,]. Inflammatory cells are not usually prominent although a sparse mononuclear cell infiltrate can be observed. There is no vascular inflammation [1].
Clinical features
Presentation
Patients present with an oval or circular depressed plaque at the site of prior injection (Figure 100.4). The overlying epidermis is usually normal, although telangiectasia, hypopigmentation or alopecia may occur [3]. There is no associated erythema or tenderness. Atrophy generally begins within weeks to 3 months after injection. The time course and extent of the atrophy depend on several factors, including the solubility and concentration of the corticosteroid used and the depth and anatomical location of the injection [5].
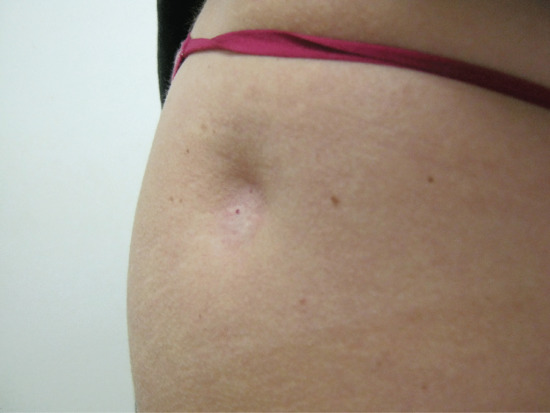
Figure 100.4 Delling of the skin over the right hip due to subcutaneous fat atrophy at the site of depot corticosteroid injection.
Differential diagnosis
Other forms of localized lipoatrophy should be considered.
Disease course and prognosis
The lipoatrophy may resolve spontaneously over the course of 1–2 years [11–14], although it may persist for longer in some cases [15].
Management
There is no established management guideline. In one small case series, four patients were treated by infiltration of the affected area with normal saline. All four patients demonstrated complete resolution of lipoatrophy and restoration of surface contour after 4–8 weekly injections. The injected volume ranged from 5 to 20 cm3 per treatment session, depending on the size to be treated. The authors speculated that the efficacy of the treatment may be due to resuspension and redistribution of the poorly soluble corticosteroid crystals by saline solution [3].
Localized lipodystrophy secondary to panniculitis
Localized lipodystrophy may be secondary to inflammation of the subcutaneous fat, of which there are several aetiologies (see Chapter 99).
Centrifugal lipodystrophy
Definition and nomenclature
Centrifugal lipodystrophy (CLD) is a form of localized lipodystrophy in which atrophic plaques extend centrifugally.
Introduction and general description
Centrifugal lipodystrophy is characterized by localized lipoatrophy that expands centrifugally. Most cases of CLD have been reported from a single region of Japan. The condition predominantly affects children who are otherwise well. Because there have been a few cases of adult onset as well as of involvement of areas other than the abdomen, the term CLD may be more appropriate than lipodystrophia centrifugalis abdominalis infantilis.
Epidemiology
Incidence and prevalence
Centrifugal lipodystrophy is rare: approximately 170 cases have been described [1] since its initial report in 1971 by Imamura et al. [2].
Age
The majority (90%) of CLD patients develop the condition in the first 4 years of life with a mean of 2.4 years and a median of 2 years [1]. Very few patients have developed their initial lesions as an adult [1, 3].
Sex
Girls outnumber boys by approximately 2 : 1 [1].
Ethnicity
Centrifugal lipodystrophy is reported almost exclusively among children of Asian descent, especially of Japanese origin [1, 4, 5, 6]. There have been a few white patients reported from the USA [7], the UK [8, 9], France [10], Italy [11], Germany [12] and Spain [13].
Associated diseases
Most CLD patients exhibit no other signs or symptoms of systemic illness. However, immunological abnormalities that have been reported include positive antinuclear antibodies [1], rheumatoid factor [10], antigliadin antibodies [12] and partial IgA deficiency [10, 12].
Pathophysiology
The cause of CLD is unknown. It has been suggested that apoptosis may play a part in the fatty tissue degeneration of CLD but it is unclear if this is a primary event [14]. Fibrous long-spacing collagen has been observed on ultrastructural studies of lesional skin, although the relation of their presence to disease pathogenesis remains to be elucidated [15]. So far, no abnormalities of serum leptin levels have been reported [1].
Pathology
The epidermis and dermis in CLD are unaffected but there is markedly reduced or absent subcutaneous tissue in the affected areas. In the inflamed periphery of the plaque, the loss of subcutaneous fat is accompanied by a moderate mononuclear cell infiltrate. Rarely, multinucleated giant cells, foam cells, swelling of blood vessels, vasculitis and thrombosis have been observed [1].
Genetics
Centrifugal lipodystrophy has occurred in one pair of dizygotic twins and in one pair of affected siblings, which has raised the possibility of a genetic component of the disease [1].
Environmental factors
Although various external factors such as hernia, congenital dislocation of the hip, external pressure, injection or previous skin infection at the sites were reported in about 10% of the cases, no clear relationship has been established between CLD and environmental insults [1].
Clinical features
Presentation
Most cases of CLD begin with a single depressed plaque involving the groin (80%), axillae (20%) or neighbouring regions. Interestingly, in all reported non-Japanese cases, the initial plaque developed exclusively in or near the groin [1]. Less common areas of involvement include the neck, lower chin, retroauricular area or scalp [16, 17]. The periphery of the atrophic plaque is typically erythematous while the centre is of normal colour or may have a violaceous or bluish hue. Ulceration of the plaques has been reported to occur rarely [1, 18]. Plaques beginning in the groin area may expand to involve the genital region, abdominal wall or even the chest wall. Plaques developing in the axillary region may extend to the chest wall and abdominal wall. During the initial enlargement period, the atrophy may be multifocal. Although most lesions are asymptomatic, some patients report mild discomfort or tenderness [1]. Regional lymph nodes are enlarged in 65% of cases. When the lesion stops expanding, the erythematous rim and lymphadenopathy tend to resolve.
Differential diagnosis
Differential diagnosis of CLD includes other localized lipodystrophies.
Complications and co-morbidities
Patients with CLD usually do not suffer from any complications and co-morbidities, except for rare instances of ulceration involving plaques [1, 18]. A case of angioblastoma developing in a non-regressing CLD lesion lasting into adulthood has been reported. The angioblastoma responded to resection followed by radiation therapy [19].
Disease course and prognosis
The atrophic area slowly enlarges centrifugally for 3–8 years, often stopping with the onset of puberty. By 13 years, 82% of cases show no residual inflammation at the periphery and 65% no longer have regional lymphadenopathy.
The atrophic portion may subsequently improve but may remain depressed [1]. Hair regrowth has been observed in cases of scalp involvement [16, 17].
Investigations
Results of routine laboratory tests are unremarkable.
Management
There is no established guideline for the management of CLD. Many topical therapies have been tried including topical steroids, pimecrolimus, vitamin A cream and dimethyl sulfoxide. Various systemic therapies have been attempted including corticosteroids, chloroquine, penicillin, vitamin E and ibuprofen. None of these have been effective in preventing enlargement, although topical and systemic steroids occasionally reduced the inflammation at the periphery of the plaque [1]. Psoralen with UVA was reported to be effective in softening the skin and preventing further enlargement in one case [4].
FAT HYPERTROPHY
Fat hypertrophy is a circumscribed expansion of subcutaneous fat and is particularly associated with insulin injection sites. Fat hypertrophy is distinguished from lipomatosis in that the latter involves the formation of multiple foci of proliferating adipocytes.
Insulin-induced localized fat hypertrophy
Definition and nomenclature
Insulin-induced fat hypertrophy is a circumscribed increase in subcutaneous fat at the site or sites of repeated insulin injection by insulin-dependent diabetics.
Introduction and general description
Insulin-induced fat hypertrophy was, and still is, the most common cutaneous complication of insulin therapy [1]. It is thought to be due to the direct local anabolic and lipogenic effects of insulin on adipocytes.
Epidemiology
Incidence and prevalence
With highly purified human insulin, the prevalence of fat hypertrophy is approximately 30% in patients with type 1 diabetes and less than 5% in patients with type 2 diabetes [2].
Age
No specific age of onset has been identified, although the condition is generally believed to be associated with a young age [2].
Pathophysiology
The fat hypertrophy is thought to be secondary to the anabolic and lipogenic effect of insulin [1, 2]. Although insulin antibodies are associated with fat hypertrophy in children and adolescents with type 1 diabetes, it is not known whether they play a role in its pathogenesis [3, 4].
Predisposing factors
Several factors have been associated with the development of the fat hypertrophy. These include recent insulin initiation, low body mass index (BMI), use of the abdomen as the injection site and infrequent rotation of the injection site [2].
Pathology
Histological examination shows lobules of large mature adipocytes. Electron microscopy has revealed discrete populations of small adipocytes, suggesting differentiation or proliferation [5].
Clinical features
Presentation
The disorder presents as soft subcutaneous swellings at the sites of insulin injection (Figure 100.5) The overlying epidermis is normal. Because the involved skin tends to be hypoaesthetic relative to uninvolved skin, patients often continue to inject insulin preferentially into affected sites [1].
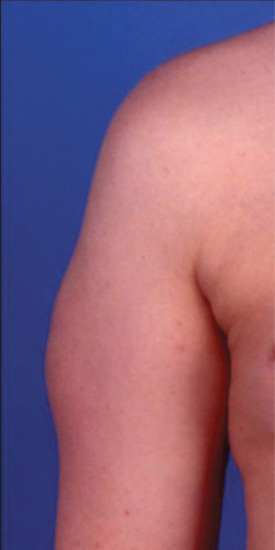
Figure 100.5 Insulin-induced fat hypertrophy of the lateral aspect of the upper arm.
Differential diagnosis
Localized cutaneous amyloidosis has been associated with insulin injection sites and may be mistaken for insulin-induced fat hypertrophy [6]. Furthermore, insulin per se has been identified as one of the proteins that can form amyloid fibrils [7]. Affected skin feels firmer on palpation than that overlying insulin-induced fat hypertrophy and is unlikely to improve after changing the insulin injection site [1].
Complications and co-morbidities
Because the lipohypertrophic nodules have decreased vascularity, impaired insulin absorption at these sites may result in poor glycaemic control [1, 8–10].
Disease course and prognosis
Regression of insulin-induced lipohypertrophy is possible once management is instituted.
Management
To prevent the development of fat hypertrophy, patients requiring insulin injections should be advised of the importance of changing injection sites regularly [2]. No universal treatment guideline has been established in the management of this problem. Rotation of injection sites is recommended with the anticipation that existing nodules will regress [1]. Most patients requiring insulin will nowadays be prescribed biosynthetic human insulin analogues, which carry a lower risk of inducing fat hypertrophy. If they are not, changing to human insulin, particularly the short-acting, rapidly absorbed preparations, may help to reverse the condition [11]. Changing the insulin delivery method from injection to continuous subcutaneous infusion using a pump has also been advocated. This method of administration reduces overall insulin requirements and is therefore thought to lessen the overall stimulus to lipogenesis [1]. If the above strategies fail, some patients will demand removal of the excess fat. Liposuction has achieved good cosmetic results [12, 13].
SUBCUTANEOUS LIPOMATOSIS
Benign symmetrical lipomatosis
Definition and nomenclature
Benign symmetrical lipomatosis (BSL) is an uncommon condition characterized by multiple, symmetrical, unencapsulated fatty tissue deposits throughout the body.
Introduction and general description
What subsequently came to be known as Madelung disease was first mentioned by Brodie in 1846 [1]. Madelung described a series of patients with the lipomatosis in 1888 [2] and Launois and Bensaude reported it again 10 years later [3]. More recently, the term benign symmetrical lipomatosis has been preferred for this uncommon condition. It is characterized by multiple, symmetrical, unencapsulated fatty tissue deposits involving multiple areas of the body, particularly the neck, shoulder girdle and the proximal upper and lower extremities.
Epidemiology
Incidence and prevalence
The exact incidence and prevalence of BSL is not known. The incidence in Italy has been estimated to be 1 : 25 000 [4]. There are, however, less than 300 cases reported in the literature since its initial description [5].
Age
The age of onset ranges from 30 to 60 years old [6]. However, a few paediatric cases have been reported [7, 8].
Sex
Benign symmetrical lipomatosis is a disorder that predominantly affects males, with a reported male : female ratio as high as 31 : 1 [1, 6, 9–11].
Ethnicity
Historically, BSL has been linked to people of Mediterranean descent [1]. However, cases from non-Mediterranean ethnicities have been described [5, 12, 13].
Associated diseases
Many conditions are associated with BSL. These include alcohol abuse, chronic hepatic disorders possibly related to the underlying alcohol abuse, lipid abnormalities, arterial hypertension, chronic obstructive pulmonary disease (COPD), gynaecomastia, hypothyroidism, peripheral neuropathy, diabetes, hyperuricaemia and myoclonic epilepsy and ragged red fibres (MERRF) syndrome [1, 7, 8, 10, 14, 15, 16]. In one study of 22 patients from Spain, 95.5% of the patients had high alcohol intake and 59.1% had some form of hepatic disease. Furthermore, 77% of these patients were reported to be regular smokers. Statistics of reported associations include 41% with dyslipidaemia, 27% with gynaecomastia, 23% with arterial hypertension, 23% with COPD, 14% with hyperuricaemia, 9% with hypothyroidism and 4.5% with type 2 diabetes [1].
Peripheral neuropathy, polyneuropathy and autonomic dysfunction are also common findings among BSL patients [11, 17–19, 20] and it may be the presenting symptom in some [21]. However, there is controversy as to whether these neurological manifestations are directly related to BSL or are the result of alcoholism.
Compression symptoms affecting the respiratory and digestive tracts are thought to be directly due to fatty masses in the anterior neck and, in one case series, were the symptoms that in all cases occasioned their patients’ initial consultation [1].
Pathophysiology
While many theories on the pathogenesis of BSL exist, a role for brown fat in BSL has garnered attention. Although, grossly and histologically, the accumulated adipose tissue in BSL is indistinguishable from mature fat [1], ultrastructural analysis of adipose tissue deposits in BSL suggests that it resembles brown fat [22]. Further support for the role of brown fat in BSL came from another study that demonstrated mRNA expression of brown adipose tissue-specific uncoupling protein-1 (UCP-1) in a lipoma of a patient with BSL, but not in the normal subcutaneous fat from the same patient [23]. Brown fat hypertrophy has been proposed to result from functional sympathetic denervation [22], or from alterations in the synthesis of intracellular cyclic adenosine monophosphate (cAMP) under noradrenergic stimulation [24].
Pathology
Lipomatous tissue in BSL consists of small adipocytes with no atypical features and a slight increase in vascular and fibrous elements. The lipomas are typically not encapsulated and may infiltrate across fasciomuscular and neurovascular planes [25].
Genetics
Genetic alternations in mitochondrial DNA have been demonstrated in BSL patients [18, 26]. Most reported BSL cases have been sporadic, although a very small number of familial cases exist for which autosomal recessive inheritance has been proposed [27].
Clinical features
Presentation
Fatty masses develop most commonly on the neck (Figure 100.6), shoulder girdle and proximal upper and lower extremities. Other less common locations include the retroauricular and submandibular areas [28], tongue [21], mammary region [22], abdomen [7] and perineum or scrotum [29]. Although rare, laryngeal involvement resulting in a compromised airway has been described [12, 30–33]. The fatty deposits are slow growing and progressive over many years. In contrast to Dercum disease, the fatty growths in BSL are not tender or painful.

Figure 100.6 Non-tender fatty deposits around the neck in a patient with benign symmetrical lipomatosis. (Courtesy of Professor L. Requena, Universidad Autónoma de Madrid, Spain.)
Clinical variants
Enzi's classification of BSL is the most widely accepted [6]. In type I BSL, fatty deposits are mainly distributed around the neck, shoulders, supraclavicular fossa and proximal regions of the upper extremities. In type II BSL, more caudal regions of the body are involved, while the neck is unaffected. As a consequence, type II BSL patients may appear similar to obese patients.
Differential diagnosis
Differential diagnosis of BSL includes obesity, Dercum disease and familial multiple lipomatosis. Genetic syndromes presenting with multiple lipomas such as Cowden syndrome, Bannayan–Riley–Ruvalcaba syndrome, multiple endocrine neoplasia type 1 (MEN1) and Gardner syndrome can be differentiated from BSL by the presence of other disease-specific clinical features.
Complications and co-morbidities
Respiratory or digestive symptoms may develop if the lipomatosis from the anterior neck is significant enough to compress vital structures. There have been a few cases of head and neck cancer, especially squamous cell carcinoma, reported in patients with BSL [9, 34]. However, it is possible that this simply reflects the increased alcohol and tobacco consumption observed among BSL patients. Malignant transformation of lipoma into liposarcomas in BSL has been reported only twice [35, 36].
Disease course and prognosis
The lipomatous masses are slow growing and progressive. There are few long-term follow-up data on BSL patients. In one study, however, in which 31 BSL patients were observed for up to 26 years, three of the eight deaths observed were sudden and occurred in patients with autonomic neuropathy in the absence of coronary artery disease or history of cardiac disease [20].
Investigations
The diagnosis of BSL is based on clinical presentation and disease history. Chest radiographs may show abnormal symmetrical mass lesions due to accumulations of adipose tissue. MRI is the best diagnostic tool in evaluation of the spread of adipose tissue, presence of tracheal compression, vascular topography within the fat mass and exclusion of synchronous malignant disease [36, 37].
Management
There is no established guideline for the management of BSL. A thorough clinical review, especially for respiratory symptoms, should be carried out at each visit to check for signs of compression or for the rare occurrence of malignant transformation.
Medical treatments have been tried with generally poor results. Oral salbutamol, which induces lipolysis by adrenergic stimulation, has been tested on BSL patients but the results have been disappointing [24]. Fibrate has been claimed to stop the growth or even reduce the volume of fatty masses in BSL patients [38]. Among BSL patients who reported alcohol abuse, abstinence from alcohol has had mixed results in preventing the progression or recurrence of the lesions after surgical treatment [1, 39]. General weight loss does not seem to be effective in reducing fatty masses [1].
Open surgery, lipectomy and liposuction, with or without ultrasound assistance, have been tried with varying success rates. Liposuction is less traumatic, has a lower complication rate and produces less scarring than open surgery [40, 41, 42, 43, 44, 45]. However, because the fatty deposits are unencapsulated and diffusely infiltrate muscles and vessels without a clear plane for dissection, open surgery with direct observation of important structures is a safer approach in certain anatomical regions where liposuction may be too dangerous [1, 45]. Some authors advocate ultrasound-assisted liposuction which can treat multiple areas in a single session and produces limited scarring.
Regardless of the mode of surgical intervention, multiple procedures are often required, especially given the high rates of recurrence [1, 46, 47]. In one study, BSL patients underwent an average of 3.3 open surgical procedures with a 51% recurrence rate. Isolated lipoaspiration had a 95% recurrence rate in the same study. Recurrence rates vary by anatomical site and are highest following procedures on the arms and pectoral region. The average time taken for fat to reaccumulate after a surgical intervention is approximately 21 months [1].
Emergency intervention is needed when compression symptoms develop. In rare cases where the fatty tissue deposits extend to the mediastinum and compress the trachea, a tracheostomy may be required [20].
Dercum disease
Definition and nomenclature
Dercum disease is a rare disease characterized by generalized overweight status or obesity and pronounced pain in the adipose tissue with or without the presence of lipomas.
Introduction and general description
Dercum disease was first described in 1888 [1]. The disease is characterized by diffuse or localized pain involving adipose tissue, usually affecting those who are overweight or obese. Patients experience a number of other somatoform symptoms, and management of the condition is a challenge.
Epidemiology
Incidence and prevalence
The exact incidence and prevalence of Dercum disease is not known.
Age
Dercum disease most commonly presents between the ages 35 and 50 years [2, 3]. While it was initially proposed that Dercum disease affects postmenopausal women, a recent survey revealed that 86% of patients developed their symptoms prior to the menopause [3].
Sex
Dercum disease is 5–30 times more common in women than in men [3, 4].
Ethnicity
No known ethnic predilection has been identified.
Associated diseases
Although various symptoms or diseases have been observed in these patients, none are consistently associated. These include easy bruising, sleep disturbances, impaired memory, depression, difficulty concentrating, anxiety, rapid heart beat, shortness of breath, diabetes, bloating, constipation, fatigue, weakness and joint and muscle aches [3, 5].
Pathophysiology
While the exact aetiology of Dercum disease is not known, many theories have been proposed. A local defect in lipid metabolism has been considered. One study showed a difference in the formation of long-chain mono-unsaturated fatty acids between the painful adipose tissue and the unaffected adipose tissue in the same patient [6]. Another study showed that the proportion of mono-saturated fatty acids was significantly higher in Dercum disease patients than in healthy controls [7].
Painful adipose tissue from a Dercum disease subject was found to have significantly lower conversion rate of glucose to neutral glycerides than non-painful adipose tissue from the same subject [8]. In vitro analysis demonstrated that painful adipose tissue had reduced responsiveness to norepinephrine and lack of response to the antilipolytic effect of insulin compared with non-painful adipose tissue [9].
Roles of various inflammatory factors have also been investigated. Higher levels of IL-13 and significantly lower macrophage inflammatory protein 1β and fractalkine expression were seen in Dercum disease patients. Fractalkine is a unique chemokine that is constitutively expressed by neurons. When fractalkine receptors are occupied, pain and resistance to opioid analgesia are promoted, which is in concordance with symptoms in Dercum disease [10].
Other theories for pain in Dercum disease have included endocrine dysfunction [9, 11, 12, 13, 14, 15, 16] as well as autonomic nervous system dysfunction [17] and stretching of and pressure on nerves by the growing fatty masses [18, 19].
The significance of these observations is, however, unclear and their contribution to disease pathogenesis remains to be elucidated.
Pathology
When lipomas are present, they show the typical histological features of lipomas without inflammation or vascular abnormalities [20].
Genetics
The majority of cases occur sporadically [5]. An autosomal dominant inheritance with variable expression has also been reported [2, 21, 22, 23, 24, 25]. HLA typing has not been revealing [9]. A to G mutation at position A8344 of mitochondrial DNA, which is sometimes associated with familial multiple lipomas, is not detected [24, 26].
Clinical features
Presentation
The main symptom in Dercum disease is the either abrupt or indolent onset of pain in adipose tissue [1]. Lipomas may or may not be present in these patients, who are typically obese. Patients have most commonly described the pain as burning or aching of the subcutaneous tissue. Some experience spontaneous paroxysmal attacks of pain [5, 27]. The most common locations of painful fat and lipomas are the extremities (Figure 100.7), trunk, pelvic area and buttocks [3]. The head and neck are spared [27]. Interestingly, patients experience more pain in the medial aspects of the involved extremities [5]. Lipomas, when present, vary in size and firmness [3].

Figure 100.7 Painful lipomas on the lower extremity of a patient with Dercum disease. (Courtesy of Professor L. Requena, Universidad Autónoma de Madrid, Spain.)
While fatigue and psychiatric manifestations were initially included as cardinal symptoms of Dercum disease [28], subsequent literature has not supported this claim. Certainly, not all patients with Dercum disease exhibit psychiatric symptoms [29, 30]. More recently, an association between BMI and anxiety and personality disorders has been noted [31]. It has also been proposed that pain and obesity in Dercum disease may contribute to the psychiatric manifestations seen in some patients [5].
Clinical variants
Various classifications have been proposed over the years [5, 16, 27, 28]. In 2012, Hansson and colleagues proposed the following classification [5]:
- I Generalized diffuse form: widespread painful adipose tissue without clear lipomas.
- II Generalized nodular form: general pain in adipose tissue and intense pain in and around multiple lipomas.
- III Localized nodular form: pain in and around multiple lipomas.
- IV Juxta-articular form: pain in solitary deposits of excess fat, for example at the medial aspect of the knee.
Differential diagnosis
Many diseases have similar symptoms to those of Dercum disease. For the generalized diffuse form, fibromyalgia, lipoedema, panniculitis and primary psychiatric disorders may be considered in the differential.
Other types of Dercum disease must be differentiated from conditions that may include solitary or multiple lipomas, as they may also sometimes be painful. These include multiple symmetrical lipomatosis, familial multiple lipomatosis, adipocytic tumours, neurofibromatosis type 1, MEN1 and MERRF syndrome. MERRF syndrome is a disease of mitochondria which is sometimes accompanied by non-painful lipomas.
Complications and co-morbidities
The lipomas in Dercum disease may in rare instances become necrotic [32] or may compress visceral organs [33, 34]. Other co-morbidities are mostly related to the associated obesity or the psychiatric morbidity.
Disease course and prognosis
Very little is known about the natural progression of Dercum disease. Several case reports suggest that the pain gets worse over time [25]. However, in one long-term study of patients with the condition, pain seemed to be relatively constant over the 5-year study period [35].
Investigations
Diagnosis is based on clinical criteria after a thorough physical examination and exclusion of the differential diagnoses discussed earlier [5]. There are no laboratory markers for the condition and laboratory tests for inflammatory and autoimmune disease are typically negative [3, 36–38].
Management
Overall, there is little evidence on which to base treatment of Dercum disease. Management may be best achieved through a multidisciplinary approach involving multiple specialists including dermatologists, surgeons, pain specialists, psychiatrists and psychologists.
Pain in this setting has been observed to be refractory to traditional analgesics and non-steroidal anti-inflammatory drugs (NSAIDs) [8, 20, 23, 39, 40, 41, 42, 43, 44, 45, 46]. However, this experience has been challenged recently after a study found that pain diminished in 89% of patients when treated with NSAIDs and in 97% when treated with narcotics [3].
Topical lidocaine with or without prilocaine [38, 47, 48], intralesional lidocaine [49] and intravenous lidocaine [8, 36, 40, 42, 50, 51, 52] have also been tried with varying degrees of success. Pain relief from intravenous lidocaine infusion lasted from 10 h [50] to 12 months [40].
Systemic corticosteroids have been reported to improve pain in some [15, 23, 53], while worsening it in other patients [54]. Two patients with juxta-articular Dercum disease treated with intralesional corticosteroids experienced dramatic improvement of pain [55]. Methotrexate alone [25] and in combination with infliximab [56], pregabalin or oxacarbazepine [16, 37, 57, 58] has also been used. Two patients with concurrent Dercum disease and hepatitis C were successfully treated with interferon α-2b [59].
A more recent small pilot study involving 10 patients showed that rapid cycling hypobaric pressure may decrease pain [60].
Procedure-based approaches with suction-assisted liposuction [35] and lipectomy [4, 18, 19, 20, 61, 62, 63] have also been tried. However, pain often recurs after these procedures.
Infiltrating lipomatosis of the face
Definition
Infiltrating lipomatosis of the face is a disorder where the unilateral overgrowth of unencapsulated benign and mature but invasive adipocytes involves the lower two-thirds of the face.
Introduction and general description
Infiltrating lipomatosis of the face (IL-F) is a disorder characterized by unilateral overgrowth of unencapsulated benign and mature but invasive adipocytes involving the lower two-thirds of the face. Although it usually presents at birth (congenital infiltrating lipomatosis of the face) it may present in adolescence or early adulthood. It is associated with soft tissue and bony hypertrophy [1, 2]. IL-F patients have normal psychomotor development [3].
Epidemiology
Incidence and prevalence
Infiltrating lipomatosis of the face is a rare disorder with approximately 50 cases reported in the world literature [1–4, 5–12, 13, 14–26].
Age
Although most cases present at birth, adolescent and early adulthood presentations have also been reported. All reported cases presented during the first three decades of life [23].
Sex
There is no gender predilection [3].
Ethnicity
There is no racial predilection.
Pathophysiology
The pathogenesis of IL-F is unknown. Given the tissue overgrowth, IL-F has been postulated to be part of the spectrum of overgrowth syndromes. PTEN and RET mutations have been excluded. Though no mutation has been found, IL-F is generally considered to be the result of secondary somatic mutation at some point in early development [13].
A study evaluating the expression of angiogenic and vasculogenic factors refuted a role for neovascularization in the pathogenesis of IL-F [27].
Pathology
In the initial case description, Slavin et al. described the histopathological features of IL-F, which include infiltration of mature benign adipocytes into adjacent soft tissue with hypertrophy of the underlying skeleton. They also emphasized the absence of malignant characteristics and lipoblasts but found an increase in fibrous elements with focal fibrosis affecting nerve bundles and vessels with unifocally thickened muscular walls [2].
Causative organisms
Two patients with IL-F have been found to have cytomegalovirus inclusions within the secretory cells of the parotid gland. However, the relationship between cytomegalovirus and IL-F remains unclear [6, 8].
Clinical features
Presentation
Patients with IL-F present with unilateral facial swelling that is typically present at birth or in early childhood although it may be delayed until early adult life [23]. The facial swelling is due to the proliferation of unencapsulated benign and mature adipocytes as well as associated soft tissue and bony hypertrophy. There is a wide range of clinical presentations depending on the extent of involvement of the underlying tissue. The lower two-thirds of the face is most commonly involved [1].
Macroglossia and mucosal neuromas on the tongue and buccal mucosa have also been reported [13, 19, 21], as have dental abnormalities such as abnormal tooth formation[7], root hypoplasia [23] and early eruption of deciduous and permanent teeth on the affected side [13, 20, 21, 23]. A cutaneous capillary blush, usually occurring after resection, has also been reported [13].
Differential diagnosis
Differential diagnosis of IL-F includes other hamartomatous or overgrowth syndromes such as Proteus syndrome, vascular or lymphatic malformations and trauma, as well as benign and malignant neoplasms of the bone and/or soft tissue. MEN2b, Bannayan–Riley–Ruvalcaba syndrome and Cowden syndrome should be considered since they are also associated with mucosal neuromas [13, 21]. Congenital hemifacial hyperplasia shares some features with IL-F but this condition does not involve lipocytic infiltration [13, 21]. Conditions causing contralateral hypoplasia, such as hemifacial microsomia and progressive hemifacial atrophy (Romberg syndrome) should be excluded [20]. Other disorders of fat tissue infiltration such as liposarcoma or lipoblastomatosis may be ruled out based on histological findings [15, 16].
Complications and co-morbidities
Complications and co-morbidities arising from IL-F depend on the extent of involvement of the underlying soft tissue and bone. The lipomatosis itself is benign, although cosmesis may be significantly altered. Resection of IL-F risks injury to vital structures such as the facial nerve.
Investigations
Imaging with MRI provides the best delineation [13].
Management
Management of IL-F involves resection of the tumour followed by reconstruction. The resection is almost always subtotal due to the infiltrative nature of the tumour into vital structures and carries a risk of injury to the facial nerve. On average, IL-F patients undergo at least three surgical procedures [9] with a recurrence rate that is as high as 62.5% with surgical resection alone [14].
The timing of surgery remains controversial. While earlier reports advocated early and wide local excision to prevent extensive lipomatous infiltration [2, 9], more recent literature favours delayed resection with temporizing measures such as liposuction, excision of mucosal neuromas and surgery to the upper lip to restore facial symmetry [13]. By delaying the resection until early adulthood it is believed that the chances of facial nerve damage and the total number of debulking procedures required are diminished [5]. It has also been conjectured that growth hormone may play a role in recurrences, implying that mass reduction attempts prior to the end of adolescence may be more likely to fail [13]. Recently, a case of IL-F demonstrating C-KIT and platelet-derived growth factor receptor oncogene expression was treated with imatinib after subtotal surgical resection. At 18 months follow-up, the patient showed no disease progression [1].
Encephalocraniocutaneous lipomatosis
Definition and nomenclature
Encephalocraniocutaneous lipomatosis (ECCL) is a rare, neurocutaneous syndrome characterized by profound mental retardation, early onset of seizures, unilateral temporofrontal lipomatosis, ipsilateral cerebral and leptomeningeal lipomatosis, cerebral malformation and calcification, and lipomas of the skull, eye and heart.
Introduction and general description
Encephalocraniocutaneous lipomatosis is a rare, sporadic, neurocutaneous syndrome involving tissues of ectodermal and mesodermal origin. It is associated with profound mental retardation, early onset of seizures, unilateral temporofrontal lipomatosis, ipsilateral cerebral and leptomeningeal lipomatosis, cerebral malformation and calcification, and lipomas of the skull, eye and heart [1, 2]. The hallmark skin finding is naevus psiloliparus, a fatty hamartomatous malformation, of the scalp [3]. The condition is typically present at birth or shortly after birth.
Epidemiology
Incidence and prevalence
Encephalocraniocutaneous lipomatosis is a rare disorder with about 60 cases reported in the English literature.
Age
Cutaneous and eye findings are usually present at or shortly after birth. Neurological manifestations may present at a later time.
Sex
There is no gender predilection.
Ethnicity
There is no racial or ethnic predilection.
Pathophysiology
The pathogenesis of ECCL is unknown. Most tissues affected in ECCL are of neural crest origin. In addition, because all the CNS anomalies are caused by a mesenchymal defect affecting tissues surrounding the brain or the vessels, and since no primary structural brain malformation is observed, it is postulated that a gene involved in vasculogenesis and the development of multiple mesenchymal tumours may be involved [4, 5].
Pathology
The histopathology of naevus psiloliparus shows focal dermal fibrosis with subcutaneous fat in the reticular dermis.
Clinical features
Presentation
Patients with ECCL present with a wide spectrum of clinical manifestations. Naevus psiloliparus of the scalp is the most common skin finding and was present in 44/54 patients studied [6]. Unilateral or bilateral subcutaneous fatty masses are often seen in the frontotemporal or zygomatic region, and they are rarely seen outside the craniofacial region. Patchy or linear alopecia which is typically non-scarring is commonly observed: it may follow the lines of Blaschko or occasionally be scarring. Fibromas, lipomas and fibrolipomas present typically as ipsilateral skin tag-like cutaneous polyps on the eyelid or following a line from the outer canthus to the tragus. Less common cutaneous manifestations include irregular or disrupted eyebrows and café-au-lait macules [6].
Choristomas, with or without other eye anomalies, were observed in 43/54 patients [6] but other ocular abnormalities may also be seen.
Brain anomalies primarily involve the tissue surrounding the brain including blood vessels [4]. Intracranial lipomas are the most prominent feature; other features include meningeal and meningovascular anomalies and spinal cord lipomas, which may be extensive. CNS findings are frequently confined to the same body side as the cutaneous lesions. The spectrum of neurological manifestations of ECCL patients is broad, ranging from normal development to mental impairment of various degrees to intractable seizures. Less commonly, skeletal system involvement with jaw tumours or lytic bone lesions may be present; these may be progressive [5, 7–10].
Congenital heart malformation, in particular aortic coarctation, is seen in about 10% of ECCL patients [6].
Differential diagnosis
This includes the Proteus syndrome, oculoectodermal syndrome, oculocerebrocutaneous syndrome and epidermal naevus syndrome [11–15].
Complications and co-morbidities
Complications and co-morbidities depend on the extent of organ system involvement.
Investigations
Investigations for suspected ECCL include a biopsy of suspected naevus psiloliparus lesions or ‘skin tags’ around the eye. Ophthalmological examination is required to detect associated ocular anomalies. Neuroimaging studies may characterize and assess the extent of CNS involvement [6]. Since fatty masses can involve the spinal cord, it is recommended that MRI of the spine be performed [6]. An electrocardiogram and an echocardiogram may help screen for cardiac problems.
Management
Treatment is symptomatic and requires multidisciplinary care.
LIPOEDEMA
Lipoedema is the term used to describe swelling of tissues due to abnormal accumulation of subcutaneous fat rather than fluid as seen in lymphoedema, with which it may be confused or coexist (lipo-lymphoedema). In the majority of cases it affects the lower extremities but it may also affect the upper extremities and the scalp. Hereafter the term lipoedema, if unqualified, will be used to denote lipoedema of the lower limbs.
Lipoedema of the lower limbs
Definition
Lipoedema is characterized by progressive, symmetrical and bilateral enlargement of involved areas due to subcutaneous deposition of fat.
Introduction and general description
Lipoedema, first described in 1940 [1], is characterized by progressive, symmetrical and bilateral enlargement due to subcutaneous deposition of fat occurring anywhere from the buttocks to the ankles with sparing of the feet [1, 2]. Lipoedema occurs independently of lymphatic stasis or venous insufficiency. Imaging studies verify that oedema is minimal and the swelling is in fact due to homogenous enlargement of the subcutaneous compartment [3, 4, 5, 6, 7, 8]. Its principal clinical features are summarized in Box 100.1 [2].
Epidemiology
Incidence and prevalence
The exact incidence and prevalence of lipoedema are unknown. It is probably underreported and often misdiagnosed due to unfamiliarity with this disorder [9, 10, 11, 12]. In lymphoedema clinics, up to 15% of referred patients were diagnosed with lipoedema [13, 14].
Age
Onset occurs during or soon after puberty [15].
Sex
Lipoedema occurs almost exclusively in women [1, 2, 9, 16]. The condition has been reported only rarely in men [9, 17].
Ethnicity
There is no racial or ethnic predilection.
Associated diseases
Most lipoedema patients do not have elevated lipids or other abnormal laboratory values [9, 18]. One group of investigators found elevated plasma lipids as well as an abnormal fatty acid composition of tissue triglycerides in lipoedema patients relative to controls. However, the clinical significance of this finding is unclear [19].
Pathophysiology
The aetiology of lipoedema is unknown. Hormones appear to play a role in the development of lipoedema, given that it occurs almost exclusively in women with a typical onset near puberty or during periods of significant hormonal changes [1, 2, 9, 16]. Moreover, male lipoedema cases occurred in those receiving hormonal therapy for prostate carcinoma or with hepatic cirrhosis [12, 20].
The anatomy of the lymphatic vessel system in lipoedema patients has been found to be normal or sufficient, as far as the large lymph vessels are concerned [15]. The degree of insufficiency in lipoedema patients, if present, never however reached the level of true chronic venous insufficiency or lymphoedema [10, 11, 21, 22]. The lymphatic transport in lipoedema decreases as the body ages and the fibrosis increases [15]. As a result, in longstanding cases, lipoedema and lymphoedema often coexist [12].
Predisposing factors
Although obese patients may be overrepresented, individuals with normal weight are also affected [1, 2, 9, 16].
Pathology
No histological abnormalities have been identified by haematoxylin and eosin (H&E) stains, fat stains or electron microscopy [9, 10, 13, 14, 16, 17, 18, 19.
Genetics
A significant portion (16–64%) of affected women have a self-reported positive family history of lipoedema [1, 11, 14]. Pit-1 mutation was identified in female members of one family with multiple anterior pituitary hormone deficiency and lipoedema [23]. However, no other genetic mutations have been identified.
Clinical features
Presentation
The onset of symmetrical and bilateral gradual fat deposition starts around puberty in most cases. However, it may also develop during other periods of hormonal changes such as pregnancy or menopause [24].
Enlargement of the lower extremities is disproportionate in relation to the trunk, upper extremities, face and neck [15], even among obese patients [2, 13]. Feet are generally spared. Fat deposition of lipoedema begins abruptly above the malleoli, causing a sharp demarcation between the normal and abnormal tissue at the ankles, which is known as the ‘cuff sign’. The only sign during early stages of disease may be the disappearance of the retromalleolar sulcus. As the disease progress, this characteristic physical sign becomes more prominent. If the upper extremities are affected, a similar cuff sign that ends sharply above the wrists is seen with sparing of the hands [15]. Although the non to minimal pitting oedema component is mild initially, prominent oedema may eventually develop [15].
Patients often complain of heaviness and discomfort of the involved areas with sensitivity to pressure. The swelling and aching is aggravated by exercise and warm weather [15].
Clinical variants
A classification based on the location of involved areas has been proposed [25]:
- I Mostly buttocks.
- II Buttocks to knees.
- III Buttocks to malleoli.
- IV Mainly arms.
- V Mainly lower legs.
Differential diagnosis
Differential diagnosis of lipoedema includes lymphoedema, obesity, chronic venous insufficiency and lipohypertrophy (Table 100.1) [12, 15, 26]. In contrast to lipoedema, patients with lymphoedema have a positive Stemmer's sign where there is an inability to pinch a fold of skin at the base of the second toe due to thickening and fibrosis of the subcutaneous tissue. In obesity, the increase in subcutaneous fat is generalized and pain is not usually a feature; furthermore, the typical sparing of the feet is not seen in obese patients. Chronic venous insufficiency is associated with hyperpigmentation and, initially, pitting oedema that can be relieved by leg elevation; with time, however, lymphoedema may supervene. The main difference between lipohypertrophy and lipoedema is the absence of oedema and pain in lipohypertrophy [15]. Other differential diagnoses include Dercum disease and benign symmetrical lipomatosis.
Table 100.1 Differential diagnosis of lipoedema.
| Lipoedema | Lymphoedema | Obesity | Chronic venous insufficiency | |
| Sex | Female | Both | Both | Both |
| Family history | Frequent | Rare (except for Milroy disease) | Frequent | In some cases |
| Effect of leg elevation | Little to none | Initially with moderate improvementa | No effect | Marked improvementb |
| History of cellulitis | None | Frequent in secondary lymphoedema | None | Frequent |
| History of ulcers | None | None | None | Very frequent |
| Symmetry | Present | Absent | Present | Present or absent |
| Involvement of feet | No | Yes | Yes | Yes |
| Tenderness to palpation | Yes | No | No | Yes |
| Pitting oedema | None or minimal initiallyc | Present with varying severity | Absent | Markedb |
| Tissue consistency | May be soft, fat, or firm | Doughy, similar to gelatin | Soft, like fat | Soft, pitting |
aLongstanding lymphoedema may not show improvement with leg elevation.
bLongstanding venous insufficiency may be complicated by lymphoedema, in which case this may not apply.
cLongstanding lipoedema may have pitting oedema.
Complications and co-morbidities
Mobility can be significantly impaired.
Disease course and prognosis
Lipoedema is a progressive lifelong disorder.
Investigations
Lipoedema remains a clinical diagnosis. MRI may be used to verify that the swelling is due to homogenous enlargement of the subcutaneous compartment [3, 4, 5, 6, 7, 8]. Dynamic lymphoscintigraphy can rule out true lymphoedema. Since venous disease can present concomitantly, duplex ultrasound is advocated by some if patients’ complaints cannot be fully explained by lipoedema [15]. No specific abnormalities have been found in phlebograms or arteriograms from lipoedema patients [16, 18, 19].
Management
Management includes strategies to address the physical as well as psychological impact of the disease, and, as such, a multidisciplinary approach is necessary [15].
Management of diet is important because additional fat laid down in lipoedematous limbs may be abnormally resistant to control by dieting and exercise and will also compound the difficulties of taking adequate exercise, causing further frustration and low self-esteem [15, 20, 27]. Because BMI and total body weight may not accurately reflect obesity status in lipoedema patients, some authors advocate the use of waist circumference as an indicator for ‘healthy weight’ [15, 28].
Diuretics and leg elevation do not help patients with lipoedema [11, 13], although they may benefit patients with concomitant lymphoedema.
Complex physical decongestion therapy, which combines manual lymphatic drainage and compression therapy, is widely accepted as a conservative therapeutic approach [13, 20, 29, 30]. While manual lymphatic drainage reduces the actual volume, compression by stocking or bandage is used to minimize recurrence. Compression therapy may improve, in part, the symptoms of lipoedema and also mitigate the progression of the lymphatic component. Certainly, patients with concomitant chronic venous insufficiency and/or lymphoedema will benefit from compression therapy [15]. In lipoedema patients, however, despite lifelong decongestion therapy, the amount of subcutaneous tissue increases and the disease overall progresses over time.
Tumescent liposuction has become an integral part in the management of lipoedema. Ideally, it should be performed early, and multiple sessions are often required [15]. Patients experience significant improvement in swelling, pain, mobility, appearance and quality of life [24, 25, 31–33]. Decongestion therapy should remain an integral part of post-liposuction management [24].
Lipo-lymphoedema
Definition
Lipo-lymphoedema is the coexistence of lipoedema and secondary lymphoedema.
Introduction and general description
Secondary lymphoedema may coexist in patients with longstanding lipoedema and the distinction between the two entities can be a challenge. The increased pressure from expansion of fat tissues in lipoedema contributes to the development of lipo-lymphoedema by causing mechanical obstruction of small lymphatic vessels.
Pathophysiology
The lymphatic system in lipoedema is thought to be normal or sufficient, at least as far as the large lymph vessels are concerned. However, the increased pressure from the expansion of fat tissues may cause mechanical obstruction of the small lymphatic vessels in the septa, resulting in mild lymphostasis and oedema of the subcutaneous tissue. Furthermore, lymphatic transport in lipoedema also decreases as fibrosis increases with age, and this can exacerbate the secondary lymphoedema [1].
Pathology
No distinct histological abnormalities have been identified in lipo-lymphoedema by H&E, fat stains or electron microscopy [2, 3, 4].
Clinical features
Presentation
In addition to the physical examination findings in lipoedema, patients with lipo-lymphoedema may have a positive Stemmer's sign – the inability to pinch a skin fold at the base of the second toe due to oedema or fibrosis of the skin. Stemmer's sign is often present in lymphoedema patients but is usally absent in lipoedema patients [5].
Disease course and prognosis
Lipo-lymphoedema has a chronic and progressive course.
Investigations
Lipo-lymphoedema is a clinical diagnosis. Table 100.2 describes imaging modalities that may assist in identifying lipo-lymphoedema [5].
Table 100.2 Imaging characteristics of lipoedema and lymphoedema.
| Imaging modality | Lipoedema | Lymphoedema |
| Lymphangiogram | Normal | Abnormal |
| Lymphoscintigram | Normal, with mild lymphatic delay in some | Abnormal with delayed lymphatic flow, subdermal collateralization and dermal backflow |
| Computed tomography | Diffuse and homogenous lipomatous hypertrophy of subcutaneous tissue | Thickening of the calf subcutaneous tissues and perimuscular aponeurosis. Increased fat density. Honeycomb appearance due to fibrous and oedematous stranding of fat |
| Magnetic resonance imaging | Normal skin thickness, increased fatty tissue | Thickening of the skin and subcutaneous tissue. Honeycomb appearance |
Management
The lymphatic oedema component of lipo-lymphoedema may improve initially with leg elevation and compression. See also the management section for lipoedema of the lower limbs.
Lipoedema of the scalp and lipoedematous alopecia
Definition and nomenclature
Lipoedema of the scalp (LS) and lipoedematous alopecia (LA) are characterized by the presence of a thick, boggy scalp due to a prominent increase in subcutaneous adipose tissue, which may be associated with varying degrees of hair loss.
Introduction and general description
Lipoedema of the scalp is a rare condition of unknown aetiology where a thick, boggy scalp develops due to a prominent increase in subcutaneous adipose tissue. It can be associated with scarring or non-scarring hair loss, when it is known as lipoedematous alopecia [1]. LS was first described by Cornbleet in 1935 [2] and LA was first described by Coskey et al. in 1961 [3]. There is some debate as to whether LS and LA represent clinical variants of the same entity or two different disorders [1]. Since they are both very rare, they will be grouped together for the purposes of this discussion.
Epidemiology
Incidence and prevalence
These are rare conditions with only approximately 45 cases of LS and 30 cases of LA reported in the literature [1, 4].
Age
Lipoedema of the scalp and LA are reported most commonly in adults [2, 3, 5–11, 12, 13–26, 27, 28–31].
Sex
Among reported cases, there is an approximate female to male ratio of 5 : 1 [2, 3, 4, 5–11, 12, 13–26, 27, 28, 29].
Ethnicity
Both LS and LA have been described in all ethnicities [2, 3, 4, 5–11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29].
Associated diseases
Several conditions have been reported in association with LS or LA but there is no consistent link with any of them [3, 5, 8, 12, 13, 15, 16–19, 21, 23].
Pathophysiology
The exact aetiology is unknown. A failure to maintain the integrity of the dermal–subcutaneous interface with an expansion and invasion of subcutaneous fat into the dermis is likely to be of relevance [27]. Some have speculated that female sex hormones may play a role, since these conditions have a predilection for women [29].
Hair cycle disturbance could occur as a result of lymphangiectasia [1, 16, 21, 24] or oedema [1, 9], both of which have been observed histologically in LS and LA. Alternatively, the fat itself might invade and destroy the hair follicle [27].
Pathology
The main pathological feature in both LS and LA is an approximate doubling in scalp thickness resulting from expansion of the subcutaneous fat layer [1]. Dilated lymphatic vessels have been observed in both LA and LS [1, 16, 21, 24]. In LA there is also a loss of hair follicles without any inflammation. Perifollicular fat cells in direct continuity with underlying subcutaneous fat lobules are noted in the dermis, together with fibrous tracks [27] and fragmentation of dermal elastic fibres [1, 11].
Genetics
No genetic mutation has been identified in patients with LS and LA. However, an LS mother–LA daughter pair has been reported [23].
Environmental factors
One group of Egyptian investigators observed that all of their patients wore a special tight head scarf called a mandil [19]. The associations between LS, LA and other headwear have not been reported by others.
Clinical features
Presentation
Although LS and LA are usually asymptomatic, some patients may experience pain, pruritus, paraesthesiae or headache [27]. While LA patients may present earlier due to alopecia, most LS patients were not aware of their condition and presented for other reasons [1].
Patients with LS and LA present with boggy thickening of the scalp, predominantly over the vertex, parietal and occipital scalp [12]. The bogginess may gradually extend to the entire scalp. The condition is more easily palpable than visible [1]. The consistency of the scalp has been likened to cotton wadding as used in quilting and upholstering [27]. The involved scalp is easily pressed down to the underlying bone, but returns to its original form immediately pressure is removed [19].
Hair loss in LA is variable and both scarring and non-scarring alopecia have been reported. Hairs do not exceed 2 cm in length and tend to break off easily [3]. Sometimes the hair in the affected area can have a lighter colour than hair from the surrounding unaffected scalp [4].
Of note is the fact that none of the LS and LA patients reported to date have had concurrent lipoedema of the legs.
Differential diagnosis
Differential diagnosis of LS and LA includes naevus lipomatosus superficialis of the scalp, cutis verticis gyrata and encephalocraniocutaneous lipomatosis. Naevus lipomatosus superficialis is usually present at birth and may have solitary or multiple lesions [20]. The scalp in cutis verticis gyrata is furrowed and on biopsy demonstrates closely packed hair follicles and considerable thickening of the collagen bundles of the dermis and subcutaneous tissue [25]. Encephalocraniocutaneous lipomatosis is associated with ophthalmological and neurological abnormalities.
Disease course and prognosis
Both LS and LA are chronic and persistent conditions.
Investigations
Ultrasound or MRI of the involved scalp area can confirm subcutaneous tissue thickening [27]. The average scalp thickness in healthy individuals was 5.8 ± 0.12 mm [32]. In contrast, the scalp thickness in LA and LS patients ranged from 8.5 to 16 mm with an average of 11.4 mm [25]. Biopsy of the affected area may also support the diagnosis.
Management
There is no specific and effective management guideline for LS and LA. Pain, pruritus and paraesthesia are managed symptomatically. Surgical excision of LS has been reported in one patient. At 12-month follow-up, there was no recurrence of LS although residual alopecia remained [27]. Topical minoxidil solutions have not been helpful in regrowing hair [27].
MISCELLANEOUS DISORDERS OF SUBCUTANEOUS FAT
Cellulite
Definition and nomenclature
Cellulite is an architectural disorder of human adipose tissue. It is characterized by the dimpled and nodular appearance of the skin in cellulite-prone areas in postpubertal females [1].
Introduction and general description
The term cellulite was first introduced in the French literature 150 years ago [2]. Cellulite is nowadays among the more common concerns of patients presenting to dermatologists. It is a localized metabolic disorder of subcutaneous tissue that results in a readily visible alteration in the appearance of the female body. Cellulite presents as a change of skin topography evident by skin with dimpling and nodularity that principally involves the pelvic region, lower limbs and abdomen of women. The topographical alteration is caused by herniations of subcutaneous fat through its fibrous connective tissue support matrix.
Epidemiology
Incidence and prevalence
It is estimated that between 85% and 98% of postpubertal females display some degree of cellulite [1].
Age
It first appears after puberty.
Sex
It is almost exclusive to women [1].
Ethnicity
White women are affected more frequently than women of other races [3].
Pathophysiology
The exact pathophysiology of cellulite is not fully understood; many theories seek to explain the structural, architectural, metabolic and biochemical differences between cellulite and non-cellulite fat. In a study performed by Rosenbaum et al. [4], biopsies from women with and without cellulite showed an irregular and discontinuous dermal–subcutaneous interface that was characterized by fat protrusion into the dermis. In contrast, the dermal–subcutaneous and connective tissue interface was smooth and continuous in male subjects. These findings suggest that there is a sexual dimorphism in the structural characteristics of subdermal connective tissue that predisposes women to the development of cellulite. Women with cellulite have a higher percentage of thinner, perpendicularly orientated, dermal septa: this appears to facilitate herniation of adipose tissue into the reticular dermis [5, 6].
There are also regional differences in both the hormonal responsiveness and metabolic activity of human adipose tissue. For example, adipocytes in the gluteal–femoral region are larger and are influenced by female sex hormones. The gluteal–femoral adipocytes are also metabolically more stable and resistant to lipolysis [1]. Although it has been conjectured that deterioration of the dermal vasculature with oedema and deposition of glycosoaminoglycans in the dermal capillary walls could result in cellulite [3], this observation has not been supported by others [5, 6, 7]. Similarly, the role of inflammation in the pathogenesis of cellulite remains controversial [2, 5, 7, 8, 9, 10].
Predisposing factors
Excessively high carbohydrate diets provoke hyperinsulinaemia and lipogenesis, leading to an increase in total body fat content, and thereby predispose to cellulite [1, 11]. Prolonged periods of sitting or standing may impede normal blood flow and lead to stasis, which alters microcirculation and may increase the risk of cellulite. Finally, fluid retention and the hormonal environment in pregnancy may also be contributory [1].
Pathology
Cellulite is a clinical diagnosis. Tissue sampling is not required for the diagnosis. Histopathological specimens demonstrate indentations of subcutaneous fat into the dermis [5].
Genetics
There seems to be a genetic predisposition to the development of cellulite as most women with cellulite report its occurrence in other family members [12]. However, no specific genetic mutation has been identified.
Clinical features
Presentation
Cellulite presents almost exclusively in postpubertal females and involves the skin of the pelvic region, lower limbs and abdomen. The skin appears dimpled, like the surface of a mattress or of orange peel [1, 5]. This surface irregularity is especially apparent when the skin is pinched. Cellulite can affect individuals with both high and low BMI [1].
Differential diagnosis
Obesity is differentiated from cellulite in that obesity is characterized by hypertrophy and hyperplasia of adipose tissue that is not necessarily limited to the pelvis, thighs and abdomen [1]. The skin is not dimpled in obesity.
Classification of severity
Cellulite has been divided into three grades of severity [1]:
- Grade I Skin dimpling is apparent on pinching but not otherwise.
- Grade II Skin dimpling is apparent on standing but not on lying down.
- Grade III Skin dimpling is apparent both on standing and on lying down.
Complications and co-morbidities
There is no associated morbidity or mortality [1].
Disease course and prognosis
Cellulite can be progressive.
Management
Although cellulite is a very common complaint, treatments proposed for it have lacked substantial proof of efficacy. There is no single treatment or treatment combination that has been shown to be reliably effective. The best currently available treatments have resulted in mild to moderate improvement at best. Most of any improvement obtained is not sustained over time and maintenance treatment is often needed.
Weight gain may accentuate the visibility of cellulite. Weight reduction may reduce but, ironically, may also sometimes increase its prominence. One group of investigators has shown, however, that, on average, cellulite severity decreases following weight loss. This is especially true for those with higher BMI and greater cellulite severity grading [13].
Various over-the-counter and prescription topical therapies have been advocated for cellulite. Topical application of 0.3% retinol for 6 months or more has been claimed to improve cellulite [14]. So-called mesotherapy, whereby a variety of substances including phosphatidylcholine, caffeine, theophylline and herbal extracts are injected into subcutaneous tissue in an attempt to dissolve fat, has been widely promoted but, not surprisingly, has not been shown to have any useful place in managing cellulite [14, 15, 16].
Many non-invasive devices have been promoted for the treatment of cellulite. These include a skin-kneading device, unipolar and bipolar radiofrequency devices, ultrasound devices and selective cryolysis [14].
More invasive procedures such as subcision and liposuction have also been attempted. While subcision may temporarily improve cellulite appearance, long-term efficacy remains to be demonstrated. Liposuction for the treatment of cellulite carries risk. While liposuction reduces fat deposits deeper in the subcutaneous fat, cellulite adipose tissue is deposited more superficially. When liposuction is performed at more superficial levels, there is an increased risk of necrosis and poor cosmetic outcome. Laser-assisted liposuction and/or laser-assisted lipoplasty, which are less invasive than traditional liposuction and offer simultaneous skin tightening, may be the preferred treatment [1].
In the search for a novel approach to managing cellulite, various investigators have looked at peroxisome proliferator-activated receptors (PPARs), which are found on adipocytes and are thought to have an effect on the extracellular matrix, as possible targets for the treatment of cellulite but there is no evidence to date that they are likely to be effective [14, 17–21].
The ideal management for this condition, which so many women find distressing and for the treatment of which considerable sums have been expended, has yet to be found.
Obesity and the skin
Rates of people being overweight and obese (BMI >25 kg/m2) have been rising at an alarming rate in many countries of the world in recent years: in England, for instance, 62.1% of adults were overweight or obese in 2013 [1]. The prevalence of obesity (BMI >30 kg/m2 in adult men and women rose over the 20 years from 1993 to 2013 from 13.2% and 16.4% to 26.0% and 23.8%, respectively [1]. Nearly 10% of all children entering English schools (at age 4–5 years) were classified as obese (weight ≥95th centile for age) [1].
Obesity exposes people not only to well-known metabolic and cardiovascular consequences including type 2 diabetes but also has significant effects on other body systems including the skin [2].
Physiological consequences of obesity on the skin. Subcutaneous fat insulates the body core from the external environment. Whilst this may be of benefit in protecting the body from the effects of exposure to cold it also means that it is harder for the obese to dissipate heat from the core out to the skin surface. This increases the reliance on sweating for thermoregulation in obese people, who tend to sweat more profusely than those who are not overweight [3]. Paradoxically, however, transepidermal water loss is increased in obese people, who are prone to xerosis [4].
Mechanical problems contributing to skin disease in the obese. Friction, sweating and maceration within body folds frequently lead to a painful erosive intertriginous dermatitis with secondary candidosis. Obese women with restricted mobility are more likely to have problems with urinary continence, which can contribute further to inflammation, with an irritant contact dermatitis affecting the genito-crural folds [5]. Stretch marks (striae distensae) are common, particularly if weight gain has been rapid [2, 5, 6]. They tend to be located in the axillary folds, upper thighs, buttocks and abdomen.
The mechanical effects of obesity may impede lymphatic drainage not only in the lower extremities but also elsewhere, for example in abdominal apron folds, resulting in lymphoedema [3]. The chronic high pressure exerted on the skin of the soles of the feet may result in plantar hyperkeratosis and postmenopausal plantar keratoderma (keratoderma climactericum) [2, 6].
Peripheral vascular and lymphovascular disorders in the obese. Obesity puts strain on the vascular system. Lower limb venous hypertension from whatever cause may be exacerbated in the obese by high intra-abdominal pressures and by immobility. The risks of venous eczema and venous ulceration are increased and they may be more difficult to manage. Chronic lymphoedema frequently supervenes (Figure 100.8) [2].
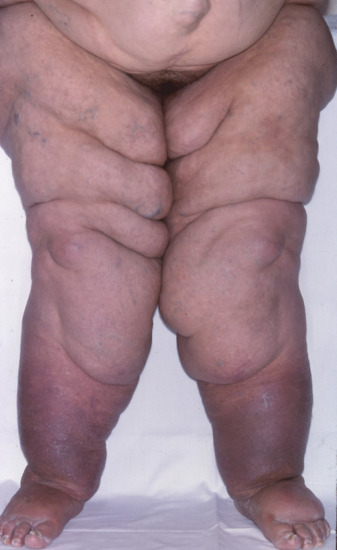
Figure 100.8 Gross obesity with bilateral lymphoedema of lower legs.
Cutaneous infections and obesity. The prevalence of superficial skin infections such as candidosis, dermatophytosis and erythrasma is raised in the obese [2]. Pyogenic infections such as folliculitis and furunculosis are also more frequently seen and may be recurrent. In a large study of patients requiring operative treatment of skin and soft tissue infections by community-acquired meticillin-resistant Staphylococcus aureus (MRSA), obese patients were more than three times likely to need further surgical intervention within a year than non-obese patients [7]. Lymphoedema in the obese is commonly complicated by streptococcal cellulitis. Obesity also increases the risk of surgical wound infection and of necrotizing fasciitis [8].
Cutaneous consequences of immunological and endocrine dysregulation in the obese. It is increasingly recognized that adipose tissue has multiple, complex regulatory and hormonal functions in addition to its role as an energy store (see Chapter 99); these functions may be disturbed in the obese. Altered adipocytokine secretion may have an effect on inflammation in the obese [9, 10]: for instance, delayed-type hypersensitivity responses are increased in obesity and decline with weight reduction [11]. Adipocytokines may also play a role in the well-recognized link between severe psoriasis and obesity, with evidence that weight reduction, including that achieved by bariatric surgery, can produce significant improvement in psoriasis [2, 12, 13]. There is also some evidence of a link between obesity and atopy including atopic eczema [2, 13].
The hyperinsulinaemia associated with obesity increases androgen production from adipose tissue and reduces the production of sex-hormone-binding globulin, thus increasing circulating free androgen levels. This manifests as increased risks of acne, hirsutism, androgenetic alopecia and polycystic ovary syndrome in the obese [2, 15]. Type 2 diabetes is a common complication of obesity and is associated with a variety of specific skin disorders (see Chapter 64).
Genetic disorders associated with obesity. Obesity is a component of a number of genetic disorders of which Prader–Willi syndrome is one of the best known. These are discussed in Chapter 74.
Other skin disorders in the obese. Dercum disease (see earlier in this chapter) is strongly associated with obesity. The presence of obesity may aggravate a variety of other skin disorders including hidradenitis suppurativa and pilonidal sinus. Surgical wound healing is impaired in the obese with an increased risk of incisional hernia [16].
References
Acquired lipodystrophy
Acquired generalized lipodystrophy
- Garg A. Clinical review: lipodystrophies: genetic and acquired body fat disorders. J Clin Endocrinol Metab 2011;96:3313–25.
- Ziegler L. Lipodystrophies: report of seven cases. Brain 1928; 51:147–67.
- Lawrence RD. Lipodystrophy and hepatomegaly, with diabetes, lipaemia, and other metabolic disturbances; a case throwing new light on the action of insulin. Lancet 1946;1(6403):724 passim.
- Iglesias P, Alvarez Fidalgo P, Codoceo R, Diez JJ. Lipoatrophic diabetes in an elderly woman: clinical course and serum adipocytokine concentrations. Endocr J 2004;51:279–86.
- Llamas-Velasco M, Dauden E, Martinez-Penas G, Garcia-Diez A. Late-onset acquired generalized lipodystrophy with muscle involvement. Actas Dermosifiliogr 2011;103:729–32.
- Wachslicht-Rodbard H, Muggeo M, Kahn CR, Saviolakis GA, Harrison LC, Flier JS. Heterogeneity of the insulin-receptor interaction in lipoatropic diabetes. J Clin Endocrinol Metab 1981;52:416–25.
- Franklin B, Ginsberg H, Haque WU, et al. Very low-density lipoprotein metabolism in an unusual case of lipoatrophic diabetes. Metabolism 1984; 33:814–19.
- Stacpoole PW, Alig J, Kilgore LL, et al. Lipodystrophic diabetes mellitus. Investigations of lipoprotein metabolism and the effects of omega-3 fatty acid administration in two patients. Metabolism 1988; 37:944–51.
- Klein S, Jahoor F, Wolfe RR, Stuart CA. Generalized lipodystrophy: in vivo evidence for hypermetabolism and insulin-resistant lipid, glucose, and amino acid kinetics. Metabolism 1992;41:893–6.
- Misra A, Garg A. Clinical features and metabolic derangements in acquired generalized lipodystrophy: case reports and review of the literature. Medicine (Baltimore) 2003;82:129–46.
- Chao PJ, Tsai JC, Chang DM, Shin SJ, Lee YJ. A case of acquired generalized lipodystrophy with cerebellar degeneration and type 2 diabetes mellitus. Rev Diabet Stud 2004;1:193–7.
- Kikkawa R, Hoshi M, Shigeta Y, Izumi K. Lack of ketosis in lipoatrophic diabetes. Diabetes 1972;21:827–31.
- Lupsa BC, Sachdev V, Lungu AO, Rosing DR, Gorden P. Cardiomyopathy in congenital and acquired generalized lipodystrophy: a clinical assessment. Medicine (Baltimore) 2010; 89:245–50.
- Dolberg BK, Lenhard MJ. Successful outcome of pregnancy in a patient with generalized lipoatrophic diabetes mellitus. Endocr Pract 2000;6:34–6.
- Huemer C, Kitson H, Malleson PN, et al. Lipodystrophy in patients with juvenile dermatomyositis – evaluation of clinical and metabolic abnormalities. J Rheumatol 2001;28:610–15.
- Wilson DE, Chan IF, Stevenson KB, Horton SC, Schipke C. Eucaloric substitution of medium chain triglycerides for dietary long chain fatty acids in acquired total lipodystrophy: effects on hyperlipoproteinemia and endogenous insulin resistance. J Clin Endocrinol Metab 1983;57:517–23.
- Goebel FD, Dorfler H, Goebel HH, Zollner N. Muscle capillary basement membrane thickness in lipoatrophic diabetes. Res Exp Med (Berl) 1977;171:271–6.
- Coelho PC, Jordao A, Andre O, de Queiroz MV, Eurico-Lisboa P. Rheumatological manifestations of lipoatrophic diabetes. Clin Rheumatol 1995;14:229–30.
- Griebel M, Mallory SB. Generalized weight loss in a child. Generalized lipodystrophy. Arch Dermatol 1988;124:573-6.
- Seip M, Trygstad O. Generalized lipodystrophy, congenital and acquired (lipoatrophy). Acta Paediatr Suppl 1996;413:2–28.
- Davis J, Tizard J. Generalized lipodystrophy, infectious mononucleosis, mild infantile hemiplegia. Proc R Soc Med 1954;47:128–9.
- Garg A. Lipodystrophies. Am J Med. 2000;108:143–52.
- Savage DB, Semple RK, Clatworthy MR, et al. Complement abnormalities in acquired lipodystrophy revisited. J Clin Endocrinol Metab 2009;94:10–16.
- Eren E, Ozkan TB, Cakir ED, Saglam H, Tarim O. Acquired generalized lipodystrophy associated with autoimmune hepatitis and low serum C4 level. J Clin Res Pediatr Endocrinol 2010;2:39–42.
- Hubler A, Abendroth K, Keiner T, et al. Dysregulation of insulin-like growth factors in a case of generalized acquired lipoatrophic diabetes mellitus (Lawrence syndrome) connected with autoantibodies against adipocyte membranes. Exp Clin Endocrinol Diabetes 1998;106:79–84.
- Lewis GF, Carpentier A, Adeli K, Giacca A. Disordered fat storage and mobilization in the pathogenesis of insulin resistance and type 2 diabetes. Endocr Rev 2002;23:201–29.
- McGarry JD. Banting lecture 2001: dysregulation of fatty acid metabolism in the etiology of type 2 diabetes. Diabetes 2002;51:7–18.
- Ravussin E, Smith SR. Increased fat intake, impaired fat oxidation, and failure of fat cell proliferation result in ectopic fat storage, insulin resistance, and type 2 diabetes mellitus. Ann NY Acad Sci 2002;967:363–78.
- Bays H, Mandarino L, DeFronzo RA. Role of the adipocyte, free fatty acids, and ectopic fat in pathogenesis of type 2 diabetes mellitus: peroxisomal proliferator-activated receptor agonists provide a rational therapeutic approach. J Clin Endocrinol Metab 2004;89:463–78.
- Pardini VC, Victoria IM, Rocha SM, et al. Leptin levels, beta-cell function, and insulin sensitivity in families with congenital and acquired generalized lipoatropic diabetes. J Clin Endocrinol Metab 1998;83:503–8.
- Reitman ML, Arioglu E, Gavrilova O, Taylor SI. Lipoatrophy revisited. Trends Endocrinol Metab 2000;11:410–16.
- Weyer C, Funahashi T, Tanaka S, et al. Hypoadiponectinemia in obesity and type 2 diabetes: close association with insulin resistance and hyperinsulinemia. J Clin Endocrinol Metab 2001;86:1930–5.
- Haque WA, Shimomura I, Matsuzawa Y, Garg A. Serum adiponectin and leptin levels in patients with lipodystrophies. J Clin Endocrinol Metab 2002;87:2395.
- Taylor WB, Honeycutt WM. Progressive lipodystrophy and lipoatrophic diabetes. Review of the literature and case reports. Arch Dermatol 1961;84:31–6.
- Smith JP, Burns TW, Leigh CC. Diabetes mellitus and lipoatrophy. South Med J 1990;83:573–6.
- Sasaki T, Ono H, Nakajima H, Sugimoto J. Lipoatrophic diabetes. J Dermatol 1992;19:246–9.
- Senior B, Gellis SS. The syndromes of total lipodystrophy and of partial lipodystrophy. Pediatrics 1964;33:593–612.
- Ipp MM, Howard NJ, Tervo RC, Gelfand EW. Sicca syndrome and total lipodystrophy: a case in a fifteen-year-old female patient. Ann Intern Med 1976;85:443–6.
- Wesenberg RL, Gwinn JL, Barnes GR, Jr. The roentgenographic findings in total lipodystrophy. Am J Roentgenol Radium Ther Nucl Med 1968;103:154–64.
- Golden MP, Charles MA, Arquilla ER, et al. Insulin resistance in total lipodystrophy: evidence for a pre-receptor defect in insulin action. Metabolism 1985;34:330–5.
- Vantyghem MC, Vigouroux C, Magre J, et al. Late-onset lipoatrophic diabetes. Phenotypic and genotypic familial studies and effect of treatment with metformin and lispro insulin analog. Diabetes Care 1999;22:1374–6.
- Simha V, Garg A. Body fat distribution and metabolic derangements in patients with familial partial lipodystrophy associated with mandibuloacral dysplasia. J Clin Endocrinol Metab 2002;87:776–85.
- Edge J, Dunger DB, Dillon MJ. Weber-Christian panniculitis and chronic active hepatitis. Eur J Pediatr 1986;145:227–9.
- Billings JK, Milgraum SS, Gupta AK, Headington JT, Rasmussen JE. Lipoatrophic panniculitis: a possible autoimmune inflammatory disease of fat. Report of three cases. Arch Dermatol 1987;123:1662–6.
- Catalano PM, Capeless EL, Simmons GM, Robbins DC, Horton ES. Successful pregnancy outcome in association with lipoatrophic diabetes mellitus. Obstet Gynecol 1990;76(5 Pt 2):978–9.
- Garg A, Fleckenstein JL, Peshock RM, Grundy SM. Peculiar distribution of adipose tissue in patients with congenital generalized lipodystrophy. J Clin Endocrinol Metab 1992;75:358–61.
- Lutz BS, Toussaint S, Wei FC. Bilateral facial lipoatrophy secondary to connective tissue panniculitis treated with two microsurgically transplanted latissimus dorsi muscles. Ann Plast Surg 1998;40:302–7.
- Van der Wal KG, Mulder JW. Facial contour reconstruction in partial lipodystrophy using two temporalis muscle flaps. A case report. Int J Oral Maxillofac Surg 1998;27:14–16.
- Oral EA, Simha V, Ruiz E, et al. Leptin-replacement therapy for lipodystrophy. N Engl J Med 2002;346:570–8.
- Petersen KF, Oral EA, Dufour S, et al. Leptin reverses insulin resistance and hepatic steatosis in patients with severe lipodystrophy. J Clin Invest 2002;109:1345–50.
Acquired partial lipodystrophy
- Garg A. Lipodystrophies. Am J Med 2000;108:143–52.
- Garg A. Clinical review: lipodystrophies: genetic and acquired body fat disorders. J Clin Endocrinol Metab 2011;96:3313–25.
- Mitchell S. Singular case of absence of adipose matter in the upper half of the body. Am J Med Sci 1885;90:105–6.
- Barraquer L. Histoire clinique d'un cas d'atrophie du tissu celluloadipeux. Neurolog Zentralblatt 1907;26:1072.
- Simons A. Eine seltnen Trophoneurose: “Lipodystrophia progressiva”. Z Ges Neurol Psychiat 1911;5:29–38.
- Poley JR, Stickler GB. Progressive lipodystrophy. a clinical study of 50 patients. Am J Dis Child 1963;106:356–63.
- Ipp MM, Minta JO, Gelfand EW. Disorders of the complement system in lipodystrophy. Clin Immunol Immunopathol 1977;7:281–7.
- Jasin HE. Systemic lupus erythematosus, partial lipodystrophy and hypocomplementemia. J Rheumatol 1979;6:43–50.
- McLean RH, Hoefnagel D. Partial lipodystrophy and familial C3 deficiency. Hum Hered 1980;30:149–54.
- Torrelo A, Espana A, Boixeda P, Ledo A. Partial lipodystrophy and dermatomyositis. Arch Dermatol 1991;127:1846–7.
- Senior B, Gellis SS. The syndromes of total lipodystrophy and of partial lipodystrophy. Pediatrics 1964;33:593–612.
- O'Mahony D, O'Mahony S, Whelton MJ, McKiernan J. Partial lipodystrophy in coeliac disease. Gut 1990;31:717–18.
- Smak Gregoor PJ, van Bommel EF, Ketterings C, van Saase JL, Kramer P. Progressive lipodystrophy, a diagnosis at a glance. Nephrol Dial Transplant 1998;13:507–9.
- Spranger S, Spranger M, Tasman AJ, Reith W, Voigtlander T, Voigtlander V. Barraquer-Simons syndrome (with sensorineural deafness): a contribution to the differential diagnosis of lipodystrophy syndromes. Am J Med Genet 1997;71:397–400.
- Caramaschi P, Biasi D, Lestani M, Chilosi M. A case of acquired partial lipodystrophy associated with POEMS syndrome. Rheumatology (Oxford) 2003;42:488–90.
- Rooney DP, Ryan MF. Diabetes with partial lipodystrophy following sclerodermatous chronic graft vs host disease. Diabet Med 2006;23:436–40.
- Winhoven SM, Hafejee A, Coulson IH. An unusual case of an acquired acral partial lipodystrophy (Barraquer-Simons syndrome) in a patient with extrinsic allergic alveolitis. Clin Exp Dermatol 2006;31:594–6.
- Williams DG, Bartlett A, Duffus P. Identification of nephritic factor as an immunoglobulin. Clin Exp Immunol 1978;33:425–9.
- Mathieson PW, Peters K. Are nephritic factors nephritogenic? Am J Kidney Dis 1994;24:964–66.
- Misra A, Peethambaram A, Garg A. Clinical features and metabolic and autoimmune derangements in acquired partial lipodystrophy: report of 35 cases and review of the literature. Medicine (Baltimore) 2004;83:18–34.
- Sissons JG, West RJ, Fallows J, et al. The complement abnormalities of lipodystrophy. N Engl J Med 1976 26;294:461–5.
- Mathieson PW, Wurzner R, Oliveria DB, Lachmann PJ, Peters DK. Complement-mediated adipocyte lysis by nephritic factor sera. J Exp Med 1993;177:1827–31.
- Mathieson PW, Peters DK. Lipodystrophy in MCGN type II: the clue to links between the adipocyte and the complement system. Nephrol Dial Transplant 1997;12:1804–6.
- Peake PW, O'Grady S, Pussell BA, Charlesworth JA. Detection and quantification of the control proteins of the alternative pathway of complement in 3T3-L1 adipocytes. Eur J Clin Invest 1997;27:922–7.
- Oswiecimska J, Ziora K, Geisler G, Dyduch A. Acquired partial lipodystrophy in an 11-year-old girl. Pediatr Int 2008;50(5):714–16.
- Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 1993;259:87–91.
- Van Hall G, Steensberg A, Sacchetti M, et al. Interleukin-6 stimulates lipolysis and fat oxidation in humans. J Clin Endocrinol Metab 2003;88:3005–10.
- Taylor WB, Honeycutt WM. Progressive lipodystrophy and lipoatrophic diabetes. Review of the literature and case reports. Arch Dermatol 1961;84:31–6.
- Hegele RA, Cao H, Liu DM, et al. Sequencing of the reannotated LMNB2 gene reveals novel mutations in patients with acquired partial lipodystrophy. Am J Hum Genet 2006;79:383–9.
- Baccjo VF, Maillet F, Berian L, et al. Neutralising antibodies against C3NeF in intravenous immunoglobulin. Lancet 1992;340:63–4.
- Levy Y, George J, Yona E, et al. Partial lipodystrophy, mesangiocapillary glomerulonephritis, and completment dysregulation. An autoimmune phenomenon. Immunol Res 1998:18:55–60.
HIV-associated lipodystrophy
- Hengel RL, Watts NB, Lennox JL. Benign symmetric lipomatosis associated with protease inhibitors. Lancet 1997;350:1596.
- Garg A. Clinical review: lipodystrophies: genetic and acquired body fat disorders. J Clin Endocrinol Metab 2011;96:3313–25.
- Domingo P, Estrada V, Lopez-Aldeguer J, Villaroya F, Martinez E. Fat redistribution syndromes associated with HIV-1 infection and combination antiretroviral therapy. AIDS Rev 2012;14:112–23.
- Carr A, Emery S, Law M, Puls R, Lundgren JD, Powderly WG. An objective case definition of lipodystrophy in HIV-infected adults: a case-control study. Lancet 2003;361:726–35.
- Law M, Puls R, Cheng AK, Cooper DA, Carr A. Evaluation of the HIV lipodystrophy case definition in a placebo-controlled, 144-week study in antiretroviral-naive adults. Antivir Ther 2006;11:179–86.
- Chen D, Misra A, Garg A. Clinical review 153: lipodystrophy in human immunodeficiency virus-infected patients. J Clin Endocrinol Metab 2002;87:4845–56.
- Martinez E, Mocroft A, Garcia-Viejo MA, et al. Risk of lipodystrophy in HIV-1-infected patients treated with protease inhibitors: a prospective cohort study. Lancet 2001;357:592–8.
- Narciso P, Tozzi V, D'Offizi G, et al. Metabolic and morphologic disorders in patients treated with highly active antiretroviral therapy since primary HIV infection. Ann NY Acad Sci 2001;946:214–22.
- Arribas JR, Pozniak AL, Gallant JE, et al. Tenofovir disoproxil fumarate, emtricitabine, and efavirenz compared with zidovudine/lamivudine and efavirenz in treatment-naive patients: 144-week analysis. J Acquir Immune Defic Syndr 2008;47:74–8.
- Babl FE, Regan AM, Pelton SI. Abnormal body-fat distribution in HIV-1-infected children on antiretrovirals. Lancet 1999;353:1243–4.
- Jaquet D, Levine M, Ortega-Rodriguez E, et al. Clinical and metabolic presentation of the lipodystrophic syndrome in HIV-infected children. AIDS 2000;14:2123–8.
- Arpadi SM, Cuff PA, Horlick M, Wang J, Kotler DP. Lipodystrophy in HIV-infected children is associated with high viral load and low CD4+-lymphocyte count and CD4+-lymphocyte percentage at baseline and use of protease inhibitors and stavudine. J Acquir Immune Defic Syndr 2001;27:30–4.
- Brambilla P, Bricalli D, Sala N, et al. Highly active antiretroviral-treated HIV-infected children show fat distribution changes even in absence of lipodystrophy. AIDS 2001;15:2415–22.
- McComsey G, Rightmire A, Wirtz V, Yang R, Mathew M, McGrath D. Changes in body composition with ritonavir-boosted and unboosted atazanavir treatment in combination with lamivudine and stavudine: a 96-week randomized, controlled study. Clin Infect Dis 2009;48:1323–6.
- Grinspoon S, Carr A. Cardiovascular risk and body-fat abnormalities in HIV-infected adults. N Engl J Med 2005;352:48–62.
- Villarroya F, Domingo P, Giralt M. Lipodystrophy in HIV 1-infected patients: lessons for obesity research. Int J Obes (Lond) 2007;31:1763–76.
- Murata H, Hruz PW, Mueckler M. The mechanism of insulin resistance caused by HIV protease inhibitor therapy. J Biol Chem 2000;275:20251–4.
- Carr A, Miller J, Law M, Cooper DA. A syndrome of lipoatrophy, lactic acidaemia and liver dysfunction associated with HIV nucleoside analogue therapy: contribution to protease inhibitor-related lipodystrophy syndrome. AIDS 2000;14:F25–32.
- Lee H, Hanes J, Johnson KA. Toxicity of nucleoside analogues used to treat AIDS and the selectivity of the mitochondrial DNA polymerase. Biochemistry 2003;42(50):14711–19.
- Caron-Debarle M, Lagathu C, Boccara F, Vigouroux C, Capeau J. HIV-associated lipodystrophy: from fat injury to premature aging. Trends Mol Med 2010;16:218–29.
- Domingo P, Matias-Guiu X, Pujol RM, et al. Subcutaneous adipocyte apoptosis in HIV-1 protease inhibitor-associated lipodystrophy. AIDS 1999;13:2261–7.
- Lafontan M, Berlan M. Do regional differences in adipocyte biology provide new pathophysiological insights? Trends Pharmacol Sci 2003;24:276–83.
- Heath KV, Hogg RS, Singer J, Chan KJ, O'Shaughnessy MV, Montaner JS. Antiretroviral treatment patterns and incident HIV-associated morphologic and lipid abnormalities in a population-based chort. J Acquir Immune Defic Syndr 2002;30:440–7.
- Lichtenstein KA, Delaney KM, Armon C, et al. Incidence of and risk factors for lipoatrophy (abnormal fat loss) in ambulatory HIV-1-infected patients. J Acquir Immune Defic Syndr 2003;32:48–56.
- Podzamczer D, Ferrer E, Sanchez P, et al. Less lipoatrophy and better lipid profile with abacavir as compared to stavudine: 96-week results of a randomized study. J Acquir Immune Defic Syndr 2007;44:139–47.
- Haubrich RH, Riddler SA, DiRienzo AG, et al. Metabolic outcomes in a randomized trial of nucleoside, nonnucleoside and protease inhibitor-sparing regimens for initial HIV treatment. AIDS 2009;23:1109–18.
- Funk E, Brissett AE, Friedman CD, Bressler FJ. HIV-associated facial lipoatrophy: establishment of a validated grading scale. Laryngoscope 2007;117:1349–53.
- Hansen BR, Haugaard SB, Iversen J, Nielsen JO, Andersen O. Impact of switching antiretroviral therapy on lipodystrophy and other metabolic complications: a review. Scand J Infect Dis 2004;36:244–53.
- Leyes P, Martinez E, Forga Mde T. Use of diet, nutritional supplements and exercise in HIV-infected patients receiving combination antiretroviral therapies: a systematic review. Antivir Ther 2008;13:149–59.
- Hadigan C. Dietary habits and their association with metabolic abnormalities in human immunodeficiency virus-related lipodystrophy. Clin Infect Dis 2003;37(Suppl. 2):S101–4.
- Malita FM, Karelis AD, Toma E, Rabasa-Lhoret R. Effects of different types of exercise on body composition and fat distribution in HIV-infected patients: a brief review. Can J Appl Physiol 2005;30:233–45.
- Yki-Jarvinen H. Thiazolidinediones. N Engl J Med 2004;351:1106–18.
- Calmy A, Bloch M, Wand H, et al. No significant effect of uridine or pravastatin treatment for HIV lipoatrophy in men who have ceased thymidine analogue nucleoside reverse transcriptase inhibitor therapy: a randomized trial. HIV Med 2010;11:493–501.
- McComsey GA, Walker UA, Budhathoki CB, et al. Uridine supplementation in the treatment of HIV lipoatrophy: results of ACTG 5229. AIDS 2010;24:2507–15.
- Fontdevila J, Serra-Renom JM, Raigosa M, et al. Assessing the long-term viability of facial fat grafts: an objective measure using computed tomography. Aesthet Surg J 2008;28:380–6.
- Guaraldi G, Fontdevila J, Christensen LH, et al. Surgical correction of HIV-associated facial lipoatrophy. AIDS 2011;25:1–12.
- Martinez E, Visnegarwala F, Grund B, et al. The effects of intermittent, CD4-guided antiretroviral therapy on body composition and metabolic parameters. AIDS 2010;24:353–63.
- Moyle G, Moutschen M, Martinez E, et al. Epidemiology, assessment, and management of excess abdominal fat in persons with HIV infection. AIDS Rev 2010;12:3–14.
- Snel YE, Brummer RJ, Doerga ME, et al. Adipose tissue assessed by magnetic resonance imaging in growth hormone-deficient adults: the effect of growth hormone replacement and a comparison with control subjects. Am J Clin Nutr 1995;61:1290–4.
- Rietschel P, Hadigan C, Corcoran C, et al. Assessment of growth hormone dynamics in human immunodeficiency virus-related lipodystrophy. J Clin Endocrinol Metab 2001;86:504–10.
- Kotler DP, Muurahainen N, Grunfeld C, et al. Effects of growth hormone on abnormal visceral adipose tissue accumulation and dyslipidemia in HIV-infected patients. J Acquir Immune Defic Syndr 2004;35:239–52.
- Grunfeld C, Thompson M, Brown SJ, et al. Recombinant human growth hormone to treat HIV-associated adipose redistribution syndrome: 12 week induction and 24-week maintenance therapy. J Acquir Immune Defic Syndr 2007;45:286–97.
- Falutz J, Allas S, Blot K, et al. Metabolic effects of a growth hormone-releasing factor in patients with HIV. N Engl J Med 2007;357:2359–70.
- Johnson RM. Reduction mammoplasty in an HIV-positive woman. AIDS Patient Care STDS 2005;19:353–5.
- Hultman CS, McPhail LE, Donaldson JH, Wohl DA. Surgical management of HIV-associated lipodystrophy: role of ultrasonic-assisted liposuction and suction-assisted lipectomy in the treatment of lipohypertrophy. Ann Plast Surg 2007;58:255–63.
- Nelson L, Stewart KJ. Plastic surgical options for HIV-associated lipodystrophy. J Plast Reconstr Aesthet Surg 2008;61:359–65.
Localized lipoatrophy and/or lipodystrophy
Semicircular lipoatrophy
- Gschwandtner WR, Munzberger H. [Lipoatrophia semicircularis. Linear and circular atrophy of the subcutaneous fat in the extremities.] Hautarzt 1974;25:222–7.
- De Groot AC. Is lipoatrophia semicircularis induced by pressure? Br J Dermatol 1994;131:887–90.
- Herane MI, Urbina F, Sudy E. Lipoatrophia semicircularis: a compressive lipoatrophy consecutive to persistent mechanical pressure. J Dermatol 2007;34:390–3.
- Gomez-Espejo C, Perez-Bernal A, Camacho-Martinez F. A new case of semicircular lipoatrophy associated with repeated external microtraumas and review of the literature. J Eur Acad Dermatol Venereol 2005;19:459–61.
- Nagore E, Sanchez-Motilla JM, Rodriguez-Serna M, Vilata JJ, Aliaga A. Lipoatrophia semicircularis – a traumatic panniculitis: report of seven cases and review of the literature. J Am Acad Dermatol 1998;39:879–81.
- Reinoso-Barbero L, Gonzalez-Gomez MF, Belanger-Quintana D, et al. Case-control study of semicircular lipoatrophy, a new occupational disease in office workers. J Occup Health 2013;55(3):149–57.
- Bloch PH, Runne U. [Lipoatrophia semicircularis in the male. Coincidence of arterial variations and micro-traumas as a possible disease cause.] Hautarzt 1978;29:270–2.
- Schnitzler L, Verret JL, Titon JP. [Lipoatrophia semi-circularis of the thigh.] Ann Dermatol Venereol 1980;107:421–6.
- Gschwandtner WR, Munzberger H. [Lipoatrophia semicircularis (author's translation).] Wien Klin Wochenschr 1975;87:164–8.
- Mallett R, Champion R. Lipoatrophia semicircularis. Br J Dermatol 1989;121:94–5.
- Betti R, Urbani CE, Inselvini E, Crosti C. Semicircular lipoatrophy. Clin Exp Dermatol 1992;17:382–3.
- Senecal S, Victor V, Choudat D, Hornez-Davin S, Conso F. Semicircular lipoatrophy: 18 cases in the same company. Contact Dermatitis 2000;42:101–2.
- Mascaro JM, Ferrando J. Lipoatrophia semicircularis: the perils of wearing jeans? Int J Dermatol 1982;21:138–9.
- Mascaro JM, Ferrando J. The perils of wearing jeans; lipoatrophia semicircularis. Int J Dermatol 1983;22:333.
- Ogino J, Saga K, Tamagawa M, Akutsu Y. Magnetic resonance imaging of semicircular lipoatrophy. Dermatology 2004;209:340–1.
- Gruber PC, Fuller LC. Lipoatrophy semicircularis induced by trauma. Clin Exp Dermatol 2001;26:269–71.
- Rongioletti F, Rebora A. Annular and semicircular lipoatrophies. Report of three cases and review of the literature. J Am Acad Dermatol 1989;20:433–6.
- Hodak E, David M, Sandbank M. Semicircular lipoatrophy – a pressure-induced lipoatrophy? Clin Exp Dermatol 1990;15:464–5.
- Thiele B, Ippen H. [Multilocular progressive lipoatrophia semicircularis.] Hautarzt 1983;34:292–2.
- Egli M, Rufli T. Lipoatrophia semicircularis. Dermatologica 1978;157:300–1.
Localized lipoatrophy due to injected drugs
- Underwood LE, Voina SJ, Van Wyk JJ. Restoration of growth by human growth hormone (Roos) in hypopituitary dwarfs immunized by other human growth hormone preparations: clinical and immunological studies. J Clin Endocrinol Metab 1974;38:288–97.
- Dahl PR, Zalla MJ, Winkelmann RK. Localized involutional lipoatrophy: a clinicopathologic study of 16 patients. J Am Acad Dermatol 1996;35:523–8.
- Kayikcioglu A, Akyurek M, Erk Y. Semicircular lipoatrophy after intragluteal injection of benzathine penicillin. J Pediatr 1996;129:166–7.
- Bauerschmitz J, Bork K. Multifocal disseminated lipoatrophy secondary to intravenous corticosteroid administration in a patient with adrenal insufficiency. J Am Acad Dermatol 2002;46(Suppl. 5):S130–2.
Insulin-induced localized lipoatrophy
- Richardson T, Kerr D. Skin-related complications of insulin therapy: epidemiology and emerging management strategies. Am J Clin Dermatol 2003;4:661–7.
- Hulst SG. Treatment of insulin-induced lipoatrophy. Diabetes 1976;25:1052–4.
- Reeves WG, Allen BR, Tattersall RB. Insulin-induced lipoatrophy: evidence for an immune pathogenesis. Br Med J 1980;280:1500–3.
- Ramos AJ, Farias MA. Human insulin-induced lipoatrophy: a successful treatment with glucocorticoid. Diabetes Care 2006;29:926–7.
- Wilson RM, Douglas CA, Tattersall RB, Reeves WG. Immunogenicity of highly purified bovine insulin: a comparison with conventional bovine and highly purified human insulins. Diabetologia 1985;28:667–70.
- McNally PG, Jowett NI, Kurinczuk JJ, Peck RW, Hearnshaw JR. Lipohypertrophy and lipoatrophy complicating treatment with highly purified bovine and porcine insulins. Postgrad Med J 1988;64:850–3.
- Griffin ME, Feder A, Tamborlane WV. Lipoatrophy associated with lispro insulin in insulin pump therapy: an old complication, a new cause? Diabetes Care 2001;24:174.
- Ampudia-Blasco FJ, Hasbum B, Carmena R. A new case of lipoatrophy with lispro insulin in insulin pump therapy: is there any insulin preparation free of complications? Diabetes Care 2003;26:953–4.
- Felner EI. Human insulin-induced lipoatrophy. J Pediatr 2003;142:448.
- Arranz A, Andia V, Lopez-Guzman A. A case of lipoatrophy with Lispro insulin without insulin pump therapy. Diabetes Care 2004;27:625–6.
- Ampudia-Blasco FJ, Girbes J, Carmena R. A case of lipoatrophy with insulin glargine: long-acting insulin analogs are not exempt from this complication. Diabetes Care 2005;28:2983.
- Lopez X, Castells M, Ricker A, Velazquez EF, Mun E, Goldfine AB. Human insulin analog-induced lipoatrophy. Diabetes Care 2008;31:442–4.
- Bloom A. Fat atrophy due to insulin. BMJ 1972;4(5836):366.
- Atlan-Gepner C, Bongrand P, Farnarier C, et al. Insulin-induced lipoatrophy in type I diabetes. A possible tumor necrosis factor-alpha-mediated dedifferentiation of adipocytes. Diabetes Care 1996;19:1283–5.
- Jaap AJ, Horn HM, Tidman MJ, Walker JD. Lipoatrophy with human insulin. Diabetes Care 1996;19:1289–90.
- Fisherman EW, Feinberg AR, Feinberg SM. Local subcutaneous atrophy. JAMA 1962;179:971–2.
- Dahl PR, Zalla MJ, Winkelmann RK. Localized involutional lipoatrophy: a clinicopathologic study of 16 patients. J Am Acad Dermatol 1996;35:523–8.
- Jermendy G, Nadas J, Sapi Z. “Lipoblastoma-like” lipoatrophy induced by human insulin: morphological evidence for local dedifferentiation of adipocytes? Diabetologia 2000;43:955–6.
- Edidin DV. Cutaneous manifestations of diabetes mellitus in children. Pediatr Dermatol 1985;2:161–79.
- Kahn CR, Rosenthal AS. Immunologic reactions to insulin: insulin allergy, insulin resistance, and the autoimmune insulin syndrome. Diabetes Care 1979;2:283–95.
- Kim KT, Ahn SK, Choi EH, Lee SH. Membranous lipodystrophy associated with insulin lipoatrophy. Int J Dermatol 1997;36:299–301.
- Radermecker RP, Pierard GE, Scheen AJ. Lipodystrophy reactions to insulin: effects of continuous insulin infusion and new insulin analogs. Am J Clin Dermatol 2007;8:21–8.
- Morgan AM. Localized reactions to injected therapeutic materials. Part 1. Medical agents. J Cutan Pathol 1995;22:193–214.
- Whitley TH, Lawrence PA, Smith CL. Amelioration of insulin lipoatrophy by dexamethasone injection. JAMA 1976;235:839–40.
- Valenta LJ, Elias AN. Insulin-induced lipodystrophy in diabetic patients resolved by treatment with human insulin. Ann Intern Med 1985;102(6):790–1.
- Kumar O, Miller L, Mehtalia S. Use of dexamethasone in treatment of insulin lipoatrophy. Diabetes 1977;26:296–9.
- Galloway JA, Bressler R. Insulin treatment in diabetes. Med Clin North Am 1978;62(4):663–80.
- Logwin S, Conget I, Jansa M, Vidal M, Nicolau C, Gomis R. Human insulin-induced lipoatrophy. Successful treatment using a jet-injection device. Diabetes Care 1996;19:255–6.
- Chantelau E, Reuter M, Schotes S, Starke AA. Severe lipoatrophy with human insulin: successfully treated by CSII. Diabet Med 1993;10:580–1.
Localized lipoatrophy due to injected corticosteroid
- Dahl PR, Zalla MJ, Winkelmann RK. Localized involutional lipoatrophy: a clinicopathologic study of 16 patients. J Am Acad Dermatol 1996;35:523–8.
- Goldman L. Reactions following intralesional and sublesional injections of corticosteroids. JAMA 1962;182:613–16.
- Shumaker PR, Rao J, Goldman MP. Treatment of local, persistent cutaneous atrophy following corticosteroid injection with normal saline infiltration. Dermatol Surg 2005;31:1340–3.
- Fisherman EW, Feinberg AR, Feinberg SM. Local subcutaneous atrophy. JAMA 1962;179:971–2.
- Schetman D, Hambrick GW, Jr, Wilson CE. Cutaneous changes following local injection of triamcinolone. Arch Dermatol 1963;88:820–8.
- Iuel J, Kryger J. Local cutaneous atrophy following corticosteroid injection. Acta Rheumatol Scand 1965;11:137–44.
- Goldman L. Histological effects of hydrocortisone in the skin of man. Ann NY Acad Sci 1955;61:520–33.
- Fulop E. Mechanism of local skin atrophy caused by intradermally injected corticosteroids. Dermatologica 1976;152(Suppl. 1):139–46.
- Pariser H, Murray PF. Intralesional injections of triamcinolone. Effects of different concentrations on psoriatic lesions. Arch Dermatol 1963;87:183–7.
- Reddy S, Ananthakrishnan S, Garg A. A prospective observational study evaluating hypothalamic-pituitary-adrenal axis alteration and efficacy of intramuscular triamcinolone acetononide for steroid responsive dermatologic disease. J Am Acad Dermatol 2013;69:226–31.
- Cassidy JT, Bole GG. Cutaneous atrophy secondary to intra-articular corticosteroid administration. Ann Intern Med 1966;65:1008–18.
- Dyment PG. Local atrophy following triamcinolone injection. Pediatrics 1970;46:136–7.
- Jacobs MB. Local subcutaneous atrophy after corticosteroid injection. Postgrad Med 1986;80:159–60.
- Louis DS, Hankin FM, Eckenrode JF. Cutaneous atrophy after corticosteroid injection. Am Fam Physician 1986;33:183–6.
- Lund IM, Donde R, Knudsen EA. Persistent local cutaneous atrophy following corticosteroid injection for tendinitis. Rheumatol Rehabil 1979;18:91–3.
Centrifugal lipodystrophy
- Imamura S. Lipodystrophia centrifugalis abdominalis infantilis: statistical analysis of 168 cases. Pediatr Dermatol 2012;29:437–41.
- Imamura S, Yamada M, Ikeda T. Lipodystrophia centrifugalis abdominalis infantilis. Arch Dermatol 1971;104:291–8.
- Imamura S, Taniguchi S. Lipoatrophic lesions preceded by pain and erythema – a new clinical entity? Eur J Dermatol 2000;10:540–1.
- Chang SE, Bae GY, Jeong YI, Lee HW, Choi JH, Moon KC. Lipodystrophia centrifugalis abdominalis in Korea. Pediatr Dermatol 2004;21:538–41.
- Mak KH, Ho HF, Chan LY, Chong LY. Lipodystrophia centrifugalis abdominalis infantilis: two cases from China. J Dermatol 2001;28:320–3.
- Wu MC, Hsu CK, Lee JY. Lipodystrophia centrifugalis abdominalis infantilis: report of four cases. Pediatr Dermatol 2012;29:308–10.
- Sarpa HG, Vanderhooft S, Bowen AR. Lipodystrophia centrifugalis abdominalis infantilis in a Caucasian girl: report of a case with perieccrine inflammation. J Cutan Pathol 2008;35:971–4.
- Zachary CB, Wells RS. Centrifugal lipodystrophy. Br J Dermatol 1984;110:107–10.
- Franks A, Verbov JL. Unilateral localized idiopathic lipoatrophy. Clin Exp Dermatol 1993;18:468–9.
- Tremeau-Martinage C, Bayle-Lebey P, Grouteau E, Bazex J. Localized lipoatrophia with persistent circulating autoantibodies and partial immunoglobulin A deficiency in a child. Dermatology 1996;192:353–7.
- Caputo R. Lipodystrophia centrifugalis sacralis infantilis. A 15-year follow-up observation. Acta Derm Venereol 1989;69:442–3.
- Muller S, Beissert S, Metze D, Luger TA, Bonsmann G. Lipodystrophia centrifugalis abdominalis infantilis in a 4-year-old Caucasian girl: association with partial IgA deficiency and autoantibodies. Br J Dermatol 1999;140:1161–4.
- Llistosella E, Puig L, Perez F. Lipodystrophia centrifugalis abdominalis infantilis: a case report. Pediatr Dermatol 1997;14:216–18.
- Okita H, Ohtsuka T, Yamakage A, Yamazaki S. Lipodystrophia centrifugalis abdominalis infantilis – immunohistochemical demonstration of an apoptotic process in the degenerating fatty tissue. Dermatology 2000;201:370–2.
- Kagoura M, Toyoda M, Matsui C, Higaki S, Morohashi M. An ultrastructural study of lipodystrophia centrifugalis abdominalis infantilis, with special reference to fibrous long-spacing collagen. Pediatr Dermatol 2001;18:13–16.
- Hagari Y, Ikehara A, Nuno K, Mihara M. Centrifugal lipodystrophy presenting with serpiginous erythema and alopecia. Cutis 2002;69:281–3.
- Fukumoto D, Kubo Y, Saito M, Arase S. Centrifugal lipodystrophy of the scalp presenting with an arch-form alopecia: a 10-year follow-up observation. J Dermatol 2009;36:499–503.
- Higashi Y, Kamiwada R, Kanekura T. Lipodystrophia centrifugalis abdominalis infantilis manifesting as recurrent skin ulcerations. Int J Dermatol 2009;48:1113–15.
- Hiraiwa A, Takai K, Fukui Y, Adachi A, Fujii H. Nonregressing lipodystrophia centrifugalis abdominalis with angioblastoma (Nakagawa). Arch Dermatol 1990;126:206–9.
Fat hypertrophy
Insulin-induced localized fat hypertrophy
- Richardson T, Kerr D. Skin-related complications of insulin therapy: epidemiology and emerging management strategies. Am J Clin Dermatol 2003;4:661–7.
- Hauner H, Stockamp B, Haastert B. Prevalence of lipohypertrophy in insulin-treated diabetic patients and predisposing factors. Exp Clin Endocrinol Diabetes 1996;104:106–10.
- Raile K, Noelle V, Landgraf R, Schwarz HP. Insulin antibodies are associated with lipoatrophy but also with lipohypertrophy in children and adolescents with type 1 diabetes. Exp Clin Endocrinol Diabetes 2001;109:393–6.
- Radermecker RP, Pierard GE, Scheen AJ. Lipodystrophy reactions to insulin: effects of continuous insulin infusion and new insulin analogs. Am J Clin Dermatol 2007;8:21–8.
- Fujikura J, Fujimoto M, Yasue S, et al. Insulin-induced lipohypertrophy: report of a case with histopathology. Endocr J 2005;52:623–8.
- Swift B. Examination of insulin injection sites: an unexpected finding of localized amyloidosis. Diabet Med 2002;19:881–2.
- Dische FE, Wernstedt C, Westermark GT, et al. Insulin as an amyloid-fibril protein at sites of repeated insulin injections in a diabetic patient. Diabetologia 1988;31:158–61.
- Young RJ, Hannan WJ, Frier BM, Steel JM, Duncan LJ. Diabetic lipohypertrophy delays insulin absorption. Diabetes Care 1984;7:479–80.
- Turner BC, Hejlesen OK, Kerr D, Cavan DA. Impaired absorption and omission of insulin: a novel method of detection using the diabetes advisory system computer model. Diabetes Technol Ther 2001;3:99–109.
- Chowdhury TA, Escudier V. Poor glycaemic control caused by insulin induced lipohypertrophy. BMJ 2003;327:383–4.
- Roper NA, Bilous RW. Resolution of lipohypertrophy following change of short-acting insulin to insulin lispro (Humalog). Diabet Med 1998;15:1063–4.
- Hardy KJ, Gill GV, Bryson JR. Severe insulin-induced lipohypertrophy successfully treated by liposuction. Diabetes Care 1993;16:929–30.
- Samdal F, Amland PF, Sandsmark M, Birkeland KI. Diabetic lipohypertrophy treated with suction-assisted lipectomy. J Intern Med 1993;234:489–92.
Subcutaneous lipomatosis
Benign symmetrical lipomatosis
- Brea-Garcia B, Cameselle-Teijeiro J, Couto-Gonzalez I, Taboada-Suarez A, Gonzalez-Alvarez E. Madelung's disease: comorbidities, fatty mass distribution, and response to treatment of 22 patients. Aesthetic Plast Surg 2013;37:409–16.
- Madelung OW. Ueber den Fetthals. Arch Klin Chir 1888;37:106–30.
- Launois PE, Bensaude R. De l'adenolipomatose symetrique. Soc Med Hosp Paris Bull Mem 1898;15:298–318.
- Enzi G, Biondetti PR, Fiore D, Mazzoleni F. Computed tomography of deep fat masses in multiple symmetrical lipomatosis. Radiology 1982;144:121–4.
- Chan HF, Sun Y, Lin CH, Chen RC. Madelung's disease associated with polyneuropathy and symptomatic hypokalemia. J Formos Med Assoc 2013;112:283–6.
- Enzi G. Multiple symmetric lipomatosis: an updated clinical report. Medicine (Baltimore) 1984;63:56–64.
- Kratz C, Lenard HG, Ruzicka T, Gartner J. Multiple symmetric lipomatosis: an unusual cause of childhood obesity and mental retardation. Eur J Paediatr Neurol 2000;4:63–7.
- Shetty C, Avinash KR, Auluck A, Mupparapu M. Multiple symmetric lipomatosis (MSL) of neck in a child (Madelung's disease): report of a rare presentation. Dentomaxillofac Radiol 2007, 36:51–4.
- Ruzicka T, Vieluf D, Landthaler M, Braun-Falco O. Benign symmetric lipomatosis Launois-Bensaude. Report of ten cases and review of the literature. J Am Acad Dermatol 1987;17:663–74.
- Semenou D, Coeugniet E, Segard M, Martinot-Duquennoy V, Delaporte E. [Launois-Bensaude's disease: report of 17 cases.] Ann Chir Plast Esthet 2008;53:399–407.
- Lee HW, Kim TH, Cho JW, Ryu BY, Kim HK, Choi CS. Multiple symmetric lipomatosis: Korean experience. Dermatol Surg 2003;29:235–40.
- Lee CH, Kang KT, Ko JY, Chang WH. Laryngeal involvement in Madelung's disease with acute airway compromise. Kaohsiung J Med Sci 2012;28:462–3.
- Sia KJ, Tang IP, Tan TY. Multiple symmetrical lipomatosis: case report and literature review. J Laryngol Otol 2012;126:756–8
- Ahuja AT, King AD, Chan ES, et al. Madelung disease: distribution of cervical fat and preoperative findings at sonography, MR, and CT. Am J Neuroradiol 1998;19:707–10.
- Iglesias L, Perez-Llantada E, Saro G, Pino M, Hernandez JL. Benign symmetric lipomatosis (Madelung's disease). Eur J Intern Med 2000;11:171–3.
- Kobayashi J, Nagao M, Miyamoto K, Matsubara S. MERRF syndrome presenting with multiple symmetric lipomatosis in a Japanese patient. Intern Med 2010;49:479–82.
- Pollock M, Nicholson GI, Nukada H, Cameron S, Frankish P. Neuropathy in multiple symmetric lipomatosis. Madelung's disease. Brain 1988;111(5):1157–71.
- Klopstock T, Naumann M, Schalke B, Bischof F, et al. Multiple symmetric lipomatosis: abnormalities in complex IV and multiple deletions in mitochondrial DNA. Neurology 1994;44:862–6.
- Saiz Hervas E, Martin Llorens M, Lopez Alvarez J. Peripheral neuropathy as the first manifestation of Madelung's disease. Br J Dermatol 2000;143:684–6.
- Enzi G, Busetto L, Ceschin E, Coin A, Digito M, Pigozzo S. Multiple symmetric lipomatosis: clinical aspects and outcome in a long-term longitudinal study. Int J Obes Relat Metab Disord 2002;26:253–61.
- Vargas-Diez E, Dauden E, Jones-Caballero M, Garcia-Diez A. Madelung's disease involving the tongue. J Am Acad Dermatol 2000;42:511–13.
- Zancanaro C, Sbarbati A, Morroni M, et al. Multiple symmetric lipomatosis. Ultrastructural investigation of the tissue and preadipocytes in primary culture. Lab Invest 1990;63:253–8.
- Vila MR, Gamez J, Solano A, et al. Uncoupling protein-1 mRNA expression in lipomas from patients bearing pathogenic mitochondrial DNA mutations. Biochem Biophys Res Commun 2000;278:800–2.
- Leung NW, Gaer J, Beggs D, Kark AE, Holloway B, Peters TJ. Multiple symmetric lipomatosis (Launois-Bensaude syndrome): effect of oral salbutamol. Clin Endocrinol (Oxf) 1987;27:601–6.
- Bassetto F, Scarpa C, De Stefano F, Busetto L. Surgical treatment of multiple symmetric lipomatosis with ultrasound-assisted liposuction. Ann Plast Surg 2014;73:559–62.
- Klopstock T, Naumann M, Seibel P, Shalke B, Reiners K, Reichmann H. Mitochondrial DNA mutations in multiple symmetric lipomatosis. Mol Cell Biochem 1997;174:271–5.
- Chalk CH, Mills KR, Jacobs JM, Donaghy M. Familial multiple symmetric lipomatosis with peripheral neuropathy. Neurology 1990;40:1246–50.
- Gonzalez-Garcia R, Rodriguez-Campo FJ, Sastre-Perez J, Munoz-Guerra MF. Benign symmetric lipomatosis (Madelung's disease): case reports and current management. Aesthetic Plast Surg 2004;28:108–12; discussion 13.
- Poggi G, Moro G, Teragni C, Delmonte A, Saini G, Bernardo G. Scrotal involvement in Madelung disease: clinical, ultrasound and MR findings. Abdom Imaging 2006;31:503–5.
- Birnholz JC, Macmillan AS, Jr. Advanced laryngeal compression due to diffuse, symmetric lipomatosis (Madelung's disease). Br J Radiol 1973;46:245–9.
- Moretti JA, Miller D. Laryngeal involvement in benign symmetric lipomatosis. Arch Otolaryngol 1973;97:495–6.
- Durr ML, Agrawal N, Saunders JR, Ha PK. Laryngeal lipoma associated with diffuse lipomatosis: case report and literature review. Ear Nose Throat J 2010;89:34–7.
- Lee DH, Lim SC, Lee JK. Laryngeal involvement in Madelung disease. Otolaryngol Head Neck Surg 2011;144:481–2.
- Chan ES, Ahuja AT, King AD, Lau WY. Head and neck cancers associated with Madelung's disease. Ann Surg Oncol 1999;6:395–7.
- Tizian C, Berger A, Vykoupil KF. Malignant degeneration in Madelung's disease (benign lipomatosis of the neck): case report. Br J Plast Surg 1983;36:187–9.
- Borriello M, Lucidi A, Carbone A, Iannone V, Ferrandina G. Malignant transformation of Madelung's disease in a patient with a coincidental diagnosis of breast cancer: a case report. Diagn Pathol 2012;7:116.
- Mimica M, Pravdic D, Nakas-Icindic E, et al. Multiple symmetric lipomatosis: a diagnostic dilemma. Case Rep Med 2013;2013:836903.
- Heike Z, Gudrun UM, Frank RD, Vetter H, Walger P. Multiple benign symmetric lipomatosis – a differential diagnosis of obesity: is there a rationale for fibrate treatment? Obes Surg 2008;18:240–2.
- Basse P, Lohman M, Hovgaard C, Alsbjorn B. Multiple symmetric lipomatosis. Acta Chir Plast 1991;33:204–9.
- Adamo C, Vescio G, Battaglia M, Gallelli G, Musella S. Madelung's disease: case report and discussion of treatment options. Ann Plast Surg 2001;46:43–5.
- Faga A, Valdatta LA, Thione A, Buoro M. Ultrasound assisted liposuction for the palliative treatment of Madelung's disease: a case report. Aesthetic Plast Surg 2001;25:181–3.
- Verhelle NA, Nizet JL, Van den Hof B, Guelinckx P, Heymans O. Liposuction in benign symmetric lipomatosis: sense or senseless? Aesthetic Plast Surg 2003;27:319–21.
- Grassegger A, Haussler R, Schmalzl F. Tumescent liposuction in a patient with Launois-Bensaude syndrome and severe hepatopathy. Dermatol Surg 2007;33:982–5.
- Ramos S, Pinheiro S, Diogo C, Cabral L, Cruzeiro C. Madelung disease: a not-so-rare disorder. Ann Plast Surg 2010;64:122–4.
- Martinez-Escribano JA, Gonzalez R, Quecedo E, Febrer I. Efficacy of lipectomy and liposuction in the treatment of multiple symmetric lipomatosis. Int J Dermatol 1999;38:551–4.
- Ujpal M, Nemeth ZS, Reichwein A, Szabo GY. Long-term results following surgical treatment of benign symmetric lipomatosis (BSL). Int J Oral Maxillofac Surg 2001;30:479–83.
- Constantinidis J, Steinhart H, Zenk J, Bohlender J, Iro H. [Surgical therapy of Madelung's disease in the head and neck area]. HNO 2003;51:216–20.
Dercum disease
- Dercum FX. A subcutaneous connective tissue dystrophy of the arms and back, associated with symptoms resembling myxoedema. Univ Med Mag (Philadelphia) 1888;1:1–11.
- Wortham NC, Tomlinson IP. Dercum's disease. Skinmed 2005;4:157–62; quiz 63–4.
- Herbst KL, Asare-Bediako S. Adiposis dolorosa is more than painful fat. Endocrinologist 2007;17:326–34.
- Bonatus TJ, Alexander AH. Dercum's disease (adiposis dolorosa). A case report and review of the literature. Clin Orthop Relat Res 1986:251–3.
- Hansson E, Svensson H, Brorson H. Review of Dercum's disease and proposal of diagnostic criteria, diagnostic methods, classification and management. Orphanet J Rare Dis 2012;7:23.
- Blomstrand R, Juhlin L, Nordenstam H, Ohlsson R, Werner B, Engstrom J. Adiposis dolorosa associated with defects of lipid metabolism. Acta Derm Venereol 1971;51:243–50.
- Fagher B, Monti M, Nilsson-Ehle P, Akesson B. Fat-cell heat production, adipose tissue fatty acids, lipoprotein lipase activity and plasma lipoproteins in adiposis dolorosa. Clin Sci (Lond) 1991;81:793–8.
- Taniguchi A, Okuda H, Mishima Y, et al. A case of adiposis dolorosa: lipid metabolism and hormone secretion. Int J Obes 1986;10:277–81.
- Pimenta WP, Paula FJ, Dick-de-Paula I, et al. Hormonal and metabolic study of a case of adiposis dolorosa (Dercum's disease). Braz J Med Biol Res 1992;25:889–93.
- Herbst KL, Coviello AD, Chang A, Boyle DL. Lipomatosis-associated inflammation and excess collagen may contribute to lower relative resting energy expenditure in women with adiposis dolorosa. Int J Obes (Lond) 2009;33:1031–8.
- Dercum FX. Three cases of a hitherto unclassified affection resembling in its grosser aspects obesity, but associated with special nervous symptoms-adiposis dolorosa. Am J Med Sci 1892;101:521–3.
- Waldorp NW. An original clinical interpretation of Dercum's disease (adiposis dolorosa). Endocrinology 1924;8:51–60.
- Winkelman N, Eckel JL. Adiposis dolorosa (Dercum's disease): a clinicopathological study. JAMA 1925;85:1935–9.
- Steiger WA, Litvin H, Lasche EM, Durant TM. Adiposis dolorsa (Dercum's disease). N Engl J Med 1952;247:393–6.
- Palmer ED. Dercum's disease: adiposis dolorosa. Am Fam Physician 1981;24:155–7.
- Tiesmeier J, Warnecke H, Schuppert F. [An uncommon cause of recurrent abdominal pain in a 63-year-old obese woman.] Dtsch Med Wochenschr 2006;131:434–7.
- Dalziel K. The nervous system and adipose tissue. Clin Dermatol 1989;7:62–77.
- Mella BA. Adiposis dolorosa. Univ Mich Med Cent J 1967;33:79–81.
- Lemont H, Picciotti J, Pruzansky J. Dercum's disease. J Am Podiatry Assoc 1979;69:389–91.
- Amine B, Leguilchard F, Benhamou CL. Dercum's disease (adiposis dolorosa): a new case-report. Joint Bone Spine 2004;71:147–9.
- Lynch HT, Harlan WL. Hereditary factors in adiposis dolorosa (Dercum's disease). Am J Hum Genet 1963;15:184–90.
- Cantu JM, Ruiz-Barquin E, Jimenez M, Castillo L, Macotela-Ruiz E. Autosomal dominant inheritance in adiposis dolorosa (Dercum's disease). Humangenetik 1973;18:89–91.
- Brodovsky S, Westreich M, Leibowitz A, Schwartz Y. Adiposis dolorosa (Dercum's disease): 10-year follow-up. Ann Plast Surg 1994;33:664–8.
- Campen R, Mankin H, Louis DN, Hirano M, Maccollin M. Familial occurrence of adiposis dolorosa. J Am Acad Dermatol 2001;44:132–6.
- Moi L, Canu C, Pirari P, Mura MN, Piludu G, Del Giacco GS. [Dercum's disease: a case report.].Ann Ital Med Int 2005;20:187–91.
- Kyllerman M, Brandberg G, Wiklund LM, Mansson JE. Dysarthria, progressive parkinsonian features and symmetric necrosis of putamen in a family with painful lipomas (Dercum disease variant). Neuropediatrics 2002;33:69–72.
- Brorson H, Fagher B. [Dercum's disease. Fatty tissue rheumatism caused by immune defense reaction?] Lakartidningen 1996;93:1430–6.
- Roux J, Vitaut M. Maladie de Dercum (adiposis dolorosa). Revue Neurol (Paris) 1901;9:881–8.
- Labbe M, Boulin R. Lipomatose douloureuse at maladie de Dercum. Bull Mem Soc Med Hop Paris 1927;51:687–95.
- Gram HC. A symptom-triad of the post-climacteric period (adipositas dolorosa-arthritis genuum-hypertensio arterialis). Acta Med Scand 1930;73:139.
- Petry NM, Barry D, Pietrzak RH, Wagner JA. Overweight and obesity are associated with psychiatric disorders: results from the National Epidemiologic Survey on Alcohol and Related Conditions. Psychosom Med 2008;70:288–97.
- Haddad D, Athmani B, Costa A, Cartier S. [Dercum's disease: a severe complication in a rare disease. A case report.] Ann Chir Plast Esthet 2005;50:247–50.
- Dercum FX, McCarthy DJ. Autopsy in a case of adiposis dolorosa. Am J Med Sci 1902;124:994–1006.
- Rosenberg B, Hurwitz A, Hermann H. Dercum's disease with unusual retroperitoneal and paravesical fatty infiltration. Surgery 1963;54:451–5.
- Hansson E, Svensson H, Brorson H. Liposuction may reduce pain in Dercum's disease (adiposis dolorosa). Pain Med 2011;12:942–52.
- Steiner J, Schiltz K, Heidenreich F, Weissenborn K. [Lipomatosis dolorosa – a frequently overlooked disease picture.] Nervenarzt 2002;73:183–7.
- Lange U, Oelzner P, Uhlemann C. Dercum's disease (lipomatosis dolorosa): successful therapy with pregabalin and manual lymphatic drainage and a current overview. Rheumatol Int 2008;29:17–22.
- Kosseifi S, Anaya E, Dronovalli G, Leicht S. Dercum's disease: an unusual presentation. Pain Med 2010;11:1430–4.
- Eisman J, Swezey RL. Juxta-articular adiposis dolorosa: what is it? Report of 2 cases. Ann Rheum Dis 1979;38:479–82.
- Atkinson RL. Intravenous lidocaine for the treatment of intractable pain of adiposis dolorosa. Int J Obes 1982;6:351–7.
- Nahir AM, Schapira D, Scharf Y. Juxta-articular adiposis dolorosa – a neglected disease. Isr J Med Sci 1983;19:858–9.
- Juhlin L. Long-standing pain relief of adiposis dolorosa (Dercum's disease) after intravenous infusion of lidocaine. J Am Acad Dermatol 1986;15(2 Pt 2):383–5.
- Scheinberg MA, Diniz R, Diamant J. Improvement of juxtaarticular adiposis dolorosa by fat suction. Arthritis Rheum 1987;30:1436–7.
- De Silva M, Earley MJ. Liposuction in the treatment of juxta-articular adiposis dolorosa. Ann Rheum Dis 1990;49:403–4.
- Chopra A, Walia P, Chopra D, Jassal JS. Adiposis dolorosa. Indian J Dermatol Venereol Leprol 2000;66:101–2.
- George R, Poonnoose P, Isaiah R. Dercum's disease (adiposis dolorosa): delayed diagnosis in a patient with cervical cancer. J Palliat Care 2004;20:324–5.
- Reggiani M, Errani A, Staffa M, Schianchi S. Is EMLA effective in Dercum's disease? Acta Derm Venereol 1996;76:170–1.
- Desai MJ, Siriki R, Wang D. Treatment of pain in Dercum's disease with Lidoderm (lidocaine 5% patch): a case report. Pain Med 2008;9:1224–6.
- Boller R. Die Novocainbehandling des morbus Dercum. Klin Wochenschr 1934;13:1786–9.
- Iwane T, Maruyama M, Matsuki M, Ito Y, Shimoji K. Management of intractable pain in adiposis dolorosa with intravenous administration of lidocaine. Anesth Analg 1976;55:257–9.
- Skagen K, Petersen P, Kastrup J, Norgaard T. The regulation of subcutaneous blood flow in patient with Dercum's disease. Acta Derm Venereol 1986; 66:337–9.
- Petersen P, Kastrup J. Dercum's disease (adiposis dolorosa). Treatment of the severe pain with intravenous lidocaine. Pain 1987;28:77–80.
- Spota BB, Brage D. [Cortisone therapy of Dercum's disease.] Dia Med 1952;24:1930–2.
- Greenbaum SS, Varga J. Corticosteroid-induced juxta-articular adiposis dolorosa. Arch Dermatol 1991;127:231–3.
- Weinberger A, Wysenbeec AJ, Pinkhas J. Juxta-articular adiposis dolorosa associated with rheumatoid arthritis. Report of 2 cases with good response to local corticosteroid injection. Clin Rheumatol 1987;6:446–8.
- Singal A, Janiga JJ, Bossenbroek NM, Lim HW. Dercum's disease (adiposis dolorosa): a report of improvement with infliximab and methotrexate. J Eur Acad Dermatol Venereol 2007;21:717.
- Pardal Refoyo JL. [Adiposis dolorosa (Dercum's disease).] An Otorrinolaringol Ibero Am 1996;23:435–40.
- Campen RB, Sang CN, Duncan LM. Case records of the Massachusetts General Hospital. Case 25-2006. A 41-year-old woman with painful subcutaneous nodules. N Engl J Med 2006;355:714–22.
- Gonciarz Z, Mazur W, Hartleb J, et al. Interferon alfa-2b induced long-term relief of pain in two patients with adiposis dolorosa and chronic hepatitis C. J Hepatol 1997;27:1141.
- Herbst KL, Rutledge T. Pilot study: rapidly cycling hypobaric pressure improves pain after 5 days in adiposis dolorosa. J Pain Res 2010;3:147–53.
- Kling D. Juxa-articular adiposis dolorosa. Its significance and relation to Dercum;s disease and osteo-arthritis. Arch Surg 1937;34:599–630.
- Held JL, Andrew JA, Kohn SR. Surgical amelioration of Dercum's disease: a report and review. J Dermatol Surg Oncol 1989;15:1294–6.
- Devillers AC, Oranje AP. Treatment of pain in adiposis dolorosa (Dercum's disease) with intravenous lidocaine: a case report with a 10-year follow-up. Clin Exp Dermatol 1999;24:240–1.
Infiltrating lipomatosis of the face
- Tracy JC, Klement GL, Scott AR. Interdisciplinary management of congenital infiltrating lipomatosis. Int J Pediatr Otorhinolaryngol 2013;77(12):2071–4.
- Slavin SA, Baker DC, McCarthy JG, Mufarrij A. Congenital infiltrating lipomatosis of the face: clinicopathologic evaluation and treatment. Plast Reconstr Surg 1983;72:158–64.
- Urs AB, Augustine J, Kumar P, Arora S, Aggarwal N, Sultana N. Infiltrating lipomatosis of the face: a case series. J Nat Sci Biol Med 2013;4:252–7.
- De Rosa G, Cozzolino A, Guarino M, Giardino C. Congenital infiltrating lipomatosis of the face: report of cases and review of the literature. J Oral Maxillofac Surg 1987;45:879–83.
- Van Wingerden JJ, Erlank JD, Becker JH. Liposuction for congenital infiltrating lipomatosis of the face. Plast Reconstr Surg 1988;81:989.
- Donati L, Candiani P, Grappolini S, Klinger M, Signorini M. Congenital infiltrating lipomatosis of the face related to cytomegalovirus infection. Br J Plast Surg 1990;43:124–6.
- MacMillan AR, Oliver AJ, Reade PC, Marshall DR. Regional macrodontia and regional bony enlargement associated with congenital infiltrating lipomatosis of the face presenting as unilateral facial hyperplasia. Brief review and case report. Int J Oral Maxillofac Surg 1990;19:283–6.
- Patel RV, Gondalia JS. Congenital infiltrating lipomatosis of the face. Br J Plast Surg 1991;44:157–8.
- Kang N, Ross D, Harrison D. Unilateral hypertrophy of the face associated with infiltrating lipomatosis. J Oral Maxillofac Surg 1998;56:885–7.
- Gorken C, Alper M, Bilkay U, Celik N, Songur E. Congenital infiltrating lipomatosis of the face. J Craniofac Surg 1999;10:365–8.
- Bouletreau P, Breton P, Freidel M. Congenital infiltrating lipomatosis of the face: case report. J Oral Maxillofac Surg 2000;58:807–10.
- Ergun SS, Kural S, Ulay M, Bilgic B. Infiltrating lipomatosis of the face. Ann Plast Surg 2001;47:346–8.
- Padwa BL, Mulliken JB. Facial infiltrating lipomatosis. Plast Reconstr Surg 2001; 108:1544–54.
- Chen CM, Lo LJ, Wong HF. Congenital infiltrating lipomatosis of the face: case report and literature review. Chang Gung Med J 2002;25:194–200.
- Unal S, Demirkan F, Arslan E, Cinel L. Infiltrating lipomatosis of the face: a case report and review of the literature. J Oral Maxillofac Surg 2003;61:1098–101.
- Malik A, Jagmohan P, Thukral BB, Khanna G, Rajni. Congenital infiltrating lipomatosis of the face and neck. Acta Radiol 2004;45:556–60.
- Rajeswaran R, Murthy J, Chandrasekharan A, Joseph S. Case report: congenital infiltrating lipomatosis of face. Indian J Radiol Imaging 2008;18:306–9.
- Alkan O, Yildirim T, Seyhan T, Erbay G, Erol I. Congenital infiltrating lipomatosis of the face with hemimegalencephaly. Neuropediatrics 2009;40:141–3.
- Pires Fraga MF, Mello D, Jorge D, Perin LF, Helene A. Congenital infiltrating lipomatosis. J Plast Reconstr Aesthet Surg 2009;62:e561–4.
- Kamal D, Breton P, Bouletreau P. Congenital infiltrating lipomatosis of the face: report of three cases and review of the literature. J Craniomaxillofac Surg 2010;38:610–14.
- Kim JE, Gottschall JA, Bachman RP, Nemzer L, Puligandla B, Schauer G. Facial infiltrating lipomatosis: physical, radiological, and histopathological findings. Arch Otolaryngol Head Neck Surg 2010;136:301–3.
- Maruyama K, Okumura A, Negoro T, Watanabe K. Congenital infiltrating lipomatosis of the face with ipsilateral hemimegalencephaly, band heterotopia, and hypertrophy of brainstem and cerebellum. Neuropediatrics 2010;41:147–50.
- Singh K, Sen P, Musgrove BT, Thakker N. Facial infiltrating lipomatosis: a case report and review of literature. Int J Surg Case Rep 2011;2:201–15.
- Balaji SM. Congenital diffuse infiltrating facial lipomatosis. Ann Maxillofac Surg 2012;2:190–6.
- Keramidas T, Lagogiannis G, Vlachou V, Katsikeris N. Congenital infiltrating lipomatosis of the face with associated involvement of the TMJ structures. Case report and review of the literature. J Craniomaxillofac Surg 2012;40:750–6.
- Sun L, Sun Z, Zhu J, Ma X. Tooth abnormalities in congenital infiltrating lipomatosis of the face. Oral Surg Oral Med Oral Pathol Oral Radiol 2013;115:e52–62.
- Couto RA, Mulliken JB, Padwa BL, et al. Facial infiltrating lipomatosis: expression of angiogenic and vasculogenic factors. J Craniofac Surg 2011;22:2405–8.
Encephalocraniocutaneous lipomatosis
- Delfino LN, Fariello G, Quattrocchi CC, et al. Encephalocraniocutaneous lipomatosis (ECCL): neuroradiological findings in three patients and a new association with fibrous dysplasia. Am J Med Genet A 2011;155A:1690–6.
- Haberland C, Perou M. Encephalocraniocutaneous lipomatosis. A new example of ectomesodermal dysgenesis. Arch Neurol 1970;22:144–55.
- Pardo IA, Nicolas ME. A Filipino male with encephalocraniocutaneous lipomatosis (Haberland's syndrome). J Dermatol Case Rep 2013;7:46–8.
- Moog U, Jones MC, Viskochil DH, Verloes A, Van Allen MI, Dobyns WB. Brain anomalies in encephalocraniocutaneous lipomatosis. Am J Med Genet A 2007;143A:2963–72.
- Moog U, Roelens F, Mortier GR, et al. Encephalocraniocutaneous lipomatosis accompanied by the formation of bone cysts: Harboring clues to pathogenesis? Am J Med Genet A 2007;143A:2973–80.
- Moog U. Encephalocraniocutaneous lipomatosis. J Med Genet 2009;46:721–9.
- Savage MG, Heldt L, Dann JJ, Bump RL. Encephalocraniocutaneous lipomatosis and mixed odontogenic tumors. J Oral Maxillofac Surg 1985;43:617–20.
- Hauber K, Warmuth-Metz M, Rose C, Brocker EB, Hamm H. Encephalocraniocutaneous lipomatosis: a case with unilateral odontomas and review of the literature. Eur J Pediatr 2003;162:589–93.
- Andreadis DA, Rizos CB, Belazi M, Peneva M, Antoniades DZ. Encephalocraniocutaneous lipomatosis accompanied by maxillary compound odontoma and juvenile angiofibroma: report of a case. Birth Defects Res A Clin Mol Teratol 2004;70:889–91.
- Zielinska-Kazmierska B, Grodecka J, Jablonska-Polakowska L, Arkuszewski P. Mandibular osteoma in the encephalocraniocutaneous lipomatosis. J Craniomaxillofac Surg 2005;33:286–9.
- Cohen MM, Jr. Proteus syndrome: an update. Am J Med Genet C Semin Med Genet 2005;137C:38–52.
- Turner JT, Cohen MM, Jr., Biesecker LG. Reassessment of the Proteus syndrome literature: application of diagnostic criteria to published cases. Am J Med Genet A 2004;130A:111–22.
- Ardinger HH, Horii KA, Begleiter ML. Expanding the phenotype of oculoectodermal syndrome: possible relationship to encephalocraniocutaneous lipomatosis. Am J Med Genet A 2007;143A:2959–62.
- Hunter AG. Oculocerebrocutaneous and encephalocraniocutaneous lipomatosis syndromes: blind men and an elephant or separate syndromes? Am J Med Genet A 2006;140:709–26.
- Moog U, Jones MC, Bird LM, Dobyns WB. Oculocerebrocutaneous syndrome: the brain malformation defines a core phenotype. J Med Genet 2005;42:913–21.
Lipoedema
Lipoedema of the lower limbs
- Allen EV, Hines EA, Jr. Lipedema of the legs: a syndrome characterized by fat legs and orthostatic edema. Proc Staff Meet Mayo Clin 1940;15:184–7.
- Wold LE, Hines EA, Jr, Allen EV. Lipedema of the legs; a syndrome characterized by fat legs and edema. Ann Intern Med 1951;34:1243–50.
- Hadjis NS, Carr DH, Banks L, Pflug JJ. The role of CT in the diagnosis of primary lymphedema of the lower limb. Am J Roentgenol 1985;144:361–4.
- Vaughan BF. CT of swollen legs. Clin Radiol 1990;41:24–30.
- Duewell S, Hagspiel KD, Zuber J, von Schulthess GK, Bollinger A, Fuchs WA. Swollen lower extremity: role of MR imaging. Radiology 1992;184:227–31.
- Werner GT, Rodiek SO. [Value of nuclear magnetic resonance tomography in leg edema of unknown origin. Preliminary report.] Z Lymphol 1993;17:2–5.
- Dimakakos PB, Stefanopoulos T, Antoniades P, Antoniou A, Gouliamos A, Rizos D. MRI and ultrasonographic findings in the investigation of lymphedema and lipedema. Int Surg 1997;82:411–16.
- Monnin-Delhom ED, Gallix BP, Achard C, Bruel JM, Janbon C. High resolution unenhanced computed tomography in patients with swollen legs. Lymphology 2002;35:121–8.
- Beninson J, Edelglass JW. Lipedema – the non-lymphatic masquerader. Angiology 1984;35:506–10.
- Bilancini S, Lucchi M, Tucci S, Eleuteri P. Functional lymphatic alterations in patients suffering from lipedema. Angiology 1995;46:333–9.
- Harwood CA, Bull RH, Evans J, Mortimer PS. Lymphatic and venous function in lipoedema. Br J Dermatol 1996;134:1–6.
- Fonder MA, Loveless JW, Lazarus GS. Lipedema, a frequently unrecognized problem. J Am Acad Dermatol 2007;57(Suppl. 2):S1–3.
- Herpertz U. [Lipedema.] Z Lymphol 1995;19:1–11.
- Gregl A. [Lipedema.] Z Lymphol 1987;11:41–3.
- Langendoen SI, Habbema L, Nijsten TE, Neumann HA. Lipoedema: from clinical presentation to therapy. A review of the literature. Br J Dermatol 2009;161:980–6.
- Rudkin GH, Miller TA. Lipedema: a clinical entity distinct from lymphedema. Plast Reconstr Surg 1994;94:841–7; discussion 8–9.
- Chen SG, Hsu SD, Chen TM, Wang HJ. Painful fat syndrome in a male patient. Br J Plast Surg 2004;57:282–6.
- Greer K. Lipedema of the legs. Cutis 1974;14:98–100.
- Stallworth JM, Hennigar GR, Jonsson HT, Jr, Rodriguez O. The chronically swollen painful extremity. A detailed study for possible etiological factors. JAMA 1974;228:1656–9.
- Macdonald JM, Sims N, Mayrovitz HN. Lymphedema, lipedema, and the open wound: the role of compression therapy. Surg Clin North Am 2003;83:639–58.
- Weissleder H, Brauer JW, Schuchhardt C, Herpertz U. [Value of functional lymphoscintigraphy and indirect lymphangiography in lipedema syndrome.] Z Lymphol 1995;19:38–41.
- Boursier V, Pecking A, Vignes S. [Comparative analysis of lymphoscintigraphy between lipedema and lower limb lymphedema.] J Mal Vasc 2004;29:257–61.
- Bano G, Mansour S, Brice G, et al. Pit-1 mutation and lipoedema in a family. Exp Clin Endocrinol Diabetes 2010;118:377–80.
- Schmeller W, Hueppe M, Meier-Vollrath I. Tumescent liposuction in lipoedema yields good long-term results. Br J Dermatol 2011;166:161–8.
- Meier-Vollrath I, Schmeller W. [Lipoedema – current status, new perspectives.] J Dtsch Dermatol Ges 2004;2:181–6.
- Koss T, Lanatra N, Stiller MJ, Grossman ME. An unusual combination: lipedema with myiasis. J Am Acad Dermatol 2004;50:969–72.
- Brunner U. [Vascular diseases in lipedema of the legs. Special symptoms, common therapeutic results, viewpoint on vascular surgery.] Schweiz Med Wochenschr 1982;112:1130–7.
- Zhu S, Heshka S, Wang Z, et al. Combination of BMI and waist circumference for identifying cardiovascular risk factors in whites. Obes Res 2004;12:633–45.
- Rank BK, Wong GS. Lipoedema. Aust NZ J Surg 1966;35:166–9.
- Foldi M, Idiazabal G. The role of operative management of varicose veins in patients with lymphedema and/or lipedema of the legs. Lymphology 2000;33:167–71.
- Rapprich S, Loehnert M, Hagedorn M. Therapy of lipoedema syndrome by liposuction under tumescent local anaesthesia. Ann Dermatol Venereol 2002;129:1S711.
- Schmeller W, Meier-Vollrath I. Tumescent liposuction: a new and successful therapy for lipedema. J Cutan Med Surg 2006;10:7–10.
- Rapprich S, Dingler A, Podda M. Liposuction is an effective treatment for lipedema-results of a study with 25 patients. J Dtsch Dermatol Ges 2010;9:33–40.
Lipo-lymphoedema
- Langendoen SI, Habbema L, Nijsten TE, Neumann HA. Lipoedema: from clinical presentation to therapy. A review of the literature. Br J Dermatol 2009;161:980–6.
- Stallworth JM, Hennigar GR, Jonsson HT, Jr, Rodriguez O. The chronically swollen painful extremity. A detailed study for possible etiological factors. JAMA 1974;228:1656–9.
- Beninson J, Edelglass JW. Lipedema – the non-lymphatic masquerader. Angiology 1984;35:506–10.
- Rudkin GH, Miller TA. Lipedema: a clinical entity distinct from lymphedema. Plast Reconstr Surg 1994;94:841–7; discussion 8–9.
- Warren AG, Janz BA, Borud LJ, Slavin SA. Evaluation and management of the fat leg syndrome. Plast Reconstr Surg 2007;119:9e–15e.
Lipoedema of the scalp and lipoedematous alopecia
- Yasar S, Gunes P, Serdar ZA, Tosun I. Clinical and pathological features of 31 cases of lipedematous scalp and lipedematous alopecia. Eur J Dermatol 2011;21:520–8.
- Cornbleet T. Cutis verticis gyrata? lipoma? Arch Dermatol Syphilol 1935;32:688.
- Coskey RJ, Fosnaugh RP, Fine G. Lipedematous alopecia. Arch Dermatol 1961;84:619–22.
- Zeng YP, Ma DL, Wang BX. Lipedematous scalp with heterochromia of scalp hair in a boy. Eur J Dermatol 2011;21:144–5.
- Curtis JW, Heising RA. Lipedematous alopecia associated with skin hyperelasticity. Arch Dermatol 1964;89:819–20.
- Lee JH, Sung YH, Yoon JS, Park JK. Lipedematous scalp. Arch Dermatol 1994;130:802–3.
- Kane KS, Kwan T, Baden HP, Bigby M. Woman with new-onset boggy scalp. Arch Dermatol 1998;134:499–500,2–3.
- Bridges AG, von Kuster LC, Estes SA. Lipedematous alopecia. Cutis 2000;65:199–202.
- Fair KP, Knoell KA, Patterson JW, Rudd RJ, Greer KE. Lipedematous alopecia: a clinicopathologic, histologic and ultrastructural study. J Cutan Pathol 2000;27:49–53.
- Ikejima A, Yamashita M, Ikeda S, Ogawa H. A case of lipedematous alopecia occurring in a male patient. Dermatology 2000;201:168–70.
- Tiscornia JE, Molezzi A, Hernandez MI, Kien MC, Chouela EN. Lipedematous alopecia in a white woman. Arch Dermatol 2002;138:1517–18.
- Scheufler O, Kania NM, Heinrichs CM, Exner K. Hyperplasia of the subcutaneous adipose tissue is the primary histopathologic abnormality in lipedematous scalp. Am J Dermatopathol 2003;25:248–52.
- Bukhari I, Al Mulhim F, Al Hoqail R. Hyperlipidemia and lipedematous scalp. Ann Saudi Med 2004;24:484–5.
- Mohsin RM. Lipedematous scalp, a rare form of diffuse hair loss. J Am Acad Dermatol 2004;50:P90.
- High WA, Hoang MP. Lipedematous alopecia: an unusual sequela of discoid lupus, or other co-conspirators at work? J Am Acad Dermatol 2005;53(2 Suppl. 1):S157–61.
- Martin JM, Monteagudo C, Montesinos E, Guijarro J, Llombart B, Jorda E. Lipedematous scalp and lipedematous alopecia: a clinical and histologic analysis of 3 cases. J Am Acad Dermatol 2005;52:152–6.
- Piraccini BM, Voudouris S, Pazzaglia M, Rech G, Vicenzi C, Tosti A. Lipedematous alopecia of the scalp. Dermatol Online J 2006;12:6.
- Rowan DM, Simpson A, Wong KP. Lipedematous scalp in a child. Pediatr Dermatol 2006;23:276–8.
- El Darouti MA, Marzouk SA, Mashaly HM, et al. Lipedema and lipedematous alopecia: report of 10 new cases. Eur J Dermatol 2007;17:351–2.
- Mansur AT, Yasar S, Aydingoz IE, Goktay F, Ozdemir N, Sungurlu F. Colocalization of lipedematous scalp and nevus lipomatosus superficialis: a case report. J Cutan Pathol 2007;34:342–5.
- Moravvej-Farshi H, Mohtasham N. An unusual case of lipedematous alopecia. Arch Iran Med 2007;10:532–4.
- Scott MJ, Jr, Scott MJ, 3rd. Lipedematous alopecia: spongy scalp syndrome. Cutis 2007;80:321–4.
- Yasar S, Mansur AT, Goktay F, Sungurlu F, Vardar Aker F, Ozkara S. Lipedematous scalp and lipedematous alopecia: report of three cases in white adults. J Dermatol 2007;34:124–30.
- Fernandez-Torres R, Verea Hernando MM, Castro JM, Alvarez R, Fonseca E. Lipedematous scalp: report of a case. J Dermatol 2008;35:550–1.
- Gonzalez-Guerra E, Haro R, Angulo J, Del Carmen Farina M, Martin L, Requena L. Lipedematous alopecia: an uncommon clinicopathologic variant of nonscarring but permanent alopecia. Int J Dermatol 2008;47:605–9.
- Kavak A, Yuceer D, Yildirim U, Baykal C, Sarisoy HT. Lipedematous scalp: a rare entity. J Dermatol 2008;35:102–5.
- Yip L, Mason G, Pohl M, Sinclair R. Successful surgical management of lipoedematous alopecia. Australas J Dermatol 2008;49:52–4.
- Martinez-Moran C, Sanz-Munoz C, Miranda-Sivelo A, Torne I, Miranda-Romero A. [Lipedematous scalp.] Actas Dermosifiliogr 2009;100:69–72.
- Cunha Filho RR, Almeida HL, Jr, Cartell A. [Lipedematous scalp with early onset.] An Bras Dermatol 2010;85:81–3.
- Lee WS, Lee IW, Ahn SK. Diffuse heterochromia of scalp hair. J Am Acad Dermatol 1996;35(5 Pt 2):823–5.
- Garn SM, Selby S, Young R. Scalp thickness and the fat-loss theory of balding. AMA Arch Derm Syphilol 1954;70:601–8.
Miscellaneous disorders of subcutaneous fat
Cellulite
- Khan MH, Victor F, Rao B, Sadick NS. Treatment of cellulite. Part I. Pathophysiology. J Am Acad Dermatol 2010;62:361–70; quiz 71–2.
- Scherwitz C, Braun-Falco O. So-called cellulite. J Dermatol Surg Oncol 1978;4:230–4.
- Rossi AB, Vergnanini AL. Cellulite: a review. J Eur Acad Dermatol Venereol 2000;14:251–62.
- Rosenbaum M, Prieto V, Hellmer J, et al. An exploratory investigation of the morphology and biochemistry of cellulite. Plast Reconstr Surg 1998;101:1934–9.
- Nurnberger F, Muller G. So-called cellulite: an invented disease. J Dermatol Surg Oncol 1978;4:221–9.
- Querleux B, Cornillon C, Jolivet O, Bittoun J. Anatomy and physiology of subcutaneous adipose tissue by in vivo magnetic resonance imaging and spectroscopy: relationships with sex and presence of cellulite. Skin Res Technol 2002;8:118–24.
- Pierard GE, Nizet JL, Pierard-Franchimont C. Cellulite: from standing fat herniation to hypodermal stretch marks. Am J Dermatopathol 2000;22:34–7.
- Draelos ZD, Marenus KD. Cellulite. Etiology and purported treatment. Dermatol Surg 1997;23:1177–81.
- Kligman AM. Cellulite: facts and fiction. J Geriatr Dermatol 1997:136–9.
- Mirrashed F, Sharp JC, Krause V, Morgan J, Tomanek B. Pilot study of dermal and subcutaneous fat structures by MRI in individuals who differ in gender, BMI, and cellulite grading. Skin Res Technol 2004;10:161–8.
- Peterson JD, Goldman MP. Laser, light, and energy devices for cellulite and lipodystrophy. Clin Plast Surg 2011;38:463–74, vii.
- Gold MH. Cellulite – an overview of non-invasive therapy with energy-based systems. J Dtsch Dermatol Ges 2012;10:553–8.
- Smalls LK, Hicks M, Passeretti D, et al. Effect of weight loss on cellulite: gynoid lypodystrophy. Plast Reconstr Surg 2006;118:510–16.
- Khan MH, Victor F, Rao B, Sadick NS. Treatment of cellulite. Part II. Advances and controversies. J Am Acad Dermatol 2010;62:373–84; quiz 85–6.
- Brandi C, D'Aniello C, Grimaldi L, et al. Carbon dioxide therapy in the treatment of localized adiposities: clinical study and histopathological correlations. Aesthetic Plast Surg 2001;25:170–4.
- Brandi C, D'Aniello C, Grimaldi L, Caiazzo E, Stanghellini E. Carbon dioxide therapy: effects on skin irregularity and its use as a complement to liposuction. Aesthetic Plast Surg 2004;28:222–5.
- Westergaard M, Henningsen J, Svendsen ML, et al. Modulation of keratinocyte gene expression and differentiation by PPAR-selective ligands and tetradecylthioacetic acid. J Invest Dermatol 2001;116:702–12.
- Rawlings AV. Cellulite and its treatment. Int J Cosmet Sci 2006;28:175–90.
- Luo J, Sladek R, Carrier J, Bader JA, Richard D, Giguere V. Reduced fat mass in mice lacking orphan nuclear receptor estrogen-related receptor alpha. Mol Cell Biol 2003;23:7947–56.
- Sztalryd C, Xu G, Dorward H, et al. Perilipin A is essential for the translocation of hormone-sensitive lipase during lipolytic activation. J Cell Biol 2003;161:1093–103.
- Akter MH, Yamaguchi T, Hirose F, Osumi T. Perilipin, a critical regulator of fat storage and breakdown, is a target gene of estrogen receptor-related receptor alpha. Biochem Biophys Res Commun 2008;368:563–8.
Obesity and the skin
- Public Health England (PHE). PHE Obesity. https://www.noo.org.uk (last accessed August 2015).
- Shipman AR, Millington G. Obesity and the skin. Br J Dermatol 2011;165:743–50.
- Yosipovitch G, DeVore A, Dawn A. Obesity and the skin: skin physiology and skin manifestations of obesity. J Am Acad Dermatol 2007;56:901–16.
- Guida B, Nino M, Perrino NR, et al. The impact of obesity on skin disease and epidermal permeability barrier status. J Eur Acad Dermatol Venereol 2009;24:191–5.
- Garcia-Hidalgo L, Orozco-Topete R, Gonzalez-Barranco J, et al. Dermatoses in 156 obese adults. Obes Res 1999;7:299–302.
- Scheinfeld NS. Obesity and dermatology. Clin Dermatol 2004;22:303–9.
- Sreeramoju P, Porbandarwalla NS, Arango J, et al. Recurrent skin and soft tissue infections due to methicillin-resistant Staphylococcus aureus requiring operative debridement. Am J Surg 2011;201:216–20.
- Roujeau JC. Necrotizing fasciitis. Clinical criteria and risk factors. Ann Dermatol Venereol 2001;128:376–81.
- Guzik TJ, Mangalat D, Korbut R. Adipocytokines – novel link between inflammation and vascular function? J Physiol Pharmacol 2006;57:505–28.
- Wang P, Mariman E, Renes J, Keijer J. The secretory function of adipocytes in the physiology of white adipose tissue. J Cell Physiol 2008;216:3–13.
- Stallone DD, Stunkard AJ, Zweiman B, et al. Decline in delayed type hypersensitivity response in obese women following weight reduction. Clin Diagn Lab Immunol 1994;1:1202–5.
- Hercogova J, Ricceri F, Tripo L, et al. Psoriasis and body mass index. Dermatol Ther 2010;23:152–4.
- Perez-Perez I, Allegue F, Caeiro JI, et al. Severe psoriasis, morbid obesity and bariatric surgery. Clin Exp Dermatol 2009;34:e421–2.
- Murray CS, Canoy D, Buchan I, et al. Body mass index in young children and allergic disease: gender differences in a longitudinal study. Clin Exp Allergy 2011;41:78–85.
- Labrie F, Luu-The V, Labrie C, et al. Intracrinology and the skin. Horm Res 2000;54:218–29.
- Bellon JM, Duran HJ. Biological factors involved in the genesis of incisional hernia. Cir Esp 2008;83:3–7.